
Sperling Nội tiết học Nhi khoa, Ấn bản thứ 5 – Biên dịch: Ths.Bs. Lê Đình Sáng
Sperling Pediatric Endocrinology, Fifth Edition
Tác giả: Sperling, Mark A., MD – Nhà xuất bản: Elsevier Inc.
PHẦN III. NỘI TIẾT HỌC TRẺ EM VÀ THANH THIẾU NIÊN
Chương 23. Hạ đường huyết ở trẻ nhỏ và trẻ em
Disorders of Mineral Metabolism II. Abnormalities of Mineral Homeostasis in the Newborn, Infant, Child, and Adolescent
Allen W. Root; Michael A. Levine
Sperling Pediatric Endocrinology, 20, 705-813
MỤC LỤC CHƯƠNG
|
GIỚI THIỆU
Trong điều kiện sinh lý, glucose là nhiên liệu bắt buộc cho não bộ. Não không thể tự tổng hợp glucose cũng như không thể dự trữ glycogen quá 20 phút; do đó, sự sống của não đòi hỏi một nguồn cung cấp glucose liên tục. Não có thể sử dụng các nhiên liệu thay thế từ tuần hoàn, miễn là nồng độ của chúng đủ cao để đi vào não với lượng đủ; ví dụ, thể ceton tăng cao khi nhịn đói kéo dài hoặc lactate trong lúc vận động gắng sức và trong bệnh dự trữ glycogen (GSD) type 1 không được điều trị. Việc vận chuyển glucose từ máu vào não qua trung gian chất vận chuyển glucose 1 (GLUT-1) là một hàm số trực tiếp của nồng độ glucose huyết tương động mạch, và các cơ chế sinh lý bình thường duy trì glucose huyết tương ở mức đảm bảo cung cấp đủ glucose cho não. Ở nồng độ glucose huyết tương sinh lý (70-100 mg/dL, tương đương 3,9-5,6 mmol/L), tốc độ vận chuyển glucose từ máu vào não vượt quá tốc độ chuyển hóa glucose của não. Tuy nhiên, ở nồng độ glucose huyết tương thấp hơn 54 mg/dL (3,0 mmol/L), tốc độ chuyển hóa glucose của não giảm; và ở nồng độ glucose huyết tương thấp hơn nữa, suy chức năng não xảy ra, và hạ đường huyết sâu và kéo dài gây tổn thương não vĩnh viễn và cuối cùng là chết não.
Ngoài giai đoạn sơ sinh và đầu thời kỳ nhũ nhi, hạ đường huyết không phổ biến và thường do một rối loạn mắc phải của hệ nội tiết, nhịn đói kéo dài ở những cá nhân nhạy cảm (ví dụ, một bệnh lý đường tiêu hóa xen kẽ), các bất thường bẩm sinh như tăng insulin máu (HI) hoặc các lỗi bẩm sinh của chuyển hóa, hoặc phơi nhiễm tình cờ và hiếm khi là cố ý với thuốc hoặc độc tố. Khi khoảng cách giữa các cữ bú tăng lên ở trẻ đang lớn, các quá trình nội tiết và chuyển hóa sinh lý bình thường đảm bảo duy trì đường huyết ổn định; tuy nhiên, hạ đường huyết có thể biểu hiện lần đầu tiên ở giai đoạn sau của thời kỳ nhũ nhi hoặc đầu thời thơ ấu khi có các khiếm khuyết bẩm sinh nhẹ của các hệ thống này. Ngược lại, hạ đường huyết xuất hiện ở trẻ lớn và người lớn thường do một rối loạn mắc phải.
Chương này mô tả một phương pháp tiếp cận chẩn đoán dựa trên việc xác định nguyên nhân cụ thể của sự thất bại trong việc duy trì cân bằng nội môi glucose bình thường. Thông tin chẩn đoán quan trọng thường được thu thập tốt nhất từ các mẫu máu và nước tiểu (được gọi là các mẫu xét nghiệm trọng yếu) lấy tại thời điểm hạ đường huyết và ngay trước khi điều trị đảo ngược tình trạng hạ đường huyết.
SỰ PHÁT TRIỂN SINH LÝ CỦA CHUYỂN HÓA GLUCOSE TRONG GIAI ĐOẠN NHŨ NHI VÀ TRẺ EM
Sản xuất và sử dụng Glucose
Tốc độ dòng glucose vào và ra khỏi tuần hoàn thường được điều hòa chặt chẽ và cân bằng glucose toàn thân được duy trì, đồng thời đảm bảo cung cấp glucose liên tục cho não. Sau một bữa ăn chứa carbohydrate điển hình, sự gia tăng tiết insulin (cùng với việc ức chế tiết hormone đối kháng) dẫn đến việc xử lý nhanh chóng glucose ăn vào, hoặc cho nhu cầu năng lượng ngay lập tức hoặc lưu trữ qua việc lắng đọng glycogen và chuyển đổi thành chất béo, dẫn đến việc phục hồi nồng độ glucose huyết tương về mức cơ bản trong vòng 2 đến 3 giờ. Bier và cộng sự đã chỉ ra rằng ở trẻ em, từ trẻ sinh non đến 6 tuổi, tốc độ sản xuất glucose trung bình là 5 đến 8 mg/kg/phút; sau đó, sản xuất glucose theo trọng lượng cơ thể giảm dần về giá trị trung bình ở người lớn là 2,3 mg/kg/phút. Hơn nữa, tốc độ sản xuất glucose tương quan tuyến tính với trọng lượng não ước tính ở mọi lứa tuổi. Vì não chiếm phần lớn lượng glucose sử dụng hàng ngày, không có gì ngạc nhiên khi tốc độ sản xuất glucose của người lớn đạt được vào giữa thời thơ ấu (trọng lượng cơ thể ~30 kg), khi não của trẻ nặng khoảng 90% so với não người lớn (Hình 23.1).
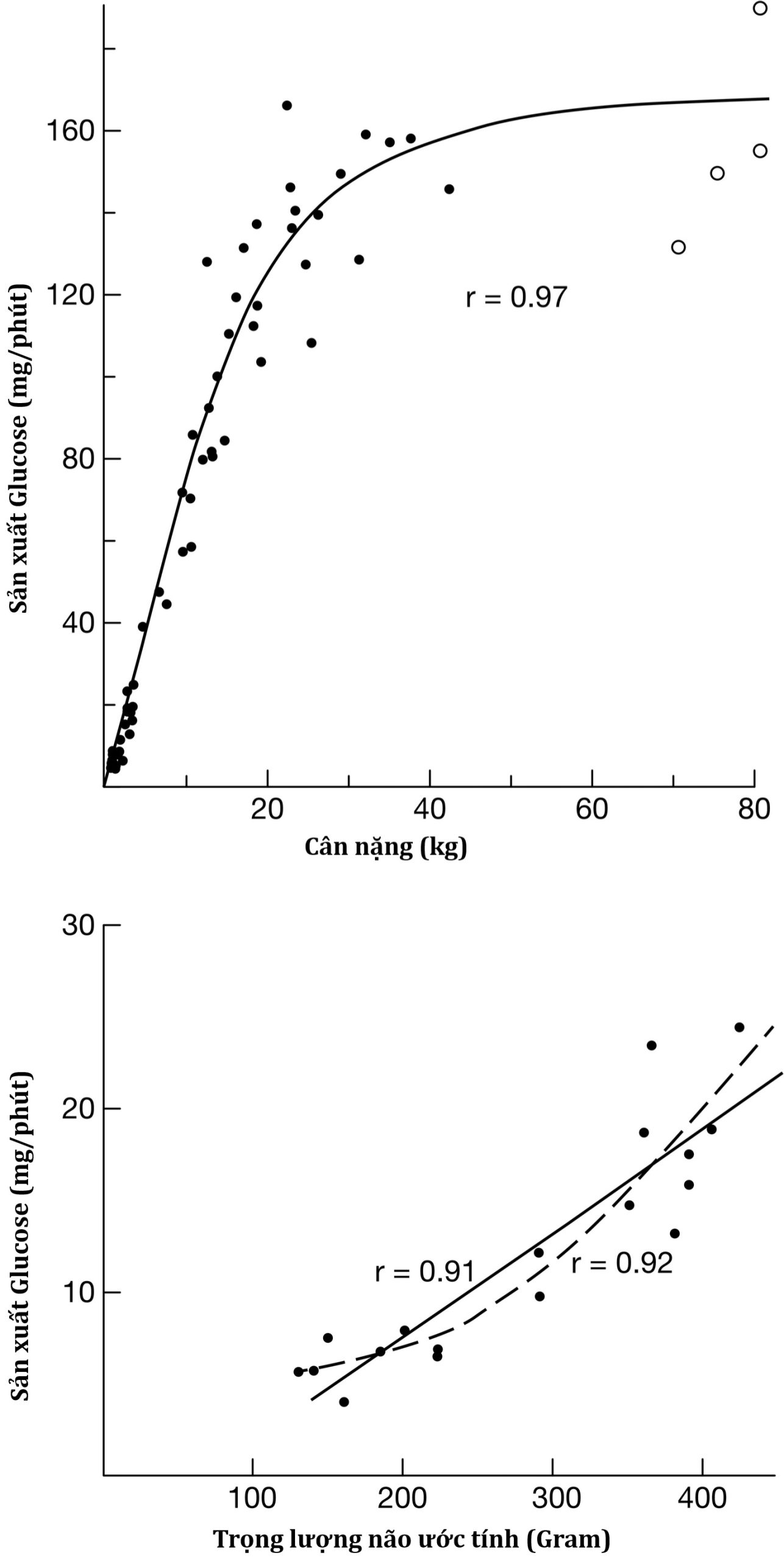
Hình 23.1 Sản xuất glucose theo trọng lượng cơ thể (trên) và trọng lượng não ước tính (dưới). Lưu ý sự thay đổi độ dốc ở khoảng 30 đến 40 kg trọng lượng cơ thể, khi sự phát triển của não đã hoàn tất.
Khi nhịn đói, ban đầu não sử dụng gần như hoàn toàn glucose được cung cấp từ sự kết hợp của quá trình ly giải glycogen và tân tạo glucose ở gan. Khi kho dự trữ glycogen ở gan giảm, quá trình ly giải lipid ở mô mỡ được kích hoạt để tăng cường cung cấp các acid béo tự do (FFA) làm nhiên liệu cho các mô ngoại vi, chẳng hạn như cơ, và cho quá trình sinh ceton ở gan, tạo ra các thể ceton làm nhiên liệu cho não và thay thế một phần việc sử dụng glucose. Lưu ý rằng thời gian để đạt đến giai đoạn tăng ceton máu khi nhịn đói ở trẻ sơ sinh và trẻ nhỏ ngắn hơn đáng kể so với người lớn, do tỷ lệ não so với khối lượng cơ lớn hơn. Như thể hiện trong Hình 23.2, sự thay đổi nguồn nhiên liệu cho não được phản ánh qua sự thay đổi nồng độ trong huyết tương của các nhiên liệu chuyển hóa chính. Khối lượng cơ nhỏ hơn đáng kể của trẻ sơ sinh và trẻ nhỏ so với khối lượng não hạn chế khả năng chịu đựng việc nhịn đói kéo dài của chúng. Vì sự phát triển của não gần như hoàn tất vào 10 đến 12 tuổi, glucose huyết tương có thể được duy trì trên 70 mg/dL (3,9 mmol/L) trong thời gian nhịn đói ngày càng dài hơn ở thanh thiếu niên và người lớn. Tuy nhiên, hầu hết trẻ sơ sinh đủ tháng, từ khoảng 1 tuần đến 1 tuổi, có thể chịu đựng nhịn đói trong 15 đến 18 giờ trước khi glucose huyết tương giảm xuống dưới 70 mg/dL (3,9 mmol/L) và đến 1 tuổi, hầu hết trẻ em bình thường có thể nhịn đói tới 24 giờ. Đến 5 tuổi, trẻ có thể chịu được nhịn đói tới 36 giờ, và hầu hết người lớn có thể duy trì glucose huyết tương khi đói trên 70 mg/dL (3,9 mmol/L) trong 48 đến 72 giờ (Hình 23.3). Khi hạ đường huyết được gây ra bởi việc nhịn đói trong thời gian ngắn hơn so với dự kiến theo tuổi của trẻ, cần xem xét khả năng có một rối loạn hạ đường huyết tiềm ẩn.
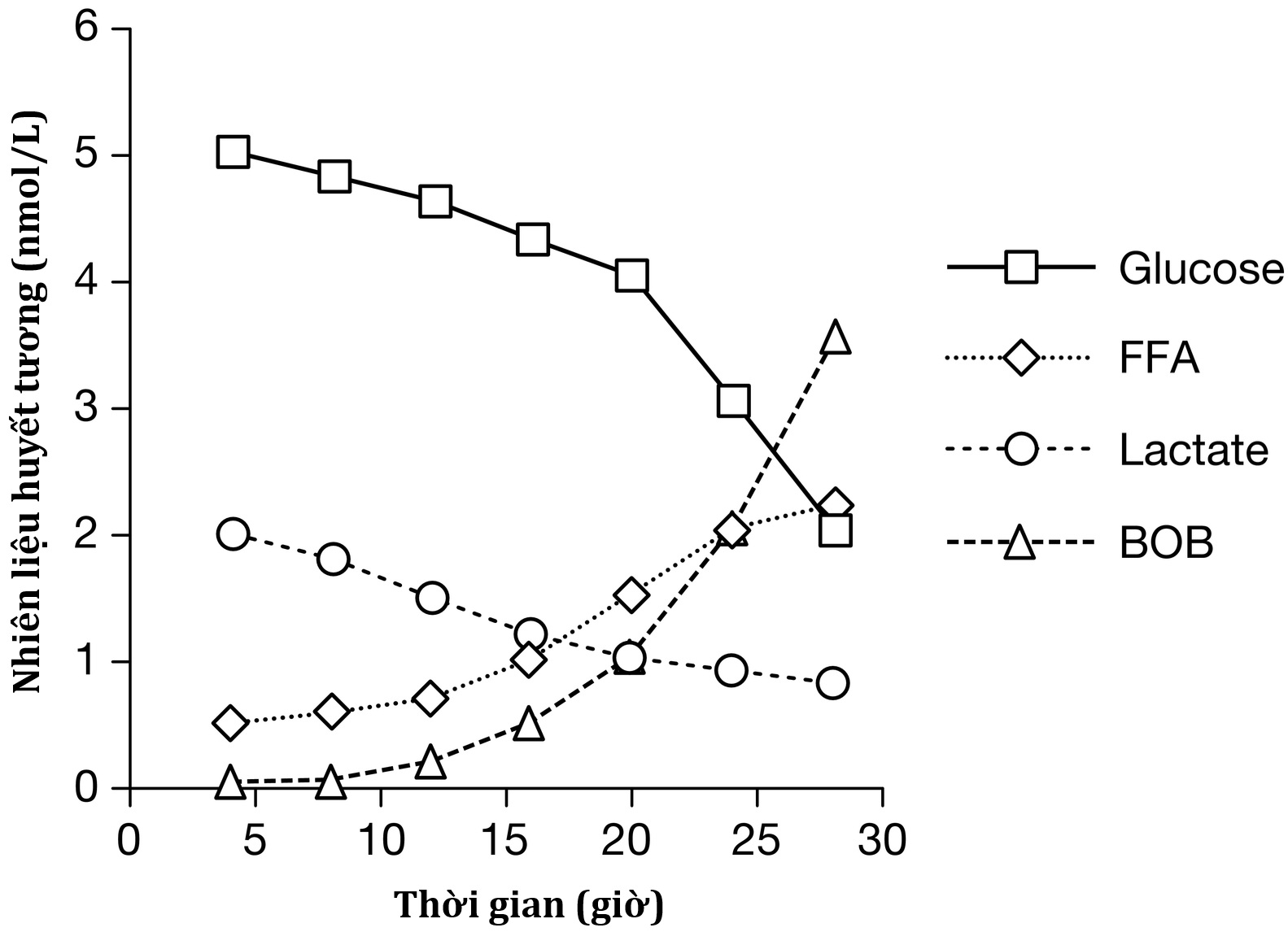
Hình 23.2 Những thay đổi nồng độ trong huyết tương của glucose và các nhiên liệu chuyển hóa chính trong thời gian nhịn đói ở một đứa trẻ bình thường. Lưu ý rằng nồng độ glucose huyết tương giảm về phía giá trị hạ đường huyết sau 24 giờ, khi dự trữ glycogen gan cạn kiệt. Nồng độ lactate, một cơ chất tân tạo glucose tiêu biểu, giảm dần trong quá trình nhịn đói. Cuối giai đoạn nhịn đói, nồng độ acid béo tự do trong huyết tương tăng lên khi quá trình ly giải lipid được kích hoạt—tiếp theo là sự gia tăng beta-hydroxybutyrate khi tốc độ oxy hóa acid béo ở gan và quá trình sinh ceton tăng lên. BOB, beta-hydroxybutyrate; FFA, acid béo tự do.
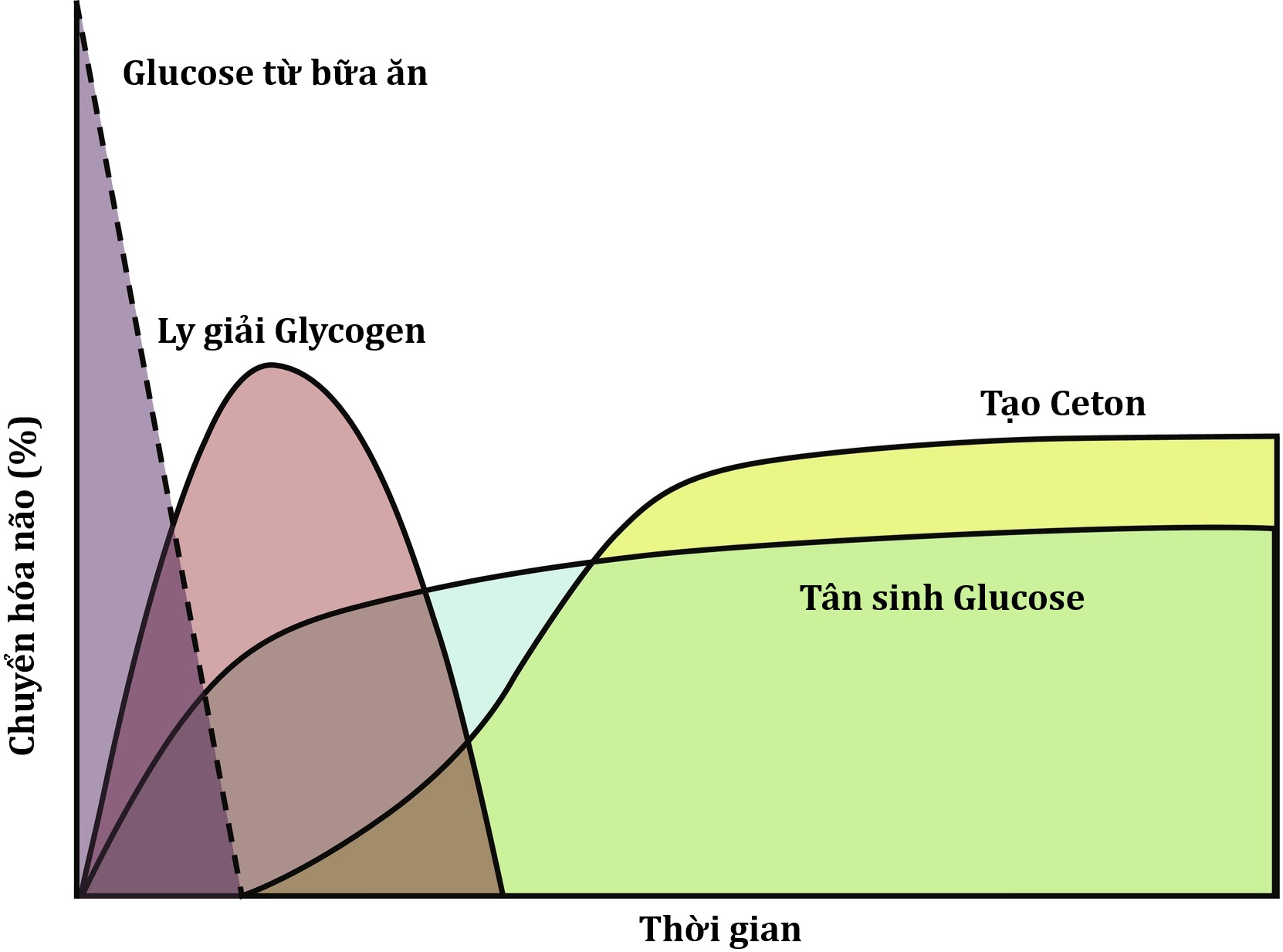
Hình 23.3 Sự thay đổi nguồn nhiên liệu trong quá trình nhịn đói. Trẻ 1 tháng tuổi—nhịn đói 14 đến 18 giờ; trẻ 1 đến 5 tuổi—nhịn đói 16 đến 24 giờ; trẻ 6 đến 12 tuổi—nhịn đói 24 đến 36 giờ, người lớn—nhịn đói 30 đến 48 giờ.
Thích ứng với khoảng cách giữa các cữ bú dài hơn
Sau khi ăn, nồng độ insulin huyết tương ở trẻ em có cân nặng bình thường tăng từ mức cơ bản 3 đến 10 µU/mL lên mức đỉnh 20 đến 50 µU/mL, có tác dụng kích thích tổng hợp glycogen gan, ức chế ly giải glycogen và tân tạo glucose ở gan, và tăng cường hấp thu và sử dụng glucose ở ngoại vi (cơ và mô mỡ) (Bảng 23.1). Đồng thời, quá trình tổng hợp triglyceride được kích hoạt và quá trình ly giải lipid và sinh ceton bị ức chế. Trong trạng thái sau hấp thu, một chuỗi các phản ứng sinh lý đặc trưng tham gia vào việc ngăn ngừa hoặc điều chỉnh hạ đường huyết. Phản ứng sớm nhất đối với việc giảm nồng độ glucose huyết tương là giảm tiết insulin, khi glucose huyết tương giảm trong phạm vi sinh lý (80-85 mg/dL, tương đương 4,4-4,7 mmol/L), tiếp theo là tăng tiết các hormone đối kháng điều hòa glucose (glucagon, epinephrine, cortisol và hormone tăng trưởng [GH]), khi glucose huyết tương giảm xuống ngay dưới phạm vi sinh lý sau hấp thu (65-70 mg/dL, tương đương 3,6-3,9 mmol/L), và kích hoạt thần kinh giao cảm xảy ra ở mức 55 mg/dL (3,1 mmol/L) (Hình 23.4). Các tác động kết hợp của việc ức chế tiết insulin, tăng nồng độ các hormone đối kháng và kích hoạt thần kinh giao cảm sẽ huy động các nhiên liệu dự trữ và giảm sử dụng glucose, do đó ngăn ngừa hạ đường huyết. Với nồng độ insulin bị ức chế, sự tăng tiết glucagon và kích hoạt thần kinh giao cảm sẽ kích hoạt quá trình ly giải glycogen, giai đoạn đầu tiên trong cơ chế bảo vệ chuyển hóa chống lại hạ đường huyết. Ở trẻ sơ sinh, kho dự trữ glycogen gan có thể cung cấp glucose trong tối đa 4 giờ (xem Hình 23.3). Khi trẻ lớn lên, hàm lượng glycogen gan so với lượng glucose não sử dụng lớn hơn và có thể cung cấp glucose cho đến 8 giờ nhịn đói. Thiếu hụt glucagon đơn độc cực kỳ hiếm và, ngoại trừ trẻ em dùng thuốc chẹn beta, thiếu hụt hoạt động thần kinh giao cảm cũng hiếm không kém. Do đó, hạ đường huyết xảy ra trong vòng 2 đến 4 giờ sau bữa ăn cho thấy hoặc là một rối loạn nguyên phát của quá trình ly giải glycogen hoặc tiết insulin quá mức (rối loạn điều hòa). Khi kho dự trữ glycogen cạn kiệt, việc duy trì nồng độ glucose huyết tương phụ thuộc vào quá trình tân tạo glucose và giảm sử dụng glucose ở mô (do tăng quá trình oxy hóa FFA và các thể ceton) (xem Hình 23.3). Các tiền chất tân tạo glucose chính là các acid amin, đặc biệt là alanin và glutamin, từ cơ xương, và glycerol từ quá trình ly giải lipid ở mô mỡ (Hình 23.5). FFA trở thành nguồn nhiên liệu chính của cơ thể khi thời gian nhịn đói kéo dài hơn.
Quá trình oxy hóa acid béo trong ty thể ở gan tạo ra các thể ceton (acetoacetate và β-hydroxybutyrate [BOHB]), được não và cơ xương, bao gồm cả cơ tim, sử dụng để sản xuất năng lượng (xem Hình 23.2). Quá trình oxy hóa FFA và sử dụng thể ceton làm giảm việc sử dụng glucose ở ngoại vi, giúp đảm bảo cung cấp đủ glucose cho não và các mô chỉ có thể sử dụng glucose làm nhiên liệu (ví dụ, tế bào hồng cầu và tủy thận), đồng thời ngăn ngừa sự phân hủy quá mức protein cơ. Quá trình ly giải triglyceride trong mô mỡ được kích hoạt bởi nồng độ insulin thấp cùng với sự tăng tiết của các hormone đối kháng (đặc biệt là GH) và kích hoạt thần kinh giao cảm. Ở trẻ sơ sinh, sự gia tăng nồng độ ceton huyết tương bắt đầu trong vòng 12 đến 18 giờ nhịn đói; trong khi đó, ở trẻ lớn hơn, tình trạng tăng ceton máu đáng kể có thể không xuất hiện cho đến 18 đến 24 giờ nhịn đói. Cortisol, được tiết ra để đáp ứng với stress, làm tăng tốc quá trình tân tạo glucose và ly giải lipid và giảm sử dụng glucose.
Các khiếm khuyết trong quá trình tân tạo glucose (ví dụ, thiếu hụt fructose-1,6-bisphosphatase; xem Hình 23.5) thường chỉ biểu hiện sau khi kho dự trữ glycogen đã cạn kiệt; theo đó, hạ đường huyết thường không xảy ra ở trạng thái mới ăn no. Các rối loạn oxy hóa acid béo cũng thường biểu hiện sau khi nhịn đói kéo dài hơn. Trong vài tháng đầu đời, khoảng cách giữa các cữ bú ở trẻ sơ sinh bú mẹ theo nhu cầu dần dần tăng từ khoảng 2 đến 3 giờ lên 4 giờ hoặc hơn, và cuối cùng là 8 đến 12 giờ khi các cữ bú đêm được bỏ qua. Do đó, các rối loạn tân tạo glucose và oxy hóa acid béo hiếm khi biểu hiện dưới dạng hạ đường huyết trong giai đoạn sơ sinh khi việc cho ăn diễn ra thường xuyên; thay vào đó, chúng biểu hiện muộn hơn trong thời kỳ nhũ nhi khi khoảng cách giữa các cữ bú trở nên kéo dài hơn. Tuy nhiên, chúng có thể xuất hiện ngay sau khi sinh trước khi sữa mẹ về. Các thiếu hụt bẩm sinh hoặc mắc phải của cortisol và GH cũng có thể gây hạ đường huyết trong giai đoạn sơ sinh, nếu là bẩm sinh và nặng, hoặc muộn hơn trong thời kỳ nhũ nhi khi các giai đoạn nhịn đói dài hơn thường xảy ra. Thiếu hụt kết hợp cortisol (hormone vỏ thượng thận [ACTH]) và GH trong suy tuyến yên có thể gây ra hạ đường huyết khởi phát sớm hơn và nặng hơn so với thiếu hụt đơn độc GH hoặc cortisol.
Bảng 23.1 Điều hòa nội tiết của chuyển hóa nhiên liệu
| Insulin | Glucagon | Epinephrine | Cortisol | Hormone tăng trưởng | |
|---|---|---|---|---|---|
| Hấp thu glucose | + | + | |||
| Ly giải glycogen | – | + | + | + | + |
| Tân tạo glucose | – | + | + | + | + |
| Ly giải lipid | – | + | + | + | + |
| Sinh ceton | – | + | + | + |
+ biểu thị kích thích, – ức chế; “bao gồm cả kích hoạt hệ thần kinh giao cảm; cortisol kích thích tổng hợp glycogen, và cả cortisol và hormone tăng trưởng đều có tác dụng cho phép đối với các hiệu ứng tân tạo glucose và ly giải glycogen của glucagon và epinephrine.
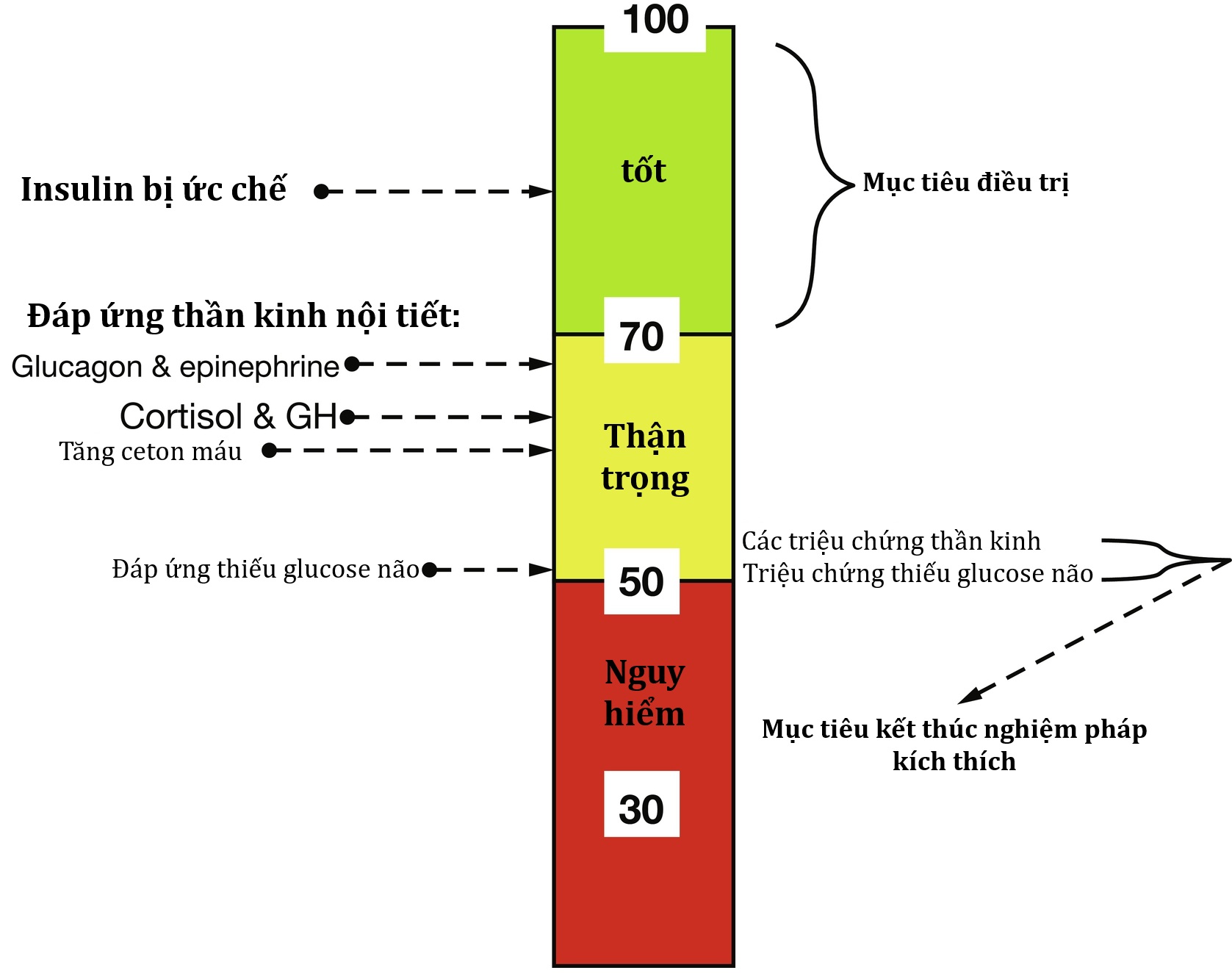
Hình 23.4 Diễn giải các mức glucose huyết tương (mg/dL) và các mục tiêu glucose huyết tương để điều trị hạ đường huyết và để kết thúc các nghiệm pháp kích thích. GH, hormone tăng trưởng.
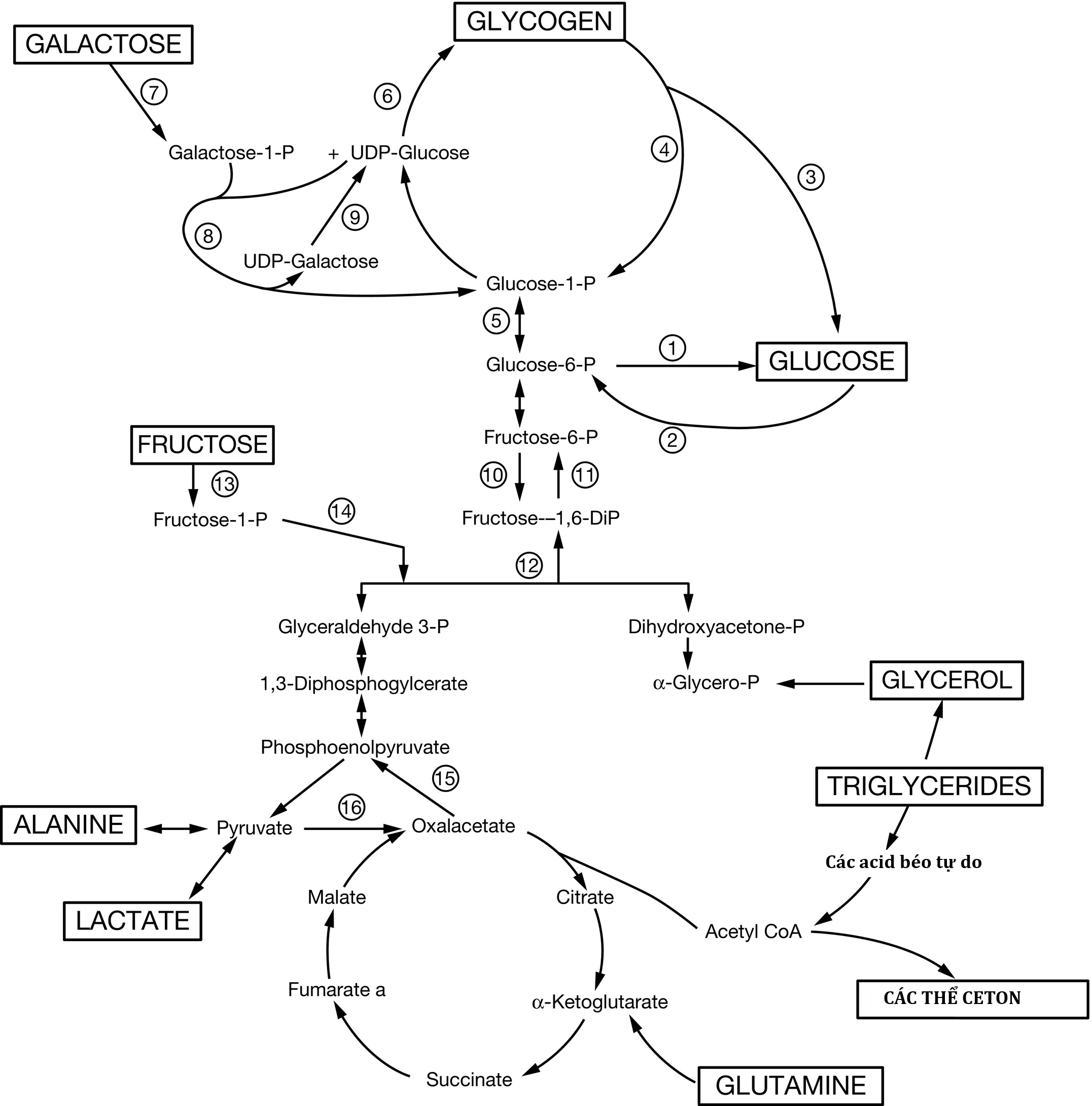
Hình 23.5 Các con đường chuyển hóa chính của chuyển hóa trung gian. Sự gián đoạn của các yếu tố trong các con đường này có thể là nguyên nhân gây ra hạ đường huyết. Không hiển thị sự kiểm soát của hormone đối với các con đường này. Các enzyme được chỉ định là: (1) glucose 6-phosphatase, (2) glucokinase, (3) amylo-1,6-glucosidase, (4) phosphorylase, (5) phosphoglucomutase, (6) glycogen synthetase, (7) galactokinase, (8) galactose 1-phosphate uridyl transferase, (9) uridine diphosphogalactose-4-epimerase, (10) phosphofructokinase, (11) fructose 1,6-diphosphatase, (12) fructose 1,6-diphosphate aldolase, (13) fructokinase, (14) fructose 1-phosphate aldolase, (15) phosphoenolpyruvate carboxykinase, và (16) pyruvate carboxylase. UDP, Uridine diphosphate.
TRIỆU CHỨNG, DẤU HIỆU VÀ ẢNH HƯỞNG CỦA HẠ ĐƯỜNG HUYẾT
Việc kiểm soát các phản ứng đối kháng ít nhất một phần là tại chỗ: nồng độ glucose thấp và sự giảm tiết insulin của tế bào β trong tiểu đảo tụy sẽ kích thích tiết glucagon. Nồng độ glucose huyết tương thấp được cảm nhận ở tĩnh mạch cửa gan, cũng như ở ruột, thể cảnh, và khoang miệng (và được truyền đến não) và cũng được cảm nhận trực tiếp ở hành não và vùng dưới đồi, khởi đầu sự gia tăng hoạt động giao cảm-thượng thận.
Tác nhân kích hoạt các phản ứng đối kháng là chính nồng độ glucose huyết tương. Tốc độ giảm glucose và mức độ insulin tuyệt đối có ít ảnh hưởng. Các triệu chứng của hạ đường huyết, phản ánh phản ứng của não đối với sự thiếu hụt glucose, đã được nghiên cứu rộng rãi và mô tả rõ ràng ở người lớn (Hộp 23.1). Các triệu chứng thần kinh tự chủ (neurogenic) chủ yếu phát sinh từ nhận thức về những thay đổi sinh lý gây ra bởi sự kích hoạt thần kinh giao cảm (không phải tủy thượng thận) và bao gồm cả các phản ứng adrenergic (run, hồi hộp, lo lắng/bồn chồn) và cholinergic (đổ mồ hôi, đói, dị cảm). Nhận biết về hạ đường huyết chủ yếu phụ thuộc vào nhận thức về các tác động trung ương và ngoại vi của các phản ứng thần kinh tự chủ (trái ngược với các phản ứng do thiếu glucose não – neuroglycopenic) đối với hạ đường huyết. Các triệu chứng thần kinh tự chủ được nhận thấy ở mức glucose huyết tương dưới 54 mg/dL (3.0 mmol/L), ngưỡng mà tại đó việc sử dụng glucose của não trở nên hạn chế. Ở trẻ lớn và người lớn, việc tìm kiếm thức ăn hoặc sự giúp đỡ là một cơ chế bảo vệ cực kỳ quan trọng chống lại tình trạng hạ đường huyết nặng hơn. Glucose huyết tương dưới 50 mg/dL (2.8 mmol/L) gây ra các biểu hiện thiếu glucose não (kết quả của việc không cung cấp đủ glucose để duy trì năng lượng cho não), chẳng hạn như suy giảm nhận thức, mất phối hợp vận động, lú lẫn, hôn mê và co giật. Hạ thân nhiệt thường xảy ra khi hạ đường huyết kéo dài ở trẻ lớn và người lớn và được cho là kết quả của một cơ chế thần kinh tự chủ. Đáng chú ý là trẻ nhỏ (6-11 tuổi) mắc đái tháo đường type 1 có khả năng phát hiện mức đường huyết thấp kém.
