
Sperling Nội tiết học Nhi khoa, Ấn bản thứ 5 – Biên dịch: Ths.Bs. Lê Đình Sáng
Sperling Pediatric Endocrinology, Fifth Edition
Tác giả: Sperling, Mark A., MD – Nhà xuất bản: Elsevier Inc.
PHẦN II: CÁC RỐI LOẠN NỘI TIẾT Ở TRẺ SƠ SINH
Chương 6. Cơ quan sinh dục không rõ ràng
Selma Feldman Witchel; Peter A. Lee
Sperling Pediatric Endocrinology, 6, 123-174
Mở đầu
Trong suốt lịch sử, con người đã luôn tìm cách thấu hiểu sự khác biệt giữa các giới tính sinh học và các cơ chế chịu trách nhiệm cho những khác biệt này. Sự biệt hóa tuyến sinh dục và phát triển giới tính bình thường phụ thuộc vào một sự sắp xếp tỉ mỉ và đồng bộ của một mạng lưới các con đường tín hiệu nội tiết, cận tiết và tự tiết. Mạng lưới này bao gồm các hoạt động và tương tác của các gen đặc hiệu, các ảnh hưởng biểu sinh, các yếu tố phiên mã và hormone. Những xáo trộn trong mạng lưới phức tạp của quá trình điều hòa gen và biểu hiện gen chi phối sự phát triển tuyến sinh dục của thai nhi dẫn đến các rối loạn phát triển giới tính (disorders of sex development – DSD). Ước tính có khoảng một trong 4000 trẻ sơ sinh mắc phải DSD. Tuy nhiên, con số này sẽ tăng lên nếu bao gồm tất cả các biến thể của sự phát triển giới tính, kể cả tinh hoàn ẩn và mọi dạng lỗ tiểu lệch thấp. Tỷ lệ mắc bệnh của một số phân nhóm chỉ mới được báo cáo một cách chính xác, chẳng hạn như nữ giới 46,XY là 6,4/100.000; hội chứng không nhạy cảm androgen (androgen insensitivity syndrome – AIS) là 4,1/100.000; và loạn sản tuyến sinh dục là 1,5/100.000.
DSD bao gồm một phổ các rối loạn trong đó các khía cạnh về nhiễm sắc thể, di truyền, tuyến sinh dục, nội tiết tố hoặc giải phẫu của giới tính không điển hình. Việc hiểu rõ sinh học phát triển và phôi thai học của hệ niệu-sinh dục là rất quan trọng để phân loại và xác định cơ sở phân tử của rối loạn ở từng bệnh nhân. Ngày càng rõ ràng rằng có nhiều gen, yếu tố biểu sinh, yếu tố môi trường và tương tác protein tham gia vào quá trình biệt hóa tuyến sinh dục và phát triển giới tính. Vai trò của bác sĩ nội tiết nhi trong việc chăm sóc một trẻ sơ sinh có cơ quan sinh dục không rõ ràng bắt đầu từ thời điểm trình diện ban đầu—khi sinh, thời thơ ấu, tuổi vị thành niên, hoặc trước khi sinh đối với những trẻ được xác định qua xét nghiệm tiền sản. Trách nhiệm của bác sĩ trong việc quản lý đứa trẻ này bắt đầu sớm và tiếp tục cho đến khi việc chuyển giao cho các nhà cung cấp dịch vụ chăm sóc người lớn được coi là phù hợp.
Sau Tuyên bố Đồng thuận năm 2006 về quản lý các rối loạn liên giới tính, kiến thức về cơ sở phân tử của sự biệt hóa/phát triển giới tính đã được mở rộng. Số lượng và khả năng của các công cụ di truyền có sẵn để xác định cơ sở phân tử đã tăng lên. Các cân nhắc bao gồm một cách tiếp cận toàn diện đối với trẻ bị DSD, sự tham gia của cha mẹ, sự công nhận về sức khỏe tâm lý, vai trò của các nhóm hỗ trợ và nhu cầu chăm sóc theo chiều dọc toàn diện. Việc thu thập dữ liệu kết quả, mặc dù vẫn còn hạn chế, giúp mở rộng kiến thức y học và cải thiện chất lượng. Việc phát triển và đánh giá các mô hình chăm sóc đa ngành tập trung vào chất lượng chăm sóc đang được tiến hành.
Trò chuyện với cha mẹ
Đối với cha mẹ, sự ra đời của đứa con là một sự kiện được mong đợi từ lâu và đầy phấn khích. Do tần suất siêu âm trước sinh ngày càng tăng, cha mẹ thường đã được thông báo về giới tính của con mình, có thể đã chọn tên cho con và thường tổ chức một buổi tiệc “tiết lộ giới tính”. Trong một số trường hợp, sự phát triển bất thường của cơ quan sinh dục đã được xác định qua siêu âm và cha mẹ đã được thông báo về tình trạng này. Trong kịch bản này, cha mẹ và các nhà cung cấp dịch vụ chăm sóc sức khỏe có thể trao đổi về các xét nghiệm chẩn đoán và khuyến nghị điều trị có khả năng xảy ra. Trong trường hợp không có thông tin trước, cha mẹ đột nhiên phải đối mặt với một đứa trẻ sơ sinh bị dị tật bẩm sinh liên quan đến cơ quan sinh dục ngoài và sự không chắc chắn về giới tính của con mình. Sự lo lắng và e ngại về sức khỏe và giới tính của em bé đặc biệt gây tổn thương khi cha mẹ không hề hay biết trước về sự phát triển bất thường của cơ quan sinh dục.
Ban đầu, cha mẹ cần được chúc mừng về sự ra đời của đứa con. Họ cần được nghe rằng con họ có một tình trạng ảnh hưởng đến sự phát triển giới tính và tình trạng này sẽ được giải quyết một cách cẩn thận và toàn diện. Các cuộc thảo luận phù hợp bao gồm việc chia sẻ rằng các bất thường về phát triển giới tính liên quan đến hệ thống phức tạp điều khiển sự phát triển của hệ sinh sản, bao gồm cả sự phát triển của cơ quan sinh dục ngoài. Việc giải thích rằng giới tính của con họ không thể được xác định chỉ bằng cách kiểm tra cơ quan sinh dục ngoài là điều cần thiết. Điều quan trọng là phải nhấn mạnh rằng sự phát triển không điển hình không phải là lỗi của cha mẹ và họ không nên cảm thấy tội lỗi hay xấu hổ. Bác sĩ và các chuyên gia y tế không nên suy đoán hoặc đưa ra các chẩn đoán giả định. Cha mẹ có thể được trấn an rằng tất cả các nghiên cứu phù hợp sẽ được thực hiện để cung cấp thông tin toàn diện nhằm xác định giới tính nuôi dưỡng phù hợp nhất cho con họ. Việc sử dụng các công cụ di truyền có thể xác định nguyên nhân phân tử cụ thể của DSD, với lưu ý rằng nguyên nhân phân tử cụ thể có thể vẫn chưa được biết.
Mục tiêu là chăm sóc lấy bệnh nhân làm trung tâm trong bối cảnh gia đình và một đội ngũ đa chuyên khoa bao gồm các bác sĩ nội tiết nhi, bác sĩ tiết niệu/phẫu thuật nhi, chuyên gia di truyền, bác sĩ sơ sinh, bác sĩ chẩn đoán hình ảnh, các nhà cung cấp dịch vụ sức khỏe hành vi và các y tá giáo dục nội tiết nhi. Một mục tiêu điều trị ban đầu là xác định xem có tình trạng đe dọa tính mạng tiềm ẩn hoặc liên quan cần điều trị khẩn cấp cụ thể hay không. Một thành viên trong đội ngũ nên đóng vai trò là người giao tiếp chính với gia đình.
Mặc dù xã hội đương đại có những đề cập cởi mở về giới tính và tình dục, cha mẹ có thể gặp khó khăn khi nghĩ về con mình như một sinh thể có tính dục và cảm thấy xấu hổ khi thảo luận về giới tính, bản dạng giới và xu hướng tính dục trong tương lai của con. Thái độ văn hóa, những kỳ vọng có từ trước và hệ thống hỗ trợ gia đình ảnh hưởng đến phản ứng của cha mẹ và có thể ảnh hưởng đến các lựa chọn của họ cho đứa trẻ. Cha mẹ cần được thông báo rằng bản dạng giới của đứa trẻ là trải nghiệm cá nhân về giới tính của một người. Đội ngũ y tế cần thúc đẩy một mạng lưới cởi mở và quan tâm để hỗ trợ cha mẹ. Quan trọng là, đội ngũ y tế cần thu hút cha mẹ tham gia vào quá trình ra quyết định y tế và thảo luận về các thông tin liên quan. Điều quan trọng là phải nhận thức rằng việc các bác sĩ lâm sàng sử dụng sự không chắc chắn trong lâm sàng có thể làm cha mẹ lạc hướng và làm trầm trọng thêm sự lo lắng của họ. Đối với việc ra quyết định phẫu thuật, các bác sĩ lâm sàng nên cố gắng cân bằng giữa sự thận trọng và lợi ích, và xem xét các lựa chọn vượt ra ngoài sự phân đôi “phẫu thuật” và “không phẫu thuật”.
Nếu giới tính của đứa trẻ vẫn chưa rõ ràng, cần thu thập thông tin để hỗ trợ cha mẹ và đội ngũ chăm sóc sức khỏe xác định giới tính nuôi dưỡng phù hợp nhất. Thông thường, việc này có thể được thực hiện trong vòng vài giờ hoặc vài ngày. Trong những trường hợp phức tạp hơn, quá trình chẩn đoán có thể mất nhiều thời gian hơn. Trong những tình huống không thể xác định được nguyên nhân cụ thể, việc phân loại DSD chung (xem phần sau) cung cấp cơ sở cho việc ra quyết định.
Các yếu tố liên quan đến quá trình ra quyết định y tế bao gồm mức độ phát triển của hệ thống sinh sản bên trong và bên ngoài, bằng chứng về chức năng tuyến sinh dục (tiềm năng tiết hormone tuổi dậy thì và khả năng sinh sản), và khả năng đáp ứng với hormone. Trong một số trường hợp, những yếu tố này còn quan trọng hơn cả bộ nhiễm sắc thể đồ. Các gen và sản phẩm gen liên quan đến sự phát triển giới tính được lập bản đồ trên các nhiễm sắc thể thường và nhiễm sắc thể giới tính. Các biến thể di truyền được lập bản đồ trên các vùng mã hóa và không mã hóa ảnh hưởng đến sự biệt hóa giới tính và phát triển giới tính của thai nhi và trẻ em.
Khi đã đạt được sự đồng thuận về một loại chẩn đoán, cần xem xét các thông tin kết quả có sẵn. Kiến thức về một nguyên nhân cụ thể, bao gồm các chi tiết tức thời và kết quả lâu dài, cho phép lập kế hoạch tối ưu cho các can thiệp điều trị và tư vấn di truyền cho các lần mang thai trong tương lai. Các nhà cung cấp dịch vụ chăm sóc sức khỏe cần nhận thức rằng dữ liệu kết quả có sẵn để hỗ trợ quá trình ra quyết định còn hạn chế. Thông tin hiện có trong các báo cáo đã công bố phần lớn dựa trên các nghiên cứu hồi cứu thu được bằng các phương pháp và chiến lược đa dạng. Mức độ, thời điểm và thời gian tiếp xúc với androgen trước khi sinh, chỉ có thể được ước tính, có khả năng ảnh hưởng đến sự phát triển của hệ thần kinh trung ương (CNS) và các mô khác. Tiếp xúc với androgen trước khi sinh có thể ảnh hưởng đến sự phát triển của bản dạng giới.
Một nghiên cứu cắt ngang đa trung tâm đã báo cáo dữ liệu kết quả ngắn hạn trên 92 trẻ được phát hiện có cơ quan sinh dục không rõ ràng với sự phân bố nhiễm sắc thể đồ là 57% 46,XX; 34% 46,XY; và 9% bất thường nhiễm sắc thể giới tính. Hơn 90% cá thể 46,XX bị tăng sản tuyến thượng thận bẩm sinh (CAH); 65% cá thể 46,XY không có chẩn đoán phân tử; và hầu hết các cá thể 46,XY được nuôi dưỡng như nam giới. Các quan niệm thay đổi về can thiệp y tế và phẫu thuật đòi hỏi sự thận trọng trong việc ra quyết định, trong khi các nghiên cứu kết quả bổ sung đang được tiến hành. Các nghiên cứu có thể giải quyết các khía cạnh cụ thể, chẳng hạn như tuân thủ liệu pháp hormone và sự hài lòng của bệnh nhân hoặc sức khỏe tâm lý, chất lượng cuộc sống và nhận thức bản thân ở nam giới bị CAH.
Cuộc trò chuyện đầu tiên với cha mẹ nên củng cố sự gắn kết của cha mẹ với con mình. Càng nhiều càng tốt, một giọng điệu tích cực và lạc quan sẽ hữu ích. Thực tế, giọng điệu cảm xúc của cuộc trò chuyện ban đầu này có ý nghĩa hơn thông tin thực tế được cung cấp và được cha mẹ ghi nhớ trong nhiều năm. Sự tôn trọng quan điểm của gia đình và cá nhân, cùng với sự sẵn lòng lặp lại hoặc trì hoãn các giải thích chi tiết, là rất quan trọng. Giữa sự đau khổ về mặt cảm xúc liên quan đến sự không chắc chắn về giới tính của con mình, cha mẹ có thể không thể tiếp thu được lượng lớn thông tin mà cuối cùng cần được chia sẻ. Các cuộc thảo luận lặp đi lặp lại với cha mẹ sẽ giúp họ thừa nhận những lo ngại về mặt cảm xúc và trí tuệ của mình đối với con. Sự quen thuộc và thấu hiểu sẽ giúp cha mẹ gắn kết với con và tương tác với các thành viên gia đình, bạn bè và đồng nghiệp.
Trừ khi giới tính khi sinh đã rõ ràng vào thời điểm này, đội ngũ chăm sóc sức khỏe nên đề nghị cha mẹ trì hoãn việc đặt tên cho trẻ, thông báo về sự ra đời của em bé và đăng ký khai sinh cho đến khi có thêm thông tin. Thông điệp cần phải rõ ràng rằng họ sẽ được tích cực tham gia vào quá trình xác định giới tính nuôi dưỡng cho con mình. Cho đến khi giới tính nuôi dưỡng được xác định, tốt nhất là gọi đứa trẻ là “em bé của bạn” hoặc “con của bạn”. Các từ như anh ấy, cô ấy, và nó nên được tránh. Đội ngũ đa chuyên khoa có trách nhiệm giáo dục nhân viên phụ trợ về cách gọi đứa trẻ là “em bé của bạn”.
Các giải thích thực tế về quá trình biệt hóa giới tính tập trung vào tình hình của con họ nên được phác thảo ban đầu. Mục tiêu chính vào thời điểm này là cung cấp cho cha mẹ một sự hiểu biết cơ bản rằng các cấu trúc sinh dục bên trong và bên ngoài của cả nam và nữ đều phát triển từ cùng một mô nguyên thủy. Cũng hữu ích khi giải thích rằng không có hormone nào dành riêng cho nam và nữ. Thay vào đó, môi trường mà thai nhi nam và nữ phát triển được đặc trưng bởi lượng tương đối khác nhau của các hormone này. Sử dụng các bản phác thảo, hình ảnh và sơ đồ đơn giản có thể hữu ích để giải thích phôi thai học về sự phát triển của cơ quan sinh dục cho cha mẹ. Một số cha mẹ có thể được hưởng lợi từ việc thực hành những từ họ sẽ sử dụng để thảo luận về sức khỏe của trẻ với các thành viên khác trong gia đình. Các giải thích chi tiết có thể được xem lại nhiều lần khi trẻ lớn lên, đặc biệt là vì trẻ không thể tích cực tham gia vào các cuộc thảo luận ban đầu.
Trong các cuộc trò chuyện ban đầu, việc kiểm tra trẻ cùng với cha mẹ để xác định các phát hiện thể chất cụ thể của con họ thường có lợi. Việc nhìn thấy cơ quan sinh dục ngoài của con họ có thể làm giảm sự e ngại và củng cố nhận thức rằng nhu cầu của con họ cũng tương tự như tất cả các trẻ sơ sinh khác. Thông tin có thể được trình bày để giảm thiểu lo lắng và trang bị tốt hơn cho cha mẹ để tham gia vào quá trình ra quyết định. Thảo luận về nhiều mối quan tâm (đặc biệt là những mối quan tâm liên quan đến bản dạng giới, sự phát triển dậy thì, xu hướng tính dục, chức năng tình dục và khả năng sinh sản) có thể hữu ích. Việc giải quyết một cách trung thực các mối quan tâm của cha mẹ cuối cùng sẽ tạo ra sự tin tưởng và những cảm xúc tích cực để giúp cha mẹ thúc đẩy lòng tự trọng của con mình. Lý tưởng nhất, mỗi bậc cha mẹ đạt được một giải pháp cá nhân với cam kết về một viễn cảnh tích cực cho tương lai của đứa trẻ.
Thuật ngữ
Dưới sự bảo trợ của Hiệp hội Nội tiết Nhi khoa (Bắc Mỹ) và Hiệp hội Nội tiết Nhi khoa Châu Âu, một tuyên bố đồng thuận quốc tế đã được xây dựng, khuyến nghị một phân loại sửa đổi của thuật ngữ y học được sử dụng cho các rối loạn phát triển giới tính để tránh các thuật ngữ gây nhầm lẫn và miệt thị. Phân loại mô tả này cố gắng nhạy cảm với các mối quan tâm của cha mẹ và đủ linh hoạt để kết hợp thông tin di truyền phân tử mới. Hệ thống phân loại cập nhật tích hợp các cân nhắc về di truyền phân tử vào danh pháp cho “rối loạn phát triển giới tính (DSD)” và cung cấp một cách tiếp cận để đánh giá chẩn đoán. Có những phản đối việc sử dụng từ “rối loạn” vì điều này ngụ ý bệnh lý, với từ “khác biệt” đôi khi được sử dụng thay thế.
Các thuật ngữ như lưỡng tính giả, lưỡng tính thật, và ghi nhãn giới tính trong chẩn đoán nên được loại bỏ. Tuy nhiên, một số bệnh nhân vẫn thích sử dụng thuật ngữ liên giới tính. Để bao hàm tất cả các loại DSD, hệ thống phân loại này rất rộng và bao gồm một số tình trạng không biểu hiện với những bất thường rõ ràng của sự phát triển cơ quan sinh dục (Bảng 6.1). Mục tiêu chính của hệ thống phân loại này là cung cấp một khuôn khổ cho chẩn đoán, đánh giá và quản lý chăm sóc dựa trên tình trạng nhiễm sắc thể giới tính. Hiện tại, microarray, phân tích gen ứng viên và giải trình tự toàn bộ exome/genome ngày càng được sử dụng. Các loại DSD bao gồm DSD do nhiễm sắc thể giới tính, chẳng hạn như 45,X/46,XY; DSD thể noãn tinh; DSD 46,XY, chẳng hạn như rối loạn phát triển tinh hoàn, rối loạn tổng hợp và hoạt động của androgen, đảo ngược giới tính 46,XY và DSD 46,XX; và đảo ngược giới tính 46,XX. Một số chẩn đoán được bao gồm trong nhiều hơn một loại do sự phức tạp của sự phát triển nhiễm sắc thể và tuyến sinh dục. Số lượng gen được xác định có liên quan đến sự phát triển giới tính tiếp tục tăng lên. Tuy nhiên, bất chấp nhiều tiến bộ gần đây, nguyên nhân phân tử cụ thể của cơ quan sinh dục không rõ ràng ở một cá thể không phải lúc nào cũng có thể được xác định, đặc biệt là ở những người có DSD 46,XY.
Bảng 6.1. Rối loạn phát triển giới tính do nhiễm sắc thể giới tính
| Hội chứng | Kiểu nhân | Khiếm khuyết nhiễm sắc thể |
| Hội chứng Turner | 45,X thể khảm 45,X/46,XX |
Monosomy X Monosomy X thể khảm |
| Hội chứng Turner với sự sắp xếp lại cấu trúc nhiễm sắc thể X | 46,X,i(Xq) 46,X,del(Xp) 46,X,+mar |
isochromosome Xq mất đoạn Xp nhiễm sắc thể marker |
| Loạn sản tuyến sinh dục | 45,X/46,XY
XX/XY |
Mất nhiễm sắc thể Y thể khảm ở XY, thể khảm |
| Nam XX dương tính với gen SRY
Loạn sản tuyến sinh dục |
46,XX hoặc
46,X,der(X)t(X;Y) |
Chuyển vị Yp (gen SRY) sang nhiễm sắc thể X hoặc nhiễm sắc thể thường |
| Nam XX âm tính với gen SRY | 46,XX | gen SRY không có mặt |
| Hội chứng Klinefelter và các biến thể của nó | XXY
thể khảm 46,XXY/46,XY XXYY XX/XXY |
Disomy X
Disomy X thể khảm Disomy X và Y Mất nhiễm sắc thể Y thể khảm ở XXY |
Xác định giới tính
Thông qua các thí nghiệm của Alfred Jost với thỏ thai nhi trong những năm 1940 và 1950, các yêu cầu quan trọng của tinh hoàn và testosterone đối với sự biệt hóa giới tính nam đã được thiết lập. Thành phần nhiễm sắc thể của phôi người, XX hoặc XY, quyết định giới tính tuyến sinh dục. Nghiên cứu về sự tiến hóa của nhiễm sắc thể giới tính ở người cho thấy rằng các nhiễm sắc thể giới tính của con người đã tiến hóa từ một cặp nhiễm sắc thể thường tổ tiên khoảng 300 triệu năm trước. Sự xuất hiện của locus SRY, một số lần đảo đoạn trên nhiễm sắc thể Y và sự mất đi của quá trình trao đổi chéo X-Y đã dẫn đến tình trạng hiện tại của con người.
Các nghiên cứu về bệnh nhân bị rối loạn phát triển giới tính cuối cùng đã dẫn đến việc xác định locus di truyền chịu trách nhiệm chính cho sự chuyển đổi nhị phân này, đó là vùng xác định giới tính trên nhiễm sắc thể Y (SRY) trên nhiễm sắc thể Y. Các nghiên cứu liên quan đến việc tạo ra chuột chuyển gen SRY+ đã xác nhận vai trò thiết yếu của SRY và cung cấp thêm sự hiểu biết phân tử về sự biệt hóa tinh hoàn.
Giới tính di truyền được thiết lập tại thời điểm thụ tinh, tiếp theo là xác định giới tính, sự chuyển đổi nhị phân khởi động vận mệnh phát triển của tuyến sinh dục phôi thai để trở thành tinh hoàn hoặc buồng trứng. Việc xác định giới tính phần lớn bị ảnh hưởng bởi sự điều hòa phiên mã, trong khi các hormone được tiết ra và các thụ thể hormone ảnh hưởng đến sự phát triển kiểu hình. Biệt hóa giới tính đề cập đến quá trình mà qua đó kiểu hình nam hoặc nữ phát triển.
Tuyến sinh dục, các ống sinh dục trong và các cấu trúc sinh dục ngoài đều phát triển từ các mô lưỡng tiềm năng. Mỗi tế bào trong tuyến sinh dục đang phát triển đều có tiềm năng biệt hóa thành tế bào tinh hoàn hoặc tế bào buồng trứng, tùy thuộc vào cách hệ phiên mã của tế bào chưa biệt hóa thực hiện con đường của nó để phát triển thành buồng trứng hoặc tinh hoàn. Trong trạng thái lưỡng tiềm năng này, tồn tại tính đa năng với các gen sẵn sàng cho việc kích hoạt hoặc kìm hãm. Nói cách khác, việc xác định giới tính của tuyến sinh dục lưỡng tiềm năng phụ thuộc vào sự cam kết số phận của tế bào đối với một con đường tại một thời điểm chính xác trong quá trình phát triển, đồng thời duy trì sự kìm hãm tích cực con đường phát triển thay thế.
Trong tình huống thông thường, kiểu nhân (46,XY hoặc 46,XX) của tuyến sinh dục nguyên thủy quyết định xem nó sẽ biệt hóa thành tinh hoàn hay buồng trứng, tương ứng. Có sự khác biệt cố hữu giữa các tế bào XX và XY; khoảng 85% nhiễm sắc thể X thứ hai trong một tế bào XX trải qua quá trình bất hoạt X và các gen trên nhiễm sắc thể Y liên quan đến quá trình sinh tinh được biểu hiện trong các tế bào XY. Các yếu tố cục bộ (chẳng hạn như hormone do tuyến sinh dục đang phát triển tiết ra hoặc các yếu tố phiên mã đặc hiệu cho mô) ảnh hưởng đến sự biệt hóa tiếp theo của các cấu trúc sinh dục bên trong và bên ngoài. Quá trình này tích hợp các tín hiệu con đường đặc hiệu cho giới tính dường như đối kháng lẫn nhau.
Cấu hình chromatin và cấu trúc ba chiều không gian của nó ảnh hưởng đến biểu hiện gen bằng cách điều chỉnh khả năng của các yếu tố phiên mã liên kết với axit deoxyribonucleic (DNA). Sự sửa đổi histone và methyl hóa DNA ảnh hưởng đến tổ chức chromatin. Bối cảnh chromatin tích cực điều chỉnh sự đối kháng không ngừng giữa buồng trứng và tinh hoàn. Các gen liên quan đến sự phát triển con đường nam giới mất đi các dấu hiệu kìm hãm của chúng khi con đường tinh hoàn được kích hoạt và ngược lại đối với các gen liên quan đến con đường buồng trứng. Các yếu tố điều hòa cis, chẳng hạn như các yếu tố làm im lặng và tăng cường, phối hợp biểu hiện không-thời gian cụ thể của các gen trong mạng lưới phiên mã; các yếu tố điều hòa đặc hiệu cho giới tính được hình thành trong quá trình phát triển. Các protein polycomb là các chất điều hòa phiên mã được bảo tồn, phối hợp cấu trúc chromatin và cấu trúc nhiễm sắc thể. Các chất điều hòa chromatin, ví dụ như các protein polycomb, đại diện cho các nút quan trọng nơi các tín hiệu sinh học điều chỉnh biểu hiện gen.
Sự chệch hướng khỏi chuỗi sự kiện bình thường dẫn đến các rối loạn phát triển giới tính có thể biểu hiện dưới dạng sự biệt hóa tuyến sinh dục bất thường, sự biệt hóa sinh dục bên trong không nhất quán, hoặc sự không rõ ràng của cơ quan sinh dục ngoài. Mặc dù cơ quan sinh dục không rõ ràng thường không được coi là một trường hợp cấp cứu y tế, loại dị tật bẩm sinh này thường gây ra sự đau khổ tột cùng cho cha mẹ và gia đình mở rộng. Khi suy thượng thận đi kèm với cơ quan sinh dục không rõ ràng, việc đánh giá và điều trị ngay lập tức là cần thiết. Dù thế nào đi nữa, việc giới thiệu và đánh giá kịp thời bởi một đội ngũ đa chuyên khoa có chuyên môn về rối loạn biệt hóa giới tính được khuyến nghị mạnh mẽ.
Sự phát triển của hệ sinh sản
Mào niệu-sinh dục và sự phát triển tuyến sinh dục lưỡng tiềm năng
Mào niệu-sinh dục tạo ra tuyến sinh dục, vỏ thượng thận, thận và đường sinh sản. Tuyến sinh dục có nguồn gốc từ trung bì trung gian và phụ thuộc vào sự xâm nhập, tăng sinh và định hướng chính xác của các tế bào biểu mô khoang cơ thể (Hình 6.1). Ở người, vào tuần thứ 4 đến thứ 6 của thai kỳ, các mào niệu-sinh dục phát triển thành các cặp lồi ra của biểu mô khoang cơ thể (trung biểu mô) ở mặt bụng của trung thận. Khi biểu mô khoang cơ thể tăng sinh, màng đáy của nó tan rã để cho phép các tế bào khoang cơ thể xâm nhập vào để hình thành tuyến sinh dục đang phát triển. Tín hiệu Notch đảm bảo sự phân cực đúng đắn của các tế bào này. Trong tinh hoàn đang phát triển, các tế bào xâm nhập ban đầu hình thành các tế bào Sertoli, trong khi các tế bào xâm nhập sau đó phát triển thành các tế bào kẽ, bao gồm cả các tế bào Leydig. Trong buồng trứng, các tế bào xâm nhập tạo ra cả tế bào vỏ và tế bào hạt.
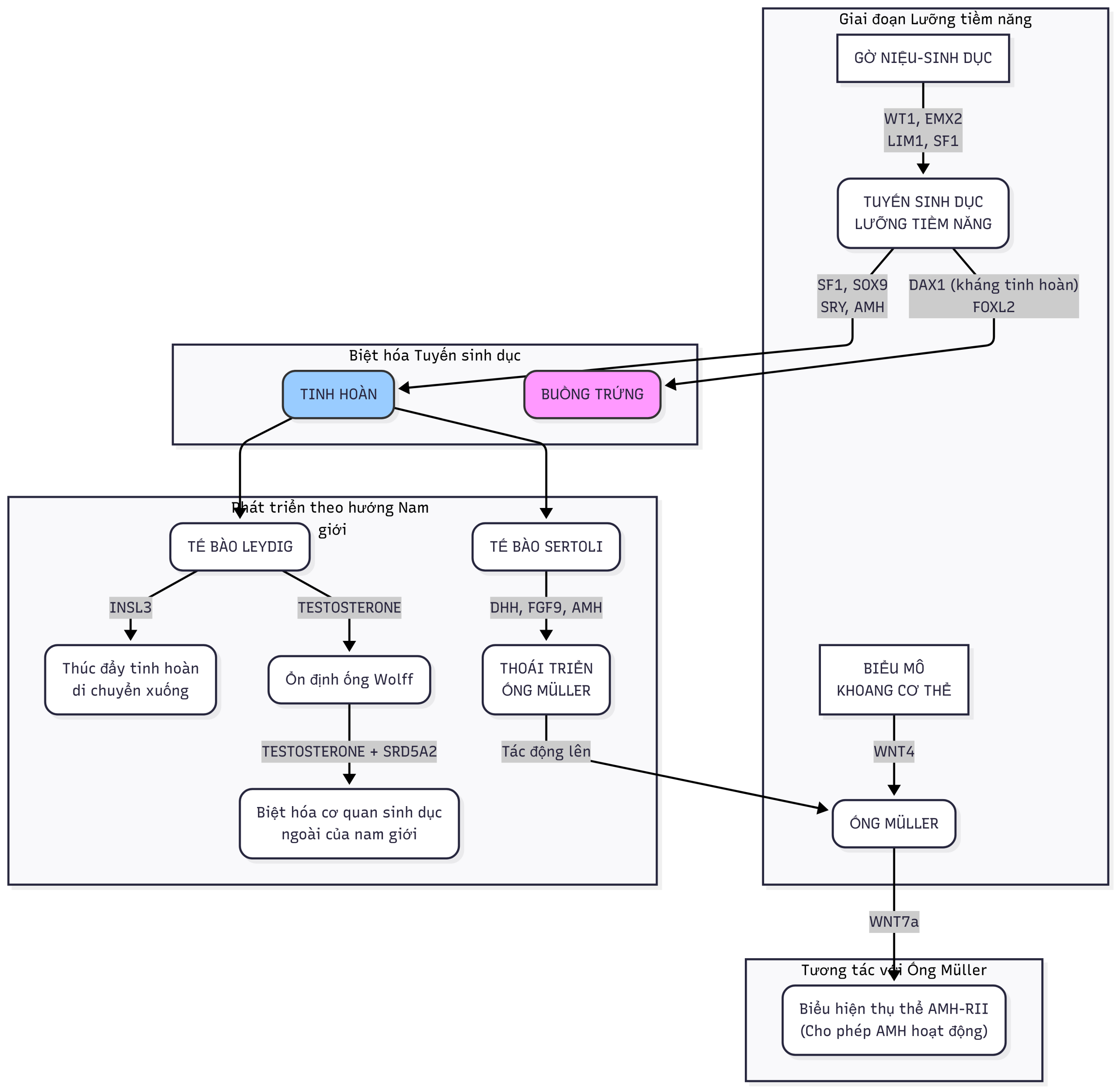
Hình 6.1. Các gen liên quan đến quá trình biệt hóa giới tính (Lược đồ đã được người dịch vẽ lại cho trực quan hơn). Khối u Wilms (WT1), EMX2, LIM1, CBX2 và yếu tố tạo steroid-1 (SF1) đóng vai trò trong sự biệt hóa của tuyến sinh dục từ mào niệu-sinh dục. Các gen liên quan đến sự biệt hóa tinh hoàn bao gồm SF-1, SOX9, vùng xác định giới tính trên Y (SRY) và hormone kháng Müller (AMH). Vùng nguy cấp bẩm sinh suy tuyến thượng thận-giới tính nhạy cảm với liều lượng trên X (DAX1) dường như có chức năng như một yếu tố kháng tinh hoàn. Wnt4 thúc đẩy sự phát triển của ống Müller, trong khi Wnt7a thúc đẩy sự biểu hiện của thụ thể AMH (AMH-RII). Tế bào Sertoli tiết ra AMH, (hoạt động thông qua thụ thể tương ứng của nó [AMH-RII]) thúc đẩy sự thoái triển của ống Müller. Tế bào Leydig tiết ra testosterone và hormone giống insulin-3 (INSL3). Testosterone ổn định ống Wolff và được chuyển đổi thành dihydrotestosterone (DHT) bởi 5α-reductase trong các mô đích để thúc đẩy sự biệt hóa của tuyến tiền liệt và sự phát triển của cơ quan sinh dục ngoài nam. INSL3 tham gia vào sự di chuyển của tinh hoàn qua ổ bụng. FOXL2, RSPO1, WNT4 và β-catenin tham gia vào sự phát triển của buồng trứng.
Các tế bào soma của tuyến sinh dục lưỡng tiềm năng đòi hỏi sự biểu hiện của các gen khối u Wilms (WT1), protein gắn GATA 4 (GATA4), và yếu tố tạo steroid 1 (NR5A1/SF1). Gen WT1 mã hóa một yếu tố phiên mã kẽm, được biểu hiện trong các mô trung bì phôi thai và dường như ảnh hưởng đến các tương tác trung bì-biểu mô. GATA4 được biểu hiện trong các tế bào soma của mào niệu-sinh dục và tuyến sinh dục lưỡng tiềm năng trước khi biểu hiện đặc hiệu cho giới tính. Chromobox homolog 2 (CBX2) được biểu hiện trong tuyến sinh dục đang phát triển ở tuần thứ 7 của thai kỳ và đóng một vai trò trong sự phát triển sớm của tuyến sinh dục. CBX2 hoạt động ở thượng nguồn của SRY, thúc đẩy sự chuyển hoạt của NR5A1 và SOX9, và kìm hãm yếu tố phiên mã forkhead 2 (FOXL2). NR5A1 được biểu hiện trong mào niệu-sinh dục và dường như điều hòa tăng cường biểu hiện SRY, thúc đẩy sự phát triển của tinh hoàn. FOXL2, một yếu tố phiên mã forkhead được biểu hiện trong buồng trứng, điều phối các quyết định về số phận tế bào; FOXL2 ủng hộ sự phát triển của buồng trứng và kìm hãm sự phát triển của tinh hoàn. Ngoài các yếu tố phiên mã và các yếu tố tiết ra cụ thể (hormone), sự tiếp xúc vật lý với trung thận dường như quan trọng đối với sự biệt hóa tuyến sinh dục sau đó.
Do nguồn gốc của chúng trong hệ niệu-sinh dục đang phát triển, buồng trứng và tinh hoàn ban đầu được đặt ở vị trí cao trong ổ bụng gần thận. Các phân tử tín hiệu cụ thể kích hoạt hoặc kìm hãm việc xác định tuyến sinh dục cho cả tinh hoàn và buồng trứng. Như đã lưu ý trước đó, sự đối kháng hoặc cạnh tranh lẫn nhau liên tục giữa các gen và protein cụ thể ảnh hưởng đến các quyết định về số phận tế bào trong sự phát triển của tuyến sinh dục. Các yếu tố phiên mã điều chỉnh “sự cạnh tranh” này bằng cách ảnh hưởng đến biểu hiện gen. Các ví dụ cụ thể bao gồm FOXL2 so với SRY-box 9 (SOX9) và SOX9 so với Wingless-type MMMTV integration site family member 4 (WNT4)/β-catenin, sẽ được thảo luận sau. Do đó, việc xác định và biệt hóa giới tính phản ánh sự đối kháng lẫn nhau giữa các yếu tố xác định tinh hoàn, SRY-SOX9-FGF9, và các yếu tố xác định buồng trứng, roof plate-specific spondin-1 (RSPO1)-WNT -β-catenin-FOXL2. Điều kỳ lạ là sự đối kháng này vẫn tồn tại sau khi sinh (xem phần sau). Như dự đoán, các gen enzym tạo steroid cho thấy sự biểu hiện cao hơn trong tinh hoàn người đang phát triển so với buồng trứng.
Sự phát triển tinh hoàn ở người
Sự biệt hóa tinh hoàn bắt đầu sớm hơn sự phát triển của buồng trứng. Bằng chứng đầu tiên về sự biệt hóa tinh hoàn là sự xuất hiện của các tế bào Sertoli nguyên thủy vào tuần thứ 6 đến 7 của thai kỳ trong tinh hoàn thai nhi người. Các tế bào, chủ yếu là tế bào nội mô, di chuyển từ trung thận và tương tác với các tế bào tiền-Sertoli để thúc đẩy sự phát triển của các dây tinh hoàn, bắt đầu vào khoảng tuần thứ 7 đến 8 của thai kỳ với các dây có thể nhận dạng được vào tuần thứ 9 đến 10 của thai kỳ. Các dây tinh hoàn là tiền thân của các ống sinh tinh sẽ chứa các tế bào Sertoli và tế bào mầm. Tương tác giữa các tế bào nội mô và trung mô dường như ảnh hưởng đến sự phát triển của các dây tinh hoàn.
Công tắc nhị phân chịu trách nhiệm cho sự phát triển của tinh hoàn là gen SRY nằm trên nhánh ngắn của nhiễm sắc thể Y. Cơ chế chính xác chịu trách nhiệm kích hoạt biểu hiện SRY vẫn chưa được xác định hoàn toàn. Trong tinh hoàn người, SRY được biểu hiện trong các tế bào hỗ trợ vào khoảng tuần thứ 6 của thai kỳ; biểu hiện ở mức độ thấp vẫn tồn tại trong suốt thai kỳ. Protein SRY chứa một miền nhóm di động cao (HMG) và được mã hóa bởi một gen exon đơn. Hai tín hiệu định vị hạt nhân được đặt trong miền HMG. Protein SRY được biểu hiện trong các tế bào tiền-Sertoli, nơi nó kích hoạt một công tắc phân tử để gây ra sự biệt hóa tế bào Sertoli, do đó bắt đầu quá trình biệt hóa giới tính nam. Miền HMG của protein SRY liên kết với rãnh nhỏ của DNA, nơi nó hoạt động như một yếu tố phiên mã bằng cách uốn cong DNA để có thể cho phép các protein khác tiếp cận các vùng điều hòa và thúc đẩy sự lắp ráp của các phức hợp phiên mã nucleoprotein. Một mức SRY ngưỡng phải đạt được tại một thời điểm quan trọng trong thai kỳ để thiết lập sự biệt hóa giới tính nam. Nếu không, con đường biệt hóa buồng trứng sẽ được kích hoạt.
Biểu hiện SRY độc lập với sự hiện diện của các tế bào mầm. SRY làm tăng biểu hiện của gen SRY-related HMG box-containing-9 (SOX9). Các nghiên cứu kiểu hình-kiểu gen ở người và chuột cho thấy biểu hiện SOX9 là một bước quan trọng, sau SRY, trong sự phát triển của tinh hoàn. Ở thượng nguồn của điểm bắt đầu phiên mã SOX9, có ít nhất ba yếu tố tăng cường, eSR-A, eSR-B và eALDI, phối hợp để thúc đẩy biểu hiện SOX9. Dữ liệu có sẵn cho thấy SRY và NR5A1 liên kết tại eALDI để thúc đẩy biểu hiện SOX9 ban đầu. Một khi biểu hiện đã được khởi xướng, SOX9 tự điều hòa dương tính biểu hiện của chính nó. Sau đó, SOX9 và NR5A1 liên kết với các vùng trong cả ba locus tăng cường để khuếch đại biểu hiện SOX9 của tinh hoàn. Sử dụng cả cơ chế trực tiếp và gián tiếp, SOX9 can thiệp vào các gen thúc đẩy sự biệt hóa buồng trứng. Những dữ liệu này phù hợp với các gợi ý rằng sự điều hòa gen của động vật có vú phụ thuộc vào nhiều yếu tố tăng cường dư thừa và các biến thể số lượng bản sao trong vùng này có thể gây ra sự đảo ngược giới tính.
Ngoài SRY, SOX9, và NR5A1, biểu hiện của các gen khác là cần thiết cho sự biệt hóa giới tính nam bình thường. Yếu tố tăng trưởng nguyên bào sợi (FGF9) hoạt động thông qua các thụ thể FGF (FGFR) thúc đẩy biểu hiện của SOX9 và kìm hãm WNT4 và FOXL2. Các gen được phát hiện có kiểu biểu hiện tương tự như SOX9 trong quá trình phát triển tinh hoàn bao gồm CITED1, ANKRD18A, G6PD, SLC52A3, KEL, ZNF280B, PRPS2, và INHBB. Ngoài việc kích hoạt con đường biệt hóa tinh hoàn, tinh hoàn đang phát triển còn biểu hiện các yếu tố để kìm hãm tích cực sự phát triển của buồng trứng. Cụ thể, tinh hoàn đang phát triển kìm hãm con đường tín hiệu R-spondin-WNT. Các E3 ubiquitin ligase xuyên màng, ZNRF3 và RNF43, ức chế tín hiệu WNT bằng cách nhắm mục tiêu thụ thể Frizzled để phân hủy.
Bằng phương pháp hóa mô miễn dịch, các protein NR5A1 và SOX9 có thể được phát hiện trong mô tuyến sinh dục phôi người ở tuần thứ 6 đến 7 của thai kỳ. Tại thời điểm này, biểu hiện SOX9 trở nên giới hạn ở nhân của các tế bào Sertoli trong một thai nhi 46,XY nhưng vẫn ở dạng tế bào chất trong một thai nhi 46,XX. Sau đó, hormone kháng Müller (AMH) được biểu hiện; với sự gia tăng biểu hiện protein AMH, biểu hiện protein khối u Wilms (WT1) và GATA-4 tăng lên trong tinh hoàn thai nhi. Biểu hiện giới hạn ở nam của yếu tố phiên mã liên quan đến doublesex và Mab-3 1 (DMRT1) đã được tìm thấy ở các thai nhi người 6 và 7 tuần tuổi. DMRT1 dường như cần thiết để duy trì các dây tinh hoàn và ngăn chặn sự chuyển biệt hóa sang kiểu hình nữ. Sự tăng trưởng nhanh nhất về số lượng tế bào Sertoli dường như xảy ra trong nửa sau của tam cá nguyệt thứ nhất và tam cá nguyệt thứ hai. Sau khi các tế bào Sertoli đã phát triển, các tế bào Leydig của thai nhi xuất hiện vào khoảng tuần thứ bảy đến thứ tám của thai kỳ và sản xuất androgen để thúc đẩy các cấu trúc sinh dục nam bên trong và bên ngoài.
Các tế bào Leydig bao gồm hai quần thể riêng biệt; các tế bào Leydig của thai nhi biệt hóa trong tử cung và các tế bào Leydig trưởng thành xuất hiện sau khi sinh. Sự biệt hóa của tế bào Leydig thai nhi phụ thuộc vào các tín hiệu cận tiết, chẳng hạn như thụ thể yếu tố tăng trưởng có nguồn gốc từ tiểu cầu-alpha (PDGFR-α), Desert hedgehog (DHH), PTCH1, và Aristaless-related homeobox (ARX). NR5A1 được biểu hiện trong các tế bào Leydig để thúc đẩy biểu hiện gen enzym tạo steroid. Số lượng tế bào Leydig của thai nhi phản ánh sự kích thích của gonadotropin vì số lượng này giảm ở thai nhi nam vô não và tăng ở thai nhi 46,XY, với nồng độ gonadotropin tăng cao thứ phát do không nhạy cảm androgen hoàn toàn.
Biểu hiện của các gen liên quan đến quá trình tạo steroid của tinh hoàn tăng lên vào khoảng tuần thứ 8 của thai kỳ. Gonadotropin màng đệm người (hCG) của nhau thai kích thích sự tiết androgen sớm của tinh hoàn. Về sau trong thai kỳ, sự tiết LH của tuyến yên thúc đẩy sự tiết androgen của tinh hoàn. Đến tuần thứ 11 của thai kỳ, các khoang tinh hoàn, các thành phần ống và kẽ, và các loại tế bào quan tâm (Leydig, Sertoli, và tế bào mầm) có thể được nhìn thấy. Trong tinh hoàn thai nhi người, mức độ axit ribonucleic thông tin (mRNA) của HSD17B3, CYP11A1, và PTC1 tăng đáng kể trong suốt tam cá nguyệt thứ hai mà không có những thay đổi đáng kể về mức độ CYP17A1, LHR, hoặc INSL3.
Sự phát triển buồng trứng ở người
Thay vì sự biệt hóa buồng trứng là con đường mặc định xảy ra khi không có biểu hiện gen SRY, rõ ràng là các gen cụ thể ảnh hưởng đến sự biệt hóa buồng trứng. Các gen liên quan đến sự biệt hóa buồng trứng bao gồm WNT4, FOXL2, follistatin (FST), protein hình thái xương-2 (BMP2), GATA4/FOG2, và RSPO1. RSPO1 là một yếu tố tiết ra kích hoạt con đường tín hiệu WNT β-catenin. FOXL2 kìm hãm các gen đặc hiệu cho nam, đặc biệt là SOX9 bắt đầu từ trong thai nhi; sự kìm hãm tích cực này tiếp tục trong suốt tuổi trưởng thành. Do đó, biểu hiện của FOXL2 và SOX9 loại trừ lẫn nhau trong các tuyến sinh dục đang phát triển, cũng như buồng trứng và tinh hoàn sau khi sinh.
