
Sperling Nội tiết học Nhi khoa, Ấn bản thứ 5 – Biên dịch: Ths.Bs. Lê Đình Sáng
Sperling Pediatric Endocrinology, Fifth Edition
Tác giả: Sperling, Mark A., MD – Nhà xuất bản: Elsevier Inc.
CHƯƠNG 3: Các Con Đường Dẫn Truyền Tín Hiệu Thụ Thể Trung Gian Cho Hoạt Động Của Hormone
Bassil Kublaoui; Michael A. Levine Sperling Pediatric Endocrinology, 3, 30-85
Mở đầu
Hormone thực hiện hoạt động của chúng bằng cách gắn vào các protein thụ thể đặc hiệu, một quá trình gây ra những thay đổi về cấu hình hoặc tái phân bố trong các ngăn của các protein này. Thụ thể được hoạt hóa giờ đây có khả năng tạo ra các hiệu ứng nội bào tích cực (hoặc tiêu cực) mà cuối cùng được ghi nhận là một đáp ứng sinh lý. Tính đặc hiệu của hoạt động hormone được quyết định bởi ái lực của hormone đối với các thụ thể khác nhau, sự biểu hiện đặc hiệu của thụ thể trên từng loại tế bào, và các đáp ứng duy nhất được tạo ra khi phối tử chiếm giữ thụ thể.
Kể từ đầu những năm 2000, sự hiểu biết của chúng ta về hoạt động của hormone đã tiến bộ nhanh chóng nhờ vào thành công của hệ gen học (genomics) và các kỹ thuật sinh học phân tử tiên tiến. Cách tiếp cận kết hợp này đã dẫn đến việc phát hiện và phân loại một số lượng lớn bất ngờ các thụ thể, một số khá mới lạ và một số khác thậm chí không được lường trước, là thành viên của các họ protein lớn được bảo tồn về mặt di truyền. Hơn nữa, sự hiểu biết của chúng ta về hoạt động của thụ thể đã được làm sáng tỏ nhờ việc xác định và mô tả chi tiết các protein truyền tín hiệu sau thụ thể và các cơ chế truyền tín hiệu. Bốn siêu họ thụ thể chính đã được xác định, được phân biệt bởi cấu trúc protein, vị trí tế bào và các hệ thống tác động. Các họ này bao gồm các thụ thể kết cặp G-protein (GPCRs), thụ thể cytokine, thụ thể tyrosine kinase (RTKs), và các thụ thể nhân (Bảng 3.1). Chương này tổng quan các đặc điểm chính của các họ thụ thể quan trọng này. Các đột biến ảnh hưởng đến chức năng thụ thể dẫn đến các rối loạn nội tiết cũng được nhấn mạnh.
Bảng 3.1 Các Loại Thụ Thể Hormone Chính
| Loại Thụ Thể | Các Thụ Thể Hormone |
|---|---|
| Thụ thể kết cặp G-protein | ACTH và các melanocortin khác, vasopressin V2, LH, FSH, TSH, GnRH, TRH, GHRH, yếu tố giải phóng corticotropin, somatostatin, glucagon, oxytocin, peptide ức chế dạ dày, PTH loại 1, acid béo tự do, GPR54, orexin, ghrelin, melanin-concentrating, calcitonin, glucagon-like peptide-1, và các thụ thể cảm nhận calci |
| Thụ thể cytokine loại 1 | Hormone tăng trưởng, prolactin, và các thụ thể leptin |
| Thụ thể tyrosine kinase | Insulin, IGF-1, và các thụ thể yếu tố tăng trưởng nguyên bào sợi |
| Thụ thể nhân | Hormone tuyến giáp, vitamin D3, PPARγ, HNF4A, glucocorticoid, androgen, estrogen, mineralocorticoid, và các thụ thể DAX1 |
ACTH, hormone vỏ thượng thận; DAX1, gen 1 vùng tương đồng quan trọng gây bất sản thượng thận bẩm sinh đảo ngược giới tính nhạy cảm với liều lượng trên nhiễm sắc thể X; FSH, hormone kích thích nang trứng; GHRH, hormone giải phóng hormone tăng trưởng; GnRH, hormone giải phóng gonadotropin; GPR54, thụ thể 54 của peptide có nguồn gốc từ KISS-1; HNF4A, yếu tố nhân tế bào gan-4α; IGF-1, yếu tố tăng trưởng giống insulin-1; LH, hormone tạo hoàng thể; PPARγ, thụ thể kích hoạt tăng sinh peroxisome γ; PTH, hormone cận giáp; TRH, hormone giải phóng thyrotropin; TSH, hormone kích thích tuyến giáp.
Các nguyên tắc cơ bản của hoạt động thụ thể
Một phân tử gắn vào một thụ thể được gọi là phối tử (ligand). Khi sự gắn kết của phối tử dẫn đến việc hoạt hóa các quá trình truyền tín hiệu bên trong tế bào, phối tử đó được gọi là chất chủ vận (agonist). Đáp ứng của một thụ thể đối với phối tử của nó thường được đánh giá bằng hai đặc tính của phối tử: hiệu lực và hiệu quả. Hiệu lực mô tả nồng độ phối tử cần thiết để gây ra hiệu ứng sinh học bằng cách gắn vào thụ thể. Một chất chủ vận có hiệu lực mạnh sẽ hoạt hóa các thụ thể ở nồng độ thấp, thường ở khoảng nanomol. Hiệu quả mô tả hiệu ứng tối đa do một phối tử gây ra. Hiệu lực thường được mô tả bằng cách sử dụng EC50, là nồng độ phối tử gây ra một nửa hiệu ứng tối đa. Khi một chất chủ vận tổng hợp vượt quá hiệu quả của phối tử tự nhiên, nó được gọi là siêu chủ vận (super agonist). Khi các phối tử của thụ thể (tự nhiên hoặc tổng hợp) không gây ra sự hoạt hóa hoàn toàn, chúng được gọi là chất chủ vận một phần (partial agonists). Một chất đối kháng (antagonist) là một phân tử ngăn chặn phối tử tự nhiên gắn vào thụ thể của nó. Nếu nó gắn vào cùng vị trí với chất chủ vận tự nhiên, nó được gọi là chất đối kháng cùng vị trí (orthosteric antagonist). Nếu nó gắn vào một vị trí khác với chất chủ vận tự nhiên, nó được gọi là chất đối kháng dị lập thể (allosteric antagonist). Hiệu lực của chất đối kháng được mô tả bằng IC50, là nồng độ gây ra một nửa sự ức chế tối đa. Một thụ thể có hoạt tính nền cấu thành có thể được gắn bởi một phối tử ức chế hoạt động của thụ thể khi không có phối tử tự nhiên. Trong trường hợp này, phối tử được gọi là chất chủ vận đảo nghịch (inverse agonist). Một thang đo đã được xây dựng để thể hiện tính liên tục trong chức năng của phối tử-thụ thể—từ –1 (đại diện cho một chất chủ vận đảo nghịch hoàn toàn), đến 0 (đại diện cho một chất đối kháng trung tính), đến +1 (đại diện cho một chất chủ vận hoàn toàn).
Các thụ thể có nhiều cấu hình khả dĩ đang liên tục thay đổi. Các thụ thể tồn tại ở một số cấu hình nhiều hơn các cấu hình khác, dựa trên năng lượng tự do của mỗi cấu hình. Các chất chủ vận hoạt động như những chất điều biến làm ổn định một cấu hình nhất định của thụ thể bằng cách giảm năng lượng cần thiết để đi vào cấu hình đó. Cấu hình này liên quan đến việc hoạt hóa tín hiệu xuôi dòng. Các chất chủ vận một phần làm ổn định thụ thể ở các cấu hình kém hiệu quả hơn trong việc hoạt hóa tín hiệu xuôi dòng. Các siêu chủ vận làm ổn định thụ thể ở các cấu hình hiệu quả hơn trong việc hoạt hóa tín hiệu xuôi dòng. Những thay đổi về cấu hình trong một thụ thể cũng có thể ảnh hưởng đến số lượng và loại tín hiệu mà một thụ thể tạo ra. Trong mô hình cổ điển, một GPCR được cho là hoạt động như một công tắc nhị phân có thể được hoạt hóa bằng cách gắn chất chủ vận hoặc bị ức chế bằng cách chất đối kháng ngăn chặn sự gắn kết của chất chủ vận. Bây giờ chúng ta biết rằng tín hiệu GPCR phức tạp hơn một mô hình công tắc nhị phân đơn giản (tức là “bật” và “tắt”), và các phối tử khác nhau có thể gắn vào cùng một GPCR và lựa chọn hoạt hóa một con đường xuôi dòng này thay vì một con đường khác. Khả năng các phối tử khác nhau gây ra các cấu hình khác nhau của một thụ thể mà hoạt hóa (hoặc ức chế) các tín hiệu xuôi dòng chọn lọc được gọi là tín hiệu thiên lệch (biased signaling). Các phối tử gắn vào vị trí tự nhiên của một thụ thể nhưng tạo ra các sự kiện truyền tín hiệu khác nhau được gọi là phối tử cùng vị trí (orthostatic ligands). Một ví dụ về điều này là thụ thể của hormone kích thích nang trứng (FSH), một GPCR cổ điển. FSH được glycosyl hóa hoàn toàn có tính acid hơn và hoạt động như một chất chủ vận hoàn toàn tại các thụ thể FSH, hoạt hóa tín hiệu kết cặp Gs và tạo ra cyclic adenosine monophosphate (AMP). FSH được glycosyl hóa một phần có tính kiềm hơn và hoạt động như một chất chủ vận một phần hoặc một phối tử thiên lệch vì nó hoạt hóa cả hai con đường Gs và Gi, chúng cạnh tranh với nhau. FSH đã bị loại bỏ gốc đường không có tác dụng lên tín hiệu nhưng lại gắn vào thụ thể và do đó hoạt động như một chất đối kháng cạnh tranh. Các biến thể được glycosyl hóa khác nhau này tồn tại trong tuần hoàn và là một phương tiện để tinh chỉnh tín hiệu từ tuyến yên đến tuyến sinh dục. Trong một số trường hợp, một phối tử trung tính có thể gắn vào thụ thể tại một vị trí khác với vị trí gắn của phối tử tự nhiên và bằng cách đó, ảnh hưởng đến cấu hình của thụ thể sao cho hiệu quả của phối tử được tăng lên, giảm đi, hoặc bị thiên lệch. Chúng được gọi là chất điều biến dị lập thể (allosteric modulators). Chúng không tự hoạt hóa hay ức chế các thụ thể. Tương tự, có những phối tử tổng hợp dị lập thể cho thụ thể FSH thể hiện sự thiên lệch tín hiệu từ chất chủ vận hoàn toàn đến chất chủ vận đảo nghịch.
Các thụ thể thường được tìm thấy liên kết với các protein khác dù ở bề mặt tế bào hay trong bào tương. Các protein liên kết này ảnh hưởng đến cấu hình của thụ thể. Một ví dụ là các protein điều chỉnh hoạt động của thụ thể hay RAMPs. Thụ thể calcitonin và thụ thể giống calcitonin là các GPCR có thể gắn với một số phối tử (calcitonin, adrenomedullin, amylin, và peptide liên quan đến gen calcitonin). RAMP1, RAMP2, và RAMP3 liên kết với cả thụ thể calcitonin và thụ thể giống calcitonin, và tùy thuộc vào RAMP nào được liên kết sẽ quyết định tính chọn lọc đối với một trong các phối tử nêu trên.
Tính linh hoạt về cấu hình của các thụ thể và sự tương tác với nhiều phối tử và chất điều biến dẫn đến sự phức tạp, đặc hiệu, và tinh chỉnh cao hơn, cũng như hiệu quả tổng thể.
Thụ thể kết cặp G-protein
Hơn 1% hệ gen của động vật có xương sống mã hóa một họ protein lớn gồm các thụ thể cảm nhận các phân tử bên ngoài tế bào và hoạt hóa các con đường dẫn truyền tín hiệu và, cuối cùng, là các đáp ứng tế bào. Các protein thụ thể này được nhúng trong màng sinh chất và được kết cặp với các hệ thống tạo tín hiệu nội bào bởi các protein G dị tam thể (heterotrimeric G proteins) (tức là GPCRs). GPCRs còn được gọi là thụ thể có bảy vùng xuyên màng (seven-transmembrane domain receptors), thụ thể 7TM (7TM receptors), thụ thể bảy xoắn (heptahelical receptors), và thụ thể xoắn ốc (serpentine receptors). Chúng được gọi là thụ thể xuyên màng vì chúng đi qua màng tế bào, và chúng được gọi là thụ thể bảy lần xuyên màng vì chúng có các vùng xoắn alpha đi qua màng tế bào 7 lần. Hệ gen người mã hóa khoảng 950 GPCR. GPCR liên quan đến nhiều bệnh và cũng là mục tiêu của khoảng 40% tất cả các loại thuốc hiện đại. Khoảng 150 trong số các GPCR được tìm thấy trong hệ gen người có chức năng chưa được biết. Hầu hết các GPCR là thụ thể mùi và pheromone. Cũng cần lưu ý rằng hầu hết các hormone đều gắn với GPCR, và do đó, sự dẫn truyền tín hiệu phụ thuộc G-protein là cơ chế phổ biến nhất cho hoạt động của hormone (Bảng 3.2).
Bảng 3.2 Thụ Thể Kết Cặp G-protein và Các Tình Trạng Lâm Sàng Liên Quan Đến Đột Biến Thụ Thể
| Thụ Thể | Đột Biến Dòng Mầm | Rối Loạn Nội Tiết |
|---|---|---|
| Thụ thể ACTH/melanocortin-2 | Đột biến bất hoạt (đồng hợp tử, dị hợp tử kép) | Suy giảm glucocorticoid gia đình loại 1 |
| Thụ thể melanocortin-4 | Đột biến bất hoạt (hầu hết dị hợp tử, một số đồng hợp tử) | Béo phì |
| Thụ thể vasopressin V2 | Đột biến bất hoạt (hầu hết lặn liên kết X, hiếm khi trội liên kết X) | Đái tháo nhạt do thận liên kết X |
| Thụ thể LH | Đột biến bất hoạt (đồng hợp tử, dị hợp tử kép) Đột biến hoạt hóa (dị hợp tử) | Nam: thiểu sản tế bào Leydig loại I và II Nữ: không triệu chứng hoặc suy sinh dục tăng gonadotropin với vô kinh nguyên phát Nam: dậy thì sớm giới hạn ở nam |
| Thụ thể FSH | Đột biến bất hoạt (đồng hợp tử, dị hợp tử kép) | Nữ: loạn sản buồng trứng tăng gonadotropin di truyền lặn trên NST thường hoặc suy sinh dục tăng gonadotropin nhẹ hơn Nam: suy giảm sinh tinh biến đổi |
| Thụ thể TSH | Đột biến bất hoạt (hầu hết đồng hợp tử hoặc dị hợp tử kép, hiếm khi dị hợp tử) Đột biến hoạt hóa (dị hợp tử) | Đề kháng TSH Cường giáp không tự miễn di truyền trội trên NST thường/u tuyến độc |
| Thụ thể GnRH | Đột biến bất hoạt (đồng hợp tử hoặc dị hợp tử kép) | Suy sinh dục giảm gonadotropin đơn độc |
| Thụ thể TRH | Đột biến bất hoạt (dị hợp tử kép) | Suy giáp trung ương |
| GPR54 | Đột biến bất hoạt (đồng hợp tử, dị hợp tử kép) | Suy sinh dục giảm gonadotropin đơn độc, khứu giác bình thường |
| Ghrelin | Đột biến bất hoạt (đồng hợp tử, có thể dị hợp tử) | Tầm vóc thấp do giảm tiết hormone tăng trưởng |
| Thụ thể GHRH | Đột biến bất hoạt (đồng hợp tử/dị hợp tử kép) | Thiếu hụt hormone tăng trưởng đơn độc |
| Thụ thể PTH loại 1 | Đột biến bất hoạt (đồng hợp tử, dị hợp tử) Đột biến hoạt hóa (dị hợp tử) |
Loạn sản sụn Blomstrand nếu đồng hợp tử và hiếm khi nếu dị hợp tử; u sụn nếu dị hợp tử Loạn sản sụn hành xương Jansen |
| Thụ thể cảm nhận calci | Đột biến bất hoạt (dị hợp tử, đồng hợp tử)<br>Đột biến hoạt hóa (dị hợp tử) | Tăng calci máu giảm calci niệu lành tính gia đình điển hình nếu dị hợp tử, tăng năng cận giáp nặng ở trẻ sơ sinh hiếm khi nếu dị hợp tử, điển hình nếu đồng hợp tử Giảm calci máu giảm calci niệu di truyền trội trên NST thường, hội chứng Bartter loại V |
ACTH, hormone vỏ thượng thận; FSH, hormone kích thích nang trứng; GHRH, hormone giải phóng hormone tăng trưởng; GnRH, hormone giải phóng gonadotropin; GPR54, thụ thể 54 của peptide có nguồn gốc từ KISS-1; LH, hormone tạo hoàng thể; PTH, hormone cận giáp; TRH, hormone giải phóng thyrotropin; TSH, hormone kích thích tuyến giáp.
Siêu họ GPCR được chia thành tám loại chính. Các thụ thể này chứa một miền ngoại bào đầu tận amino, thường được gọi là miền ngoại bào (ectodomain) hoặc miền ngoài (exodomain). Các thụ thể này cũng chứa bảy chuỗi xoắn alpha được cho là xuyên màng (TM-I đến TM-VII). Các chuỗi xoắn alpha được nối với nhau bởi ba vòng nội bào (i1–i3) và ba vòng ngoại bào (e1–e3), thường được gọi chung là vùng xoắn ốc (serpentine region) (Hình 3.1). Vùng nội bào đầu tận carboxy thường được gọi là miền nội bào (endodomain).
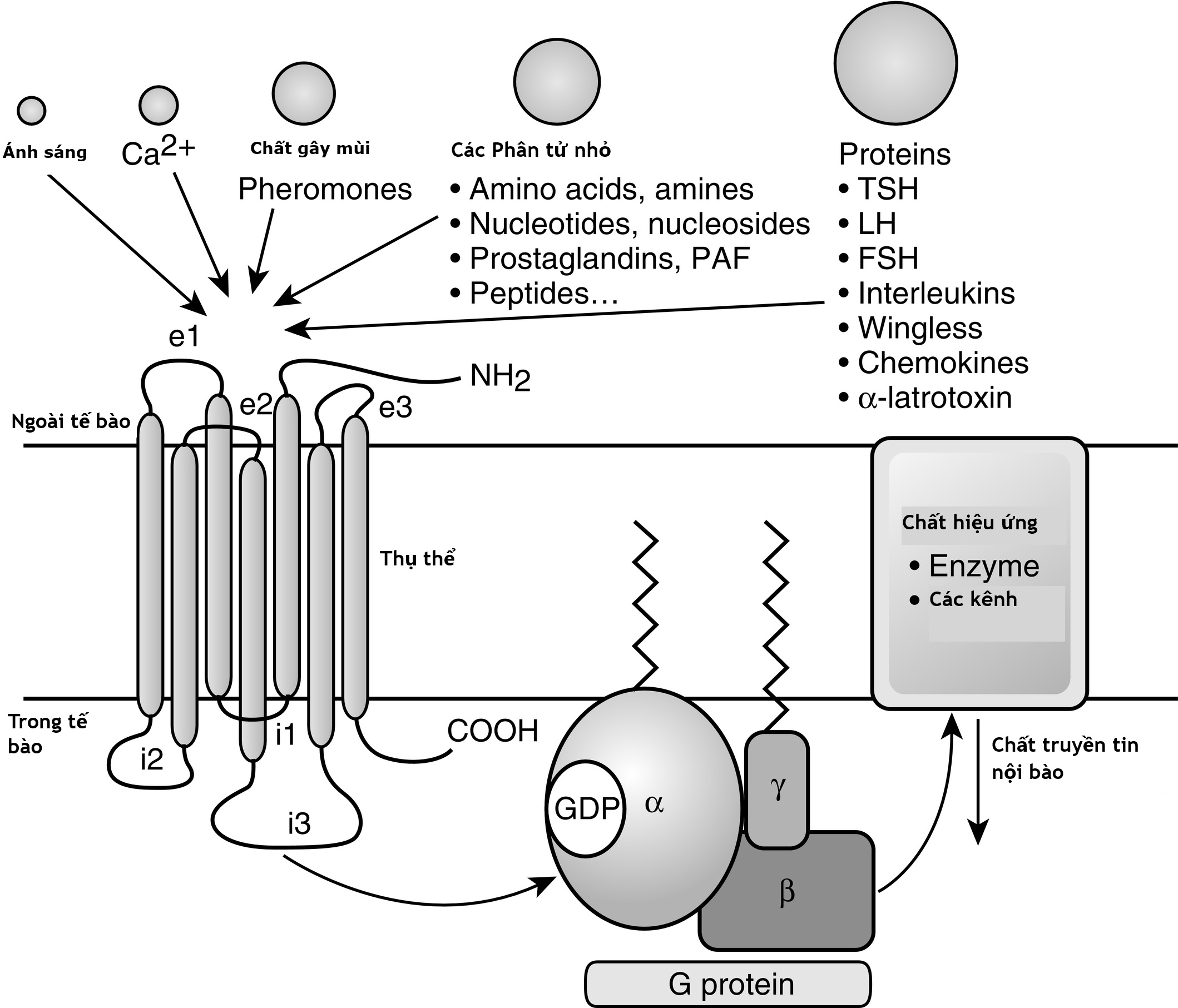
Hình 3.1 Cấu trúc và chức năng của thụ thể kết cặp G-protein (GPCR). GPCR có một miền ngoại bào đầu tận amino, bảy miền xuyên màng giả định được ngăn cách bởi ba vòng ngoại bào (e1-e3) và ba vòng nội bào (i1-i3), và một miền nội bào đầu tận carboxy. Sự gắn kết của phối tử dẫn đến việc trao đổi guanosine 5’-triphosphate (GTP) cho guanosine 5’-diphosphate (GDP), gây ra sự phân ly của G-protein thành một tiểu đơn vị GTPα và một tiểu đơn vị βγ. Sau đó, các tiểu đơn vị này làm thay đổi hoạt động của các enzyme tác động nội bào và các kênh xuyên màng, dẫn đến sự thay đổi nồng độ nội bào của các chất truyền tin thứ hai có thể bao gồm cyclic adenosine monophosphate và calci.
GPCR được hoạt hóa bởi một loạt các tín hiệu đa dạng, bao gồm protein, nucleotide, gốc acid amin, Ca2+, photon ánh sáng, và các chất tạo mùi (xem Hình 3.1). Người ta giả định rằng sự gắn kết của phối tử làm thay đổi cấu hình của các miền xuyên màng và các vòng nội bào, làm tăng ái lực của thụ thể đối với các protein gắn guanosine nucleotide dị tam thể (G-protein) đặc hiệu (xem Hình 3.1). Các G-protein có chung một cấu trúc dị tam thể bao gồm một tiểu đơn vị α và một phức hợp βγ liên kết chặt chẽ. Tiểu đơn vị α tương tác với các phân tử dò tìm và tác động, gắn guanosine 5’-triphosphate (GTP), và có hoạt tính GTPase nội tại. Có 16 gen ở động vật có vú mã hóa cho khoảng 20 chuỗi α khác nhau. Các tiểu đơn vị Gα được phân loại thành bốn nhóm và bao gồm Gsα (G kích thích), Giα (G ức chế) và Goα (G khác), Gq/11α, và G12/13α. Chúng hoạt động khác nhau trong việc nhận biết chất tác động nhưng có cấu trúc và cơ chế hoạt hóa tương tự. Các tiểu đơn vị Gα bao gồm hai miền: một miền gắn GTP và một miền chèn xoắn ốc. Miền gắn GTP tương đồng với các GTPase nhỏ giống Ras và bao gồm các vùng chuyển đổi I và II, chúng thay đổi cấu hình trong quá trình hoạt hóa. Các vùng chuyển đổi là các vòng của chuỗi xoắn alpha với các cấu hình nhạy cảm với guanine nucleotide. Miền chèn xoắn ốc được chèn vào miền gắn GTP trước vùng chuyển đổi I và là duy nhất cho các G-protein dị tam thể. Miền chèn xoắn ốc này cô lập guanine nucleotide tại giao diện với miền gắn GTP và phải được dịch chuyển để cho phép sự phân ly nucleotide.
Các tiểu đơn vị α liên kết với một nhóm nhỏ hơn các tiểu đơn vị β(5) và γ(12). Tính đặc hiệu tổ hợp trong các liên kết giữa các tiểu đơn vị G-protein khác nhau tạo ra tiềm năng cho sự đa dạng to lớn và có thể cho phép các dị tam thể riêng biệt tương tác chọn lọc chỉ với một số lượng hạn chế các GPCR và protein tác động.
Có hai con đường dẫn truyền tín hiệu chính liên quan đến GPCR: con đường tín hiệu cyclic AMP và con đường tín hiệu phosphatidylinositol. Việc tạo tín hiệu do G-protein gây ra được điều chỉnh bởi một “đồng hồ phân tử” được xác định bởi tốc độ trao đổi và thủy phân GTP. Ở trạng thái không hoạt động, G-protein tồn tại ở dạng dị tam thể với guanosine 5’-diphosphate (GDP) gắn vào chuỗi α. Sự tương tác của một thụ thể đã gắn phối tử với một G-protein dẫn đến việc giải phóng GDP, sau đó là sự gắn kết của GTP vào chuỗi α. Sự gắn kết của GTP vào chuỗi α dẫn đến sự phân ly của chuỗi α khỏi phức hợp βγ, cho phép chuỗi α-GTP tự do tương tác với các enzyme đích và các kênh ion. Các phức hợp βγ cũng tham gia vào các sự kiện truyền tín hiệu xuôi dòng thông qua sự tương tác với một loạt các mục tiêu ngày càng mở rộng, bao gồm một số dạng adenylyl cyclase và phospholipase C, các kênh kali, và các kinase của GPCR.
Tín hiệu G-protein bị chấm dứt bởi sự thủy phân α-GTP thành α-GDP bởi một GTPase nội tại. Một nhóm protein, được gọi là chất điều hòa tín hiệu G-protein (regulators of G protein signaling – RGSs), hoạt động như các protein hoạt hóa GTPase (GAPs), đặc hiệu cho các tiểu đơn vị Gα. Các protein này làm tăng tốc độ thủy phân GTP thành GDP và chấm dứt tín hiệu được truyền đi. Trong một số trường hợp, chính chất tác động có thể có hoạt tính GAP nội tại, giúp vô hiệu hóa con đường. Điều này đúng trong trường hợp của phospholipase C β, có hoạt tính GAP trong vùng đầu tận carboxy của nó. Đây là một hình thức điều hòa thay thế cho tiểu đơn vị Gα. Tuy nhiên, cần lưu ý rằng các GAP không có các gốc xúc tác để hoạt hóa G-protein α. Thay vào đó, GAPs làm giảm năng lượng hoạt hóa cần thiết để phản ứng diễn ra. Sau khi GTP bị thủy phân, chuỗi Gα-GDP tái liên kết với phức hợp βγ; G-protein dị tam thể tái liên kết giờ đây có khả năng tham gia vào một chu trình khác của tín hiệu được hoạt hóa bởi thụ thể.
Tính đặc hiệu trong việc gắn phối tử được tạo ra bởi sự biến đổi trong cấu trúc bậc một của các miền ngoại bào và nội bào. Tính đặc hiệu của các đáp ứng tác động được tạo ra bởi sự biến đổi trong cấu trúc bậc một của các miền nội bào và các dạng đồng phân của các tiểu đơn vị Gα của G-protein. Một số GPCR kết cặp chủ yếu với các tiểu đơn vị Gαi/Gαo, chúng hoạt động chủ yếu để làm giảm hoạt tính adenylyl cyclase. Các GPCR khác kết cặp chủ yếu với các tiểu đơn vị Gαs làm tăng hoạt tính adenylyl cyclase hoặc các tiểu đơn vị Gαq/Gα18202122 làm tăng hoạt tính phospholipase C.
Thật thú vị, dữ liệu cho thấy rằng các protein khung xương tế bào có thể điều chỉnh sự kết cặp thụ thể-G-protein. Ví dụ, protein khung xương tế bào màng hồng cầu 4.1G có thể can thiệp vào sự dẫn truyền tín hiệu của thụ thể adenosine A1. 4.1G cũng ảnh hưởng đến sự tích lũy cyclic AMP qua trung gian của thụ thể glutamate chuyển hóa 1α, làm tăng khả năng gắn phối tử của thụ thể glutamate chuyển hóa 1α, và làm thay đổi sự phân bố tế bào của nó. 4.1G cũng có thể đóng một vai trò trong sự dimer hóa thụ thể-thụ thể.
Sự homo- và heterodimer hóa của thụ thể không phụ thuộc chất chủ vận và do chất chủ vận gây ra ngày càng được công nhận là những yếu tố quyết định quan trọng của chức năng GPCR. Ví dụ, GPCR thụ thể somatostatin 5 (SSTR5) chủ yếu tồn tại dưới dạng monomer khi không có chất chủ vận. Tuy nhiên, chúng tạo thành homodimer khi có chất chủ vận. Hơn nữa, đã được chứng minh rằng SSTR5 có thể tạo thành heterodimer với thụ thể dopamine loại 2 (DRD2)—một GPCR khác—khi có chất chủ vận hsst2 hoặc dopamine. Sự hoạt hóa do chất chủ vận gây ra của heterodimer SSTR5-DRD2 trong các tế bào buồng trứng chuột hamster Trung Quốc (CHO) biểu hiện SSTR5 và DRD2 được tăng lên, so với sự hoạt hóa do chất chủ vận gây ra của các monomer và homodimer trong các tế bào CHO chỉ biểu hiện SSTR5 hoặc DRD2. Sự heterodimer hóa của các thụ thể cũng có thể dẫn đến sự bất hoạt của một trong các thụ thể trong phức hợp. Ví dụ, sự heterodimer hóa của thụ thể somatostatin 2A (sst2A) với thụ thể somatostatin 3 (SSTR3) dường như dẫn đến sự bất hoạt của SSTR3 đã được heterodimer hóa, mà không làm bất hoạt SSTR2 đã được heterodimer hóa.
GPCR có thể tạo thành heterodimer với các protein xuyên màng không phải là thụ thể. Cả thụ thể calcitonin (CALCR) và protein giống thụ thể calcitonin (CALCRL) đều có thể tạo thành heterodimer với ba protein phụ kiện khác nhau được gọi là “RAMPs“: RAMP1, RAMP2, và RAMP3. Trong khi CALCR có thể được hoạt hóa bởi phối tử khi không có sự heterodimer hóa với RAMP, CALCRL chỉ được hoạt hóa bởi phối tử nếu được heterodimer hóa với một RAMP. Các RAMP làm thay đổi tính đặc hiệu của phối tử của thụ thể đã được heterodimer hóa.
Các CALCR không ở dạng heterodimer với RAMPs được hoạt hóa bởi calcitonin và do đó tạo thành CALCR cổ điển. Tuy nhiên, các CALCR được heterodimer hóa với RAMP1, RAMP2, và RAMP3 gắn với amylin và lần lượt tạo thành các thụ thể amylin1, amylin2, và amylin3. Các CALCRL được dimer hóa với RAMP1 gắn với peptide liên quan đến gen calcitonin và tạo thành thụ thể peptide liên quan đến gen calcitonin. Các CALCRL được dimer hóa với RAMP2 và RAMP3 gắn với adrenomedullin và lần lượt tạo thành các thụ thể adrenomedullin1 và adrenomedullin2. Các RAMP làm thay đổi chức năng của các GPCR khác truyền tín hiệu hormone. Sự phân bố và chức năng của các thụ thể hormone cận giáp 1 và 2 bị thay đổi bởi sự gắn kết lần lượt với RAMP2 và RAMP3. Sự phân bố và chức năng của thụ thể glucagon bị thay đổi bởi sự gắn kết với RAMP2. Sự dimer hóa/heterodimer hóa có thể xảy ra trong lưới nội chất (ER), ngay sau khi quá trình tổng hợp protein diễn ra. ER đóng một vai trò trong việc xác định liệu một protein có được biểu hiện ở nơi khác trong tế bào hay không, do đó bảo vệ tế bào khỏi các protein bị gấp sai và (có khả năng) bị đột biến. CALCRL không được heterodimer hóa là một thụ thể mồ côi vì các CALCRL không thể rời khỏi ER để đến màng tế bào, trừ khi được heterodimer hóa với RAMPs.
Các thụ thể melanocortin cũng sử dụng các protein phụ kiện. Hormone vỏ thượng thận (ACTH) trong tuần hoàn gắn với năm dạng khác nhau của thụ thể melanocortin (loại 1–5), nhưng chỉ có thụ thể melanocortin 2 (MC2R) ở vỏ thượng thận mới dẫn đến việc giải phóng steroid thượng thận. MC2R tương tác với Gs, dẫn đến việc hoạt hóa adenylyl cyclase và hình thành cyclic AMP. MC2R là GPCR nhỏ nhất được biết đến cho đến nay và thuộc về một họ các thụ thể melanocortin (loại 1–5) gắn với các dẫn xuất khác nhau của proopiomelanocortin, đặc biệt là hormone kích thích tế bào hắc tố α (α-MSH). Protein phụ kiện protein liên quan đến thụ thể melanocortin 2 (MRAP) là cần thiết cho chức năng của MC2R, vì nó rất quan trọng cho việc vận chuyển thụ thể từ ER đến bề mặt tế bào. Hơn nữa, MRAP tạo điều kiện cho việc truyền tín hiệu của MC2R. Do đó, việc mất chức năng của MRAP ngăn cản sự biểu hiện của MC2R trên màng và hoàn toàn ngăn chặn tín hiệu ACTH. Chuột thiếu MRAP chết khi sinh, trừ khi được cứu bằng glucocorticoid, nhưng có sản xuất mineralocorticoid và catecholamine bình thường. Tuyến thượng thận của chuột trưởng thành thiếu MRAP nhỏ với hình thái thượng thận bất thường, phân vùng vỏ bất thường, và sự biệt hóa tế bào tiền thân thượng thận bất thường. Thật thú vị, MRAP tạo thành một homodimer đối song song độc đáo ở gần MC2R. Protein phụ kiện MRAP cũng có thể tương tác với các thụ thể melanocortin khác, đặc biệt là MC5R, nhưng lại có tác động tiêu cực đến tín hiệu của chúng. Sự biểu hiện của MRAP được cho là chủ yếu có mặt ở lớp bó trong tuyến thượng thận của chuột, phù hợp với vai trò hỗ trợ của nó trong sản xuất glucocorticoid. Do đó, các đột biến trong MC2R hoặc MRAP có thể dẫn đến suy giảm glucocorticoid gia đình thứ phát sau đề kháng ACTH. Ngược lại, MRAP2, một protein có 39% tương đồng acid amin với MRAP, chia sẻ chức năng vận chuyển MC2R của MRAP nhưng dường như không đóng vai trò hỗ trợ chính trong tín hiệu ACTH của vỏ thượng thận. Ngược lại, các nghiên cứu in vitro đã chỉ ra rằng sự biểu hiện quá mức của MRAP2 có thể ức chế sự hoạt hóa MC2R. MRAP2 dường như đóng một vai trò trong cân bằng nội môi năng lượng vì chuột KO MRAP2 phát triển béo phì. MRAP2 tương tác với MC4R trong nhân cạnh não thất của vùng dưới đồi (PVN), nơi mà KO MRAP2 đặc hiệu cho PVN lặp lại kiểu hình KO MRAP2 toàn thể.
Việc ER không xuất khẩu được các homodimer GPCR đột biến và heterodimer GPCR kiểu dại-GPCR đột biến ra màng tế bào đã được phát hiện là nguyên nhân của các tình trạng nội tiết trội âm. Một đột biến trội âm là một đột biến dị hợp tử dẫn đến một kiểu hình mà người ta mong đợi do mất chức năng ở cả hai alen. Một số đột biến MC4R dị hợp tử gây ra béo phì di truyền trội do sự tương tác của MC4R kiểu dại với thụ thể đột biến, và hiệu ứng đặc hiệu này của tương tác protein-protein dẫn đến một hiệu ứng trội âm. Ngoài ra, một số đột biến dị hợp tử trong gen mã hóa thụ thể vasopressin V2 gây ra bệnh đái tháo nhạt do thận thông qua việc sản xuất các protein đột biến cản trở sự di chuyển của các thụ thể bình thường đến màng tế bào. Các thụ thể đột biến này cản trở sự biểu hiện trên bề mặt tế bào của các thụ thể kiểu dại bằng cách tạo thành heterodimer với các thụ thể kiểu dại mà không thể được xuất khẩu từ ER ra màng tế bào. Phát hiện này giải thích tại sao những phụ nữ dị hợp tử đối với các đột biến gen thụ thể vasopressin V2 này không cô đặc được nước tiểu ngay cả với liều cao desmopressin, một chất chủ vận thụ thể vasopressin V2 tổng hợp, mặc dù có khả năng sản xuất các thụ thể vasopressin V2 kiểu dại. Một hiện tượng tương tự giải thích sự di truyền trội của tình trạng đề kháng thụ thể hormone kích thích tuyến giáp (TSH) một phần ở những bệnh nhân dị hợp tử đối với một số đột biến TSH bất hoạt. Ở những bệnh nhân này, các thụ thể TSH đột biến tạo thành các oligomer với các thụ thể kiểu dại và ngăn chặn sự xuất khẩu của các thụ thể kiểu dại từ ER ra màng tế bào.
Tương tự, sự gấp sai và định tuyến sai của một số thụ thể hormone giải phóng gonadotropin (GnRH) đột biến trong ER (cũng như sự oligomer hóa của các thụ thể GnRH đột biến này với các thụ thể GnRH kiểu dại) làm giảm sự biểu hiện trên màng tế bào của các thụ thể GnRH kiểu dại. Tuy nhiên, hiện tượng này chưa được phát hiện có ý nghĩa lâm sàng ở những bệnh nhân dị hợp tử đối với các đột biến gây ra suy sinh dục giảm gonadotropin đơn độc (IHH) di truyền lặn trên nhiễm sắc thể thường, vì những cá thể dị hợp tử này thể hiện một trục GnRH-gonadotropin nguyên vẹn và không có dấu hiệu lâm sàng của IHH. Do đó, ở những cá thể này, đủ số lượng thụ thể GnRH kiểu dại không bị oligomer hóa với các thụ thể GnRH đột biến và được vận chuyển đến màng tế bào để duy trì các tương tác GnRH-thụ thể GnRH đủ bình thường để tránh sự phát triển của IHH.
Hầu hết các GPCR đều hoạt hóa G-protein ở mức độ rất thấp khi không có sự gắn kết của phối tử. Một số GPCR có hoạt tính cấu thành (tức là không phụ thuộc phối tử) cao hơn nhiều, chẳng hạn như thụ thể hormone tạo hoàng thể, TSH, hormone giải phóng thyrotropin (TRH), glucagon-like peptide-1, melanocortin, và cannabinoid. Các thụ thể này có thể hoạt hóa G-protein khi không có sự gắn kết của phối tử, thể hiện hoạt tính cấu thành tăng tuyến tính với sự gia tăng biểu hiện của các thụ thể trên bề mặt tế bào. Như đã mô tả trước đó, các chất chủ vận đảo nghịch làm giảm hoạt tính của các thụ thể này. Ở các thụ thể không có hoạt tính cấu thành, các đột biến di truyền dẫn đến sự thay thế một acid amin duy nhất cũng có thể làm tăng đáng kể tốc độ tương tác của thụ thể không gắn phối tử với G-protein của nó. Có thể các chất chủ vận đảo nghịch sẽ đóng một vai trò trong việc điều trị các tình trạng y tế gây ra bởi các đột biến GPCR dẫn đến tăng hoạt hóa cấu thành của thụ thể.
Sự giải mẫn cảm và tái mẫn cảm của thụ thể đóng một vai trò trong hoạt động của GPCR. Ba quá trình giải mẫn cảm thụ thể đã được mô tả. Quá trình giải mẫn cảm thụ thể đầu tiên là sự tách cặp nhanh chóng của G-protein khỏi GPCR. Quá trình này xảy ra trong vòng vài giây đến vài phút sau khi bắt đầu và xảy ra do sự phosphoryl hóa GPCR. Các kinase của thụ thể G-protein (GRKs) ngày càng được công nhận đóng một vai trò chính khi quá trình này liên quan đến sự giải mẫn cảm tương đồng. Sự phosphoryl hóa qua trung gian GRK của các gốc serine và threonine trong vòng nội bào thứ ba, hoặc miền nội bào đầu tận carboxy dẫn đến sự hoạt hóa của β-arrestin, từ đó làm bất hoạt adenylyl cyclase (Hình 3.2). Các protein kinase phụ thuộc chất truyền tin thứ hai cũng góp phần vào sự giải mẫn cảm của thụ thể khi quá trình này liên quan đến sự giải mẫn cảm tương đồng, nhưng chúng cũng tham gia vào sự giải mẫn cảm của thụ thể khi sự giải mẫn cảm liên quan đến sự giải mẫn cảm dị đồng. Sự giải mẫn cảm dị đồng hay không phụ thuộc chất chủ vận xảy ra do sự hoạt hóa của một thụ thể khác với thụ thể bị giải mẫn cảm.

Hình 3.2 Sự giải mẫn cảm và tái chế của các thụ thể kết cặp G-protein (GPCR). Ngay sau khi một chất chủ vận gắn vào một GPCR, sự phosphoryl hóa của các kinase thụ thể G-protein đối với các gốc serine và threonine trong vòng nội bào thứ ba hoặc miền nội bào đầu tận carboxy dẫn đến sự hoạt hóa của β-arrestin. Sự hoạt hóa của β-arrestin làm bất hoạt adenylyl cyclase và khởi đầu sự biệt trí của GPCR trong các túi được bao bọc bởi clathrin. Sự dephosphoryl hóa của thụ thể bị biệt trí và sự phân ly sau đó của thụ thể khỏi β-arrestin được theo sau bởi sự tái chế của GPCR đến màng tế bào. Ngoài ra, một khi bị biệt trí, GPCR có thể bị phá hủy trong lysosome.
Quá trình giải mẫn cảm thụ thể thứ hai là sự nội hóa/biệt trí (internalization/sequestration) của GPCR. Quá trình này chậm hơn so với sự tách cặp do phosphoryl hóa thụ thể của G-protein khỏi GPCR và xảy ra trong vòng vài phút đến vài giờ sau khi bắt đầu. Ngoài sự phosphoryl hóa, cả GPCR và β-arrestin đều được biến đổi sau dịch mã theo nhiều cách, bao gồm cả bằng cách ubiquitin hóa. Quá trình này có thể đảo ngược vì các thụ thể có thể được tái chế trở lại bề mặt tế bào (xem Hình 3.2). GRKs và β-arrestin đóng một vai trò trong việc khởi đầu sự nội hóa/biệt trí của các GPCR β2-adrenergic, hormone tạo hoàng thể (LH), FSH, TSH, TRH, vasopressin V2, angiotensin II loại 1A, và các GPCR khác trong các túi được bao bọc bởi clathrin (xem Hình 3.2). Sự dephosphoryl hóa của thụ thể bị biệt trí, sau đó là sự phân ly của thụ thể khỏi β-arrestin, là cần thiết để thụ thể được tái chế trở lại màng tế bào và được tái mẫn cảm (xem Hình 3.2).
Quá trình giải mẫn cảm thụ thể thứ ba là sự điều hòa giảm (downregulation). Với sự điều hòa giảm, số lượng GPCR nội bào giảm do sự thoái biến lysosome tăng lên và sự tổng hợp thụ thể giảm đi do sự thay đổi của các cơ chế điều hòa phiên mã và sau phiên mã (xem Hình 3.2). Sự điều hòa giảm là một quá trình chậm xảy ra trong vòng vài giờ đến vài ngày sau khi bắt đầu các quá trình dẫn đến sự phát triển của nó. Đối với nhiều GPCR, sự ubiquitin hóa thụ thể thúc đẩy sự thoái biến của các thụ thể được hoạt hóa bởi chất chủ vận trong lysosome. Các protein khác cũng đóng vai trò quan trọng trong sự giải mẫn cảm, bao gồm phosphodiesterase, các protein họ RGS, và các protein neo A-kinase. Cùng nhau, mạng lưới phức tạp gồm các kinase, ubiquitin ligase, và các protein thích ứng này điều phối sự giải mẫn cảm cấp tính và kéo dài của GPCR.
Một trong những cách mà đột biến thụ thể vasopressin V2 Arg137His can thiệp vào chức năng của thụ thể đột biến và gây ra bệnh đái tháo nhạt do thận liên kết X là bằng cách làm thay đổi sự giải mẫn cảm và tái chế của thụ thể đột biến. Các nghiên cứu in vitro đã tiết lộ rằng thụ thể đột biến được phosphoryl hóa một cách cấu thành. Do đó, ngay cả khi không có sự gắn kết của phối tử, thụ thể đột biến vẫn bị β-arrestin gắn vào—từ đó dẫn đến sự biệt trí của thụ thể đột biến trong các túi được bao bọc bởi clathrin. Việc tái chế thụ thể đột biến trở lại màng tế bào đòi hỏi thụ thể đột biến phải được dephosphoryl hóa và tách khỏi β-arrestin. Tuy nhiên, thụ thể đột biến không trải qua quá trình dephosphoryl hóa trong khi bị biệt trí, và do đó không thể tách khỏi β-arrestin và được tái chế trở lại màng tế bào—do đó làm giảm sự biểu hiện của thụ thể đột biến trên màng tế bào.