Ngưỡng kích hoạt cho các phản ứng thần kinh-nội tiết đã được xác định rõ nhất cho người trẻ khỏe mạnh và chỉ thay đổi nhẹ theo giới tính, tuổi tác, tập thể dục, giấc ngủ và tình trạng dinh dưỡng. Không có bằng chứng nào cho thấy định nghĩa về hạ đường huyết nên thấp hơn ở trẻ sơ sinh và trẻ nhỏ. Thực tế, cả epinephrine và GH đều được giải phóng ở mức glucose huyết tương cao hơn ở trẻ em so với người lớn, cho thấy rằng ngưỡng cho các phản ứng thần kinh-nội tiết đối với việc giảm nồng độ glucose huyết tương ở trẻ em không thấp hơn và thực sự có thể cao hơn so với người lớn. Ở các ngưỡng glucose huyết tương tương đương, nồng độ epinephrine cũng tăng rõ rệt ở trẻ em so với người lớn. Thuốc làm thay đổi các phản ứng thần kinh-nội tiết đối với hạ đường huyết, ví dụ, thuốc chẹn beta làm giảm phản ứng, trong khi caffeine làm tăng phản ứng. Việc tiếp xúc trước đó với hạ đường huyết và tăng đường huyết gây ra những thay đổi quan trọng trên lâm sàng đối với ngưỡng phản ứng thần kinh-nội tiết. Ngay cả một cơn hạ đường huyết trung bình nặng cũng có thể làm giảm hoặc hạ thấp ngưỡng kích hoạt trong 24 giờ hoặc hơn; sau khi hạ đường huyết kéo dài hoặc tái phát, các phản ứng tự chủ có thể bị suy giảm đủ để các tác động thiếu glucose não có thể là biểu hiện lâm sàng duy nhất của hạ đường huyết nặng, một tình trạng được gọi là suy giảm tự chủ liên quan đến hạ đường huyết (HAAF). HAAF đã được chứng minh ở người lớn và trẻ em mắc đái tháo đường type 1, ở người lớn không mắc đái tháo đường có u tiết insulin, và ở trẻ sơ sinh bị hạ đường huyết tái phát do tăng insulin máu bẩm sinh. Ngược lại, tăng đường huyết mạn tính có liên quan đến ngưỡng glucose cao hơn cho các phản ứng đối kháng.
Các ảnh hưởng đến nhận thức, hành vi và mức độ ý thức thường được đảo ngược hoàn toàn khi mức glucose được nâng lên, mặc dù sự suy giảm tâm lý thần kinh tinh vi có thể đo được nhiều ngày sau đó. Tuy nhiên, tình trạng thiếu glucose não nặng và kéo dài gây tổn thương não và chết tế bào thần kinh. Trong các thí nghiệm trên động vật linh trưởng, glucose huyết tương dưới 20 mg/dL (1,1 mmol/L) trong 5 đến 6 giờ chắc chắn gây ra tổn thương nghiêm trọng. Ở cả trẻ sơ sinh và người lớn, chụp cộng hưởng từ (MRI) cho thấy những thay đổi đặc trưng do tổn thương não do hạ đường huyết gây ra. Các thay đổi bệnh lý được quan sát đặc biệt ở mô vỏ não và chất trắng; trong khi tiểu não và thân não có xu hướng được bảo tồn. Suy giảm nhận thức vĩnh viễn là phổ biến ở cả trẻ em và người lớn sau các cơn hạ đường huyết nặng, tái phát.
| HỘP 23.1 Triệu chứng của Hạ đường huyết
TRIỆU CHỨNG THẦN KINH TỰ CHỦ DO KÍCH HOẠT HỆ THẦN KINH TỰ CHỦ
TRIỆU CHỨNG THIẾU GLUCOSE NÃO DO GIẢM SỬ DỤNG GLUCOSE Ở NÃO
|
ĐỊNH NGHĨA HẠ ĐƯỜNG HUYẾT
Hạ đường huyết lâm sàng được định nghĩa là nồng độ glucose huyết tương đủ thấp để gây ra các phản ứng thần kinh-nội tiết phòng vệ, gây ra các triệu chứng và/hoặc dấu hiệu, hoặc làm suy giảm chức năng não. Vì các dấu hiệu và triệu chứng không đặc hiệu, hạ đường huyết có thể khó nhận biết, và một nồng độ glucose huyết tương thấp duy nhất có thể là một sai số. Ví dụ, khi máu được lấy nhưng huyết tương không được tách ngay lập tức khỏi các thành phần tế bào, quá trình glycolysis trong hồng cầu làm cho nồng độ glucose giảm với tốc độ 6 mg/dL/giờ. Đây là một nguyên nhân phổ biến của các mức glucose thấp giả tạo được báo cáo trong các bảng xét nghiệm chuyển hóa đo tại các phòng thí nghiệm thương mại. Máy đo đường huyết tại giường, ban đầu được thiết kế để quản lý bệnh đái tháo đường, hữu ích cho mục đích sàng lọc, nhưng độ chính xác của chúng bị giới hạn ở khoảng ±10 đến 15 mg/dL (0,6-0,8 mmol/L) trong khoảng hạ đường huyết. Mức đường huyết toàn phần thấp hơn khoảng 15% so với nồng độ huyết tương, và tốt hơn là nên nhất quán tham chiếu đến nồng độ glucose huyết tương. Trước khi xác định chẩn đoán hạ đường huyết và tiến hành đánh giá chẩn đoán, cần phải xác nhận nồng độ glucose huyết tương thấp bằng phương pháp xét nghiệm lâm sàng. Vì những lý do này, các hướng dẫn nhấn mạnh giá trị của tam chứng Whipple để xác nhận hạ đường huyết: (1) các triệu chứng và/hoặc dấu hiệu phù hợp với hạ đường huyết, (2) một nồng độ glucose huyết tương thấp được ghi nhận, và (3) sự thuyên giảm của các triệu chứng và dấu hiệu khi glucose huyết tương được phục hồi về mức bình thường. Bởi vì trẻ sơ sinh và trẻ nhỏ không thể nhận biết hoặc thông báo các triệu chứng của mình một cách đáng tin cậy, việc nhận biết hạ đường huyết có thể cần được xác nhận bằng các phép đo lặp lại nồng độ glucose huyết tương và các nghiệm pháp kích thích chính thức.
Điều quan trọng cần nhận thấy là hạ đường huyết không thể được định nghĩa bằng một nồng độ glucose huyết tương cụ thể duy nhất vì ngưỡng cho các phản ứng não cụ thể đối với hạ đường huyết xảy ra trên một phạm vi nồng độ glucose huyết tương, và các ngưỡng này có thể bị thay đổi bởi sự hiện diện của các nhiên liệu thay thế, chẳng hạn như ceton, và cũng bởi tình trạng hạ đường huyết trước đó gần đây. Cũng không thể xác định một giá trị glucose huyết tương duy nhất gây tổn thương não, và mức độ tổn thương bị ảnh hưởng bởi các yếu tố khác, chẳng hạn như thời gian và mức độ hạ đường huyết, sự sẵn có của các nhiên liệu thay thế, và các sai số và yếu tố kỹ thuật tiềm ẩn dẫn đến sự không chính xác trong các phép đo glucose.
Mặc dù hạ đường huyết thường không đi kèm với các triệu chứng rõ ràng ở trẻ sơ sinh, bằng chứng về tổn thương não do hạ đường huyết kéo dài hoặc tái phát cho thấy các ngưỡng lâm sàng và mục tiêu điều trị tương tự được áp dụng bất kể tuổi tác sau giai đoạn sơ sinh ngay sau sinh (48-72 giờ). Hai ngưỡng hạ đường huyết khác nhau được khuyến nghị sử dụng trong thực hành lâm sàng. Để xác định nguyên nhân của hạ đường huyết và kết thúc một nghiệm pháp nhịn đói chẩn đoán, lấy các mẫu xét nghiệm trọng yếu khi glucose huyết tương dưới 55 mg/dL (3,1 mmol/L). Tuy nhiên, với mục đích điều trị, giới hạn dưới của phạm vi glucose huyết tương nên là 70 mg/dL (3,9 mmol/L) (xem Hình 23.4). Mục tiêu này rất quan trọng để tránh các giai đoạn đường huyết thấp có thể làm giảm các phản ứng thần kinh-nội tiết và triệu chứng đối với hạ đường huyết và dẫn đến nguy cơ cao hơn cho các đợt hạ đường huyết nặng hơn sau này.
Như đã lưu ý trước đó, tác động của các mức glucose huyết tương cụ thể lên hệ thần kinh trung ương có thể khác nhau giữa các bệnh nhân, đặc biệt là sau khi trải qua các đợt hạ đường huyết trước đó. Bởi vì không có sự tương ứng chính xác giữa nguy cơ gây hại (bao gồm cả tổn thương não) và mức độ nghiêm trọng của các triệu chứng hoặc các mức glucose huyết tương cụ thể, hạ đường huyết phải luôn được điều trị khẩn cấp.
TIẾP CẬN CHẨN ĐOÁN HẠ ĐƯỜNG HUYẾT THEO HỆ THỐNG KHI NHỊN ĐÓI
Để cung cấp điều trị thích hợp cho một rối loạn hạ đường huyết, điều cần thiết là phải thiết lập một chẩn đoán cụ thể về nguyên nhân. Bởi vì hầu hết các rối loạn hạ đường huyết, đặc biệt là ở trẻ sơ sinh và trẻ em, liên quan đến sự xáo trộn của thích ứng nhịn đói, phương pháp tiếp cận chẩn đoán tốt nhất là một nghiệm pháp nhịn đói được theo dõi chặt chẽ, vừa tái tạo tình trạng hạ đường huyết vừa khảo sát tính toàn vẹn của các hệ thống chuyển hóa và nội tiết chính được trình bày trong Hình 23.3 và 23.5 và trong Bảng 23.1. Các “hệ thống nhịn đói” này bao gồm bốn hệ thống chuyển hóa (ly giải glycogen gan, tân tạo glucose gan, ly giải lipid mô mỡ, và sinh ceton gan) và sự điều hòa của chúng bởi hệ thống nội tiết. Hệ thống nội tiết bao gồm: (1) insulin, ức chế hoạt động của cả bốn hệ thống chuyển hóa; và (2) bốn hormone đối kháng chống lại hoạt động của insulin trong các mô cụ thể: (a) glucagon tác động cấp tính lên gan để kích thích ly giải glycogen và tân tạo glucose và tạo điều kiện cho quá trình sinh ceton; (b) kích hoạt hệ thần kinh adrenergic (phản ánh qua sự gia tăng nồng độ epinephrine huyết tương) tác động cấp tính lên mô mỡ và gan để kích thích ly giải lipid, sinh ceton, ly giải glycogen, và tân tạo glucose; (c) cortisol có tác dụng lâu dài hơn cần thiết để duy trì kho dự trữ glycogen gan và thúc đẩy tân tạo glucose; và (d) GH, có tác dụng lâu dài hơn đối với khả năng ly giải lipid của mô mỡ (xem Bảng 23.1).
Hoạt động của các hệ thống chuyển hóa và nội tiết này được phản ánh bởi những thay đổi trong các nhiên liệu chuyển hóa chính trong tuần hoàn trong quá trình nhịn đói bình thường, như được trình bày trong Hình 23.2. Khi dự trữ glycogen gan cạn kiệt, glucose huyết tương giảm về mức hạ đường huyết, lactate huyết tương giảm 25% đến 50% (một dấu hiệu của tân tạo glucose tăng cường), FFA huyết tương tăng 4 đến 6 lần (một dấu hiệu của ly giải lipid), và ceton huyết tương tăng 20 đến 40 lần (một dấu hiệu của sinh ceton, thường được đo bằng BOHB là thể ceton chính). Việc đo lường các nhiên liệu này tại thời điểm hạ đường huyết (mẫu xét nghiệm trọng yếu) cung cấp thông tin quan trọng về thành phần nào của hệ thống nhịn đói chịu trách nhiệm gây ra hạ đường huyết (Hình 23.6). Ví dụ, sự tăng bất thường của lactate huyết tương cho thấy một khiếm khuyết trong tân tạo glucose; sự ức chế bất thường của BOHB, với sự gia tăng bình thường hoặc quá mức của FFA, cho thấy một khiếm khuyết trong oxy hóa acid béo; sự ức chế bất thường của cả FFA và BOHB cho thấy hoạt động của insulin quá mức; và sự giảm bình thường của lactate huyết tương đi kèm với sự gia tăng nhanh chóng của nồng độ FFA và ceton xảy ra sau một thời gian nhịn đói tương đối ngắn (tức là, hạ đường huyết do ceton) cho thấy một khiếm khuyết trong ly giải glycogen. Thiếu hụt cortisol hoặc GH thường biểu hiện bằng hạ đường huyết do ceton; tuy nhiên, trong giai đoạn sơ sinh, thiếu hụt hormone tuyến yên kết hợp, đặc biệt, có thể biểu hiện dưới dạng hạ đường huyết không do ceton, giống như HI. Thiếu hụt hoạt động thần kinh giao cảm (ví dụ, thứ phát sau thuốc chẹn beta) có thể biểu hiện bằng hạ đường huyết không do ceton do suy giảm ly giải lipid. Thiếu hụt glucagon như một bệnh nguyên phát chưa được mô tả, trong khi thiếu hụt thứ phát là phổ biến ở những bệnh nhân bị hạ đường huyết tái phát, chẳng hạn như tăng insulin máu bẩm sinh hoặc đái tháo đường type 1 (HAAF).
Các xét nghiệm bổ sung có thể được thực hiện riêng để đánh giá thêm các rối loạn cụ thể hoặc có thể được thêm vào một cách thuận tiện vào cuối nghiệm pháp nhịn đói bằng cách sử dụng máu lấy thêm cùng với mẫu xét nghiệm trọng yếu. Nồng độ insulin huyết thanh thường không đủ cao để chẩn đoán các rối loạn HI; do đó, nồng độ insulin trong giới hạn bình thường không loại trừ khả năng HI; nồng độ C-peptide huyết tương có thể phản ánh chính xác hơn sự tiết insulin của tụy vì insulin được đưa qua tĩnh mạch cửa bị gan chiết xuất một cách biến đổi, trong khi C-peptide đi qua gan mà không bị chiết xuất. tăng insulin máu có thể được xác nhận một cách thuận tiện bằng nghiệm pháp kích thích glucagon: ở trẻ em bình thường, có rất ít hoặc không có phản ứng tại thời điểm hạ đường huyết, vì kho dự trữ glycogen gan đã cạn kiệt; trong khi ở HI, có một phản ứng đường huyết tăng lớn bất thường do sự ức chế ly giải glycogen. Nồng độ cortisol hoặc GH trong huyết tương đôi khi tăng đến giá trị đủ cao trong cơn hạ đường huyết để loại trừ sự thiếu hụt của một hoặc cả hai; tuy nhiên, trước khi chẩn đoán thiếu hụt cortisol hoặc GH, các giá trị thấp phải được xác nhận bằng các nghiệm pháp kích thích đặc hiệu bổ sung cho các hormone này. Nồng độ carnitine tự do và toàn phần trong huyết tương, cùng với hồ sơ acylcarnitine huyết tương, hữu ích cho việc chẩn đoán hầu hết, nhưng không phải tất cả, các khiếm khuyết oxy hóa acid béo.
Các xét nghiệm khác có thể được xem xét nếu nghi ngờ các rối loạn cụ thể. Nồng độ amoniac huyết tương tăng 3 đến 5 lần ở những bệnh nhân mắc hội chứng tăng amoniac máu tăng insulin máu do các đột biến kích hoạt của GLUD1. Việc ức chế C-peptide huyết tương khi nồng độ insulin tăng cao có thể hữu ích trong việc chứng minh việc sử dụng insulin ngoại sinh. Nồng độ protein gắn yếu tố tăng trưởng giống insulin-1 (IGFBP-1) trong huyết tương thường tăng 5 đến 10 lần khi nồng độ insulin giảm trong quá trình nhịn đói, do đó giá trị IGFB-1 thấp hỗ trợ chẩn đoán HI. Các mẫu để xét nghiệm thuốc hoặc độc tố có thể được xem xét trong các trường hợp nghi ngờ nuốt phải thuốc hạ đường huyết đường uống, độc tố thực vật (trái ackee-ackee chưa chín, trái vải), hoặc sử dụng insulin lén lút (hạ đường huyết giả hoặc Munchausen by proxy).
Các phương pháp chi tiết để thực hiện các nghiệm pháp nhịn đói và các xét nghiệm khác hữu ích cho việc chẩn đoán hạ đường huyết được cung cấp ở cuối chương này.
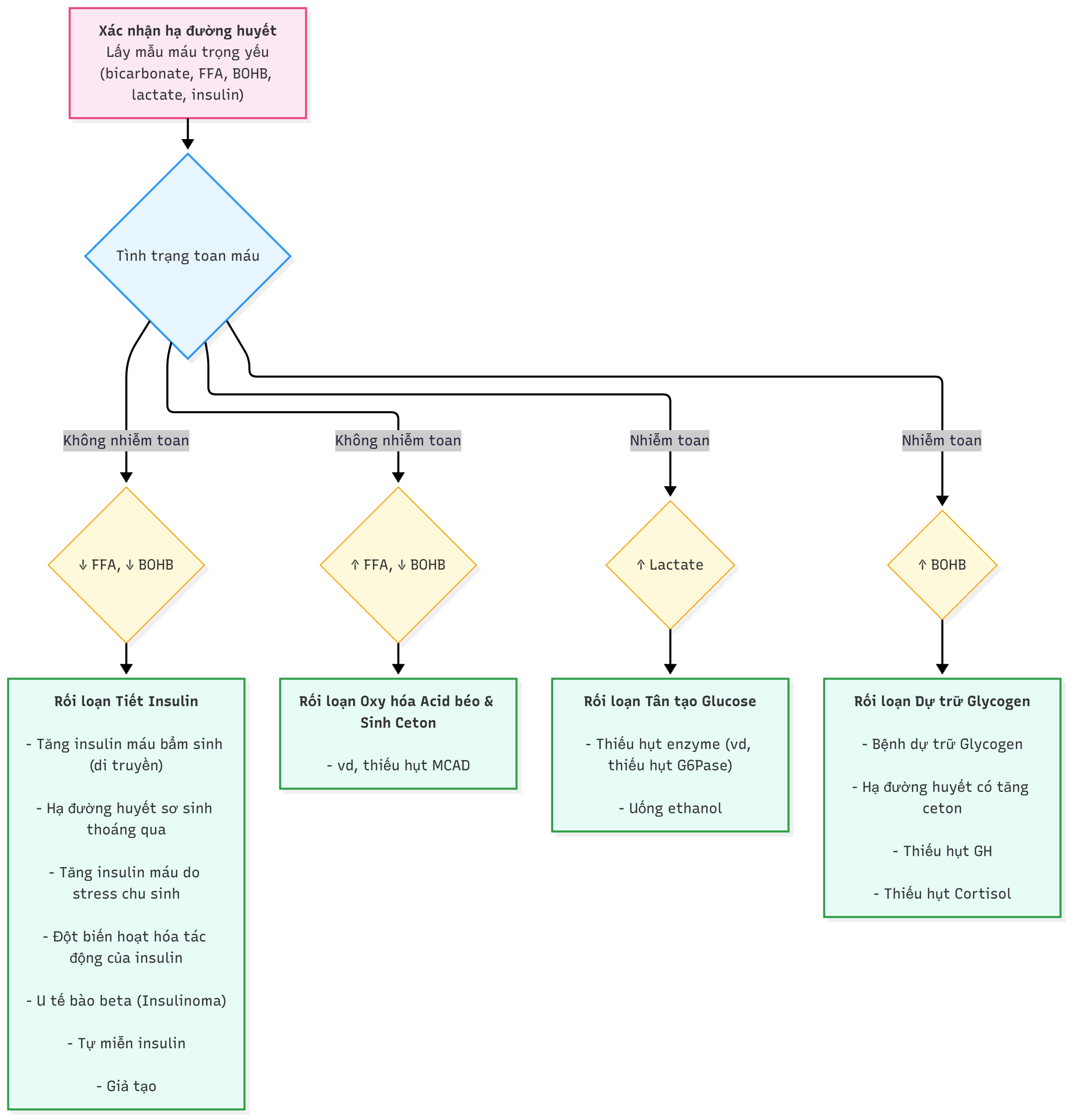
Hình 23.6 Một phương pháp tiếp cận theo lưu đồ đối với hạ đường huyết. BOHB, Beta-hydroxybutyrate; FFA, acid béo tự do; G-6-Pase, glucose 6-phosphatase; GH, hormone tăng trưởng; MCAD, medium chain acylCoA dehydrogenase.
CÁC NGUYÊN NHÂN CHÍNH GÂY HẠ ĐƯỜNG HUYẾT Ở TRẺ NHỎ VÀ TRẺ EM
Tăng insulin máu
Tăng insulin máu (HI) là dạng hạ đường huyết phổ biến nhất và nặng nhất ở trẻ sơ sinh và trẻ nhỏ, và cũng là một trong những nguyên nhân phổ biến nhất gây hạ đường huyết ở trẻ mới biết đi và trẻ lớn hơn. Hạ đường huyết do tăng insulin máu đặc biệt nguy hiểm cho não, vì sự thiếu hụt glucose không thể được bù đắp bằng việc tăng sản xuất ceton để hỗ trợ chuyển hóa não. Mặc dù nhiều trẻ sơ sinh mắc tăng insulin máu bẩm sinh nặng biểu hiện trong tuần đầu tiên của cuộc đời, các dạng di truyền nhẹ hơn của tăng insulin máu vẫn tiếp tục là một nguyên nhân quan trọng gây hạ đường huyết trong suốt thời kỳ nhũ nhi. Trẻ lớn hơn và thanh thiếu niên mắc tăng insulin máu có thể có u tuyến tụy tiết insulin mắc phải.
HI bẩm sinh có thể biểu hiện sau những tháng đầu đời khi khoảng cách giữa các cữ bú kéo dài và các cữ bú đêm được bỏ qua. Những trẻ này có thể bị lơ mơ hoặc co giật vào buổi sáng sớm và có thể có tiền sử co giật trước đó và chậm phát triển không giải thích được, hoặc hạ đường huyết trong giai đoạn sơ sinh không được đánh giá hoặc điều trị đầy đủ. Trong các dạng tăng insulin máu di truyền trội, có thể có tiền sử các triệu chứng hạ đường huyết ở cha hoặc mẹ hoặc các họ hàng khác; tuy nhiên, người mang gen thường có thể không nhận ra các triệu chứng. Như đã nêu trong Chương 7, tất cả các nguyên nhân di truyền của tăng insulin máu đều có thể biểu hiện ban đầu trong giai đoạn sơ sinh.
Chẩn đoán tăng insulin máu được gợi ý khi hạ đường huyết không đi kèm với tăng ceton máu và có thể được xác nhận khi tiêm glucagon gây ra sự gia tăng lớn (>30-40 mg/dL, tương đương 1,7-2,2 mmol/L so với ban đầu) trong nồng độ glucose huyết tương. tăng insulin máu có thể được gợi ý bởi tốc độ sử dụng glucose tăng (tốc độ truyền glucose [GIR] > 8 mg/kg/phút), nhưng điều này ít nhất quán hơn ở trẻ lớn so với trẻ sơ sinh. Một mẫu xét nghiệm trọng yếu lấy trong cơn hạ đường huyết, cho thấy nồng độ insulin hoặc C-peptide tăng, có thể là kết luận; tuy nhiên, thường không thể chứng minh rõ ràng nồng độ insulin tăng cao. Bảng 23.2 cho thấy các tiêu chuẩn chẩn đoán hạ đường huyết do tăng insulin máu. Trong các trường hợp sử dụng insulin lén lút, các xét nghiệm miễn dịch insulin tiêu chuẩn có thể không phát hiện được các chất tương tự insulin được tiêm từ bên ngoài, chẳng hạn như lispro, aspart, glargine, v.v. Trong các trường hợp nghi ngờ, phải yêu cầu các xét nghiệm cụ thể.
Bảng 23.2 Tiêu chuẩn chẩn đoán hạ đường huyết do tăng insulin máu
| Tiêu chuẩn |
|---|
| Tốc độ truyền glucose > 8 mg/kg/phút |
| Glucose huyết tương xét nghiệm ≤ 50 mg/dL (2,8 mmol/L) |
| Insulin huyết thanh có thể phát hiện được |
| C-peptide ≥ 0,5 ng/mL |
| BOHB < 1,8 mmol/L |
| FFA < 1,7 mmol/L |
| Đáp ứng với glucagon ≥ 30 mg/dL (1,7 mmol/L) |
| IGFBP-1 < 110 ng/mL |
BOHB, Beta-hydroxybutyrate; FFA, acid béo tự do; IGFBP-1, protein gắn yếu tố tăng trưởng giống insulin 1.
Tăng insulin máu do kênh Kali nhạy cảm với Adenosine Triphosphate
Đột biến trong các gen của kênh kali nhạy cảm với adenosine triphosphate (KATP) là các khiếm khuyết di truyền được xác định phổ biến nhất ở những bệnh nhân mắc tăng insulin máu bẩm sinh. Các gen ABCC8 và KCNJ11 trên nhiễm sắc thể 11p mã hóa hai tiểu đơn vị, SUR1 và Kir6.2, của kênh KATP trong màng sinh chất của tế bào beta. Dòng kali đi ra qua kênh này làm tăng phân cực màng sinh chất của tế bào beta và là một yếu tố điều hòa âm quan trọng của sự tiết insulin; các khiếm khuyết di truyền làm suy giảm hoạt động của kênh KATP và ngăn cản việc mở kênh gây ra sự khử cực liên tục của màng sinh chất và sự tiết insulin không kiểm soát. Như được mô tả trong Chương 7, có ba dạng KATP-HI riêng biệt:
- Di truyền hai alen lặn của đột biến KATP gây ra tăng insulin máu lan tỏa với khởi phát sơ sinh nặng và không đáp ứng với diazoxide.
- Đột biến kênh KATP lặn cũng có thể gây ra tăng insulin máu bẩm sinh khu trú theo cơ chế “hai đòn tấn công”, liên quan đến một đột biến KATP lặn di truyền từ cha kết hợp với một đột biến xôma trong phôi ở gen tương ứng của mẹ, dẫn đến hiện tượng đồng phân lưỡng bội đơn cha 11p (paternal 11p uniparental isodisomy) hiệu quả cho vùng chứa cả gen KATP và các gen in dấu Beckwith-Wiedemann. Vùng u tuyến tế bào tiểu đảo khu trú kết quả chứa các tế bào beta với các kênh KATP khiếm khuyết, dẫn đến HI.
- Đột biến kênh KATP di truyền trội hoạt động theo kiểu trội-âm để tạo ra tăng insulin máu lan tỏa với các kênh KATP có mức độ suy giảm chức năng kênh khác nhau. Hầu hết các đột biến KATP trội đều đáp ứng với diazoxide, nhưng cũng có các đột biến không đáp ứng với diazoxide.
Các dạng KATP-HI lan tỏa và khu trú chiếm hơn 90% các trường hợp tăng insulin máu bẩm sinh không đáp ứng với diazoxide trong giai đoạn đầu của thời kỳ nhũ nhi, và có biểu hiện lâm sàng tương tự nhau. Cả hai đều có thể cần phẫu thuật để duy trì mức glucose an toàn.
Ngoài giai đoạn sơ sinh, các đột biến KATP trội phổ biến hơn và thường đáp ứng với diazoxide.
Diazoxide (5-15 mg/kg/ngày) là thuốc đầu tay để điều trị HI, nhưng thường không hiệu quả đối với KATP-HI, vì nó hoạt động như một chất mở kênh KATP. Octreotide có thể được tiêm dưới da với liều 5 đến 15 mcg/kg/ngày. Các chất tương tự somatostatin tác dụng kéo dài hơn, như lanreotide, gần đây đã được giới thiệu, với một số thành công ở trẻ nhũ nhi lớn hơn. Thuốc chẹn kênh canxi không hiệu quả. Nếu điều trị nội khoa không thể duy trì nồng độ glucose huyết tương lớn hơn 70 mg/dL (3,9 mmol/L), với lịch ăn bình thường, phẫu thuật có thể là cần thiết. Điều trị phẫu thuật đối với KATP-HI khu trú có thể chữa khỏi, trong khi đối với bệnh lan tỏa, ngay cả phẫu thuật cắt bỏ 95% đến 99% tuyến tụy cũng có thể không chữa khỏi hạ đường huyết, và đái tháo đường phụ thuộc insulin là một hậu quả thường xuyên. Chụp cắt lớp phát xạ positron 18F-fluorodopa (18F-L-DOPA PET) đã được chứng minh là chính xác trong việc định vị tổn thương khu trú trước phẫu thuật.
Tăng insulin máu do Glutamate Dehydrogenase
HI có thể được gây ra bởi các đột biến hoạt hóa trội trong gen GLUD1 trên nhiễm sắc thể 10q, mã hóa cho glutamate dehydrogenase (GDH). Rối loạn này còn được gọi là hội chứng tăng amoniac máu HI, vì hoạt động enzyme GDH tăng trong ống thận dẫn đến tăng sinh amoniac ở thận. Hạ đường huyết trong GDH-HI thường nhẹ hơn so với KATP-HI, và ít có khả năng biểu hiện trong giai đoạn sơ sinh. Như thường thấy với các rối loạn di truyền trội, có tới 80% các trường hợp mắc GDH-HI có đột biến de novo. Tăng amoniac máu trong GDH-HI dường như không có triệu chứng. Tuy nhiên, trẻ em bị ảnh hưởng thường có các biểu hiện ở não, có thể do hoạt động GDH tăng trong các tế bào thần kinh và nồng độ glutamate hoặc các chất dẫn truyền thần kinh khác bị thay đổi: bao gồm co giật cơn vắng (động kinh toàn thể), rối loạn hành vi và chậm phát triển từ nhẹ đến trung bình.
Trong GDH-HI, các đột biến hoạt hóa của GDH trong tế bào beta khuếch đại quá trình sản xuất ATP do leucine kích hoạt, dẫn đến việc đóng các kênh KATP và giải phóng insulin, độc lập với nồng độ glucose. Ở trẻ em bị ảnh hưởng, việc ăn các bữa ăn giàu protein có thể gây hạ đường huyết (hạ đường huyết nhạy cảm với leucine hoặc protein). Ở thận, các đột biến tương tự làm tăng quá trình oxy hóa glutamate thành alpha-ketoglutarate với sự giải phóng amoniac.
Chẩn đoán GDH-HI tương tự như các dạng tăng insulin máu khác; tuy nhiên, nồng độ amoniac huyết tương tăng liên tục (thường là 80-120 µmol/L) là đặc hiệu cho rối loạn này. Bởi vì tăng amoniac máu có nguồn gốc từ thận, nồng độ amoniac huyết tương không liên quan đến sự tiết insulin hoặc nồng độ glucose huyết tương và cũng không liên quan đến việc ăn protein. Do đó, nồng độ amoniac tăng cao có thể dễ dàng được chứng minh trong các mẫu huyết tương ngẫu nhiên. Hầu hết bệnh nhân đáp ứng tốt với diazoxide và không cần phẫu thuật.
Tăng insulin máu do Glucokinase
Glucokinase (được mã hóa bởi GCK) là enzyme đóng vai trò cảm biến glucose trong các tế bào beta của tuyến tụy. Các đột biến tăng chức năng của glucokinase dẫn đến ngưỡng glucose thấp hơn để tiết insulin, dẫn đến hạ đường huyết dai dẳng (ngược lại, các đột biến mất chức năng của GCK tạo ra ngưỡng glucose cao hơn để giải phóng insulin, gây ra một dạng đái tháo đường đơn gen nhẹ phổ biến (MODY2)). Ít nhất 15 đột biến GCK-HI biểu hiện trội khác nhau đã được báo cáo. Những thay đổi trong động học enzyme của các đột biến riêng lẻ dự đoán các mức độ hạ thấp ngưỡng glucose rất khác nhau, nhưng không tương quan với mức độ nghiêm trọng và diễn biến lâm sàng. Trong một số trường hợp, đột biến GCK-HI biểu hiện bằng hạ đường huyết sơ sinh, đủ nặng để cần phẫu thuật cắt bỏ tụy để kiểm soát hạ đường huyết; tuy nhiên, một số người mang đột biến đã thoát khỏi sự nhận biết ngay cả khi trưởng thành.
GCK-HI có thể đặc biệt khó chẩn đoán, vì nồng độ glucose huyết tương khi đói có xu hướng ổn định ở ngưỡng glucose từ 50 đến 65 mg/dL (2,8-3,6 mmol/L). Trong khi nhịn đói, nếu glucose huyết tương vẫn ở ngưỡng này trong một thời gian dài, nồng độ FFA và BOHB có thể tăng lên, gợi ý một dạng hạ đường huyết do ceton thay vì HI. Nồng độ amoniac huyết tương bình thường ở bệnh nhân GCK-HI và họ không bị hạ đường huyết nhạy cảm với protein. Phản ứng với diazoxide chỉ là một phần và thoáng qua ở hầu hết các bệnh nhân mắc GCK-HI và một số đã phải phẫu thuật cắt bỏ tụy. Một báo cáo đã mô tả kết quả tốt khi sử dụng chế độ ăn ketogenic để ngăn ngừa hạ đường huyết có triệu chứng; chất tương tự somatostatin tác dụng kéo dài, lanreotide, có thể hữu ích ở những bệnh nhân lớn tuổi hơn, nhưng kinh nghiệm còn hạn chế.
Tăng insulin máu do Hexokinase 1
Hexokinase 1 (HK1) có liên quan chặt chẽ với glucokinase và thực hiện bước đầu tiên trong quá trình oxy hóa glucose bằng cách phosphoryl hóa glucose thành glucose-6-phosphate ở hầu hết các mô của cơ thể, ngoại trừ các tế bào beta. Trái ngược với glucokinase, có ái lực thấp với glucose (Km~5 mM) đặt ngưỡng kích thích tiết insulin bằng glucose vào khoảng 4 đến 4,5 mM (70-80 mg/dL, tương đương 3,9-4,4 mmol/L), HK1 có ái lực rất cao với glucose (Km~1 mM), và do đó sự biểu hiện của nó trong các tế bào beta thường không được phép. Hai báo cáo về tăng insulin máu bẩm sinh có thể do sự biểu hiện của HK1 ở tế bào beta gần đây đã được công bố. Báo cáo đầu tiên mô tả một phả hệ lớn, di truyền trội với tăng insulin máu bẩm sinh đáp ứng với diazoxide, trong đó phân tích liên kết đã xác định một haplotype chung trên nhiễm sắc thể 10 bao gồm 3 đột biến không mã hóa trong locus HK1. Gia đình này nằm trong một trong những mô tả ban đầu về tăng insulin máu bẩm sinh của McQuarrie vào năm 1953. Người ta cho rằng một hoặc nhiều đột biến này đã khiến HK1 được biểu hiện trong các tế bào beta và làm giảm ngưỡng glucose để tiết insulin xuống khoảng 55 đến 60 mg/dL (3,1-3,3 mmol/L). Trong báo cáo thứ hai, sự biểu hiện HK1 ở tế bào beta tăng đã được phát hiện bằng cách nhuộm hóa mô miễn dịch các tiểu đảo tụy từ một số trẻ sơ sinh đã trải qua phẫu thuật vì tăng insulin máu bẩm sinh không đáp ứng với diazoxide; những trẻ sơ sinh này cũng được cho là mắc tăng insulin máu do sự thất bại trong việc im lặng HK1 trong các tế bào beta. Như được mô tả sau này, sự thất bại trong việc im lặng biểu hiện của một gen tế bào beta “không được phép” cũng đã được đề xuất là cơ chế của tăng insulin máu do tập thể dục: biểu hiện của chất vận chuyển pyruvate qua màng sinh chất, MCT1.
Tăng insulin máu do Short Chain 3-Hydroxyacyl-CoA Dehydrogenase
Short chain 3-hydroxyacyl-CoA dehydrogenase ([SCHAD], được mã hóa bởi HADH trên nhiễm sắc thể 4 tại q22-26) là một enzyme oxy hóa acid béo trong ty thể xúc tác quá trình oxy hóa các short chain 3-hydroxyacyl-CoA. Nồng độ protein SCHAD tương đối cao trong các tế bào beta tuyến tụy, nơi nó dường như điều hòa âm hoạt động của GDH, mục tiêu của các đột biến tăng chức năng chịu trách nhiệm cho GDH-HI (hội chứng tăng amoniac máu HI). Các đột biến mất hoạt tính đồng hợp tử của gen HADH có liên quan đến hoạt động GDH của tế bào beta tăng và gây ra một dạng tăng insulin máu tương tự như GDH-HI, nhưng không có tăng amoniac máu hoặc các khiếm khuyết não cụ thể. Khiếm khuyết toàn thân trong quá trình oxy hóa acid béo trong thiếu hụt SCHAD dẫn đến sự tích tụ các cơ chất ngược dòng dẫn đến sự tăng cao chẩn đoán của các acid hữu cơ trong nước tiểu (3-hydroxyglutarate) và acylcarnitine huyết tương (3-hydroxybutyryl-carnitine). Bệnh nhân mắc tăng insulin máu do thiếu hụt SCHAD đã đáp ứng tốt với điều trị bằng diazoxide.
Tăng insulin máu do Yếu tố hạt nhân tế bào gan 4a
Yếu tố hạt nhân tế bào gan 4a (HNF4A) là một yếu tố phiên mã quan trọng cho sự phát triển của tế bào beta tuyến tụy và sự tiết insulin. Các đột biến mất hoạt tính dị hợp tử của HNF4A là một nguyên nhân được công nhận rõ ràng của bệnh đái tháo đường đơn gen (MODY1). Gần đây, người ta đã nhận ra rằng chính những đột biến này cũng gây ra sự tiết insulin quá mức trong giai đoạn đầu đời, biểu hiện bằng thai to và hạ đường huyết tăng insulin máu sơ sinh dai dẳng đáp ứng với diazoxide. tăng insulin máu thường thoáng qua, chỉ kéo dài vài tuần đến vài tháng sau khi sinh; tuy nhiên, nó có thể kéo dài trong vài năm của thời thơ ấu. Trong thập kỷ thứ hai và thứ ba, những đứa trẻ này có thể phát triển dạng đái tháo đường MODY1, đáp ứng với điều trị bằng sulfonylureas. Trong hầu hết các trường hợp HNF4A HI, cân nặng khi sinh trên mức trung bình, hạ đường huyết xảy ra trong những ngày đầu đời, và một bên cha hoặc mẹ có tiền sử mắc bệnh đái tháo đường type MODY. Trẻ em có đột biến HNF4A cụ thể, p.Arg76Trp, có thêm các biểu hiện, bao gồm hội chứng Fanconi ở thận và gan to và men gan tăng, cho thấy rằng sự biểu hiện của chất vận chuyển GLUT2 trong ống thận và gan bị suy giảm do khiếm khuyết trong HNF4A.
Tăng insulin máu do Yếu tố hạt nhân tế bào gan 1A
Yếu tố hạt nhân tế bào gan 1a (HNFIA) là một yếu tố phiên mã quan trọng khác cho sự phát triển của tế bào beta. Các đột biến mất hoạt tính dị hợp tử của HNFIA gây ra dạng đái tháo đường đơn gen phổ biến nhất, MODY3. Một số trường hợp đột biến HNFIA gây ra tăng insulin máu bẩm sinh đã được báo cáo, một số trong đó cũng có liên quan đến bệnh đái tháo đường MODY ở những bệnh nhân lớn tuổi hơn. Một sự khác biệt thú vị, không giải thích được giữa HNF4A và HNFIA tăng insulin máu là cân nặng khi sinh lớn so với tuổi thai chỉ liên quan đến đột biến HNF4A, nhưng không phải HNFIA.
Tăng insulin máu do Protein tách cặp 2
Protein tách cặp 2 (UCP2) chuyển các chất trung gian của chu trình Krebs ra khỏi ty thể và ưu tiên quá trình oxy hóa acid amin so với glucose trong các tế bào beta, gan và các mô khác. Các đột biến mất chức năng của UCP2 có thể tăng cường tạo ATP từ glucose và do đó khuếch đại các phản ứng insulin đối với kích thích glucose, đủ để gây ra hạ đường huyết tăng insulin máu. Một số trường hợp đã được báo cáo, thường đáp ứng với điều trị bằng diazoxide.