Sự phát triển buồng trứng bình thường đòi hỏi các tín hiệu FOXL2, RSPO1 và WNT/β-catenin kinh điển. RSPO1 hoạt động thông qua các thụ thể bề mặt tế bào LGR4/5 cô lập các E3 ubiquitin ligase xuyên màng ZNRF3 và RNF43, dẫn đến tăng tín hiệu WNT. RSPO1 và WNT4 ổn định và khuếch đại tín hiệu β-catenin để kích hoạt phiên mã gen đích. WNT4 ức chế các tế bào kẽ tiết androgen, ức chế sự hình thành mạch máu khoang cơ thể và hỗ trợ các dẫn xuất của ống Müller.
Hai gen có vai trò trong sự phát triển dòng mầm, alpha (FIGLA) và homeobox buồng trứng sơ sinh (NOBOX), được biểu hiện trong các tế bào mầm của thai nhi hoặc trẻ sơ sinh, nơi chúng tuyển mộ các tế bào hạt đang phát triển để hình thành các nang trứng nguyên thủy và sơ cấp. Các yếu tố phiên mã bổ sung liên quan đến sự phát triển buồng trứng bao gồm SOHLH1, SOHLH2, NOBOX, LHX8, FIGLA, và LHX9. Buồng trứng thai nhi người trong tam cá nguyệt thứ hai biểu hiện các protein cần thiết để tổng hợp và đáp ứng với các tín hiệu estrogen, progestogen và androgen.
Sự phát triển tế bào mầm
Mặc dù là điều kiện tiên quyết cho sự phát triển của tế bào mầm để duy trì loài, tế bào mầm không cần thiết cho sự phát triển ban đầu của buồng trứng hoặc tinh hoàn. Các tế bào mầm nguyên thủy có nguồn gốc từ lá phôi trên. Vào khoảng tuần thứ 6 của thai kỳ ở người, các tế bào mầm nguyên thủy tăng sinh và di chuyển từ ruột sau dọc theo các sợi thần kinh để chiếm cứ các mào sinh dục. SOX17 là một yếu tố đa năng rất quan trọng cho sự biệt hóa của tế bào mầm nguyên thủy ở người thông qua tác động của nó để thúc đẩy biểu hiện gen đặc hiệu cho tế bào mầm. SOX17 dường như thúc đẩy biểu hiện BLIMP, dường như ức chế các gen nội bì và trung bì.
Khi đến tuyến sinh dục đang phát triển, môi trường cục bộ sẽ định hướng số phận của các tế bào mầm nguyên thủy theo hướng phát triển nam hoặc nữ. Do đó, các con đường biệt hóa cuối cùng cho các tế bào mầm là lưỡng hình giới tính. Khi quá trình di chuyển này bị sai lệch, quần thể tế bào mầm của tuyến sinh dục có thể bị thiếu hụt. Sự di chuyển sai lệch có thể dẫn đến di chuyển lạc chỗ vào các cơ quan khác. Các tế bào mầm lạc chỗ chủ yếu nằm ở hệ thần kinh trung ương, nhưng cũng có thể được tìm thấy ở trung thất, lồng ngực và vùng chậu. Các khối u tế bào mầm có thể phát triển trong các tế bào mầm lạc chỗ khi các tế bào mầm không bị loại bỏ bằng quá trình chết theo chương trình.
Các giai đoạn phát triển của tế bào mầm nữ bao gồm sự khởi đầu của quá trình giảm phân, sự hình thành và phân rã của các ổ tế bào mầm, và sự tập hợp của các noãn bào đơn lẻ thành các nang trứng nguyên thủy. Đối với các tế bào mầm nam, các giai đoạn đặc trưng là di chuyển, nguyên phân và ngừng chu kỳ tế bào. Noãn bào trải qua quá trình giảm phân, trong khi quá trình giảm phân bị ức chế tích cực trong các tế bào mầm nam. Protein liên kết RNA deleted in azoospermia-like (DAZL) dường như cho phép đi vào quá trình giảm phân; biểu hiện DAZL tăng lên từ tuần thứ 9 đến 14 của thai kỳ. Một số protein liên kết RNA, chẳng hạn như LIN28, DAZL và BOLL, được biểu hiện trong quá trình tạo noãn. Một protein khác, meiosis specific with coiled-coil domain (MEIOC) ổn định các mRNA mã hóa các protein liên quan đến giảm phân; sự biểu hiện của nó dường như độc lập với axit retinoic. DAZL ức chế biểu hiện SOX17 hạn chế tính đa năng của tế bào mầm. Do đó, DAZL đóng vai trò kép: nó khởi đầu quá trình giảm phân trong buồng trứng và kìm hãm các yếu tố đa năng.
Buồng trứng thai nhi được đặc trưng bởi một gradient phát triển với sự tồn tại của nhiều quần thể phụ của tế bào mầm ở các giai đoạn phát triển khác nhau; các tế bào mầm biệt hóa hơn nằm ở trung tâm của buồng trứng đang phát triển. Trong buồng trứng đang phát triển, các tế bào mầm ban đầu hình thành các cụm noãn nguyên bào được nối với nhau bằng các cầu nối nội bào. Các noãn nguyên bào được chọn lọc sẽ đi vào quá trình giảm phân và tiến triển qua kỳ đầu I của giảm phân (MPI). Các tế bào soma bao quanh các noãn bào riêng lẻ để tạo thành các nang trứng. Khoảng tuần thứ 16 đến 20 của thai kỳ, các cụm noãn nguyên bào phân rã để tạo thành các nang trứng nguyên thủy. Các noãn bào trong các nang trứng nguyên thủy sẽ ngừng tăng trưởng ở giai đoạn diplotene, kéo dài cho đến khi sự tăng trưởng của noãn bào được khởi động lại khi bắt đầu dậy thì. Số lượng nang trứng lớn nhất tồn tại vào giữa thai kỳ. Từ mức cao nhất là 6,8 triệu noãn bào vào khoảng tháng thứ 5 của thai kỳ, khoảng 2 triệu còn lại khi sinh. Tín hiệu Notch làm trung gian cho các tương tác giữa noãn bào và tế bào hạt, điều chỉnh sự tồn tại của noãn bào và thúc đẩy sự phân rã của các ổ noãn nguyên bào. Môi trường bên trong trong suốt quá trình phát triển buồng trứng của thai nhi có khả năng ảnh hưởng trực tiếp đến khả năng sinh sản của thai nhi nữ đang phát triển (kiểm soát kích thước dự trữ buồng trứng) và chất lượng của các noãn bào mà cuối cùng sẽ trở thành con của cô ấy (bằng cách ảnh hưởng đến mức độ chọn lọc và chết theo chương trình).
Các nang trứng nguyên thủy tạo thành dự trữ buồng trứng. Sau khi sinh, việc tuyển mộ và trưởng thành của các nang trứng nguyên thủy phần lớn được chi phối bởi sự tiết AMH, androgen, hormone tạo hoàng thể (LH) và hormone kích thích nang trứng (FSH). Mất tế bào mầm sớm có thể dẫn đến loạn sản tuyến sinh dục hoặc suy buồng trứng sớm (POI). Sự thoái hóa nang trứng nhanh hơn góp phần vào sự cạn kiệt nang trứng đặc trưng của các tuyến sinh dục dạng dải trong monosomy X. Mặc dù thường không liên quan đến cơ quan sinh dục không rõ ràng, các đột biến trong các gen chi phối sự phát triển của noãn bào và buồng trứng gây ra các DSD đặc trưng bởi dậy thì muộn hoặc POI. Các gen này ảnh hưởng đến quá trình nguyên phân của tế bào mầm, giảm phân của tế bào mầm và sửa chữa DNA.
Ngược lại, sự phát triển của tế bào mầm tinh hoàn ít năng động hơn. Các tế bào Sertoli bao bọc các tế bào mầm để tạo thành các dây sinh tinh vào khoảng tuần thứ 7 đến 9 của thai kỳ trong tuyến sinh dục XY của người. Các tế bào mầm trong tinh hoàn đang phát triển sẽ đi vào trạng thái ngừng nguyên phân cho đến khi bắt đầu dậy thì.
Vào giai đoạn đầu của thai kỳ, các tế bào mầm nguyên thủy trải qua quá trình tái lập trình biểu sinh, làm thay đổi cấu trúc chromatin. Nói cách khác, các dấu ấn di truyền được xóa bỏ và sự methyl hóa DNA giảm xuống tại các đảo CpG, các vị trí bắt đầu phiên mã, các gen và các vùng giữa các gen. Quá trình khử methyl ảnh hưởng đến gần như toàn bộ bộ gen, bao gồm cả nhiễm sắc thể X bất hoạt vào tuần thứ 10 đến 11 của thai kỳ. Trong quá trình giảm phân, cả hai nhiễm sắc thể X đều cần phải hoạt động để cho phép ghép cặp hiệu quả. Sau đó, các allele của mẹ và cha trải qua quá trình in dấu lưỡng hình giới tính, sao cho các noãn bào và tinh trùng đang phát triển được đánh dấu khác nhau, phản ánh các kiểu methyl hóa đặc hiệu theo “nguồn gốc cha mẹ”. Quá trình này xảy ra muộn trong quá trình phát triển của thai nhi ở nam và sau khi sinh ở tế bào mầm nữ. Nghiên cứu các rối loạn phụ thuộc vào nguồn gốc cha mẹ, chẳng hạn như hội chứng Beckwith-Wiedemann, Prader-Willi và Angelman và đái tháo đường sơ sinh đã làm sáng tỏ tầm quan trọng của quá trình in dấu này.
Sự phát triển tuyến thượng thận ở người
Đến ngày thứ 33 sau khi thụ thai, vỏ thượng thận của thai nhi người đã khác biệt với tuyến sinh dục đang phát triển. Do vai trò là nguồn cung cấp dehydroepiandrosterone sulfate (DHEA-S) cho quá trình sinh tổng hợp estrogen của nhau thai, vỏ thượng thận của thai nhi phát triển nhanh chóng. Đến ngày thứ 50 đến 52 sau khi thụ thai, sự biểu hiện của một số enzym tạo steroid, protein điều hòa cấp tính tạo steroid (StAR), 11β-hydroxylase (CYP11B1), 17α-hydroxylase/ 17,20-lyase (CYP17A1), và 21-hydroxylase (CYP21A2) trong vỏ thượng thận của thai nhi đã được chứng minh bằng phương pháp hóa mô miễn dịch. Quá trình sinh tổng hợp cortisol của tuyến thượng thận thai nhi tạm thời đạt đỉnh vào tuần thứ 8 đến 9 của thai kỳ, trùng với sự biểu hiện tạm thời của cả nerve growth factor IB-like (NGFI-B) và 3β-hydroxysteroid dehydrogenase-2 (HSD3B2) ở tuyến thượng thận. Đồng thời, hormone vỏ thượng thận (ACTH) có thể được phát hiện ở thùy trước tuyến yên, cho thấy sự hiện diện của cơ chế phản hồi âm trong tam cá nguyệt thứ nhất. Trong thời gian biệt hóa giới tính nam bắt đầu, cơ chế phản hồi âm này có thể phục vụ để ngăn chặn sự nam hóa của thai nhi nữ nhằm đảm bảo sự biệt hóa giới tính nữ bình thường. Các hồ sơ biểu hiện riêng biệt cho các enzym tạo steroid của tuyến thượng thận thai nhi đã được mô tả với sự khác biệt giữa tam cá nguyệt thứ nhất và thứ hai. Các tế bào chromaffin có nguồn gốc từ hệ thần kinh giao cảm di chuyển vào vỏ thượng thận để tạo thành tủy thượng thận.
Sự phát triển của các cấu trúc sinh dục trong
Ống Wolff có nguồn gốc là ống bài tiết của trung thận và phát triển thành mào tinh, ống dẫn tinh, ống phóng tinh và túi tinh. Ống Müller hay ống cận trung thận có nguồn gốc là một sự lõm vào của biểu mô khoang cơ thể và phát triển thành vòi trứng, tử cung và một phần ba trên của âm đạo.
Các tế bào Sertoli của tinh hoàn đang phát triển tiết ra AMH, còn được gọi là hormone ức chế Müller (MIH). Ở thai nhi người 46,XY, biểu hiện AMH có thể được phát hiện vào tuần thứ 7 của thai kỳ, không phụ thuộc vào sự hiện diện của các tế bào mầm trong tinh hoàn và thúc đẩy sự thoái triển của ống Müller. AMH, một thành viên của họ yếu tố tăng trưởng biến đổi-β (TGF-β), trải qua quá trình phân cắt protein để trở nên có hoạt tính sinh học. AMH liên kết với thụ thể của nó, AMH-RII, trên bề mặt của các tế bào trung mô ống Müller để gây ra sự gia tăng biểu hiện metalloproteinase 2 của chất nền. Kết quả cuối cùng là sự thoái hóa và mất tính toàn vẹn của màng đáy của các tế bào biểu mô và trung mô ống Müller, dẫn đến sự thoái triển của ống Müller.
Biểu hiện AMH được điều hòa chặt chẽ; biểu hiện không phù hợp ở thai nhi 46,XX sẽ dẫn đến bất sản tử cung. Ở thai nhi 46,XX không có cả AMH và testosterone, các dẫn xuất của ống Müller vẫn tồn tại và ống Wolff thoái triển. Khi một thai nhi nữ bị tiếp xúc không phù hợp với AMH (như ở bò freemartin), sự thoái triển ống Müller và sự nam hóa buồng trứng xảy ra. Các ống Müller cặp đôi phát sinh dưới dạng các chỗ lõm vào của biểu mô khoang cơ thể. Khoảng tuần thứ tám của thai kỳ, các ống Müller hợp nhất, tiếp theo là sự thoái hóa của một vách ngăn biểu mô ở đường giữa. Sự tồn tại thay đổi của vách ngăn này có thể dẫn đến các dị tật tử cung. Nhiều yếu tố ảnh hưởng đến sự phát triển của tử cung, cổ tử cung và âm đạo. Các gen HOX xác định chương trình phát triển cho các vùng tử cung riêng biệt; HOXA9 xác định ống tử cung, HOXA10 và HOXA11 xác định tử cung, và HOXA13 xác định âm đạo. Tấm âm đạo xuất hiện trong tuần thứ 11 của thai kỳ thông qua sự tắc nghẽn của ống tử cung-âm đạo có nguồn gốc từ ống Müller và cuối cùng biến mất khi tấm âm đạo dài ra và tạo ống.
Trục hạ đồi-tuyến yên-tuyến sinh dục (HPG) của thai nhi hoạt động vào giữa thai kỳ, với nồng độ testosterone của thai nhi đạt đỉnh vào khoảng tuần thứ 15 đến 16 của thai kỳ. Trước thời điểm này, hCG của nhau thai kích thích sản xuất testosterone bởi các tế bào Leydig của thai nhi. Sự tiết testosterone của các tế bào Leydig của thai nhi ổn định ống Wolff ở thai nhi 46,XY. Các phân tử tín hiệu đặc hiệu cho vùng, chẳng hạn như BMP, homeobox (HOXA10 và HOXA11), yếu tố tăng trưởng biệt hóa 7 (GDF7), relaxin, một thụ thể mồ côi kết hợp với protein G (LGR4), yếu tố tăng trưởng có nguồn gốc từ tiểu cầu A (PDGFA) và thụ thể tương ứng của nó (PDGFRA) ảnh hưởng đến sự phát triển của mào tinh và túi tinh.
Tuyến tiền liệt, một tuyến sinh dục phụ của nam giới, góp phần vào huyết tương dịch tinh và phát triển từ xoang niệu-sinh dục. Sau khi cảm ứng ban đầu phụ thuộc vào testosterone của sự biệt hóa tuyến tiền liệt, sự phát triển tiếp theo liên quan đến các tương tác biểu mô-trung mô dẫn đến sự biệt hóa tế bào và hình thái phân nhánh. Các phân tử tín hiệu cần thiết, FGF, sonic hedgehog (SHH), BMP, HOXA13 và HOXD13, tương tự như những phân tử cần thiết cho sự biệt hóa cơ quan sinh dục ngoài.
Sự phát triển của các cấu trúc sinh dục ngoài
Củ sinh dục, các nếp gấp niệu đạo và các chỗ phồng môi-bìu tạo ra cơ quan sinh dục ngoài. Androgen đóng một vai trò phụ thuộc vào thời gian trong sự hình thành, biệt hóa và tăng trưởng của cơ quan sinh dục ngoài của thai nhi nam vào khoảng tuần thứ 8 đến 14 của thai kỳ, trong cửa sổ lập trình nam hóa (MPW).
Trong giai đoạn này, một dương vật hình trụ 2 mm với các chỗ phồng sinh dục phát triển vào tuần thứ 9 của thai kỳ. Trong dương vật đang phát triển, niệu đạo hình thành bằng cách tạo ống của tấm niệu đạo với sự hợp nhất sau đó để tạo thành niệu đạo dương vật vào tuần thứ 12 đến 14 của thai kỳ. Củ sinh dục phát triển thành thể hang của dương vật, và các nếp gấp môi-bìu hợp nhất để tạo thành bìu. Đến tuần thứ 14, cơ quan sinh dục ngoài rõ ràng là nam tính, ngoài vị trí của tinh hoàn. Sau đó, dương vật phát triển với tốc độ 0,7 mm/tuần từ tuần thứ 14 đến đủ tháng. Sự thiếu hụt androgen hoặc hoạt động của androgen trong MPW làm giảm chiều dài dương vật, một tác dụng không thể được cứu vãn hoàn toàn bằng liệu pháp T sau khi sinh. MPW xác định trước kích thước dương vật tiềm năng, trong khi hoạt động của androgen sau khi sinh là cần thiết để hiện thực hóa tiềm năng bình thường.
Ở thai nhi 46,XX, trong trường hợp không có androgen, các nếp gấp niệu đạo và các chỗ phồng môi-bìu không hợp nhất và phát triển thành môi bé và môi lớn, tương ứng. Củ sinh dục tạo thành âm vật, và việc tạo ống của tấm âm đạo tạo ra phần dưới của âm đạo. Đến tuần thứ 11 của thai kỳ, âm vật nổi bật và các ranh giới bên của rãnh niệu-sinh dục đã tách ra. Sự phát triển tối thiểu của âm vật, môi lớn được xác định rõ, môi bé kém phát triển, và các lỗ âm đạo và niệu đạo đáy chậu riêng biệt có mặt vào tuần thứ 20 của thai kỳ.
Khoảng cách hậu môn-sinh dục
Khoảng cách hậu môn-sinh dục (AGD), khoảng cách từ hậu môn đến củ sinh dục, cho thấy hoạt động của androgen trước khi sinh và, như dự đoán, là lưỡng hình giới tính. Các nhà điều tra đã sử dụng các mốc giải phẫu khác nhau để đánh giá AGD ở người. Salazar-Martinez và các đồng nghiệp đã định nghĩa AGD là từ hậu môn đến chỗ nối bìu-đáy chậu (khoảng cách hậu môn-bìu) ở nam và từ hậu môn đến mép trước (khoảng cách hậu môn-mép) ở nữ; định nghĩa này dường như cải thiện độ tin cậy giữa các người quan sát. Salazar-Martinez và các đồng nghiệp đã báo cáo các phạm vi tiêu chuẩn cho khoảng cách hậu môn-bìu và hậu môn-mép là 21 ± 3,0 mm ở nam và 11 ± 2 mm ở nữ. Ở cả hai giới, AGD tăng lên đến 12 tháng và duy trì kiểu lưỡng hình giới tính. AGD cũng cho thấy mối tương quan dương với chiều dài dương vật khi sinh và với sự gia tăng AGD từ khi sinh đến 3 tháng tuổi. AGD cung cấp một dấu ấn sinh học về sự tiếp xúc sớm của thai nhi với androgen và sự nam hóa. Đo lường AGD có thể được sử dụng như một công cụ chẩn đoán cho sự tiếp xúc với androgen trong tử cung và đánh giá tác động của các chất gây rối loạn nội tiết môi trường tiềm tàng đối với sự phát triển của cơ quan sinh dục ngoài. AGD khi sinh dự đoán khoảng cách hậu môn-sinh dục ở người lớn, nhưng các mối tương quan với các thông số sinh sản của nam giới trưởng thành không nhất quán.
Biệt hóa giới tính của não bộ
Các nghiên cứu lâm sàng cho thấy não bộ có tính lưỡng hình giới tính và testosterone là một hormone nam hóa ở người. Nam giới bị thiếu hụt aromatase biểu hiện hành vi tâm lý tình dục và bản dạng giới nam. Ngược lại, các cá thể 46,XY mắc hội chứng không nhạy cảm androgen hoàn toàn (CAIS) phát triển bản dạng giới nữ. Tuy nhiên, dữ liệu sơ bộ cho thấy sự khác biệt di truyền, độc lập với sự tiếp xúc với steroid giới tính, là cơ sở phân tử cho một số khía cạnh của sự lưỡng hình giới tính của não và các mô không phải tuyến sinh dục khác. Androgen có tác dụng tổ chức và kích hoạt chức năng của hệ thần kinh trung ương. Các tương tác phức tạp giữa các yếu tố di truyền và hormone góp phần vào sự khác biệt giới tính của não người.
Các mô hình chuột
Đối với nhiều bệnh nhân bị DSD, nguyên nhân phân tử cụ thể vẫn chưa được biết. Do đó, việc nghiên cứu các mô hình động vật, đặc biệt là các mô hình chuột chuyển gen, đã hữu ích để đi sâu vào các quá trình liên quan đến sự phát triển giới tính. Nghiên cứu chuột bình thường và chuyển gen đã xác nhận vai trò quan trọng của gen vùng xác định giới tính trên Y (SRY) trong sự biệt hóa nam khi những con chuột XX chỉ mang một đoạn 14 kb của nhiễm sắc thể Y cho thấy một kiểu hình nam. Tuy nhiên, các cơ chế phân tử chịu trách nhiệm cho sự biệt hóa/phát triển giới tính và sự phát triển sau khi làm tổ khác nhau giữa loài gặm nhấm và con người. Bất chấp những hạn chế này, thông tin thu thập được từ các mô hình tiền lâm sàng cung cấp thông tin hữu ích về các cơ chế phân tử của sự phát triển giới tính.
Việc hiểu rõ sự điều hòa gen, các ảnh hưởng biểu sinh và các tương tác protein-protein là cần thiết để làm rõ sự điều hòa của các con đường phát triển giới tính; kiến thức này có thể xác định các yếu tố liên quan đến bệnh nhân bị DSD. Kết quả của các quyết định về số phận tế bào liên quan đến sự đối kháng giữa các chương trình phát triển nam và nữ. Trong giai đoạn lưỡng tiềm năng, cả các gen đặc hiệu cho nam và nữ đều được biểu hiện. Sau đó, sự biểu hiện không-thời gian chính xác của các gen cụ thể diễn ra. Sự phát triển tuyến sinh dục bất thường đã được mô tả ở những con chuột đồng hợp tử về việc xóa các gen liên quan đến sự biệt hóa niệu-sinh dục, tức là, Wt1, Sf1, Emx2, Cbx2/M33, Six1/Six4, và Lim1/Lhx9. Kiểu hình của chuột knockout Wt1 bao gồm tử vong phôi, thất bại trong sự phát triển của tuyến sinh dục và thận, và sự phát triển bất thường của trung biểu mô, tim và phổi. Việc xóa đồng hợp tử của Emx2, một yếu tố phiên mã homeodomain, dẫn đến một kiểu hình gây chết phôi liên quan đến sự vắng mặt của thận, niệu quản, tuyến sinh dục và đường sinh dục. Vì biểu hiện Wt1 ban đầu là bình thường trong mầm trung thận của chuột knockout Emx2, Emx2 có khả năng nằm ở hạ nguồn của Wt1. Điều thú vị là sự phát triển của tuyến thượng thận và bàng quang là bình thường ở chuột knockout Emx2. Mặc dù kiểu hình của các con chuột knockout cụ thể đã được báo cáo, các chi tiết về cơ chế điều hòa chi phối biểu hiện gen thường bị thiếu.
Các cơ chế biểu sinh sửa đổi cấu trúc chromatin để điều hòa thuận nghịch biểu hiện gen theo cách phát triển. Các cơ chế này bao gồm các sửa đổi histone, tức là, methyl hóa/khử methyl, acetyl hóa/khử acetyl, ubiquitin hóa, và methyl hóa DNA. Các protein nhóm polycomb, bao gồm nhiều tiểu đơn vị protein, có chức năng sửa đổi cấu trúc chromatin. Một protein polycomb là Cbx2/M33. Kiểu hình của chuột knockout Cbx2 bao gồm sự đảo ngược giới tính từ nam sang nữ và tuyến sinh dục kém phát triển ở cả hai giới. Mặc dù tinh hoàn nhỏ, sự biểu hiện quá mức của Sry hoặc Sox9 ở chuột knockout Cbx2 đã cứu vãn sự đảo ngược giới tính ở chuột XY. Dựa trên các nghiên cứu động vật có sẵn, Cbx2 điều chỉnh biểu hiện của Sf1/Nr5a1, Sry, Sox9, Dax1, Gata4, Arx, và Dmrt1 ở cả hai giới, và ảnh hưởng đến sự tăng sinh và kích thước của tuyến sinh dục.
Mặc dù các kiểu biểu hiện theo thời gian của SRY ở người và Sry ở chuột khác nhau, việc hiểu rõ các cơ chế điều hòa chịu trách nhiệm cho biểu hiện Sry của chuột đã có liên quan đến SRY của người. Các yếu tố biểu sinh ảnh hưởng đến biểu hiện của gen Sry của chuột. Cả quá trình khử methyl và sửa đổi histone đều cần thiết cho biểu hiện Sry. Histone demethylase JMJD1A ảnh hưởng tích cực đến biểu hiện Sry bằng cách điều chỉnh các dấu H3K9me2; những con chuột thiếu Jmjd1a cho thấy sự đảo ngược giới tính một phần từ nam sang nữ. Một cơ chế khác liên quan đến protein GADD4G, làm trung gian cho quá trình khử methyl DNA và tín hiệu kinase protein được kích hoạt bởi mitogen (MAPK). Chuột đột biến mất chức năng Gadd45g biểu hiện sự đảo ngược giới tính từ nam sang nữ; GADD45G dường như kích hoạt con đường MAPK bằng cách phosphoryl hóa GATA4 để kích hoạt biểu hiện Sry. Chuột bị đột biến mất chức năng Gata4 cho thấy kiểu hình tương tự như chuột Gadd4g với biểu hiện Sry giảm và đảo ngược giới tính XY.
Một cơ chế biểu sinh khác liên quan đến việc điều hòa Sry liên quan đến protein liên kết CREB (CBP, CREBBP, KAT3A) và p300 (EP300, KAT3B). Các protein này là các histone/lysine acetyl-transferase sửa đổi các protein liên quan đến chromatin để điều hòa biểu hiện gen. Chuột bị đột biến mất chức năng biểu hiện sự đảo ngược giới tính từ nam sang nữ. Các protein này có thể hoạt động như các trung tâm mạng lưới tương tác với các protein khác để ảnh hưởng đến phiên mã gen.
Việc nghiên cứu các mô hình chuột đã được sử dụng để tìm hiểu thêm về sự phát triển của ống Wolff và ống Müller. Sự phát triển của biểu mô ống Wolff phụ thuộc vào các tín hiệu từ trung mô ống Wolff. Testosterone của tinh hoàn kích hoạt các thụ thể androgen trung mô để khởi xướng sự trao đổi chéo trung mô-biểu mô, tăng sản xuất yếu tố tăng trưởng biểu bì và đối kháng với COUP-TFII để thúc đẩy sự ổn định của ống Wolff. Các yếu tố liên quan đến sự thoái triển của ống Müller bao gồm AMH, AMHR2, và SMAD; tuy nhiên, cơ chế phân tử mà AMH sử dụng để gây ra sự thoái triển của ống Müller vẫn cần được xác định.
Rối loạn nhiễm sắc thể giới tính
Thể khảm 45,X/46,XY
Hầu hết các rối loạn nhiễm sắc thể giới tính, chẳng hạn như 45,X và các biến thể tế bào học và 47,XXY và các biến thể tế bào học, không liên quan đến sự phát triển bất thường của cơ quan sinh dục ngoài và sẽ không được thảo luận trong chương này (xem Bảng 6.1). Ngược lại, các cá thể có kiểu nhân 45,X/46,XY và 46,XX/46,XY biểu hiện một loạt các kiểu hình. Trong số các cá thể có kiểu nhân 45,X/46,XY, các cấu trúc sinh dục trong và ngoài có thể từ nam bình thường đến không rõ ràng đến nữ. Trong khi các đặc điểm mô học điển hình bao gồm các ống sinh tinh kém phát triển được bao quanh bởi mô đệm buồng trứng gợn sóng, sự biệt hóa tuyến sinh dục có thể từ tinh hoàn bình thường đến tuyến sinh dục dạng dải. Tại thời điểm dậy thì, sự nam hóa có thể xảy ra.
Phần lớn các cá thể được xác định bằng kiểu nhân trước sinh là 45,X/46,XY dường như là nam giới được androgen hóa bình thường; tuy nhiên, các cá thể được chẩn đoán sau sinh có xu hướng có nhiều dấu hiệu lâm sàng hơn. Một kiểu nhân bình thường của tế bào lympho máu ngoại vi ở các cá thể bị loạn sản tuyến sinh dục cho thấy sự hiện diện của thể khảm nhiễm sắc thể giới tính trong (các) tuyến sinh dục. Các cá thể bị DSD do nhiễm sắc thể giới tính do loạn sản tuyến sinh dục có nguy cơ cao phát triển các khối u tuyến sinh dục, chẳng hạn như u nguyên bào sinh dục hoặc u tế bào mầm, vì một tuyến sinh dục bị loạn sản mang nhiễm sắc thể Y có nguy cơ cao bị biến đổi tân sinh. Mặc dù các khối u tuyến sinh dục thường không phát triển cho đến thập kỷ thứ hai của cuộc đời, chúng có thể xảy ra ở độ tuổi trẻ hơn.
Trong một loạt 63 nam giới có kiểu nhân 45,X/46,XY, các đối tượng được phân thành hai nhóm; một nhóm có bất thường sinh dục và nhóm thứ hai có các lý do khác, chẳng hạn như tầm vóc thấp hoặc vô sinh. Các cá thể được xác định bởi các bất thường sinh dục có xu hướng có tỷ lệ phát triển dậy thì tự phát thấp hơn, tầm vóc thấp hơn và khả năng bị tân sinh tế bào mầm cao hơn. Mặc dù hầu hết các tuyến sinh dục được phân loại là tinh hoàn loạn sản, một số tinh hoàn dường như tương đối bình thường với bằng chứng về quá trình sinh tinh. Không có nang trứng nào được quan sát, cho thấy rằng các tuyến sinh dục được dán nhãn là noãn tinh đã bị dán nhãn sai và bao gồm các mô không biệt hóa/giống dải. Dựa trên những phát hiện mô học này, người ta đã gợi ý rằng nguyên nhân của kiểu nhân 45,X/46,XY là sự mất một nhiễm sắc thể Y ở một số tế bào. Sinh tinh khu trú đã được xác định ở khoảng 25% các đối tượng này. Do đó, khi xem xét cắt bỏ tuyến sinh dục do nguy cơ tân sinh, nên xem xét việc bảo tồn khả năng sinh sản.
Các rối loạn liên quan đến rối loạn phát triển giới tính và các bất thường bẩm sinh khác (Bảng 6.2)
ARX (Lissencephaly liên kết X loại 2)
Lissencephaly liên kết X loại 2 được đặc trưng bởi cơ quan sinh dục không rõ ràng, lissencephaly (não nhẵn), co giật khởi phát sớm, không có thể chai, khuyết tật trí tuệ, và rối loạn chức năng vùng dưới đồi, và các đột biến gen Aristaless-related homeobox (ARX). Gen này là một thành viên của họ yếu tố phiên mã homeodomain loại cặp đôi và được lập bản đồ trên Xp21.3. ARX góp phần vào nhiều khía cạnh của sự phát triển não bộ bao gồm sự tạo hình, tăng sinh và di chuyển của tế bào thần kinh, và sự trưởng thành và biệt hóa của tế bào, đặc biệt là sự tạo ra và di chuyển của các tế bào thần kinh GABAergic. Các phát hiện ở chuột knockout Arx cho thấy không có biểu hiện trong buồng trứng đang phát triển, nhưng sự biệt hóa tế bào Leydig bị suy giảm. Dữ liệu có sẵn cho thấy Arx là một chất điều hòa dương tính cho sự biệt hóa của các tế bào Leydig thai nhi ở giai đoạn tiền thân.
Bảng 6.2. Các hội chứng rối loạn liên quan đến rối loạn phát triển giới tính
| Gen (Băng NST) | Hội chứng | Kiểu nhân | Khiếm khuyết di truyền | Kiểu di truyền | Bất thường bẩm sinh đặc trưng |
| ARX (Xp21.3) | Hydranencephaly với CƠ QUAN SINH DỤC BẤT THƯỜNG (OMIM #300215) | XY | đột biến điểm, mất/lặp đoạn | Liên kết X | lissencephaly, không có thể chai, co giật khó chữa khởi phát sớm, thân nhiệt không ổn định |
| ATRX (Xq13.1) | Hội chứng chậm phát triển trí tuệ alpha-thalassemia (OMIM #301040) | XY | đột biến điểm | Liên kết X | alpha-thalassemia, chậm phát triển trí tuệ nặng, và các bất thường sinh dục |
| CDKN1C (11p15) | Hội chứng IMAGe (OMIM #614732) | XY | đột biến điểm | Trội NST thường, truyền từ mẹ | chậm tăng trưởng trong tử cung (IUGR), loạn sản hành xương, suy thượng thận |
| CHD7 (8q12.2) | Hội chứng CHARGE (OMIM#214800) | XY, XX | đột biến điểm (70%), mất/lặp đoạn (hiếm) | Trội NST thường | coloboma, dị tật tim, hẹp cửa mũi sau, bất thường tai |
| DHCR7 (11q13.4) | Hội chứng Smith-Lemli-Opitz (OMIM #270400) | XY | đột biến điểm (96%), mất/lặp đoạn (4%) | Lặn NST thường | đặc điểm khuôn mặt đặc trưng, thừa ngón sau trục, dính ngón chân 2-3, hở hàm ếch |
| EMX2 (10q26.11) | (OMIM #600035) | XY | vi mất đoạn | đảo ngược giới tính, thận, chậm phát triển, schizencephaly | |
| GLI3 (7p14.1) | Hội chứng Pallister-Hall (OMIM #146510) | XY, XX | đột biến điểm (20%–25%), mất/lặp đoạn (hiếm) | Trội NST thường | hamartoma vùng dưới đồi, thừa ngón giữa trục, nắp thanh quản chẻ đôi |
| HHAT (1q32.2) | loạn sản sụn (PMID: 24784881) | XY | đột biến điểm | Lặn NST thường | lùn với loạn sản sụn toàn thể |
| HOXA13 (7p15.2) | Hội chứng tay-chân-sinh dục (OMIM#140000) | XY, XX | mở rộng polyalanine (60%), đột biến điểm (35%), mất/lặp đoạn (2%–5%) | Trội NST thường | ngón tay cái và ngón chân cái ngắn, tử cung hai sừng |
| HSD17B4 ᵃ (5q23.1) | Hội chứng Perrault 1 (OMIM #233400) | XX | đột biến điểm | Lặn NST thường | điếc thần kinh giác quan |
| KAT6B (10q22.2) | Hội chứng xương bánh chè-sinh dục (OMIM #606170) | XY | đột biến điểm, mất/lặp đoạn (hiếm) | Trội NST thường | thiểu sản xương bánh chè, bàn chân khoèo, đầu nhỏ, co cứng gập, bất sản thể chai |
| POR (7q11.2) | Hội chứng Antley-Bixler (OMIM#201750) | XY, XX | đột biến điểm, mất/lặp đoạn (hiếm) | Lặn NST thường | dính khớp quay-cánh tay, thiểu sản giữa mặt, hẹp cửa mũi sau, co cứng khớp |
| RSPO1 (1p34.3) | Dày sừng lòng bàn tay-chân/đảo ngược giới tính (OMIM #610644) | XX | đột biến điểm | Trội NST thường | dày sừng da lòng bàn tay-chân, khuynh hướng ung thư biểu mô tế bào vảy của da |
| SAMD9 (7q21.2) | Hội chứng MIRAGE (OMIM #610456) | XY | đột biến điểm | Trội NST thường | hội chứng loạn sản tủy, IUGR, suy thượng thận, tiêu chảy mạn tính, nhiễm trùng phổi |
| SOX9 (17q24.3) | Loạn sản campomelic (OMIM #114290) | XY | đột biến điểm (90%), chuyển vị (5%), mất/lặp đoạn (2%) | Trội NST thường | chuỗi Pierre Robin, hở hàm ếch, chi ngắn cong, 11 cặp xương sườn |
| TBX3 (12q24.1) | Hội chứng trụ-vú (OMIM #181450) | XX | đột biến điểm | Trội NST thường | khiếm khuyết chi bên trụ, thiểu sản tuyến vú, bất thường răng |
| TP63 (3q28) | Hội chứng chi-vú (OMIM #603543) | XX | đột biến điểm | Trội NST thường | bất thường bàn tay/bàn chân, tuyến vú và núm vú |
| TSPYL1 (6q22.1) | SIDDT (OMIM #608800) | XY | đột biến điểm | Lặn NST thường | nhịp tim chậm, hạ thân nhiệt, co thắt thanh quản, co thắt phế quản |
| WT1 (11p13) | Các hội chứng Denys-Drash, Frasier, và Meacham (OMIM #194080) | XY, XX | đột biến điểm | Trội NST thường | khối u Wilms, bệnh thận, u nguyên bào sinh dục, thoát vị hoành bẩm sinh |
| ZEB2 (2q22.3) | Hội chứng Mowat-Wilson (OMIM#235730) | XY | đột biến điểm (80%–85%), mất/lặp đoạn (15%–17%), chuyển vị (1%–2%) | Trội NST thường | bệnh Hirschsprung, dị tật tim bẩm sinh, bất sản thể chai, mắt nhỏ, bất thường Axenfeld |
Trội NST thường (AD), Lặn NST thường (AR), Chậm tăng trưởng trong tử cung (IUGR), Hội chứng đột tử ở trẻ sơ sinh với loạn sản tinh hoàn (SIDDT).
CDKN1C (Hội chứng IMAGe)
Hội chứng IMAGe được đặc trưng bởi chậm tăng trưởng trong tử cung, loạn sản hành xương, thiểu sản tuyến thượng thận, tinh hoàn ẩn và dương vật nhỏ. Các bất thường sinh dục dường như chỉ giới hạn ở nam giới bị ảnh hưởng. Cả các trường hợp lẻ tẻ và gia đình đều đã được mô tả. Các đột biến sai nghĩa dị hợp tử trong gen CDKN1C đã được phát hiện trong một số trường hợp. Gen này nằm ở nhiễm sắc thể 11p15.5, mã hóa một protein 316 axit amin, p57 Kip2, điều hòa âm tính sự tăng sinh tế bào và ức chế sự tiến triển của chu kỳ tế bào. Gen này nằm trong một locus di truyền phức tạp chứa một số gen bao gồm, IGF2 và H19, chịu sự điều chỉnh in dấu thông qua các yếu tố tác động cis; CDKN1C được biểu hiện từ mẹ. Các đột biến liên quan đến hội chứng IMAGe hoặc Silver-Russell dường như khu trú ở miền liên kết kháng nguyên nhân tế bào tăng sinh, dẫn đến tăng chức năng với sự ức chế quá mức sự tăng trưởng và biệt hóa. Điều kỳ lạ là, các đột biến mất chức năng ảnh hưởng đến miền liên kết kinase phụ thuộc cyclin của protein này lại liên quan đến các hội chứng phát triển quá mức, chẳng hạn như hội chứng Beckwith-Wiedemann.
CHD7 (Hội chứng CHARGE)
Hội chứng CHARGE liên quan đến các đột biến trong gen protein liên kết DNA helicase chromodomain-7 (CHD7). Các đặc điểm của hội chứng này bao gồm coloboma mắt, dị tật tim, hẹp cửa mũi sau, tầm vóc thấp, bất thường sinh dục, bất thường tai và mất thính giác. Dương vật nhỏ và tinh hoàn ẩn được tìm thấy ở nam giới. Suy sinh dục do suy gonadotropin có thể xảy ra. Mặc dù thường là lẻ tẻ, các trường hợp di truyền trội trên nhiễm sắc thể thường đã được mô tả. Gen CHD7, nằm ở nhiễm sắc thể 8q12.1-q12.2, mã hóa một protein lớn tham gia vào việc tái tạo chromatin và phiên mã.
DHCR7 (Hội chứng Smith-Lemli-Opitz)
Một số enzym xúc tác quá trình chuyển đổi lanosterol thành cholesterol. Hoạt động giảm của các enzym này dẫn đến thiếu hụt cholesterol. Enzym, 7-dehydrocholesterol reductase, được mã hóa bởi gen 7-dehydrocholesterol reductase (DHCR7), nằm ở nhiễm sắc thể 11q12-q13, xúc tác bước cuối cùng trong quá trình sinh tổng hợp cholesterol. Smith-Lemli-Opitz (SLO) là một rối loạn di truyền lặn trên nhiễm sắc thể thường gây ra bởi các đột biến mất chức năng của DHCR7. Như dự đoán, nồng độ 7-dehydroxycholesterol tăng cao là cần thiết để xác nhận chẩn đoán.
Các đặc điểm lâm sàng bao gồm bất thường niệu-sinh dục, chậm phát triển trí tuệ, chậm phát triển, bất thường khuôn mặt, chậm phát triển vận động, bất thường hành vi và các triệu chứng tự kỷ. Các bất thường niệu-sinh dục phổ biến nhất bao gồm đảo ngược giới tính từ nam sang nữ, lỗ tiểu lệch thấp và tinh hoàn ẩn. Các bất thường trên khuôn mặt bao gồm đầu nhỏ, mũi rộng, lỗ mũi hếch, cằm nhỏ, cổ ngắn và hở hàm ếch. Ngón tay cái ngắn, dính ngón chân thứ hai và thứ ba, và thừa ngón sau trục là phổ biến. Các dị tật hệ thần kinh trung ương bao gồm bất thường vách trong suốt, bất sản thể chai, hoặc holoprosencephaly trong các trường hợp nặng.
Trẻ sơ sinh thường bị giảm trương lực cơ, khó ăn và chậm phát triển. Có thể cần nuôi ăn qua ống thông do tăng cân kém. Suy giảm sinh tổng hợp cholesterol có thể liên quan đến suy thượng thận. Tuy nhiên, suy thượng thận rõ ràng là không phổ biến. Tuy nhiên, suy thượng thận, đặc biệt là trong khi bị stress, có thể xảy ra. Do đó, chức năng trục hạ đồi-tuyến yên-thượng thận nên được đánh giá và liều glucocorticoid khi stress có thể có lợi. Mặc dù hiệu quả vẫn chưa rõ ràng, việc bổ sung cholesterol trong chế độ ăn thường được kê đơn.
Các đột biến DHCR7 liên quan đến giảm cholesterol và tích tụ các chất trung gian sterol ở phía trước của enzym bị khiếm khuyết. Nồng độ cholesterol giảm dẫn đến nồng độ steroid giảm, với sự giảm sau đó trong quá trình sinh tổng hợp glucocorticoid, mineralocorticoid và steroid giới tính. Ngoài việc là tiền chất cho vitamin D và axit mật, cholesterol còn tham gia vào tín hiệu SHH và màng tế bào. Dữ liệu có sẵn cho thấy sự tích tụ của các chất trung gian sterol chứ không phải là sự thiếu hụt cholesterol dẫn đến một số phát hiện lâm sàng. Tín hiệu SHH bị suy giảm góp phần vào các dị tật, chẳng hạn như bất sản thể chai, holoprosencephaly và thừa ngón sau trục. Sử dụng các tế bào gốc đa năng cảm ứng từ các đối tượng bị SLOS, dữ liệu cho thấy sự tích tụ của 7-dehydrocholesterol và đồng phân của nó, 8-dehydrocholesterol, chứ không phải là sự thiếu hụt cholesterol, có khả năng góp phần vào các biểu hiện thần kinh.
Chẩn đoán trước sinh có thể được thực hiện bằng cách đo nồng độ 7-dehydrocholesterol trong nước ối. Phụ nữ mang thai bị ảnh hưởng có nồng độ estriol huyết tương thấp; nồng độ 16α-hydroxyestrogen trong máu tăng cao; và nồng độ dehydrosteroid C18, C19, và C21 không bão hòa Δ7 và Δ8 trong nước tiểu tăng cao, có lẽ là do sản xuất cholesterol của thai nhi bị suy giảm. Tỷ lệ mắc SLO được xác nhận về mặt sinh hóa ước tính là 1/20.000 đến 1/60.000 ca sinh sống. Tỷ lệ dị hợp tử cho các đột biến DHCR7 cao một cách đáng ngạc nhiên. Vì tỷ lệ mắc SLO ở tuần thứ 16 của thai kỳ tương đương với tỷ lệ mắc khi sinh, nên có khả năng xảy ra tình trạng mất thai sớm và/hoặc giảm khả năng sinh sản của các cặp vợ chồng mang gen.
Gen EMX2
Một bệnh nhân nam có DSD 46,XY, một thận duy nhất và khuyết tật trí tuệ; anh ta có một vi mất đoạn nhỏ liên quan đến EMX2.