Trong số tám loại GPCR, chỉ có các loại A, B, và C chứa các thụ thể cho các hormone và chất dẫn truyền thần kinh của động vật có vú (Hình 3.3). Các thụ thể loại A chứa các thụ thể giống rhodopsin và được chia thành ít nhất 15 nhóm. Bốn trong số các nhóm này chứa các thụ thể được hoạt hóa bởi hormone. Đó là các nhóm thụ thể peptide, thụ thể protein hormone, thụ thể GnRH, và nhóm thụ thể TRH và chất kích thích tiết.
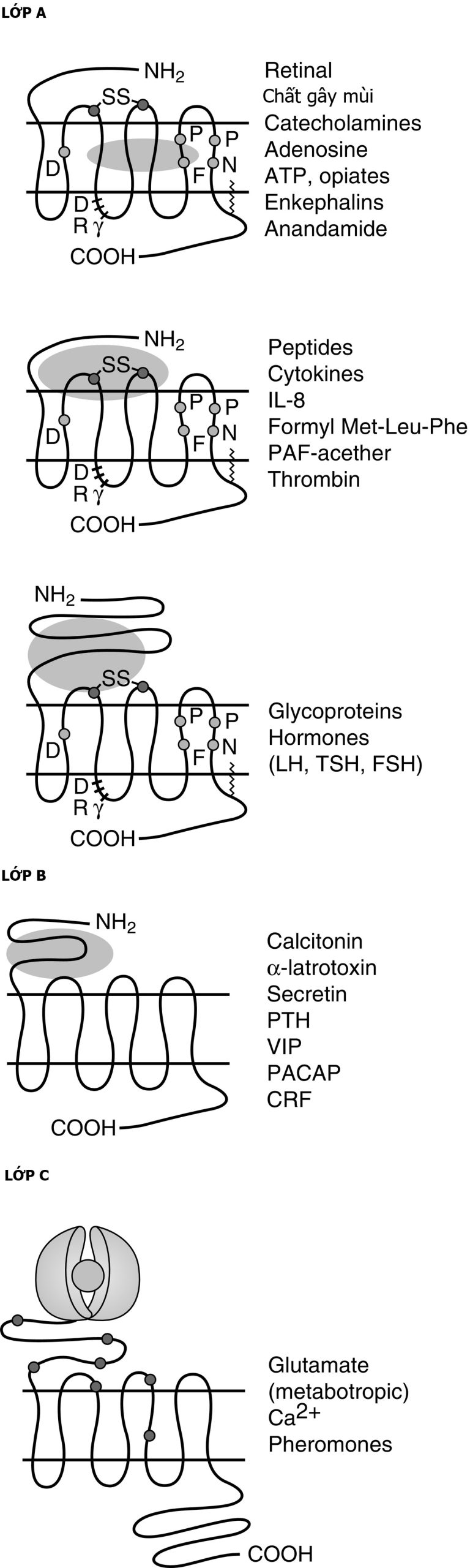
Hình 3.3 Ví dụ về các thụ thể kết cặp G-protein loại A, B và C. Hình bầu dục đại diện cho phối tử. Các thụ thể này có thể khác nhau về trình tự acid amin, về chiều dài của miền ngoại bào đầu tận amino và miền nội bào đầu tận carboxy, và về các vùng thụ thể liên quan đến tương tác phối tử-thụ thể.
Nhóm thụ thể peptide bao gồm các thụ thể angiotensin, ACTH/melanocortin, oxytocin, somatostatin, và vasopressin. Nhóm thụ thể protein hormone bao gồm các thụ thể cho các hormone glycoprotein, bao gồm các thụ thể FSH, LH, và thyrotropin (TSH). Các thụ thể này có miền ngoại bào đầu tận amino lớn và các vị trí gắn phối tử bao gồm vòng ngoại bào thứ nhất và thứ ba (xem Hình 3.3). Cũng có nhiều sự tương đồng trong trình tự acid amin giữa các thụ thể này (xem Hình 3.3). Nhóm thụ thể GnRH chỉ chứa thụ thể GnRH. Nhóm thụ thể TRH và chất kích thích tiết bao gồm thụ thể TRH và thụ thể chất kích thích tiết hormone tăng trưởng (GH).
Các GPCR loại B có cấu trúc tương tự như các thành viên của nhóm thụ thể protein hormone (xem Hình 3.3). Tuy nhiên, không giống như các thụ thể hormone glycoprotein, các GPCR loại B không có trình tự acid amin tương tự. Họ này chứa các thụ thể cho các hormone có trọng lượng phân tử cao hơn, bao gồm calcitonin, glucagon, peptide ức chế dạ dày, hormone cận giáp (PTH), và yếu tố giải phóng corticotrophin (CRF).
Các thụ thể loại C có một miền ngoại bào lớn, với hai thùy được ngăn cách bởi một vùng bản lề đóng lại trên phối tử (xem Hình 3.3). Vùng này còn được gọi là miền hoặc mô-đun bẫy ruồi Venus (Venus flytrap) do cơ chế bẫy của vùng bản lề. Họ này bao gồm thụ thể cảm nhận calci (CASR).
Hầu hết các đột biến bất hoạt GPCR có thể được phân loại vào một trong năm loại. Các đột biến bất hoạt loại I cản trở quá trình sinh tổng hợp thụ thể. Các đột biến bất hoạt loại II cản trở việc vận chuyển thụ thể đến bề mặt tế bào. Các đột biến bất hoạt loại III cản trở việc gắn phối tử. Các đột biến bất hoạt loại IV cản trở sự hoạt hóa thụ thể. Các đột biến bất hoạt loại V không gây ra các khiếm khuyết rõ ràng trong quá trình sinh tổng hợp, vận chuyển, gắn phối tử, hoặc hoạt hóa thụ thể, nhưng có thể gây ra các rối loạn y tế. Cũng có những đột biến bất hoạt can thiệp vào chức năng thụ thể thông qua nhiều cơ chế và do đó không thể được xếp vào một loại.
Các thụ thể loại A truyền tín hiệu hormone
Nhóm Thụ Thể Peptide
Thụ Thể Adrenocorticotropin và Melanocortin-2
Một tên gọi khác của thụ thể ACTH là thụ thể melanocortin-2 (MC2R) vì thụ thể ACTH là một trong năm thành viên của họ thụ thể melanocortin của GPCR, tất cả đều kết cặp với Gs để hoạt hóa việc tạo ra cyclic AMP nội bào. Để rõ ràng, khi thảo luận về các tương tác giữa ACTH và thụ thể của nó, tên cũ sẽ được sử dụng trong phần còn lại của chương này. Gen thụ thể ACTH nằm trên nhánh ngắn của nhiễm sắc thể 18 (18p11.2). Thụ thể ACTH có một miền ngoại bào và nội bào tương nhỏ. Sự hoạt hóa do Adrenocorticotropin gây ra của thụ thể ACTH trong lớp bó và lớp lưới của vỏ thượng thận sẽ kích thích Gs, dẫn đến tăng nồng độ cyclic AMP nội bào, từ đó kích thích quá trình tạo steroid bằng cách hoạt hóa các kinase phụ thuộc cyclic AMP.
Thiếu hụt glucocorticoid đơn độc di truyền, đề kháng ACTH, và thiếu hụt glucocorticoid gia đình (FGD) là những tên gọi khác nhau cho cùng một hội chứng di truyền lặn trên nhiễm sắc thể thường bao gồm thiếu hụt glucocorticoid kèm theo bài tiết mineralocorticoid bình thường. FGD đã được phân loại thêm thành FGD loại 1 và 2 và hội chứng triple A. Bệnh nhân bị FGD loại 1 có các đột biến MC2R trên cả hai alen, dẫn đến các thụ thể ACTH có chức năng bất thường, và chiếm 25% các trường hợp FGD. Ngược lại, bệnh nhân bị FGD loại 2 có đề kháng ACTH do các đột biến trong MRAP. Triple A (hội chứng Allgrove) là một hội chứng di truyền lặn trên nhiễm sắc thể thường được đặc trưng bởi suy thượng thận đề kháng ACTH, co thắt tâm vị, và không có nước mắt—gây ra bởi các đột biến trong gen hội chứng co thắt tâm vị-suy vỏ thượng thận-không có nước mắt (AAAS) mã hóa protein ALADIN. ALADIN được cho là điều hòa các phức hợp lỗ nhân, và sự vận chuyển nhân-bào tương đóng một vai trò trong việc điều chỉnh đáp ứng stress oxy hóa trong các tế bào thượng thận.
Phổ bệnh ở bệnh nhân FGD loại 1 thay đổi từ biểu hiện ở trẻ sơ sinh với hạ đường huyết, co giật, hoặc trụy tuần hoàn đến biểu hiện ở thời thơ ấu với mệt mỏi và tăng nhạy cảm với nhiễm trùng. Ít phổ biến hơn, bệnh nhân có thể biểu hiện hen suyễn thời thơ ấu mà giải quyết bằng điều trị với liều sinh lý của glucocorticoid. Tăng sắc tố da được cho là do tăng nồng độ ACTH tác động lên MC1R có thể được thấy ngay từ tháng đầu đời, nhưng thường trở nên rõ ràng sau tháng thứ tư. Có một trường hợp được báo cáo về FGD loại 1 không có tăng sắc tố da, mặc dù nồng độ ACTH tăng cao, ở một bệnh nhân có đột biến đồng hợp tử ở cả MC2R và MC1R. Giả định rằng tăng sắc tố da là do tăng nồng độ ACTH tác động lên MC1R (cả trong FGD loại 1 và bệnh Addison) đã được chứng thực ở bệnh nhân này, người có đột biến MC1R trước đây đã được liên hệ với kiểu hình tóc đỏ và da nhợt. Trẻ sơ sinh bị FGD loại 1 cũng có thể bị vàng da. Tầm vóc cao kèm theo tuổi xương tiến triển hoặc không tương xứng, mặc dù tuổi dậy thì bình thường, dường như phổ biến ở trẻ em bị FGD loại 1. Sinh lý bệnh của tầm vóc cao vẫn chưa được làm sáng tỏ dứt khoát. Một giả thuyết là tác dụng đồng hóa của GH không bị cortisol đối kháng.
Bệnh nhân FGD1 không có giai đoạn phát dục thượng thận, xác nhận tầm quan trọng của ACTH trong việc khởi phát và duy trì giai đoạn phát dục thượng thận. Khi biểu hiện, nồng độ cortisol, androstenedione, và dihydroepiandrosterone trong huyết tương thấp hoặc bình thường thấp—và nồng độ ACTH trong huyết tương tăng cao. Khi nằm ngửa, bệnh nhân FGD loại 1 có nồng độ renin và aldosterone gần như bình thường. Về mặt mô học, lớp bó và lớp lưới bị teo trong FGD. Tuy nhiên, chứng tỏ ACTH không có vai trò thiết yếu trong sự phát triển phôi thai và duy trì lớp cầu, vỏ thượng thận ở bệnh nhân mắc tất cả các loại FGD đều chứa các tế bào lớp cầu.
FGD loại 2 là do các đột biến trong MRAP1 như đã mô tả trước đó và chiếm 20% tổng số các trường hợp FGD. Bệnh nhân FGD loại 2 biểu hiện với các triệu chứng nặng ở độ tuổi sớm hơn so với bệnh nhân FGD loại 1 (tuổi trung vị 0,08 tuổi so với 2 tuổi). Bệnh nhân có thể biểu hiện khuyết tật thần kinh nặng, co giật, và tật đầu nhỏ được cho là do hạ đường huyết không được nhận biết. Bệnh nhân FGD loại 2 có chiều cao bình thường. Điều này được cho là do điều trị sớm bằng glucocorticoid. Các đột biến là đột biến vị trí nối hoặc đột biến vô nghĩa được dự đoán sẽ tạo ra các protein không có miền xuyên màng, vốn cần thiết cho sự tương tác với MC2R.
Những bất thường trong biểu hiện thụ thể ACTH có thể được thấy trong các tình trạng khác. Bằng chứng cho thấy chuỗi tín hiệu ACTH-thụ thể-Gαs-adenylyl cyclase-cyclic AMP duy trì sự biệt hóa của các tế bào vỏ thượng thận và sự suy giảm của chuỗi này dẫn đến sự mất biệt hóa và tăng sinh của các tế bào vỏ thượng thận. Ung thư biểu mô vỏ thượng thận từ một số bệnh nhân đã được phát hiện có mất dị hợp tử (LOH) đối với gen thụ thể ACTH, dẫn đến biểu hiện messenger ribonucleic acid (mRNA) của thụ thể ACTH giảm rõ rệt. Sự phát triển của các khối u có LOH đối với gen thụ thể ACTH cũng có thể hung hãn hơn so với các khối u khác. Một đột biến hoạt hóa của Gi2 làm ức chế hoạt tính adenylyl cyclase một cách cấu thành cũng đã được tìm thấy trong các khối u vỏ thượng thận. Do đó, giảm hoạt tính thụ thể ACTH có thể liên quan đến sự hình thành khối u.
Thật thú vị, nhiều bệnh nhân bị tăng sản thượng thận dạng nốt lớn không phụ thuộc ACTH (AIMAH)—một nguyên nhân của hội chứng Cushing không phụ thuộc ACTH do các đột biến bất hoạt của gen ức chế khối u giả định ARMC5 —biểu hiện tăng nồng độ glucocorticoid để đáp ứng với các hormone không phải corticotropin mà bình thường không gây giải phóng glucocorticoid. Các hormone này bao gồm peptide ức chế dạ dày, arginine và lysine vasopressin ngoại sinh, LH, gonadotropin màng đệm người (HCG), angiotensin II, catecholamine, leptin, và các chất chủ vận thụ thể serotonin. Sự tăng biểu hiện của các thụ thể cho các phối tử này trong các tuyến thượng thận bất thường đã được xem là một lời giải thích khả dĩ cho sự cảm ứng bất thường của việc giải phóng glucocorticoid bởi các phối tử không phải corticotropin này. Tuy nhiên, các thụ thể cho một số phối tử này được biểu hiện trong các tuyến thượng thận bình thường. Do đó, cơ chế của hiện tượng này vẫn cần được làm sáng tỏ hoàn toàn.
Các Thụ Thể Melanocortin Khác
Các nghiên cứu trên chuột cho thấy thụ thể melanocortin-3 (MC3R), một trong năm thành viên khác của họ thụ thể melanocortin, điều chỉnh sự lắng đọng chất béo, vì những con chuột thiếu MC3R hoặc có các thụ thể hoạt động một phần có chi tiêu năng lượng khi nghỉ ngơi bình thường và khối lượng mỡ tăng và khối lượng không mỡ giảm, bao gồm cả sự hình thành xương giảm. Vai trò của MC3R ở người ít rõ ràng hơn. Hơn 24 biến thể MC3R ở người đã được xác định mà không có bằng chứng về béo phì. Tuy nhiên, những bệnh nhân có các biến thể này không được xác định kiểu hình về khối lượng mỡ để đánh giá xem họ có sao chép kiểu hình của mô hình chuột hay không, mô hình này biểu hiện trọng lượng cơ thể bình thường nhưng lượng năng lượng hấp thụ lớn hơn và sự phân chia năng lượng bị thay đổi, thiên về các tế bào tích lũy lipid. Một số biến thể mã hóa (p.D158Y, p.T280S, p.I183N), có khả năng gây bệnh do sản xuất cyclic AMP giảm đáng kể, cũng như các biến thể không mã hóa, cho thấy béo phì khởi phát sớm ở tất cả các cá nhân bị ảnh hưởng. Tình trạng đồng hợp tử đối với một cặp đa hình đơn nucleotide của gen MC3R (p.T6K + p.V81I) dẫn đến việc sản xuất các MC3R hoạt động một phần đã được phát hiện có liên quan đến béo phì khởi phát ở trẻ em da trắng Mỹ và trẻ em người Mỹ gốc Phi. Các đối tượng đồng hợp tử đối với đột biến kép có chỉ số khối cơ thể (BMI)-z, khối lượng mỡ và phần trăm khối lượng mỡ cao hơn, cũng như chu vi vòng eo lớn hơn. Điều này bất chấp việc không tìm thấy sự khác biệt về lượng năng lượng hấp thụ, chi tiêu năng lượng khi nghỉ ngơi, tổng chi tiêu năng lượng, thương số hô hấp, hoạt động thể chất. Một mô hình chuột mang các biến thể MC3R T6K + V81I của người cho thấy đột biến kép biểu hiện khối lượng mỡ và hiệu quả cho ăn lớn hơn với khối lượng không mỡ giảm. Những phát hiện này tương tự như chuột knockout MC3R.
Thụ thể melanocortin-4 (MC4R) là một thành viên khác của họ thụ thể melanocortin và đóng một vai trò trong việc kiểm soát sự thèm ăn và cân nặng. MC4R có hoạt tính cấu thành cơ bản (tức là không phụ thuộc phối tử) có thể bị ức chế bởi chất chủ vận đảo nghịch peptide liên quan đến agouti (AgRP). Sự hoạt hóa của MC4R bởi chất chủ vận tự nhiên của nó là α-MSH tạo ra các hiệu ứng gây chán ăn. Hơn 150 đột biến MC4R tự nhiên đã được xác định, gây ra béo phì do ăn nhiều, tăng khối lượng nạc, tăng mật độ xương, và tăng sự phát triển theo chiều dọc. Bệnh nhân có đột biến đồng hợp tử dường như bị béo phì nặng hơn so với những người thân dị hợp tử của họ, phù hợp với sự di truyền đồng trội.
Các đột biến MC4R được cho là nguyên nhân đơn gen phổ biến nhất của béo phì ở người. Tỷ lệ lưu hành của các đột biến MC4R gây bệnh trong các quần thể béo phì thay đổi rộng rãi, từ 0,5% đến 5,8%, tùy thuộc vào tiêu chí sàng lọc và dân số. Các đa hình gen AgRP dường như có liên quan đến chứng chán ăn tâm thần.
Người ta biết rất ít về các thụ thể melanocortin-5 (MC5Rs) ở động vật và người. Chỉ có bằng chứng yếu từ một nghiên cứu liên kết và liên hợp duy nhất của các gia đình ở Quebec cho thấy MC5Rs cũng có thể đóng một vai trò trong việc điều chỉnh trọng lượng cơ thể và khối lượng mỡ.
Một thành viên khác của họ thụ thể melanocortin, MC1R, kiểm soát sắc tố da và tóc. Sự hoạt hóa của MC1R trong các tế bào hắc tố của da và nang lông bởi các peptide có nguồn gốc từ proopiomelanocortin (POMC) là α-MSH và ACTH sẽ kích thích sự tổng hợp eumelanin, một sắc tố màu nâu-đen. Sự ức chế hoạt tính cấu thành cơ bản của MC1R bởi protein agouti, hoặc các đột biến cụ thể, dẫn đến việc giải phóng pheomelanin, một sắc tố màu đỏ-vàng, từ các tế bào hắc tố.
Các đột biến đồng hợp tử bất hoạt của gen POMC gây ra suy thượng thận, tóc đỏ, da trắng, và béo phì khởi phát sớm. Suy thượng thận được đặc trưng bởi thiếu hụt glucocorticoid do thiếu sản xuất ACTH từ tiền chất POMC. Da trắng và tóc đỏ là do thiếu sự giải phóng eumelanin từ tế bào hắc tố do ACTH và α-MSH gây ra, vốn là kết quả của sự hoạt hóa MC1R. Đáng chú ý, những bệnh nhân không phải da trắng có đột biến POMC đồng hợp tử dường như không có kiểu hình da trắng và tóc đỏ. Ở những người da trắng, sự tổng hợp eumelanin dường như phụ thuộc vào các peptide có nguồn gốc từ POMC, trong khi ở những người da sẫm màu hơn, các gen khác có thể kiểm soát sự tổng hợp eumelanin. Béo phì là do thiếu các hiệu ứng gây chán ăn do α-MSH gây ra, thường là kết quả khi α-MSH hoạt hóa MC4R. Tình trạng dị hợp tử đối với các đột biến gen POMC đã được liên kết với chứng ăn nhiều, béo phì khởi phát sớm, và tăng sự phát triển theo chiều dọc. Cả đột biến đồng hợp tử và dị hợp tử của prohormone convertase 1 đều gây béo phì ở người. Prohormone convertase 1 tác động lên POMC, proinsulin, và proglucagon. Bệnh nhân thiếu prohormone convertase 1 cũng bị bệnh ruột sơ sinh và hạ đường huyết sau ăn. Nguyên nhân của bệnh ruột chưa được biết nhưng được giả thuyết là liên quan đến việc xử lý GLP-2 bởi prohormone convertase 1. GLP-2 được biết là kích thích sự tăng sinh và sửa chữa của biểu mô ruột.
Thụ Thể Vasopressin
Đái tháo nhạt do thận (NDI) là kết quả của sự giảm đáp ứng của ống thận với arginine vasopressin (AVP), dẫn đến mất nước tự do quá mức. NDI được đặc trưng bởi chứng uống nhiều và đi tiểu nhiều không đáp ứng với vasopressin và các chất tương tự vasopressin. Vasopressin gắn vào thụ thể vasopressin V2 (AVPR2), một thụ thể kết cặp Gs, ở màng đáy bên của các tế bào chính ống góp ở thận và hoạt hóa sự di chuyển của các kênh nước aquaporin-2 (AQP2) đến màng đỉnh, do đó gây ra tính thấm nước. NDI liên kết X là do các đột biến bất hoạt của gen thụ thể vasopressin V2 (AVPR2) nằm ở Xq28 và chiếm khoảng 90% các trường hợp NDI do di truyền. Hơn 200 đột biến AVPR2 đã được mô tả, bao gồm đột biến sai nghĩa, vô nghĩa, chèn, xóa, và sắp xếp lại phức tạp. Các đột biến đã được phân loại thành năm loại dựa trên cơ chế, bao gồm phiên mã bất thường, xử lý mRNA, dịch mã, gấp sai và giữ lại trong tế bào, mất vị trí gắn G-protein, mất vị trí gắn AVP, và các khiếm khuyết trong vận chuyển nội bào. Một số bệnh nhân bị NDI liên kết X đáp ứng với liều cao desmopressin. NDI di truyền lặn trên nhiễm sắc thể thường (ARNDI) là do các đột biến mất chức năng trong gen của kênh nước AQP2 và chiếm khoảng 10% các dạng NDI di truyền. Hơn 50 đột biến đã biết gây ra ARNDI. Các dạng NDI di truyền trội trên nhiễm sắc thể thường cũng do các đột biến trong AQP2 có chức năng, nhưng không được vận chuyển đến màng đỉnh. Mười một đột biến đã được mô tả, chiếm ít hơn 1% các dạng NDI di truyền và thường có kiểu hình nhẹ hơn so với ARNDI hoặc NDI liên kết X.
Các đột biến tăng chức năng trong thụ thể vasopressin V2 cũng đã được báo cáo. Việc giải trình tự DNA của gen V2R của hai bệnh nhân đã xác định được các đột biến sai nghĩa dị hợp tử ở cả hai, dẫn đến sự thay đổi codon 137 từ arginine thành cysteine (p.R137C) hoặc leucine (p.R137L). Những đột biến này dẫn đến sự hoạt hóa cấu thành của thụ thể và các đặc điểm lâm sàng của hội chứng tiết hormone chống bài niệu không phù hợp (SIADH), được gọi là hội chứng thận tiết hormone chống bài niệu không phù hợp (NSIAD). Cho đến nay, khoảng 30 trường hợp NSIAD đã được báo cáo, hầu hết được mô tả ở nam giới vì tình trạng này liên kết X. Bệnh nhân có thể biểu hiện ở giai đoạn sơ sinh nhưng đôi khi không biểu hiện cho đến khi trưởng thành. Đã có một số báo cáo về các bệnh nhân nữ bị NSIAD, một số được chẩn đoán ở giai đoạn sơ sinh và những người khác ở tuổi trưởng thành. Bệnh nhân có đột biến p.R137L đã thể hiện sự giảm nồng độ AVP như mong đợi với một thử nghiệm nạp nước, nhưng nồng độ AQP2 trong nước tiểu vẫn tăng cao một cách không phù hợp.
Nhóm Thụ Thể Hormone Glycoprotein
Các hormone glycoprotein bao gồm TSH, FSH, LH, và HCG. Các hormone này có chung các tiểu đơn vị α dimer hóa với các tiểu đơn vị β đặc hiệu cho từng hormone. TSH, FSH, và LH gắn vào miền ngoại bào đầu tận amino của lần lượt các thụ thể TSH, FSH, và LH. Tác dụng của HCG được trung gian bởi thụ thể LH, còn được gọi là thụ thể hormone tạo hoàng thể/choriogonadotropin (LHCGR).
Các thụ thể hormone glycoprotein có một miền ngoại bào đầu tận amino lớn (350 đến 400 gốc), còn được gọi là miền ngoại bào (ectodomain), tham gia vào việc gắn phối tử (xem Hình 3.3). Miền ngoại bào bao gồm các đoạn lặp lại giàu leucine được bảo tồn cao giữa các thụ thể hormone glycoprotein. Có sự tương đồng từ 39% đến 46% của miền ngoại bào và 68% đến 72% của miền xuyên màng hay vùng xoắn ốc giữa ba thụ thể hormone glycoprotein.
Các thụ thể hormone glycoprotein được kết cặp với Gs, và sự gắn kết của hormone sẽ kích thích adenylyl cyclase, dẫn đến tăng nồng độ cyclic AMP nội bào và hoạt hóa protein kinase A (PKA). Các đột biến dẫn đến rối loạn chức năng nội tiết đã được báo cáo cho mỗi loại thụ thể hormone glycoprotein.
Thụ Thể Hormone Tạo Hoàng Thể/Choriogonadotropin
Cả đột biến bất hoạt và hoạt hóa của thụ thể LH đều đã được tìm thấy ở người. Gen thụ thể LH nằm trên nhiễm sắc thể 2p21 và bao gồm 11 exon. Exon 1 mã hóa một peptide hướng thụ thể LH đến màng sinh chất. Các exon từ 2 đến 10 mã hóa miền ngoại bào. Exon cuối cùng mã hóa các miền xuyên màng còn được gọi là vùng xoắn ốc (serpentine regions). Các đột biến vô nghĩa đơn lẻ, thay đổi acid amin, và xóa một phần gen đã được mô tả tạo ra các thụ thể LH có hoạt tính giảm. Các thay đổi acid amin đơn lẻ cũng đã được tìm thấy dẫn đến sự hoạt hóa Gs khi không có sự gắn kết của phối tử.
Sự phát triển của đề kháng LH đòi hỏi các đột biến trên cả hai alen làm bất hoạt gen thụ thể LH, vì một alen thụ thể bình thường có khả năng sản xuất đủ protein thụ thể để đảm bảo tín hiệu sinh lý. Ngược lại, các đột biến hoạt hóa của gen thụ thể LH gây ra các rối loạn nội tiết ở trạng thái dị hợp tử.
Trong thai nhi, các thụ thể LH chủ yếu được hoạt hóa bởi HCG. Các tế bào Leydig bắt đầu biểu hiện thụ thể LH ngay sau khi biệt hóa tinh hoàn ở tuần thứ 8 của thai kỳ. Sau đó, việc sản xuất androgen, do sự hoạt hóa của các thụ thể này bởi HCG, đóng một vai trò quan trọng trong sự phát triển của bộ phận sinh dục nam và sự di chuyển của tinh hoàn. Do đó, trẻ sơ sinh nam, với các đột biến bất hoạt của thụ thể LH, có thể biểu hiện bộ phận sinh dục phát triển bất thường—bao gồm dương vật nhỏ, tinh hoàn ẩn, và rối loạn phát triển giới tính XY.
Nam giới có các đột biến làm bất hoạt hoàn toàn thụ thể LH biểu hiện sự thất bại trong biệt hóa tế bào Leydig của tinh hoàn thai nhi. Kiểu hình này, được biết đến là thiểu sản tế bào Leydig loại 1, bao gồm bộ phận sinh dục ngoài giống nữ với âm đạo cụt, không có các dẫn xuất Müllerian, và tinh hoàn ở bẹn với các tế bào Leydig không có hoặc chưa trưởng thành. Ngoài ra, bệnh nhân có nồng độ LH huyết thanh tăng cao, nồng độ FSH huyết thanh bình thường, và nồng độ testosterone huyết thanh giảm không tăng đáp ứng với việc sử dụng HCG. Các đột biến dẫn đến kiểu hình này bao gồm một đột biến vô nghĩa (Arg545Stop) dẫn đến một thụ thể thiếu TM4-7, một thay đổi p.Ala593Pro, và một sự xóa bỏ TM7 (p.Leu608del, p.Val609) làm giảm sự biểu hiện trên bề mặt tế bào của thụ thể LH. Các thụ thể đột biến này không thể kết cặp với Gs.
Nam giới có các đột biến không làm bất hoạt hoàn toàn thụ thể LH biểu hiện với thiểu sản tế bào Leydig loại 2, được đặc trưng bởi dương vật nhỏ và giảm nam hóa. Một đột biến dẫn đến kiểu hình này bao gồm việc chèn một lysine mang điện tích ở vị trí 625 của TM7 thay cho isoleucine kỵ nước làm gián đoạn sự dẫn truyền tín hiệu. Một đột biến khác (p.Ser616Tyr, được tìm thấy ở những bệnh nhân bị thiểu sản tế bào Leydig nhẹ) có liên quan đến sự giảm biểu hiện trên bề mặt tế bào của thụ thể LH. Các đột biến xóa và vô nghĩa khác cũng đã được tìm thấy gây ra thiểu sản tế bào Leydig nhẹ.
Nam giới có các đột biến bất hoạt của thụ thể LH cũng có thể biểu hiện với một kiểu hình có mức độ nghiêm trọng trung gian giữa thiểu sản tế bào Leydig loại 1 và loại 2. Một bệnh nhân dị hợp tử kép với p.Ser616Tyr trên một alen và một đột biến xóa bất hoạt (△ exon 8) trên alen kia, đã biểu hiện thiểu sản tế bào Leydig, dương vật nhỏ, và lỗ tiểu lệch thấp. Đột biến Cys131Arg cũng đã được tìm thấy ở những bệnh nhân bị thiểu sản tế bào Leydig, dương vật nhỏ, và lỗ tiểu lệch thấp. Đột biến này nằm trong đoạn lặp lại giàu leucine của miền ngoại bào thụ thể LH và cản trở sự gắn kết phối tử ái lực cao.
Sự xóa bỏ exon 10 của gen thụ thể LH dẫn đến một thụ thể LH gắn LH và HCG bình thường. Thật thú vị, trong khi sự gắn kết HCG có thể gây ra tín hiệu xuyên màng bình thường, sự gắn kết LH lại không hoạt hóa được thụ thể. Vì HCG là hormone chính trong tử cung hoạt hóa thụ thể LH, và đáp ứng chất truyền tin thứ hai của các thụ thể đột biến đối với HCG không bị suy giảm, không có gì đáng ngạc nhiên khi một bệnh nhân nam được phát hiện là đồng hợp tử đối với đột biến này đã được sinh ra với bộ phận sinh dục nam bình thường. Tuy nhiên, quá trình dậy thì và chức năng sinh dục sau này phụ thuộc vào sự hoạt hóa của LH đối với thụ thể LH.
Vì sự xóa bỏ exon 10 của gen thụ thể LH dẫn đến một thụ thể LH đột biến, với tín hiệu nội bào giảm sút để đáp ứng với LH, cũng không có gì đáng ngạc nhiên khi bệnh nhân đồng hợp tử đối với đột biến này được phát hiện có sự phát triển dậy thì chậm, tinh hoàn nhỏ, và suy sinh dục tăng gonadotropin, khi được đánh giá ở tuổi 18. Liệu pháp HCG kéo dài đã dẫn đến việc bình thường hóa sản xuất testosterone của tinh hoàn, tăng kích thước tinh hoàn, và sự xuất hiện của tinh trùng trong tinh dịch. Tương tự, các đột biến bất hoạt của tiểu đơn vị β LH gây ra sự phát triển dậy thì bất thường, thiếu hụt testosterone nghiêm trọng, và không có tinh trùng, nhưng bộ phận sinh dục ngoài bình thường ở nam giới. Ở nữ giới, các đột biến bất hoạt của tiểu đơn vị β LH có liên quan đến sự phát triển dậy thì và hành kinh bình thường, sau đó là kinh nguyệt thưa, buồng trứng đa nang to và vô sinh.
Nữ giới có các đột biến mất chức năng của thụ thể LH có thể không có triệu chứng hoặc biểu hiện vô kinh hoặc kinh nguyệt thưa. Nữ giới có các đột biến thụ thể LH bất hoạt hoàn toàn thường bị vô kinh nguyên phát, không có khả năng rụng trứng, và nồng độ estrogen và progesterone giảm, kèm theo nồng độ LH và FSH tăng cao. Những cá nhân bị ảnh hưởng có thể có các dấu hiệu của nồng độ estrogen thấp, bao gồm tử cung thiểu sản, âm đạo có thành mỏng, giảm tiết dịch âm đạo, và giảm khối lượng xương, mặc dù sự phát triển vú ở tuổi dậy thì là bình thường. Các đột biến đồng hợp tử của thụ thể LH (p.N400S, và p.Ala449Thr) đã được liên kết với hội chứng nang trứng rỗng, một rối loạn trong đó không có noãn nào được lấy ra trong quá trình thụ tinh trong ống nghiệm.
Các đột biến hoạt hóa cấu thành thụ thể LH gây ra dậy thì sớm giới hạn ở nam (MLPP), còn được gọi là nhiễm độc tinh hoàn (testotoxicosis) —có thể là gia đình hoặc lẻ tẻ. Các bé trai mắc tình trạng này phát triển dậy thì sớm không phụ thuộc GnRH trước 4 tuổi, khi có đột biến p.Asp578Gly, và sớm nhất là trong năm đầu đời, khi có đột biến p.Asp578Tyr. Bệnh nhân mắc tình trạng này cũng có thể có dương vật to khi sinh.
Trong 5 năm đầu đời, bệnh nhân MLPP có nồng độ LH và FSH rất thấp nhưng nồng độ testosterone ở mức dậy thì. Trong giai đoạn thiếu niên và trưởng thành, nồng độ testosterone không tăng trên mức nồng độ phù hợp với lứa tuổi và nồng độ gonadotropin trở lại bình thường. Do đó, thanh thiếu niên và người lớn mắc MLPP thường không biểu hiện các dấu hiệu thừa androgen (như rậm lông hoặc mụn trứng cá nặng). Hầu hết các đột biến gây ra MLPP nằm ở TM6 và i3, những vùng tham gia vào sự kết cặp thụ thể-protein Gs. Một kiểu hình nhẹ hơn đã được báo cáo ở một bệnh nhân có đột biến hoạt hóa dị hợp tử (p.C617Y) trong TM7. Đột biến này được di truyền từ mẹ của bệnh nhân, người dường như không bị ảnh hưởng. Các đột biến hoạt hóa soma gây ra u tuyến tế bào Leydig lẻ tẻ.
Các đột biến hoạt hóa của thụ thể LH dường như không gây ra các rối loạn lâm sàng ở nữ giới. Ở các bé gái trước tuổi dậy thì, điều này có thể là do sự biểu hiện thụ thể LH thấp hoặc không có hoặc do sự biểu hiện aromatase không đủ trong các tế bào hạt trước tuổi dậy thì. Trong tuổi dậy thì, sự hoạt hóa của các thụ thể LH trên các tế bào vỏ trong buồng trứng dẫn đến việc sản xuất androgen được chuyển đổi thành estrogen bởi aromatase trong các tế bào hạt. LH, cùng với FSH, cũng đóng một vai trò trong việc gây ra sự biệt hóa của các nang trứng thành nang Graafian và kích hoạt sự rụng trứng và giải phóng noãn. Việc xác định kiểu hình chi tiết của người mẹ mang gen của một nam giới MLPP có đột biến hoạt hóa p.Asp578Gly của thụ thể LH đã không cho thấy bất kỳ bất thường nào trong chu kỳ kinh nguyệt hoặc khả năng sinh sản của bà. Động học LH, nồng độ androgen, và FSH, cũng như đáp ứng với các chất chủ vận GnRH, đều bình thường.
Thụ Thể Hormone Kích Thích Nang Trứng
Các đột biến bất hoạt và hoạt hóa của thụ thể FSH đã được mô tả, nhưng chúng ít phổ biến hơn nhiều so với các đột biến của thụ thể LH. Gen thụ thể FSH nằm trên nhiễm sắc thể 2 tại vị trí p21 và chứa 10 exon. Exon cuối cùng của gen thụ thể FSH mã hóa cho các miền xuyên màng và nội bào.
Ở nữ giới, FSH cần thiết cho sự trưởng thành bình thường của nang trứng và điều hòa sản xuất estrogen bởi các tế bào hạt của buồng trứng. Ở nam giới tuổi dậy thì, FSH cần thiết cho sự tăng sinh của tế bào Sertoli, sự phát triển của tinh hoàn và duy trì quá trình sinh tinh.
Đột biến bất hoạt đầu tiên của thụ thể FSH được tìm thấy ở những phụ nữ Phần Lan mắc chứng loạn sản buồng trứng tăng gonadotropin di truyền lặn trên nhiễm sắc thể thường (ODG). ODG được đặc trưng bởi vô kinh nguyên phát, vô sinh, và buồng trứng dạng dải hoặc thiểu sản với kiểu nhân 46XX và nồng độ gonadotropin tăng cao. Hai mươi hai trong số 75 bệnh nhân Phần Lan mắc ODG được phát hiện là đồng hợp tử đối với một đột biến điểm c.C566T trong exon 7 của gen thụ thể FSH. Đột biến này dẫn đến việc sản xuất một thụ thể FSH với sự thay thế p.Ala189Val trong một vùng của miền gắn phối tử ngoại bào được cho là có vai trò trong việc luân chuyển thụ thể hoặc hướng thụ thể đến màng sinh chất. Thụ thể bị đột biến thể hiện ái lực gắn phối tử bình thường nhưng có khả năng gắn kết giảm và sự dẫn truyền tín hiệu bị suy giảm khi được nghiên cứu trong các tế bào Sertoli của chuột đã được biến nạp gen. Nam giới đồng hợp tử đối với đột biến này có sự suy giảm khả năng sinh tinh thay đổi và thể tích tinh hoàn từ thấp đến bình thường thấp, nhưng không bị vô tinh và có thể có khả năng sinh sản. Đột biến điểm c.C556T không phổ biến bên ngoài Phần Lan, nơi tần suất người mang gen là 0,96%. Các đột biến khác làm thay đổi sự dẫn truyền tín hiệu nhưng không ảnh hưởng đến sự biểu hiện hoặc gắn kết của thụ thể bao gồm p.Ala189Val, p.Asn191Ile, p.Ala419Thr, và p.Phe591Ser. Đột biến p.Ala189Val gây ra vô kinh tăng gonadotropin nguyên phát ở phụ nữ và không sinh tinh ở nam giới ở trạng thái đồng hợp tử và vô kinh thứ phát ở trạng thái dị hợp tử. Đột biến Asn191Ile gần đó cũng gây ra vô kinh tăng gonadotropin ở trạng thái đồng hợp tử, nhưng không có kiểu hình lâm sàng ở trạng thái dị hợp tử. Đột biến Ala419Thr được xác định ở một phụ nữ dị hợp tử bị vô kinh nguyên phát. Đột biến Phe591Ser gây ra vô kinh nguyên phát và suy buồng trứng sớm (POF) ở trạng thái đồng hợp tử và một khuynh hướng phát triển các khối u dây sinh dục buồng trứng ở trạng thái dị hợp tử. Vô kinh nguyên phát và POF đã được mô tả ở những phụ nữ có đột biến đồng hợp tử làm suy giảm hoàn toàn khả năng gắn kết của thụ thể với FSH hoặc dẫn đến giảm sự biểu hiện của thụ thể FSH trên bề mặt tế bào.
Một loạt bài báo được công bố gần đây hơn đã mô tả 13 gia đình người Trung Quốc bị suy buồng trứng sớm không hội chứng. Họ đã tìm thấy ba đột biến mới và 11 đột biến đồng hợp tử đã được mô tả trước đó cùng 3 đột biến dị hợp tử kép đã được mô tả trước đó. Các biểu hiện lâm sàng dao động từ vô kinh nguyên phát đến có kinh nguyệt bình thường với kinh nguyệt thưa hoặc vô kinh thứ phát. Đây là nghiên cứu đầu tiên xác định một đột biến dịch khung (p.Lys140Argfs*16 trong miền ngoại bào) và một đột biến sai nghĩa trong peptide tín hiệu của thụ thể FSH (p.Gly15Asp). Đột biến mới còn lại là p.Pro504Ser trong miền xuyên màng.
Tình trạng dị hợp tử kép đối với các đột biến gây mất một phần chức năng của thụ thể FSH có thể gây ra rối loạn chức năng nội tiết ở phụ nữ. Phụ nữ có thể biểu hiện vô sinh, vô kinh thứ phát, loãng xương, và tiền sử dậy thì bình thường hoặc muộn, kèm theo LH và FSH tăng, estradiol huyết tương bình thường thấp, nồng độ inhibin B huyết tương thấp, buồng trứng hơi to với các nang trứng chưa trưởng thành, và tử cung nhỏ. Điều này có thể do các đột biến gen thụ thể FSH dẫn đến một đột biến p.Ile160Thr trong miền ngoại bào làm suy giảm sự biểu hiện trên bề mặt tế bào và một đột biến p.Arg573Cys trong e3 cản trở sự dẫn truyền tín hiệu. Những phụ nữ khác biểu hiện vô kinh nguyên phát và gonadotropin rất cao, nồng độ estradiol và inhibin B huyết tương thấp, buồng trứng kích thước bình thường với các nang trứng chưa trưởng thành, và tử cung kích thước bình thường. Tình trạng này có liên quan đến sự thay thế p.Asp224Val trong miền ngoại bào, dẫn đến suy giảm sự biểu hiện trên bề mặt tế bào và sự thay thế p.Leu601Val trong e3 làm suy giảm sự dẫn truyền tín hiệu.
Các đột biến hoạt hóa của thụ thể FSH cũng đã được mô tả. Đáng ngạc nhiên, một nam giới đã cắt bỏ tuyến yên được phát hiện có khả năng sinh sản và có nồng độ testosterone huyết thanh trên 4,9 nmol/L (141 ng/dL) và thể tích tinh hoàn bình thường, mặc dù nồng độ gonadotropin không thể phát hiện được. Bệnh nhân này được phát hiện là dị hợp tử đối với một đột biến c.A1700G trong exon 10 của gen thụ thể FSH dẫn đến sự thay thế p.Asp567Gly trong một vùng của vòng nội bào tương thứ ba được bảo tồn cao giữa các thụ thể FSH, LH, và TSH. Sự thay thế tương tự trong các vùng tương ứng của thụ thể LH và TSH cũng dẫn đến các thụ thể hoạt động cấu thành và được tìm thấy tương ứng trong MLPP và u tuyến giáp. Các đột biến hoạt hóa khác đã được xác định gây ra hội chứng quá kích buồng trứng tự phát (OHSS). OHSS là một biến chứng phổ biến của các phác đồ điều trị được sử dụng để kích thích trứng cho thụ tinh trong ống nghiệm và được đặc trưng bởi nhiều nang noãn được lót bởi các tế bào hoàng thể hóa, có thể dẫn đến khó chịu và chướng bụng, cũng như buồng trứng to và ứ dịch. Một đột biến như vậy là p.Asp567Asn, được tìm thấy ở một phụ nữ bị OHSS tự phát tái phát. Các đột biến p.Thr449Ile và p.Thr449Ala gây ra một sự thay đổi cấu hình dẫn đến mất tính đặc hiệu đối với FSH, dẫn đến sự nhạy cảm với HCG và TSH, gây ra OHSS tự phát trong thai kỳ hoặc với suy giáp. Đột biến p.Ile545Thr gây ra OHSS tự phát ở một phụ nữ trong ba tháng đầu của thai kỳ, mặc dù nồng độ HCG bình thường. Thụ thể đột biến này thể hiện hoạt động cấu thành có thể phát hiện được, cũng như sự hoạt hóa lộn xộn bởi HCG và TSH.
Thụ Thể Hormone Kích Thích Tuyến Giáp
Gen thụ thể TSH nằm trên nhiễm sắc thể 14 và chứa 10 exon, với chín exon đầu tiên mã hóa miền ngoại bào lớn và exon thứ 10 mã hóa phần còn lại của thụ thể. Ở nồng độ TSH ngoại bào thấp, sự hoạt hóa thụ thể TSH dẫn đến sự kích thích Gαs — hoạt hóa adenylyl cyclase, dẫn đến tăng nồng độ cyclic AMP nội bào. Ở nồng độ TSH ngoại bào cao hơn, sự hoạt hóa của thụ thể TSH cũng kích thích các protein Gq và G11 — hoạt hóa phospholipase C và dẫn đến việc sản xuất diacylglycerol và inositol phosphate.