Tăng insulin máu do tập thể dục
Tổng cộng có 13 bệnh nhân thuộc ba gia đình đã được phát hiện mắc chứng hạ đường huyết tăng insulin máu do tập thể dục (EIHI) di truyền trội sau các giai đoạn tập thể dục kỵ khí cường độ cao. Các biểu hiện lâm sàng đã thay đổi, với một số trường hợp có các cơn hạ đường huyết nặng từ thời thơ ấu; một số biểu hiện ngất do hạ đường huyết sau khi tập thể dục, khi còn là thanh thiếu niên; và một số chỉ bị ảnh hưởng nhẹ, khi trưởng thành. Hầu hết các bệnh nhân duy trì mức glucose bình thường trong khi nhịn đói kéo dài. Thử nghiệm kích thích, với việc đạp xe cường độ cao trong thời gian ngắn, đã gây ra sự gia tăng dự kiến của nồng độ lactate và pyruvate trong huyết tương, nhưng làm tăng rõ rệt nồng độ insulin trong huyết tương trong khoảng 10 phút sau khi tập thể dục, gây hạ đường huyết trong 45 phút tiếp theo. Truyền tĩnh mạch pyruvate đã kích thích một sự gia tăng tương tự của insulin, cho thấy rằng khiếm khuyết phản ánh một sự đáp ứng bất thường của tế bào beta đối với pyruvate. Hạ đường huyết tái phát chỉ có thể phòng ngừa được một phần bằng điều trị diazoxide.
Sự tiết insulin quá mức trong EIHI xảy ra do các đột biến ở phía trên vùng promoter của SLC16A1 cho phép chất vận chuyển monocarboxylate MCT1 được biểu hiện trên màng sinh chất của tế bào beta. Thông thường, sự biểu hiện của MCTI bị “ngăn cấm” trong các tế bào beta để ngăn pyruvate và lactate hoạt động như các chất kích thích tiết insulin.
Các dạng Tăng insulin máu bẩm sinh có hội chứng
Hội chứng Beckwith-Wiedemann. Hội chứng Beckwith-Wiedemann (BWS) là một hội chứng phát triển quá mức gây ra bởi các đột biến khảm trong phôi của một vùng in dấu trên nhiễm sắc thể 11p, gần vị trí của các gen kênh KATP của tế bào beta, SUR1 và Kir6.2. Locus BWS bao gồm các gen ức chế tăng trưởng được biểu hiện trên nhiễm sắc thể của mẹ (H19, P57) và các gen thúc đẩy tăng trưởng (IGF2) được biểu hiện trên nhiễm sắc thể của cha; sự biểu hiện của các gen này được kiểm soát bởi hai vị trí kiểm soát in dấu. Mặc dù BWS đã được liên kết với tăng insulin máu trong nhiều năm, gần đây người ta đã phát hiện ra rằng hầu hết các trường hợp BWS có tăng insulin máu dai dẳng đều liên quan đến đồng phân lưỡng bội đơn cha cho vùng 11p (11pUPD), là nguyên nhân của khoảng 20% các trường hợp BWS. Mô bệnh học trong những trường hợp này cho thấy sự phát triển quá mức của tế bào tiểu đảo (adenomatosis), có thể liên quan đến gần như toàn bộ tuyến tụy. Một số trường hợp đáp ứng với diazoxide, nhưng hạ đường huyết ở một số trường hợp có thể không kiểm soát được bằng diazoxide và có thể cần điều trị nội khoa tích cực hơn với octreotide và cho ăn liên tục hoặc phẫu thuật cắt bỏ tụy. Một số trường hợp 11pUPD BWS cũng có một đột biến kênh KATP lặn di truyền từ cha; những trường hợp này có thể có tăng insulin máu nặng và nhu cầu glucose cao bất thường. Khả năng mắc 11pUPD BWS nên được nghi ngờ ở trẻ sơ sinh mắc tăng insulin máu có các đặc điểm của BWS, chẳng hạn như phì đại nửa người, lưỡi to, hoặc thoát vị rốn.
Hội chứng Kabuki. Hội chứng Kabuki có liên quan đến các đặc điểm dị dạng dễ nhận biết bao gồm các đặc điểm trên khuôn mặt giống mặt nạ của diễn viên Kabuki và gây ra bởi các đột biến khảm xôma trong MLL2 (KMT2D) hoặc KDM6A, hai gen điều hòa quá trình methyl hóa chromatin. Một tỷ lệ trẻ sơ sinh mắc hội chứng Kabuki có hạ đường huyết dai dẳng do HI, có thể đáp ứng hoặc không đáp ứng với điều trị bằng diazoxide.
Hội chứng Turner. Hội chứng Turner đã được báo cáo là có liên quan đến tăng insulin máu bẩm sinh trong nhiều thập kỷ. Một nghiên cứu gần đây cho thấy tần suất hội chứng Turner trong một loạt lớn trẻ em mắc tăng insulin máu đã tăng ~50 lần, cho thấy nguy cơ mắc tăng insulin máu trong hội chứng Turner là khoảng 1/1000 so với nguy cơ mắc tăng insulin máu trong dân số chung là 1/40.000. Một số trường hợp hội chứng Turner đáp ứng với diazoxide, nhưng những trường hợp khác không đáp ứng với diazoxide và cần phẫu thuật cắt bỏ tụy. Bất thường nhiễm sắc thể nhất quán duy nhất ở các bé gái mắc hội chứng Turner bị tăng insulin máu là kiểu nhân 45,XO, cho thấy rằng việc mất một gen trên nhiễm sắc thể X là nguyên nhân của HI. Một gen ứng cử viên có thể là KDM6A, một gen giả nhiễm sắc thể thường trên nhiễm sắc thể X, vì các đột biến mất hoạt tính của KDM6A là một trong những nguyên nhân của hội chứng Kabuki, trong đó cũng có tần suất tăng insulin máu tăng.
Tăng insulin máu do Glycoprotein thiếu Carbohydrate
Các rối loạn glycosyl hóa bẩm sinh (CDG) được gây ra bởi các đột biến mất chức năng di truyền lặn trong các gen điều hòa quá trình glycosyl hóa protein. Hầu hết gây ra các bệnh có hội chứng ảnh hưởng đến nhiều hệ cơ quan, bao gồm não, xương, gan và thận. Hạ đường huyết do tăng insulin máu đã được báo cáo ở trẻ em mắc ba trong số các khiếm khuyết N-glycosyl hóa đã được xác định: CDG la, lb và Id (OMIM IDs: 601785, 602579, và 601110). Hầu hết các trường hợp CDG-HI đã được xác định trong giai đoạn sơ sinh, nhưng một số đã được chẩn đoán muộn hơn trong thời kỳ nhũ nhi hoặc đầu thời thơ ấu. CDG-HI thường được nghi ngờ ở một đứa trẻ có khuôn mặt dị dạng và các biểu hiện ảnh hưởng đến nhiều hệ cơ quan khác, bao gồm đặc biệt là não, gan, ruột và xương. Trong cả ba loại, các trường hợp đã được báo cáo trong đó hạ đường huyết tăng insulin máu là vấn đề biểu hiện hoặc chủ đạo. Cơ chế sản xuất insulin quá mức chưa được xác định.
Phosphomannomutase 2 (PMM2)-CDG (CDG-la) là loại CDG phổ biến nhất và liên quan đến hoạt động thiếu hụt của phosphomannomutase 2 do các đột biến của PMM2. Hầu hết các bệnh nhân đều bị chậm phát triển nặng, thiểu sản tiểu não, giảm trương lực cơ và co giật. Bệnh ruột mất protein và bệnh gan góp phần vào việc không phát triển. Mức độ antithrombin III thiếu hụt có thể gây huyết khối. Các đặc điểm dị dạng là phổ biến và có thể tinh vi hoặc rõ ràng, bao gồm phân bố mỡ bất thường và núm vú ngược. Hạ đường huyết tăng insulin máu chỉ xảy ra ở một số ít bệnh nhân, nhưng có thể nhẹ hoặc đủ nặng để phải phẫu thuật cắt bỏ tụy.
Phosphomannose isomerase (MPI)-CDG (CDG-Ib) liên quan đến hoạt động thiếu hụt của phosphomannose isomerase do các đột biến của MPI. Không giống như CDG-1a, hệ thần kinh trung ương được bảo tồn và hạ đường huyết tăng insulin máu là một đặc điểm phổ biến, biểu hiện trong những ngày đầu đời hoặc muộn hơn trong năm đầu tiên. Bệnh gan và bệnh ruột mất protein thường là các vấn đề lâm sàng chủ đạo. Một số trẻ em bị nôn theo chu kỳ. Mức độ nghiêm trọng lâm sàng có thể thay đổi, và người lớn bị ảnh hưởng nhẹ đã được chẩn đoán. Mannose đường uống với liều lên đến 150 mg/kg/ngày có thể khắc phục hầu hết các bất thường lâm sàng, làm cho MPI-CDG trở thành CGD duy nhất có phương pháp điều trị cụ thể.
ALD3-CDG (CDG-Id) liên quan đến hoạt động thiếu hụt của mannosyltransferase 6 do các đột biến của ALD3. Các đặc điểm lâm sàng giống với PMM2-CDG, với tổn thương hệ thần kinh trung ương nghiêm trọng. Một trường hợp hạ đường huyết tăng insulin máu sơ sinh nặng đã được báo cáo, nhưng không rõ liệu đó là kết quả trực tiếp của CDG hay hậu quả của stress chu sinh.
PGM-1 chuyển đổi thuận nghịch glucose-6-P thành glucose-1-P và cần thiết cho cả quá trình hình thành và phân hủy glycogen. Các đột biến mất hoạt tính lặn của PGM1 gây ra một hội chứng gồm các đặc điểm dị dạng, tầm vóc thấp và hạ đường huyết tăng ceton máu khi đói. Ngoài các bất thường trong quá trình glycosyl hóa protein (do đó được chỉ định là CDG-It), hạ đường huyết tăng insulin máu không ceton xảy ra khi bệnh nhân được cho ăn. Thiếu hụt PGM1 làm suy giảm cả quá trình tổng hợp và phân hủy glycogen gan (do đó gây ra hạ đường huyết tăng ceton máu khi đói và việc dán nhãn rối loạn là GSD type 14). Tuy nhiên, thiếu hụt PGM1 trong tế bào beta dường như gây ra phản ứng insulin phóng đại đối với tải glucose, dẫn đến hạ đường huyết tăng insulin máu sau ăn. Chẩn đoán có thể được thực hiện bằng các xét nghiệm sàng lọc các bất thường trong quá trình glycosyl hóa transferrin. Điều trị bằng diazoxide không hiệu quả trong việc kiểm soát hạ đường huyết tăng insulin máu sau ăn, nhưng việc cho ăn có chỉ số đường huyết thấp, bột ngô sống (UCS), hoặc truyền glucose liều thấp liên tục đã thành công hơn. Bởi vì khiếm khuyết này có khả năng làm suy giảm quá trình tổng hợp galactose, điều trị bằng bổ sung galactose đã được thử nghiệm với một số thành công trong việc khắc phục các bất thường của quá trình glycosyl hóa transferrin.
Hạ đường huyết do AKT2
Ba trẻ em bị hạ đường huyết do các đột biến tăng chức năng của AKT2 đã được mô tả. AKT2 là một serine/threonine kinase là một trong những bước xuôi dòng trong quá trình truyền tín hiệu sau thụ thể insulin; mất hoạt tính AKT2 có liên quan đến kháng insulin, trong khi kích hoạt AKT2 gây ra sự gia tăng độ nhạy insulin. Bắt đầu từ khoảng 6 tháng tuổi, tất cả các trường hợp được báo cáo đều có các đợt hạ đường huyết có triệu chứng, không ceton, nặng giống như HI, nhưng liên quan đến nồng độ insulin, proinsulin và FFA thấp. Hạ đường huyết chủ yếu là sau ăn và có thể được kiểm soát bằng cách cho ăn dextrose liều thấp liên tục. Các đặc điểm lâm sàng khác bao gồm cân nặng khi sinh lớn và phát triển quá mức không đối xứng sau sinh của cơ thể hoặc khuôn mặt. Mỗi bệnh nhân trong ba bệnh nhân không có quan hệ họ hàng đều có cùng một đột biến de novo của AKT2. Các đột biến này là sau hợp tử và khảm, điều này có thể giải thích bản chất cục bộ của sự phát triển quá mức.
Hạ đường huyết do Phospatidylinositol-3-Kinase
Phospatidylinositol-3-Kinase (PI3K) là một bước enzyme trong con đường truyền tín hiệu sau thụ thể insulin ở thượng nguồn từ AKT2. Các đột biến tăng chức năng sau hợp tử của hai enzyme liên quan đến P13-kinase, PIK3CA và PIK3R2, gần đây đã được báo cáo là gây ra một dạng hạ đường huyết không ceton có hội chứng với nồng độ insulin thấp, tương tự như các đột biến tăng chức năng của AKT2 được mô tả trước đó. Các đặc điểm bao gồm phát triển quá mức không đối xứng, não to và chậm phát triển. Diazoxide không hiệu quả và cần điều trị bằng dextrose liên tục với tốc độ truyền glucose thấp để kiểm soát hạ đường huyết. Hạ đường huyết dường như chủ yếu do hoạt động sau thụ thể insulin tăng ở gan.
Hạ đường huyết tăng insulin máu liên quan đến khiếm khuyết thụ thể Insulin
Các đột biến mất hoạt tính của thụ thể insulin (INSR), gây kháng insulin và đái tháo đường, cũng có thể liên quan đến hạ đường huyết tăng insulin máu sau ăn với mức độ nghiêm trọng khác nhau. Các khiếm khuyết nghiêm trọng trong INSR biểu hiện trong thời kỳ nhũ nhi và thời thơ ấu với các đặc điểm dị dạng do thiếu hụt rõ rệt mô mỡ (loạn dưỡng mỡ) và, theo thứ tự mức độ nghiêm trọng, bao gồm hội chứng Donohue (leprechaunism) (OMIM ID: 246200) và hội chứng Rabson-Mendenhall (OMIM ID: 262190); các khiếm khuyết nhẹ hơn có thể biểu hiện muộn hơn trong thời thơ ấu hoặc đầu tuổi trưởng thành dưới dạng kháng insulin type A (OMIM ID: 610549). Các dạng kháng insulin nghiêm trọng hơn có thể do các đột biến INSR hai alen, và các khiếm khuyết đơn alen có thể liên quan đến các dạng kháng insulin nhẹ hơn; các đột biến ở tiểu đơn vị beta của INSR có xu hướng nhẹ hơn các đột biến ở tiểu đơn vị alpha. Nồng độ insulin huyết tương tăng gấp 10 đến 100 lần so với bình thường. Hạ đường huyết ở những bệnh nhân này thường là sau ăn và được cho là do sự thanh thải insulin chậm (sự chậm trễ tương tự trong thanh thải insulin cũng được cho là chịu trách nhiệm cho hạ đường huyết sau ăn trong hạ đường huyết tự miễn do kháng thể kháng insulin). Hạ đường huyết khi đói có liên quan đến tình trạng kháng insulin nghiêm trọng hơn được thấy ở trẻ sơ sinh và trẻ nhũ nhi mắc hội chứng Donohue và được cho là do nồng độ insulin rất cao, có tác dụng giống insulin bằng cách phản ứng chéo với thụ thể yếu tố tăng trưởng giống insulin-1 (IGF-1), mặc dù một báo cáo trước đó đã chứng minh một mô hình thích ứng nhịn đói tăng ceton máu nhanh, cho thấy có thể có sự suy giảm trong việc dự trữ glycogen gan. Trẻ sơ sinh mắc hội chứng Donohue biểu hiện trong giai đoạn sơ sinh và hầu hết tử vong do nhiễm trùng trong những năm đầu đời. Các đặc điểm bao gồm chậm tăng trưởng trong tử cung, mỡ dưới da giảm rõ rệt, ngoại hình đặc trưng gợi nhớ đến một yêu tinh (“leprechaunism”), ngực và âm vật hoặc dương vật to, rậm lông, gai đen, và tăng đường huyết sau ăn. Hạ đường huyết, vài giờ sau bữa ăn, có thể được ghi nhận trong những ngày đầu sau khi sinh và tồn tại suốt đời. Cho ăn thường xuyên là phương pháp quản lý chính cho hạ đường huyết; diazoxide nói chung không hiệu quả.
Trẻ em mắc hội chứng Rabson-Mendenhall bị ảnh hưởng ít nghiêm trọng hơn so với những trẻ mắc hội chứng Donohue, nhưng có chung nhiều đặc điểm, bao gồm giảm mô mỡ, gai đen, rậm lông, và cơ quan sinh dục to ở cả nam và nữ. Nhiều trẻ em này cũng bị hạ đường huyết sau ăn. Bệnh đái tháo đường có xu hướng tiến triển và kháng insulin có thể nghiêm trọng. Nữ thanh thiếu niên có thể có bằng chứng về tăng androgen tương tự như hội chứng buồng trứng đa nang. Tử vong trong thời thơ ấu do nhiễm toan ceton đái tháo đường là phổ biến. Điều trị bằng leptin (metreleptin) đã được báo cáo là cải thiện khiêm tốn việc kiểm soát glucose ở một số bệnh nhân mắc hội chứng Rabson-Mendenhall.
Hạ đường huyết sau ăn (phản ứng) cũng đã được báo cáo ở thanh thiếu niên mắc kháng insulin type A, trong đó bệnh đái tháo đường phát triển dần dần hơn, đôi khi khi thanh thiếu niên đến tuổi trưởng thành. Các đặc điểm có thể bao gồm giảm mô mỡ, gai đen, và nồng độ triglyceride tăng. Kháng insulin có thể dẫn đến suy tế bào beta cuối cùng và thiếu hụt insulin.
Hạ đường huyết phản ứng cũng đã được báo cáo trong các tình trạng kháng insulin do các rối loạn sau thụ thể, và nên được xem xét trong các hội chứng kháng insulin khác, chẳng hạn như loạn dưỡng mỡ bẩm sinh (hội chứng Berardinelli-Seip), do các khiếm khuyết lặn trong AGPAT2, BSCL2, hoặc PTRF.
Tăng insulin máu giả
Các trường hợp tăng insulin máu giả đã được báo cáo ở trẻ sơ sinh và trẻ em, thường là kết quả của việc cha mẹ hoặc người chăm sóc khác cho dùng insulin hoặc một chất kích thích tiết insulin. Dạng lạm dụng trẻ em này được gọi là Munchausen by proxy và có thể gây ra phẫu thuật tụy không cần thiết và có thể gây tử vong. Trong hầu hết các trường hợp được báo cáo, cha mẹ (thường là mẹ) là y tá hoặc chuyên gia y tế khác hoặc có quyền truy cập vào insulin hoặc sulfonylureas đang được sử dụng tại nhà để điều trị bệnh đái tháo đường. Thông thường, các triệu chứng kinh điển của tăng insulin máu đều có mặt, nhưng có thể xảy ra không đều. Như trong các dạng tăng insulin máu khác, hạ đường huyết đi kèm với sự ức chế ceton và FFA huyết tương và một phản ứng dương tính bất thường với glucagon. Các nghiệm pháp nhịn đói khi không có người chăm sóc sẽ bình thường; tuy nhiên, cần phải hết sức thận trọng để đảm bảo không có sự can thiệp của tác nhân bị nghi ngờ. Nồng độ insulin huyết tương cực cao (ví dụ, >100 µU/mL) đôi khi là một manh mối (đặc biệt có khả năng được tìm thấy sau khi dùng insulin tác dụng ngắn hoặc trung gian, chẳng hạn như regular hoặc isophane (NPH); ngược lại, nồng độ insulin huyết tương thường chỉ tăng nhẹ trong tăng insulin máu do nguyên nhân nội sinh). Việc phát hiện insulin ngoại sinh ngày càng trở nên khó khăn bằng các xét nghiệm insulin tiêu chuẩn vì chúng có thể khác nhau về khả năng phát hiện các chất tương tự insulin (lispro, aspart, glargine, glulisine, detemir, degludec).
Bằng chứng kết luận nhất của insulin ngoại sinh là sự chênh lệch giữa insulin huyết tương tăng cao, nhưng nồng độ C-peptide huyết tương rất thấp, cho thấy sản xuất insulin nội sinh đã bị ức chế. U tiết insulin có thể liên quan đến insulin huyết tương tăng, nhưng các giá trị hiếm khi lớn hơn 100 µU/ml và nồng độ C-peptide và proinsulin phải tăng tương ứng.
Việc dùng lén lút (hoặc, phổ biến hơn là vô tình) các thuốc hạ đường huyết đường uống tác dụng kéo dài gây tiết insulin, chẳng hạn như glyburide hoặc các sulfonylureas khác, gây tăng cả insulin và C-peptide và có thể gây hạ đường huyết dai dẳng trong 24 giờ hoặc lâu hơn. Các xét nghiệm sàng lọc độc chất thông thường trong máu và nước tiểu có thể không phát hiện ra sulfonylureas, nhưng có thể sắp xếp xét nghiệm cụ thể nếu nghi ngờ một loại thuốc cụ thể.
Nếu nghi ngờ sử dụng insulin hoặc thuốc hạ đường huyết đường uống lén lút, nên lấy các mẫu huyết tương/huyết thanh, từ thời điểm hạ đường huyết, để xét nghiệm nồng độ insulin và C-peptide huyết tương và bảo quản cho các xét nghiệm khác có thể trở nên cần thiết để ghi nhận sự liên quan của một tác nhân ngoại sinh. Tham khảo ý kiến phòng thí nghiệm sẽ hữu ích trong việc giải thích và lựa chọn các xét nghiệm phù hợp. Điều trị hạ đường huyết bằng glucose tĩnh mạch thường là đủ, nhưng có thể cần glucagon tĩnh mạch liên tục trong các trường hợp nặng và diazoxide hoặc octreotide có thể hữu ích trong các trường hợp tăng tiết insulin kéo dài.
Hạ đường huyết tự miễn
Hai loại hạ đường huyết tự miễn đã được báo cáo: một loại qua trung gian kháng thể kháng insulin và loại kia qua trung gian kháng thể kháng thụ thể insulin. Hạ đường huyết do kháng thể kháng insulin đã được báo cáo thường xuyên nhất từ Nhật Bản (“bệnh Hirata”), nhưng các trường hợp đã được mô tả ở cả hai giới, mọi lứa tuổi và từ nhiều khu vực trên thế giới. Gần như tất cả các trường hợp hạ đường huyết tự miễn ở trẻ sơ sinh và trẻ em đều thuộc loại này. Cơ chế hạ đường huyết được cho là do sự phân ly chậm của insulin khỏi kháng thể trong giai đoạn sau ăn hoặc sau hấp thu. Hạ đường huyết thường là sau ăn, nhưng có thể là khi đói, và đôi khi đủ nặng để gây co giật hoặc hôn mê. Các đặc điểm chuyển hóa là của HI, với nồng độ ceton và FFA huyết tương thấp. Các xét nghiệm insulin huyết tương có thể cho giá trị rất cao và duy trì cao liên tục theo thời gian do sự can thiệp của các kháng thể gắn insulin nội sinh (điều này có thể thay đổi, tùy thuộc vào phương pháp xét nghiệm); ngược lại, giá trị C-peptide huyết tương có thể bị ức chế, gợi ý khả năng sử dụng insulin hoặc sulfonylurea lén lút. Rối loạn này có thể được kích hoạt bởi các bệnh nhiễm virus thông thường hoặc do tiếp xúc với các loại thuốc chứa nhóm sulfhydryl hoặc thiol, chẳng hạn như methimazole hoặc acid lipoic, và dường như có liên quan đến các rối loạn nội tiết tự miễn khác, chẳng hạn như cường giáp. Ở Nhật Bản, một mối liên quan với alen HLA HLA-DRB1 04:06 đã được báo cáo; một tỷ lệ cao HLA-DRB1*04:03 đã được mô tả ở người châu Âu mắc chứng rối loạn này. Trong số các phương pháp điều trị khác nhau được báo cáo là cải thiện hạ đường huyết có các đợt dùng glucocorticoid, lọc huyết tương và truyền immunoglobulin tĩnh mạch. Rối loạn này thường tự khỏi theo thời gian khi nồng độ tự kháng thể giảm.
Dạng hạ đường huyết tự miễn thứ hai là do các kháng thể gắn vào và kích hoạt thụ thể insulin (tương tự như các immunoglobulin kích thích tuyến giáp trong bệnh Graves). Khởi phát ở trẻ em là hiếm, nhưng các trường hợp trẻ nhất là 8 tháng tuổi đã được ghi nhận. Ở trẻ em, có thể có hạ đường huyết khi đói nghiêm trọng với tốc độ sử dụng glucose rất cao và không đáp ứng với diazoxide hoặc octreotide. Ở người lớn, rối loạn này có liên quan đến kháng insulin type B (tự miễn), hoặc một khối u ác tính hoặc một rối loạn viêm nghiêm trọng. Một số bệnh nhân có cả kháng thể kháng insulin và kháng thụ thể. Nồng độ C-peptide và insulin thường không thể phát hiện được và diazoxide và octreotide không hiệu quả trong việc kiểm soát hạ đường huyết. Điều trị bằng glucocorticoid, lọc huyết tương, hoặc immunoglobulin tĩnh mạch có thể được xem xét.
U tiết insulin (Insulinoma)
U tiết insulin là nguyên nhân phổ biến nhất của hạ đường huyết dai dẳng mắc phải ở người lớn và trẻ lớn và hiếm khi có thể xảy ra ở trẻ em từ 2 tuổi. Thường thì, u tiết insulin là các khối u đơn độc, nhỏ, kích thước 0,5 đến 1 cm, phát triển chậm và hiếm khi trở thành ác tính hoặc di căn; các khối u đa ổ và u tiết insulin tái phát có thể xảy ra, đặc biệt ở những bệnh nhân mắc hội chứng đa u tuyến nội tiết type 1 (MEN1) do các đột biến mất hoạt tính trội trong gen MENIN trên nhiễm sắc thể 11q. Trẻ em mắc u tiết insulin biểu hiện với các đợt hạ đường huyết có triệu chứng tái phát, thường ban đầu nhẹ và có thể không được nhận ra trong vài tháng trước khi các biểu hiện nặng, chẳng hạn như co giật, xảy ra. Chẩn đoán tăng insulin máu dễ dàng nhất được thực hiện bằng một nghiệm pháp nhịn đói để chứng minh hạ đường huyết không ceton, cùng với một phản ứng đường huyết tăng lớn không phù hợp với glucagon. Ở trẻ nhỏ hơn, đặc biệt, có thể không thể phân biệt lâm sàng giữa u tiết insulin và một dạng di truyền của tăng insulin máu lan tỏa.
Phần lớn các u tiết insulin ở trẻ em dường như có liên quan đến đồng phân lưỡng bội đơn cha 11p, mặc dù điều này dường như không phổ biến ở các u tiết insulin ở người lớn. Tần suất đột biến di truyền trong gen MENIN dường như cao hơn ở trẻ em (42% các u tiết insulin ở trẻ em tại Bệnh viện Nhi Philadelphia) so với ở người lớn mắc u tiết insulin (~8%). Điều này rất quan trọng, vì sự hiện diện của nhiều khối u trong lần phẫu thuật đầu tiên và sự tái phát của các khối u có nhiều khả năng xảy ra ở những bệnh nhân mắc MEN1 hơn là trong các trường hợp u tiết insulin lẻ tẻ.
Ban đầu, diazoxide có thể kiểm soát tốt hạ đường huyết ở những bệnh nhân có u tiết insulin, đặc biệt là trong giai đoạn đầu, khi khối u còn nhỏ; nhưng diazoxide có thể trở nên không hiệu quả khi khối u phát triển. Cắt bỏ bằng phẫu thuật là phương pháp điều trị được lựa chọn; do đó, việc chẩn đoán hình ảnh cẩn thận trước phẫu thuật để xác định vị trí tổn thương (hoặc các tổn thương) trước khi phẫu thuật là rất quan trọng. Trong một loạt tám trường hợp ở trẻ em, chẩn đoán hình ảnh đã thành công trong việc xác định vị trí u tiết insulin trong 40% bằng MRI, 40% bằng siêu âm nội soi, 30% bằng chụp cắt lớp vi tính, và chỉ 20% bằng siêu âm bụng. Xạ hình Octreotide và chụp PET F-DOPA đôi khi cung cấp thêm tiện ích chẩn đoán. Tại phẫu thuật, sờ nắn và kiểm tra và siêu âm trong mổ nói chung đã thành công trong việc xác định và cắt bỏ u tiết insulin. Bệnh nhân mắc MEN1, do các đột biến trội của MENIN, có nguy cơ mắc các khối u nội tiết khác, bao gồm các u tuyến hoạt động hoặc không hoạt động của tuyến cận giáp, tuyến yên, và tuyến tụy và cần được theo dõi thường xuyên, cũng như xét nghiệm để xác định các thành viên gia đình bị ảnh hưởng.
Hạ đường huyết sau ăn sau phẫu thuật đường tiêu hóa (“Hội chứng dumping muộn”)
Các hormone và tín hiệu đường tiêu hóa—trục ruột-tiểu đảo—đóng một vai trò quan trọng trong việc điều hòa sự tiết insulin để đáp ứng với các bữa ăn. Sự gián đoạn của hệ thống này do phẫu thuật dạ dày có thể dẫn đến hạ đường huyết do giải phóng insulin sau ăn quá mức. Ở trẻ em, nguyên nhân phổ biến nhất là phẫu thuật dạ dày cho bệnh trào ngược dạ dày-thực quản (ví dụ, phẫu thuật Nissen); ở người lớn, nguyên nhân phổ biến nhất là phẫu thuật nối tắt dạ dày để điều trị béo phì nặng. “Hạ đường huyết do dinh dưỡng” này chỉ xảy ra sau ăn (tức là, phản ứng), chứ không phải khi đói, và được gây ra bởi sự giải phóng khuếch đại của peptide-1 giống glucagon từ ruột non. Rối loạn này đôi khi được gọi là hội chứng dumping muộn để phân biệt với hội chứng dumping sớm, được gây ra bởi sự làm rỗng dạ dày nhanh chóng gây ra các thay đổi thẩm thấu và các triệu chứng, chẳng hạn như hạ huyết áp, đổ mồ hôi, và tiêu chảy, ngay sau khi ăn.
Ở trẻ nhỏ, hạ đường huyết sau ăn sau phẫu thuật Nissen có thể đi kèm với các triệu chứng đường ruột của hội chứng dumping sớm, nhưng thường biểu hiện dưới dạng các triệu chứng thiếu glucose não và tự chủ đơn độc từ 1 đến 3 giờ sau bữa ăn. Hạ đường huyết có thể được xác nhận liên quan đến các triệu chứng sau một bữa ăn thông thường hoặc có thể được gây ra bởi một bữa ăn hỗn hợp chính thức hoặc nghiệm pháp dung nạp glucose đường uống. Mặc dù nghiệm pháp dung nạp glucose có phần không sinh lý, và những người bình thường có thể bị hạ đường huyết 3 đến 4 giờ sau khi tải glucose, nghiệm pháp dung nạp glucose được một số người ưa thích hơn vì là một xét nghiệm được tiêu chuẩn hóa hơn. Bệnh nhân mắc hội chứng dumping muộn hạ đường huyết có sự gia tăng bất thường của insulin huyết tương lên đến 200 đến 300 µU/mL trong 30 đến 60 phút đầu tiên sau khi uống glucose, tiếp theo là hạ đường huyết sau 3 đến 4 giờ. Thường cũng có một đỉnh tăng đường huyết rõ rệt ngay sau khi uống glucose do làm rỗng dạ dày nhanh chóng; tuy nhiên, điều này không phải lúc nào cũng xảy ra và không phải là nguyên nhân của hạ đường huyết.
Điều trị hạ đường huyết hội chứng dumping muộn có thể bao gồm các biện pháp ăn kiêng làm chậm quá trình làm rỗng dạ dày và giải phóng glucose tự do vào ruột non, chẳng hạn như ăn các bữa nhỏ thường xuyên gồm carbohydrate phức tạp, các bữa ăn nhẹ giữa các bữa ăn, tránh đường đơn, và cung cấp chất béo và protein cùng với carbohydrate. Trong các trường hợp nặng hơn, acarbose, một chất ức chế alpha-glucosidase làm chậm quá trình tiêu hóa tinh bột và các carbohydrate phức tạp khác, có thể hiệu quả với liều từ 25 đến 100 mg, với mỗi bữa ăn. Trong một số trường hợp, cần phải cho ăn qua ống thông dạ dày hoặc ống thông hỗng tràng liên tục.
Hạ đường huyết tăng insulin máu như một tiền triệu của khởi phát đái tháo đường.
Các trường hợp hạ đường huyết khi đói, nhưng phổ biến hơn là sau ăn, thỉnh thoảng đã được mô tả trước khi khởi phát đái tháo đường type 1 hoặc trong giai đoạn thuyên giảm “tuần trăng mật” khi nhu cầu insulin thấp. Trong Thử nghiệm Phòng ngừa Đái tháo đường-Type 1, những người họ hàng bậc một và bậc hai được theo dõi chặt chẽ của các bệnh nhân mắc đái tháo đường type 1 có nguy cơ mắc bệnh đái tháo đường cao (tức là, những người bị tiền đái tháo đường) được phát hiện có hạ đường huyết hóa học được phát hiện bằng hồ sơ glucose hàng quý với tỷ lệ 7,5 đợt trên 100 người-năm. Các đợt hạ đường huyết, với đường huyết được ghi nhận thấp hơn 50 mg/dL (2,8 mmol/L), xảy ra với tỷ lệ 2,6 đợt trên 100 người-năm. Không có báo cáo nào về các đợt hạ đường huyết nặng. Cơ chế rất có thể liên quan đến sự giải phóng insulin giai đoạn hai quá mức là hậu quả của một phản ứng insulin giai đoạn một khiếm khuyết; các cơ chế tiềm năng khác cũng đã được đề xuất, bao gồm tác động chậm của insulin, do các kháng thể kháng insulin hoặc giải phóng insulin thứ phát sau sự phá hủy viêm của các tế bào beta. Mối quan hệ giữa hạ đường huyết phản ứng và khởi phát đái tháo đường type 2 (“rối loạn insulin máu”) đã được giả định từ đầu những năm 1930, và được công nhận rộng rãi trong giới đái tháo đường học vào giữa thế kỷ 20. Đái tháo đường type 2 sớm cũng thường được đặc trưng bởi mất phản ứng insulin giai đoạn một với thức ăn, dẫn đến các đỉnh glucose cao hơn, tiếp theo là các đáy glucose thấp hơn. Trong hầu hết các trường hợp hạ đường huyết tiền triệu trong cả đái tháo đường type 1 và type 2, glucose huyết tương không đạt đến mức đủ thấp để gây ra các triệu chứng thiếu glucose não. Nếu cần, điều trị để tránh tăng đường huyết sau ăn quá mức, chẳng hạn như các bữa ăn có chỉ số đường huyết thấp, có thể hiệu quả. Trong đái tháo đường type 1 trong giai đoạn thuyên giảm, việc tăng liều insulin để giảm tăng đường huyết sau ăn có thể hữu ích.
Hạ đường huyết trong Đái tháo đường liên quan đến Xơ nang.
Trong bệnh xơ nang (CF), sự tiết insulin giai đoạn một bị suy giảm và sự tiết glucagon giảm khi dung nạp glucose xấu đi. Cả hạ đường huyết khi đói và phản ứng trong một nghiệm pháp dung nạp glucose thông thường là tương đối phổ biến trước khi khởi phát đái tháo đường liên quan đến xơ nang (CFRD); tuy nhiên, tần suất hạ đường huyết phản ứng có thể không phổ biến hơn so với những người không mắc CF. Trong một nghiên cứu, sự xuất hiện của hạ đường huyết sau một nghiệm pháp dung nạp glucose đường uống (OGTT) có liên quan đến nguy cơ tiến triển thành CFRD trong 10 năm thấp hơn, so với những người không bị hạ đường huyết.
Hạ đường huyết do u không phải tiểu đảo
Một số khối u không tiết insulin đôi khi có liên quan đến hạ đường huyết cận ung thư. Hầu hết các trường hợp liên quan đến các khối u ác tính lớn có nguồn gốc trung mô, biểu mô, hoặc tạo máu. Các trường hợp ở trẻ em là hiếm, nhưng đã được báo cáo với u nguyên bào thần kinh và u Wilms. Các cơn hạ đường huyết thường xảy ra khi đói và có liên quan đến nồng độ ceton và FFA huyết tương thấp không phù hợp, và với việc sử dụng glucose tăng, bắt chước sự dư thừa insulin. Nồng độ insulin, proinsulin, và C-peptide trong huyết tương thấp, và các loại thuốc ức chế tiết insulin (diazoxide, somatostatin) không hiệu quả, cho thấy sự kích hoạt của các thụ thể insulin bởi các yếu tố tuần hoàn khác. Trong hầu hết các trường hợp, nồng độ IGF-2 đã được phát hiện là tăng và được giả định là gây ra hạ đường huyết bằng cách phản ứng chéo với cả thụ thể insulin và IGF-1. IGF-2 thường được sản xuất ở gan và một lượng hạn chế được tiết vào tuần hoàn gắn với IGFBP-3 và tiểu đơn vị acid-labile (ALS). IGF-2 do khối u sản xuất có thể hoạt động mạnh hơn do quá trình xử lý không hoàn chỉnh và liên kết kém hiệu quả với IGFBP-3 và ALS hoặc có thể có cấu trúc bất thường—một dạng phân tử lớn được gọi là big IGF-2. Ngoài việc kích thích thụ thể insulin, việc kích thích thụ thể IGF-2 có thể điều hòa giảm sự tiết GH, dẫn đến nồng độ IGFBP-3 thấp.
Việc chứng minh nồng độ IGF-2 huyết tương tăng với sự ức chế của cả nồng độ IGF-1 và insulin xác nhận hạ đường huyết do khối u loại này. Nếu khối u không thể được loại bỏ hoàn toàn, một can thiệp có thể hiệu quả là điều trị bằng GH để tăng nồng độ IGFBP-3 và giảm lượng IGF tự do, do đó cải thiện tình trạng hạ đường huyết.
Cần lưu ý rằng không phải tất cả các trường hợp hạ đường huyết cận ung thư đều qua trung gian IGF-2. Hạ đường huyết đi kèm với nhiễm toan lactic, được cho là do tăng chuyển hóa glucose kỵ khí, đã được báo cáo trong các bệnh u lympho và bệnh bạch cầu.
Các bệnh dự trữ Glycogen
Các bệnh dự trữ glycogen (GSD) hay glycogenoses bao gồm một số bệnh di truyền gây ra bởi sự thiếu hụt các enzyme điều hòa quá trình tổng hợp hoặc phân hủy glycogen, dẫn đến tăng dự trữ glycogen ở một số mô, đặc biệt là gan và cơ. Glycogen là dạng dự trữ carbohydrate ở người: glycogen gan đóng vai trò là một kho dự trữ glucose quan trọng trong khoảng thời gian giữa các bữa ăn; glycogen trong cơ và các mô khác không thể được giải phóng dưới dạng glucose tự do, nhưng được sử dụng cho các nhu cầu nội sinh. Quá trình ly giải glycogen gan cung cấp một phần lớn glucose đi vào tuần hoàn, bắt đầu trong vòng vài giờ sau khi ăn, và tiếp tục cho đến khi kho dự trữ glycogen cạn kiệt.
Glycogen là một polymer phân nhánh cao của các gốc glucose, hầu hết trong số đó tạo thành các chuỗi thẳng được liên kết bằng liên kết α-1,4-glycosidic. Các nhánh được tạo ra bởi các liên kết α-1,6-glycosidic, xảy ra trung bình cứ 10 gốc một lần. Sau khi ăn carbohydrate, nồng độ glucose và insulin trong huyết tương tăng lên và glycogen mới được tổng hợp.