GLI3 (Hội chứng Pallister-Hall)
Hội chứng Pallister-Hall được đặc trưng bởi các bất thường sinh dục, hamartoma vùng dưới đồi, thừa ngón sau trục và hậu môn không thủng. Lỗ tiểu lệch thấp, dương vật nhỏ và bìu chẻ đôi hoặc kém phát triển được mô tả ở bệnh nhân XY trong khi ứ dịch tử cung-âm đạo và/hoặc bất sản âm đạo được mô tả ở bệnh nhân XX. Rối loạn di truyền trội trên nhiễm sắc thể thường này là do các đột biến trong gen Gli-Kruppel Family Member 3 (GLI3), nằm ở nhiễm sắc thể 7p14.1. Protein này hoạt động như cả một chất kích hoạt phiên mã và một chất kìm hãm phiên mã của các mục tiêu hạ nguồn trong con đường SHH. Hội chứng Pallister-Hall thường do các đột biến dịch khung/vô nghĩa và đột biến nối nằm ở một phần ba giữa của gen, được dự đoán sẽ tạo ra một protein kìm hãm chức năng bị cắt ngắn. Điều kỳ lạ là, các đột biến bên ngoài vùng này có liên quan đến hội chứng Greigs cephalopolysyndactyly. Dữ liệu tiền lâm sàng sử dụng các mô hình knockout ở chuột cho thấy sự nảy chồi niệu quản bất thường, ứ nước niệu quản, ứ nước thận và thiểu sản thận.
HHAT (Hội chứng Nivelon-Nivelon-Mabille)
Hội chứng Nivelon-Nivelon-Mabille là một rối loạn cực kỳ hiếm gặp được đặc trưng bởi tầm vóc thấp nguyên thủy, loạn sản sụn toàn thể, đầu nhỏ, ngón ngắn, dị dạng khuôn mặt và đảo ngược giới tính XY. Hội chứng này là do các đột biến mất chức năng di truyền lặn trên nhiễm sắc thể thường trong gen O-acyl-transferase hedgehog acyl-transferase (HHAT) được lập bản đồ trên nhiễm sắc thể 1q32.2. Protein HHAT palmitoyl hóa miền N-terminal của các protein hedgehog. Các protein hedgehog, SHH, DHH và Indian hedgehog (IHH), đóng vai trò chính trong việc tạo hình và biệt hóa phôi. Dựa trên kiểu hình của chuột knockout, việc mất Hhat dẫn đến loạn sản tinh hoàn đặc trưng bởi kích thước tinh hoàn giảm, dây tinh hoàn giảm và gần như hoàn toàn không có tế bào Leydig của thai nhi. Điều kỳ lạ là, kiểu hình được mô tả cho bệnh nhân XY có đột biến DHH và chuột knockout Dhh giống với kiểu hình được báo cáo cho bệnh nhân HHAT và chuột knockout Hhat.
HOXA13 (Hội chứng Tay-Chân-Sinh dục)
Các gen HOX là một nhóm các yếu tố phiên mã liên quan đến sự phát triển và tạo hình phôi. Các gen này được tổ chức thành các cụm đặc trưng bởi các kiểu biểu hiện không-thời gian cụ thể. Hội chứng tay–chân–sinh dục (HFGS) (OMIM #140000) là một rối loạn di truyền trội trên nhiễm sắc thể thường, hiếm gặp, liên quan đến các đột biến hoặc mất đoạn của gen HOXA13 trên nhiễm sắc thể 7p15. Hội chứng này được đặc trưng bởi các bất thường niệu-sinh dục bẩm sinh và các bất thường xương biến đổi. Các bất thường xương chủ yếu ảnh hưởng đến bàn tay và bàn chân, chẳng hạn như ngón tay cái và ngón chân cái ngắn, các đốt giữa ngắn lại, ngón út vẹo, và sự hợp nhất của các đốt xa và giữa của các ngón chân. Mặc dù khoảng 50% số người bị ảnh hưởng có bất thường niệu-sinh dục, nhiều người vẫn có khả năng sinh sản bình thường. Nam giới có thể bị lỗ tiểu lệch thấp hoặc tinh hoàn ẩn. Nữ giới có thể bị hợp nhất ống Müller không hoàn toàn hoặc tử cung hai sừng liên quan đến nguy cơ sẩy thai tự nhiên, sinh non hoặc thai chết lưu tăng lên. Trong một số trường hợp, kiểu hình xương có thể nhẹ. Các bệnh đi kèm khác bao gồm nhiễm trùng đường tiết niệu mạn tính, lỗ niệu quản lạc chỗ, viêm bể thận mạn tính và suy thận.
KAT6B (Hội chứng Xương bánh chè-Sinh dục)
Hội chứng xương bánh chè-sinh dục là một rối loạn hiếm gặp được đặc trưng bởi loạn sản xương, co cứng gập, bất thường sinh dục, dị tật sọ mặt và chậm phát triển liên quan đến các đột biến gen lysine acetyltransferase 6 (KAT6B). Các đặc điểm xương bao gồm xương bánh chè kém phát triển hoặc không có, xương thái dương phẳng và ngón ngắn. Các bất thường sinh dục bao gồm môi bé kém phát triển, phì đại âm vật, bìu kém phát triển và tinh hoàn ẩn. Các bất thường tim, ứ nước thận và chậm phát triển đã được mô tả. Gen này nằm ở nhiễm sắc thể 10q22.2 và chứa 20 exon. Protein KAT6B là một thành phần của phức hợp histone H3 acetyltransferase và có chức năng như một histone acetyltransferase. Protein KAT6B có chức năng như một chất điều chỉnh chromatin bằng cách xúc tác quá trình acetyl hóa các lysine cụ thể. Hầu hết các đột biến là đột biến de novo. Hội chứng Say-Barber-Biececker-Young-Simpson cũng liên quan đến các đột biến KAT6B; các đặc điểm khuôn mặt cụ thể bao gồm blepharphimosis/sụp mí và một khuôn mặt bất động giống mặt nạ phân biệt các hội chứng này.
Gen NR2F2
Gen này mã hóa yếu tố phiên mã promoter ngược dòng của ovalbumin gà (COUP-TF2), một thụ thể hạt nhân. Protein này dường như có chức năng như một yếu tố ủng hộ buồng trứng và chống tinh hoàn. Các đột biến trong gen này có liên quan đến một DSD 46,XX hội chứng được đặc trưng bởi sự nam hóa sinh dục, bệnh tim bẩm sinh và các phát hiện soma khác nhau. Các đột biến dịch khung đã được báo cáo ở ba bệnh nhân 46,XX được xác định bởi cơ quan sinh dục không rõ ràng. Cả ba bệnh nhân đều bị bệnh tim bẩm sinh. Một trẻ sơ sinh chết vì tim trái thiểu sản; không có mô học tuyến sinh dục. Hai bệnh nhân còn lại mang đột biến de novo c.97_103delCCGCCCG được dự đoán sẽ tạo ra đột biến dịch khung, p.Pro33Alafs*77; họ cũng có hội chứng blepharophimosis ptosis epicanthus inversus (BPES) và tử cung. Một bệnh nhân bị rối loạn noãn tinh; mô học tuyến sinh dục không có sẵn cho bệnh nhân còn lại.
Các nghiên cứu hóa mô miễn dịch được thực hiện trên buồng trứng người cho thấy sự nhuộm màu COUP-TF2 dương tính ở các khu vực mô đệm âm tính với FOXL2 phù hợp với việc COUP-TF2 có chức năng như một yếu tố ủng hộ buồng trứng và chống tinh hoàn trong các tuyến sinh dục nữ đang phát triển. Các nghiên cứu tiền lâm sàng ở chuột cho thấy phôi cái thiếu Coup-tfII sở hữu cả ống Wolff và ống Müller; việc duy trì ống Wolff độc lập với androgen và có khả năng là do tín hiệu kinase điều hòa tín hiệu ngoại bào được phosphoryl hóa tăng cường trong biểu mô ống Wolff. Những dữ liệu này cho thấy COUP-TFII tích cực thúc đẩy sự thoái triển của ống Wolff ở phôi cái.
RSPO1
Gen R-spondin 1 (RSPO1) mã hóa một protein miền giống furin tiết ra giúp ổn định β-catenin trong con đường tín hiệu Wnt. Các đột biến trong gen này, nằm ở nhiễm sắc thể 1p34, có liên quan đến sự đảo ngược giới tính 46, XX. Gen này ban đầu được xác định bằng cách điều tra ở những cá thể bị dày sừng lòng bàn tay-chân với ung thư biểu mô tế bào vảy của da và đảo ngược giới tính trong một gia đình. Các cá thể XX bị ảnh hưởng, bị đảo ngược giới tính không có cấu trúc Müller. Một cá thể 46,XY, đồng hợp tử về các đột biến RSPO1, đã có hai con. Một cá thể XX bị rối loạn noãn tinh và dày sừng da lòng bàn tay-chân được phát hiện là đồng hợp tử về một đột biến nối trong gen RSPO1. Ngoài sự đảo ngược giới tính và dày sừng lòng bàn tay-chân, các đặc điểm được báo cáo bao gồm mắt nhỏ và loạn dưỡng móng.
SAMD9 (Hội chứng MIRAGE)
Hội chứng Mirage được đặc trưng bởi loạn sản tủy, nhiễm trùng, chậm tăng trưởng trong tử cung (IUGR), thiểu sản tuyến thượng thận, đảo ngược giới tính từ nam sang nữ XY, và bệnh đường ruột. Bệnh phổi, nhiễm trùng nặng và loạn sản tủy góp phần gây tử vong trong 2 năm đầu đời. Rối loạn này có liên quan đến các đột biến dị hợp tử trong gen sterile α motif domain–containing protein 9 (SAMD9) nằm ở nhiễm sắc thể 7q21.2. Protein 1589 axit amin được biểu hiện cao ở tuyến thượng thận của thai nhi; hóa mô miễn dịch cho thấy sự định vị của SAMD9 trong tế bào chất của các tế bào vùng dứt khoát và vùng thai nhi của tuyến thượng thận, cùng vị trí với các tế bào dương tính với biểu hiện NR5A1. SAMD9 được coi là một gen ức chế khối u; các đột biến là đột biến tăng chức năng, dẫn đến IUGR và hậu quả đối với sự biệt hóa của tuyến thượng thận và tinh hoàn. Điều kỳ lạ là, các đột biến thứ hai, chẳng hạn như monosomy mắc phải đối với nhiễm sắc thể 7, làm thay đổi kiểu hình và kéo dài sự sống. Buonocore và các đồng nghiệp cho rằng SAMD9 cần thiết để kiểm soát sự cân bằng giữa tăng sinh tế bào và biệt hóa tế bào; việc kích hoạt các đột biến SAMD9 can thiệp vào sự tăng sinh tế bào dẫn đến các mô kém phát triển.
SOX9
SOX9 là một thành viên của họ gen miền HMG liên quan đến SRY nằm ở nhiễm sắc thể 17q24.3-17q25.1 mã hóa một protein 508 axit amin. Các đột biến mất chức năng dị hợp tử của SOX9 có liên quan đến bệnh lùn campomelic di truyền trội trên nhiễm sắc thể thường và đảo ngược giới tính từ nam sang nữ. Các đặc điểm của bệnh lùn campomelic bao gồm cong bẩm sinh của các xương dài, xương vai kém phát triển, 11 cặp xương sườn, ngực hẹp, trật khớp háng bẩm sinh và bàn chân khoèo. Các đặc điểm trên khuôn mặt bao gồm cằm nhỏ, hở hàm ếch, đầu to, sống mũi phẳng và tai thấp dị dạng. Khi các dị tật xương nghiêm trọng, khả năng sống sót sau khi sinh có thể bị hạn chế. Các thai nhi 46,XY bị ảnh hưởng biểu hiện sự đảo ngược giới tính, với sự biệt hóa cơ quan sinh dục ngoài, từ không rõ ràng đến nữ. Loạn sản tuyến sinh dục và sự tồn tại của các dẫn xuất ống Müller là điển hình. Sự không đồng nhất về kiểu hình với các kiểu hình khác nhau, bao gồm DSD noãn tinh và đảo ngược giới tính hoàn toàn, đã được mô tả ở các anh chị em bị ảnh hưởng. Loạn sản acampomelic đã được mô tả trong đó các bất thường chi điển hình không có. Các bệnh nhân bị lùn acampomelic và đảo ngược giới tính đã được báo cáo. Loạn sản campomelic gia đình liên quan đến một sự mất đoạn ở thượng nguồn của SOX9 đã được mô tả ở một người mẹ và một đứa trẻ 46,XY có cơ quan sinh dục ngoài nữ, tử cung bình thường và tuyến sinh dục dạng dải.
Các đột biến SOX9 có thể ảnh hưởng đến ái lực liên kết DNA, khả năng uốn cong DNA, nhập khẩu hạt nhân, chuyển hoạt và xuất khẩu hạt nhân. Suy giảm đơn bội là cơ chế chịu trách nhiệm cho nhiều hậu quả của các đột biến SOX9. Thể khảm tế bào soma, đột biến dòng mầm de novo và các sự kiện chuyển đổi gen nguyên phân đã được mô tả. Các đột biến nằm ở thượng nguồn của SOX9 trong các vùng điều hòa đã được xác định ở những bệnh nhân có các mức độ đảo ngược giới tính khác nhau. Các lặp đoạn của locus eSR-A có liên quan đến rối loạn tinh hoàn 46,XX và rối loạn noãn tinh 46,XX, trong khi các mất đoạn đã được xác định ở những bệnh nhân bị đảo ngược giới tính 46,XY.
TBX3 (Hội chứng Trụ-Vú)
Hội chứng Trụ-Vú được đặc trưng bởi tinh hoàn ẩn, dương vật nhỏ, thiểu sản/bất sản vú, thiểu sản/bất sản núm vú, dậy thì muộn, không có lông nách và các bất thường xương. Các bất thường xương bao gồm thiểu sản xương cánh tay và xương trụ, thiếu ngón, thừa ngón và các bất thường ngón tay. Gen TBX3 đóng một vai trò trong sự phát triển của trục lưng-bụng của chi. Các đặc điểm nội tiết bổ sung bao gồm suy sinh dục do suy gonadotropin, tầm vóc thấp, thiếu hụt hormone tăng trưởng và béo phì. Các nghiên cứu chẩn đoán hình ảnh thần kinh đã xác định các dị tật tuyến yên và não.
WT1
Hội chứng Denys-Drash được đặc trưng bởi các bất thường niệu-sinh dục, khối u Wilms, bệnh thận và các đột biến gen ức chế khối u Wilms (WT1). Trong số các cá thể 46,XY bị ảnh hưởng, cơ quan sinh dục ngoài có thể từ không rõ ràng đến nữ bình thường. Sự biệt hóa sinh dục trong thay đổi do sự phát triển và thoái triển không nhất quán của cấu trúc Wolff và/hoặc Müller. Thông thường, bệnh thận bắt đầu trong vài năm đầu đời, biểu hiện bằng protein niệu, và dẫn đến suy thận giai đoạn cuối do xơ cứng màng treo cầu thận khu trú hoặc lan tỏa. Thông thường, các tuyến sinh dục thường bị loạn sản ở các cá thể 46,XY. Các cá thể 46,XX bị ảnh hưởng thường có sự phát triển cơ quan sinh dục ngoài nữ bình thường.
Tuy nhiên, một đột biến WT1 mới đã được báo cáo ở một cá thể XX được xác định khi sinh do phì đại âm vật và một lỗ đáy chậu duy nhất. Cô được phát hiện có một ống tử cung phải chưa trưởng thành; mô học tuyến sinh dục cho thấy tinh hoàn hai bên với các ống sinh tinh chứa chủ yếu là tế bào Sertoli và hiếm tế bào mầm. Kiểu nhân của cả hai tuyến sinh dục đều là XX. Phân tích di truyền cho thấy một biến thể dịch khung de novo ở exon 10, mã hóa ngón tay DNA thứ tư. Biến thể này được dự đoán sẽ tạo ra một đột biến có hại p.Arg485Glyfs*14, dường như là một đột biến tăng chức năng làm tăng ái lực liên kết DNA, thúc đẩy sự biểu hiện quá mức của NR5A1 và điều hòa tăng cường SOX9, và cuối cùng dẫn đến sự phát triển của tinh hoàn.
Gen ức chế khối u Wilms (WT1), nằm ở nhiễm sắc thể 11p13, đóng một vai trò quan trọng trong cả sự biệt hóa của thận và tuyến sinh dục. Thông qua việc nối xen kẽ, nhiều vị trí bắt đầu dịch mã và chỉnh sửa RNA sau phiên mã, hơn 30 dạng đồng phân được tạo ra từ một gen này. Miền carboxyl cuối của protein WT1 chứa bốn ngón tay kẽm đóng vai trò là miền liên kết axit nucleic.
Hai dạng đồng phân chính khác nhau bởi sự bao gồm hoặc loại trừ ba axit amin, lysine, threonine và serine (KTS), giữa miền ngón tay kẽm thứ ba và thứ tư. Các nghiên cứu định vị dưới nhân đã chỉ ra rằng dạng –KTS chủ yếu đồng định vị với các yếu tố phiên mã và ưu tiên liên kết với DNA, trong khi dạng + KTS chủ yếu đồng định vị với các yếu tố nối và đóng vai trò trong quá trình xử lý RNA. Tỷ lệ của các dạng đồng phân + KTS/–KTS dường như được điều hòa chặt chẽ. Do đó, tùy thuộc vào bối cảnh tế bào, WT1 có thể hoạt động như một chất kích hoạt phiên mã, một chất kìm hãm phiên mã, hoặc một chất ức chế khối u. WT1 đóng một vai trò trong sự cân bằng giữa các quá trình chuyển đổi trung mô-biểu mô.
Bệnh nhân bị đột biến WT1 biểu hiện sự không đồng nhất về kiểu hình. Mặc dù khối u Wilms và các bất thường niệu-sinh dục có thể liên quan đến các mất đoạn dị hợp tử của WT1, chỉ 6% đến 15% các khối u Wilms lẻ tẻ có liên quan đến các đột biến WT1. Các mất đoạn dị hợp tử ở nhiễm sắc thể 11p13 có thể là một phần của một hội chứng mất gen liền kề được gọi là hội chứng WAGR (khối u Wilms, vô mống mắt, bất thường niệu-sinh dục, u nguyên bào sinh dục và chậm phát triển trí tuệ). Nói chung, các đột biến sai nghĩa ở các exon 6–9 có liên quan đến loạn sản tuyến sinh dục nặng và bệnh thận khởi phát sớm. Dạng (–) KTS dường như có tác dụng bảo vệ khỏi sự phát triển của khối u Wilms, trong khi các đột biến nằm ở miền kìm hãm N-terminal có liên quan đến sự phát triển của khối u Wilms.
Hội chứng Frasier được đặc trưng bởi loạn sản tuyến sinh dục, bệnh cầu thận tiến triển, và nguy cơ cao bị u nguyên bào sinh dục. Khối u Wilms cực kỳ hiếm gặp trong hội chứng Frasier. Tổn thương thận điển hình là xơ hóa cầu thận khu trú. Phần lớn các trường hợp có liên quan đến một đột biến điểm WT1 cụ thể trong intron 9, liên quan đến việc nối bị thay đổi và giảm lượng dạng đồng phân + KTS.
Hội chứng Meacham được đặc trưng bởi các bất thường sinh dục, dị tật tim bẩm sinh tím tái và thiểu sản phổi thứ phát do các bất thường cơ hoành. Ở trẻ sơ sinh 46,XY, phổ các bất thường sinh dục trong kéo dài từ sự hiện diện của tử cung đến một túi âm đạo bịt kín. Cơ quan sinh dục ngoài đã được mô tả là từ nữ bình thường đến không rõ ràng.
XH2 (Hội chứng ATRX)
Hội chứng ATRX (α-thalassemia, chậm phát triển trí tuệ, protein liên kết X) là một rối loạn liên kết X được đặc trưng bởi alpha-thalassemia nhẹ, chậm phát triển trí tuệ nặng và các bất thường sinh dục do các đột biến trong gen XH2 nằm ở nhiễm sắc thể Xq13.3. Các bất thường niệu-sinh dục, bao gồm cơ quan sinh dục không rõ ràng, tinh hoàn ẩn, bìu kém phát triển, lỗ tiểu lệch thấp, bìu khăn choàng và dương vật nhỏ xảy ra ở khoảng 80% bệnh nhân. Các đặc điểm bổ sung bao gồm tầm vóc thấp, chậm phát triển tâm thần vận động, đầu nhỏ, co giật, bàn chân khoèo, và các vấn đề về đường tiêu hóa. Khuôn mặt được mô tả là thô với thiểu sản giữa mặt, mũi ngắn và răng cửa cách xa nhau. Các thể vùi hemoglobin H liên quan đến alpha-thalassemia có thể được chứng minh trên các phết máu ngoại vi nhuộm xanh cresyl rực rỡ. Thông thường, các cấu trúc ống Wolff có mặt, trong khi các cấu trúc ống Müller và tế bào mầm không có. Các mối tương quan kiểu hình-kiểu gen không nhất quán. Hầu hết các trường hợp được di truyền từ các bà mẹ mang gen và hầu hết, nhưng không phải tất cả, các phụ nữ mang gen cho thấy sự bất hoạt ưu tiên của nhiễm sắc thể X mang đột biến ATRX.
Rối loạn này, còn được gọi là hội chứng Carpenter-Waziri, là do các đột biến trong gen ATRX (còn được gọi là XH2 hoặc XHP) nằm ở Xq13.3. Protein ATRX là một thành viên của họ DNA helicase SWI/SNF và bao gồm ba miền: một miền N-terminal chứa một miền giống homeodomain thực vật (PHD), một họa tiết cuộn-xoắn, và một miền helicase C-terminal. Các bất thường niệu-sinh dục thường liên quan đến các đột biến nằm trong miền PHD và được dự đoán sẽ tạo ra một protein bị cắt ngắn. Trong quá trình giảm phân, ATRX duy trì tính toàn vẹn của bộ gen bằng cách điều chỉnh các cơ chế đáp ứng tổn thương DNA. Các đột biến ATRX có liên quan đến sự methyl hóa bất thường của các chuỗi DNA lặp lại, thất bại trong việc thiết lập sự sửa đổi biểu sinh cần thiết cho sự bất hoạt nhiễm sắc thể X và sự im lặng bị suy giảm của các gen in dấu.
Chuột knockout ATRX ở tế bào Sertoli có tinh hoàn nhỏ, thể tích ống sinh tinh giảm, số lượng tế bào Sertoli giảm và chết theo chương trình của tế bào Sertoli tăng liên quan đến pha G2/M kéo dài trong đời sống thai nhi. Phát hiện này phù hợp với quan niệm rằng biểu hiện protein ATRX ảnh hưởng đến sự tiến triển G2-M với những hậu quả cuối cùng đối với sự tồn tại của tế bào. Hậu quả đặc hiệu cho mô của sự điều hòa biểu sinh và sự tiến triển chu kỳ tế bào bị gián đoạn có thể giải thích các đặc điểm khác nhau liên quan đến các đột biến ATRX.
Các rối loạn liên quan đến rối loạn phát triển giới tính 46,XX và rối loạn phát triển giới tính 46,XY (Bảng 6.3)
Gen SF1/NR5A1
Gen SF1/NR5A1, nằm ở nhiễm sắc thể 9q33, mã hóa một protein 461 axit amin. Protein này là một chất điều hòa phiên mã thụ thể hạt nhân liên quan đến sự phát triển và chức năng của tuyến thượng thận, tuyến sinh dục và vùng dưới đồi. Protein chứa một miền liên kết DNA N-terminal chứa hai ngón tay kẽm, một miền bản lề và miền liên kết phối tử. Protein điều chỉnh biểu hiện của SRY, SOX9, AMH, và AMHR. Trong các tế bào Leydig, NR5A1 thúc đẩy biểu hiện của LHCGR, STAR, CYP11A1, và CYP17A1, cần thiết cho quá trình sinh tổng hợp testosterone.
Bảng 6.3. Các rối loạn phát triển giới tính không kèm hội chứng ở cá thể XY
| Gen | Vị trí | Kiểu hình | Khiếm khuyết di truyền | Kiểu di truyền |
| AMH | 19p13.3 | Hội chứng ống Müller tồn tại, loạn sản tuyến sinh dục (OMIM #261550) | Đột biến điểm | AR |
| AMHR2 | 12q13.13 | Hội chứng ống Müller tồn tại (OMIM #261550) | Đột biến điểm | AR |
| AR | Xq12 | Kháng androgen, loạn sản tuyến sinh dục (OMIM #300068) | Mất đoạn, đột biến điểm | Liên kết X |
| AXIN1 | 16p13.3 | Tinh hoàn ẩn, bất thường nhân đôi đuôi (OMIM #603816) | Đột biến điểm | không rõ |
| CBX2 | 17q25 | Loạn sản tuyến sinh dục hoàn toàn, DSD noãn tinh (OMIM #613080) | Đột biến điểm | AR |
| DHH | 12q13.12 | Loạn sản tuyến sinh dục, một phần hoặc hoàn toàn (OMIM #233420) | Đột biến điểm | AR |
| DMRT1 | 9p24.3 | Loạn sản tuyến sinh dục, một phần hoặc hoàn toàn. Bất thường bổ sung ở bệnh nhân mất đoạn 9p24.3 (OMIM #154230) | Đột biến điểm, mất đoạn 9p24.3 (hội chứng mất gen liền kề) | AD |
| ESR2 | 14q23.2-q23.3 | Đảo ngược giới tính XY (OMIM #601663) | Đột biến điểm | AD, AR |
| FGFR1, nhiều | 8p11.23, nhiều | Suy sinh dục do suy gonadotropin có hoặc không có mất khứu giác, xem (OMIM #147950) để biết thêm các locus | Mất/lặp đoạn, đột biến điểm | AD, AR, đa gen |
| GATA4 | 8p23.1 | Bất thường tinh hoàn có/không có bệnh tim bẩm sinh (OMIM# 615542) | Mất/lặp đoạn, đột biến điểm | AD |
| KAL1 | Xp22.31 | Suy sinh dục do suy gonadotropin với mất khứu giác (OMIM #308700) | Mất/lặp đoạn, đột biến điểm | Liên kết X |
| LHCGR | 2p16.3 | Thiểu sản tế bào Leydig (OMIM #238320) | Đột biến điểm | AR |
| MAMLD1 | Xq28 | Dương vật nhỏ, bìu chẻ đôi, lỗ tiểu lệch thấp dương vật-bìu (OMIM #300758) | Đột biến điểm | Liên kết X |
| MAP3K1 | 5q11.2 | Loạn sản tuyến sinh dục (OMIM #613762) | Đột biến điểm | AD |
| NR0B1(DAX1) | Xp21.2 | Loạn sản tuyến sinh dục hoàn toàn, đảo ngược giới tính (OMIM #300018) | Lặp đoạn, chuyển vị X-nhiễm sắc thể thường, mất đoạn thượng nguồn | Liên kết X |
| NR5A1 (SF1) | 9q33.3 | Thiếu androgen, loạn sản tuyến sinh dục hoàn toàn, suy thượng thận (OMIM #612965) | Mất/lặp đoạn, đột biến điểm | AD |
| SOX8 | 16p13.3 | Cơ quan sinh dục không rõ ràng, loạn sản tuyến sinh dục hoàn toàn (PMID: 21373258) (28) | Đột biến điểm, lặp đoạn thượng nguồn | AD |
| SRY | Yp11.2 | Loạn sản tuyến sinh dục (OMIM #400044) | Mất/lặp đoạn, đột biến điểm | Liên kết Y |
| WWOX | 16q23.1-q23.2 | Loạn sản tuyến sinh dục (PMID: 22071891) | Mất đoạn | AD |
| ZFPM2 (FOG2) | 8q23.1 | Loạn sản tuyến sinh dục có/không có bệnh tim bẩm sinh (OMIM #616067) | Đột biến điểm | AR |
| ZNRF3 | 22q12.1 | Loạn sản tuyến sinh dục (OMIM# 612062) | Đột biến điểm | AD |
Trội NST thường (AD), Lặn NST thường (AR).
Sự không đồng nhất về kiểu hình là đặc trưng của bệnh nhân có đột biến NR5A1. Trong số các cá thể 46,XY, các kiểu hình được báo cáo bao gồm từ sự phát triển tinh hoàn bị suy giảm đến vô sinh nam. Trong số các cá thể 46,XX, các kiểu hình được báo cáo bao gồm từ rối loạn noãn tinh 46,XX đến suy buồng trứng nguyên phát. Suy thượng thận có thể là đặc điểm chính ở một số bệnh nhân. Các thay đổi vô nghĩa, dịch khung và sai nghĩa đã được xác định. Hầu hết các đột biến là dị hợp tử. Các trường hợp gia đình đã được báo cáo. Các kiểu hình khác nhau đã được mô tả trong một gia đình duy nhất.
Do nhiều hoạt động của NR5A1 trong sự biệt hóa và chức năng của tuyến sinh dục, một số cơ chế hoạt động tiềm năng có thể xảy ra với các đột biến NR5A1. Trong số các cá thể 46,XY, các đột biến NR5A1 không thể kích hoạt đầy đủ con đường nam giới dẫn đến tình trạng thiếu nam hóa. Trong số các bệnh nhân XX (âm tính với SRY) bị nam hóa, dựa trên các nghiên cứu trong ống nghiệm, các đột biến NR5A1 dường như không kích hoạt bất thường con đường nam giới. Thay vào đó, các nghiên cứu trong ống nghiệm cho thấy các đột biến sai nghĩa p.Arg92Trp hoặc p.Ala260Val đã kìm hãm tín hiệu Wnt, do đó cản trở sự biệt hóa buồng trứng ở các bệnh nhân 46,XX (âm tính với SRY) bị nam hóa. Nói cách khác, việc điều hòa giảm các gen “chống tinh hoàn” của nữ làm giảm sự ức chế con đường phát triển của nam, chuyển hướng xác định số phận tế bào để thúc đẩy sự biệt hóa tinh hoàn ở các cá thể XX. Điều kỳ lạ là, các cá thể 46,XX có một đột biến sai nghĩa khác, p.Arg92Gln, đã được báo cáo là bị suy thượng thận và có hoặc không có DSD noãn tinh.
Độ thấm không hoàn toàn và biểu hiện biến đổi là những đặc điểm được công nhận của các đột biến NR5A1. Các đột biến NR5A1 minh họa cách các biến thể cụ thể trong một yếu tố phiên mã hoạt động như một công tắc tế bào/cơ quan để định hướng kết quả phát triển. Với giải trình tự toàn bộ exome (WES), các biến thể trong các gen khác đã được chứng minh. Do đó, tính đa gen hoặc các gen điều chỉnh có thể ảnh hưởng đến biểu hiện kiểu hình của các đột biến NR5A1.
SOX8
SOX8 nằm ở nhiễm sắc thể 16p.13.3. Các đột biến SOX8 có liên quan đến sự đảo ngược giới tính 46,XY và vô sinh ở cả các cá thể 46,XX và 46,XY. Các đột biến sai nghĩa dị hợp tử của SOX8 đã được xác định ở những cá thể bị đảo ngược giới tính 46,XY.
Các rối loạn phát triển giới tính 46,XY không kèm hội chứng (Loạn sản tuyến sinh dục) (xem Bảng 6.3)
Chromobox Homolog 2 (CBX2)
Gen CBX2, được lập bản đồ trên nhiễm sắc thể 17q25, là một tiểu đơn vị của phức hợp kìm hãm polycomb liên quan đến chromatin 1. Như đã lưu ý trước đó, các protein nhóm polycomb điều chỉnh cấu trúc chromatin, cuối cùng ảnh hưởng đến biểu hiện gen. Tồn tại hai dạng đồng phân, CBX2.1 và dạng ngắn hơn CBX2.2. Các đột biến sai nghĩa mất chức năng của CBX2.1 đã được xác định ở một trẻ sơ sinh 46,XY được xác định bởi sự không khớp giữa kiểu nhân trước sinh và ngoại hình cơ quan sinh dục nữ khi sinh. Đánh giá sau đó đã xác định một tử cung bình thường và mô giống buồng trứng trên mô học. Hai bệnh nhân có đột biến CBX2.2 đã được báo cáo; cả hai bệnh nhân đều có kiểu nhân 46,XY. Một bệnh nhân có tinh hoàn loạn sản, trong khi không có mô tuyến sinh dục nào có thể được xác định ở bệnh nhân thứ hai. Dữ liệu có sẵn cho thấy CBX2.1 liên quan đến việc xác định tinh hoàn, trong khi CBX2.2 liên quan đến sự phát triển sớm của tuyến sinh dục. Các yếu tố điều chỉnh việc nối xen kẽ của gen này vẫn chưa được biết.
Desert Hedgehog
Gen Desert hedgehog (DHH) nằm trên nhiễm sắc thể 12q12-q13.1 và mã hóa một protein bao gồm 396 axit amin. DHH là một thành viên của họ Hedgehog và cũng được biểu hiện trong các tế bào Schwann của hệ thần kinh ngoại biên. Đây là một rối loạn di truyền lặn trên nhiễm sắc thể thường liên quan đến loạn sản tuyến sinh dục XY và bệnh thần kinh bó nhỏ. Tuy nhiên, bệnh thần kinh là một đặc điểm không nhất quán. DHH thúc đẩy sự hình thành của vỏ mô liên kết xung quanh các dây thần kinh ngoại biên và cần thiết cho sự tồn tại của chúng.
Một bệnh nhân 46,XY có cơ quan sinh dục ngoài nữ, bệnh đa dây thần kinh, một tinh hoàn ở một bên và một tuyến sinh dục dạng dải ở bên kia. Phân tích di truyền cho thấy tính đồng hợp tử đối với một sự thay thế nucleotide đơn tại codon khởi đầu, được dự đoán sẽ loại bỏ sự khởi đầu dịch mã tại vị trí bắt đầu bình thường. Phân tích mô học của dây thần kinh bắp chân cho thấy sự hình thành rộng rãi của các bó nhỏ trong nội thần kinh. Bệnh thần kinh bó nhỏ liên quan đã được cho là do dị tật phát triển của các dây thần kinh ngoại biên và được đặc trưng bởi sự hiện diện của nhiều bó nhỏ với sự suy giảm cảm giác kiểu găng tay và tất. Các khối u tuyến sinh dục, bao gồm seminoma tại chỗ, u nguyên bào sinh dục và u tế bào mầm đã được báo cáo.
Doublesex and Mab-3–Related Transcription Factor 1 (DMRT1) and Doublesex and Mab-3–Related Transcription Factor 2 (DMRT2)
Monosomy đối với nhiễm sắc thể 9p xa đã được báo cáo trong sự đảo ngược giới tính từ nam sang nữ. Hầu hết các mất đoạn liên quan đến đảo ngược giới tính liên quan đến vùng nhiễm sắc thể 9p24.3 nơi có ba gen DMRT. DMRT1 dường như có liên quan đến sự biệt hóa của tế bào Sertoli và tế bào. Cơ quan sinh dục ngoài đã được mô tả là không rõ ràng hoặc nữ. Cơ quan sinh dục ngoài có thể đối xứng hoặc không đối xứng. Sự biệt hóa của cơ quan sinh dục trong rất thay đổi, với sự hiện diện của các di tích ống Müller và Wolff đã được báo cáo. Ngoài sự đảo ngược giới tính, các đặc điểm lâm sàng bao gồm chậm phát triển trí tuệ, tai thấp, đầu hình tam giác, sống mũi rộng, nếp gấp lòng bàn tay đơn, dị tật tim, động kinh và vẹo cột sống. Các bất thường sinh dục được báo cáo ở các cá thể XY bao gồm loạn sản tuyến sinh dục, noãn tinh, lỗ tiểu lệch thấp, đảo ngược dương vật-bìu và tinh hoàn ẩn. U nguyên bào sinh dục đã được báo cáo. Mặc dù các mối tương quan kiểu hình/kiểu gen không rõ ràng, suy giảm đơn bội đối với DMRT1 dường như là đủ để gây ra loạn sản tuyến sinh dục. Cơ quan sinh dục ngoài nữ và tầm vóc thấp đã được mô tả ở một bệnh nhân có kiểu nhân 46,XY thể khảm đặc trưng bởi nhiễm sắc thể vòng 9 và suy giảm đơn bội DMRT1.
GATA4
Các protein GATA bao gồm một họ các yếu tố phiên mã đặc hiệu cho mô chứa hai miền ngón tay kẽm được bảo tồn. Ngón tay kẽm C-terminal là cần thiết để nhận dạng DNA và ngón tay kẽm N-terminal góp phần vào sự ổn định; cả hai ngón tay kẽm đều cần thiết cho các tương tác protein-protein với các yếu tố phiên mã khác. Gen GATA4 được lập bản đồ trên nhiễm sắc thể 8p23.1. Các đột biến dị hợp tử của GATA4 có liên quan đến bệnh tim bẩm sinh và các rối loạn phát triển giới tính XY. Sự không đồng nhất về kiểu hình quan sát được đã được cho là do độ thấm không hoàn toàn và tính đa gen. Ba nam giới trong một gia đình được báo cáo là biểu hiện bệnh tim bẩm sinh và các bất thường khác nhau trong sự phát triển giới tính nam, trong khi người mang gen nữ bị bệnh tim bẩm sinh mà không có kiểu hình buồng trứng. Đột biến cụ thể này, p.Gly221Arg, đã loại bỏ khả năng của protein GATA4 tương tác với protein ZFPM2/FOG2 và làm gián đoạn sự kích hoạt hiệp đồng của promoter AMH bởi GATA4 và NR5A1.
Mastermind-Like Domain Containing 1 (MAMLD1)
MAMLD1 đã được xác định trong các nghiên cứu điều tra cơ sở di truyền của bệnh cơ ống liên kết X. Gen này còn được gọi là khung đọc mở 6 của nhiễm sắc thể X (CXorf6). Tuy nhiên, một số biến thể đã được phát hiện ở những người bình thường. Trong ống nghiệm, hầu hết các biến thể MAMLD1 hoạt động tương tự như loại hoang dã ngoại trừ biến thể L210X. Sự thiếu vắng một kiểu hình sinh sản đáng kể ở chuột knockout chuyển gen làm dấy lên nghi ngờ liệu MAMLD1 có đủ để gây ra một DSD hay không. Do đó, những phát hiện này cho thấy các biến thể MAMLD1 là không đủ để giải thích cho kiểu hình DSD của bệnh nhân.
Mitogen-Activated Kinase Kinase Kinase 1 (MAP3K1)
Gen MAP3K1 nằm ở nhiễm sắc thể 5q11.2. Gen này mã hóa một protein liên quan đến tín hiệu MAPK. Các kiểu hình của các cá thể bị ảnh hưởng bao gồm loạn sản tuyến sinh dục hoàn toàn 46,XY, lỗ tiểu lệch thấp đáy chậu-bìu, tinh hoàn ẩn và sự phát triển ống Müller thay đổi. U nguyên bào sinh dục và u tế bào mầm đã được báo cáo. Di truyền dường như là trội trên nhiễm sắc thể thường giới hạn giới tính. Các biến thể liên quan đến loạn sản tuyến sinh dục làm tăng sự phosphoryl hóa của các mục tiêu hạ nguồn, MAPK11 (p38), MAP3K (ERK1), và MAPK1 (ERK2) và làm tăng sự liên kết của các đồng yếu tố RHOA và MAP3K4 (AXIN1); kết quả cuối cùng là giảm biểu hiện SOX9. Do đó, các đột biến tăng chức năng này kìm hãm các yếu tố thúc đẩy tinh hoàn và ổn định beta-catenin, ủng hộ con đường biệt hóa buồng trứng.
Nuclear Receptor Subfamily 0, Group B, Member 1 (NROB1)
Gen hai exon này, NROB1, nằm ở Xp21.2 mã hóa DAX1. Protein 460 axit amin là một thụ thể hạt nhân mồ côi không có miền liên kết DNA ngón tay kẽm điển hình. N-terminal của protein 470 axit amin chứa một miền liên kết DNA mới, trong khi C-terminal cho thấy các đặc điểm của một miền liên kết phối tử của thụ thể hormone hạt nhân. NROB1 được biểu hiện trên toàn trục HPG. Protein DAX1 có chức năng như một chất kìm hãm phiên mã của nhiều gen, bao gồm NR5A1 và một số gen enzym tạo steroid.
Sự lặp đoạn của locus DAX1/NROB1 có liên quan đến sự đảo ngược giới tính từ nam sang nữ. Sự biệt hóa cơ quan sinh dục ngoài bao gồm từ nữ đến không rõ ràng. Mô tả về cơ quan sinh dục trong bao gồm sự hiện diện của các cấu trúc Müller và Wolff. Tuyến sinh dục thường được mô tả là dạng dải. Sử dụng lai so sánh bộ gen trên mảng (CGH) độ phân giải cao, một sự lặp đoạn kẽ dưới kính hiển vi của gen DAX1/NROB1 đã được phát hiện trong quá trình đánh giá kỹ lưỡng hai chị em. Sự lặp đoạn 637 kb này bao gồm NROB1, bốn gen MAGEB, CXorf21, glycerol kinase (GK), và một phần của gen MAP3K7IP3. Chị gái có biểu hiện vô kinh nguyên phát, cơ quan sinh dục ngoài nữ, kiểu nhân 46,XY và loạn sản tuyến sinh dục; em gái có kiểu hình nữ chưa dậy thì, nhưng được phát hiện có kiểu nhân 46,XY.
Các đột biến mất chức năng có liên quan đến thiểu sản tuyến thượng thận bẩm sinh liên kết X (AHC). Trong rối loạn này, sự phát triển của vỏ thượng thận thai nhi là bình thường. Tuy nhiên, vỏ thượng thận trưởng thành hoặc dứt khoát không phát triển. Suy thượng thận có thể không rõ ràng trong giai đoạn sơ sinh ngay lập tức nhưng có thể trở nên rõ ràng trong thời kỳ sơ sinh sớm. Mặc dù suy thượng thận thường biểu hiện ở trẻ sơ sinh hoặc trẻ nhỏ, sự không đồng nhất về kiểu hình về mức độ nghiêm trọng và tuổi khởi phát xảy ra. Tinh hoàn ẩn một hoặc hai bên cũng có thể xảy ra. Ở tuổi dậy thì dự kiến, suy sinh dục do suy gonadotropin gây ra bởi rối loạn chức năng vùng dưới đồi và tuyến yên có thể xảy ra ở nam giới bị ảnh hưởng. Dậy thì muộn đã được ghi nhận ở nữ giới dị hợp tử. Một phụ nữ đồng hợp tử về các đột biến NROB1 đã được báo cáo. Kiểu hình của cô là suy sinh dục do suy gonadotropin. Là một phần của hội chứng mất gen liền kề, AHC liên kết X có thể liên quan đến thiếu hụt glycerol kinase, loạn dưỡng cơ Duchenne, thiếu hụt ornithine transcarbamylase và chậm phát triển trí tuệ.
Các đột biến vô nghĩa đã được xác định trên toàn gen. Các đột biến sai nghĩa chiếm 20% các đột biến liên quan đến AHC. Các đột biến này có xu hướng tập trung ở C-terminal của protein tương ứng với miền liên kết phối tử giả định và làm suy giảm hoạt động kìm hãm phiên mã của protein. Một đột biến điểm sai nghĩa, nằm ở vùng bản lề của protein, đã được xác định ở một bé gái 8 tuổi có các đặc điểm lâm sàng và xét nghiệm cho thấy suy thượng thận. Các nghiên cứu bổ sung cho thấy đột biến này cản trở sự định vị hạt nhân của protein. Điều kỳ lạ là, người cha bán hợp tử và em gái dị hợp tử của bệnh nhân khởi phát không biểu hiện kiểu hình AHC.
PBX1 (Pre-B-Cell-Leukemia Factor 1)
Yếu tố tiền-B-cell-leukemia 1 (PBX1), nằm ở nhiễm sắc thể 1q23.3, là một thành viên của họ protein Three Amino Acid Loop Extension (TALE). PBX1 dimer hóa với các protein khác để tạo ra các phức hợp hạt nhân thúc đẩy tính đặc hiệu liên kết của các protein HOX với DNA. Các đột biến trong PBX1 đã được báo cáo ở những bệnh nhân bị bất thường bẩm sinh ảnh hưởng đến thận và đường tiết niệu, kết hợp với chậm phát triển, bất thường tai và bệnh tim bẩm sinh. Một đột biến PBX1 de novo, p.Arg235Gln, nằm trong homeodomain được bảo tồn, đã được xác định ở một đứa trẻ 46,XY có cơ quan sinh dục ngoài nữ; tuyến sinh dục phải bao gồm các ống sinh tinh thưa thớt với mô sợi và tuyến sinh dục trái chỉ bao gồm mô sợi. Các nghiên cứu biểu hiện cho thấy đột biến sai nghĩa này không định vị được trong nhân, ngụ ý PBX1 là cần thiết cho sự phát triển của tinh hoàn và đột biến này là một đột biến mất chức năng.
Protein Phosphatase Two Regulatory Subunit B”Gamma (PPP2R3C)
Gen này, được lập bản đồ trên nhiễm sắc thể 14q13.2, có 13 exon mã hóa một protein 453 axit amin. Cùng với các tiểu đơn vị giàn giáo A và xúc tác C, ba protein này tạo thành một protein phosphatase 2A dị tam phân, có chức năng khử phosphoryl hóa thuận nghịch của các phosphoprotein. Bốn bệnh nhân nữ có kiểu hình với loạn sản tuyến sinh dục 46,XY, mặt phẳng, nếp gấp mi hai bên, tai thấp xoay ra sau và nếp gấp lòng bàn tay đơn đã được mô tả. Cả bốn người đều đồng hợp tử về các đột biến sai nghĩa; cha mẹ là những người mang gen dị hợp tử. Điều kỳ lạ là, ba trong số bốn người cha dị hợp tử được phát hiện có quá trình sinh tinh bị suy giảm đặc trưng bởi tinh trùng dị dạng với các bất thường nặng ở đầu, thể đỉnh và nhân.