Các thụ thể TSH khác với các thụ thể hormone glycoprotein khác ở chỗ chúng tồn tại ở hai dạng hoạt động như nhau. Đó là dạng chuỗi đơn và dạng hai tiểu đơn vị của thụ thể TSH (Hình 3.4). Dạng chuỗi đơn của thụ thể TSH được tạo thành từ ba tiểu đơn vị liền kề: tiểu đơn vị A, peptide C, và tiểu đơn vị B. Tiểu đơn vị A bắt đầu ở đầu tận amino của miền ngoại bào và chứa hầu hết miền ngoại bào. Peptide C được kết nối với đầu tận carboxy của tiểu đơn vị A và tiếp tục miền ngoại bào. Peptide C chứa một chuỗi 50 acid amin chỉ được tìm thấy trong các thụ thể TSH. Tiểu đơn vị B được kết nối với đầu C của peptide C và chứa các TM và phần nội bào tương đầu tận carboxy của thụ thể. Dạng hai tiểu đơn vị của thụ thể bị thiếu peptide C, được cắt ra khỏi protein trong quá trình xử lý nội bào và bao gồm các tiểu đơn vị A và B được gắn với nhau bằng các liên kết disulfide. Đáng ngạc nhiên là cả hai dạng thụ thể đều được hoạt hóa như nhau bởi TSH vì peptide C và các vùng lân cận của tiểu đơn vị A và B tham gia vào quá trình dẫn truyền tín hiệu.
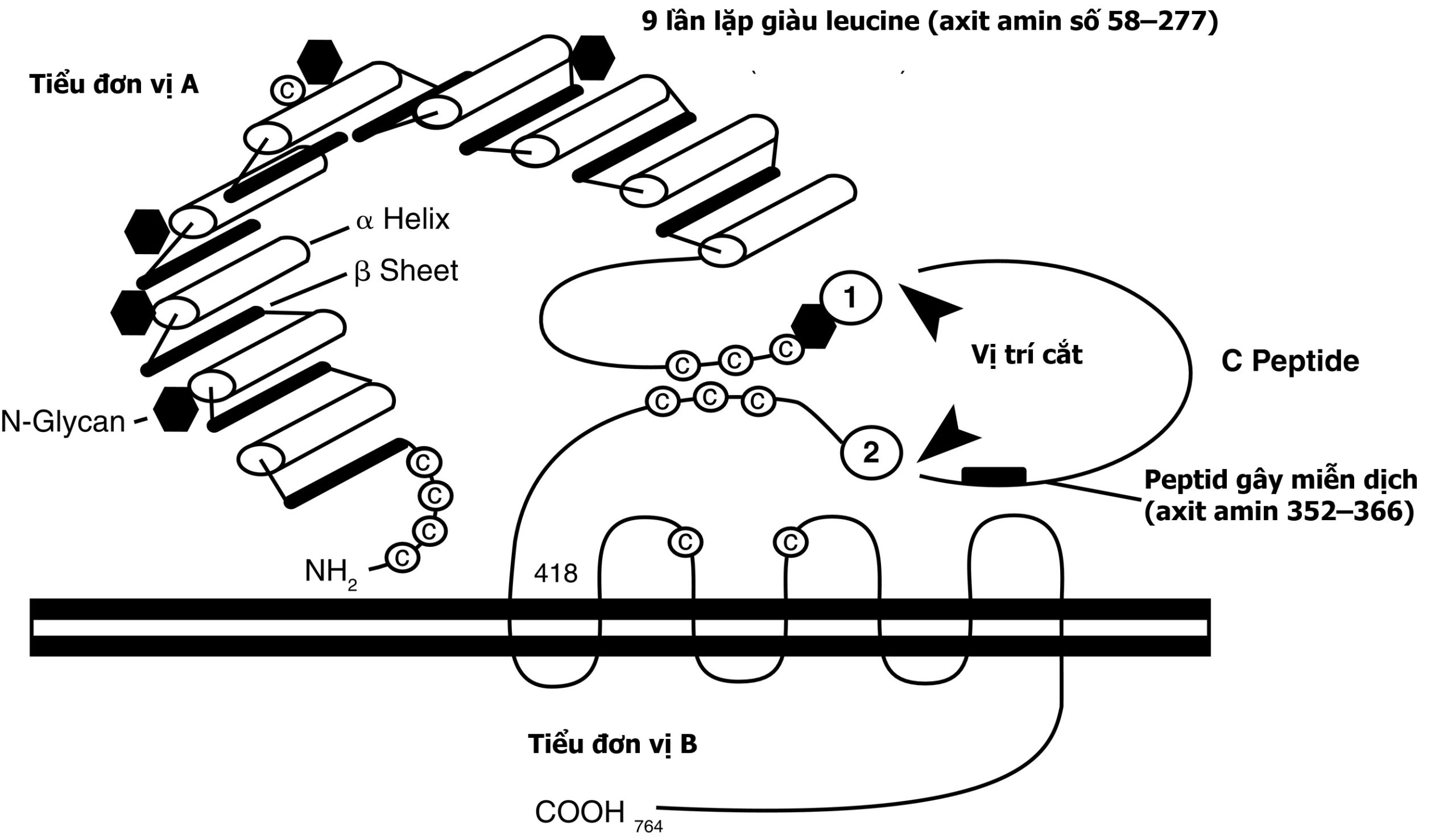
Hình 3.4 Thụ thể hormone kích thích tuyến giáp (TSH). Có hai dạng thụ thể TSH. Dạng chuỗi đơn được tạo thành từ một tiểu đơn vị A, peptide C, và một tiểu đơn vị B. Sự cắt bỏ sau dịch mã của peptide C khỏi dạng chuỗi đơn dẫn đến dạng hai tiểu đơn vị. Dạng này bao gồm tiểu đơn vị A được nối với tiểu đơn vị B bằng các liên kết disulfide giữa các gốc cysteine đầu tận carboxy của tiểu đơn vị A và các gốc cysteine đầu tận amino của tiểu đơn vị B.
Các đột biến sai nghĩa của gen thụ thể TSH, dẫn đến sự thay thế Ser-281 gần đầu tận carboxy của tiểu đơn vị A bằng Ile, Thr, hoặc Asn, dẫn đến một thụ thể TSH hoạt động cấu thành có thể gây ra cường giáp trong tử cung hoặc bẩm sinh, hoặc các u tuyến độc. Các đột biến soma hoạt hóa gây ra các u tuyến độc cũng đã được tìm thấy trong các miền xuyên màng khác nhau của thụ thể TSH. Cụ thể hơn, các cụm đột biến nằm ở vùng i3 và TM6 — được phát hiện có liên quan đến sự dẫn truyền tín hiệu trong tất cả các thụ thể hormone glycoprotein. Tỷ lệ lưu hành của các đột biến hoạt hóa của thụ thể TSH trong các u tuyến độc đã được ước tính dao động từ 2,5% ở Nhật Bản đến 86% ở Brazil.
Các đột biến soma hoạt hóa của thụ thể TSH cũng đã được tìm thấy trong các bướu cổ đa nhân. Thật thú vị, các đột biến hoạt hóa khác nhau đã được tìm thấy trong các nhân riêng biệt ở cùng một cá nhân. Một số ung thư biểu mô tuyến giáp biệt hóa tốt có các đột biến hoạt hóa của thụ thể TSH. Các đột biến soma hoạt hóa của gen GNAS mã hóa Gαs cũng đã được tìm thấy trong một số u tuyến độc và ung thư biểu mô tuyến giáp biệt hóa. Các đột biến dòng mầm hoạt hóa của thụ thể TSH có thể gây ra cường giáp không tự miễn di truyền lẻ tẻ hoặc trội trên nhiễm sắc thể thường, biểu hiện trong tử cung, trong giai đoạn sơ sinh, thời thơ ấu, và trong một số trường hợp ở tuổi trưởng thành. Những đột biến này đã được tìm thấy trong các miền ngoại bào đầu tận amino và xuyên màng.
Bệnh nhân có các đột biến dị hợp tử dẫn đến các thụ thể TSH hoạt động cấu thành thường phát triển cường giáp. Ngược lại, các đột biến mất chức năng trên cả hai alen trong gen thụ thể TSH gây ra suy giáp. Hầu hết các đột biến mất chức năng của thụ thể TSH đã biết đều nằm ở miền ngoại bào đầu tận amino. Một sự thay thế p.Asp410Asn tự phát, gần đầu tận carboxy của peptide C, dẫn đến một thụ thể TSH, với ái lực gắn phối tử bình thường và sự dẫn truyền tín hiệu qua trung gian Gαs bị suy giảm. Bệnh nhân đồng hợp tử đối với đột biến này biểu hiện với suy giáp bù.
Bệnh nhân đồng hợp tử hoặc dị hợp tử kép đối với các đột biến mất chức năng của thụ thể TSH biểu hiện với hội chứng đề kháng TSH (RTSH). Các đột biến mất chức năng của thụ thể TSH gây ra RTSH đã được xác định trong miền ngoại bào đầu tận amino, TM4, TM6, i2, e1, và e3. Mức độ nghiêm trọng lâm sàng của RTSH có thể dao động từ trạng thái bình giáp kèm theo nồng độ TSH tăng cao (RTSH được bù hoàn toàn), đến suy giáp nhẹ không kèm theo bướu cổ (suy giáp được bù một phần), đến thiểu sản tuyến giáp bẩm sinh kèm theo suy giáp sâu (RTSH không được bù). Ở những bệnh nhân bị RTSH không được bù, một tuyến giáp hai thùy nhỏ nằm ở vị trí bình thường. Các đột biến mất chức năng của thụ thể TSH là một nguyên nhân hiếm gặp của suy giáp bẩm sinh, phổ biến hơn ở Nhật Bản và Đài Loan (≤ 7% trẻ em), nơi đột biến p.R450H đặc biệt thường gặp. Vì sự biểu hiện của symporter natri-iodide phụ thuộc vào TSH, sự hấp thu iod 125 và pertechnetate 99m của tuyến giáp bị giảm hoặc không có ở những bệnh nhân bị RTSH. Trong những trường hợp hiếm hoi, sự hấp thu iod ở mức bình thường cao. Các đột biến dị hợp tử kép này của thụ thể TSH có một số hoạt động của Gαs và không có hoạt động của Gq, cho thấy rằng sự hấp thu iod chỉ được kiểm soát bởi hoạt động của Gαs chứ không phải hoạt động của Gq. Một số gia đình đã được phát hiện có một dạng RTSH di truyền trội trên nhiễm sắc thể thường không phải do đột biến của thụ thể TSH.
Thụ Thể Gonadotropin Màng Đệm Người và Hormone Kích Thích Tuyến Giáp trong Thai Kỳ
Do sự tương đồng về cấu trúc với TSH, ở nồng độ rất cao, HCG có thể hoạt hóa thụ thể TSH. Trong thai kỳ, sự hoạt hóa của HCG đối với các thụ thể TSH dẫn đến sự gia tăng hormone tuyến giáp sau tuần thứ chín của thai kỳ — và giảm nồng độ TSH trong khoảng từ tuần thứ 9 đến tuần thứ 12 của thai kỳ. Hiện tượng này thường không dẫn đến cường giáp ở mẹ (nhiễm độc giáp thai kỳ). Tuy nhiên, khi nồng độ HCG tăng cao bất thường do bệnh nguyên bào nuôi thai kỳ gây ra bởi thai trứng hoặc ung thư màng đệm, cường giáp có thể xảy ra. Tỷ lệ nhiễm độc giáp trong bệnh nguyên bào nuôi thai kỳ tương quan với nồng độ HCG. Trong một nghiên cứu trên 196 bệnh nhân được điều trị hóa chất cho khối u nguyên bào nuôi thai kỳ, tỷ lệ nhiễm độc giáp là 7%. Nhiễm độc giáp sinh hóa chỉ xảy ra ở những bệnh nhân có nồng độ HCG > 10^5 và nhiễm độc giáp lâm sàng chỉ xảy ra ở những bệnh nhân có nồng độ HCG lớn hơn 10^6. TSH huyết thanh bị ức chế một cách nhất quán khi nồng độ HCG trên 4 × 10^5 mIU/mL.
Một người mẹ và con gái đã được xác định mắc chứng cường giáp thai kỳ tái phát và nồng độ HCG huyết thanh bình thường. Những cá nhân này được phát hiện là dị hợp tử đối với một đột biến sai nghĩa trong gen thụ thể TSH, dẫn đến sự thay thế p.Lys183Arg trong miền ngoại bào của thụ thể. Người ta tin rằng sự thay thế này làm tăng độ nhạy cảm của thụ thể đối với sự hoạt hóa bởi HCG, gây ra cường giáp thai kỳ.
Nhóm Thụ Thể Hormone Giải Phóng Gonadotropin
Thụ Thể Hormone Giải Phóng Gonadotropin
Gen thụ thể GnRH nằm trên 4q13 và bao gồm ba exon. Không giống như các thụ thể hormone glycoprotein, các thụ thể GnRH thiếu một miền nội bào đầu tận carboxy. Trái ngược với hầu hết các GPCR, thụ thể GnRH được kết cặp với Gq/G11 và do đó sự gắn kết của phối tử dẫn đến sự kích thích phospholipase C chứ không phải adenylyl cyclase. Phospholipase C phân cắt phosphatidylinositol-4,5-diphosphate (PIP2) thành inositol 1,4,5-triphosphate (IP3) và diacylglycerol, dẫn đến tăng hoạt tính của protein kinase C.
Một số bệnh nhân bị IHH là đồng hợp tử hoặc dị hợp tử kép đối với các đột biến mất chức năng trong gen thụ thể GnRH. Không giống như những bệnh nhân mắc hội chứng Kallmann (KS), họ có khứu giác bình thường. Các đột biến thụ thể GnRH được tìm thấy ở khoảng 5% bệnh nhân bị suy sinh dục giảm gonadotropin bẩm sinh có khứu giác bình thường. Các đột biến thụ thể GnRH gây ra IHH dẫn đến giảm sự gắn kết của GnRH hoặc suy giảm sự dẫn truyền tín hiệu của thụ thể GnRH, hoặc giảm sự biểu hiện của thụ thể GnRH trên màng tế bào do sự định tuyến sai của các oligomer thụ thể GnRH từ ER. Một số đột biến, bao gồm p.E90K, p.L266R, và p.S168R, gây ra sự gấp sai và giữ lại trong ER, thể hiện một hiệu ứng trội âm do sự giữ lại các thụ thể kiểu dại.
Bệnh nhân nữ có các đột biến làm suy giảm một phần chức năng của thụ thể GnRH có thể biểu hiện vô kinh nguyên phát và vô sinh, liên quan đến sự phát triển vú bình thường, tử cung bình thường hoặc nhỏ, và buồng trứng nhỏ với các nang trứng chưa trưởng thành. Nam giới có cùng các đột biến có thể biểu hiện suy sinh dục giảm gonadotropin không hoàn toàn (đặc trưng bởi dậy thì muộn và không hoàn toàn) hoặc với suy sinh dục giảm gonadotropin hoàn toàn (đặc trưng bởi không dậy thì).
Một số bệnh nhân bị IHH do các thụ thể GnRH đột biến có đáp ứng gonadotropin một phần hoặc bình thường với GnRH ngoại sinh. Tuy nhiên, biên độ giảm trong sự tiết LH theo nhịp có thể được quan sát thấy ở những bệnh nhân này. Nữ giới có đáp ứng gonadotropin một phần hoặc bình thường với GnRH ngoại sinh có nhiều khả năng có khả năng sinh sản hơn để đáp ứng với GnRH ngoại sinh theo nhịp so với những người không đáp ứng.
Các đột biến hoạt hóa của thụ thể GnRH chưa được mô tả trong dòng mầm hoặc trong các u tuyến yên.
Nhóm Thụ Thể Hormone Giải Phóng Thyrotropin và Chất Kích Thích Tiết
Thụ Thể Hormone Giải Phóng Thyrotropin
Giống như thụ thể GnRH, sự hoạt hóa của thụ thể TRH dẫn đến tăng hoạt tính của phospholipase C. Cho đến nay, chỉ có các đột biến bất hoạt gây rối loạn chức năng nội tiết đã được báo cáo cho thụ thể TRH. Một bệnh nhân đã được xác định bị suy giáp trung ương do các thụ thể TRH bị đột biến. Anh ta biểu hiện trong năm thứ chín của cuộc đời với tầm vóc thấp (−2,6 SD), kèm theo tuổi xương chậm (−4,1 SD), nồng độ thyroxine huyết tương thấp, và nồng độ TSH huyết tương bình thường. TRH ngoại sinh không gây ra sự gia tăng nồng độ TSH và prolactin trong huyết tương. Anh ta được phát hiện là dị hợp tử kép đối với các đột biến gen thụ thể TRH, dẫn đến các thụ thể không thể gắn TRH hoặc gây ra sản xuất IP3. Một gia đình khác đã được xác định có sự đề kháng hoàn toàn với TRH do một đột biến vô nghĩa trong TRHR (p.R17X) tạo ra một thụ thể TRH thiếu toàn bộ miền xuyên màng. Người khởi phát là đồng hợp tử và biểu hiện tầm vóc thấp, suy giảm tăng trưởng, và mệt mỏi ở tuổi 11. Anh ta có T4 tự do thấp, với TSH bình thường thấp. Thử nghiệm kích thích TRH đã không kích thích được TSH hoặc prolactin. Đáng ngạc nhiên là người chị 33 tuổi của anh ta, cũng là đồng hợp tử, đã thoát khỏi sự phát hiện mặc dù đã có hai lần mang thai đủ tháng bình thường. Bà không có dấu hiệu hoặc triệu chứng của suy giáp nhưng có các xét nghiệm chức năng tuyến giáp tương tự như người khởi phát. Bà cho con bú bình thường. Cả người khởi phát và chị gái của anh ta đều có chức năng nhận thức bình thường. Báo cáo này cho thấy rằng thụ thể TRH không cần thiết cho chức năng nhận thức bình thường hoặc khả năng sinh sản và cho con bú của phụ nữ. Mô hình chuột chứng thực những phát hiện này.
Các Thụ Thể Loại A Khác Truyền Tín Hiệu Hormone
Thụ Thể Acid Béo Tự Do 1
Tại thời điểm một GPCR mới được phát hiện, phối tử cho thụ thể mới được phát hiện thường chưa được biết. Do đó, cho đến khi một phối tử cụ thể được phát hiện, các GPCR này được gọi là thụ thể mồ côi (orphan receptors). Theo Ủy ban Danh pháp Gen của Tổ chức Hệ gen Người (HUGO), các thụ thể mồ côi kết cặp G-protein này nên được đặt tên bằng chữ và số GPR theo sau là một con số, cho đến khi phối tử của chúng được biết. Một khi một phối tử cụ thể được xác định, một tên cụ thể hơn sẽ được đặt cho thụ thể.
Các phối tử cho GPR40 chưa được biết khi thụ thể lần đầu tiên được phát hiện. Ủy ban Danh pháp Gen HUGO đã đổi tên thụ thể thành thụ thể acid béo tự do 1 (FFAR1) khi các phối tử được xác định là các acid béo chuỗi trung bình và dài. Với những trường hợp ngoại lệ hiếm hoi được xác định rõ ràng, chương này tuân theo các khuyến nghị của Ủy ban Danh pháp Gen HUGO (xem www.gene.ucl.ac.uk/nomenclature/index.html để biết thêm thông tin về danh pháp thụ thể).
FFAR1 là một trong số các GRCR cho các chất trung gian lipid. Các chất trung gian lipid là các chất truyền tin lipid giữa các tế bào bao gồm sphingosine 1-phosphate, sphingosylphosphorylcholine, dioleoyl phosphatidic acid, lysophosphatidic acid, eicosatetraenoic acid, các acid mật, và các acid béo tự do. FFAR1 được hoạt hóa bởi các acid béo chuỗi trung bình và dài, trong khi FFAR2 (trước đây được gọi là GPR43) và FFAR3 (trước đây được gọi là GPR41) được hoạt hóa bởi các acid béo chuỗi ngắn hơn. Hiện có bằng chứng cho thấy sự hoạt hóa FFAR1 bởi các acid béo chuỗi trung bình và dài có ý nghĩa nội tiết. FFAR1 được biểu hiện trong các tế bào β-tiểu đảo tụy của người. FFAR1 tham gia vào sự bài tiết cholecystokinin từ các tế bào I để đáp ứng với các acid béo. Nó cũng có liên quan đến sự bài tiết GLP-1 và GIP được kích thích bởi acid béo từ các tế bào L và K. GPR120 được biểu hiện trong các tế bào nội tiết ruột và có vai trò sinh lý trong sự bài tiết GLP-1. FFAR2 và FFAR3 được biểu hiện trong mô mỡ và FFAR3 có liên quan đến sản xuất leptin.
Sự kích thích do acid béo gây ra của FFAR1 trong các tế bào β-tiểu đảo dẫn đến sự hoạt hóa của con đường truyền tin thứ hai Gαq-phospholipase C, từ đó dẫn đến sự giải phóng calci từ ER làm tăng sự gia tăng nồng độ calci nội bào qua trung gian insulin do sự hoạt hóa gây ra bởi glucose của các kênh calci phụ thuộc điện thế. Bởi vì nồng độ calci nội bào tăng lên gây ra sự giải phóng insulin, sự tăng cường qua trung gian FFAR1 của sự gia tăng nồng độ calci nội bào qua trung gian glucose dẫn đến sự khuếch đại của sự giải phóng insulin được kích thích bởi glucose.
Một biến thể trong FFAR1 (p.Gly180Ser), được tìm thấy trong một quần thể người Sicilia, đã dẫn đến béo phì, suy giảm dung nạp glucose, và sự bài tiết insulin được kích thích bởi lipid. Hai biến thể khác, p.Arg211His và p.Asp175Asn, không liên quan đến sự thay đổi trong việc giải phóng insulin. TAK-875, một chất chủ vận FFAR1, đã được chứng minh là làm giảm hemoglobin A1c ở những bệnh nhân mắc bệnh tiểu đường loại 2, trong một thử nghiệm lâm sàng giai đoạn 2. Chuột kiểu dại được cho ăn chế độ ăn nhiều chất béo trong 8 tuần phát triển tình trạng không dung nạp glucose, kháng insulin, tăng triglyceride máu, và gan nhiễm mỡ — trong khi chuột knockout FFAR1 trên cùng chế độ ăn không phát triển các tình trạng này. Sự liên quan lâm sàng đối với bệnh nhân vẫn chưa rõ ràng. Tuy nhiên, một đa hình Arg211His trong gen FFAR1 có thể giải thích một số sự thay đổi trong khả năng tiết insulin được tìm thấy ở nam giới Nhật Bản: những người đồng hợp tử Arg/Arg có nồng độ insulin huyết thanh, mô hình đánh giá kháng insulin tại nhà, và mô hình đánh giá chức năng tế bào beta tại nhà thấp hơn so với những người đồng hợp tử His/His.
Thụ Thể KISS1/GPR54
Các nghiên cứu trên các mô hình động vật cho thấy rằng các tế bào thần kinh biểu hiện Kiss1 ở vùng dưới đồi điều chỉnh các tế bào thần kinh biểu hiện GnRH để bắt đầu dậy thì và điều chỉnh phản hồi steroid sinh dục đối với sự giải phóng GnRH. Các đột biến bất hoạt đồng hợp tử trong gen mã hóa thụ thể KISS1 (GPR54) ban đầu được mô tả ở các bệnh nhân người Pháp và Ả Rập Xê Út bị IHH; trong cả hai trường hợp, các đối tượng bị ảnh hưởng đều đến từ các gia đình có quan hệ huyết thống. Các bệnh nhân Ả Rập Xê Út mang đột biến p.Leu148Ser, trong khi các bệnh nhân Pháp mang một đoạn xóa 155bp. Leu148 được bảo tồn cao giữa các GPCR loại A. Đột biến không ảnh hưởng đến sự biểu hiện, gắn kết phối tử, hoặc liên kết với Gs, nhưng làm suy giảm sự hoạt hóa xúc tác do phối tử gây ra của Gs. Cùng lúc đó, một bệnh nhân người Mỹ gốc Phi bị IHH đã được mô tả là dị hợp tử kép đối với các đột biến GPR54 bất hoạt. Kể từ khi công bố các báo cáo ban đầu này, các bệnh nhân bổ sung đã được mô tả. Một cậu bé có cha là người Jamaica và mẹ là người Síp gốc Thổ Nhĩ Kỳ, bị tinh hoàn ẩn và dương vật nhỏ khi sinh, và nồng độ LH và FSH không thể phát hiện được ở 2 tháng tuổi, đã được phát hiện có các đột biến GPR54 dị hợp tử kép. Một đột biến sai nghĩa khác (p.Leu102Pro) thể hiện sự bất hoạt hoàn toàn của tín hiệu GPR54 đã được xác định. Đáng ngạc nhiên, các bệnh nhân có đột biến này thể hiện sự tiết LH và FSH theo nhịp tự phát với tần suất bình thường, và một biên độ bị cùn và các thành viên trong gia đình có sự phát triển dậy thì một phần. Các đột biến mất chức năng trên cả hai alen trong GPR54 là một nguyên nhân hiếm gặp của IHH có khứu giác bình thường. Một nghiên cứu về các nguyên nhân di truyền của suy sinh dục giảm gonadotropin bẩm sinh có khứu giác bình thường cho thấy rằng các đột biến KISS1R chiếm 2% các biến thể.
Không giống như các bệnh nhân bị KS, nhưng tương tự như các bệnh nhân có đột biến GnRH, các bệnh nhân có đột biến GPR54 có khứu giác nguyên vẹn. Trái ngược với các bệnh nhân bị IHH do đột biến GnRH, các bệnh nhân có đột biến GPR54 tăng nồng độ gonadotropin huyết thanh để đáp ứng với GnRH ngoại sinh.
Các phối tử cho GPR54 có nguồn gốc từ một protein tiền chất duy nhất, kisspeptin-1. Protein dẫn xuất dài nhất hoạt động như một phối tử cho GPR54 là metastin, được gọi như vậy vì nó là một gen ức chế di căn trong các tế bào u ác tính. Metastin bao gồm kisspeptin-1 69-121. Tuy nhiên, các peptide đầu tận carboxy ngắn hơn có nguồn gốc từ kisspeptin-1 gắn và hoạt hóa GPR54. Việc sử dụng metastin cho các tình nguyện viên nam trưởng thành làm tăng nồng độ LH, FSH, và testosterone.
Một đột biến hoạt hóa trong GPR54 đã được xác định ở một bệnh nhân bị dậy thì sớm trung ương. Cô gái được nhận làm con nuôi được phát hiện có một đột biến Arg386Pro, dẫn đến sự hoạt hóa kéo dài của tín hiệu để đáp ứng với kisspeptin. Phân tích đột biến của 28 đối tượng bị dậy thì sớm trung ương vô căn đã không tìm thấy bất kỳ biến thể nào trong KISS1 hoặc KISS1R. Một nghiên cứu lớn hơn cho thấy một sự liên quan giữa một số biến thể và dậy thì sớm trung ương ở các cô gái Hàn Quốc.
Thụ Thể Orexin
Orexin tác động lên các thụ thể đặc hiệu nằm chủ yếu ở vùng dưới đồi để kiểm soát lượng thức ăn và đóng một vai trò trong việc điều hòa giấc ngủ/thức. Có hai loại thụ thể orexin: thụ thể orexin-1 và thụ thể orexin-2. Cũng có hai loại orexin, orexin A và orexin B, được hình thành từ peptide tiền chất preproorexin. Orexin còn được gọi là hypocretin, và orexin A đồng nghĩa với hypocretin-1 và orexin B với hypocretin-2. Orexin A tác động lên các thụ thể orexin-1 và orexin-2, trong khi orexin B chỉ tác động lên các thụ thể orexin-2.
Giống như hầu hết các GPCR loại A, các thụ thể orexin kết cặp với Gq/11 và Gi/Go để lần lượt hoạt hóa phospholipase C và ức chế adenylyl cyclase. Đáng ngạc nhiên, bằng chứng cho thấy rằng các thụ thể orexin cũng kết cặp với Gs — làm tăng hoạt tính adenylyl cyclase. Orexin làm tăng lượng thức ăn và thời gian thức. Orexin A và sự hoạt hóa của thụ thể orexin-1 có tác dụng gây thèm ăn lớn hơn so với orexin B và sự hoạt hóa của thụ thể orexin-2. Thụ thể orexin-2 trung gian cho hiệu ứng gây tỉnh táo của orexin. Hầu hết các bệnh nhân mắc chứng ngủ rũ có mất trương lực cơ có nồng độ orexin A giảm trong dịch não tủy và thiếu các tế bào thần kinh chứa orexin. Điều này được cho là do sự chết tế bào sau sinh của các tế bào thần kinh orexin ở vùng dưới đồi. HLA DQB1*0602 có liên quan đến chứng ngủ rũ có mất trương lực cơ và một quá trình tự miễn đã được đề xuất, nhưng không có kháng thể tự miễn nào được xác định. Cho đến nay, không có đột biến nào trong các thụ thể orexin được tìm thấy ở người. Một đột biến trong thụ thể orexin-2 gây ra chứng ngủ rũ ở chó. Có một đột biến được mô tả (p.leu16ARG) trong gen HCRT ở một đứa trẻ mắc chứng ngủ rũ khởi phát sớm có mất trương lực cơ. Đột biến này đã được chứng minh là làm suy giảm quá trình xử lý và vận chuyển của orexin đột biến, dẫn đến nồng độ orexin A không thể phát hiện được trong dịch não tủy.
Thụ Thể Ghrelin
Thụ thể ghrelin còn được gọi là thụ thể chất kích thích tiết GH loại 1a vì sự hoạt hóa của các thụ thể ở vùng dưới đồi và các tế bào hướng sinh dục tuyến yên làm tăng cường sự bài tiết GH. Ghrelin là một sản phẩm của sự biến đổi sau dịch mã của sản phẩm gen ghrelin là proghrelin. Ghrelin chủ yếu được sản xuất ở dạ dày. Sự hoạt hóa của các thụ thể ghrelin nằm ở vùng dưới đồi gây ra sự bài tiết hormone giải phóng hormone tăng trưởng (GHRH). Ở cấp độ các tế bào hướng sinh dục tuyến yên, nó kích thích sự bài tiết GH. Thụ thể ghrelin cũng kích thích sự thèm ăn (tức là vai trò gây thèm ăn).
Nồng độ ghrelin trong huyết tương tăng cao ngay trước khi ăn và giảm nhanh sau khi ăn. Ngoài ra, việc tiêm ghrelin vào tĩnh mạch cho người làm tăng sự thèm ăn và lượng thức ăn. Nồng độ ghrelin trong huyết tương tăng cao ở những người mắc hội chứng Prader Willi. Do đó, chứng ăn nhiều ở những bệnh nhân mắc hội chứng Prader Willi có thể ít nhất một phần là do sự hoạt hóa quá mức của các thụ thể ghrelin bởi ghrelin. Sàng lọc 184 trẻ em và thanh thiếu niên cực kỳ béo phì để tìm các đột biến của gen thụ thể ghrelin đã không xác định được một đột biến nào có khả năng gây béo phì. Ngược lại, các nghiên cứu sàng lọc quy mô lớn đã xác định các đột biến và các đa hình đơn nucleotide (SNP) rải rác khắp thụ thể ghrelin, và hai SNP và một đột biến trong vùng promoter, liên quan đến tăng hoạt tính phiên mã, cùng phân ly với béo phì.
Những người có tầm vóc thấp trong hai dòng họ Ma-rốc không có quan hệ họ hàng đã được phát hiện có một sự chuyển vị C thành A ở vị trí 611 trong exon đầu tiên của gen thụ thể ghrelin. Sự chuyển vị này dẫn đến sự thay thế acid amin alanin không phân cực và trung tính ở vị trí 204 của thụ thể bằng acid amin glutamate phân cực và mang điện tích (p.ala204glu). Đột biến này cản trở hoạt động cấu thành bình thường của thụ thể và làm giảm sự biểu hiện của thụ thể trên màng tế bào. Tuy nhiên, sự hoạt hóa của thụ thể bởi ghrelin vẫn được bảo tồn. Hai phần ba số cá nhân dị hợp tử trong các dòng họ được nghiên cứu có tầm vóc thấp, với chiều cao lớn hơn hoặc bằng 2 SD dưới mức trung bình. Chiều cao của một cá nhân dị hợp tử là –3,7 SD dưới mức trung bình. Trước khi bắt đầu liệu pháp GH, cá nhân duy nhất trong các dòng họ đồng hợp tử đối với đột biến này có chiều cao –3,7 SD, và trở nên thừa cân trong tuổi dậy thì. Cân nặng của các bệnh nhân dị hợp tử đối với đột biến này thay đổi từ thiếu cân đến thừa cân. Một bệnh nhân khác biểu hiện tầm vóc thấp nghiêm trọng (−3 SD), nôn mửa, nhiễm ceton, hạ đường huyết, và BMI thấp đã được xác định là một dị hợp tử kép đối với một đột biến p.W2X và một đột biến p.R237W trong thụ thể ghrelin. Nồng độ yếu tố tăng trưởng giống insulin-1 (IGF-1) trong huyết thanh của anh ta thấp ở mức 44 ng/mL và anh ta đã thất bại trong thử nghiệm kích thích GH, nhưng có một thử nghiệm tạo IGF-1 bình thường. Bệnh nhân này đã cải thiện được tốc độ tăng trưởng chiều cao và giải quyết được tình trạng hạ đường huyết sau khi điều trị bằng GH. Bốn đột biến thụ thể ghrelin khác đã được xác định trong một nhóm thuần tập người Nhật có tầm vóc thấp (p.Q36del, p.P108L, p.C173R, và p.D246A). p.Q36del cho thấy một sự giảm nhỏ trong hoạt động. C173R dẫn đến sự giữ lại trong tế bào. D246 gây ra tín hiệu bị suy giảm, và P108L dẫn đến giảm ái lực gắn với ghrelin. Hai đột biến khác đã được xác định trong một nhóm thuần tập các bệnh nhân bị chậm tăng trưởng và dậy thì thể tạng ở Brazil (Ser84Ile và Val182Ala). Cả hai đều dẫn đến giảm hoạt động cơ bản. Các bệnh nhân có tầm vóc thấp khi biểu hiện (−2,4 và −2,3 SD) nhưng đã đạt được chiều cao trưởng thành bình thường mà không cần điều trị.
Một sản phẩm khác của sự biến đổi sau dịch mã của proghrelin, obestatin, dường như đóng một vai trò trong việc kiểm soát sự thèm ăn và cân nặng. Sự hoạt hóa của thụ thể obestatin, trước đây được gọi là GPR39, ở chuột dẫn đến giảm lượng thức ăn và cân nặng.
Thụ Thể Hormone Tập Trung Melanin
Trước đây được gọi là SLC-1 hoặc GPR24, thụ thể hormone tập trung melanin (MCH) loại 1 (MCHR1) — và thụ thể MCH loại 2 được phát hiện gần đây hơn (MCHR2), trước đây được gọi là SLT hoặc GPR145 — có thể đóng một vai trò trong việc điều hòa việc ăn uống và chuyển hóa năng lượng ở người. Khi được hoạt hóa, MCHR1 kết cặp với Gq/11 và Gi/o để lần lượt tăng hoạt tính phospholipase C và ức chế hoạt tính adenylyl cyclase. MCHR2 kết cặp với Gq/11, và sự gắn kết MCH dẫn đến tăng hoạt tính phospholipase C.
Các nghiên cứu trên động vật gặm nhấm cho thấy MCH là một hormone gây thèm ăn, và việc điều trị động vật gặm nhấm bằng các chất đối kháng MCHR1 làm giảm lượng thức ăn, cân nặng, và mỡ cơ thể. MCHR2 không được biểu hiện ở động vật gặm nhấm. Việc xóa bỏ MCHR1 ở chuột ngăn chặn việc ăn quá nhiều để đáp ứng với các tín hiệu thức ăn trong điều kiện no. Hai đột biến mất chức năng đã được xác định trong MCHR1 ở người (R210H và P377S). Các tế bào được biến nạp với một trong hai thụ thể đột biến này đã không đáp ứng với MCH, mặc dù sự biểu hiện trên bề mặt tế bào của thụ thể là bình thường, cho thấy một khiếm khuyết trong sự hoạt hóa của thụ thể. Những đột biến này đã được xác định ở hai cá nhân bị thiếu cân rõ rệt và không được tìm thấy trong một nhóm thuần tập béo phì. Phân tích gen MCHR1 ở hơn 4000 trẻ em và thanh thiếu niên béo phì người Đức, Đan Mạch, Pháp, và Mỹ đã tiết lộ một số SNP và các biến thể gen ở trẻ em và thanh thiếu niên Đức có thể liên quan đến béo phì. Một nghiên cứu khác trên 106 đối tượng người Mỹ bị béo phì khởi phát sớm đã không xác định được dứt khoát các đột biến MCHR1 và MCHR2 là nguyên nhân gây béo phì.
Các thụ thể loại B truyền tín hiệu hormone
Thụ Thể Hormone Giải Phóng Hormone Tăng Trưởng
Gen thụ thể GHRH nằm ở 7p14. Các thụ thể GHRH tương tác với Gs để kích thích adenylyl cyclase, dẫn đến tăng nồng độ cyclic AMP nội bào, từ đó dẫn đến sự tăng sinh của các tế bào hướng sinh dục và sự bài tiết GH. Do đó, không có gì đáng ngạc nhiên khi các đột biến hoạt hóa trong Gαs dẫn đến sự hoạt hóa cấu thành của adenylyl cyclase đã được tìm thấy trong một số u tuyến yên tiết GH ở người.
Nhiều đột biến trong thụ thể GHRH gây ra thiếu hụt GH đơn độc đã được xác định. Chúng bao gồm sáu đột biến vị trí nối, hai vi xóa, hai đột biến vô nghĩa, một đột biến dịch khung, 10 đột biến sai nghĩa, và một đột biến trong vùng promoter. Đột biến tự nhiên đầu tiên trong thụ thể GHRH (p.D60G) được tìm thấy ở chuột nhỏ, có kiểu hình lùn. Đột biến này trong một acid amin được bảo tồn trong miền ngoại bào làm suy giảm khả năng gắn GHRH. Đột biến đầu tiên ở người trong thụ thể GHRH (p.Glu72X) được xác định trong một gia đình Ấn Độ có quan hệ huyết thống. Cùng một đột biến đã được tìm thấy trong ba dòng họ dường như không có quan hệ huyết thống từ Ấn Độ, Pakistan, và Sri Lanka. Một đột biến khác (đột biến vị trí nối 5′ trong intron 1) đã được xác định trong một dòng họ lớn ở Brazil với hơn 100 cá nhân. Cả hai đột biến đều dẫn đến việc sản xuất các protein bị cắt ngắn rõ rệt không có hoạt tính thụ thể. Một đột biến dịch khung đã được xác định ở một bệnh nhân bị tầm vóc thấp nghiêm trọng và là trường hợp được ghi nhận đầu tiên về thiểu sản tuyến yên trước khởi phát sớm. Trong một gia đình khác, hai anh chị em bị thiếu hụt GH đơn độc được phát hiện là dị hợp tử kép đối với các đột biến gen thụ thể GHRH bất hoạt. Ba đột biến mới khác đã được xác định trong các gia đình có tầm vóc thấp nghiêm trọng ở Vương quốc Anh (p.W273S, p.R94L, và p.R162W). Đột biến duy nhất được tìm thấy trong vùng promoter của GHRH ảnh hưởng đến một trong các vị trí gắn Pit-1.
Các nghiên cứu trên các đối tượng trong các dòng họ lớn này là đồng hợp tử hoặc dị hợp tử kép đối với các đột biến gen thụ thể GHRH bất hoạt đã cho thấy rằng trẻ em bị ảnh hưởng trải qua sự suy giảm tăng trưởng sau sinh nghiêm trọng với tầm vóc thấp cân đối. Nam giới có giọng nói cao và dậy thì chậm vừa phải. Không giống như trẻ sơ sinh bị thiếu hụt GH hoàn toàn, chúng không có trán dô, dương vật nhỏ, hoặc hạ đường huyết. Các đặc điểm khác bao gồm khuôn mặt búp bê, giảm khối lượng cơ, béo phì trung tâm, da mỏng nhăn nheo, và chậm sắc tố của tóc ở trẻ em và thanh thiếu niên. Tuổi xương chậm so với tuổi đời, nhưng tiến triển so với tuổi chiều cao. Một số bệnh nhân đã được phát hiện có thiểu sản tuyến yên. Tốc độ tăng trưởng tăng lên với liệu pháp GH ngoại sinh. Đáng chú ý, điều trị GH cho hai anh chị em từ Thổ Nhĩ Kỳ, với đột biến p.E72X, đã cho phép họ đạt được chiều cao trưởng thành bình thường, mặc dù chiều cao trước điều trị là −6,7 và –8,6 SD và bắt đầu GH vào khoảng 14 tuổi.
Các nghiên cứu sâu hơn trong nhóm thuần tập Brazil cho thấy những người đồng hợp tử có béo phì bụng tăng, lipoprotein mật độ thấp (LDL), và cholesterol toàn phần cao hơn nhưng độ dày thành động mạch cảnh bình thường và không có bằng chứng về xơ vữa động mạch sớm. Điều trị những bệnh nhân này bằng GH trong 6 tháng đã cải thiện thành phần cơ thể, giảm LDL và cholesterol toàn phần, và tăng lipoprotein mật độ cao (HDL). Đáng ngạc nhiên, điều này có liên quan đến sự gia tăng độ dày nội trung mạc động mạch cảnh và các mảng xơ vữa. Đánh giá lại, 5 năm sau khi ngừng GH, cho thấy sự trở lại mức ban đầu đối với các chỉ số này. Bệnh nhân có đột biến vô nghĩa ảnh hưởng đến thụ thể GHRH cũng biểu hiện các kiểu ngủ bị thay đổi với những bất thường trong cả giấc ngủ chuyển động mắt nhanh và không chuyển động mắt nhanh. Những người dị hợp tử đối với đột biến vô nghĩa có chiều cao trưởng thành và điểm SD IGF-1 bình thường nhưng biểu hiện giảm trọng lượng cơ thể, BMI, khối lượng nạc, khối lượng mỡ, và tăng độ nhạy cảm với insulin.
Các SNP trong thụ thể GHRH đã được chứng minh là góp phần vào sự thay đổi chiều cao-SDS, nhưng các đột biến vẫn là một nguyên nhân hiếm gặp của thiếu hụt GH đơn độc.
Thụ Thể Polypeptide Ức Chế Dạ Dày
Gen thụ thể peptide ức chế dạ dày (GIPR) nằm trên nhánh dài của nhiễm sắc thể 19. Hai dạng đồng phân chức năng tồn tại ở người do sự nối ghép thay thế. Sự hoạt hóa GIPR gây ra sự hoạt hóa Gαs của adenylyl cyclase. Peptide ức chế dạ dày (GIP) còn được gọi là polypeptide insulinotropic phụ thuộc glucose và được giải phóng bởi các tế bào K trong ruột non để đáp ứng với thức ăn. GIP có nhiều tác động sinh lý, bao gồm kích thích giải phóng glucagon, somatostatin, và insulin bởi các tế bào tiểu đảo tụy. Các đột biến ở người trong GIPR chưa được xác định cho đến nay. Một nghiên cứu đã xác định một SNP trong GIPR có liên quan đến kháng insulin ở trẻ em béo phì người Đức. Một nghiên cứu khác đã xác định một SNP trong GIPR có liên quan đến giảm nồng độ C-peptide lúc đói và khi được kích thích.
GIPR có liên quan đến hội chứng Cushing phụ thuộc vào thức ăn. Nồng độ cortisol trong tuần hoàn ở những bệnh nhân bị hội chứng Cushing phụ thuộc vào thức ăn hoặc phụ thuộc vào GIP tăng lên bất thường để đáp ứng với việc ăn uống. GIP thường không gây ra sự giải phóng cortisol từ các tế bào vỏ thượng thận. Những bệnh nhân này có thể có các u tuyến thượng thận hoặc tăng sản thượng thận hai bên dạng nốt làm biểu hiện quá mức các GIPR, từ đó kích thích bất thường sự bài tiết cortisol khi được hoạt hóa. Do đó, ở những bệnh nhân này, sự giải phóng GIP sau ăn dẫn đến sự hoạt hóa của các GIPR thượng thận được biểu hiện và hoạt động bất thường này, dẫn đến sự bài tiết cortisol thượng thận quá mức.
Thụ Thể Hormone Cận Giáp và Peptide Liên Quan Đến Hormone Cận Giáp
Hai loại thụ thể PTH đã được xác định. Thụ thể PTH loại 1 (PTHR1) được hoạt hóa bởi PTH và peptide liên quan đến hormone cận giáp (PTHrP) và trung gian cho các tác dụng của PTH trong xương và thận. Mặc dù có 51% tương đồng với PTHR1, thụ thể PTH loại 2 (PTHR2) được hoạt hóa bởi PTH nhưng không phải PTHrP. PTHR2 đặc biệt phong phú trong não và tuyến tụy nhưng cũng được biểu hiện trong đĩa tăng trưởng; phối tử tự nhiên của nó là TIP39. Chức năng của PTHR2 phần lớn chưa được biết.
PTHR1 có một miền ngoại bào đầu tận amino lớn chứa sáu gốc cysteine được bảo tồn. Sự gắn kết của phối tử gây ra sự tương tác của PTHR1 với các protein Gs và Gq, dẫn đến sự hoạt hóa của các con đường truyền tin thứ hai adenylyl cyclase/PKA và phospholipase C/protein kinase C. Thật thú vị, các đột biến trong i2 cản trở sự kết cặp của PTHR1 với Gq, mà không cản trở sự kết cặp với Gs — trong khi các đột biến trong i3 phá vỡ sự kết cặp của thụ thể với cả hai G protein. Sự gắn kết của PTH với PTHR1 dẫn đến sự nội hóa của một phần màng sinh chất chứa một phức hợp tam phân của thụ thể được hoạt hóa-Gsα-adenylyl cyclase thể hiện sự sản xuất cyclic AMP bền vững. Ngược lại, sự gắn kết của PTHrP với thụ thể PTHR1 dẫn đến sự hình thành một phức hợp vẫn còn trên bề mặt tế bào và tạo ra cyclic AMP chỉ trong một khoảng thời gian ngắn.