Quá trình tổng hợp và phân hủy glycogen trong gan theo các con đường riêng biệt bắt đầu và kết thúc bằng glucose 1-phosphate (xem Hình 23.5). Gan có khả năng thấm tự do với glucose, đầu tiên được chuyển đổi thành glucose 6-phosphate trước khi có thể đi vào một trong nhiều con đường chuyển hóa. Glucose 6-phosphate có thể được chuyển đổi thuận nghịch thành glucose-1-phosphate, là điểm khởi đầu cho quá trình tổng hợp glycogen. Ngoài ra, glucose-6-phosphate có thể được thủy phân thành glucose bởi glucose-6-phosphatase (G6Pase), hoặc nó có thể được chuyển hóa qua con đường đường phân thành pyruvate và lactate, hoặc qua con đường pentose phosphate, thành ribose-5-phosphate, một tiền chất của quá trình tổng hợp nucleotide. Glycogen synthase xúc tác sự hình thành các liên kết α-1,4. Một enzyme tạo nhánh hình thành các liên kết α-1,6 làm cho glycogen trở thành một polymer phân nhánh.
Quá trình phân hủy glycogen (ly giải glycogen) đòi hỏi sự tương tác tuần tự của một số enzyme. Đầu tiên, phosphorylase gan liên tiếp cắt các liên kết 1,4 cho đến khi còn bốn đơn vị glucosyl của điểm nhánh. Sau đó, 4-α-glucanotransferase bộc lộ các điểm nhánh liên kết 1,6 bằng cách chuyển ba gốc glucosyl đến một nơi khác trên phân tử glycogen. Amylo-1,6-glucosidase, enzyme khử nhánh, sau đó tách các đơn vị glucosyl liên kết 1,6. Do đó, các hành động tuần tự của phosphorylase và enzyme khử nhánh giải phóng các đơn vị glucose dự trữ; hoạt động của phosphorylase tạo ra glucose 1-phosphate và enzyme khử nhánh giải phóng glucose tự do. Trong khi nhịn đói, enzyme khử nhánh huy động khoảng 8% glycogen gan dưới dạng glucose tự do; phần còn lại đòi hỏi hoạt động của G6Pase gan.
Dấu hiệu đặc trưng của các bệnh glycogenosis gan là hạ đường huyết khi đói. Các loại glycogenosis gan, sự thiếu hụt enzyme cụ thể của chúng, các mô bị ảnh hưởng, phương thức di truyền và vị trí nhiễm sắc thể của các gen liên quan được trình bày trong Bảng 23.3. Bảng 23.4 tóm tắt các đặc điểm sinh hóa chính của các bệnh glycogenosis gan (loại 0, I, III, VI và IX) thường gây hạ đường huyết.
Bảng 23.3 Các bệnh dự trữ Glycogen ở gan
| Rối loạn | Mô bị ảnh hưởng | Enzyme | Di truyền | Gen | Nhiễm sắc thể | Hạ đường huyết khi đói |
|---|---|---|---|---|---|---|
| GSD Type 0 | Gan | Glycogen synthase | Lặn NST thường | GYS2 | 12p12.2 | Nhẹ đến trung bình |
| GSD Type la | Gan, thận, ruột | Glucose 6-phosphatase | Lặn NST thường | G6PC | 17q21 | Nặng |
| GSD Type Ib | Gan | Chất vận chuyển Glucose 6-phosphate (T1) | Lặn NST thường | SLC37A | 11q23 | Nặng |
| GSD Type Illa | Gan, cơ, tim | Enzyme khử nhánh Glycogen | Lặn NST thường | AGL | 1p21 | Nhẹ đến trung bình |
| GSD Type Illb | Gan | Enzyme khử nhánh Glycogen | Lặn NST thường | AGL | 1p21 | Nhẹ đến trung bình |
| GSD Type IV | Gan | Enzyme tạo nhánh | Lặn NST thường | GBE | 3p12.2 | Kèm suy gan |
| GSD Type VI | Gan | Glycogen phosphorylase | Lặn NST thường | PYGL | 14q21-22 | Nhẹ |
| GSD Type IXa | Gan, hồng cầu, bạch cầu | Dạng gan của tiểu đơn vị α của phosphorylase kinase | Liên kết X | PHKA2 | Xp22.1-p22.2 | Nhẹ đến trung bình |
| GSD Type IXb | Gan, cơ, hồng cầu, bạch cầu | Tiểu đơn vị β của phosphorylase kinase gan và cơ | Lặn NST thường | PHKB | 16q12-q13 | Nhẹ đến trung bình |
| GSD Type IXC | Gan | Dạng tinh hoàn/gan của tiểu đơn vị γ của phosphorylase kinase | Lặn NST thường | PHKG2 | 16p11-p12 | Nhẹ |
| GSD Type XI | Gan, tụy, ruột, và thận | Chất vận chuyển Glucose 2 (GLUT2) | Lặn NST thường | SCL2A2 | 3q26.1-q26.3 | Nhẹ |
Bảng 23.4 Đặc điểm sinh hóa của các bệnh Glycogenosis gan phổ biến nhất
| Type | Triglyceride | Acid uric | Lactate | Đáp ứng với Glucose đường uống (Lactate) | Đáp ứng với Glucose đường uống (Glucose) | Đáp ứng với Glucagon 4-8 giờ sau bữa ăn (Glucose) | Đáp ứng với Glucagon 4-8 giờ sau bữa ăn (Lactate) | Đáp ứng với Glucagon 2 giờ sau bữa ăn (Glucose) | Đáp ứng với Glucagon 2 giờ sau bữa ăn (Lactate) |
|---|---|---|---|---|---|---|---|---|---|
| GSD-0 | N | N | N | ↓↓ | ↑↑ | 0 | 0-↑ | ↑ | ↑↑ |
| GSD-1 | ↑↑↑ | ↑↑↑ | ↑↑ | ↑↑ | ↑ | 0 | ↑↑↑ | 0 | ↑↑ |
| GSD-III | ↑↑ | N | N | ↓ | ↑ | 0 | 0 | 0 | ↑ |
| GSD-VI, IX | ↑ | N | N | ↓ | ↑ | 0-↑ | 0 | ↑ | ↑ |
0, không tăng; 0-↑, tăng không ổn định; ↑, tăng nhẹ; ↑↑, tăng trung bình; ↑↑↑, tăng rõ rệt; ↓, giảm nhẹ; ↓↓, giảm trung bình; N, bình thường. Những đối tượng nghi ngờ mắc GSD-I không được phép nhịn đói quá 4 giờ. Sau một bữa ăn chứa glucose. Xem văn bản để giải thích tại sao có đáp ứng đường huyết nhẹ ngay sau bữa ăn ở các loại GSD được liệt kê.
Thiếu hụt Glycogen Synthase (GSD Type 0)
Đột biến trong gen glycogen synthase (GYS2) trên nhiễm sắc thể 12p12.2 gây ra một rối loạn di truyền lặn trên nhiễm sắc thể thường hiếm gặp, đặc trưng bởi không có khả năng dự trữ glycogen gan, dẫn đến hàm lượng glycogen gan giảm rõ rệt 4 đến 6 giờ sau bữa ăn (~0,5 g/100 g trọng lượng ướt của gan so với 5 g/100 g ở trẻ bình thường), nhưng hàm lượng glycogen cơ bình thường. Glucose ăn vào được chuyển đổi ưu tiên thành lactate. Các triệu chứng hạ đường huyết buổi sáng xuất hiện khi ngừng cho ăn vào ban đêm. GSD type 0 có một kiểu hình sinh hóa đặc trưng: hạ đường huyết khi đói và tăng ceton máu xen kẽ với tăng đường huyết thoáng qua kèm theo glucose niệu và tăng acid lactic máu vào ban ngày, đặc biệt là sau các bữa ăn nhiều carbohydrate. Trong cơn hạ đường huyết khi đói, nồng độ các hormone đối kháng tăng phù hợp, nồng độ insulin thấp phù hợp, và ceton tăng.
Không giống như các GSD gan khác, gan không to; do đó, việc gọi thiếu hụt glycogen synthase là một bệnh dự trữ là một cách gọi sai. Mặc dù hiếm gặp, rối loạn này nên được xem xét trong chẩn đoán phân biệt “hạ đường huyết do ceton”. Sau một đêm nhịn đói, glucose đường uống (1,75 g/kg) gây tăng đường huyết và tăng acid lactic máu, trong khi glucagon (0,03 mg/kg tiêm bắp [IM]) thường không có tác dụng đáng kể lên nồng độ glucose huyết tương. Xét nghiệm di truyền các đột biến trong GYS2 có sẵn trên thị trường.
Mục tiêu điều trị là ngăn ngừa hạ đường huyết và tăng ceton máu vào ban đêm, và tăng đường huyết và tăng acid lactic máu vào ban ngày. Hạ đường huyết khi đói và nhiễm ceton có thể được ngăn ngừa bằng cách cho ăn bột ngô sống (uncooked cornstarch – UCS) trước khi đi ngủ, 1 đến 1,5 g/kg. Trong khi bị bệnh, có thể dùng một liều bột ngô tương tự mỗi 6 giờ để ngăn ngừa hạ đường huyết. Vào ban ngày, bệnh nhân được cho ăn thường xuyên (ví dụ, mỗi 4 giờ); vì quá trình tân tạo glucose còn nguyên vẹn, chế độ ăn nên chứa một lượng protein tăng lên để cung cấp cơ chất cho quá trình tân tạo glucose và giảm lượng carbohydrate (chủ yếu là carbohydrate phức tạp, chỉ số đường huyết thấp) để giảm thiểu tăng đường huyết và tăng acid lactic máu sau ăn. Chế độ ăn kiêng này làm giảm các triệu chứng, đảo ngược các bất thường sinh hóa và cải thiện sự tăng trưởng.
Thiếu hụt Glucose-6-Phosphatase (GSD Type I, Bệnh Von Gierke, Bệnh glycogenosis gan-thận)
Lần đầu tiên được mô tả vào năm 1929, GSD type I là một rối loạn di truyền lặn trên nhiễm sắc thể thường gây ra bởi sự thiếu hoạt động của enzyme gan G6Pase, hoặc do thiếu chính G6Pase hoặc do thiếu enzyme vận chuyển G6P translocase (G6PT). G6Pase xúc tác bước cuối cùng trong quá trình sản xuất glucose từ glucose-6-phosphate. Sự thiếu hụt enzyme này làm suy giảm sản xuất glucose từ cả quá trình ly giải glycogen và tân tạo glucose (xem Hình 23.5). Giảm sản xuất glucose gây hạ đường huyết trong khoảng thời gian giữa các bữa ăn, và tăng sản xuất lactate, acid uric và triglyceride. Glycogen và triglyceride tích tụ trong gan, dẫn đến gan to rõ rệt; glycogen cũng tích tụ trong thận và niêm mạc ruột.
Hệ thống enzyme G6Pase nằm trong màng lưới nội chất (ER) và bao gồm nhiều tiểu đơn vị. Tiểu đơn vị xúc tác, chuyển đổi glucose-6-phosphate thành glucose, hướng vào bên trong ER. Ba hệ thống vận chuyển vận chuyển cơ chất, Glucose-6-phosphate, và các sản phẩm, phosphate, orthophosphate vô cơ và glucose, qua màng ER. chất vận chuyển glucose-6-phosphate vận chuyển glucose-6-phosphate vào và phosphate ra khỏi ER; GLUT2 vận chuyển glucose ra khỏi ER.
Khoảng 80% bệnh nhân mắc GSD-I có hoạt động xúc tác của hệ thống G6Pase bị thiếu hụt dẫn đến GSD type la (GSD-la). Khoảng 100 đột biến khác nhau đã được tìm thấy trong gen G6Pase, G6PC, nằm trên nhiễm sắc thể 17q21. Bệnh nhân mắc GSD type Ib, gây ra bởi sự thất bại trong việc vận chuyển glucose-6-phosphate vào lòng ER, có các đột biến (khoảng 80 đột biến đã được mô tả) trong gen G6PT, SLC37A4, trên nhiễm sắc thể 11q23.
Tỷ lệ mắc GSD-1 ước tính là một trong 100.000 ca sinh trong dân số chung; tỷ lệ hiện mắc là một trong 20.000 ở người Do Thái Ashkenazi. Các triệu chứng biểu hiện thay đổi theo độ tuổi. Hạ đường huyết có triệu chứng có thể xuất hiện ngay sau khi sinh; tuy nhiên, hầu hết các bệnh nhân không có triệu chứng, miễn là họ được cho ăn thường xuyên chứa đủ glucose để ngăn ngừa hạ đường huyết. Các triệu chứng hạ đường huyết thường chỉ xuất hiện khi khoảng cách giữa các cữ ăn tăng lên, chẳng hạn như khi trẻ bắt đầu ngủ suốt đêm hoặc khi một bệnh xen kẽ làm gián đoạn các kiểu ăn uống bình thường. Tình trạng này có thể không được nhận ra cho đến khi trẻ được vài tháng tuổi và gan to và bụng chướng được ghi nhận trong một cuộc kiểm tra thể chất định kỳ. Bệnh nhân có thể biểu hiện thở nhanh (do nhiễm toan lactic) và sốt nhẹ không có nhiễm trùng rõ ràng. Bệnh nhân không được điều trị có thể có ngoại hình giống Cushing (khuôn mặt tròn như thiên thần hoặc “giống búp bê”), chậm tăng trưởng và chậm phát triển vận động. Phát triển xã hội và nhận thức thường không bị ảnh hưởng trừ khi trẻ bị tổn thương não do hạ đường huyết nặng tái phát.
Trong thời kỳ nhũ nhi, nồng độ đường huyết thường giảm xuống dưới 50 mg/dL (2,8 mmol/L) trong vòng 3 đến 4 giờ sau khi ăn. Khoảng thời gian nhịn đói dài hơn gây hạ đường huyết nặng hơn kèm theo tăng acid lactic máu và nhiễm toan chuyển hóa. Mặc dù giá trị glucose huyết tương trong khoảng 20 đến 50 mg/dL (1,1-2,8 mmol/L), bệnh nhân trải qua ít triệu chứng tự chủ hoặc thiếu glucose não của hạ đường huyết, phản ánh khả năng của não sử dụng lactate làm nhiên liệu thay thế và sự điều hòa giảm các phản ứng tự chủ sau hạ đường huyết kéo dài tái phát. Insulin huyết thanh bị ức chế phù hợp và các hormone đối kháng tăng lên trong một nỗ lực vô ích để kích thích ly giải glycogen và tân tạo glucose, thay vào đó dẫn đến nhiễm toan lactic và ly giải lipid quá mức, gây ra tăng acid béo máu rõ rệt. Thiếu hụt tose-1-6-bisphosphatase ở trẻ sơ sinh có thể giống GSD I, với hạ đường huyết và nhiễm toan lactic tái phát (xem Hình 23.6); tuy nhiên, trái ngược với GSD-1, việc sử dụng glucagon gây ra một phản ứng đường huyết nhanh chóng ở trạng thái mới ăn và xét nghiệm di truyền FBP1 xác nhận chẩn đoán (xem Bảng 23.4).
Huyết thanh của bệnh nhân không được điều trị có thể có lipid, với nồng độ triglyceride cực cao (thường >500 mg/dL) và nồng độ phospholipid và cholesterol lipoprotein tổng và mật độ thấp tăng vừa phải; nồng độ cholesterol lipoprotein mật độ cao thấp. Nồng độ triglyceride cao là do tăng tổng hợp ở gan (huy động FFA từ mô mỡ để đáp ứng với hạ đường huyết), suy giảm quá trình sinh ceton, và giảm thanh thải triglyceride, thứ phát sau giảm hoạt động của lipoprotein lipase. U vàng phát ban có thể xuất hiện trên các bề mặt duỗi của các chi và trên mông và tăng triglyceride máu nặng (>1000 mg/dL) có thể gây viêm tụy cấp.
GSD I không được điều trị hoặc kiểm soát kém có liên quan đến sự tăng rõ rệt của acid uric huyết tương. Tăng acid uric máu là do tăng phân hủy adenosine monophosphate (AMP) thành inosine và acid uric do mất ức chế AMP deaminase là hậu quả của sự tích tụ glucose-6-phosphate và giảm cả ATP và phosphate vô cơ trong tế bào gan. Một cơ chế tương tự gây tăng acid uric máu trong thiếu hụt fructose-1,6-bisphosphatase (xem phần sau về Rối loạn di truyền của tân tạo Glucose) do sự tích tụ của fructose-1,6-phosphate và là lý do fructose không được sử dụng trong dịch truyền tĩnh mạch.
Mặc dù hạ đường huyết có triệu chứng trở nên ít nghiêm trọng hơn theo tuổi tác, tuy nhiên, nếu không có liệu pháp đầy đủ, sự tăng trưởng sẽ bị còi cọc và dậy thì bị trì hoãn. Tuy nhiên, khi liệu pháp glucose liên tục được bắt đầu sớm trong đời và kiểm soát chuyển hóa tốt lâu dài được duy trì, bệnh nhân có thể tăng trưởng và phát triển bình thường.
Suy giảm chức năng tiểu cầu gây ra xu hướng chảy máu biểu hiện bằng chảy máu cam tái phát hoặc rỉ máu sau phẫu thuật nha khoa hoặc phẫu thuật khác. Giảm độ bám dính của tiểu cầu, kết tập tiểu cầu bất thường và suy giảm giải phóng adenosine diphosphate (ADP) để đáp ứng với collagen và epinephrine đã được quan sát thấy. Các khiếm khuyết trong chức năng tiểu cầu là thứ phát sau các bất thường chuyển hóa toàn thân và được khắc phục bằng cách cải thiện trạng thái chuyển hóa.
Thiếu máu là phổ biến ở trẻ em mắc GSD I. Nguyên nhân của thiếu máu là đa yếu tố và bao gồm thiếu sắt và các chất dinh dưỡng khác, nhiễm toan lactic mạn tính, mất máu do rong kinh, bệnh thận giai đoạn cuối (ESRD), và viêm ruột kết (ở bệnh nhân mắc GSD Ib). Bệnh nhân có u tuyến gan có thể bị thiếu máu kháng sắt thứ phát sau biểu hiện hepcidin bất thường. Các triệu chứng đường ruột thường không phải là một đặc điểm nổi bật nhưng có thể bao gồm tiêu chảy.
Thận to dễ dàng được chứng minh bằng siêu âm. Thiếu hụt G6Pase dẫn đến lắng đọng glycogen trong thận và làm xáo trộn quá trình chuyển hóa của các tế bào ống thận, dẫn đến thiếu hụt năng lượng tương đối. Tăng lưu lượng máu thận và tốc độ lọc cầu thận (GFR) có thể là một cơ chế bù trừ cho sự thiếu hụt năng lượng nội bào. Các biểu hiện ở thận bao gồm rối loạn chức năng ống thận gần và xa, cũng như tổn thương cầu thận có thể dẫn đến ESRD. Rối loạn chức năng ống thận gần (glucose niệu, phosphaturia, hạ kali máu và acid amin niệu toàn thể) có thể hồi phục khi kiểm soát sinh hóa của bệnh được cải thiện. Một số bệnh nhân có khiếm khuyết toan hóa ống thận xa liên quan đến giảm citrat niệu và tăng calci niệu, dẫn đến sỏi thận và nhiễm calcin thận. Tăng bài tiết albumin qua nước tiểu có thể được quan sát thấy ở thanh thiếu niên. Tổn thương thận tiến triển nặng với protein niệu, tăng huyết áp và giảm độ thanh thải creatinine do xơ cứng cầu thận cục bộ phân đoạn và xơ hóa kẽ, có thể được thấy ở người trẻ tuổi. Bệnh nhân có nồng độ lactate máu, lipid huyết thanh và acid uric tăng liên tục dường như có nguy cơ cao phát triển bệnh thận. Việc bình thường hóa các thông số chuyển hóa làm giảm protein niệu, và liệu pháp tối ưu được thực hiện tại hoặc trước 1 tuổi có thể trì hoãn, ngăn ngừa hoặc làm chậm sự tiến triển của bệnh thận.
Sự phát triển của u tuyến gan là một biến chứng phổ biến, xảy ra ở 75% bệnh nhân khi họ đến tuổi trưởng thành. Mặc dù các u tuyến thường được quan sát thấy lần đầu tiên trong thập kỷ thứ hai và thứ ba của cuộc đời, chúng có thể xuất hiện trước tuổi dậy thì. Các u tuyến có thể xuất huyết hoặc trở thành ung thư biểu mô tế bào gan ác tính. Siêu âm là phương pháp sàng lọc ưa thích cho các u tuyến gan. Khi nghi ngờ ác tính, nên thực hiện MRI và đo alpha-fetoprotein huyết thanh.
Các nghiên cứu X-quang đã chứng minh tình trạng thiếu xương, và các nghiên cứu bệnh học đã cho thấy loãng xương, không có bằng chứng về các bất thường trong chuyển hóa canxi, phosphate, cận giáp hoặc vitamin D. Hàm lượng khoáng chất trong xương giảm so với trẻ em bình thường cùng tuổi. Các rối loạn nội tiết và chuyển hóa, bao gồm tăng cortisol máu, kháng GH, dậy thì muộn và nhiễm toan lactic, có thể giải thích cho việc giảm khoáng hóa xương.
Kinh nguyệt không đều và rậm lông do tăng androgen là không phổ biến; tuy nhiên, ở tất cả các loại GSD gan, siêu âm đã cho thấy tỷ lệ cao buồng trứng đa nang về mặt hình thái (ngay cả ở trẻ em trước tuổi dậy thì), ý nghĩa lâm sàng của nó vẫn chưa rõ ràng.
Mặc dù bản thân tim không bị ảnh hưởng trong GSD 1 (không giống như GSD III), tăng huyết áp có thể phát triển liên quan đến bệnh thận. Tăng huyết áp phổi biểu hiện trong thập kỷ thứ hai hoặc thứ ba của cuộc đời và dẫn đến tử vong do suy tim tiến triển cũng đã được mô tả.
Bệnh nhân mắc GSD Ib có các triệu chứng tương tự như GSD la, nhưng có thêm giảm bạch cầu trung tính liên tục hoặc theo chu kỳ với mức độ nghiêm trọng khác nhau, từ nhẹ đến mất bạch cầu hạt hoàn toàn, và có liên quan đến các bệnh nhiễm khuẩn tái phát. Giảm bạch cầu trung tính là hậu quả của sự trưởng thành tủy xương bị xáo trộn và đi kèm với các khiếm khuyết chức năng của bạch cầu trung tính và bạch cầu đơn nhân lưu hành. Một loạt trường hợp gần đây cho thấy việc bổ sung vitamin E có thể cải thiện tình trạng giảm bạch cầu trung tính và giảm tỷ lệ nhiễm trùng. Bệnh nhân thường phát triển một bệnh viêm ruột giống bệnh Crohn, đáp ứng với điều trị bằng yếu tố kích thích dòng bạch cầu hạt (G-CSF). Trẻ em mắc GSD Ib dễ bị các biến chứng răng miệng (loét niêm mạc tái phát, viêm nướu và bệnh nha chu tiến triển nhanh), áp xe quanh hậu môn đau đớn, và có tỷ lệ mắc bệnh tự miễn tuyến giáp và suy giáp nguyên phát tăng.
Nghiên cứu chẩn đoán. Trong thời kỳ nhũ nhi, glucose huyết tương thường giảm xuống dưới 50 mg/dL (2,8 mmol/L) trong vòng 3 đến 4 giờ sau khi ăn và đi kèm với tăng acid lactic máu và nhiễm toan chuyển hóa. Huyết thanh có thể đục hoặc trắng sữa, với triglyceride rất cao và nồng độ cholesterol tăng vừa phải. Nồng độ acid uric huyết thanh, aspartate aminotransferase và alanine aminotransferase thường tăng. Phương pháp đơn giản nhất để xác định bản chất có thể có của sự thiếu hụt enzyme ở một đứa trẻ bị nghi ngờ mắc GSD I là lấy các mẫu máu nối tiếp để đo các chất chuyển hóa (glucose, lactate, FFA, ceton và acid uric) trong tối đa 6 giờ (hoặc cho đến khi glucose huyết tương giảm xuống ≤50 mg/dL), sau khi uống glucose (1,75 g/kg), hoặc 30 phút sau khi ngừng truyền glucose tĩnh mạch hoặc qua sonde dạ dày liên tục qua đêm. Hạ đường huyết, cùng với nồng độ lactate máu tăng, kết hợp với nghiệm pháp kích thích glucagon khi no bất thường, sẽ xác định chẩn đoán. Ở trẻ bình thường, nghiệm pháp kích thích glucagon sau ăn 2 giờ làm cho glucose huyết tương tăng thêm 30 mg/dL (1,7 mmol/L) hoặc hơn trong vòng 30 phút mà không có sự thay đổi nồng độ lactate, trong khi ở GSD 1, phản ứng glucose là tối thiểu và có sự gia tăng rõ rệt của lactate. Vì việc sử dụng glucagon có thể dẫn đến tình trạng nhiễm toan chuyển hóa và mất bù trầm trọng hơn ở GSD I, nó không còn được khuyến cáo trong đánh giá chẩn đoán GSD I nghi ngờ. Phân tích đột biến của các gen G6PC (GSD la) và SLC37A4 (GSD lb) được khuyến nghị để xác nhận GSD type I. Vì GSD la phổ biến hơn, trừ khi có giảm bạch cầu trung tính, nên giải trình tự toàn bộ G6PC được khuyến nghị trước tiên. Lưu ý rằng mặc dù giảm bạch cầu trung tính gợi ý GSD Ib, nó cũng đôi khi được thấy ở GSD la và số lượng bạch cầu trung tính có thể bình thường ở bệnh nhân mắc GSD Ib trong những năm đầu đời. Sinh thiết gan để đo hoạt độ G6Pase có thể cần thiết ở những bệnh nhân hiếm gặp không có đột biến gen có thể xác định được. Ở hầu hết các cá nhân mắc GSD Ia, hoạt độ enzyme G6Pase <10% so với bình thường. Hoạt độ G6Pase phải được phân tích ở cả vi thể nguyên vẹn và bị phá vỡ hoàn toàn, vì hoạt độ enzyme bình thường hóa khi đông lạnh làm phá vỡ tính toàn vẹn của ER. Vì lý do này, hoạt độ enzyme G6Pase trên mô sinh thiết gan đông lạnh nhanh sẽ không phát hiện được GSD Ib, và cần có mô sinh thiết gan tươi (chưa đông lạnh) để phân tích chính xác hoạt độ G6PT. Bản thân glycogen có hình dạng bình thường. Siêu âm bụng cho thấy gan to và thận to không có lách to. Thận to là đặc điểm của GSD I và không phải là một đặc điểm của các GSD khác.
Điều trị bao gồm cung cấp một nguồn glucose liên tục trong chế độ ăn để ngăn glucose huyết tương giảm xuống dưới ngưỡng điều hòa ngược glucose. Khi hạ đường huyết được ngăn ngừa bằng cách cung cấp một lượng glucose thích hợp trong suốt cả ngày và đêm, các bất thường sinh hóa được cải thiện, kích thước gan giảm, xu hướng chảy máu được đảo ngược và sự tăng trưởng được cải thiện. Các mục tiêu sinh hóa được khuyến nghị: glucose huyết tương lớn hơn 70 đến 75 mg/dL (3,9-4,2 mmol/L), triglyceride dưới 500 mg/dL, và acid uric dưới 7,5 mg/dL.
Các phương pháp khác nhau có thể được sử dụng để cung cấp một nguồn glucose liên tục với tốc độ đủ để đáp ứng nhu cầu glucose trong khoảng thời gian giữa các bữa ăn: tiêm tĩnh mạch, qua đường tiêu hóa bằng cách truyền qua sonde dạ dày (hoặc sonde mũi-dạ dày hoặc sonde mở dạ dày), hoặc bằng cách sử dụng thực phẩm có chỉ số đường huyết thấp. Trong số các loại thực phẩm sau, bột ngô sống (UCS) có các đặc tính phù hợp nhất được mô tả cho đến nay. Lượng glucose tối thiểu cần thiết ở trạng thái cơ bản có thể được tính bằng công thức tính tốc độ sản xuất glucose cơ bản: y = 0.0014x³ - 0.214x² + 10.411x - 9.084, trong đó y = mg glucose mỗi phút và x = trọng lượng cơ thể lý tưởng tính bằng kilogam. Lượng và/hoặc lịch trình sử dụng glucose hoặc UCS được điều chỉnh, nếu cần, dựa trên kết quả theo dõi lâm sàng và sinh hóa.
Trẻ sơ sinh mắc GSD I thường cần được cho ăn mỗi 2 đến 3 giờ bằng một loại sữa công thức không chứa lactose hoặc sucrose. Sữa công thức phải chứa một polymer của glucose (chất rắn xi-rô ngô, maltodextrin) sẽ cung cấp một lượng glucose bằng với tốc độ sản xuất glucose được tính toán. Nếu việc cho ăn vào ban đêm gặp khó khăn, có thể cho ăn liên tục qua đêm, sử dụng cùng một loại sữa công thức, qua sonde mũi-dạ dày hoặc sonde mở dạ dày được kiểm soát bằng bơm truyền.
Bột ngô sống (UCS) dùng đường uống hoạt động như một kho dự trữ glucose trong ruột được hấp thu chậm vào tuần hoàn. Ở nhiều trung tâm, UCS đã thay thế việc cho ăn thường xuyên ban ngày bằng glucose hoặc các polymer glucose và truyền glucose qua sonde dạ dày liên tục qua đêm. Nó đã được sử dụng thành công ở trẻ sơ sinh từ 8 tháng tuổi. UCS được cho dưới dạng hỗn dịch trong nước hoặc đồ uống có chất làm ngọt nhân tạo (ví dụ: Kool-Aid) hoặc trong sữa công thức cho trẻ sơ sinh, cách nhau 3 đến 5 giờ vào ban ngày, và cách nhau 4 đến 6 giờ qua đêm. Lượng cho được xác định bằng cách nhân khoảng thời gian giữa các cữ ăn với nhu cầu glucose hàng giờ được tính toán cho trọng lượng cơ thể lý tưởng. Một muỗng canh (8 g) UCS chứa 7,3 g carbohydrate. Lịch trình và lượng UCS cho ăn gián đoạn tối ưu cho bệnh nhân ở các độ tuổi khác nhau được xác định theo kinh nghiệm bằng cách theo dõi chuyển hóa để đảm bảo rằng các mục tiêu sinh hóa đang được đạt được.
Bột ngô sáp giải phóng kéo dài (Glycosade, Vitaflo, International Ltd., Liverpool, Anh) đã được phê duyệt ở Anh vào năm 2009 để quản lý GSD 1 và được phát hành tại Hoa Kỳ dưới dạng thực phẩm y tế vào năm 2012. Các nghiên cứu hiệu quả ở trẻ em từ 5 tuổi đã cho thấy sự kéo dài khả năng chịu đói từ trung bình 4,1 đến 7,8 giờ, sử dụng bột ngô giải phóng kéo dài so với UCS tiêu chuẩn. Không dung nạp đường tiêu hóa và làm trầm trọng thêm bệnh viêm là các tác dụng phụ phổ biến của công thức giải phóng kéo dài.
Khi cung cấp đủ glucose ngoại sinh, tăng acid uric máu và tăng lipid máu đáng kể thường được phục hồi về gần mức bình thường. Nếu tăng acid uric máu nặng vẫn tồn tại, allopurinol, một chất ức chế xanthine oxidase (5-10 mg/kg/ngày dưới dạng một liều duy nhất hàng ngày hoặc chia mỗi 12 giờ), làm giảm hiệu quả acid uric huyết thanh về mức bình thường. Các thuốc hạ lipid (gemfibrozil hoặc fenofibrate) được chỉ định khi tăng lipid máu nặng dai dẳng, mặc dù đã điều trị glucose tối ưu, gây ra nguy cơ viêm tụy cấp đáng kể.
Chất béo trong chế độ ăn nên được giới hạn ở mức dưới 20% tổng năng lượng nạp vào, được phân bổ đều giữa các chất béo không bão hòa đơn, không bão hòa đa và bão hòa. Cholesterol trong chế độ ăn được giới hạn ở mức dưới 300 mg/ngày. Carbohydrate thường cung cấp 60% đến 65% lượng calo hàng ngày. Trong tổng số calo hàng ngày, 30% đến 45% được kê đơn (cả lượng và lịch trình) dưới dạng UCS. Hầu hết carbohydrate còn lại trong chế độ ăn, lý tưởng, nên là tinh bột có chỉ số đường huyết thấp. Với nhu cầu glucose được kê đơn, tổng lượng calo nạp vào được quyết định bởi sự thèm ăn của trẻ, miễn là tốc độ tăng cân không quá mức. Chuyên gia dinh dưỡng phải đảm bảo rằng bệnh nhân đang tiêu thụ đủ lượng protein, chất béo, khoáng chất và vitamin để hỗ trợ tăng trưởng tối ưu. Khi đủ glucose được kê đơn để duy trì đường huyết bình thường, các sản phẩm sữa và trái cây, mặc dù chứa galactose và fructose, tương ứng, có thể được sử dụng một cách tiết kiệm để cung cấp các chất dinh dưỡng, khoáng chất và vitamin thiết yếu.
Bệnh thận. Thuốc ức chế men chuyển angiotensin (ACE) và thuốc chẹn thụ thể angiotensin đã được báo cáo là cải thiện GFR ở những bệnh nhân có tình trạng tăng lọc. Thuốc lợi tiểu thiazide và citrate đường uống có thể giúp ngăn ngừa nhiễm calcin thận và sỏi thận. Nên tránh dùng thuốc lợi tiểu quai vì nguy cơ tăng calci niệu.
Thiếu máu. Nên tối ưu hóa lượng sắt, vitamin B12 và acid folic. Bệnh nhân có u tuyến gan có thể phát triển thiếu máu kháng sắt do biểu hiện bất thường của hepcidin, một chất điều hòa chính của chuyển hóa sắt, trong khối u. Bổ sung Erythropoietin có thể cần thiết ở những bệnh nhân bị ESRD.
Giảm bạch cầu trung tính. G-CSF có hiệu quả trong điều trị giảm bạch cầu trung tính và nhiễm trùng ở bệnh nhân GSD-Ib; nên theo dõi lách to vì đây là biến chứng nghiêm trọng nhất của liệu pháp G-CSF ở nhóm dân số này.
Theo dõi. Theo dõi glucose huyết tương thường xuyên trước bữa ăn, trước khi dùng bột ngô, và trước và sau khi tập thể dục là điều cần thiết để thiết lập kiểm soát chuyển hóa tốt. Nếu liều bột ngô được thay đổi, glucose huyết tương cũng nên được theo dõi thường xuyên sau khi dùng bột ngô để xác định thời gian tác dụng hiệu quả. Các nghiên cứu lâm sàng về cảm biến glucose liên tục, ở cả người lớn và trẻ em, cho thấy chúng có thể được sử dụng an toàn và đáng tin cậy ở những bệnh nhân mắc GSD 1, và có thể giúp giảm thời gian hạ đường huyết, cũng như phát hiện hạ đường huyết không triệu chứng.
Theo dõi định kỳ nên bao gồm: công thức máu toàn phần với công thức bạch cầu, acid uric huyết thanh, triglyceride và cholesterol. Nồng độ triglyceride được coi là thông số hữu ích nhất để kiểm soát chuyển hóa, do sự biến động nhanh chóng của nồng độ glucose huyết tương, lactate và transaminase. Sự tiến triển của u tuyến tăng đáng kể đã được báo cáo ở những bệnh nhân có triglyceride trung bình lớn hơn 500 mg/dL.
Đại học Di truyền và Genomics Y khoa Hoa Kỳ đã cung cấp các hướng dẫn sàng lọc bệnh nhân GSD I cho các biến chứng. Chúng bao gồm đo creatinin huyết thanh, urê, điện giải, canxi, phosphate, xét nghiệm chức năng gan, protein và albumin mỗi 6 tháng. Microalbumin niệu, protein, creatinin, canxi và citrat nên được kiểm tra mỗi năm từ 0 đến 5 tuổi và mỗi 6 tháng nếu lớn hơn 5 tuổi. Độ thanh thải creatinin (ước tính GFR) nên được thực hiện hàng năm cho những người lớn hơn 5 tuổi. Nên thực hiện siêu âm bụng để đánh giá gan, thận, lách và buồng trứng hàng năm nếu dưới 10 tuổi, và mỗi 6 tháng sau 10 tuổi. Đo mật độ xương nên được thực hiện mỗi 1 đến 2 năm cho những người lớn hơn 5 tuổi. Siêu âm tim (ECHO) và điện tâm đồ (ECG) định kỳ nên được thực hiện để sàng lọc tăng huyết áp phổi bắt đầu từ 10 tuổi và lặp lại ít nhất mỗi 3 năm.
Một nghiên cứu lâm sàng để đánh giá hiệu quả và sự an toàn của liệu pháp gen để thay thế G6Pase gan ở người, sử dụng virus adeno liên kết mang gen G6PC của người, hiện đang được tiến hành.
Thiếu hụt Amylo-1,6-Glucosidase (GSD Type III, Thiếu hụt Enzyme khử nhánh, Bệnh giới hạn Dextrin, Bệnh Cori, Bệnh Forbes)
GSD Type III gây ra bởi sự thiếu hụt hoạt động của enzyme khử nhánh glycogen (GDE) (xem Hình 23.5), dẫn đến suy giảm phân hủy glycogen, giảm ly giải glycogen, và tích tụ glycogen bất thường (giới hạn dextrin). Đây là một bệnh di truyền lặn trên nhiễm sắc thể thường do các đột biến của gen AGL trên nhiễm sắc thể 1p21, mã hóa cho bốn đồng dạng GDE. Đồng dạng 1, được phân bố rộng rãi nhất và là dạng chủ yếu trong gan, bị khiếm khuyết trong GSD-IIIa. Các đồng dạng 2, 3 và 4 chỉ nằm ở cơ xương và cơ tim. GDE có hai chức năng enzyme: một transferase bộc lộ liên kết 1,6 và amylo-1,6-glucosidase, tạo ra glucose tự do. Không có GDE, quá trình phân hủy glycogen không thể tiến hành qua các điểm nhánh ngoài cùng, dẫn đến giới hạn dextrin, một dạng glycogen bất thường có chuỗi bên ngắn. Chỉ có 5% đến 10% các gốc glucose bên ngoài có thể được giải phóng bởi phosphorylase. Tại Hoa Kỳ, 80% đến 85% bệnh nhân mắc GSD-III thiếu hoạt động GDE ở cả gan và cơ (GSD-IIIa), 15% thiếu hoạt động GDE chỉ ở gan (GSD-IIIb); hiếm khi, bệnh nhân thiếu hoạt động chỉ ở cơ (GSD-IIIc). Ngược lại, ở Israel, phần lớn bệnh nhân thiếu hoạt động enzyme chỉ ở gan (GSD-IIIb).
Sự thay đổi về lâm sàng và enzyme là một đặc điểm của thiếu hụt GDE. Trong thời kỳ nhũ nhi và thời thơ ấu, biểu hiện có thể không thể phân biệt được với GSD-I. Gan to, hạ đường huyết khi đói kèm theo nhiễm ceton, tăng lipid máu và chậm phát triển là những đặc điểm chủ yếu. Khoảng 70% bệnh nhân bị yếu cơ, nhưng điều này thường không có ý nghĩa lâm sàng ở thời thơ ấu.
Bởi vì một lượng nhỏ glucose có thể được sản xuất từ các đoạn 1,4 ngoài các điểm nhánh glycogen ngoài cùng và từ quá trình tân tạo glucose, bệnh nhân thiếu hụt GDE có thể chịu được thời gian nhịn đói dài hơn, và hạ đường huyết thường ít nghiêm trọng hơn so với những người bị thiếu hụt G6Pase. Trẻ sơ sinh mắc GSD-III có thể không có triệu chứng theo lịch ăn thường xuyên của chúng và thường không bị bệnh nặng khi bị nhiễm trùng và các căng thẳng khác làm gián đoạn việc ăn uống như trẻ em mắc GSD-I.