SRY
SRY là một gen exon đơn mã hóa một protein 204 axit amin. Protein này chứa một miền liên kết DNA HMG được bao quanh bởi các tín hiệu định vị hạt nhân (NLS). Phần lớn các đột biến SRY gây đảo ngược giới tính nằm ở miền HMG/NLS và ảnh hưởng đến ái lực liên kết DNA, khả năng uốn cong DNA hoặc định vị hạt nhân. Các đột biến nằm ở miền N-terminal và C-terminal đã được xác định ở những cá thể bị đảo ngược giới tính 46,XY. Các đột biến ở các miền không phải HMG có thể ảnh hưởng đến sự kích hoạt phiên mã, liên kết DNA hoặc các tương tác protein-protein. Thể khảm của cha đối với các đột biến SRY trong đó các tế bào khác nhau mang các gen SRY khác nhau đã được mô tả. Đáng bối rối hơn là các phả hệ trong đó những người cha và những người anh em không bị ảnh hưởng mang cùng một allele SRY đột biến như người khởi phát. Những phát hiện nghịch lý này cho thấy sự tham gia của các gen khác, các tương tác gen-gen và các tương tác gen-môi trường trong quá trình biệt hóa giới tính. Tuy nhiên, chỉ 15% đến 20% các trường hợp DSD 46,XY do loạn sản tuyến sinh dục có thể được quy cho các đột biến SRY. Các đột biến trong gen SRY cũng đã được báo cáo ở những bệnh nhân có các dấu hiệu điển hình của hội chứng Turner và kiểu nhân 45,X/46XY.
WW Domain-Containing Oxidoreductase (WWOX)
Gen WWOX được lập bản đồ trên 16q23.1-16q23.2. Gen này mã hóa một protein tế bào chất đóng vai trò trong sự tăng trưởng, biệt hóa và ức chế khối u. Một bệnh nhân bị đảo ngược giới tính 46,XY được phát hiện mang một mất đoạn dị hợp tử của các exon 6 đến 8 (axit amin 173–352). Bệnh nhân này có các nếp gấp môi-bìu không hợp nhất, tuyến sinh dục hai bên không sờ thấy, phì đại âm vật, một lỗ đáy chậu duy nhất và tuyến sinh dục loạn sản trong ổ bụng. Các đột biến WWOX đã được xác định trong bệnh thất điều tiểu não di truyền lặn trên nhiễm sắc thể thường với động kinh và chậm phát triển trí tuệ.
Zinc Finger and Ring Finger Protein 3 (ZNRF3)
ZNRF3 được lập bản đồ trên nhiễm sắc thể 22q12.1 mã hóa một E3 ubiquitin ligase xuyên màng thường can thiệp vào tín hiệu WNT trong tuyến sinh dục để thúc đẩy sự biệt hóa tinh hoàn. Các đột biến dị hợp tử đã được báo cáo ở 5 bệnh nhân XY bị đảo ngược giới tính; bốn trong số năm bệnh nhân có biểu hiện dậy thì muộn ở cuối tuổi vị thành niên.
Zinc Finger Protein, Multitype 2 (ZFPM2)
Protein Friend of GATA 2 (FOG2), được mã hóa bởi ZFPM2 và được lập bản đồ trên nhiễm sắc thể 8q23.1, là một đồng yếu tố phiên mã điều chỉnh hoạt động của GATA4 bằng cách liên kết với ngón tay kẽm N-terminal của nó. Một cậu bé có chuyển vị cân bằng và điểm gãy với intron 4 của gen FOG2 bị bệnh tim bẩm sinh, tinh hoàn co rút và suy sinh dục do tăng gonadotropin.
Rối loạn phát triển giới tính dạng tinh hoàn-buồng trứng
Rối loạn phát triển giới tính (DSD) dạng tinh hoàn-buồng trứng được định nghĩa là sự hiện diện của mô buồng trứng chứa nang noãn và mô tinh hoàn chứa ống sinh tinh trong cùng một cá thể. Hình thái cơ quan sinh dục ngoài không thể dùng để dự đoán mô học của tuyến sinh dục. Tuyến sinh dục có thể là một tuyến lưỡng tính (ovotestis), buồng trứng và/hoặc tinh hoàn. Về mặt mô học, dạng phổ biến nhất là tuyến lưỡng tính. Tuyến lưỡng tính thường nằm ở bên phải và trong ổ bụng. Về mặt đại thể, tuyến lưỡng tính có thể có dạng hình trứng hoặc hai thùy. Trong hầu hết các tuyến lưỡng tính, mô buồng trứng và mô tinh hoàn được phân tách rõ rệt theo kiểu sắp xếp đầu-cuối. Trong trường hợp này, vùng tinh hoàn thường cho thấy sự biệt hóa kém của lớp áo trắng (tunica albuginea) với mô kẽ không điển hình. Trong một số trường hợp, các tế bào trứng (oocyte) nằm xen kẽ giữa các dây/ống sinh tinh. Tỷ lệ mô buồng trứng và mô tinh hoàn khác nhau giữa các bệnh nhân; sự phát triển của tử cung cũng thay đổi.
Bộ nhiễm sắc thể phổ biến nhất là 46,XX. Các bộ nhiễm sắc thể thể khảm như 46,XX/46,XY, 46,XX/47,XXY, 45,X/46,XY, 47,XYY/46, và XY/45,X cũng đã được mô tả. Trong một số trường hợp, lượng vật liệu nhiễm sắc thể Y trong tế bào lympho máu ngoại vi rất hạn chế, ví dụ như gen SRY chỉ có thể được phát hiện bằng phương pháp khuếch đại chuỗi polymerase (PCR). Trong các trường hợp khác, SRY dường như không có trong tế bào lympho máu ngoại vi nhưng lại được phát hiện trong mô tinh hoàn. Một trường hợp mất đoạn vùng khởi động (promoter) của gen SRY đã được xác định trong mô tinh hoàn của một bệnh nhân 46,XX bị rối loạn tinh hoàn-buồng trứng. Tuy nhiên, DSD dạng tinh hoàn-buồng trứng vẫn có thể xảy ra khi không có vật liệu nhiễm sắc thể Y. Như đã đề cập ở các phần khác trong chương này, các đột biến đặc hiệu, ví dụ như ở các gen NR5A1, NR2F2, RSPO1, và SOX9, đã được xác định ở các bệnh nhân bị rối loạn tinh hoàn-buồng trứng.
Hầu hết các trường hợp là ngẫu nhiên (sporadic). Tuy nhiên, đã có mô tả về các phả hệ trong đó cùng tồn tại cả nam giới XX và cá thể XX bị rối loạn tinh hoàn-buồng trứng. Mặc dù hầu hết bệnh nhân được chẩn đoán ở tuổi sơ sinh hoặc thời thơ ấu, nam giới về kiểu hình có thể biểu hiện triệu chứng vú to hai bên (gynecomastia). Một bệnh nhân có bộ nhiễm sắc thể 46,XX/46,XY được chẩn đoán lúc sơ sinh với lỗ tiểu lệch thấp ở dương vật-bìu, tuyến sinh dục bên trái đã xuống và tuyến sinh dục bên phải chưa xuống; sinh thiết cho thấy tinh hoàn trước tuổi dậy thì bình thường ở bên trái và mô buồng trứng bình thường ở bên phải. Việc đánh giá và quản lý sâu hơn cho bệnh nhân này đã dẫn đến quyết định nuôi dưỡng theo giới tính nam, kèm theo phẫu thuật cắt bỏ vòi trứng-buồng trứng phải và cắt bỏ tử cung. Bệnh nhân sau đó tái khám ở tuổi 17 vì một khối u không đau ở bìu, khối u này đã được cắt bỏ và xác định là mô buồng trứng bình thường. Trường hợp này minh họa cho sự không nhất quán tiềm tàng trong sự phát triển tuyến sinh dục ở những bệnh nhân bị rối loạn tinh hoàn-buồng trứng, cùng với khả năng xảy ra sai sót trong việc lấy mẫu sinh thiết tuyến sinh dục.
Dữ liệu về khả năng sinh sản của các bệnh nhân này còn hạn chế. Trong số 33 bệnh nhân được theo dõi dọc, các tế bào mầm được xác định trong mô tinh hoàn ở giai đoạn sơ sinh đã bị thoái hóa, dẫn đến tình trạng không có tinh trùng (azoospermia). Các phát hiện mô học ở người lớn mô tả một hình ảnh loạn sản chỉ có tế bào Sertoli với sự hyalin hóa các ống sinh tinh. Tuy nhiên, các kỹ thuật hỗ trợ sinh sản đã được sử dụng thành công để giúp những người đàn ông bị rối loạn tinh hoàn-buồng trứng có con. Một vài trường hợp mang thai cũng đã được báo cáo ở những phụ nữ bị rối loạn này.
Rối loạn phát triển giới tính XX không hội chứng (Bảng 6.4)
Rối loạn phát triển giới tính dạng tinh hoàn 46,XX
DSD dạng tinh hoàn được đặc trưng bởi kiểu hình nam với bộ nhiễm sắc thể 46,XX. Tần suất của hội chứng nam XX là khoảng 1 trên 25.000 nam giới. Dạng DSD này có thể được phân loại thành các nhóm SRY dương tính và SRY âm tính. Nam giới 46,XX SRY dương tính thường có cơ quan sinh dục ngoài của nam giới bình thường, tinh hoàn nhỏ không có tinh trùng, suy sinh dục tăng gonadotropin, và thường biểu hiện với tình trạng vô sinh. Trong hầu hết các trường hợp, gen SRY nằm trên nhiễm sắc thể X do sự tái tổ hợp giữa nhiễm sắc thể X và Y. Tuy nhiên, sự chuyển đoạn sang một nhiễm sắc thể thường cũng có thể xảy ra. Một ví dụ là trường hợp phát hiện tình cờ tinh hoàn nhỏ, không có tinh trùng và sự chuyển đoạn của gen SRY lên đầu cuối của nhiễm sắc thể 16q ở một người đàn ông 46,XX 61 tuổi.
Bảng 6.4. Rối loạn phát triển giới tính và Loạn sản tuyến sinh dục ở các cá thể XX (SRY-âm tính)
| Gen, Vị trí | Kiểu hình | Khiếm khuyết di truyền | Kiểu di truyền |
| BMP15, Xp11.22 | Loạn sản buồng trứngª, vô kinh nguyên phát (OMIM #300510) | Đột biến điểm (di truyền từ cha), mất đoạn | Liên kết X |
| DCAF17, 2q31.1 | Suy sinh dục, loạn sản buồng trứng, rụng tóc một phần, Hội chứng Woodhouse-Sakati (OMIM #241080) | Đột biến điểm | Lặn NST thường |
| DIAPH2, Xq21.33 | Vô kinh nguyên phát hoặc thứ phát (OMIM #300511) | Đột biến điểm, mất đoạn | Liên kết X |
| EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5 | 12q24.31, 14q24.3, 1p34.1, 2p23.3, 3q27.1 | Bệnh não chất trắng với chất trắng biến mất, loạn sản tuyến sinh dục, vô kinh nguyên phát hoặc thứ phát (OMIM #603896) | Đột biến điểm |
| FIGLA, 2p13.3 | Suy buồng trứng (OMIM #612310) | Đột biến điểm | Trội NST thường |
| FMR1, X | Tiền đột biến Fragile X (OMIM #311360/#300869) | Mở rộng lặp lại bộ ba nucleotide | Liên kết X |
| FOXL2, 3q22.3 | Hẹp khe mi với loạn sản buồng trứng (OMIM #110100) | Đột biến điểm, chuyển đoạn, mất đoạn/nhân đoạn | Trội NST thường |
| HFM1, 1p22.2 | Loạn sản buồng trứng, suy buồng trứng (PMID: 27603904) (29) | Đột biến điểm | Không rõ |
| LMNA, 1q22 | Loạn sản buồng trứng với bệnh cơ tim (OMIM #212112) | Đột biến điểm | Lặn NST thường |
| MCM8, 20p12.3 | Loạn sản buồng trứng, vô kinh nguyên phát (OMIM #236700) | Đột biến điểm | Lặn NST thường |
| MCM9, 6q22.31 | Loạn sản buồng trứng, vô kinh nguyên phát (OMIM #616185) | Đột biến điểm | Lặn NST thường |
| MKKS, 20p12.2 | Hội chứng McKusick-Kaufman, thiểu sản sinh dục, ứ dịch trong tử cung và âm đạo (OMIM #236700) | Đột biến điểm | Lặn NST thường |
| NOBOX, 7q35 | Loạn sản buồng trứng, suy buồng trứng nguyên phát (OMIM #611548) | Đột biến điểm | Trội & Lặn NST thường |
| NR5A1 (SF1), 9q33.3 | Loạn sản tuyến sinh dục, DSD tinh hoàn-buồng trứng, suy buồng trứng thứ phát, đảo ngược giới tính (OMIM #612964) | Mất đoạn/nhân đoạn, Đột biến điểm | Trội NST thường |
| NUP107, 12q15 | Loạn sản buồng trứng, vô kinh nguyên phát (OMIM #607617) | Đột biến điểm | Lặn NST thường |
| PMM2, 16p13.2 | Loạn sản buồng trứng, teo tiểu não, suy giảm trí tuệ, bệnh võng mạc sắc tố (OMIM #212065) | Đột biến điểm | Lặn NST thường |
| POF1B, | Suy buồng trứng (OMIM #300604) | ||
| PSMC3IP, 17q21.2 | Loạn sản buồng trứng, vô kinh nguyên phát (OMIM #614324) | Đột biến điểm | Lặn NST thường |
| REC8, 14q12 | Loạn sản buồng trứng, suy buồng trứng (PMID: 27603904) (29) | Đột biến điểm | Không rõ |
| SMC1B, 22q12.31 | Loạn sản buồng trứng, suy buồng trứng (PMID: 27603904) (29) | Đột biến điểm | Không rõ |
| SOHLH1, 9q34.3 | Loạn sản buồng trứng, suy buồng trứng (PMID: 27603904) (29) | Đột biến điểm | Lặn NST thường |
| SOHLH2, | Loạn sản buồng trứng, suy buồng trứng (OMIM #616066) | Trội NST thường | |
| SOX3, Xq26.3 | Loạn sản tuyến sinh dục, nam hóa đảo ngược giới tính 46,XX (OMIM #300833) | Mất đoạn/nhân đoạn | Liên kết X |
| SOX8, 16p13.3 | Loạn sản buồng trứng, vô kinh nguyên phát hoặc thứ phát | Đột biến điểm | Trội NST thường |
| SOX9, 17q24.3-q25.1 | Đảo ngược giới tính 46,XX, hội chứng nhân đoạn nhiễm sắc thể 17q24 gây nam hóa (OMIM #278850) | Nhân đoạn 584–516 kb ngược dòng của SOX9 | Trội NST thường |
| SOX10, 22q11.2q13 | Đảo ngược giới tính 46,XX, hội chứng Waardenburg, bệnh thần kinh ngoại biên mất myelin | Nhân đoạn | Trội NST thường |
| SPIDR, 8q11.21 | Loạn sản buồng trứng, vô kinh nguyên phát hoặc thứ phát (OMIM #615384) | Đột biến điểm | Lặn NST thường |
| STAG3, 7q22.1 | Loạn sản buồng trứng, suy buồng trứng (OMIM #615723) | Đột biến điểm | Lặn NST thường |
| SYCE1, 10q26.3 | Loạn sản buồng trứng, suy buồng trứng (PMID: 27603904) (29) | Đột biến điểm | Không rõ |
| WNT4, 1p34.12 | Bất sản Müller, tăng androgen, vô kinh nguyên phát (OMIM #158330) | Đột biến điểm | Trội NST thường |
AD, Di truyền trội trên nhiễm sắc thể thường; AR, Di truyền lặn trên nhiễm sắc thể thường.
Khoảng 10% trong số các bệnh nhân này là SRY-âm tính. Phổ kiểu hình dao động từ cơ quan sinh dục không điển hình đến cơ quan sinh dục ngoài của nam giới bình thường. Tinh hoàn thường nhỏ do ngừng giảm phân và thoái hóa tế bào mầm. Nguyên nhân phân tử của nam giới 46,XX SRY-âm tính đa dạng hơn. Nhân đoạn gen SOX9 đã được mô tả trong một gia đình; tất cả các thành viên bị ảnh hưởng đều có đặc điểm sinh dục thứ phát của nam giới bình thường và không có tinh trùng. Các trường hợp nhân đoạn khoảng 600 kb ngược dòng của gen SOX9 đã được xác định ở một số nam giới XX SRY-âm tính. Sự biểu hiện quá mức của gen SOX10 tại vị trí 22q13 được tìm thấy ở một bệnh nhân đảo ngược giới tính 46,XX kèm theo nhiều bất thường bẩm sinh.
SOX3 là một gen có một exon duy nhất được xác định trên Xq27. Nó đóng vai trò trong sự phát triển của não, tuyến yên và sọ mặt. Một bệnh nhân 46,XX SRY-âm tính được giới thiệu để đánh giá tình trạng lỗ tiểu lệch thấp và tinh hoàn ẩn hai bên được phát hiện có tuyến lưỡng tính hai bên và nhân đoạn gen SOX3. Một số bệnh nhân khác cũng đã được báo cáo có nhân đoạn gen SOX3. Sự biểu hiện quá mức của SOX3 trong một mô hình chuột XX chuyển gen tiền lâm sàng đã dẫn đến sự phát triển tinh hoàn và gợi ý giả thuyết rằng SOX3 hoạt động như một gen thay thế cho SRY trong tình huống này. Thiểu sản thùy trước tuyến yên và vị trí lạc chỗ của thùy sau tuyến yên đã được mô tả ở hai bệnh nhân.
Rối loạn phát triển giới tính XX/Suy buồng trứng sớm
Loạn sản tuyến sinh dục 46,XX là một rối loạn hiếm gặp liên quan đến dậy thì muộn và mãn kinh sớm kèm theo suy sinh dục tăng gonadotropin (xem Bảng 6.4). Các cá thể bị ảnh hưởng thường có biểu hiện không phát triển dậy thì tự nhiên, vô kinh nguyên phát và thiểu sản tử cung.
Như đã đề cập trước đó, sự phát triển của buồng trứng là một quá trình tích cực liên quan đến nhiều gen. Tuy nhiên, các đột biến của gen liên quan đến sự phát triển buồng trứng thường không đi kèm với cơ quan sinh dục không điển hình; sự phát triển cơ quan sinh dục ngoài thường là của nữ giới. Thay vào đó, các rối loạn này thường biểu hiện với dậy thì muộn, vô kinh nguyên phát và suy buồng trứng nguyên phát (POI). Trong các rối loạn này, các nang noãn có thể không phát triển hoặc có thể bị thoái hóa sớm. Các đột biến trong các gen liên quan đến sự phát triển buồng trứng như Forkhead box L2 (FOXL2), Newborn ovary homeobox (NOBOX), bone morphogenetic protein 15 (BMP15), và Factor in germline alpha (FIGLA) đã được mô tả. BMP15, một thành viên của siêu họ TGF-β, ảnh hưởng đến sự tăng trưởng và biệt hóa của tế bào hạt. Các đột biến sai nghĩa dị hợp tử của gen BMP15 đã được xác định ở những phụ nữ bị POI và liên quan đến loạn sản tuyến sinh dục XX liên kết X. Loạn dưỡng chất trắng buồng trứng là một bệnh não chất trắng đặc trưng bởi sự biến mất chất trắng và loạn sản buồng trứng liên quan đến các đột biến trong các gen EIF2B2, EIF2B4, và EIF2B5. Một đột biến trong gen protein tương tác 3, adenosine triphosphatase (ATPase), tiểu đơn vị 26 của proteasome (PSMC3IP) đã được mô tả trong loạn sản tuyến sinh dục 46,XX di truyền lặn trên nhiễm sắc thể thường; đột biến mất chức năng này làm giảm sự hoạt hóa phiên mã do estrogen của protein được mã hóa bởi gen này. Hội chứng Fragile X gây ra bởi sự bất hoạt của gen FMR1 do sự tăng methyl hóa DNA của vùng khởi động và vùng lặp lại CGG 5’-UTR mở rộng; alen tiền đột biến chứa 55-200 lần lặp lại CGG có liên quan đến POI. Tỷ lệ alen tiền đột biến trong dân số chung được ước tính là 1 trên 150 đến 300 nữ giới.
FOXL2
Forkhead box L2 (FOXL2) là một thành viên của họ phiên mã winged helix/forkhead. Protein này chứa một vùng liên kết DNA, chủ yếu nằm trong nhân. Protein chứa ba chuỗi xoắn α. Các đột biến trong FOXL2, nằm ở nhiễm sắc thể 3q23, có liên quan đến hội chứng hẹp khe mi-teo mi-lộn mi (BPES) di truyền trội trên nhiễm sắc thể thường. BPES được đặc trưng bởi loạn sản mí mắt bao gồm khe mi nhỏ (blepharophimosis), sụp mi (ptosis), nếp quạt ngược (epicanthus inversus), và sống mũi rộng. Kiểu hình của BPES loại 1 bao gồm các dấu hiệu ở mắt và suy buồng trứng sớm. Bệnh nhân BPES loại 2 chỉ biểu hiện các đặc điểm ở mắt. Bệnh nhân dị hợp tử với đột biến FOXL2 thường trải qua quá trình phát triển dậy thì tự nhiên và có kinh nguyệt, nhưng sau đó bị suy giảm nang noãn sớm, dẫn đến suy buồng trứng sớm. Hình ảnh mô học của buồng trứng ở bệnh nhân BPES loại 1 được báo cáo là thay đổi từ sự hiện diện của các nang noãn nguyên thủy với một số nang noãn thoái hóa đến sự vắng mặt hoàn toàn của các nang noãn. Các đột biến vô nghĩa, được dự đoán sẽ tạo ra các protein bị cắt ngắn thiếu vùng ức chế phiên mã, có liên quan đến BPES loại 1. Sự mở rộng của chuỗi polyalanine có liên quan đến BPES loại 2.
Một vai trò chính của FOXL2 trong buồng trứng dường như là ức chế phiên mã các gen liên quan đến quá trình sinh tổng hợp steroid, chẳng hạn như StAR, P450scc, và aromatase để ngăn chặn sự biệt hóa và tăng sinh sớm của các tế bào hạt. Mất hoạt động ức chế phiên mã do các đột biến vô nghĩa có thể góp phần vào sự suy giảm sớm của các nang noãn và suy buồng trứng sớm. Một bệnh nhân bị rối loạn tinh hoàn-buồng trứng được phát hiện có biểu hiện cả FOXL2 và SOX9. Tuy nhiên, nói chung, sự biểu hiện của các protein này là loại trừ lẫn nhau.
Điều kỳ lạ là ở dê, các đột biến tại locus này gây ra kiểu hình di truyền trội trên nhiễm sắc thể thường đặc trưng bởi sự không có sừng ở cả dê đực và dê cái (polledness) và đảo ngược giới tính từ cái sang đực ở dê XX theo kiểu di truyền lặn.
NOBOX
Gen Newborn Ovary Homeobox (NOBOX) được biểu hiện trong các tế bào mầm và các tế bào trứng nguyên thủy. NOBOX mã hóa một yếu tố phiên mã basic helix-loop-helix tham gia vào quá trình chuyển đổi từ nang noãn nguyên thủy sang nang noãn sơ cấp. Protein này được biểu hiện ở buồng trứng của thai nhi và người trưởng thành. Gen NOBOX nằm ở nhiễm sắc thể 7q35. Kiểu hình của bệnh nhân có đột biến NOBOX bao gồm dậy thì muộn, với vô kinh nguyên phát và vô kinh thứ phát. POI liên quan đến đột biến NOBOX cho thấy cả kiểu di truyền trội và lặn trên nhiễm sắc thể thường.
FIGLA
Factor in germline alpha (FIGLA) là một yếu tố phiên mã đặc hiệu cho tế bào mầm nằm ở nhiễm sắc thể 2p13.3. FIGLA được biểu hiện trong buồng trứng của thai nhi người. Kiểu hình của phụ nữ có đột biến FIGLA bao gồm vô kinh thứ phát và buồng trứng không có nang noãn. Cả hai kiểu di truyền trội và lặn trên nhiễm sắc thể thường đã được mô tả.
SPIDR
Scaffolding protein involved in DNA repair (SPIDR), được xác định vị trí trên nhiễm sắc thể 8q11.21, mã hóa một protein tham gia vào quá trình sửa chữa DNA tương đồng. Các chị em gái có biểu hiện vô kinh nguyên phát được phát hiện bị suy sinh dục tăng gonadotropin liên quan đến đột biến đồng hợp tử.
NUP107
Nucleoporin tạo thành các phức hợp lỗ nhân đóng vai trò là cổng kiểm soát vận chuyển giữa nhân và tế bào chất. Một trong những protein đó là nucleoporin-107, được mã hóa bởi NUP107, nằm ở nhiễm sắc thể 12q15. Các biến thể sai nghĩa trong NUP107 phân ly với dậy thì muộn và suy sinh dục tăng gonadotropin liên quan đến POI trong hai gia đình có dân tộc khác nhau. Các nữ giới bị ảnh hưởng vẫn khỏe mạnh; không có kiểu hình nam nào rõ ràng.
Gen WNT4
WNT4 là một phân tử được tiết ra, liên kết với các thành viên của họ thụ thể frizzled, dẫn đến sự điều hòa phiên mã của các gen đích. WNT4 làm tăng biểu hiện follistatin, ức chế sự hình thành mạch máu thể xoang (tác dụng chống tinh hoàn) và hỗ trợ sự sống còn của tế bào mầm buồng trứng (tác dụng hỗ trợ buồng trứng). Nhân đoạn gen WNT4, nằm ở nhiễm sắc thể 1p31-1p35, có liên quan đến đảo ngược giới tính từ nam sang nữ 46,XY. Một bệnh nhân như vậy có biểu hiện cơ quan sinh dục ngoài không điển hình kèm theo lỗ tiểu lệch thấp nghiêm trọng, tuyến sinh dục dạng sợi, di tích của cả cấu trúc Müller và Wolff, sứt môi, đầu nhỏ và chậm phát triển trong tử cung (IUGR). Một bệnh nhân đảo ngược giới tính từ nữ sang nam bị loạn sản thận, tuyến thượng thận và phổi được phát hiện có đột biến mất chức năng đồng hợp tử của WNT4. Các đột biến mất chức năng của WNT4 đã được phát hiện ở những phụ nữ 46,XX bị vô kinh nguyên phát, thứ phát sau các bất thường ống Müller và dư thừa androgen. Các kiểu hình của những bệnh nhân này ủng hộ giả thuyết rằng WNT4 đóng một vai trò trong sự biệt hóa buồng trứng.
Tinh hoàn biến mất (Vanishing testes)
Các thuật ngữ hội chứng thoái triển tinh hoàn và tinh hoàn biến mất được sử dụng để mô tả sự vắng mặt tinh hoàn ở các bé trai bị tinh hoàn ẩn. Trong một số trường hợp, tình trạng này đi kèm với cơ quan sinh dục không điển hình và nam hóa không hoàn toàn, có lẽ là do sự thoái triển của mô tinh hoàn xảy ra trong khoảng từ 8 đến 14 tuần của thai kỳ. Các dấu hiệu thực thể phản ánh thời gian hoạt động của tinh hoàn. Khi phẫu thuật, có thể xác định được một thừng tinh thô sơ và một mẩu mô tinh hoàn. Khám nghiệm mô học của các mẩu tinh hoàn thường cho thấy các đại thực bào chứa hemosiderin và vôi hóa loạn dưỡng. Một cơ chế tiềm tàng là tai biến mạch máu trước sinh, liên quan đến xoắn tinh hoàn trước sinh, dẫn đến thoái triển tinh hoàn. Một khả năng khác là sự phát triển mạch máu bất thường. Mặc dù thường là ngẫu nhiên, thoái triển tinh hoàn có tính gia đình cũng đã được mô tả.
Rối loạn sinh tổng hợp cholesterol và steroid
Các bất thường ảnh hưởng đến các con đường sinh tổng hợp cholesterol, cortisol và steroid sinh dục có thể dẫn đến cơ quan sinh dục không điển hình (Bảng 6.5). Sinh tổng hợp steroid (steroidogenesis) là các quá trình qua trung gian nhiều enzyme mà qua đó cholesterol được chuyển đổi thành các hormone steroid có hoạt tính sinh học (Hình 6.2). Quá trình này phụ thuộc vào sự điều hòa phối hợp của việc hấp thu cholesterol vào tế bào, vận chuyển vào ty thể và hoạt động của các enzyme đặc hiệu cho từng mô. Các chi tiết về sự phức tạp của các quá trình sinh tổng hợp steroid vẫn đang tiếp tục được làm sáng tỏ. Ba con đường đã được mô tả: (1) kinh điển/cổ điển, (2) “cửa sau thay thế” (alternative backdoor), và (3) androgen C-19 được 11-oxy hóa.
Bảng 6.5. Các rối loạn sinh tổng hợp steroid
| Gen, Vị trí | Kiểu hình | Khiếm khuyết di truyền | Kiểu di truyền |
| AKR1C2 và AKR1C4, 10p15.1 | Loạn sản tuyến sinh dục hoàn toàn, Thiếu hụt Isozyme 3α-Hydroxysteroid Dehydrogenase (OMIM #614279) | Đột biến điểm | Lặn NST thường |
| CYP5A, 18q22.3 | Nam hóa không hoàn toàn, dậy thì muộn, methemoglobin huyết (OMIM #250790) | Lặn NST thường | |
| CYP11A1, 15q24.1 | Suy thượng thận, đảo ngược giới tính, một phần hoặc hoàn toàn (OMIM #613743) | Đột biến điểm | Lặn/Trội NST thường?, đa gen |
| CYP11B1, 8q24.3 | Tăng sản thượng thận bẩm sinh nam hóa do thiếu 11β-hydroxylase, tăng huyết áp (OMIM #202010) | Đột biến điểm, mất đoạn | Lặn NST thường |
| CYP17A1, 10q24.32 | Thiếu 17α-hydroxylase/17,20-lyase, cơ quan sinh dục không điển hình, dậy thì muộn (OMIM#202110) | Lặn NST thường | |
| CYP19A1, 15q21 | Tăng sản thượng thận bẩm sinh nam hóa do thiếu aromatase nhau thai (OMIM #613546) | Đột biến điểm | Lặn NST thường |
| CYP21A2, 6p21.33 | Tăng sản thượng thận bẩm sinh nam hóa do thiếu 21-hydroxylase (OMIM #201910) | Mất đoạn, đột biến điểm | Lặn NST thường |
| HSD3B2, 1p12 | Thiếu 3β-Hydroxysteroid dehydrogenase, cơ quan sinh dục không điển hình (OMIM #201810) | Đột biến điểm | Lặn NST thường |
| HSD17B3, 9q22.32 | Thiếu 17β-Hydroxysteroid dehydrogenase, loạn sản tuyến sinh dục hoàn toàn hoặc nam hóa kém và vú to ở nam giới (OMIM# 264300) | Đột biến điểm | Lặn NST thường |
| POR, 7q11.23 | Tăng sản bẩm sinh nam hóa do thiếu P450 oxidoreductase, (OMIM #613571) | Đột biến điểm, mất đoạn | Lặn NST thường |
| SRD5A2, 2p23.1 | Cơ quan sinh dục không điển hình, thiếu 5α-reductase, (OMIM# 264600) | Đột biến điểm, mất đoạn trong gen | Lặn NST thường |
| STAR, 8p11.23 | Tăng sản thượng thận bẩm sinh dạng lipid (OMIM #201710) | Đột biến điểm | Lặn NST thường |
AD, Di truyền trội trên nhiễm sắc thể thường; AR, Di truyền lặn trên nhiễm sắc thể thường.
Hình 6.2 Cơ quan sinh dục không điển hình với sự bất đối xứng ở một bệnh nhân bị rối loạn phát triển giới tính 46,XY (loạn sản tuyến sinh dục hỗn hợp).
Nhiều enzyme sinh tổng hợp steroid là các protein chứa heme cytochrome P450 hấp thụ ánh sáng ở bước sóng 450 nm ở trạng thái khử hoặc là các hydroxysteroid dehydrogenase. Các enzyme P450 nhận electron từ dạng khử của nicotinamide adenine dinucleotide phosphate (NADPH) và sử dụng trung tâm heme của chúng để xúc tác. Các enzyme loại 1 nằm trong ty thể và nhận electron từ NADPH từ một flavoprotein, ferredoxin reductase, và một protein sắt-lưu huỳnh, ferredoxin. Các enzyme loại 2 nằm trong lưới nội chất và nhận electron từ NADPH từ một protein khác, P450 oxidoreductase.
Các enzyme hydroxysteroid dehydrogenase sử dụng NAD+ và NADP+ làm đồng yếu tố; các enzyme này không chứa nhóm heme. Các hydroxysteroid dehydrogenase được phân thành hai nhóm. Một nhóm là họ short-chain dehydrogenase reductase. Nhóm còn lại là họ aldo-keto reductase. Các enzyme này hoạt động như dehydrogenase và reductase. Mặc dù các enzyme này có thể thực hiện cả hai hoạt động dehydrogenase và reductase trong ống nghiệm, hầu hết chúng hoạt động theo một chiều.
Trong giai đoạn bào thai, các enzyme sinh tổng hợp steroid được biểu hiện ở nhau thai, tinh hoàn và tuyến thượng thận. Các lỗi bẩm sinh trong sinh tổng hợp testosterone có thể dẫn đến nam hóa không hoàn toàn và cơ quan sinh dục không điển hình ở thai nhi 46,XY. Các đột biến mất chức năng trong sinh tổng hợp cortisol ở tuyến thượng thận dẫn đến tăng nồng độ androgen ở thai nhi XX với hậu quả là nam hóa cơ quan sinh dục ngoài.
Trong con đường androgen kinh điển/cổ điển, tinh hoàn của thai nhi tiết ra testosterone, được chuyển đổi thành dihydrotestosterone (DHT) ở các mô đích, chẳng hạn như tuyến tiền liệt và cơ quan sinh dục ngoài. Trong tinh hoàn, cholesterol được chuyển đổi thành pregnenolone bởi enzyme cắt chuỗi bên cholesterol P450scc được mã hóa bởi CYP11A1. Pregnenolone được chuyển đổi thành 17-hydroxypregnenolone bởi hoạt động 17α-hydroxylase của P450c17 được mã hóa bởi CYP17A1 và sau đó thành DHEA bởi hoạt động 17,20-lyase của P450c17. DHEA được chuyển đổi thành androstenedione bởi 3β-hydroxysteroid dehydrogenase loại 2 được mã hóa bởi HSD3B2. Androstenedione được chuyển đổi thành testosterone bởi 17β-hydroxysteroid dehydrogenase loại 3 được mã hóa bởi HSD17B3. Ở da cơ quan sinh dục, testosterone được chuyển đổi thành DHT bởi 5α-reductase loại 2 được mã hóa bởi SRD5A2.
Thông qua các nghiên cứu về chuột túi tammar và các bệnh nhân bị rối loạn sinh tổng hợp steroid, sự hiện diện của một con đường khác dẫn đến tổng hợp DHT đã được phát hiện. Trong “con đường cửa sau thay thế” này, quá trình sinh tổng hợp steroid bỏ qua DHEA, androstenedione và testosterone (Hình 6.3). Thay vào đó, 17-hydroxyprogesterone (17-OHP) trải qua quá trình khử 3α- và 5α- sau đó là các bước 17,20-lyase, 17β-hydroxysteroid dehydrogenase và oxy hóa 3α để tạo ra DHT.
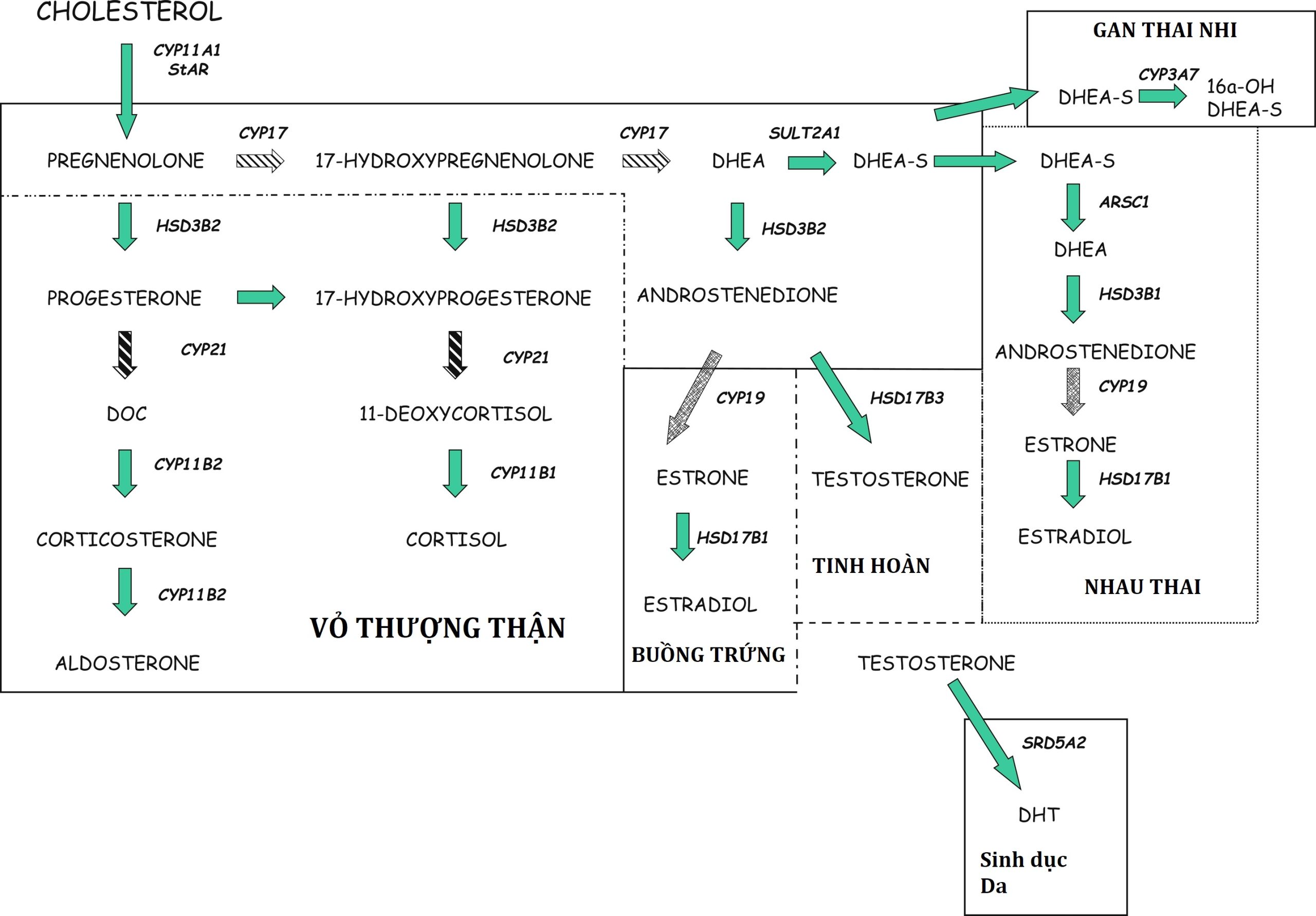
Hình 6.3 Sơ đồ các con đường sinh tổng hợp steroid cổ điển. Các chất nền, sản phẩm và gen liên quan đến sinh tổng hợp steroid ở tuyến thượng thận, buồng trứng, tinh hoàn và nhau thai được chỉ ra. Các gen là 17α-hydroxylase/17,20-lyase (CYP17), 3β-hydroxysteroid dehydrogenase (HSD3B2), 21-hydroxylase (CYP21), 11β-hydroxylase (CYP11B1), aldosterone synthase (CYP11B2), aromatase (CYP19), 17β-hydroxysteroid dehydrogenase loại 1 (HSD17B1), 17β-hydroxysteroid dehydrogenase loại 3 (HSD17B3), 5α-reductase loại 2 (SRD5A2), sulfotransferase (SULT2A1), và steroid sulfatase/arylsulfatase C (ARSC1). CYP3A7 là một enzyme cytochrome P450 được biểu hiện ở gan thai nhi, nơi nó xúc tác cho quá trình 16α-hydroxyl hóa estrone (E1) và dehydroepiandrosterone (DHEA). Biểu hiện của nó giảm sau khi sinh. Các enzyme sinh tổng hợp steroid sử dụng P450 oxidoreductase (POR), một flavoprotein được mã hóa bởi POR, để chuyển electron được chỉ ra bằng các mũi tên gạch chéo. DHEA-S, Dehydroepiandrosterone sulfate.
Con đường sinh tổng hợp steroid thứ ba liên quan đến các steroid C-19 được 11-oxy hóa, từ lâu đã được công nhận là các androgen chính ở cá xương. Sự liên quan của các steroid được 11-oxy hóa đối với sức khỏe con người đã được khám phá (xem Hình 6.3). Loại steroid này phụ thuộc vào hoạt động của 11β-hydroxylase (P450c11B1) được mã hóa bởi CYP11B1 và được biểu hiện ở lớp bó và lớp lưới của tuyến thượng thận. Androstenedione và testosterone là chất nền cho enzyme này và có thể được chuyển đổi thành 11β-hydroxyandrostenedione và 11β-hydroxytestosterone, tương ứng. Các bước enzyme tiếp theo có thể tạo ra 11-ketotestosterone và 11-dihydroketotestosterone; cả hai đều là các androgen mạnh kích hoạt thụ thể androgen. Trong khi các steroid C-19 được 11-oxy hóa được xác định là có liên quan đến sinh lý sau sinh, sự đóng góp của chúng, nếu có, vào quá trình sinh tổng hợp steroid của thai nhi vẫn chưa được biết.
Cả con đường sinh tổng hợp androgen kinh điển và “con đường cửa sau thay thế” đều rất quan trọng để thúc đẩy sự nam hóa bình thường của thai nhi XY. Các protein cần thiết cho sinh tổng hợp testosterone bao gồm NR5A1, thụ thể LH, protein điều hòa cấp tính sinh tổng hợp steroid (StAR), P450scc, P450c17, 3β-hydroxysteroid dehydrogenase loại 2, 17β-hydroxysteroid dehydrogenase loại 3, P450-oxidoreductase, 5α-reductase loại 1 và 2, aldo-keto reductase loại 1C2 (AKR1C2), và aldo-keto reductase loại 1C4 (AKR1C4) (xem Hình 6.3). Các enzyme, ví dụ như SRD5A1, liên quan đến con đường cửa sau thay thế chủ yếu được biểu hiện ở các mô ngoài tuyến sinh dục, chẳng hạn như gan và nhau thai. Dựa trên một số ít thai nhi nam ở giữa thai kỳ, androsterone được báo cáo là androgen cửa sau chính trong tuần hoàn của thai nhi nam; những dữ liệu này cũng cho thấy progesterone của nhau thai cung cấp chất nền cho sinh tổng hợp androsterone.
Các lỗi bẩm sinh trong sinh tổng hợp glucocorticoid thường liên quan đến các dạng tăng sản thượng thận bẩm sinh (CAH) nam hóa. Vỏ thượng thận của thai nhi có nguồn gốc từ biểu mô thể xoang và bao gồm hai vùng chính: vùng thai nhi và vùng người lớn. Vùng thai nhi chủ yếu chịu trách nhiệm tổng hợp DHEA, sau đó được sulfat hóa để cung cấp chất nền cho sinh tổng hợp estrogen của nhau thai (xem Hình 6.3). Vùng người lớn, sau khi sinh sẽ biệt hóa thành ba lớp của vỏ thượng thận người lớn, chủ yếu chịu trách nhiệm sinh tổng hợp cortisol. Vào tuần thứ 10 của thai kỳ, tuyến thượng thận tiết ra DHEA-S và trục hạ đồi-tuyến yên-tuyến thượng thận đã hoạt động. Mức độ biểu hiện phiên mã CYP17A1 của nhau thai vẫn chưa được giải quyết, nhưng hầu hết các dữ liệu gần đây cho thấy biểu hiện ở mức tối thiểu.
Gen Thụ thể Luteinizing Hormone Choriogonadotropin
Thiểu sản tế bào Leydig là một rối loạn di truyền lặn trên nhiễm sắc thể thường đặc trưng bởi sự thất bại trong biệt hóa tế bào Leydig của tinh hoàn, thứ phát sau các đột biến bất hoạt của LHCGR, dẫn đến sự đề kháng của tế bào đích với LH. Gen LHCGR, được xác định vị trí trên nhiễm sắc thể 2p21, mã hóa một protein 674 axit amin. LHCGR là một thụ thể bảy miền xuyên màng kết hợp với protein G. Các cơ chế cụ thể mà qua đó các đột biến mất chức năng gây ra sự đề kháng LH bao gồm giảm protein thụ thể, giảm liên kết với phối tử, thay đổi quá trình vận chuyển thụ thể và suy giảm khả năng kích hoạt Gs.
Không có khả năng đáp ứng với hCG hoặc LH làm giảm sinh tổng hợp testosterone của tế bào Leydig. Kiểu hình của trẻ sơ sinh 46,XY bị ảnh hưởng dao động từ nam hóa không hoàn toàn đến cơ quan sinh dục ngoài của nữ giới. Các cá thể bị ảnh hưởng được nuôi dưỡng như nữ giới thường tìm đến bác sĩ vì chậm phát triển vú. Các dẫn xuất ống Müller không có vì AMH được tiết ra bởi các tế bào Sertoli không bị ảnh hưởng. Tinh hoàn thường ở bẹn hoặc trong ổ bụng. Các xét nghiệm cho thấy LH tăng, testosterone thấp và nồng độ FSH bình thường. Không có đáp ứng testosterone đáng kể với kích thích hCG. Mô học tinh hoàn cho thấy không có tế bào Leydig; các ống sinh tinh và thể tích tinh hoàn được bảo tồn do sự tiết và đáp ứng FSH bình thường. Nam hóa không hoàn toàn với lỗ tiểu lệch thấp, dương vật nhỏ và tinh hoàn ẩn có thể xảy ra với các đột biến sai nghĩa mất chức năng không hoàn toàn.
Các nữ giới di truyền, chị em của các cá thể 46,XY bị ảnh hưởng, mang các đột biến giống hệt nhau cho thấy sự biệt hóa sinh dục nữ bình thường và phát triển dậy thì bình thường nhưng bị vô kinh và vô sinh. Tử cung có kích thước từ nhỏ đến bình thường. U nang buồng trứng có thể phát triển. Nồng độ estrogen thường không đạt được ngưỡng cần thiết cho sự rụng trứng. Phân tích di truyền có thể hữu ích để phân biệt các đột biến LHCGR với các rối loạn khác ảnh hưởng đến sinh tổng hợp testosterone.
