
Sperling Nội tiết học Nhi khoa, Ấn bản thứ 5 – Biên dịch: Ths.Bs. Lê Đình Sáng
Sperling Pediatric Endocrinology, Fifth Edition
Tác giả: Sperling, Mark A., MD – Nhà xuất bản: Elsevier Inc.
PHẦN III. NỘI TIẾT HỌC TRẺ EM VÀ THANH THIẾU NIÊN
Chương 15. U tế bào ưa crôm/U cận hạch thần kinh, Ung thư biểu mô tuyến giáp thể tủy, và các Hội chứng Đa u tuyến Nội tiết Di truyền
Steven G. Waguespack; Lauren Fishbein
Pheochromocytoma/Paraganglioma, Medullary Thyroid Carcinoma, and Hereditary Endocrine Neoplasia Syndromes
Sperling Pediatric Endocrinology, 15, 491-527
Giới thiệu
U tân sinh nội tiết bao gồm một loạt các khối u lành tính và ác tính phát sinh từ các tuyến nội tiết hoặc các mô thần kinh nội tiết khác, chẳng hạn như các cận hạch. Hầu hết các khối u nội tiết ở trẻ em, điển hình là ung thư biểu mô tuyến giáp thể nhú, là các trường hợp lẻ tẻ và không do một đột biến dòng mầm có thể xác định được; trong khi đó, những khối u khác, tiêu biểu là các khối u sản xuất catecholamin (Bảng 15.1) và ung thư biểu mô tuyến giáp thể tủy (MTC), lại có tính gia đình và xuất hiện trong bối cảnh của một hội chứng khối u di truyền rộng hơn. Trong các bệnh lý u tân sinh nội tiết có tính gia đình, phương thức di truyền là trội trên nhiễm sắc thể thường và các đột biến chủ yếu là đột biến bất hoạt gây mất chức năng của một gen đè nén khối u. Những tiến bộ trong xét nghiệm và nghiên cứu di truyền đã dẫn đến việc liên tục phát hiện ra các gen nhạy cảm với khối u mới, bên cạnh sự hiểu biết rõ hơn về sinh lý bệnh cơ bản của những rối loạn đặc biệt này. Kiến thức về mối quan hệ kiểu gen-kiểu hình tiếp tục phát triển, cũng như thực hành lâm sàng liên quan đến độ tuổi thực hiện xét nghiệm di truyền tiên đoán, tầm soát các khối u nội tiết ở người mang gen không triệu chứng và thời điểm can thiệp điều trị. Với lĩnh vực thay đổi nhanh chóng này, bệnh nhân có khối u nội tiết nên được đánh giá tại các chương trình có chuyên môn đa ngành đã được công nhận. Ngoài ra, kết quả xét nghiệm di truyền và tư vấn di truyền chính thức cần được tích hợp đầy đủ vào kế hoạch điều trị và theo dõi dài hạn. Chương này tổng quan về sinh lý bệnh, chẩn đoán và quản lý các khối u nội tiết ở trẻ em và các hội chứng di truyền phổ biến nhất liên quan đến chẩn đoán của chúng.
Bảng 15.1 Các Rối Loạn và Gen Chính Liên Quan Đến U Tế Bào Ưa Crôm và U Cận Hạch Thần Kinh
| Hội Chứng Di Truyền | Gen (Nhiễm Sắc Thể) | Loại Khối U | Kiểu Hình Lâm Sàng a | Tuổi Sớm Nhất b | Hướng Dẫn Tầm Soát c |
|---|---|---|---|---|---|
| Đa u tuyến nội tiết loại 2A (MEN2A) | RET (10q11.21) | PHEO/PGL hiếm gặp & PHEO hỗn hợp d | • Adrenergic • Hiếm khi ác tính • Nền tăng sản tủy thượng thận |
8 | Bắt đầu trước 11 tuổi đối với đột biến codon 634; trước 16 tuổi đối với các codon khác |
| Đa u tuyến nội tiết loại 2B (MEN2B) | RET (10q11.21) | PHEO/PGL hiếm gặp | • Adrenergic • Hiếm khi ác tính • Nền tăng sản tủy thượng thận |
10 | Bắt đầu trước 11 tuổi đối với đột biến codon 918 và 883 |
| U xơ thần kinh loại 1 (NF1) | NF1 (17q11.2) | PHEO/PGL hiếm gặp & PHEO hỗn hợp d | • Adrenergic • Ít gặp ác tính |
7 | Tầm soát bất kỳ bệnh nhân NF1 nào có tăng huyết áp, dấu hiệu/triệu chứng của thừa catecholamin, hoặc khối u thượng thận hay cạnh động mạch chủ phát hiện tình cờ |
| Hội chứng u cận hạch thần kinh gia đình loại 1 (PGL1) | SDHD (11q23.1) | PGL (chủ yếu đầu & cổ > ngực/bụng/chậu)/PHEO | • Không chức năng hoặc noradrenergic • Hiếm khi ác tính |
5 | Bắt đầu trước 10 tuổi ở bệnh nhân có di truyền bệnh từ cha e |
| Hội chứng u cận hạch thần kinh gia đình loại 3 (PGL3) | SDHC (1q23.3) | PGL (chủ yếu đầu & cổ/ngực) | • Không chức năng hoặc noradrenergic • Hiếm khi ác tính |
13 | Bắt đầu trước 10 tuổi f |
| Hội chứng u cận hạch thần kinh gia đình loại 4 (PGL4) | SDHB (1p36.13) | PGL (Bụng/chậu > đầu & cổ)/PHEO | • Noradrenergic hoặc không chức năng • Tỷ lệ ác tính ~ 25% |
3 | Bắt đầu trước 5 tuổi g |
| Hội chứng u cận hạch thần kinh gia đình loại 5 (PGL5) | SDHA (5p15.33) | PGL/PHEO | • Không chức năng hoặc noradrenergic • Ít gặp ác tính |
8 | Bắt đầu trước 10 tuổi h |
| Bệnh von Hippel-Lindau (VHL) | VHL (3p25.3) | PHEO/PGL | • Noradrenergic • Hiếm khi ác tính |
2 | Bắt đầu trước 5 tuổi |
HTN, Tăng huyết áp; PGL, u cận hạch thần kinh; PHEO, u tế bào ưa crôm.
Tư vấn và xét nghiệm di truyền
Chẩn đoán một khối u nội tiết ở trẻ em, đặc biệt là khối u có nguồn gốc thần kinh nội tiết, cần đặt ra nghi ngờ về khả năng có một khuynh hướng di truyền tiềm ẩn. Tư vấn di truyền là một quá trình giao tiếp nhằm thúc đẩy sự hiểu biết, ra quyết định và đối phó liên quan đến tác động của bệnh di truyền. Quá trình này nên được tích hợp vào tất cả các giai đoạn chăm sóc, cả tại thời điểm chẩn đoán và trong quá trình theo dõi dài hạn, vì nhu cầu của bệnh nhân thay đổi theo thời gian và cũng vì các khuyến nghị về xét nghiệm di truyền và quản lý có khả năng sẽ phát triển.
Xét nghiệm di truyền, được minh họa rõ nét trong bệnh đa u tuyến nội tiết (MEN) loại 2, là một quá trình nhiều bước bắt đầu với một bệnh nhân đã có biểu hiện lâm sàng của bệnh. Trong hầu hết các trường hợp, chẳng hạn như trong MEN1 và MEN2A, đây có thể là cha mẹ hoặc một người thân cùng huyết thống khác, nhưng trong các rối loạn di truyền khác có độ xâm nhập bệnh thấp, chẳng hạn như hội chứng u cận hạch thần kinh (PGL) có tính gia đình, việc xét nghiệm di truyền của gia đình thực sự có thể bắt đầu với đứa trẻ là ca bệnh chỉ điểm. Xét nghiệm di truyền phục vụ nhiều mục đích, bao gồm xác nhận nguyên nhân di truyền của một khối u nội tiết, xác định các bệnh nhân không có triệu chứng nhưng có nguy cơ mắc bệnh lâm sàng, và hướng dẫn về quản lý lâm sàng và kế hoạch hóa gia đình. Khi chỉ định xét nghiệm di truyền tiên đoán, điều quan trọng là phải hiểu kết quả sẽ được sử dụng như thế nào trong việc quản lý bệnh nhân. Trong một số trường hợp (ví dụ, MEN2), việc biết được kiểu gen sẽ dẫn đến một can thiệp phòng ngừa bệnh (cắt tuyến giáp sớm), trong khi ở các hội chứng di truyền khác, chẳng hạn như MEN1 và bệnh von Hippel-Lindau (VHL), nó sẽ chỉ dẫn đến chẩn đoán sớm hơn.
Quá trình xét nghiệm di truyền ở trẻ em gắn liền với nhiều hàm ý về đạo đức, pháp lý và tâm lý xã hội. Mặc dù có những lợi ích y tế rõ ràng từ việc chẩn đoán sớm, xét nghiệm di truyền ở trẻ em có khả năng gây hại cho bệnh nhân và gia đình: thay đổi hình ảnh bản thân của trẻ và nhận thức của cha mẹ về trẻ, thay đổi quan điểm sống của bệnh nhân, lo lắng về khả năng bị phân biệt đối xử do di truyền, thay đổi các mối quan hệ gia đình, “y tế hóa” sớm một đứa trẻ vốn khỏe mạnh, và những lo ngại về các vấn đề sinh sản trong tương lai. Lợi ích y tế kịp thời cho trẻ nên là lý do chính để thực hiện xét nghiệm di truyền. Trong mọi trường hợp mà rủi ro so với lợi ích của xét nghiệm di truyền không rõ ràng, nhà cung cấp dịch vụ y tế nên tôn trọng quyết định của gia đình. Có một số tài nguyên trực tuyến về tư vấn và xét nghiệm di truyền, bao gồm Hiệp hội Tư vấn Di truyền Quốc gia (National Society of Genetic Counselors), trang web “The Genetics of Cancer” của Viện Ung thư Quốc gia (National Cancer Institute), và trang web GeneReviews®.
U tế bào ưa crôm và U cận hạch thần kinh
U tế bào ưa crôm (PHEO) và u cận hạch thần kinh (PGL) là các khối u thần kinh nội tiết (NET) phát sinh từ các tế bào cận hạch có nguồn gốc từ mào thần kinh. PHEO (Hình 15.1) là thuật ngữ được sử dụng cho một khối u sản xuất catecholamin phát triển từ các tế bào ưa crôm trong tủy thượng thận, trong khi PGL (Hình 15.2) là các khối u ngoài tuyến thượng thận phát sinh từ cả cận hạch giao cảm và đối giao cảm nằm trong hệ thần kinh tự chủ bên ngoài trục não tủy. Trước đây, thuật ngữ PHEO được sử dụng thay thế cho PGL, nhưng tốt nhất nên duy trì sự phân biệt giữa hai loại u tân sinh này vì những khác biệt cơ bản về di truyền, biểu hiện lâm sàng và tiềm năng ác tính (xem Bảng 15.1).
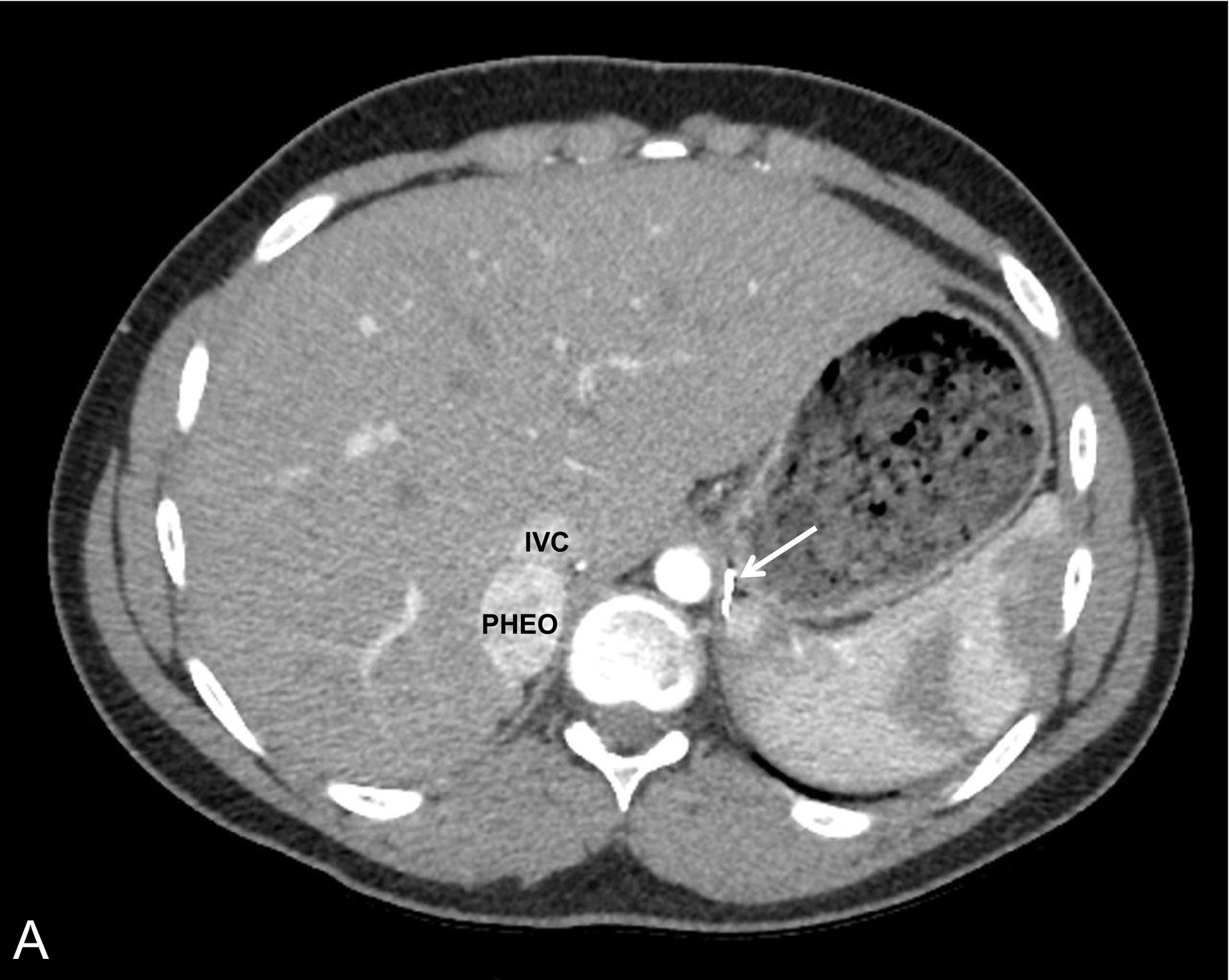
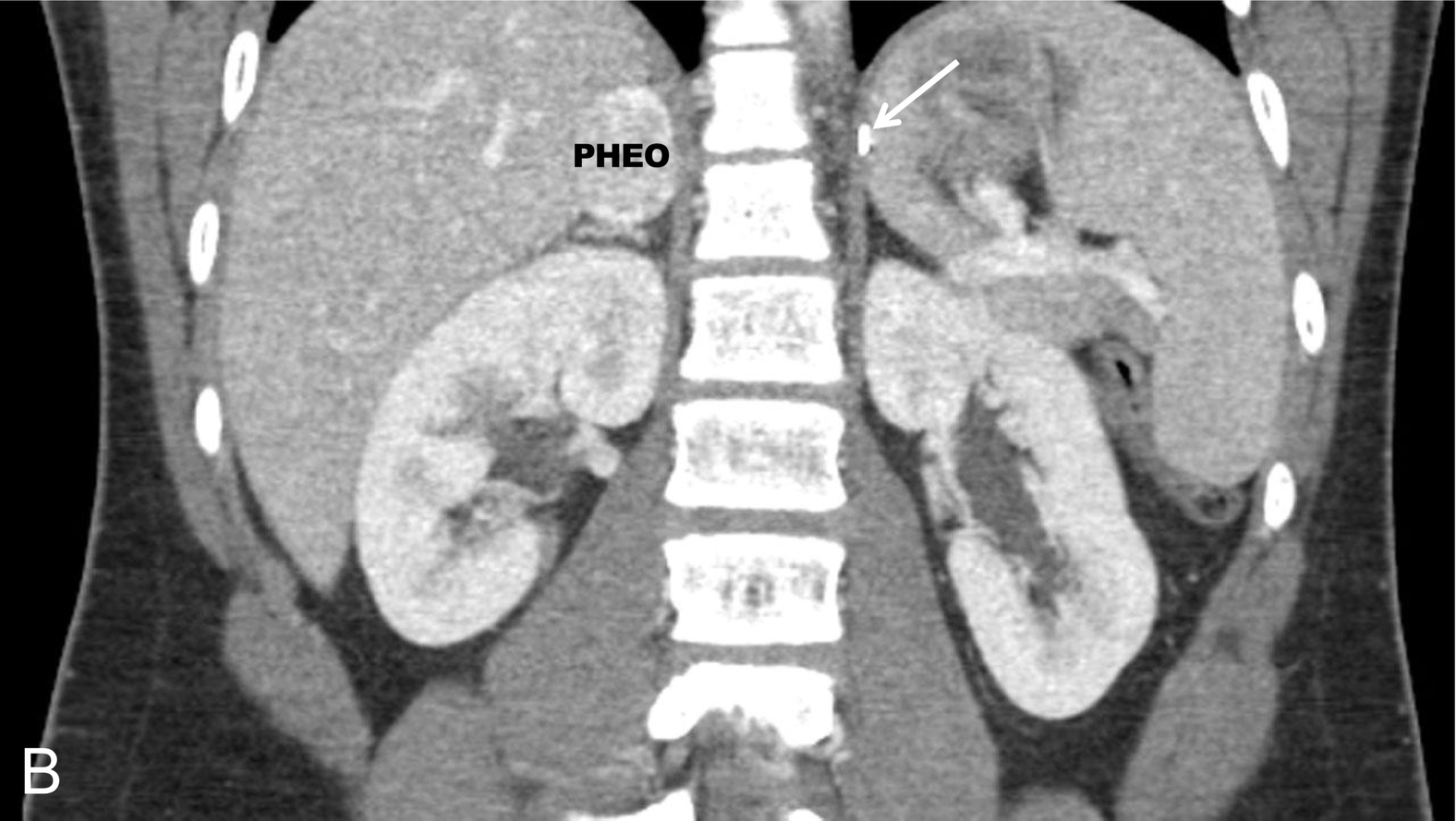
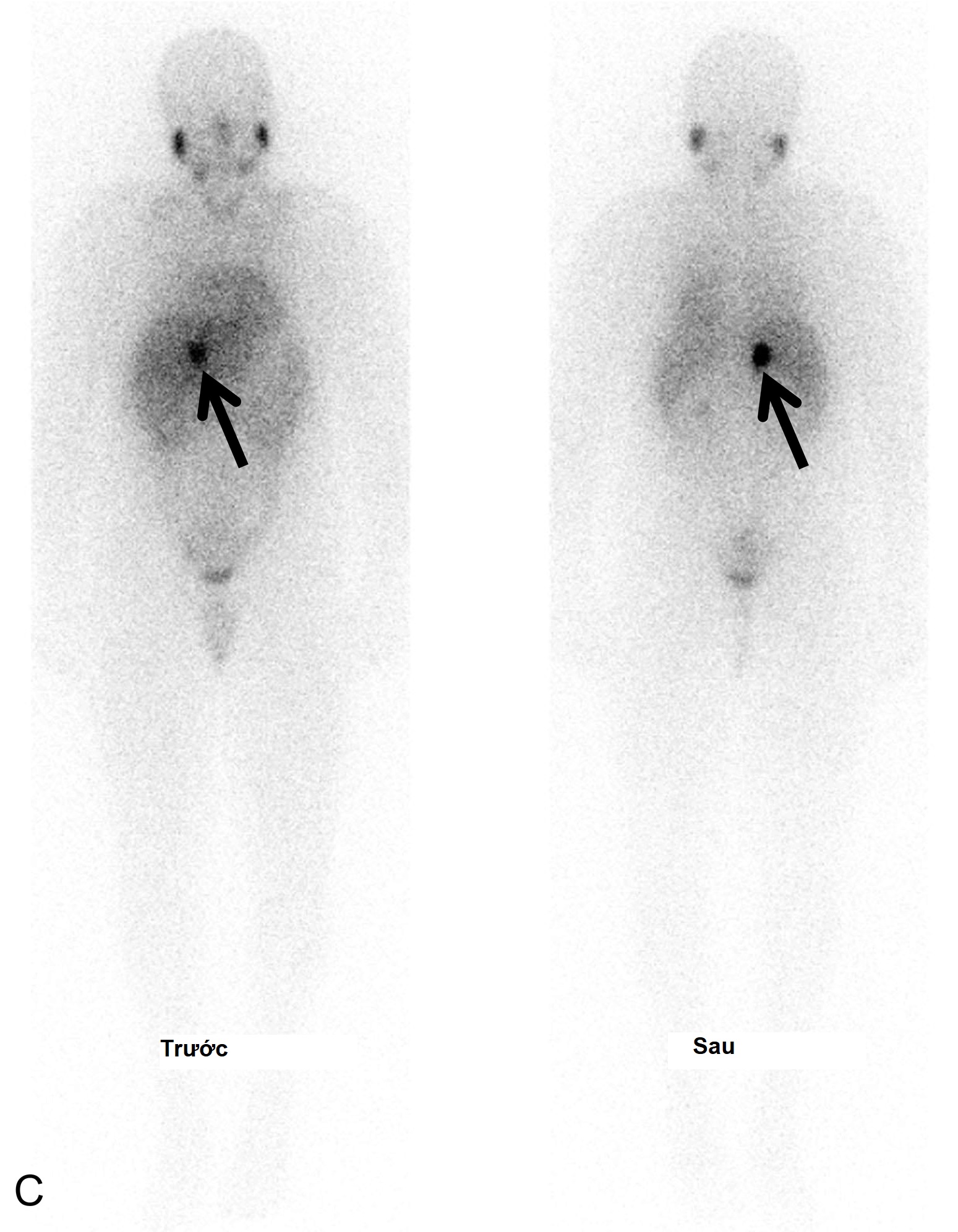
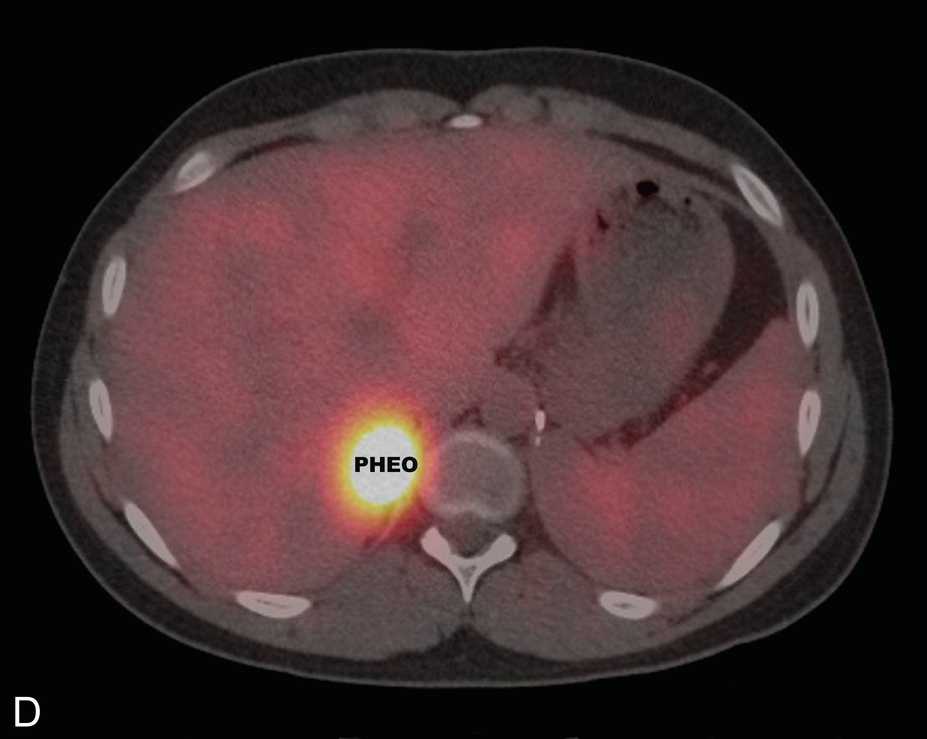
Hình 15.1 Một bệnh nhi nam 14 tuổi, huyết áp bình thường, mắc bệnh von Hippel-Lindau và có tiền sử u tế bào ưa crôm bên trái và u cận hạch thần kinh ở bụng được chẩn đoán lúc 7 tuổi, được phát hiện có nồng độ normetanephrin tăng cao sau khi tầm soát. (A) Chụp cắt lớp vi tính (CT) có cản quang, mặt cắt ngang và (B) tái tạo mặt cắt đứng, đã xác định một khối u tân sinh giàu mạch máu phát sinh từ cực trên của tuyến thượng thận phải (mũi tên, kẹp phẫu thuật từ lần cắt tuyến thượng thận trước). Xạ hình Metaiodobenzylguanidine (MIBG) đã xác nhận bản chất chức năng của khối u và loại trừ các khối u đồng bộ khác. (C) Hình ảnh phẳng và (D) hình ảnh hợp nhất CT/SPECT cắt ngang ở cùng bệnh nhân, 24 giờ sau khi tiêm 123I MIBG. IVC, tĩnh mạch chủ dưới; PHEO, u tế bào ưa crôm.
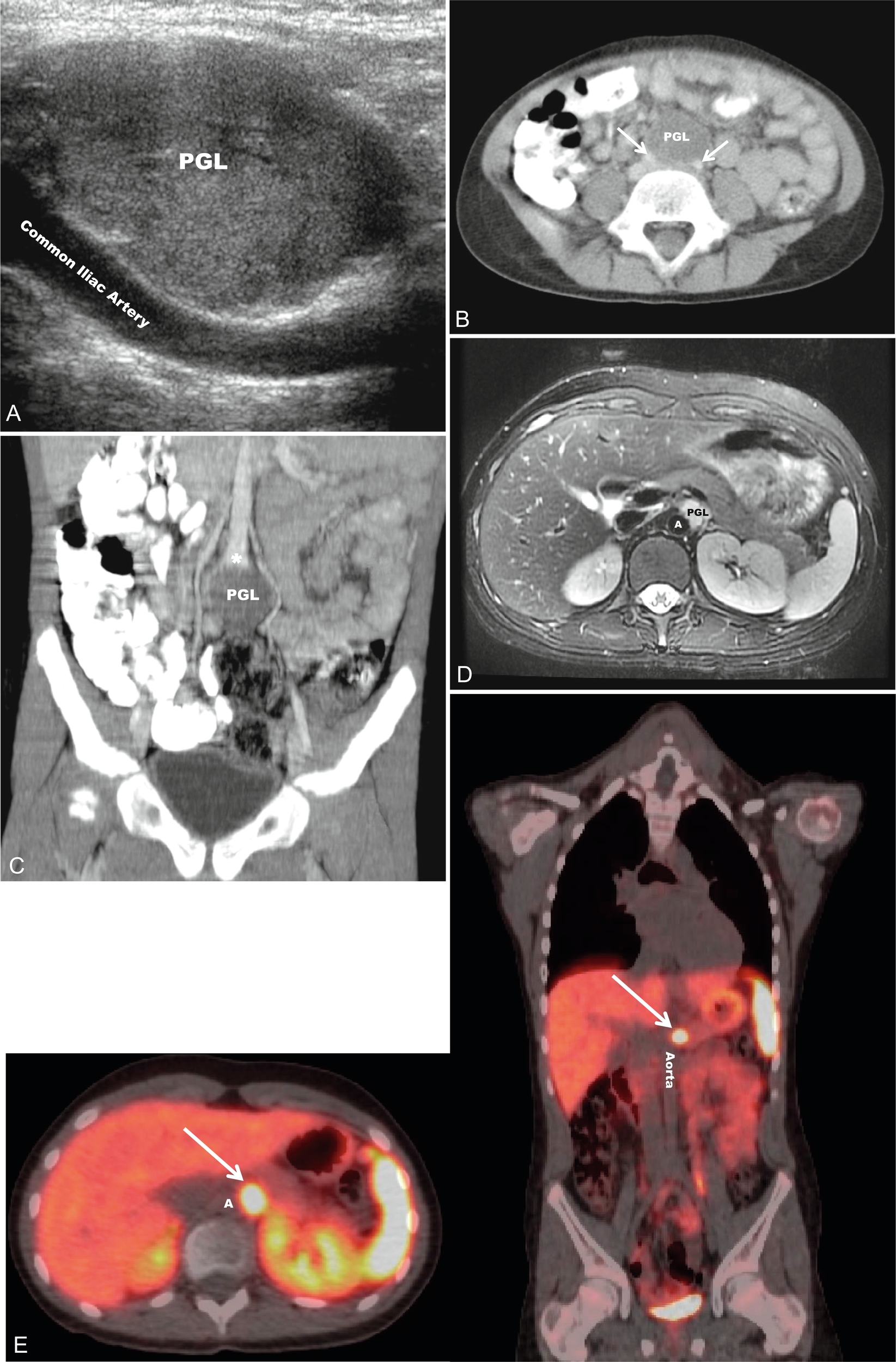
Hình 15.2 Một bệnh nhi nữ 6 tuổi có u cận hạch thần kinh (PGL) và đột biến SDHB, biểu hiện với tăng huyết áp nặng trong một buổi khám sức khỏe định kỳ. (A) Siêu âm bụng (mặt cắt dọc) cho thấy một khối u 3,3 cm đồng nhất, tăng sinh mạch bên cạnh các động mạch chậu chung. (B) Chụp cắt lớp vi tính (CT) có cản quang, mặt cắt ngang (mũi tên, các động mạch chậu chung) và (C) tái tạo mặt cắt đứng, xác nhận một khối u ở vị trí phân đôi động mạch chủ (dấu sao). Một bệnh nhi nam 13 tuổi có đột biến SDHB bị tăng huyết áp và nồng độ norepinephrine, normetanephrin, và chromogranin A bất thường trong quá trình tầm soát định kỳ. (D) Hình ảnh T2-weighted cắt ngang từ chụp cộng hưởng từ bụng cho thấy một khối u 2 cm cạnh động mạch chủ (PGL) giữa gốc động mạch thân tạng và động mạch mạc treo tràng trên, có tín hiệu T2 tăng cao. Điều này sau đó đã được xác nhận là một u cận hạch thần kinh (mũi tên) thông qua hình ảnh chức năng với chụp cắt lớp phát xạ positron/CT 68Ga-DOTATATE (E). A, động mạch chủ. Common Iliac Artery = Động mạch chậu chung, Aorta = Động mạch chủ
PHEO/PGL là những khối u hiếm gặp với tỷ lệ mắc hàng năm ở Hoa Kỳ là 500 đến 1600 ca mỗi năm. Khoảng 10% PHEO/PGL được chẩn đoán ở trẻ em với độ tuổi trung bình là 13 tuổi; có sự chiếm ưu thế nhẹ ở bé trai, đặc biệt khi được chẩn đoán dưới 10 tuổi. Ít hơn 2% trẻ em được chẩn đoán tăng huyết áp sẽ có một khối u tân sinh sản xuất catecholamin.
PGL xảy ra ở tất cả các vị trí có cận hạch (từ nền sọ đến khung chậu) và là các khối u tân sinh có chức năng (giao cảm) hoặc không có chức năng (đối giao cảm), tùy thuộc vào vị trí xuất phát và sinh lý bệnh cơ bản (xem Bảng 15.1). PGL phát sinh ở vùng đầu và cổ, hiếm gặp hơn ở trẻ em, hầu như chỉ là các khối u đối giao cảm không chức năng, trong khi hầu hết các PGL trong ổ bụng (thường xuất hiện trong cơ quan Zuckerkandl [xem Hình 15.2] ở vị trí cạnh thượng thận, hoặc xung quanh rốn thận) là các khối u giao cảm và do đó là các khối u bài tiết. Phần lớn PHEO/PGL được chẩn đoán ở trẻ em là có chức năng, tổng hợp và bài tiết catecholamin (dopamine, norepinephrine, và/hoặc epinephrine) và các chất chuyển hóa của chúng (bao gồm 3-methoxytyramine, normetanephrin, và metanephrin, tương ứng) (Hình 15.3). Tất cả các PHEO/PGL có chức năng đều chứa mô ưa crôm, đề cập đến màu nâu-đen do quá trình oxy hóa catecholamin sau khi nhuộm bằng muối crôm. Các khối u đa trung tâm thường gặp trong các trường hợp PHEO/PGL ở trẻ em.