Trẻ sơ sinh mắc GSD-III phát triển tình trạng tăng ceton máu khi đói là kết quả của sự chuyển đổi nhanh sang trạng thái đói. Nồng độ lactate và acid uric trong máu bình thường, vì con đường tân tạo glucose còn nguyên vẹn và quá trình đường phân ở gan không tăng (xem Hình 23.5). Tăng lipid máu ít nghiêm trọng hơn ở GSD-III so với GSD-I. Phát hiện lâm sàng ban đầu có thể là gan to và chậm phát triển. Lách to có thể được thấy ở trẻ 4 đến 6 tuổi ở những bệnh nhân phát triển xơ gan. Thận không to và rối loạn chức năng thận không xảy ra.
Trẻ sơ sinh và trẻ em không được điều trị tăng trưởng chậm và dậy thì muộn. Bệnh nhân thiếu GDE trong cơ thường có yếu cơ tối thiểu, không có ý nghĩa lâm sàng trong thời thơ ấu. Ngoại trừ bệnh cơ, các triệu chứng và dấu hiệu đặc trưng giảm dần theo tuổi tác. Tuy nhiên, bệnh cơ thường trở nên nổi bật trong thập kỷ thứ ba hoặc thứ tư của cuộc đời, biểu hiện bằng yếu cơ tiến triển chậm liên quan đến các cơ gần. Ở một số bệnh nhân, các cơ nhỏ của bàn tay cũng bị ảnh hưởng. Tổn thương tim là phổ biến do sự tích tụ giới hạn dextrin trong tim gây ra một bệnh cơ tim giống như bệnh cơ tim phì đại vô căn. Phì đại thất trái đồng tâm đáng kể thường phát triển sau tuổi dậy thì và biểu hiện bằng phì đại thất trên ECG và tăng khối lượng và độ dày thành thất trái trên siêu âm tim. Những thay đổi ở thất thường được thấy ở bệnh nhân mắc GSD-Illa, nhưng cũng đã được báo cáo ở bệnh nhân mắc GSD-IIIb. Bệnh cơ tim có triệu chứng có thể xảy ra và đột tử đã được cho là do rối loạn nhịp tim; sự tích tụ glycogen trong hệ thống dẫn truyền tim đã được quan sát thấy khi khám nghiệm tử thi.
Kích thước của gan có xu hướng giảm trong tuổi dậy thì. Sinh thiết thường cho thấy xơ gan, và một số bệnh nhân trưởng thành phát triển xơ gan. U tuyến gan đã được tìm thấy ở 4% đến 25% bệnh nhân, so với 22% đến 78% bệnh nhân GSD-I. Sự phát triển của ung thư biểu mô tế bào gan là hiếm, nhưng đã được mô tả ở những bệnh nhân trên 30 tuổi.
Các đặc điểm sinh hóa điển hình là hạ đường huyết khi đói kèm theo nhiễm ceton (tức là, hạ đường huyết do ceton), nhưng không có sự gia tăng nồng độ lactate máu hoặc acid uric huyết thanh (xem Hình 23.6). Tăng triglyceride máu và tăng cholesterol máu xảy ra ở khoảng hai phần ba và một phần ba bệnh nhân, tương ứng. Tăng triglyceride máu thường được tìm thấy ở trẻ nhỏ (<3 tuổi) và không đạt đến mức đủ cao để gây viêm tụy. Men gan luôn tăng ở trẻ em (ít nhất gấp đôi giới hạn trên của mức bình thường, và thường >500 IU/L), nhưng giảm ở tuổi dậy thì và có thể bình thường ở người lớn. Creatine kinase (CK) thường tăng rõ rệt ở bệnh nhân mắc GSD-IIIa.
Sau một đêm nhịn đói, nồng độ glucose và lactate huyết tương không tăng sau khi dùng glucagon (xem Bảng 23.4). Tuy nhiên, khi xét nghiệm được thực hiện 2 giờ sau một bữa ăn nhiều carbohydrate (trước khi các nhánh ngoài của glycogen bị phân hủy), một phản ứng đường huyết có thể được gây ra. Chẩn đoán GSD-III được thực hiện bằng phân tích đột biến gen AGL hoặc, cách khác, bằng sinh thiết gan và/hoặc cơ. Kết quả sinh thiết cho thấy hàm lượng glycogen có cấu trúc bất thường (phân nhánh cao) tăng gấp 3 đến 5 lần trong mô bị ảnh hưởng. Việc phân loại phụ chính xác của GSD-III đòi hỏi sinh thiết cả gan và cơ. Mặc dù tổn thương cơ có thể được suy ra từ sự hiện diện của nồng độ CK huyết thanh cao, một mức bình thường không loại trừ sự thiếu hụt enzyme cơ. Tại thời điểm chẩn đoán, sinh thiết cơ là cách duy nhất để dự đoán chính xác liệu bệnh cơ xương hoặc tổn thương cơ tim có khả năng phát triển trong tương lai hay không, trừ khi một đột biến đặc hiệu cho GSD-IIIb được xác định.
Bởi vì chỉ một lượng glucose hạn chế có thể được huy động từ glycogen, hạ đường huyết phát triển trong một đêm nhịn đói ở thời kỳ nhũ nhi và đầu thời thơ ấu. Điều này xảy ra mặc dù tăng tân tạo glucose và tăng cường hấp thu các acid amin tân tạo glucose ở gan, dẫn đến nồng độ một số acid amin trong huyết tương thấp, chẳng hạn như alanin. Như trong GSD-I, việc cung cấp liên tục một lượng glucose đầy đủ, sử dụng UCS, kết hợp với lượng calo tổng, protein và các chất dinh dưỡng khác bình thường, sẽ khắc phục rối loạn lâm sàng và sinh hóa và phục hồi sự tăng trưởng bình thường. UCS 1 đến 1,75 g/kg cách nhau 6 giờ (ví dụ: vào nửa đêm, 6 giờ sáng, v.v.) duy trì đường huyết bình thường, tăng tốc độ tăng trưởng và giảm nồng độ aminotransferase huyết thanh. Trẻ sơ sinh dưới 12 tháng tuổi có thể không sản xuất đủ amylase để tiêu hóa UCS và bị chướng bụng, đầy hơi và tiêu chảy. Nếu các tác dụng phụ này xảy ra, việc giới thiệu UCS từ từ hoặc bổ sung pancrelipase (chứa amylase) có thể cải thiện khả năng dung nạp. UCS có thể được trộn trong bất kỳ loại đồ uống nào, mặc dù việc thêm vào sữa hoặc sữa chua được ưu tiên hơn, vì nó bổ sung protein và chất béo. Việc bổ sung protein whey được khuyến nghị để kéo dài thời gian đường huyết bình thường. Nên tránh dùng UCS quá mức vì nó dẫn đến lắng đọng glycogen dư thừa và tăng cân. Đối với những bệnh nhân bị chậm phát triển và bệnh cơ đáng kể, việc cho ăn liên tục qua đêm bằng hỗn hợp dinh dưỡng bao gồm glucose, oligosaccharide glucose và acid amin kết hợp với việc cho ăn gián đoạn vào ban ngày bằng các bữa ăn có hàm lượng protein cao có thể có lợi. Thanh thiếu niên và người lớn mắc GSD IIIa nên hướng tới một chế độ ăn giàu protein (25% tổng lượng calo) và carbohydrate phức tạp thấp (<50% tổng lượng calo) và nên tránh đường đơn và nhịn đói. Chế độ ăn giàu protein có thể cải thiện chức năng cơ bằng cách tăng cường tổng hợp protein cơ và giảm dự trữ glycogen. Cũng có báo cáo rằng chế độ ăn giàu protein (~30% tổng lượng calo) có thể đảo ngược và có thể ngăn ngừa bệnh cơ tim. Bệnh nhân mắc GSD-IIIb có thể chuyển sang chế độ ăn cân bằng thông thường.
Theo dõi glucose huyết tương nên được thực hiện trước bữa ăn hoặc UCS, trước khi đi ngủ và bữa sáng, với việc bổ sung theo dõi thêm trong thời gian bị bệnh. Xét nghiệm chức năng gan nên được thực hiện mỗi 6 tháng. Như ở bệnh nhân GSD-1, nên thực hiện siêu âm gan hàng năm để sàng lọc u tuyến gan. Nên thực hiện MRI gan mỗi 6 đến 12 tháng ở bệnh nhân lớn tuổi và ở trẻ em, khi siêu âm cho thấy một u tuyến đã tăng kích thước. Nên thực hiện ECG và ECHO định kỳ để đánh giá rối loạn nhịp tim, phì đại thất, và rối loạn chức năng tâm thu hoặc tâm trương. Siêu âm tim nên được thực hiện mỗi 12 đến 24 tháng ở bệnh nhân GSD-IIIa và mỗi 5 năm ở những người mắc GSD-IIIb. ECG nên được thực hiện mỗi hai năm ở bệnh nhân GSD-IIIa. Không có hạn chế tập thể dục nào được khuyến nghị cho bệnh nhân GSD-III, trừ khi họ có bệnh tim đáng kể. Tập thể dục giúp ngăn ngừa bệnh cơ nặng hơn và mật độ khoáng xương thấp. Đánh giá vật lý trị liệu được khuyến nghị mỗi 6 tháng.
Thiếu hụt phức hợp Phosphorylase gan (GSD Type VI, Thiếu hụt Phosphorylase gan, Bệnh Hers; GSD Type IX, Thiếu hụt Phosphorylase Kinase)
Phosphorylase gan, enzyme giới hạn tốc độ của quá trình ly giải glycogen, được kích hoạt bởi một chuỗi các phản ứng enzyme được kích hoạt bởi glucagon và epinephrine. Đầu tiên, adenylate cyclase xúc tác sự hình thành cyclic adenosine monophosphate (CAMP), sau đó kích hoạt một protein kinase phụ thuộc CAMP. Protein kinase sau đó phosphoryl hóa một phosphorylase kinase (PHK), chuyển đổi phosphorylase gan không hoạt động thành dạng hoạt động của nó. Phosphorylase hoạt động thủy phân các liên kết α-1,4 và huy động glucose từ glycogen (xem Hình 23.5).
Các GSD gây ra bởi sự giảm hoạt động của phosphorylase gan là một nhóm các rối loạn không đồng nhất (xem Bảng 23.3). PHK gan hoặc GSD type IX, xảy ra ở khoảng một trong 100.000 ca sinh và chiếm khoảng 25% tất cả các trường hợp GSD, là dạng phổ biến nhất; trong khi đó, thiếu hụt phosphorylase gan (GSD type VI) là hiếm.
PHK của gan và cơ là một enzyme phức tạp bao gồm bốn tiểu đơn vị: α, β, γ, δ. Enzyme được điều hòa bằng cách phosphoryl hóa các gốc serine cụ thể của các tiểu đơn vị α và β và bằng canxi thông qua tiểu đơn vị δ, một thành viên của họ calmodulin. Tiểu đơn vị γ có hoạt tính xúc tác. Đột biến trong ba gen khác nhau của các tiểu đơn vị PHK (PHKA2, PHKB và PHKG2) có thể dẫn đến thiếu hụt hoạt động của phosphorylase gan. GSD IXa, do các đột biến trong PHKA2, là biến thể phổ biến nhất (xem Bảng 23.3).
GSD IXa là một rối loạn liên kết X chỉ giới hạn ở bé trai. Bệnh nhân hiếm khi có hạ đường huyết có triệu chứng trong thời kỳ nhũ nhi, trừ khi họ nhịn đói trong một thời gian dài. Họ có thể phát triển tăng ceton máu tương tự, nhưng thường nhẹ hơn so với GSD type III. Nhiễm toan chuyển hóa là hiếm. Rối loạn thường được phát hiện khi gan to và bụng chướng được ghi nhận trong một cuộc kiểm tra thể chất. Tăng trưởng thể chất thường bị suy giảm, và phát triển vận động có thể bị trì hoãn do hậu quả của giảm trương lực cơ. Với tuổi tác ngày càng tăng, các bất thường lâm sàng và sinh hóa dần dần cải thiện, và có thể xảy ra tăng trưởng bù, hầu hết bệnh nhân trưởng thành đều không có triệu chứng. GSD IXC, do các đột biến PHKG2, có xu hướng có một kiểu hình nghiêm trọng hơn bao gồm xơ gan và xơ gan ở thời thơ ấu. Các báo cáo gần đây cho thấy xơ gan cũng có thể xảy ra ở GSD VI và IXa. Bệnh cơ tim nhẹ đã được xác định bằng ECHO khi theo dõi lâu dài bệnh nhân GSD VI và IXb.
Hạ đường huyết là bất thường và nồng độ lactate và acid uric trong máu bình thường. Có thể có tăng triglyceride máu nhẹ, tăng cholesterol máu và nồng độ transaminase tăng. Nhiễm ceton xảy ra khi đói. Các xét nghiệm chức năng không đặc biệt hữu ích trong việc đánh giá những bệnh nhân này. Sau một đêm nhịn đói, nồng độ lactate máu bình thường, và việc sử dụng glucagon gây ra một phản ứng đường huyết nhanh chóng, không có sự gia tăng nồng độ lactate máu. Phản ứng đường huyết với glucagon không thể được sử dụng để phân biệt giữa thiếu hụt phosphorylase kinase và thiếu chính phosphorylase. Do đó, chẩn đoán xác định GSD type IX đòi hỏi xét nghiệm di truyền hoặc xác định hoạt động của phosphorylase kinase trong hồng cầu hoặc bạch cầu. Chẩn đoán GSD type VI cũng có thể được thiết lập bằng xét nghiệm di truyền hoặc, cách khác, bằng cách phân tích hoạt động của phosphorylase trong các phân đoạn tế bào máu tinh khiết, và thường không yêu cầu sinh thiết gan. Hoạt động của phosphorylase cơ bình thường; mô học cơ và hàm lượng glycogen bình thường.
Trong quá khứ, điều trị cụ thể ngoài việc tránh nhịn đói kéo dài được coi là không cần thiết. Tuy nhiên, nghiên cứu gần đây cho thấy rằng liệu pháp với chế độ ăn giàu protein và UCS ở bệnh nhân GSD IX cải thiện sự tăng trưởng tuyến tính, sức khỏe chung, giảm gan to và có thể liên quan đến sự thoái triển của các phát hiện siêu âm về xơ gan. UCS, 2 g/kg trước khi đi ngủ, thường ngăn ngừa hạ đường huyết và nhiễm ceton ở những bệnh nhân này.
Theo dõi glucose và ceton huyết tương nên được thực hiện thường xuyên. Sự tăng trưởng và tiến triển dậy thì nên được theo dõi cẩn thận. Nên thực hiện siêu âm gan hàng năm bắt đầu từ 5 tuổi. Vì có nguy cơ loãng xương tăng, nên đo mật độ xương một lần khi tăng trưởng hoàn tất.
Thiếu hụt Enzyme tạo nhánh Glycogen (GSD Type IV, Bệnh Andersen)
Có sự suy giảm tối thiểu của quá trình ly giải glycogen và tân tạo glucose là bình thường; theo đó, hạ đường huyết là hiếm; tuy nhiên, hạ đường huyết khi đói có thể xảy ra khi xơ gan nặng phát triển.
Hội chứng Fanconi-Bickel (GSD Type XI)
GSD XI là một rối loạn di truyền lặn trên nhiễm sắc thể thường hiếm gặp, đặc trưng bởi sự tích tụ glycogen gan-thận do thiếu hụt chất vận chuyển glucose thuận lợi GLUT2, chất trung gian vận chuyển hai chiều glucose và galactose trong tế bào gan, tế bào beta tuyến tụy, tế bào ruột và trong các ống lượn gần của thận.
Bệnh nhân thường biểu hiện trong thời kỳ nhũ nhi với chậm phát triển, tiêu chảy mạn tính do kém hấp thu carbohydrate, gan to và hạ đường huyết khi đói, khi khoảng cách giữa các cữ ăn tăng lên. Một bệnh lý ống lượn gần toàn thể với glucose niệu nặng và còi xương hạ phosphat máu là những đặc điểm đặc trưng. Bệnh nhân lớn tuổi có khuôn mặt hình mặt trăng, bụng chướng, tầm vóc thấp và dậy thì muộn. Thận to (có thể phát hiện bằng siêu âm) là kết quả của sự tích tụ glycogen. Các phát hiện lâm sàng có mức độ nghiêm trọng khác nhau.
Hạ đường huyết do ceton khi đói và tăng đường huyết và tăng galactose máu sau ăn (do suy giảm hấp thu hai loại đường ở gan) là những đặc điểm sinh hóa đặc trưng. Giảm tiết insulin (do suy giảm cảm nhận glucose của tế bào beta) cũng có thể góp phần làm suy giảm hấp thu glucose ở gan và tăng đường huyết sau ăn. Các kết quả xét nghiệm khác bao gồm tăng galactose máu, glucose niệu, mất bicarbonate ở thận, protein niệu, phosphaturia, acid amin niệu toàn thể, và nồng độ phosphatase kiềm huyết thanh tăng. Phân tích đột biến của gen GLUT2 xác nhận chẩn đoán.
Khuyến nghị cho ăn thường xuyên bằng carbohydrate hấp thu chậm và hạn chế galactose. Bổ sung UCS có tác dụng có lợi đối với kiểm soát chuyển hóa và tăng trưởng. Chuyển hóa fructose không bị ảnh hưởng; do đó, monosaccharide này có thể được sử dụng như một nguồn carbohydrate thay thế. Nước và điện giải phải được thay thế. Có thể cần kiềm để bù đắp cho nhiễm toan ống thận, và còi xương hạ phosphat máu cần bổ sung phosphate và vitamin D.
Hạ đường huyết do Ceton
Hạ đường huyết do ceton đã được công nhận trong gần một thế kỷ là loại hạ đường huyết phổ biến nhất ở trẻ em từ 1 đến 6 tuổi, với một biểu hiện và diễn biến được mô tả rõ ràng, nhưng một nguyên nhân chưa được hiểu đầy đủ. Thông thường, tình trạng này biểu hiện dưới dạng các đợt hạ đường huyết buổi sáng tái phát trong năm thứ hai hoặc thứ ba của cuộc đời—nhưng khởi phát sớm nhất là 6 tháng đã được báo cáo. Tình trạng này thường tự khỏi vào lúc 8 đến 9 tuổi. Bệnh sử kinh điển là của một đứa trẻ, thường là nam có tiền sử cân nặng khi sinh thấp, đã ăn kém vào ngày hôm trước hoặc bỏ bữa tối, khó đánh thức vào buổi sáng hôm sau, và biểu hiện các triệu chứng thiếu glucose não có thể từ lơ mơ đến co giật. Các đợt hạ đường huyết đặc biệt có khả năng xảy ra trong khi bị bệnh, khi lượng thức ăn bị hạn chế. Điều trị để tránh nhịn đói qua đêm kéo dài thường giới hạn các cơn tái phát xuống dưới một đến hai lần một năm.
Tại thời điểm hạ đường huyết, nồng độ ceton cao được tìm thấy trong huyết tương và nước tiểu, và nồng độ insulin huyết tương bị ức chế, cho thấy tăng insulin máu không chịu trách nhiệm cho hạ đường huyết. Vẫn chưa được giải quyết liệu những đứa trẻ này có biểu hiện một phản ứng chuyển hóa nhanh nhưng về chất lượng là bình thường đối với việc nhịn đói (tức là, đầu dưới của phân bố bình thường về khả năng chịu đói, “đói nhanh”) hay liệu hạ đường huyết do ceton đại diện cho một nhóm các rối loạn không đồng nhất về sự sẵn có của cơ chất hạn chế đang chờ được phân định thêm. Ví dụ, khả năng chịu đói ngắn hơn nhiều ở trẻ sơ sinh nhỏ do tỷ lệ não so với khối lượng cơ thể lớn hơn và do đó dễ bị hạ đường huyết hơn, khi nhịn đói hơn 12 giờ, đặc biệt nếu việc ăn uống bình thường đã bị suy giảm do chán ăn, nôn mửa hoặc tiêu chảy do một bệnh xen kẽ. Mặt khác, ở những đứa trẻ trước đây được chẩn đoán mắc hạ đường huyết do ceton, một số lượng ngày càng tăng các khiếm khuyết chuyển hóa cụ thể đã được công nhận, chẳng hạn như các dạng nhẹ của các rối loạn dự trữ glycogen (GSD Type IXa) hoặc sử dụng ceton. Do đó, hạ đường huyết do ceton nên được coi là một chẩn đoán loại trừ và nên xem xét điều tra thêm ở bất kỳ đứa trẻ nào bị hạ đường huyết do ceton có các đợt tái phát.
Một số nghiên cứu đã chỉ ra rằng hạ đường huyết do ceton phản ánh sự sản xuất dưới mức hơn là sử dụng quá mức glucose. Truyền alanine, fructose, hoặc glycerol tạo ra sự gia tăng nồng độ glucose huyết tương, không có sự thay đổi đáng kể trong nồng độ lactate hoặc pyruvate máu, cho thấy toàn bộ con đường tân tạo glucose từ mức pyruvate còn nguyên vẹn, và gợi ý rằng sự thiếu hụt cơ chất chứ không phải là một khiếm khuyết trong tân tạo glucose có liên quan.
Glucagon gây ra một phản ứng đường huyết bình thường ở trẻ em bị ảnh hưởng trong trạng thái no, nhưng không phải tại thời điểm hạ đường huyết, cho thấy các con đường ly giải glycogen cũng còn nguyên vẹn. Nồng độ glycerol huyết tương bình thường ở những đứa trẻ này, ở cả trạng thái no và đói. Phản ứng chuyển hóa với việc truyền BOHB không khác với trẻ em bình thường. Cuối cùng, nồng độ các hormone chống lại hạ đường huyết tăng phù hợp, trong khi nồng độ insulin thấp phù hợp.
Trẻ em mắc hạ đường huyết do ceton có nồng độ alanin huyết tương giảm ở trạng thái cơ bản, sau một đêm nhịn đói, và giảm sâu hơn nữa khi nhịn đói kéo dài. Alanin là acid amin chính được sử dụng để tân tạo glucose. Sự hình thành và giải phóng của nó từ cơ trong các giai đoạn hạn chế calo được tăng cường bởi sự hiện diện của chu trình glucose-alanin, cũng như bởi sự hình thành de novo từ các cơ chất khác, chẳng hạn như các acid amin chuỗi nhánh. Giảm alanin máu trong hạ đường huyết do ceton có thể phản ánh “đói nhanh,” hơn là một khiếm khuyết cụ thể trong chuyển hóa alanin. Như đã chỉ ra trong mô tả ban đầu về hạ đường huyết do ceton, những đứa trẻ này thường nhỏ hơn so với các bạn cùng tuổi và thường có tiền sử hạ đường huyết sơ sinh thoáng qua. Do đó, hạ đường huyết do ceton có thể chỉ đơn giản phản ánh sự dự trữ giảm của một khối lượng cơ nhỏ ở độ tuổi mà nhu cầu glucose trên một đơn vị trọng lượng cơ thể để hỗ trợ chuyển hóa não tương đối cao; trẻ em mắc hạ đường huyết do ceton có thể đại diện cho đầu dưới của phạm vi bình thường về khả năng chịu đói. Sự thuyên giảm tự phát của hạ đường huyết do ceton vào lúc 8 đến 9 tuổi có thể được giải thích bằng sự gia tăng khối lượng cơ so với kích thước não, với kết quả là tăng cung cấp cơ chất nội sinh, và giảm tương đối nhu cầu glucose trên một đơn vị khối lượng cơ thể, theo tuổi tác.
Chẩn đoán hạ đường huyết do ceton được xác nhận bằng một nghiệm pháp nhịn đói có giám sát. Hạ đường huyết với FFA, BOHB và acetoacetate huyết tương tăng phát triển trong vòng 14 đến 24 giờ ở hầu hết những đứa trẻ này, trong khi trẻ em bình thường cùng tuổi có thể chịu đựng nhịn đói mà không phát triển hạ đường huyết trong ít nhất 24 giờ.
Các đợt hạ đường huyết do ceton có thể được ngăn ngừa hoặc giảm thiểu bằng cách tránh nhịn đói kéo dài. Thời gian nhịn đói qua đêm nên được rút ngắn xuống dưới 10 đến 12 giờ, với một bữa ăn nhẹ chứa carbohydrate trước khi đi ngủ và ăn sáng ngay. Khi các đợt được kích hoạt bởi bệnh tật, cha mẹ có thể xét nghiệm nước tiểu của trẻ để tìm ceton hoặc, tốt hơn là, BOHB huyết tương. Sự xuất hiện của tăng ceton máu trước hạ đường huyết vài giờ và cho thấy cần phải có chất lỏng giàu carbohydrate. Nếu những chất này không thể được dung nạp, đứa trẻ nên được đưa đến phòng cấp cứu để truyền glucose tĩnh mạch. Một lá thư giải thích và khuyến nghị điều trị có thể đẩy nhanh phản ứng của phòng cấp cứu.
Hạ đường huyết do ceton chỉ nên được coi là một chẩn đoán loại trừ, vì hạ đường huyết từng đợt kèm theo nhiễm ceton có thể xảy ra với sự thiếu hụt của một số hormone hoặc một loạt các khiếm khuyết của tân tạo glucose hoặc chuyển hóa glycogen, đặc biệt là suy tuyến yên và GSD. Các đợt hạ đường huyết do ceton tái phát không thể giải thích được bằng bệnh xen kẽ nên kích hoạt việc đánh giá lại một rối loạn tiềm ẩn có thể đã bị bỏ sót trước đây. Trong số các rối loạn đáng được xem xét đặc biệt là các rối loạn dự trữ glycogen nhẹ hơn (ví dụ, GSD Type IXa do thiếu hụt phosphorylase kinase liên kết X, PHKA2); suy thượng thận do glucocorticoid dạng hít hoặc xịt mũi; hoặc các khiếm khuyết trong việc sử dụng ceton, chẳng hạn như thiếu hụt MCT1 được mã hóa bởi SLC16A1.
Thiếu hụt Hormone
Cả GH và cortisol đều làm tăng sản xuất glucose, giảm sử dụng glucose, và tăng tốc quá trình ly giải lipid và sinh ceton. Trái ngược với glucagon và epinephrine, làm tăng nồng độ glucose huyết tương trong vòng vài phút, tác động của GH và cortisol bị trì hoãn trong vài giờ và cả hai đều không có vai trò chính trong việc bảo vệ chống lại hạ đường huyết cấp tính do insulin gây ra. Tuy nhiên, chúng rất quan trọng trong việc ngăn ngừa hạ đường huyết trong khi nhịn đói kéo dài.
Thiếu hụt Hormone Tăng trưởng và Suy tuyến yên
Thiếu hụt GH là một nguyên nhân quan trọng gây hạ đường huyết khi đói, đặc biệt là ở trẻ sơ sinh và trẻ nhỏ. Mặc dù ít phổ biến hơn, hạ đường huyết có triệu chứng và không có triệu chứng, thường bị thúc đẩy bởi stress, đói, hoặc tập thể dục, cũng có thể xảy ra ở trẻ lớn và người lớn bị thiếu hụt GH đơn độc hoặc thiếu hụt nhiều hormone tuyến yên.
Suy tuyến yên trong giai đoạn sơ sinh có thể gây hạ đường huyết nặng dai dẳng hoặc tái phát, và một số trẻ sơ sinh có thể cần tốc độ truyền glucose tương đương với tốc độ cần thiết để điều trị tăng insulin máu bẩm sinh, ngoài ra còn giống tăng insulin máu bởi có nồng độ FFA và ceton bị ức chế, và phản ứng đường huyết với glucagon. Dương vật nhỏ gợi ý thiếu hụt gonadotropin cùng tồn tại. Trẻ sơ sinh bị suy tuyến yên bẩm sinh có thể bị vàng da ứ mật với men gan tăng phù hợp với viêm gan sơ sinh. Chẩn đoán thiếu hụt GH không thể dựa trên một giá trị GH duy nhất thu được tại thời điểm hạ đường huyết tự phát hoặc do nhịn đói gây ra. Các nghiên cứu đã chỉ ra rằng một giá trị GH thấp duy nhất, tại thời điểm hạ đường huyết khi đói, có độ đặc hiệu kém đối với chẩn đoán thiếu hụt GH. Việc xác nhận chẩn đoán nên dựa trên một nền tảng vững chắc của bằng chứng lâm sàng, bao gồm việc chứng minh bằng hình ảnh về sự phát triển tuyến yên bất thường hoặc các khiếm khuyết đường giữa khác.
Thiếu hụt GH đơn độc ở trẻ nhỏ có thể gây hạ đường huyết buổi sáng kèm theo tăng ceton máu (“hạ đường huyết do ceton”). Nồng độ insulin huyết tương bị ức chế phù hợp và phản ứng đường huyết với glucagon trong cơn hạ đường huyết tự phát là kém. Mức độ nhiễm ceton tại thời điểm hạ đường huyết có thể thấp hơn dự kiến trong loại hạ đường huyết do ceton vô căn phổ biến hơn, có lẽ phản ánh sự tăng nhạy cảm với insulin và suy giảm ly giải lipid. Người ta cho rằng tình trạng giảm ceton máu tương đối (tức là, suy giảm sản xuất nhiên liệu thay thế) có thể góp phần vào sự phát triển của hạ đường huyết khi đói ở trẻ nhỏ bị thiếu hụt GH. Sau giai đoạn đầu của thời kỳ nhũ nhi, tăng trưởng chậm và nồng độ IGF-1 thấp là những chỉ số đáng tin cậy hơn của thiếu hụt GH, và chẩn đoán xác định thiếu hụt GH nên được xác nhận bằng cách chứng minh các phản ứng GH thấp đối với các kích thích tiêu chuẩn.
Kháng Hormone Tăng trưởng và Thiếu hụt Yếu tố Tăng trưởng giống Insulin-1
Hạ đường huyết khi đói tái phát là phổ biến trong tình trạng kháng GH do các khiếm khuyết phân tử trong thụ thể GH, dẫn đến không có khả năng tạo ra IGF-1, thường được gọi là bệnh lùn Laron. Hạ đường huyết có xu hướng cải thiện ở tuổi vị thành niên, mặc dù vẫn có thể xảy ra khi nhịn đói kéo dài. Tần suất hạ đường huyết tương tự (≥45%) đã được báo cáo ở hai quần thể lớn nhất mắc tình trạng này, ở Israel và Ecuador. Khả năng bị hạ đường huyết khi đói cải thiện khi điều trị bằng IGF-1 tổng hợp; tuy nhiên, một trong những tác dụng giống insulin của việc điều trị IGF-1 là hạ đường huyết, có thể xảy ra trong vòng một giờ sau khi tiêm, nếu trẻ không ăn.
Thiếu hụt Cortisol và Adrenocorticotropin
Cortisol có vai trò cực kỳ quan trọng trong việc hỗ trợ sản xuất glucose trong khi đói bằng cách tăng dự trữ glycogen và tân tạo glucose, và huy động FFA; cortisol cũng có vai trò gián tiếp trong việc bảo vệ chống lại hạ đường huyết bằng cách cho phép epinephrine được tổng hợp bình thường trong tủy thượng thận. Hoạt động của phenylethanolamine N-methyl transferase, enzyme chuyển đổi norepinephrine thành epinephrine, phụ thuộc vào cortisol trong thượng thận.
Thiếu hụt cortisol làm tăng nhạy cảm với insulin; ví dụ, tần suất hạ đường huyết tăng và nhu cầu insulin giảm là những biểu hiện đặc trưng của bệnh Addison ở bệnh đái tháo đường type 1.
Hạ đường huyết khi đói có thể xảy ra ở những bệnh nhân bị thiếu hụt glucocorticoid cả nguyên phát và thứ phát và phổ biến hơn ở trẻ sơ sinh, trẻ nhũ nhi, và trẻ nhỏ so với thanh thiếu niên và người lớn, và ở những người bị thiếu hụt cortisol nghiêm trọng hơn. Hạ đường huyết rất có thể xảy ra sau khi nhịn đói kéo dài hoặc trong tình trạng căng thẳng do bệnh tật, đặc biệt nếu việc thay thế glucocorticoid hàng ngày thông thường đã bị gián đoạn do bệnh tật.
Thiếu hụt ACTH kết hợp với thiếu hụt GH, đặc biệt là trong suy tuyến yên bẩm sinh, có nhiều khả năng gây hạ đường huyết hơn là suy thượng thận nguyên phát. Thiếu hụt ACTH bẩm sinh, biểu hiện bằng hạ đường huyết sớm, đã được báo cáo với các đột biến của một số gen liên quan đến sự phát triển tuyến yên (POU1F1, PROP1, TPIT). Thiếu hụt ACTH không phải lúc nào cũng rõ ràng trên lâm sàng trong giai đoạn đầu của thời kỳ nhũ nhi nhưng có thể biểu hiện muộn hơn trong thời thơ ấu.
Ngày càng có nhiều khiếm khuyết di truyền cụ thể dẫn đến thiếu hụt hoặc kháng ACTH đơn độc đã được xác định. Tóc đỏ và béo phì khởi phát sớm nghiêm trọng là những đặc điểm đặc trưng của trẻ em bị thiếu hụt ACTH do đột biến proopiomelanocortin (POMC). Hạ đường huyết ở trẻ sơ sinh do thiếu hụt ACTH đã được cho là do khiếm khuyết trong quá trình phân cắt prohormone. Thiếu hụt ACTH vô căn có thể mắc phải ở thời thơ ấu, thanh thiếu niên, hoặc sau này trong cuộc đời trưởng thành. Bằng chứng gián tiếp, chẳng hạn như mối liên quan với viêm tuyến giáp tự miễn, cho thấy viêm tuyến yên tự miễn là một nguyên nhân phổ biến và trong một số trường hợp, kháng thể kháng tuyến yên đã được chứng minh. Trong tình trạng kháng ACTH, tăng sắc tố da đi kèm với các biểu hiện khác của suy thượng thận.
Ức chế chức năng thượng thận do điều trị là một nguyên nhân phổ biến của suy glucocorticoid, nhưng nó có thể bị bỏ qua vì các bất thường điện giải điển hình của thiếu hụt mineralocorticoid không có mặt. Một cơn khủng hoảng thượng thận với tình trạng kiệt sức, nôn mửa, hạ huyết áp và hạ đường huyết có thể được kích hoạt bởi một sự kiện căng thẳng, ở một đứa trẻ gần đây đã ngừng điều trị glucocorticoid liều cao, và cũng đã được báo cáo trong liệu pháp liều thấp cách ngày. Thường xuyên hơn, hạ đường huyết là do các chế phẩm glucocorticoid dạng hít và xịt mũi là phương pháp điều trị chính cho bệnh hen suyễn và dị ứng cho hàng triệu trẻ em. Nồng độ cortisol huyết thanh dưới 3 mcg/dL trong khoảng từ 7 đến 9 giờ sáng ở một đứa trẻ đang dùng glucocorticoid tại chỗ hoặc dạng hít là chẩn đoán suy thượng thận; nếu nồng độ từ 3 mcg/dL trở lên, cần phải làm nghiệm pháp kích thích ACTH để đánh giá chức năng thượng thận.
Hạ đường huyết có thể là một đặc điểm biểu hiện của suy thượng thận nguyên phát, và thường xảy ra trong khi bị bệnh hoặc đói ở những bệnh nhân đang được điều trị bằng glucocorticoid thay thế. Suy thượng thận nguyên phát có liên quan đến các phản ứng epinephrine thiếu hụt đối với hạ đường huyết, và hạ đường huyết biểu hiện bằng các triệu chứng thiếu glucose não hơn là các triệu chứng adrenergic có thể ít được nhận ra hơn. Hạ đường huyết tương đối thường xuyên ở những bệnh nhân được điều trị mắc tăng sản thượng thận bẩm sinh.
Như đã lưu ý trước đó đối với thiếu hụt GH, chẩn đoán thiếu hụt cortisol hoặc ACTH, là nguyên nhân gây hạ đường huyết của một đứa trẻ, không thể dựa trên một giá trị cortisol thấp duy nhất trong cơn hạ đường huyết, và đòi hỏi bằng chứng lâm sàng bổ sung (chẳng hạn như nồng độ ACTH huyết tương tăng rõ rệt đồng thời trong trường hợp suy thượng thận nguyên phát) hoặc một nghiệm pháp kích thích thượng thận tiêu chuẩn.
Thiếu hụt Glucagon và Epinephrine
Tầm quan trọng của glucagon và epinephrine trong các phản ứng đối kháng điều hòa với hạ đường huyết cho thấy rằng sự thiếu hụt đơn độc của một trong hai hormone có khả năng gây hạ đường huyết, nhưng chưa có trường hợp nào hạ đường huyết do thiếu hụt nguyên phát đơn độc của một trong hai hormone được ghi nhận rõ ràng ở trẻ em.
Mặc dù đã có các báo cáo trường hợp về việc giảm bài tiết epinephrine ở bệnh nhân bị hạ đường huyết, bằng chứng từ các bệnh nhân đái tháo đường type 1 và u tiết insulin cho thấy rằng việc giảm bài tiết epinephrine có khả năng là hậu quả hơn là nguyên nhân của hạ đường huyết tái phát. Phản ứng epinephrine khiếm khuyết đi kèm với suy thượng thận có thể được phục hồi ít nhất một phần bằng cách thay thế glucocorticoid. Hạ đường huyết khi đói nghiêm trọng đã được báo cáo ở một đứa trẻ nhỏ đang dùng thuốc chẹn beta, propranolol.
Mặc dù có tầm quan trọng sống còn trong việc đối kháng điều hòa glucose cấp tính, chưa có trường hợp nào được ghi nhận rõ ràng về hạ đường huyết ở trẻ em do thiếu hụt glucagon đơn độc. Một trường hợp, thường được trích dẫn như một ví dụ về thiếu hụt glucagon đơn độc, sau đó đã được chứng minh là một trường hợp SCHAD-HI gia đình.
Rối loạn di truyền trong tân tạo Glucose
Trong giai đoạn sau hấp thu sớm, nồng độ glucose huyết tương được duy trì bởi cả quá trình ly giải glycogen và tân tạo glucose. Khi nhịn đói tiếp tục kéo dài hơn 6 đến 10 giờ và glycogen gan cạn kiệt, tân tạo glucose đóng góp một tỷ lệ ngày càng tăng của glucose lưu hành (xem Hình 23.3). Alanin từ cơ là một cơ chất chính cho tân tạo glucose, nhưng lactate có nguồn gốc từ quá trình đường phân ở mô ngoại vi cũng góp phần vào sản xuất glucose thông qua chu trình Cori, và trong các giai đoạn sau của quá trình nhịn đói, glycerol, có nguồn gốc từ quá trình ly giải triglyceride của mô mỡ, trở thành một nguồn cơ chất tân tạo glucose chính (FFA không thể được chuyển đổi thành glucose). Các khiếm khuyết di truyền của bốn enzyme tân tạo glucose cần thiết để vượt qua các bước không thể đảo ngược trong quá trình đường phân (pyruvate carboxylase, phosphoenolpyruvate carboxykinase, fructose 1,6-diphosphatase [FDPase], và G6Pase) gây hạ đường huyết, đặc trưng bởi sự tăng rõ rệt của lactate (xem Hình 23.5; thiếu hụt G6Pase được mô tả trước đó trong phần Các bệnh dự trữ Glycogen).
Thiếu hụt Pyruvate Carboxylase
Pyruvate carboxylase là một protein chứa biotin bao gồm bốn tiểu đơn vị chuyển đổi pyruvate thành oxaloacetate, là bước đầu tiên trong con đường tân tạo glucose. Sự kích hoạt phụ thuộc vào acetyl CoA và xảy ra chủ yếu trong quá trình huy động FFA trong khi đói. Một số dạng thiếu hụt đã được mô tả, thường biểu hiện dưới dạng nhiễm toan lactic nặng và bệnh não trong giai đoạn đầu của thời kỳ nhũ nhi, thường được kích hoạt bởi sự mất bù chuyển hóa trong khi bị bệnh. Gan to là phổ biến. Hạ đường huyết không phải lúc nào cũng có mặt trong thiếu hụt pyruvate carboxylase. Nước tiểu chứa một lượng lớn alpha-ketoglutarate. Chẩn đoán có thể được xác nhận bằng cách giải trình tự trực tiếp gen PC trên nhiễm sắc thể 11q13. Quản lý bao gồm các bữa ăn carbohydrate thường xuyên và hỗ trợ dextrose tĩnh mạch trong khi bị bệnh.