Tăng sản thượng thận bẩm sinh dạng Lipid
Rối loạn di truyền lặn trên nhiễm sắc thể thường này được đặc trưng bởi một khiếm khuyết nghiêm trọng trong việc chuyển đổi cholesterol thành pregnenolone, dẫn đến suy giảm tổng hợp tất cả các hormone steroid của tuyến thượng thận và tuyến sinh dục. Suy giảm sinh tổng hợp testosterone trong tử cung ngăn cản sự biệt hóa giới tính nam. Do đó, tất cả các thai nhi bị ảnh hưởng (46,XY hoặc 46,XX) đều có cơ quan sinh dục ngoài của nữ giới. Ở thai nhi XY, các tế bào Sertoli còn nguyên vẹn, sự tiết AMH không bị ảnh hưởng và các yếu tố ống Müller thoái triển. Nồng độ hormone steroid thấp hoặc không thể phát hiện, nồng độ ACTH tăng và hoạt độ renin huyết tương (PRA) tăng phù hợp với chẩn đoán này. Nồng độ 17-hydroxypregnenolone hoặc pregnenolone tăng giúp phân biệt thiếu hụt 3β-hydroxysteroid dehydrogenase với tăng sản thượng thận bẩm sinh dạng lipid.
Tăng sản thượng thận bẩm sinh dạng lipid là do các đột biến trong gen StAR được xác định vị trí trên nhiễm sắc thể 8p11.23. Protein StAR tạo điều kiện cho việc vận chuyển cholesterol qua ty thể đến P450scc. Trong tăng sản thượng thận bẩm sinh dạng lipid, việc vận chuyển cholesterol vào ty thể bị suy giảm dẫn đến tích tụ các este cholesterol và các sản phẩm tự oxy hóa sterol. Cuối cùng, sự tích tụ lipid làm thay đổi cấu trúc tế bào gây ra sự phá hủy tế bào và mất hoàn toàn quá trình sinh tổng hợp steroid phụ thuộc StAR. Do đó, tăng sản thượng thận bẩm sinh dạng lipid có “hai tác động”: một là khiếm khuyết sinh tổng hợp steroid và hai là phá hủy tế bào sinh steroid. Vì vùng xác định của vỏ thượng thận tương đối im lặng trong thai kỳ, thiếu hụt aldosterone có thể không rõ ràng trong giai đoạn sơ sinh ngay lập tức. Cơ chế hai tác động này cho phép sự phát triển dậy thì tự nhiên ở các bé gái bị ảnh hưởng vì quá trình tổng hợp estrogen đáng kể không diễn ra cho đến tuổi dậy thì. Các tế bào nang buồng trứng được tuyển chọn và bị tổn thương tuần tự, dẫn đến suy sinh dục tăng gonadotropin.
CAH dạng lipid không cổ điển là một dạng nhẹ hơn với chức năng tuyến thượng thận và tuyến sinh dục thay đổi, biểu hiện ở thời thơ ấu và thanh thiếu niên. CAH dạng lipid không cổ điển có thể biểu hiện với thiếu hụt glucocorticoid đơn độc và giống với thiếu hụt glucocorticoid có tính gia đình do các đột biến trong gen thụ thể ACTH (MC2R).
Ở người, vì 17-OHP không phải là chất nền được ưa chuộng cho phản ứng 17,20-lyase, con đường này có tầm quan trọng chức năng trong các rối loạn sinh tổng hợp steroid liên quan đến tăng nồng độ 17-OHP.
Enzyme Cytochrome P450 cắt chuỗi bên
Enzyme cắt chuỗi bên (còn được gọi là cholesterol desmolase) là một enzyme cytochrome P450 được mã hóa bởi gen CYP11A1 nằm ở nhiễm sắc thể 15q23-q24. Enzyme này chuyển đổi cholesterol thành pregnenolone, rất cần thiết cho quá trình sinh tổng hợp steroid và đóng một vai trò quan trọng trong tổng hợp progesterone của nhau thai. Trẻ em bị ảnh hưởng bởi rối loạn di truyền lặn trên nhiễm sắc thể thường này bị suy thượng thận và có cơ quan sinh dục ngoài của nữ giới bất kể bộ nhiễm sắc thể. Sau khi sinh, sự phì đại tuyến thượng thận thường không rõ ràng trên siêu âm hoặc hình ảnh cộng hưởng từ. Sự vắng mặt của mô tuyến thượng thận/tuyến sinh dục được cho là do sự tích tụ lipid tương tự như sự phá hủy tế bào được quan sát thấy ở bệnh nhân có đột biến StAR. Phổ kiểu hình bao gồm suy thượng thận với lỗ tiểu lệch thấp, phát triển cơ quan sinh dục ngoài bình thường, tiến triển dậy thì bình thường hoặc suy tuyến sinh dục. Các đột biến CYP11A1 đã được phát hiện ở những bệnh nhân bị suy thượng thận nguyên phát. Phân tích di truyền có thể cần thiết để phân biệt rối loạn này với tăng sản thượng thận bẩm sinh dạng lipid.
Tăng sản thượng thận bẩm sinh nam hóa
Các dạng CAH nam hóa là một nhóm các rối loạn gây ra bởi các đột biến trong các gen enzyme sinh tổng hợp steroid liên quan đến sinh tổng hợp cortisol. Các gen này là 3β-hydroxysteroid dehydrogenase loại 2 (HSD3B2), 21-hydroxylase (CYP21A2), và 11β-hydroxylase (CYP11B1). Ba rối loạn này có chung một sinh lý bệnh. Sản xuất cortisol không đủ dẫn đến giảm ức chế phản hồi ngược, tăng sản xuất ACTH, tích tụ các chất trung gian steroid trước enzyme thiếu hụt và tăng nồng độ androgen. Các biểu hiện cụ thể và các bất thường xét nghiệm thay đổi tùy thuộc vào gen enzyme nào bị liên quan. Thật vậy, mức độ thiếu hụt glucocorticoid và mineralocorticoid thường thay đổi tương ứng với mức độ nghiêm trọng của sự thiếu hụt enzyme.
Sự tích tụ các chất trung gian steroid, chẳng hạn như 17-OHP, dẫn đến tăng nồng độ androgen. Ở nữ giới bị ảnh hưởng, việc tiếp xúc với androgen tăng lên sẽ thúc đẩy sự nam hóa của cơ quan sinh dục ngoài. 17-OHP dư thừa có thể được chuyển đổi qua con đường cửa sau thay thế thành DHT. Con đường thay thế này bao gồm khử 5α và 3α của 17-OHP thành 5α-pregnane-3α,17α-diol-20-one (pdiol), cuối cùng tạo ra androstanediol, là chất nền cho quá trình oxy hóa 3α và chuyển đổi thành DHT (Hình 6.4). Trong cuộc sống bào thai, sự tích tụ 17-OHP do các đột biến trong CYP21A2, CYP11B2, hoặc P450-oxidoreductase (POR) có thể làm tăng dòng chảy qua “con đường cửa sau” này, dẫn đến tăng nồng độ DHT.
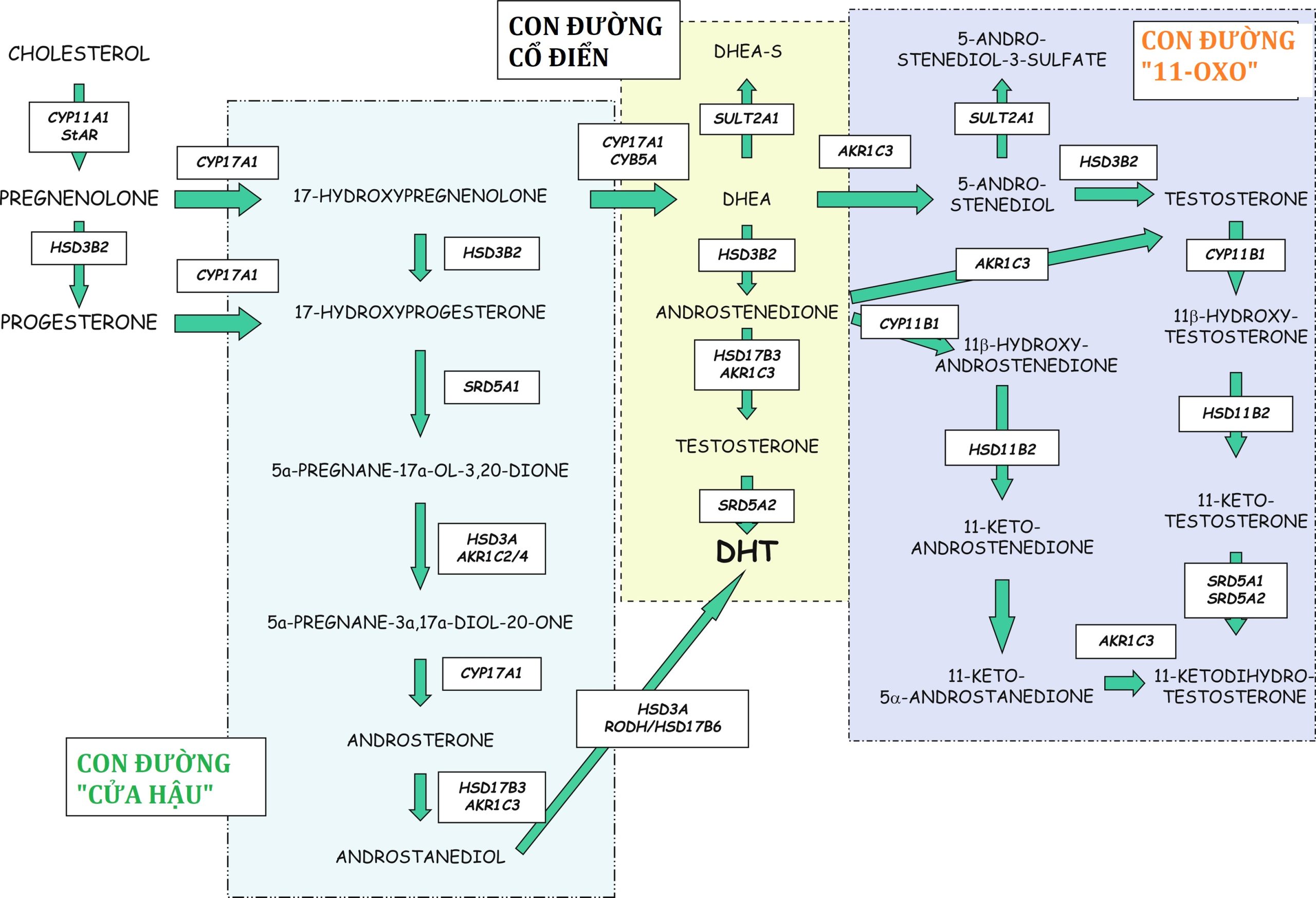
Hình 6.4 Các con đường sinh tổng hợp steroid cổ điển, cửa sau và 11-oxo-C19. Con đường sinh tổng hợp steroid cổ điển liên quan đến tinh hoàn (nền sáng), “con đường cửa sau” (nền tối), và các androgen 11-oxo-C19 (nền tím) được chỉ ra. Khi có nồng độ ACTH và 17-OHP tăng do đột biến CYP21, CYP11B1, hoặc POR, con đường cửa sau có thể góp phần vào nồng độ androgen quá mức gây ra sự nam hóa của thai nhi XX.
Thiếu hụt 21-Hydroxylase
Loại CAH phổ biến nhất (chiếm 90%–95% các trường hợp) là thiếu hụt 21-hydroxylase do các đột biến trong gen 21-hydroxylase (CYP21A2) nằm ở nhiễm sắc thể 6p21.3 trong vùng kháng nguyên bạch cầu người (HLA) lớp III. Tỷ lệ mắc các dạng cổ điển được báo cáo dao động từ 1:5000 đến 1:15.000 và thay đổi giữa các nhóm dân tộc/chủng tộc. Hoạt động 21-hydroxylase giảm làm suy giảm quá trình chuyển đổi 17-OHP thành 11-deoxycortisol ở lớp bó (nơi chủ yếu sinh tổng hợp cortisol) và chuyển đổi progesterone thành deoxycorticosterone ở lớp cầu, nơi chủ yếu sinh tổng hợp aldosterone. Một hướng dẫn thực hành lâm sàng sâu rộng gần đây đã được công bố.
Các bé gái sơ sinh bị thiếu hụt 21-hydroxylase thể mất muối cổ điển thường có biểu hiện ngay trong giai đoạn sơ sinh do cơ quan sinh dục không điển hình (Hình 6.5). Khi chẩn đoán bị trì hoãn, các bé gái bị ảnh hưởng sẽ phát triển tình trạng mất nước, hạ natri máu và tăng kali máu do thiếu hụt glucocorticoid và mineralocorticoid. Ở các nữ giới bị ảnh hưởng, phổ nam hóa sinh dục thay đổi từ phì đại âm vật đến lỗ tiểu lệch thấp ở đáy chậu đến sự hợp nhất hoàn toàn của các nếp gấp môi-niệu đạo và môi-bìu, tạo ra một dương vật với dây cong và lỗ niệu đạo ở đầu dương vật. Mức độ nam hóa cơ quan sinh dục ngoài có thể lớn đến mức các bé gái sơ sinh bị ảnh hưởng trông giống như bé trai với tinh hoàn ẩn hai bên. Trừ khi được xác định bằng sàng lọc sơ sinh, các bé trai thường có biểu hiện ở 2 đến 3 tuần tuổi với chậm lớn, bú kém, lờ đờ, mất nước, hạ huyết áp, hạ natri máu và tăng kali máu. Khi chẩn đoán bị trì hoãn hoặc bỏ sót, CAH có khả năng gây tử vong. Các chương trình sàng lọc sơ sinh làm giảm tỷ lệ mắc bệnh và tử vong liên quan đến suy thượng thận cấp.
Hình 6.5 Cơ quan sinh dục không điển hình ở nữ giới bị nam hóa do thiếu 21-hydroxylase. Các nếp gấp môi-bìu hợp nhất và âm vật phì đại.
Ở trẻ sơ sinh bị ảnh hưởng, nồng độ 17-OHP ngẫu nhiên thường tăng cao. Nồng độ lớn hơn 5000 ng/dL và có thể cao hơn nhiều. Nồng độ androstenedione và progesterone cũng thường tăng. Trong một số trường hợp, PRA có thể hữu ích để đánh giá tình trạng mineralocorticoid. Việc đo 21-deoxycortisol cực kỳ hữu ích, nhưng sự sẵn có của xét nghiệm hormone này còn hạn chế. Đối với trẻ sơ sinh nữ, có tử cung bình thường và có thể được xác định trên siêu âm. Buồng trứng có thể quá nhỏ để có thể dễ dàng xác định trên siêu âm. Mặc dù tiếp xúc với androgen trước sinh quá mức, vị trí buồng trứng vẫn bình thường và các cấu trúc Wolffian bên trong không được giữ lại.
Phổ hoạt động 21-hydroxylase bị suy giảm dao động từ thiếu hụt glucocorticoid và mineralocorticoid hoàn toàn đến các thiếu hụt nhẹ biểu hiện chủ yếu bằng sự tiết androgen thượng thận quá mức bù trừ. Trẻ sơ sinh có khả năng tổng hợp aldosterone đầy đủ thường không biểu hiện mất muối rõ ràng. Trẻ sơ sinh nữ có khả năng tổng hợp aldosterone đầy đủ vẫn có thể có đủ phơi nhiễm androgen trong tử cung để nam hóa cơ quan sinh dục ngoài. Trong trường hợp không có các chương trình sàng lọc sơ sinh, các bé trai bị ảnh hưởng có khả năng sinh tổng hợp aldosterone có thể không được xác định cho đến khi chúng có biểu hiện phát triển sinh dục quá mức hoặc dậy thì sớm. Trẻ sơ sinh với các dạng CAH nhẹ hơn thường không được xác định bởi hầu hết các chương trình sàng lọc sơ sinh. Phân tích sắc ký khí/khối phổ hormone steroid trong nước tiểu cho thấy tỷ lệ pdiol so với các chất chuyển hóa của con đường Δ4 và Δ5 tăng, điều này cho thấy hoạt động sau sinh của con đường cửa sau, đặc biệt là trong giai đoạn đầu của trẻ sơ sinh.
CYP21A2 nằm cách một gen giả có độ tương đồng cao, CYP21A1P, khoảng 30 kilobase. Gen tenascin-XB (TNXB) mã hóa một protein chất nền ngoại bào nằm trên sợi DNA đối diện với CYP21A2. Hiện tại, hơn 200 đột biến CYP21A2 đã được báo cáo. Tuy nhiên, chỉ một vài đột biến chiếm phần lớn các alen bị ảnh hưởng. Hầu hết các đột biến phổ biến đại diện cho các sự kiện chuyển đổi gen trong đó CYP21A2 đã thu được các trình tự CYP21A1P có hại. Tần suất của các đột biến cụ thể thay đổi giữa các nhóm dân tộc. Việc xác định kiểu gen phân tử có thể là một phương pháp bổ trợ hữu ích cho sàng lọc sơ sinh. Cần lưu ý rằng nhiều đột biến có thể xảy ra trên một alen duy nhất và các đột biến CYP21A2 khác nhau có thể xảy ra trong một gia đình.
Thiếu hụt 11β-Hydroxylase
CAH do thiếu hụt 11β-hydroxylase được đặc trưng bởi thiếu hụt glucocorticoid, tiết androgen quá mức và tăng huyết áp. Dạng CAH này là do các đột biến trong gen 11β-hydroxylase (CYP11B1). Enzyme này được biểu hiện ở lớp bó, nơi nó chuyển đổi 11-deoxycortisol thành cortisol. CAH, do các đột biến CYP11B1, là hiếm gặp (3%–5% các trường hợp) ngoại trừ tỷ lệ mắc cao ở người Do Thái Ma-rốc, với tỷ lệ mắc bệnh gần một trong 6000. Mặc dù có cùng một đột biến (R448H), sự không đồng nhất về kiểu hình đối với mức độ nam hóa và tăng huyết áp xảy ra ngay cả trong một gia đình. Các nữ giới bị ảnh hưởng có thể có cơ quan sinh dục không điển hình. Phát hiện xét nghiệm điển hình là nồng độ 11-deoxycortisol tăng. Nồng độ 17-hydroxyprogesterone, androstenedione và testosterone trong huyết thanh có thể tăng nhẹ. Nồng độ PRA thấp hoặc bị ức chế. Tuy nhiên, trẻ sơ sinh có thể bị mất muối có lẽ do kháng mineralocorticoid. Bệnh nhân với các dạng không cổ điển đã được xác định. Mặc dù các đáp ứng hormone được kích thích bằng ACTH ở những người mang gen dị hợp tử thường bình thường, nhưng nồng độ 11-deoxycortisol và 11-deoxycorticosterone tăng đã được báo cáo.
Gen CYP11B1 nằm ở nhiễm sắc thể 8q22 gần với một gen có độ tương đồng cao là CYP11B2, mã hóa cho aldosterone synthase. Các đột biến mới liên quan đến thiếu hụt 11β-hydroxylase cổ điển và không cổ điển đã được xác định. CYP11B1 được biểu hiện ở lớp bó, trong khi CYP11B2 được biểu hiện chủ yếu ở lớp cầu.
Thiếu hụt 3β-Hydroxysteroid Dehydrogenase
CAH do thiếu hụt 3β-hydroxysteroid dehydrogenase loại 2, liên quan đến các đột biến trong gen 3β-hydroxysteroid dehydrogenase loại 2 (HSD3B2), dẫn đến sự nam hóa cơ quan sinh dục ngoài của thai nhi 46,XX do tăng tổng hợp DHEA. Các thai nhi 46,XY bị ảnh hưởng có cơ quan sinh dục không điển hình đặc trưng bởi sự nam hóa không hoàn toàn của cơ quan sinh dục ngoài thứ phát sau thiếu hụt testosterone. Mặc dù giảm tổng hợp testosterone, các thai nhi 46,XY bị ảnh hưởng thường có các cấu trúc ống Wolff còn nguyên vẹn (bao gồm cả ống dẫn tinh). Enzyme phụ thuộc NAD+ 3β-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase xúc tác quá trình chuyển đổi các tiền chất steroid Δ5, pregnenolone, 17-hydroxypregnenolone và DHEA thành các Δ4-ketosteroid tương ứng, progesterone, 17-OHP và androstenedione.
Hai isozyme được mã hóa bởi hai gen khác nhau có độ tương đồng cao đã được xác định và định vị trên nhiễm sắc thể 1p13.1. Gen loại 1 (HSD3B1) được biểu hiện chủ yếu ở da, nhau thai, tuyến tiền liệt và các mô ngoại vi khác, trong khi HSD3B2 được biểu hiện chủ yếu ở vỏ thượng thận và tuyến sinh dục. Các đột biến trong HSD3B2, nhưng không phải HSD3B1, đã được phát hiện ở những bệnh nhân bị CAH do thiếu 3β-hydroxysteroid dehydrogenase. Suy thượng thận cấp xảy ra trong giai đoạn sơ sinh khi các đột biến mất chức năng hoàn toàn làm suy giảm sinh tổng hợp mineralocorticoid, glucocorticoid và steroid sinh dục. Các biểu hiện điển hình cho các dạng không mất muối bao gồm dậy thì sớm và (ở trẻ sơ sinh 46,XY) lỗ tiểu lệch thấp ở đáy chậu. Các phát hiện xét nghiệm xác nhận bao gồm nồng độ pregnenolone, 17-hydroxypregnenolone và DHEA tăng với tỷ lệ steroid Δ5 so với Δ4 tăng. Vì hoạt động enzyme của isozyme loại 1 không bị suy giảm, nồng độ 17-OHP và androstenedione tăng có thể được tìm thấy.
Khiếm khuyết trong sinh tổng hợp steroid sinh dục
Thiếu hụt 17α-Hydroxylase/17,20-Lyase
Sự tổng hợp mineralocorticoid, glucocorticoid và steroid sinh dục được điều chỉnh bởi enzyme, 17α-hydroxylase/17-20-lyase. Enzyme này, được mã hóa bởi một gen duy nhất, CYP17A1 được định vị trên nhiễm sắc thể 10q24.3, xúc tác hai bước riêng biệt trong quá trình sinh tổng hợp steroid, pregnenolone được 17α-hydroxyl hóa thành 17α-hydroxypregnenolone, sau đó trải qua hoạt động 17,20-lyase để tạo thành DHEA. Các yếu tố thuận lợi cho phản ứng 17,20-lyase bao gồm sự sẵn có của P450 oxidoreductase và cytochrome b5 và sự phosphoryl hóa serine/threonine của protein P450c17.
Các đột biến mất chức năng cản trở sinh tổng hợp glucocorticoid và steroid sinh dục. Nam giới bị ảnh hưởng có biểu hiện nam hóa không hoàn toàn của cơ quan sinh dục ngoài. Nữ giới bị ảnh hưởng có sự phát triển sinh dục bình thường và biểu hiện dậy thì muộn. Bệnh nhân thường bị tăng huyết áp do tăng tổng hợp mineralocorticoid được kích thích bởi ACTH. Nồng độ progesterone, deoxycortisone, corticosterone, 18-hydroxycorticosterone và 18-hydroxy deoxycorticosterone (DOC) trong huyết thanh tăng và có thể được ức chế bằng liệu pháp thay thế glucocorticoid. Nồng độ glucocorticoid và steroid sinh dục thấp hoặc không có với đáp ứng kém khi kích thích ACTH phù hợp với chẩn đoán này. Các đột biến chỉ ảnh hưởng đến hoạt động 17,20-lyase là cực kỳ hiếm. Kiểu hình thiếu 17,20-lyase đơn độc rõ ràng có thể do các đột biến trong các gen khác, chẳng hạn như cytochrome b5, P450 oxidoreductase và một số enzyme aldo-keto reductase, như được mô tả sau này.
Thiếu hụt Cytochrome b5
Cytochrome b5 tham gia vào quá trình chuyển electron cho một số phản ứng cytochrome P450. Mặc dù nó không phải là một chất cho electron hiệu quả cho P450c17, protein này cho phép sự tương tác giữa P450c17 và oxidoreductase để thúc đẩy phản ứng 17,20-lyase cần thiết cho tổng hợp steroid sinh dục. Một trẻ sơ sinh 46,XY được phát hiện đồng hợp tử với một đột biến mất chức năng trong gen cytochrome b5 (CYB5) được mô tả là có dương vật nhỏ, bìu chẻ đôi, lỗ tiểu lệch thấp ở bìu, nồng độ DHEA-S không thể phát hiện, nồng độ testosterone thấp và nồng độ methemoglobin tăng. Một đột biến sai nghĩa liên quan đến methemoglobin huyết nhẹ và các anh chị em 46,XY nam hóa không hoàn toàn đã được mô tả.
Thiếu hụt Isozyme 3α-Hydroxysteroid Dehydrogenase
Việc đánh giá lại các gia đình ban đầu được báo cáo là có thiếu hụt 17,20-lyase đơn độc đã dẫn đến việc xác định các con đường thay thế cho tổng hợp androgen. Điều tra một gia đình cho thấy không có đột biến nào trong CYP17A1, NR5A1, POR, hoặc CYB5. Thay vào đó, các đột biến đã được phát hiện trong gen 3α-hydroxysteroid dehydrogenase loại III (AKR1C2), được định vị trên nhiễm sắc thể 10p15.1. Kiểu hình, nam hóa không hoàn toàn của các cá thể 46,XY bị ảnh hưởng, tuân theo kiểu di truyền lặn liên kết giới tính. Một đột biến ghép nối trong một gen 3α-HSD liên quan chặt chẽ khác, AKR1C4, đã được tìm thấy trong một gia đình thứ hai. Di truyền hai gen liên quan đến các đột biến ở cả AKR1C2 và AKR1C4 đã được tìm thấy trong gia đình thứ hai này. Do đó, cả hai con đường cổ điển và cửa sau dường như đều cần thiết cho sự phát triển cơ quan sinh dục ngoài bình thường của nam giới. Tuy nhiên, các chi tiết về vai trò cụ thể của các enzyme này vẫn cần được làm sáng tỏ.
Thiếu hụt Cytochrome P450 Oxidoreductase
Năm 1985, một rối loạn với bằng chứng sinh hóa, cho thấy hoạt động 17α-hydroxylase và 21-hydroxylase giảm, đã được báo cáo lần đầu tiên. Rối loạn này được phát hiện có liên quan đến các đột biến trong gen cytochrome P450 oxidoreductase (POR), được định vị trên nhiễm sắc thể 7q11-12. POR mã hóa một protein hoạt động như một chất cho electron bắt buộc cho các enzyme P450 sinh steroid và gan ở microsome. Protein POR đóng một vai trò quan trọng trong tổng hợp glucocorticoid và steroid sinh dục.
Các đặc điểm lâm sàng bao gồm cơ quan sinh dục không điển hình, dính khớp sọ, thiểu sản giữa mặt và dính khớp quay-cánh tay. Khi sinh, cơ quan sinh dục không điển hình được ghi nhận ở cả trẻ sơ sinh nam và nữ. Tuy nhiên, sự nam hóa tiến triển sau sinh không xảy ra. Tổng hợp testosterone không đủ có khả năng gây ra sự nam hóa không hoàn toàn của trẻ sơ sinh nam. Sự nam hóa của thai nhi nữ được cho là do sự chuyển hướng của 17-OHP dư thừa sang “con đường cửa sau”, dẫn đến tăng tổng hợp DHT. Trong thai kỳ, một số bà mẹ phát triển các dấu hiệu liên quan đến dư thừa androgen, chẳng hạn như mụn trứng cá, rậm lông và phì đại âm vật. Các phát hiện xét nghiệm điển hình bao gồm 17-OHP tăng, steroid sinh dục thấp và nồng độ mineralocorticoid bình thường. Một số cá nhân bị ảnh hưởng có thể được hưởng lợi từ liệu pháp thay thế glucocorticoid hàng ngày. Những người khác có thể cần điều trị glucocorticoid chỉ cho liều stress.
Các dị tật xương giống với những dị tật được tìm thấy trong hội chứng Antley-Bixler, một rối loạn di truyền trội trên nhiễm sắc thể thường liên quan đến các đột biến trong gen FGFR2. Bệnh nhân mắc hội chứng Antley-Bixler do đột biến FGFR2 có quá trình sinh tổng hợp steroid bình thường, trong khi bệnh nhân có đột biến POR có quá trình sinh tổng hợp steroid bất thường. Cơ sở phân tử của các bất thường xương không rõ ràng nhưng bị nghi ngờ là do suy giảm hoạt động của các enzyme liên quan đến sinh tổng hợp sterol, chẳng hạn như 14α-lanosterol demethylase (CYP51A1) và squalene epoxidase hoặc chuyển hóa axit retinoic.
Thiếu hụt 17β-Hydroxysteroid Dehydrogenase
Rối loạn này là do các đột biến của gen 17β-hydroxysteroid dehydrogenase loại 3 (HSD17B3) nằm ở nhiễm sắc thể 9q22. Enzyme này được biểu hiện gần như độc quyền trong tinh hoàn, nơi nó chuyển đổi androstenedione thành testosterone. Các đột biến mất chức năng dẫn đến thiếu hụt testosterone và sau đó là sự nam hóa không hoàn toàn của thai nhi 46,XY. Trong rối loạn di truyền lặn trên nhiễm sắc thể thường này, cơ quan sinh dục ngoài dao động từ nữ giới với lỗ tiểu lệch thấp ở đáy chậu và túi âm đạo cụt đến không điển hình với sự hợp nhất môi-bìu đến lỗ tiểu lệch thấp. Tinh hoàn có mặt và có thể sờ thấy trong các nếp gấp môi-bìu hoặc xuống không hoàn toàn. Mặc dù có cơ quan sinh dục ngoài của nữ giới, các cấu trúc Wolffian thường có mặt.
Khi không được nhận biết, bệnh nhân thường được coi là nữ giới khi sinh. Ở tuổi dậy thì, sự nam hóa tiến triển xảy ra. Mức độ nam hóa dao động từ rậm lông, giọng nói trầm và phì đại âm vật. Sự nam hóa được cho là do sự chuyển đổi ngoại tinh hoàn của androstenedione thành testosterone. Một số cá nhân bị ảnh hưởng thay đổi vai trò giới từ nữ sang nam. Do nguy cơ tân sinh tuyến sinh dục dường như thấp, không có chống chỉ định nào đối với việc thay đổi vai trò giới, chỉ dấu giới và giữ lại tinh hoàn ở vị trí bìu. Sự chuyển đổi tăng của androstenedione thành estrogen có thể gây ra vú to ở nam giới.
Việc xác định giới tính nam phù hợp có thể được thực hiện trong giai đoạn sơ sinh khi chẩn đoán được nghi ngờ và xác nhận. Các đặc điểm xét nghiệm chẩn đoán bao gồm tỷ lệ androstenedione/testosterone cơ bản và sau khi kích thích hCG tăng. Các đặc điểm lâm sàng tương tự như các đặc điểm của thiếu 5α-reductase và không nhạy cảm với androgen; phân tích di truyền phân tử có thể có lợi để xác nhận chẩn đoán. Các bé gái 46,XX bị ảnh hưởng thường không có triệu chứng, có cơ quan sinh dục trong và ngoài của nữ giới bình thường và phát triển dậy thì bình thường; vô sinh đã được báo cáo ở một số trường hợp.
Thiếu hụt 5α-Reductase
Rối loạn di truyền lặn trên nhiễm sắc thể thường này là do các đột biến trong gen 5α-reductase loại 2 (SRD5A2), nằm ở nhiễm sắc thể 2p23. Gen này được biểu hiện chủ yếu ở các mô đích của androgen, nơi nó chuyển đổi testosterone thành DHT. Các cụm cá nhân có đột biến SRD5A2 đã được mô tả ở các vùng của Cộng hòa Dominica, Papua New Guinea, Thổ Nhĩ Kỳ và Trung Đông.
Các cá thể 46,XY bị ảnh hưởng có cơ quan sinh dục không điển hình đặc trưng bởi sự biệt hóa của các cấu trúc Wolffian, không có các cấu trúc có nguồn gốc từ ống Müller, dương vật nhỏ, xoang niệu-sinh dục với lỗ tiểu lệch thấp ở đáy chậu và túi âm đạo cụt. Bệnh nhân được nuôi dưỡng như nữ giới có thể có biểu hiện vô kinh nguyên phát thường liên quan đến cơ quan sinh dục không điển hình. Các trường hợp mất đoạn, đột biến sai nghĩa và dị nhiễm sắc thể đơn bội từ một phía đã được báo cáo. Do sự không đồng nhất rộng rãi trong các đặc điểm lâm sàng, mối tương quan kiểu hình/kiểu gen không được thiết lập rõ ràng. Tỷ lệ testosterone/DHT ngẫu nhiên có thể không xác định được các cá nhân bị ảnh hưởng; tỷ lệ testosterone/DHT được kích thích bằng hCG có thể cần thiết. Cần lưu ý về chẩn đoán này bao gồm sự cần thiết của kích thích hCG ở bệnh nhân trước dậy thì và xác định hormone chính xác trong đó phản ứng chéo giữa testosterone và DHT được giảm thiểu.
Ở tuổi dậy thì, sự nam hóa tiến triển xảy ra với sự phát triển cơ bắp, thay đổi giọng nói và phì đại dương vật. Các đặc điểm này được cho là do hoạt động của 5α-reductase loại 1 (SRD5A1). SRD5A1, nằm ở nhiễm sắc thể 5p15, được biểu hiện ở da và da đầu sau dậy thì. Sự không đồng nhất về kiểu hình xảy ra thường xuyên. Một số cá nhân 46,XY mắc chứng rối loạn này được nuôi dưỡng như con gái đã phát triển bản dạng giới nam và thay đổi vai trò giới ở tuổi vị thành niên hoặc tuổi trưởng thành. Mặc dù có sự nam hóa, lông mặt có xu hướng thưa thớt, tuyến tiền liệt bị thiểu sản, tinh dịch có xu hướng nhớt và lượng xuất tinh thấp. Nam giới bị ảnh hưởng thường bị không có tinh trùng hoặc ít tinh trùng. Thụ tinh trong tử cung bằng tinh trùng của một nam giới bị ảnh hưởng đã dẫn đến mang thai.
Thiếu hụt Aromatase nhau thai
Thiếu hụt aromatase nhau thai là một rối loạn di truyền lặn trên nhiễm sắc thể thường hiếm gặp phát sinh từ đột biến trong gen aromatase, CYP19A1, nằm ở nhiễm sắc thể 15q21.2 và mã hóa một protein 503 axit amin. Các đột biến bất hoạt CYP19A1 làm suy giảm quá trình chuyển đổi androgen thành estrogen, dẫn đến tăng androgen. Trong các thai kỳ có thai nhi bị ảnh hưởng, sự nam hóa tiến triển của người mẹ đặc trưng bởi rậm lông, phì đại âm vật, mụn trứng cá và hói trán xảy ra. Trong thai kỳ, nồng độ testosterone, DHT và androstenedione tăng và nồng độ estradiol, estrone và estriol thấp. Trong thời kỳ hậu sản, một số đặc điểm lâm sàng của tình trạng dư thừa androgen thoái lui và nồng độ androgen tăng cao trở lại mức bình thường.
Khi sinh, trẻ sơ sinh 46,XX bị nam hóa ở các mức độ khác nhau với sự hợp nhất môi-bìu, phì đại âm vật và lỗ tiểu lệch thấp ở đáy chậu. Các cá thể 46,XX bị ảnh hưởng thường biểu hiện dậy thì muộn đặc trưng bởi sự phát triển vú tối thiểu hoặc không có, vô kinh nguyên phát, suy sinh dục tăng gonadotropin, buồng trứng đa nang và giảm mật độ khoáng của xương. Khi sinh, trẻ sơ sinh 46,XY bị ảnh hưởng có sự phát triển sinh dục trong và ngoài bình thường. Nam giới bị ảnh hưởng thường có biểu hiện sau dậy thì với tầm vóc cao, đau xương, trưởng thành xương chậm và vô sinh. Điều tra những người đàn ông thiếu hụt aromatase cho thấy thiếu hụt estrogen có liên quan đến béo bụng, kháng insulin, rối loạn lipid máu và vô sinh tương đối. Bệnh nhân có các đặc điểm kiểu hình ít nghiêm trọng hơn đã được mô tả.
Aromatase là một enzyme cytochrome P450 đóng vai trò quan trọng trong sinh tổng hợp estrogen (steroid C18) từ androgen (steroid C19). Ngoài vai trò trong sinh tổng hợp estrogen ở thanh thiếu niên và người lớn, aromatase nằm trong nhau thai người chuyển đổi androgen thượng thận của thai nhi thành estrogen và bảo vệ người mẹ khỏi các tác động nam hóa tiềm tàng của androgen của thai nhi.
Tăng androgen huyết ở mẹ
Trong thai kỳ, tăng androgen huyết ở mẹ có thể xảy ra thứ phát sau u tế bào lutein của thai kỳ, các khối u tiết androgen và tiếp xúc với androgen ngoại sinh. Nồng độ androgen quá mức của người mẹ có thể gây ra sự nam hóa cơ quan sinh dục ngoài của thai nhi 46,XX. Các chất gây rối loạn nội tiết là các hóa chất hoặc hỗn hợp hóa chất ngoại sinh cản trở bất kỳ khía cạnh nào của hoạt động hormone. Thuốc trừ sâu organochlorine, biphenyl đa clo hóa (PCB) và alkylpolyethoxylate được coi là “chất gây rối loạn nội tiết” do các đặc tính estrogen và/hoặc kháng androgen của chúng.
Rối loạn hoạt động của androgen
Trong quá trình phát triển giới tính, hoạt động của androgen là cần thiết để thúc đẩy sự duy trì các dẫn xuất ống Wolff, sự phát triển của tuyến tiền liệt và sự biệt hóa của cơ quan sinh dục ngoài của nam giới. Hoạt động của androgen được trung gian bởi thụ thể androgen (AR, còn được gọi là NR3C4), một thành viên của họ thụ thể hormone steroid/thyroid. Tương tự như các thành viên khác của họ thụ thể này, thụ thể androgen là một yếu tố phiên mã phụ thuộc vào phối tử với cấu trúc mô-đun đặc trưng. Các mô-đun chính của protein bao gồm miền kích hoạt phiên mã đầu amino (AF1), miền liên kết DNA và miền liên kết phối tử. Các đặc điểm khác bao gồm tín hiệu định vị hạt nhân, một miền kích hoạt phiên mã khác (AF2) trong miền liên kết phối tử đầu carboxy và vùng bản lề. Protein AR có hai vùng lặp lại trinucleotide đa hình nằm ở miền đầu amino mã hóa cho các vùng lặp lại polyglutamine (CAG) và polyglycine (GCN). Ngoài con đường cổ điển, các hoạt động androgen nhanh không qua gen cũng xảy ra.
Không nhạy cảm với androgen là một rối loạn di truyền lặn liên kết X do các đột biến AR; AR nằm gần tâm động ở Xq11-12. Khoảng 30% các trường hợp là đột biến mới. Thể khảm tế bào soma, khi đột biến phát sinh ở giai đoạn sau hợp tử, có liên quan đến nguy cơ tái phát thấp hơn. Không nhạy cảm với androgen hoàn toàn (CAIS) được đặc trưng bởi cơ quan sinh dục ngoài của nữ giới, không có các dẫn xuất ống Müller, lông mu thưa thớt, các khối u ở bẹn và vô kinh nguyên phát ở các cô gái vị thành niên. Các dẫn xuất ống Wolff (ví dụ: ống dẫn tinh và mào tinh hoàn) không có do thiếu hoạt động của androgen. Mặc dù các trường hợp ngoại lệ hiếm gặp đã được mô tả, các cấu trúc có nguồn gốc từ ống Müller thường không có vì chức năng của tế bào Sertoli là bình thường với sự tiết AMH trong tử cung. Người ta cho rằng 1% đến 2% các bé gái bị thoát vị bẹn hai bên có thể bị không nhạy cảm với androgen. Việc tìm thấy một tuyến sinh dục trong túi thoát vị nên được chỉ định xét nghiệm di truyền tế bào. Sự tăng vọt LH dự kiến trong nồng độ testosterone trong vài tháng đầu đời có thể không có ở một số trẻ sơ sinh bị CAIS. Sự phát triển vú tự nhiên ở tuổi dậy thì do sự thơm hóa của androgen thành estrogen xảy ra nếu các tuyến sinh dục còn tại chỗ.
Không nhạy cảm với androgen một phần (PAIS) được đặc trưng bởi các đặc điểm lâm sàng, cho thấy một phản ứng sinh học một phần với androgen. Các đặc điểm điển hình bao gồm cơ quan sinh dục không điển hình với lỗ tiểu lệch thấp ở đáy chậu, dương vật nhỏ và bìu chẻ đôi. Vị trí tinh hoàn thay đổi, từ không xuống đến có thể sờ thấy trong bìu. Trẻ sơ sinh bị PAIS thường biểu hiện sự tăng vọt testosterone sơ sinh dự kiến, cho thấy rằng khả năng đáp ứng với androgen trước sinh đóng một vai trò trong việc ghi dấu của trục HPG. Các đặc điểm của không nhạy cảm với androgen nhẹ bao gồm vú to ở nam giới và vô sinh ở nam giới bình thường. Tuổi theo thời gian khi có biểu hiện muộn hơn là điển hình. Trong mọi trường hợp, bộ nhiễm sắc thể là 46,XY.
Các phát hiện xét nghiệm điển hình là nồng độ LH và testosterone tăng vì tổng hợp testosterone ở tinh hoàn không bị cản trở. Tuy nhiên, có sự mất ức chế phản hồi ngược của gonadotropin. Nồng độ LH thường cao hơn nồng độ FSH vì sự tiết inhibin của tinh hoàn không bị cản trở. Nồng độ FSH có thể tăng hoặc bình thường. Trẻ sơ sinh bị PAIS có thể cần các xét nghiệm nội tiết động để đánh giá sự tiết testosterone của tế bào Leydig được kích thích bằng hCG và quan trọng là khả năng đáp ứng của cơ quan đích với androgen.
Khi không có phối tử, protein AR chủ yếu nằm trong tế bào chất, nơi nó liên kết với các protein chaperone. Khi liên kết với phối tử, cấu dạng của thụ thể androgen thay đổi. Các phức hợp phối tử-thụ thể nhị phân hóa và di chuyển đến nhân. Một đặc điểm chính của sự nhị phân hóa thụ thể androgen là sự tương tác nội phân tử giữa các miền đầu N và đầu C. Sự liên kết của phối tử ổn định thụ thể androgen và làm chậm quá trình thoái hóa của nó. Hiệu lực lớn hơn của DHT được cho là do sự ổn định lớn hơn của phức hợp DHT-thụ thể so với phức hợp testosterone-thụ thể. Trong nhân, phức hợp liên kết với các yếu tố đáp ứng androgen và làm thay đổi sự phiên mã của gen đích.
Hơn 500 đột biến AR khác nhau đã được mô tả. Nói chung, kiểu hình tương quan với mức độ suy giảm hoạt động của androgen. Tuy nhiên, các đặc điểm lâm sàng có thể khác nhau mặc dù có cùng một đột biến (ngay cả trong cùng một gia đình). CAIS hoặc PAIS liên quan đến cùng một đột biến AR có thể xảy ra ở anh chị em ruột. Các đột biến sai nghĩa khác nhau ở cùng một vị trí cũng có thể liên quan đến các kiểu hình khác nhau. Các đột biến mất chức năng hoàn toàn và các codon kết thúc sớm thường liên quan đến CAIS. Các đột biến mất chức năng một phần thường là các đột biến sai nghĩa. Các thụ thể có đột biến trong miền liên kết DNA liên kết với phối tử bình thường nhưng không thể kích hoạt phiên mã các gen đích. Các đột biến trong miền liên kết phối tử có thể liên quan đến giảm ái lực với phối tử và/hoặc tăng sự bất ổn của phức hợp hormone-thụ thể. Ngoài việc xác định hormone, đánh giá chẩn đoán có thể bao gồm phân tích trình tự DNA của gen AR.
Các đột biến có thể nằm ở các vùng không mã hóa của gen; WES đã xác định được một đột biến trong intron dẫn đến một phiên mã được ghép nối thay thế trong một gia đình lớn bị PAIS liên quan đến lỗ tiểu lệch thấp sơ sinh và vú to ở nam giới tuổi dậy thì. Hai bé gái 46,XY không liên quan được chẩn đoán mắc CAIS được phát hiện có cùng một đột biến dòng mầm c.-547C > T trong AR -5’-UTR. Đột biến này đã đưa ra một khung đọc mở ngược dòng được dịch trong 5′UTR, dẫn đến giảm protein AR. Hai chị em bị CAIS hoàn toàn được phát hiện có một đột biến điểm kích hoạt pseudoexon trong intron giữa các exon 6 và 7 của AR; đột biến này dẫn đến sự ghép nối mRNA bất thường và giảm sản xuất protein.
Hoạt động phiên mã của AR phụ thuộc vào các protein khác, bao gồm các chất đồng kích hoạt và đồng ức chế. Các protein khác này có lẽ điều chỉnh các tương tác vật lý, liên kết bộ máy phiên mã cơ bản, phức hợp phối tử-thụ thể và chromatin. Ví dụ, một đột biến sai nghĩa của AR đã làm thay đổi sự tương tác của protein AR với protein melanoma antigen A-11 (MAGE-11) bằng cách cản trở các tác động kích thích của chất đồng điều hòa.
Ngoài tình trạng không nhạy cảm với androgen, bệnh Kennedy (còn được gọi là teo cơ tủy và hành não) được định vị tại locus AR. Bệnh Kennedy, một rối loạn thoái hóa thần kinh tiến triển khởi phát ở độ tuổi 30 hoặc 40, có liên quan đến sự mở rộng quá mức của vùng lặp lại trinucleotide polyglutamine CAG trong exon 1 của AR. Chiều dài lặp lại lớn hơn 35 có liên quan đến teo cơ tủy và hành não. Cơ chế được cho là gây ra sự thoái hóa thần kinh có liên quan đến vai trò của AR trong chức năng của phức hợp thúc đẩy kỳ sau/cyclosome ubiquitin ligase, hoạt động với protein adaptor của nó để ảnh hưởng đến sự ngừng chu kỳ tế bào và kiến trúc thần kinh. Khi có sự lặp lại CAG mở rộng, AR ức chế phức hợp này, dẫn đến sự tái hoạt động chu kỳ tế bào bất thường. Các triệu chứng nhẹ của tình trạng không nhạy cảm với androgen có thể được phát hiện với sự giảm nhẹ nồng độ mRNA và protein của AR.