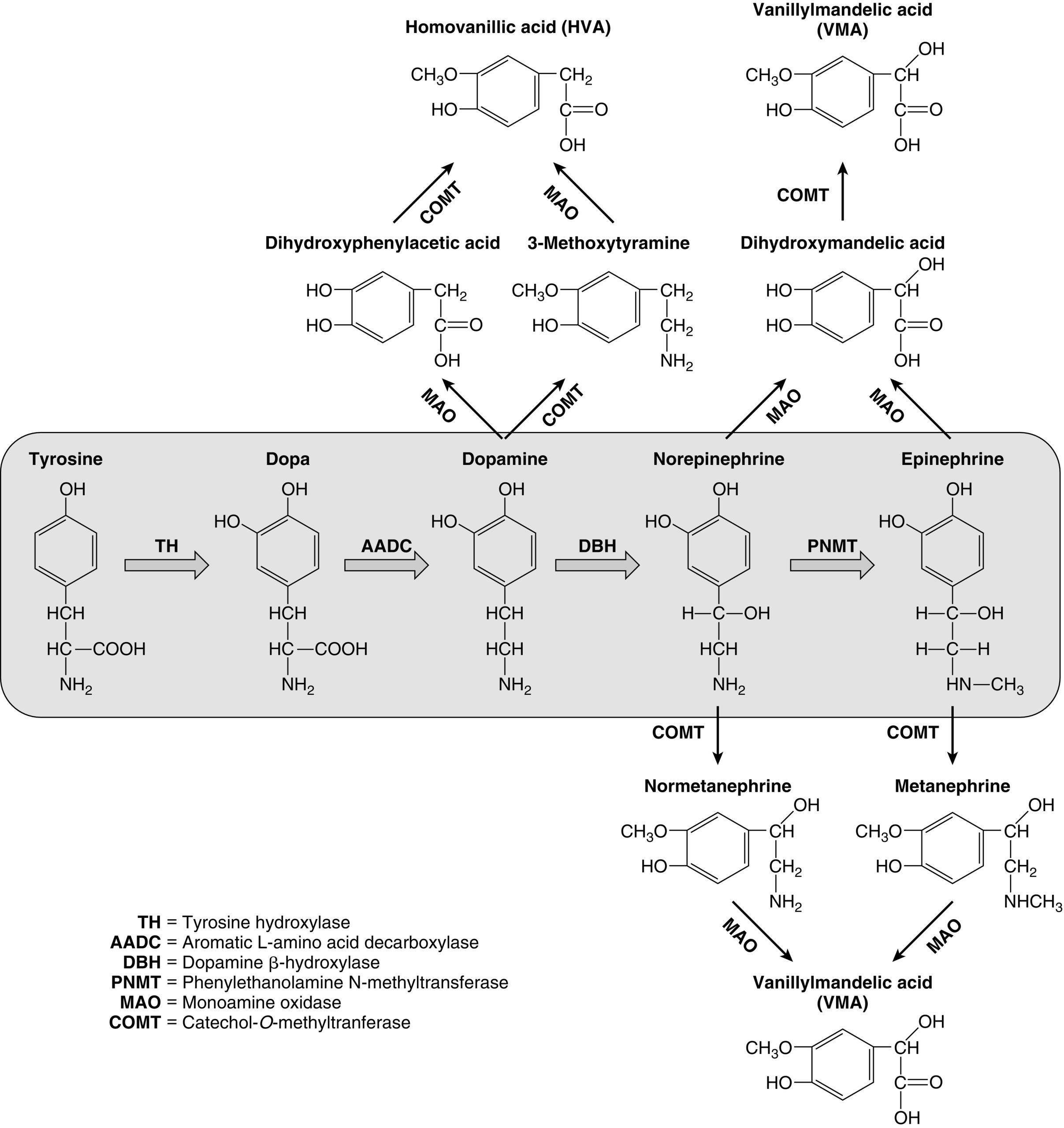
Hình 15.3 Các catecholamin được tổng hợp từ axit amin tyrosine, được chuyển đổi thành 3,4-dihydroxyphenylalanine (dopa) bởi enzyme tyrosine hydroxylase (TH), đây là bước giới hạn tốc độ trong quá trình sinh tổng hợp catecholamin. Các quá trình khử carboxyl (aromatic L-amino acid decarboxylase; AADC) và hydroxyl hóa (dopamine β-hydroxylase; DBH) sau đó tạo ra dopamine và norepinephrine tương ứng, và norepinephrine sau đó được chuyển đổi thành epinephrine thông qua enzyme cytosolic phenylethanolamine N-methyltransferase (PNMT). Các catecholamin được chuyển hóa bởi hai enzyme chính: monoamine oxidase (MAO) và catechol-O-methyltransferase (COMT).
Sinh tổng hợp và tác dụng của Catecholamin
Dopamine, norepinephrine, và epinephrine (gọi chung là catecholamin) là các chất dẫn truyền thần kinh hóa học và hormone đóng vai trò quan trọng trong việc điều hòa nhiều quá trình sinh lý và sự phát triển của các bệnh lý thần kinh, tâm thần, nội tiết và tim mạch. Các catecholamin bao gồm một nhóm catechol (1,2-dihydroxybenzene) và một nhóm amin ở chuỗi bên. Chúng được tổng hợp từ axit amin tyrosine, được chuyển đổi thành 3,4-dihydroxyphenylalanine (dopa) bởi enzyme tyrosine hydroxylase, bước giới hạn tốc độ trong quá trình sinh tổng hợp catecholamin (xem Hình 15.3). Quá trình khử carboxyl và hydroxyl hóa enzyme của dopa sau đó tạo ra dopamine và norepinephrine tương ứng, và norepinephrine sau đó được chuyển đổi thành epinephrine thông qua enzyme cytosolic phenylethanolamine N-methyltransferase (PNMT).
Các catecholamin được tổng hợp và lưu trữ trong các hạt bên trong tủy thượng thận, nơi chúng được giải phóng qua quá trình xuất bào vào hệ tuần hoàn để đáp ứng với các kích thích căng thẳng. Dopamine và norepinephrine cũng được sản xuất bởi các tế bào thần kinh sau hạch trong hệ thần kinh giao cảm. Epinephrine chỉ được tạo ra trong tủy thượng thận, nơi nó chiếm phần lớn catecholamin (~80%) vì sự biểu hiện của PNMT phụ thuộc và được điều hòa bởi nồng độ glucocorticoid cao tại chỗ (chỉ xảy ra ở tủy thượng thận, được bao quanh bởi vỏ thượng thận tổng hợp cortisol, với một gradient nồng độ rõ rệt về phía tủy thượng thận). Tác dụng của catecholamin bị chấm dứt thông qua việc tái hấp thu nhanh chóng vào các đầu dây thần kinh bởi chất vận chuyển norepinephrine và qua quá trình chuyển hóa bởi hai enzyme chính: monoamine oxidase (MAO) và catechol-O-methyltransferase (COMT) (xem Hình 15.3).
Các tác động phức tạp của norepinephrine và epinephrine được trung gian bởi các thụ thể α- và β-adrenergic kết hợp với G-protein, trong khi dopamine liên kết với một loại thụ thể dopamine kết hợp với G-protein khác (năm thụ thể riêng biệt [D1–D5] được chia thành hai họ: D1-like và D2-like) (Bảng 15.2). Phân loại ban đầu của các thụ thể adrenergic dựa trên khả năng của epinephrine vừa kích thích (thụ thể α) vừa ức chế (thụ thể β) cơ trơn. Các chất chủ vận và đối kháng đặc hiệu xác định phân loại thụ thể adrenergic (α1, α2, β1, β2, và β3) và có thể được sử dụng làm tác nhân điều trị. Thụ thể D2 là thụ thể dopamine chính được nhắm đến trong liệu pháp dùng thuốc.
Bảng 15.2 Phân Loại, Chức Năng và Dược Lý của Thụ Thể Catecholamin
| Loại Thụ Thể | Chất Chủ Vận Dược Lý Chính | Chất Đối Kháng Dược Lý Chính | Tác Dụng Sinh Học Chính |
|---|---|---|---|
| α1 | Phenylephrine, midodrine | Doxazosin, prazosin, terazosin, v.v. | Gây co mạch; thúc đẩy tăng trưởng và cấu trúc tim |
| α2 | Clonidine, methyldopa, tizanidine | Yohimbine | Ức chế giải phóng norepinephrine; gây co mạch; ức chế giải phóng catecholamin thượng thận và điều hòa dẫn truyền thần kinh dopamine ở TKTW |
| β1 | Dobutamine | Atenolol, bisoprolol, esmolol, metoprolol, v.v. | Tăng nhịp tim và sức co bóp |
| β2 | Albuterol, levalbuterol, salmeterol, terbutaline, v.v. | Propranolol (chất đối kháng β2 nguyên mẫu cũng là chất đối kháng β1) và các loại khác | Giãn cơ trơn (giãn tiểu động mạch và tĩnh mạch; giãn cơ khí-phế quản) |
| β3 | Mirabegron | Đang nghiên cứu | Tác dụng chuyển hóa trong mô mỡ và cơ xương |
| D1-like (D1 và D5) | Levodopa | Không có tác nhân chính | Gây giãn mạch; tăng tiết renin; thúc đẩy giải phóng norepinephrine và epinephrine; các chức năng TKTW khác nhau |
| D2-like (D2, D3, và D4) | Bromocriptine, cabergoline, pramipexole, ropinirole | Aripiprazole, chlorpromazine, haloperidol, metoclopramide, prochlorperazine, v.v. | Giảm tiết prolactin; giảm tiết renin; ức chế giải phóng norepinephrine và epinephrine; các chức năng TKTW khác nhau |
TKTW, hệ thần kinh trung ương.
(Nguồn: Westfall, T.C., Westfall, D.P. (2011). Neurotransmission: the autonomic and somatic motor nervous systems. In: Brunton, L.L., ed. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 12 ed. New York, NY, McGraw-Hill, 171–218; Westfall, T.C., Westfall, D.P. (2011). Catecholamines and sympathomimetic drugs. In: Brunton, L.L., ed. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 12 ed. New York, NY: McGraw-Hill, 277–333; Sanders-Bush, E., Hazelwood, L. (2011). 5-Hydroxytryptamine (Serotonin) and Dopamine. In: Brunton, L.L., ed. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 12 ed. New York, NY, McGraw-Hill, 335–361.)
Biểu hiện lâm sàng
Biểu hiện lâm sàng của PHEO/PGL ở trẻ em rất đa dạng. Trẻ em có các khối u này có thể được phát hiện do các triệu chứng tăng tiết catecholamin, triệu chứng do hiệu ứng khối của khối u (ví dụ, đau), một phát hiện hình ảnh tình cờ, hoặc do tầm soát định kỳ trong một trong các hội chứng khối u di truyền liên quan (xem Bảng 15.1). PHEO/PGL cũng có thể phát sinh trong bối cảnh bệnh tim bẩm sinh tím. Do nguồn gốc thần kinh nội tiết, PHEO/PGL rất hiếm khi có thể đồng tiết các hormone khác gây ra hội chứng lâm sàng thừa hormone lạc chỗ, chủ yếu là hội chứng Cushing, do sản xuất quá mức hormone vỏ thượng thận (ACTH).
Biểu hiện lâm sàng của một PHEO/PGL có chức năng phụ thuộc vào sự khác biệt cố hữu trong sản xuất catecholamin, cũng như độ nhạy cảm của từng bệnh nhân đối với catecholamin. Các dấu hiệu và triệu chứng của thừa catecholamin bao gồm: tăng huyết áp, thường là kéo dài ở đa số các trường hợp ở trẻ em; đau đầu dữ dội; các cơn kịch phát với bộ ba kinh điển là đau đầu, đánh trống ngực và vã mồ hôi (ít phổ biến hơn ở trẻ em); hạ huyết áp tư thế và ngất; da tái; run; và/hoặc lo âu. PHEO/PGL ở trẻ em cũng có thể gây ra các dấu hiệu và triệu chứng không đặc hiệu, chẳng hạn như nhìn mờ; đau bụng, táo bón, tiêu chảy và các triệu chứng tiêu hóa khác; sụt cân; tăng đường huyết; đa niệu và khát nhiều; sốt nhẹ; và các vấn đề về hành vi, rối loạn tăng động giảm chú ý, và/hoặc sa sút trong học tập. PGL bàng quang có thể biểu hiện bằng tiểu máu và các triệu chứng kịch phát trong khi đi tiểu.
Các biến chứng của thừa catecholamin có thể bao gồm cơn tăng huyết áp, bệnh cơ tim (bệnh cơ tim takotsubo), rối loạn nhịp tim, viêm tụy, táo bón nặng và giả tắc ruột, đột quỵ, co giật, và thậm chí là khủng hoảng đa cơ quan và tử vong. Các triệu chứng của PGL đối giao cảm (thường nằm ở vùng đầu và cổ) bao gồm mất thính lực, ù tai theo nhịp đập, khối u ở cổ, và các triệu chứng khác của hiệu ứng khối, chẳng hạn như khàn giọng, cảm giác đầy họng, và khó nuốt.
Lên đến 80% dân số trẻ em mắc PHEO/PGL có một hội chứng khuynh hướng di truyền (xem Bảng 15.1 và phần sau), và một phần trong số này sẽ chỉ được chẩn đoán sau khi một khối u được xác định trong một chương trình tầm soát. So với bệnh lẻ tẻ, PHEO/PGL di truyền được xác định trong quá trình tầm soát tiền triệu chứng định kỳ có kích thước nhỏ hơn và ít triệu chứng hơn (hoặc thậm chí không có triệu chứng). Trong mọi trường hợp PHEO/PGL ở trẻ em, cần tiến hành tư vấn và xét nghiệm di truyền; việc xác định một đột biến dòng mầm sớm sau chẩn đoán dường như có tác động tích cực đến việc quản lý và kết quả lâm sàng của bệnh nhân mắc bệnh di truyền. Hiệp hội Nghiên cứu Ung thư Hoa Kỳ đã phát triển các hướng dẫn tầm soát cho dân số trẻ em mắc các hội chứng PHEO/PGL di truyền, và các tác giả khác cũng đã công bố các khuyến nghị tầm soát ở các quần thể trẻ em mắc MEN2 và bệnh VHL.
Đánh giá
Chẩn đoán sinh hóa
Chẩn đoán PHEO/PGL đã được đơn giản hóa nhờ những tiến bộ trong các phương pháp xét nghiệm được sử dụng để phát hiện và định lượng nồng độ catecholamin và các chất chuyển hóa của chúng trong máu và nước tiểu. Việc đo lường metanephrin phân đoạn trong huyết tương và/hoặc nước tiểu (metanephrin và normetanephrin) là xét nghiệm nhạy nhất (độ nhạy gần 100%) để chẩn đoán PHEO/PGL và nên là xét nghiệm chẩn đoán chính (Hình 15.4).
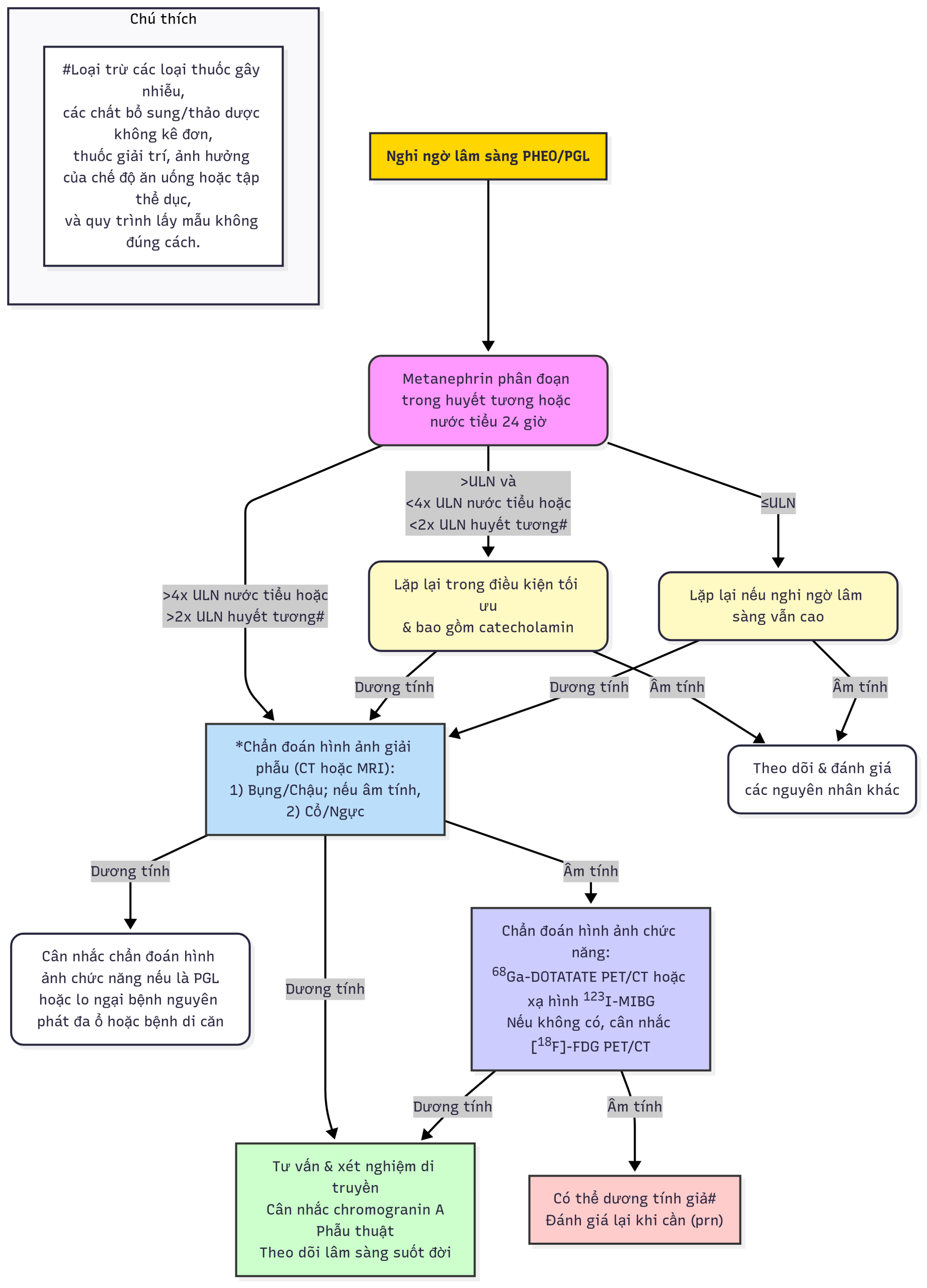
Hình 15.4 Chẩn đoán u tế bào ưa crôm (PHEO)/u cận hạch thần kinh (PGL) ở trẻ em. ULN, giới hạn trên của mức bình thường.
(Sửa đổi từ Waguespack, S.G., Rich, T., Grubbs, E., và cộng sự. (2010). A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metabol, 95(5), 2023–2037.)
Độ nhạy cao của xét nghiệm metanephrin dựa trên thực tế là có sự chuyển hóa catecholamin liên tục bên trong khối u, một quá trình xảy ra độc lập với việc giải phóng catecholamin, vốn có thể xảy ra không liên tục hoặc ở mức độ thấp. Sự gia tăng metanephrin lớn hơn 4 lần so với giới hạn trên của khoảng tham chiếu có liên quan đến xác suất gần 100% về sự hiện diện của một khối u tiết catecholamin; tuy nhiên, một PHEO/PGL vẫn có thể hiện diện với nồng độ metanephrin cao hơn hai lần giới hạn trên của mức bình thường, đặc biệt là trong bối cảnh bệnh di truyền. Bất kỳ loại thuốc nào được biết là gây nhiễu cho các xét nghiệm này (ví dụ, acetaminophen, thuốc chống trầm cảm ba vòng, phenoxybenzamine, và thuốc thông mũi, cùng những loại khác) nên được ngưng trước khi xét nghiệm. Việc hạn chế chế độ ăn uống không cần thiết phải được áp dụng thường quy nhưng nên được xem xét nếu xét nghiệm chỉ đo lường normetanephrin không liên hợp hoặc nếu nghi ngờ có khối u tiết dopamine. Việc đo lường các chất chuyển hóa catecholamin là axit vanillylmandelic (VMA) và axit homovanillic (HVA) không còn được khuyến nghị để đánh giá PHEO/PGL. Tuy nhiên, xét nghiệm HVA và VMA trong các mẫu nước tiểu ngẫu nhiên vẫn là một thành phần quan trọng trong việc đánh giá u nguyên bào thần kinh, nơi các chất phân tích này có độ nhạy và độ đặc hiệu cao để phát hiện khối u.
Ở những bệnh nhân có nồng độ metanephrin tăng nhẹ, trong đó nghi ngờ kết quả xét nghiệm dương tính giả, cần xem xét việc đo lường các chất phân tích này trong huyết tương được lấy từ bệnh nhân ở tư thế nằm ngửa, 30 phút sau khi đặt kim hoặc catheter lưu vào tĩnh mạch. Các nghiệm pháp ức chế clonidine và kích thích glucagon đã là một phần của quy trình chẩn đoán ở người lớn nhưng hiếm khi được yêu cầu và chưa được xác thực trong chẩn đoán PHEO/PGL ở trẻ em.
Các khối u sản xuất catecholamin có thể được phân loại nhỏ thành loại noradrenergic hoặc adrenergic dựa trên kiểu giải phóng catecholamin của chúng. PHEO/PGL noradrenergic tiết norepinephrine và normetanephrin, như được thấy trong bệnh VHL và trong các khối u liên quan đến các hội chứng PGL/PHEO di truyền. Các khối u adrenergic tiết cả epinephrine và norepinephrine và các chất chuyển hóa của chúng, và những khối u này thường là PHEO phát sinh lẻ tẻ hoặc trong bối cảnh lâm sàng của MEN2 hoặc u xơ thần kinh loại 1 (NF1). Sự khác biệt trong việc bài tiết catecholamin này là do sự giảm biểu hiện của PNMT trong các khối u noradrenergic thứ phát do sự tăng methyl hóa promoter của PNMT.
Các khối u tiết dopamine rất hiếm và thường là các PGL ngoài thượng thận liên quan đến succinate dehydrogenase (SDH)x, cũng như các PGL ở đầu và cổ. Việc đo lường methoxytyramine, một chất chuyển hóa của dopamine, (xem Hình 15.3) có thể giúp xác định các khối u xâm lấn hơn, đặc biệt trong bối cảnh đột biến SDHx, nhưng xét nghiệm này không phổ biến. Cần xem xét một khối u tiết dopamine ở những bệnh nhân huyết áp bình thường được xác định có một khối u có vẻ phù hợp với PHEO/PGL, trong trường hợp đó nên đo dopamine và các chất chuyển hóa của nó, HVA và methoxytyramine (nếu có). Để tầm soát định kỳ ở những bệnh nhân có đột biến SDHx đã biết, nên kiểm tra tổng catecholamin ngoài tổng metanephrin vì khả năng này.
Chromogranin A là một protein bài tiết chính có trong chất nền hòa tan của các hạt ưa crôm, đóng vai trò như một chất chỉ điểm khối u có thể tương quan với kích thước và tiềm năng ác tính của PHEO/PGL. Chromogranin A cũng có vẻ là một chất chỉ điểm hữu ích trong các PGL liên quan đến SDHB im lặng về mặt sinh hóa, làm cho nó trở thành một xét nghiệm tiềm năng hữu ích trong việc tầm soát những người mang đột biến SDHx không triệu chứng. Tuy nhiên, chromogranin A có thể tăng giả trong nhiều trường hợp, bao gồm việc sử dụng đồng thời thuốc ức chế bơm proton và thuốc ức chế tái hấp thu serotonin, suy thận, và các rối loạn tuyến tụy, do đó cần thận trọng trong việc giải thích.
Các nghiên cứu hình ảnh học
Một khi chẩn đoán thừa catecholamin được xác định từ xét nghiệm sinh hóa, các nghiên cứu hình ảnh học nên được thực hiện để xác định vị trí của (các) khối u (xem Hình 15.1, 15.2, và 15.4). Các nghiên cứu hình ảnh ban đầu nên bao gồm hình ảnh cắt lớp ngang của bụng và chậu (chụp cắt lớp vi tính [CT] hoặc chụp cộng hưởng từ [MRI]), sau đó là hình ảnh cổ và ngực nếu các nghiên cứu ban đầu không cho kết quả (xem Hình 15.4). CT và MRI có độ nhạy chẩn đoán tương tự, do đó việc lựa chọn phương pháp hình ảnh tốt nhất được xác định bởi thực hành tại địa phương và sở thích của bệnh nhân. Siêu âm bụng (US) cũng có thể được xem xét ở trẻ nhỏ, nếu chuyên môn tại địa phương cho phép (xem Hình 15.2). PHEO/PGL là những khối u tân sinh giàu mạch máu (và do đó ngấm thuốc cản quang) thường chứa các vùng hoại tử, nang, và/hoặc xuất huyết. Trên CT, đơn vị Hounsfield không cản quang hầu như luôn lớn hơn 10, và trên MRI, PHEO/PGL biểu hiện một hình ảnh tăng tín hiệu kinh điển trên hình ảnh T2-weighted (gọi là dấu hiệu bóng đèn) và không mất cường độ tín hiệu trên hình ảnh in-phase và out-of-phase (xem Hình 15.2).
Y học hạt nhân, hay hình ảnh chức năng, có thể hữu ích trong một số trường hợp nhất định để chẩn đoán PHEO/PGL (đặc biệt để xác nhận chẩn đoán ở một khối u ngoài thượng thận) hoặc để đánh giá bệnh nhân về bệnh đa ổ hoặc di căn, đặc biệt ở những bệnh nhân có các yếu tố nguy cơ ác tính và kiểu hình noradrenergic. Có một số phương pháp xạ hình y học hạt nhân có thể hữu ích. Metaiodobenzylguanidine (MIBG) là một hợp chất tổng hợp có cấu trúc tương tự norepinephrine (ngoại trừ nó cũng có một chuỗi bên guanidine chống lại sự chuyển hóa), và nó tích tụ ưu tiên trong các mô adrenergic, chủ yếu do sự tái hấp thu qua hệ thống vận chuyển norepinephrine. MIBG được sử dụng cho mục đích chẩn đoán được đánh dấu phóng xạ bằng 123I hoặc 131I, mặc dù 123I MIBG là tác nhân được lựa chọn vì các đặc tính hình ảnh vượt trội và liều bức xạ thấp hơn đáng kể vì nó không phải là một chất phát tia β. Xạ hình 123I MIBG (xem Hình 15.1) có thể xác nhận bản chất sản xuất catecholamin của một khối u, định vị các khối u không nhìn thấy bằng hình ảnh cắt lớp, và có khả năng xác định các vị trí bệnh khác, mặc dù độ nhạy của nó không cao và do đó việc sử dụng nó bị hạn chế vì lý do này, trừ khi đang xem xét điều trị bằng 131I-MIBG cho bệnh di căn. Trước khi xạ hình 123I-MIBG, cần cẩn thận để đảm bảo rằng bệnh nhân không dùng các loại thuốc (ví dụ, thuốc thông mũi, thuốc chẹn kênh canxi, hoặc labetalol) được biết là làm giảm sự hấp thu MIBG, và nên dùng kali iodua để ngăn chặn sự hấp thu iốt phóng xạ của tuyến giáp. Do những hạn chế của xét nghiệm MIBG, việc sử dụng các phương thức hình ảnh hạt nhân khác kết hợp chụp cắt lớp phát xạ positron (PET) với hình ảnh CT đã trở nên phổ biến hơn, đặc biệt là [18F] fluorodeoxyglucose (FDG) PET/CT và 68Ga-DOTATATE PET/CT (xem Hình 15.2). 68Ga-DOTATATE PET/CT đã trở thành phương thức hình ảnh chức năng được lựa chọn, do tính đặc hiệu của nó đối với các thụ thể somatostatin thường được tìm thấy trên các khối u thần kinh nội tiết, chẳng hạn như PHEO/PGL và độ nhạy vượt trội của nó so với FDG-PET/CT và xạ hình 123I-MIBG.
Các vấn đề di truyền
Kiến thức về các nguyên nhân di truyền của các khối u sản xuất catecholamin đang nhanh chóng mở rộng. Các hội chứng thường liên quan nhất đến PHEO/PGL ở trẻ em là bệnh VHL, tiếp theo là các hội chứng PGL/PHEO di truyền, đặc biệt là những hội chứng do đột biến ở SDHB (PGL4) và SDHD (PGL1), sau đó là NF1 và MEN2 (xem Bảng 15.1 và các phần riêng sau trong chương). Các gen bổ sung liên quan đến sự phát triển của PHEO/PGL, nhưng với tỷ lệ mắc thấp hơn nhiều, bao gồm fumarate hydratase (FH); yếu tố cảm ứng thiếu oxy 2 alpha (HIF2A, còn được biết đến là [aka] EPAS1); thành viên họ kinesin 1B (KIF1B); MEN1; yếu tố X liên kết với MYC (MAX); prolyl hydroxylase domain 1 (PHD1, aka EGLN2) và 2 (PHD2, aka EGLN1); yếu tố lắp ráp phức hợp succinate dehydrogenase 2 (SDHAF2); phức hợp succinate dehydrogenase, tiểu đơn vị A (SDHA); phức hợp succinate dehydrogenase, tiểu đơn vị C (SDHC); và protein xuyên màng 127 (TMEM127). Gần đây, nhiều gen hơn đã được mô tả là có liên quan đến PHEO/PGL và cần nghiên cứu thêm để xác định mối quan hệ nhân quả. Chúng bao gồm protein-1 liên kết với BRCA1 (BAP1); malate dehydrogenase 2 (MDH2); proto-oncogene MER, tyrosine kinase (MERTK); proto-oncogene MET (MET); thành viên 11 họ chất mang hòa tan 25 (SLC25A11); và các gen khác. Tiền sử gia đình, biểu hiện lâm sàng của bệnh nhân, và sự khác biệt trong kiểu hình sinh hóa (noradrenergic so với adrenergic) giúp ưu tiên việc xét nghiệm di truyền, nhưng sự sẵn có của giải trình tự thế hệ tiếp theo đã dẫn đến sự phát triển của các bảng gen mục tiêu đã được xác thực hiện nay được sử dụng phổ biến hơn, đặc biệt là khi xét nghiệm một ca bệnh chỉ điểm.
Dựa trên phân tích bộ gen, các gen nhạy cảm với PHEO/PGL hiện được chia thành hai cụm chính: cụm 1 (một phân nhóm giả thiếu oxy, được chia nhỏ thành phụ thuộc chu trình axit tricarboxylic [TCA] và phụ thuộc VHL/EPAS1) và cụm 2 (một phân nhóm tín hiệu kinase). Hầu hết các PHEO/PGL ở trẻ em sẽ thuộc cụm 1 (SDHx, VHL) thay vì cụm 2 (NF1, RET). PHEO/PGL phát sinh trong bối cảnh của VHL hoặc các hội chứng PGL di truyền (cụm 1) xảy ra ở độ tuổi trẻ hơn và thường là các khối u noradrenergic, sản xuất gần như độc quyền norepinephrine và normetanephrin. Các khối u adrenergic (tiết epinephrine và metanephrin, ngoài norepinephrine và normetanephrin) được thấy ở cụm 2 (MEN2, NF1) và các trường hợp lẻ tẻ.
Quản lý
Điều trị phẫu thuật
Cắt bỏ phẫu thuật là phương pháp điều trị chính cho PHEO/PGL. Sinh thiết trước phẫu thuật không được chỉ định và có khả năng nguy hiểm. Phương pháp được lựa chọn cho hầu hết các PHEO là cắt tuyến thượng thận qua nội soi, bằng đường qua phúc mạc hoặc sau phúc mạc. Nên xem xét mở bụng ở những bệnh nhân có PHEO lớn và/hoặc lo ngại về bệnh ác tính tiềm ẩn dựa trên biểu hiện lâm sàng, nền tảng di truyền, hoặc hình ảnh của khối u trên phim chụp. Trong trường hợp PHEO hai bên hoặc PHEO di truyền đã biết, nên thực hiện phẫu thuật bảo tồn vỏ thượng thận để giảm thiểu nguy cơ phải thay thế glucocorticoid và mineralocorticoid suốt đời và các rủi ro đi kèm của suy thượng thận nguyên phát. Bởi vì rất khó để bảo tồn một phần vỏ thượng thận có mạch máu đủ để ngăn chặn sự phụ thuộc vào corticosteroid mà không để lại một ít tủy thượng thận còn sót lại, nên có nguy cơ tái phát PHEO trong phần còn lại. Dữ liệu gần đây cho thấy tỷ lệ tái phát sau phẫu thuật bảo tồn vỏ là 10% hoặc thấp hơn. Phương pháp phẫu thuật để loại bỏ PGL phụ thuộc vào vị trí của khối u, nhưng trong các trường hợp bệnh ở bụng được chọn lọc cũng có thể được thực hiện qua nội soi. PGL ở đầu và cổ có thể được theo dõi chờ đợi, nếu nhỏ, hoặc được điều trị bằng phẫu thuật hoặc xạ trị.
Điều quan trọng là bác sĩ gây mê phải có kinh nghiệm trong việc quản lý PHEO/PGL trong mổ vì có thể xảy ra rối loạn nhịp tim và huyết áp có thể rất không ổn định. Cả thuốc hạ huyết áp tiêm tĩnh mạch (ví dụ, nitroprusside, phentolamine, esmolol, labetalol, v.v.) và thuốc co mạch (ví dụ, phenylephrine và norepinephrine) nên có sẵn để sử dụng trong mổ. Nguy cơ tăng huyết áp lớn nhất xảy ra trong quá trình khởi mê và thao tác khối u, trong khi hạ huyết áp có nhiều khả năng xảy ra sau khi thắt tĩnh mạch thượng thận, khi sự sụt giảm đột ngột nồng độ catecholamin dẫn đến giãn mạch. Sau phẫu thuật, bệnh nhân nên được theo dõi hai biến chứng chính là hạ huyết áp và hạ đường huyết. Ở những bệnh nhân đã được cắt tuyến thượng thận bảo tồn vỏ trong bối cảnh cắt PHEO hai bên, nên cung cấp glucocorticoid liều stress trong giai đoạn chu phẫu và thực hiện nghiệm pháp kích thích cosyntropin liều cao để xác định nhu cầu thay thế steroid thượng thận liên tục.