Thiếu hụt Phosphoenolpyruvate Carboxykinase
Phosphoenolpyruvate carboxykinase (PEPCK) xúc tác quá trình chuyển đổi oxaloacetate thành phosphoenolpyruvate là bước quan trọng thứ hai trong tân tạo glucose. Các đồng dạng tế bào chất và ty thể được mã hóa bởi PEPCK1 và PEPCK2, tương ứng. Sự thiếu hụt của cả hai dạng đã được báo cáo ở những bệnh nhân hiếm gặp, nhưng kiểu hình lâm sàng vẫn chưa chắc chắn. Gần đây, các bệnh nhân bị thiếu hụt dạng tế bào chất của PEPCK mới được xác nhận ở cấp độ phân tử. Những bệnh nhân này biểu hiện với các đợt hạ đường huyết gián đoạn, không có sự tăng rõ rệt của lactate huyết tương, nhưng có nồng độ các chất trung gian của chu trình acid tricarboxylic trong nước tiểu tăng. Một số bệnh nhân bị thiếu hụt dạng ty thể của PEPCK đã được mô tả dựa trên dữ liệu enzyme, nhưng chưa được xác nhận ở cấp độ phân tử. Những bệnh nhân này biểu hiện với giảm trương lực cơ, gan to, chậm phát triển, nhiễm toan lactic và hạ đường huyết.
Thiếu hụt Fructose 1,6-Diphosphatase
FDPase, hay fructose 1,6-bisphosphatase, xúc tác quá trình chuyển đổi fructose 1,6-diphosphate thành fructose-6-phosphate. FDPase được mã hóa bởi FBP1 trên nhiễm sắc thể 9q22.32. Thiếu hụt FDPase làm suy giảm sự hình thành glucose từ lactate; glycerol; các acid amin tân tạo glucose, chẳng hạn như alanin; và fructose.
Hạ đường huyết do thiếu hụt FDPase lần đầu tiên được mô tả vào năm 1970 bởi Baker và Winegrad. Thiếu hụt FDPase có thể biểu hiện bằng hạ đường huyết và nhiễm toan lactic trong những ngày đầu đời, nhưng có tới 50% các trường hợp biểu hiện muộn hơn. Các biểu hiện điển hình bao gồm thở nhanh do nhiễm toan lactic và nhiễm toan ceton, co giật và hôn mê do hạ đường huyết, và gan to. Các cơn được thúc đẩy ở trẻ sơ sinh và trẻ nhỏ bởi việc nhịn đói kéo dài trong các bệnh xen kẽ. Acid uric tăng tương tự như GSD type 1, thứ phát sau sự cô lập phosphate dưới dạng các phosphate fructose trong gan, và tăng phân hủy các nucleotide adenin. Chẩn đoán nên được nghi ngờ khi nhịn đói dẫn đến hạ đường huyết và nhiễm toan lactic, với phản ứng đường huyết kém với glucagon. Chẩn đoán có thể được xác nhận bằng phân tích đột biến di truyền của FBP1. Điều trị các cơn hạ đường huyết cấp tính là truyền glucose và bicarbonate tĩnh mạch. Điều trị mạn tính là tránh nhịn đói kéo dài và giảm, nhưng không loại bỏ hoàn toàn, fructose khỏi chế độ ăn.
Không dung nạp Fructose di truyền
Không dung nạp fructose di truyền (HFI) lần đầu tiên được mô tả như một đặc ứng với việc ăn fructose, và sau đó được xác định là do các đột biến lặn của aldolase B (ALDOB tại 9q31.1). Aldolase B đóng một vai trò thiết yếu trong chuyển hóa fructose bằng cách chuyển đổi fructose-1-phosphate thành dihydroxyacetone phosphate và glyceraldehyde, là các cơ chất cho cả quá trình tân tạo glucose thông qua fructose 1,6-diphosphate hoặc quá trình oxy hóa thông qua chuyển đổi thành pyruvate. Việc ăn fructose (ví dụ, dưới dạng sucrose trong trái cây) ở những bệnh nhân bị thiếu hụt aldolase B gây ra sự tích tụ fructose-1-phosphate trong gan, thận và các tế bào ruột, dẫn đến sự suy giảm phosphate nội bào và cả các triệu chứng cấp tính và tổn thương cơ quan lâu dài tiềm tàng. Sự suy giảm phosphate gây hạ đường huyết bằng cách làm suy giảm quá trình ly giải glycogen và, như đã lưu ý trước đó trong thiếu hụt G6Pase và thiếu hụt FDPase, cũng gây tăng acid uric máu do tăng phân hủy nucleotide adenin. Hạ đường huyết của HFI xảy ra ngay sau khi ăn fructose, thay vì khi đói.
HFI biểu hiện ở trẻ sơ sinh, sau khi fructose được đưa vào chế độ ăn trong trái cây hoặc dưới dạng sucrose (đường ăn). Việc ăn fructose gây nôn, đau bụng, tiêu chảy và hạ đường huyết. Với việc ăn một lượng lớn fructose, nồng độ acid lactic và acid uric trong huyết tương tăng và nồng độ phốt pho, kali và bicarbonate giảm. Mức độ nghiêm trọng của các triệu chứng tỷ lệ thuận với lượng fructose ăn vào; ở trẻ sơ sinh, lượng lớn (ví dụ, trong sữa hoặc sữa công thức được làm bằng sucrose) có thể gây sốc, suy gan cấp và tử vong. Trẻ lớn hơn ít có khả năng ăn phải lượng độc hại do đau bụng do fructose gây ra. Điều trị cấp tính hạ đường huyết bằng glucose tĩnh mạch nhanh chóng đảo ngược các triệu chứng và bệnh nhân bị ảnh hưởng vẫn khỏe mạnh nếu ngừng tiếp xúc với fructose. Việc ăn fructose mạn tính gây chậm phát triển và rối loạn chức năng gan ngày càng tăng. Tác động sớm nhất đến thận là tổn thương ống lượn gần với glucose niệu và phosphaturia; tiếp xúc kéo dài với fructose có thể dẫn đến suy thận.
Thử thách Fructose có khả năng nguy hiểm và không cần thiết để chẩn đoán, vì xét nghiệm đột biến di truyền của ALDOB đã có sẵn. Điều trị mạn tính là tránh tất cả các loại thực phẩm chứa fructose (trái cây, sucrose, xi-rô ngô fructose cao); vì sorbitol được chuyển hóa thành fructose, nó cũng cần được loại trừ.
Khiếm khuyết Oxy hóa Acid béo
Oxy hóa acid béo là nguồn năng lượng chính cho tim và cơ xương hoạt động và, ngoài não, trở thành nhiên liệu chính của cơ thể trong khi nhịn đói kéo dài. Quá trình ly giải các kho dự trữ triglyceride của mô mỡ giải phóng glycerol, làm cơ chất cho tân tạo glucose gan, và FFA, được sử dụng trực tiếp làm nhiên liệu cho các mô ngoại vi và, gián tiếp, để tiết kiệm tiêu thụ glucose. FFA được chuyển đổi bằng quá trình β-oxy hóa trong ty thể ở gan thành các thể ceton (BOHB và acetoacetate), là một nhiên liệu thay thế cho não. Các bước chính trong vận chuyển acid béo vào ty thể, chu trình β-oxy hóa, và con đường tổng hợp ceton được phác thảo trong Chương 7. Các rối loạn di truyền trong tất cả các bước này đã được xác định, nói chung, biểu hiện với các cơn hạ đường huyết không ceton đe dọa tính mạng, hôn mê và suy sụp tim mạch do stress nhịn đói gây ra. Các khiếm khuyết cản trở các bước đầu trong quá trình oxy hóa acid béo chuỗi dài có xu hướng có nhiều đặc điểm toàn thân hơn, bao gồm bệnh cơ tim mạn tính và yếu cơ xương, trong khi các khiếm khuyết ở các bước sau ảnh hưởng đến quá trình oxy hóa acid béo chuỗi ngắn hơn, chẳng hạn như thiếu hụt medium chain acyl-CoA dehydrogenase (MCAD), có thể biểu hiện chủ yếu bằng các đợt bệnh cấp tính do nhịn đói gây ra. Là một nhóm, các rối loạn di truyền của quá trình oxy hóa acid béo tương đối phổ biến (tỷ lệ mắc bệnh thiếu hụt MCAD là ~1/15.000 ca sinh ở Hoa Kỳ, và 1/5000 ở Bắc Đức), và phải được xem xét trong chẩn đoán phân biệt của bất kỳ đứa trẻ nào bị hạ đường huyết. Các chương trình sàng lọc sơ sinh tồn tại ở hầu hết các quốc gia, có thể xác định hầu hết các khiếm khuyết di truyền trong quá trình oxy hóa acid béo, dựa trên phân tích hồ sơ acylcarnitine (ngoại lệ bao gồm thiếu hụt HMG-CoA synthase). Tuy nhiên, các bác sĩ nội tiết nhi cũng phải có khả năng nhận biết các rối loạn oxy hóa acid béo này, vì nhiều trường hợp có thể biểu hiện dưới dạng hạ đường huyết khởi phát mới ở các độ tuổi từ giai đoạn sơ sinh đến thời kỳ nhũ nhi, và đến vài năm tuổi.
Hạ đường huyết có thể biểu hiện trong những ngày đầu đời (xem Chương 7) ở những em bé tiếp xúc với stress nhịn đói sớm do bú mẹ thất bại. Tuy nhiên, thông thường, bệnh nhân có thể không biểu hiện cho đến vài tháng tuổi, khi khoảng cách cho ăn qua đêm kéo dài đến 8 đến 12 giờ nhịn đói. Các rối loạn di truyền của quá trình oxy hóa acid béo đặc biệt có khả năng trở nên rõ ràng trên lâm sàng trong quá trình đói nhanh của bệnh đường tiêu hóa. Bởi vì các rối loạn oxy hóa acid béo làm suy giảm quá trình sinh ceton, hạ đường huyết liên quan đến các rối loạn này là không ceton; tuy nhiên, trái ngược với các rối loạn HI, nồng độ FFA trong huyết tương tăng. Các cơn bệnh cấp tính có thể đi kèm với hôn mê, gan nhiễm mỡ và bệnh não, bắt chước hội chứng Reye, và có thể không đáp ứng ngay với glucose. Các đợt có thể gây tử vong nhanh chóng và có thể giống với hội chứng đột tử ở trẻ sơ sinh. Bệnh cơ tim phì đại hoặc giãn và yếu cơ xương là những đặc điểm nổi bật của một số khiếm khuyết nhất định, chẳng hạn như thiếu hụt long chain acyl CoA dehydrogenase, thiếu hụt vận chuyển carnitine, và thiếu hụt carnitine-palmitoyl transferase 2. Ở những bệnh nhân bị thiếu hụt carnitine nguyên phát do chất vận chuyển carnitine màng sinh chất (SLC22A5), cần điều trị bằng bổ sung carnitine liều cao; giá trị của việc bổ sung carnitine trong các rối loạn khác còn gây tranh cãi. Chẩn đoán có thể dựa trên hồ sơ acylcarnitine huyết tương bất thường và được xác nhận bằng phân tích đột biến di truyền.
Điều trị chính của các rối loạn oxy hóa acid béo này là tránh nhịn đói. Ở trẻ nhỏ hơn, nhịn đói nên được giới hạn dưới 8 đến 10 giờ; trẻ lớn hơn có thể chịu được nhịn đói đến 10 đến 12 giờ. Các đợt bệnh cấp tính kèm theo nhịn đói nên được điều trị bằng cách nhanh chóng truyền dextrose tĩnh mạch để nâng glucose huyết tương lên mức bình thường cao.
Hạ đường huyết do các tác nhân ngoại sinh
Ở trẻ em, hạ đường huyết do các tác nhân ngoại sinh thường là do vô tình nuốt phải. Ethanol, salicylat, quinin và thuốc chẹn beta, chẳng hạn như propranolol, là những tác nhân thường xuyên nhất liên quan đến hạ đường huyết ở trẻ em.
Hạ đường huyết do Rượu
Ethanol là một nguyên nhân khét tiếng của hạ đường huyết khi đói nghiêm trọng ở trẻ nhỏ và người lớn nghiện rượu suy dinh dưỡng, và đôi khi ở thanh thiếu niên khỏe mạnh bị say. Quá trình oxy hóa ethanol trong gan gây ra sự gia tăng tỷ lệ nicotinamide adenine dinucleotide (NAD)H:NAD làm suy giảm quá trình chuyển đổi lactate thành pyruvate và, do đó, ức chế tân tạo glucose. Sự ức chế tân tạo glucose này có thể dẫn đến hạ đường huyết trong bối cảnh nhịn đói đủ lâu để làm cạn kiệt kho dự trữ glycogen gan. Trẻ nhỏ nhịn đói qua đêm đặc biệt dễ bị tổn thương, cũng như thanh thiếu niên bị say rượu mà không ăn. Hạ đường huyết có liên quan đến nhiễm toan lactic và tăng ceton máu. Cần điều trị bằng dextrose tĩnh mạch để đảo ngược tình trạng hạ đường huyết.
Ngộ độc Salicylat
Ở liều cao, aspirin và các salicylat khác đã được báo cáo là gây hạ đường huyết ở cả trẻ em và người lớn. Các cơ chế được đề xuất bao gồm kích thích giải phóng insulin hoặc ức chế tân tạo glucose gan. Trẻ sơ sinh dường như dễ bị hạ đường huyết do salicylat hơn trẻ lớn. Do những lo ngại về vai trò có thể có của salicylat trong việc gây ra hội chứng Reye, việc sử dụng thường quy của chúng ở trẻ nhỏ phần lớn đã bị từ bỏ.
Hypoglycin A và Bệnh nôn mửa Jamaica
Hạ đường huyết do ăn phải quả ackee chưa chín lần đầu tiên được mô tả là bệnh nôn mửa Jamaica xảy ra ở trẻ nhỏ đang tìm kiếm thức ăn tiềm năng do suy dinh dưỡng/đói. Độc tố gây ra được xác định là hypoglycin A, được chuyển hóa thành một hợp chất ba vòng là cơ chất tự sát cho medium chain acyl-CoA dehydrogenase. Một bệnh tương tự gần đây đã được liên kết với việc ăn quả vải chưa chín, chứa một độc tố liên quan (methylene cyclopropyl-glycine). Các độc tố trong cả hai loại quả này tạo ra một tình trạng tương tự như thiếu hụt MCAD (được mô tả trước đó và trong Chương 7).
Hạ đường huyết trong Nhịn đói, Thiếu ăn, Bệnh tật và Stress
Trong khi nhịn đói kéo dài, FFA và ceton cung cấp một tỷ lệ nhiên liệu ngày càng lớn và nồng độ glucose huyết tương có thể giảm xuống dưới 50 mg/dL (2,8 mM), không có triệu chứng tự chủ hoặc thiếu glucose não, hoặc các ảnh hưởng xấu rõ ràng vì nồng độ ceton tăng cao cung cấp đủ nhiên liệu thay thế cho não (xem Hình 23.2). Điều này đặc biệt có khả năng được thấy ở trẻ nhỏ khỏe mạnh và phụ nữ trẻ, do các kho dự trữ nhiên liệu cơ thể tương đối nhỏ hơn và khối lượng cơ thể nhỏ hơn so với kích thước não. Hạ đường huyết sinh hóa, ví dụ, đôi khi được quan sát thấy ở trẻ nhỏ hơn nhịn đói trước phẫu thuật quá mức.
Nguy cơ hạ đường huyết sinh hóa, đôi khi có triệu chứng, còn tăng hơn nữa ở trẻ em trong một loạt các bệnh căng thẳng, chẳng hạn như sưởi ấm lại sau khi hạ thân nhiệt sâu và gần chết đuối.
Hạ đường huyết trong Tình trạng Đói và Suy dinh dưỡng
Suy dinh dưỡng mạn tính làm tổn hại khả năng duy trì đường huyết bình thường, và hạ đường huyết ở trẻ sơ sinh và người lớn suy dinh dưỡng có thể khó đảo ngược. Ở trẻ em châu Phi mắc bệnh kwashiorkor, hạ đường huyết đã được báo cáo là một biến cố cuối cùng phổ biến. Ở các nước phát triển, chán ăn tâm thần là một nguyên nhân quan trọng của suy dinh dưỡng nặng, và hạ đường huyết là phổ biến và đôi khi có thể nghiêm trọng.
Hạ đường huyết khi Tập thể dục Kéo dài
Cơ sử dụng glucose với tốc độ cao trong khi tập thể dục; tuy nhiên, hạ đường huyết thường được ngăn ngừa trong khi tập thể dục kéo dài bằng cách giảm tiết insulin và tăng tiết glucagon và catecholamine. Tuy nhiên, hạ đường huyết đôi khi xảy ra trong khi tập thể dục cường độ cao, ngay cả ở người lớn được cho là khỏe mạnh, và có thể được ngăn ngừa bằng cách ăn carbohydrate phức hợp, nhưng không phải là đường bổ sung. Các trường hợp hạ đường huyết đủ nặng để gây co giật sau một cuộc chạy marathon hoặc các bài tập sức bền tương đương đã được báo cáo. tăng insulin máu do tập thể dục vì các đột biến promoter của MCT1 (được mô tả trước đây) có thể giải thích ít nhất một số trường hợp hạ đường huyết liên quan đến tập thể dục.
Bệnh tiêu chảy
Hạ đường huyết không phổ biến ở trẻ em khỏe mạnh trước đó bị viêm dạ dày ruột cấp do virus, trừ khi đã có một thời gian đói (như xảy ra sau khi tiêu thụ kéo dài nước hoặc chất lỏng không đường) hoặc tiêu chảy nặng. Hạ đường huyết trong những trường hợp này thường là do ceton và có kết quả tốt. Bởi vì hạ đường huyết không phổ biến ở trẻ em khỏe mạnh, và các rối loạn chuyển hóa hoặc nội tiết tố chưa được nghi ngờ trước đó có thể biểu hiện tại phòng cấp cứu với hạ đường huyết, điều quan trọng là phải xác nhận tiền sử đói kéo dài và loại trừ các tình trạng tiềm ẩn, đặc biệt là các rối loạn oxy hóa acid béo.
Khi tiêu chảy (ví dụ, dịch tả hoặc shigella) phát triển ở trẻ em suy dinh dưỡng nặng, hạ đường huyết là một dấu hiệu tiên lượng xấu và có thể gây tử vong do cạn kiệt các cơ chất tân tạo glucose và các nhiên liệu thay thế.
Nhiễm khuẩn huyết
Các bệnh truyền nhiễm hiếm khi gây hạ đường huyết; thực tế, tăng đường huyết do các phản ứng stress adrenergic có nhiều khả năng xảy ra hơn. Tuy nhiên, nhiễm khuẩn huyết, đặc biệt là nhiễm não mô cầu (không liên quan đến suy thượng thận), có thể biểu hiện bằng hạ đường huyết nặng. Nghiên cứu trên động vật cho thấy các cytokine, chẳng hạn như yếu tố hoại tử khối u có thể khuếch đại sự hấp thu glucose. Dữ liệu hạn chế ở người cho thấy sự ức chế phù hợp của insulin và sự gia tăng của các hormone đối kháng điều hòa, cũng như các cytokine.
Hạ đường huyết trong các bệnh nhiễm trùng cụ thể
Hạ đường huyết đã được báo cáo xảy ra ở khoảng một phần ba trẻ em bị sốt rét nặng và là một yếu tố nguy cơ làm tăng tỷ lệ tử vong. Nồng độ insulin thấp phù hợp, trong khi nồng độ lactate và alanin cao, cho thấy tân tạo glucose gan bị suy giảm. Quản lý nên bao gồm cung cấp glucose và theo dõi nồng độ glucose huyết tương. Ngoài ra, các loại thuốc điều trị sốt rét (đặc biệt là quinin) có thể làm trầm trọng thêm tình trạng hạ đường huyết do khả năng kích thích giải phóng insulin của chúng.
Hạ đường huyết đã được báo cáo nhiều lần ở trẻ sơ sinh bị nhiễm ho gà, gây ra bởi tăng insulin máu và không chỉ đơn giản là do nhịn đói. Tăng insulin máu, nhưng không có hạ đường huyết, cũng đã được chứng minh là xảy ra ở chuột sau khi tiêm phòng ho gà. Bằng chứng cho thấy độc tố ho gà khuếch đại phản ứng sinh insulin đối với glucose hơn là kích thích tiết insulin.
Hạ đường huyết trong Bệnh hệ thống cơ quan
Hạ đường huyết là phổ biến ở những bệnh nhân nặng ở mọi lứa tuổi và có thể xảy ra trong tình trạng suy hoặc bệnh nặng của gần như mọi hệ cơ quan chính. Gan là nguồn cung cấp glucose chính trong giai đoạn sau hấp thu, và trong điều kiện thí nghiệm, hạ đường huyết xảy ra sau khi mất 80% gan. Hạ đường huyết trong bệnh gan ở người ít dự đoán được hơn. Trong xơ gan và suy gan tiến triển, nồng độ glucose thường vẫn bình thường, ngay cả ở những bệnh nhân bị hôn mê gan. Tuy nhiên, hạ đường huyết khi đói có thể xảy ra lẻ tẻ trong nhiều dạng bệnh gan, rất có thể là do suy giảm dự trữ glycogen, ít phụ thuộc vào mức độ suy gan theo các thước đo khác. Hạ đường huyết đã được báo cáo ở người lớn và trẻ em, với tổn thương gan do thuốc, ngộ độc, và ở những bệnh nhân bị viêm gan truyền nhiễm và các nguyên nhân khác gây tổn thương gan. Hạ đường huyết cũng có thể xảy ra như một biến chứng của các bệnh di truyền hiếm gặp khác nhau ảnh hưởng đến gan. Ví dụ, thiếu hụt citrin có một dạng khởi phát trung gian ở trẻ em, đặc trưng bởi chậm phát triển, các bất thường thần kinh và hành vi từng đợt kèm theo tăng amoniac máu, và đôi khi là hạ đường huyết.
Tân tạo glucose ở thận cũng thường góp phần duy trì đường huyết trong khi đói, và hạ đường huyết có thể xảy ra ở những bệnh nhân suy thận mạn giai đoạn cuối. Bởi vì các phản ứng tự chủ đối với hạ đường huyết thường bị suy giảm trong suy thận mạn, các biểu hiện thiếu glucose não thường chiếm ưu thế. Trong nhiều trường hợp hạ đường huyết với bệnh thận, hạ đường huyết phản ánh một biến chứng của lọc máu hoặc các thủ thuật khác và sự tồn tại của một “hạ đường huyết do urê huyết” cụ thể đã bị thách thức.
Nhiều trường hợp hạ đường huyết khi đói liên quan đến cả viêm tụy cấp và mạn tính đã được báo cáo ở người lớn và trẻ em. Cả hạ đường huyết và tăng đường huyết đều được công nhận là các biến chứng của viêm tụy do quai bị ở trẻ em.
Nguyên nhân của hạ đường huyết liên quan đến bệnh tim nặng, cả bệnh tim bẩm sinh tím ở trẻ nhỏ, và suy tim sung huyết ở trẻ lớn và người lớn là phức tạp, với bằng chứng về cả sự hấp thu glucose tăng và suy giảm tân tạo glucose. Ở trẻ sơ sinh, hạ đường huyết có thể gây suy tim và cải thiện khi phục hồi mức glucose bình thường.
Cơ xương đóng góp các cơ chất cho tân tạo glucose trong khi đói, và hạ đường huyết khi đói xảy ra với một số dạng loạn dưỡng cơ và teo cơ tủy sống đặc trưng bởi khối lượng cơ giảm. Một cơ chế mới cho hạ đường huyết, do bệnh nội sọ, sẽ là sự thay đổi của các nhánh hướng tâm hoặc ly tâm của cảm nhận glucose vùng dưới đồi; điều này đã được chứng minh ở loài gặm nhấm và đã được viện dẫn trong các trường hợp hiếm gặp hạ đường huyết đi kèm với khối u não và chấn thương.
Hạ đường huyết đi kèm với nhiễm toan lactic đã được báo cáo là cả những biểu hiện không phổ biến và các biến cố giai đoạn cuối ở những bệnh nhân mắc bệnh bạch cầu cấp và mạn tính, và u lympho. Trẻ em theo các chế độ hóa trị liệu kéo dài đã được phát hiện có tỷ lệ hạ đường huyết khi đói cao do cạn kiệt glycogen và các tiền chất tân tạo glucose và do các tác động trực tiếp của các chất tương tự purine đường uống.
Hạ đường huyết trong Đơn vị Chăm sóc Tích cực
Hạ đường huyết là phổ biến ở trẻ em bị bệnh nặng, thường không có triệu chứng và, đặc biệt khi liên quan đến sự biến thiên glucose tăng, có liên quan đến kết quả kém hơn (tăng tỷ lệ mắc bệnh, thời gian nằm viện và tỷ lệ tử vong). Ngoài các quá trình cụ thể của các bệnh tiềm ẩn, các yếu tố góp phần khác có thể bao gồm cạn kiệt cơ chất, tiêu thụ glucose nhanh, suy dinh dưỡng, suy giảm tân tạo glucose, tác động của cytokine và suy thượng thận. Các yếu tố do điều trị, chẳng hạn như đường truyền bị đặt sai vị trí, thay đổi tốc độ truyền dextrose tĩnh mạch, hoặc tác dụng của thuốc cần được xem xét. Việc ngừng đột ngột dinh dưỡng đường tiêm có thể dẫn đến hạ đường huyết, đặc biệt ở trẻ em dưới 3 tuổi; theo đó, nên giảm dần tốc độ truyền để giảm nguy cơ hạ đường huyết phản ứng. Các yếu tố ảnh hưởng đến độ tin cậy của việc đo glucose (ví dụ, hematocrit thay đổi, oxy hóa, lấy máu từ đường truyền, thuốc) phổ biến hơn trong đơn vị chăm sóc tích cực (ICU). Như đã lưu ý trước đó, điều quan trọng là không quên khả năng một bệnh cấp tính đang bộc lộ một rối loạn chuyển hóa glucose được bù trừ trước đó, đặc biệt là ở một đứa trẻ nhỏ.
Việc sử dụng insulin nhằm mục đích kiểm soát đường huyết chặt chẽ để cải thiện kết quả ở trẻ em bị bệnh nặng gần đây đã trở thành nguyên nhân phổ biến nhất của hạ đường huyết trong ICU. Tuy nhiên, một thử nghiệm lâm sàng ngẫu nhiên đa trung tâm gần đây cho thấy trẻ em bị bệnh nặng có tăng đường huyết không được hưởng lợi từ việc kiểm soát chặt chẽ, từ 80 đến 110 mg/dL so với mức glucose huyết tương mục tiêu từ 150 đến 180 mg/dL.
Hạ đường huyết phản ứng và “Các cơn”
Hạ đường huyết phản ứng đề cập đến hạ đường huyết có triệu chứng xảy ra từ 1 đến 4 giờ sau bữa ăn, và hiếm gặp ở trẻ em, ngoài những trẻ đã phẫu thuật dạ dày (phẫu thuật Nissen cho trào ngược dạ dày thực quản hoặc phẫu thuật nối tắt dạ dày để điều trị béo phì). Tuy nhiên, trẻ em có các triệu chứng được cho là do hạ đường huyết không hiếm khi được giới thiệu đến các phòng khám nội tiết. Tam chứng Whipple (các triệu chứng điển hình của hạ đường huyết, liên quan đến mức glucose huyết tương thấp được ghi nhận và thuyên giảm khi điều trị để tăng glucose) được đề xuất bởi các hướng dẫn từ Hiệp hội Nội tiết Nhi khoa và Hiệp hội Nội tiết là cần thiết trước khi xem xét chẩn đoán hạ đường huyết, vì các triệu chứng của hạ đường huyết rất không đặc hiệu. Mặc dù máy đo đường huyết tại nhà có thể hữu ích trong việc kiểm tra khả năng hạ đường huyết, cần thận trọng khi xem xét việc sử dụng chúng vì các vấn đề về độ chính xác của kết quả (xem phần sau về Hạ đường huyết giả). Không nên dựa vào OGTT để chẩn đoán hạ đường huyết phản ứng, vì nhiều người bình thường phát triển glucose thấp không triệu chứng từ 2 đến 4 giờ sau khi tải glucose. Các sửa đổi chế độ ăn nhỏ để tránh thực phẩm có chỉ số đường huyết cao và bao gồm các loại thực phẩm được tiêu hóa chậm hơn, cùng với protein và chất béo, thường đủ để kiểm soát các triệu chứng trong hạ đường huyết phản ứng.
Hạ đường huyết giả
Một mức glucose huyết tương thấp được phát hiện bất ngờ ở một đứa trẻ dường như “khỏe mạnh”, đã làm xét nghiệm hóa sinh ngoại trú vì những lý do không liên quan đến chuyển hóa carbohydrate, thường là một kết quả giả. Nguyên nhân phổ biến nhất của hạ đường huyết giả là xử lý mẫu không đúng cách trước khi chúng đến phòng thí nghiệm. Nếu máu được lấy và để yên, glucose sẽ bị tiêu thụ bởi cả hồng cầu và bạch cầu. Nếu một mẫu máu toàn phần được để đông ở nhiệt độ phòng, nồng độ glucose huyết thanh có thể giảm từ 7 đến 20 mg/dL/giờ. Tốc độ đường phân phụ thuộc vào nhiệt độ và việc làm lạnh ngay các mẫu làm giảm nhưng không loại bỏ hoàn toàn việc tiêu thụ glucose. Việc sử dụng các ống thu thập tách tế bào khỏi huyết thanh cũng ngăn chặn quá trình đường phân. Fluoride ức chế nhưng không loại bỏ hoàn toàn quá trình đường phân, và nồng độ glucose có thể giảm 20% hoặc hơn khi quá trình xử lý bị trì hoãn dù chỉ vài giờ. Quá trình đường phân sau khi lấy máu tiêu thụ một tỷ lệ glucose tương đối cao hơn khi mẫu chứa mức glucose thấp tại thời điểm lấy, và hạ đường huyết giả phổ biến hơn khi số lượng hồng cầu hoặc bạch cầu tăng, và cũng đã được mô tả trong một cơn tan máu với số lượng hồng cầu có nhân cao.
Năm 2013, Tổ chức Tiêu chuẩn hóa Quốc tế (ISO) đã tuyên bố rằng máy đo đường huyết được chấp nhận là chính xác nếu 95% hoặc nhiều hơn kết quả đo nằm trong khoảng ±15 mg/dL hoặc ±15% (tùy theo giá trị nào lớn hơn) so với các giá trị tham chiếu. Do đó, theo ISO, nếu đường huyết thực là 60 mg/dL, glucose đo được có thể là 45 đến 75 mg/dL. Hướng dẫn năm 2014 của Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA) cho các máy đo đường huyết không kê đơn yêu cầu 95% hoặc nhiều hơn kết quả nằm trong khoảng ±15% so với các giá trị tham chiếu và 99% hoặc nhiều hơn kết quả nằm trong khoảng ±20% trên toàn bộ phạm vi đường huyết. Dựa trên các tiêu chí của FDA, nếu đường huyết thực là 60 mg/dL, glucose đo được sẽ được coi là chính xác chấp nhận được trong khoảng từ 51 đến 69 mg/dL. Các đánh giá gần đây về các máy đo đường huyết có bán trên thị trường cho thấy sự khác biệt đáng kể về hiệu suất trong phạm vi glucose thấp. Hơn nữa, độ chính xác trong phạm vi glucose thấp thay đổi đáng kể giữa các máy đo; ví dụ, một nghiên cứu cho thấy khi glucose huyết tương tham chiếu nhỏ hơn 70 mg/dL, sự khác biệt tuyệt đối trung bình của các máy đo khác nhau dao động từ 2 đến 11 mg/dL và sự khác biệt tương đối tuyệt đối trung bình (MARD) dao động từ 4,3% đến 24,2%. Trong một nghiên cứu khác, MARD dao động từ 8,9% đến 39,2%, khi glucose huyết tương tham chiếu nhỏ hơn 70 mg/dL.
Các thiết bị theo dõi glucose liên tục (CGM) ngày càng được sử dụng để theo dõi các kiểu glucose, nhưng các điểm đáy tuyệt đối có thể không chính xác và tính hữu dụng của chúng trong việc đánh giá hạ đường huyết vẫn chưa chắc chắn. Có những lo ngại về độ chính xác của CGM ở nồng độ glucose thấp; tuy nhiên, trong một nghiên cứu sử dụng hệ thống theo dõi glucose, phần lớn các bậc cha mẹ của trẻ em bị tăng insulin máu bẩm sinh thấy xu hướng glucose là hữu ích. CGM cũng đã được sử dụng để báo hiệu hạ đường huyết sắp xảy ra trong các nghiên cứu về kiểm soát đường huyết chặt chẽ, ở trẻ em bị bệnh nặng, và mặc dù có khả năng hữu ích, việc sử dụng chúng chưa được đánh giá một cách có hệ thống trong chẩn đoán và quản lý các rối loạn hạ đường huyết đa dạng. Một số báo cáo đã gợi ý rằng việc sử dụng CGM có thể làm giảm tần suất và thời gian hạ đường huyết ở trẻ sơ sinh có nguy cơ cao. Cần có các thử nghiệm ở những bệnh nhân bị tăng insulin máu bẩm sinh trước khi CGM có thể được khuyến nghị như một phương pháp thay thế thường quy để theo dõi nồng độ glucose trong quá trình điều trị. Cũng không có dữ liệu về việc sử dụng CGM để chẩn đoán hạ đường huyết, chẳng hạn như sử dụng nó trong môi trường ngoại trú như một phương pháp thay thế cho nghiệm pháp nhịn đói nội trú.
Mặc dù có những cân nhắc đã nói ở trên, điều quan trọng là phải nhấn mạnh rằng không nên bỏ qua các mức glucose thấp bất ngờ vì khả năng một xét nghiệm phòng thí nghiệm “tình cờ” có thể phát hiện ra một tình trạng hạ đường huyết mạn tính không được nhận biết.
ĐIỀU TRỊ CẤP CỨU HẠ ĐƯỜNG HUYẾT
Sau khi lấy được mẫu xét nghiệm trọng yếu, nên tiêm tĩnh mạch một liều nhỏ 0,2 g/kg dextrose trong vòng 1 phút (2 mL/kg dextrose 10%). Sau đó là truyền tĩnh mạch liên tục 8 mg/kg/phút bằng dung dịch dextrose 10%. Tốc độ truyền glucose này có thể được tính toán nhanh chóng và thuận tiện bằng cách sử dụng công thức đơn giản là 5 mL/kg/giờ dung dịch dextrose 10% cung cấp khoảng 8 mg/kg/phút glucose. Nồng độ glucose nên được xác định 15 phút sau khi tiêm bolus và trong khi đang truyền glucose duy trì. Nếu hạ đường huyết tái phát, có thể tiêm bolus 0,5 g/kg (5 ml/kg dextrose 10%) và tăng tốc độ truyền glucose lên 25% đến 50%. Để biết chi tiết về các phương pháp điều trị cụ thể của từng rối loạn, hãy xem các phần về từng tình trạng.
PHỤ LỤC: CÁC XÉT NGHIỆM HỮU ÍCH ĐỂ CHẨN ĐOÁN CÁC RỐI LOẠN HẠ ĐƯỜNG HUYẾT
Nghiệm pháp nhịn đói chẩn đoán: Thời gian nhịn đói tối đa dựa trên tuổi: dưới 1 tháng 18 giờ, 1-12 tháng 24 giờ, 1-12 tuổi 36 giờ, trên 13 tuổi 72 giờ. Khoảng thời gian giữa các lần lấy mẫu dựa trên nồng độ glucose huyết tương (PG): theo dõi PG và BOHB huyết tương bằng các máy đo tại giường tương ứng mỗi 3 giờ cho đến khi PG dưới 70 mg/dL (3,9 mmol/L), sau đó mỗi giờ cho đến khi PG dưới 60 mg/dL (3,3 mmol/L), sau đó mỗi 30 phút cho đến khi PG dưới 50 mg/dL (2,8 mmol/L). Tiêu chí kết thúc nhịn đói bao gồm: thời gian tối đa (dựa trên tuổi bệnh nhân), glucose huyết tương được xác nhận dưới 50 mg/dL, BOHB ≥ 2,0 mmol/L x 2 hoặc có triệu chứng nặng. Sau khi xác nhận PG dưới 50 mg/dL (ví dụ, trong phép đo phòng thí nghiệm STAT), lấy các mẫu máu trọng yếu để xét nghiệm (theo dõi tại các thời điểm trung gian của insulin, lactate, BOHB, FFA thường hữu ích, đặc biệt nếu có thể xảy ra các thay đổi cấp tính để đáp ứng với việc bệnh nhân phát triển các triệu chứng adrenergic; Bảng 23.5).
Mẫu xét nghiệm trọng yếu: đề cập đến các xét nghiệm được lấy tại thời điểm PG dưới 50 mg/dL để giúp xác định nguyên nhân của hạ đường huyết. Mẫu trọng yếu có thể bao gồm một bảng chuyển hóa toàn diện, insulin, BOHB, FFA, lactate, GH, cortisol, C-peptide, amoniac, hồ sơ acylcarnitine, carnitine tự do và toàn phần, IGFBP-1, ceton niệu, acid hữu cơ niệu. Các phản ứng bình thường đối với hạ đường huyết khi đói (PG < 50 mg/dL) ở trẻ sơ sinh, trẻ em và người lớn là tương tự nhau: insulin dưới 2 đến 3 µU/mL (giới hạn dưới của xét nghiệm), lactate dưới 1,5 mmol/L, BOHB lớn hơn 2,0 mmol/L, FFA lớn hơn 1,0 mmol/L (xem Ferrara và cộng sự).
Nghiệm pháp kích thích Glucagon: Vào cuối nghiệm pháp nhịn đói, sau khi đã lấy được các mẫu trọng yếu (và nếu nhịn đói được kết thúc vì PG < 50 mg/dL và BOHB < 2,0 mmol/L), tiêm 1 mg glucagon tiêm bắp hoặc tiêm tĩnh mạch. PG trên máy đo tại giường nên là 50 đến 55 (hoặc thấp hơn) mg/dL trước khi dùng glucagon (thời điểm 0). Kiểm tra PG tại thời điểm 0, 10, 20, 30 và 40 phút. Nếu PG không tăng hơn 20 mg/dL sau 20 phút, hãy xem xét kết thúc nghiên cứu. Một phản ứng dương tính (bất thường) là sự gia tăng PG ít nhất 30 mg/dL trong vòng 40 phút, cho thấy rằng tác động insulin dư thừa đã kìm hãm sự phân hủy glycogen và đã được khắc phục bởi tác động của liều glucagon cao.
Nghiệm pháp dung nạp Protein đường uống cho Tăng insulin máu nhạy cảm với Protein
- Bệnh nhân phải nhịn đói (không ăn uống gì) trong hơn 3 giờ
- Xác minh dị ứng của bệnh nhân vì bột protein chứa protein sữa bò (whey)
- Bột protein Beneprotein 1,16 g/kg (trộn bột protein trong 4 ounce Crystal Light)
- Cho uống dung dịch protein bằng miệng hoặc qua sonde mũi-dạ dày (NG) trong vòng chưa đầy 5 phút
- Mẫu xét nghiệm: PG, insulin tại các thời điểm -15, 0, +15, +30, +45, +60, +90, +120, +150 và +180 phút
- Chấm dứt nghiệm pháp nếu PG dưới 50 mg/dL trong quá trình nghiệm pháp dung nạp protein đường uống
- Diễn giải: Phản ứng dương tính (bất thường) là sự sụt giảm PG hơn 12 mg/dL và dưới 70 mg/dL.
Nghiệm pháp dung nạp Glucose đường uống cho Hạ đường huyết sau ăn
- Bệnh nhân phải nhịn ăn 4 giờ trước khi bắt đầu nghiên cứu
- Dùng dextrose (ví dụ, Glucola) 1,75 g/kg uống trong vòng 15 đến 20 phút hoặc qua ống NG—tối đa 75 g.