Các đột biến mất chức năng trên cả hai alen trong gen PTHR1 gây ra loạn sản sụn Blomstrand. Rối loạn gây chết người này được đặc trưng bởi sự biệt hóa tế bào sụn tăng tốc, dẫn đến bệnh lùn chi ngắn, thiểu sản hàm dưới, không phát triển vú và núm vú, và răng bị ảnh hưởng nghiêm trọng. Một bệnh nhân mắc tình trạng hiếm gặp này được phát hiện là đồng hợp tử đối với một đột biến điểm dẫn đến sự thay thế p.Pro132Leu trong miền đầu tận amino cản trở sự gắn kết của phối tử. Một bệnh nhân khác được phát hiện là đồng hợp tử đối với một đột biến dịch khung dẫn đến một thụ thể bị cắt ngắn thiếu TM5-7, và các miền nội bào và ngoại bào liền kề.
Một bệnh nhân thứ ba được phát hiện có một đột biến di truyền từ mẹ làm thay đổi sự nối ghép của mRNA mẹ, dẫn đến một PTHR1 có một đoạn xóa các gốc từ 373 đến 383 trong TM5 (cũng cản trở sự gắn kết của phối tử). Mặc dù dị hợp tử đối với đột biến này, bệnh nhân không thể sản xuất các PTHR1 bình thường, vì vì những lý do chưa rõ, alen của cha không được biểu hiện. Tình trạng dị hợp tử đối với các đột biến sai nghĩa p.Arg150Cys soma hoặc dòng mầm trong PTHR1 đã được xác định ở hai trong số sáu bệnh nhân bị u sụn, một tình trạng thường là lẻ tẻ và được cho là do một đột biến tế bào soma sau hợp tử. U sụn là các khối u sụn lành tính phát triển trong các hành xương và có thể được kết hợp vào các thân xương của các xương ống dài, ở gần sụn đĩa tăng trưởng; có một nguy cơ tăng lên của sự chuyển dạng ác tính thành sarcoma xương. Bệnh nhân bị u sụn đa phát (OMIM ID: 166000) mắc bệnh Ollier (thuật ngữ của Tổ chức Y tế Thế giới), một rối loạn được đặc trưng bởi sự hiện diện của nhiều u sụn với sự phân bố không đối xứng của các tổn thương thay đổi về kích thước, số lượng, và vị trí. Khi u sụn đa phát xảy ra với u máu mô mềm, rối loạn này được gọi là hội chứng Maffucci. Các nghiên cứu in vitro cho thấy đột biến p.Arg150Cys có hoạt tính kích hoạt nhẹ nhưng dẫn đến sự kích thích phospholipase C thay vì adenylyl cyclase. Một con chuột chuyển gen knockin biểu hiện PTHR1 đột biến, dưới sự kiểm soát của promoter collagen loại 2, cho thấy sự phát triển của các khối u tương tự như những khối u được quan sát thấy trong u sụn ở người. Ý nghĩa lâm sàng của những quan sát này là không chắc chắn, vì hầu hết các trường hợp u sụn là do các đột biến trong gen IDH1 và IDH2.
Các đột biến mất chức năng trên cả hai alen trong PTHR1 cũng là nguyên nhân của hội chứng Eiken, được đặc trưng bởi một loạn sản xương với sự cốt hóa bị chậm lại nghiêm trọng, chủ yếu ở các đầu xương, xương chậu, bàn tay, và bàn chân, cũng như sự tạo hình bất thường của xương. Duchatelet và cộng sự đã lập bản đồ hội chứng Eiken đến nhiễm sắc thể 3p gần gen PTHR1. Các cá nhân bị ảnh hưởng là đồng hợp tử đối với một đột biến vô nghĩa trong đuôi nội bào tương đầu tận carboxy của gen PTHR1 (p.R485X). Ở một cậu bé 7 tuổi mắc hội chứng Eiken, sinh ra từ cha mẹ là anh em họ đời đầu không bị ảnh hưởng, Moirangthem và cộng sự đã xác định một đột biến sai nghĩa đồng hợp tử tại một gốc được bảo tồn trong gen PTHR1 (p.E35K). Cuối cùng, sự thất bại nguyên phát trong việc mọc răng không hội chứng (PFE) là do các đột biến dị hợp tử trong gen PTHR1. Ba đột biến riêng biệt, cụ thể là c.1050-3C > G, c.543 + 1G > A, và c.463G > T, đã được xác định ở 15 cá nhân bị ảnh hưởng từ bốn gia hệ đa thành viên. Tất cả các đột biến đều làm cắt ngắn protein trưởng thành và do đó sẽ dẫn đến một thụ thể không có chức năng.
Một số trường hợp loạn sản sụn hành xương Jansen đã được phát hiện là do các đột biến hoạt hóa cấu thành của gen PTHR1. Rối loạn di truyền trội trên nhiễm sắc thể thường này được đặc trưng bởi bệnh lùn chi ngắn do sự biệt hóa tế bào sụn cuối cùng bị suy giảm và sự khoáng hóa chậm, kèm theo tăng calci máu. Thật thú vị, sự hoạt hóa cấu thành dường như dẫn đến chủ yếu là hoạt động Gαs quá mức vì hoạt động adenylyl cyclase tăng lên và hoạt động phospholipase C không thay đổi trong các tế bào COS-7 biểu hiện các thụ thể bị đột biến.
Các Thụ Thể Loại B Khác Truyền Tín Hiệu Hormone
Các thụ thể loại B khác truyền tín hiệu hormone bao gồm các thụ thể glucagon-like peptide-1, glucagon, calcitonin, và yếu tố giải phóng corticotrophin. Các thụ thể loại B thường kết cặp với các protein Gs dị tam thể, dẫn đến sự hoạt hóa của adenylyl cyclase — từ đó dẫn đến nồng độ cyclic AMP nội bào tăng cao. (Xem chương về Đái tháo đường để thảo luận về vai trò của GLP1 trong việc thúc đẩy bài tiết insulin và việc sử dụng các chất tương tự GLP1 hoặc các chất ức chế sự phân hủy GLP1 trong điều trị.)
Các thụ thể loại C truyền tín hiệu hormone
Thụ Thể Cảm Nhận Calci
Thụ thể cảm nhận calci (CaSR) nằm trên nhánh dài của nhiễm sắc thể 3 (3q21.1). CaSR có một miền đầu tận amino lớn chứa chín vị trí glycosyl hóa tiềm năng. Sự gắn kết của calci ion hóa với CaSR dẫn đến sự hoạt hóa của phospholipase C thông qua sự hoạt hóa của các protein Gq/11.
CaSR là một thành phần không thể thiếu của một hệ thống phản hồi sử dụng PTH và sự tái hấp thu calci ở ống thận để giữ nồng độ calci ion hóa trong huyết thanh trong một phạm vi sinh lý hẹp. Nồng độ calci ion hóa ngoại bào tăng lên sẽ hoạt hóa các CaSR trong các tế bào chính của tuyến cận giáp và các tế bào biểu mô ống thận, dẫn đến giảm giải phóng PTH và tái hấp thu calci ở ống thận. Khi nồng độ calci ion hóa giảm, sự hoạt hóa CaSR giảm — dẫn đến tăng giải phóng PTH và tăng cường tái hấp thu calci ở ống thận.
CaSR cũng gắn với magiê, và do đó sự bài tiết PTH có thể bị ức chế bởi nồng độ magiê huyết thanh tăng cao với hậu quả là hạ calci máu. CaSR có thể tham gia vào cân bằng nội môi magiê bằng cách thay đổi sự tái hấp thu magiê ở nhánh lên dày của quai Henle trong thận. Có khả năng là nồng độ magiê quanh ống tăng lên sẽ hoạt hóa các CaSR của thận, dẫn đến sự ức chế tái hấp thu magiê từ nhánh lên dày của quai Henle — từ đó dẫn đến tăng bài tiết magiê qua thận.
Cả đột biến mất chức năng và tăng chức năng trong CaSR đều đã được mô tả ở những bệnh nhân bị hạ calci máu và tăng calci máu. Tăng calci máu giảm calci niệu gia đình (lành tính) loại 1 (FHH1) và tăng năng cận giáp nặng ở trẻ sơ sinh (NSHPT) là do các đột biến mất chức năng của gen CaSR. Hầu hết các đột biến này nằm ở miền ngoại bào đầu tận amino. Với một vài trường hợp ngoại lệ, những người dị hợp tử đối với các đột biến mất chức năng có FHH1, một tình trạng lành tính được đặc trưng bởi sự bài tiết calci qua nước tiểu rất thấp, tăng calci máu nhẹ, nồng độ PTH huyết thanh bình thường hoặc hơi tăng, và ít hoặc không có triệu chứng của tăng calci máu hoặc tăng năng cận giáp. Ngược lại, những người đồng hợp tử đối với các đột biến như vậy sẽ phát triển NSHPT, một tình trạng đe dọa tính mạng được đặc trưng bởi tăng calci máu nghiêm trọng, nồng độ PTH huyết thanh tăng rõ rệt và các khuyết tật xương. Do đó, con của các bậc cha mẹ FHH có quan hệ huyết thống có nguy cơ bị NSHPT. Đôi khi, trẻ sơ sinh bị NSHPT là dị hợp tử đối với các đột biến gen CaSR mã hóa một protein thụ thể trội âm. Trong hầu hết các trường hợp, FHH1 được di truyền theo kiểu trội trên nhiễm sắc thể thường, nhưng sự di truyền lặn trên nhiễm sắc thể thường đã được mô tả trong một dòng họ trong đó đột biến CaSR chỉ bất hoạt yếu.
Chức năng CaSR giảm cản trở sự ức chế của ion calci đối với sự giải phóng PTH và sự tái hấp thu calci ở ống thận. Do đó, FHH1 được đặc trưng bởi tăng calci máu nhẹ kèm theo nồng độ PTH huyết thanh bình thường hoặc tăng một cách không phù hợp và bởi sự bài tiết calci qua nước tiểu tương đối thấp. Những người bị FHH1 cũng có thể bị tăng magiê máu do giảm sự ức chế quanh ống của sự tái hấp thu magiê từ các quai lên dày của thận bởi CaSR. Ngoài FHH1, còn có hai biến thể khác, FBH loại 2 (FBH2) và FBH loại 3 (FBH3), là do các đột biến mất chức năng dị hợp tử lần lượt trong GNA11 và AP2S1.
FHH1 là do các đột biến mất chức năng dị hợp tử của gen CaSR trên 3q21.1. Hai locus nhiễm sắc thể khác đã được xác định ở những bệnh nhân bị FBH không có đột biến gen CaSR. FHH2 đã được lập bản đồ đến GNA11, mã hóa tiểu đơn vị α của G11, nằm ở 19p13.3 và về mặt sinh hóa và lâm sàng tương tự như FHH1. FBH3, còn được gọi là biến thể Oklahoma (FHHOK), là do các đột biến trong gen AP2S1 nằm ở 19q13. Người lớn bị FHH3 có hạ phosphat máu, nồng độ PTH huyết thanh tăng, và nhuyễn xương, ngoài các phát hiện lâm sàng và sinh hóa được tìm thấy ở những người bị FHH1 và FHH2. NSHPT được đặc trưng bởi tăng calci máu nghiêm trọng kèm theo nồng độ PTH trong tuần hoàn tăng, sự khoáng hóa xương kém, biến dạng lồng ngực, và nhiều gãy xương dài và xương sườn.
Các đột biến hoạt hóa của gen CaSR gây ra hạ calci máu di truyền trội trên nhiễm sắc thể thường loại 1 (ADH1), vì chức năng CaSR tăng lên dẫn đến tăng sự ức chế của ion calci đối với sự giải phóng PTH và ức chế sự tái hấp thu calci ở ống thận. ADH1 được đặc trưng bởi hạ calci máu và hạ magiê máu, kèm theo sự bài tiết calci qua nước tiểu bình thường hoặc tăng một cách không phù hợp và nồng độ PTH huyết thanh bình thường hoặc thấp một cách không phù hợp. Bệnh nhân bị ADH1 có thể không có triệu chứng hoặc có thể biểu hiện co cứng, chuột rút, hoặc co giật trong giai đoạn sơ sinh hoặc thời thơ ấu. Tương tự như các đột biến bất hoạt, hầu hết các đột biến hoạt hóa đều nằm ở miền ngoại bào đầu tận amino. Điều trị bệnh nhân bị ADH1 bằng các dạng hoạt hóa của vitamin D (ví dụ: calcitriol) rất phức tạp, vì việc bình thường hóa nồng độ calci huyết thanh có liên quan đến sự xấu đi của tăng calci niệu, do đó làm tăng thêm nguy cơ sỏi thận, nhiễm calci thận, và suy thận. Ngược lại, sự bài tiết calci qua nước tiểu không quá mức ở những bệnh nhân bị ADH2, những người có các đột biến hoạt hóa trong GNA11. GNA11 mã hóa tiểu đơn vị α của G11, G protein chính kết cặp với CaSR trong các tế bào cận giáp nhưng không phải trong thận.
Một số bệnh nhân có các đột biến hoạt hóa của gen CaSR sẽ phát triển hội chứng Bartter loại V, tương tự như các loại hội chứng Bartter khác, được đặc trưng bởi nhiễm kiềm chuyển hóa hạ kali máu và bởi tăng aldosteron do nồng độ renin tăng. Bệnh nhân bị hội chứng Bartter loại V, không giống như bệnh nhân bị các loại hội chứng Bartter khác, cũng có thể bị hạ calci máu có triệu chứng và có nguy cơ phát triển nhiễm calci thận do tăng calci niệu. Bằng chứng từ các nghiên cứu biểu hiện chức năng in vitro cho thấy rằng bệnh nhân có các đột biến tăng chức năng dị hợp tử nhẹ hoặc trung bình của CaSR phát triển ADH1, trong khi những người có các đột biến tăng chức năng dị hợp tử nghiêm trọng của CaSR cũng sẽ phát triển hội chứng Bartter loại V.
Một số đa hình acid amin đơn của gen CaSR dường như dự đoán nồng độ calci ion hóa trong máu toàn phần và calci toàn phần trong huyết thanh và có thể làm tăng nguy cơ mắc các rối loạn chuyển hóa xương và khoáng chất ở những người có các yếu tố nguy cơ di truyền và môi trường khác đối với các rối loạn này. Những người dị hợp tử hoặc đồng hợp tử đối với một đa hình gen CaSR Gln1011Glu có xu hướng có nồng độ calci cao hơn những người có đa hình này. 15,4% trong số 387 phụ nữ trẻ khỏe mạnh người Canada, có ít nhất một alen gen CaSR với một đa hình Ala986Ser, được phát hiện có nồng độ calci toàn phần cao hơn so với phần còn lại của những phụ nữ không có đa hình này.
Một nghiên cứu khác trên 377 nam và nữ trưởng thành khỏe mạnh người Ý không có quan hệ họ hàng đã phát hiện ra rằng 24% đối tượng nghiên cứu là dị hợp tử hoặc đồng hợp tử đối với đa hình p.Ala986Ser và xác nhận phát hiện rằng những người không có đa hình này có nồng độ calci ion hóa trong máu toàn phần thấp hơn những người có đa hình này. Đa hình p.Ala986Ser cũng đã được liên kết với bệnh Paget và tăng năng cận giáp nguyên phát. Những người có một đa hình Arg990Gly ít phổ biến hơn có xu hướng có nồng độ calci ion hóa trong máu toàn phần thấp hơn những người không có đa hình này. Đa hình p.Arg990Gly đã được phát hiện có liên quan đến tăng calci niệu và sỏi thận.
Các tự kháng thể chống lại CaSR cản trở sự gắn kết của calci với thụ thể có thể gây ra tăng calci máu giảm calci niệu tự miễn. Những bệnh nhân này có tăng năng cận giáp nguyên phát với các đặc điểm lâm sàng và sinh hóa của bệnh nhân FHH1. Ngược lại, các tự kháng thể hoạt hóa CaSR là một nguyên nhân của suy cận giáp mắc phải tự miễn. Cả hai tình trạng này có thể xảy ra liên quan đến các tình trạng tự miễn khác (như viêm tuyến giáp tự miễn), với bệnh celiac ở những bệnh nhân bị tăng calci máu giảm calci niệu tự miễn, và với viêm tuyến giáp tự miễn và hội chứng đa tuyến tự miễn loại 1 và 2 ở những bệnh nhân bị suy cận giáp mắc phải tự miễn. Các tự kháng thể hoạt hóa CaSR được tìm thấy ở khoảng một phần ba số người bị suy cận giáp mắc phải.
Rối loạn gen G protein
Ngày càng có nhiều rối loạn ở người có liên quan đến các đột biến soma hoặc dòng mầm trong các gen mã hóa các tiểu đơn vị của G protein và dẫn đến tăng chức năng hoặc mất chức năng trong protein truyền tín hiệu. Ở đây, chúng tôi sẽ giới hạn thảo luận về các rối loạn liên quan đến các đột biến của gen GNAS mã hóa Gαs, vì đây là G protein phổ biến nhất gây ra các rối loạn nội tiết.
Đột Biến Bất Hoạt của Gen GNAS
Giả suy cận giáp loại 1a (PHP1a; loạn dưỡng xương di truyền Albright [AHO]) và giả giả suy cận giáp (PPHP) là do các đột biến bất hoạt dị hợp tử của gen GNAS mã hóa Gαs. PHP1a được đặc trưng bởi sự đề kháng của cơ quan đích đối với PTH với hậu quả là hạ calci máu, tăng phosphat máu, và nồng độ PTH trong tuần hoàn tăng cao. Ngoài ra, bệnh nhân cũng biểu hiện sự đề kháng với các hormone khác có thụ thể kết cặp với Gs, chẳng hạn như GHRH, TSH, gonadotropin, calcitonin, và các chất dẫn truyền thần kinh vùng dưới đồi. Bệnh nhân PHP1a cũng bị suy giảm nhận thức thần kinh và béo phì phản ánh tác động của Gαs trong não. Ngoài ra, bệnh nhân biểu hiện một tập hợp các khuyết tật phát triển đã được gọi là loạn dưỡng xương di truyền Albright và bao gồm sự hóa xương dị chỗ, tầm vóc thấp, dị thường sọ mặt, và tật ngắn ngón D/E của bàn tay và bàn chân, được đặc trưng bởi các ngón tay bị rút ngắn và xương bàn tay thứ tư và thứ năm ngắn. Bệnh nhân mắc tình trạng liên quan PPHP có chung các đặc điểm của AHO như PHP1a, nhưng không bị đề kháng hormone. Sự phân biệt giữa hai biểu hiện này của cùng một khiếm khuyết gen không phải là ngẫu nhiên mà là kết quả của một cơ chế phức tạp của sự ghi dấu gen kiểm soát sự phiên mã của gen GNAS. Do đó, bệnh nhân có đột biến GNAS trên một alen của mẹ sẽ phát triển một dạng thiếu hụt Gsα nghiêm trọng hơn với sự đề kháng hormone (tức là PHP1a), trong khi bệnh nhân có các đột biến giống hệt trên alen GNAS của cha sẽ có một tình trạng nhẹ hơn với sự đáp ứng hormone bình thường (tức là PPHP).
Mặc dù hầu hết các tế bào biểu hiện Gsα từ cả hai alen của cha mẹ, trong một số tế bào, sự biểu hiện Gsα bị ức chế từ alen GNAS của cha. Do đó, bệnh nhân bị PHP1a phát triển sự đề kháng hormone chỉ giới hạn ở tuyến giáp, các tế bào hướng sinh dục tuyến yên, và các tế bào ống lượn gần của thận vì trong các tế bào này, Gαs chủ yếu có nguồn gốc từ alen GNAS của mẹ. Do đó, bệnh nhân có các đột biến gen GNAS bất hoạt di truyền từ mẹ biểu hiện rất ít Gαs trong các tế bào này và phát triển sự đề kháng hormone. Bởi vì Gsα không được biểu hiện từ alen của cha trong các mô bị ghi dấu, bệnh nhân PPHP có các đột biến gen GNAS bất hoạt di truyền từ cha không bị thiếu hụt protein Gsα trong các tế bào này, vì họ sẽ có một lượng protein Gsα bình thường được sản xuất từ alen GNAS kiểu dại của mẹ. Bệnh nhân bị PHP1a hoặc PPHP chỉ biểu hiện 50% lượng protein Gsα bình thường trong các tế bào trong đó sự phiên mã Gsα không được kiểm soát bởi cơ chế ghi dấu, dẫn đến sự thiếu hụt đơn bội, và có khả năng giải thích cho các đặc điểm tương tự của AHO xảy ra trong hai tình trạng này. Các đối tượng có các đột biến GNAS di truyền từ cha có các đặc điểm thay đổi của AHO mà không có sự đề kháng hormone và đã được mô tả là mắc PPHP, loạn sản xương tiến triển (POH), hoặc u xương ở da, dựa trên kiểu hình lâm sàng. Cơ sở cho những sự phân biệt này vẫn chưa được biết.
Các đối tượng bị PHP loại 1b (PHP1b; MIM 603233) thiếu các đặc điểm điển hình của AHO, nhưng có thể bị tật ngắn ngón nhẹ. Sự đề kháng PTH là biểu hiện chính của sự đề kháng hormone, nhưng một số bệnh nhân có nồng độ TSH huyết thanh hơi tăng và nồng độ hormone tuyến giáp trong huyết thanh bình thường là bằng chứng của sự đề kháng TSH một phần. Ban đầu người ta cho rằng PHP1b là do các đột biến bất hoạt trong gen PTHR1. Tuy nhiên, không có đột biến gen PTHR1 có hại nào được tìm thấy ở những bệnh nhân bị PHP1b.
Các khiếm khuyết biểu sinh trong sự ghi dấu của GNAS là nguyên nhân của PHP1b. Có ba exon đầu tiên thay thế cho GNAS (tức là NESP55, XLαs, và exon A/B) nối với các exon từ 2 đến 13 và có liên quan đến các vùng methyl hóa khác biệt (DMR). Quan trọng nhất, DMR exon A/B được methyl hóa trong các tế bào mầm của mẹ và dường như là yếu tố kiểm soát chính cho sự phiên mã từ exon 1. Mất methyl hóa trong DMR ở thượng nguồn của exon A/B của alen mẹ là một phát hiện nhất quán ở những bệnh nhân bị PHP1b, và khiếm khuyết biểu sinh này giải thích cho sự biểu hiện Gsα giảm từ alen bị ảnh hưởng. Hầu hết các trường hợp PHP1b di truyền trội trên nhiễm sắc thể thường là do các vi xóa trên alen của mẹ bao gồm các exon từ 3 đến 5 hoặc 4 đến 6 của gen mã hóa syntaxin-16 (STX16). Ba vi xóa di truyền từ mẹ khác liên quan đến NESP55 và AS (delNESP55/ASdel3-4) hoặc chỉ AS (delAS3-4) đã được xác định, và những lần xóa này tạo ra một sự phá vỡ methyl hóa toàn cầu hơn bao gồm ba DMR của GNAS (A/B, XL/AS, và NESP55). Cơ sở di truyền cho hầu hết các trường hợp PHP1b lẻ tẻ vẫn chưa được biết và dường như không liên quan đến locus GNAS. Những bệnh nhân này có các khiếm khuyết biểu sinh toàn cầu trong sự methyl hóa ảnh hưởng đến cả ba DMR. Trong một số trường hợp, lưỡng bội đơn cha một phần hoặc hoàn toàn đối với nhiễm sắc thể 20 đã được xác định, trong đó hai bản sao bình thường của GNAS đều có nguồn gốc từ người cha. Lưỡng bội đơn cha sẽ dự đoán một sự thiếu hụt gần như hoàn toàn của Gαs trong các tế bào và mô bị ghi dấu trong đó Gαs không được phiên mã từ alen của cha.
Gsα không được biểu hiện trong các ống lượn gần của thận của những bệnh nhân này do thiếu một alen GNAS có kiểu biểu sinh của mẹ. Ngược lại, cả hai alen GNAS có kiểu biểu sinh của cha đều được biểu hiện bình thường trong các tế bào không bị ghi dấu trong đó sự biểu hiện của Gsα từ alen của cha không bị ức chế.
Đột Biến Hoạt Hóa của Gen GNAS
Khi một GPCR được hoạt hóa bởi một phối tử, thụ thể được hoạt hóa sẽ tương tác với G protein dị tam thể và cho phép giải phóng GDP đã gắn khỏi tiểu đơn vị Gα bằng cách thay thế bằng GTP. Gα gắn GTP sẽ phân ly khỏi phức hợp Gβγ, và cả hai phức hợp đều tự do liên kết với các phân tử tạo tín hiệu xuôi dòng. Việc tạo ra các chất truyền tin thứ hai bị chấm dứt bởi một GTPase nội tại trong tiểu đơn vị Gα hoạt động như một bộ đếm thời gian; sự thủy phân GTP thành GDP sẽ làm bất hoạt Gα và tăng ái lực của nó đối với Gβγ — dẫn đến sự tái liên kết của một G protein dị tam thể không hoạt động sẵn sàng cho một chu kỳ hoạt hóa do thụ thể gây ra khác. Đối với Gsα, các acid amin Arg201 và Gln227 là rất quan trọng đối với hoạt động của GTPase. Các đột biến GNAS dẫn đến sự thay thế các gốc acid amin này dẫn đến sự hủy bỏ hoạt động của GTPase và do đó kéo dài trạng thái hoạt động của Gαs, từ đó dẫn đến sự kích thích cấu thành (tức là không phụ thuộc vào phối tử của thụ thể) của adenylyl cyclase. Các đột biến soma của các gốc này làm gián đoạn hoạt động của GTPase có mặt ở khoảng 40% các khối u tuyến yên tiết GH và một số khối u tiết ACTH và không tiết; trong một số khối u tuyến cận giáp, buồng trứng, tinh hoàn, tuyến giáp, và tuyến thượng thận; và trong một số u nhầy trong cơ.
Các đột biến Gαs Arg201 khảm lan rộng hơn làm giảm hoạt động của GTPase gây ra loạn sản xơ hoặc (khi sự phân bố của đột biến trong mô rất lan rộng) hội chứng McCune-Albright, được đặc trưng bởi bộ ba đốm café-au-lait, loạn sản xơ đa xương, và tăng chức năng nội tiết nguyên phát, đặc biệt là dậy thì sớm không phụ thuộc gonadotropin.
Bệnh nhân mắc hội chứng McCune-Albright cũng có thể có sản xuất GH quá mức, cường giáp, và tăng cortisol máu, cũng như các nốt ở tuyến yên, tuyến giáp, và tuyến thượng thận do tác dụng thúc đẩy tăng trưởng của cyclic AMP quá mức trong các mô này. Hạ phosphat máu, không phải là hiếm ở những bệnh nhân mắc hội chứng McCune-Albright, dường như là do sự sản xuất quá mức của yếu tố tăng trưởng nguyên bào sợi (FGF)-23 phosphatonin bởi các tổn thương xương loạn sản xơ. Bệnh nhân mắc hội chứng McCune-Albright cũng có thể có các vấn đề không phải nội tiết, chẳng hạn như bất thường gan mật, bệnh cơ tim, bệnh thần kinh thị giác, và đột tử.
Thụ thể cytokine
Cytokine là các phân tử được sản xuất bởi một tế bào tác động lên một tế bào khác. Do đó, thuật ngữ cytokine có thể áp dụng không chỉ cho các phân tử có chức năng miễn dịch, mà còn cho các hormone. Do đó, GH, prolactin, và leptin được phân loại là các cytokine loại 1. Các cytokine loại 1 này và các cytokine loại 1 khác (bao gồm các interleukin [IL] 2–9, 11–13, và 15; erythropoietin; thrombopoietin; và yếu tố kích thích dòng bạch cầu hạt) được đặc trưng bởi một cấu trúc bó bốn xoắn α và truyền tín hiệu thông qua các thụ thể cytokine loại 1. Các cytokine loại 2 bao gồm các interferon và IL-10 và không bao gồm các hormone.
Các cytokine loại 1 được chia thành các cytokine chuỗi dài và chuỗi ngắn. Prolactin, leptin, và GH thuộc phân lớp chuỗi dài của các cytokine loại 1 vì các xoắn của chúng dài 25 acid amin. Các cytokine loại 1 chuỗi ngắn, bao gồm IL-2 và yếu tố tế bào gốc, có các xoắn dài khoảng 15 acid amin.
Cấu Trúc và Chức Năng của Thụ Thể Cytokine Loại 1
Tất cả các thụ thể cytokine loại 1 đều có bốn gốc cysteine được bảo tồn, các mô-đun fibronectin loại 2, một mô-típ Trp-Ser-X-Trp-Ser trong miền ngoại bào, và một vùng Box 1/Box 2 giàu proline trong miền nội bào tương. Ngoại trừ yếu tố tế bào gốc, các thụ thể cytokine loại 1 không chứa các miền xúc tác, chẳng hạn như các kinase.
Các thụ thể cytokine loại 1 cho các cytokine loại 1 chuỗi dài cần sự homodimer hóa để hoạt hóa. Đầu tiên, phối tử gắn vào một thụ thể đơn phân. Sau đó, phối tử tương tác với một thụ thể thứ hai để gây ra sự dimer hóa và hoạt hóa của thụ thể. Các thụ thể được hoạt hóa sau đó kích thích các thành viên của họ kinase tyrosine Janus (kinase Jak) để phosphoryl hóa các gốc tyrosine trên cả chính kinase và vùng nội bào tương của các thụ thể. Các chất dẫn truyền tín hiệu và chất hoạt hóa phiên mã (STAT) sau đó gắn vào các miền thụ thể nội bào tương đã được phosphoryl hóa hoặc các kinase Jak thông qua một miền SH2, và được phosphoryl hóa tyrosine. Các STAT đã được phosphoryl hóa sau đó phân ly khỏi các thụ thể hoặc các kinase Jak, tạo thành các homodimer hoặc heterodimer, và di chuyển vào nhân. Trong nhân, các dimer STAT gắn và làm thay đổi hoạt động của các vùng điều hòa của DNA đích.
Có bốn kinase Jak. Jak3 chỉ được biểu hiện trong các tế bào lympho-tạo máu, trong khi Jak1, Jak2, và Tyk2 được biểu hiện trong mọi tế bào. Có bảy STAT (Stat1, Stat2, Stat3, Stat4, Stat5a, Stat5b, và Stat6), có các trình tự miền SH2 khác nhau mang lại các tính đặc hiệu thụ thể khác nhau.
Các Thụ Thể Cytokine Truyền Tín Hiệu Hormone
Các hoạt động của GH, prolactin, và leptin được trung gian thông qua các thụ thể cytokine loại 1 đặc hiệu. Các đột biến của thụ thể hormone tăng trưởng (GHR) và thụ thể leptin đã được xác định là cơ sở của các rối loạn nội tiết cụ thể (Bảng 3.3).
Bảng 3.3 Các Thụ Thể Cytokine và Các Tình Trạng Lâm Sàng Liên Quan Đến Đột Biến Thụ Thể
| Thụ Thể | Đột Biến Dòng Mầm | Rối Loạn Nội Tiết |
|---|---|---|
| Thụ thể hormone tăng trưởng | Một số bất hoạt (dị hợp tử) Bất hoạt (đồng hợp tử, dị hợp tử kép) | Mẫn cảm hormone tăng trưởng một phần với suy giảm tăng trưởng nhẹ đến trung bình Không mẫn cảm hormone tăng trưởng/hội chứng Laron với suy giảm tăng trưởng sau sinh từ trung bình đến nặng |
| Thụ thể leptin | Bất hoạt (đồng hợp tử) | Béo phì và suy sinh dục giảm gonadotropin |
Thụ Thể Hormone Tăng Trưởng
Gen GHR nằm trên nhánh ngắn của nhiễm sắc thể 5 (5p13.1-p12), và 9 trong số 13 exon của gen mã hóa cho thụ thể. Một trình tự tín hiệu bài tiết được mã hóa bởi exon 2, miền gắn phối tử ngoại bào đầu tận amino được mã hóa bởi các exon từ 3 đến 7, miền xuyên màng đơn được mã hóa bởi exon 9, và miền nội bào tương đầu tận carboxy được mã hóa bởi các exon 9 và 10. Protein gắn hormone tăng trưởng (GHBP) được sản xuất bằng cách cắt bỏ miền ngoại bào của GHR khỏi phần còn lại của thụ thể. Khoảng 50% GH trong tuần hoàn được gắn với GHBP.
Sự gắn kết của GH với thụ thể của nó gây ra sự dimer hóa của thụ thể và liên kết với JAK2, một thành viên của họ kinase Janus, dẫn đến sự tự phosphoryl hóa của JAK2 và một loạt các sự phosphoryl hóa của các protein tế bào, bao gồm Stat1, Stat3, và Stat5. Quan trọng nhất trong số các protein này là STAT5b, kết nối sự gắn kết của GH với sự hoạt hóa của biểu hiện gen dẫn đến các tác dụng nội bào của GH, bao gồm sự tổng hợp của IGF-1, protein gắn yếu tố tăng trưởng giống insulin 3 (IGFBP3), và tiểu đơn vị không ổn định với acid (ALS). Các STAT đã được phosphoryl hóa di chuyển vào nhân, nơi chúng điều hòa các gen đáp ứng với GH. Cụ thể, GH gián tiếp kiểm soát sự tăng trưởng bằng cách điều hòa sản xuất IGF-1 — có tác dụng trực tiếp lên sự tăng sinh và phì đại tế bào. Jak2 cũng hoạt hóa con đường kinase protein được hoạt hóa bởi mitogen (MAP) và các con đường chất nền của thụ thể insulin. Tuy nhiên, mức độ mà các con đường này góp phần vào hoạt động của GH vẫn chưa được biết.
Bệnh nhân được coi là không mẫn cảm với hormone tăng trưởng (GHI) nếu họ không thể hiện các đáp ứng tăng trưởng và chuyển hóa phù hợp với nồng độ GH sinh lý. Kiểu hình của GHI có thể thay đổi và dao động từ suy giảm tăng trưởng sau sinh trung bình đơn độc đến suy giảm tăng trưởng sau sinh nghiêm trọng, kèm theo các đặc điểm kinh điển của hội chứng Laron ở những bệnh nhân Ecuador bị thiếu hụt GHR. Các đặc điểm của thiếu hụt GHR bao gồm đường chân tóc thái dương trán lùi, trán dô, giảm kích thước dọc của khuôn mặt, sống mũi thiểu sản, hốc mắt nông, củng mạc xanh, dương vật nhỏ trước tuổi dậy thì, răng vĩnh viễn chen chúc, không có răng hàm thứ ba, bàn tay và bàn chân nhỏ, móng tay thiểu sản, thiểu cơ, tuổi bắt đầu đi bộ chậm, giọng nói cao, cholesterol toàn phần và LDL tăng, và hạ đường huyết lúc đói. Tất cả các bệnh nhân bị GHI đều có nồng độ GH trong tuần hoàn bình thường hoặc tăng cao, nồng độ IGF-1 trong tuần hoàn giảm rõ rệt, và tuổi xương chậm.
Bệnh nhân đồng hợp tử hoặc dị hợp tử kép đối với sự xóa bỏ các exon 5 và 6 — hoặc đồng hợp tử hoặc dị hợp tử kép đối với nhiều đột biến vô nghĩa, sai nghĩa, dịch khung, và vị trí nối trong suốt gen GHR — đã được phát hiện bị GHI đặc trưng bởi suy giảm tăng trưởng sau sinh nghiêm trọng và thường có nồng độ GHBP trong tuần hoàn thấp hoặc không có. Bệnh nhân đồng hợp tử hoặc dị hợp tử kép đối với các đột biến nối p.Arg274Thr hoặc p.Gly223Gly dẫn đến một thụ thể bị cắt ngắn không thể neo vào màng sinh chất (hoặc dẫn đến đột biến sai nghĩa p.Asp152His cản trở sự dimer hóa GHR) có nồng độ GHBP trong tuần hoàn bình thường.
Bệnh nhân dị hợp tử đối với các đột biến làm thay đổi GHR có các phức hợp dimer hóa bao gồm hai thụ thể kiểu dại, một thụ thể kiểu dại và một thụ thể đột biến, và hai thụ thể đột biến. Do đó, tình trạng dị hợp tử đối với các đột biến gen GHR mất chức năng có thể có một hiệu ứng trội âm vì các dimer thụ thể kiểu dại/thụ thể đột biến có thể không thể hoạt động bình thường. Như mong đợi từ hiện tượng này, một số bệnh nhân bị suy giảm tăng trưởng từ trung bình đến nặng đã được phát hiện là dị hợp tử đối với các đột biến điểm hoặc nối mất chức năng của gen GHR làm thay đổi các miền nội bào tương hoặc ngoại bào. Trong một số trường hợp, sự phân rã mRNA qua trung gian vô nghĩa có thể dẫn đến sự thoái biến của mRNA bất thường và ngăn chặn một hiệu ứng trội âm tiềm tàng.
Một số bệnh nhân bị tầm vóc thấp nghiêm trọng và GHI không có đột biến GHR. Thay vào đó, họ có các khiếm khuyết trong sự dẫn truyền tín hiệu nội bào qua trung gian GHR — bao gồm sự hoạt hóa STAT bị suy giảm. Một số bệnh nhân bị GHI được phát hiện có các đột biến STAT5b. Không giống như các bệnh nhân có đột biến GHR, bệnh nhân có đột biến STAT5b cũng biểu hiện chậm phát triển thần kinh nhận thức nghiêm trọng, bệnh phổi mãn tính (xơ phổi và/hoặc viêm phổi kẽ, bạch huyết), chàm nặng, giảm tế bào lympho T, và chức năng tế bào T bất thường. Bệnh nhân bị GHI hiện có thể được điều trị thành công bằng IGF-1 tái tổ hợp, nhưng điều này không dẫn đến chiều cao trưởng thành bình thường, trái ngược với thiếu hụt GH trong đó liệu pháp GH có thể đạt được chiều cao trưởng thành bình thường.
Thụ thể leptin
Gen thụ thể leptin (LEPR, còn được gọi là Ob-R) nằm ở 1p31. Có năm dạng đồng phân của LEPR do sự nối ghép thay thế của bản phiên mã gen LEPR (Hình 3.5). Chỉ có dạng đồng phân Ob-Rb chứa cả mô-típ gắn kinase Jak và mô-típ STAT cần thiết để truyền tín hiệu tối đa các tác dụng của leptin. Các dạng đồng phân Ob-Ra, Ob-Rc, và Ob-Rd chứa các miền ngoại bào và xuyên màng nguyên vẹn nhưng thiếu mô-típ STAT từ các miền nội bào tương của chúng. Dạng đồng phân Ob-Re thiếu các miền xuyên màng và nội bào tương. Do đó, dạng đồng phân Ob-Rb được cho là dạng đồng phân chính tham gia vào việc trung gian cho các tác dụng của leptin.
Hình 3.5 Các dạng đồng phân của thụ thể leptin. Có năm dạng đồng phân của thụ thể leptin. Hộp 1 đại diện cho mô-típ gắn kinase Jak, và hộp 2 đại diện cho mô-típ chất dẫn truyền tín hiệu và chất hoạt hóa phiên mã (STAT). Dạng đồng phân Ob-Rb là dạng đồng phân duy nhất chứa các mô-típ gắn kinase Jak và STAT, và do đó nó được cho là dạng đồng phân chính tham gia vào việc trung gian cho các tác dụng của leptin. Các dạng đồng phân Ob-Ra, Ob-Rc, và Ob-Rd thiếu mô-típ STAT. Dạng đồng phân Ob-Re thiếu các miền xuyên màng (TM) và nội bào tương.
Ba chị em gái từ một dòng họ có quan hệ huyết thống được phát hiện là đồng hợp tử đối với một đột biến nối trong gen LEPR dẫn đến sự biểu hiện của một protein 831 acid amin (Ob-Rhd) thiếu các miền xuyên màng và nội bào tương. Họ đã bị ăn nhiều và béo phì bệnh lý từ khi sinh ra. Họ được phát hiện có nồng độ leptin trong tuần hoàn tăng cao, sự bài tiết TSH và GH giảm, và không phát triển dậy thì do suy sinh dục giảm gonadotropin. Những người mang gen dị hợp tử của đột biến này không bị béo phì bệnh lý và không bị dậy thì muộn hoặc không dậy thì.
Các đột biến vô nghĩa hoặc sai nghĩa của LEPR đã được xác định ở 3% trong một nhóm thuần tập được chọn gồm 300 đối tượng bị béo phì khởi phát sớm. Những người có đột biến bị ăn nhiều, béo phì nghiêm trọng bắt đầu trong năm đầu đời với BMISDS trung bình là +5,1, tỷ lệ nhiễm trùng thời thơ ấu cao, chức năng miễn dịch bị thay đổi, với số lượng CD4 giảm vừa phải (988 so với 1100 tế bào/mL3), và dậy thì muộn do suy sinh dục giảm gonadotropin. Sự tăng trưởng thời thơ ấu bình thường nhưng chiều cao cuối cùng ở mức –1,7 đến –2 SD do thiếu sự bứt phá tăng trưởng ở tuổi dậy thì. Quan trọng là, nồng độ leptin trong tuần hoàn nằm trong phạm vi được dự đoán bởi khối lượng mỡ tăng cao, và các đặc điểm lâm sàng ít nghiêm trọng hơn so với những đối tượng bị thiếu hụt leptin bẩm sinh. Các thành viên trong gia đình dị hợp tử có BMISDS trung bình là +0,6 tương tự như của các thành viên trong gia đình có thụ thể leptin bình thường. Đặc điểm chức năng của các đột biến sai nghĩa này cho thấy các khiếm khuyết gây ra sự giữ lại trong tế bào, gấp sai, hoặc không truyền tín hiệu đến các con đường xuôi dòng. Các đột biến mới bổ sung đã được báo cáo gây ra các kiểu hình tương tự.
Thụ thể tyrosine kinase
Siêu họ thụ thể tyrosine kinase (RTK) bao gồm 15 họ thụ thể tyrosine kinase (Hình 3.6). Với một ngoại lệ, các họ này bao gồm các thụ thể có một miền xuyên màng (xem Hình 3.6). Các thụ thể xuyên màng đơn thường chứa một phần ngoại bào đầu tận amino, một xoắn xuyên màng, một vùng cạnh màng, một miền tyrosine kinase (TK), và một vùng đầu tận carboxy (xem Hình 3.6). Các thụ thể này cần sự dimer hóa để được hoạt hóa tối đa. Các thụ thể thuộc họ RTK insulin khác với các RTK khác, vì chúng chứa hai chuỗi polypeptide xuyên màng, được liên kết bởi các liên kết disulfide, với hai chuỗi peptide ngoại bào xen kẽ và do đó không dimer hóa (xem Hình 3.6).
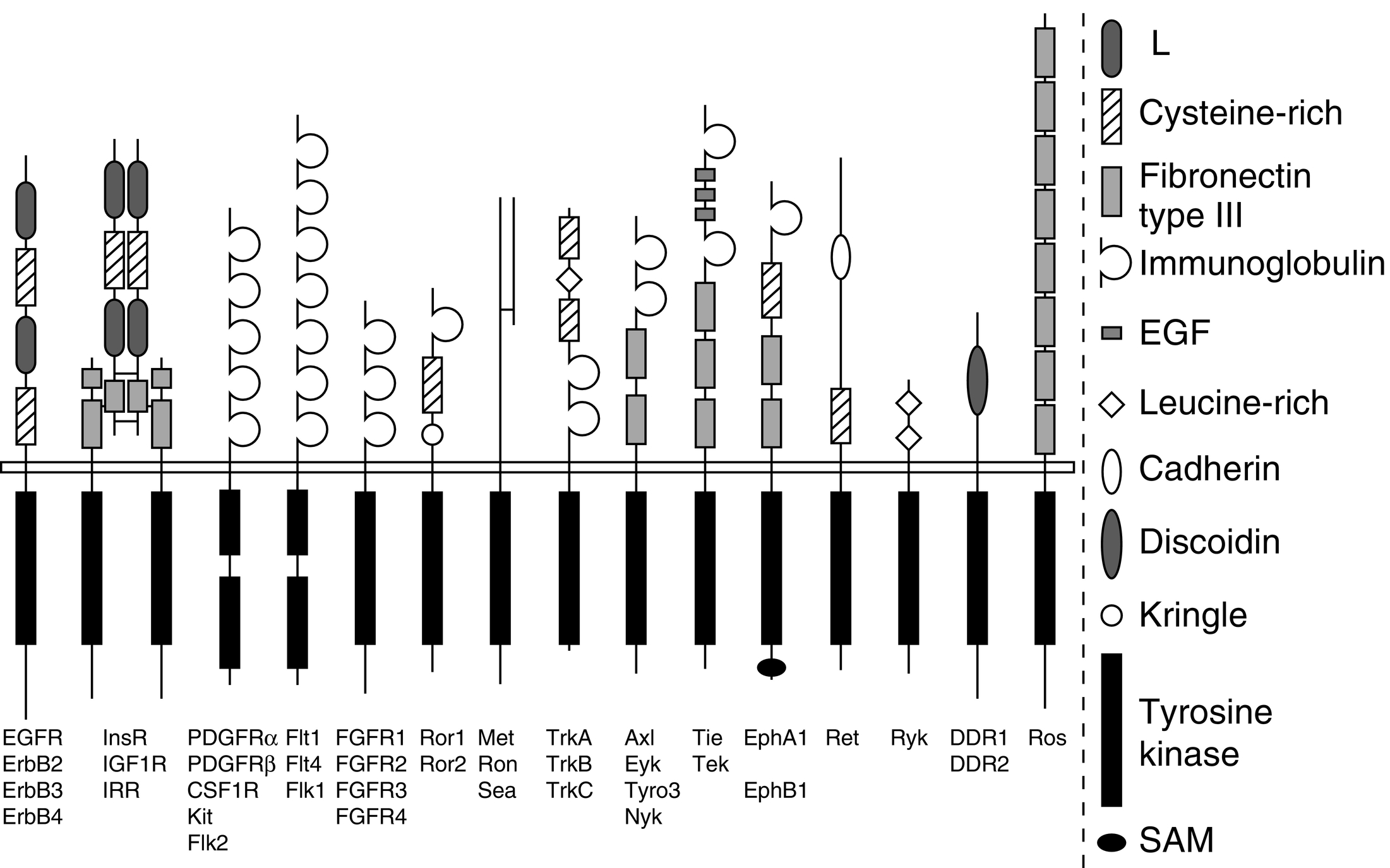
Hình 3.6 15 họ thụ thể tyrosine kinase. Mỗi họ có một phần ngoại bào đặc trưng và một phần nội bào tương chứa một miền tyrosine kinase.