Chuẩn bị nội khoa cho phẫu thuật
Một khi chẩn đoán PHEO/PGL có chức năng đã được xác nhận, nên bắt đầu điều trị nội khoa để bình thường hóa huyết áp và giảm bớt các dấu hiệu và triệu chứng của thừa catecholamin (Bảng 15.3). Nếu phẫu thuật được lên kế hoạch, điều trị nội khoa nên được thực hiện ít nhất 10 đến 14 ngày trước phẫu thuật để giảm thiểu các biến chứng tiềm tàng có thể phát sinh từ các đợt tăng catecholamin cấp tính. Không có một quy trình chung nào cho việc quản lý nội khoa của PHEO/PGL trước phẫu thuật. Đối với người lớn, hướng dẫn của Hiệp hội Nội tiết khuyến nghị liệu pháp hàng đầu bằng thuốc chẹn thụ thể α-adrenergic và liệu pháp hàng thứ hai bằng thuốc chẹn kênh canxi. Dữ liệu ở trẻ em còn hạn chế, nhưng các nghiên cứu nhỏ cho thấy các phác đồ này là an toàn.
Bảng 15.3 Quản Lý Nội Khoa Trước Phẫu Thuật U Tế Bào Ưa Crôm/U Cận Hạch Thần Kinh Giao Cảm
| Nhóm Thuốc | Thuốc | Cơ Chế Tác Dụng | Liều Khởi Đầu ở Trẻ Em |
|---|---|---|---|
| Thuốc chẹn thụ thể α-adrenergic | Doxazosin | Đối kháng α1 | 0.5–1 mg mỗi ngày |
| Phenoxybenzamine | Đối kháng α1 và α2 | 0.2–0.25 mg/kg/ngày chia 2 lần (tối đa 10 mg 2 lần/ngày) | |
| Prazosin | Đối kháng α1 | 0.05–0.1 mg/kg/ngày chia 3 lần (tối đa 1 mg 3 lần/ngày) | |
| Thuốc chẹn thụ thể β-adrenergic | Atenolol | Đối kháng β1 | 0.5–1 mg/kg/ngày 1 lần hoặc chia 2 lần (tối đa 50 mg mỗi ngày) |
| Metoprolol | Đối kháng β1 | 1-2 mg/kg/ngày chia 2 lần (tối đa 50 mg 2 lần/ngày) | |
| Propranolol | Đối kháng β1 và β2 | 1–2 mg/kg/ngày chia 2 hoặc 3 lần (tối đa 80 mg mỗi ngày) | |
| Thuốc chẹn kênh canxi | Amlodipine | Chẹn kênh canxi | 0.1 mg/kg/ngày (< 6 tuổi); 2.5–5 mg mỗi ngày (tuổi ≥ 6) |
| Nifedipine (phóng thích kéo dài) | Chẹn kênh canxi | 0.25–0.5 mg/kg/ngày 1 lần hoặc chia 2 lần (tối đa 60 mg tổng liều hàng ngày) | |
| Thuốc ức chế tổng hợp catecholamin | Metyrosine | Ức chế tyrosine hydroxylase | 125–250 mg 4 lần/ngày |
BID, hai lần mỗi ngày; QID, bốn lần một ngày; TID, ba lần mỗi ngày.
Việc chẹn thụ thể α hiệu quả giúp cải thiện triệu chứng, hạ huyết áp, và làm giãn lòng mạch và tăng thể tích máu. Nhịp tim nhanh mà không có hạ huyết áp là một dấu hiệu của việc chẹn alpha tốt và nên được điều trị bằng cách bổ sung một thuốc chẹn thụ thể β-adrenergic thay vì giảm liều thuốc chẹn alpha. Không nên bắt đầu dùng thuốc chẹn beta khi chưa chẹn alpha tốt vì nguy cơ lý thuyết về tăng huyết áp phản xạ do hoạt động thụ thể α-adrenergic không được kiểm soát. Phenoxybenzamine là một chất đối kháng thụ thể α không cạnh tranh có thời gian bán hủy dài; do đó nó cũng có thể làm tăng nguy cơ hạ huyết áp sau phẫu thuật. Các thuốc chẹn thụ thể α1 chọn lọc, chẳng hạn như prazosin và doxazosin, cũng có thể được sử dụng. Mặc dù labetalol và carvedilol, các loại thuốc có cả hoạt tính đối kháng thụ thể α và β, có vẻ là những lựa chọn hấp dẫn để chẹn trước phẫu thuật, chúng không được khuyến nghị cho điều trị nội khoa chính vì khả năng chẹn thụ thể α-adrenergic thấp hơn so với hoạt tính đối kháng β của chúng. Hạ huyết áp tư thế có triệu chứng có thể được thấy khi bắt đầu điều trị nội khoa, đặc biệt với các khối u lớn có hoạt tính sinh hóa mạnh, vì vậy bắt buộc phải bắt đầu với liều thấp và tăng liều và/hoặc tần suất vài ngày một lần, cho đến khi huyết áp bình thường theo tuổi và chiều cao và bệnh nhân ít bị hạ huyết áp tư thế. Phenoxybenzamine chỉ được cung cấp dưới dạng một liều duy nhất (viên nang 10 mg) và do đó nó sẽ cần được dược phòng pha chế để cho phép sử dụng các liều thấp hơn cần thiết ở trẻ nhỏ. Phenoxybenzamine cũng là một loại thuốc đắt tiền, điều này làm cho các thuốc chẹn thụ thể α1 chọn lọc trở thành một lựa chọn hấp dẫn hơn trong nhiều trường hợp.
Metyrosine là một chất ức chế cạnh tranh của tyrosine hydroxylase, bước giới hạn tốc độ của quá trình sinh tổng hợp catecholamin (xem Hình 15.3) và nó cũng có thể được sử dụng như một phần của phác đồ chuẩn bị trước phẫu thuật, mặc dù dữ liệu ở trẻ em còn hạn chế. Tuy nhiên, không phải tất cả các trung tâm đều sử dụng metyrosine thường quy vì chi phí và các tác dụng phụ tiềm ẩn đáng kể (an thần, tiêu chảy, và các biểu hiện ngoại tháp) và lợi ích không rõ ràng trong hầu hết các trường hợp. Vài ngày trước phẫu thuật, nên bổ sung muối bằng đường uống (hoặc qua tăng cường khẩu phần ăn hoặc bằng viên natri clorua) để tăng thể tích máu nhằm giảm thiểu hạ huyết áp sau phẫu thuật. Một số trung tâm cũng thường xuyên cho bệnh nhân nhập viện để truyền dịch tĩnh mạch trước khi cắt PHEO/PGL, và điều này có thể được xem xét cho những trẻ có triệu chứng nặng với khối u lớn.
Tiên lượng và theo dõi
Tiên lượng dài hạn của hầu hết trẻ em được chẩn đoán mắc PHEO/PGL là rất tốt. Tuy nhiên, trong mọi trường hợp, đều có nguy cơ phát triển các khối u đồng bộ và/hoặc dị bộ ở cả hai tuyến thượng thận, cũng như ở các vị trí ngoài thượng thận. Thực tế, có tới 50% bệnh nhân được chẩn đoán mắc PHEO/PGL ở thời thơ ấu sẽ phát triển thêm một khối u nguyên phát trong đời (25% sau 9 năm và 50% sau 31 năm kể từ khi chẩn đoán), nhấn mạnh sự cần thiết của việc theo dõi suốt đời và tư vấn và xét nghiệm di truyền phù hợp.
Bệnh ác tính trong PHEO/PGL được định nghĩa là sự hiện diện của di căn ở một vị trí mà các tế bào ưa crôm và cận hạch không thường trú (chủ yếu là xương nhưng cũng có thể là hạch bạch huyết, gan, và/hoặc phổi). Thực tế, hướng dẫn gần đây nhất của Tổ chức Y tế Thế giới (WHO) đã loại bỏ thuật ngữ PHEO/PGL ác tính và thay bằng PHEO/PGL di căn. Không có một đặc điểm mô học hoặc hồ sơ hóa mô miễn dịch nào có thể độc lập dự đoán tiềm năng di căn trong một PHEO/PGL đã được cắt bỏ, nhưng các hệ thống chấm điểm mô bệnh học (điểm Phaeochromocytoma and the Adrenal Gland Scaled và hệ thống phân độ cho PHEO và PGL thượng thận) đã cố gắng tạo ra các dự đoán cho các đặc điểm thường được ghi nhận hơn trong các khối u di căn. Thật không may, các hệ thống chấm điểm này bị hạn chế bởi rất nhiều sự thay đổi giữa các quan sát viên và trong cùng một quan sát viên. Các đặc điểm đáng tin cậy nhất của nguy cơ tăng tiềm năng di căn bao gồm vị trí ngoài thượng thận, kích thước lớn hơn 5 cm, và đột biến dòng mầm SDHB. Nguy cơ cao nhất về ác tính và tử vong ở trẻ em là ở PGL giao cảm liên quan đến SDHB, chiếm phần lớn các khối u di căn.
Dựa trên dữ liệu từ một cơ quan đăng ký khối u của Anh, tỷ lệ mắc bệnh ác tính ở trẻ em được ước tính là 0,02 trên một triệu mỗi năm. Khoảng 9% đến 16% PHEO/PGL ở trẻ em trở thành di căn, mặc dù một số trung tâm chuyển tuyến báo cáo tỷ lệ ác tính cao hơn nhiều (lên đến 65%) ở PHEO/PGL trẻ em, có thể phản ánh sự sai lệch do chuyển tuyến. Mặt khác, thời gian tiềm ẩn giữa chẩn đoán và xác nhận bệnh di căn trung bình là 9 năm; do đó, tỷ lệ ác tính thực sự có thể cao hơn so với nhận định trước đây vì nó chỉ có thể được xác định trong quá trình theo dõi hệ thống dài hạn. Trẻ em mắc bệnh di căn thường có diễn biến lâm sàng âm thầm hơn với thời gian sống sót toàn bộ trung bình hơn 6 năm sau khi chẩn đoán bệnh di căn.
Bởi vì PHEO/PGL có thể có diễn biến lâm sàng không thể đoán trước và bởi vì trẻ em có nguy cơ phát triển các khối u nguyên phát dị bộ, bệnh di căn muộn, và tái phát tại chỗ, việc theo dõi dài hạn bằng tầm soát sinh hóa và các nghiên cứu hình ảnh định kỳ là cần thiết cho tất cả trẻ em mắc PHEO/PGL. Đối với trẻ em không triệu chứng có một đột biến di truyền được xác định làm tăng nguy cơ phát triển PHEO/PGL, nên tầm soát sinh hóa hàng năm, với độ tuổi bắt đầu tầm soát được xác định bởi đột biến gen cụ thể (xem Bảng 15.1). Hơn nữa, nên thực hiện hình ảnh cắt lớp ngang định kỳ, thường là MRI do không có phơi nhiễm bức xạ, để theo dõi các bệnh nhân có nguy cơ tái phát hoặc bệnh ác tính cao (ví dụ, PGL ở bụng) hoặc có nguy cơ phát triển một PHEO/PGL có thể không được xác định chỉ bằng xét nghiệm sinh hóa (ví dụ, các hội chứng PGL-PHEO di truyền).
Ung thư biểu mô tuyến giáp thể tủy
Ung thư biểu mô tuyến giáp thể tủy (MTC) là một khối u thần kinh nội tiết (NET) ác tính phát sinh từ các tế bào C cạnh nang sản xuất calcitonin của tuyến giáp. Ở trẻ em, MTC chiếm 5% hoặc ít hơn các khối u ác tính của tuyến giáp và có tỷ lệ mắc hàng năm là 0,3 ca/triệu/năm; đây là loại ung thư tuyến giáp phổ biến nhất ở bệnh nhân dưới 5 tuổi. Trong dân số nhi khoa, MTC có tỷ lệ nữ:nam khá tương đương, không giống như các ung thư biểu mô tuyến giáp biệt hóa, vốn thường gặp ở bé gái hơn bé trai. MTC ở trẻ em hầu như luôn là kết quả của một đột biến hoạt hóa di truyền hoặc mới (de novo) trong tiền-oncogen rearranged during transfection (RET) và được chẩn đoán trong bối cảnh của MEN2A hoặc MEN2B, tùy thuộc vào đột biến cụ thể. Do mối liên quan chặt chẽ của MTC với một biến thể axit deoxyribonucleic (DNA) RET dòng mầm, xét nghiệm di truyền được khuyến nghị trong mọi trường hợp. Mặc dù các khối u không di truyền lẻ tẻ chiếm 70% đến 75% các trường hợp MTC ở người lớn, những khối u như vậy hiếm gặp ở trẻ em. MTC lẻ tẻ chủ yếu liên quan đến các đột biến soma (không phải dòng mầm) ở RET (cụ thể là đột biến p.M918T) và RAS.
RET là một thành viên của siêu họ cadherin và mã hóa cho một receptor tyrosine kinase có một miền liên kết ngoại bào và một miền tyrosine kinase nội bào (Hình 15.5). Các phối tử nội sinh (kích hoạt RET thông qua một đồng thụ thể liên kết phối tử có ái lực cao, GFRα) là thành viên của họ yếu tố dinh dưỡng thần kinh có nguồn gốc từ tế bào đệm (GDNF), tham gia vào việc điều hòa sự phát triển của mô thần kinh. Do đó, protein RET đóng một vai trò quan trọng trong sự phát triển của các tế bào có nguồn gốc từ mào thần kinh, hệ niệu dục, và hệ thần kinh trung ương và ngoại biên, đặc biệt là hệ thần kinh ruột. Trong MEN2, sự kích hoạt cấu thành độc lập với phối tử của receptor tyrosine kinase RET, dưới dạng dimer (do đột biến ở miền giàu cysteine ngoại bào; MEN2A) hoặc monomer (do đột biến ở miền tyrosine kinase nội bào; MEN2B), kích thích nhiều con đường tín hiệu xuôi dòng thúc đẩy sự tăng trưởng, tăng sinh, tồn tại và biệt hóa của tế bào.
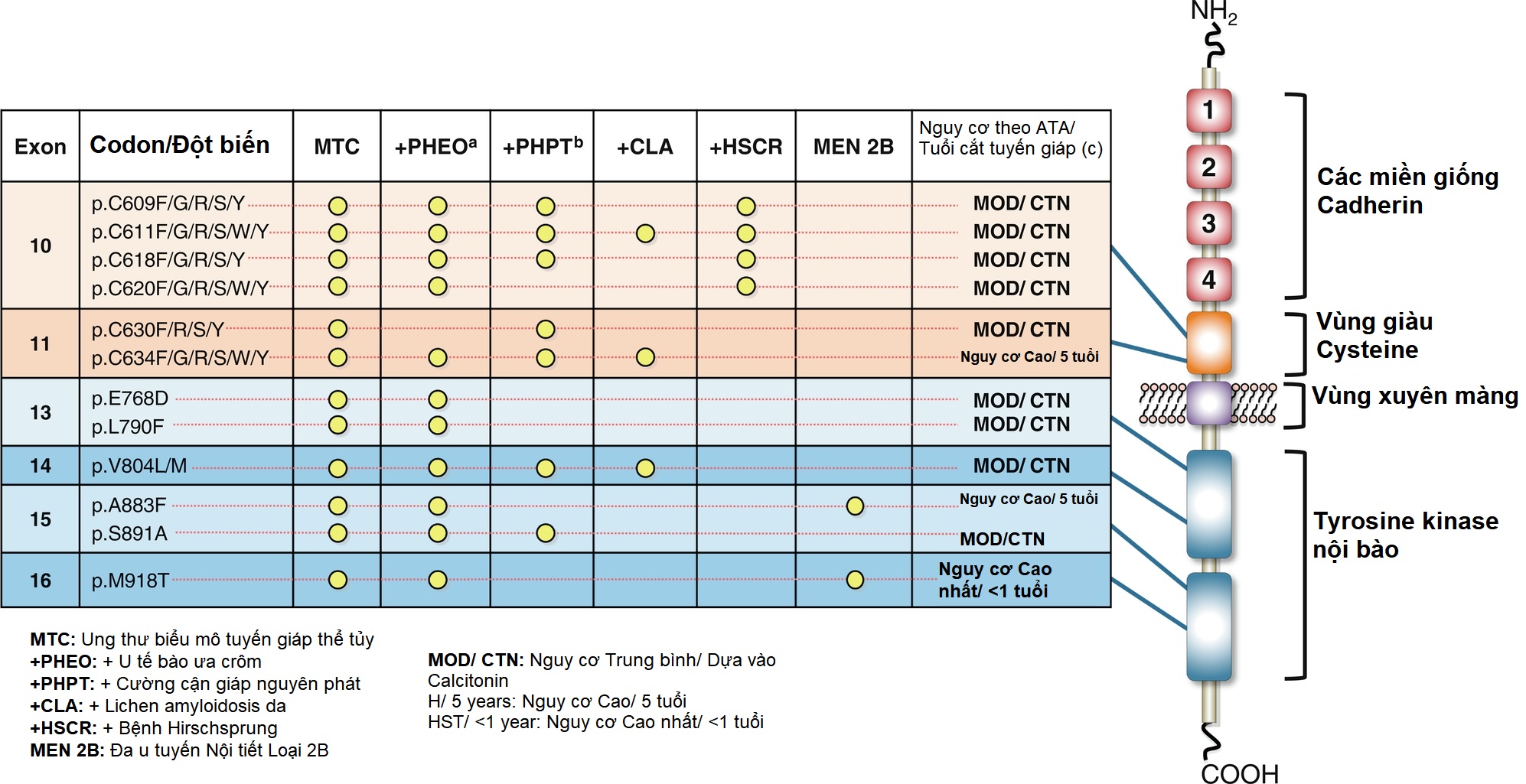
Hình 15.5 Receptor tyrosine kinase RET, các codon RET thường bị đột biến, các kiểu hình MEN2 liên quan, và phân tầng nguy cơ và khuyến nghị của ATA năm 2015 về thời điểm cắt tuyến giáp và tầm soát định kỳ các biểu hiện khác của MEN2.
ATA, Hiệp hội Tuyến giáp Hoa Kỳ; CTN, nồng độ calcitonin huyết thanh cơ bản; CLA, lichen amyloidosis da; H, nguy cơ cao theo ATA; HSCR, bệnh Hirschsprung; HST, nguy cơ cao nhất theo ATA; MEN2, Đa u tuyến Nội tiết Loại 2; MOD, nguy cơ trung bình theo ATA; MTC, ung thư biểu mô tuyến giáp thể tủy; PHEO, u tế bào ưa crôm; PHPT, cường cận giáp nguyên phát. a Tầm soát PHEO hàng năm được khuyến nghị ở tuổi 11 đối với các đột biến ở codon RET 634, 883, và 918 và ở tuổi 16 đối với các codon RET 609, 611, 618, 620, 768, 790, 804, và 891. Bệnh nhân có các đột biến chưa được biết là có liên quan đến PHEO có thể được tầm soát định kỳ. b Tầm soát PHPT hàng năm được khuyến nghị ở tuổi 11 đối với các đột biến ở codon RET 634 và ở tuổi 16 đối với các codon RET 609, 611, 618, 630, 804, và 891. Bệnh nhân có các đột biến chưa được biết là có liên quan đến PHPT có thể được tầm soát định kỳ. c Tuổi cắt tuyến giáp là độ tuổi mà phẫu thuật nên được xem xét cho những người mang gen RET tiền triệu chứng. Tuổi xét nghiệm sự hiện diện của đột biến RET nên được tiến hành ở độ tuổi sớm hơn sau khi được tư vấn di truyền thích hợp và dựa trên mong muốn của gia đình. CTN chỉ ra rằng thời điểm cắt tuyến giáp có thể được xác định bởi nồng độ calcitonin huyết thanh cơ bản, ngoài sự ưu tiên của cha mẹ/người giám hộ sau khi có ý kiến đa ngành. Để có danh sách toàn diện hơn về các đột biến RET, người đọc được giới thiệu đến các tài liệu khác. (Sửa đổi từ Waguespack, S.G., Rich, T.A., Perrier, N.D., Jimenez, C., Cote, G.J. (2011). Management of medullary thyroid carcinoma and MEN2 syndromes in childhood. Nat Rev Endocrinol, 7(10), 596–607.)
MTC di truyền thường đa ổ, hai bên, và nằm ở vùng giữa đến trên của các thùy tuyến giáp (Hình 15.6), một khu vực có nồng độ tế bào C cao nhất. Khám nghiệm vi thể và nhuộm calcitonin của tuyến giáp thường sẽ xác định được tăng sản tế bào C, giai đoạn ban đầu trong một chuỗi biến đổi ung thư dẫn đến sự phát triển của MTC vi thể không xâm lấn và cuối cùng là di căn hạch bạch huyết và di căn xa do ung thư biểu mô xâm lấn rõ ràng (xem Hình 15.6). Bệnh nhân mắc MTC có tính gia đình có sự tiến triển bệnh ác tính liên quan đến tuổi, với di căn hạch và di căn xa thường xảy ra nhiều năm sau khi bắt đầu tăng sản tế bào C. Các khoang hạch cổ và trung thất là những vị trí di căn phổ biến nhất, trong khi các vị trí xa cho sự lan rộng của MTC bao gồm phổi, gan, và xương/tủy xương.
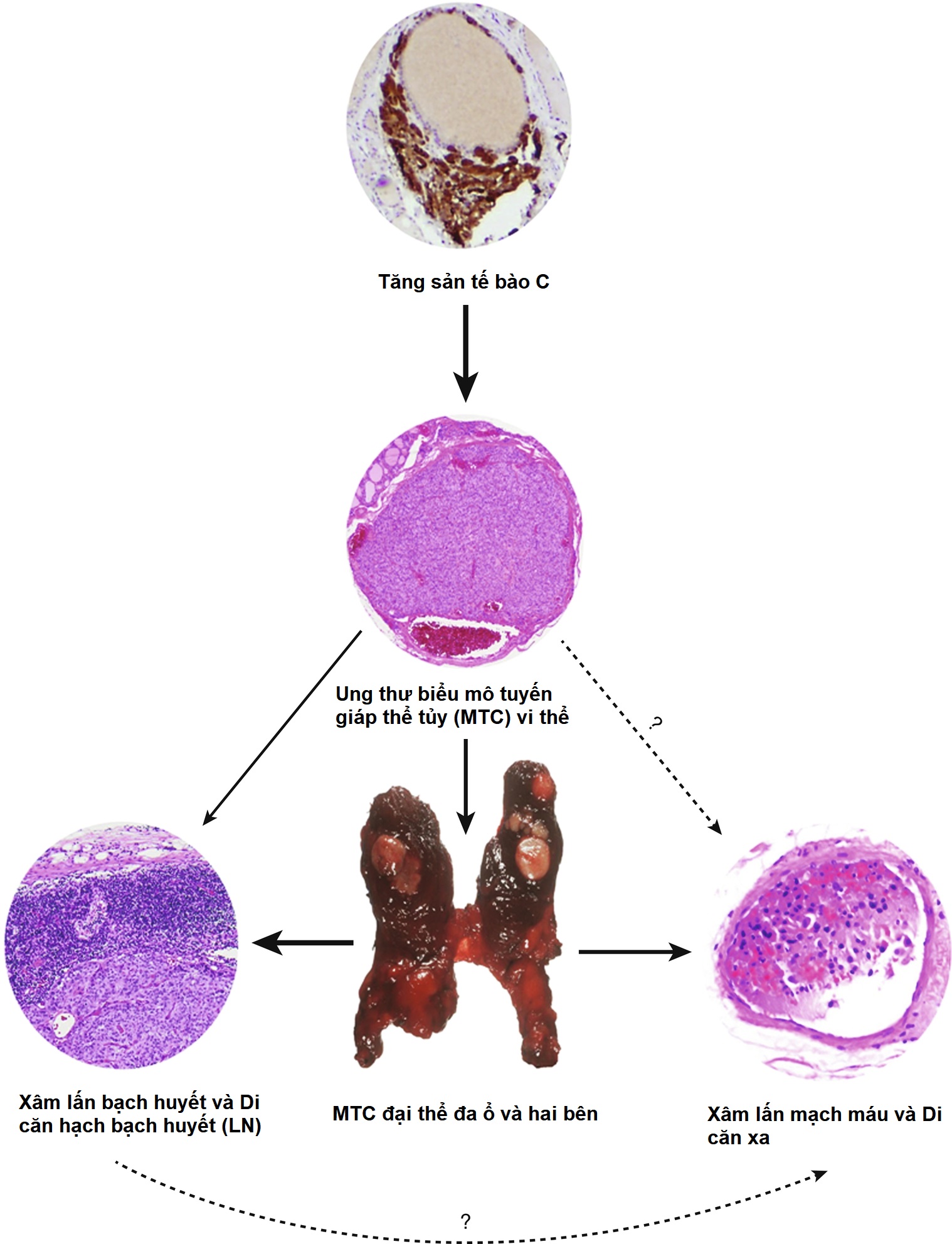
Hình 15.6 Một đột biến RET dòng mầm gây ra tăng sản tế bào C, giai đoạn ban đầu trong một chuỗi biến đổi ung thư cuối cùng dẫn đến sự phát triển của MTC vi thể không xâm lấn và có thể là di căn hạch và di căn xa do ung thư biểu mô xâm lấn rõ ràng.
Độ đậm của các mũi tên biểu thị xác suất giả định của sự kiện xảy ra ở bệnh nhân nhi điển hình có đột biến codon 634 của RET. LN, hạch bạch huyết; MTC, ung thư biểu mô tuyến giáp thể tủy. (Nguồn: Waguespack, S.G., Rich, T.A., Perrier, N.D., Jimenez, C., Cote, G.J. (2011). Management of medullary thyroid carcinoma and MEN2 syndromes in childhood. Nat Rev Endocrinol, 7(10), 596–607.)
Biểu hiện lâm sàng
Trẻ em mắc MTC lẻ tẻ có biểu hiện tương tự như các khối u ác tính khác của tuyến giáp, thường là với một nốt tuyến giáp sờ thấy được và/hoặc hạch cổ; tiêu chảy cũng hiếm khi có thể là biểu hiện lâm sàng ban đầu (chủ yếu do nồng độ calcitonin cực cao). Mặt khác, trẻ em mắc bệnh di truyền do đột biến RET dòng mầm ít khi có biểu hiện bệnh lâm sàng rõ ràng, ngoại trừ MTC liên quan đến MEN2B, một chẩn đoán vẫn thường bị trì hoãn. Do đó, trong kỷ nguyên hiện tại, biểu hiện chủ yếu của MTC ở trẻ em là việc xác định tiền triệu chứng một đột biến RET và cuối cùng là xác định MTC vi thể sau khi cắt tuyến giáp sớm.
Đánh giá và Quản lý
Có một số hướng dẫn và bài tổng quan có sẵn để hỗ trợ bác sĩ lâm sàng trong việc đánh giá và quản lý cụ thể trẻ em bị nghi ngờ hoặc đã được chứng minh mắc MTC. Tương tự như các khối u ác tính khác của tuyến giáp ở trẻ em, phẫu thuật là nền tảng của điều trị và là cơ hội duy nhất để chữa khỏi MTC. Do tỷ lệ biến chứng phẫu thuật cao hơn ở trẻ em và các khía cạnh độc đáo của việc chăm sóc y tế và phẫu thuật cho bệnh nhân MTC, việc điều trị nên được tiến hành tại các trung tâm chăm sóc cấp ba với chuyên môn đa ngành, có khối lượng bệnh nhân lớn. Ngoài quyết định về thời điểm can thiệp, bác sĩ phẫu thuật tuyến giáp phải kết hợp kiến thức về kiểu gen RET của bệnh nhân và dữ liệu lâm sàng vào quá trình ra quyết định để xác định phương pháp phẫu thuật tối ưu. Việc loại bỏ cẩn thận và an toàn tất cả các mô tuyến giáp, bao gồm cả bao sau, là mục tiêu của một cuộc phẫu thuật cắt tuyến giáp dự phòng/sớm cho bệnh nhân MEN2 không có MTC lâm sàng. Nạo vét hạch khoang trung tâm (mức VI) thường quy không được thực hiện trong bối cảnh một cuộc phẫu thuật hoàn toàn dự phòng vì di căn hạch cực kỳ hiếm trong bối cảnh đó. Tuy nhiên, nếu phẫu thuật dành cho một khối u di truyền có cấu trúc rõ ràng (đặc biệt nếu nồng độ calcitonin vượt quá 40 pg/mL) hoặc cho MTC lẻ tẻ, nên thực hiện cắt toàn bộ tuyến giáp và nạo vét hạch cổ trung tâm đồng thời. Nạo vét các khoang hạch cổ bên (mức IIA–V) thường chỉ được thực hiện trong các trường hợp có bằng chứng lâm sàng về sự liên quan của hạch cổ bên, mặc dù một số trung tâm dựa trên quyết định này vào nồng độ calcitonin trước phẫu thuật.
May mắn thay, MTC đe dọa tính mạng hiếm khi xảy ra ở trẻ em và chủ yếu chỉ trong bối cảnh lâm sàng của MEN2B. Trong MTC tiến triển, tỷ lệ đáp ứng với hóa trị độc tế bào truyền thống (dacarbazine, vincristine, và cyclophosphamide) là thấp. Gần đây hơn, hai liệu pháp phân tử nhắm mục tiêu (vandetanib và cabozantinib) ức chế RET và các receptor tyrosine kinase khác đã được phê duyệt ở người lớn để điều trị MTC di căn. Các nghiên cứu sử dụng các loại thuốc này ở trẻ em mắc MEN2B đã cho thấy hiệu quả và độ an toàn tương đương. Các liệu pháp mới ức chế chọn lọc kinase RET trong các khối u có đột biến hoặc dung hợp RET đang cho thấy hứa hẹn trong các thử nghiệm lâm sàng bắt đầu vào năm 2017.
Tiên lượng
Nhìn chung, tiên lượng của MTC ở trẻ em là tốt, với tỷ lệ sống sót sau 5 và 15 năm lần lượt là 97% và 89%. Nói chung, tình trạng hạch dương tính và giai đoạn cao hơn tại thời điểm chẩn đoán dự đoán khả năng sống không bệnh thấp hơn và tỷ lệ tử vong cao hơn trong MTC. Khi giai đoạn khối u tăng lên, nguy cơ di căn tại chỗ và di căn xa đều tăng, mặc dù di căn hạch vẫn có thể xảy ra khi khối u có kích thước nhỏ hơn 1 cm. Bệnh nhân mắc MEN2B có tiên lượng đặc biệt dè dặt, với một phần năm bệnh nhân tử vong ở độ tuổi trung bình là 25 tuổi (khoảng < 1–59 tuổi), mặc dù không phải lúc nào cũng do MTC.
Nhiều trẻ em có biểu hiện MTC lâm sàng đã có bệnh di căn tại thời điểm chẩn đoán. Do đó, phần lớn các trường hợp MTC ở trẻ em không được chẩn đoán trước khi xảy ra di căn hạch là những bệnh ung thư không thể chữa khỏi nhưng diễn biến âm thầm. Mức độ xâm lấn của diễn biến lâm sàng có thể được dự đoán bởi đột biến RET cụ thể (xem phần MEN2 sau), biểu hiện lâm sàng của trẻ, và bằng cách sử dụng các chất chỉ điểm khối u huyết thanh, calcitonin và kháng nguyên carcinoembryonic (CEA). Sự mất biểu hiện calcitonin, nồng độ CEA không tương xứng với calcitonin, và thời gian nhân đôi CEA và/hoặc calcitonin nhanh chóng đều là những điềm báo của một diễn biến bệnh xâm lấn.
Các hội chứng tân sinh nội tiết di truyền
Phức hợp Carney
Lần đầu tiên được mô tả vào năm 1985, phức hợp Carney (CNC) là một hội chứng đốm sắc tố có tính gia đình, không đồng nhất về mặt di truyền và lâm sàng với phương thức di truyền trội trên nhiễm sắc thể thường. Nó được đặc trưng bởi các nốt ruồi xanh và/hoặc sắc tố da dạng đốm ảnh hưởng đến môi, kết mạc, và các bề mặt niêm mạc khác (Hình 15.7); u nhầy ở vú, tim, và da; các khối u và/hoặc tăng hoạt động nội tiết, kinh điển là bệnh vỏ thượng thận dạng nốt tăng sắc tố nguyên phát (PPNAD) và tăng tiết hormone tăng trưởng/prolactin của tuyến yên; và u schwann tế bào hắc tố thể cát, cùng các biểu hiện lâm sàng khác. Hai hoặc nhiều locus di truyền riêng biệt có liên quan đến CNC: gen PRKAR1A trên nhiễm sắc thể 17q24.2 và một gen chưa xác định trên nhiễm sắc thể 2p16. PRKAR1A mã hóa cho tiểu đơn vị RI-α của protein kinase A, chất trung gian chính của tín hiệu cyclic adenosine monophosphate (cAMP) nội bào. Hầu hết các đột biến PRKAR1A là các đột biến điểm bất hoạt, và khoảng 30% bệnh nhân CNC mắc bệnh mới (de novo). Ở những bệnh nhân mắc CNC, tỷ lệ phát hiện đột biến PRKAR1A tổng thể là 62%.