- Mẫu xét nghiệm tại các thời điểm -15, 0, +30, +60, +90, +120, +150, +180, +210 và +240 phút cho PG và insulin
- Diễn giải: Trẻ em bị hạ đường huyết dumping muộn do phẫu thuật Nissen có mức tăng insulin quá mức (220 ± 310 µIU/mL [1610 ± 2250 pmol/L]), sau đó là mức PG đáy hạ đường huyết (47 ± 11 mg/dL [2,6 ± 0,6 mmol/L]) sau khi uống glucose. Chúng cũng thường, nhưng không phải luôn luôn, có sự gia tăng PG quá mức (180 ± 87 mg/dL [10 ± 4,9 mmol/L]) so với nhóm chứng: tăng insulin 36 ± 22 µIU/mL (255 ± 160 pmol/L); PG đáy 86 ± 21 mg/dL (4,8 ± 1,2 mmol/L); PG tăng 58 ± 40 mg/dL (3,2 ± 2,2 mmol/L).
Bảng 23.5 Quy trình cho các nghiên cứu nhịn đói
| Loại nhịn đói | Thời gian nhịn đói dựa trên tuổi, Mục đích | Theo dõi Glucose huyết tương và BOHB | Kết thúc nhịn đói |
|---|---|---|---|
| Nhịn đói chẩn đoán | <1 tháng: 18 giờ, 1-12 tháng: 24 giờ, >12 tháng: 36 giờ, >13 tuổi: 72 giờ. Mục đích: Thiết lập chẩn đoán cơ bản gây hạ đường huyết của bệnh nhân | Máy đo glucose và BOHB tại giường: Mỗi 3 giờ cho đến khi PG <70, sau đó mỗi 1 giờ cho đến khi <60, sau đó mỗi 30 phút cho đến khi <50 mg/dL. | Kết thúc nhịn đói vì: hết thời gian, PG < 50 mg/dL, BOHB ≥ 2,0 mmol/L x 2, hoặc có triệu chứng. GST: khi PG < 50 mg/dL, tiêm 1 mg glucagon IV/IM và sau đó theo dõi PG tại 10, 20, 30, 40 phút (bỏ qua nếu BOHB > 2 mmol/L). |
| Nhịn đói an toàn | Diazoxide <1 tháng: 8-12 giờ; 1-12 tháng: 12 giờ; >1 năm: 18 giờ. Octreotide: 6-8 giờ (bỏ một cữ ăn). Lanreotide: 18 giờ nếu không dùng dextrose. Mục đích: Kiểm tra hiệu quả của phác đồ hiện có | Máy đo glucose và BOHB tại giường: Mỗi 3 giờ cho đến khi PG < 70 mg/dL. | Kết thúc nhịn đói vì: hết thời gian, PG được xác nhận < 70 mg/dL, BOHB ≥ 2,5-3 mmol/L x 2. |
| Nhịn đói chứng minh khỏi bệnh | 12 giờ nếu đang dùng dextrose qua sonde dạ dày liên tục. <1 tháng: 18 giờ; 1-12 tháng: 24 giờ; >1 năm: 36 giờ; >13 tuổi: 48 giờ. Mục đích: Chứng minh bệnh đã khỏi ở những người đã được điều trị dứt điểm hoặc đã ngưng thuốc | Máy đo glucose và BOHB tại giường: Mỗi 3 giờ cho đến khi PG <70, sau đó mỗi 1 giờ cho đến khi <60, sau đó mỗi 30 phút cho đến khi <50 mg/dL. | Kết thúc nhịn đói vì: hết thời gian, PG được xác nhận < 50 mg/dL, BOHB ≥ 3 mmol/L x 2, hoặc có triệu chứng. Lấy mẫu HI: PG, FFA, BOHB, insulin, IGFBP-1, C-peptide. GST: khi PG < 50 mg/dL (nếu BOHB < 3 mmol/L) tiêm 1 mg glucagon IM/IV và theo dõi PG tại 10, 20, 30, 40 phút. |
| U tiết insulin (Insulinoma) | >13 tuổi: 72 giờ. |
Vào cuối quá trình nhịn đói chẩn đoán, lấy mẫu xét nghiệm trọng yếu. Nếu PG < 50 mg/dL và BOHB < 1,5 mmol/L, điều này phù hợp nhất với HI. Do đó, các xét nghiệm tăng insulin máu là quan trọng nhất để lấy trước tiên: PG, FFA, BOHB, insulin (cân nhắc thêm C-peptide, cortisol, GH, amoniac, lactate, hồ sơ acylcarnitine, carnitine, IGFBP-1). GST được chỉ định nếu nghi ngờ HI. Nếu BOHB ≥ 2 mmol/L khi kết thúc nhịn đói, điều này cho thấy một dạng hạ đường huyết do ceton. Do đó, các xét nghiệm hạ đường huyết do ceton là quan trọng để lấy trước tiên: PG, FFA, BOHB, cortisol, GH, lactate, hồ sơ acylcarnitine, carnitine, insulin, C-peptide, amoniac, IGFBP-1. GST KHÔNG được chỉ định. BOHB, Beta-hydroxybutyrate; FFA, acid béo tự do; GST, nghiệm pháp kích thích glucagon; HI, tăng insulin máu; IGFBP-1, protein gắn yếu tố tăng trưởng giống insulin 1; IM, tiêm bắp; IV, tiêm tĩnh mạch; PG, glucose huyết tương.
BẢNG CHÚ GIẢI THUẬT NGỮ ANH VIỆT – CHƯƠNG 23
| STT | Thuật ngữ tiếng Anh | Phiên âm IPA | Nghĩa Tiếng Việt |
|---|---|---|---|
| 1 | Hypoglycemia | /ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết |
| 2 | Toddler | /ˈtɑːd.lɚ/ | Trẻ nhỏ (tuổi tập đi) |
| 3 | Child | /tʃaɪld/ | Trẻ em |
| 4 | Glucose Metabolism | /ˈɡluː.koʊs məˈtæb.ə.lɪ.zəm/ | Chuyển hóa glucose |
| 5 | Infancy | /ˈɪn.fən.si/ | Giai đoạn nhũ nhi |
| 6 | Childhood | /ˈtʃaɪld.hʊd/ | Thời thơ ấu |
| 7 | Glucose Production | /ˈɡluː.koʊs prəˈdʌk.ʃən/ | Sản xuất glucose |
| 8 | Glucose Utilization | /ˈɡluː.koʊs ˌjuː.t̬əl.əˈzeɪ.ʃən/ | Sử dụng glucose |
| 9 | Feeding Intervals | /ˈfiː.dɪŋ ˈɪn.tɚ.vəlz/ | Khoảng cách giữa các cữ ăn |
| 10 | Symptoms | /ˈsɪmp.təmz/ | Triệu chứng |
| 11 | Signs | /saɪnz/ | Dấu hiệu |
| 12 | Fasting | /ˈfæs.tɪŋ/ | Nhịn đói |
| 13 | Hyperinsulinism (HI) | /ˌhaɪ.pɚˈɪn.sə.lɪ.nɪ.zəm/ | Tăng insulin máu |
| 14 | Glycogen Storage Diseases (GSD) | /ˈɡlaɪ.kə.dʒən ˈstɔːr.ɪdʒ dɪˌziː.zɪz/ | Bệnh dự trữ glycogen |
| 15 | Hormone Deficiency | /ˈhɔːr.moʊn dɪˈfɪʃ.ən.si/ | Thiếu hụt hormone |
| 16 | Gluconeogenesis | /ˌɡluː.koʊ.niː.oʊˈdʒen.ə.sɪs/ | Tân tạo glucose |
| 17 | Exogenous Agents | /ɛkˈsɑː.dʒə.nəs ˈeɪ.dʒənts/ | Tác nhân ngoại sinh |
| 18 | Hypoglycin A | /ˌhaɪ.poʊˈɡlaɪ.sɪn eɪ/ | Hypoglycin A |
| 19 | Jamaican Vomiting Sickness | /dʒəˈmeɪ.kən ˈvɑː.mɪ.tɪŋ ˈsɪk.nəs/ | Bệnh nôn mửa Jamaica |
| 20 | Starvation | /stɑːrˈveɪ.ʃən/ | Tình trạng đói |
| 21 | Reactive Hypoglycemia | /riˈæk.tɪv ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết phản ứng |
| 22 | Artifactual Hypoglycemia | /ˌɑːr.t̬əˈfæk.tʃu.əl ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết giả |
| 23 | Emergency Treatment | /ɪˈmɝː.dʒən.si ˈtriːt.mənt/ | Điều trị cấp cứu |
| 24 | Obligate fuel | /ˈɑː.blɪ.ɡət fjuːl/ | Nhiên liệu bắt buộc |
| 25 | Brain | /breɪn/ | Não |
| 26 | Physiological conditions | /ˌfɪz.i.əˈlɑː.dʒɪ.kəl kənˈdɪʃ.ənz/ | Điều kiện sinh lý |
| 27 | Synthesize | /ˈsɪn.θə.saɪz/ | Tổng hợp |
| 28 | Glycogen | /ˈɡlaɪ.kə.dʒən/ | Glycogen |
| 29 | Alternative fuels | /ɔːlˈtɝː.nə.t̬ɪv fjuːlz/ | Nhiên liệu thay thế |
| 30 | Circulation | /ˌsɝː.kjəˈleɪ.ʃən/ | Tuần hoàn |
| 31 | Concentration | /ˌkɑːn.sənˈtreɪ.ʃən/ | Nồng độ |
| 32 | Ketones | /ˈkiː.toʊnz/ | Ceton |
| 33 | Lactate | /ˈlæk.teɪt/ | Lactate |
| 34 | Vigorous exercise | /ˈvɪɡ.ɚ.əs ˈek.sɚ.saɪz/ | Tập thể dục gắng sức |
| 35 | Blood-to-brain glucose transport | /blʌd tə breɪn ˈɡluː.koʊs ˈtræn.spɔːrt/ | Vận chuyển glucose từ máu vào não |
| 36 | Glucose transporter 1 (GLUT-1) | /ˈɡluː.koʊs trænsˈpɔːr.t̬ɚ wʌn/ | Chất vận chuyển glucose 1 |
| 37 | Arterial plasma | /ɑːrˈtɪr.i.əl ˈplæz.mə/ | Huyết tương động mạch |
| 38 | Cerebral metabolic rate | /səˈriː.brəl ˌmet̬.əˈbɑː.lɪk reɪt/ | Tốc độ chuyển hóa não |
| 39 | Functional brain failure | /ˈfʌŋk.ʃən.əl breɪn ˈfeɪl.jɚ/ | Suy chức năng não |
| 40 | Permanent brain injury | /ˈpɝː.mə.nənt breɪn ˈɪn.dʒər.i/ -> Tổn thương não vĩnh viễn | |
| 41 | Brain death | /breɪn deθ/ | Chết não |
| 42 | Newborn period | /ˈnuː.bɔːrn ˈpɪr.i.əd/ | Giai đoạn sơ sinh |
| 43 | Acquired disorder | /əˈkwaɪɚd dɪˈsɔːr.dɚ/ | Rối loạn mắc phải |
| 44 | Endocrine system | /ˈen.də.krɪn ˈsɪs.təm/ | Hệ nội tiết |
| 45 | Intercurrent illness | /ˌɪn.t̬ɚˈkɝː.ənt ˈɪl.nəs/ | Bệnh xen kẽ |
| 46 | Congenital abnormalities | /kənˈdʒen.ə.t̬əl ˌæb.nɔːrˈmæl.ə.t̬iz/ | Dị tật bẩm sinh |
| 47 | Inborn errors of metabolism | /ˈɪn.bɔːrn ˈer.ɚz əv məˈtæb.ə.lɪ.zəm/ | Rối loạn chuyển hóa bẩm sinh |
| 48 | Toxins | /ˈtɑːk.sɪnz/ | Độc tố |
| 49 | Normoglycemia | /ˌnɔːr.moʊ.ɡlaɪˈsiː.mi.ə/ | Đường huyết bình thường |
| 50 | Glucose homeostasis | /ˈɡluː.koʊs ˌhoʊ.mi.oʊˈsteɪ.sɪs/ | Cân bằng nội môi glucose |
| 51 | Critical samples | /ˈkrɪt̬.ɪ.kəl ˈsæm.pəlz/ | Mẫu xét nghiệm trọng yếu |
| 52 | Glucose flux | /ˈɡluː.koʊs flʌks/ | Luồng glucose |
| 53 | Systemic glucose balance | /sɪˈstem.ɪk ˈɡluː.koʊs ˈbæl.əns/ | Cân bằng glucose toàn thân |
| 54 | Carbohydrate-containing meal | /ˌkɑːr.boʊˈhaɪ.dreɪt kənˈteɪ.nɪŋ miːl/ | Bữa ăn chứa carbohydrate |
| 55 | Insulin secretion | /ˈɪn.sə.lɪn sɪˈkriː.ʃən/ | Sự tiết insulin |
| 56 | Inhibition | /ˌɪn.hɪˈbɪʃ.ən/ | Sự ức chế |
| 57 | Counterregulatory hormone | /ˌkaʊn.t̬ɚˈreɡ.jə.lə.tɔːr.i ˈhɔːr.moʊn/ | Hormone đối kháng điều hòa |
| 58 | Basal levels | /ˈbeɪ.zəl ˈlev.əlz/ | Mức cơ bản/nền |
| 59 | Premature infants | /ˌpriː.məˈtʃʊr ˈɪn.fənts/ | Trẻ sinh non |
| 60 | Glycogenolysis | /ˌɡlaɪ.kə.dʒəˈnɑː.lə.sɪs/ | Ly giải glycogen |
| 61 | Liver | /ˈlɪv.ɚ/ | Gan |
| 62 | Adipose tissue | /ˈæd.ə.poʊs ˈtɪʃ.uː/ | Mô mỡ |
| 63 | Lipolysis | /laɪˈpɑː.lə.sɪs/ | Ly giải lipid |
| 64 | Free fatty acids (FFA) | /friː ˌfæt̬.i ˈæs.ɪdz/ | Acid béo tự do |
| 65 | Peripheral tissues | /pəˈrɪf.ɚ.əl ˈtɪʃ.uːz/ | Mô ngoại vi |
| 66 | Ketogenesis | /ˌkiː.t̬oʊˈdʒen.ə.sɪs/ | Sinh ceton |
| 67 | Hyperketonemia | /ˌhaɪ.pɚ.kiː.t̬oʊˈniː.mi.ə/ | Tăng ceton máu |
| 68 | Metabolic fuels | /ˌmet̬.əˈbɑː.lɪk fjuːlz/ | Nhiên liệu chuyển hóa |
| 69 | Beta-hydroxybutyrate (BOHB) | /ˈbeɪ.t̬ə haɪˌdrɑːk.siˈbjuː.t̬ə.reɪt/ | Beta-hydroxybutyrate |
| 70 | Postabsorptive state | /poʊst.æbˈsɔːrp.tɪv steɪt/ | Trạng thái sau hấp thu |
| 71 | Glucagon | /ˈɡluː.kə.ɡɑːn/ | Glucagon |
| 72 | Epinephrine | /ˌep.əˈnef.rɪn/ | Epinephrine (Adrenaline) |
| 73 | Cortisol | /ˈkɔːr.t̬ə.sɔːl/ | Cortisol |
| 74 | Growth hormone (GH) | /ɡroʊθ ˈhɔːr.moʊn/ | Hormone tăng trưởng |
| 75 | Sympathetic neural activation | /ˌsɪm.pəˈθet̬.ɪk ˈnʊr.əl ˌæk.təˈveɪ.ʃən/ | Kích hoạt thần kinh giao cảm |
| 76 | Skeletal muscle | /ˈskel.ə.t̬əl ˈmʌs.əl/ | Cơ xương |
| 77 | Gluconeogenic precursors | /ˌɡluː.koʊ.niː.oʊˈdʒen.ɪk prɪˈkɝː.sɚz/ | Tiền chất tân tạo glucose |
| 78 | Amino acids | /əˌmiː.noʊ ˈæs.ɪdz/ | Acid amin |
| 79 | Alanine | /ˈæl.ə.niːn/ | Alanine |
| 80 | Glutamine | /ˈɡluː.tə.miːn/ | Glutamine |
| 81 | Glycerol | /ˈɡlɪs.ə.rɔːl/ | Glycerol |
| 82 | Triglycerides | /traɪˈɡlɪs.ə.raɪdz/ | Triglyceride |
| 83 | Mitochondrial fatty acid oxidation | /ˌmaɪ.t̬əˈkɑːn.dri.əl ˌfæt̬.i ˈæs.ɪd ˌɑːk.səˈdeɪ.ʃən/ | Oxy hóa acid béo trong ty thể |
| 84 | Acetoacetate | /ˌæs.ə.toʊˈæs.ə.teɪt/ | Acetoacetate |
| 85 | Red blood cells | /red blʌd selz/ | Hồng cầu |
| 86 | Renal medullae | /ˈriː.nəl məˈdʌl.iː/ | Tủy thận |
| 87 | Fructose-1,6-bisphosphatase deficiency | /ˈfrʌk.toʊs wʌn sɪks bɪsˈfɑːs.fə.teɪs dɪˈfɪʃ.ən.si/ | Thiếu hụt Fructose-1,6-bisphosphatase |
| 88 | Lactation | /lækˈteɪ.ʃən/ | Sự tiết sữa |
| 89 | Adrenocorticotropic hormone (ACTH) | /əˌdriː.noʊˌkɔːr.t̬ɪ.koʊˈtroʊ.pɪk ˈhɔːr.moʊn/ | Hormone vỏ thượng thận |
| 90 | Hypopituitarism | /ˌhaɪ.poʊ.pɪˈtuː.ɪ.tə.rɪ.zəm/ | Suy tuyến yên |
| 91 | Intraislet | /ˌɪn.trəˈaɪ.lət/ | Nội tiểu đảo (tụy) |
| 92 | Hepatic portal vein | /hɪˈpæt̬.ɪk ˈpɔːr.t̬əl veɪn/ | Tĩnh mạch cửa gan |
| 93 | Hindbrain | /ˈhaɪnd.breɪn/ | Não sau |
| 94 | Hypothalamus | /ˌhaɪ.poʊˈθæl.ə.məs/ | Vùng dưới đồi |
| 95 | Sympathoadrenal activity | /ˌsɪm.pə.θoʊ.əˈdriː.nəl ækˈtɪv.ə.t̬i/ | Hoạt động giao cảm-thượng thận |
| 96 | Neuroglycopenia | /ˌnʊr.oʊ.ɡlaɪ.koʊˈpiː.ni.ə/ | Thiếu glucose não |
| 97 | Autonomic (neurogenic) symptoms | /ˌɔː.t̬əˈnɑː.mɪk (ˌnʊr.oʊˈdʒen.ɪk) ˈsɪmp.təmz/ | Triệu chứng thần kinh tự chủ (thần kinh) |
| 98 | Adrenomedullary | /əˌdriː.noʊˈmed.jə.ler.i/ | Thuộc tủy thượng thận |
| 99 | Adrenergic responses | /ˌæd.rəˈnɝː.dʒɪk rɪˈspɑːnsɪz/ | Phản ứng adrenergic |
| 100 | Cholinergic responses | /ˌkoʊ.lɪˈnɝː.dʒɪk rɪˈspɑːnsɪz/ | Phản ứng cholinergic |
| 101 | Sweating | /ˈswet̬.ɪŋ/ | Đổ mồ hôi |
| 102 | Shakiness | /ˈʃeɪ.ki.nəs/ | Run rẩy |
| 103 | Trembling | /ˈtrem.blɪŋ/ | Run |
| 104 | Tachycardia | /ˌtæk.ɪˈkɑːr.di.ə/ | Nhịp tim nhanh |
| 105 | Anxiety | /æŋˈzaɪ.ə.t̬i/ | Lo lắng |
| 106 | Weakness | /ˈwiːk.nəs/ | Yếu sức |
| 107 | Hunger | /ˈhʌŋ.ɡɚ/ | Đói |
| 108 | Nausea | /ˈnɑː.zi.ə/ | Buồn nôn |
| 109 | Vomiting | /ˈvɑː.mɪ.t̬ɪŋ/ | Nôn |
| 110 | Pallor | /ˈpæl.ɚ/ | Xanh xao |
| 111 | Hypothermia | /ˌhaɪ.poʊˈθɝː.mi.ə/ | Hạ thân nhiệt |
| 112 | Neuroglycopenic symptoms | /ˌnʊr.oʊ.ɡlaɪ.koʊˈpiː.nɪk ˈsɪmp.təmz/ | Triệu chứng thiếu glucose não |
| 113 | Headache | /ˈhed.eɪk/ | Đau đầu |
| 114 | Visual disturbances | /ˈvɪʒ.u.əl dɪˈstɝː.bənsɪz/ | Rối loạn thị giác |
| 115 | Lethargy | /ˈleθ.ɚ.dʒi/ | Lơ mơ |
| 116 | Lassitude | /ˈlæs.ə.tuːd/ | Mệt mỏi, uể oải |
| 117 | Restlessness | /ˈrest.ləs.nəs/ | Bồn chồn |
| 118 | Irritability | /ˌɪr.ə.t̬əˈbɪl.ə.t̬i/ | Dễ cáu kỉnh |
| 119 | Difficulty with speech | /ˈdɪf.ɪ.kəl.ti wɪθ spiːtʃ/ | Khó nói |
| 120 | Inability to concentrate | /ˌɪn.əˈbɪl.ə.t̬i tə ˈkɑːn.sən.treɪt/ | Không thể tập trung |
| 121 | Mental confusion | /ˈmen.təl kənˈfjuː.ʒən/ | Lú lẫn tâm thần |
| 122 | Somnolence | /ˈsɑːm.nə.ləns/ | Buồn ngủ |
| 123 | Stupor | /ˈstuː.pɚ/ | Sững sờ |
| 124 | Loss of consciousness | /lɔːs əv ˈkɑːn.ʃəs.nəs/ | Mất ý thức |
| 125 | Coma | /ˈkoʊ.mə/ | Hôn mê |
| 126 | Twitching | /ˈtwɪtʃ.ɪŋ/ | Co giật cơ |
| 127 | Convulsions | /kənˈvʌl.ʃənz/ | Co giật toàn thể |
| 128 | Epilepsy | /ˈep.ə.lep.si/ | Động kinh |
| 129 | Bizarre neurologic signs | /bɪˈzɑːr ˌnʊr.əˈlɑː.dʒɪk saɪnz/ | Dấu hiệu thần kinh kỳ lạ |
| 130 | Motor disturbances | /ˈmoʊ.t̬ɚ dɪˈstɝː.bənsɪz/ | Rối loạn vận động |
| 131 | Sensory disturbances | /ˈsen.sər.i dɪˈstɝː.bənsɪz/ | Rối loạn cảm giác |
| 132 | Loss of intellectual ability | /lɔːs əv ˌɪn.t̬əˈlek.tʃu.əl əˈbɪl.ə.t̬i/ | Mất khả năng trí tuệ |
| 133 | Personality changes | /ˌpɝː.səˈnæl.ə.t̬i ˈtʃeɪn.dʒɪz/ | Thay đổi nhân cách |
| 134 | Bizarre behavior | /bɪˈzɑːr bɪˈheɪ.vjɚ/ | Hành vi kỳ lạ |
| 135 | Outburst of temper | /ˈaʊt.bɝːst əv ˈtem.pɚ/ | Cơn nóng giận bộc phát |
| 136 | Psychologic disintegration | /ˌsaɪ.kəˈlɑː.dʒɪk dɪˌsɪn.t̬əˈɡreɪ.ʃən/ | Tan rã tâm lý |
| 137 | Manic behavior | /ˈmæn.ɪk bɪˈheɪ.vjɚ/ | Hành vi hưng cảm |
| 138 | Depression | /dɪˈpreʃ.ən/ | Trầm cảm |
| 139 | Psychoses | /saɪˈkoʊ.siːz/ | Loạn thần |
| 140 | Paresthesias | /ˌpær.əsˈθiː.ʒəz/ | Dị cảm |
| 141 | Cognition | /kɑːɡˈnɪʃ.ən/ | Nhận thức |
| 142 | Seizures | /ˈsiː.ʒɚz/ | Cơn co giật |
| 143 | Beta-blockers | /ˈbeɪ.t̬ə ˈblɑː.kɚz/ | Thuốc chẹn beta |
| 144 | Caffeine | /kæˈfiːn/ | Caffein |
| 145 | Hyperglycemia | /ˌhaɪ.pɚ.ɡlaɪˈsiː.mi.ə/ | Tăng đường huyết |
| 146 | Hypoglycemia-associated autonomic failure (HAAF) | /ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə əˈsoʊ.ʃi.eɪ.t̬ɪd ˌɔː.t̬əˈnɑː.mɪk ˈfeɪl.jɚ/ | Suy giảm thần kinh tự chủ liên quan đến hạ đường huyết |
| 147 | Insulinoma | /ˌɪn.sə.lɪˈnoʊ.mə/ | U tiết insulin |
| 148 | Neuropsychological impairment | /ˌnʊr.oʊ.saɪ.kəˈlɑː.dʒɪ.kəl ɪmˈper.mənt/ | Suy giảm thần kinh-tâm lý |
| 149 | Neuronal death | /nʊˈroʊ.nəl deθ/ | Chết tế bào thần kinh |
| 150 | Magnetic resonance imaging (MRI) | /mæɡˌnet̬.ɪk ˈrez.ə.nəns ˈɪm.ɪ.dʒɪŋ/ | Chụp cộng hưởng từ |
| 151 | Cortical tissue | /ˈkɔːr.t̬ɪ.kəl ˈtɪʃ.uː/ | Mô vỏ não |
| 152 | White matter | /waɪt ˈmæt̬.ɚ/ | Chất trắng |
| 153 | Cerebellum | /ˌser.əˈbel.əm/ | Tiểu não |
| 154 | Brainstem | /ˈbreɪn.stem/ | Thân não |
| 155 | Cognitive impairment | /ˈkɑːɡ.nə.t̬ɪv ɪmˈper.mənt/ | Suy giảm nhận thức |
| 156 | Clinical laboratory method | /ˈklɪn.ɪ.kəl ˈlæb.rə.tɔːr.i ˈmeθ.əd/ | Phương pháp xét nghiệm lâm sàng |
| 157 | Whipple’s triad | /ˈwɪp.əlz ˈtraɪ.æd/ | Tam chứng Whipple |
| 158 | Provocative testing | /prəˈvɑː.kə.t̬ɪv ˈtes.tɪŋ/ | Nghiệm pháp kích thích |
| 159 | Point-of-care glucose meters | /pɔɪnt əv ker ˈɡluː.koʊs ˈmiː.t̬ɚz/ | Máy đo đường huyết tại điểm chăm sóc |
| 160 | Whole blood | /hoʊl blʌd/ | Máu toàn phần |
| 161 | Glycolysis | /ɡlaɪˈkɑː.lə.sɪs/ | Đường phân |
| 162 | Metabolic systems | /ˌmet̬.əˈbɑː.lɪk ˈsɪs.təmz/ | Các hệ thống chuyển hóa |
| 163 | Endocrine system | /ˈen.də.krɪn ˈsɪs.təm/ | Hệ nội tiết |
| 164 | C-peptide | /ˈsiː ˈpep.taɪd/ | C-peptide |
| 165 | Glucagon stimulation test | /ˈɡluː.kə.ɡɑːn ˌstɪm.jəˈleɪ.ʃən test/ | Nghiệm pháp kích thích glucagon |
| 166 | Acylcarnitines | /ˌeɪ.səlˈkɑːr.nə.tiːnz/ | Acylcarnitine |
| 167 | Ammonia | /əˈmoʊ.ni.ə/ | Amoniac |
| 168 | Hyperammonemia syndrome | /ˌhaɪ.pɚˌæm.oʊˈniː.mi.ə ˈsɪn.droʊm/ | Hội chứng tăng amoniac máu |
| 169 | GLUD1 gene | /ˌdʒiː.el.juː.diːˈwʌn dʒiːn/ | Gen GLUD1 |
| 170 | Exogenous insulin administration | /ɛkˈsɑː.dʒə.nəs ˈɪn.sə.lɪn ədˌmɪn.ɪˈstreɪ.ʃən/ | Sử dụng insulin ngoại sinh |
| 171 | Insulin-like growth factor binding protein-1 (IGFBP-1) | /ˈɪn.sə.lɪn laɪk ɡroʊθ ˈfæk.tɚ ˈbaɪn.dɪŋ ˈproʊ.tiːn wʌn/ | Protein gắn yếu tố tăng trưởng giống insulin-1 |
| 172 | Oral hypoglycemic drugs | /ˈɔːr.əl ˌhaɪ.poʊ.ɡlaɪˈsiː.mɪk drʌɡz/ | Thuốc hạ đường huyết đường uống |
| 173 | Surreptitious insulin administration | /ˌsɝː.əpˈtɪʃ.əs ˈɪn.sə.lɪn ədˌmɪn.ɪˈstreɪ.ʃən/ | Sử dụng insulin lén lút |
| 174 | Factitious hypoglycemia | /fækˈtɪʃ.əs ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết giả tạo |
| 175 | Munchausen by proxy | /ˈmʌn.tʃaʊ.zən baɪ ˈprɑːk.si/ | Hội chứng Munchausen by proxy |
| 176 | Glucose infusion rate (GIR) | /ˈɡluː.koʊs ɪnˈfjuː.ʒən reɪt/ | Tốc độ truyền glucose |
| 177 | Insulin analogs | /ˈɪn.sə.lɪn ˈæn.ə.lɔːɡz/ | Các chất tương tự insulin |
| 178 | Lispro | /ˈlɪs.proʊ/ | Lispro |
| 179 | Aspart | /ˈæs.pɑːrt/ | Aspart |
| 180 | Glargine | /ˈɡlɑːr.dʒiːn/ | Glargine |
| 181 | Adenosine triphosphate-sensitive potassium (KATP) channel | /əˈden.ə.siːn traɪˈfɑːs.feɪt ˈsen.sə.t̬ɪv pəˈtæs.i.əm ˈtʃæn.əl/ | Kênh kali nhạy cảm với ATP |
| 182 | ABCC8 gene | /ˌeɪ.biː.siː.siːˈeɪt dʒiːn/ | Gen ABCC8 |
| 183 | KCNJ11 gene | /ˌkeɪ.siː.en.dʒeɪ.ɪˈlev.ən dʒiːn/ | Gen KCNJ11 |
| 184 | Chromosome | /ˈkroʊ.mə.soʊm/ | Nhiễm sắc thể |
| 185 | SUR1 subunit | /ˌes.juː.ɑːrˈwʌn ˈsʌb.juː.nɪt/ | Tiểu đơn vị SUR1 |
| 186 | Kir6.2 subunit | /ˌkeɪ.aɪ.ɑːr.sɪks.pɔɪntˈtuː ˈsʌb.juː.nɪt/ | Tiểu đơn vị Kir6.2 |
| 187 | Beta-cell | /ˈbeɪ.t̬ə sel/ | Tế bào beta |
| 188 | Potassium efflux | /pəˈtæs.i.əm ˈef.lʌks/ | Dòng kali ra ngoài |
| 189 | Hyperpolarizes | /ˌhaɪ.pɚˈpoʊ.lə.raɪzɪz/ | Tăng phân cực |
| 190 | Depolarization | /diːˌpoʊ.lər.əˈzeɪ.ʃən/ | Khử cực |
| 191 | Biallelic inheritance | /baɪ.əˈliː.lɪk ɪnˈher.ɪ.təns/ | Di truyền hai alen |
| 192 | Recessive mutations | /rɪˈses.ɪv mjuːˈteɪ.ʃənz/ | Đột biến lặn |
| 193 | Diazoxide | /ˌdaɪ.əˈzɑːk.saɪd/ | Diazoxide |
| 194 | Focal congenital HI | /ˈfoʊ.kəl kənˈdʒen.ə.t̬əl ˌeɪtʃˈaɪ/ | Tăng insulin máu bẩm sinh khu trú |
| 195 | Two-hit mechanism | /tuː hɪt ˈmek.ə.nɪ.zəm/ | Cơ chế hai đòn |
| 196 | Paternally transmitted | /pəˈtɝː.nəl.i trænsˈmɪt̬.ɪd/ | Di truyền từ cha |
| 197 | Somatic mutation | /soʊˈmæt̬.ɪk mjuːˈteɪ.ʃən/ | Đột biến soma |
| 198 | Uniparental isodisomy | /ˌjuː.nɪ.pəˈren.təl ˌaɪ.soʊˈdaɪ.sə.mi/ | Đồng ly thể đơn cha mẹ |
| 199 | Beckwith-Wiedemann imprinted genes | /ˈbek.wɪθ ˈviː.də.mæn ɪmˈprɪn.t̬ɪd dʒiːnz/ | Gen in dấu Beckwith-Wiedemann |
| 200 | Islet cell adenomatosis | /ˈaɪ.lət sel ˌæd.ə.noʊ.məˈtoʊ.sɪs/ | Bệnh u tuyến tế bào tiểu đảo |
| 201 | Dominantly inherited | /ˈdɑː.mə.nənt.li ɪnˈher.ɪ.t̬ɪd/ | Di truyền trội |
| 202 | Dominant-negative fashion | /ˈdɑː.mə.nənt ˈneɡ.ə.t̬ɪv ˈfæʃ.ən/ | Theo kiểu trội âm tính |
| 203 | Octreotide | /ɑːkˈtriː.oʊ.taɪd/ | Octreotide |
| 204 | Subcutaneous injection | /ˌsʌb.kjuːˈteɪ.ni.əs ɪnˈdʒek.ʃən/ | Tiêm dưới da |
| 205 | Somatostatin analogs | /soʊˌmæt̬.oʊˈstæt̬.ɪn ˈæn.ə.lɔːɡz/ | Các chất tương tự somatostatin |
| 206 | Lanreotide | /lænˈriː.oʊ.taɪd/ | Lanreotide |
| 207 | Calcium-channel blockers | /ˈkæl.si.əm ˈtʃæn.əl ˈblɑː.kɚz/ | Thuốc chẹn kênh canxi |
| 208 | Pancreatectomy | /ˌpæŋ.kri.əˈtek.tə.mi/ | Phẫu thuật cắt tụy |
| 209 | Insulin-dependent diabetes | /ˈɪn.sə.lɪn dɪˈpen.dənt ˌdaɪ.əˈbiː.t̬iːz/ | Đái tháo đường phụ thuộc insulin |
| 210 | 18F-fluorodopa positron-emission tomography (18F-L-DOPA PET) | /eɪˈtiːn ɛf ˈflʊər.oʊˌdoʊ.pə ˈpɑː.zɪ.trɑːn ɪˈmɪʃ.ən təˈmɑː.ɡrə.fi/ | Chụp cắt lớp phát xạ positron với 18F-L-DOPA |
| 211 | Preoperative localization | /priːˈɑː.pɚ.ə.t̬ɪv ˌloʊ.kəl.əˈzeɪ.ʃən/ | Định vị trước phẫu thuật |
| 212 | Glutamate Dehydrogenase (GDH) | /ˈɡluː.t̬ə.meɪt ˌdiː.haɪˈdrɑː.dʒə.neɪs/ | Glutamate Dehydrogenase |
| 213 | Activating mutations | /ˈæk.təˌveɪ.t̬ɪŋ mjuːˈteɪ.ʃənz/ | Đột biến hoạt hóa |
| 214 | Renal ammoniagenesis | /ˈriː.nəl əˌmoʊ.ni.əˈdʒen.ə.sɪs/ | Sinh amoniac ở thận |
| 215 | De novo mutation | /diː ˈnoʊ.voʊ mjuːˈteɪ.ʃən/ | Đột biến mới |
| 216 | Absence seizures | /ˈæb.səns ˈsiː.ʒɚz/ | Cơn vắng ý thức |
| 217 | Generalized epilepsy | /ˈdʒen.ə.rə.laɪzd ˈep.ə.lep.si/ | Động kinh toàn thể |
| 218 | Neurotransmitters | /ˌnʊr.oʊ.trænsˈmɪt̬.ɚz/ | Chất dẫn truyền thần kinh |
| 219 | Leucine-sensitive hypoglycemia | /ˈluː.siːn ˈsen.sə.t̬ɪv ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết nhạy cảm với leucin |
| 220 | Protein-sensitive hypoglycemia | /ˈproʊ.tiːn ˈsen.sə.t̬ɪv ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết nhạy cảm với protein |
| 221 | Alpha-ketoglutarate | /ˈæl.fə ˌkiː.t̬oʊˈɡluː.tə.reɪt/ | Alpha-ketoglutarate |
| 222 | Glucokinase | /ˌɡluː.koʊˈkaɪ.neɪs/ | Glucokinase |
| 223 | Gain-of-function mutation | /ɡeɪn əv ˈfʌŋk.ʃən mjuːˈteɪ.ʃən/ | Đột biến tăng chức năng |
| 224 | Loss-of-function mutation | /lɔːs əv ˈfʌŋk.ʃən mjuːˈteɪ.ʃən/ | Đột biến mất chức năng |
| 225 | Monogenic diabetes (MODY2) | /ˌmɑː.noʊˈdʒen.ɪk ˌdaɪ.əˈbiː.t̬iːz/ | Đái tháo đường đơn gen (MODY2) |
| 226 | Enzyme kinetics | /ˈen.zaɪm kɪˈnet̬.ɪks/ | Động học enzyme |
| 227 | Ketogenic diet | /ˌkiː.t̬oʊˈdʒen.ɪk ˈdaɪ.ət/ | Chế độ ăn ketogenic |
| 228 | Hexokinase 1 (HK1) | /ˌhek.soʊˈkaɪ.neɪs wʌn/ | Hexokinase 1 |
| 229 | Phosphorylating | /fɑːsˈfɔːr.əˌleɪ.t̬ɪŋ/ | Phosphoryl hóa |
| 230 | Pedigree | /ˈped.ə.ɡriː/ | Phả hệ |
| 231 | Linkage analysis | /ˈlɪŋ.kɪdʒ əˈnæl.ə.sɪs/ | Phân tích liên kết |
| 232 | Haplotype | /ˈhæp.loʊ.taɪp/ | Haplotype (Kiểu đơn bội) |
| 233 | Immunohistochemical staining | /ˌɪm.juː.noʊˌhɪs.toʊˈkem.ɪ.kəl ˈsteɪ.nɪŋ/ | Nhuộm hóa mô miễn dịch |
| 234 | Pyruvate | /paɪˈruː.veɪt/ | Pyruvate |
| 235 | Plasma membrane transporter, MCT1 | /ˈplæz.mə ˈmem.breɪn trænsˈpɔːr.t̬ɚ ˌem.siː.tiːˈwʌn/ | Chất vận chuyển màng sinh chất, MCT1 |
| 236 | Short chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD) | /ʃɔːrt tʃeɪn θriː haɪˌdrɑːk.siˈæs.əl ˌkoʊˈeɪ ˌdiː.haɪˈdrɑː.dʒə.neɪs/ | Short chain 3-hydroxyacyl-CoA dehydrogenase |
| 237 | Urinary organic acids | /ˈjʊr.əˌner.i ɔːrˈɡæn.ɪk ˈæs.ɪdz/ | Acid hữu cơ trong nước tiểu |