Bất thường ống Müller
Hội chứng ống Müller tồn tại
Hội chứng ống Müller tồn tại (PMDS) là một rối loạn di truyền lặn trên nhiễm sắc thể thường do các đột biến trong gen AMH nằm ở nhiễm sắc thể 19p13.3 hoặc gen thụ thể của nó (AMH-RII) nằm ở nhiễm sắc thể 12q13.13. Các kiểu hình của bệnh nhân có đột biến AMH hoặc AMH-RII là tương đương nhau. AMH là một thành viên của họ TGF-β và truyền tín hiệu thông qua hai thụ thể serine/threonine gắn màng tương tác khác nhau. Phối tử, AMH, liên kết với thụ thể loại II, dẫn đến việc tuyển dụng và phosphoryl hóa một thụ thể loại I. Thụ thể loại II đặc hiệu cho AMH, trong khi có nhiều phân nhóm của thụ thể loại 1. Nồng độ AMH thấp ở những bệnh nhân có đột biến trong gen AMH. Ở những bệnh nhân có đột biến AMH-RII, nồng độ AMH bình thường hoặc tăng. Nữ giới mang đột biến trên cả hai alen AMH dường như có khả năng sinh sản bình thường.
Các đặc điểm lâm sàng điển hình của PMDS bao gồm tinh hoàn ẩn, tinh hoàn lạc chỗ liên quan đến thoát vị bẹn, và thoát vị tử cung bẹn. Sự biệt hóa tinh hoàn thường bình thường, nhưng các ống bài xuất của nam giới có thể được nhúng vào trong các di tích ống Müller hoặc phát triển không hoàn toàn. Vô sinh có thể xảy ra thứ phát sau tinh hoàn ẩn, sự quấn vào nhau của ống dẫn tinh và thành tử cung, hoặc thiếu sự thông thương thích hợp giữa tinh hoàn và các ống bài xuất. Xoắn tinh hoàn không phải là hiếm vì tinh hoàn có thể không được neo đúng vào đáy của mỏm bọc tinh hoàn.
Bất thường ống Müller ở các cá thể 46,XX
Hội chứng Mayer-Rokitansky-Küster-Hauser (MRKH) là tình trạng không có tử cung và hai phần ba trên của âm đạo bẩm sinh; phần dưới của âm đạo thường không bị ảnh hưởng. Sự phát triển buồng trứng thường bình thường. Rối loạn này xảy ra ở khoảng 1:4500 bé gái. Biểu hiện điển hình là một cô gái vị thành niên bị vô kinh nguyên phát kèm theo sự phát triển vú và lông mu phù hợp.
MRKH có thể được phân loại thành loại I hoặc loại II. MRKH loại I được đặc trưng bởi bất sản tử cung đơn độc. MRKH loại II được đặc trưng bởi bất sản tử cung kèm theo dị tật ngoài sinh dục, thường là dị tật thận và xương. Các phát hiện ở thận, được xác định ở khoảng 40%, bao gồm bất sản thận một bên, thận hình móng ngựa, thận lạc chỗ, thiểu sản thận và ứ nước ở thận. Hội chứng MURCS được định nghĩa là bất sản ống Müller, bất sản thận và loạn sản đốt sống cổ-ngực; các bất thường về tim và hệ thần kinh cũng có thể có mặt.
Ứ dịch trong tử cung và âm đạo, thiểu sản Müller và tật thừa ngón có liên quan đến cả hội chứng McKusick-Kaufman và Bardet-Biedel loại 6. Cả hai rối loạn di truyền lặn trên nhiễm sắc thể thường này đều liên quan đến các đột biến trong gen MKKS-BBS6 nằm ở nhiễm sắc thể 20p12. Hội chứng McKusick-Kaufman và Bardet-Biedel loại 6 tương tự nhau; đặc điểm phân biệt chính là sự xuất hiện của viêm võng mạc sắc tố ở Bardet Biedel loại 6.
Không có một khiếm khuyết gen đơn lẻ nào được xác định cho MRKH. Các biến thể số lượng bản sao tái phát trong các vùng nhiễm sắc thể cụ thể, chẳng hạn như 17q12, 16p11.2, 1q21.1 và 22q11.21, có liên quan đến MRKH. Các đột biến trong WNT4, LHX1, HNF1B, TBX6, RBM8A và WNT9B có liên quan đến bất sản Müller. Bệnh nhân MRKH liên quan đến các đột biến dị hợp tử WNT4 được báo cáo là có tăng androgen huyết lâm sàng hoặc sinh hóa. Thiểu sản ống Müller có liên quan đến các bất thường mặt-tai-đốt sống, chẳng hạn như hội chứng Goldenhar và hội chứng DiGeorge. Nếu không có cấy ghép tử cung hoặc mang thai hộ, các cá nhân bị ảnh hưởng không thể mang thai. Hầu hết các trường hợp là ngẫu nhiên. Do đó, việc xác định sự di truyền đã gặp nhiều thách thức.
Siêu âm vùng chậu và hình ảnh cộng hưởng từ (MRI) cho thấy sự phát triển bất thường của tử cung, cổ tử cung và phần trên âm đạo. Các nghiên cứu hình ảnh nên bao gồm đánh giá các bất thường ở thận và dị tật xương. Hình thái buồng trứng nên phù hợp với giai đoạn dậy thì. Do tần suất cao của các bất thường liên quan, khám thực thể cẩn thận để tìm các dị tật xương nên được bao gồm trong đánh giá chẩn đoán của phụ nữ có sự phát triển bất thường của hệ thống ống Müller.
Dương vật nhỏ, lỗ tiểu lệch thấp và tinh hoàn ẩn
Lỗ tiểu lệch thấp
Lỗ tiểu lệch thấp là một tình trạng thiểu sản bẩm sinh của dương vật đặc trưng bởi sự dịch chuyển về phía bụng của lỗ niệu đạo, dây cong và thiếu da quy đầu ở mặt bụng. Lỗ tiểu lệch thấp ở đầu xa thường gặp hơn nhiều so với ở gốc và chiếm khoảng 70% các trường hợp. Lỗ tiểu lệch thấp ở gốc có thể liên quan đến dây cong, tinh hoàn ẩn và các bất thường bẩm sinh khác. Tỷ lệ mắc được báo cáo dao động từ 1:100 đến 1:1000. Lỗ tiểu lệch thấp có tính gia đình đã được mô tả. Các gen liên quan đến việc hình thành củ sinh dục sớm bao gồm BMP4, BMP7, HOXA4, HOXB6, FGF8, FGF10 và FGFR2. Các mất đoạn liên quan đến 19q13 có liên quan đến IUGR, đầu nhỏ, chậm phát triển sau sinh, tật dính ngón và lỗ tiểu lệch thấp; gen protein tương tác với khối u Wilms (WTIP) nằm trong vùng bị mất đoạn.
Tinh hoàn ẩn
Tinh hoàn ẩn là rối loạn biệt hóa giới tính phổ biến nhất, ảnh hưởng đến 3% trẻ sơ sinh nam. Tinh hoàn ẩn có thể được phân loại là không hội chứng/đơn độc hoặc có hội chứng (Bảng 6.6).
Bảng 6.6. Các rối loạn liên quan đến tinh hoàn ẩn hoặc dương vật nhỏ ở nam giới
| Tên | Gen | Vị trí |
| Hội chứng Aarskog | FGD1 (OMIM #305400) FGD1 FGD1 | Xp11.22, “Tầm vóc thấp, hai mắt xa nhau, bìu hình khăn choàng, tật ngón ngắn” |
| Beckwith-Wiedemann | CDKN1C (OMIM# 130650) | 11p15.4, Hội chứng Beckwith-Wiedemann |
| Hội chứng Börjeson-Forssman-Lehman | PHF6 (OMIM #301900) | Xq26.2, “Suy sinh dục, co giật, chậm phát triển, béo phì” |
| Hội chứng Carnevale | COLEC11 (OMIM #265050) | 2p25.3, “Lỗ tiểu lệch thấp, thận hình móng ngựa, dính khớp sọ, hai mắt xa nhau” |
| Hội chứng Carpenter-1 | RAB23 (OMIM #201000) | 6p12.1-p11.2, “Đầu nhọn, dính ngón, bệnh tim bẩm sinh, tai đóng thấp, béo phì” |
| Hội chứng Carpenter-2 | MEGF8 (OMIM #614976) | 19q13.2, “Dính khớp sọ, dính nhiều ngón ở tay và chân, đảo ngược phủ tạng, bệnh tim bẩm sinh” |
| Hội chứng Cornelia de Lange | NIPBL (OMIM #122470) | 5p13.2, “Thiểu sản cơ quan sinh dục ngoài nam, tinh hoàn ẩn, chậm phát triển trí tuệ, giao chân mày” |
| Hội chứng Costello | HRAS (OMIM #218040) | 11p15.5, “Tầm vóc thấp, mặt thô, hai mắt xa nhau, bệnh tim bẩm sinh, các dấu hiệu ở da, chậm phát triển” |
| Hội chứng Hajdu-Cheney | NOTCH2 (OMIM #102500) | 1p12, “Tầm vóc thấp, mặt thô, cong xương dài, bất thường đốt sống, hai mắt xa nhau, lông mày rậm, cằm nhỏ” |
| Hội chứng Juberg-Marsidi | HUWE1 (OMIM #309590) | Xp11.22, “Hai mắt gần nhau, khe mi nhỏ, tai loạn sản, to hoặc đóng thấp, mặt dài, vòm miệng cao, môi trên mỏng, tật ngón ngắn, chậm phát triển” |
| Hội chứng Johanson-Blizzard | UBR1 (OMIM #243800) | 15q15.2, “Tầm vóc thấp, chậm lớn, suy giáp, điếc thần kinh giác quan, không có hậu môn và suy tụy ngoại tiết” |
| Hội chứng tăng xương Lenz-Majewski | PTDSS1 (OMIM #151050) | 8q22.1, “Loạn sản xương xơ cứng, da lỏng lẻo, tật ngón ngắn, tăng xương lan tỏa tiến triển, chậm phát triển” |
| Hội chứng Leopard | PTPN11 (OMIM #151100) | 12q24.13, “Tầm vóc thấp, sụp mi, hai mắt xa nhau, tai đóng thấp, cổ có màng, bệnh tim bẩm sinh, nhiều nốt ruồi” |
| Hội chứng Lowe | OCRL (OMIM #309000) | Xq26.1, “Glôcôm bẩm sinh, đục thủy tinh thể, chậm phát triển trí tuệ, còi xương kháng vitamin D, amino acid niệu” |
| 3MC1 | MASP1 (OMIM #257920) | 3q27.3, “Hai mắt xa nhau, hẹp khe mi, sụp mi và lông mày cong cao” |
| 3MC2 | COLEC11 (OMIM #265050) | 2p25.2, “Tầm vóc thấp, mất thính lực, sứt môi/hở hàm ếch” |
| 3MC3 | COLEC10 (OMIM #265050) | 8q24.12, “Tầm vóc thấp, sụp mi, sứt môi/hở hàm ếch” |
| Hội chứng McKusick-Kaufman | MKKS (OMIM #236700) | 20p12.2, “Bệnh tim bẩm sinh, thiểu sản phổi, tật thừa ngón” |
| Meckel-Gruber, loại 1 | MKS1 (OMIM #249000) | 17q22, “Tật thừa ngón, thận đa nang” |
| Hội chứng Meier-Gorlinª | ORC6 (OMIM #607213) | 16q11.1, “IUGR, tầm vóc thấp, tai nhỏ, đầu nhỏ, tinh hoàn ẩn” |
| Hội chứng MEND | EBP (OMIM #300960) | Xp11.23, “Tầm vóc thấp, đục thủy tinh thể, dính ngón 2-3 ở chân, co giật, chậm phát triển” |
| Hội chứng Miller-Dieker | Mất đoạn gen tiếp giáp (OMIM #247200) | Mất đoạn 17p13.3, “Não phẳng, đầu nhỏ, khe mi xếch xuống, mũi và cằm nhỏ, dị tật tim, tầm vóc thấp, chậm phát triển” |
| Mowat-Wilson | ZEB2 (OMIM #235730) | 2q22.3, “Đầu nhỏ, chậm phát triển” |
| Hội chứng tim-sinh dục Najjar | LMNA (OMIM #212112) | 1q22, “Bệnh cơ tim, suy sinh dục tăng gonadotropin” |
| Hội chứng Noonan | PTPN11 (OMIM #163950) | 12q24.13, “Tầm vóc thấp, sụp mi, hai mắt xa nhau, cổ có màng, bệnh tim bẩm sinh, giảm tiểu cầu, bệnh von Willebrand” |
| Hội chứng ống Müller tồn tại | AMH (OMIM #600957) | 19p.13.3, Hội chứng ống Müller tồn tại |
| Hội chứng ống Müller tồn tại | AMHR (OMIM #600956) | 12q13.13, Hội chứng ống Müller tồn tại |
| Pfeiffer | FGFR1 (OMIM #101600) | 8p11.23, “Dính khớp sọ, ngón tay cái rộng, tật ngón ngắn, dính khớp sọ, thiểu sản giữa mặt, ngón tay cái rộng, ngón chân cái rộng, tật ngón ngắn” |
| Pfeiffer | FGFR2 (OMIM #101600) | 10q26.13, “Dính khớp sọ, ngón tay cái rộng, tật ngón ngắn, thiểu sản giữa mặt, ngón tay cái rộng, ngón chân cái rộng” |
| Hội chứng Prader-Willi | NDN/SNRPN (OMIM #17620) | 15q11.2, Hội chứng Prader-Willi |
| Hội chứng Robinow-RRS1 | ROR2 (OMIM #268310) | 9q22.31, “Loạn sản xương, trán dô, hai mắt xa nhau, lùn chi ngắn, phân đoạn đốt sống, chậm phát triển” |
| Hội chứng Robinow-RRS2 | NXN (OMIM #618529) | 17p13.3, “Loạn sản xương, tầm vóc thấp trung chi sau sinh, đầu tương đối to” |
| Hội chứng Robinow DRS1 | WNT5A (OMIM #180700) | 3p14.3, “Đầu to, tầm vóc thấp trung chi sau sinh” |
| Hội chứng Rubinstein-Taybi, loại 1 | CREBBP (OMIM #180849) | 16p13.3, “Đầu nhỏ, chậm phát triển trí tuệ, lông mày cong cao, lông mi dài” |
| Hội chứng Rubinstein-Taybi, loại 2 | EP300 (OMIM #602700) | 22q13.2, “Đầu nhỏ, chậm phát triển trí tuệ, lông mày cong cao, lông mi dài” |
| Hội chứng Sotos | NSD1 (OMIM #117550) | 5q35.3, “To não, điếc dẫn truyền, chậm phát triển, tuổi xương cao” |
| Hội chứng Seckel, loại 1 | ATR (OMIM #210600) | 3q23, Vẻ mặt đầu chim |
| Hội chứng Seckel, loại 2 | RBBP8 (OMIM #606744) | 18q11.2, Vẻ mặt đầu chim |
| Hội chứng Seckel, loại 4 | CENPJ (OMIM #613676) | 13q12.12-q12.13, Vẻ mặt đầu chim |
| Hội chứng Seckel, loại 5 | CEP152 (OMIM #613823) | 15q21.1, Vẻ mặt đầu chim |
| Shprintzen-Goldberg | KIAA1279 (OMIM #609460) | 10q22.1, “Đầu nhỏ, đặc điểm dị dạng, chậm phát triển, bệnh Hirschsprung, thiểu năng trí tuệ, đầu nhỏ, và đặc điểm dị dạng trên khuôn mặt. Hầu hết bệnh nhân cũng có bất thường hồi não” |
| Simpson-Golabi-Behmel, loại 1 | GPC3 (OMIM #312870) | Xq26.2, “Tầm vóc cao, mặt thô, bệnh tim bẩm sinh” |
| Hội chứng Townes-Brocks | SALL1 (OMIM #107480) | 16q12.1, “Lỗ tiểu lệch thấp, tinh hoàn ẩn, thiểu sản thận, tử cung hai sừng” |
| Hội chứng Varadi-Papp (hội chứng miệng-mặt-ngón, loại VI) | CPLANE1 (OMIM #277170) | 5p13.2, “Bất sản/loạn sản thận, tầm vóc thấp, tinh hoàn ẩn, tật thừa ngón” |
| Hội chứng Warburg Micro-1 (WARBM1) | RAB3GAP1 (OMIM #600118) | 2q21.3, “Đầu nhỏ, mắt nhỏ, giác mạc nhỏ, đục thủy tinh thể bẩm sinh, teo thần kinh thị, thiểu sản thể chai, chậm phát triển trí tuệ nặng, liệt hai chi co cứng” |
| Weaver | EZH2 (OMIM #117550) | 7q36.1, “Đầu to, hai mắt xa nhau, chậm phát triển, nhân trung rộng nổi bật, cằm nhỏ, rãnh cằm ngang sâu và móng mọc sâu” |
IUGR, Chậm phát triển trong tử cung.
Do sự xuống tự nhiên, tỷ lệ mắc giảm xuống còn 1% vào 6 tháng tuổi. Tinh hoàn ẩn có thể liên quan đến giảm số lượng tế bào mầm, suy giảm sự trưởng thành của tế bào mầm và giảm số lượng tế bào Leydig. Trong một số trường hợp tinh hoàn ẩn một bên, mô học bất thường có thể rõ ràng ở tinh hoàn đối bên đã xuống bình thường. Thường thì tinh hoàn ẩn là một phát hiện đơn độc, nhưng nó có thể liên quan đến suy sinh dục do vùng dưới đồi, biệt hóa tinh hoàn bất thường, suy giảm sinh tổng hợp testosterone, không nhạy cảm với androgen, não trước không phân chia, sản xuất hoặc hoạt động AMH bất thường, các bất thường ảnh hưởng đến chức năng INSL3/LGR8 và có thể là các yếu tố môi trường. Các mối liên quan khác bao gồm hội chứng bụng quả mận, lộn bàng quang và các bất thường ở thận. Đái tháo đường ở mẹ, bao gồm cả đái tháo đường thai kỳ, có thể là một yếu tố nguy cơ.
Trong quá trình biệt hóa giới tính, các tuyến sinh dục được định vị giữa hai cấu trúc: dây chằng treo dương vật và dây chằng bìu-tinh hoàn (gubernaculum). Gubernaculum là một cấu trúc trung mô đóng vai trò chính trong việc xuống tinh hoàn. Ở nữ giới, dây chằng treo dương vật tồn tại dưới dạng dây chằng treo buồng trứng. Hoạt động của androgen trong giai đoạn trong ổ bụng thúc đẩy sự thoái triển của dây chằng treo dương vật. Việc xuống tinh hoàn được chia thành hai giai đoạn: trong ổ bụng và bẹn-bìu. Các yếu tố liên quan đến sự phát triển của gubernaculum trong giai đoạn trong ổ bụng bao gồm INSL3 và thụ thể của nó, LGR8/RXFP2. INSL3 được tiết ra bởi các tế bào Leydig. Thụ thể của nó, LGR8/RXFP2, là một thụ thể kết hợp với protein G giàu leucine được biểu hiện bởi gubernaculum. Vào tuần thứ 13 hoặc 14 của thai kỳ, gubernaculum neo tinh hoàn vào vòng bẹn trong. Vào khoảng 22 đến 25 tuần của thai kỳ, tinh hoàn và mào tinh hoàn được định vị tại các vòng trong của ống bẹn. Việc xuống tinh hoàn bị ảnh hưởng bởi androgen qua ống bẹn thường được hoàn thành vào cuối tháng thứ bảy của thai kỳ, với việc hoàn thành giai đoạn bẹn-bìu vào cuối tuần 35.
Các mối quan tâm về kết quả bao gồm nguy cơ vô sinh và tân sinh tinh hoàn. Trong một nghiên cứu ngẫu nhiên tiền cứu, phẫu thuật cố định tinh hoàn (orchidopexy) ở 9 tháng tuổi dẫn đến thể tích tinh hoàn lớn hơn và tăng số lượng tế bào mầm so với phẫu thuật ở 3 tuổi. Do đó, việc phẫu thuật điều chỉnh tinh hoàn ẩn ở giai đoạn đầu của trẻ sơ sinh được ưu tiên.
Các đột biến sai nghĩa dị hợp tử của INSL3 đã được xác định ở những bệnh nhân bị tinh hoàn ẩn. Các đột biến cũng đã được xác định trong gen LGR8 ở nam giới bị tinh hoàn ẩn. Các biến thể trình tự đã được xác định trong gen HOXA10 ở các bé trai bị tinh hoàn ẩn. Tuy nhiên, các đột biến trong INSL3, LGR8, hoặc HOXA10 là những nguyên nhân hiếm gặp của tinh hoàn ẩn đơn độc. Thông thường, tinh hoàn của bệnh nhân không nhạy cảm với androgen đã hoàn thành giai đoạn trong ổ bụng nhưng không thể trải qua quá trình xuống bẹn-bìu vì giai đoạn thứ hai này phụ thuộc vào androgen. Tuy nhiên, tình trạng không nhạy cảm với androgen càng hoàn toàn thì khả năng tìm thấy tinh hoàn trong ổ bụng càng lớn. Việc phơi nhiễm của thai nhi XX với androgen không thúc đẩy sự xuống buồng trứng đáng kể ở người, bằng chứng là vị trí buồng trứng bình thường ở nữ giới bị CAH.
Suy sinh dục do giảm Gonadotropin
Suy sinh dục do giảm gonadotropin liên quan đến thiếu hụt GnRH được đặc trưng bởi một phổ các đặc điểm lâm sàng về sinh sản, khứu giác và phi sinh sản. Các kiểu di truyền bao gồm liên kết X, trội trên nhiễm sắc thể thường và lặn trên nhiễm sắc thể thường. Trẻ sơ sinh nam bị suy sinh dục do giảm gonadotropin có thể được xác định trong giai đoạn sơ sinh với dương vật nhỏ và/hoặc tinh hoàn ẩn. Cơ quan sinh dục không điển hình thường không xảy ra vì sự tiết hCG của nhau thai không bị ảnh hưởng. Giảm tiết gonadotropin gây giảm tiết testosterone, dẫn đến dương vật nhỏ và tinh hoàn ẩn. Có thể có thêm các thiếu hụt hormone tuyến yên trước khác.
Khoảng 1000 đến 2000 tế bào thần kinh GnRH cư trú trong vùng dưới đồi sau sinh được sinh ra trong mảng khứu giác và di chuyển trong giai đoạn đầu của sự phát triển bào thai từ mũi qua não trước đến vùng dưới đồi. Việc điều tra các gia đình bị suy sinh dục do giảm gonadotropin di truyền đã dẫn đến việc xác định các gen cụ thể liên quan đến quá trình này (Bảng 6.7). Hội chứng Kallmann là tên gọi được sử dụng cho dạng suy sinh dục do giảm gonadotropin di truyền lặn liên kết X liên quan đến mất khứu giác do sự thất bại trong việc di chuyển của các tế bào thần kinh GnRH từ mảng khứu giác vào não trước dọc theo các nhánh của dây thần kinh lá mía-mũi. Thiểu sản hoặc bất sản đường khứu giác đã được tìm thấy trên MRI. Cơ sở phân tử của dạng liên kết X này là các đột biến trong gen Kallmann (ANOS1) (nằm ở Xp22.3). Gen này thoát khỏi sự bất hoạt X, mã hóa một protein 680 axit amin và giúp nhắm mục tiêu các tế bào thần kinh GnRH đến vùng dưới đồi.
Bảng 6.7. Các rối loạn đơn gen liên quan đến suy sinh dục do vùng dưới đồi
| Gen | Vị trí | Kiểu hình | Kiểu di truyền |
| ANOS1 (KAL1) | Xp22.31 | Suy sinh dục do vùng dưới đồi có mất khứu giác | Liên kết X |
| CCDC141 | 2q31.2 | Suy sinh dục do vùng dưới đồi có/không có mất khứu giác | Lặn NST thường, đa gen |
| FEZF1 | 7q31.32 | Suy sinh dục do vùng dưới đồi có mất khứu giác | Lặn NST thường, đa gen |
| FGFR1 | 8p11.23 | Suy sinh dục do vùng dưới đồi có/không có mất khứu giác; não trước không phân chia; sứt môi/hở hàm ếch | Trội/Lặn NST thường, đa gen |
| FGF8 | 10q24.32 | Suy sinh dục do vùng dưới đồi có/không có mất khứu giác | Trội NST thường, đa gen |
| FGF17 | 8p21.3 | Suy sinh dục do vùng dưới đồi có/không có mất khứu giác | Trội NST thường, đa gen, mới |
| FSHβ | 11p14.1 | Suy buồng trứng/không có tinh trùng | Lặn NST thường |
| FSHR | 2p16.3 | Vô kinh nguyên phát | Lặn NST thường |
| GNRH1 | 8p21.2 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Lặn NST thường, đa gen |
| GNRHR | 4q13.2 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Lặn NST thường, đa gen |
| HESX1 | 3p14.3 | Suy sinh dục do vùng dưới đồi có mất khứu giác | Trội NST thường |
| HS6ST1 | 2q14.3 | Suy sinh dục do vùng dưới đồi có/không có mất khứu giác | Trội NST thường, đa gen |
| IGSF10 | 3q25.1 | Dậy thì muộn | Trội NST thường |
| IL17RD | 3p14.3 | Suy sinh dục do vùng dưới đồi có mất khứu giác | Lặn/Trội NST thường, đa gen |
| KISS1 | 1q32.1 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Lặn NST thường |
| KISSR1 | 19p13.3 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Trội/Lặn NST thường, đa gen |
| LHB | 19q13.33 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Lặn NST thường |
| LHX4 | 1q25.2 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Trội NST thường |
| NSMF | 9q34.3 | Suy sinh dục do vùng dưới đồi có/không có mất khứu giác | Đa gen |
| PROK2 | 3p13 | Suy sinh dục do vùng dưới đồi có/không có mất khứu giác | Trội NST thường, đa gen |
| PROKR2 | 20p12.3 | Suy sinh dục do vùng dưới đồi có/không có mất khứu giác | Trội/Lặn NST thường, đa gen |
| PROP1 | 5q35.3 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Lặn NST thường |
| RNF216 | 7p22.1 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Lặn NST thường, đa gen |
| SEMA3A | 7q21.11 | Suy sinh dục do vùng dưới đồi có mất khứu giác | Trội NST thường, đa gen |
| SEMA3E | 7q21.11 | Suy sinh dục do vùng dưới đồi có mất khứu giác | Trội NST thường |
| SEMA7A | 15q24.1 | Suy sinh dục do vùng dưới đồi có/không có mất khứu giác | Đa gen |
| SOX2 | 3q26.33 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Lặn NST thường |
| SOX10 | 22q13.1 | Suy sinh dục do vùng dưới đồi có mất khứu giác | Trội NST thường |
| TAC3 | 12q13.3 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Lặn NST thường, đa gen |
| TACR3 | 4q24 | Suy sinh dục do vùng dưới đồi không có mất khứu giác | Lặn NST thường, đa gen |
| WDR11 | 10q26.12 | Suy sinh dục do vùng dưới đồi có/không có mất khứu giác | Trội NST thường |
AD, Di truyền trội trên nhiễm sắc thể thường; AR, Di truyền lặn trên nhiễm sắc thể thường.
Các đột biến trong các gen GNRHR, FGFR1, FGF8, NSMF, GNRH1, KISS1, KISS1R, PROK2, và PROKR2 cũng có liên quan đến suy sinh dục do giảm gonadotropin. Các đột biến trong TAC3 và thụ thể của nó TACR3 có liên quan đến suy sinh dục do giảm gonadotropin; tỷ lệ dương vật nhỏ ở nam giới mang đột biến TACR3 là cao. Các khiếm khuyết tuyến sinh dục có thể xảy ra trong hội chứng CHARGE do các đột biến CHD7; các đặc điểm bổ sung bao gồm khuyết mống mắt, khuyết tật tim, hẹp lỗ mũi sau, tầm vóc thấp và mất thính giác. Thay vì là một rối loạn đơn gen, suy sinh dục do giảm gonadotropin dường như là một rối loạn đa gen.
Các chất gây rối loạn môi trường
Điều trị trước sinh bằng diethylstilbesterol (DES), một estrogen tổng hợp không steroid, có liên quan đến các bất thường niệu-sinh dục của cả thai nhi nam và nữ với tinh hoàn ẩn xảy ra ở thai nhi 46,XY. Cơ quan sinh dục không điển hình, được mô tả ở ba trẻ sơ sinh 46,XY sinh ra ở các khu vực nông nghiệp nặng, được cho là do phơi nhiễm của thai nhi với các chất gây rối loạn nội tiết, đặc biệt là vì không có đột biến nào được phát hiện trong các gen SRY hoặc SRD5A2. Mặc dù không có nguyên nhân xác định nào được tìm thấy, các chất môi trường khác nhau, chẳng hạn như thuốc diệt cỏ, thuốc diệt nấm, thuốc trừ sâu và chất làm dẻo, đã được coi là các chất gây rối loạn nội tiết tiềm tàng. Các cơ chế tiềm tàng bao gồm liên kết với các thụ thể hormone hạt nhân điều chỉnh biểu hiện gen hoặc các thay đổi biểu sinh. Bằng chứng đáng tin cậy xác nhận các tác động có hại của môi trường đối với sự phát triển sinh dục còn thiếu.
Các rối loạn khác
Tổ hợp VACTERL (VA) được đặc trưng bởi một số bất thường. Các bất thường này bao gồm bất thường đốt sống, không có hậu môn, dị tật tim mạch, rò khí quản-thực quản và/hoặc teo thực quản, bất thường thận, bất thường chi và/hoặc cơ quan sinh dục không điển hình. Thiểu sản/bất sản Müller, bất sản thận và loạn sản đốt sống cổ-ngực (tổ hợp MURCS) được đặc trưng bởi vô kinh nguyên phát. Các đặc điểm của rối loạn này có thể bao gồm không có tử cung và vòi trứng, các bất thường cột sống cổ và các bất thường ở thận. Tình trạng không có dương vật hoàn toàn còn được gọi là aphallia là hiếm gặp và có thể liên quan đến các bất thường bẩm sinh khác của hệ niệu-sinh dục và hệ tiêu hóa.
Lộn bàng quang
Phức hợp lộn bàng quang-lỗ tiểu lệch trên (BEEC) mô tả một khiếm khuyết trường nguyên phát liên quan đến xương chậu, đường tiết niệu và cơ quan sinh dục ngoài. Phổ dị tật này bao gồm lỗ tiểu lệch trên, lộn bàng quang và tồn tại ổ nhớp. Một biến thể khác là phức hợp OEIS (thoát vị rốn, lộn bàng quang, không có hậu môn và các khuyết tật cột sống). Các phát hiện bổ sung có thể bao gồm thận lạc chỗ và bất sản thận cũng như các bất thường ảnh hưởng đến hệ xương, hệ niệu-sinh dục và hệ tiêu hóa. Sự xuất hiện trong gia đình hiếm khi được mô tả cho rối loạn được cho là đa yếu tố này. Lộn ổ nhớp được đặc trưng bởi sự tồn tại của một ổ nhớp chung liên quan đến sự thất bại trong việc hợp nhất của các củ sinh dục. Nguyên nhân cơ bản vẫn chưa được xác định. Tuy nhiên, vùng vi nhân đoạn 22q11.2 đã được phát hiện nhiều lần trên phép lai so sánh bộ gen trên mảng ở một số bệnh nhân; một gen ứng cử viên là gen điều hòa phiên mã giống khóa kéo leucine (LZTR1) nằm ở vùng này.
Chẩn đoán
Một phương pháp tiếp cận cá nhân hóa toàn diện là phù hợp cho một đứa trẻ được phát hiện có cơ quan sinh dục không điển hình. Trong một số trường hợp, chẩn đoán lần đầu tiên được xem xét khi trẻ có biểu hiện dậy thì muộn. Ngoài tiền sử bệnh kỹ lưỡng và khám thực thể hoàn chỉnh, đánh giá nội tiết và đánh giá di truyền cung cấp các phương pháp tiếp cận có giá trị. Danh sách các nguyên nhân tiềm tàng đặc biệt đối với các cá nhân 46,XY tiếp tục mở rộng. Tuy nhiên, mặc dù đã xác định được các gen mới và công nghệ di truyền tiên tiến, một chẩn đoán phân tử xác định có thể không được thiết lập cho tất cả mọi người, đặc biệt là đối với các cá nhân 46,XY. Ahmed và các đồng nghiệp đề xuất các khuyến nghị cho các bé trai bị DSD.
Tiền sử
Cần khai thác tiền sử gia đình chi tiết. Tiền sử gia đình nên bao gồm việc xác định các trường hợp tử vong ở trẻ sơ sinh không rõ nguyên nhân, hôn nhân cận huyết và vô sinh. Trẻ sơ sinh bị CAH có thể đã tử vong trước khi được chẩn đoán. Tiền sử gia đình có thể âm tính trong trường hợp các rối loạn di truyền lặn trên nhiễm sắc thể thường. Vô sinh, POI và vú to ở nam giới có thể đại diện cho các kiểu hình nhẹ hơn của một số DSD. Đối với các rối loạn liên kết X, chẳng hạn như không nhạy cảm với androgen, có thể có các thành viên gia đình bên mẹ bị ảnh hưởng (tức là, các dì bị vô kinh hoặc vô sinh hoặc các chú bị nam hóa một phần). Các câu hỏi thích hợp bao gồm phơi nhiễm trước sinh với androgen, estrogen ngoại sinh hoặc nội sinh, hoặc các chất gây rối loạn nội tiết tiềm tàng. Cần hỏi về tình trạng nam hóa của mẹ trong thai kỳ.
Khám thực thể
DSD bao gồm một phổ các phát hiện thực thể. Sự phát triển sinh dục không điển hình có thể xảy ra như một thành phần của một hội chứng. Do đó, cần khám cẩn thận khuôn mặt, tim, các chi và các ngón tay để đánh giá các đặc điểm dị dạng. Các phát hiện thực thể cụ thể bao gồm từ dương vật nhỏ, lỗ tiểu lệch thấp, tinh hoàn ẩn, phì đại âm vật tối thiểu và bìu hóa môi lớn đến các dạng cơ quan sinh dục không điển hình rộng hơn. Phì đại âm vật nghiêm trọng với sự hợp nhất môi lớn phía sau ở một bệnh nhân 46,XX có thể phân biệt được với lỗ tiểu lệch thấp ở đáy chậu, tinh hoàn ẩn và bìu chẻ đôi ở một cá nhân 46,XY.
Trong quá trình khám thực thể, cần tập trung vào kích thước và hình dạng của dương vật, sự đối xứng của cơ quan sinh dục ngoài, và sự hiện diện và vị trí của các tuyến sinh dục có thể sờ thấy. Cần lưu ý vị trí của niệu đạo và liệu có một hay hai lỗ mở ở đáy chậu. Mức độ nam hóa cần được ghi nhận cẩn thận, ghi lại hình dạng, chiều dài lưng khi kéo dài và đường kính của dương vật (bao gồm cả quy đầu). Vị trí của lỗ niệu đạo, mức độ hợp nhất của các nếp gấp môi-niệu đạo, và mức độ hợp nhất của nếp gấp môi-bìu và khoảng cách hậu môn-sinh dục cũng cần được lưu ý. Các nếp gấp môi-bìu hợp nhất từ sau ra trước; hình thái có thể dao động từ sự hợp nhất môi lớn phía sau, đến bán bìu hợp nhất một phần, hoặc đến bìu hợp nhất hoàn toàn với sự hợp nhất môi-niệu đạo, kéo dài đến một lỗ niệu đạo ở đường giữa.
Các cấu trúc tuyến sinh dục hoặc phần phụ có thể được xác định khi sờ nắn cẩn thận các cấu trúc môi-bìu, bìu hoặc môi lớn, vùng bẹn và bụng dưới. Vùng bẹn có thể được “vắt” từ trước ra sau để đưa tinh hoàn vào bìu. Việc không sờ thấy tinh hoàn có thể cho thấy một trẻ sơ sinh XX bị nam hóa với tăng sản thượng thận hoặc một trẻ sơ sinh XY với tinh hoàn ẩn hoặc không có tinh hoàn. Các cấu trúc được sờ thấy trong các nếp gấp môi-bìu thường là tinh hoàn thường có cấu trúc hình trứng đặc trưng. Hiếm khi, buồng trứng, tuyến lưỡng tính, hoặc thậm chí cổ tử cung có thể được tìm thấy trong các nếp gấp môi-bìu.
Sự đối xứng hoặc bất đối xứng của sự biệt hóa sinh dục ngoài cung cấp manh mối về nguyên nhân của cơ quan sinh dục không điển hình (Hình 6.6 và Hình 6.7). Các cấu trúc một bên với sự bất đối xứng của các cấu trúc sinh dục khác cho thấy DSD dạng tinh hoàn-buồng trứng hoặc 45X/46,XY và thường liên quan đến sự bất thường trong việc xuống của tuyến sinh dục một bên. Sự bất đối xứng ngụ ý các ảnh hưởng tại chỗ khác nhau, thường phản ánh các bất thường trong sự biệt hóa tuyến sinh dục (xem Hình 6.6 và Hình 6.7). Việc khám lại nhiều lần có thể có giá trị.
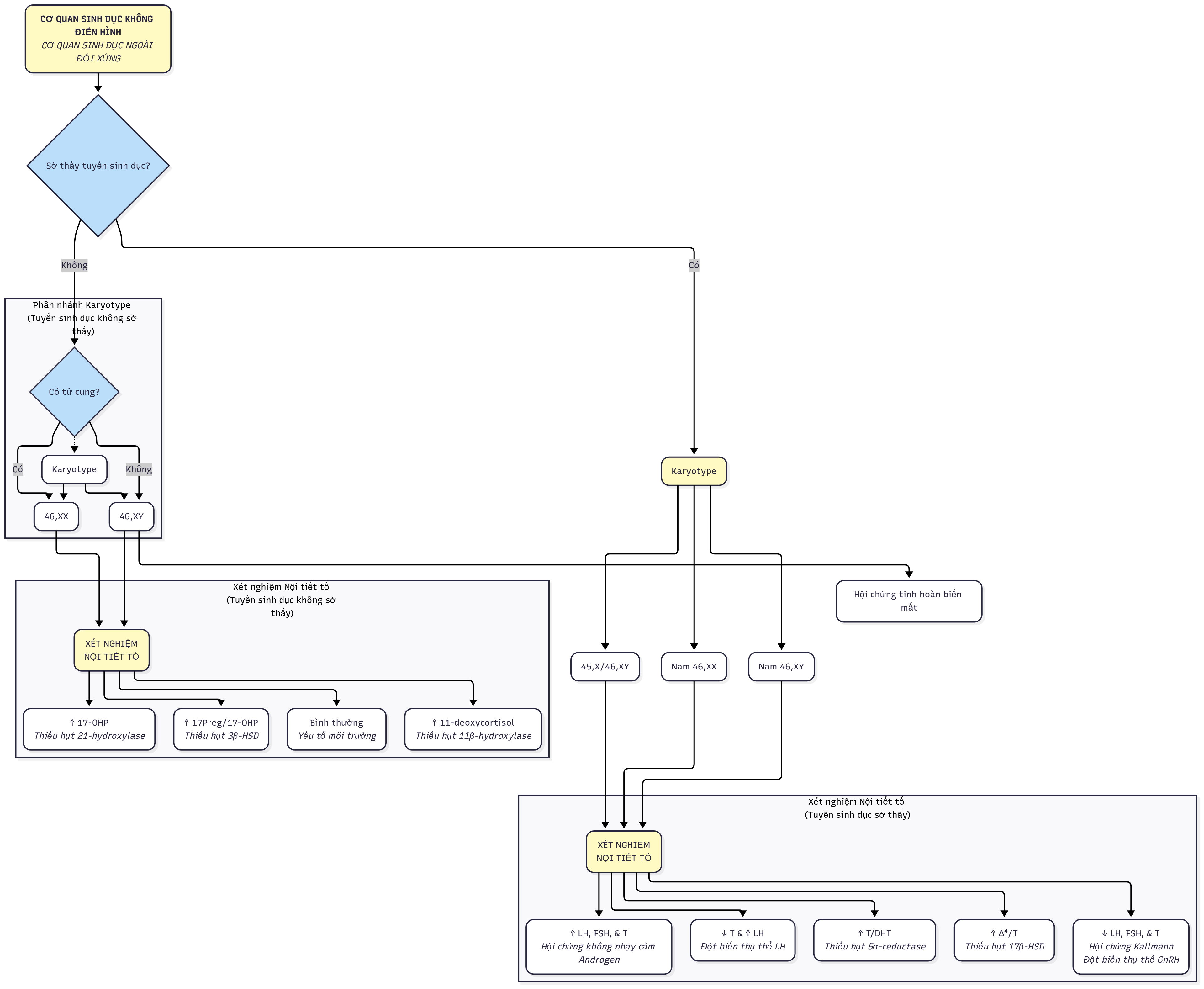
Hình 6.6 Lưu đồ tiếp cận trẻ có cơ quan sinh dục không điển hình đối xứng. Hình dạng của các nếp gấp môi-bìu và sự hiện diện/vắng mặt của các tuyến sinh dục có thể sờ thấy là tương đương ở cả hai bên. Sự hiện diện hoặc vắng mặt của các tuyến sinh dục có thể sờ thấy sẽ định hướng cho việc đánh giá xét nghiệm ban đầu. Khám siêu âm để xác định xem có tử cung hay không là hữu ích. Ví dụ, sự hợp nhất đối xứng của các nếp gấp môi-bìu, không sờ thấy tuyến sinh dục và sự hiện diện của tử cung cung cấp bằng chứng gián tiếp mạnh mẽ cho chẩn đoán một nữ giới bị nam hóa với tăng sản thượng thận bẩm sinh.
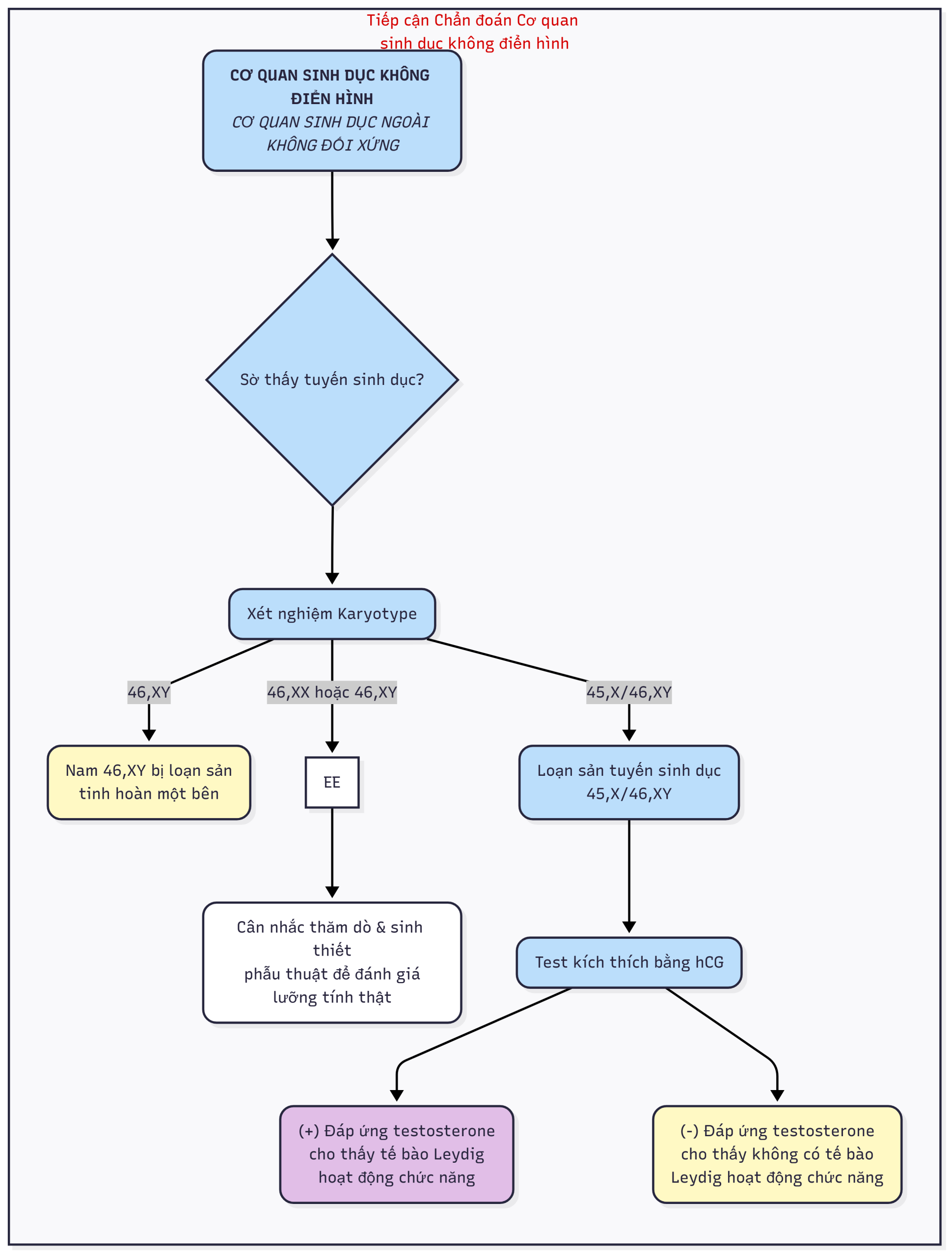
Hình 6.7 Lưu đồ tiếp cận trẻ có cơ quan sinh dục không điển hình bất đối xứng. Trong trường hợp này, các nếp gấp môi-bìu có thể có hình dạng khác nhau hoặc chỉ sờ thấy tuyến sinh dục ở một bên.