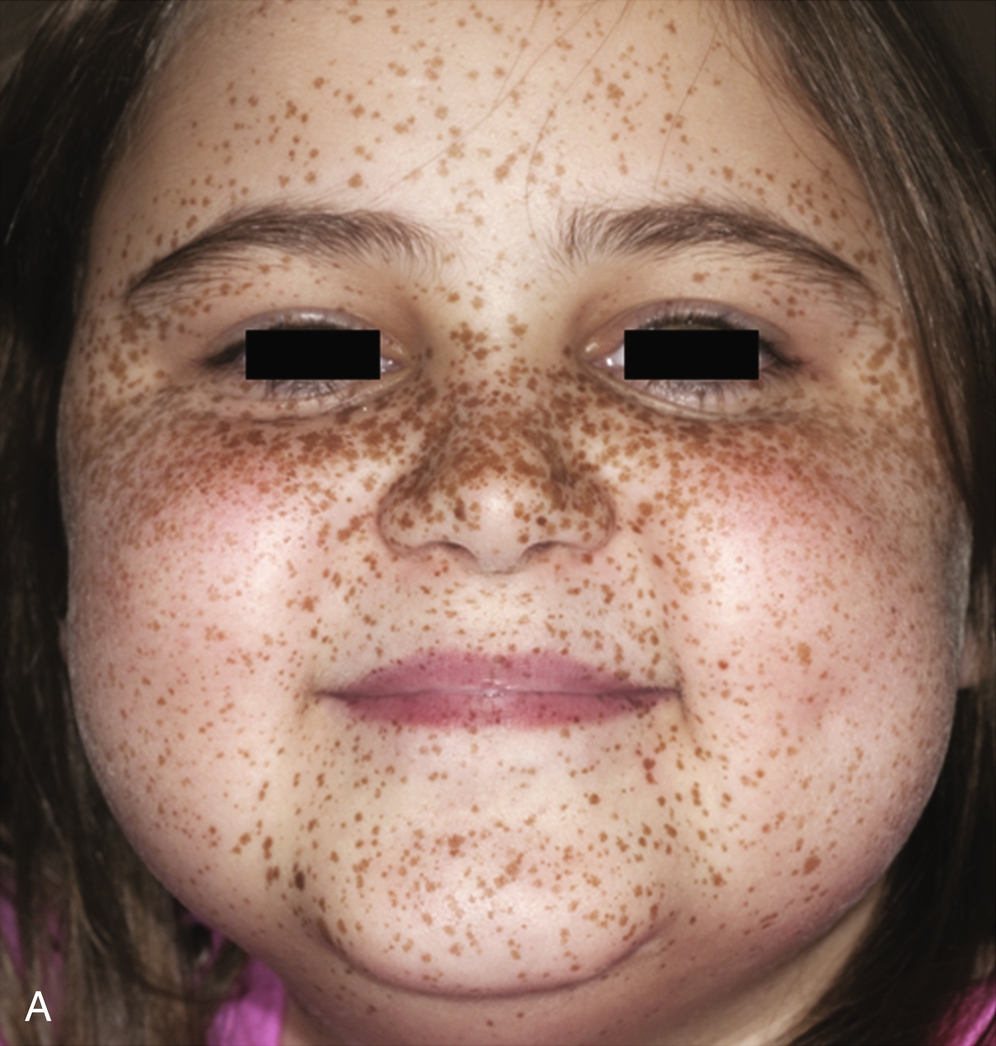
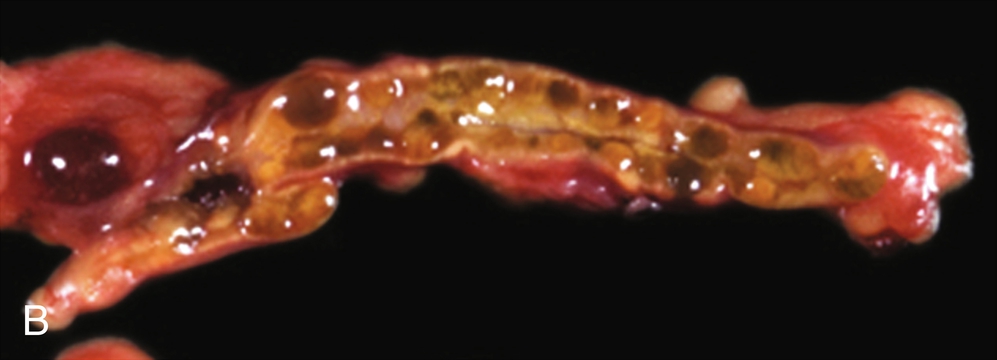
Hình 15.7 Phức hợp Carney và bệnh vỏ thượng thận dạng nốt tăng sắc tố nguyên phát (PPNAD). (A) Một bệnh nhi nữ 10 tuổi mắc hội chứng Cushing độc lập với hormone vỏ thượng thận (ACTH) và phức hợp Carney cho thấy khuôn mặt tròn, đỏ bừng, và các đốm sắc tố kinh điển trên mặt. (B) Mẫu bệnh phẩm đại thể của tuyến thượng thận phải ở một bệnh nhân khác mắc PPNAD và phức hợp Carney.
Kiểu hình nội tiết chính của CNC là PPNAD, xảy ra đơn độc hoặc cùng với các biểu hiện khác của CNC, trong 60% các trường hợp; ung thư biểu mô vỏ thượng thận cũng hiếm khi được báo cáo. PPNAD là một dạng hiếm của tăng cortisol độc lập với ACTH, về mặt bệnh học liên quan đến nhiều nốt nhỏ (< 1 cm) màu đen hoặc nâu (chứa lipofuscin) trong một vỏ thượng thận teo đét (xem Hình 15.7). Tuổi khởi phát trung bình là 34 tuổi (với khoảng 20% các trường hợp xảy ra ở trẻ em), và có sự chiếm ưu thế ở nữ sau tuổi dậy thì. Tình trạng tăng cortisol thường khởi phát âm thầm, và một phát hiện đặc trưng trong hội chứng Cushing liên quan đến PPNAD là sự gia tăng nghịch lý trong bài tiết glucocorticoid vào ngày thứ hai của việc sử dụng dexamethasone liều cao trong nghiệm pháp Liddle. Hình ảnh tuyến thượng thận có giá trị hạn chế để xác nhận PPNAD vì các tổn thương nhỏ, nhưng nó có thể gợi ý bệnh vi nốt lan tỏa trên CT có cản quang. Điều trị PPNAD có triệu chứng thường là cắt bỏ hai bên tuyến thượng thận, mặc dù điều này có thể không cần thiết ở mọi bệnh nhân.
Tăng tiết hormone tăng trưởng (gây ra bệnh khổng lồ hoặc bệnh to đầu chi, tùy thuộc vào tuổi khởi phát) là kiểu hình tuyến yên chính của CNC, xảy ra ở 10% đến 12% bệnh nhân CNC. Về mặt bệnh học, điều này có thể liên quan đến một u tuyến hoặc tăng sản. Tăng prolactin máu đồng thời là phổ biến và cũng do tăng sản tiềm ẩn hoặc u tuyến rõ ràng trong các trường hợp hiếm gặp. Các bất thường tinh vi trong bài tiết hormone tăng trưởng và prolactin có thể được xác định ở tới 75% bệnh nhân mắc CNC. U tuyến corticotroph cũng đã được mô tả trong các trường hợp hiếm gặp. U tân sinh tuyến giáp xảy ra ở 25% bệnh nhân CNC với hầu hết là các nốt lành tính, mặc dù ung thư biểu mô tuyến giáp biệt hóa (cả ung thư biểu mô nhú và nang) đã được xác định trong 2,5% các trường hợp. Sự liên quan của tuyến giáp xảy ra ở độ tuổi trung bình khoảng 33 tuổi (khoảng 12–57 tuổi) và có thể liên quan đến nhiễm độc giáp. Nam giới mắc CNC có nguy cơ bị u tinh hoàn, chủ yếu là u tế bào Sertoli vôi hóa tế bào lớn hai bên (LCCSCT), được xác định lâm sàng ở 33% đến 41% bệnh nhân. Tương tự như hội chứng Peutz-Jeghers (xem sau), nữ hóa tuyến vú và/hoặc tinh hoàn to có thể là một biểu hiện lâm sàng của LCCSCT, có thể là ác tính trong những trường hợp rất hiếm.
Một khi CNC được chẩn đoán lâm sàng hoặc một đứa trẻ không triệu chứng được phát hiện mang đột biến PRKAR1A dòng mầm, việc theo dõi định kỳ sự phát triển của các khối u và rối loạn nội tiết đặc trưng của hội chứng có thể được thực hiện bằng cách sử dụng các khuyến nghị đã công bố để tầm soát.
U tuyến yên đơn độc có tính gia đình
U tuyến yên di truyền có thể xảy ra như một thực thể riêng biệt trong các gia đình không mắc MEN1, và thuật ngữ u tuyến yên đơn độc có tính gia đình (FIPA) lần đầu tiên được đề xuất vào năm 2006. FIPA được định nghĩa bởi sự xuất hiện của u tuyến yên ở hai hoặc nhiều thành viên trong cùng một gia đình mà không có các biểu hiện lâm sàng khác gợi ý MEN1 hoặc CNC. Với tất cả các phân nhóm hormone được đại diện, các khối u tuyến yên xảy ra trong các gia đình FIPA có biểu hiện tương tự như các u tuyến yên lẻ tẻ, và có thể có sự không đồng nhất lâm sàng đáng kể trong cùng một gia đình.
U tuyến yên đơn độc có tính gia đình liên quan đến AIP
Trong một nhóm nhỏ các gia đình FIPA, rối loạn cuối cùng được xác định là do các đột biến dòng mầm trong gen protein tương tác với thụ thể aryl hydrocarbon (AIP) (nhiễm sắc thể 11q13.2). Đột biến ở AIP chỉ chiếm 15% đến 30% các trường hợp FIPA và độ xâm nhập của bệnh không hoàn toàn, ước tính dưới 30%. Những cá nhân FIPA có đột biến AIP có các khối u lớn hơn (chủ yếu là macroadenoma) và được chẩn đoán ở độ tuổi trẻ hơn nhiều so với các bệnh nhân âm tính với AIP. Tuổi chẩn đoán trung bình là 16 đến 23 tuổi và 50% đến 60% các trường hợp biểu hiện ở trẻ em hoặc thanh thiếu niên. Phần lớn các u tuyến đột biến AIP là u tuyến somatotroph hoặc mammosomatotroph, thường biểu hiện bằng bệnh khổng lồ ở tới 30% các trường hợp (đặc biệt là nam giới) và kháng trị hơn với các liệu pháp điển hình được kê đơn cho bệnh khổng lồ/bệnh to đầu chi. Ở những bệnh nhân dưới 18 tuổi có biểu hiện u tuyến yên macroadenoma dường như lẻ tẻ, đặc biệt là u tuyến somatotroph, 20% có thể mang đột biến AIP dòng mầm. Trong một loạt lớn các bệnh nhân nhi mắc macroprolactinoma, 9% các trường hợp được xét nghiệm được phát hiện có đột biến AIP, và những trẻ này đáp ứng tốt tương tự với chất chủ vận dopamine như các trường hợp lẻ tẻ.
Do việc mô tả đặc điểm AIP-liên quan FIPA tương đối gần đây, không có hướng dẫn đồng thuận dựa trên bằng chứng về thời điểm xét nghiệm di truyền cho các đột biến AIP trong các trường hợp u tuyến yên lẻ tẻ hoặc cho các thành viên có nguy cơ của một gia đình bị ảnh hưởng có đột biến AIP. Các khuyến nghị về tầm soát lâm sàng định kỳ ở những người mang đột biến AIP không triệu chứng cũng chưa được thiết lập tốt, nhưng một số chuyên gia đã đề xuất một phương pháp chăm sóc bao gồm giáo dục bệnh nhân về các triệu chứng của bệnh tuyến yên và thực hiện đánh giá nội tiết và tăng trưởng hàng năm. Do độ xâm nhập lâm sàng thấp của các đột biến AIP và khả năng cao của các khối u tuyến yên chức năng, vốn rất có thể sẽ được xác định thông qua xét nghiệm sinh hóa và xem xét biểu đồ tăng trưởng ở trẻ em, không rõ liệu việc tầm soát MRI định kỳ có được chỉ định hay không.
Các hội chứng u cận hạch-u tế bào ưa crôm di truyền
Các hội chứng u cận hạch-u tế bào ưa crôm di truyền (HPS) được đặc trưng chủ yếu bởi sự phát triển của PGL đối giao cảm và giao cảm và PHEO. Năm hội chứng HPS di truyền trội trên nhiễm sắc thể thường (PGL1-5) đã được mô tả (xem Bảng 15.1) và là do các đột biến dòng mầm trong các gen riêng biệt mã hóa cho bất kỳ tiểu đơn vị nào của gen enzyme succinate dehydrogenase (SDHA, SDHB, SDHC, và SDHD) hoặc một protein cần thiết cho quá trình flavin hóa của SDHA (SDHAF2): PGL1(SDHD), PGL2 (SDHAF2), PGL3 (SDHC), PGL4 (SDHB), và PGL5 (SDHA).
Các gen SDHx bao gồm các tiểu đơn vị cho phức hợp II của chuỗi hô hấp ty thể, khi bị đột biến, dẫn đến sự ổn định của yếu tố cảm ứng thiếu oxy 1 với trạng thái giả thiếu oxy sau đó. Kiểu hình lâm sàng có phần khác nhau giữa năm hội chứng và vẫn chưa được mô tả rõ ràng trong một số trường hợp, chẳng hạn như PGL5 (SDHA). Ví dụ, các đột biến ở SDHD chủ yếu gây ra PGL đầu và cổ đối giao cảm lành tính (còn được gọi là u cuộn cảnh hoặc u thể cảnh), trong khi các đột biến ở SDHB có liên quan nhiều hơn đến PGL bụng và nguy cơ ác tính cao hơn (xem Bảng 15.1 và Hình 15.2). Trong mọi trường hợp, độ xâm nhập của kiểu hình lâm sàng dưới 100% và tăng theo tuổi. Các đột biến ở SDHD (và cả SDHAF2) thể hiện hiệu ứng nguồn gốc từ cha mẹ, với bệnh chỉ xảy ra khi đột biến được di truyền từ người cha, mặc dù các báo cáo gần đây về sự phát triển PHEO/PGL ở những cá nhân có di truyền đột biến SDHD từ mẹ đã được công bố. Ngoài PGL và PHEO, các khối u liên quan đến SDHx bao gồm u mô đệm đường tiêu hóa (GIST), một phát hiện cũng đã được gọi là hội chứng Carney-Stratakis hoặc cặp đôi Carney. Các khối u tân sinh không phải cận hạch khác được cho là do đột biến SDHx bao gồm ung thư biểu mô tế bào thận sáng, u tuyến yên, và các NET khác. Sự kết hợp của PHEO/PGL và u tuyến yên xảy ra trong bối cảnh đột biến SDHx dòng mầm đã được đặt tên là “3PAs”. Chương trình tầm soát tối ưu cho trẻ em mắc HPS (xem Bảng 15.1) vẫn chưa được xác định rõ ràng nhưng thường bao gồm khám sức khỏe/theo dõi huyết áp hàng năm, đo metanephrin huyết tương hoặc nước tiểu hàng năm, và MRI toàn thân hai năm một lần. Tương tự như tầm soát tiền triệu chứng trong các hội chứng khuynh hướng khối u khác, MRI là phương thức hình ảnh được ưu tiên trong HPS.
Hội chứng cường cận giáp-u xương hàm
Hội chứng cường cận giáp-u xương hàm (HPT-JT) là một rối loạn di truyền có độ xâm nhập không hoàn toàn, được đặc trưng bởi u tân sinh cận giáp/cường cận giáp nguyên phát (PHPT) liên quan đến u xơ hóa xương ở hàm trên và/hoặc hàm dưới. Lần đầu tiên được báo cáo vào năm 2002, CDC73 (nhiễm sắc thể 1q31.2; trước đây được gọi là HRPT2) là gen duy nhất được biết đến có đột biến gây ra hội chứng HPT-JT, và các đột biến CDC73 được xác định ở khoảng 60% các gia đình mắc hội chứng HPT-JT. PHPT do một u tuyến cận giáp lành tính đơn độc là biểu hiện lâm sàng phổ biến nhất, nhưng ung thư biểu mô cận giáp là phổ biến, chiếm 10% đến 15% các trường hợp. Độ tuổi chẩn đoán PHPT trẻ nhất là 7 tuổi. U xơ hóa xương, còn được gọi là u xơ hóa xi măng, có thể xâm lấn tại chỗ và xảy ra ở 30% đến 40% các cá nhân mắc hội chứng HPT-JT; chúng chủ yếu được điều trị bằng phẫu thuật cắt bỏ. Các biểu hiện lâm sàng khác của hội chứng HPT-JT bao gồm các tổn thương thận (nang, hamartoma, và nephroblastoma [u Wilms]) và các khối u tử cung. Theo dõi lâm sàng định kỳ những cá nhân không triệu chứng có đột biến CDC73 được khuyến nghị (bắt đầu từ 5–10 tuổi) và bao gồm các xét nghiệm sinh hóa hàng năm để đánh giá PHPT và hình ảnh (chụp X-quang toàn cảnh răng và siêu âm thận) mỗi 5 năm; các thanh thiếu niên nữ trong độ tuổi sinh sản nên được siêu âm vùng chậu nếu có bất kỳ chảy máu tử cung bất thường nào.
Đa u tuyến Nội tiết Loại 1
MEN1 là một hội chứng khối u di truyền được đặc trưng bởi tăng sản tuyến và các khối u tân sinh lành tính hoặc ác tính xảy ra ở hai hoặc nhiều tuyến nội tiết, kinh điển là tuyến cận giáp, tuyến yên, và tụy nội tiết (Bảng 15.4). Các khối u MEN1 có thể hoạt động nội tiết hoặc không chức năng và tính đa ổ là phổ biến, ngoại trừ các khối u tuyến yên. Bệnh nhân cũng có nguy cơ phát triển các khối u vỏ thượng thận (ACT), PHEO, NET ngoài ổ bụng (tuyến ức và phế quản-phổi), các khối u lành tính của da (u xơ mạch, u collagen, u mỡ), các khối u hệ thần kinh trung ương (TKTW) (u màng não và u màng não thất), và/hoặc u cơ trơn tử cung. Nguy cơ ung thư vú ở phụ nữ mắc MEN1 có thể tăng cao.
Bảng 15.4 Các Biểu Hiện Lâm Sàng Sớm Nhất và Quy Trình Tầm Soát Sinh Hóa/Hình Ảnh Được Đề Xuất cho Các Khối U Thần Kinh Nội Tiết và Vỏ Thượng Thận ở Trẻ Em Mắc Đa U Tuyến Nội Tiết Loại 1 a
| Khối U | Tuổi Chẩn Đoán Sớm Nhất (Năm) và Tham Khảo | Tuổi Bắt Đầu Tầm Soát (Năm) | Dấu Hiệu/Triệu Chứng Lâm Sàng của Bệnh | Các Xét Nghiệm Sinh Hóa b và Hình Ảnh Được Sử Dụng để Tầm Soát Bệnh Nhân Không Triệu Chứng |
|---|---|---|---|---|
| Tuyến yên | 5 | 5–10 | Không triệu chứng, đau đầu, mất thị lực, tăng trưởng tuyến tính bất thường, dậy thì muộn, tiết sữa, kinh nguyệt không đều | Prolactin, IGF-1 |
| Cận giáp | 4 | 5–10 | Không triệu chứng, sỏi thận, đa niệu, khó tập trung, mệt mỏi, chán ăn, đau bụng, táo bón | Canxi toàn phần, PTH nguyên vẹn |
| Tuyến ức & Phế quản | 16, 15 | 15 | Không triệu chứng, đau ngực/vai, ho, khó thở, thở khò khè, ho ra máu | Không xét nghiệm sinh hóa định kỳ (trừ khi xác định được tổn thương hoặc hội chứng lâm sàng) |
| Dạ dày | KA | KA | Không triệu chứng | Không xét nghiệm sinh hóa định kỳ |
| Tá tràng/tụy | ||||
| -U insulin | 5 | 5 | Lú lẫn, mất ý thức, các triệu chứng giảm đường huyết thần kinh, yếu, co giật | Glucose, insulin |
| -U gastrin | 7 | 15 | Bệnh loét dạ dày tá tràng, khó tiêu, đau bụng, tiêu chảy | Gastrin |
| -Không chức năng | 12 | 15 | Không triệu chứng | Chromogranin A, polypeptide tụy |
| -Chức năng khác | KA | KA | Đái tháo đường, phát ban da, sụt cân, viêm miệng, thiếu máu (u glucagon), Tiêu chảy nước, hạ kali máu, vô toan (u VIPoma) | Không xét nghiệm sinh hóa định kỳ |
| Vỏ thượng thận | 3 | 15 | Nam hóa (dấu hiệu chính), hội chứng Cushing, tăng huyết áp, đau bụng/lưng | Không xét nghiệm sinh hóa định kỳ (trừ khi xác định được tổn thương hoặc hội chứng lâm sàng) |
IGF-1, yếu tố tăng trưởng giống insulin 1; MEN, đa u tuyến nội tiết; MRI, chụp cộng hưởng từ; NETs, u thần kinh nội tiết; PTH, hormone cận giáp; PHPT, cường cận giáp nguyên phát.
Bản chất di truyền của các u tuyến nội tiết đa phát và mối liên hệ với một rối loạn đơn gen lần đầu tiên được Wermer đề xuất vào năm 1954. Nguyên nhân di truyền chính xác của hội chứng đã được xác định và xác nhận vào năm 1997. Gen MEN1 là một gen đè nén khối u mã hóa cho một protein hạt nhân phổ biến (menin), đóng vai trò trong điều hòa phiên mã, ổn định bộ gen, phân chia và tăng sinh tế bào. Hầu hết bệnh nhân mắc MEN1 được di truyền đột biến từ một phụ huynh bị ảnh hưởng, nhưng khoảng 10% các cá nhân mắc MEN1 là các ca bệnh mới (de novo); khoảng một phần tư bệnh nhân dưới 21 tuổi sẽ là các ca bệnh chỉ điểm.
MEN1 là một ví dụ điển hình của cơ chế “hai đột biến” của bệnh (Hình 15.8), lần đầu tiên được Knudson mô tả trong bệnh u nguyên bào võng mạc di truyền và có thể áp dụng cho bất kỳ bệnh nào do các đột biến bất hoạt của một gen đè nén khối u. Một đột biến MEN1 dòng mầm dị hợp tử di truyền là không đủ để gây ra sự hình thành khối u. Do đó, một đột biến soma (đột biến thứ hai) trong alen MEN1 kiểu dại là cần thiết để gây bệnh. Đột biến thứ hai này thường xóa đi gen MEN1 hoạt động bình thường duy nhất, dẫn đến mất tính dị hợp tử tại locus MEN1 trong DNA khối u và làm suy yếu các ràng buộc thông thường của menin đối với sự tăng trưởng tế bào. Hơn 90% các khối u từ bệnh nhân MEN1 có mất tính dị hợp tử do sự sắp xếp lại dưới nhiễm sắc thể hoặc xóa toàn bộ nhiễm sắc thể; các cơ chế khác cho đột biến thứ hai bao gồm các đột biến điểm và các đoạn chèn/xóa nhỏ trong gen MEN1 kiểu dại. Cho đến nay, đã có vài trăm đột biến dòng mầm duy nhất của gen MEN1 được mô tả, và chúng xảy ra thông qua nhiều cơ chế và phân bố khắp gen MEN1 mà không có các điểm nóng. Xét nghiệm bằng giải trình tự DNA trực tiếp xác định hầu hết các đột biến MEN1, nhưng khoảng 5% đến 10% người mắc MEN1 không có đột biến trong vùng mã hóa hoặc các vị trí nối của MEN1. Những bệnh nhân như vậy có thể là một kiểu hình tương tự (phenocopy) (xem phần về MEN4) hoặc họ có thể mang một đột biến ở các vùng không được dịch mã hoặc intron hoặc một đoạn xóa gen lớn, loại sau này đòi hỏi các công nghệ khác, chẳng hạn như khuếch đại đầu dò phụ thuộc vào sự nối đa kênh (MLPA) để phát hiện.
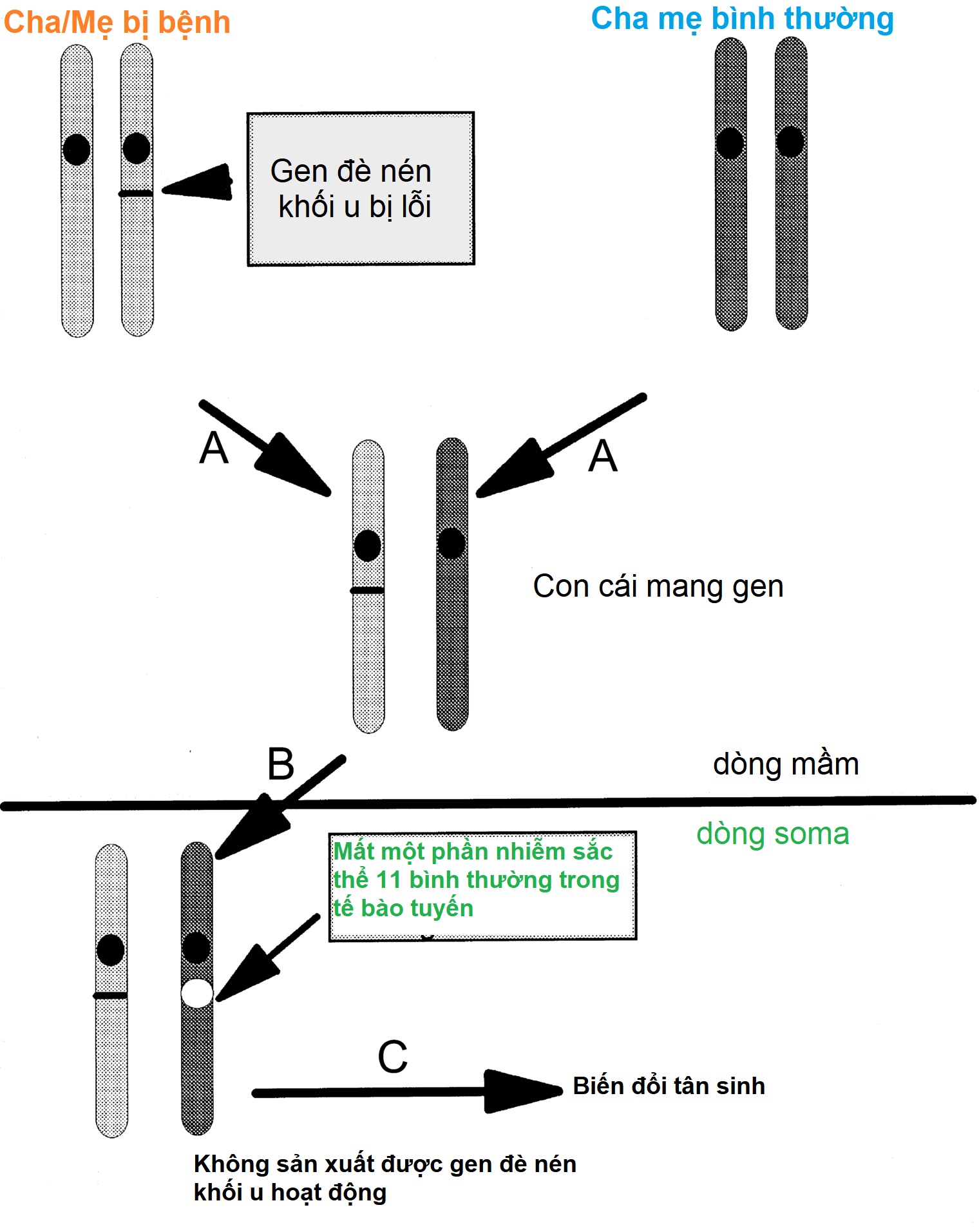
Hình 15.8 Thuyết “hai đột biến” về sự hình thành khối u được minh họa bởi đa u tuyến nội tiết (MEN)1. (A) Một phụ huynh bị ảnh hưởng truyền một gen MEN1 đột biến cho con cái, người này được thừa hưởng một gen bình thường (kiểu dại) từ người phụ huynh còn lại. (B) Mất tính dị hợp tử trong một tế bào soma (một “đột biến thứ hai” thường xảy ra thông qua sự sắp xếp lại dưới nhiễm sắc thể hoặc xóa toàn bộ nhiễm sắc thể) làm gián đoạn chức năng của alen MEN1 bình thường còn lại. (C) Sự vắng mặt của hoạt động đè nén khối u của menin trong một tế bào sau đó dẫn đến sự hình thành khối u.
Biểu hiện lâm sàng và Quản lý
Hầu hết trẻ em sẽ được chẩn đoán mắc MEN1 sau khi xác định được đột biến MEN1 dòng mầm, là kết quả của xét nghiệm di truyền theo tầng trong một gia đình MEN1 đã biết. Biểu hiện lâm sàng của bệnh liên quan đến MEN1 rất đa dạng, ngay cả giữa các thành viên trong cùng một gia đình, và sẽ phụ thuộc vào vị trí và chức năng của (các) khối u tiềm ẩn. Không có gì ngạc nhiên, các khối u chức năng thường biểu hiện sớm hơn 5 đến 10 năm so với các khối u không bài tiết. Mặc dù có tính biểu hiện thay đổi, nhưng độ xâm nhập của kiểu hình gần như hoàn toàn, sao cho các biểu hiện lâm sàng và sinh hóa của MEN1 thường sẽ phát triển ở 80% và hơn 98% bệnh nhân, tương ứng, vào thập kỷ thứ năm của cuộc đời. Độ xâm nhập của kiểu hình lâm sàng ở trẻ em mắc MEN1 trước đây được ước tính là dưới 1% trước 5 tuổi, 7% vào 10 tuổi, 28% vào 15 tuổi, và 52% vào 20 tuổi (Hình 15.9). Hầu hết các nghiên cứu gần đây hơn đều tìm thấy độ xâm nhập cao tương tự ở trẻ em. Trong hai nghiên cứu, 73% đến 78% bệnh nhân dưới 21 tuổi có ít nhất một biểu hiện lâm sàng của MEN1. Trong một loạt nghiên cứu khác, 54,5% trẻ em được theo dõi bằng một chương trình tầm soát cụ thể đã phát triển ít nhất một biểu hiện lâm sàng của MEN1 ở độ tuổi trung bình là 22,5 tuổi (khoảng 12–31 tuổi), 83% trong số đó không có triệu chứng lâm sàng. Tuy nhiên, một nghiên cứu khác đã ghi nhận độ xâm nhập thấp hơn nhiều (12%) của kiểu hình lâm sàng MEN1 trước 19 tuổi. Trái ngược với MEN2, như sẽ được thảo luận sau, không có mối tương quan kiểu gen-kiểu hình rõ ràng trong MEN1, vì vậy nhà cung cấp dịch vụ y tế không thể dựa vào đột biến cụ thể hoặc tiền sử gia đình để dự đoán tuổi khởi phát, mức độ nghiêm trọng, hoặc loại biểu hiện lâm sàng.
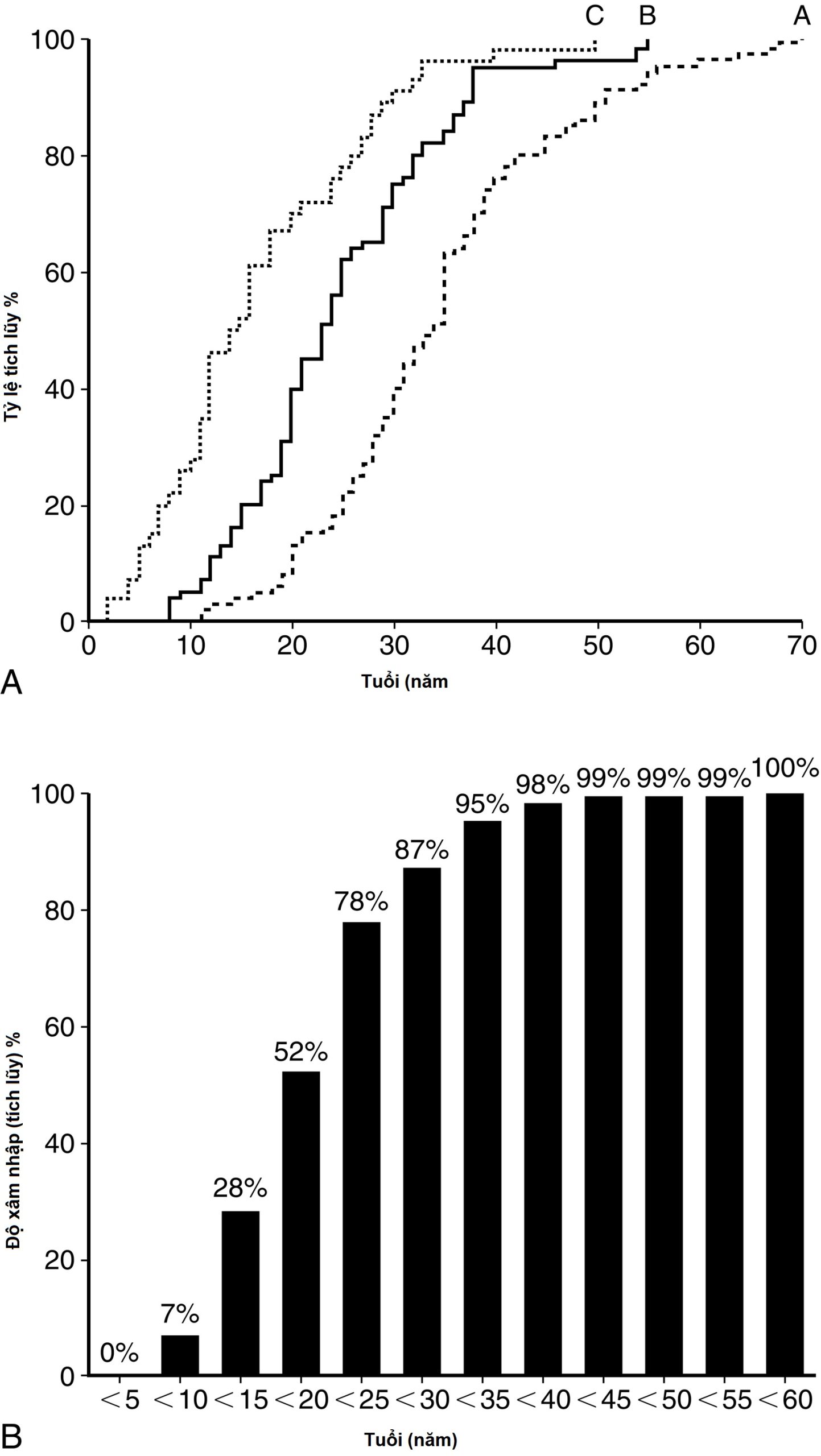
Hình 15.9 Độ xâm nhập của kiểu hình đa u tuyến nội tiết (MEN)1. (A) Phân bố tuổi và (B) độ xâm nhập liên quan đến tuổi của các biểu hiện lâm sàng MEN1 ở 201 người mang gen MEN1 đột biến. Nhóm A là những bệnh nhân có triệu chứng lâm sàng ở độ tuổi được mô tả. Nhóm B là những bệnh nhân không có triệu chứng nhưng có kết quả tầm soát sinh hóa dương tính ở độ tuổi được mô tả. Nhóm C là những bệnh nhân không có triệu chứng với kết quả tầm soát sinh hóa âm tính với độ tuổi của lần xét nghiệm sinh hóa cuối cùng được hiển thị. Bệnh nhân ở Nhóm B và C trẻ hơn đáng kể so với Nhóm A (P < 0,001). Độ xâm nhập liên quan đến tuổi (B) là tỷ lệ những người mang đột biến đã phát triển một biểu hiện lâm sàng theo một độ tuổi nhất định. (Nguồn: Bassett, J.H., Forbes, S.A., Pannett, A.A., và cộng sự. (1998). Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet, 62(2), 232–244).