Sự hoạt hóa của RTK dẫn đến sự phosphoryl hóa của các gốc tyrosine trong vòng hoạt hóa (vòng A) trong (các) miền TK, dẫn đến sự hoạt hóa của (các) TK. Sự hoạt hóa của (các) TK, lần lượt, gây ra sự chuyển phosphate từ adenosine triphosphate (ATP) đến các gốc tyrosine trong phần nội bào của thụ thể và trong các protein nội bào đóng vai trò là các vị trí gắn cho các chất truyền tin thứ hai.
Ngày càng có nhiều bằng chứng cho thấy các thành viên của siêu họ thụ thể TK có thể tương tác trực tiếp và gián tiếp với các G protein dị tam thể. Các thụ thể insulin, IGF-1, và IGF-2 dường như tương tác trực tiếp lần lượt với Gi/o và Gq/11, Gi/o, và Gi. Các thụ thể FGF (FGFR) dường như tương tác trực tiếp và gián tiếp với Gs. Sự thay đổi chức năng bẩm sinh của các thụ thể trong các họ RTK insulin và FGF dẫn đến các rối loạn nội tiết (Bảng 3.4).
Bảng 3.4 Các Thụ Thể Tyrosine Kinase và Các Tình Trạng Lâm Sàng Liên Quan Đến Đột Biến Thụ Thể
| Thụ Thể | Đột Biến Dòng Mầm | Rối Loạn Nội Tiết |
|---|---|---|
| Thụ thể insulin | Bất hoạt (dị hợp tử) | Một số trường hợp hội chứng loại A |
| Bất hoạt (đồng hợp tử, dị hợp tử kép) | Các hội chứng Rabson-Mendenhall, Donohue (leprechaunism), và một số trường hợp hội chứng loại A | |
| Thụ thể IGF-1 | Xóa gen (dị hợp tử) | Suy giảm tăng trưởng trước và sau sinh |
| FGFR1 | Đột biến bất hoạt (dị hợp tử) | Hội chứng Kallmann, thiếu răng, hở hàm ếch |
| FGFR2 | Đột biến bất hoạt (dị hợp tử) | Các hội chứng Apert, Pfeiffer, Crouzon |
| FGFR3 | Đột biến hoạt hóa (dị hợp tử) | Loạn sản sụn, loạn sản sụn nặng với chậm phát triển và gai đen, loạn sản tử vong loại I và II, và các loạn sản xương gây chết người có đốt sống dẹt (các loại San Diego) |
FGFR, thụ thể yếu tố tăng trưởng nguyên bào sợi; IGF-1, yếu tố tăng trưởng giống insulin-1.
Họ tyrosine kinase thụ thể insulin
Họ RTK insulin bao gồm thụ thể insulin (INSR) và thụ thể IFG-1 (IGF1R). Các thụ thể này là các dị tứ phân bao gồm hai tiểu đơn vị α và β trong một cấu hình αββα (Hình 3.7). Các tiểu đơn vị α ngoại bào giàu cysteine được liên kết bởi các liên kết disulfide, và mỗi tiểu đơn vị α được liên kết với một tiểu đơn vị β xuyên màng sinh chất và nội bào bởi các liên kết disulfide. Mỗi tiểu đơn vị β chứa một miền TK và một vùng đầu tận carboxy chứa các gốc tyrosine.
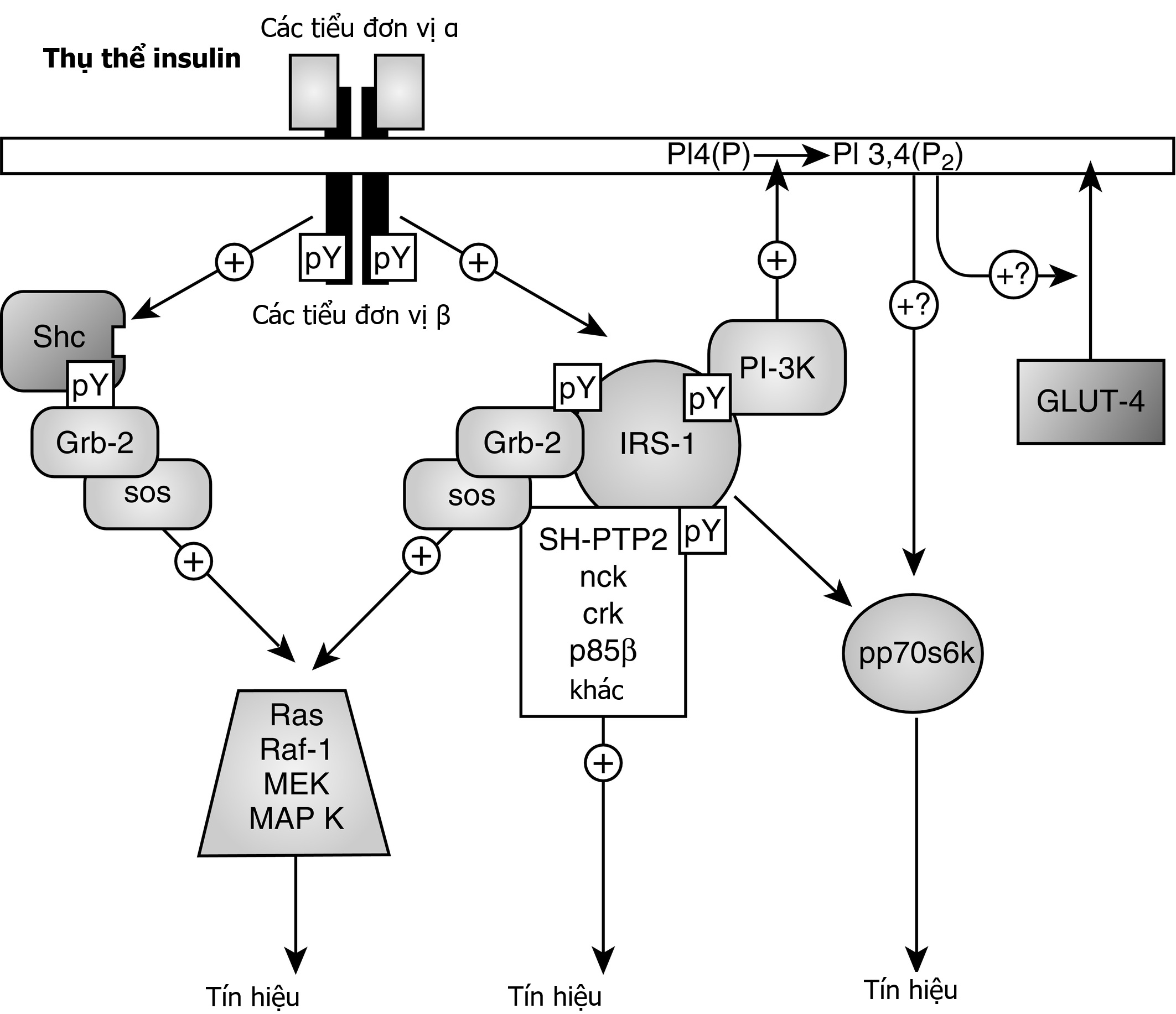
Hình 3.7 Tín hiệu thụ thể insulin. Các protein chất nền của thụ thể insulin, các protein chứa miền SH2 (bao gồm Grb-2 và Shc), và các protein khác (bao gồm SOS) tương tác để hoạt hóa chuỗi Ras/Raf-1/MAP K, chuỗi PI-3K/PKB, và các enzyme khác — bao gồm SH-PTP2 (SHP2) và p70(s6k).
Cả insulin và IGF-1 đều có thể gắn với các INSR và IGF1R. Tuy nhiên, insulin có ái lực lớn hơn đối với INSR và IGF-1 có ái lực lớn hơn đối với IGF1R. Sự gắn kết của phối tử làm thay đổi cấu hình của thụ thể, dẫn đến sự tự phosphoryl hóa trans của các gốc tyrosine đầu tận carboxy trên một tiểu đơn vị β bởi TK trên tiểu đơn vị β kia. Các gốc tyrosine đã được phosphoryl hóa tạo ra các mô-típ có thể được gắn bởi các protein chứa miền Src homology 2 (SH2), bao gồm Shc, Grb-2, SHP2, nck, phosphatidylinositol-3-kinase (PI3K), và Crk. TK của thụ thể cũng phosphoryl hóa các gốc tyrosine trong các protein chất nền của thụ thể insulin (IRS), bao gồm IRS-1 và IRS-2, gắn với các INSR và IGF1R. Khi được phosphoryl hóa, các gốc tyrosine này tạo ra các mô-típ được gắn bởi các protein chứa miền SH2. Do đó, các chất nền của thụ thể insulin có thể đóng vai trò là các protein gắn — cho phép các protein chứa miền SH2 tương tác gián tiếp với các INSR và IGF1R, khi các ràng buộc không gian không cho phép các tương tác trực tiếp giữa các protein và các thụ thể. Cuối cùng, các IRS, các protein chứa miền SH2, và các protein khác (bao gồm mSOS) tương tác để hoạt hóa các chuỗi Ras/Raf/MAPKK/MAPK và PI3K/protein kinase B (PKB) (xem Hình 3.7).
Sự hoạt hóa của chuỗi Ras/Raf/MAPKK/MAPK làm tăng sự nguyên phân và tăng sinh, và sự hoạt hóa của chuỗi PI3K/PKB làm tăng sự hấp thu glucose và tổng hợp glycogen. Bằng chứng cho thấy IGF-1 có tác dụng lớn hơn lên sự tăng trưởng tế bào so với chuyển hóa glucose vì sự hoạt hóa của IGF1R kích thích chuỗi Ras/MAPK nhiều hơn so với sự hoạt hóa của INSR. Ngược lại, dường như insulin có tác dụng lớn hơn lên chuyển hóa glucose vì sự hoạt hóa của INSR kích thích chuỗi PI3K/PKB nhiều hơn so với sự hoạt hóa của IGF1R.
Thụ thể insulin
Gen INSR nằm trên 19p và chứa 22 exon. Các tiền chất nửa thụ thể αβ có nguồn gốc từ sự phân giải protein của một proreceptor duy nhất bao gồm các tiểu đơn vị α và β nối tiếp nhau và sự liên kết disulfide của các tiểu đơn vị này. Các tiền chất nửa thụ thể αβ này sau đó kết hợp để tạo thành một thụ thể insulin dị tứ phân αββα duy nhất. Thật thú vị, các tiền chất nửa thụ thể αβ được mã hóa bởi một alen có thể kết hợp với các tiền chất nửa thụ thể αβ được mã hóa bởi alen kia để tạo thành một thụ thể insulin duy nhất. Hiện tượng này giải thích tại sao các đột biến dị hợp tử dẫn đến hoạt động TK của tiểu đơn vị β bị suy giảm có thể có một hiệu ứng trội âm vì sự hoạt hóa của INSR đòi hỏi sự tự phosphoryl hóa trans của một tiểu đơn vị β bởi tiểu đơn vị β kia.
Các đột biến trong thụ thể insulin dẫn đến hội chứng Donohue (leprechaunism), hội chứng Rabson-Mendenhall, hoặc hội chứng kháng insulin loại A. Bệnh nhân bị leprechaunism hoặc hội chứng Donohue kháng insulin nghiêm trọng. Họ biểu hiện trong giai đoạn sơ sinh với sự chậm tăng trưởng nghiêm trọng trong tử cung và sau sinh, teo mỡ, và gai đen. Họ cũng có các đặc điểm dị dạng bao gồm mắt hình cầu, cằm nhỏ, và tai to. Trẻ sơ sinh nam bị ảnh hưởng thường có dương vật to, trong khi trẻ sơ sinh nữ bị ảnh hưởng thường có phì đại âm vật và rậm lông. Mặc dù tăng insulin máu liên quan đến không dung nạp glucose hoặc đái tháo đường, vấn đề chuyển hóa glucose chính đối với những bệnh nhân này là hạ đường huyết lúc đói. Nhiều bệnh nhân mắc tình trạng này không sống sót qua năm đầu đời. Không giống như các bệnh nhân mắc hội chứng Rabson-Mendenhall, bệnh nhân bị leprechaunism không biểu hiện nhiễm toan ceton do đái tháo đường.
Bệnh nhân mắc hội chứng Rabson-Mendenhall biểu hiện trong thời thơ ấu với sự kháng insulin nghiêm trọng. Mặc dù bệnh nhân mắc rối loạn này ban đầu có thể biểu hiện hạ đường huyết lúc đói, cuối cùng họ sẽ phát triển nhiễm toan ceton do đái tháo đường nghiêm trọng kháng với liệu pháp insulin. Bệnh nhân mắc tình trạng này cũng bị gai đen, tăng trưởng theo chiều dọc nhanh, móng loạn dưỡng, răng sớm và loạn sản, đặc điểm khuôn mặt thô, và tăng sản tuyến tùng.
Bệnh nhân mắc hội chứng kháng insulin loại A bị gai đen và kháng insulin di truyền nghiêm trọng khi không có các tự kháng thể INSR. Bệnh nhân mắc hội chứng này có xu hướng gầy và phát triển không dung nạp glucose. Nữ giới mắc hội chứng này cũng biểu hiện các dấu hiệu của tăng androgen buồng trứng, bao gồm rậm lông, mụn trứng cá nặng, phì đại âm vật, kinh nguyệt thưa, và vô sinh.
Bệnh nhân mắc hội chứng kháng insulin loại B được phân biệt với bệnh nhân mắc hội chứng kháng insulin loại A bởi sự hiện diện của các kháng thể kháng INSR trong huyết tương ngăn chặn sự gắn kết của insulin. Bệnh nhân mắc hội chứng kháng insulin loại B biểu hiện trong tuổi trưởng thành với gai đen, tăng androgen buồng trứng, và kháng insulin nghiêm trọng liên quan đến các dấu hiệu của bệnh tự miễn — bao gồm rụng tóc từng mảng, bạch biến, xơ gan mật nguyên phát, viêm khớp, và viêm thận. Đáng ngạc nhiên, những bệnh nhân này có thể biểu hiện hạ đường huyết lúc đói có thể có hoặc không kèm theo tăng đường huyết sau ăn. Bệnh Hodgkin và thất điều-giãn mạch cũng có liên quan đến hội chứng này. Thuật ngữ HAIR-AN (tăng androgen, kháng insulin, và gai đen) cũng đã được sử dụng để mô tả những phụ nữ có các đặc điểm của hội chứng kháng insulin loại A và B liên quan đến béo phì. Tuy nhiên, thuật ngữ này không chính xác vì nhiều phụ nữ đã được gán nhãn là mắc HAIR-AN thực sự có thể mắc hội chứng kháng insulin loại A hoặc B hoặc hội chứng buồng trứng đa nang nghiêm trọng.
Các đột biến trong INSR đã được tìm thấy ở tất cả các bệnh nhân bị leprechaunism và hội chứng Rabson-Mendenhall, và ở 10% đến 15% bệnh nhân mắc hội chứng kháng insulin loại A. Những đột biến này được chia thành năm loại. Các đột biến loại I là các đột biến dịch khung hoặc vô nghĩa làm chấm dứt dịch mã sớm và do đó cản trở sự tổng hợp INSR. Các đột biến loại II cản trở quá trình xử lý sau dịch mã và vận chuyển nội bào của INSR. Các đột biến loại III làm giảm sự gắn kết của insulin với INSR. Các đột biến loại IV là các đột biến điểm thường nằm ở vùng nội bào của tiểu đơn vị β làm giảm hoạt động TK của INSR. Các đột biến loại V làm tăng sự thoái biến của INSR bằng cách tăng sự nội bào hóa và thoái biến của các thụ thể do insulin gây ra.
Bệnh nhân mắc hội chứng Rabson-Mendenhall và leprechaunism là đồng hợp tử hoặc dị hợp tử kép đối với các đột biến này. Một số bệnh nhân mắc hội chứng loại A đã được phát hiện là dị hợp tử đối với các đột biến tiểu đơn vị β trội âm làm giảm hoạt động TK 75%. Các bệnh nhân khác mắc hội chứng loại A đã được phát hiện là đồng hợp tử hoặc dị hợp tử kép đối với các đột biến tiểu đơn vị α cản trở sự vận chuyển của thụ thể đến màng sinh chất, các đột biến tiểu đơn vị β cản trở hoạt động TK, hoặc các đột biến cản trở sự phân cắt proreceptor thành các tiểu đơn vị α và β. Vẫn còn những bệnh nhân khác đã được phát hiện có nồng độ mRNA INSR giảm có thể do một đột biến mất chức năng trong vùng promoter của gen INSR. Thật thú vị, một bệnh nhân bị leprechaunism có cha mẹ mắc hội chứng loại A đã được mô tả. Người khởi phát được phát hiện là đồng hợp tử đối với một đột biến INSR làm giảm hoạt động TK, và cha mẹ được phát hiện là dị hợp tử đối với đột biến này.
Thụ thể yếu tố tăng trưởng giống insulin-1
Các tác dụng thúc đẩy tăng trưởng của IGF-1 được trung gian bởi các thụ thể IGF1R. Các tiểu đơn vị αβ của IGF1R được mã hóa bởi một gen duy nhất. Giống như thụ thể insulin, một tiền chất nửa thụ thể αβ được sản xuất, sau đó kết hợp với một tiền chất nửa thụ thể có thể được mã hóa từ alen kia để tạo thành một IGF1R dị tứ phân hoàn chỉnh. IGF1R có ái lực với insulin thấp hơn 100 lần so với IGF-1.
Những bệnh nhân dị hợp tử đối với nhiễm sắc thể 15 dạng vòng, dẫn đến việc xóa gen IGF1R, bị chậm tăng trưởng trong tử cung (IUGR) và suy giảm tăng trưởng sau sinh. Các đặc điểm liên quan khác bao gồm tuổi xương chậm, chậm phát triển tâm thần, bất thường tim, tinh hoàn ẩn, và các đặc điểm dị dạng bao gồm tật đầu nhỏ, mặt tam giác, trán dô, hai hốc mắt xa nhau, và tật ngắn ngón. Tương tự, IUGR và suy giảm tăng trưởng sau sinh thường được tìm thấy ở những bệnh nhân dị hợp tử đối với việc xóa đoạn xa của 15q dẫn đến việc xóa gen IGF1R. Bệnh nhân bị xóa đoạn xa của 15q thường có tật đầu nhỏ, khuôn mặt tam giác, hai hốc mắt xa nhau, vòm miệng cao, cằm nhỏ, thận nang, và thiểu sản hoặc loạn sản phổi. Tuy nhiên, nhiễm sắc thể vòng và vùng bị xóa của đoạn xa 15q có thể dẫn đến mất các gen khác — và người ta không biết mức độ mà sự vắng mặt của gen IGF1R góp phần vào kiểu hình phức tạp của những bệnh nhân này.
Mất hoàn toàn chức năng của IGF1R gây chết trong mô hình chuột. Tuy nhiên, sàng lọc di truyền 42 trẻ em lùn, có tiền sử IUGR (khoảng 10% trẻ sơ sinh bị IUGR vẫn nhỏ), đã phát hiện một bé gái là dị hợp tử kép đối với các đột biến sai nghĩa trong IGF1R (p.R108Q và p.K115N). Các nguyên bào sợi được nuôi cấy từ bệnh nhân có chức năng thụ thể IGF-1 giảm so với các nguyên bào sợi đối chứng. Hơn nữa, trong một nhóm thuần tập thứ hai gồm 50 trẻ em có tầm vóc thấp có nồng độ IGF-1 trong tuần hoàn tăng cao, Abuzzahab và cộng sự đã xác định một bé trai có đột biến vô nghĩa dị hợp tử (R59X) trong IGF1R làm giảm số lượng thụ thể IGF-1 trên các nguyên bào sợi. Trẻ em có đột biến IGF1R bị IUGR nghiêm trọng, tật đầu nhỏ, và tầm vóc thấp, với các đột biến trên cả hai alen có kiểu hình nghiêm trọng hơn.
Ngoài chức năng INSR bị khiếm khuyết, một số bệnh nhân bị leprechaunism và hội chứng Rabson-Mendenhall đề kháng với tác dụng hạ đường huyết hoặc thúc đẩy tăng trưởng của IGF-1 và có chức năng IGF1R bất thường — dẫn đến giảm sự gắn kết của phối tử hoặc thay đổi tín hiệu nội bào. Không có đột biến gen IGF1R có hại nào được xác định ở những bệnh nhân mắc các hội chứng này, và nhiều bệnh nhân bị leprechaunism và hội chứng Rabson-Mendenhall có các IGF1R hoạt động bình thường và không có bằng chứng về sự đề kháng IGF-1.
Cho đến gần đây, không có đột biến hoạt hóa nào của IGF1R được tìm thấy. Vi nhân đôi 15q26.3, bao gồm cả IGF1R, được cho là nguyên nhân của một kiểu hình tăng trưởng quá mức. Gần đây, một đột biến hoạt hóa dị hợp tử của IGF1R đã được phát hiện ở một bệnh nhân có tầm vóc cực cao ở mức +3,06 SD, cao 6’8”. Người khởi phát có chiều dài khi sinh ở mức +1,2 SD. Giai đoạn bứt phá tăng trưởng của anh bắt đầu ở tuổi 16 và anh tiếp tục phát triển cho đến 20 tuổi. Anh có nồng độ IGF-1 rất thấp (-3 SD), với giai đoạn bứt phá tăng trưởng chậm và kéo dài liên quan đến giảm nồng độ testosterone và SHBG trong bối cảnh nồng độ LH và FSH bình thường. Chiều cao của mẹ và cha anh lần lượt ở mức +1,1 SD và -0,3 SD. Biến thể này đã được nghiên cứu trong các nguyên bào sợi của chuột thiếu IGF1R và cho thấy sự kích thích quá mức các gen được biết là do IGF1R điều hòa so với IGF1R kiểu dại. Nó cũng dẫn đến sự gia tăng biểu hiện của thụ thể androgen từ 50 đến 70 lần. Bệnh nhân đã phát triển một u màng não ở xương bướm đã được cắt bỏ ở tuổi 40.
Họ thụ thể yếu tố tăng trưởng nguyên bào sợi
Có bốn thành viên trong họ TK FGFR. Đó là FGFR1, FGFR2, FGFR3, và FGFR4. Các thụ thể này bao gồm một chuỗi polypeptide duy nhất chứa một vùng ngoại bào đầu tận amino, một vùng xuyên màng, và một vùng nội bào (xem Hình 3.6). Vùng ngoại bào chứa ba miền giống immunoglobulin: IgI, IgII, và IgIII (xem Hình 3.6). Vùng nội bào chứa một miền TK được chia thành hai đoạn (TK1 và TK2) bởi một đoạn acid amin xen kẽ.
FGF là một họ các yếu tố tăng trưởng liên quan đến sự hình thành mạch, chữa lành vết thương, và phát triển phôi thai. Các FGF là các protein gắn heparin và các tương tác với các proteoglycan heparan sulfat liên kết trên bề mặt tế bào đã được chứng minh là cần thiết cho sự dẫn truyền tín hiệu FGF. FGF là những nhân tố chính trong các quá trình tăng sinh và biệt hóa của nhiều loại tế bào và mô. Ở người, 22 thành viên của họ FGF đã được xác định, tất cả đều là các phân tử truyền tín hiệu có liên quan về mặt cấu trúc. Dưới dạng monomer, FGF chỉ có thể gắn vào một FGFR duy nhất — tạo thành một phức hợp 1:1 không hoạt động. Sự hoạt hóa FGFR bằng cách dimer hóa xảy ra khi hai hoặc nhiều phân tử FGF trong các phức hợp 1:1 được liên kết bởi các proteoglycan heparan sulfat.
Sự hoạt hóa của FGFR làm tăng hoạt tính TK của thụ thể. Hoạt tính TK tăng lên dẫn đến sự tự phosphoryl hóa của một gốc tyrosine trong vùng đầu tận carboxy, tạo ra một vị trí gắn cho miền SH2 của phospholipase Cγ (PLCγ). Một khi PLCγ được gắn vào vị trí này, nó sẽ được phosphoryl hóa và hoạt hóa. Ở các tế bào sụn, sự hoạt hóa của FGFR3 cũng gây ra sự hoạt hóa của STAT1.
Thụ Thể Yếu Tố Tăng Trưởng Nguyên Bào Sợi 1
Các đột biến bất hoạt của gen FGFR1 là một nguyên nhân của hội chứng Kallmann di truyền trội trên nhiễm sắc thể thường. Những người mắc hội chứng Kallmann bị mất khứu giác và suy sinh dục giảm gonadotropin đơn độc. FGFR1, nằm trên 8p12, đóng một vai trò trong sự di cư của các tế bào thần kinh khứu giác và GnRH từ mảng mũi đến hành khứu, và trong sự di cư tiếp theo của các tế bào thần kinh GnRH đến vùng dưới đồi. Trước khi xác định được các đột biến gen FGFR1 này, hội chứng Kallmann liên kết X được cho là chỉ do các đột biến bất hoạt của gen KAL1. KAL1 mã hóa anosmin-1, phối tử cho thụ thể FGFR1. Giống như FGFR1, anosmin-1 đóng một vai trò trong sự di cư của các tế bào thần kinh khứu giác và GnRH đến mảng mũi, và trong sự di cư tiếp theo của các tế bào thần kinh GnRH đến vùng dưới đồi.
Có một tỷ lệ xâm nhập cao đối với chứng mất khứu giác và các dấu hiệu của suy sinh dục giảm gonadotropin (bao gồm không dậy thì, dương vật nhỏ, và tinh hoàn ẩn) ở 10% bệnh nhân KS bị KS liên kết X do các đột biến gen KAL1. Những người mang gen nữ của các đột biến gen KAL1 không bị mất khứu giác hoặc suy sinh dục giảm gonadotropin đơn độc. Trái ngược với các bệnh nhân bị KS do các đột biến gen KAL1, khoảng 10% bệnh nhân KS có các đột biến gen FGFR1 (ngay cả trong cùng một dòng họ) biểu hiện các kiểu hình thay đổi, từ mất khứu giác và suy sinh dục giảm gonadotropin hoàn toàn (đặc trưng bởi tinh hoàn ẩn và dương vật nhỏ ở nam giới và không phát triển dậy thì ở cả hai giới) đến mất khứu giác hoặc dậy thì muộn.
Cũng cần lưu ý rằng trong hầu hết các dòng họ có các đột biến gen FGFR1, nữ giới biểu hiện các kiểu hình KS nhẹ hơn nam giới. Những người mang gen nữ thậm chí có thể không có triệu chứng. Bởi vì gen KAL1 nằm trên nhiễm sắc thể X, nữ giới có thể sản xuất nhiều anosmin-1 hơn nam giới. Do đó, một lời giải thích khả dĩ cho các kiểu hình KS nhẹ hơn ở nữ giới có các đột biến gen FGFR1 có thể là do nồng độ anosmin-1 tăng lên ở nữ giới có thể dẫn đến sự hoạt hóa do anosmin-1 gây ra của các FGFR1 đột biến, từ đó có thể bù đắp một phần cho đột biến. Thật thú vị, thiếu răng và hở hàm ếch không phải là một phát hiện hiếm gặp ở những người bị KS do các đột biến gen FGFR1, trong khi bất sản thận một bên và cử động đồng bộ hai bên có liên quan đến KS phát sinh từ các đột biến gen KAL1.
Thụ Thể Yếu Tố Tăng Trưởng Nguyên Bào Sợi 2–4
Một nhóm đa dạng các rối loạn xương là do các đột biến hoạt hóa trong gen FGFR1, cũng như trong các gen liên quan mã hóa FGFR2 và FGFR3. Nói chung, các đột biến tăng chức năng trong FGFR1 và FGFR2 gây ra hầu hết các hội chứng liên quan đến hẹp sọ, trong khi các hội chứng lùn phần lớn liên quan đến các đột biến FGFR3. Loạn sản xương răng là một rối loạn “chéo” có các kiểu hình xương thường liên quan đến các đột biến FGFR1, FGFR2, và FGFR3. Loạn sản xương răng là do các đột biến sai nghĩa trong các gốc được bảo tồn cao bao gồm các màng gắn phối tử và xuyên màng của FGFR1, do đó xác định các vai trò mới cho thụ thể này như một chất điều hòa âm tính của sự phát triển xương dài. Các đột biến tăng chức năng trong các gen mã hóa các thụ thể FGF khác gây ra các rối loạn xương liên quan: hội chứng Pfeiffer (các đột biến hoạt hóa của FGFR1 và FGFR2), hội chứng Crouzon (các đột biến tăng chức năng FGFR2), hội chứng Crouzon với gai đen (một đột biến FGFR3), hội chứng Apert (các đột biến FGFR2), và hẹp sọ (các đột biến tăng chức năng FGFR3). Một số hội chứng lùn chi ngắn di truyền trội trên nhiễm sắc thể thường — bao gồm loạn sản sụn, loạn sản sụn nặng với chậm phát triển và gai đen (SADDAN), thiểu sản sụn, và ba loại loạn sản xương gây chết người có đốt sống dẹt (PLSD) (loạn sản tử vong I [TDI], loạn sản tử vong II [TDII], và các loại San Diego [PLSD-SD]) — thường do các đột biến gen FGFR3 hoạt hóa cấu thành dị hợp tử gây ra.
Những người bị loạn sản sụn có các đột biến hoạt hóa trong miền xuyên màng của FGFR3, với đột biến p.Gly380Asn được tìm thấy ở hơn 95% bệnh nhân loạn sản sụn. Từ 40% đến 70% những người bị thiểu sản sụn có một đột biến hoạt hóa p.Asn540Lys trong miền TK1. Tất cả những người bị TDII đều có một đột biến hoạt hóa p.Lys650Glu trong vòng hoạt hóa của miền TK2, và hơn 90% những người bị TDI và PLSD-SD có các đột biến FGFR3. Bệnh nhân bị SADDAN có một đột biến hoạt hóa trong cùng một codon như bệnh nhân bị TDII. Thay vì đột biến p.Lys650Glu liên quan đến TDII, bệnh nhân bị SADDAN có một đột biến Lys650Met. Tuy nhiên, không giống như bệnh nhân bị TDII, bệnh nhân bị SADDAN không bị hẹp sọ và hộp sọ hình lá chẻ — và thường sống sót qua thời thơ ấu.
Gen FGFR3 chủ yếu được biểu hiện ở các đĩa tăng trưởng sụn trong của xương dài, não, và da trước và sau sinh. Sự hoạt hóa cấu thành của FGFR3 trong các tế bào sụn dẫn đến sự ngừng tăng trưởng và apoptosis. Ngoài ra, sự hoạt hóa cấu thành của FGFR3 cũng được giả định là làm thay đổi sự di cư của tế bào thần kinh vì bệnh nhân bị SADDAN, TDI, và TDII có các bất thường thần kinh có thể bao gồm chậm phát triển, ít chất trắng, đa hồi não nhỏ, vỏ não thái dương loạn sản, loạn sản nhân, và lạc chỗ tế bào thần kinh. Hơn nữa, sự hoạt hóa cấu thành của FGFR trong các nguyên bào sợi và tế bào sừng của da được cho là gây ra gai đen được thấy ở những bệnh nhân bị SADDAN và hội chứng Crouzon với gai đen. Tuy nhiên, người ta vẫn chưa biết tại sao một số đột biến hoạt hóa FGFR3 ảnh hưởng đến hệ xương, hệ thần kinh trung ương, và da, trong khi các đột biến hoạt hóa FGFR3 khác chỉ ảnh hưởng đến hệ xương.
Các đột biến mất chức năng trong FGFR3 cũng đã được liên kết với bệnh ở người. Một hội chứng không phổ biến được đặc trưng bởi tật co ngón, tầm vóc cao, vẹo cột sống, và mất thính giác (hội chứng CATSHL) đã được liên kết với một đột biến sai nghĩa dị hợp tử (p.R621H) trong miền TK và mất một phần chức năng của FGFR3. Những phát hiện này cho thấy rằng tín hiệu FGFR3 bất thường có thể gây ra các dị tật ở người bằng cách thúc đẩy, cũng như ức chế, sự phát triển xương sụn trong.
Thụ thể nhân
Sử dụng một cây phát sinh chủng loại dựa trên sự tiến hóa của hai miền thụ thể nhân được bảo tồn cao (miền C gắn DNA và miền E gắn phối tử), Laudet đã chia các thụ thể nhân thành sáu phân họ có liên quan. Phân họ 0 chứa các thụ thể, chẳng hạn như thụ thể tuyến sinh dục phôi (EGON) và DAX1, không có miền C hoặc E được bảo tồn (Hình 3.8). Phân họ 1 bao gồm các thụ thể acid retinoic được hoạt hóa bởi chất tăng sinh peroxisome, hormone tuyến giáp, và vitamin D3. Phân họ 2 bao gồm yếu tố nhân tế bào gan-4α (HNF4A) và các thụ thể retinoid X (RXR). Phân họ 3 chứa các thụ thể steroid. Bằng chứng cho thấy phân họ 3 (bao gồm các thụ thể glucocorticoid, androgen, progesterone, và mineralocorticoid) đã tiến hóa nhanh chóng từ một gen thụ thể steroid chung khoảng 500 triệu năm trước.
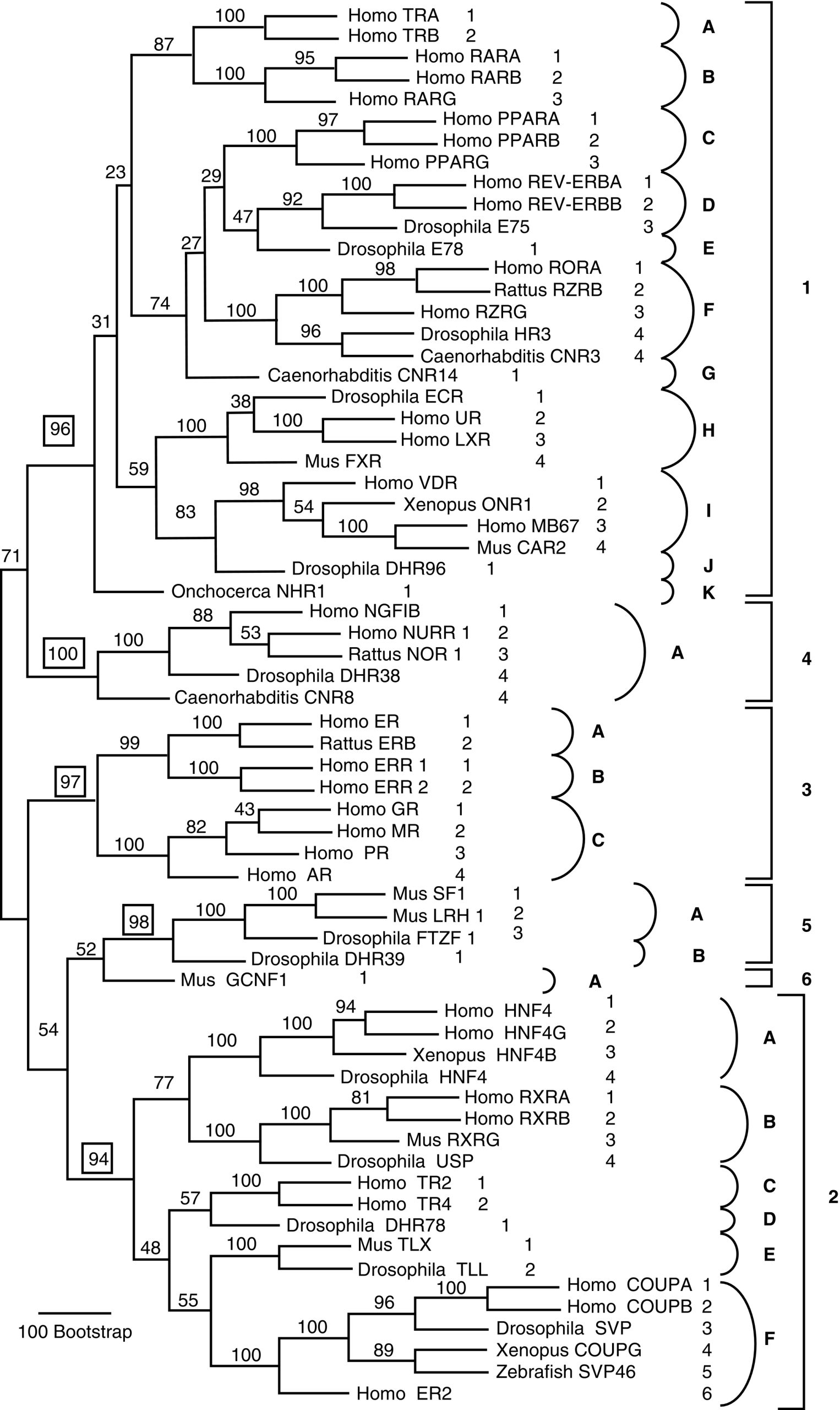
Hình 3.8 Cây phát sinh chủng loại của các thụ thể nhân dựa trên sự tiến hóa của các miền C và E được bảo tồn cao. Các số ở phía bên phải của hình đại diện cho các phân họ, và các chữ cái viết hoa đại diện cho các nhóm thụ thể có liên quan chặt chẽ hơn. Các số nhỏ ở bên phải tên thụ thể được sử dụng kết hợp với các chữ cái phân họ và chữ cái nhóm trong một danh pháp thụ thể nhân được đề xuất. Danh pháp này đề xuất rằng các thụ thể nhân nên được đặt tên là NR, theo sau là số phân họ, chữ cái nhóm, và số thụ thể riêng lẻ. Do đó, thụ thể mineralocorticoid được đặt tên là NR3C2 và thụ thể γ được hoạt hóa bởi chất tăng sinh peroxisome được đặt tên là NR1C3 theo danh pháp này. Các số ở bên trái tên thụ thể đại diện cho các giá trị bootstrap. Các giá trị xác định các phân họ có nhiều hơn một thành viên được đóng khung.
Các phân họ từ 4 đến 6 chứa một số thụ thể nhân, cụ thể là NR4A1-3, NR5A1-2, và NR6A1. Danh pháp như sau: phân họ thụ thể nhân 4, nhóm A, và thành viên 1 cho NR4A1. NR5A1 còn được gọi là yếu tố tạo steroid-1 hoặc SF-1.
Cấu Trúc Chung của các Thụ Thể Nhân
Các thụ thể nhân được tạo thành từ bốn miền: A/B, C, D, và E (Hình 3.9). Ủng hộ quan điểm rằng các phân họ thụ thể nhân có nguồn gốc từ một thụ thể mồ côi tổ tiên chung, các miền C và E được bảo tồn cao giữa các phân họ. Các đột biến của một số thụ thể nhân có liên quan đến các rối loạn nội tiết (Bảng 3.5).
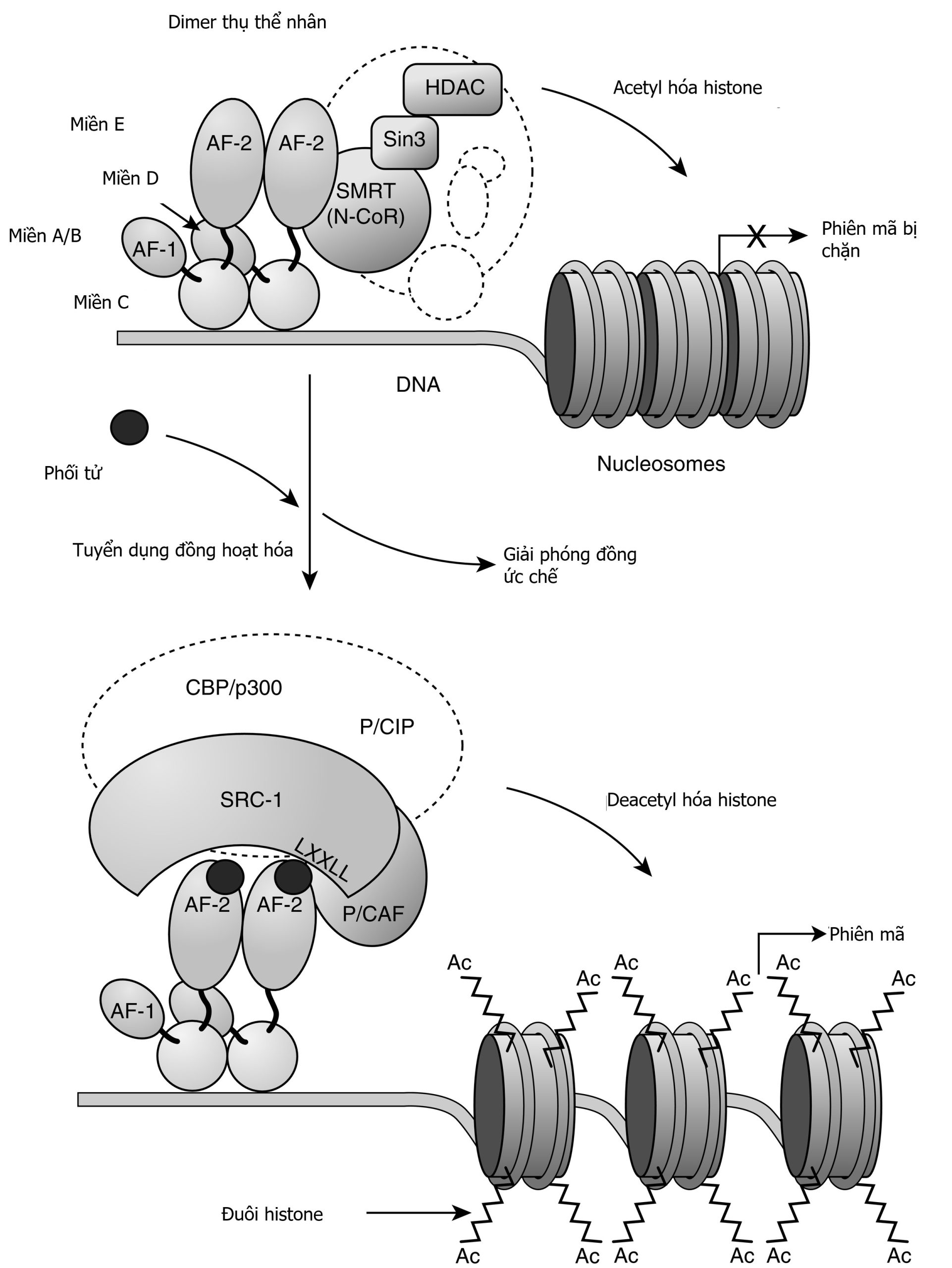
Hình 3.9 Sự hoạt hóa phiên mã do phối tử gây ra bởi các thụ thể nhân. Thường thì, các đồng ức chế, bao gồm chất trung gian ức chế của các thụ thể retinoid và hormone tuyến giáp (SMRT) và đồng ức chế thụ thể nhân (N-CoR), gắn vào một thụ thể nhân không được gắn bởi phối tử của nó. Các đồng ức chế này sau đó liên kết với Sin3, từ đó liên kết với một histone deacetylase (HDAC). Sau đó, HDAC ức chế phiên mã bằng cách deacetyl hóa các đuôi histone — dẫn đến sự nén chặt của các nucleosome thành các cấu trúc không thể tiếp cận bởi các yếu tố phiên mã. Sự gắn kết của phối tử gây ra những thay đổi cấu trúc trong miền E dẫn đến sự giải phóng các phức hợp đồng ức chế/Sin3/HDAC khỏi thụ thể và sự gắn kết của các phức hợp đồng hoạt hóa có thể bao gồm đồng hoạt hóa thụ thể steroid 1 (SRC-1), p300/protein gắn yếu tố đáp ứng cyclic adenosine monophosphate (CBP), yếu tố liên quan đến p300/CBP (P/CAF), hoặc protein liên quan đến đồng tích hợp p300/CBP (pCIP) với mô-típ LXXLL của AF2-AD. Sau đó, các phức hợp đồng hoạt hóa gây ra phiên mã bằng cách acetyl hóa (Ac) các đuôi histone — dẫn đến sự giải nén của các nucleosome thành các cấu trúc có thể tiếp cận bởi các yếu tố phiên mã. Các đường đứt nét được sử dụng để đại diện cho các phức hợp đồng hoạt hóa và đồng ức chế vì thành phần của chúng trong cơ thể sống vẫn chưa được biết.