| 238 | 3-hydroxyglutarate | /θriː haɪˌdrɑːk.siˈɡluː.tə.reɪt/ | 3-hydroxyglutarate |
| 239 | 3-hydroxybutyryl-carnitine | /θriː haɪˌdrɑːk.siˈbjuː.tə.rəlˈkɑːr.nə.tiːn/ | 3-hydroxybutyryl-carnitine |
| 240 | Hepatocyte nuclear factor 4a (HNF4A) | /hɪˈpæt̬.oʊ.saɪt ˈnuː.kli.ɚ ˈfæk.tɚ fɔːr eɪ/ | Yếu tố nhân tế bào gan 4a |
| 241 | Transcription factor | /trænsˈkrɪp.ʃən ˈfæk.tɚ/ | Yếu tố phiên mã |
| 242 | Heterozygous | /ˌhet̬.ɚ.oʊˈzaɪ.ɡəs/ | Dị hợp tử |
| 243 | Fetal macrosomia | /ˈfiː.təl ˌmæk.roʊˈsoʊ.mi.ə/ | Thai to |
| 244 | Sulfonylureas | /ˌsʌl.fə.nɪl.jʊˈriː.əz/ | Sulfonylurea |
| 245 | Renal Fanconi syndrome | /ˈriː.nəl fænˈkoʊ.ni ˈsɪn.droʊm/ | Hội chứng Fanconi thận |
| 246 | Hepatomegaly | /ˌhep.ə.toʊˈmeɡ.ə.li/ | Gan to |
| 247 | Transaminases | /trænsˈæm.əˌneɪ.sɪz/ | Transaminase |
| 248 | GLUT2 transporter | /ˌdʒiː.el.juː.tiːˈtuː trænsˈpɔːr.t̬ɚ/ | Chất vận chuyển GLUT2 |
| 249 | Hepatocyte nuclear factor 1a (HNF1A) | /hɪˈpæt̬.oʊ.saɪt ˈnuː.kli.ɚ ˈfæk.tɚ wʌn eɪ/ | Yếu tố nhân tế bào gan 1a |
| 250 | Uncoupling protein 2 (UCP2) | /ʌnˈkʌp.lɪŋ ˈproʊ.tiːn tuː/ | Protein tách cặp 2 |
| 251 | Krebs cycle | /krebz ˈsaɪ.kəl/ | Chu trình Krebs |
| 252 | Exercise-Induced Hyperinsulinism (EIHI) | /ˈek.sɚ.saɪz ɪnˈduːst ˌhaɪ.pɚˈɪn.sə.lɪ.nɪ.zəm/ | Tăng insulin máu do tập thể dục |
| 253 | Anaerobic exercise | /ˌæn.əˈroʊ.bɪk ˈek.sɚ.saɪz/ | Tập thể dục kỵ khí |
| 254 | Syncope | /ˈsɪŋ.kə.pi/ | Ngất |
| 255 | Provocative testing | /prəˈvɑː.kə.t̬ɪv ˈtes.tɪŋ/ | Nghiệm pháp kích thích |
| 256 | Intravenous infusion | /ˌɪn.trəˈviː.nəs ɪnˈfjuː.ʒən/ | Truyền tĩnh mạch |
| 257 | Insulin secretagogues | /ˈɪn.sə.lɪn sɪˈkriː.tə.ɡɔːɡz/ | Thuốc kích thích tiết insulin |
| 258 | Monocarboxylate transporter (MCT1) | /ˌmɑː.noʊ.kɑːrˈbɑːk.sə.leɪt trænsˈpɔːr.t̬ɚ/ | Chất vận chuyển monocarboxylate |
| 259 | Syndromic forms | /sɪnˈdroʊ.mɪk fɔːrmz/ | Các thể hội chứng |
| 260 | Beckwith-Wiedemann Syndrome (BWS) | /ˈbek.wɪθ ˈviː.də.mæn ˈsɪn.droʊm/ | Hội chứng Beckwith-Wiedemann |
| 261 | Overgrowth syndrome | /ˈoʊ.vɚ.ɡroʊθ ˈsɪn.droʊm/ | Hội chứng phát triển quá mức |
| 262 | Embryonic mosaic mutations | /ˌem.briˈɑː.nɪk moʊˈzeɪ.ɪk mjuːˈteɪ.ʃənz/ | Đột biến khảm phôi |
| 263 | Imprinted region | /ɪmˈprɪn.t̬ɪd ˈriː.dʒən/ | Vùng in dấu di truyền |
| 264 | Paternal isodisomy | /pəˈtɝː.nəl ˌaɪ.soʊˈdaɪ.sə.mi/ | Đồng ly thể từ cha |
| 265 | Hemihypertrophy | /ˌhem.i.haɪˈpɝː.trə.fi/ | Phì đại nửa người |
| 266 | Macroglossia | /ˌmæk.roʊˈɡlɑː.si.ə/ | Lưỡi to |
| 267 | Umbilical hernia | /ʌmˈbɪl.ɪ.kəl ˈhɝː.ni.ə/ | Thoát vị rốn |
| 268 | Kabuki Syndrome | /kəˈbuː.ki ˈsɪn.droʊm/ | Hội chứng Kabuki |
| 269 | Dysmorphic features | /dɪsˈmɔːr.fɪk ˈfiː.tʃɚz/ | Đặc điểm dị dạng |
| 270 | Chromatin methylation | /ˈkroʊ.mə.tɪn ˌmeθ.əˈleɪ.ʃən/ | Methyl hóa chromatin |
| 271 | Turner Syndrome | /ˈtɝː.nɚ ˈsɪn.droʊm/ | Hội chứng Turner |
| 272 | Karyotype | /ˈker.i.oʊ.taɪp/ | Kiểu nhân (nhiễm sắc thể đồ) |
| 273 | Pseudoautosomal | /ˌsuː.doʊ.ɔː.təˈsoʊ.məl/ | Giả nhiễm sắc thể thường |
| 274 | Congenital disorders of glycosylation (CDG) | /kənˈdʒen.ə.t̬əl dɪˈsɔːr.dɚz əv ˌɡlaɪ.koʊ.səˈleɪ.ʃən/ | Rối loạn glycosyl hóa bẩm sinh |
| 275 | N-glycosylation | /en ˌɡlaɪ.koʊ.səˈleɪ.ʃən/ | N-glycosyl hóa |
| 276 | Dysmorphic facies | /dɪsˈmɔːr.fɪk ˈfeɪ.ʃi.iːz/ | Diện mạo dị dạng |
| 277 | Phosphomannomutase 2 (PMM2) | /ˌfɑːs.foʊˌmæn.oʊˈmjuː.teɪs tuː/ | Phosphomannomutase 2 |
| 278 | Cerebellar hypoplasia | /ˌser.əˈbel.ɚ ˌhaɪ.poʊˈpleɪ.ʒə/ | Thiểu sản tiểu não |
| 279 | Hypotonia | /ˌhaɪ.poʊˈtoʊ.ni.ə/ | Giảm trương lực cơ |
| 280 | Protein-losing enteropathy | /ˈproʊ.tiːn ˈluː.zɪŋ ˌen.təˈrɑː.pə.θi/ | Bệnh ruột mất protein |
| 281 | Antithrombin III | /ˌæn.tiˈθrɑːm.bɪn θriː/ | Antithrombin III |
| 282 | Thromboses | /θrɑːmˈboʊ.siːz/ | Huyết khối |
| 283 | Inverted nipples | /ɪnˈvɝː.t̬ɪd ˈnɪp.əlz/ | Núm vú tụt vào trong |
| 284 | Phosphomannose isomerase (MPI) | /ˌfɑːs.foʊˈmæn.oʊs aɪˈsɑː.mə.reɪs/ | Phosphomannose isomerase |
| 285 | Cyclic vomiting | /ˈsaɪ.klɪk ˈvɑː.mɪ.t̬ɪŋ/ | Nôn chu kỳ |
| 286 | Mannose | /ˈmæn.oʊs/ | Mannose |
| 287 | Mannosyltransferase 6 | /ˌmæn.oʊ.səlˈtræns.fə.reɪs sɪks/ | Mannosyltransferase 6 |
| 288 | Perinatal stress | /ˌper.ɪˈneɪ.t̬əl stres/ | Stress chu sinh |
| 289 | Phosphoglucomutase 1 (PGM1) | /ˌfɑːs.foʊˌɡluː.koʊˈmjuː.teɪs wʌn/ | Phosphoglucomutase 1 |
| 290 | Transferrin glycosylation | /trænsˈfer.ɪn ˌɡlaɪ.koʊ.səˈleɪ.ʃən/ | Glycosyl hóa Transferrin |
| 291 | Uncooked cornstarch (UCS) | /ʌnˈkʊkt kɔːrn.stɑːrtʃ/ | Bột ngô sống |
| 292 | Low glycemic index | /loʊ ɡlaɪˈsiː.mɪk ˈɪn.deks/ | Chỉ số đường huyết thấp |
| 293 | Galactose | /ɡəˈlæk.toʊs/ | Galactose |
| 294 | AKT2 | /ˌeɪ.keɪ.tiːˈtuː/ | AKT2 (Protein kinase B beta) |
| 295 | Serine/threonine kinase | /ˈser.iːn ˈθriː.ə.niːn ˈkaɪ.neɪs/ | Serine/threonine kinase |
| 296 | Post-insulin receptor signaling | /poʊst ˈɪn.sə.lɪn rɪˈsep.tɚ ˈsɪɡ.nə.lɪŋ/ | Tín hiệu sau thụ thể insulin |
| 297 | Insulin resistance | /ˈɪn.sə.lɪn rɪˈzɪs.təns/ | Kháng insulin |
| 298 | Insulin sensitivity | /ˈɪn.sə.lɪn ˌsen.səˈtɪv.ə.t̬i/ | Nhạy cảm với insulin |
| 299 | Postprandial | /ˌpoʊstˈpræn.di.əl/ | Sau ăn |
| 300 | Asymmetric overgrowth | /ˌeɪ.səˈmet.rɪk ˈoʊ.vɚ.ɡroʊθ/ | Phát triển quá mức không đối xứng |
| 301 | Postzygotic mutation | /ˌpoʊst.zaɪˈɡɑː.t̬ɪk mjuːˈteɪ.ʃən/ | Đột biến sau hợp tử |
| 302 | Mosaic | /moʊˈzeɪ.ɪk/ | Thể khảm |
| 303 | Phospatidylinositol-3-Kinase (PI3K) | /ˌfɑːs.fəˌtaɪ.dəl.ɪˈnoʊ.sɪ.tɔːl θriː ˈkaɪ.neɪs/ | Phospatidylinositol-3-Kinase |
| 304 | PIK3CA gene | /ˌpiː.aɪ.keɪ.θriː.siːˈeɪ dʒiːn/ | Gen PIK3CA |
| 305 | PIK3R2 gene | /ˌpiː.aɪ.keɪ.θriː.ɑːrˈtuː dʒiːn/ | Gen PIK3R2 |
| 306 | Megalencephaly | /ˌmeɡ.ə.lenˈsef.ə.li/ | Não to |
| 307 | Developmental delay | /dɪˌvel.əpˈmen.təl dɪˈleɪ/ | Chậm phát triển |
| 308 | Insulin receptor (INSR) | /ˈɪn.sə.lɪn rɪˈsep.tɚ/ | Thụ thể insulin |
| 309 | Diabetes mellitus | /ˌdaɪ.əˈbiː.t̬iːz ˈmel.ɪ.təs/ | Đái tháo đường |
| 310 | Lipodystrophy | /ˌlɪp.oʊˈdɪs.trə.fi/ | Loạn dưỡng mỡ |
| 311 | Donohue syndrome (leprechaunism) | /ˈdɑː.nə.hjuː ˈsɪn.droʊm (ˈlep.rəˌkɑː.nɪ.zəm)/ | Hội chứng Donohue (người lùn) |
| 312 | Rabson-Mendenhall syndrome | /ˈræb.sən ˈmen.dən.hɔːl ˈsɪn.droʊm/ | Hội chứng Rabson-Mendenhall |
| 313 | Type A insulin resistance | /taɪp eɪ ˈɪn.sə.lɪn rɪˈzɪs.təns/ | Kháng insulin type A |
| 314 | Biallelic | /baɪ.əˈliː.lɪk/ | Hai alen |
| 315 | Monoallelic | /ˌmɑː.noʊ.əˈliː.lɪk/ | Một alen |
| 316 | Insulin clearance | /ˈɪn.sə.lɪn ˈklɪr.əns/ | Độ thanh thải insulin |
| 317 | Antiinsulin antibodies | /ˌæn.tiˈɪn.sə.lɪn ˈæn.t̬iˌbɑː.diz/ | Kháng thể kháng insulin |
| 318 | Insulin-like growth factor-1 (IGF-1) receptor | /ˈɪn.sə.lɪn laɪk ɡroʊθ ˈfæk.tɚ wʌn rɪˈsep.tɚ/ | Thụ thể yếu tố tăng trưởng giống insulin-1 |
| 319 | Intrauterine growth retardation | /ˌɪn.trəˈjuː.t̬ə.rɪn ɡroʊθ ˌriː.tɑːrˈdeɪ.ʃən/ | Chậm tăng trưởng trong tử cung |
| 320 | Subcutaneous fat | /ˌsʌb.kjuːˈteɪ.ni.əs fæt/ | Mỡ dưới da |
| 321 | Hypertrichosis | /ˌhaɪ.pɚ.trɪˈkoʊ.sɪs/ | Rậm lông |
| 322 | Acanthosis nigricans | /ˌæ.kænˈθoʊ.sɪs ˈnaɪ.ɡrɪ.kænz/ | Bệnh gai đen |
| 323 | Diabetic ketoacidosis | /ˌdaɪ.əˈbet̬.ɪk ˌkiː.t̬oʊˌæs.ɪˈdoʊ.sɪs/ | Nhiễm toan ceton do đái tháo đường |
| 324 | Leptin (metreleptin) | /ˈlep.tɪn (ˌme.trəˈlep.tɪn)/ | Leptin (metreleptin) |
| 325 | Hyperandrogenism | /ˌhaɪ.pɚˈæn.droʊ.dʒə.nɪ.zəm/ | Tăng androgen |
| 326 | Polycystic ovarian syndrome | /ˌpɑː.liˈsɪs.tɪk oʊˈver.i.ən ˈsɪn.droʊm/ | Hội chứng buồng trứng đa nang |
| 327 | Postreceptor disorders | /poʊst.rɪˈsep.tɚ dɪˈsɔːr.dɚz/ | Rối loạn sau thụ thể |
| 328 | Congenital lipodystrophy (Berardinelli-Seip syndrome) | /kənˈdʒen.ə.t̬əl ˌlɪp.oʊˈdɪs.trə.fi (ˌber.ɑːr.dɪˈnel.i saɪp ˈsɪn.droʊm)/ | Loạn dưỡng mỡ bẩm sinh (Hội chứng Berardinelli-Seip) |
| 329 | Insulin secretagogue | /ˈɪn.sə.lɪn sɪˈkriː.tə.ɡɔːɡ/ | Thuốc kích thích tiết insulin |
| 330 | Isophane (NPH) insulin | /ˈaɪ.soʊ.feɪn ɪn.sə.lɪn/ | Insulin Isophane (NPH) |
| 331 | Glulisine | /ɡluːˈlɪ.siːn/ | Glulisine |
| 332 | Detemir | /ˈde.tə.mɪr/ | Detemir |
| 333 | Degludec | /ˈdeɡ.lu.dek/ | Degludec |
| 334 | Proinsulin | /ˌproʊˈɪn.sə.lɪn/ | Proinsulin |
| 335 | Glyburide | /ˈɡlaɪ.bjʊ.raɪd/ | Glyburide |
| 336 | Toxicology screens | /ˌtɑːk.səˈkɑː.lə.dʒi skriːnz/ | Sàng lọc độc chất |
| 337 | Autoimmune hypoglycemia | /ˌɔː.t̬oʊ.ɪˈmjuːn ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết tự miễn |
| 338 | Hirata disease | /hɪˈrɑː.tə dɪˈziːz/ | Bệnh Hirata |
| 339 | Sulfhydryl groups | /ˌsʌlfˈhaɪ.drəl ɡruːps/ | Nhóm sulfhydryl |
| 340 | Thiol groups | /ˈθaɪ.ɔːl ɡruːps/ | Nhóm thiol |
| 341 | Methimazole | /məˈθɪm.ə.zoʊl/ | Methimazole |
| 342 | Lipoic acid | /ˌlɪp.oʊ.ɪk ˈæs.ɪd/ | Acid lipoic |
| 343 | Hyperthyroidism | /ˌhaɪ.pɚˈθaɪ.rɔɪ.dɪ.zəm/ | Cường giáp |
| 344 | HLA allele | /ˌeɪtʃ.elˈeɪ əˈliːl/ | Alen HLA |
| 345 | Glucocorticoids | /ˌɡluː.koʊˈkɔːr.t̬ɪ.kɔɪdz/ | Glucocorticoid |
| 346 | Plasmapheresis | /ˌplæz.mə.fəˈriː.sɪs/ | Thay huyết tương |
| 347 | Intravenous immunoglobulin | /ˌɪn.trəˈviː.nəs ɪˌmjuː.noʊˈɡlɑː.bjə.lɪn/ | Globulin miễn dịch tiêm tĩnh mạch |
| 348 | Autoantibody titers | /ˌɔː.t̬oʊˈæn.t̬iˌbɑː.di ˈtaɪ.t̬ɚz/ | Hiệu giá tự kháng thể |
| 349 | Thyroid-stimulating immunoglobulins | /ˈθaɪ.rɔɪd ˈstɪm.jə.leɪ.t̬ɪŋ ɪˌmjuː.noʊˈɡlɑː.bjə.lɪnz/ | Globulin miễn dịch kích thích tuyến giáp |
| 350 | Graves disease | /ɡreɪvz dɪˈziːz/ | Bệnh Graves |
| 351 | Malignancy | /məˈlɪɡ.nən.si/ | Bệnh ác tính |
| 352 | Multiple endocrine neoplasia type 1 (MEN1) syndrome | /ˈmʌl.tə.pəl ˈen.də.krɪn ˌniː.oʊˈpleɪ.ʒə taɪp wʌn ˈsɪn.droʊm/ | Hội chứng đa u tuyến nội tiết type 1 |
| 353 | MENIN gene | /ˈmen.ɪn dʒiːn/ | Gen MENIN |
| 354 | Endoscopic ultrasound | /ˌen.doʊˈskɑː.pɪk ˈʌl.trə.saʊnd/ | Siêu âm nội soi |
| 355 | Computed tomography | /kəmˈpjuː.t̬ɪd təˈmɑː.ɡrə.fi/ | Chụp cắt lớp vi tính |
| 356 | Intraoperative ultrasound | /ˌɪn.trəˈɑː.pɚ.ə.t̬ɪv ˈʌl.trə.saʊnd/ | Siêu âm trong mổ |
| 357 | Parathyroids | /ˌpær.əˈθaɪ.rɔɪdz/ | Tuyến cận giáp |
| 358 | Pituitary | /pɪˈtuː.ɪ.ter.i/ | Tuyến yên |
| 359 | Pancreas | /ˈpæŋ.kri.əs/ | Tụy |
| 360 | Gastrointestinal surgery | /ˌɡæs.troʊ.ɪnˈtes.tə.nəl ˈsɝː.dʒər.i/ | Phẫu thuật đường tiêu hóa |
| 361 | Enteroinsular axis | /ˌen.tə.roʊˈɪn.sjə.lɚ ˈæk.sɪs/ | Trục ruột-tiểu đảo |
| 362 | Gastroesophageal reflux | /ˌɡæs.troʊ.ɪˌsɑː.fəˈdʒiː.əl ˈriː.flʌks/ | Trào ngược dạ dày thực quản |
| 363 | Nissen fundoplication | /ˈnɪs.ən ˌfʌn.doʊ.plɪˈkeɪ.ʃən/ | Phẫu thuật Nissen (tạo nếp gấp đáy vị) |
| 364 | Gastric bypass bariatric surgery | /ˈɡæs.trɪk ˈbaɪ.pæs ˌber.iˈæt.rɪk ˈsɝː.dʒər.i/ | Phẫu thuật nối tắt dạ dày giảm béo |
| 365 | Alimentary hypoglycemia | /ˌæl.əˈmen.t̬ɚ.i ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết do ăn uống |
| 366 | Glucagon-like peptide 1 | /ˈɡluː.kə.ɡɑːn laɪk ˈpep.taɪd wʌn/ | Peptide giống glucagon 1 |
| 367 | Late dumping syndrome | /leɪt ˈdʌmp.ɪŋ ˈsɪn.droʊm/ | Hội chứng dumping muộn |
| 368 | Early dumping syndrome | /ˈɝː.li ˈdʌmp.ɪŋ ˈsɪn.droʊm/ | Hội chứng dumping sớm |
| 369 | Osmotic shifts | /ɑːzˈmɑː.t̬ɪk ʃɪfts/ | Thay đổi thẩm thấu |
| 370 | Hypotension | /ˌhaɪ.poʊˈten.ʃən/ | Hạ huyết áp |
| 371 | Diarrhea | /ˌdaɪ.əˈriː.ə/ | Tiêu chảy |
| 372 | Mixed meal test | /mɪkst miːl test/ | Nghiệm pháp bữa ăn hỗn hợp |
| 373 | Oral glucose tolerance test (OGTT) | /ˈɔːr.əl ˈɡluː.koʊs ˈtɑː.lɚ.əns test/ | Nghiệm pháp dung nạp glucose đường uống |
| 374 | Hyperglycemic spike | /ˌhaɪ.pɚ.ɡlaɪˈsiː.mɪk spaɪk/ | Tăng đường huyết đột ngột |
| 375 | Acarbose | /əˈkɑːr.boʊs/ | Acarbose |
| 376 | Alpha-glucosidase inhibitor | /ˈæl.fə ɡluːˈkoʊ.sə.deɪs ɪnˈhɪb.ə.t̬ɚ/ | Thuốc ức chế alpha-glucosidase |
| 377 | Intragastric tube feedings | /ˌɪn.trəˈɡæs.trɪk tuːb ˈfiː.dɪŋz/ | Cho ăn qua sonde dạ dày |
| 378 | Intrajejunal tube feedings | /ˌɪn.trə.dʒəˈdʒuː.nəl tuːb ˈfiː.dɪŋz/ | Cho ăn qua sonde hỗng tràng |
| 379 | Prodrome | /ˈproʊ.droʊm/ | Tiền triệu |
| 380 | Honeymoon remission phase | /ˈhʌn.i.muːn rɪˈmɪʃ.ən feɪz/ | Giai đoạn lui bệnh (tuần trăng mật) |
| 381 | Prediabetes | /ˌpriː.daɪ.əˈbiː.t̬iːz/ | Tiền đái tháo đường |
| 382 | Dysinsulinism | /dɪsˈɪn.sjə.lɪ.nɪ.zəm/ | Loạn tiết insulin |
| 383 | Cystic Fibrosis (CF) | /ˈsɪs.tɪk faɪˈbroʊ.sɪs/ | Xơ nang |
| 384 | Cystic fibrosis-related diabetes (CFRD) | /ˈsɪs.tɪk faɪˈbroʊ.sɪs rɪˈleɪ.t̬ɪd ˌdaɪ.əˈbiː.t̬iːz/ | Đái tháo đường liên quan đến xơ nang |
| 385 | Nonislet tumor | /nɑːnˈaɪ.lət ˈtuː.mɚ/ | Khối u không phải tiểu đảo |
| 386 | Paraneoplastic hypoglycemia | /ˌpær.əˌniː.oʊˈplæs.tɪk ˌhaɪ.poʊ.ɡlaɪˈsiː.mi.ə/ | Hạ đường huyết cận ung thư |
| 387 | Mesenchymal origin | /ˌmes.əŋˈkaɪ.məl ˈɔːr.ə.dʒɪn/ | Nguồn gốc trung mô |
| 388 | Epithelial origin | /ˌep.əˈθiː.li.əl ˈɔːr.ə.dʒɪn/ | Nguồn gốc biểu mô |
| 389 | Hematopoietic origin | /hɪˌmæt̬.oʊ.pɔɪˈet̬.ɪk ˈɔːr.ə.dʒɪn/ | Nguồn gốc tạo máu |
| 390 | Neuroblastoma | /ˌnʊr.oʊ.blæsˈtoʊ.mə/ | U nguyên bào thần kinh |
| 391 | Wilms tumor | /wɪlmz ˈtuː.mɚ/ | U Wilms |
| 392 | Big IGF-2 | /bɪɡ ˌaɪ.dʒiːˈɛf tuː/ | IGF-2 lớn |
| 393 | Lactic acidemia | /ˈlæk.tɪk ˌæs.ɪˈdiː.mi.ə/ | Nhiễm acid lactic máu |
| 394 | Anaerobic glucose metabolism | /ˌæn.əˈroʊ.bɪk ˈɡluː.koʊs məˈtæb.ə.lɪ.zəm/ | Chuyển hóa glucose kỵ khí |
| 395 | Lymphomas | /lɪmˈfoʊ.məz/ | U lympho |
| 396 | Leukemias | /luːˈkiː.mi.əz/ | Bệnh bạch cầu |
| 397 | Glycogenoses | /ˌɡlaɪ.kə.dʒəˈnoʊ.siːz/ | Các bệnh dự trữ glycogen |
| 398 | a-1,4-glycosidic bonds | /ˈæl.fə wʌn fɔːr ˌɡlaɪ.koʊˈsɪd.ɪk bɑːndz/ | Liên kết α-1,4-glycosidic |
| 399 | a-1,6-glycosidic bonds | /ˈæl.fə wʌn sɪks ˌɡlaɪ.koʊˈsɪd.ɪk bɑːndz/ | Liên kết α-1,6-glycosidic |
| 400 | Glucose 1-phosphate | /ˈɡluː.koʊs wʌn ˈfɑːs.feɪt/ | Glucose 1-phosphate |
| 401 | Glucose 6-phosphate | /ˈɡluː.koʊs sɪks ˈfɑːs.feɪt/ | Glucose 6-phosphate |
| 402 | Glucose-6-phosphatase (G6Pase) | /ˌɡluː.koʊs sɪks ˈfɑːs.fə.teɪs/ | Glucose-6-phosphatase |
| 403 | Glycolytic pathway | /ˌɡlaɪ.koʊˈlɪt̬.ɪk ˈpæθ.weɪ/ | Con đường đường phân |
| 404 | Pentose phosphate pathway | /ˈpen.toʊs ˈfɑːs.feɪt ˈpæθ.weɪ/ | Con đường pentose phosphate |
| 405 | Ribose-5-phosphate | /ˈraɪ.boʊs faɪv ˈfɑːs.feɪt/ | Ribose-5-phosphate |
| 406 | Nucleotide synthesis | /ˈnuː.kli.ə.taɪd ˈsɪn.θə.sɪs/ | Tổng hợp nucleotide |
| 407 | Glycogen synthase | /ˈɡlaɪ.kə.dʒən ˈsɪn.θeɪs/ | Glycogen synthase |
| 408 | Branching enzyme | /ˈbræn.tʃɪŋ ˈen.zaɪm/ | Enzyme tạo nhánh |
| 409 | Hepatic phosphorylase | /hɪˈpæt̬.ɪk fɑːsˈfɔːr.ə.leɪs/ | Phosphorylase gan |
| 410 | 4-a-glucanotransferase | /fɔːr ˈæl.fə ˌɡluː.kæn.oʊˈtræns.fə.reɪs/ | 4-α-glucanotransferase |
| 411 | Amylo-1,6-glucosidase | /ˌæm.ɪ.loʊ wʌn sɪks ɡluːˈkoʊ.sə.deɪs/ | Amylo-1,6-glucosidase |
| 412 | Debranching enzyme | /diːˈbræn.tʃɪŋ ˈen.zaɪm/ | Enzyme khử nhánh |
| 413 | Glucosuria | /ˌɡluː.koʊsˈjʊə.ri.ə/ | Glucose niệu |
| 414 | Hyperlacticacidemia | /ˌhaɪ.pɚ.læk.tɪk.æs.ɪˈdiː.mi.ə/ | Tăng acid lactic máu |
| 415 | Von Gierke Disease | /fɔn ˈɡiːr.kə dɪˈziːz/ | Bệnh Von Gierke |
| 416 | Hepatorenal glycogenosis | /hɪˌpæt̬.oʊˈriː.nəl ˌɡlaɪ.kə.dʒəˈnoʊ.sɪs/ | Bệnh glycogenosis gan-thận |
| 417 | G6P translocase (G6PT) | /ˌdʒiː.sɪks.piː trænsˈloʊ.keɪs/ | G6P translocase |
| 418 | Endoplasmic reticulum (ER) | /ˌen.doʊˈplæz.mɪk rɪˈtɪk.jə.ləm/ | Lưới nội chất |
| 419 | Ashkenazi Jews | /ˌɑːʃ.kəˈnɑː.zi dʒuːz/ | Người Do Thái Ashkenazi |
| 420 | Hyperpnea | /ˌhaɪ.pɚpˈniː.ə/ | Thở nhanh sâu |
| 421 | Cushingoid appearance | /ˈkʊʃ.ɪŋ.ɔɪd əˈpɪr.əns/ | Dạng Cushing |
| 422 | Phospholipids | /ˌfɑːs.foʊˈlɪp.ɪdz/ | Phospholipid |
| 423 | Low-density lipoprotein-cholesterol | /loʊ ˈden.sə.t̬i ˌlaɪ.poʊˈproʊ.tiːn kəˈles.tə.rɔːl/ | Cholesterol lipoprotein mật độ thấp |
| 424 | High-density lipoprotein-cholesterol | /haɪ ˈden.sə.t̬i ˌlaɪ.poʊˈproʊ.tiːn kəˈles.tə.rɔːl/ | Cholesterol lipoprotein mật độ cao |
| 425 | Lipoprotein lipase | /ˌlaɪ.poʊˈproʊ.tiːn ˈlaɪ.peɪs/ | Lipoprotein lipase |
| 426 | Eruptive xanthomata | /ɪˈrʌp.tɪv zænˈθoʊ.mə.tə/ | U vàng phát ban |
| 427 | Pancreatitis | /ˌpæŋ.kri.əˈtaɪ.t̬ɪs/ | Viêm tụy |
| 428 | Hyperuricemia | /ˌhaɪ.pɚ.jʊə.rɪˈsiː.mi.ə/ | Tăng acid uric máu |
| 429 | Adenosine monophosphate (AMP) | /əˈden.ə.siːn ˌmɑː.noʊˈfɑːs.feɪt/ | Adenosine monophosphate |
| 430 | Platelet function | /ˈpleɪt.lət ˈfʌŋk.ʃən/ | Chức năng tiểu cầu |
| 431 | Epistaxis | /ˌep.əˈstæk.sɪs/ | Chảy máu cam |
| 432 | Adenosine diphosphate (ADP) | /əˈden.ə.siːn daɪˈfɑːs.feɪt/ | Adenosine diphosphate |
| 433 | Anemia | /əˈniː.mi.ə/ | Thiếu máu |
| 434 | Menorrhagia | /ˌmen.əˈreɪ.dʒi.ə/ | Rong kinh |
| 435 | End-stage renal disease (ESRD) | /end steɪdʒ ˈriː.nəl dɪˈziːz/ | Bệnh thận giai đoạn cuối |
| 436 | Enterocolitis | /ˌen.tə.roʊ.koʊˈlaɪ.t̬ɪs/ | Viêm ruột-đại tràng |
| 437 | Hepcidin | /hepˈsaɪ.dɪn/ | Hepcidin |
| 438 | Glomerular filtration rate (GFR) | /ɡloʊˈmer.jə.lɚ fɪlˈtreɪ.ʃən reɪt/ | Tốc độ lọc cầu thận |
| 439 | Phosphaturia | /ˌfɑːs.fəˈtʊə.ri.ə/ | Phosphat niệu |
| 440 | Hypokalemia | /ˌhaɪ.poʊ.kəˈliː.mi.ə/ | Hạ kali máu |
| 441 | Aminoaciduria | /əˌmiː.noʊ.æs.ɪˈdʊə.ri.ə/ | Acid amin niệu |
| 442 | Hypocitraturia | /ˌhaɪ.poʊ.sɪ.trəˈtʊə.ri.ə/ | Giảm citrat niệu |
| 443 | Hypercalciuria | /ˌhaɪ.pɚ.kæl.siˈʊə.ri.ə/ | Tăng calci niệu |
| 444 | Renal calculi | /ˈriː.nəl ˈkæl.kjə.laɪ/ | Sỏi thận |
| 445 | Nephrocalcinosis | /ˌnef.roʊ.kæl.səˈnoʊ.sɪs/ | Nhiễm calcin thận |
| 446 | Proteinuria | /ˌproʊ.təˈnʊə.ri.ə/ | Protein niệu |
| 447 | Hypertension | /ˌhaɪ.pɚˈten.ʃən/ | Tăng huyết áp |
| 448 | Creatinine clearance | /kriˈæt̬.ə.nɪn ˈklɪr.əns/ | Độ thanh thải creatinin |
| 449 | Focal segmental glomerulosclerosis | /ˈfoʊ.kəl seɡˈmen.təl ɡloʊˌmer.jə.loʊ.skləˈroʊ.sɪs/ | Xơ cứng cầu thận cục bộ phân đoạn |
| 450 | Interstitial fibrosis | /ˌɪn.tɚˈstɪʃ.əl faɪˈbroʊ.sɪs/ | Xơ hóa kẽ |
| 451 | Nephropathy | /nəˈfrɑː.pə.θi/ | Bệnh thận |
| 452 | Hepatic adenomas | /hɪˈpæt̬.ɪk ˌæd.əˈnoʊ.məz/ | U tuyến gan |
| 453 | Hepatocellular carcinomas | /hɪˌpæt̬.oʊˈsel.jə.lɚ ˌkɑːr.səˈnoʊ.məz/ | Ung thư biểu mô tế bào gan |
| 454 | Alpha-fetoprotein | /ˈæl.fə ˌfiː.t̬oʊˈproʊ.tiːn/ | Alpha-fetoprotein |
| 455 | Osteopenia | /ˌɑːs.ti.oʊˈpiː.ni.ə/ | Thiếu xương |
| 456 | Osteoporosis | /ˌɑːs.ti.oʊ.pəˈroʊ.sɪs/ | Loãng xương |
| 457 | Parathyroid | /ˌpær.əˈθaɪ.rɔɪd/ | Tuyến cận giáp |
| 458 | Vitamin D | /ˈvaɪ.t̬ə.mɪn diː/ | Vitamin D |
| 459 | Bone mineral content | /boʊn ˈmɪn.ə.rəl ˈkɑːn.tent/ | Hàm lượng khoáng chất trong xương |
| 460 | Hypercortisolemia | /ˌhaɪ.pɚ.kɔːr.t̬ɪ.soʊˈliː.mi.ə/ | Tăng cortisol máu |
| 461 | Hirsutism | /ˈhɝː.suː.tɪ.zəm/ | Rậm lông |
| 462 | Pulmonary hypertension | /ˈpʊl.məˌner.i ˌhaɪ.pɚˈten.ʃən/ | Tăng huyết áp phổi |
| 463 | Agranulocytosis | /eɪˌɡræn.jə.loʊ.saɪˈtoʊ.sɪs/ | Mất bạch cầu hạt |
| 464 | Myeloid maturation | /ˈmaɪ.ə.lɔɪd ˌmætʃ.əˈreɪ.ʃən/ | Sự trưởng thành dòng tủy |
| 465 | Neutrophils | /ˈnuː.trə.fɪlz/ | Bạch cầu trung tính |
| 466 | Monocytes | /ˈmɑː.noʊ.saɪts/ | Bạch cầu đơn nhân |
| 467 | Inflammatory bowel disease | /ɪnˈflæm.ə.tɔːr.i ˈbaʊ.əl dɪˈziːz/ | Bệnh viêm ruột |
| 468 | Crohn disease | /kroʊnz dɪˈziːz/ | Bệnh Crohn |
| 469 | Granulocyte colony-stimulating factor (G-CSF) | /ˈɡræn.jə.loʊ.saɪt ˈkɑː.lə.ni ˈstɪm.jə.leɪ.t̬ɪŋ ˈfæk.tɚ/ | Yếu tố kích thích dòng bạch cầu hạt |
| 470 | Mucosal ulceration | /mjuːˈkoʊ.səl ˌʌl.səˈreɪ.ʃən/ | Loét niêm mạc |
| 471 | Gingivitis | /ˌdʒɪn.dʒəˈvaɪ.t̬ɪs/ | Viêm nướu |
| 472 | Periodontal disease | /ˌper.i.oʊˈdɑːn.təl dɪˈziːz/ | Bệnh nha chu |
| 473 | Perianal abscesses | /ˌper.iˈeɪ.nəl ˈæb.se.sɪz/ | Áp xe quanh hậu môn |
| 474 | Thyroid autoimmunity | /ˈθaɪ.rɔɪd ˌɔː.t̬oʊ.ɪˈmjuː.nə.t̬i/ | Tự miễn tuyến giáp |
| 475 | Primary hypothyroidism | /ˈpraɪ.mer.i ˌhaɪ.poʊˈθaɪ.rɔɪ.dɪ.zəm/ | Suy giáp nguyên phát |
| 476 | Aspartate aminotransferase | /əˈspɑːr.teɪt əˌmiː.noʊˈtræns.fə.reɪs/ | Aspartate aminotransferase |
| 477 | Alanine aminotransferase | /ˈæl.ə.niːn əˌmiː.noʊˈtræns.fə.reɪs/ | Alanine aminotransferase |
| 478 | Microsomes | /ˈmaɪ.kroʊ.soʊmz/ | Vi thể (microsome) |
| 479 | Splenomegaly | /ˌspliː.noʊˈmeɡ.ə.li/ | Lách to |
| 480 | Nephromegaly | /ˌnef.roʊˈmeɡ.ə.li/ | Thận to |
| 481 | Angiotensin converting enzyme (ACE) inhibitors | /ˌæn.dʒi.oʊˈten.sɪn kənˈvɝː.t̬ɪŋ ˈen.zaɪm ɪnˈhɪb.ə.t̬ɚz/ | Thuốc ức chế men chuyển angiotensin |
| 482 | Angiotensin receptor blockers | /ˌæn.dʒi.oʊˈten.sɪn rɪˈsep.tɚ ˈblɑː.kɚz/ | Thuốc chẹn thụ thể angiotensin |
| 483 | Thiazide diuretics | /ˈθaɪ.ə.zaɪd ˌdaɪ.jʊˈret̬.ɪks/ | Thuốc lợi tiểu thiazide |
| 484 | Loop diuretics | /luːp ˌdaɪ.jʊˈret̬.ɪks/ | Thuốc lợi tiểu quai |
| 485 | Erythropoietin | /ɪˌrɪθ.roʊˈpɔɪ.ə.tɪn/ | Erythropoietin |
| 486 | Splenomegaly | /ˌspliː.noʊˈmeɡ.ə.li/ | Lách to |
| 487 | Microalbumin | /ˌmaɪ.kroʊ.ælˈbjuː.mɪn/ | Microalbumin |
| 488 | Bone densitometry | /boʊn ˌden.sɪˈtɑː.mə.tri/ | Đo mật độ xương |
| 489 | Echocardiography (ECHO) | /ˌek.oʊˌkɑːr.diˈɑː.ɡrə.fi/ | Siêu âm tim |
| 490 | Electrocardiogram (ECG) | /ɪˌlek.troʊˈkɑːr.di.oʊ.ɡræm/ | Điện tâm đồ |
| 491 | Gene therapy | /dʒiːn ˈθer.ə.pi/ | Liệu pháp gen |
| 492 | Adeno-associated virus | /ˈæd.ə.noʊ əˈsoʊ.ʃi.eɪ.t̬ɪd ˈvaɪ.rəs/ | Virus adeno liên kết |
| 493 | Limit Dextrinosis | /ˌlɪm.ɪt ˌdeks.trəˈnoʊ.sɪs/ | Bệnh giới hạn Dextrin |
| 494 | Cori Disease | /ˈkɔːr.i dɪˈziːz/ | Bệnh Cori |
| 495 | Forbes Disease | /fɔːrbz dɪˈziːz/ | Bệnh Forbes |
| 496 | Limit dextrin | /ˈlɪm.ɪt ˈdeks.trɪn/ | Giới hạn dextrin |
| 497 | Isoforms | /ˈaɪ.soʊ.fɔːrmz/ | Dạng đồng phân (isoform) |
| 498 | Myopathy | /maɪˈɑː.pə.θi/ | Bệnh cơ |
| 499 | Fibrosis | /faɪˈbroʊ.sɪs/ | Xơ hóa |
| 500 | Idiopathic hypertrophic cardiomyopathy | /ˌɪd.i.oʊˈpæθ.ɪk ˌhaɪ.pɚˈtroʊ.fɪk ˌkɑːr.di.oʊ.maɪˈɑː.pə.θi/ | Bệnh cơ tim phì đại vô căn |