Trong trường hợp không được điều trị, các khối u nội tiết liên quan đến MEN1 có liên quan đến tỷ lệ tử vong cao hơn (50% xác suất tử vong vào 50 tuổi), và nguyên nhân tử vong ở 50% đến 70% bệnh nhân mắc MEN1 thường là một quá trình khối u ác tính hoặc di chứng của bệnh. Hiện tại, nguy cơ tử vong lớn nhất trong MEN1 chủ yếu đến từ các NET ác tính ở tá tràng-tụy và tuyến ức.
Việc quản lý cụ thể của từng khối u tân sinh nội tiết liên quan đến MEN1 thường tương tự như đối với các khối u lẻ tẻ tương ứng xảy ra ở những bệnh nhân không mắc MEN1, với những ngoại lệ đáng chú ý đối với tuyến cận giáp và tụy (xem sau). Do đó, phần này sẽ tập trung nhiều hơn vào các khía cạnh độc đáo của những rối loạn này khi chúng liên quan đến MEN1 ở trẻ em. Cũng như hầu hết các hội chứng khối u di truyền, bệnh nhân mắc MEN1 và gia đình của họ nên được quản lý bởi một đội ngũ đa ngành bao gồm các chuyên gia có liên quan có kinh nghiệm trong việc quản lý các khối u nội tiết.
Cường cận giáp nguyên phát
Cường cận giáp nguyên phát (PHPT) là biểu hiện nội tiết phổ biến và sớm nhất của MEN1. Nó được chẩn đoán ở 75% đến 91% các trường hợp bệnh nhân MEN1 có biểu hiện lâm sàng trước 21 tuổi, có hoặc không có các biểu hiện MEN1 khác; hầu hết các chẩn đoán được thực hiện sau 10 tuổi. Bệnh nhân MEN1 có tăng sản tuyến cận giáp đa tuyến chứ không phải là u tuyến đơn tuyến. Phần lớn trẻ em mắc PHPT liên quan đến MEN1 không có triệu chứng, và sỏi thận là dấu hiệu biểu hiện chính của bệnh có triệu chứng.
Chẩn đoán PHPT dựa trên nồng độ canxi huyết thanh tăng cao trong bối cảnh nồng độ PTH bình thường một cách không phù hợp hoặc tăng rõ rệt; đôi khi, PTH có thể tăng với nồng độ canxi bình thường. Do tăng sản cận giáp đa ổ ở bệnh nhân MEN1, hình ảnh trước phẫu thuật thường không có lợi. Tuy nhiên, trong trường hợp phẫu thuật lại, các nghiên cứu định vị, chẳng hạn như xạ hình technetium (99mTc) sestamibi (thường có hình ảnh SPECT đồng thời), siêu âm, CT có cản quang, CT bốn chiều (4D), hoặc ít phổ biến hơn là MRI có thể hữu ích cho việc lập kế hoạch phẫu thuật.
Phẫu thuật cắt bỏ các tuyến cận giáp tăng chức năng là phương pháp điều trị dứt điểm PHPT ở bệnh nhân MEN1, nhưng phương pháp phẫu thuật vẫn còn gây tranh cãi. Cắt bỏ cận giáp bán phần (3,5 tuyến) kèm cắt tuyến ức qua đường cổ hoặc cắt bỏ cận giáp toàn phần (bốn tuyến) kèm cắt tuyến ức qua đường cổ và tự ghép tuyến cận giáp vào cẳng tay không thuận là những lựa chọn chính cho liệu pháp phẫu thuật. Cắt bỏ dưới ba tuyến dẫn đến nguy cơ cao nhất về bệnh dai dẳng và tái phát (tỷ số chênh [OR], 3,11; khoảng tin cậy [CI] 95% = 2,00–4,84). Mặc dù nguy cơ HPT dai dẳng hoặc tái phát không khác biệt đáng kể giữa phẫu thuật bán phần và toàn phần, nguy cơ suy cận giáp vĩnh viễn thấp hơn đáng kể với phẫu thuật cắt bỏ cận giáp bán phần, điều này làm cho đây trở thành phương pháp được ưu tiên. Tuy nhiên, một số nghiên cứu đã cho thấy kết quả tương đương giữa hai thủ thuật ở bệnh nhân trẻ tuổi. Ở những bệnh nhân được phẫu thuật PHPT trước 21 tuổi, 67% vẫn duy trì canxi máu bình thường ở lần theo dõi cuối cùng, trong khi 22% có tăng canxi máu dai dẳng và 11% bị suy cận giáp. Tại cuộc phẫu thuật ban đầu, nên thực hiện cắt tuyến ức qua đường cổ đồng thời để loại bỏ bất kỳ tuyến cận giáp lạc chỗ tiềm năng nào và để dự phòng loại bỏ phần lớn tuyến ức, vốn có nguy cơ phát triển NET tuyến ức.
Chỉ định phẫu thuật cho PHPT không triệu chứng ở trẻ em mắc MEN1 vẫn chưa được phát triển đầy đủ, và nguy cơ suy cận giáp vĩnh viễn phải được cân bằng với các rủi ro của PHPT. Ngoài nồng độ canxi, các yếu tố, chẳng hạn như kinh nghiệm phẫu thuật, sở thích của bệnh nhân hoặc phụ huynh, và sự sẵn có của việc theo dõi nồng độ canxi dài hạn, nên được xem xét trong thời điểm phẫu thuật. Ở những bệnh nhân MEN1 được chọn lọc mắc PHPT mà phẫu thuật không khả thi, có thể xem xét điều trị nội khoa bằng tác nhân calcimimetic, cinacalcet.
U tuyến yên
U tuyến yên là biểu hiện lâm sàng phổ biến thứ hai ở trẻ em, xảy ra ở 29% đến 55% bệnh nhân MEN1 có độ xâm nhập kiểu hình MEN1 dưới 21 tuổi, hơn một nửa trong số đó sẽ có triệu chứng. Chẩn đoán trước 10 tuổi là không có khả năng. Tương tự như người lớn, u tiết prolactin là phổ biến nhất, mặc dù tất cả các phân nhóm của u tuyến yên đã được báo cáo.
Trong MEN1, dường như có tỷ lệ mắc u tuyến yên cao hơn ở nữ giới, tỷ lệ xâm lấn và u lớn (đặc biệt ở nam giới) cao hơn, và nhiều khối u đa tiềm năng hơn. Các khối u tuyến yên liên quan đến MEN1 tăng tiết có thể kháng trị hơn với các liệu pháp nội khoa tiêu chuẩn, mặc dù các dữ liệu khác cho thấy rằng điều trị nội khoa có hiệu quả tương đương so với dân số chung. Không có nguy cơ tử vong tăng lên do có khối u tuyến yên liên quan đến MEN1, mặc dù một trường hợp ung thư biểu mô tuyến yên xảy ra trong bối cảnh MEN1 đã được công bố.
U Thần kinh Nội tiết Tá tràng-Tụy
Các NET phát sinh từ các tế bào thần kinh nội tiết của tá tràng và tụy là biểu hiện lâm sàng phổ biến thứ ba của MEN1 ở trẻ em, được báo cáo có mặt ở 9% đến 42% tất cả bệnh nhân có biểu hiện MEN1 xảy ra trước 21 tuổi. Các NET đường tiêu hóa (GI) này thường có thể đa ổ trong bối cảnh MEN1. U gastrin chủ yếu xảy ra ở tá tràng (trong cái gọi là tam giác gastrinoma), trong khi các khối u hoạt động nội tiết khác phát sinh từ các tế bào tiểu đảo tụy ở nhiều vị trí khác nhau trên khắp tụy. Hơn nữa, các NET tụy có thể tiết lạc chỗ hormone giải phóng hormone tăng trưởng và ACTH. Các NET tụy không chức năng theo định nghĩa không liên quan đến các triệu chứng lâm sàng do tăng tiết hormone và do đó được chẩn đoán thông qua hình ảnh định kỳ. Những khối u như vậy có thể sản xuất quá mức các hormone im lặng về mặt lâm sàng, chẳng hạn như polypeptide tụy và chromogranin A, mặc dù các chất chỉ điểm huyết thanh này có độ chính xác chẩn đoán thấp.
Bởi vì các NET có thể nhỏ và phát triển chậm, việc định vị có thể khó khăn và thường cần sử dụng nhiều kỹ thuật. Các hướng dẫn khuyến nghị CT đa pha, MRI, và/hoặc siêu âm nội soi (EUS) để đánh giá các NET tụy. EUS mang lại độ nhạy chẩn đoán cao nhất, đặc biệt với các khối u nhỏ hơn, nhưng độ nhạy cải thiện này có thể không thay đổi việc quản lý vì các khối u nhỏ dưới 2 cm thường được theo dõi trong trường hợp không có tăng tiết hormone có triệu chứng. Các NET thường biểu hiện các thụ thể somatostatin. Do đó, xạ hình thụ thể somatostatin, đặc biệt là 68Ga-DOTATATE PET/CT, cũng đóng một vai trò quan trọng trong việc quản lý các NET tụy, đặc biệt là trong việc xác định bệnh di căn. FDG-PET không được khuyến nghị thường quy do tốc độ tăng sinh thấp của các khối u tân sinh này, nhưng các khối u biệt hóa kém có thể được định vị bằng FDG-PET. Chẩn đoán một NET ác tính sẽ cực kỳ hiếm ở trẻ em. Các hướng dẫn về chẩn đoán và quản lý các NET đã có sẵn.
U insulin
U insulin là NET tụy chức năng thường gặp nhất ở trẻ em mắc MEN1, đã được báo cáo chiếm 54% đến 78% các NET tụy trong các loạt nghiên cứu lớn được công bố và được chẩn đoán ở 12% đến 25% tất cả bệnh nhân có biểu hiện MEN1 xảy ra ở độ tuổi dưới 21. Bệnh nhân có các triệu chứng hạ đường huyết cải thiện khi ăn đường, do đó đáp ứng bộ ba Whipple. Hai triệu chứng chính được cho là do hạ đường huyết bao gồm mất ý thức hoặc hôn mê (55%) và yếu (45%), liên quan đến co giật trong một số trường hợp. Xét nghiệm chẩn đoán tiêu chuẩn vàng là nhịn ăn 72 giờ. Chẩn đoán được xác định nếu glucose huyết thanh dưới 55 mg/dL với insulin đồng thời từ 3 μU/mL trở lên bằng xét nghiệm miễn dịch hóa phát quang (ICMA), c-peptide từ 0,2 nmol/L trở lên, và không phát hiện được thuốc hạ đường huyết uống trong máu. Một phần ba bệnh nhân u insulin sẽ bị hạ đường huyết sau 12 giờ, 80% sau 24 giờ, 90% sau 48 giờ, và 100% sau 72 giờ. Bởi vì điều trị nội khoa bằng các bữa ăn carbohydrate thường xuyên, diazoxide, và/hoặc các chất tương tự somatostatin không phải lúc nào cũng thành công, phẫu thuật được coi là tiêu chuẩn chăm sóc. Trái ngược với các NET chức năng khác, u insulin có tiềm năng ác tính thấp hơn.
U gastrin
U gastrin xảy ra chủ yếu ở tá tràng và thường là các khối u nhỏ (< 1 cm), đa ổ, dẫn đến tăng sản xuất axit dạ dày và bệnh loét dạ dày tá tràng tái phát, được gọi là hội chứng Zollinger-Ellison (ZES). Các triệu chứng khác có thể bao gồm tiêu chảy và phân mỡ. Chúng là khối u chức năng phổ biến nhất ở người lớn mắc MEN1. Cực kỳ hiếm ở trẻ em và xảy ra ở 0% đến 7% bệnh nhân (0% đến 22% các NET tụy) trong các loạt nghiên cứu bao gồm trẻ em, u gastrin thường được chẩn đoán trong thập kỷ thứ hai của cuộc đời ở bệnh nhân nhi.
Chẩn đoán được xác định bởi nồng độ gastrin lúc đói trong huyết thanh tăng (thường ⩾ 1000 pg/mL) và tăng tiết axit dạ dày cơ bản (pH dạ dày < 2). Bệnh nhân có tăng gastrin máu giới hạn nên được xem xét nghiệm pháp kích thích secretin; sau một liều tiêm tĩnh mạch 2 U/kg, sự gia tăng gastrin từ 120 pg/mL trở lên so với mức cơ bản mang lại độ nhạy và độ đặc hiệu chẩn đoán cao nhất. Tăng gastrin dương tính giả có thể xảy ra do sử dụng thuốc ức chế bơm proton hoặc thuốc kháng axit, các tình trạng viêm dạ dày teo mạn tính tự miễn với vô toan, và/hoặc suy thận mạn tính. Do đó, bệnh nhân nên ngưng thuốc ức chế bơm proton ít nhất 1 tuần trước khi xét nghiệm, nhưng điều này có thể không thực hiện được ở những bệnh nhân có triệu chứng nặng. Tăng canxi máu cũng có thể kích thích tiết gastrin.
Điều trị nội khoa bằng thuốc ức chế bơm proton đường uống mạnh là phương pháp điều trị chính cho u gastrin ở bệnh nhân MEN1. Một số bệnh nhân cũng có thể cần bổ sung một chất đối kháng thụ thể H2. Vai trò của phẫu thuật trong điều trị bệnh nhân MEN1 mắc u gastrin còn gây tranh cãi. Các khối u thường đa ổ và ở tá tràng, vì vậy khả năng chữa khỏi ít hơn, trừ khi thực hiện phẫu thuật rộng rãi. Nếu thực hiện phẫu thuật, cần phải kiểm tra tá tràng và nạo vét hạch. Đối với các u gastrin tụy hoặc các tổn thương lớn hơn 2 cm, nên xem xét phẫu thuật để giảm nguy cơ di căn hạch hoặc gan.
Các khối u thần kinh nội tiết tụy chức năng khác
U glucagon tiết glucagon và trước đây chưa được mô tả ở trẻ em mắc MEN1, ngoại trừ một trường hợp duy nhất là u glucagon dường như im lặng. Hội chứng glucagonoma kinh điển được đặc trưng bởi phát ban da ở bẹn và tứ chi (gọi là ban đỏ di chuyển hoại tử) ở 70% bệnh nhân, đái tháo đường nhẹ ở 87%, sụt cân ở hầu hết bệnh nhân, viêm miệng và thiếu máu.
U VIPoma, cũng chưa được báo cáo ở trẻ em, là những khối u rất hiếm tiết peptide ruột vận mạch (VIP) và có liên quan đến hội chứng Verner-Morrison gồm tiêu chảy nước, hạ kali máu, và vô toan (tức là, tả tụy).
Các khối u thần kinh nội tiết tụy không chức năng
Ở trẻ em mắc MEN1, các NET tụy không chức năng là NET phổ biến thứ hai phát sinh ở tá tràng/tụy (40% các NET tụy; 9% tất cả các trường hợp) và được xác định chỉ bằng hình ảnh. Với các phương pháp tầm soát cải tiến, chúng đã được xác định ở 42% thanh thiếu niên được xét nghiệm và chiếm 75% các NET tụy, và do đó, độ xâm nhập thực sự của chúng có thể bị đánh giá thấp trong các nghiên cứu cũ hơn chủ yếu bao gồm các khối u chức năng, và do đó, có triệu chứng. Các nghiên cứu dịch tễ học đã cho thấy rằng 27% bệnh nhân có các NET tụy không chức năng có kích thước từ 2,1 đến 3,0 cm có di căn so với 11% ở những người có kích thước từ 2 cm trở xuống. Quan trọng hơn, các khối u không chức năng hiện được hiểu là mang lại nguy cơ tử vong tăng lên ở bệnh nhân MEN1, tương đương với các NET tuyến ức và các NET tụy chức năng, ngoại trừ u insulin. Mặc dù vẫn còn những tranh cãi về chỉ định điều trị phẫu thuật, nhưng sự đồng thuận chung là các khối u từ 2 cm (hoặc lớn hơn) hoặc các khối u có tốc độ tăng trưởng tương đối nhanh nên được xem xét để cắt bỏ.
Các biểu hiện lâm sàng khác
Các khối u thần kinh nội tiết tuyến ức, phế quản và dạ dày
Trong MEN1, các NET cũng xảy ra bên ngoài tá tràng và tụy và có thể được tìm thấy ở tuyến ức, hệ thống phế quản-phổi, và dạ dày. Trước đây được gọi là u carcinoid, thuật ngữ được ưu tiên là NET. Chẩn đoán các khối u như vậy hầu như chỉ xảy ra ở người lớn, nhưng một trường hợp NET tuyến ức ác tính đã được báo cáo ở một bệnh nhân MEN1 16 tuổi, và một NET phế quản đã được tìm thấy ở một bé gái 15 tuổi.
Các NET tuyến ức phổ biến hơn ở những người hút thuốc và nam giới (tỷ lệ nam/nữ 20:1), trong khi các NET phế quản phổ biến hơn ở phụ nữ (tỷ lệ nam/nữ 1:4). Các NET phế quản có diễn biến âm thầm hơn so với các khối u tuyến ức, vốn mang lại nguy cơ tử vong tăng đáng kể ở bệnh nhân MEN1. Các NET tuyến ức vẫn có thể phát triển mặc dù đã cắt bỏ tuyến ức dự phòng qua đường cổ trước đó, có lẽ vì toàn bộ tuyến ức không được loại bỏ qua đường mổ ở cổ, điều này nhấn mạnh tầm quan trọng của việc tiếp tục tầm soát định kỳ ngay cả khi đã thực hiện cắt tuyến ức. Các NET tuyến ức và phế quản có thể tiết lạc chỗ hormone giải phóng hormone tăng trưởng và ACTH.
Các NET dạ dày là các khối u biệt hóa tốt có tiềm năng ác tính phát sinh từ các tế bào giống tế bào ưa crôm của niêm mạc dạ dày. Các NET dạ dày loại II là những khối u xảy ra trong MEN1, phát triển thứ phát sau tăng gastrin máu mạn tính trong bối cảnh u gastrin/ZES. Do sự hiếm gặp của u gastrin ở trẻ em, các NET dạ dày loại II chưa được báo cáo ở bệnh nhân nhi.
Các khối u vỏ thượng thận
Các khối u vỏ thượng thận (ACT) một bên và hai bên được báo cáo ở 10% đến 26% bệnh nhân MEN1, với độ tuổi trung bình tại thời điểm chẩn đoán một tổn thương thượng thận là khoảng 45 tuổi; ở trẻ em mắc MEN1 lâm sàng dưới 21 tuổi, tỷ lệ mắc dao động từ 0% đến 4%. Tăng sản vỏ thượng thận, u tuyến, và ung thư biểu mô vỏ thượng thận là các loại tổn thương thượng thận được thấy; PHEO cũng đã được báo cáo ở người lớn mắc MEN1. Hầu hết các ACT là không chức năng, và các khối u chức năng chủ yếu có biểu hiện tăng tiết aldosterone hoặc cortisol, mặc dù tăng androgen/nam hóa được thấy ở trẻ em. Trong loạt nghiên cứu lớn về MEN1 do Goudet và cộng sự công bố và trong nghiên cứu của Gatta-Cherifi và các đồng nghiệp, tất cả các trường hợp ACT phát sinh ở trẻ em trước 20 tuổi đều có biểu hiện tăng androgen và được phân loại là ác tính.
Các biểu hiện da
Thường bị đánh giá thấp như một thành phần của kiểu hình MEN1, các biểu hiện da (Hình 15.10) bao gồm u xơ mạch, u collagen, và u mỡ. Thường được xác định phổ biến hơn ở bệnh nhân MEN1 lớn tuổi, các biểu hiện da liễu cũng có thể được tìm thấy ở trẻ em, thậm chí là một biểu hiện ban đầu của bệnh. U xơ mạch là các khối u lành tính của các mạch máu và mô liên kết, tương tự như những khối u được thấy trong bệnh xơ cứng củ. Chúng có dạng các sẩn hình vòm màu hồng đến nâu, xuất hiện trên mặt, đặc biệt là mũi. U collagen là các sẩn da có ranh giới rõ ràng, màu da, có xu hướng xuất hiện ở cổ và thân mình. Tỷ lệ mắc u xơ mạch, u collagen, và u mỡ ở bệnh nhân MEN1 lần lượt là 64% đến 88%, 63% đến 72%, và 3% đến 34%. Nhìn chung, các tổn thương da liên quan đến MEN1 không cần điều trị, mặc dù u mỡ có thể phát triển khá lớn và có thể cần phải được cắt bỏ do các triệu chứng tại chỗ.
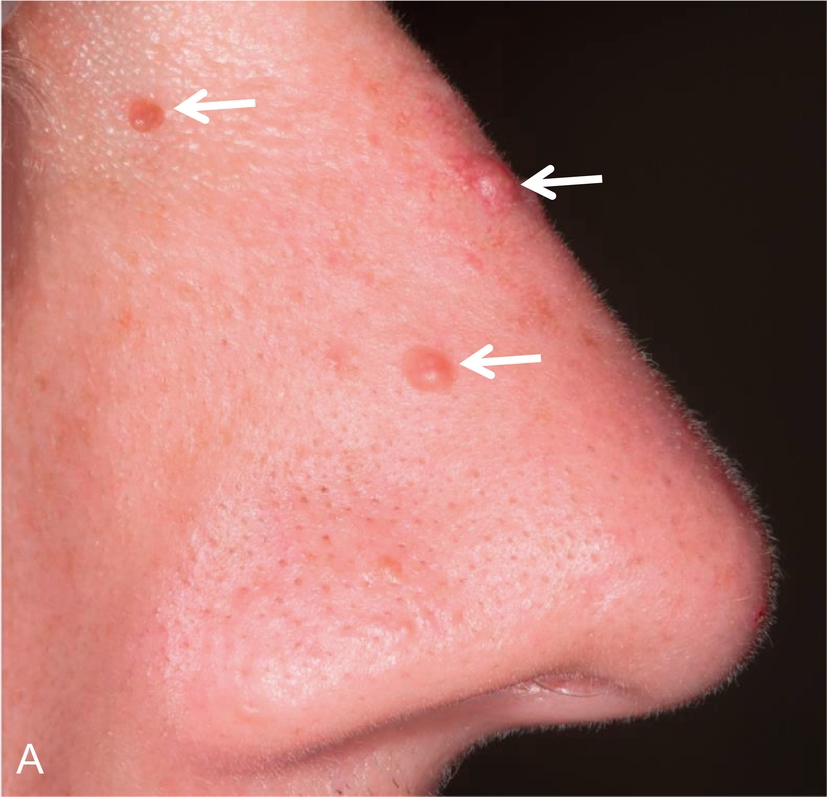
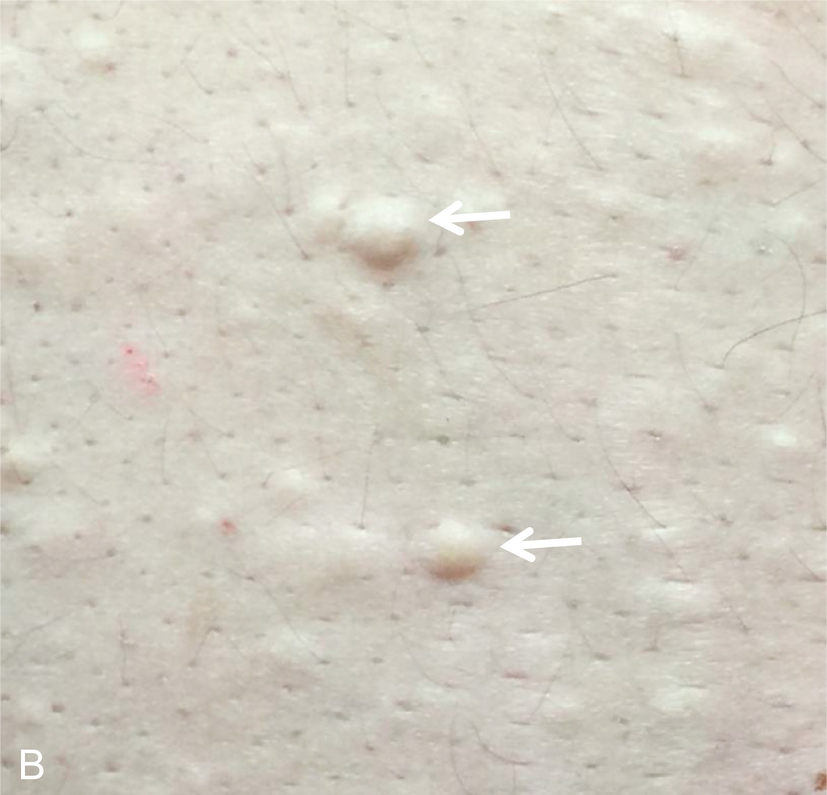
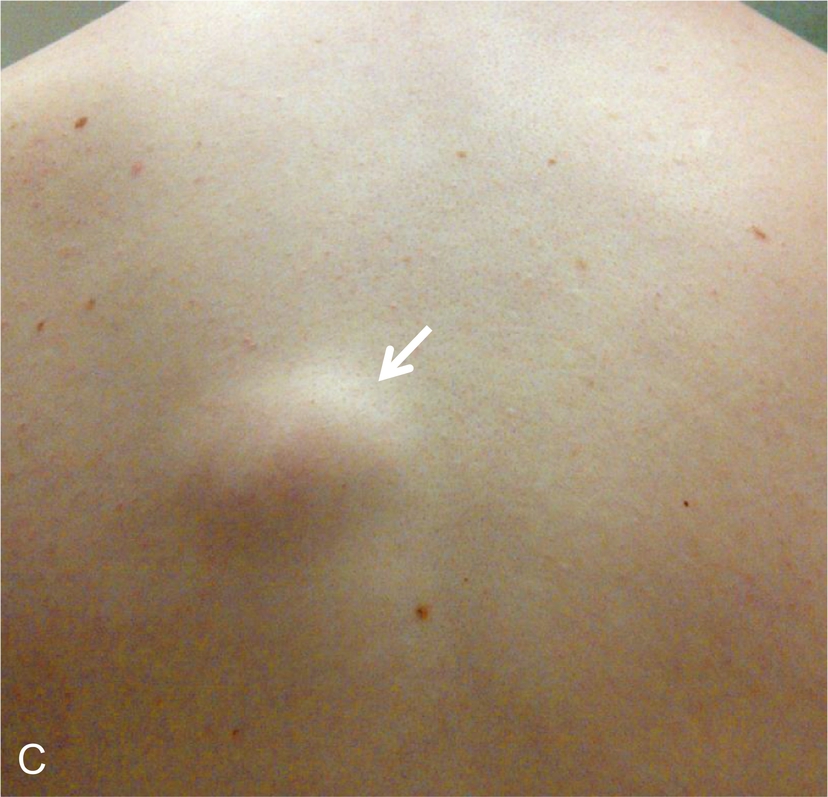
Hình 15.10 Các biểu hiện da liễu của đa u tuyến nội tiết (MEN)1. U xơ mạch (A), u collagen (B), và u mỡ (C) thường được xác định phổ biến hơn ở bệnh nhân MEN1 trưởng thành, nhưng chúng cũng có thể phát triển ở trẻ em, thậm chí là một biểu hiện ban đầu của bệnh. U xơ mạch là các sẩn giãn mạch được đặc trưng bởi mô xơ và tăng sinh mạch máu và phân bố chủ yếu trên mũi và các má lân cận. U collagen là các tổn thương mô đệm da đại diện cho các vùng collagen dư thừa và có xu hướng xảy ra ở phần thân trên, cổ và vai. U mỡ là các khối u tân sinh lành tính chứa các tế bào mỡ trưởng thành được bao bọc bởi các bao xơ mỏng.
Xét nghiệm di truyền và Tầm soát tiền triệu chứng
Một chương trình tích hợp cả tư vấn/xét nghiệm di truyền cho các cá nhân có nguy cơ và tầm soát lâm sàng cho những người mang đột biến MEN1 được thực hiện với giả định rằng chẩn đoán và điều trị sớm hơn có thể giúp giảm tỷ lệ mắc bệnh và tử vong. Các hướng dẫn chăm sóc bệnh nhân MEN1 dựa trên các tổng quan hệ thống của y văn và được phát triển bởi các chuyên gia trong lĩnh vực này đã được giới thiệu vào năm 2001 và cập nhật vào năm 2012. Các ấn phẩm sau đó đã đề xuất những thay đổi đối với hướng dẫn tầm soát năm 2012, đặc biệt là liên quan đến các NET tụy không chức năng.
Hiện tại, không có liệu pháp cụ thể nào có thể ngăn ngừa bệnh liên quan đến MEN1 cũng như không có liệu pháp dự phòng nào được đảm bảo. Ngoài ra, việc xác định sớm bệnh không triệu chứng có thể dẫn đến điều trị sớm và không cần thiết, điều này thực tế có thể có tác động tiêu cực lâu dài. Do đó, mục tiêu của việc tầm soát trong MEN1 chủ yếu là để phát hiện một biểu hiện lâm sàng ở giai đoạn sớm hơn, đặc biệt để có thể điều trị trước khi bệnh ác tính phát triển ở các cơ quan có nguy cơ cao hoặc xảy ra các hậu quả nghiêm trọng do tăng tiết hormone. Do thời gian tiềm ẩn dài cho các biểu hiện bệnh, nhiều cá nhân không bị ảnh hưởng có thể sẽ trải qua nhiều năm xét nghiệm định kỳ, điều này có thể có những hậu quả tâm lý xã hội và tài chính, và điều này phải được ghi nhớ khi một chương trình tầm soát định kỳ được lên kế hoạch cho bất kỳ bệnh nhân MEN1 nào. Trong mọi trường hợp, sở thích của bệnh nhân/phụ huynh, rủi ro của việc tầm soát, nguồn lực tại chỗ, và đánh giá lâm sàng nên được xem xét khi theo dõi định kỳ một bệnh nhân mắc MEN1.
Xét nghiệm đột biến dòng mầm MEN1 nên được đề nghị cho tất cả các bệnh nhân chỉ điểm và những người thân cấp một của những cá nhân có đột biến MEN1 đã biết, bất kể có triệu chứng hay không. Bởi vì bệnh MEN1 liên quan đã được chẩn đoán ở trẻ em chỉ mới 5 tuổi (xem Bảng 15.4), thực hành hiện tại là đề nghị xét nghiệm ở cơ hội sớm nhất, nhưng chỉ sau khi tư vấn di truyền toàn diện để bệnh nhân và gia đình mắc MEN1 nhận được thông tin y tế và di truyền mà họ cần.
Sau khi chẩn đoán di truyền, một chương trình tầm soát cụ thể nên được thiết kế để phát hiện sự phát triển của các khối u ở những bệnh nhân tiền triệu chứng, hiểu rằng độ tuổi tối ưu để bắt đầu tầm soát bệnh vẫn chưa được xác định rõ ràng. Bảng 15.4 tóm tắt một chương trình tầm soát MEN1 được đề xuất dựa trên các hướng dẫn đồng thuận và được điều chỉnh từ các ấn phẩm gần đây về chủ đề này. Vẫn còn tranh cãi về các quy trình tầm soát tối ưu, có thể ít chuyên sâu hơn so với những gì hiện đang được quy định. Mặc dù đánh giá xét nghiệm hàng năm được sử dụng bởi hầu hết các trung tâm lâm sàng, không phải tất cả các trung tâm đều chỉ định tầm soát hình ảnh cho trẻ em và thanh niên tiền triệu chứng, như các hướng dẫn năm 2012 đã đề xuất, và tần suất và loại hình ảnh (CT, MRI, EUS) là một lĩnh vực quan trọng cần được nghiên cứu thêm. Điều quan trọng cần lưu ý là những rủi ro thực sự của việc sử dụng CT (làm cho bệnh nhân MEN1 tiếp xúc với một liều bức xạ ion hóa đáng kể trong suốt cuộc đời của họ) trong một rối loạn khối u di truyền, đặc biệt là một rối loạn đòi hỏi một đột biến thứ hai ở tế bào soma để quá trình hình thành khối u xảy ra. Do đó, với thời gian theo dõi dài cần thiết ở bệnh nhân MEN1, MRI thường được ưu tiên hơn CT để giảm thiểu rủi ro từ bức xạ ion hóa, mặc dù CT vẫn đóng một vai trò quan trọng, đặc biệt là trong việc lập kế hoạch phẫu thuật.