Bảng 3.5 Các Thụ Thể Nhân và Các Tình Trạng Lâm Sàng Liên Quan Đến Đột Biến Thụ Thể
| Thụ Thể | Đột Biến Dòng Mầm | Rối Loạn Nội Tiết |
|---|---|---|
| Thụ Thể Hormone Tuyến Giáp β (TRβ) | Các đột biến bất hoạt (dị hợp tử và đồng hợp tử) | Đề kháng toàn thể với các hormone tuyến giáp |
| Thụ thể Vitamin D3 | Các đột biến bất hoạt (đồng hợp tử) | Đề kháng Vitamin D3 |
| PPARγ2 | Các đột biến bất hoạt (dị hợp tử) | Béo phì hoặc đái tháo đường týp 2 khởi phát sớm |
| HNF-4 | Các đột biến bất hoạt (dị hợp tử) | Đái tháo đường khởi phát ở tuổi trưởng thành của người trẻ (MODY) loại 1 Hạ đường huyết tăng insulin máu trong giai đoạn sơ sinh sau đó là MODY1 trong cuộc sống sau này ở một số bệnh nhân |
| Thụ thể Glucocorticoid | Các đột biến bất hoạt (dị hợp tử) | Đề kháng Glucocorticoid |
| Thụ thể Androgen | Các đột biến bất hoạt (lặn liên kết X) | Hội chứng không nhạy cảm với androgen, bệnh Kennedy |
| Thụ thể Estrogen α (ER α) | Các đột biến bất hoạt (đồng hợp tử) | Tầm vóc cao và hợp nhất đầu xương không hoàn toàn |
| Thụ thể Mineralocorticoid | Các đột biến bất hoạt (dị hợp tử, đồng hợp tử) | Giả suy aldosteron loại 1 |
| Các đột biến hoạt hóa (đồng hợp tử) | Hội chứng thừa mineralocorticoid biểu kiến | |
| DAX1 | Các đột biến bất hoạt (lặn liên kết X) | Thiểu sản thượng thận bẩm sinh liên kết X |
DAX1, gen 1 vùng tương đồng quan trọng gây bất sản thượng thận bẩm sinh đảo ngược giới tính nhạy cảm với liều lượng trên nhiễm sắc thể X; HNF-4, yếu tố nhân tế bào gan-4; PPARγ2, thụ thể γ 2 được hoạt hóa bởi chất tăng sinh peroxisome.
Miền A/B nằm ở đầu tận amino và chứa miền chức năng hoạt hóa 1 (AF-1)/τ 1. Miền AF-1/τ 1 điều hòa phiên mã gen bằng cách tương tác với các protein (chẳng hạn như các phức hợp Ada và TFIID) gây ra sự phiên mã. Chức năng hoạt hóa chuyển mã này của miền AF-1/τ 1 không phụ thuộc vào sự gắn kết của thụ thể hormone nhân với phối tử của nó và không đặc hiệu trong việc lựa chọn các trình tự DNA đích. Do đó, tính đặc hiệu của hoạt động của thụ thể hormone nhân được quyết định bởi chức năng của các miền thụ thể hormone nhân khác.
Miền C có các đặc điểm giúp mang lại tính đặc hiệu của hoạt động cho mỗi thụ thể hormone nhân. Miền này bao gồm hai mô-típ ngón tay kẽm chịu trách nhiệm cho hoạt động gắn DNA của thụ thể và việc lựa chọn các đối tác dimer hóa. Mỗi mô-đun ngón tay kẽm bao gồm một ion kẽm được bao quanh bởi các lưu huỳnh của bốn gốc cysteine, tạo ra một cấu trúc bậc ba chứa các xoắn. Hộp P nằm gần các cysteine của ngón tay kẽm đầu tiên và chứa ba đến bốn acid amin chịu trách nhiệm cho tính đặc hiệu của việc gắn vào các yếu tố đáp ứng. Hộp D bao gồm một vòng năm acid amin được gắn vào hai cysteine đầu tiên của ngón tay kẽm thứ hai cung cấp giao diện cho sự dimer hóa của thụ thể nhân.
Miền D “bản lề” chứa các tín hiệu định vị hạt nhân và góp phần vào chức năng của các miền C và E liền kề. Do đó, phần đầu tận amino của miền góp phần vào việc gắn DNA và heterodimer hóa và phần đầu tận carboxy góp phần vào việc gắn phối tử. Tín hiệu định vị hạt nhân đóng một vai trò đặc biệt quan trọng trong chức năng của các thụ thể glucocorticoid và mineralocorticoid vì các thụ thể này gắn phối tử của chúng trong bào tương và sau đó phải định vị đến nhân để thay đổi sự phiên mã gen.
Miền E được gọi là miền gắn phối tử (LBD) hoặc miền gắn hormone. Ngoài việc gắn phối tử, miền E còn có tác dụng lên sự dimer hóa và hoạt hóa chuyển mã. LBD bao gồm 11 đến 12 xoắn α (được đặt tên là H1 đến H12) và chứa một túi gắn phối tử được tạo thành từ các phần của một số xoắn khác nhau. Ví dụ, LBD của thụ thể tuyến giáp (TR) có một khoang gắn phối tử bao gồm các thành phần từ H2, H7, H8, H11, và H12. Sự đóng góp của các phần khác nhau của LBD vào túi gắn phối tử giải thích cho phát hiện rằng đột biến của các phân tử acid amin đơn trong các xoắn khác nhau của LBD có thể cản trở sự gắn kết của phối tử.
Không giống như yếu tố hoạt hóa phiên mã AF-1/τ 1, yếu tố hoạt hóa 2 của miền E (AF2-AD) cần sự gắn kết của phối tử để hoạt động (xem Hình 3.9). Thường thì, khi thụ thể không được gắn bởi phối tử của nó, các phức hợp đồng ức chế đồng thời gắn vào LBD và bộ máy phiên mã bao gồm các phức hợp protein đặt các yếu tố phiên mã vào các vị trí gắn nucleosome (xem Hình 3.9). Các phức hợp đồng ức chế sau đó ức chế phiên mã gen bằng cách sử dụng các histone deacetylase để nén chặt các nucleosome thành các cấu trúc không thể tiếp cận (xem Hình 3.9). Sự gắn kết của phối tử gây ra sự sắp xếp lại cấu trúc trong miền E dẫn đến sự giải phóng các phức hợp đồng ức chế này khỏi bộ máy phiên mã và LBD, và sự tiếp xúc của bộ máy phiên mã và mô-típ LXXLL của AF2-AD với các phức hợp đồng hoạt hóa (xem Hình 3.9). Các phức hợp đồng hoạt hóa này có hoạt tính histone acetyltransferase tác động để làm lỏng lẻo các cấu trúc nucleosome, cho phép các yếu tố phiên mã tiếp cận các vị trí gắn nucleosome (xem Hình 3.9).
Hầu hết các thụ thể nhân đều có khả năng gắn vào yếu tố đáp ứng hormone của chúng và ức chế phiên mã khi chúng không được gắn bởi phối tử của chúng. Tuy nhiên, khi không có phối tử, các thụ thể steroid được gắn vào một phức hợp các protein sốc nhiệt thay vì yếu tố đáp ứng của chúng và dường như không ức chế phiên mã.
Các chất chủ vận và đối kháng có các tác dụng khác nhau lên sự tương tác giữa túi gắn phối tử và AF2-AD. Ví dụ, khi 17β-estradiol gắn vào thụ thể estrogen, vị trí của AF2-AD chứa H12 bị thay đổi để các đồng hoạt hóa có thể tiếp cận vị trí gắn đồng hoạt hóa gắn LBD. Tuy nhiên, khi chất đối kháng estrogen raloxifene gắn vào cùng một vị trí, vị trí gắn đồng hoạt hóa trên H12 vẫn bị chặn bởi các phần khác của H12.
Mặc dù một số thụ thể nhân hoạt động hoàn toàn khi được gắn dưới dạng monomer vào DNA, các thụ thể hormone trong siêu họ thụ thể nhân hoạt động mạnh nhất khi được gắn dưới dạng homodimer hoặc heterodimer (xem Hình 3.9). RXR, HNF 4, và các thụ thể hormone steroid có thể gắn DNA dưới dạng homodimer hoặc heterodimer. Dạng đồng phân α của thụ thể estrogen (ESR1) đặc biệt lộn xộn, và có khả năng heterodimer hóa với HNF4A và các thụ thể acid retinoic, dạng đồng phân β của thụ thể estrogen (ESR2), RXR, và các thụ thể hormone tuyến giáp. Dưới dạng homodimer, RXR gắn vào đoạn lặp lại trực tiếp 1 (DR1). Nó cũng có thể kết hợp với các thụ thể tuyến giáp, vitamin D3, và các thụ thể được hoạt hóa bởi chất tăng sinh peroxisome để tạo thành các heterodimer.
Thật thú vị, người ta cũng đã đề xuất rằng một số hormone steroid cũng tác động lên các thụ thể xuyên màng — và những tương tác này có thể chịu trách nhiệm cho các tác dụng tế bào cấp tính của steroid. Progesterone đã được chứng minh là tương tác với các thụ thể oxytocin tử cung, acetylcholine nicotinic, γ-aminobutyric acid A, N-methyl-D aspartate, và các thụ thể progesterone màng tế bào tinh trùng kết cặp G-protein. Các thụ thể estrogen và glucocorticoid màng tế bào cũng đã được xác định.
Các thụ thể nhân phân họ 1: các thụ thể hormone tuyến giáp, vitamin d3, và được hoạt hóa bởi chất tăng sinh peroxisome
Thụ Thể Hormone Tuyến Giáp
Hai dạng đồng phân của thụ thể hormone tuyến giáp (THR) — thụ thể hormone tuyến giáp α (THRA) và thụ thể hormone tuyến giáp β (THRB) — được mã hóa bởi các gen c-erbA khác nhau lần lượt trên các nhiễm sắc thể 17 và 3. Sự nối ghép thay thế dẫn đến sự biểu hiện của TRα1, TRα2, TRβ1, và TRβ2 với sự phân bố mô khác nhau. TRα2 không gắn hormone tuyến giáp và chức năng của nó chưa được hiểu rõ. TRα1 là phân nhóm chiếm ưu thế trong cơ tim và cơ xương, xương, đường tiêu hóa, và hệ thần kinh trung ương. TRβ1 là phân nhóm chiếm ưu thế trong thận và gan, trong khi TRβ2 được biểu hiện ở vùng dưới đồi và tuyến yên, cũng như võng mạc và ốc tai. Các THR không được chiếm giữ bởi hormone tuyến giáp triiodothyronine (T3) tồn tại dưới dạng homodimer hoặc heterodimer, với các RXR được gắn vào các yếu tố đáp ứng hormone tuyến giáp của DNA kết hợp với các protein đồng ức chế. Sự gắn kết của hormone tuyến giáp gây ra sự giải phóng các đồng ức chế khỏi THR. Một đồng hoạt hóa, đồng hoạt hóa thụ thể steroid-1 (SRC-1), sau đó có thể gắn vào THR — cho phép hoạt hóa phiên mã.
Đề kháng toàn thể với các hormone tuyến giáp (GRTH) có thể di truyền lặn hoặc trội trên nhiễm sắc thể thường, vì sự hiện diện của một alen THRB bình thường duy nhất là đủ cho chức năng thụ thể bình thường. GRTH di truyền trội trên nhiễm sắc thể thường là do sự hiện diện của một THRB bất thường cản trở chức năng của thụ thể bình thường theo kiểu trội âm. Tỷ lệ lưu hành của GRTH trội, dựa trên sàng lọc sơ sinh, được báo cáo là 1:40.000 ca sinh sống. Bệnh nhân mắc hội chứng này có đáp ứng thụ thể bị suy giảm đối với T3. Họ có nồng độ T3, T3 đảo ngược, và thyroxine (T4) tăng cao với nồng độ TSH hơi cao. Phù hợp với sự gia tăng TSH và bướu cổ, nồng độ thyroglobulin huyết thanh có xu hướng tăng và sự hấp thu iod phóng xạ cao. Không giống như nhiễm độc giáp tự miễn, tỷ lệ T3:T4 là bình thường. Các biểu hiện lâm sàng có thể thay đổi bao gồm một số triệu chứng cường giáp và một số triệu chứng suy giáp. Các phát hiện bao gồm bướu cổ ở 65% đến 85% những người bị ảnh hưởng, tăng động ở 33% đến 68%, nhịp tim nhanh ở 33% đến 75%, chậm phát triển, tăng tỷ lệ trao đổi chất, mật độ xương thấp, khiếm thính, khuyết tật học tập, chậm phát triển, và tuổi xương chậm. GRTH có thể được phát hiện bằng sàng lọc sơ sinh nếu chương trình đo T4, ngoài hoặc thay vì chỉ TSH, vì T4 sẽ tăng và TSH sẽ tăng. Sự kết hợp của các phát hiện thường liên quan đến cường giáp và suy giáp là do sự biểu hiện khác biệt của TRα so với TRβ. Vùng dưới đồi và tuyến yên biểu hiện TRβ và điều này dẫn đến nồng độ TSH tăng cao. Các mô tim biểu hiện TRα và điều này dẫn đến nhịp tim nhanh vì T4 và T3 tăng cao tác động lên thụ thể TRα bình thường.
Các đột biến đã được tìm thấy trong các miền D và E của THRB của bệnh nhân GRTH. Những đột biến này làm thay đổi sự gắn kết của phối tử hoặc hoạt hóa chuyển mã. Tuy nhiên, hầu hết các THRB đột biến vẫn giữ được khả năng ức chế hoạt hóa chuyển mã của các gen đích thông qua các tương tác với các đồng ức chế. Một số thụ thể đột biến GRTH tiếp tục liên kết với các đồng ức chế và không thể gắn vào đồng hoạt hóa SRC-1 ngay cả khi được gắn bởi T3. Do đó, các THRB đột biến có một hiệu ứng trội âm ở trạng thái dị hợp tử vì chúng có thể cản trở chức năng của các thụ thể kiểu dại bằng cách ức chế phiên mã của DNA đích.
Bệnh nhân mang các đột biến mất chức năng trên cả hai alen của THRB thể hiện các bất thường lâm sàng nghiêm trọng hơn so với bệnh nhân có các đột biến trội âm dị hợp tử. Một bệnh nhân bị xóa cả hai alen THRB đã biểu hiện câm điếc, các đặc điểm dị dạng, và các đầu xương lốm đốm. Một bệnh nhân khác đồng hợp tử đối với một đột biến THRB (“thụ thể dòng họ S”), với một sự xóa acid amin trong miền gắn phối tử, đã biểu hiện chậm phát triển tâm thần, tuổi xương rất chậm, và nồng độ T3 và T4 rất cao. Những người mang gen dị hợp tử của đột biến thụ thể dòng họ S có các biểu hiện lâm sàng nhẹ hơn của GRTH vì THRB đột biến này vẫn giữ được hoạt tính đồng ức chế và do đó có các tác dụng trội âm.
Các đột biến trong THRA chưa được xác định cho đến gần đây. Người khởi phát được mô tả đầu tiên là dị hợp tử đối với một đột biến vô nghĩa (p.E403X) ức chế thụ thể kiểu dại theo kiểu trội âm. Bệnh nhân có các đặc điểm suy giáp (chậm tăng trưởng, chậm phát triển, loạn sản xương, và táo bón) phản ánh sự phân bố của các mô đích trong đó THRA được biểu hiện. Điều này có liên quan đến T4 và T4 tự do bình thường hoặc giới hạn thấp, T3 đảo ngược thấp, T3 và T3 tự do bình thường hoặc giới hạn cao, tỷ lệ T4:T3 thấp, và TSH bình thường. Sau khi điều trị bằng thyroxine, bình thường hóa nồng độ T4, nồng độ T3 trở nên tăng cao. Người ta đã đề xuất rằng điều này có thể liên quan đến tăng hoạt tính của deiodinase loại 1 (chuyển đổi T4 thành T3) và giảm hoạt tính của deiodinase loại 3 (chuyển đổi T4 thành T3 đảo ngược và T3 thành T2). Thụ thể đột biến không gắn được T3 được đánh dấu phóng xạ và không hoạt hóa được một gen báo cáo đáp ứng với hormone tuyến giáp và ức chế hoạt động của thụ thể kiểu dại. Báo cáo thứ hai mô tả một sự chèn một base ở cha và con gái, gây ra một sự dịch khung và chấm dứt sớm ở codon 406. Phân tích thụ thể đột biến in vitro, sau khi biểu hiện trong các tế bào nuôi cấy, cho thấy rằng thụ thể đột biến không đáp ứng với sự kích thích của T3 và có một tác dụng trội âm mạnh lên thụ thể kiểu dại. Báo cáo này và các báo cáo tiếp theo khác đã xác định thêm các đặc điểm lâm sàng, chẳng hạn như đầu to, khuôn mặt dị dạng (mặt tròn, dẹt với các đặc điểm thô), chiều dài và cân nặng khi sinh tăng, thiếu máu, và cholesterol hơi tăng. Một số báo cáo gần đây đã ghi nhận các đột biến khác nhau, bao gồm các đột biến sai nghĩa với các thụ thể hoạt động một phần. Những nghiên cứu này đã chứng minh rằng mức độ nghiêm trọng của các phát hiện lâm sàng tương quan với mức độ suy giảm của thụ thể. Đột biến p.A263V biểu hiện phiên mã bị suy giảm ở nồng độ T3 thấp, nhưng hoạt động ở nồng độ T3 cao hơn và có liên quan đến một kiểu hình nhẹ hơn. Một đột biến khác (p.N359Y), ảnh hưởng đến cả TRα1 và TRα2, có liên quan đến các đặc điểm lâm sàng bổ sung, cụ thể là, cằm nhỏ, bất sản xương đòn, hợp nhất xương bàn tay, dính ngón, tăng năng cận giáp, và tiêu chảy mãn tính.
Thụ Thể Vitamin D
Còi xương nặng, hạ calci máu, tăng năng cận giáp thứ phát, và nồng độ 1,25-dihydroxyvitamin D (calcitriol) tăng xảy ra ở những bệnh nhân mắc hội chứng di truyền lặn trên nhiễm sắc thể thường của “đề kháng vitamin D”. Những bệnh nhân này có các thụ thể vitamin D (VDR) bị khiếm khuyết. Các đột biến gây ra hội chứng này đã được tìm thấy trong các ngón tay kẽm của miền gắn DNA (miền C), dẫn đến giảm hoặc mất khả năng gắn của thụ thể vào các yếu tố điều hòa của các gen đích. Các đột biến gây bệnh cũng đã được tìm thấy dẫn đến việc sản xuất các thụ thể có khả năng gắn calcitriol và heterodimer hóa với RXR bị giảm hoặc mất, vốn cần thiết để VDR hoạt hóa chuyển mã tối đa các gen đích.
Các đột biến ít nghiêm trọng hơn trong VDR có liên quan đến giảm hấp thu calci đường tiêu hóa và mật độ khoáng của xương, ngay cả trong thời thơ ấu, và tăng nguy cơ loãng xương và gãy xương. Tuy nhiên, không thể sao chép những phát hiện này ở một số nhóm dân tộc. Do đó, các yếu tố khác (chẳng hạn như kiểu gen thụ thể estrogen, calci trong chế độ ăn uống, và tuổi tác) có thể góp phần vào tác dụng của các đa hình VDR đối với chuyển hóa khoáng của xương. Một số đa hình VDR nhất định có liên quan đến giảm tăng trưởng theo chiều dọc trước và sau sinh.
Các mối liên quan quan trọng khác đã được tìm thấy với các đa hình VDR. Các đa hình đồng hợp tử đã được báo cáo là có liên quan đến tăng năng cận giáp nguyên phát. Ngoài ra, sự hiện diện của một số alen VDR nhất định có liên quan đến tăng nguy cơ phát triển bệnh nha chu khởi phát sớm. Ngược lại, sự vắng mặt của các alen như vậy đã được liên kết với sỏi thận calci gia đình. Sự vắng mặt của một số alen nhất định có thể là một yếu tố nguy cơ cho sự phát triển của bệnh sarcoidosis. Tuy nhiên, sự hiện diện của các alen này có liên quan đến tăng calci niệu và sỏi thận và tăng nguy cơ ở phụ nữ đối với sự phát triển của ung thư vú di căn. Các đa hình VDR cũng đã được liên kết với tăng tính nhạy cảm với bệnh vẩy nến, bệnh lao, bệnh phong, và các bệnh nhiễm trùng khác.
Thụ Thể γ Được Hoạt Hóa Bởi Chất Tăng Sinh Peroxisome
Thụ thể γ được hoạt hóa bởi chất tăng sinh peroxisome (PPARγ) có một vai trò trong việc điều hòa sự biệt hóa và chuyển hóa của tế bào mỡ. Thụ thể mồ côi trước đây này đã được phối tử prostaglandin J2 nhận làm của mình. Các đột biến trong gen mã hóa PPARγ gây ra loạn dưỡng mỡ một phần gia đình loại 3 (FPLD3).
Bệnh nhân bị FPLD3 biểu hiện mất mỡ ở tay và chân, kháng insulin nghiêm trọng, đái tháo đường týp 2 khởi phát sớm, và tăng triglyceride máu nghiêm trọng. Một số đột biến dị hợp tử có một tác dụng trội âm lên thụ thể kiểu dại (V290M và P467L). Những đột biến này dẫn đến sự thay thế acid amin làm rối loạn sự định hướng của H12 trong miền E, dẫn đến giảm hoạt hóa chuyển mã phụ thuộc phối tử bởi AF-2/AD và sự tuyển dụng đồng hoạt hóa. Các đột biến khác là các đột biến điểm gây ra FPLD3 đơn giản là do sự thiếu hụt đơn bội mà không có tác dụng trội âm. Ngoài ra, một đột biến dịch khung gây ra một kiểu hình tương tự đã được xác định. Một đột biến khác trong PPARγ đã mở rộng kiểu hình bao gồm các đặc điểm cơ, miễn dịch, và huyết học. Đột biến này được cho là gây ra một sự thay đổi cấu hình ảnh hưởng đến sự hoạt hóa phiên mã bởi thụ thể.
Một đột biến sai nghĩa dẫn đến sự thay thế p.Pro115Gln gần một vị trí phosphoryl hóa serine ở vị trí 114 ức chế sự hoạt hóa phiên mã trong PPARγ2 đã được tìm thấy ở một số bệnh nhân béo phì bệnh lý. Sự thay thế p.Pro115Gln cản trở sự phosphoryl hóa của serine ở vị trí 114, dẫn đến tăng hoạt hóa phiên mã bởi PPARγ2 — từ đó dẫn đến tăng sự biệt hóa của tế bào mỡ và tích lũy triglyceride.
Các thụ thể nhân phân họ 2: các yếu tố nhân tế bào gan và các thụ thể retinoid x
Phân họ này bao gồm các thụ thể HNF và các RXR. RXR tạo thành các heterodimer với các thụ thể nhân khác (bao gồm các thụ thể estrogen, vitamin D, và hormone tuyến giáp) và với PPARγ (RXR được thảo luận ở nơi khác trong chương này).
Thụ Thể Yếu Tố Nhân Tế Bào Gan
Sự thay đổi của một thụ thể nhân mồ côi khác, HNF4A, cũng gây ra một rối loạn nội tiết. Các đột biến của gen HNF4A trên nhiễm sắc thể 20 làm thay đổi miền gắn phối tử (miền E) hoặc miền gắn DNA (miền C) đã được tìm thấy ở những bệnh nhân mắc một dạng đái tháo đường đơn gen, còn được gọi là đái tháo đường khởi phát ở tuổi trưởng thành của người trẻ loại 1 (MODY1). Bệnh nhân bị MODY thường phát triển đái tháo đường vào cuối thập kỷ thứ ba của cuộc đời. Họ có một khiếm khuyết trong sự kích thích bài tiết insulin qua trung gian glucose. Một số nghiên cứu hiện đã mô tả một kiểu hình hai pha của tăng insulin máu sơ sinh và thai to, với sự khởi phát muộn hơn của đái tháo đường ở những bệnh nhân có các đột biến HNF4A. Trong hầu hết các trường hợp, tăng insulin máu là tạm thời nhưng có thể kéo dài, cần điều trị bằng diazoxide.
Bệnh nhân dị hợp tử đối với đột biến HNF4A p.R76W cũng phát triển một hội chứng giống Fanconi-Bickel ngoài tăng insulin máu sơ sinh và thai to. Các bệnh nhân biểu hiện bệnh ống thận gần điển hình của hội chứng Fanconi-Bickel, bao gồm acid amin niệu toàn thể, protein niệu trọng lượng phân tử thấp, glucose niệu, tăng phosphat niệu, và hạ acid uric máu, cộng với các đặc điểm bổ sung, bao gồm nhiễm calci thận, suy thận, tăng calci niệu với hạ calci máu tương đối, và tăng magiê máu. Kiểu hình giống Fanconi-Bickel đã được tái tạo trong một mô hình chuột trong đó sự cắt bỏ di truyền của Hnf4a được nhắm mục tiêu đến thận, cho thấy rằng Hnf4a là cần thiết cho sự hình thành của các ống lượn gần biệt hóa và việc mất Hnf4a làm giảm sự biểu hiện của các gen đặc hiệu cho ống lượn gần. Đột biến p.R76W xảy ra trong miền gắn DNA của HNF4A, và đã được giả thuyết là gây ra sự tương tác khiếm khuyết với các gen điều hòa chính; vẫn chưa biết tại sao bệnh nhân có các đột biến HNF4A khác không phát triển cùng một kiểu hình thận.
Những người mang một sự thay thế glycine thành serine trong codon 115 trong miền gắn DNA (miền C) dường như có nguy cơ cao phát triển đái tháo đường insulin thấp. Các yếu tố nhân tế bào gan 3 (HNF-3α, -3β, và -3γ) cũng là các chất điều hòa của các gen đái tháo đường týp 2 khởi phát sớm HNF1A, HNF4A, và IPF-1/PDX-1 — có liên quan lần lượt đến các loại MODY 3, 1, và 4.
Các thụ thể nhân phân họ 3: các thụ thể steroid và các thụ thể glucocorticoid, androgen, estrogen, và mineralocorticoid
Thụ Thể Glucocorticoid
Glucocorticoid ảnh hưởng mạnh mẽ đến trương lực tim mạch; có tác dụng lên gan, cơ, và mô mỡ; và có tác dụng chống viêm và ức chế miễn dịch mạnh. Glucocorticoid quan trọng trong sự tăng trưởng và phát triển, cũng như hành vi và nhận thức. Chúng ảnh hưởng đến nhiều quá trình tế bào, bao gồm tăng sinh, biệt hóa, và apoptosis. Tất cả các tác động này đều được trung gian bởi thụ thể glucocorticoid (GR), là một yếu tố phiên mã được hoạt hóa bởi phối tử ảnh hưởng đến sự phiên mã của khoảng 20% hệ gen. Gen của thụ thể glucocorticoid được gọi là NR3C1. NR3C1 nằm trên nhiễm sắc thể 5 và bao gồm 10 exon. Nhiều dạng đồng phân của GR tồn tại do sự nối ghép thay thế, chèn, xóa, và khởi đầu dịch mã thay thế. Hai dạng chính là hGRα và hGRβ. hGRβ không gắn glucocorticoid tự nhiên hoặc tổng hợp; được biểu hiện ở một số loại tế bào, chẳng hạn như bạch cầu trung tính và tế bào biểu mô; và có thể có tác dụng ức chế lên thụ thể glucocorticoid chính hGRα. hGRα được biểu hiện khắp nơi trong tất cả các mô trừ các nhân trên chéo của vùng dưới đồi.
Các đột biến trong các thụ thể glucocorticoid gây ra đề kháng glucocorticoid toàn thể nguyên phát (PGGR) hoặc quá mẫn glucocorticoid toàn thể nguyên phát (PGGH). PGGR được đặc trưng lâm sàng bởi sự hiện diện của nồng độ cortisol và ACTH huyết tương tăng cao, kèm theo các tác dụng của tăng aldosteron và tăng androgen khi không có các vết rạn da hoặc lắng đọng mỡ trung tâm. Sự dư thừa ACTH dẫn đến tăng sản vỏ thượng thận, tăng bài tiết cortisol, và tăng sản xuất các steroid thượng thận có hoạt tính androgen (androstenedione, dehydroepiandrosterone [DHEA], DHEA-S) và mineralocorticoid (cortisol, deoxycorticosterone và corticosterone). Phổ lâm sàng rộng, từ không có triệu chứng đến tăng androgen nghiêm trọng (đặc trưng bởi mụn trứng cá nặng, rậm lông, kinh nguyệt không đều, và vô sinh), thừa mineralocorticoid (đặc trưng bởi nhiễm kiềm hạ kali máu và tăng huyết áp), và mệt mỏi. Các bé gái có thể biểu hiện bộ phận sinh dục không rõ ràng và cả bé gái và bé trai đều có thể biểu hiện dậy thì sớm cùng giới. Các biểu hiện lâm sàng của thiếu hụt glucocorticoid không thường xuyên và phần lớn chỉ giới hạn ở sự mệt mỏi. Tuy nhiên, đã có những báo cáo về hạ đường huyết thời thơ ấu.
Cả các đột biến đồng hợp tử và dị hợp tử đều đã được mô tả. Các đột biến dị hợp tử gây ra PGGR thường làm như vậy bằng cách có một tác dụng trội âm lên thụ thể kiểu dại.
Các u tuyến yên lớn tiết ACTH cũng có thể do một đột biến dịch khung trong gen GR cản trở sự dẫn truyền tín hiệu gây ra. Bệnh nhân có đột biến này biểu hiện các triệu chứng của đề kháng glucocorticoid. Khối u phát triển do sự suy giảm điều hòa phản hồi âm tính bởi glucocorticoid lên trục dưới đồi-tuyến yên.
PGGH đã được mô tả ở một phụ nữ 43 tuổi biểu hiện béo phì nội tạng, tăng huyết áp, tăng lipid máu, và đái tháo đường týp 2, và được phát hiện có một đột biến trong GR (D401H) thể hiện sự hoạt hóa chuyển mã tăng của các gen đáp ứng với glucocorticoid. Bệnh nhân có bằng chứng về đề kháng glucocorticoid trong trục dưới đồi–tuyến yên–tuyến thượng thận và quá mẫn ở mạch máu, mô mỡ, và gan. Điều này có liên quan đến ACTH và cortisol buổi sáng tăng nhưng cortisol tự do trong nước tiểu bình thường.
Thụ Thể Androgen
Gen thụ thể androgen (AR) của người nằm trên nhiễm sắc thể X. Các rối loạn đã biết được đặc trưng bởi rối loạn chức năng AR do các đột biến gen AR chỉ được biểu hiện ở những bệnh nhân có kiểu nhân 46 XY. Những đột biến này có thể được truyền từ một người mẹ mang gen không có triệu chứng hoặc có thể là de novo.
Hơn 1000 đột biến đã được mô tả trong gen AR, với hơn 500 đột biến gây ra hội chứng không nhạy cảm với androgen (AIS). Kiểu hình của hội chứng này có thể thay đổi về mức độ nghiêm trọng và đã được chia thành các dạng nhẹ, một phần, và hoàn toàn (MAIS, PAIS, và CAIS). CAIS được đặc trưng bởi tinh hoàn trong ổ bụng, không có các cấu trúc mullerian, không có lông cơ thể do androgen gây ra, chẳng hạn như lông mu và lông nách, và ngoại hình nữ. PAIS đề cập đến những người có bộ phận sinh dục ngoài không rõ ràng với âm vật to hoặc dương vật nhỏ và bệnh nhân mắc hội chứng Reifenstein. Hội chứng Reifenstein được đặc trưng bởi lỗ tiểu lệch thấp nghiêm trọng với sự phát triển của bìu và nữ hóa tuyến vú nghiêm trọng. MAIS đề cập đến những bệnh nhân có các đột biến AR là những nam giới có kiểu hình bình thường khác, biểu hiện nữ hóa tuyến vú ở tuổi vị thành niên hoặc vô sinh sau này.
Sự không đồng nhất về kiểu hình của AIS là do các vị trí khác nhau của các đột biến gây ra AIS. Hậu quả chức năng của mỗi đột biến gây ra AIS liên quan đến chức năng của miền trong đó đột biến được định vị. Tuy nhiên, mức độ suy giảm chức năng của thụ thể bị đột biến trong các nghiên cứu in-vitro không phải lúc nào cũng tương quan với mức độ nghiêm trọng về kiểu hình của hội chứng.
Các đột biến trong các exon mã hóa cho miền gắn hormone của AR làm giảm ái lực gắn hormone. Tuy nhiên, những đột biến này không loại bỏ khả năng gắn hormone của thụ thể. Do đó, bệnh nhân có những đột biến này thường biểu hiện với PAIS hoặc đôi khi với CAIS. Bệnh nhân có các đột biến trong miền gắn hormone dường như không đáp ứng với điều trị bằng liều cao testosterone. Các đột biến trong miền gắn DNA dẫn đến thất bại trong việc điều hòa gen đích. Do đó, bệnh nhân có những đột biến này thường biểu hiện CAIS.
CAIS cũng do một đột biến điểm dẫn đến một codon chấm dứt sớm (p.Gln340ter), với sự khởi đầu dịch mã rõ ràng nằm ở phía hạ nguồn của codon chấm dứt. Các nghiên cứu in vitro cho thấy rằng AR bị cắt ngắn được biểu hiện ở mức độ giảm. Nhiều đột biến khác đã được mô tả gây ra sự cắt ngắn hoặc xóa bỏ AR và AIS hoàn toàn. Hai bệnh nhân có bộ phận sinh dục không rõ ràng và nam hóa một phần được phát hiện là khảm đối với các AR đột biến.
Một số bệnh nhân mắc hội chứng Reifenstein đã được phát hiện có một đột biến trong miền gắn DNA loại bỏ sự dimer hóa của thụ thể. Các bệnh nhân khác đã được phát hiện có một đột biến trong một vùng khác của miền gắn DNA không ảnh hưởng đến sự dimer hóa của thụ thể, hoặc có các đột biến trong miền gắn hormone trong miền E. Đột biến Ala596Thr trong vùng hộp D của miền gắn DNA đã được liên kết với tăng nguy cơ ung thư vú.
AIS cũng là một đặc điểm của bệnh Kennedy, là một tình trạng di truyền lặn liên kết X gây teo tủy sống và cơ. Tình trạng này là do sự mở rộng của một đoạn poly-CAG trong exon gen AR mã hóa cho đầu N của AR, dẫn đến tăng số lượng gốc glutamine trong miền A/B. Các AR có một vùng polyQ tăng lên 48 gốc glutamine tích tụ bất thường trong các tế bào được biến nạp do sự gấp sai và quá trình phân giải protein bất thường. Bởi vì sự mở rộng polyQ không loại bỏ hoàn toàn sự hoạt hóa chuyển mã, bệnh nhân mắc bệnh Kennedy biểu hiện một kiểu hình AIS một phần nhẹ bao gồm sự nam hóa bình thường kèm theo teo tinh hoàn, nữ hóa tuyến vú, và vô sinh.
Các đột biến tăng chức năng trong AR đã được mô tả trong các bệnh ung thư tuyến tiền liệt, vú, tinh hoàn, gan, và thanh quản.
Thụ Thể Estrogen
Hai dạng đồng phân ESR dài đầy đủ chính đã được xác định ở động vật có vú. Thụ thể estrogen α (ESR1) được phát hiện đầu tiên và trung gian cho hầu hết các hoạt động đã biết của estrogen. ESR1 được biểu hiện chủ yếu ở tử cung, buồng trứng, tinh hoàn, mào tinh, vỏ thượng thận, và thận. Thụ thể estrogen β (ESR2) được phát hiện vào năm 1996. ESR1 và ESR2 có chung 95% và 50% tương đồng lần lượt trong miền gắn DNA (DBD) và LBD. Có rất ít sự tương đồng ở đầu tận amino giữa hai dạng đồng phân. ESR2 được biểu hiện chủ yếu ở tử cung, buồng trứng, tinh hoàn, tuyến tiền liệt, bàng quang, phổi, và não. Mặc dù ESR2 có ái lực cao với estrogen, nó có khả năng hoạt hóa chuyển mã thấp hơn ESR1 và vẫn chưa được phát hiện có liên quan đến bất kỳ tình trạng bệnh lý nào.
Có bằng chứng ủng hộ sự tồn tại của các ESR chức năng khác. Một số ESR giả định này định vị đến màng tế bào thay vì, hoặc ngoài, nhân. Một sản phẩm 46-kDa bị cắt ngắn ở đầu tận amino của ESR1, có tên là ER46, định vị đến màng tế bào và trung gian cho các hoạt động của estrogen được khởi đầu tại màng tế bào. Một trong những ESR giả định khác này đã được đặt tên là ER-X và được giả định là một GPCR định vị đến màng tế bào. Một thụ thể giả định khác là thụ thể estrogen giả định heterodimeric (pER) được đặt tên khéo léo, đã được tìm thấy trên màng tế bào và màng nhân. pER hoạt động như một serine phosphatase. Năm protein gắn estrogen khác cũng đã được xác định, và ít nhất ba trong số chúng định vị đến màng tế bào.
Người ta đã nghĩ rằng androgen cung cấp các tín hiệu chính cho sự đóng lại của các đầu xương trong tuổi dậy thì. Tuy nhiên, vào năm 1994, các nghiên cứu sâu rộng về một người đàn ông 28 tuổi, với sự đóng lại không hoàn toàn của các đầu xương và tầm vóc cao, đã chứng minh rằng anh ta đồng hợp tử đối với một đột biến chấm dứt sớm của codon 157 trong exon 2 của gen ESR1. Bệnh nhân đã tiếp tục tăng trưởng theo chiều dọc mặc dù sự phát triển dậy thì khác là bình thường, chứng tỏ rằng ESR trung gian cho sự đóng lại của đầu xương. Sự biểu hiện của gen này dẫn đến việc sản xuất một ESR1 không chức năng thiếu cả miền gắn DNA và hormone. Anh ta cũng được phát hiện có nồng độ estradiol tăng, suy giảm dung nạp glucose với tăng insulin máu, và mật độ xương giảm.
Củng cố thêm mối liên quan giữa ESR1 và sự đóng lại của đầu xương là quan sát thấy rằng phụ nữ bị ung thư vú dương tính với ESR1 và có một đột biến trong miền B (alen B’) của ESR1 có tỷ lệ sẩy thai tự nhiên và tầm vóc cao tăng lên. Những mối liên quan này không được tìm thấy ở những người mang gen nữ của alen này không bị ung thư vú hoặc bị ung thư vú âm tính với ESR1. Do đó, một đột biến thứ hai (chưa được phát hiện) có khả năng đóng một vai trò trong sự phát triển của tầm vóc cao và sẩy thai tự nhiên ở những người mang gen nữ bị ung thư vú dương tính với ESR1.
Thụ Thể Mineralocorticoid
Đề kháng mineralocorticoid còn được gọi là giả suy aldosteron (PHA). Cả các trường hợp lẻ tẻ và gia đình với các trường hợp di truyền trội hoặc lặn trên nhiễm sắc thể thường đều đã được báo cáo. Biểu hiện lâm sàng của bệnh nhân bị PHA dao động từ mất muối không có triệu chứng; đến suy giảm tăng trưởng; đến chậm phát triển mãn tính, lơ mơ, và nôn mửa; đến mất nước đe dọa tính mạng kèm theo mất muối nghiêm trọng. Bệnh nhân bị các dạng PHA nghiêm trọng thường biểu hiện trong vòng một năm sau khi sinh và thậm chí có thể biểu hiện trong tử cung với đa ối do đa niệu. Về mặt sinh hóa, tình trạng này được đặc trưng bởi mất muối qua nước tiểu, hạ natri máu, tăng kali huyết tương, hoạt động của aldosterone và renin, và chuyển hóa aldosterone trong nước tiểu không đáp ứng với điều trị bằng mineralocorticoid.
Hai dạng PHA được công nhận (loại I và II). PHAI di truyền lặn trên nhiễm sắc thể thường (toàn thể) là kết quả của các đột biến trong kênh natri biểu mô (ENaC), trong khi PHAI di truyền trội trên nhiễm sắc thể thường (thận) là do các đột biến trong thụ thể mineralocorticoid (MR). PHAII là kết quả của các đột biến trong một họ các kinase serine-threonine được gọi là WNK1 và WNK4, nằm ở phía hạ nguồn của hoạt động của aldosterone. PHAI được đặc trưng bởi đề kháng mineralocorticoid ở ống thận, trong khi PHAII là kết quả của sự đề kháng mineralocorticoid ở thận, ruột, hoặc tuyến nước bọt hoặc tuyến mồ hôi và còn được gọi là hội chứng Gordon. Bệnh nhân mắc các tình trạng này biểu hiện tăng kali máu chỉ đáp ứng với điều trị bằng các ion không phải clorua, chẳng hạn như bicarbonate hoặc sulfat, làm tăng việc cung cấp natri đến ống lượn xa. Nói chung, bệnh nhân bị PHA1 di truyền trội trên nhiễm sắc thể thường có một hội chứng mất muối nhẹ và cải thiện theo tuổi tác, trong khi bệnh nhân bị PHA1 di truyền lặn trên nhiễm sắc thể thường bị mất muối và tăng kali máu nghiêm trọng, cũng như tăng natri trong mồ hôi và nước bọt và nhiễm trùng đường hô hấp thường xuyên không cải thiện theo tuổi tác.
Hơn 50 đột biến khác nhau của MR gây ra PHAI di truyền trội trên nhiễm sắc thể thường đã được mô tả (tất cả đều là dị hợp tử). Có sự không đồng nhất rõ ràng về các biểu hiện lâm sàng trong các gia đình. Sự thiếu hụt đơn bội do sự thoái biến của mRNA hoặc protein rõ ràng là đủ để gây ra PHAI di truyền trội trên nhiễm sắc thể thường. Tuy nhiên, một số đột biến đã được chứng minh là có tác dụng trội âm lên thụ thể kiểu dại.
Các rối loạn di truyền có liên quan đến tăng hoạt động của MR gây ra tăng huyết áp khởi phát sớm nghiêm trọng. Một tình trạng như vậy, được đặc trưng bởi hoạt động MR quá mức, được gọi là hội chứng thừa mineralocorticoid biểu kiến. Bệnh nhân mắc tình trạng di truyền lặn trên nhiễm sắc thể thường này có thể biểu hiện suy giảm tăng trưởng trước và sau sinh, tăng huyết áp do tăng thể tích, nhiễm calci tủy thận, và nhiễm kiềm chuyển hóa hạ kali máu kèm theo hạ renin máu và hạ aldosteron máu. Bệnh nhân cũng có thể không có triệu chứng và chỉ biểu hiện các bất thường sinh hóa. Bệnh nhân mắc hội chứng này cũng có tỷ lệ cortisol-cortisone trong huyết thanh và nước tiểu tăng. Hội chứng này là do các đột biến trong gen 11 β-hydroxysteroid dehydrogenase loại 2 (HSD11B2) làm giảm hoạt tính enzym của protein. 11 β-HSD2 chuyển đổi glucocorticoid hoạt động cortisol thành cortisone không hoạt động; ngược lại, dạng đồng phân loại I, được mã hóa bởi gen HSD11B1 riêng biệt, có cả hoạt tính 11-β dehydrogenase và hoạt tính 11-oxoreductase và cũng có thể chuyển đổi cortisone thành cortisol. Dạng đồng phân loại I được biểu hiện khắp nơi, trong khi dạng đồng phân loại II có sự biểu hiện hạn chế hơn và được biểu hiện cao ở thận, nơi nó cần thiết để bảo vệ MR của thận khỏi nồng độ cortisol huyết thanh thường cao hơn, cho phép aldosterone điều hòa cân bằng nội môi natri. Do đó, hoạt động giảm của 11 β-HSD2 làm tăng nồng độ cortisol trong các tế bào thận biểu hiện các mô MR, dẫn đến tăng sự gắn kết và hoạt hóa của MR bởi cortisol.
Một dạng đề kháng mineralocorticoid thoáng qua, có thể do sự trưởng thành bất thường của chức năng thụ thể aldosterone, cũng tồn tại. Biến thể PHA này được gọi là hội chứng tăng kali máu thời thơ ấu. Trẻ em mắc rối loạn này biểu hiện chậm phát triển hoặc suy giảm tăng trưởng theo chiều dọc kèm theo tăng kali máu và nhiễm toan chuyển hóa. Tình trạng này tự khỏi vào nửa sau của thập kỷ đầu tiên của cuộc đời.
Các thụ thể nhân phân họ 0: dax1
Các thụ thể nhân phân họ 0 bao gồm DAX1. DAX1 đóng một vai trò trong việc điều hòa sản xuất steroid, chất ức chế mullerian, và gonadotropin.