Các phép đo chiều dài dương vật được kéo dài từ đầu dương vật đến xương mu. Như dự đoán, chiều dài dương vật phụ thuộc vào tuổi thai. Đối với trẻ sơ sinh nam đủ tháng, chiều dài dương vật kéo dài trung bình là 3,5 ± 0,7 cm và đường kính trung bình là 1,1 ± 0,2 cm; giới hạn dưới (khoảng –2,5 độ lệch chuẩn [SD]) lúc đủ tháng là 2,0 cm. Một dương vật nhỏ đơn độc có thể là hậu quả của việc giảm tiếp xúc với testosterone trong nửa sau của thai kỳ do suy tế bào Leydig, thiếu hụt LH, hội chứng không nhạy cảm với androgen, đột biến LHCGR hoặc thiếu hụt GH. Việc tìm thấy cả dương vật nhỏ và lỗ tiểu lệch thấp có thể được phân loại là một DSD; việc đánh giá thêm thường được bảo đảm.
Việc đo âm vật đòi hỏi một ước tính cẩn thận của đầu gần với việc loại trừ da bao phủ. Chiều dài âm vật thường nhỏ hơn 1,0 cm, mặc dù có những biến thể hiếm gặp. Chiều dài và chiều rộng của âm vật có thể được đo. Đối với trẻ sơ sinh nữ đủ tháng, chiều dài âm vật trung bình là 4,0 ± 1,24 mm và chiều rộng âm vật trung bình là 3,32 ± 0,78 mm. Chỉ số âm vật là tích của chiều dài nhân với chiều rộng; chỉ số âm vật trung bình là 13,3 mm. Vị trí của lỗ niệu đạo cần được xác định bằng cách quan sát, chứng kiến dòng nước tiểu hoặc bằng cách cẩn thận đặt một ống thông cứng. Nếu quan sát thấy đi tiểu, cần lưu ý lực, đường kính và hướng của dòng nước tiểu. Vị trí của ống thông được đặt cũng có thể cung cấp thông tin ban đầu quan trọng. Nếu hướng về phía lỗ hậu môn và có thể sờ thấy dưới da đáy chậu, ống thông có khả năng nằm trong một xoang niệu-sinh dục, như thường xảy ra với sự nam hóa của thai nhi 46,XX thứ phát sau thiếu 21-hydroxylase. Tuy nhiên, một niệu đạo dương vật được dự đoán nếu ống thông hướng về phía trước và không thể sờ thấy.
Tỷ lệ hậu môn-sinh dục được đo bằng khoảng cách giữa hậu môn và mép sau âm hộ, chia cho khoảng cách giữa hậu môn và gốc dương vật. Vì siêu âm vùng chậu là một phần của đánh giá xét nghiệm ban đầu, việc khám trực tràng có thể không cần thiết. Nếu có, một cổ tử cung ở đường giữa thường có thể được sờ thấy khi khám trực tràng.
Thang đo Prader thường được sử dụng để phân loại hình thái của cơ quan sinh dục ngoài: (1) cơ quan sinh dục nữ bình thường với phì đại âm vật; (2) hợp nhất môi lớn một phần và phì đại âm vật; (3) hợp nhất môi-bìu, sao cho có một lỗ mở duy nhất từ xoang niệu-sinh dục và phì đại âm vật; (4) hợp nhất các nếp gấp môi-bìu sao cho lỗ mở duy nhất nằm ở gốc của cấu trúc dương vật; và (5) nam hóa hoàn toàn với dương vật có kích thước dương vật, hợp nhất môi lớn hoàn toàn và lỗ niệu đạo trên quy đầu. Một thang điểm nam hóa bên ngoài rất hữu ích để đánh giá sự nam hóa ở trẻ sơ sinh nam; hệ thống tính điểm này đánh giá sự hợp nhất của bìu, kích thước dương vật, vị trí của lỗ niệu đạo và vị trí của các tuyến sinh dục.
Ngoài việc khám sinh dục, việc khám nên bao gồm cân nặng, chiều dài và các đặc điểm khác để xác định xem các phát hiện có phù hợp với tuổi thai hay không, đặc biệt là ở trẻ có vẻ là nữ vì âm vật nổi bật hơn ở trẻ sinh non. Hình thái này là do lượng mỡ dưới da ít và sự hoàn thành tăng trưởng của âm vật trước tam cá nguyệt cuối của cuộc sống bào thai. Một cuộc khám cẩn thận bao gồm việc kiểm tra các đặc điểm dị dạng bổ sung vì cơ quan sinh dục không điển hình có thể xảy ra cùng với các bất thường khác. Chúng bao gồm các khuyết tật đường giữa mặt, kích thước đầu, và các bất thường tai và ngón tay. Lộn bàng quang và lỗ tiểu lệch trên đại diện cho một dị tật không do nội tiết của hệ thống tiết niệu. Trẻ sơ sinh bị CAH, cả nam và nữ, có thể biểu hiện tăng sắc tố của cơ quan sinh dục và núm vú do suy thượng thận và tăng tiết ACTH.
Trong một số trường hợp, một đứa trẻ lớn hơn hoặc một thanh thiếu niên được giới thiệu để đánh giá. Việc khám sinh dục ở trẻ em có thể được thực hiện một cách tinh tế và nhanh chóng để tránh gây tổn thương cho trẻ. Việc sờ nắn chỉ nên được thực hiện nếu thực sự cần thiết. Trước khi khám trẻ, đặc biệt là trẻ nhỏ, điều quan trọng là phải nhắc nhở trẻ rằng đây là một khu vực riêng tư, giải thích lý do khám và xin phép cả trẻ và cha mẹ.
Các xét nghiệm
Các xét nghiệm ban đầu để đánh giá cơ quan sinh dục không điển hình nên bao gồm bộ nhiễm sắc thể, xác định hormone và các nghiên cứu hình ảnh. Siêu âm là phương pháp hình ảnh điển hình vì nó không cần gây mê, không xâm lấn và không cần tia xạ hoặc chất cản quang. Siêu âm có thể đánh giá sự hiện diện và vị trí của các tuyến sinh dục, thận, tuyến thượng thận và tử cung. Các cấu trúc tuyến sinh dục có thể quá nhỏ để xác định chắc chắn bằng siêu âm. Do đó, việc không quan sát thấy các tuyến sinh dục trên siêu âm không chứng minh được sự vắng mặt của các tuyến sinh dục. Trong một số trường hợp, MRI có thể giúp đánh giá các cấu trúc Müller và các bất thường trong đường tiết niệu. Hạn chế của MRI bao gồm sự không chính xác trong việc phát hiện các tuyến sinh dục trong ổ bụng, các tuyến sinh dục dạng dải hoặc tử cung trước dậy thì. Sau khi hoàn thành các đánh giá khác, nội soi ổ bụng có thể cần thiết để đánh giá chính xác các cấu trúc trong ổ bụng và các tuyến sinh dục, trong khi nội soi bàng quang có thể cần thiết để đánh giá niệu đạo và âm đạo. Khi chẩn đoán vẫn chưa chắc chắn, nội soi ổ bụng để quan sát các cấu trúc sinh dục trong và lấy sinh thiết mô tuyến sinh dục có thể hỗ trợ các khuyến nghị quản lý. Mức độ biệt hóa tuyến sinh dục và tình trạng tế bào mầm có thể dự đoán chức năng tuyến sinh dục và làm rõ nguy cơ khối u tế bào mầm.
Khi không sờ thấy tuyến sinh dục, cơ quan sinh dục ngoài không điển hình đối xứng, và có tử cung và có thể có buồng trứng, chẩn đoán có khả năng nhất là một thai nhi 46,XX bị nam hóa với CAH. Tuy nhiên, không thể loại trừ khả năng các tinh hoàn bị loạn sản rõ rệt. Nếu chẩn đoán phân biệt dựa trên biểu hiện bao gồm CAH, các xét nghiệm ban đầu nên bao gồm điện giải đồ, PRA, nồng độ 17-OHP, AMH và cortisol trong huyết thanh.
Các xét nghiệm di truyền
Bộ nhiễm sắc thể xác nhận giới tính nhiễm sắc thể, ngay cả khi xét nghiệm nhiễm sắc thể trước sinh đã được thực hiện. Xét nghiệm di truyền tế bào bằng phương pháp lập karyotype G-banded có thể phát hiện sự hiện diện của hai hoặc nhiều dòng tế bào. Do thời gian quay vòng nhanh hơn, lai tại chỗ phát huỳnh quang (FISH) hoặc PCR với các dấu hiệu đặc hiệu cho nhiễm sắc thể X và Y được khuyến nghị. Nói chung, karyotype máu ngoại vi là đủ để xác định bộ nhiễm sắc thể. Tuy nhiên, bệnh nhân có thể có bộ nhiễm sắc thể thể khảm với một hoặc nhiều dòng tế bào bổ sung chỉ giới hạn ở mô tuyến sinh dục. Phân tích vi mảng CGH ngày càng có sẵn và có thể xác định sự mất cân bằng DNA bộ gen dưới kính hiển vi hoặc các biến thể số lượng bản sao. Tuy nhiên, các xét nghiệm CGH và đa hình đơn nucleotide (SNP) có thể không phát hiện được các chuyển đoạn nhiễm sắc thể cân bằng và thể khảm ở mức độ thấp.
Các phân tích di truyền phân tử ngày càng có sẵn để xác định và xác nhận cơ sở phân tử của cơ quan sinh dục không điển hình và có thể được coi là một phần của quá trình chẩn đoán được tiến hành song song với việc xác định kiểu hình lâm sàng và phân tích hormone. Việc xác định đột biến cụ thể giúp cải thiện độ chính xác của tư vấn di truyền và dự đoán về nguy cơ tái phát. Phòng thí nghiệm di truyền thường có thể thực hiện FISH cho gen SRY, hoặc các xét nghiệm nhiễm sắc thể chi tiết khác, chẳng hạn như vi mảng CGH (Hình 6.8). Đối với một số DSD, các phân tích di truyền phân tử của các gen cụ thể có sẵn thông qua các phòng thí nghiệm thương mại. Thông tin về các chi tiết cụ thể có thể được lấy từ một nguồn tài nguyên dựa trên web do Viện Y tế Quốc gia tài trợ, Genetests. Trong một số trường hợp, xét nghiệm di truyền có sẵn thông qua các phòng thí nghiệm nghiên cứu.
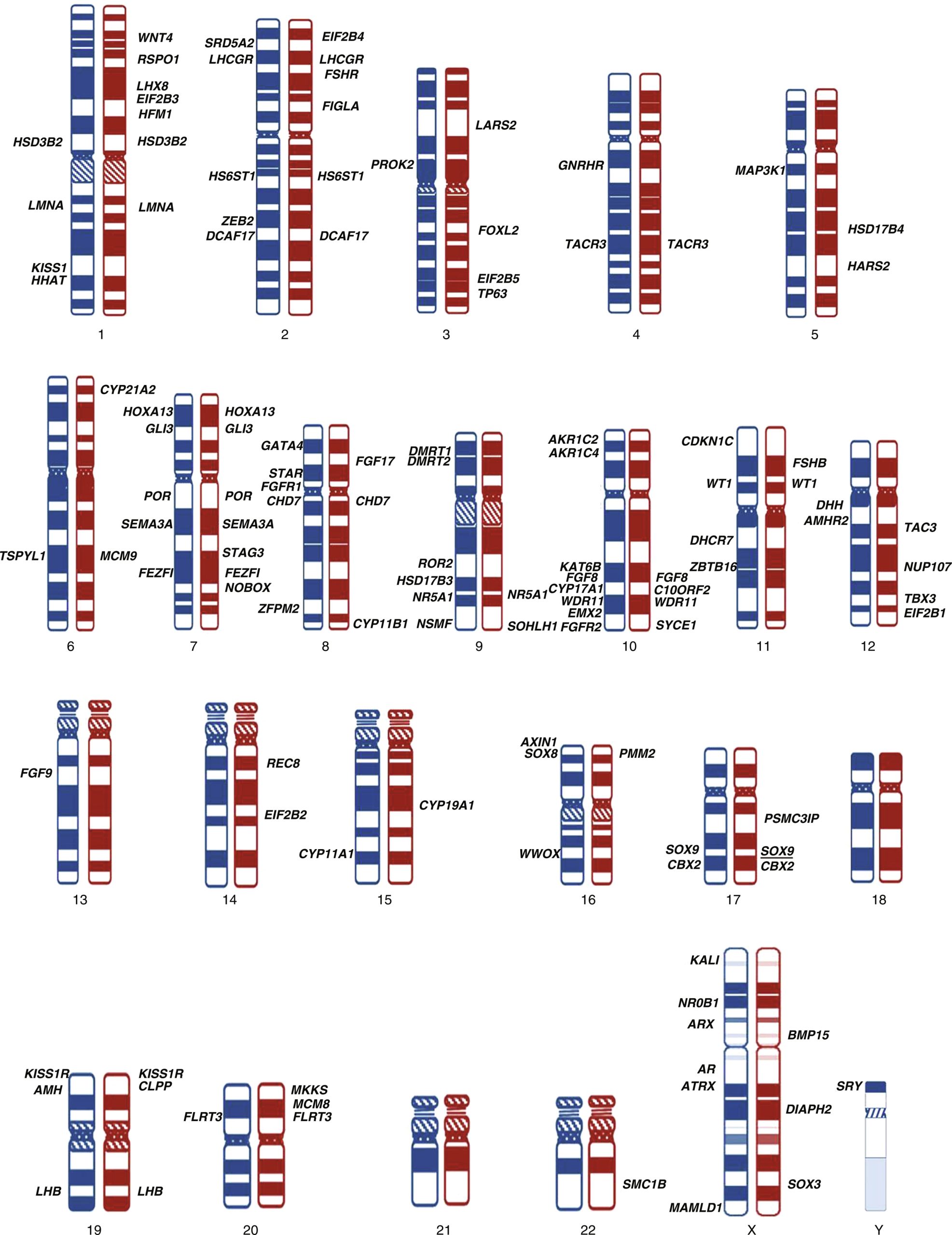
Hình 6.8. Bản đồ các gen liên quan đến các rối loạn phát triển giới tính (DSD). Các gen được biết là gây ra loạn sản tuyến sinh dục, suy sinh dục do giảm gonadotropin, rối loạn sinh tổng hợp steroid, các bất thường niệu-sinh dục đơn độc, các tình trạng hội chứng liên quan đến cơ quan sinh dục không điển hình và suy tuyến sinh dục nguyên phát và thứ phát được liệt kê dọc theo mỗi nhiễm sắc thể. Bên trái, các gen liên quan đến các cá thể XY, bên phải, các gen được biết là gây ra loạn sản buồng trứng và DSD ở các cá thể XX.(Từ Yatsenko, S.A., Witchel, S.F. (2017). Cách tiếp cận di truyền đối với cơ quan sinh dục không điển hình và các rối loạn phát triển giới tính: Những gì bác sĩ lâm sàng cần biết. Semin Perinatol, 41(4), 232–243.)
Giải trình tự thế hệ tiếp theo (NGS) có thể mang lại lợi ích cho việc quản lý bệnh nhân bằng cách xác định các rối loạn cụ thể và các kiểu di truyền. Giải trình tự toàn bộ exome (WES) và giải trình tự toàn bộ bộ gen (WGS) là các kỹ thuật để phân tích đồng thời toàn bộ exome và toàn bộ bộ gen, tương ứng. Phân tích di truyền toàn bộ exome ngày càng có sẵn. WES không xác định được các biến thể trong các vùng không mã hóa. WGS có thể phát hiện các đột biến trong các vùng không mã hóa, bao gồm các intron, các vùng không được dịch, và các vùng điều hòa cis gần và xa; các biến thể như vậy có thể sửa đổi cấu dạng chromatin làm thay đổi vị trí cấu trúc giữa các vùng điều hòa với hậu quả là sự biểu hiện sai của các gen cụ thể. Cả hai kỹ thuật đều không phát hiện đầy đủ các biến thể số lượng bản sao (CNV) lớn hơn, các trình tự lặp lại, lệch bội hoặc các sửa đổi biểu sinh. Sau đó, việc xác nhận các biến thể di truyền được xác định bằng PCR, sau đó là giải trình tự Sanger hoặc đa hình chiều dài đoạn giới hạn là cần thiết.
Các phương pháp NGS này tạo ra nhiều thông tin; WES thường phát hiện 15.000 đến 20.000 biến thể và WGS phát hiện 3 đến 4 triệu biến thể, khác với bộ gen tham chiếu của con người. Các công cụ này tạo ra lượng dữ liệu di truyền khổng lồ, đòi hỏi sự hỗ trợ tin sinh học có hiểu biết. Với việc xác định nhiều biến thể, việc phân tích các biến thể có ý nghĩa từ các biến thể lành tính là rất quan trọng. Các biến thể có thể được xác định là gây bệnh rõ ràng bởi đột biến cụ thể, kiểu di truyền, kết quả của nhiều phần mềm dự đoán mô hình hóa in silico, các xét nghiệm chức năng và sự bảo tồn giữa các loài. Trường Cao đẳng Di truyền và Gen y học Hoa Kỳ cung cấp các hướng dẫn để giải thích các biến thể trong bối cảnh thuật ngữ tiêu chuẩn (Bảng 6.8). Do tốc độ khám phá gen đang diễn ra, việc phân tích lại các biến thể có ý nghĩa không chắc chắn có thể được bảo đảm. Sự không đồng nhất về kiểu hình và độ xâm nhập không hoàn toàn làm phức tạp việc giải thích các phân tích di truyền. Các bác sĩ lâm sàng cũng cần nhận thức được các vấn đề đạo đức tiềm tàng, chẳng hạn như phát hiện một đột biến gây bệnh trong một gen không liên quan đến DSD (phát hiện tình cờ). Tư vấn trước khi xét nghiệm và sự đồng ý sau khi được thông báo là bắt buộc trước khi thực hiện các nghiên cứu di truyền.
Bảng 6.8. Các thuật ngữ được sử dụng để mô tả các biến thể trình tự
| Diễn giải | Ý nghĩa |
| Gây bệnh (Pathogenic) | Góp phần gây ra bệnh |
| Có khả năng gây bệnh/liên quan | Dữ liệu phù hợp với vai trò gây bệnh với một mức độ tin cậy xác định |
| Lành tính (Benign) | Dữ liệu có sẵn cho thấy biến thể không góp phần gây ra bệnh |
| Có khả năng lành tính | Dữ liệu phù hợp với vai trò không gây bệnh với một mức độ tin cậy xác định |
| Biến thể có ý nghĩa không chắc chắn (VUS) | Dữ liệu không đủ để phân loại biến thể |
| Liên quan (Associated) | Biến thể được làm giàu đáng kể ở các cá nhân bị ảnh hưởng so với các nhóm chứng phù hợp |
| Gây tổn hại (Damaging) | Biến thể ảnh hưởng đến mức độ bình thường hoặc chức năng sinh hóa của một sản phẩm gen |
| Có hại (Deleterious) | Biến thể làm giảm khả năng sinh sản của người mang gen |
Trong một số trường hợp, trẻ sơ sinh bị DSD được phát hiện thông qua xét nghiệm tiền sản không xâm lấn (NIPT) sử dụng DNA của thai nhi tự do trong huyết thanh của mẹ. Kỹ thuật này bao gồm giải trình tự song song hàng loạt hoặc phân tích SNP của các đoạn DNA của thai nhi dài 150 đến 180 cặp bazơ và tránh được các rủi ro do lấy mẫu nhung mao màng đệm và chọc ối. Hiện tại, các báo cáo dương tính giả và âm tính giả hạn chế tính hữu dụng của kỹ thuật này. Xét nghiệm di truyền chẩn đoán xâm lấn truyền thống là cần thiết nếu kết quả NIPT cho thấy một bất thường di truyền hoặc khác với các phát hiện siêu âm.
Xác định Hormone
Các nghiên cứu ban đầu khác phụ thuộc vào các phát hiện thực thể. Nếu cơ quan sinh dục ngoài bị nam hóa đối xứng ở bất kỳ mức độ nào mà không có các tuyến sinh dục có thể sờ thấy, đặc biệt nếu có tử cung bình thường, các nghiên cứu bổ sung nên hướng đến các nguyên nhân gây nam hóa của một trẻ sơ sinh nữ. Vì thiếu 21-hydroxylase là nguyên nhân phổ biến nhất của sự nam hóa và cơ quan sinh dục không điển hình ở trẻ sơ sinh 46,XX, các xét nghiệm ban đầu nên bao gồm việc xác định nồng độ 17- hydroxyprogesterone (17-OHP). Nói chung, nồng độ 17-OHP nên được đo không sớm hơn 48 đến 72 giờ tuổi vì có khả năng cho kết quả dương tính giả có thể do các steroid sulfate phản ứng chéo. Tuy nhiên, nồng độ 17-OHP cực kỳ cao có thể được phát hiện ở các thời điểm sớm hơn ở trẻ sơ sinh bị CAH cổ điển. Nếu một hoặc cả hai tuyến sinh dục có thể được sờ thấy, mục đích của các nghiên cứu sàng lọc là để xác định sự đầy đủ của quá trình tổng hợp androgen và hoạt động của androgen ở một trẻ sơ sinh nam. Việc xác định nồng độ LH, FSH và testosterone ở trẻ sơ sinh cung cấp thông tin về chức năng của tinh hoàn và trục HPG (xem Bảng 6.4).
Mẫu nồng độ hormone steroid cung cấp bằng chứng cho các khiếm khuyết cụ thể trong quá trình sinh tổng hợp steroid (xem Bảng 6.4). Chẩn đoán CAH phát sinh từ thiếu 21-hydroxylase được xác nhận bằng cách tìm thấy nồng độ 17-OHP tăng. Thông thường, nồng độ 17-OHP lớn hơn 10.000 ng/dL (300 nmol/L) ở trẻ sơ sinh bị ảnh hưởng. Đối với thiếu 11β-hydroxylase, nồng độ 11-deoxycortisol và 17-OHP tăng. Đối với thiếu 3β-hydroxysteroid dehydrogenase, nồng độ pregnenolone, 17-hydroxypregnenolone và DHEA thường tăng. Nồng độ steroid thấp đối với bệnh nhân bị tăng sản thượng thận bẩm sinh dạng lipid và đột biến CYP11A1. Khi các dạng mất muối của CAH được bao gồm trong chẩn đoán phân biệt, điện giải đồ huyết thanh và PRA nên được theo dõi. Thông thường, mặc dù thiếu mineralocorticoid, hạ natri máu và tăng kali máu không có mặt khi sinh và phát triển trong tuần đầu tiên của cuộc đời.
Đối với các dạng CAH nhẹ hơn hoặc các rối loạn khác ảnh hưởng đến sinh tổng hợp steroid thượng thận, xét nghiệm kích thích ACTH có thể cần thiết để xác nhận chẩn đoán. Sau khi lấy một mẫu máu cơ bản, ACTH tổng hợp (0,25 mg) có thể được tiêm tĩnh mạch nhanh hoặc tiêm bắp. Một mẫu máu thứ hai để đo đáp ứng hormone được kích thích bằng ACTH có thể được lấy sau 30 hoặc 60 phút. Các dạng CAH nhẹ hơn thường không ảnh hưởng đến sự biệt hóa sinh dục ngoài và do đó thường không liên quan đến cơ quan sinh dục không điển hình. Trẻ sơ sinh bị CAH khởi phát muộn thường không được phát hiện thông qua các chương trình sàng lọc sơ sinh, có lẽ vì nồng độ 17-OHP trong máu toàn phần được xác định từ giấy lọc sàng lọc sơ sinh thấp hơn các giá trị được sử dụng làm ngưỡng.
Ngoài việc đánh giá chẩn đoán cho các rối loạn sinh tổng hợp steroid, các phép đo hormone trong giai đoạn sơ sinh ngay lập tức cung cấp một chỉ số về chức năng của trục HPG. Nồng độ testosterone thấp và gonadotropin tăng ở một trẻ sơ sinh 46,XY có cơ quan sinh dục không điển hình cho thấy sinh tổng hợp testosterone không đủ. Nồng độ testosterone và gonadotropin tăng ở một trẻ sơ sinh có cơ quan sinh dục ngoài của nữ giới, các khối u ở môi lớn hai bên và bộ nhiễm sắc thể 46,XY phù hợp với chẩn đoán không nhạy cảm với androgen (xem Bảng 6.5).
Nồng độ AMH phản ánh chức năng của tế bào Sertoli; nồng độ có sự khác biệt về giới tính, với các giá trị cao ở bé trai (20–80 ng/mL) trong 6 năm đầu đời và các giá trị thấp ở bé gái. Nồng độ AMH có thể giúp phân biệt giữa tình trạng không có tinh hoàn (anorchia) và tinh hoàn ẩn (cryptorchidism). Ngoài ra, nồng độ AMH có thể hữu ích trong các rối loạn loạn sản tinh hoàn hoặc ở nữ giới bị nam hóa để xác định sự hiện diện của mô tinh hoàn. Nồng độ inhibin B, thấp hơn ở nữ giới so với nam giới, cung cấp một dấu hiệu khác về chức năng của tế bào Sertoli.
Việc đánh giá sự tiết testosterone có thể hữu ích, đặc biệt đối với bệnh nhân có bằng chứng về mô tinh hoàn qua sờ nắn hoặc siêu âm và nồng độ AMH cho thấy có mô tinh hoàn. Một phương pháp là sử dụng hCG và đo đáp ứng hormone. So với LH, 24 axit amin bổ sung ở đầu carboxy của protein làm tăng hoạt tính sinh học của hCG và sự hiện diện của một axit sialic ở đầu cuối trên chuỗi carbohydrate kéo dài thời gian bán hủy của nó. Các chế phẩm hCG đặc hiệu bao gồm hCG chiết xuất từ nước tiểu của phụ nữ mang thai và hCG tái tổ hợp. Mặc dù hCG tái tổ hợp thường đắt hơn, nó cho thấy sự nhất quán tốt giữa các lô, hoạt tính đặc hiệu cao và độ tinh khiết cao hơn. Nhiều phác đồ đã được sử dụng với sự thay đổi về liều lượng và tần suất dùng thuốc để đánh giá loạn sản tuyến sinh dục, thiểu sản tế bào Leydig, các khiếm khuyết trong sinh tổng hợp testosterone và khả năng đáp ứng với androgen. Một phác đồ bao gồm việc tiêm dưới da 1000 đến 1500 đơn vị, hàng ngày hoặc cách ngày, trong 1 đến 5 ngày, với việc lấy mẫu máu vào ngày sau mũi tiêm cuối cùng. Có thể tiêm ngắt quãng trong tối đa 3 tuần để kích thích sự phát triển của dương vật nhằm chứng minh cả sự tiết testosterone và khả năng đáp ứng của mô đích. Tổng liều không được vượt quá 15.000 đơn vị hCG. Việc xác định hormone nên bao gồm androstenedione, testosterone và DHT. Nồng độ testosterone nên tăng hơn gấp đôi. Tỷ lệ hormone sau kích thích được sử dụng để đánh giá các khiếm khuyết trong sinh tổng hợp testosterone. Sau kích thích, tỷ lệ testosterone/androstenedione dưới 0,8 ở trẻ sơ sinh và dưới 1,0 ở bệnh nhân trước dậy thì và vị thành niên gợi ý thiếu hụt 17β-hydroxysteroid dehydrogenase-3. Tương tự, tỷ lệ testosterone/DHT cơ bản lớn hơn 10 và sau kích thích hCG lớn hơn 20 gợi ý thiếu hụt 5α-reductase. Tuy nhiên, có thể cần xét nghiệm đột biến để phân biệt chính xác giữa các rối loạn này.
Sàng lọc sơ sinh
Các chương trình sàng lọc sơ sinh (NBS) đã được thiết lập ở tất cả 50 tiểu bang của Mỹ và nhiều quốc gia để xác định trẻ sơ sinh bị CAH thể cổ điển. Thông thường, các chương trình sàng lọc đo nồng độ 17-OHP trong máu toàn phần được chiết xuất từ một đốm máu khô trên giấy lọc bằng xét nghiệm miễn dịch có sẵn trên thị trường, chẳng hạn như xét nghiệm miễn dịch huỳnh quang lanthanide tăng cường phân ly (DELFIA, Perkin-Elmer, Waltham, MA). Nồng độ 17-OHP trong máu toàn phần tăng nhẹ được phát hiện khá thường xuyên (đặc biệt ở trẻ sinh non) làm phức tạp việc ra quyết định lâm sàng về tình trạng bệnh và nhu cầu bắt đầu điều trị bằng glucocorticoid. Nguyên nhân của các kết quả dương tính giả này bao gồm sinh non, các steroid phản ứng chéo, lấy mẫu trước 48 đến 72 giờ tuổi, dị hợp tử đối với thiếu hụt 21-hydroxylase, trẻ bị stress và CAH khởi phát muộn. Kết quả âm tính giả có thể xảy ra; với kết quả âm tính giả phổ biến hơn ở bé gái so với bé trai. Việc mẹ được điều trị bằng glucocorticoid cũng có thể dẫn đến kết quả âm tính giả. Nếu chẩn đoán phân biệt bao gồm CAH, cần thực hiện xét nghiệm chuyên biệt ngay cả khi kết quả sàng lọc sơ sinh được báo cáo là âm tính đối với thiếu hụt 21-hydroxylase.
Để tránh có quá nhiều kết quả sàng lọc dương tính giả, các mức ngưỡng thường được chọn để xác định tất cả trẻ sơ sinh bị các dạng mất muối cổ điển hoặc nam hóa đơn thuần. Trẻ sơ sinh bị CAH khởi phát muộn thường không được phát hiện qua NBS. Độ đặc hiệu có thể được cải thiện bằng cách sử dụng các quy trình bổ sung, chẳng hạn như chiết xuất hữu cơ, sắc ký hoặc phân tích sắc ký lỏng-khối phổ song song (LC/MS/MS). Kỹ thuật này, LC/MS/MS, có thể có lợi để xác nhận hoặc loại trừ chẩn đoán CAH và cũng có thể được sử dụng để chẩn đoán các rối loạn sinh tổng hợp steroid khác.
Một thách thức đối với NBS là đảm bảo độ nhạy và độ đặc hiệu tối đa. Các chiến lược để cải thiện việc xác định trẻ em bị ảnh hưởng bao gồm lấy mẫu máu lặp lại hoặc phân tích di truyền phân tử. Hiện tại, khoảng 22% trẻ sơ sinh ở Mỹ thường được sàng lọc lần thứ hai. Các phương pháp tiếp cận khác bao gồm sử dụng các điểm cắt dựa trên tuổi thai, mẫu thứ hai cho xét nghiệm miễn dịch 17-OHP, nồng độ 21-deoxycortisol, phân tích sắc ký khí-khối phổ, hoặc tỷ lệ hormone, chẳng hạn như tỷ lệ 17-OHP/11-deoxycortisol.
Chẩn đoán hình ảnh, Nội soi và Nội soi ổ bụng
Các nghiên cứu nội soi có thể được sử dụng để xác định vị trí hợp lưu âm đạo-niệu đạo so với cổ bàng quang và lỗ mở duy nhất của xoang niệu-sinh dục. Bằng cách sử dụng ống soi bàng quang và đặt ống thông, các khoảng cách này có thể được xác định trước hoặc kết hợp với các nghiên cứu cản quang ngược dòng được thực hiện để phác thảo niệu đạo và chứng minh (nếu có) âm đạo, tử cung, cổ tử cung và khoang tử cung. Thông tin như vậy là cần thiết để lập kế hoạch phẫu thuật tạo hình, đánh giá nguy cơ biến chứng y tế, và trong một số trường hợp cung cấp thông tin để xác định giới tính nuôi dưỡng. Nếu các thủ thuật đó không hữu ích, có thể cần nội soi ổ bụng để quan sát và sinh thiết các cấu trúc sinh dục trong và tuyến sinh dục. MRI có thể hữu ích để xác định các mối quan hệ giải phẫu.
Điều trị
Trong khi chờ đợi kết quả của các nghiên cứu xét nghiệm ban đầu, và khi cần thiết, sự chú ý được tập trung vào quyết định chính: giới tính nuôi dưỡng. Trong tình huống lý tưởng, một đội ngũ chuyên gia DSD sẽ gặp gỡ gia đình. Đội ngũ DSD thường bao gồm một bác sĩ nội tiết nhi, một bác sĩ phẫu thuật nhi hoặc tiết niệu nhi có chuyên môn về phẫu thuật tạo hình niệu-sinh dục, một chuyên gia sức khỏe hành vi (nhà tâm lý học, nhân viên xã hội, hoặc bác sĩ tâm thần), và một bác sĩ sơ sinh. Ý kiến từ một nhà tư vấn đạo đức có thể hữu ích. Sử dụng tất cả các dữ liệu có sẵn, đội ngũ DSD chia sẻ thông tin của trẻ với gia đình và giáo dục họ. Tại thời điểm chẩn đoán và các tương tác sau đó, gia đình được hưởng lợi từ việc có cơ hội bày tỏ nỗi sợ hãi và lo lắng của họ trong môi trường không đe dọa để chuẩn bị cho việc ra quyết định liên quan.
Các gia đình phải vật lộn để hiểu thông tin y tế phức tạp và đối phó với sự khác biệt về giải phẫu của con mình. Cân nhắc bổ sung bao gồm khả năng bị kỳ thị bởi cộng đồng, lo lắng về bản dạng giới của con, đau khổ về khả năng sinh sản của con, không chắc chắn về nguy cơ u tuyến sinh dục và những bất tiện vốn có với nhiều lần đi khám bệnh. Sự tham gia ban đầu của cha mẹ trong các cuộc thảo luận và ra quyết định liên quan đến các lựa chọn về giới tính nuôi dưỡng, điều trị y tế và các can thiệp phẫu thuật có thể sẽ xác định cách tiếp cận cho các cuộc thảo luận tiếp theo. Các cuộc thảo luận trung thực, nhạy cảm và thẳng thắn với cha mẹ chỉ giúp ích cho đứa trẻ. Mục tiêu cao nhất là một chiến lược tích cực để thúc đẩy lòng tự trọng tích cực của trẻ.
Thông thường, bác sĩ sơ sinh là bác sĩ đầu tiên giao tiếp với cha mẹ về tình trạng cơ quan sinh dục không điển hình. Ngoài việc nói chuyện với cha mẹ, bác sĩ sơ sinh còn hỗ trợ điều phối chăm sóc và bắt đầu các nghiên cứu xét nghiệm. Trong một số trường hợp, bác sĩ sơ sinh điều phối việc chuyển trẻ đến một cơ sở chăm sóc tuyến ba. Cần phải vạch ra kế hoạch quản lý lâu dài.
Trong nhiều trường hợp, giới tính nuôi dưỡng là rõ ràng dựa trên khám thực thể ban đầu và kết quả xét nghiệm. Tuy nhiên, đối với một số trẻ sơ sinh, cần phải đưa ra quyết định về giới tính nuôi dưỡng. Thảo luận về bản dạng giới trong tương lai và khả năng sinh sản là rất có giá trị. Việc xem xét các truyền thống và phong tục của gia đình trong quá trình ra quyết định sẽ trấn an đứa trẻ và gia đình. Sau một cuộc thảo luận có đầy đủ thông tin, cha mẹ và các nhà cung cấp dịch vụ chăm sóc sức khỏe sẽ chọn giới tính nuôi dưỡng.
Sức khỏe tâm thần/hành vi/tâm lý cần được xem xét ngay từ những tương tác ban đầu với gia đình. Tác động tâm lý có thể được đánh giá và giải quyết bằng các mối quan hệ liên quan đến các thành viên của đội ngũ DSD. Chuyên gia sức khỏe hành vi nên được đào tạo kỹ lưỡng và có kiến thức về các nguồn lực sẵn có cho bệnh nhân và gia đình họ. Ngoài các cuộc hẹn cần thiết cho nhu cầu y tế, phẫu thuật và tâm lý của bệnh nhân, các buổi gặp gỡ bổ sung có thể mang lại lợi ích cho cha mẹ để giải quyết các câu hỏi và mối quan tâm của họ về con mình. Các đánh giá này nên được thực hiện kịp thời.
Khi đứa trẻ trưởng thành, những giải thích trung thực về tình trạng bệnh là rất cần thiết. Tất cả những người liên quan nên cảnh giác về tác động tổng thể của tình trạng này, cũng như nhu cầu tư vấn tâm lý. Đứa trẻ nên tiến từ một đối tác thầm lặng thành một thành viên tham gia đầy đủ vào quá trình ra quyết định này, với mong muốn của đứa trẻ được ưu tiên khi thích hợp.
Khi một chẩn đoán cụ thể đã được xác nhận, việc ra quyết định và điều trị có thể được hướng dẫn tương ứng. Các cân nhắc về chăm sóc y tế bao gồm các quyết định liên quan đến giới tính nuôi dưỡng, nhu cầu phẫu thuật có thể có và thời gian biểu cho phẫu thuật theo kế hoạch, kế hoạch điều trị y tế, và tư vấn và hỗ trợ tâm lý kịp thời và phù hợp.
Quyết định giới tính nuôi dưỡng
Các quyết định liên quan đến giới tính nuôi dưỡng phù hợp dựa trên sinh lý bệnh cụ thể, đặc điểm cơ quan sinh dục ngoài, giải phẫu hệ thống sinh sản bên trong, tiên lượng về sự phát triển dậy thì tự nhiên, khả năng hoạt động tình dục và cực khoái, và tiềm năng sinh sản. Với việc sử dụng bảo tồn khả năng sinh sản và các kỹ thuật hỗ trợ sinh sản, tiềm năng sinh sản trong tương lai lớn hơn so với trước đây.
Trong những trường hợp cần đưa ra quyết định về giới tính nuôi dưỡng, mỗi đứa trẻ đều cần được xem xét cẩn thận các phát hiện thực thể, thông tin xét nghiệm và dữ liệu kết quả có sẵn. Cha mẹ thường lo lắng vì sợ bị xã hội kỳ thị và lo lắng rằng đứa trẻ sẽ từ chối giới tính được chỉ định khi lớn lên. Thật vậy, sự không chắc chắn về đứa con sơ sinh của họ có thể gây đau khổ và tổn thương đến mức khả năng ra quyết định của họ cho đứa trẻ bị phá vỡ. Trong hầu hết các trường hợp, trẻ em được nuôi dưỡng là nam hoặc nữ phù hợp với bộ nhiễm sắc thể, thừa nhận các trường hợp ngoại lệ hiếm gặp ủng hộ việc đảo ngược giới tính (khi việc tái chỉ định giới tính tự khởi xướng là phù hợp). Mặc dù bộ nhiễm sắc thể cung cấp thông tin, kiểu hình của bệnh nhân phản ánh sự tương tác của nhiều yếu tố. Các nghiên cứu kết quả theo chiều dọc ngày càng có sẵn liên quan đến chức năng của người trưởng thành, chất lượng cuộc sống và bản dạng giới.
Bản dạng giới đề cập đến trải nghiệm của một người về bản thân. Trong hầu hết các trường hợp, bản dạng giới phù hợp với giới tính nuôi dưỡng (giới tính khai sinh). Các yếu tố chính, các bước quan trọng và trình tự phát triển tâm lý tình dục góp phần vào bản dạng giới vẫn chưa chắc chắn. Phát triển tâm lý tình dục có ba thành phần: bản dạng giới, vai trò giới và xu hướng tính dục. Bản dạng giới được thiết lập ở tuổi sơ sinh. Vai trò giới, bị ảnh hưởng bởi phơi nhiễm hormone trước sinh, thường phát triển trong thời thơ ấu nhưng có thể thay đổi trong suốt cuộc đời do phản ứng của mỗi cá nhân đối với quan điểm và kỳ vọng của xã hội. Xu hướng tính dục thường không được biết trước tuổi dậy thì và không thể được xem xét khi cân nhắc giới tính nuôi dưỡng.
Dự báo tốt nhất về bản dạng giới ở bệnh nhân DSD dường như là giới tính nuôi dưỡng. Tuy nhiên, như có thể dự đoán, một cá nhân bị DSD có thể có những cảm xúc, câu hỏi và suy nghĩ phức tạp về bản dạng giới của chính mình. Ngoài ra, xã hội ngày càng nhận ra rằng giới tính tồn tại trên một phổ phi nhị nguyên. Dựa trên nghiên cứu DSD-LIFE quy mô lớn, số lượng cá nhân thay đổi giới tính cao hơn ở một số chẩn đoán nhất định. Tuy nhiên, nhìn chung con số này là thấp.
Bản dạng giới ở hầu hết bệnh nhân 46,XY bị CAIS là nữ, trong khi bản dạng giới đối với những người bị PAIS có thể là nữ hoặc nam. Các quyết định liên quan đến giới tính nuôi dưỡng đối với các cá nhân bị PAIS có thể là một thách thức vì tác động của androgen lên sự phát triển của hệ thần kinh trung ương và khả năng đáp ứng của mô là khác nhau. Đáp ứng lâm sàng với testosterone ngoại sinh có thể giúp ích cho quá trình ra quyết định. Khả năng đáp ứng với testosterone có thể được xác định bằng cách đánh giá sự phát triển của dương vật, sau một hoặc nhiều lần tiêm bắp testosterone. Tuy nhiên, khoảng một phần tư bệnh nhân PAIS không hài lòng với việc chỉ định giới tính, bất kể họ được nuôi dưỡng là nam hay nữ.
Phần lớn bệnh nhân bị thiếu hụt 5α-reductase tự nhận mình là nam, và khoảng một nửa số người được chẩn đoán thiếu hụt 17β-hydroxysteroid tự tái chỉ định từ nữ sang nam. Nếu có câu hỏi liên quan đến giới tính nuôi dưỡng đối với trẻ sơ sinh 46,XX bị nam hóa, thường khuyến nghị giới tính nuôi dưỡng là nữ vì hầu hết các cá nhân 46,XX bị nam hóa với CAH đều tự nhận mình là nữ. Thông thường, một vấn đề chính đối với nữ giới 46,XX bị nam hóa là nó liên quan đến các câu hỏi về phẫu thuật bộ phận sinh dục. Tuy nhiên, trong số những người bị nam hóa hoàn toàn khi sinh và được nuôi dưỡng là nam với chẩn đoán chậm trễ, sự phát triển giới tính nam với kết quả lâu dài tốt đã được báo cáo. Một báo cáo xem xét lựa chọn giới tính nuôi dưỡng là nam cho bệnh nhân 46,XX bị CAH nam hóa cực độ.
Phơi nhiễm androgen trước sinh được cho là một yếu tố ảnh hưởng đến bản dạng giới. Các bé gái bị CAH có xu hướng chọn bạn chơi là nam và ưa thích các trò chơi điển hình của nam giới. Trong số các cá nhân 46,XX bị nam hóa với CAH, tỷ lệ xu hướng đồng tính luyến ái tăng đã được báo cáo chủ yếu dựa trên hình ảnh tình dục và sức hấp dẫn tình dục tự báo cáo, với các báo cáo về sự tham gia đồng tính luyến ái thực tế được ghi nhận kém hơn. Nhìn chung, sự điều chỉnh tâm lý đã được phát hiện là tương đương đối với nữ giới bị CAH và anh chị em không bị ảnh hưởng của họ.
Mặc dù có bằng chứng cho thấy phơi nhiễm androgen với hệ thần kinh trung ương làm thay đổi hành vi chung và nhận thức, nhưng vẫn còn nhiều điều cần tìm hiểu về ảnh hưởng của bản chất và sự nuôi dưỡng đối với bản dạng giới. Nhìn chung, ngoại trừ CAIS, các cá nhân 46,XY có phơi nhiễm androgen của thai nhi biểu hiện dưới dạng nam hóa bộ phận sinh dục một phần nên được nuôi dưỡng là nam, trừ khi có các trường hợp cá biệt. Lý tưởng nhất, trẻ sơ sinh 46,XY bị thiếu hụt 17β-HSD3 hoặc 5α-reductase sẽ được chẩn đoán trong giai đoạn sơ sinh và được nuôi dưỡng là nam. Cần thận trọng, suy ngẫm và đối thoại trước khi những người có bộ nhiễm sắc thể 46,XY được chỉ định giới tính nuôi dưỡng là nữ vì phơi nhiễm androgen của thai nhi dường như ảnh hưởng mạnh mẽ đến quan niệm bản thân là nam mặc dù sự phát triển cơ quan sinh dục ngoài bị ảnh hưởng.
Trong số các cá nhân bị DSD liên quan đến loạn sản tuyến sinh dục, sự tiết testosterone được kích thích bằng hCG và đáp ứng lâm sàng tạo điều kiện cho các quyết định liên quan đến giới tính nuôi dưỡng. Kiểu hình sinh dục, khả năng của tế bào Leydig tiết ra testosterone, và mức độ nam hóa sinh dục đáp ứng với kích thích androgen là những chỉ số liên quan đến tiềm năng phát triển dậy thì tự nhiên và nhu cầu điều trị thay thế hormone. Tuy nhiên, trong loạn sản tuyến sinh dục, tác động của phơi nhiễm androgen trước sinh đối với quá trình phát triển giới tính thường không xác định được và không thể dự đoán một cách đáng tin cậy từ các nghiên cứu chẩn đoán thường quy trong giai đoạn sơ sinh.
Trong thập kỷ qua, số lượng cá nhân bị bồn chồn giới tính (sự không phù hợp giữa giới tính trải nghiệm và giới tính được chỉ định dựa trên giới tính sinh học) đã tăng lên ở những người có sự phát triển giới tính nhị nguyên điển hình. Các khía cạnh của việc chăm sóc người chuyển giới có thể được áp dụng cho những người sinh ra bị DSD và phát triển chứng bồn chồn giới tính.
Điều trị nội khoa
Một phương pháp tiếp cận nhóm đa ngành được khuyến nghị để chăm sóc bệnh nhân. Ngoại trừ các rối loạn ảnh hưởng đến sinh tổng hợp glucocorticoid và mineralocorticoid, hầu hết các tình trạng DSD không cần điều trị y tế cụ thể ở tuổi sơ sinh. Tại thời điểm dậy thì dự kiến, bệnh nhân suy sinh dục sẽ cần liệu pháp thay thế hormone phù hợp. Nhìn chung, liệu pháp thay thế hormone được bắt đầu bằng liều thấp của hormone steroid sinh dục phù hợp với việc tăng dần được thiết kế để phản ánh sự phát triển dậy thì tự nhiên. Sẽ hữu ích khi xem xét cùng gia đình tần suất dự kiến của các lần khám ngoại trú và cách đánh giá sự đầy đủ của liệu pháp thay thế.