Đa u tuyến Nội tiết Loại 2
Mô tả ban đầu về MEN2A được cho là của Sipple khi ông báo cáo trường hợp một người đàn ông 33 tuổi chết vì xuất huyết nội sọ và được phát hiện khi khám nghiệm tử thi có PHEO hai bên, MTC hai bên, và có thể là tăng sản cận giáp. Năm 1962, Cushman đã đề xuất một mối liên quan giữa các khối u nội tiết này, và MEN2 sau đó được Steiner và cộng sự đặt tên là một hội chứng lâm sàng riêng biệt. Các nghiên cứu phân tích liên kết ở các gia đình lớn mắc MEN2 đã dẫn đến việc định vị gen giả định đến nhiễm sắc thể 10. Cuối cùng, các đột biến trong tiền-oncogen RET (nhiễm sắc thể 10q11.21) được phát hiện là nguyên nhân gây ra MEN2A.
Đặc điểm ban đầu của kiểu hình MEN2B trong y văn tiếng Anh được Williams và Pollock đưa ra vào năm 1966, và MEN2B sau đó được phân biệt là một biến thể của MTC di truyền với kiểu hình u thần kinh niêm mạc. Năm 1994, MEN2B được báo cáo cũng là thứ phát sau các đột biến hoạt hóa ở RET.
Là kết quả của những nỗ lực hợp tác ban đầu của Hiệp hội Đột biến RET Quốc tế, nhanh chóng trở nên rõ ràng rằng chỉ có một số lượng hạn chế các đột biến có liên quan đến MEN2 và có sự tương quan kiểu gen-kiểu hình mạnh mẽ (xem Hình 15.5). Do độ xâm nhập rất cao của MTC ở những cá nhân có đột biến RET, đã có sự tích hợp nhanh chóng của xét nghiệm di truyền vào các quy trình quản lý cho bệnh nhân MTC và gia đình của họ. Do đó, xét nghiệm di truyền tiên đoán đã mở ra kỷ nguyên thực hiện cắt toàn bộ tuyến giáp dự phòng cho những cá nhân tiền triệu chứng mang đột biến dòng mầm RET nguy cơ cao.
Đa u tuyến Nội tiết 2A
MEN2A chiếm 90% đến 95% các trường hợp MEN2 và được đặc trưng bởi sự phát triển MTC trong suốt cuộc đời ở hơn 90% những người mang đột biến RET. Tùy thuộc vào đột biến cụ thể, PHEO và/hoặc PHPT xảy ra ở 0% đến 50% và 0% đến 20% các cá nhân mắc MEN2A, tương ứng; các trường hợp hiếm gặp của u hạch thần kinh thượng thận cũng đã được mô tả. Hơn nữa, các đột biến hoạt hóa ở RET cũng có liên quan đến bệnh Hirschsprung và lichen amyloidosis da, một rối loạn da liễu gây ngứa dữ dội và các thay đổi da thứ phát thường nằm ở vùng gian vai của lưng. Mặc dù trước đây được thảo luận như một hội chứng riêng biệt, ung thư biểu mô tuyến giáp thể tủy có tính gia đình (FMTC), trong đó MTC là biểu hiện lâm sàng duy nhất của một đột biến RET dòng mầm, hiện được coi là một biến thể của MEN2A.
Trong MEN2A, các đột biến chủ yếu nằm ở miền giàu cysteine ngoại bào của tiền-oncogen RET, thường ở exon 10 (codon 609, 611, 618, hoặc 620) hoặc exon 11 (codon 634), nhưng các đột biến cũng có thể được tìm thấy ở miền tyrosine kinase nội bào (xem Hình 15.5). Các đột biến codon 634 chiếm phần lớn các trường hợp MEN2A kinh điển, nhưng gần đây hơn, tỷ lệ các đột biến ở các codon khác (đặc biệt là codon 804) đã tăng lên. Khi xét nghiệm phân tử trở nên phổ biến ở bệnh nhân MTC, nhiều đột biến và biến thể DNA RET hơn đang được xác định, điều này đang góp phần vào một phổ ngày càng thay đổi của kiểu gen và kiểu hình MEN2A.
Ung thư biểu mô tuyến giáp thể tủy trong Đa u tuyến Nội tiết 2A
MTC liên quan đến MEN2A, khi được xác định ở trẻ em, thường được chẩn đoán sau khi cắt tuyến giáp sớm theo chỉ dẫn của kết quả xét nghiệm di truyền và thường là một microcarcinoma có kích thước < 1 cm. Hầu hết trẻ em mắc MEN2A sẽ có tiền sử gia đình dương tính; các đột biến RET mới (de novo) chỉ chiếm 2% đến 9% các trường hợp. Có một mối quan hệ kiểu gen-kiểu hình mạnh mẽ, sao cho có thể ước tính được tốc độ mà một người mang đột biến sẽ phát triển MTC, nhưng lịch sử tự nhiên của MTC trong MEN2A có thể rất đa dạng, ngay cả giữa các thành viên trong cùng một gia đình. Bệnh nhân có đột biến ở codon RET 634 có nguy cơ MTC cao nhất, tiếp theo là những người có đột biến ở codon 609, 611, 618, 620, hoặc 630, trong khi các đột biến ở các codon khác mang lại nguy cơ MTC khởi phát sớm thấp nhất. Tuy nhiên, một khi MTC được chẩn đoán, tỷ lệ sống sót toàn bộ và sự phát triển của bệnh di căn xa là tương tự giữa các bệnh nhân có đột biến RET nguy cơ cao và trung bình, điều này cho thấy thêm rằng kiểu gen quyết định tuổi khởi phát của MTC nhiều hơn là mức độ xâm lấn lâm sàng của nó sau khi chẩn đoán.
U tế bào ưa crôm trong Đa u tuyến Nội tiết 2A
PHEO liên quan đến MEN2 là một khối u adrenergic thường phát sinh trên nền tăng sản tủy thượng thận, mặc dù một số nhóm thích thuật ngữ microPHEO để chỉ các khối tăng sinh tủy nhỏ hơn 1 cm. Với chẩn đoán rất có thể xảy ra trong thập kỷ thứ tư của cuộc đời, PHEO phát triển chủ yếu do các đột biến codon 634 và ở mức độ thấp hơn nhiều với các đột biến ở exon 10 (codon 609, 611, 618, và 620) và các codon khác. Thông thường, một PHEO được chẩn đoán đồng thời với MTC hoặc chỉ sau khi MTC được xác định; trong các nghiên cứu gần đây hơn, nó là biểu hiện lâm sàng ban đầu ở tới 15% các trường hợp. Các hướng dẫn hiện tại khuyến nghị tầm soát metanephrin định kỳ cho PHEO ở trẻ em mắc MEN2A, bắt đầu từ 11 tuổi, ở những bệnh nhân có đột biến codon 634, và ở tuổi 16 ở tất cả những người khác (xem Bảng 15.1).
Cường cận giáp nguyên phát trong Đa u tuyến Nội tiết 2A
Bệnh nhân mắc MEN2A có nguy cơ phát triển PHPT do u tuyến và tăng sản cận giáp. PHPT chủ yếu liên quan đến các đột biến codon 634 và ít được mô tả hơn ở các đột biến RET khác. Khởi phát ở trẻ em là cực kỳ hiếm nhưng đã được mô tả ở một đứa trẻ mới 5 tuổi. Thường thì PHPT được chẩn đoán trong thập kỷ thứ tư của cuộc đời, và do đó nó hiếm khi là biểu hiện đầu tiên của MEN2A. Tầm soát PHPT được khuyến nghị ở tuổi 11, ở trẻ em có đột biến codon 634, và ở tuổi 16 ở tất cả những người khác (xem Hình 15.5).
Đa u tuyến Nội tiết 2B
MEN2B là một bệnh có độ xâm nhập hoàn toàn, chiếm 5% đến 10% các trường hợp MEN2. MEN2B hầu như luôn (> 95%) là do một đột biến duy nhất ở exon 16 (p.M918T) nằm ở miền tyrosine kinase nội bào của thụ thể RET (xem Hình 15.5). Các trường hợp MEN2B hiếm gặp có thể là do các đột biến kép RET liên quan đến codon 804 hoặc một đột biến ở codon 883 (p.A883F, exon 15). Những người mang đột biến p.A883F có tuổi khởi phát bệnh muộn hơn và lịch sử tự nhiên âm thầm hơn so với những người mang đột biến p.M918T. Không giống như MEN2A, 84% đến 90% các trường hợp MEN2B phát sinh do một đột biến mới (de novo), với đứa trẻ có cha mẹ không bị ảnh hưởng. Do điều này và do sự hiếm gặp của hội chứng, chẩn đoán MEN2B hầu như luôn bị trì hoãn, ngay cả khi có các đặc điểm lâm sàng rõ ràng có thể bắt đầu ngay từ khi còn nhỏ.
MEN2B được đặc trưng bởi sự phát triển của MTC (100% các trường hợp), PHEO (lên đến 50% các trường hợp; các trường hợp hiếm gặp của u hạch thần kinh thượng thận cũng đã được mô tả), và một kiểu hình lâm sàng rất dễ xâm nhập và đặc trưng (100% các trường hợp; Hình 15.11). Mặc dù kiểu hình lâm sàng có mặt ở tất cả các bệnh nhân, nhưng các biểu hiện riêng lẻ có sự trình bày thay đổi và phụ thuộc vào độ tuổi. Nhiều u thần kinh niêm mạc là một biểu hiện lâm sàng chính và xảy ra trên môi, lưỡi, và/hoặc kết mạc, ngoài hệ tiết niệu và đường tiêu hóa. Các triệu chứng của bệnh u hạch thần kinh ruột bao gồm táo bón và các vấn đề về ăn uống ở trẻ sơ sinh và sự phát triển của phình đại tràng. Một thành phần thứ hai của kiểu hình MEN2B là các bất thường về cơ xương, bao gồm thể trạng marfanoid, khuôn mặt dài hẹp, bàn chân vòm, ngực lõm, vòm miệng cao, vẹo cột sống, và/hoặc trượt chỏm xương đùi. Các đặc điểm lâm sàng khác bao gồm lỏng lẻo khớp, giảm trương lực cơ hoặc yếu cơ gốc chi, môi dày, các phát hiện nhãn khoa (không thể khóc ra nước mắt ở trẻ sơ sinh [alacrima], mí mắt dày và lộn ra ngoài [ectropion], sụp mí nhẹ, và các dây thần kinh giác mạc nổi rõ), và dậy thì muộn. Các biểu hiện miệng của MEN2B có độ xâm nhập cao và thường dẫn đến chẩn đoán lâm sàng. PHPT không phải là một đặc điểm của MEN2B.
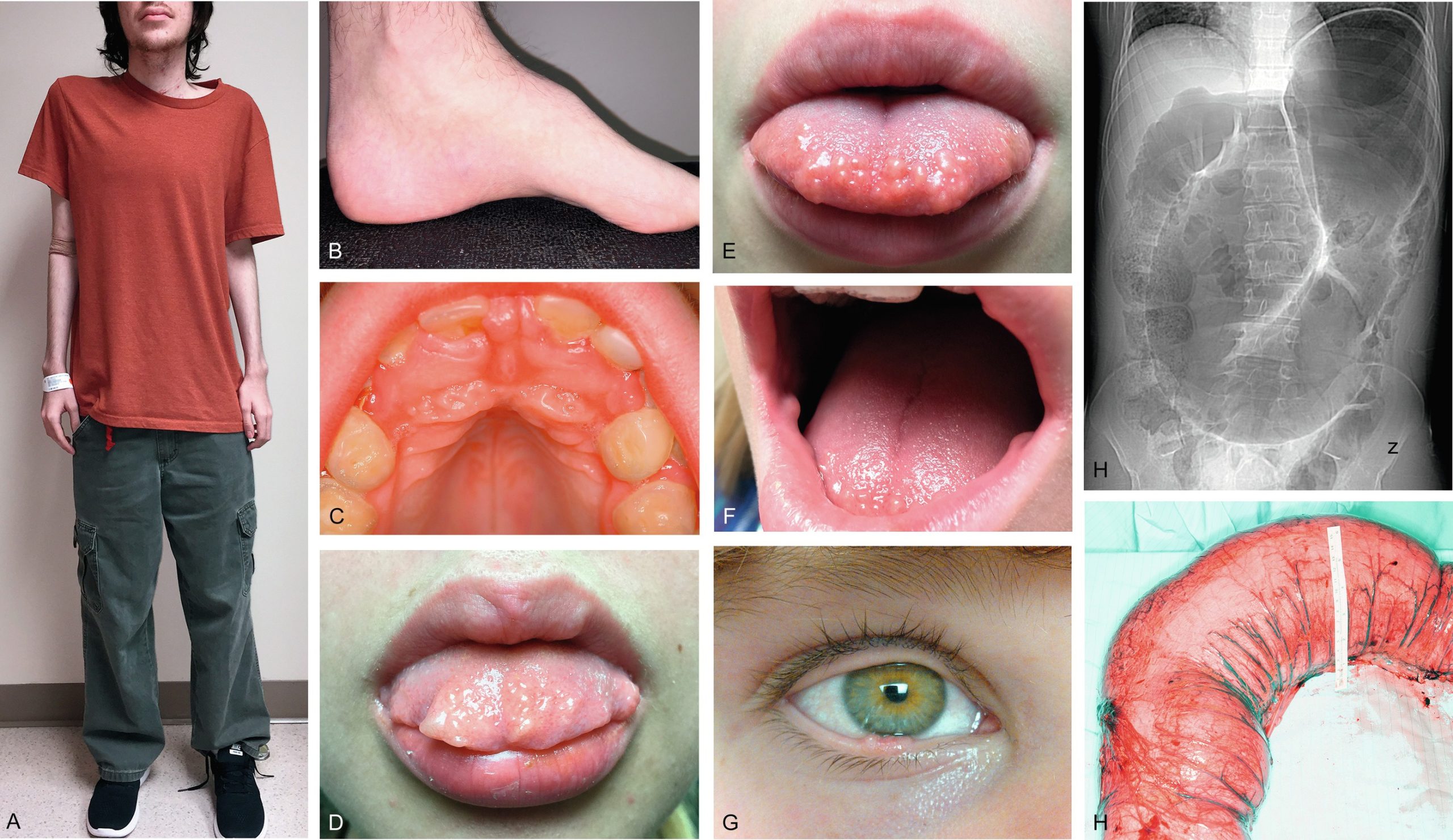
Hình 15.11 Kiểu hình lâm sàng đặc trưng của MEN2B: (1) thể trạng marfanoid (A) và các đặc điểm xương khác bao gồm vẹo cột sống (A), bàn chân vòm (B), và vòm miệng cao (C); (2) môi dày và u thần kinh ảnh hưởng đến lưỡi (D) và (E), niêm mạc miệng (F), kết mạc (G), và các bề mặt niêm mạc khác; (3) các dấu hiệu nhãn khoa bao gồm sụp mí và mí trên lộn ra ngoài (ectropion) (G); và (4) các vấn đề tiêu hóa chủ yếu liên quan đến suy giảm nhu động đại tràng do bệnh u hạch thần kinh ruột lan tỏa có thể dẫn đến phình đại tràng (H). Để biết thêm các biểu hiện lâm sàng của MEN2B, vui lòng tham khảo văn bản. (Nguồn: Castinetti, F., Moley, J., Mulligan, L., Waguespack, S.G. (2018). A comprehensive review on MEN2B. Endocr Relat Cancer, 25(2), T29–T39)
Ung thư biểu mô tuyến giáp thể tủy trong Đa u tuyến Nội tiết 2B
MTC xảy ra trong MEN2B là một bệnh ác tính xâm lấn với sự khởi phát di căn rất sớm (bệnh hạch đã được ghi nhận trong năm đầu đời), giai đoạn ung thư cao (được xác định bởi hệ thống phân loại khối u, hạch, và di căn [TNM]) tại thời điểm chẩn đoán, và tuổi khởi phát MTC trung bình (thập kỷ thứ hai của cuộc đời) sớm hơn khoảng 10 năm so với quan sát thấy ở MEN2A. Khởi phát MTC sớm hơn được quan sát thấy ở những trẻ mắc MEN2B có các biểu hiện ruột nặng, những trẻ này dường như cũng có tiên lượng kém hơn những trẻ không có các biểu hiện ruột này. Tỷ lệ mắc bệnh và tử vong cao hơn nhiều ở MEN2B, mặc dù tỷ lệ tử vong cao hơn có thể phản ánh sự khởi phát bệnh sớm hơn và giai đoạn khối u tiến triển hơn tại thời điểm trình bày, thay vì một ung thư biểu mô vốn dĩ xâm lấn hơn. Kết quả lâm sàng đang được cải thiện trong kỷ nguyên hiện tại do chẩn đoán và điều trị được cải thiện ở độ tuổi trẻ hơn và do đó ở giai đoạn MTC thấp hơn.
U tế bào ưa crôm trong Đa u tuyến Nội tiết 2B
So với dân số MEN2A không đồng nhất hơn, bệnh nhân mắc MEN2B có nguy cơ phát triển PHEO trong đời cao hơn (~ 50%). Kiểu hình lâm sàng của bệnh không khác so với MEN2A, mặc dù tuổi chẩn đoán trung bình là khoảng 25 tuổi, sớm hơn khoảng một thập kỷ so với MEN2A. Hai điểm khác biệt đáng chú ý khác giữa MEN2A và MEN2B là PHEO thậm chí còn ít có khả năng được chẩn đoán trước MTC và tỷ lệ tử vong trong lịch sử do PHEO thấp hơn so với tử vong do MTC. Mặc dù có kiểu hình MTC xâm lấn và một kinase tyrosine RET được kích hoạt cấu thành, nguy cơ PHEO ác tính ở bệnh nhân mắc MEN2B là cực kỳ hiếm (0%–3%). Cho đến nay, trường hợp PHEO liên quan đến MEN2B được ghi nhận trẻ nhất là ở một đứa trẻ 10 tuổi. Các hướng dẫn đương đại khuyến nghị bắt đầu tầm soát ở tuổi 11.
Thời điểm cắt tuyến giáp trong Đa u tuyến Nội tiết 2
Khi MTC di căn ra ngoài tuyến giáp, nó thường trở thành một căn bệnh không thể chữa khỏi và cuối cùng có thể gây ra giảm tuổi thọ. Do MEN2 có thể dễ dàng được chẩn đoán thông qua xét nghiệm di truyền tiên đoán, MTC di truyền đã trở thành một trong số ít các bệnh ác tính có thể được ngăn ngừa (thông qua cắt tuyến giáp dự phòng) hoặc chữa khỏi (thông qua cắt tuyến giáp sớm) trước khi nó trở nên rõ ràng về mặt lâm sàng. Trong tay các bác sĩ phẫu thuật có kinh nghiệm, trẻ em được thực hiện cắt toàn bộ tuyến giáp trước khi bắt đầu di căn có cơ hội tuyệt vời để không bị tái phát bệnh; chữa khỏi cũng có thể xảy ra ngay cả đối với trẻ em mắc MEN2 nguy cơ cao, mặc dù phẫu thuật không được thực hiện ở độ tuổi được quy định sớm nhất. Trong thực tế, nhiều mẫu bệnh phẩm cắt tuyến giáp từ trẻ em mắc MEN2A chứa các MTC vi thể, không di căn, thường được chữa khỏi sau khi cắt tuyến giáp sớm; ở những trẻ không được chữa khỏi sau phẫu thuật ban đầu, calcitonin có thể vẫn bất thường nhưng không có bằng chứng cấu trúc của bệnh. Đối với hầu hết trẻ em mắc MEN2B, việc chữa khỏi là không thể đạt được do thực tế là hầu hết các cuộc phẫu thuật ở nhóm dân số này là điều trị, không phải dự phòng, do chẩn đoán lâm sàng thường bị trì hoãn; tuy nhiên, ở trẻ em được chẩn đoán và điều trị trước 1 tuổi, chữa khỏi về mặt sinh hóa đạt được ở 83% các trường hợp.
Có sự đồng thuận rộng rãi rằng mục tiêu điều trị ở một đứa trẻ mắc MEN2 là loại bỏ tuyến giáp có nguy cơ trước khi xảy ra di căn MTC không thể chữa khỏi, đồng thời giảm thiểu tỷ lệ mắc bệnh y tế và phẫu thuật tiềm ẩn. Các khuyến nghị về thời điểm can thiệp phẫu thuật đã phát triển trong 25 năm qua. Trước đây chỉ dựa trên kiểu gen, thực hành đương đại là đề nghị cắt tuyến giáp sớm ở trẻ em ở độ tuổi được quyết định bởi kiểu gen, và trong trường hợp các đột biến ít có khả năng gây ra MTC ở trẻ nhỏ, nồng độ calcitonin cơ bản (xem Hình 15.5). Mặc dù siêu âm tuyến giáp thường được tích hợp vào việc ra quyết định lâm sàng, nhưng nó là một xét nghiệm không nhạy để chẩn đoán MTC và do đó không nên được sử dụng độc lập để xác định tuổi phẫu thuật.
Trong các hướng dẫn của Hiệp hội Tuyến giáp Hoa Kỳ (ATA) được công bố vào năm 2015, các đột biến RET gây bệnh được phân tầng thành ba loại nguy cơ chính: nguy cơ cao nhất (codon 918), nguy cơ cao (codon 634 và 883), và nguy cơ trung bình (tất cả các loại khác) (xem Hình 15.5). Các nghiên cứu gần đây đã cho thấy rằng các đột biến nguy cơ trung bình của ATA là một nhóm không đồng nhất và có thể cần phải có các phân nhóm nhỏ hơn trong loại này để thông báo tốt hơn về nguy cơ phát triển MTC. Cắt toàn bộ tuyến giáp được khuyến nghị trong năm đầu đời ở những người mang đột biến nguy cơ cao nhất và ở hoặc trước 5 tuổi đối với những người có đột biến nguy cơ cao. Với tất cả các đột biến RET khác (nguy cơ trung bình), việc phát hiện nồng độ calcitonin tăng cao có thể được sử dụng để giúp quyết định thời điểm phẫu thuật, lưu ý rằng một số đột biến nguy cơ trung bình của ATA có thể độc lực hơn những đột biến khác. Nồng độ calcitonin dưới 30 đến 40 pg/mL có liên quan đến nguy cơ di căn hạch cực kỳ thấp, và do đó phẫu thuật cho một đứa trẻ ngay khi calcitonin đang tăng lên sẽ dẫn đến chữa khỏi lâu dài. Cuối cùng, các quyết định về tuổi phẫu thuật nên được đưa ra bởi một đội ngũ đa ngành có kinh nghiệm trong quản lý MEN2 và cha mẹ/người giám hộ của đứa trẻ. Đáng chú ý, nồng độ calcitonin tăng cao không dự báo một cách đáng tin cậy chẩn đoán MTC; tăng giả có thể được tìm thấy trong các tình trạng không phải tân sinh, chẳng hạn như bệnh thận mạn tính, viêm tuyến giáp tự miễn, và cường cận giáp. Ngược lại, MTC có thể được xác định về mặt bệnh học ngay cả khi có nồng độ calcitonin bình thường.
Đa u tuyến Nội tiết Loại 4
MEN4 là một hội chứng gồm nhiều khối u nội tiết do các đột biến dòng mầm trong gen CDKN1B trên nhiễm sắc thể 12p13.1, mã hóa cho chất ức chế kinase phụ thuộc cyclin p27Kip1 và đóng một vai trò quan trọng trong việc điều hòa sự tăng sinh tế bào. Các đột biến đồng hợp tử ở gen Cdkn1b của chuột ban đầu được xác định là gây ra MENX, một hội chứng đa khối u ở chuột có các đặc điểm kiểu hình của cả MEN1 và MEN2. Sau đó, các nghiên cứu ở người không có đột biến MEN1 đã được chứng minh đã xác nhận rằng một nhóm nhỏ sẽ có các đột biến dị hợp tử trong gen tương đồng ở người CDKN1B, và những bệnh nhân này có kiểu hình giống MEN1, sau đó được gọi là MEN4. Các cá nhân có đột biến dòng mầm CDKN1B có nguy cơ phát triển các khối u của tuyến cận giáp (biểu hiện lâm sàng chính), tuyến yên, tuyến thượng thận, và/hoặc hệ thống thần kinh nội tiết đường tiêu hóa, cùng những khối u khác. Mặc dù MEN4 là một nguyên nhân rất hiếm của đa u tuyến nội tiết, việc tầm soát đột biến của gen CDKN1B nên được xem xét ở những bệnh nhân có chẩn đoán lâm sàng MEN1, nhưng xét nghiệm di truyền MEN1 âm tính. Chiến lược giám sát tối ưu chưa được thiết lập nhưng tối thiểu nên bao gồm xét nghiệm định kỳ cho PHPT và các khối u nội tiết chức năng khác.
Bệnh to đầu chi-khổng lồ liên kết nhiễm sắc thể X
Bệnh to đầu chi-khổng lồ liên kết nhiễm sắc thể X (X-LAG) là rối loạn di truyền được mô tả gần đây nhất có liên quan đến các khối u tuyến yên ở trẻ em. Đây là một rối loạn có di truyền liên kết X, được đặc trưng bởi bệnh khổng lồ tuyến yên với sự khởi phát trong vài năm đầu đời (< 4 tuổi). Bệnh là do các vi nhân đôi dòng mầm di truyền hoặc mới (phổ biến nhất) trên nhiễm sắc thể Xq26.3 bao gồm gen chịu trách nhiệm, GPR101, mã hóa cho một thụ thể mồ côi kết hợp với G-protein. Thể khảm soma cho vi nhân đôi Xq26.3 dẫn đến kiểu hình X-LAG cũng đã được báo cáo trong các trường hợp đơn lẻ ở nam giới. Một biến thể sai nghĩa GPR101 dòng mầm và soma (p.E308D) cũng đã được xác định trong một số trường hợp bệnh to đầu chi, mặc dù vẫn chưa chắc chắn liệu biến thể DNA này có đóng vai trò gây bệnh trong sinh bệnh học của các u tuyến somatotroph hay không.
Về mặt lâm sàng, bệnh nhân X-LAG có biểu hiện thừa hormone tăng trưởng rõ rệt, có liên quan đến tăng prolactin máu ở khoảng 80% đến 90% các trường hợp; về mặt mô học, xác định được các u tuyến somatotroph–lactotroph hỗn hợp và/hoặc tăng sản tuyến yên. Trái ngược hoàn toàn với FIPA liên quan đến AIP, X-LAG được đặc trưng bởi sự chiếm ưu thế ở nữ giới, với 24/33 trường hợp được báo cáo là nữ. Điều thú vị là tất cả các ca bệnh chỉ điểm nữ được công bố cho đến nay đều là các ca bệnh mới (de novo) và việc truyền dọc một đoạn nhân đôi GPR101 từ một phụ huynh bị ảnh hưởng sang con gái vẫn chưa được báo cáo.
Các hội chứng khối u khác liên quan đến tân sinh nội tiết
Đa polyp liên quan đến Adenomatous Polyposis Coli
Các tình trạng đa polyp liên quan đến adenomatous polyposis coli (APC) là kết quả của các đột biến dòng mầm di truyền trội trong gen đè nén khối u APC (nhiễm sắc thể 5q22.2) và bao gồm các hội chứng lâm sàng đa polyp tuyến có tính gia đình (FAP, bao gồm cả các hội chứng Gardner và Turcot hiện đã là lịch sử), FAP suy yếu, và ung thư biểu mô tuyến dạ dày và đa polyp gần của dạ dày (GAPPS). Bệnh nhân có đột biến APC có nguy cơ cao nhất bị ung thư đại tràng nhưng cũng có thể phát triển tân sinh nội tiết. So với dân số chung, các ACT phổ biến hơn ít nhất hai lần (16% bệnh nhân) trong FAP, và tương tự như tân sinh thượng thận lẻ tẻ, các ACT liên quan đến APC có thể hoạt động nội tiết hoặc không chức năng và hiếm khi có thể là ác tính. Bệnh nhân có đột biến APC cũng có nguy cơ ung thư tuyến giáp trong đời lên đến 12%. Phân nhóm chính là ung thư biểu mô tuyến giáp thể nhú, kinh điển là biến thể mô học dạng sàng-morular, và đó là một chẩn đoán hầu như chỉ được thực hiện ở phụ nữ trẻ trong thập kỷ thứ ba của cuộc đời. Ngoài việc sờ nắn tuyến giáp, các ý kiến về việc tầm soát ung thư tuyến giáp trong một tình trạng đa polyp liên quan đến APC là khác nhau; không khuyến nghị giám sát định kỳ các khối u thượng thận.
Hội chứng Beckwith-Wiedemann
Hội chứng Beckwith-Wiedemann (BWS) là một rối loạn tăng trưởng bẩm sinh gây ra bởi sự rối loạn điều hòa phiên mã gen trong một vùng in dấu trên nhiễm sắc thể 11p15 bao gồm các gen, chẳng hạn như CDKN1C và IGF2, là những chất điều hòa mạnh mẽ sự tăng trưởng của thai nhi. Các phát hiện lâm sàng rất đa dạng và bao gồm thai to và phì đại nửa người, lưỡi to, tạng to, các khối u phôi thai (ví dụ, u Wilms, u nguyên bào gan, u nguyên bào thần kinh, u nguyên bào tụy, và sarcoma cơ vân), thoát vị rốn, hạ đường huyết sơ sinh, nếp/lỗ tai, ACT/tế bào thượng thận to, và các bất thường thận (ví dụ, loạn sản tủy, vôi hóa thận, thận xốp tủy, và thận to). Một loạt các khối u ác tính, thường giới hạn ở khởi phát thời thơ ấu, đã được liên kết với BWS và xảy ra ở 7,4% tất cả các trường hợp. ACT, cả lành tính và ác tính, là một trong những khối u tân sinh phổ biến hơn được xác định trong BWS, chiếm tới 8% các khối u lành tính và 7% các khối u ác tính. Tế bào vỏ thượng thận thai nhi to (một thành phần của tạng to tiềm ẩn) và các tổn thương nang thượng thận cũng xảy ra. PHEO và ung thư biểu mô tuyến giáp là các khối u nội tiết bổ sung đã được báo cáo. Các quy trình khác nhau đã được đề xuất để giám sát khối u trong BWS và các thuật toán đã được phát triển để quản lý trẻ em có khối u thượng thận được xác định. Trẻ em có lưỡng bội một nguồn gốc từ cha dường như có nguy cơ cao nhất về sự phát triển của ACT và do đó việc tầm soát bằng siêu âm bụng nên tập trung nhiều hơn vào những trẻ này, cũng như những trẻ có xét nghiệm phân tử âm tính.
Tam chứng Carney
Tam chứng Carney ban đầu được mô tả vào năm 1977 là sự kết hợp của u cơ trơn dạng biểu mô dạ dày (sau này được đổi tên thành GIST), PGL, và u sụn phổi. Theo thời gian, kiểu hình đã mở rộng để bao gồm các ACT không chức năng lâm sàng và u cơ trơn thực quản. Tam chứng Carney ảnh hưởng chủ yếu đến phụ nữ trẻ (85%) với độ tuổi khởi phát trung bình là 20 tuổi (7–48 tuổi). Do biểu hiện không hoàn toàn của kiểu hình, PGL không có mặt ở tất cả các bệnh nhân bị nghi ngờ mắc tam chứng Carney. PHEO có thể xảy ra ở một số ít bệnh nhân, và thể động mạch chủ-phổi là một vị trí phổ biến cho sự phát triển của PGL, mặc dù PGL xảy ra tương đương ở đầu và cổ, ngực, và bụng.
Mặc dù tam chứng Carney không phải là một rối loạn di truyền theo nghĩa truyền thống, khoảng 10% bệnh nhân sẽ được xác định có một biến thể DNA dòng mầm trong các gen SDHA, SDHB, hoặc SDHC. Hơn nữa, sự tăng methyl hóa của SDHC dường như là đặc điểm phân tử chính của tam chứng Carney.
Hội chứng DICER1
Các đột biến dòng mầm trong gen DICER1 gây ra hội chứng DICER1, một rối loạn di truyền trội trên nhiễm sắc thể thường hiếm gặp có độ xâm nhập giảm, được đặc trưng bởi một loạt các khối u lành tính và ác tính, bao gồm u nguyên bào màng phổi-phổi (PPB), các khối u mô đệm dây sinh dục buồng trứng (chủ yếu là u tế bào Sertoli-Leydig nhưng cũng có u tế bào hạt vị thành niên và gynandroblastoma), u nang thận, và các khối u tân sinh tuyến giáp (bướu cổ đa nhân và ung thư tuyến giáp biệt hóa). Các khối u khác ít được quan sát hơn bao gồm u biểu mô tủy thể mi, sarcoma cơ vân dạng chùm nho của cổ tử cung hoặc các vị trí khác, hamartoma sụn-trung mô mũi, sarcoma thận, u nguyên bào tuyến yên, và u nguyên bào tuyến tùng.