DAX1
Gen 1 vùng tương đồng quan trọng gây bất sản thượng thận bẩm sinh đảo ngược giới tính nhạy cảm với liều lượng trên nhiễm sắc thể X (DAX1) là một thụ thể nhân mồ côi vì phối tử của nó vẫn chưa được xác định. Nó có sự tương đồng trong miền E với các thụ thể mồ côi khác, bao gồm cả các RXR. Tuy nhiên, DAX1 có một miền C gắn DNA bất thường chứa một đoạn lặp lại acid amin thay vì các mô-típ ngón tay kẽm. DAX1 ức chế phiên mã qua trung gian SF-1. SF-1 là một thụ thể nhân mồ côi khác điều hòa phiên mã của các steroid hydroxylase của tuyến thượng thận và tuyến sinh dục, chất ức chế mullerian, và các gen gonadotropin.
Các đột biến gen DAX1 đã được xác định gây ra thiểu sản thượng thận bẩm sinh liên kết X. Bệnh nhân mắc tình trạng này bị suy thượng thận bẩm sinh và do đó thiếu hụt sản xuất glucocorticoid, mineralocorticoid, và androgen. Khoảng 40% bệnh nhân nam bị ảnh hưởng biểu hiện trong 2 tháng đầu đời với một cơn khủng hoảng thượng thận mất muối, và phần còn lại biểu hiện muộn hơn trong thời thơ ấu với suy mineralocorticoid, giống như thiếu hụt aldosterone synthase hoặc PHA. Những người biểu hiện với cơn khủng hoảng thượng thận có thể bị tăng kali máu, hạ natri máu, hạ huyết áp, hoặc ngừng tim, do cả thiếu hụt mineralocorticoid và glucocorticoid trong bối cảnh nồng độ 17-hydroxyprogesterone bình thường. Bệnh nhân có thể có nồng độ cortisol bình thường. Chẩn đoán hình ảnh tuyến thượng thận bằng bất kỳ phương thức nào ở độ tuổi này đều không hữu ích, vì tình trạng này ảnh hưởng đến vùng vĩnh viễn chứ không phải vùng thai nhi của tuyến thượng thận. Vùng thai nhi khá lớn khi sinh và không teo lại cho đến 6 đến 12 tháng tuổi. Chẩn đoán hình ảnh không thể phân biệt được vùng thai nhi với vùng vĩnh viễn và báo cáo một tuyến thượng thận bình thường trong vài tháng đầu đời. Chẩn đoán hình ảnh sau năm đầu đời dự kiến sẽ cho thấy một tuyến thượng thận thiểu sản. Thiếu hụt gonadotropin và không có tinh trùng cũng xảy ra ở những bệnh nhân này nhưng điều này không biểu hiện cho đến tuổi dậy thì. Bệnh nhân có các phát hiện phù hợp với suy sinh dục giảm gonadotropin (do một khiếm khuyết kết hợp dưới đồi/tuyến yên) và có thể không phát triển dậy thì hoặc dậy thì ngừng lại ở khoảng giai đoạn 3 theo Prader. Những người mang gen nữ có thể bị dậy thì muộn. Tất cả các đột biến đã được tìm thấy gây ra thiểu sản thượng thận bẩm sinh liên kết X đều nằm trong hoặc ngăn chặn sự phiên mã của vùng miền E ức chế phiên mã qua trung gian SF-1. Do đó, các đột biến DAX1 có thể gây ra thiểu sản thượng thận bẩm sinh liên kết X bằng cách thay đổi sự điều hòa của SF-1 đối với sự hình thành tuyến sinh dục và tuyến thượng thận. Sự xóa bỏ DAX1 cũng có thể xảy ra trong bối cảnh hội chứng xóa gen liền kề, dẫn đến thiếu hụt glycerol kinase phức tạp (cGKD) nếu các cá nhân có các đoạn xóa kéo dài từ gen GK vào gen loạn dưỡng cơ Duchenne (DMD), hoặc liên quan đến một sự mở rộng telomeric đáng kể từ DAX1.
Sự nhân đôi của Xp bao gồm DAX1 ở nam giới di truyền có kiểu nhân XY dẫn đến sự đảo ngược giới tính. Sự biểu hiện quá mức của DAX1 ức chế tất cả các khía cạnh của sự biệt hóa nam giới bắt đầu từ gờ niệu sinh dục tiến triển đến một tuyến sinh dục lưỡng tiềm, dưới ảnh hưởng của WT1 và SF1, đến sự biệt hóa thành một tinh hoàn dưới ảnh hưởng của SF1, SOX9, SRY, và AMH và dẫn đến bộ phận sinh dục ngoài giống nữ với các cơ quan nội tạng nữ thay đổi từ không có buồng trứng, tử cung, và cổ tử cung đến buồng trứng bình thường với các cấu trúc mullerian thô sơ. Sự nhân đôi trong vùng này của nhiễm sắc thể X cũng có liên quan đến chậm phát triển, tai thấp, hở hàm ếch, tật dính ngón, giảm trương lực cơ, môi trên mỏng, nếp gấp khỉ, và các bất thường tim.
Tóm tắt
Sự hiểu biết về các thụ thể truyền tín hiệu hoặc ảnh hưởng đến hoạt động của hormone đã tăng lên đáng kể. Khi các kỹ thuật sinh học phân tử được cải thiện, người ta kỳ vọng rằng kiến thức về hoạt động của thụ thể sẽ tiếp tục tăng với tốc độ nhanh chóng. Có khả năng các khiếm khuyết tinh vi trong chức năng của thụ thể (chẳng hạn như các đột biến vùng điều hòa hoặc promoter làm tăng hoặc giảm biểu hiện gen thụ thể, hoặc các đột biến trong các protein truyền tin thứ hai) sẽ được tìm thấy gây ra các rối loạn nội tiết. Cũng có khả năng các thụ thể mới sẽ được phát hiện truyền tín hiệu hoặc ảnh hưởng đến hoạt động của hormone và các vai trò nội tiết sẽ được tìm thấy cho các thụ thể mà trước đây không được cho là trung gian hoặc thay đổi hoạt động của hormone.
Bảng chú giải các thuật ngữ y học chuyên ngành – Chương 3
| STT | Thuật ngữ tiếng Anh | Phiên âm IPA | Nghĩa Tiếng Việt |
|---|---|---|---|
| 1 | Hormone | /ˈhɔːr.moʊn/ | Hormone, nội tiết tố |
| 2 | Receptor proteins | /rɪˈsɛp.tɚ ˈproʊ.tiːnz/ | Protein thụ thể |
| 3 | Conformational changes | /ˌkɑːn.fɚˈmeɪ.ʃən.əl ˈtʃeɪndʒɪz/ | Thay đổi cấu hình |
| 4 | Compartmental redistribution | /kəmˌpɑːrtˈmen.t̬əl ˌriː.dɪs.trɪˈbjuː.ʃən/ | Tái phân bố trong các ngăn |
| 5 | Physiologic response | /ˌfɪz.i.əˈlɑː.dʒɪk rɪˈspɑːns/ | Đáp ứng sinh lý |
| 6 | Ligand | /ˈlɪɡ.ənd/ | Phối tử |
| 7 | Genomics | /dʒəˈnoʊ.mɪks/ | Hệ gen học |
| 8 | G protein–coupled receptors (GPCRs) | /dʒiː ˈproʊ.tiːn ˈkʌp.əld rɪˈsɛp.tɚz/ | Thụ thể kết cặp G-protein (GPCR) |
| 9 | Cytokine receptors | /ˈsaɪ.t̬oʊ.kaɪn rɪˈsɛp.tɚz/ | Thụ thể cytokine |
| 10 | Tyrosine kinase receptors (RTKs) | /ˈtaɪ.rə.siːn ˈkaɪ.neɪs rɪˈsɛp.tɚz/ | Thụ thể tyrosine kinase (RTK) |
| 11 | Nuclear receptors | /ˈnuː.kli.ɚ rɪˈsɛp.tɚz/ | Thụ thể nhân |
| 12 | Agonist | /ˈæɡ.ə.nɪst/ | Chất chủ vận |
| 13 | Potency | /ˈpoʊ.t̬ən.si/ | Hiệu lực |
| 14 | Efficacy | /ˈef.ə.kə.si/ | Hiệu quả |
| 15 | EC50 (Half maximal effective concentration) | /iː siː ˈfɪf.ti/ | EC50 (Nồng độ hiệu quả bán phần) |
| 16 | Super agonist | /ˈsuː.pɚ ˈæɡ.ə.nɪst/ | Siêu chủ vận |
| 17 | Partial agonists | /ˈpɑːr.ʃəl ˈæɡ.ə.nɪsts/ | Chất chủ vận một phần |
| 18 | Antagonist | /ænˈtæɡ.ə.nɪst/ | Chất đối kháng |
| 19 | Orthosteric antagonist | /ˌɔːr.θoʊˈster.ɪk ænˈtæɡ.ə.nɪst/ | Chất đối kháng cùng vị trí |
| 20 | Allosteric antagonist | /ˌæl.oʊˈster.ɪk ænˈtæɡ.ə.nɪst/ | Chất đối kháng dị lập thể |
| 21 | IC50 (Half maximal inhibitory concentration) | /aɪ siː ˈfɪf.ti/ | IC50 (Nồng độ ức chế bán phần) |
| 22 | Inverse agonist | /ɪnˈvɜːrs ˈæɡ.ə.nɪst/ | Chất chủ vận đảo nghịch |
| 23 | Biased signaling | /ˈbaɪ.əst ˈsɪɡ.nəl.ɪŋ/ | Tín hiệu thiên lệch |
| 24 | Orthostatic ligands | /ˌɔːr.θoʊˈstæt.ɪk ˈlɪɡ.əndz/ | Phối tử cùng vị trí |
| 25 | Glycosylated | /ɡlaɪˈkɑː.sə.leɪ.t̬ɪd/ | Được glycosyl hóa |
| 26 | Allosteric modulators | /ˌæl.oʊˈster.ɪk ˈmɑː.dʒə.leɪ.t̬ɚz/ | Chất điều biến dị lập thể |
| 27 | Receptor activity modifying proteins (RAMPs) | /rɪˈsɛp.tɚ ækˈtɪv.ə.t̬i ˈmɑː.dɪ.faɪ.ɪŋ ˈproʊ.tiːnz/ | Protein điều chỉnh hoạt động của thụ thể (RAMP) |
| 28 | Calcitonin | /ˌkæl.səˈtoʊ.nɪn/ | Calcitonin |
| 29 | Adrenomedullin | /əˌdriː.noʊˈmedʒ.ə.lɪn/ | Adrenomedullin |
| 30 | Amylin | /ˈæm.ə.lɪn/ | Amylin |
| 31 | Calcitonin gene-related peptide | /ˌkæl.səˈtoʊ.nɪn dʒiːn rɪˈleɪ.t̬ɪd ˈpep.taɪd/ | Peptide liên quan đến gen calcitonin |
| 32 | Signal transduction | /ˈsɪɡ.nəl trænsˈdʌk.ʃən/ | Dẫn truyền tín hiệu |
| 33 | Heterotrimeric G proteins | /ˌhet̬.ɚ.oʊ.traɪˈmer.ɪk dʒiː ˈproʊ.tiːnz/ | Protein G dị tam thể |
| 34 | Seven-transmembrane domain receptors | /ˈsev.ən trænsˈmem.breɪn doʊˈmeɪn rɪˈsɛp.tɚz/ | Thụ thể có bảy vùng xuyên màng |
| 35 | Heptahelical receptors | /ˌhep.təˈhel.ɪ.kəl rɪˈsɛp.tɚz/ | Thụ thể bảy xoắn |
| 36 | Serpentine receptors | /ˈsɜːr.pən.tiːn rɪˈsɛp.tɚz/ | Thụ thể xoắn ốc |
| 37 | Alpha helices | /ˈæl.fə ˈhiː.lɪ.siːz/ | Xoắn alpha |
| 38 | Ectodomain / Exodomain | /ˈek.toʊ.doʊ.meɪn/ / /ˈek.soʊ.doʊ.meɪn/ | Miền ngoại bào / Miền ngoài |
| 39 | Intracellular loops | /ˌɪn.trəˈsel.jə.lɚ luːps/ | Vòng nội bào |
| 40 | Extracellular loops | /ˌek.strəˈsel.jə.lɚ luːps/ | Vòng ngoại bào |
| 41 | Serpentine region | /ˈsɜːr.pən.tiːn ˈriː.dʒən/ | Vùng xoắn ốc |
| 42 | Endodomain | /ˈen.doʊ.doʊ.meɪn/ | Miền nội bào |
| 43 | Guanosine 5’-triphosphate (GTP) | /ˈɡwɑː.nə.siːn faɪv praɪm traɪˈfɑːs.feɪt/ | Guanosine 5’-triphosphate (GTP) |
| 44 | Guanosine 5’-diphosphate (GDP) | /ˈɡwɑː.nə.siːn faɪv praɪm daɪˈfɑːs.feɪt/ | Guanosine 5’-diphosphate (GDP) |
| 45 | GTPase activity | /ˌdʒiː.tiː.piˈeɪz ækˈtɪv.ə.t̬i/ | Hoạt tính GTPase |
| 46 | Phospholipase C | /ˌfɑːs.foʊˈlaɪ.peɪs siː/ | Phospholipase C |
| 47 | Regulators of G protein signaling (RGSs) | /ˈreɡ.jə.leɪ.t̬ɚz əv dʒiː ˈproʊ.tiːn ˈsɪɡ.nəl.ɪŋ/ | Chất điều hòa tín hiệu G-protein (RGS) |
| 48 | GTPase-activating proteins (GAPs) | /ˌdʒiː.tiː.piˈeɪz ˈæk.tɪ.veɪ.t̬ɪŋ ˈproʊ.tiːnz/ | Protein hoạt hóa GTPase (GAP) |
| 49 | Dimerization | /daɪˌmer.ɪˈzeɪ.ʃən/ | Sự dimer hóa |
| 50 | Endoplasmic reticulum (ER) | /ˌen.doʊˈplæz.mɪk rəˈtɪk.jə.ləm/ | Lưới nội chất (ER) |
| 51 | Orphan receptor | /ˈɔːr.fən rɪˈsɛp.tɚ/ | Thụ thể mồ côi |
| 52 | Adrenocorticotropin hormone (ACTH) | /əˌdriː.noʊˌkɔːr.t̬ɪ.koʊˈtroʊ.pɪn ˈhɔːr.moʊn/ | Hormone vỏ thượng thận (ACTH) |
| 53 | Melanocortin 2 receptor (MC2R) | /məˌlæn.əˈkɔːr.t̬ɪn tuː rɪˈsɛp.tɚ/ | Thụ thể melanocortin 2 (MC2R) |
| 54 | α-melanocyte-stimulating hormone (α-MSH) | /ˈæl.fə məˈlæn.ə.saɪt ˈstɪm.jə.leɪ.t̬ɪŋ ˈhɔːr.moʊn/ | Hormone kích thích tế bào hắc tố α (α-MSH) |
| 55 | Melanocortin 2 receptor-associated protein (MRAP) | /məˌlæn.əˈkɔːr.t̬ɪn tuː rɪˈsɛp.tɚ əˈsoʊ.ʃi.eɪ.t̬ɪd ˈproʊ.tiːn/ | Protein liên quan đến thụ thể melanocortin 2 (MRAP) |
| 56 | Dominant negative mutation | /ˈdɑː.mə.nənt ˈneɡ.ə.t̬ɪv mjuːˈteɪ.ʃən/ | Đột biến trội âm |
| 57 | Nephrogenic diabetes insipidus | /ˌnef.roʊˈdʒen.ɪk ˌdaɪ.əˈbiː.t̬iːz ɪnˈsɪp.ɪ.dəs/ | Đái tháo nhạt do thận |
| 58 | Oligomerization | /əˌlɪɡ.ə.mər.ɪˈzeɪ.ʃən/ | Sự oligomer hóa |
| 59 | Isolated hypogonadotropic hypogonadism (IHH) | /ˈaɪ.sə.leɪ.t̬ɪd ˌhaɪ.poʊ.ɡoʊˌnæd.əˈtroʊ.pɪk ˌhaɪ.poʊˈɡoʊ.næd.ɪ.zəm/ | Suy sinh dục giảm gonadotropin đơn độc (IHH) |
| 60 | Constitutive activity | /kənˈstɪtʃ.u.ə.t̬ɪv ækˈtɪv.ə.t̬i/ | Hoạt tính cấu thành |
| 61 | Receptor desensitization | /rɪˈsɛp.tɚ diːˌsen.sə.t̬əˈzeɪ.ʃən/ | Sự giải mẫn cảm của thụ thể |
| 62 | Receptor resensitization | /rɪˈsɛp.tɚ riːˌsen.sə.t̬əˈzeɪ.ʃən/ | Sự tái mẫn cảm của thụ thể |
| 63 | G protein receptor kinases (GRKs) | /dʒiː ˈproʊ.tiːn rɪˈsɛp.tɚ ˈkaɪ.neɪ.sɪz/ | Kinase của thụ thể G-protein (GRK) |
| 64 | Homologous desensitization | /həˈmɑː.lə.ɡəs diːˌsen.sə.t̬əˈzeɪ.ʃən/ | Giải mẫn cảm tương đồng |
| 65 | β-arrestins | /ˈbeɪ.t̬ə əˈres.tɪnz/ | β-arrestin |
| 66 | Heterologous desensitization | /ˌhet̬.ɚˈɑː.lə.ɡəs diːˌsen.sə.t̬əˈzeɪ.ʃən/ | Giải mẫn cảm dị đồng |
| 67 | Internalization/sequestration | /ɪnˌtɜːr.nəl.əˈzeɪ.ʃən/ /ˌsiː.kwesˈtreɪ.ʃən/ | Sự nội hóa/biệt trí |
| 68 | Downregulation | /ˌdaʊn.reɡ.jəˈleɪ.ʃən/ | Sự điều hòa giảm |
| 69 | Lysosomal degradation | /ˌlaɪ.səˈsoʊ.məl ˌdeɡ.rəˈdeɪ.ʃən/ | Thoái biến lysosome |
| 70 | Rhodopsin-like receptors | /ˌroʊˈdɑːp.sɪn laɪk rɪˈsɛp.tɚz/ | Thụ thể giống rhodopsin |
| 71 | Secretagogue | /sɪˈkriː.t̬ə.ɡɑːɡ/ | Chất kích thích tiết |
| 72 | Venus flytrap domain | /ˈviː.nəs ˈflaɪ.træp doʊˈmeɪn/ | Miền bẫy ruồi Venus |
| 73 | Familial glucocorticoid deficiency (FGD) | /fəˈmɪl.i.əl ˌɡluː.koʊˈkɔːr.tɪ.kɔɪd dɪˈfɪʃ.ən.si/ | Suy giảm glucocorticoid gia đình (FGD) |
| 74 | Triple A (Allgrove) syndrome | /ˈtrɪp.əl eɪ (ˈɔːl.ɡroʊv) ˈsɪn.droʊm/ | Hội chứng Triple A (Allgrove) |
| 75 | Achalasia | /ˌæk.əˈleɪ.ʒə/ | Co thắt tâm vị |
| 76 | Alacrima | /əˈlæk.rɪ.mə/ | Không có nước mắt |
| 77 | Hyperpigmentation | /ˌhaɪ.pɚ.pɪɡ.menˈteɪ.ʃən/ | Tăng sắc tố da |
| 78 | Adrenarche | /ˌæd.rəˈnɑːr.ki/ | Giai đoạn phát dục thượng thận |
| 79 | Zona fasciculata | /ˈzoʊ.nə fəˌsɪk.jəˈlɑː.t̬ə/ | Lớp bó |
| 80 | Zona reticularis | /ˈzoʊ.nə rəˌtɪk.jəˈler.ɪs/ | Lớp lưới |
| 81 | Zona glomerulosa | /ˈzoʊ.nə ɡloʊˌmer.jəˈloʊ.sə/ | Lớp cầu |
| 82 | Adrenocortical carcinomas | /əˌdriː.noʊˈkɔːr.tɪ.kəl ˌkɑːr.sɪˈnoʊ.məz/ | Ung thư biểu mô vỏ thượng thận |
| 83 | Loss of heterozygosity (LOH) | /lɑːs əv ˌhet̬.ɚ.oʊ.zaɪˈɡɑː.sə.t̬i/ | Mất dị hợp tử (LOH) |
| 84 | ACTH-independent macronodular adrenal hyperplasia (AIMAH) | /ˌeɪ.siː.tiː.eɪtʃ ˌɪn.dɪˈpen.dənt ˌmæk.roʊˈnɑː.dʒə.lɚ əˈdriː.nəl ˌhaɪ.pɚˈpleɪ.ʒə/ | Tăng sản thượng thận dạng nốt lớn không phụ thuộc ACTH (AIMAH) |
| 85 | Gastric inhibitory peptide | /ˈɡæs.trɪk ɪnˈhɪb.ɪ.tɔːr.i ˈpep.taɪd/ | Peptide ức chế dạ dày |
| 86 | Melanocortin-3 receptor (MC3R) | /məˌlæn.əˈkɔːr.t̬ɪn θriː rɪˈsɛp.tɚ/ | Thụ thể melanocortin-3 (MC3R) |
| 87 | Melanocortin-4 receptor (MC4R) | /məˌlæn.əˈkɔːr.t̬ɪn fɔːr rɪˈsɛp.tɚ/ | Thụ thể melanocortin-4 (MC4R) |
| 88 | Agouti-related peptide (AgRP) | /əˈɡuː.ti rɪˈleɪ.t̬ɪd ˈpep.taɪd/ | Peptide liên quan đến agouti (AgRP) |
| 89 | Anorexigenic | /ˌæn.ɔː.rekˈsɪdʒ.ə.nɪk/ | Gây chán ăn |
| 90 | Hyperphagic obesity | /ˌhaɪ.pɚˈfeɪ.dʒɪk oʊˈbiː.sə.t̬i/ | Béo phì do ăn nhiều |
| 91 | Lean body mass | /liːn ˈbɑː.di mæs/ | Khối lượng cơ thể nạc |
| 92 | Monogenic cause | /ˌmɑː.noʊˈdʒen.ɪk kɑːz/ | Nguyên nhân đơn gen |
| 93 | Anorexia nervosa | /ˌæn.əˌrek.si.ə nɚˈvoʊ.sə/ | Chán ăn tâm thần |
| 94 | Melanocortin-5 receptor (MC5R) | /məˌlæn.əˈkɔːr.t̬ɪn faɪv rɪˈsɛp.tɚ/ | Thụ thể melanocortin-5 (MC5R) |
| 95 | Melanocortin-1 receptor (MC1R) | /məˌlæn.əˈkɔːr.t̬ɪn wʌn rɪˈsɛp.tɚ/ | Thụ thể melanocortin-1 (MC1R) |
| 96 | Proopiomelanocortin (POMC) | /ˌproʊ.oʊ.pi.oʊ.məˌlæn.əˈkɔːr.tɪn/ | Proopiomelanocortin (POMC) |
| 97 | Eumelanin | /juːˈmel.ə.nɪn/ | Eumelanin (sắc tố nâu đen) |
| 98 | Pheomelanin | /ˌfiː.oʊˈmel.ə.nɪn/ | Pheomelanin (sắc tố đỏ vàng) |
| 99 | Prohormone convertase 1 | /ˌproʊˈhɔːr.moʊn ˈkɑːn.vɜːr.teɪs wʌn/ | Prohormone convertase 1 |
| 100 | Arginine vasopressin (AVP) | /ˈɑːr.dʒə.niːn ˌveɪ.zoʊˈpres.ɪn/ | Arginine vasopressin (AVP) |
| 101 | Aquaporin-2 (AQP2) | /ˌæk.wəˈpɔːr.ɪn tuː/ | Aquaporin-2 (AQP2) |
| 102 | Inappropriate antidiuretic hormone secretion (SIADH) | /ˌɪn.əˈproʊ.pri.ət ˌæn.ti.daɪ.jʊˈret.ɪk ˈhɔːr.moʊn sɪˈkriː.ʃən/ | Hội chứng tiết hormone chống bài niệu không phù hợp (SIADH) |
| 103 | Luteinizing hormone/choriogonadotropin receptor (LHCGR) | /ˈluː.ti.ə.naɪ.zɪŋ ˈhɔːr.moʊn ˌkɔːr.i.oʊ.ɡoʊˌnæd.əˈtroʊ.pɪn rɪˈsɛp.tɚ/ | Thụ thể hormone tạo hoàng thể/choriogonadotropin (LHCGR) |
| 104 | Leucine-rich repeats | /ˈluː.siːn rɪtʃ rɪˈpiːts/ | Đoạn lặp lại giàu leucine |
| 105 | Leydig cell hypoplasia | /ˈlaɪ.dɪɡ sel ˌhaɪ.poʊˈpleɪ.ʒə/ | Thiểu sản tế bào Leydig |
| 106 | Micropenis | /ˌmaɪ.kroʊˈpiː.nɪs/ | Dương vật nhỏ |
| 107 | Cryptorchidism | /krɪpˈtɔːr.kɪ.dɪ.zəm/ | Tinh hoàn ẩn |
| 108 | XY disorder of sexual differentiation | /ˌeksˈwaɪ dɪsˈɔːr.dɚ əv ˈsek.ʃu.əl ˌdɪf.ə.ren.ʃiˈeɪ.ʃən/ | Rối loạn phát triển giới tính XY |
| 109 | Müllerian derivatives | /mjuːˈlɪər.i.ən dəˈrɪv.ə.tɪvz/ | Dẫn xuất Müllerian |
| 110 | Hypospadias | /ˌhaɪ.poʊˈspeɪ.di.əs/ | Lỗ tiểu lệch thấp |
| 111 | Azoospermia | /ˌeɪ.zoʊ.oʊˈspɜːr.mi.ə/ | Không có tinh trùng |
| 112 | Oligomenorrhea | /ˌɑː.lɪ.ɡoʊˌmen.əˈriː.ə/ | Kinh nguyệt thưa |
| 113 | Empty follicle syndrome | /ˈemp.ti ˈfɑː.lɪ.kəl ˈsɪn.droʊm/ | Hội chứng nang trứng rỗng |
| 114 | Male-limited precocious puberty (MLPP) | /meɪl ˈlɪm.ɪ.t̬ɪd prɪˈkoʊ.ʃəs ˈpjuː.bɚ.t̬i/ | Dậy thì sớm giới hạn ở nam (MLPP) |
| 115 | Testotoxicosis | /ˌtes.toʊ.tɑːk.sɪˈkoʊ.sɪs/ | Nhiễm độc tinh hoàn |
| 116 | Leydig cell adenomas | /ˈlaɪ.dɪɡ sel ˌæd.əˈnoʊ.məz/ | U tuyến tế bào Leydig |
| 117 | Theca cells | /ˈθiː.kə selz/ | Tế bào vỏ trong |
| 118 | Granulosa cells | /ˌɡræn.jəˈloʊ.sə selz/ | Tế bào hạt |
| 119 | Graafian follicles | /ˈɡræf.i.ən ˈfɑː.lɪ.kəlz/ | Nang Graafian |
| 120 | Follicle-Stimulating Hormone (FSH) | /ˈfɑː.lɪ.kəl ˈstɪm.jə.leɪ.t̬ɪŋ ˈhɔːr.moʊn/ | Hormone kích thích nang trứng (FSH) |
| 121 | Sertoli cell | /sərˈtoʊ.li sel/ | Tế bào Sertoli |
| 122 | Spermatogenesis | /ˌspɜːr.mə.toʊˈdʒen.ə.sɪs/ | Quá trình sinh tinh |
| 123 | Ovarian dysgenesis (ODG) | /oʊˌver.i.ən dɪsˈdʒen.ə.sɪs/ | Loạn sản buồng trứng (ODG) |
| 124 | Primary amenorrhea | /ˈpraɪ.mer.i əˌmen.əˈriː.ə/ | Vô kinh nguyên phát |
| 125 | Streak ovaries | /striːk ˈoʊ.vər.iz/ | Buồng trứng dạng dải |
| 126 | Karyotype | /ˈker.i.oʊ.taɪp/ | Kiểu nhân |
| 127 | Premature ovarian failure (POF) | /ˌpriː.məˈtʃʊr oʊˌver.i.ən ˈfeɪl.jɚ/ | Suy buồng trứng sớm (POF) |
| 128 | Sex cord ovarian tumors | /seks kɔːrd oʊˌver.i.ən ˈtuː.mɚz/ | U dây sinh dục buồng trứng |
| 129 | Premature ovarian insufficiency | /ˌpriː.məˈtʃʊr oʊˌver.i.ən ˌɪn.səˈfɪʃ.ən.si/ | Suy buồng trứng sớm |
| 130 | Frameshift mutation | /ˈfreɪm.ʃɪft mjuːˈteɪ.ʃən/ | Đột biến dịch khung |
| 131 | Compound heterozygosity | /ˈkɑːm.paʊnd ˌhet̬.ɚ.oʊ.zaɪˈɡɑː.sə.t̬i/ | Dị hợp tử kép |
| 132 | Ovarian hyperstimulation syndrome (OHSS) | /oʊˌver.i.ən ˌhaɪ.pɚˌstɪm.jəˈleɪ.ʃən ˈsɪn.droʊm/ | Hội chứng quá kích buồng trứng (OHSS) |
| 133 | Follicular cysts | /fəˈlɪk.jə.lɚ sɪsts/ | Nang noãn |
| 134 | Luteinized cells | /ˈluː.ti.ə.naɪzd selz/ | Tế bào hoàng thể hóa |
| 135 | Thyroid-Stimulating Hormone (TSH) | /ˈθaɪ.rɔɪd ˈstɪm.jə.leɪ.t̬ɪŋ ˈhɔːr.moʊn/ | Hormone kích thích tuyến giáp (TSH) |
| 136 | Diacylglycerol | /ˌdaɪ.əˌsɪlˈɡlɪs.ə.rɔːl/ | Diacylglycerol |
| 137 | Inositol phosphate | /ɪˈnoʊ.sɪ.tɔːl ˈfɑːs.feɪt/ | Inositol phosphate |
| 138 | Toxic adenomas | /ˈtɑːk.sɪk ˌæd.əˈnoʊ.məz/ | U tuyến độc |
| 139 | Multinodular goiters | /ˌmʌl.tiˈnoʊ.dʒə.lɚ ˈɡɔɪ.t̬ɚz/ | Bướu cổ đa nhân |
| 140 | Differentiated thyroid carcinomas | /ˌdɪf.əˈren.ʃi.eɪ.t̬ɪd ˈθaɪ.rɔɪd ˌkɑːr.sɪˈnoʊ.məz/ | Ung thư biểu mô tuyến giáp biệt hóa |
| 141 | Resistance to TSH (RTSH) | /rɪˈzɪs.təns tuː ˌtiː.esˈeɪtʃ/ | Đề kháng TSH (RTSH) |
| 142 | Euthyroid state | /juːˈθaɪ.rɔɪd steɪt/ | Trạng thái bình giáp |
| 143 | Congenital thyroid hypoplasia | /kənˈdʒen.ə.t̬əl ˈθaɪ.rɔɪd ˌhaɪ.poʊˈpleɪ.ʒə/ | Thiểu sản tuyến giáp bẩm sinh |
| 144 | Sodium-iodide symporter | /ˈsoʊ.di.əm ˈaɪ.ə.daɪd ˈsɪm.pɔːr.t̬ɚ/ | Đồng vận chuyển natri-iodide |
| 145 | Human Chorionic Gonadotropin (HCG) | /ˈhjuː.mən ˌkɔːr.iˈɑː.nɪk ɡoʊˌnæd.əˈtroʊ.pɪn/ | Gonadotropin màng đệm người (HCG) |
| 146 | Gestational thyrotoxicosis | /dʒesˈteɪ.ʃən.əl ˌθaɪ.roʊ.tɑːk.sɪˈkoʊ.sɪs/ | Nhiễm độc giáp thai kỳ |
| 147 | Gestational trophoblastic disease | /dʒesˈteɪ.ʃən.əl ˌtroʊ.foʊˈblæs.tɪk dɪˈziːz/ | Bệnh nguyên bào nuôi thai kỳ |
| 148 | Molar pregnancy | /ˈmoʊ.lɚ ˈpreɡ.nən.si/ | Thai trứng |
| 149 | Choriocarcinoma | /ˌkɔːr.i.oʊˌkɑːr.sɪˈnoʊ.mə/ | Ung thư màng đệm |
| 150 | Gonadotropin-Releasing Hormone (GnRH) | /ɡoʊˌnæd.əˈtroʊ.pɪn rɪˈliː.sɪŋ ˈhɔːr.moʊn/ | Hormone giải phóng gonadotropin (GnRH) |
| 151 | Kallmann syndrome (KS) | /ˈkɑːl.mən ˈsɪn.droʊm/ | Hội chứng Kallmann (KS) |
| 152 | Thyrotropin-Releasing Hormone (TRH) | /ˌθaɪ.roʊˈtroʊ.pɪn rɪˈliː.sɪŋ ˈhɔːr.moʊn/ | Hormone giải phóng thyrotropin (TRH) |
| 153 | Central hypothyroidism | /ˈsen.trəl ˌhaɪ.poʊˈθaɪ.rɔɪ.dɪ.zəm/ | Suy giáp trung ương |
| 154 | Free fatty acid receptor 1 (FFAR1) | /friː ˌfæt̬.i ˈæs.ɪd rɪˈsɛp.tɚ wʌn/ | Thụ thể acid béo tự do 1 (FFAR1) |
| 155 | Lipid mediators | /ˈlɪp.ɪd ˈmiː.di.eɪ.t̬ɚz/ | Chất trung gian lipid |
| 156 | Pancreatic β-islet cells | /ˌpæŋ.kriˈæt̬.ɪk ˈbeɪ.t̬ə ˈaɪ.lət selz/ | Tế bào β-tiểu đảo tụy |
| 157 | Cholecystokinin | /ˌkoʊ.ləˌsɪs.təˈkaɪ.nɪn/ | Cholecystokinin |
| 158 | Glucose-stimulated insulin release | /ˈɡluː.koʊs ˈstɪm.jə.leɪ.t̬ɪd ˈɪn.sə.lɪn rɪˈliːs/ | Giải phóng insulin được kích thích bởi glucose |
| 159 | KISS1 receptor (GPR54) | /kɪs wʌn rɪˈsɛp.tɚ/ | Thụ thể KISS1 (GPR54) |
| 160 | Kisspeptin-1 | /kɪsˈpep.tɪn wʌn/ | Kisspeptin-1 |
| 161 | Metastin | /məˈtæs.tɪn/ | Metastin |
| 162 | Central precocious puberty | /ˈsen.trəl prɪˈkoʊ.ʃəs ˈpjuː.bɚ.t̬i/ | Dậy thì sớm trung ương |
| 163 | Orexin receptors | /ɔːˈrek.sɪn rɪˈsɛp.tɚz/ | Thụ thể Orexin |
| 164 | Hypocretins | /ˌhaɪ.poʊˈkriː.tɪnz/ | Hypocretin |
| 165 | Narcolepsy with cataplexy | /ˈnɑːr.kə.lep.si wɪθ ˈkæt̬.ə.plek.si/ | Chứng ngủ rũ có mất trương lực cơ |
| 166 | Ghrelin receptor | /ˈɡrel.ɪn rɪˈsɛp.tɚ/ | Thụ thể Ghrelin |
| 167 | Growth hormone secretagogue | /ɡroʊθ ˈhɔːr.moʊn sɪˈkriː.t̬ə.ɡɑːɡ/ | Chất kích thích tiết hormone tăng trưởng |
| 168 | Somatotrophs | /ˈsoʊ.mə.toʊ.trɑːfs/ | Tế bào hướng sinh dục |
| 169 | Prader Willi syndrome | /ˈprɑː.dɚ ˈwɪl.i ˈsɪn.droʊm/ | Hội chứng Prader Willi |
| 170 | Constitutional delay of growth and puberty | /ˌkɑːn.stəˈtuː.ʃən.əl dɪˈleɪ əv ɡroʊθ ænd ˈpjuː.bɚ.t̬i/ | Chậm tăng trưởng và dậy thì thể tạng |
| 171 | Obestatin | /oʊˈbes.tə.tɪn/ | Obestatin |
| 172 | Melanin-concentrating hormone (MCH) receptor | /ˈmel.ə.nɪn ˈkɑːn.sən.treɪ.t̬ɪŋ ˈhɔːr.moʊn rɪˈsɛp.tɚ/ | Thụ thể hormone tập trung melanin (MCH) |
| 173 | Growth Hormone–Releasing Hormone (GHRH) Receptor | /ɡroʊθ ˈhɔːr.moʊn rɪˈliː.sɪŋ ˈhɔːr.moʊn rɪˈsɛp.tɚ/ | Thụ thể Hormone Giải phóng Hormone Tăng trưởng (GHRH) |
| 174 | Isolated GH deficiency | /ˈaɪ.sə.leɪ.t̬ɪd ˌdʒiːˈeɪtʃ dɪˈfɪʃ.ən.si/ | Thiếu hụt GH đơn độc |
| 175 | Pituitary hypoplasia | /pɪˈtuː.ə.ter.i ˌhaɪ.poʊˈpleɪ.ʒə/ | Thiểu sản tuyến yên |
| 176 | Gastric Inhibitory Polypeptide Receptor (GIPR) | /ˈɡæs.trɪk ɪnˈhɪb.ɪ.tɔːr.i ˌpɑː.liˈpep.taɪd rɪˈsɛp.tɚ/ | Thụ thể Polypeptide Ức chế Dạ dày (GIPR) |
| 177 | Food-dependent Cushing syndrome | /fuːd dɪˈpen.dənt ˈkʊʃ.ɪŋz ˈsɪn.droʊm/ | Hội chứng Cushing phụ thuộc vào thức ăn |
| 178 | Parathyroid Hormone (PTH) | /ˌpær.əˈθaɪ.rɔɪd ˈhɔːr.moʊn/ | Hormone cận giáp (PTH) |
| 179 | Parathyroid hormone–related peptide (PTHrP) | /ˌpær.əˈθaɪ.rɔɪd ˈhɔːr.moʊn rɪˈleɪ.t̬ɪd ˈpep.taɪd/ | Peptide liên quan đến hormone cận giáp (PTHrP) |
| 180 | Blomstrand chondrodysplasia | /ˈblɑːm.strænd ˌkɑːn.droʊ.dɪsˈpleɪ.ʒə/ | Loạn sản sụn Blomstrand |
| 181 | Enchondromatosis | /enˌkɑːn.droʊ.məˈtoʊ.sɪs/ | U sụn |
| 182 | Ollier disease | /ɔːˈljeɪ dɪˈziːz/ | Bệnh Ollier |
| 183 | Maffucci syndrome | /məˈfuː.tʃi ˈsɪn.droʊm/ | Hội chứng Maffucci |
| 184 | Eiken syndrome | /ˈaɪ.kən ˈsɪn.droʊm/ | Hội chứng Eiken |
| 185 | Primary failure of tooth eruption (PFE) | /ˈpraɪ.mer.i ˈfeɪl.jɚ əv tuːθ ɪˈrʌp.ʃən/ | Thất bại nguyên phát trong việc mọc răng (PFE) |
| 186 | Jansen metaphyseal chondrodysplasia | /ˈjæn.sən ˌmet̬.əˈfɪz.i.əl ˌkɑːn.droʊ.dɪsˈpleɪ.ʒə/ | Loạn sản sụn hành xương Jansen |
| 187 | Calcium-Sensing Receptor (CaSR) | /ˈkæl.si.əm ˈsen.sɪŋ rɪˈsɛp.tɚ/ | Thụ thể cảm nhận calci (CaSR) |
| 188 | Parathyroid chief cells | /ˌpær.əˈθaɪ.rɔɪd tʃiːf selz/ | Tế bào chính của tuyến cận giáp |
| 189 | Familial hypocalciuric hypercalcemia (FHH) | /fəˈmɪl.i.əl ˌhaɪ.poʊ.kæl.sjʊˈriː.ɪk ˌhaɪ.pɚ.kælˈsiː.mi.ə/ | Tăng calci máu giảm calci niệu gia đình (FHH) |
| 190 | Neonatal severe hyperparathyroidism (NSHPT) | /ˌniː.oʊˈneɪ.t̬əl sɪˈvɪr ˌhaɪ.pɚˌpær.əˈθaɪ.rɔɪ.dɪ.zəm/ | Tăng năng cận giáp nặng ở trẻ sơ sinh (NSHPT) |
| 191 | Autosomal-dominant hypocalcemia (ADH) | /ˌɔː.t̬əˈsoʊ.məl ˈdɑː.mə.nənt ˌhaɪ.poʊ.kælˈsiː.mi.ə/ | Hạ calci máu di truyền trội trên NST thường (ADH) |
| 192 | Bartter syndrome type V | /ˈbɑːr.t̬ɚ ˈsɪn.droʊm taɪp faɪv/ | Hội chứng Bartter loại V |
| 193 | Hypokalemic metabolic alkalosis | /ˌhaɪ.poʊ.kəˈliː.mɪk ˌmet̬.əˈbɑː.lɪk ˌæl.kəˈloʊ.sɪs/ | Nhiễm kiềm chuyển hóa hạ kali máu |
| 194 | Paget disease | /ˈpædʒ.ɪt dɪˈziːz/ | Bệnh Paget |
| 195 | Nephrolithiasis | /ˌnef.roʊ.lɪˈθaɪ.ə.sɪs/ | Sỏi thận |
| 196 | Pseudohypoparathyroidism (PHP) | /ˌsuː.doʊˌhaɪ.poʊˌpær.əˈθaɪ.rɔɪ.dɪ.zəm/ | Giả suy cận giáp (PHP) |
| 197 | Albright hereditary osteodystrophy (AHO) | /ˈɔːl.braɪt həˈred.ɪ.ter.i ˌɑːs.ti.oʊˈdɪs.trə.fi/ | Loạn dưỡng xương di truyền Albright (AHO) |
| 198 | Pseudopseudohypoparathyroidism (PPHP) | /ˌsuː.doʊˌsuː.doʊˌhaɪ.poʊˌpær.əˈθaɪ.rɔɪ.dɪ.zəm/ | Giả giả suy cận giáp (PPHP) |
| 199 | Heterotopic ossifications | /ˌhet̬.ɚ.oʊˈtɑː.pɪk ˌɑː.sə.fɪˈkeɪ.ʃənz/ | Hóa xương dị chỗ |
| 200 | Brachydactyly | /ˌbræk.iˈdæk.tə.li/ | Tật ngắn ngón |
| 201 | Genomic imprinting | /dʒəˈnoʊ.mɪk ˈɪm.prɪn.tɪŋ/ | Ghi dấu gen |
| 202 | Progressive osseous heteroplasia (POH) | /prəˈɡres.ɪv ˈɑː.si.əs ˌhet̬.ɚ.oʊˈpleɪ.ʒə/ | Loạn sản xương tiến triển (POH) |
| 203 | Osteoma cutis | /ˌɑːs.tiˈoʊ.mə ˈkjuː.tɪs/ | U xương ở da |
| 204 | Paternal uniparental disomy (UPD) | /pəˈtɜːr.nəl ˌjuː.nɪ.pəˈren.təl daɪˈsoʊ.mi/ | Lưỡng bội đơn cha (UPD) |
| 205 | Fibrous dysplasia | /ˈfaɪ.brəs dɪsˈpleɪ.ʒə/ | Loạn sản xơ |
| 206 | McCune-Albright syndrome | /məˈkuːn ˈɔːl.braɪt ˈsɪn.droʊm/ | Hội chứng McCune-Albright |
| 207 | Café-au-lait spots | /kæˌfeɪ oʊ ˈleɪ spɑːts/ | Đốm café-au-lait |
| 208 | Polyostotic fibrous dysplasia | /ˌpɑː.li.ɑːsˈtɑː.t̬ɪk ˈfaɪ.brəs dɪsˈpleɪ.ʒə/ | Loạn sản xơ đa xương |
| 209 | Phosphatonin | /fɑːsˈfɑː.tə.nɪn/ | Phosphatonin |
| 210 | Cytokines | /ˈsaɪ.t̬oʊ.kaɪnz/ | Cytokine |
| 211 | Interleukins (ILs) | /ˌɪn.t̬ɚˈluː.kɪnz/ | Interleukin (IL) |
| 212 | Erythropoietin | /ɪˌrɪθ.roʊˈpɔɪ.ə.tɪn/ | Erythropoietin |
| 213 | Thrombopoietin | /ˌθrɑːm.boʊˈpɔɪ.ə.tɪn/ | Thrombopoietin |
| 214 | Granulocyte-colony–stimulating factor | /ˈɡræn.jə.loʊ.saɪt ˈkɑː.lə.ni ˈstɪm.jə.leɪ.t̬ɪŋ ˈfæk.tɚ/ | Yếu tố kích thích dòng bạch cầu hạt |
| 215 | Interferons | /ˌɪn.t̬ɚˈfɪər.ɑːnz/ | Interferon |
| 216 | Janus family of tyrosine kinases (Jak kinases) | /ˈdʒæn.əs ˈfæm.ə.li əv ˈtaɪ.rə.siːn ˈkaɪ.neɪ.sɪz/ | Họ kinase tyrosine Janus (kinase Jak) |
| 217 | Signal transducers and activators of transcription (STATs) | /ˈsɪɡ.nəl trænsˈduː.sɚz ænd ˈæk.tɪ.veɪ.t̬ɚz əv trænsˈkrɪp.ʃən/ | Chất dẫn truyền tín hiệu và chất hoạt hóa phiên mã (STAT) |
| 218 | Growth hormone receptor (GHR) | /ɡroʊθ ˈhɔːr.moʊn rɪˈsɛp.tɚ/ | Thụ thể hormone tăng trưởng (GHR) |
| 219 | Growth hormone binding protein (GHBP) | /ɡroʊθ ˈhɔːr.moʊn ˈbaɪn.dɪŋ ˈproʊ.tiːn/ | Protein gắn hormone tăng trưởng (GHBP) |
| 220 | Insulin-like growth factor binding protein 3 (IGFBP3) | /ˈɪn.sə.lɪn laɪk ɡroʊθ ˈfæk.tɚ ˈbaɪn.dɪŋ ˈproʊ.tiːn θriː/ | Protein gắn yếu tố tăng trưởng giống insulin 3 (IGFBP3) |
| 221 | Acid labile subunit (ALS) | /ˈæs.ɪd ˈleɪ.baɪl ˈsʌbˌjuː.nɪt/ | Tiểu đơn vị không ổn định với acid (ALS) |
| 222 | Mitogen activated protein (MAP) kinase | /ˈmaɪ.t̬ə.dʒən ˈæk.tɪ.veɪ.t̬ɪd ˈproʊ.tiːn ˈkaɪ.neɪs/ | Kinase protein được hoạt hóa bởi mitogen (MAP) |
| 223 | Growth hormone insensitivity (GHI) | /ɡroʊθ ˈhɔːr.moʊn ɪnˌsen.səˈtɪv.ə.t̬i/ | Không mẫn cảm với hormone tăng trưởng (GHI) |
| 224 | Laron syndrome | /ˈler.ən ˈsɪn.droʊm/ | Hội chứng Laron |
| 225 | Leptin receptor (LEPR) | /ˈlep.tɪn rɪˈsɛp.tɚ/ | Thụ thể leptin (LEPR) |
| 226 | Receptor tyrosine kinase (RTK) | /rɪˈsɛp.tɚ ˈtaɪ.rə.siːn ˈkaɪ.neɪs/ | Thụ thể tyrosine kinase (RTK) |
| 227 | Juxtamembrane region | /ˌdʒʌk.stəˈmem.breɪn ˈriː.dʒən/ | Vùng cạnh màng |
| 228 | Insulin receptor (INSR) | /ˈɪn.sə.lɪn rɪˈsɛp.tɚ/ | Thụ thể insulin (INSR) |
| 229 | IFG-1 receptor (IGF1R) | /ˌaɪ.dʒiː.ef wʌn rɪˈsɛp.tɚ/ | Thụ thể IFG-1 (IGF1R) |
| 230 | Heterotetramers | /ˌhet̬.ɚ.oʊˈte.trə.mɚz/ | Dị tứ phân |
| 231 | Trans-autophosphorylation | /træns ˌɔː.t̬oʊˌfɑːs.fɔːr.əˈleɪ.ʃən/ | Tự phosphoryl hóa trans |
| 232 | Src homology 2 (SH2)-domain | /sɑːrk həˈmɑː.lə.dʒi tuː doʊˈmeɪn/ | Miền Src homology 2 (SH2) |
| 233 | Phosphatidylinositol-3-kinase (PI3K) | /ˌfɑːs.fəˌtaɪ.dəl.ɪˈnoʊ.sɪ.tɔːl θriː ˈkaɪ.neɪs/ | Phosphatidylinositol-3-kinase (PI3K) |
| 234 | Insulin receptor substrate proteins (IRS) | /ˈɪn.sə.lɪn rɪˈsɛp.tɚ ˈsʌb.streɪt ˈproʊ.tiːnz/ | Protein chất nền của thụ thể insulin (IRS) |
| 235 | Protein kinase B (PKB) | /ˈproʊ.tiːn ˈkaɪ.neɪs biː/ | Protein kinase B (PKB) |
| 236 | Mitogenesis | /ˌmaɪ.t̬oʊˈdʒen.ə.sɪs/ | Sự nguyên phân |
| 237 | Donohue syndrome (leprechaunism) | /ˌdɑː.nəˈhjuː ˈsɪn.droʊm (ˈlep.rə.kɑː.nɪ.zəm)/ | Hội chứng Donohue (leprechaunism) |
| 238 | Rabson-Mendenhall syndrome | /ˈræb.sən ˈmen.dən.hɔːl ˈsɪn.droʊm/ | Hội chứng Rabson-Mendenhall |
| 239 | Type A insulin resistance syndrome | /taɪp eɪ ˈɪn.sə.lɪn rɪˈzɪs.təns ˈsɪn.droʊm/ | Hội chứng kháng insulin loại A |
| 240 | Lipoatrophy | /ˌlaɪ.poʊˈæ.trə.fi/ | Teo mỡ |
| 241 | Acanthosis nigricans | /ˌæk.ənˌθoʊ.sɪs ˈnaɪ.ɡrɪ.kænz/ | Gai đen |
| 242 | Micrognathia | /ˌmaɪ.kroʊˈneɪ.θi.ə/ | Cằm nhỏ |
| 243 | Clitoromegaly | /ˌklaɪ.t̬ɚ.oʊˈmeɡ.ə.li/ | Phì đại âm vật |
| 244 | Hirsutism | /ˈhɜːr.suː.tɪ.zəm/ | Rậm lông |
| 245 | Diabetic ketoacidosis | /ˌdaɪ.əˈbet̬.ɪk ˌkiː.t̬oʊˌæs.ɪˈdoʊ.sɪs/ | Nhiễm toan ceton do đái tháo đường |
| 246 | Pineal hyperplasia | /ˌpaɪˈniː.əl ˌhaɪ.pɚˈpleɪ.ʒə/ | Tăng sản tuyến tùng |
| 247 | Type B insulin resistance syndrome | /taɪp biː ˈɪn.sə.lɪn rɪˈzɪs.təns ˈsɪn.droʊm/ | Hội chứng kháng insulin loại B |
| 248 | HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) | /heər ˈæn/ | HAIR-AN (tăng androgen, kháng insulin, và gai đen) |
| 249 | Polycystic ovary syndrome | /ˌpɑː.liˈsɪs.tɪk ˈoʊ.vər.i ˈsɪn.droʊm/ | Hội chứng buồng trứng đa nang |
| 250 | Intrauterine growth retardation (IUGR) | /ˌɪn.trəˈjuː.t̬ɚ.ɪn ɡroʊθ ˌriː.tɑːrˈdeɪ.ʃən/ | Chậm tăng trưởng trong tử cung (IUGR) |
| 251 | Ring chromosome | /rɪŋ ˈkroʊ.mə.soʊm/ | Nhiễm sắc thể vòng |
| 252 | Microcephaly | /ˌmaɪ.kroʊˈsef.ə.li/ | Tật đầu nhỏ |
| 253 | Hypertelorism | /ˌhaɪ.pɚˈtel.ə.rɪ.zəm/ | Hai hốc mắt xa nhau |
| 254 | Meningioma | /məˌnɪn.dʒiˈoʊ.mə/ | U màng não |
| 255 | Fibroblast growth factor receptor (FGFR) | /ˌfaɪ.broʊˈblæst ɡroʊθ ˈfæk.tɚ rɪˈsɛp.tɚ/ | Thụ thể yếu tố tăng trưởng nguyên bào sợi (FGFR) |
| 256 | Immunoglobulin-like domains | /ɪˌmjuː.noʊˈɡlɑː.bjə.lɪn laɪk doʊˈmeɪnz/ | Miền giống immunoglobulin |
| 257 | Angiogenesis | /ˌæn.dʒi.oʊˈdʒen.ə.sɪs/ | Sự hình thành mạch |
| 258 | Heparan sulfate proteoglycans | /ˈhep.ə.ræn ˈsʌl.feɪt ˌproʊ.t̬i.oʊˈɡlaɪ.kænz/ | Proteoglycan heparan sulfat |
| 259 | Chondrocytes | /ˈkɑːn.droʊ.saɪts/ | Tế bào sụn |
| 260 | Anosmia | /əˈnɑːz.mi.ə/ | Mất khứu giác |
| 261 | Olfactory bulb | /oʊlˈfæk.tɚ.i bʌlb/ | Hành khứu |
| 262 | Synkinesia | /ˌsɪn.kɪˈniː.ʒə/ | Cử động đồng bộ |
| 263 | Craniosynostosis | /ˌkreɪ.ni.oʊˌsɪn.ɑːsˈtoʊ.sɪs/ | Hẹp sọ |
| 264 | Osteoglophonic dysplasia | /ˌɑːs.ti.oʊ.ɡləˈfɑː.nɪk dɪsˈpleɪ.ʒə/ | Loạn sản xương răng |
| 265 | Achondroplasia | /ˌeɪ.kɑːn.droʊˈpleɪ.ʒə/ | Loạn sản sụn |
| 266 | Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) | /sɪˈvɪr ˌeɪ.kɑːn.droʊˈpleɪ.ʒə wɪθ dɪˌvel.əpˈmen.təl dɪˈleɪ ænd ˌæk.ənˌθoʊ.sɪs ˈnaɪ.ɡrɪ.kænz/ | Loạn sản sụn nặng với chậm phát triển và gai đen (SADDAN) |
| 267 | Hypochondroplasia | /ˌhaɪ.poʊ.kɑːn.droʊˈpleɪ.ʒə/ | Thiểu sản sụn |
| 268 | Platyspondylic lethal skeletal dysplasias (PLSD) | /ˌplæt̬.i.spɑːnˈdɪl.ɪk ˈliː.θəl ˈskel.ə.t̬əl dɪsˈpleɪ.ʒəz/ | Loạn sản xương gây chết người có đốt sống dẹt (PLSD) |
| 269 | Thanatophoric dysplasia (TD) | /ˌθæn.ə.toʊˈfɔːr.ɪk dɪsˈpleɪ.ʒə/ | Loạn sản tử vong (TD) |
| 270 | Cloverleaf skull | /ˈkloʊ.vɚ.liːf skʌl/ | Hộp sọ hình lá chẻ |
| 271 | Polymicrogyria | /ˌpɑː.liˌmaɪ.kroʊˈdʒaɪ.ri.ə/ | Đa hồi não nhỏ |
| 272 | Neuronal heterotopia | /nʊˈrɑː.nəl ˌhet̬.ɚ.oʊˈtoʊ.pi.ə/ | Lạc chỗ tế bào thần kinh |
| 273 | Camptodactyly | /ˌkæmp.toʊˈdæk.tə.li/ | Tật co ngón |
| 274 | Scoliosis | /ˌskoʊ.liˈoʊ.sɪs/ | Vẹo cột sống |
| 275 | Peroxisome proliferator-activated retinoic acid | /pəˌrɑːk.sɪˈsoʊm proʊˌlɪf.əˈreɪ.t̬ɚ ˈæk.tɪ.veɪ.t̬ɪd ˌret̬.ɪˈnoʊ.ɪk ˈæs.ɪd/ | Acid retinoic được hoạt hóa bởi chất tăng sinh peroxisome |
| 276 | Hepatocyte nuclear factor-4α (HNF4A) | /həˈpæt̬.ə.saɪt ˈnuː.kli.ɚ ˈfæk.tɚ fɔːr ˈæl.fə/ | Yếu tố nhân tế bào gan-4α (HNF4A) |
| 277 | Retinoid X receptors (RXRs) | /ˈret̬.ə.nɔɪd eks rɪˈsɛp.tɚz/ | Thụ thể retinoid X (RXR) |
| 278 | Steroidogenic factor-1 (SF-1) | /ˌster.ɔɪ.doʊˈdʒen.ɪk ˈfæk.tɚ wʌn/ | Yếu tố tạo steroid-1 (SF-1) |
| 279 | Zinc-finger motifs | /zɪŋk ˈfɪŋ.ɡɚ moʊˈtiːfs/ | Mô-típ ngón tay kẽm |
| 280 | P-box | /ˈpiː bɑːks/ | Hộp P |
| 281 | D-box | /ˈdiː bɑːks/ | Hộp D |
| 282 | Ligand-binding domain (LBD) | /ˈlɪɡ.ənd ˈbaɪn.dɪŋ doʊˈmeɪn/ | Miền gắn phối tử (LBD) |
| 283 | Corepressor | /ˌkoʊ.rɪˈpres.ɚ/ | Đồng ức chế |
| 284 | Histone deacetylases | /ˈhɪs.toʊn diː.əˈset̬.ə.leɪ.sɪz/ | Histone deacetylase |
| 285 | Coactivator | /ˌkoʊˈæk.tɪ.veɪ.t̬ɚ/ | Đồng hoạt hóa |
| 286 | Histone acetyltransferase | /ˈhɪs.toʊn əˌset̬.əlˈtræns.fə.reɪs/ | Histone acetyltransferase |
| 287 | Heat-shock proteins | /hiːt ʃɑːk ˈproʊ.tiːnz/ | Protein sốc nhiệt |
| 288 | Homodimers | /ˌhoʊ.moʊˈdaɪ.mɚz/ | Homodimer |
| 289 | Heterodimers | /ˌhet̬.ɚ.oʊˈdaɪ.mɚz/ | Heterodimer |
| 290 | Thyroid hormone receptor α (THRA) | /ˈθaɪ.rɔɪd ˈhɔːr.moʊn rɪˈsɛp.tɚ ˈæl.fə/ | Thụ thể hormone tuyến giáp α (THRA) |
| 291 | Thyroid hormone receptor β (THRB) | /ˈθaɪ.rɔɪd ˈhɔːr.moʊn rɪˈsɛp.tɚ ˈbeɪ.t̬ə/ | Thụ thể hormone tuyến giáp β (THRB) |
| 292 | Triiodothyronine (T3) | /ˌtraɪ.aɪˌoʊ.doʊˈθaɪ.rə.niːn/ | Triiodothyronine (T3) |
| 293 | Thyroxine (T4) | /θaɪˈrɑːk.siːn/ | Thyroxine (T4) |
| 294 | Generalized resistance to thyroid hormones (GRTH) | /ˈdʒen.ər.əl.aɪzd rɪˈzɪs.təns tuː ˈθaɪ.rɔɪd ˈhɔːr.moʊnz/ | Đề kháng toàn thể với các hormone tuyến giáp (GRTH) |
| 295 | Deaf mutism | /def ˈmjuː.tɪ.zəm/ | Câm điếc |
| 296 | Stippled epiphyses | /ˈstɪp.əld ɪˈpɪf.ə.siːz/ | Đầu xương lốm đốm |
| 297 | Deiodinase | /diːˌaɪ.əˈdɪ.neɪs/ | Deiodinase |
| 298 | Skeletal dysplasia | /ˈskel.ə.t̬əl dɪsˈpleɪ.ʒə/ | Loạn sản xương |
| 299 | Macrocephaly | /ˌmæk.roʊˈsef.ə.li/ | Đầu to |
| 300 | Syndactyly | /ˌsɪnˈdæk.tə.li/ | Dính ngón |
| 301 | Vitamin D resistance | /ˈvaɪ.t̬ə.mɪn diː rɪˈzɪs.təns/ | Đề kháng vitamin D |
| 302 | Rickets | /ˈrɪk.ɪts/ | Còi xương |
| 303 | Secondary hyperparathyroidism | /ˈsek.ən.der.i ˌhaɪ.pɚˌpær.əˈθaɪ.rɔɪ.dɪ.zəm/ | Tăng năng cận giáp thứ phát |
| 304 | 1,25-dihydroxyvitamin D (calcitriol) | /wʌn ˌtwen.tiˈfaɪv daɪ.haɪˌdrɑːk.siˈvaɪ.t̬ə.mɪn diː (ˌkæl.sɪˈtraɪ.ɔːl)/ | 1,25-dihydroxyvitamin D (calcitriol) |
| 305 | Osteoporosis | /ˌɑːs.ti.oʊ.pəˈroʊ.sɪs/ | Loãng xương |
| 306 | Sarcoidosis | /ˌsɑːr.kɔɪˈdoʊ.sɪs/ | Bệnh sarcoidosis |
| 307 | Peroxisome proliferator-activating receptor γ (PPARγ) | /pəˌrɑːk.sɪˈsoʊm proʊˌlɪf.əˈreɪ.t̬ɚ ˈæk.tɪ.veɪ.t̬ɪŋ rɪˈsɛp.tɚ ˈɡæm.ə/ | Thụ thể γ được hoạt hóa bởi chất tăng sinh peroxisome (PPARγ) |
| 308 | Adipocyte | /ˈæd.ə.poʊ.saɪt/ | Tế bào mỡ |
| 309 | Prostaglandin J2 | /ˌprɑːs.təˈɡlæn.dɪn dʒeɪ tuː/ | Prostaglandin J2 |
| 310 | Familial partial lipodystrophy type 3 (FPLD3) | /fəˈmɪl.i.əl ˈpɑːr.ʃəl ˌlaɪ.poʊˈdɪs.trə.fi taɪp θriː/ | Loạn dưỡng mỡ một phần gia đình loại 3 (FPLD3) |
| 311 | Maturity-onset diabetes of the young (MODY) | /məˈtʃʊr.ə.t̬i ˌɑːnˈset ˌdaɪ.əˈbiː.t̬iːz əv ðə jʌŋ/ | Đái tháo đường khởi phát ở tuổi trưởng thành của người trẻ (MODY) |
| 312 | Neonatal hyperinsulinism | /ˌniː.oʊˈneɪ.t̬əl ˌhaɪ.pɚˈɪn.sə.lɪ.nɪ.zəm/ | Tăng insulin máu sơ sinh |
| 313 | Macrosomia | /ˌmæk.roʊˈsoʊ.mi.ə/ | Thai to |
| 314 | Fanconi-Bickel syndrome | /ˈfæŋ.koʊ.ni ˈbɪk.əl ˈsɪn.droʊm/ | Hội chứng Fanconi-Bickel |
| 315 | Proximal tubulopathy | /ˈprɑːk.sə.məl ˌtuː.bjəˈlɑː.pə.θi/ | Bệnh ống thận gần |
| 316 | Aminoaciduria | /əˌmiː.noʊˌæs.ɪˈdjʊə.ri.ə/ | Acid amin niệu |
| 317 | Glycosuria | /ˌɡlaɪ.koʊˈsjʊə.ri.ə/ | Glucose niệu |
| 318 | Hyperphosphaturia | /ˌhaɪ.pɚˌfɑːs.fəˈtjʊə.ri.ə/ | Tăng phosphat niệu |
| 319 | Hypouricemia | /ˌhaɪ.poʊ.jʊə.rɪˈsiː.mi.ə/ | Hạ acid uric máu |
| 320 | Nephrocalcinosis | /ˌnef.roʊˌkæl.səˈnoʊ.sɪs/ | Nhiễm calci thận |
| 321 | Glucocorticoid receptor (GR) | /ˌɡluː.koʊˈkɔːr.tɪ.kɔɪd rɪˈsɛp.tɚ/ | Thụ thể Glucocorticoid (GR) |
| 322 | Primary generalized glucocorticoid resistance (PGGR) | /ˈpraɪ.mer.i ˈdʒen.ər.əl.aɪzd ˌɡluː.koʊˈkɔːr.tɪ.kɔɪd rɪˈzɪs.təns/ | Đề kháng glucocorticoid toàn thể nguyên phát (PGGR) |
| 323 | Primary generalized glucocorticoid hypersensitivity (PGGH) | /ˈpraɪ.mer.i ˈdʒen.ər.əl.aɪzd ˌɡluː.koʊˈkɔːr.tɪ.kɔɪd ˌhaɪ.pɚ.sen.səˈtɪv.ə.t̬i/ | Quá mẫn glucocorticoid toàn thể nguyên phát (PGGH) |
| 324 | Hyperaldosteronism | /ˌhaɪ.pɚ.ælˈdɑːs.tə.roʊ.nɪ.zəm/ | Tăng aldosteron |
| 325 | Hyperandrogenism | /ˌhaɪ.pɚˈæn.droʊ.dʒə.nɪ.zəm/ | Tăng androgen |
| 326 | Adrenocortical hyperplasia | /əˌdriː.noʊˈkɔːr.tɪ.kəl ˌhaɪ.pɚˈpleɪ.ʒə/ | Tăng sản vỏ thượng thận |
| 327 | Dehydroepiandrosterone (DHEA) | /diːˌhaɪ.droʊˌep.i.ænˈdrɑː.stə.roʊn/ | Dehydroepiandrosterone (DHEA) |
| 328 | Isosexual precocity | /ˌaɪ.soʊˈsek.ʃu.əl prɪˈkɑː.sə.t̬i/ | Dậy thì sớm cùng giới |
| 329 | Androgen receptor (AR) | /ˈæn.droʊ.dʒən rɪˈsɛp.tɚ/ | Thụ thể Androgen (AR) |
| 330 | Androgen insensitivity syndrome (AIS) | /ˈæn.droʊ.dʒən ɪnˌsen.səˈtɪv.ə.t̬i ˈsɪn.droʊm/ | Hội chứng không nhạy cảm với androgen (AIS) |
| 331 | Reifenstein syndrome | /ˈraɪ.fən.staɪn ˈsɪn.droʊm/ | Hội chứng Reifenstein |
| 332 | Gynecomastia | /ˌɡaɪ.nə.koʊˈmæs.ti.ə/ | Nữ hóa tuyến vú |
| 333 | Kennedy disease | /ˈken.ə.di dɪˈziːz/ | Bệnh Kennedy |
| 334 | Spinal and muscular atrophy | /ˈspaɪ.nəl ænd ˈmʌs.kjə.lɚ ˈæ.trə.fi/ | Teo tủy sống và cơ |
| 335 | Estrogen receptor α (ESR1) | /ˈes.trə.dʒən rɪˈsɛp.tɚ ˈæl.fə/ | Thụ thể Estrogen α (ESR1) |
| 336 | Estrogen receptor β (ESR2) | /ˈes.trə.dʒən rɪˈsɛp.tɚ ˈbeɪ.t̬ə/ | Thụ thể Estrogen β (ESR2) |
| 337 | Epiphyseal closure | /ˌep.əˈfɪz.i.əl ˈkloʊ.ʒɚ/ | Đóng đầu xương |
| 338 | Mineralocorticoid resistance | /ˌmɪn.ər.ə.loʊˈkɔːr.tɪ.kɔɪd rɪˈzɪs.təns/ | Đề kháng mineralocorticoid |
| 339 | Pseudohypoaldosteronism (PHA) | /ˌsuː.doʊˌhaɪ.poʊ.ælˈdɑːs.tə.roʊ.nɪ.zəm/ | Giả suy aldosteron (PHA) |
| 340 | Salt wasting | /sɔːlt ˈweɪ.stɪŋ/ | Mất muối |
| 341 | Epithelial sodium channel (ENaC) | /ˌep.əˈθiː.li.əl ˈsoʊ.di.əm ˈtʃæn.əl/ | Kênh natri biểu mô (ENaC) |
| 342 | Gordon syndrome | /ˈɡɔːr.dən ˈsɪn.droʊm/ | Hội chứng Gordon |
| 343 | Syndrome of apparent mineralocorticoid excess | /ˈsɪn.droʊm əv əˈpær.ənt ˌmɪn.ər.ə.loʊˈkɔːr.tɪ.kɔɪd ɪkˈses/ | Hội chứng thừa mineralocorticoid biểu kiến |
| 344 | Hyporeninemic hypoaldosteronism | /ˌhaɪ.poʊ.riːˌnɪn.ˈiː.mɪk ˌhaɪ.poʊ.ælˈdɑːs.tə.roʊ.nɪ.zəm/ | Hạ renin máu và hạ aldosteron máu |
| 345 | 11 β-hydroxysteroid dehydrogenase type 2 (HSD11B2) | /ɪˈlev.ən ˈbeɪ.t̬ə haɪˌdrɑːk.siˈstɪər.ɔɪd diːˌhaɪˈdrɑː.dʒə.neɪs taɪp tuː/ | 11 β-hydroxysteroid dehydrogenase loại 2 (HSD11B2) |
| 346 | Dosage-sensitive sex-reversal adrenal hypoplasia congenital critical region on the X chromosome gene 1 (DAX1) | /ˈdoʊ.sɪdʒ ˈsen.sə.t̬ɪv seks rɪˈvɜːr.səl əˈdriː.nəl ˌhaɪ.poʊˈpleɪ.ʒə kənˈdʒen.ə.t̬əl ˈkrɪt̬.ɪ.kəl ˈriː.dʒən ɑːn ði eks ˈkroʊ.mə.soʊm dʒiːn wʌn/ | Gen 1 vùng tương đồng quan trọng gây thiểu sản thượng thận bẩm sinh đảo ngược giới tính nhạy cảm với liều lượng trên nhiễm sắc thể X (DAX1) |
| 347 | Mullerian-inhibiting substance | /mjuːˈlɪər.i.ən ɪnˈhɪb.ɪ.tɪŋ ˈsʌb.stəns/ | Chất ức chế Müllerian |
| 348 | X-linked adrenal hypoplasia congenita | /eks lɪŋkt əˈdriː.nəl ˌhaɪ.poʊˈpleɪ.ʒə kənˈdʒen.ɪ.tə/ | Thiểu sản thượng thận bẩm sinh liên kết X |
| 349 | Contiguous gene deletion syndrome | /kənˈtɪɡ.ju.əs dʒiːn dɪˈliː.ʃən ˈsɪn.droʊm/ | Hội chứng xóa gen liền kề |
| 350 | Glycerol kinase deficiency (GKD) | /ˈɡlɪs.ə.rɔːl ˈkaɪ.neɪs dɪˈfɪʃ.ən.si/ | Thiếu hụt glycerol kinase (GKD) |
| 351 | Duchenne muscular dystrophy (DMD) | /duːˈʃen ˈmʌs.kjə.lɚ ˈdɪs.trə.fi/ | Loạn dưỡng cơ Duchenne (DMD) |
| 352 | Bipotential gonad | /ˌbaɪ.poʊˈten.ʃəl ˈɡoʊ.næd/ | Tuyến sinh dục lưỡng tiềm |
| 353 | Clinodactyly | /ˌklaɪ.noʊˈdæk.tə.li/ | Tật vẹo ngón |
| 354 | Hypotonia | /ˌhaɪ.poʊˈtoʊ.ni.ə/ | Giảm trương lực cơ |
| 355 | Simian creases | /ˈsɪm.i.ən kriːsɪz/ | Nếp gấp khỉ |
| 356 | Fibronectin | /ˌfaɪ.broʊˈnek.tɪn/ | Fibronectin |
| 357 | Cell proliferation | /sel proʊˌlɪf.əˈreɪ.ʃən/ | Tăng sinh tế bào |
| 358 | Hypertrophy | /haɪˈpɜːr.trə.fi/ | Phì đại |
| 359 | Gonad | /ˈɡoʊ.næd/ | Tuyến sinh dục |
| 360 | Histone | /ˈhɪs.toʊn/ | Histone |
| 361 | Deacetylation | /diː.əˌset̬.əˈleɪ.ʃən/ | Deacetyl hóa |
| 362 | Acetylation | /əˌset̬.əˈleɪ.ʃən/ | Acetyl hóa |
| 363 | Adrenal crisis | /əˈdriː.nəl ˈkraɪ.sɪs/ | Cơn khủng hoảng thượng thận |
| 364 | Urogenital ridge | /ˌjʊə.roʊˈdʒen.ə.t̬əl rɪdʒ/ | Gờ niệu sinh dục |
| 365 | SRY gene | /ˌes.ɑːrˈwaɪ dʒiːn/ | Gen SRY |
| 366 | SOX9 gene | /sɑːks naɪn dʒiːn/ | Gen SOX9 |
| 367 | WT1 gene | /ˌdʌb.əl.juː.tiːˈwʌn dʒiːn/ | Gen WT1 |
| 368 | AMH (Anti-Müllerian hormone) | /ˌeɪ.emˈeɪtʃ/ | AMH (Hormone kháng Müllerian) |
| 369 | Messenger ribonucleic acid (mRNA) | /ˈmes.ən.dʒɚ ˌraɪ.boʊ.nuːˌkliː.ɪk ˈæs.ɪd/ | Acid ribonucleic thông tin (mRNA) |
| 370 | Prolactin | /proʊˈlæk.tɪn/ | Prolactin |
| 371 | Phosphodiesterases | /ˌfɑːs.foʊ.daɪˈes.tə.reɪ.sɪz/ | Phosphodiesterase |
| 372 | A-kinase-anchoring proteins | /eɪ ˈkaɪ.neɪs ˈæŋ.kər.ɪŋ ˈproʊ.tiːnz/ | Protein neo A-kinase |
| 373 | Ubiquitination | /juːˌbɪk.wɪ.tɪˈneɪ.ʃən/ | Ubiquitin hóa |
| 374 | Somatostatin | /ˌsoʊ.mə.toʊˈstæt.ɪn/ | Somatostatin |
| 375 | Oxytocin | /ˌɑːk.siˈtoʊ.sɪn/ | Oxytocin |
| 376 | Glucagon-like peptide-1 | /ˈɡluː.kə.ɡɑːn laɪk ˈpep.taɪd wʌn/ | Glucagon-like peptide-1 |
| 377 | Orexin | /ɔːˈrek.sɪn/ | Orexin |
| 378 | Mineralocorticoid | /ˌmɪn.ər.ə.loʊˈkɔːr.tɪ.kɔɪd/ | Mineralocorticoid |
| 379 | Androgen | /ˈæn.droʊ.dʒən/ | Androgen |
| 380 | Estrogen | /ˈes.trə.dʒən/ | Estrogen |
| 381 | Corticotropin-releasing factor | /ˌkɔːr.tɪ.koʊˈtroʊ.pɪn rɪˈliː.sɪŋ ˈfæk.tɚ/ | Yếu tố giải phóng corticotropin |
| 382 | Testis | /ˈtes.tɪs/ | Tinh hoàn |
| 383 | Ovary | /ˈoʊ.vər.i/ | Buồng trứng |
| 384 | Kidney | /ˈkɪd.ni/ | Thận |
| 385 | Prostate | /ˈprɑː.steɪt/ | Tuyến tiền liệt |
| 386 | Bladder | /ˈblæd.ɚ/ | Bàng quang |
| 387 | Lung | /lʌŋ/ | Phổi |
| 388 | Brain | /breɪn/ | Não |
| 389 | Uterus | /ˈjuː.t̬ɚ.əs/ | Tử cung |
| 390 | Epididymis | /ˌep.əˈdɪd.ə.mɪs/ | Mào tinh |
| 391 | Adrenal cortices | /əˈdriː.nəl ˈkɔːr.tə.siːz/ | Vỏ thượng thận |
| 392 | Phosphatase | /ˈfɑːs.fə.teɪs/ | Phosphatase |
| 393 | Spontaneous abortion | /spɑːnˈteɪ.ni.əs əˈbɔːr.ʃən/ | Sẩy thai tự nhiên |
| 394 | Polyhydramnios | /ˌpɑː.li.haɪˈdræm.ni.ɑːs/ | Đa ối |
| 395 | Salivary gland | /ˈsæl.ə.ver.i ɡlænd/ | Tuyến nước bọt |
| 396 | Sweat gland | /swet ɡlænd/ | Tuyến mồ hôi |
| 397 | Cortisone | /ˈkɔːr.t̬ə.zoʊn/ | Cortisone |
| 398 | Aldosterone synthase | /ælˈdɑː.stə.roʊn ˈsɪn.θeɪs/ | Aldosterone synthase |
| 399 | Cleft palate | /kleft ˈpæl.ət/ | Hở hàm ếch |
| 400 | Growth plate | /ɡroʊθ pleɪt/ | Đĩa tăng trưởng |
| 401 | Apoptosis | /ˌæp.əpˈtoʊ.sɪs/ | Apoptosis (Chết tế bào theo chương trình) |
| 402 | Endochondral bone growth | /ˌen.doʊˈkɑːn.drəl boʊn ɡroʊθ/ | Tăng trưởng xương sụn trong |
| 403 | Bone mineral density | /boʊn ˈmɪn.ər.əl ˈden.sə.t̬i/ | Mật độ khoáng của xương |
| 404 | Periodontal disease | /ˌper.i.oʊˈdɑːn.t̬əl dɪˈziːz/ | Bệnh nha chu |
| 405 | Psoriasis | /səˈraɪ.ə.sɪs/ | Bệnh vẩy nến |
| 406 | Tuberculosis | /tuːˌbɜːr.kjəˈloʊ.sɪs/ | Bệnh lao |
| 407 | Leprosy | /ˈlep.rə.si/ | Bệnh phong |
| 408 | Triglyceride | /traɪˈɡlɪs.ə.raɪd/ | Triglyceride |
| 409 | Hematologic | /ˌhiː.mə.t̬əˈlɑː.dʒɪk/ | Huyết học |
| 410 | IPF-1/PDX-1 gene | /… dʒiːn/ | Gen IPF-1/PDX-1 |
| 411 | HNF1A gene | /… dʒiːn/ | Gen HNF1A |
| 412 | Suprachiasmatic nuclei | /ˌsuː.prəˌkaɪ.æzˈmæt̬.ɪk ˈnuː.kli.aɪ/ | Nhân trên chéo |
| 413 | Neutrophils | /ˈnuː.trə.fɪlz/ | Bạch cầu trung tính |
| 414 | Epithelial cells | /ˌep.əˈθiː.li.əl selz/ | Tế bào biểu mô |
| 415 | Visceral obesity | /ˈvɪs.ər.əl oʊˈbiː.sə.t̬i/ | Béo phì nội tạng |
| 416 | Hyperlipidemia | /ˌhaɪ.pɚˌlɪp.ɪˈdiː.mi.ə/ | Tăng lipid máu |
| 417 | Testicular cancer | /tesˈtɪk.jə.lɚ ˈkæn.sɚ/ | Ung thư tinh hoàn |
| 418 | Liver cancer | /ˈlɪv.ɚ ˈkæn.sɚ/ | Ung thư gan |
| 419 | Laryngeal cancer | /ləˈrɪn.dʒi.əl ˈkæn.sɚ/ | Ung thư thanh quản |
| 420 | Alopecia areata | /ˌæl.əˈpiː.ʃi.ə ˌer.iˈɑː.t̬ə/ | Rụng tóc từng mảng |
| 421 | Vitiligo | /ˌvɪt̬.ɪˈlaɪ.ɡoʊ/ | Bạch biến |
| 422 | Primary biliary cirrhosis | /ˈpraɪ.mer.i ˈbɪl.i.er.i səˈroʊ.sɪs/ | Xơ gan mật nguyên phát |
| 423 | Arthritis | /ɑːrˈθraɪ.t̬ɪs/ | Viêm khớp |
| 424 | Nephritis | /nəˈfraɪ.t̬ɪs/ | Viêm thận |
| 425 | Ataxia-telangiectasia | /əˈtæk.si.ə telˌæn.dʒi.ekˈteɪ.ʒə/ | Thất điều-giãn mạch |
| 426 | Hodgkin disease | /ˈhɑːdʒ.kɪn dɪˈziːz/ | Bệnh Hodgkin |
| 427 | Postprandial hyperglycemia | /ˌpoʊstˈpræn.di.əl ˌhaɪ.pɚ.ɡlaɪˈsiː.mi.ə/ | Tăng đường huyết sau ăn |
| 428 | Fasting hypoglycemia | /ˈfæs.tɪŋ ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết lúc đói |
| 429 | STAT5b mutations | /stæt faɪv biː mjuːˈteɪ.ʃənz/ | Đột biến STAT5b |
| 430 | Neurocognitive delay | /ˌnʊər.oʊˈkɑːɡ.nə.t̬ɪv dɪˈleɪ/ | Chậm phát triển thần kinh nhận thức |
| 431 | Lung fibrosis | /lʌŋ faɪˈbroʊ.sɪs/ | Xơ phổi |
| 432 | Lymphoid interstitial pneumonia | /ˈlɪm.fɔɪd ˌɪn.t̬ɚˈstɪʃ.əl nuːˈmoʊ.ni.ə/ | Viêm phổi kẽ, bạch huyết |
| 433 | Eczema | /ˈek.sə.mə/ | Chàm |
| 434 | T-cell lymphopenia | /tiː sel ˌlɪm.foʊˈpiː.ni.ə/ | Giảm tế bào lympho T |
| 435 | Phosphorylation | /ˌfɑːs.fɔːr.əˈleɪ.ʃən/ | Sự phosphoryl hóa |
| 436 | Proteolytic cleavage | /ˌproʊ.t̬i.oʊˈlɪt̬.ɪk ˈkliː.vɪdʒ/ | Sự cắt bỏ bởi protease |
| 437 | Nonsense-mediated mRNA decay | /ˈnɑːn.sens ˈmiː.di.eɪ.t̬ɪd ˌem.ɑːr.enˈeɪ dɪˈkeɪ/ | Sự thoái biến mRNA qua trung gian vô nghĩa |
| 438 | Thyroiditis | /ˌθaɪ.rɔɪˈdaɪ.t̬ɪs/ | Viêm tuyến giáp |
| 439 | Celiac disease | /ˈsiː.li.æk dɪˈziːz/ | Bệnh celiac |
| 440 | Autoimmune polyglandular syndrome | /ˌɔː.t̬oʊ.ɪˈmjuːn ˌpɑː.liˈɡlæn.dʒə.lɚ ˈsɪn.droʊm/ | Hội chứng đa tuyến tự miễn |
| 441 | Cardiac arrest | /ˈkɑːr.di.æk əˈrest/ | Ngừng tim |
| 442 | Cardiomyopathy | /ˌkɑːr.di.oʊ.maɪˈɑː.pə.θi/ | Bệnh cơ tim |
| 443 | Optic neuropathy | /ˈɑːp.tɪk nʊˈrɑː.pə.θi/ | Bệnh thần kinh thị giác |
| 444 | Hepatobiliary abnormalities | /həˌpæt̬.oʊˈbɪl.i.er.i ˌæb.nɔːrˈmæl.ə.t̬iz/ | Bất thường gan mật |
| 445 | Haploinsufficiency | /ˌhæp.loʊˌɪn.səˈfɪʃ.ən.si/ | Thiếu hụt đơn bội |
| 446 | Syntaxin-16 (STX16) | /sɪnˈtæk.sɪn sɪksˈtiːn/ | Syntaxin-16 (STX16) |
| 447 | Hypermagnesemia | /ˌhaɪ.pɚ.mæɡ.nəˈsiː.mi.ə/ | Tăng magiê máu |
| 448 | Hypertriglyceridemia | /ˌhaɪ.pɚ.traɪˌɡlɪs.ər.ɪˈdiː.mi.ə/ | Tăng triglyceride máu |
| 449 | Hepatic steatosis | /həˈpæt̬.ɪk ˌstiː.əˈtoʊ.sɪs/ | Gan nhiễm mỡ |
| 450 | Single-nucleotide polymorphisms (SNPs) | /ˈsɪŋ.ɡəl ˈnuː.kli.ə.taɪd ˌpɑː.liˈmɔːr.fɪ.zəmz/ | Đa hình đơn nucleotide (SNP) |
| 451 | Promoter region | /prəˈmoʊ.t̬ɚ ˈriː.dʒən/ | Vùng promoter |
| 452 | Transcriptional activity | /trænsˈkrɪp.ʃən.əl ækˈtɪv.ə.t̬i/ | Hoạt tính phiên mã |
| 453 | Cosegregated | /ˌkoʊˈseɡ.rə.ɡeɪ.t̬ɪd/ | Cùng phân ly |
| 454 | Transversion | /trænsˈvɜːr.ʒən/ | Chuyển vị |
| 455 | Ketosis | /kiːˈtoʊ.sɪs/ | Nhiễm ceton |
| 456 | Insulin-like growth factor-1 (IGF-1) | /ˈɪn.sə.lɪn laɪk ɡroʊθ ˈfæk.tɚ wʌn/ | Yếu tố tăng trưởng giống insulin-1 (IGF-1) |
| 457 | Splice site | /splaɪs saɪt/ | Vị trí nối |
| 458 | Microdeletions | /ˌmaɪ.kroʊ.dɪˈliː.ʃənz/ | Vi xóa |
| 459 | Frameshift | /ˈfreɪm.ʃɪft/ | Dịch khung |
| 460 | Missense | /ˈmɪs.sens/ | Sai nghĩa |
| 461 | Nonsense | /ˈnɑːn.sens/ | Vô nghĩa |
| 462 | Pit-1 binding sites | /pɪt wʌn ˈbaɪn.dɪŋ saɪts/ | Vị trí gắn Pit-1 |
| 463 | LDL (low-density lipoprotein) | /ˌel.diːˈel/ | LDL (lipoprotein mật độ thấp) |
| 464 | HDL (high-density lipoprotein) | /ˌeɪtʃ.diːˈel/ | HDL (lipoprotein mật độ cao) |
| 465 | Carotid intima-media thickness | /kəˈrɑː.t̬ɪd ˈɪn.tə.mə ˈmiː.di.ə ˈθɪk.nəs/ | Độ dày nội trung mạc động mạch cảnh |
| 466 | Atherosclerotic plaques | /ˌæθ.ə.roʊ.skləˈrɑː.t̬ɪk plæks/ | Mảng xơ vữa |
| 467 | Insulin resistance | /ˈɪn.sə.lɪn rɪˈzɪs.təns/ | Kháng insulin |
| 468 | C-peptide | /ˈsiː ˈpep.taɪd/ | C-peptide |
| 469 | Hypophosphatemia | /ˌhaɪ.poʊˌfɑːs.fəˈtiː.mi.ə/ | Hạ phosphat máu |
| 470 | Osteomalacia | /ˌɑːs.ti.oʊ.məˈleɪ.ʃə/ | Nhuyễn xương |
| 471 | Rib cage deformity | /rɪb keɪdʒ dɪˈfɔːr.mə.t̬i/ | Biến dạng lồng ngực |
| 472 | Tetany | /ˈtet̬.ə.ni/ | Co cứng |
| 473 | Muscle cramps | /ˈmʌs.əl kræmps/ | Chuột rút |
| 474 | Seizures | /ˈsiː.ʒɚz/ | Co giật |
| 475 | Adrenal progenitor cell | /əˈdriː.nəl proʊˈdʒen.ə.t̬ɚ sel/ | Tế bào tiền thân thượng thận |
| 476 | Paraventricular nucleus of the hypothalamus (PVN) | /ˌpær.ə.venˈtrɪk.jə.lɚ ˈnuː.kli.əs əv ðə ˌhaɪ.poʊˈθæl.ə.məs/ | Nhân cạnh não thất của vùng dưới đồi (PVN) |
| 477 | Prolactin | /proʊˈlæk.tɪn/ | Prolactin |
| 478 | Glucagon | /ˈɡluː.kə.ɡɑːn/ | Glucagon |
| 479 | Dissociated bone age | /dɪˈsoʊ.ʃi.eɪ.t̬ɪd boʊn eɪdʒ/ | Tuổi xương không tương xứng |
| 480 | Anabolic effects | /ˌæn.əˈbɑː.lɪk ɪˈfekts/ | Tác dụng đồng hóa |
| 481 | Hypoglycemia | /ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết |
| 482 | Circulatory collapse | /ˈsɜːr.kjə.lə.tɔːr.i kəˈlæps/ | Trụy tuần hoàn |
| 483 | Nucleocytoplasmic transport | /ˌnuː.kli.oʊˌsaɪ.t̬oʊˈplæz.mɪk ˈtræns.pɔːrt/ | Vận chuyển nhân-bào tương |
| 484 | Oxidative stress | /ˌɑːk.sɪˈdeɪ.t̬ɪv stres/ | Stress oxy hóa |
| 485 | Body mass index (BMI) | /ˈbɑː.di mæs ˈɪn.deks/ | Chỉ số khối cơ thể (BMI) |
| 486 | Respiratory quotient | /ˈres.pɚ.ə.tɔːr.i ˈkwoʊ.ʃənt/ | Thương số hô hấp |
| 487 | Codominant inheritance | /ˌkoʊˈdɑː.mə.nənt ɪnˈher.ɪ.təns/ | Di truyền đồng trội |
| 488 | Single-chain | /ˈsɪŋ.ɡəl tʃeɪn/ | Chuỗi đơn |
| 489 | Two-subunit | /tuː ˈsʌbˌjuː.nɪt/ | Hai tiểu đơn vị |
| 490 | Disulfide bonds | /daɪˈsʌl.faɪd bɑːndz/ | Liên kết disulfide |
| 491 | Intrauterine | /ˌɪn.trəˈjuː.t̬ɚ.ɪn/ | Trong tử cung |
| 492 | Germline mutations | /ˈdʒɜːrm.laɪn mjuːˈteɪ.ʃənz/ | Đột biến dòng mầm |
| 493 | Compensated hypothyroidism | /ˈkɑːm.pən.seɪ.t̬ɪd ˌhaɪ.poʊˈθaɪ.rɔɪ.dɪ.zəm/ | Suy giáp bù |
| 494 | Bilobar thyroid gland | /baɪˈloʊ.bɑːr ˈθaɪ.rɔɪd ɡlænd/ | Tuyến giáp hai thùy |
| 495 | Peritubular | /ˌper.ɪˈtuː.bjə.lɚ/ | Quanh ống |
| 496 | Hypervolemic hypertension | /ˌhaɪ.pɚ.voʊˈliː.mɪk ˌhaɪ.pɚˈten.ʃən/ | Tăng huyết áp do tăng thể tích |
| 497 | Clavicular agenesis | /kləˈvɪk.jə.lɚ eɪˈdʒen.ə.sɪs/ | Bất sản xương đòn |
| 498 | Metacarpal fusion | /ˌmet̬.əˈkɑːr.pəl ˈfjuː.ʒən/ | Hợp nhất xương bàn tay |
| 499 | Chronic diarrhea | /ˈkrɑː.nɪk ˌdaɪ.əˈriː.ə/ | Tiêu chảy mãn tính |
| 500 | Metaphyses | /məˈtæf.ə.siːz/ | Hành xương |
| 501 | Diaphyses | /daɪˈæf.ə.siːz/ | Thân xương |