DICER1 (nhiễm sắc thể 14q32.13) là một gen mã hóa cho một endoribonuclease cắt các tiền chất axit ribonucleic (RNA) nhỏ không mã hóa để tạo ra các microRNA trưởng thành tham gia vào việc điều hòa sau phiên mã của biểu hiện gen. Các đột biến ở DICER1 lần đầu tiên được xác định trong PPB có tính gia đình và sau đó được phát hiện là nguyên nhân gây ra một hội chứng khối u đa dạng, bao gồm nhiều loại khối u khác. Hội chứng DICER1 thường là kết quả của một đột biến bất hoạt dòng mầm, theo sau là một đột biến thứ hai ở tế bào soma chủ yếu liên quan đến miền xúc tác RNase IIIb của DICER1. Hầu hết các đột biến dòng mầm là di truyền nhưng 10% đến 20% có thể phát sinh mới (de novo). Các đột biến DICER1 thể khảm soma, thường đi kèm với các đột biến cắt ngắn soma thứ hai hoặc mất tính dị hợp tử, cũng có thể gây ra hội chứng DICER1.
Các nốt tuyến giáp dường như là một trong những biểu hiện phổ biến nhất ở bệnh nhân có đột biến DICER1 dòng mầm, và có nguy cơ tăng đáng kể về bướu cổ đa nhân so với nhóm chứng gia đình và nguy cơ ung thư tuyến giáp tăng ít nhất 16 lần so với dân số chung. Bướu cổ đa nhân có tính gia đình thực tế có thể là biểu hiện lâm sàng duy nhất, và ở đứa trẻ đang được đánh giá về bướu cổ dạng nốt có thể có hội chứng, đầu to có thể là một manh mối cho sự hiện diện của một đột biến DICER1 dòng mầm. Tương tự như bệnh nốt tuyến giáp không hội chứng, nữ giới bị ảnh hưởng nhiều hơn, và đến 40 tuổi, tỷ lệ mắc tích lũy của bướu cổ đa nhân hoặc cắt tuyến giáp là 75% ở phụ nữ và 17% ở nam giới. Ung thư tuyến giáp biệt hóa xảy ra ở dưới 3% những người mang DICER1 và là một khối u nguy cơ thấp dường như không xâm lấn hơn về mặt lâm sàng. Mặc dù việc tiếp xúc với hóa trị hoặc xạ trị ban đầu được cho là góp phần vào nguy cơ ung thư tuyến giáp, một đột biến DICER1 dòng mầm đơn độc dường như là đủ để tăng nguy cơ ung thư tuyến giáp. Giám sát các khối u tuyến giáp ở trẻ em mắc hội chứng DICER1 bao gồm siêu âm tuyến giáp định kỳ, bắt đầu từ 8 tuổi và sau đó mỗi 3 năm hoặc thường xuyên hơn, nếu đang được điều trị cho một khối u khác, mặc dù một số trung tâm khuyến nghị chỉ khám sức khỏe, bắt đầu từ 10 tuổi ở những trẻ không tiếp xúc với hóa trị alkyl hóa.
U nguyên bào tuyến yên, xảy ra ở dưới 1% bệnh nhân có đột biến DICER1, là những khối u tân sinh cực kỳ hiếm và xâm lấn, phát sinh từ tuyến yên trước của thai nhi và có biểu hiện lâm sàng ở độ tuổi dưới 24 tháng với bệnh Cushing phụ thuộc ACTH và/hoặc liệt vận nhãn do hiệu ứng khối. Bởi vì khối u này rất hiếm, không rõ liệu việc giám sát định kỳ có cần thiết trong mọi trường hợp hay không, mặc dù một số trung tâm ủng hộ việc tầm soát MRI. Bất kể, việc đánh giá và nhận biết các dấu hiệu và triệu chứng của u nguyên bào tuyến yên (tăng cân nhanh, liệt vận nhãn) sẽ dẫn đến chẩn đoán sớm.
Hội chứng Li-Fraumeni
Hội chứng Li-Fraumeni (LFS) là một hội chứng khuynh hướng ung thư có độ xâm nhập cao do các đột biến dòng mầm di truyền trội trên nhiễm sắc thể thường trong gen đè nén khối u TP53 (protein khối u p53; nhiễm sắc thể 17p13.1). Sarcoma mô mềm và xương, ung thư vú tiền mãn kinh, ACT, và các khối u não là những khối u cốt lõi của LFS. Khối u nội tiết nguyên mẫu liên quan đến LFS là ACT (cả lành tính và ác tính), mặc dù một số trường hợp ung thư biểu mô tuyến giáp cũng đã được báo cáo, đặc biệt là ở các gia đình Brazil. Vỏ thượng thận là vị trí phát triển tân sinh phổ biến thứ ba trong LFS. ACT có tuổi khởi phát sớm, với chẩn đoán phổ biến nhất ở trẻ rất nhỏ, những người thực tế có thể là ca bệnh chỉ điểm cho một gia đình LFS. Ở các gia đình miền nam Brazil, đột biến TP53 p.Arg337His có liên quan đến tính nhạy cảm cao hơn với ACT đơn độc ở trẻ em (tuổi chẩn đoán trung bình 3 tuổi; khoảng 4 tháng đến 13,5 tuổi), mặc dù nguy cơ ung thư dường như cũng tăng lên đối với những người trưởng thành mang đột biến này. Trẻ em mắc ACT thường có biểu hiện nam hóa, nhưng ở mọi lứa tuổi, những khối u này có thể có các dấu hiệu/triệu chứng khác của tăng chức năng vỏ thượng thận (chẳng hạn như hội chứng Cushing) và/hoặc với các triệu chứng do hiệu ứng khối của khối u, đặc biệt là trong trường hợp các khối u không chức năng. Các khuyến nghị tầm soát ung thư, bao gồm theo dõi sự phát triển của một ACT, đã có sẵn.
Hội chứng Lynch (Ung thư đại trực tràng không polyp di truyền)
Hội chứng Lynch là một rối loạn có di truyền trội trên nhiễm sắc thể thường, thứ phát sau các đột biến trong các gen sửa chữa sai cặp DNA và được đặc trưng bởi nguy cơ tăng lên đối với ung thư đại trực tràng và nhiều khối u ác tính khác. Chẩn đoán ung thư biểu mô vỏ thượng thận đã được báo cáo ở các cá nhân và gia đình mắc hội chứng Lynch, mặc dù cho đến nay chưa có một biểu hiện ACT ở trẻ em xảy ra trong bối cảnh này.
U xơ thần kinh loại 1
NF1, còn được gọi là bệnh Von Recklinghausen, là một rối loạn di truyền trội trên nhiễm sắc thể thường do các đột biến trong gen NF1, một gen đè nén khối u nằm trên nhiễm sắc thể 17. Các đốm café-au-lait, u xơ thần kinh (dưới da và dạng đám rối), tàn nhang ở nách và bẹn, u thần kinh đệm đường thị giác, loạn sản xương, và các nốt Lisch (hamartoma lành tính) của mống mắt là những đặc điểm lâm sàng chính. Bệnh nhân NF1 có nguy cơ cao phát triển PHEO/PGL và các NET khác.
Tỷ lệ mắc một khối u sản xuất catecholamin ở bệnh nhân NF1 là 2% đến 6%. PHEO (lên đến 17% là hai bên) chiếm phần lớn các trường hợp liên quan đến NF1 nhưng PGL trong ổ bụng và khung chậu được chẩn đoán ở tới 6%. Tuổi chẩn đoán trung bình là trong thập kỷ thứ năm của cuộc đời, và các PHEO liên quan đến NF1 thường được đặc trưng bởi một kiểu hình adrenergic. Bệnh ác tính không phổ biến nhưng xảy ra ở 5% đến 12% các trường hợp. Hiếm khi, một PHEO hỗn hợp (một khối u hỗn hợp bao gồm PHEO và một khối u có nguồn gốc thần kinh liên quan về mặt phát triển, chẳng hạn như u nguyên bào thần kinh, u hạch thần kinh, hoặc u hạch nguyên bào thần kinh) có thể xảy ra trong bối cảnh NF1.
Các NET đường tiêu hóa xảy ra ở dưới 10% bệnh nhân NF1 và hầu như chỉ là các u tiết somatostatin ở tá tràng xảy ra ở vùng quanh bóng Vater và có biểu hiện lâm sàng là do chính khối u (đau, vàng da, chảy máu) chứ không phải là một hội chứng u tiết somatostatin. Các u tiết somatostatin liên quan đến NF1 thường được chẩn đoán trong thập kỷ thứ năm của cuộc đời. Các loại NET khác đã được báo cáo ở bệnh nhân NF1 bao gồm u insulin, u gastrin, và các NET tụy không chức năng. Các báo cáo trường hợp PHPT do các khối u cận giáp lành tính và ác tính và ACT cũng đã được báo cáo xảy ra trong NF1. Cuối cùng, một bệnh nội tiết độc đáo liên quan đến NF1 ở trẻ em có u thần kinh đệm đường thị giác xâm lấn là hội chứng lâm sàng của bệnh khổng lồ do tiết hormone tăng trưởng không được điều hòa.
Hội chứng Peutz-Jeghers
Hội chứng Peutz-Jeghers (PJS) là một hội chứng di truyền trội trên nhiễm sắc thể thường có độ xâm nhập không hoàn toàn, được đặc trưng bởi các polyp hamartoma trong đường tiêu hóa và tăng sắc tố niêm mạc da. Nó chủ yếu do các đột biến bất hoạt trong gen STK11 (LKB1) (nhiễm sắc thể 19p13.3). Các cá nhân mắc PJS có nguy cơ tăng lên đối với các khối u ác tính khác nhau trong suốt cuộc đời của họ, chủ yếu là ung thư đường tiêu hóa (đại trực tràng > dạ dày > ruột non) và ung thư vú, tiếp theo là ung thư tụy, các khối u ác tính phụ khoa, và ung thư phổi. Một số báo cáo trường hợp ung thư tuyến giáp biệt hóa đã được công bố, nhưng vẫn chưa rõ liệu đây là một sự xuất hiện ngẫu nhiên hay một mối liên quan trực tiếp với PJS.
Bệnh nhân mắc PJS có nguy cơ bị các khối u tuyến sinh dục độc đáo có thể có biểu hiện lâm sàng với một bệnh nội tiết ở trẻ em: u tế bào Sertoli của tinh hoàn ở nam giới và các khối u dây sinh dục buồng trứng với các ống hình khuyên (SCTAT) ở nữ giới. U tế bào Sertoli tinh hoàn trong PJS là các tổn thương lành tính thường xảy ra ở các bé trai trước tuổi dậy thì (tuổi trung bình 6,8 tuổi), có biểu hiện nữ hóa tuyến vú và tinh hoàn to hai bên (hiếm khi một bên). Các phát hiện vi thể là đặc biệt và thuật ngữ tân sinh tế bào Sertoli hyalin hóa tế bào lớn trong ống cũng đã được đề xuất. Vôi hóa là bất thường và không lan rộng trong các tổn thương này, mặc dù các u tế bào Sertoli vôi hóa tế bào lớn (tương tự như những u được xác định trong phức hợp Carney) cũng có thể xảy ra trong phổ PJS. Nữ hóa tuyến vú là do sản xuất quá mức estrogen thứ phát sau nồng độ hoạt động aromatase cao, làm cho việc sử dụng một chất ức chế aromatase trở thành phương pháp tối ưu để điều trị nội khoa các trường hợp như vậy. SCTAT là một khối u tân sinh đặc biệt có các đặc điểm hình thái nằm giữa u tế bào Sertoli và u tế bào hạt. Khi xảy ra trong PJS, những khối u này thường là các tổn thương nhỏ, vôi hóa, đa ổ, và hai bên, so với các SCTAT không phải PJS. SCTAT hầu như luôn là một khối u tân sinh lành tính với tuổi chẩn đoán trung bình là 27 tuổi (khoảng 4–57 tuổi). Biểu hiện lâm sàng thường là do sản xuất quá mức estrogen và biểu hiện bằng kinh nguyệt không đều ở phụ nữ sau tuổi dậy thì và dậy thì sớm ở các bé gái trước tuổi dậy thì. Đau bụng và một khối u phần phụ sờ thấy được cũng có thể là các đặc điểm biểu hiện, và SCTAT cũng có thể là một phát hiện tình cờ. Khám sức khỏe hàng năm (đánh giá nữ hóa tuyến vú/tinh hoàn to ở bé trai và dậy thì sớm ở bé gái) được khuyến nghị cho trẻ em mắc PJS.
Hội chứng u Hamartoma PTEN
Hội chứng u hamartoma PTEN (PHTS) là kết quả của các đột biến dòng mầm trong gen đè nén khối u PTEN (phosphatase and tensin homolog) (nhiễm sắc thể 10q23.31) và được di truyền theo kiểu trội trên nhiễm sắc thể thường. Độ xâm nhập cao (> 80%) và chẩn đoán lâm sàng có thể bị bỏ sót do biểu hiện kiểu hình thay đổi và các khối u liên quan có tỷ lệ mắc nền trong dân số cao. PHTS bao gồm phổ lâm sàng của hội chứng Cowden, hội chứng Bannayan-Riley-Ruvalcaba, hội chứng Proteus liên quan đến PTEN, và hội chứng giống Proteus. Hội chứng Cowden (CS), kiểu hình PHTS phổ biến nhất và có tiêu chuẩn chẩn đoán đã được thiết lập, là một hội chứng đa hamartoma với nguy cơ tăng lên đối với cả các khối u lành tính và ác tính phát sinh từ tế bào nang tuyến giáp và, ở phụ nữ, vú và nội mạc tử cung; các biểu hiện da đặc trưng bao gồm các sẩn dạng nhú, u nang lông mặt, và dày sừng đầu chi. Đầu to (chu vi vòng đầu > 2 độ lệch chuẩn [SD]/phân vị thứ 97,5) là một phát hiện gần như phổ biến ở trẻ em có đột biến PTEN, và các rối loạn phát triển thần kinh là phổ biến (rối loạn phổ tự kỷ ở 50%). U tân sinh tuyến giáp cuối cùng xảy ra ở phần lớn các cá nhân mắc CS và bao gồm bướu cổ đa nhân, u tuyến nang, và ung thư biểu mô tuyến giáp biệt hóa (ung thư biểu mô tuyến giáp thể nhú và nang). Ung thư biểu mô nang dường như chiếm tỷ lệ cao hơn ở những bệnh nhân dương tính với đột biến PTEN (tuổi báo cáo trẻ nhất là 7 tuổi), nhưng ung thư biểu mô nhú cũng phổ biến. Nguy cơ tân sinh tuyến giáp bắt đầu từ thời thơ ấu, và một phần tư trẻ em mắc PHTS có hình ảnh tuyến giáp bất thường. Giám sát trẻ em mắc PHTS bao gồm khám sức khỏe hàng năm và siêu âm tuyến giáp định kỳ.
Phức hợp xơ cứng củ
Phức hợp xơ cứng củ (TSC) là một rối loạn hamartoma đa cơ quan, di truyền trội trên nhiễm sắc thể thường, được đặc trưng bởi các bất thường của da (các đốm giảm sắc tố, các tổn thương da dạng hoa giấy, u xơ mạch mặt, các mảng da sần, các mảng xơ ở đầu, u xơ quanh móng); não (loạn sản vỏ não, các nốt dưới màng não thất và u sao bào tế bào khổng lồ dưới màng não thất, co giật, thiểu năng trí tuệ/chậm phát triển); thận (u cơ mỡ mạch, nang, ung thư biểu mô tế bào thận); tim (u cơ vân, rối loạn nhịp tim); và phổi (bệnh u cơ trơn bạch huyết). Hai gen được biết đến gây ra TSC bao gồm TSC1 (nhiễm sắc thể 9q34.13) và TSC2 (nhiễm sắc thể 16p13.3), mã hóa cho các protein hamartin và tuberin, tương ứng. Nhiều khối u tân sinh nội tiết xảy ra trong bối cảnh TSC đã được báo cáo, bao gồm các u tuyến yên chức năng, tăng sản và u tuyến cận giáp, các NET tụy, và các báo cáo trường hợp PHEO và ung thư tuyến giáp thể nhú. Các NET tụy có thể lành tính hoặc ác tính và chức năng hoặc không chức năng, với một tỷ lệ lớn các trường hợp này liên quan đến các đột biến gen TSC2. U insulin dường như chiếm tỷ lệ cao hơn ở bệnh nhân TSC và do đó nên có một ngưỡng thấp để đánh giá lâm sàng ở những người có các dấu hiệu hoặc triệu chứng kinh điển của hạ đường huyết hoặc các triệu chứng thần kinh xấu đi.
Bệnh Von Hippel-Lindau
Bệnh VHL là một rối loạn di truyền trội trên nhiễm sắc thể thường, có độ xâm nhập cao do các đột biến dòng mầm trong VHL (nhiễm sắc thể 3p25.3), một gen đè nén khối u lần đầu tiên được xác định vào năm 1993, và được đặc trưng bởi một kiểu hình lâm sàng thay đổi bao gồm u nguyên bào mạch máu của võng mạc (còn gọi là u mạch võng mạc) và TKTW; nang thận và ung thư biểu mô tế bào thận sáng; PHEO/PGL; nang tụy, u nang tuyến, và các NET tụy; các khối u túi nội bạch huyết; và u nang tuyến của mào tinh ở nam giới và dây chằng rộng của tử cung ở nữ giới. Khoảng 20% bệnh nhân mắc VHL không có cha mẹ bị ảnh hưởng và do đó hoặc mang một đột biến mới (de novo) hoặc thừa hưởng một đột biến VHL từ một phụ huynh có thể khảm dòng mầm; xét nghiệm di truyền rất đáng tin cậy, xác định được một đột biến ở hầu hết các cá nhân bị ảnh hưởng lâm sàng. Phần lớn các đột biến VHL là các đoạn xóa/chèn nhỏ hoặc các đột biến điểm nhưng các đoạn xóa lớn chiếm 11% các biến thể gây bệnh VHL.
Mặc dù tuổi chẩn đoán PHEO/PGL trung bình trong VHL là 28 tuổi, có tới 20% bệnh nhân nhi (tuổi trung bình 12 tuổi), chủ yếu là những cá nhân có đột biến sai nghĩa của gen VHL, sẽ được chẩn đoán mắc PHEO/PGL (xem Hình 15.1). PHEO phổ biến hơn nhiều so với PGL và bệnh ác tính là hiếm gặp. Những khối u này có thể là một trong những biểu hiện lâm sàng sớm nhất của VHL, đã được chẩn đoán ở trẻ mới 2 tuổi, và nó cũng có thể là biểu hiện lâm sàng duy nhất của VHL, như được thấy trong VHL loại 2C. Tầm soát định kỳ thường được khuyến nghị bắt đầu ở tuổi 5 ở trẻ em có đột biến sai nghĩa và/hoặc tiền sử gia đình có PHEO/PGL; những người khác đã ủng hộ việc xét nghiệm sớm hơn. Mặc dù gần như tất cả các PHEO/PGL liên quan đến VHL là các khối u noradrenergic chức năng (xem phần PHEO/PGL trước đó), các PGL đối giao cảm không chức năng xảy ra trong bối cảnh VHL cũng đã được mô tả.
Các NET tụy phát triển ở 15% bệnh nhân VHL. Rất ít trường hợp ở trẻ em đã được báo cáo trong y văn, và trường hợp trẻ nhất là ở một đứa trẻ 10 tuổi. Những khối u này không chức năng về mặt lâm sàng (do đó tầm soát sinh hóa là không cần thiết) và chúng có thể là ác tính, đặc biệt với các tổn thương lên đến 2,8 đến 3 cm hoặc với thời gian nhân đôi khối u nhanh, và những khối u ở bệnh nhân có đột biến sai nghĩa dòng mầm hoặc một đột biến ở exon 3 của VHL. Tầm soát định kỳ cho các NET tụy được thực hiện như một phần của việc xét nghiệm các biểu hiện trong ổ bụng khác của VHL và bao gồm siêu âm không liên tục (thường bắt đầu từ 8 tuổi) hoặc MRI (bắt đầu từ 16 tuổi), mặc dù một số nhóm đã khuyến nghị MRI bắt đầu từ 10 tuổi. Tương tự như MEN1, MRI được ưu tiên hơn CT để giảm thiểu phơi nhiễm bức xạ ion hóa suốt đời.
Cuối cùng, các khối u tế bào steroid buồng trứng tiết testosterone đã được mô tả, và chẩn đoán này nên được xem xét ở phụ nữ mắc VHL có biểu hiện một khối u phần phụ, vô kinh, và/hoặc rậm lông.
Tóm tắt và các phát triển trong tương lai
Mặc dù hiếm gặp, các khối u tân sinh nội tiết ở trẻ em, đặc biệt là các NET, chẳng hạn như PHEO và MTC, thường liên quan đến một hội chứng khuynh hướng khối u đã biết. Những tiến bộ trong y học bộ gen đã cải thiện sự hiểu biết của chúng ta về nguyên nhân và sinh lý bệnh của những rối loạn này, điều này đã thay đổi cách các nhà cung cấp dịch vụ y tế quản lý bệnh nhân mắc các bệnh được thảo luận trong chương này. Bởi vì đây là một lĩnh vực phát triển nhanh chóng, người đọc nên tiếp tục tìm kiếm thông tin cập nhật nhất cho các quyết định lâm sàng quan trọng liên quan đến việc chăm sóc bệnh nhân cá nhân. Nghiên cứu trong tương lai sẽ bổ sung vào kiến thức của chúng ta về mối tương quan kiểu gen-kiểu hình; các chiến lược tầm soát tối ưu cho trẻ em không triệu chứng được biết là mang một đột biến gây bệnh; thời điểm can thiệp, chẳng hạn như cắt tuyến giáp sớm trong MEN2 và cắt bỏ cận giáp trong MEN1; và việc điều trị bệnh tiến triển hoặc không thể phẫu thuật bằng các liệu pháp nhắm mục tiêu mới hơn.
Bảng Chú giải Thuật ngữ Y học Anh-Việt (Chương 15)
| STT | Thuật ngữ tiếng Anh | Phiên âm IPA | Nghĩa Tiếng Việt |
|---|---|---|---|
| 1 | Pheochromocytoma | /ˌfiː.oʊˌkroʊ.moʊ.saɪˈtoʊ.mə/ | U tế bào ưa crôm |
| 2 | Paraganglioma | /ˌpær.əˌɡæŋ.ɡliˈoʊ.mə/ | U cận hạch thần kinh |
| 3 | Medullary Thyroid Carcinoma | /ˈmɛd.jʊˌlɛr.i ˈθaɪ.rɔɪd ˌkɑːr.sɪˈnoʊ.mə/ | Ung thư biểu mô tuyến giáp thể tủy |
| 4 | Hereditary Endocrine Neoplasia | /həˈrɛd.ɪˌtɛr.i ˈɛn.də.krɪn ˌniː.oʊˈpleɪ.zi.ə/ | Đa u tuyến nội tiết di truyền |
| 5 | Endocrine neoplasms | /ˈɛn.də.krɪn ˈniː.oʊˌplæz.əmz/ | U tân sinh nội tiết |
| 6 | Benign | /bɪˈnaɪn/ | Lành tính |
| 7 | Malignant | /məˈlɪɡ.nənt/ | Ác tính |
| 8 | Endocrine glands | /ˈɛn.də.krɪn ɡlændz/ | Tuyến nội tiết |
| 9 | Neuroendocrine tissues | /ˌnʊr.oʊˈɛn.də.krɪn ˈtɪʃ.uːz/ | Mô thần kinh nội tiết |
| 10 | Paraganglia | /ˌpær.əˈɡæŋ.ɡli.ə/ | Cận hạch (thần kinh) |
| 11 | Papillary thyroid carcinoma | /ˈpæp.ɪˌlɛr.i ˈθaɪ.rɔɪd ˌkɑːr.sɪˈnoʊ.mə/ | Ung thư biểu mô tuyến giáp thể nhú |
| 12 | Sporadic | /spəˈræd.ɪk/ | Lẻ tẻ, không có tính gia đình |
| 13 | Germline mutation | /ˈdʒɜːrm.laɪn mjuːˈteɪ.ʃən/ | Đột biến dòng mầm |
| 14 | Catecholamine | /ˌkæt.əˈkoʊ.ləˌmiːn/ | Catecholamin |
| 15 | Familial | /fəˈmɪl.i.əl/ | Có tính gia đình |
| 16 | Hereditary tumor syndrome | /həˈrɛd.ɪˌtɛr.i ˈtuː.mər ˈsɪn.droʊm/ | Hội chứng khối u di truyền |
| 17 | Autosomal dominant | /ˌɔː.təˈsoʊ.məl ˈdɒm.ɪ.nənt/ | Di truyền trội trên nhiễm sắc thể thường |
| 18 | Inactivating mutations | /ɪnˈæk.tɪˌveɪ.tɪŋ mjuːˈteɪ.ʃənz/ | Đột biến bất hoạt |
| 19 | Loss of function | /lɒs əv ˈfʌŋk.ʃən/ | Mất chức năng |
| 20 | Tumor suppressor gene | /ˈtuː.mər səˈprɛs.ər dʒiːn/ | Gen đè nén khối u |
| 21 | Genetic testing | /dʒəˈnɛt.ɪk ˈtɛs.tɪŋ/ | Xét nghiệm di truyền |
| 22 | Pathophysiology | /ˌpæθ.oʊˌfɪz.iˈɒl.ə.dʒi/ | Sinh lý bệnh |
| 23 | Genotype-phenotype | /ˈdʒiː.noʊˌtaɪp – ˈfiː.noʊˌtaɪp/ | Kiểu gen – kiểu hình |
| 24 | Predictive genetic testing | /prɪˈdɪk.tɪv dʒəˈnɛt.ɪk ˈtɛs.tɪŋ/ | Xét nghiệm di truyền tiên đoán |
| 25 | Asymptomatic carrier | /ˌeɪ.sɪmp.təˈmæt.ɪk ˈkær.i.ər/ | Người mang gen không triệu chứng |
| 26 | Therapeutic intervention | /ˌθɛr.əˈpjuː.tɪk ˌɪn.tərˈvɛn.ʃən/ | Can thiệp điều trị |
| 27 | Multidisciplinary | /ˌmʌl.tiˈdɪs.ə.plɪˌnɛr.i/ | Đa chuyên khoa |
| 28 | Genetic counseling | /dʒəˈnɛt.ɪk ˈkaʊn.səl.ɪŋ/ | Tư vấn di truyền |
| 29 | Multiple endocrine neoplasia | /ˈmʌl.tɪ.pəl ˈɛn.də.krɪn ˌniː.oʊˈpleɪ.zi.ə/ | Đa u tuyến nội tiết |
| 30 | Index case | /ˈɪn.dɛks keɪs/ | Ca bệnh chỉ điểm, ca bệnh đầu tiên |
| 31 | Disease penetrance | /dɪˈziːz ˈpɛn.ɪ.trəns/ | Độ xâm nhập của bệnh |
| 32 | Ethical | /ˈɛθ.ɪ.kəl/ | (Thuộc về) đạo đức |
| 33 | Legal | /ˈliː.ɡəl/ | (Thuộc về) pháp lý |
| 34 | Psychosocial | /ˌsaɪ.koʊˈsoʊ.ʃəl/ | Tâm lý xã hội |
| 35 | Genetic discrimination | /dʒəˈnɛt.ɪk dɪˌskrɪm.ɪˈneɪ.ʃən/ | Phân biệt đối xử di truyền |
| 36 | Neuroendocrine tumors (NETs) | /ˌnʊr.oʊˈɛn.də.krɪn ˈtuː.mərz/ | U thần kinh nội tiết |
| 37 | Neural crest | /ˈnʊr.əl krɛst/ | Mào thần kinh |
| 38 | Paraganglion cells | /ˌpær.əˈɡæŋ.ɡli.ən sɛlz/ | Tế bào cận hạch |
| 39 | Chromaffin cells | /ˈkroʊ.mə.fɪn sɛlz/ | Tế bào ưa crôm |
| 40 | Adrenal medulla | /əˈdriː.nəl məˈdʌl.ə/ | Tủy thượng thận |
| 41 | Extraadrenal tumors | /ˌɛk.strə.əˈdriː.nəl ˈtuː.mərz/ | U ngoài tuyến thượng thận |
| 42 | Sympathetic (nervous system) | /ˌsɪm.pəˈθɛt.ɪk/ | (Hệ thần kinh) giao cảm |
| 43 | Parasympathetic (nervous system) | /ˌpær.əˌsɪm.pəˈθɛt.ɪk/ | (Hệ thần kinh) đối giao cảm |
| 44 | Autonomic nervous system | /ˌɔː.təˈnɒm.ɪk ˈnɜːr.vəs ˈsɪs.təm/ | Hệ thần kinh tự chủ |
| 45 | Cerebrospinal axis | /ˌsɛr.ə.broʊˈspaɪ.nəl ˈæk.sɪs/ | Trục não tủy |
| 46 | Multicentric tumors | /ˌmʌl.tiˈsɛn.trɪk ˈtuː.mərz/ | U đa trung tâm, u đa ổ |
| 47 | Annual incidence | /ˈæn.ju.əl ˈɪn.sɪ.dəns/ | Tỷ lệ mắc mới hàng năm |
| 48 | Hypertension | /ˌhaɪ.pərˈtɛn.ʃən/ | Tăng huyết áp |
| 49 | Dopamine | /ˈdoʊ.pəˌmiːn/ | Dopamin |
| 50 | Norepinephrine | /ˌnɔːrˌɛp.ɪˈnɛf.rɪn/ | Norepinephrin |
| 51 | Epinephrine | /ˌɛp.ɪˈnɛf.rɪn/ | Epinephrin |
| 52 | Metabolites | /məˈtæb.əˌlaɪts/ | Chất chuyển hóa |
| 53 | 3-methoxytyramine | /θriː mɛˌθɒk.siˈtaɪ.rəˌmiːn/ | 3-methoxytyramin |
| 54 | Normetanephrine | /ˌnɔːrˌmɛt.əˈnɛf.rɪn/ | Normetanephrin |
| 55 | Metanephrine | /ˌmɛt.əˈnɛf.rɪn/ | Metanephrin |
| 56 | Chromaffin tissue | /ˈkroʊ.mə.fɪn ˈtɪʃ.uː/ | Mô ưa crôm |
| 57 | Biosynthesis | /ˌbaɪ.oʊˈsɪn.θə.sɪs/ | Sinh tổng hợp |
| 58 | Neurotransmitters | /ˌnʊr.oʊ.trænsˈmɪt.ərz/ | Chất dẫn truyền thần kinh |
| 59 | Hormones | /ˈhɔːr.moʊnz/ | Nội tiết tố, hormon |
| 60 | Cardiovascular | /ˌkɑːr.di.oʊˈvæs.kjə.lər/ | (Thuộc về) tim mạch |
| 61 | Catechol | /ˈkæt.əˌkɒl/ | Catechol |
| 62 | Amine group | /əˈmiːn ɡruːp/ | Nhóm amin |
| 63 | Tyrosine | /ˈtaɪ.rəˌsiːn/ | Tyrosin |
| 64 | 3,4-dihydroxyphenylalanine (dopa) | /daɪˌhaɪ.drɒk.siˌfɛn.əlˈæl.əˌniːn/ | 3,4-dihydroxyphenylalanin (dopa) |
| 65 | Tyrosine hydroxylase | /ˈtaɪ.rəˌsiːn haɪˈdrɒk.sɪˌleɪs/ | Tyrosin hydroxylase |
| 66 | Rate-limiting step | /reɪt ˈlɪm.ɪ.tɪŋ stɛp/ | Bước giới hạn tốc độ |
| 67 | Decarboxylation | /diːˌkɑːr.bɒk.sɪˈleɪ.ʃən/ | Phản ứng khử carboxyl |
| 68 | Hydroxylation | /haɪˌdrɒk.sɪˈleɪ.ʃən/ | Phản ứng hydroxyl hóa |
| 69 | Phenylethanolamine N-methyltransferase (PNMT) | /ˌfɛn.əlˌɛθ.əˈnoʊ.ləˌmiːn ɛn ˈmɛθ.əlˈtræns.fəˌreɪs/ | Phenylethanolamin N-methyltransferase (PNMT) |
| 70 | Glucocorticoids | /ˌɡluː.koʊˈkɔːr.tɪˌkɔɪdz/ | Glucocorticoid |
| 71 | Adrenal cortex | /əˈdriː.nəl ˈkɔːr.tɛks/ | Vỏ thượng thận |
| 72 | Norepinephrine transporter | /ˌnɔːrˌɛp.ɪˈnɛf.rɪn trænsˈpɔːr.tər/ | Chất vận chuyển norepinephrin |
| 73 | Monoamine oxidase (MAO) | /ˌmɒn.oʊ.əˈmiːn ˈɒk.sɪˌdeɪs/ | Monoamin oxidase (MAO) |
| 74 | Catechol-O-methyltransferase (COMT) | /ˈkæt.əˌkɒl oʊ ˈmɛθ.əlˈtræns.fəˌreɪs/ | Catechol-O-methyltransferase (COMT) |
| 75 | G-protein–coupled receptors | /dʒiː ˈproʊ.tiːn ˈkʌp.əld rɪˈsɛp.tərz/ | Thụ thể kết cặp G-protein |
| 76 | α-adrenergic receptors | /ˈæl.fə ˌæd.rəˈnɜːr.dʒɪk rɪˈsɛp.tərz/ | Thụ thể adrenergic alpha |
| 77 | β-adrenergic receptors | /ˈbeɪ.tə ˌæd.rəˈnɜːr.dʒɪk rɪˈsɛp.tərz/ | Thụ thể adrenergic beta |
| 78 | Dopamine receptors | /ˈdoʊ.pəˌmiːn rɪˈsɛp.tərz/ | Thụ thể dopamin |
| 79 | Agonists | /ˈæɡ.ə.nɪsts/ | Chất chủ vận |
| 80 | Antagonists | /ænˈtæɡ.ə.nɪsts/ | Chất đối kháng |
| 81 | Clinical presentation | /ˈklɪn.ɪ.kəl ˌprɛz.ɛnˈteɪ.ʃən/ | Biểu hiện lâm sàng |
| 82 | Symptomatic | /ˌsɪmp.təˈmæt.ɪk/ | Có triệu chứng |
| 83 | Catecholamine hypersecretion | /ˌkæt.əˈkoʊ.ləˌmiːn ˌhaɪ.pər.sɪˈkriː.ʃən/ | Tăng tiết catecholamin |
| 84 | Tumor mass effect | /ˈtuː.mər mæs ɪˈfɛkt/ | Hiệu ứng khối của u |
| 85 | Incidental radiographic finding | /ˌɪn.sɪˈdɛn.təl ˌreɪ.di.oʊˈɡræf.ɪk ˈfaɪn.dɪŋ/ | Phát hiện tình cờ trên chẩn đoán hình ảnh |
| 86 | Prospective screening | /prəˈspɛk.tɪv ˈskriː.nɪŋ/ | Tầm soát tiền cứu, tầm soát chủ động |
| 87 | Cyanotic congenital heart disease | /ˌsaɪ.əˈnɒt.ɪk kənˈdʒɛn.ɪ.təl hɑːrt dɪˈziːz/ | Bệnh tim bẩm sinh có tím |
| 88 | Ectopic hormone excess | /ɛkˈtɒp.ɪk ˈhɔːr.moʊn ɛkˈsɛs/ | Tăng tiết hormon lạc chỗ |
| 89 | Cushing syndrome | /ˈkʊʃ.ɪŋ ˈsɪn.droʊm/ | Hội chứng Cushing |
| 90 | Adrenocorticotropic hormone (ACTH) | /əˌdriː.noʊˌkɔːr.tɪ.koʊˈtroʊ.pɪk ˈhɔːr.moʊn/ | Hormon vỏ thượng thận (ACTH) |
| 91 | Paroxysmal episodes | /ˌpær.əkˈsɪz.məl ˈɛp.ɪˌsoʊdz/ | Cơn kịch phát |
| 92 | Triad | /ˈtraɪ.æd/ | Bộ ba triệu chứng |
| 93 | Palpitations | /ˌpæl.pɪˈteɪ.ʃənz/ | Đánh trống ngực, hồi hộp |
| 94 | Diaphoresis | /ˌdaɪ.ə.fəˈriː.sɪs/ | Toát mồ hôi |
| 95 | Orthostatic hypotension | /ˌɔːr.θəˈstæt.ɪk ˌhaɪ.poʊˈtɛn.ʃən/ | Hạ huyết áp tư thế đứng |
| 96 | Syncope | /ˈsɪŋ.kə.pi/ | Ngất |
| 97 | Pallor | /ˈpæl.ər/ | Xanh xao |
| 98 | Tremor | /ˈtrɛm.ər/ | Run |
| 99 | Anxiety | /æŋˈzaɪ.ə.ti/ | Lo âu |
| 100 | Blurred vision | /blɜːrd ˈvɪʒ.ən/ | Nhìn mờ |
| 101 | Abdominal pain | /æbˈdɒm.ɪ.nəl peɪn/ | Đau bụng |
| 102 | Constipation | /ˌkɒn.stɪˈpeɪ.ʃən/ | Táo bón |
| 103 | Diarrhea | /ˌdaɪ.əˈriː.ə/ | Tiêu chảy |
| 104 | Gastrointestinal | /ˌɡæs.troʊ.ɪnˈtɛs.tɪ.nəl/ | (Thuộc về) tiêu hóa |
| 105 | Weight loss | /weɪt lɒs/ | Sụt cân |
| 106 | Hyperglycemia | /ˌhaɪ.pər.ɡlaɪˈsiː.mi.ə/ | Tăng đường huyết |
| 107 | Polyuria | /ˌpɒl.iˈjʊə.ri.ə/ | Đa niệu |
| 108 | Polydipsia | /ˌpɒl.iˈdɪp.si.ə/ | Uống nhiều |
| 109 | Low-grade fever | /loʊ ɡreɪd ˈfiː.vər/ | Sốt nhẹ |
| 110 | Behavioral problems | /bɪˈheɪ.vjər.əl ˈprɒb.ləmz/ | Vấn đề về hành vi |
| 111 | Attention deficit hyperactivity disorder | /əˈtɛn.ʃən ˈdɛf.ɪ.sɪt ˌhaɪ.pər.ækˈtɪv.ə.ti dɪsˈɔːr.dər/ | Rối loạn tăng động giảm chú ý |
| 112 | Micturition | /ˌmɪk.tʃəˈrɪʃ.ən/ | Sự đi tiểu |
| 113 | Hypertensive crisis | /ˌhaɪ.pərˈtɛn.sɪv ˈkraɪ.sɪs/ | Cơn tăng huyết áp cấp cứu |
| 114 | Cardiomyopathy | /ˌkɑːr.di.oʊ.maɪˈɒp.ə.θi/ | Bệnh cơ tim |
| 115 | Takotsubo cardiomyopathy | /ˌtɑː.koʊtˈsuː.boʊ ˌkɑːr.di.oʊ.maɪˈɒp.ə.θi/ | Bệnh cơ tim Takotsubo |
| 116 | Arrhythmias | /əˈrɪð.mi.əz/ | Rối loạn nhịp tim |
| 117 | Pancreatitis | /ˌpæŋ.kri.əˈtaɪ.tɪs/ | Viêm tụy |
| 118 | Intestinal pseudoobstruction | /ɪnˈtɛs.tɪ.nəl ˌsuː.doʊ.əbˈstrʌk.ʃən/ | Tắc ruột giả |
| 119 | Stroke | /stroʊk/ | Đột quỵ |
| 120 | Seizures | /ˈsiː.ʒərz/ | Co giật |
| 121 | Multisystem crisis | /ˈmʌl.tiˌsɪs.təm ˈkraɪ.sɪs/ | Cơn khủng hoảng đa hệ thống |
| 122 | Hearing loss | /ˈhɪər.ɪŋ lɒs/ | Mất thính lực |
| 123 | Pulsatile tinnitus | /ˈpʌl.sə.taɪl ˈtɪn.ɪ.təs/ | Ù tai theo nhịp đập |
| 124 | Neck mass | /nɛk mæs/ | Khối u ở cổ |
| 125 | Voice hoarseness | /vɔɪs ˈhɔːrs.nəs/ | Khàn tiếng |
| 126 | Pharyngeal fullness | /fəˈrɪn.dʒi.əl ˈfʊl.nəs/ | Cảm giác đầy ở họng |
| 127 | Dysphagia | /dɪsˈfeɪ.dʒi.ə/ | Khó nuốt |
| 128 | Hereditary predisposition | /həˈrɛd.ɪˌtɛr.i ˌpriː.dɪs.pəˈzɪʃ.ən/ | Khuynh hướng di truyền |
| 129 | Biochemical diagnosis | /ˌbaɪ.oʊˈkɛm.ɪ.kəl ˌdaɪ.əɡˈnoʊ.sɪs/ | Chẩn đoán sinh hóa |
| 130 | Fractionated plasma | /ˈfræk.ʃəˌneɪ.tɪd ˈplæz.mə/ | Huyết tương phân đoạn |
| 131 | Urine metanephrines | /ˈjʊər.ɪn ˌmɛt.əˈnɛf.rɪnz/ | Metanephrin trong nước tiểu |
| 132 | Sensitivity | /ˌsɛn.sɪˈtɪv.ə.ti/ | Độ nhạy |
| 133 | Intratumoral | /ˌɪn.trəˈtjuː.mər.əl/ | Trong khối u |
| 134 | False positive | /fɔːls ˈpɒz.ə.tɪv/ | Dương tính giả |
| 135 | Supine position | /ˈsuː.paɪn pəˈzɪʃ.ən/ | Tư thế nằm ngửa |
| 136 | Indwelling needle | /ɪnˈdwɛl.ɪŋ ˈniː.dəl/ | Kim lưu |
| 137 | Catheter | /ˈkæθ.ə.tər/ | Ống thông, catheter |
| 138 | Clonidine suppression test | /ˈklɒn.ɪ.diːn səˈprɛʃ.ən tɛst/ | Nghiệm pháp ức chế Clonidin |
| 139 | Glucagon stimulation test | /ˈɡluː.kəˌɡɒn ˌstɪm.jʊˈleɪ.ʃən tɛst/ | Nghiệm pháp kích thích Glucagon |
| 140 | Noradrenergic | /ˌnɔːrˌæd.rəˈnɜːr.dʒɪk/ | (Thuộc) hệ noradrenergic |
| 141 | Adrenergic | /ˌæd.rəˈnɜːr.dʒɪk/ | (Thuộc) hệ adrenergic |
| 142 | Promoter hypermethylation | /prəˈmoʊ.tər ˌhaɪ.pərˌmɛθ.ɪˈleɪ.ʃən/ | Tăng methyl hóa vùng khởi động |
| 143 | Succinate dehydrogenase (SDH) | /ˈsʌk.sɪˌneɪt diːˌhaɪˈdrɒdʒ.əˌneɪs/ | Succinate dehydrogenase (SDH) |
| 144 | Homovanillic acid (HVA) | /ˌhoʊ.moʊ.væˈnɪl.ɪk ˈæs.ɪd/ | Axit homovanillic (HVA) |
| 145 | Neuroblastoma | /ˌnʊr.oʊ.blæsˈtoʊ.mə/ | U nguyên bào thần kinh |
| 146 | Specificity | /ˌspɛs.ɪˈfɪs.ə.ti/ | Độ đặc hiệu |
| 147 | Chromogranin A | /ˌkroʊ.moʊˈɡræn.ɪn eɪ/ | Chromogranin A |
| 148 | Proton-pump inhibitors | /ˈproʊ.tɒn pʌmp ɪnˈhɪb.ɪ.tərz/ | Thuốc ức chế bơm proton |
| 149 | Serotonin reuptake inhibitors | /ˌsɛr.əˈtoʊ.nɪn riːˈʌp.teɪk ɪnˈhɪb.ɪ.tərz/ | Thuốc ức chế tái hấp thu serotonin |
| 150 | Renal failure | /ˈriː.nəl ˈfeɪl.jər/ | Suy thận |
| 151 | Pancreatic disorders | /ˌpæŋ.kriˈæt.ɪk dɪsˈɔːr.dərz/ | Rối loạn tuyến tụy |
| 152 | Radiographic studies | /ˌreɪ.di.oʊˈɡræf.ɪk ˈstʌd.iz/ | Nghiên cứu chẩn đoán hình ảnh |
| 153 | Cross-sectional imaging | /krɒs ˈsɛk.ʃə.nəl ˈɪm.ɪ.dʒɪŋ/ | Chẩn đoán hình ảnh cắt ngang |
| 154 | Computed tomography (CT) | /kəmˈpjuː.təd təˈmɒɡ.rə.fi/ | Chụp cắt lớp vi tính (CT) |
| 155 | Magnetic resonance imaging (MRI) | /mæɡˈnɛt.ɪk ˈrɛz.ə.nəns ˈɪm.ɪ.dʒɪŋ/ | Chụp cộng hưởng từ (MRI) |
| 156 | Ultrasound (US) | /ˈʌl.trəˌsaʊnd/ | Siêu âm |
| 157 | Hounsfield units | /ˈhaʊnz.fiːld ˈjuː.nɪts/ | Đơn vị Hounsfield |
| 158 | T2-weighted images | /tiː tuː ˈweɪ.tɪd ˈɪm.ɪ.dʒɪz/ | Hình ảnh trọng T2 |
| 159 | Nuclear medicine | /ˈnuː.kli.ər ˈmɛd.ɪ.sən/ | Y học hạt nhân |
| 160 | Functional imaging | /ˈfʌŋk.ʃə.nəl ˈɪm.ɪ.dʒɪŋ/ | Chẩn đoán hình ảnh chức năng |
| 161 | Metastatic disease | /ˌmɛt.əˈstæt.ɪk dɪˈziːz/ | Bệnh di căn |
| 162 | Malignancy | /məˈlɪɡ.nən.si/ | Bệnh ác tính |
| 163 | Metaiodobenzylguanidine (MIBG) | /ˌmɛt.aɪˌoʊ.doʊˌbɛn.zəlˈɡwɑː.nɪˌdiːn/ | Metaiodobenzylguanidin (MIBG) |
| 164 | Positron-emission tomography (PET) | /ˈpɒz.ɪ.trɒn ɪˈmɪʃ.ən təˈmɒɡ.rə.fi/ | Chụp cắt lớp phát xạ positron (PET) |
| 165 | Fluorodeoxyglucose (FDG) | /ˌflʊər.oʊ.diːˌɒk.siˈɡluː.koʊs/ | Fluorodeoxyglucose (FDG) |
| 166 | Somatostatin receptors | /ˌsoʊ.mə.toʊˈstæt.ɪn rɪˈsɛp.tərz/ | Thụ thể somatostatin |
| 167 | Fumarate hydratase (FH) | /ˈfjuː.məˌreɪt haɪˈdreɪ.teɪs/ | Fumarate hydratase (FH) |
| 168 | Hypoxia-inducible factor (HIF) | /haɪˈpɒk.si.ə ɪnˈdjuː.sə.bəl ˈfæk.tər/ | Yếu tố cảm ứng thiếu oxy (HIF) |
| 169 | Next-generation sequencing | /nɛkst ˌdʒɛn.əˈreɪ.ʃən ˈsiː.kwən.sɪŋ/ | Giải trình tự thế hệ mới |
| 170 | Genomic analysis | /dʒəˈnoʊ.mɪk əˈnæl.ə.sɪs/ | Phân tích bộ gen |
| 171 | Pseudohypoxia | /ˌsuː.doʊ.haɪˈpɒk.si.ə/ | Giả thiếu oxy |
| 172 | Tricarboxylic acid (TCA) cycle | /traɪˌkɑːr.bɒkˈsɪl.ɪk ˈæs.ɪd ˈsaɪ.kəl/ | Chu trình axit tricarboxylic (TCA) |
| 173 | Kinase signaling | /ˈkaɪ.neɪs ˈsɪɡ.nəl.ɪŋ/ | Tín hiệu kinase |
| 174 | Surgical therapy | /ˈsɜːr.dʒɪ.kəl ˈθɛr.ə.pi/ | Liệu pháp phẫu thuật |
| 175 | Surgical resection | /ˈsɜːr.dʒɪ.kəl rɪˈsɛk.ʃən/ | Cắt bỏ bằng phẫu thuật |
| 176 | Biopsy | /ˈbaɪ.ɒp.si/ | Sinh thiết |
| 177 | Laparoscopic adrenalectomy | /ˌlæp.ə.rəˈskɒp.ɪk əˌdriː.nəˈlɛk.tə.mi/ | Phẫu thuật cắt tuyến thượng thận nội soi |
| 178 | Transperitoneal | /ˌtrænsˌpɛr.ɪ.təˈniː.əl/ | Qua phúc mạc |
| 179 | Retroperitoneal | /ˌrɛt.roʊˌpɛr.ɪ.təˈniː.əl/ | Sau phúc mạc |
| 180 | Laparotomy | /ˌlæp.əˈrɒt.ə.mi/ | Mở bụng |
| 181 | Cortical-sparing | /ˈkɔːr.tɪ.kəl ˈspɛər.ɪŋ/ | Bảo tồn vỏ (thượng thận) |
| 182 | Mineralocorticoid | /ˌmɪn.ər.ə.loʊˈkɔːr.tɪˌkɔɪd/ | Mineralocorticoid |
| 183 | Primary adrenal insufficiency | /ˈpraɪ.mɛr.i əˈdriː.nəl ˌɪn.səˈfɪʃ.ən.si/ | Suy thượng thận nguyên phát |
| 184 | Remnant | /ˈrɛm.nənt/ | Phần còn lại |
| 185 | Anesthesiologist | /ˌæn.əsˌθiː.ziˈɒl.ə.dʒɪst/ | Bác sĩ gây mê |
| 186 | Intravenous | /ˌɪn.trəˈviː.nəs/ | Tiêm tĩnh mạch |
| 187 | Antihypertensive | /ˌæn.ti.haɪ.pərˈtɛn.sɪv/ | Chống tăng huyết áp |
| 188 | Nitroprusside | /ˌnaɪ.troʊˈprʌs.aɪd/ | Nitroprussid |
| 189 | Phentolamine | /fɛnˈtɒl.əˌmiːn/ | Phentolamin |
| 190 | Esmolol | /ˈɛz.məˌlɒl/ | Esmolol |
| 191 | Labetalol | /ləˈbeɪ.təˌlɒl/ | Labetalol |
| 192 | Vasopressors | /ˌveɪ.zoʊˈprɛs.ərz/ | Thuốc co mạch |
| 193 | Phenylephrine | /ˌfɛn.əlˈɛf.rɪn/ | Phenylephrin |
| 194 | Anesthesia induction | /ˌæn.əsˈθiː.zi.ə ɪnˈdʌk.ʃən/ | Khởi mê |
| 195 | Ligation | /laɪˈɡeɪ.ʃən/ | Thắt (mạch máu) |
| 196 | Adrenal vein | /əˈdriː.nəl veɪn/ | Tĩnh mạch thượng thận |
| 197 | Vasodilation | /ˌveɪ.zoʊ.daɪˈleɪ.ʃən/ | Giãn mạch |
| 198 | Cosyntropin stimulation test | /ˌkoʊ.sɪnˈtroʊ.pɪn ˌstɪm.jʊˈleɪ.ʃən tɛst/ | Nghiệm pháp kích thích Cosyntropin |
| 199 | Adrenal steroid | /əˈdriː.nəl ˈstɪə.rɔɪd/ | Steroid thượng thận |
| 200 | Medical preparation | /ˈmɛd.ɪ.kəl ˌprɛp.əˈreɪ.ʃən/ | Chuẩn bị nội khoa |
| 201 | α-adrenergic receptor blocker | /ˈæl.fə ˌæd.rəˈnɜːr.dʒɪk rɪˈsɛp.tər ˈblɒk.ər/ | Thuốc chẹn thụ thể adrenergic alpha |
| 202 | Calcium channel blocker | /ˈkæl.si.əm ˈtʃæn.əl ˈblɒk.ər/ | Thuốc chẹn kênh canxi |
| 203 | Phenoxybenzamine | /fəˌnɒk.siˈbɛn.zəˌmiːn/ | Phenoxybenzamin |
| 204 | Noncompetitive antagonist | /ˌnɒn.kəmˈpɛt.ə.tɪv ænˈtæɡ.ə.nɪst/ | Chất đối kháng không cạnh tranh |
| 205 | Half-life | /hɑːf laɪf/ | Thời gian bán thải |
| 206 | Postoperative hypotension | /ˌpoʊstˈɒp.ər.ə.tɪv ˌhaɪ.poʊˈtɛn.ʃən/ | Hạ huyết áp sau mổ |
| 207 | Prazosin | /ˈpreɪ.zə.sɪn/ | Prazosin |
| 208 | Doxazosin | /dɒkˈseɪ.zə.sɪn/ | Doxazosin |
| 209 | Carvedilol | /kɑːrˈveɪ.dɪˌlɒl/ | Carvedilol |
| 210 | Postural hypotension | /ˈpɒs.tʃər.əl ˌhaɪ.poʊˈtɛn.ʃən/ | Hạ huyết áp tư thế |
| 211 | Metyrosine | /məˈtaɪ.rəˌsiːn/ | Metyrosin |
| 212 | Extrapyramidal manifestations | /ˌɛk.strə.pɪˈræm.ɪ.dəl ˌmæn.ɪ.fɛˈsteɪ.ʃənz/ | Biểu hiện ngoại tháp |
| 213 | Oral salt loading | /ˈɔːr.əl sɔːlt ˈloʊ.dɪŋ/ | Bù muối đường uống |
| 214 | Prognosis | /prɒɡˈnoʊ.sɪs/ | Tiên lượng |
| 215 | Synchronous | /ˈsɪŋ.krə.nəs/ | Đồng thời |
| 216 | Metachronous | /məˈtæk.rə.nəs/ | Không đồng thời, kế시 |
| 217 | World Health Organization (WHO) | /wɜːrld hɛlθ ˌɔːr.ɡə.naɪˈzeɪ.ʃən/ | Tổ chức Y tế Thế giới (WHO) |
| 218 | Immunohistochemical | /ˌɪm.juː.noʊˌhɪs.toʊˈkɛm.ɪ.kəl/ | Hóa mô miễn dịch |
| 219 | Histopathologic | /ˌhɪs.toʊˌpæθ.əˈlɒdʒ.ɪk/ | Mô bệnh học |
| 220 | Interobserver variability | /ˌɪn.tər.əbˈzɜːr.vər ˌvɛər.i.əˈbɪl.ə.ti/ | Sự khác biệt giữa các người quan sát |
| 221 | Intraobserver variability | /ˌɪn.trə.əbˈzɜːr.vər ˌvɛər.i.əˈbɪl.ə.ti/ | Sự khác biệt trong cùng một người quan sát |
| 222 | British tumor registry | /ˈbrɪt.ɪʃ ˈtuː.mər ˈrɛdʒ.ɪ.stri/ | Cơ quan đăng ký khối u của Anh |
| 223 | Indolent clinical course | /ˈɪn.də.lənt ˈklɪn.ɪ.kəl kɔːrs/ | Diễn biến lâm sàng âm thầm |
| 224 | Overall survival | /ˌoʊ.vərˈɔːl sərˈvaɪ.vəl/ | Tỷ lệ sống còn toàn bộ |
| 225 | Calcitonin | /ˌkæl.sɪˈtoʊ.nɪn/ | Calcitonin |
| 226 | Parafollicular C-cells | /ˌpær.ə.fəˈlɪk.jʊ.lər siː sɛlz/ | Tế bào C cạnh nang |
| 227 | Differentiated thyroid carcinomas | /ˌdɪf.əˈrɛn.ʃiˌeɪ.tɪd ˈθaɪ.rɔɪd ˌkɑːr.sɪˈnoʊ.məz/ | Ung thư biểu mô tuyến giáp biệt hóa |
| 228 | De novo | /deɪ ˈnoʊ.voʊ/ | Mới (phát sinh) |
| 229 | Activating mutation | /ˈæk.tɪˌveɪ.tɪŋ mjuːˈteɪ.ʃən/ | Đột biến hoạt hóa |
| 230 | Protooncogene | /ˌproʊ.toʊˈɒŋ.koʊˌdʒiːn/ | Tiền-oncogen |
| 231 | Somatic mutations | /soʊˈmæt.ɪk mjuːˈteɪ.ʃənz/ | Đột biến soma |
| 232 | Cadherin superfamily | /ˈkæd.hɪər.ɪn ˌsuː.pərˈfæm.ə.li/ | Siêu họ Cadherin |
| 233 | Receptor tyrosine kinase | /rɪˈsɛp.tər ˈtaɪ.rəˌsiːn ˈkaɪ.neɪs/ | Thụ thể tyrosin kinase |
| 234 | Extracellular | /ˌɛk.strəˈsɛl.jʊ.lər/ | Ngoại bào |
| 235 | Intracellular | /ˌɪn.trəˈsɛl.jʊ.lər/ | Nội bào |
| 236 | Endogenous ligands | /ɛnˈdɒdʒ.ə.nəs ˈlaɪ.ɡændz/ | Phối tử nội sinh |
| 237 | Glial cell derived neurotrophic factor (GDNF) | /ˈɡlaɪ.əl sɛl dɪˈraɪvd ˌnʊr.oʊˈtroʊ.fɪk ˈfæk.tər/ | Yếu tố dinh dưỡng thần kinh có nguồn gốc từ tế bào đệm (GDNF) |
| 238 | Urogenital system | /ˌjʊər.oʊˈdʒɛn.ɪ.təl ˈsɪs.təm/ | Hệ niệu-dục |
| 239 | Enteric nervous system | /ɛnˈtɛr.ɪk ˈnɜːr.vəs ˈsɪs.təm/ | Hệ thần kinh ruột |
| 240 | Constitutive activation | /kənˈstɪtʃ.u.ə.tɪv ˌæk.tɪˈveɪ.ʃən/ | Kích hoạt cấu thành |
| 241 | Dimer | /ˈdaɪ.mər/ | Dimer (phân tử nhị hợp) |
| 242 | Cysteine-rich domain | /ˈsɪs.tiːn rɪtʃ doʊˈmeɪn/ | Miền giàu cystein |
| 243 | Monomer | /ˈmɒn.ə.mər/ | Monomer (phân tử đơn hợp) |
| 244 | C-cell hyperplasia | /siː sɛl ˌhaɪ.pərˈpleɪ.zi.ə/ | Tăng sản tế bào C |
| 245 | Oncologic cascade | /ˌɒŋ.kəˈlɒdʒ.ɪk kæsˈkeɪd/ | Chuỗi biến đổi ung thư |
| 246 | Noninvasive | /ˌnɒn.ɪnˈveɪ.sɪv/ | Không xâm lấn |
| 247 | Invasive carcinoma | /ɪnˈveɪ.sɪv ˌkɑːr.sɪˈnoʊ.mə/ | Ung thư biểu mô xâm lấn |
| 248 | Mediastinal | /ˌmiː.di.əˈstaɪ.nəl/ | (Thuộc) trung thất |
| 249 | Cervical lymphadenopathy | /ˈsɜːr.vɪ.kəl ˌlɪm.fæd.əˈnɒp.ə.θi/ | Hạch cổ |
| 250 | Tertiary care centers | /ˈtɜːr.ʃiˌɛr.i kɛər ˈsɛn.tərz/ | Trung tâm chăm sóc cấp ba |
| 251 | Posterior capsule | /pɒˈstɪər.i.ər ˈkæp.sjuːl/ | Bao sau |
| 252 | Central compartment neck dissection | /ˈsɛn.trəl kəmˈpɑːrt.mənt nɛk daɪˈsɛk.ʃən/ | Nạo vét hạch khoang trung tâm |
| 253 | Cytotoxic chemotherapy | /ˌsaɪ.toʊˈtɒk.sɪk ˌkiː.moʊˈθɛr.ə.pi/ | Hóa trị độc tế bào |
| 254 | Dacarbazine | /dəˈkɑːr.bəˌziːn/ | Dacarbazin |
| 255 | Vincristine | /vɪnˈkrɪs.tiːn/ | Vincristin |
| 256 | Cyclophosphamide | /ˌsaɪ.kloʊˈfɒs.fəˌmaɪd/ | Cyclophosphamid |
| 257 | Vandetanib | /vænˈdɛt.ə.nɪb/ | Vandetanib |
| 258 | Cabozantinib | /ˌkæb.oʊˈzæn.tɪ.nɪb/ | Cabozantinib |
| 259 | Disease-free survival | /dɪˈziːz friː sərˈvaɪ.vəl/ | Thời gian sống không bệnh |
| 260 | Locoregional | /ˌloʊ.koʊˈriː.dʒə.nəl/ | Tại chỗ tại vùng |
| 261 | Tumor, node, and metastasis (TNM) | /ˈtuː.mər noʊd ænd məˈtæs.tə.sɪs/ | (Phân loại) Khối u, hạch và di căn (TNM) |
| 262 | Serologic tumor markers | /ˌsɪə.rəˈlɒdʒ.ɪk ˈtuː.mər ˈmɑːr.kərz/ | Dấu ấn khối u huyết thanh học |
| 263 | Carcinoembryonic antigen (CEA) | /ˌkɑːr.sɪ.noʊˌɛm.briˈɒn.ɪk ˈæn.tɪ.dʒən/ | Kháng nguyên carcinoembryonic (CEA) |
| 264 | Calcitonin doubling time | /ˌkæl.sɪˈtoʊ.nɪn ˈdʌb.lɪŋ taɪm/ | Thời gian nhân đôi calcitonin |
| 265 | Carney complex (CNC) | /ˈkɑːr.ni ˈkɒm.plɛks/ | Phức hợp Carney (CNC) |
| 266 | Lentiginosis | /ˌlɛn.tɪdʒ.ɪˈnoʊ.sɪs/ | Bệnh đốm sắc tố |
| 267 | Blue nevi | /bluː ˈniː.vaɪ/ | Nốt ruồi xanh |
| 268 | Spotty skin pigmentation | /ˈspɒt.i skɪn ˌpɪɡ.mənˈteɪ.ʃən/ | Sắc tố da dạng đốm |
| 269 | Conjunctiva | /ˌkɒn.dʒʌŋkˈtaɪ.və/ | Kết mạc |
| 270 | Mucosal surfaces | /mjuːˈkoʊ.səl ˈsɜːr.fə.sɪz/ | Bề mặt niêm mạc |
| 271 | Myxomas | /mɪkˈsoʊ.məz/ | U nhầy |
| 272 | Primary pigmented nodular adrenocortical disease (PPNAD) | /ˈpraɪ.mɛr.i ˈpɪɡ.mən.tɪd ˈnɒdʒ.ʊ.lər əˌdriː.noʊˈkɔːr.tɪ.kəl dɪˈziːz/ | Bệnh vỏ thượng thận dạng nốt tăng sắc tố nguyên phát (PPNAD) |
| 273 | Psammomatous melanotic schwannoma | /sæˈmoʊ.mə.təs ˌmɛl.əˈnɒt.ɪk ʃwɑːˈnoʊ.mə/ | U schwann tế bào hắc tố thể cát |
| 274 | Genetic loci | /dʒəˈnɛt.ɪk ˈloʊ.saɪ/ | Locus di truyền |
| 275 | Protein kinase A | /ˈproʊ.tiːn ˈkaɪ.neɪs eɪ/ | Protein kinase A |
| 276 | Cyclic adenosine monophosphate (cAMP) | /ˈsaɪ.klɪk əˈdɛn.əˌsiːn ˌmɒn.oʊˈfɒs.feɪt/ | Cyclic adenosine monophosphat (cAMP) |
| 277 | Adrenocortical carcinoma | /əˌdriː.noʊˈkɔːr.tɪ.kəl ˌkɑːr.sɪˈnoʊ.mə/ | Ung thư biểu mô vỏ thượng thận |
| 278 | ACTH-independent | /eɪ siː tiː eɪtʃ ˌɪn.dɪˈpɛn.dənt/ | Độc lập với ACTH |
| 279 | Lipofuscin | /ˌlaɪ.poʊˈfjuː.sɪn/ | Lipofuscin |
| 280 | Atrophic cortex | /əˈtroʊ.fɪk ˈkɔːr.tɛks/ | Vỏ (thượng thận) teo |
| 281 | Insidious onset | /ɪnˈsɪd.i.əs ˈɒn.sɛt/ | Khởi phát âm thầm |
| 282 | Pathognomonic | /ˌpæθ.əɡ.nəˈmɒn.ɪk/ | Đặc trưng cho bệnh |
| 283 | Liddle test | /ˈlɪd.əl tɛst/ | Nghiệm pháp Liddle |
| 284 | Diffuse micronodular disease | /dɪˈfjuːs ˌmaɪ.kroʊˈnɒdʒ.ʊ.lər dɪˈziːz/ | Bệnh vi nốt lan tỏa |
| 285 | Gigantism | /dʒaɪˈɡæn.tɪ.zəm/ | Bệnh khổng lồ |
| 286 | Acromegaly | /ˌæk.roʊˈmɛɡ.ə.li/ | Bệnh to đầu chi |
| 287 | Adenoma | /ˌæd.əˈnoʊ.mə/ | U tuyến |
| 288 | Hyperplasia | /ˌhaɪ.pərˈpleɪ.zi.ə/ | Tăng sản |
| 289 | Hyperprolactinemia | /ˌhaɪ.pərˌproʊ.læk.tɪˈniː.mi.ə/ | Tăng prolactin máu |
| 290 | Corticotroph adenomas | /ˈkɔːr.tɪ.koʊˌtroʊf ˌæd.əˈnoʊ.məz/ | U tuyến corticotroph |
| 291 | Thyrotoxicosis | /ˌθaɪ.roʊˌtɒk.sɪˈkoʊ.sɪs/ | Nhiễm độc giáp |
| 292 | Large-cell calcifying Sertoli cell tumors (LCCSCT) | /lɑːrdʒ sɛl ˈkæl.sɪˌfaɪ.ɪŋ sɜːrˈtoʊ.li sɛl ˈtuː.mərz/ | U tế bào Sertoli vôi hóa tế bào lớn (LCCSCT) |
| 293 | Peutz-Jeghers syndrome | /pɔɪts ˈjɛɡ.ərz ˈsɪn.droʊm/ | Hội chứng Peutz-Jeghers |
| 294 | Gynecomastia | /ˌɡaɪ.nə.koʊˈmæs.ti.ə/ | Nữ hóa tuyến vú |
| 295 | Familial isolated pituitary adenomas (FIPA) | /fəˈmɪl.i.əl ˈaɪ.səˌleɪ.tɪd pɪˈtjuː.ɪˌtɛr.i ˌæd.əˈnoʊ.məz/ | U tuyến yên đơn độc có tính gia đình (FIPA) |
| 296 | Intrafamilial | /ˌɪn.trə.fəˈmɪl.i.əl/ | Trong cùng gia đình |
| 297 | Aryl hydrocarbon receptor-interacting protein (AIP) | /ˈær.ɪl ˌhaɪ.droʊˈkɑːr.bən rɪˈsɛp.tər ˌɪn.tərˈæk.tɪŋ ˈproʊ.tiːn/ | Protein tương tác với thụ thể aryl hydrocarbon (AIP) |
| 298 | Macroadenomas | /ˌmæk.roʊˌæd.əˈnoʊ.məz/ | U tuyến lớn (Macroadenoma) |
| 299 | Somatotroph | /soʊˈmæt.əˌtroʊf/ | (Tế bào/u) Somatotroph |
| 300 | Mammosomatotroph | /ˌmæm.oʊ.soʊˈmæt.əˌtroʊf/ | (Tế bào/u) Mammosomatotroph |