Đối với nữ giới, việc khởi phát dậy thì bao gồm liệu pháp estrogen liều thấp—thường được bắt đầu trong khoảng từ 10,5 đến 12 tuổi để tránh sự tăng tốc quá mức của quá trình trưởng thành xương. Liều estrogen ban đầu thường là liều thấp nhất hiện có, chẳng hạn như 0,3 mg estrogen liên hợp cách ngày hoặc 5 mcg ethinyl estradiol hàng ngày. Các chế phẩm estrogen qua da cung cấp 0,007 đến 0,025 mg mỗi ngày có thể được sử dụng và được khuyến nghị dựa trên dữ liệu kết quả ban đầu. Miếng dán qua da có thể chỉ được sử dụng vào ban đêm nhằm cố gắng bắt chước dậy thì tự nhiên. Miếng dán qua da dạng ma trận cũng có thể được cắt thành các miếng nhỏ hơn để cung cấp liều estrogen thấp hơn. Dựa trên đáp ứng lâm sàng và nhận thức của bệnh nhân, liều estrogen có thể được tăng lên trong khoảng thời gian từ 6 đến 12 tháng, sao cho đạt được liều thay thế hoàn toàn và sự phát triển trong vòng 3 năm.
Điều trị bao gồm việc bổ sung một tác nhân progestational sau 12 tháng điều trị bằng estrogen hoặc khi xảy ra chảy máu kinh nguyệt, tùy điều kiện nào xảy ra sớm hơn. Sau đó, nên sử dụng liệu pháp estrogen-progesterone theo chu kỳ. Khi đã đạt được sự phát triển dậy thì hoàn toàn, liều estrogen nên là mức tối thiểu sẽ duy trì dòng chảy kinh nguyệt bình thường và ngăn ngừa mất canxi trong xương. Các lựa chọn cho liệu pháp estrogen-progesterone theo chu kỳ bao gồm thuốc tránh thai estrogen liều thấp, estrogen qua da với progestin đường uống, hoặc miếng dán qua da estrogen-progestin.
Ví dụ, có thể sử dụng một chế độ estrogen đường uống hàng ngày hoặc dạng qua da trong 21 ngày với việc bổ sung progesterone, 5 đến 10 mg medroxyprogesterone acetate, hoặc 200 mg progesterone vi hạt hóa hàng ngày được thêm vào trong 12 ngày (ngày 10–ngày 21). Sau đó là một tuần không dùng hormone. Tại thời điểm này, chế độ thay thế có thể được kéo dài để chảy máu kinh nguyệt xảy ra ít thường xuyên hơn. Trong trường hợp không có tử cung, liệu pháp progesterone trở thành tùy chọn. Ở những bệnh nhân có nồng độ androgen lưu hành thấp, sự phát triển lông mu và ham muốn tình dục có thể được cải thiện bằng cách sử dụng liều nhỏ DHEA hoặc methyltestosterone. Gonadotropin, hCG, và/hoặc gonadotropin mãn kinh người/FSH tái tổ hợp chỉ được sử dụng để kích thích rụng trứng hoặc trong các nỗ lực hỗ trợ sinh sản.
Đối với nam giới, thay thế hormone testosterone thường bắt đầu ở tuổi 12,5 đến 14. Trong những trường hợp điều trị được chỉ định vì lý do tâm lý, liệu pháp có thể được bắt đầu sớm hơn 1 đến 2 năm. Nếu liệu pháp được bắt đầu sớm hơn để trấn an bệnh nhân về những thay đổi thể chất, tốc độ trưởng thành của xương cần được theo dõi cẩn thận. Ngược lại, việc điều trị có thể bị trì hoãn để cho phép sự trưởng thành về tâm lý hoặc cảm xúc hoặc tăng trưởng bù. Liệu pháp testosterone có thể được tiêm bắp hoặc dưới da dạng depot hoặc bôi ngoài da bằng miếng dán hoặc gel. Testosterone depot (chẳng hạn như enanthate hoặc cypionate) được bắt đầu với liều 50 mg mỗi 4 tuần, sau đó tăng liều và tần suất trong khoảng 3 năm, đến liều thay thế đầy đủ khoảng 200 mg mỗi 2 tuần. Sự sẵn có của gel trong các bơm định liều cho phép tăng dần liều lượng từ 1,25 g mỗi ngày trở lên. Trong số những người có tinh hoàn đã biệt hóa và thiếu hụt gonadotropin, các kỹ thuật hỗ trợ sinh sản có thể bao gồm lấy tế bào mầm trong tinh hoàn hoặc kích thích hFSH/hLH.
Đối với bệnh nhân bị CAH, liệu pháp thay thế hormone được theo dõi cẩn thận là rất cần thiết. Mục tiêu của liệu pháp glucocorticoid là ức chế sự tiết androgen thượng thận quá mức, trong khi vẫn duy trì sự tăng trưởng và phát triển bình thường. Thông thường, liều cortisol đường uống dao động từ 8 đến 20 mg/m²/ngày. Phạm vi này dựa trên việc cung cấp 1,5 đến 2 lần tốc độ sản xuất cortisol hàng ngày, 7 đến 12 mg/m²/ngày. Fludrocortisone đường uống (ví dụ, Florinef) thường được sử dụng để thay thế mineralocorticoid cho bệnh nhân bị CAH mất muối. Bệnh nhân bị CAH nam hóa đơn thuần có thể được hưởng lợi từ việc thay thế mineralocorticoid. Liều điển hình là 0,1 mg được dùng một lần mỗi ngày. Trẻ sơ sinh và trẻ nhỏ có thể cần liều fludrocortisone cao hơn, cũng như bổ sung muối vì kháng mineralocorticoid tương đối, tốc độ sản xuất aldosterone cao hơn và lượng natri ăn vào tương đối thấp hơn. Các chủ đề thảo luận với cha mẹ bao gồm cách nghiền và cho uống viên nén, phải làm gì nếu vô tình bỏ lỡ liều, sử dụng thẻ nhận dạng cảnh báo y tế và khi nào cần dùng liều “stress”.
Đối với trẻ em bị CAH, sự đầy đủ của liệu pháp thay thế được theo dõi bằng cách đánh giá lại định kỳ tốc độ tăng trưởng, mức độ nam hóa và cảm giác thèm muối. Theo dõi xét nghiệm có thể bao gồm nồng độ androgen và 17-OHP trong huyết thanh, sự trưởng thành của xương và bài tiết 17-ketosteroid trong nước tiểu 24 giờ. Nồng độ androstenedione hữu ích để đánh giá sự đầy đủ của việc thay thế glucocorticoid, trong khi nồng độ 17-OHP hữu ích để đánh giá tình trạng điều trị quá liều. Việc xác định nồng độ testosterone hữu ích ở các bé gái và bé trai trước dậy thì. Ở các bé gái dậy thì và sau dậy thì, chu kỳ kinh nguyệt là một chỉ số nhạy cảm của liệu pháp thay thế hormone. Sự đầy đủ của việc thay thế mineralocorticoid có thể được đánh giá bằng PRA. Các bé trai bị CAH cần được đánh giá thường xuyên bằng siêu âm để phát hiện sự phát triển của các khối u mô thượng thận lạc chỗ trong tinh hoàn; các khối u này có thể quá nhỏ để có thể sờ thấy khi khám.
Vào những thời điểm bị stress sinh lý (chẳng hạn như sốt > 38,3°C, nôn mửa dai dẳng, chấn thương đáng kể và các thủ thuật phẫu thuật), liều glucocorticoid nên được tăng lên. Nói chung, gấp 2 đến 3 lần liều thông thường là đủ để ngăn ngừa suy thượng thận. Có thể cần liều cao hơn cho các thủ thuật phẫu thuật. Tất cả các gia đình nên có ở nhà và có thể tiêm hydrocortisone (ví dụ, Solu-Cortef) vào bắp trong trường hợp khẩn cấp y tế. Liều tiêm bắp được khuyến nghị là 25 mg cho trẻ sơ sinh, 50 mg cho trẻ em dưới 4 tuổi và 100 mg cho tất cả những người khác.
Trong các thủ thuật phẫu thuật, có thể tiêm thêm hydrocortisone dưới dạng truyền tĩnh mạch liên tục hoặc tiêm bắp. Có thể sử dụng dung dịch muối sinh lý (0,9% NaCl) tiêm tĩnh mạch nếu bệnh nhân không dung nạp được việc thay thế mineralocorticoid đường uống. Trong khi đang điều trị thay thế bằng glucocorticoid (và đôi khi trong giai đoạn sơ sinh), những người bị thiếu hụt 11β-hydroxylase bị stress sinh lý có thể bị hạ natri máu và tăng kali máu và được hưởng lợi từ liệu pháp mineralocorticoid. Vào những thời điểm bị stress sinh lý, những người bị hội chứng SLO hoặc thiếu hụt P450 oxidoreductase có thể được hưởng lợi từ việc điều trị bằng glucocorticoid.
Những cân nhắc về phẫu thuật
Đối với bệnh nhân nữ bị nam hóa được nuôi dưỡng theo giới tính nữ, mức độ không điển hình kết hợp với mức độ phì đại âm vật và sự hợp nhất phía sau phải được đánh giá cẩn thận để xác định xem có nên xem xét phẫu thuật tạo hình bộ phận sinh dục, bao gồm thu nhỏ âm vật hoặc tạo hình âm vật hay không. Mặc dù các kỹ thuật phẫu thuật hiện tại bảo tồn nhiều hơn bó mạch thần kinh của dương vật, phẫu thuật chỉ nên được thực hiện sau khi cân nhắc kỹ lưỡng. Nếu lỗ niệu đạo nằm cao trong xoang niệu-sinh dục, phẫu thuật sớm có thể được chỉ định để giảm nguy cơ nhiễm trùng đường tiết niệu tái phát bằng cách cung cấp một đường thoát nước tiểu trực tiếp. Các bé gái có mức độ phì đại âm vật từ nhẹ đến trung bình thường không cần phẫu thuật vì có khả năng ảnh hưởng đến độ nhạy cảm của bộ phận sinh dục. Mặc dù đã phẫu thuật, các triệu chứng đường tiết niệu dưới, chẳng hạn như tiểu không tự chủ do gắng sức và tiểu không hết có thể tồn tại ở bệnh nhân DSD nữ và ở nam giới bị lỗ tiểu lệch thấp.
Một điểm quan trọng cần chia sẻ với cha mẹ của một bé gái bị nam hóa với CAH là các “mô sinh dục bị kích thích” sẽ thoái triển sau khi bắt đầu điều trị bằng glucocorticoid. Một mặt, cha mẹ cần được thông báo rằng có thể phù hợp để không khuyến khích phẫu thuật bộ phận sinh dục, cho đến khi đứa trẻ đủ lớn để tự đưa ra quyết định. Mặt khác, cha mẹ phải được trao quyền để đưa ra lựa chọn mà họ sẽ cảm thấy thoải mái. Điều này phải được thực hiện với sự hiểu biết rằng con gái họ có thể chỉ trích họ vì quyết định này khi cô ấy lớn hơn. Nhiều bậc cha mẹ của những cô con gái có cơ quan sinh dục không điển hình nghiêm trọng vẫn chọn phẫu thuật.
Một quy trình ra quyết định chung có thể hữu ích để khuyến khích thảo luận kỹ lưỡng giữa cha mẹ cũng như các nhà cung cấp dịch vụ chăm sóc sức khỏe. Nếu dự kiến phẫu thuật, ca mổ nên được mô tả một cách dễ hiểu cho cha mẹ. Một bác sĩ phẫu thuật có kinh nghiệm nên thảo luận về các lựa chọn, rủi ro và lợi ích của phẫu thuật, bao gồm cả sự phân bố thần kinh của âm vật hoặc dương vật và phương pháp phẫu thuật sẽ được sử dụng để cố gắng bảo tồn nguồn cung cấp mạch máu thần kinh. Mặc dù có nhiều ý kiến khác nhau về việc phẫu thuật nên được thực hiện trong một hay nhiều giai đoạn và liệu việc tái tạo âm đạo có nên được thử trong giai đoạn sơ sinh hay không, cha mẹ cần được thông báo rằng có thể cần phải tạo hình âm đạo hoặc nong âm đạo sau tuổi dậy thì. Trong số những bệnh nhân đã trải qua phẫu thuật tái tạo âm đạo trong giai đoạn sơ sinh, tần suất hẹp âm đạo sau phẫu thuật đã được báo cáo dao động từ 0% đến 77%. Các mục tiêu điều trị cho phẫu thuật tái tạo âm đạo bao gồm chức năng tình dục đầy đủ với nhu cầu tối thiểu về nong hoặc bôi trơn liên tục. Sau khi bắt đầu dậy thì, những người mắc hội chứng MRKH và những người bị CAIS thường có thể đạt được kích thước âm đạo tăng lên bằng phương pháp điều trị bằng dụng cụ nong.
Khi thảo luận về phẫu thuật cho các bé trai bị lỗ tiểu lệch thấp, các chủ đề thích hợp bao gồm những lo ngại phổ biến của nam giới về tầm quan trọng của việc có thể đứng để đi tiểu, sự phát triển đầy đủ của bộ phận sinh dục và khả năng hoạt động tình dục. Với lỗ tiểu lệch thấp, vị trí của lỗ sáo không nhất thiết cho thấy rằng sự hợp nhất đã hoàn tất đến điểm đó vì có thể có sự phát triển không đầy đủ của thể xốp và các mô xung quanh ở gần hơn. Do đó, mức độ phẫu thuật có thể không được biết cho đến khi sự hợp nhất môi-niệu đạo được quan sát rõ hơn khi phẫu thuật. Vì việc điều chỉnh lỗ tiểu lệch thấp và dây cong thường được thực hiện theo nhiều giai đoạn, cuộc thảo luận này cũng cần đề cập đến các chi tiết của phương pháp phẫu thuật, lịch trình đề xuất cho các lần tái khám, số lượng các thủ thuật phẫu thuật có thể xảy ra và độ tuổi tối ưu cho mỗi giai đoạn phẫu thuật. Bác sĩ phẫu thuật nên xem xét các lựa chọn, rủi ro và kết quả dự kiến. Nếu vị trí chính xác và tình trạng biệt hóa của tinh hoàn không được biết, cha mẹ nên biết rằng có thể cần phải thăm dò và sinh thiết. Như có thể dự đoán, các cá nhân bị DSD 46,XY có những lo ngại về tình dục, bất kể giới tính nuôi dưỡng. Ngoài phẫu thuật tạo hình bộ phận sinh dục, các yếu tố gây nhiễu tiềm tàng bao gồm lòng tự trọng, ngoại hình bộ phận sinh dục, vô sinh và nhu cầu điều trị thay thế hormone. Sự đầy đủ của tư vấn và hỗ trợ có khả năng ảnh hưởng đến kết quả ở người trưởng thành.
Các nghiên cứu kết quả phản ánh kết quả của loại phẫu thuật và thời điểm phẫu thuật. Mặc dù các kỹ thuật phẫu thuật cũ hơn dẫn đến suy giảm độ nhạy cảm của bộ phận sinh dục, nhưng vẫn chưa rõ liệu các kỹ thuật phẫu thuật hiện tại bảo tồn nguồn cung cấp mạch máu thần kinh cho âm vật có mang lại kết quả tốt hơn hay không. Người ta dự đoán (và hy vọng) rằng các kỹ thuật phẫu thuật hiện tại sẽ có kết quả tốt hơn; tuy nhiên, sẽ mất nhiều năm nữa mới có thông tin này. Trong khi chờ đợi, các nghiên cứu kết quả có sẵn có thể cung cấp thông tin.
Mục tiêu của phẫu thuật tái tạo bộ phận sinh dục đã và đang tiếp tục là một ngoại hình thẩm mỹ tốt và tiềm năng về chức năng liên quan đến độ nhạy cảm cho phản ứng tình dục. Chức năng sau phẫu thuật liên quan đến mức độ nghiêm trọng của tình trạng cơ quan sinh dục không điển hình khi sinh, mức độ phẫu thuật và các biến chứng của phẫu thuật. Ví dụ, ở nữ giới, đường kính âm đạo giảm tương quan với mức độ nam hóa trước sinh lớn hơn. Trong số nữ giới bị CAH thiếu hụt 21-hydroxylase, kết quả của phẫu thuật dường như liên quan đến các đột biến cụ thể. Những người có kiểu gen null có nhiều biến chứng phẫu thuật hơn và điểm số về chức năng tình dục kém hơn. Dữ liệu kết quả có sẵn cho thấy hầu hết bệnh nhân sau phẫu thuật có bản dạng giới nữ. Tuy nhiên, các nghiên cứu cho thấy rằng một tỷ lệ lớn hơn phụ nữ bị CAH, ngay cả những người bị CAH không cổ điển (NCAH), bị thu hút tình dục bởi những phụ nữ khác so với dân số chung. Tuy nhiên, kết quả lâu dài không được ghi nhận rõ ràng. Trong số bệnh nhân DSD 46,XX, những người sống là nữ cho biết họ thích phẫu thuật sớm và cảm thấy rằng một cuộc sống tình dục thỏa mãn là có thể. Tuy nhiên, giảm ham muốn tình dục, khó đạt cực khoái và ra mắt tình dục muộn hơn là những mối quan tâm chung. Khả năng sinh sản là có thể đối với phụ nữ bị CAH; việc giảm nồng độ progesterone là một yếu tố quan trọng để đảm bảo khả năng sinh sản cho phụ nữ bị các dạng CAH cổ điển.
Mặc dù hầu hết nam giới bị DSD 46,XY đều hài lòng với kết quả lâu dài của phẫu thuật tạo hình bộ phận sinh dục, nhưng hậu quả của dây cong và lỗ tiểu lệch thấp nghiêm trọng có thể bao gồm kích thước dương vật quá ngắn để thâm nhập vào âm đạo. Do đó, kết quả có thể kém. Các phàn nàn tái phát không chỉ bao gồm kích thước dương vật và hoạt động tình dục, mà còn cả các triệu chứng tiết niệu. Mặc dù sự hiểu biết về DSD và sự cởi mở với bệnh nhân và gia đình đã được cải thiện, các vấn đề về chất lượng cuộc sống cần được quan tâm nhiều hơn.
Nguy cơ u tuyến sinh dục
Môi trường tuyến sinh dục của thai nhi bất thường và sự biệt hóa tế bào mầm bất thường sau đó có liên quan đến nguy cơ gia tăng các khối u tế bào mầm. Sự hiện diện của vật liệu nhiễm sắc thể Y, đặc biệt là gen protein đặc hiệu cho tinh hoàn được mã hóa trên nhiễm sắc thể Y (TSPY), và một tuyến sinh dục loạn sản làm tăng nguy cơ u tuyến sinh dục. Nguy cơ tương đối đối với các khối u tuyến sinh dục chỉ đạo nhu cầu và thời điểm phẫu thuật. Tân sinh tế bào mầm tại chỗ (GCNIS) hoặc u nguyên bào sinh dục là tiền thân của các khối u tế bào mầm. Tỷ lệ hiện mắc được báo cáo đối với u nguyên bào sinh dục dao động từ 15% đến 30%, tùy thuộc vào tuổi của bệnh nhân, mô học tuyến sinh dục và các tiêu chí chẩn đoán được sử dụng cho GCNIS/u nguyên bào sinh dục. U nguyên bào sinh dục là các khối u hỗn hợp tế bào mầm-dây sinh dục-mô đệm phát sinh trong các tuyến sinh dục loạn sản, bao gồm các tế bào mầm chưa trưởng thành và các tế bào dây sinh dục-mô đệm biệt hóa không xác định, và có trước sự phát triển của các tân sinh xâm lấn hơn, chẳng hạn như u tế bào mầm, u tinh bào và u không phải tinh bào. Việc đánh giá sự biểu hiện của FOXL2 và SOX9 trong mô khối u có thể giúp phân biệt giữa những khối u có thành phần tế bào Sertoli và những khối u có thành phần tế bào hạt.
Sự biểu hiện tăng và kéo dài của các dấu hiệu hóa mô miễn dịch, POU5F1 (còn được gọi là OCT3/4) và TSPY, là phổ biến trong GCNIS và u nguyên bào sinh dục. Trong một loạt nghiên cứu, TSPY được biểu hiện nhiều trong các tế bào mầm trong tinh hoàn loạn sản và mô tuyến sinh dục không biệt hóa, cho thấy sự điều hòa tăng lên khi các tế bào mầm nằm trong một môi trường không thuận lợi. Người ta giả thuyết rằng quá trình trưởng thành bình thường của tế bào mầm bị gián đoạn, dẫn đến sự biểu hiện kéo dài của OCT3/4, xóa bỏ dấu ấn bộ gen và sau đó là sự bất tử hóa của tế bào.
Trong một loạt bệnh nhân mắc hội chứng Turner, 14/171 (8%) dương tính với vật liệu nhiễm sắc thể Y. Trong số 14 bệnh nhân này, tỷ lệ mắc u nguyên bào sinh dục là 33%. Trong một số trường hợp, vật liệu nhiễm sắc thể Y chỉ được phát hiện bằng phương pháp PCR. Hội chứng Frasier, liên quan đến một đột biến WT1 cụ thể, được đặc trưng bởi hội chứng thận hư kháng steroid, đảo ngược giới tính XY và tăng nguy cơ u tuyến sinh dục.
Do nguy cơ u tuyến sinh dục được giả thuyết là tăng lên, phẫu thuật cắt bỏ tuyến sinh dục thường được thực hiện ở bệnh nhân CAIS. Mặc dù thiếu các nghiên cứu kết quả dài hạn, dữ liệu có sẵn cho thấy nguy cơ u tuyến sinh dục là cực kỳ thấp ở bệnh nhân CAIS trong những năm vị thành niên. Một cuộc khảo sát quốc tế đã nhấn mạnh các phương pháp điều trị không nhất quán, nguy cơ thấp đối với các khối u tế bào mầm và nhấn mạnh sự cần thiết của các khuyến nghị quản lý cá nhân hóa cho bệnh nhân CAIS.
Kích thước mẫu hạn chế, sai lệch do lựa chọn, tiêu chí chẩn đoán không nhất quán đối với các tế bào ác tính và thuật ngữ khó hiểu để lại nhiều câu hỏi cần được trả lời. Ví dụ, bệnh nhân DSD nào được hưởng lợi từ sinh thiết tuyến sinh dục để đánh giá nguy cơ tân sinh? Một cân nhắc khác là cần bao nhiêu mẫu sinh thiết để đại diện cho mô học của tuyến sinh dục. Nội soi ổ bụng và phẫu thuật cắt bỏ tuyến sinh dục có hỗ trợ video là những công cụ vô giá khi nguy cơ ác tính cao. Cuối cùng, quyết định liên quan đến phẫu thuật cắt bỏ tuyến sinh dục bao gồm việc xem xét kiểu hình của bệnh nhân (giải phẫu bộ phận sinh dục trong và ngoài), bộ nhiễm sắc thể, giới tính nuôi dưỡng, các yếu tố tâm lý xã hội và mô học tuyến sinh dục.
Chuyển tiếp từ Chăm sóc Nhi khoa sang Chăm sóc Người lớn
Cha mẹ và các nhà cung cấp dịch vụ chăm sóc sức khỏe nhi khoa hình dung rằng trẻ em bị DSD sẽ trở thành những người trưởng thành hữu ích, những người sẽ tìm thấy sự thỏa mãn và hài lòng trong cuộc sống của họ. Theo báo cáo từ Anh về việc mất theo dõi bệnh nhân bị CAH, một chương trình chuyển tiếp có kế hoạch từ đội ngũ chăm sóc sức khỏe nhi khoa sang đội ngũ chăm sóc sức khỏe người lớn là rất cần thiết. Quá trình chuyển tiếp có kế hoạch này cần xem xét các nhu cầu y tế, tâm lý xã hội, giáo dục, cảm xúc, nhận thức và nghề nghiệp của người trưởng thành trẻ tuổi. Cần xem xét sự khác biệt về kỳ vọng giữa các tình huống chăm sóc sức khỏe nhi khoa và người lớn. Việc giải thích các chi tiết cụ thể về chẩn đoán của anh ấy/cô ấy với người trưởng thành trẻ tuổi là rất cần thiết. Đối với nhiều bệnh nhân này, các chi tiết cụ thể về rối loạn của họ đã được xem xét cùng cha mẹ tại thời điểm chẩn đoán khi cá nhân đó còn quá nhỏ để tham gia vào các cuộc thảo luận này. Người trưởng thành trẻ tuổi cần biết nguyên nhân, di truyền cơ bản, sinh lý bệnh của rối loạn của mình, nguy cơ tái phát và tình trạng sinh sản. Người trưởng thành trẻ tuổi, đặc biệt là những người cần điều trị thay thế bằng glucocorticoid, cần hiểu lý do điều trị.
Các nghiên cứu kết quả dài hạn
Trong số nữ giới 46,XX bị CAH, kết quả khác nhau tùy thuộc vào mức độ nghiêm trọng của tình trạng hoặc các đột biến CYP21A2 cụ thể. Kết quả phẫu thuật gần đây đã được cải thiện nhưng không phải lúc nào cũng thỏa đáng, các ảnh hưởng tâm lý khác nhau, và do đó khả năng sinh sản và sự hài lòng về tình dục cũng khác nhau. Cách tiếp cận chính bao gồm việc cá nhân hóa từng bệnh nhân. Một nghiên cứu về kết quả ở nữ giới kiểu hình bị DSD 46,XY với một nhóm chứng phù hợp theo tuổi cho thấy: (1) bệnh suất chung tăng nhưng không tăng sau khi loại trừ những người có chẩn đoán cụ thể, mang thai và sinh con; (2) tỷ lệ tử vong tương tự như nhóm chứng; và (3) giáo dục. Việc sống chung và làm mẹ đã giảm. Do đó, những cá nhân này dường như hoạt động tốt trong môi trường làm việc nhưng cuộc sống gia đình bị ảnh hưởng tiêu cực. Tình trạng sức khỏe chung là tốt, liên quan nhiều hơn đến chẩn đoán sớm và lối sống lành mạnh. Khả năng sinh sản, như có thể dự đoán, là hiếm, liên quan đến kiểu hình sinh dục ban đầu và các mối quan hệ đối tác (xảy ra ở một thiểu số). Các kỹ thuật hỗ trợ sinh sản (ART) thường được sử dụng. Khả năng sinh sản cực kỳ hiếm ở những người bị loạn sản tuyến sinh dục thuần túy hoặc hỗn hợp, DSD dạng tinh hoàn-buồng trứng và nam giới XX. Các kỹ thuật phẫu thuật tốt hơn, xã hội hóa được cải thiện, tập trung vào điều trị lâu dài và các kỹ thuật ART có thể cải thiện tiềm năng sinh sản trong tương lai. Sự hài lòng về tình dục, bao gồm các báo cáo ban đầu về chức năng tình dục ở nữ giới, do việc bảo tồn bao thần kinh mạch máu của âm vật, cho thấy kết quả tương tự với dân số chung, với các vấn đề về xã hội hóa, các mối quan hệ giữa các cá nhân và thân mật tiếp tục là một yếu tố. Các dạng lỗ tiểu lệch thấp nghiêm trọng, dù có được xác định là DSD hay không, tiếp tục là một thách thức phẫu thuật, chức năng và tâm lý xã hội dai dẳng. Kết quả lâm sàng ở các bé trai bị PAIS kém hơn so với hầu hết các DSD XY khác, do đó việc ra quyết định liên quan đến chỉ định giới tính phải đặc biệt thận trọng.
Tư vấn và Hỗ trợ Tâm lý, Di truyền và Các cân nhắc về Đạo đức
Sự liên tục chăm sóc theo chiều dọc và cung cấp các hệ thống hỗ trợ là rất cần thiết do các khía cạnh y tế và tâm lý của DSD. Nếu có một nhân viên xã hội, nhà tâm lý học hoặc bác sĩ tâm thần có kinh nghiệm, việc đánh giá và tư vấn cẩn thận là vô giá trong suốt thời thơ ấu và thanh thiếu niên. Mặc dù hầu hết các trường hợp DSD 46,XY như vậy có thể không cần điều trị y tế trong thời thơ ấu, các lần khám định kỳ với bác sĩ nội tiết nhi rất hữu ích để giải quyết các mối quan tâm của bệnh nhân và cha mẹ, cập nhật cho cha mẹ về liệu pháp mới và dữ liệu kết quả, và đảm bảo rằng các nhu cầu hỗ trợ tâm lý phù hợp đang được đáp ứng. Những lần khám này có thể thúc đẩy mối quan hệ tích cực giữa đứa trẻ và bác sĩ. Một số bậc cha mẹ có thể thấy các nhóm hỗ trợ theo từng rối loạn cụ thể (ví dụ: AIS, www.aissg.org; CAH, www.caresfoundation.org; hội chứng Turner, www.turnersyndrome.org) là hữu ích.
Nhiều tình trạng DSD là do di truyền. Các kiểu di truyền phổ biến nhất là các tính trạng lặn trên nhiễm sắc thể thường hoặc liên kết X. Tư vấn di truyền được chỉ định vì cha mẹ thường quan tâm đến việc tìm hiểu về nguy cơ tái phát. Vì sự không đồng nhất về kiểu hình xảy ra ở một số rối loạn DSD, việc xác định hormone và phân tích di truyền (khi có sẵn) cho các thành viên khác trong gia đình có thể được khuyến nghị.
Các nguyên tắc và khuyến nghị về đạo đức đã được công bố nhằm nỗ lực cung cấp một cái nhìn toàn diện về quan điểm của các bác sĩ lâm sàng, bệnh nhân và gia đình, dựa trên các nguyên tắc thúc đẩy phúc lợi của đứa trẻ và người lớn trong tương lai, duy trì quyền của họ được tham gia vào các quyết định ảnh hưởng đến họ hiện tại và sau này, và tôn trọng các mối quan hệ gia đình và cha mẹ-con cái. Các phương tiện truyền thông nói chung đã lưu ý rằng trẻ sơ sinh liên giới tính hiện có thể bị mắc kẹt trong “cuộc chiến phẫu thuật về rủi ro y tế và quyền đạo đức”. Tranh cãi này nhấn mạnh tình thế khó xử về đạo đức trong đó cha mẹ đóng vai trò là người thay thế để chăm sóc tốt nhất cho con cái vị thành niên của họ. Cha mẹ cần được giáo dục và thông báo về tất cả các thông tin có sẵn. Nhiều bài phê bình thảo luận về các chủ đề khác nhau đã được công bố; các chủ đề bao gồm cuộc đấu tranh chống lại y tế hóa, chính trị hóa và dân chủ hóa tiêu chuẩn chăm sóc, và hoạt động pháp lý/y tế.
Kết luận
Việc xác định các gen liên quan đến sự biệt hóa giới tính đã làm sáng tỏ một số sự kiện phân tử chịu trách nhiệm cho sự biệt hóa giới tính bình thường và bất thường. Kiến thức về các yếu tố di truyền, nội tiết tố và môi trường ảnh hưởng đến sự biệt hóa giới tính mang lại lợi ích cho trẻ em bị ảnh hưởng, cha mẹ của chúng và các nhà cung cấp dịch vụ chăm sóc sức khỏe của chúng. Thông tin này cho phép giáo dục cha mẹ tốt hơn về nguyên nhân, lịch sử tự nhiên và tiên lượng cho con của họ. Với sự hiểu biết về các yếu tố ảnh hưởng đến sự biệt hóa giới tính, có thể ước tính được nguy cơ tái phát.
Việc đánh giá và quản lý sau đó của một đứa trẻ có cơ quan sinh dục không điển hình dựa trên tiền sử, khám thực thể và dữ liệu xét nghiệm. Việc hiểu nền tảng và hoàn cảnh của gia đình cho phép đội ngũ y tế thảo luận về sự phát triển giới tính của đứa trẻ, sử dụng thuật ngữ mà họ có thể hiểu được tại thời điểm chẩn đoán, cũng như trong những năm tiếp theo. Chẩn đoán và quản lý ban đầu là khởi đầu của một mối quan hệ lâu dài giữa bệnh nhân, gia đình và các nhà cung cấp dịch vụ chăm sóc sức khỏe.
Mặc dù có những tiến bộ trong việc mô tả cơ sở phân tử của cơ quan sinh dục không điển hình, sự nhạy cảm và lòng trắc ẩn được thể hiện bởi đội ngũ chuyên gia y tế trong các tương tác của họ với bệnh nhân và gia đình vẫn là một khía cạnh thiết yếu trong việc quản lý trẻ sơ sinh có cơ quan sinh dục không điển hình. Các quy trình đánh giá và chẩn đoán ban đầu chỉ là khởi đầu của một mối quan hệ kéo dài với đứa trẻ và gia đình, cuối cùng đỉnh điểm là sự chuyển tiếp thành công sang các nhà cung cấp dịch vụ chăm sóc người lớn. Các tương tác tôn trọng và đồng cảm thúc đẩy sự phát triển của trẻ sơ sinh có cơ quan sinh dục không điển hình để trở thành một người trưởng thành có thể sống một cuộc sống mãn nguyện. Các nhà cung cấp dịch vụ chăm sóc sức khỏe cần phải nhận thức rằng kiến thức về tất cả các khía cạnh của việc xác định giới tính, phát triển giới tính và các yếu tố quyết định bản dạng giới vẫn cần được làm sáng tỏ, do đó việc quản lý các cá nhân bị DSD vẫn sẽ liên quan đến những điều không chắc chắn và xác suất.
Bảng chú giải thuật ngữ Y học Anh – Việt – Chương 6
| STT | Thuật ngữ tiếng Anh | Phiên âm IPA | Nghĩa Tiếng Việt |
|---|---|---|---|
| 1 | Ovotesticular disorder of sex development (DSD) | /ˌoʊvoʊtɛˈstɪkjələr dɪsˈɔrdər əv sɛks dɪˈvɛləpmənt/ | Rối loạn phát triển giới tính dạng tinh hoàn-buồng trứng |
| 2 | Ovarian tissue | /oʊˈvɛəriən ˈtɪʃuː/ | Mô buồng trứng |
| 3 | Follicles | /ˈfɒlɪkəlz/ | Nang noãn |
| 4 | Testicular tissue | /tɛˈstɪkjələr ˈtɪʃuː/ | Mô tinh hoàn |
| 5 | Seminiferous tubules | /ˌsɛmɪˈnɪfərəs ˈtuːbjuːlz/ | Ống sinh tinh |
| 6 | Individual | /ˌɪndɪˈvɪdʒuəl/ | Cá thể |
| 7 | External genital appearance | /ɪkˈstɜːrnl ˈdʒɛnɪtl əˈpɪərəns/ | Hình thái cơ quan sinh dục ngoài |
| 8 | Gonadal histology | /ɡoʊˈnædəl hɪˈstɒlədʒi/ | Mô học tuyến sinh dục |
| 9 | Gonads | /ˈɡoʊnædz/ | Tuyến sinh dục |
| 10 | Ovotestis | /ˌoʊvoʊˈtɛstɪs/ | Tuyến lưỡng tính |
| 11 | Ovary | /ˈoʊvəri/ | Buồng trứng |
| 12 | Testis | /ˈtɛstɪs/ | Tinh hoàn |
| 13 | Histology | /hɪˈstɒlədʒi/ | Mô học |
| 14 | Intraabdominal | /ˌɪntrəæbˈdɒmɪnəl/ | Trong ổ bụng |
| 15 | Ovoid | /ˈoʊvɔɪd/ | Hình trứng |
| 16 | Bilobed | /baɪˈloʊbd/ | Hai thùy |
| 17 | Gross appearance | /ɡroʊs əˈpɪərəns/ | Hình thái đại thể |
| 18 | End-to-end arrangement | /ɛnd-tu-ɛnd əˈreɪndʒmənt/ | Sắp xếp đầu-cuối |
| 19 | Testicular zone | /tɛˈstɪkjələr zoʊn/ | Vùng tinh hoàn |
| 20 | Differentiation | /ˌdɪfəˌrɛnʃiˈeɪʃən/ | Biệt hóa |
| 21 | Tunica albuginea | /ˈtuːnɪkə ˌælbjuːˈdʒɪniə/ | Lớp áo trắng |
| 22 | Atypical interstitial tissue | /eɪˈtɪpɪkəl ˌɪntərˈstɪʃəl ˈtɪʃuː/ | Mô kẽ không điển hình |
| 23 | Oocytes | /ˈoʊəsaɪts/ | Tế bào trứng |
| 24 | Seminiferous cords | /ˌsɛmɪˈnɪfərəs kɔːrdz/ | Dây sinh tinh |
| 25 | Proportion | /prəˈpɔːrʃən/ | Tỷ lệ |
| 26 | Uterine development | /ˈjuːtəraɪn dɪˈvɛləpmənt/ | Sự phát triển của tử cung |
| 27 | Karyotype | /ˈkærioʊˌtaɪp/ | Bộ nhiễm sắc thể |
| 28 | Mosaic karyotypes | /moʊˈzeɪɪk ˈkærioʊˌtaɪps/ | Bộ nhiễm sắc thể thể khảm |
| 29 | Y chromosomal material | /waɪ ˌkroʊməˈsoʊməl məˈtɪəriəl/ | Vật liệu nhiễm sắc thể Y |
| 30 | Peripheral blood lymphocytes | /pəˈrɪfərəl blʌd ˈlɪmfoʊsaɪts/ | Tế bào lympho máu ngoại vi |
| 31 | SRY gene | /ˌɛsˌɑrˈwaɪ dʒiːn/ | Gen SRY |
| 32 | Polymerase chain reaction (PCR) | /pəˈlɪməˌreɪs tʃeɪn riˈækʃən/ | Phản ứng chuỗi polymerase |
| 33 | Amplification | /ˌæmplɪfɪˈkeɪʃən/ | Khuếch đại |
| 34 | Promoter region | /prəˈmoʊtər ˈriːdʒən/ | Vùng khởi động (của gen) |
| 35 | Deletion | /dɪˈliːʃən/ | Mất đoạn |
| 36 | Specific mutations | /spəˈsɪfɪk mjuːˈteɪʃənz/ | Đột biến đặc hiệu |
| 37 | Sporadic | /spəˈrædɪk/ | Ngẫu nhiên, tản phát |
| 38 | Pedigrees | /ˈpɛdɪɡriːz/ | Phả hệ |
| 39 | Infancy | /ˈɪnfənsi/ | Tuổi sơ sinh |
| 40 | Childhood | /ˈtʃaɪldhʊd/ | Thời thơ ấu |
| 41 | Phenotypic males | /ˌfiːnoʊˈtɪpɪk meɪlz/ | Nam giới về kiểu hình |
| 42 | Bilateral gynecomastia | /baɪˈlætərəl ˌɡaɪnəkoʊˈmæstiə/ | Vú to hai bên |
| 43 | Penoscrotal hypospadias | /ˌpiːnoʊˈskroʊtəl ˌhaɪpoʊˈspeɪdiəs/ | Lỗ tiểu lệch thấp ở dương vật-bìu |
| 44 | Descended gonad | /dɪˈsɛndɪd ˈɡoʊnæd/ | Tuyến sinh dục đã xuống |
| 45 | Undescended gonad | /ˌʌndɪˈsɛndɪd ˈɡoʊnæd/ | Tuyến sinh dục chưa xuống |
| 46 | Biopsies | /ˈbaɪɒpsiz/ | Sinh thiết |
| 47 | Prepubertal testis | /ˌpriːpjuːˈbɜːrtl ˈtɛstɪs/ | Tinh hoàn trước tuổi dậy thì |
| 48 | Sex of rearing | /sɛks əv ˈrɪərɪŋ/ | Giới tính nuôi dưỡng |
| 49 | Salpingo-oophorectomy | /sælˌpɪŋɡoʊˌoʊəfəˈrɛktəmi/ | Phẫu thuật cắt bỏ vòi trứng-buồng trứng |
| 50 | Hysterectomy | /ˌhɪstəˈrɛktəmi/ | Phẫu thuật cắt bỏ tử cung |
| 51 | Painless scrotal mass | /ˈpeɪnləs ˈskroʊtəl mæs/ | Khối u không đau ở bìu |
| 52 | Excised | /ɪkˈsaɪzd/ | Được cắt bỏ |
| 53 | Inconsistency | /ˌɪnkənˈsɪstənsi/ | Sự không nhất quán |
| 54 | Gonadal development | /ɡoʊˈnædəl dɪˈvɛləpmənt/ | Sự phát triển tuyến sinh dục |
| 55 | Sampling errors | /ˈsæmplɪŋ ˈɛrərz/ | Sai sót lấy mẫu |
| 56 | Gonadal biopsies | /ɡoʊˈnædəl ˈbaɪɒpsiz/ | Sinh thiết tuyến sinh dục |
| 57 | Fertility | /fərˈtɪləti/ | Khả năng sinh sản |
| 58 | Longitudinally | /ˌlɒndʒɪˈtjuːdɪnəli/ | (Theo dõi) dọc |
| 59 | Germ cells | /dʒɜːrm sɛlz/ | Tế bào mầm |
| 60 | Degenerated | /dɪˈdʒɛnəˌreɪtɪd/ | Thoái hóa |
| 61 | Azoospermia | /eɪˌzoʊəˈspɜːrmiə/ | Không có tinh trùng |
| 62 | Histological findings | /ˌhɪstəˈlɒdʒɪkəl ˈfaɪndɪŋz/ | Các phát hiện mô học |
| 63 | Dysgenetic | /dɪsˈdʒɛnɪtɪk/ | Loạn sản |
| 64 | Sertoli-only appearance | /sərˈtoʊli-ˈoʊnli əˈpɪərəns/ | Hình ảnh chỉ có tế bào Sertoli |
| 65 | Hyalinization | /ˌhaɪəlɪnɪˈzeɪʃən/ | Sự hyalin hóa |
| 66 | Assisted reproductive techniques | /əˈsɪstɪd ˌriːprəˈdʌktɪv tɛkˈniːks/ | Kỹ thuật hỗ trợ sinh sản |
| 67 | Paternity | /pəˈtɜːrnəti/ | Vai trò làm cha, khả năng làm cha |
| 68 | Pregnancies | /ˈprɛɡnənsiz/ | Các trường hợp mang thai |
| 69 | Nonsyndromic | /ˌnɒnsɪnˈdroʊmɪk/ | Không hội chứng |
| 70 | Testicular DSD | /tɛˈstɪkjələr diː ɛs diː/ | Rối loạn phát triển giới tính dạng tinh hoàn |
| 71 | Male phenotype | /meɪl ˈfiːnoʊˌtaɪp/ | Kiểu hình nam |
| 72 | Frequency | /ˈfriːkwənsi/ | Tần suất |
| 73 | XX male syndrome | /ɛks-ɛks meɪl ˈsɪndroʊm/ | Hội chứng nam XX |
| 74 | Subclassified | /ˌsʌbˈklæsɪfaɪd/ | Phân loại nhỏ |
| 75 | SRY-positive | /ˌɛsˌɑrˈwaɪ-ˈpɒzətɪv/ | SRY dương tính |
| 76 | SRY-negative | /ˌɛsˌɑrˈwaɪ-ˈnɛɡətɪv/ | SRY âm tính |
| 77 | External genitalia | /ɪkˈstɜːrnl ˌdʒɛnɪˈteɪliə/ | Cơ quan sinh dục ngoài |
| 78 | Small azoospermic testes | /smɔːl eɪˌzoʊəˈspɜːrmɪk ˈtɛstiːz/ | Tinh hoàn nhỏ không có tinh trùng |
| 79 | Hypergonadotropic hypogonadism | /ˌhaɪpərɡoʊˌnædoʊˈtroʊpɪk ˌhaɪpoʊˈɡoʊnæˌdɪzəm/ | Suy sinh dục tăng gonadotropin |
| 80 | Infertility | /ˌɪnfɜːrˈtɪləti/ | Vô sinh |
| 81 | Recombination | /ˌriːˌkɒmbɪˈneɪʃən/ | Tái tổ hợp |
| 82 | X and Y chromosomes | /ɛks ænd waɪ ˈkroʊməsoʊmz/ | Nhiễm sắc thể X và Y |
| 83 | Translocation | /ˌtrænzloʊˈkeɪʃən/ | Chuyển đoạn |
| 84 | Autosome | /ˈɔːtəsoʊm/ | Nhiễm sắc thể thường |
| 85 | Incidental finding | /ˌɪnsɪˈdɛntəl ˈfaɪndɪŋ/ | Phát hiện tình cờ |
| 86 | Terminal end | /ˈtɜːrmɪnəl ɛnd/ | Đầu cuối |
| 87 | Chromosome | /ˈkroʊməsoʊm/ | Nhiễm sắc thể |
| 88 | Gonadal Dysgenesis | /ɡoʊˈnædəl dɪsˈdʒɛnəsɪs/ | Loạn sản tuyến sinh dục |
| 89 | Ovarian dysgenesis | /oʊˈvɛəriən dɪsˈdʒɛnəsɪs/ | Loạn sản buồng trứng |
| 90 | Primary amenorrhea | /ˈpraɪmeri əˌmɛnəˈriːə/ | Vô kinh nguyên phát |
| 91 | Point mutations | /pɔɪnt mjuːˈteɪʃənz/ | Đột biến điểm |
| 92 | Paternally transmitted | /pəˈtɜːrnəli trænsˈmɪtɪd/ | Di truyền từ cha |
| 93 | X-linked | /ɛks-lɪŋkt/ | Liên kết X |
| 94 | Hypogonadism | /ˌhaɪpoʊˈɡoʊnæˌdɪzəm/ | Suy sinh dục |
| 95 | Partial alopecia | /ˈpɑːrʃəl ˌæləˈpiːʃə/ | Rụng tóc một phần |
| 96 | Woodhouse-Sakati Syndrome | /ˈwʊdhaʊs-səˈkɑːti ˈsɪndroʊm/ | Hội chứng Woodhouse-Sakati |
| 97 | Autosomal recessive | /ˌɔːtəˈsoʊməl rɪˈsɛsɪv/ | Lặn trên nhiễm sắc thể thường |
| 98 | Secondary amenorrhea | /ˈsɛkənˌdɛri əˌmɛnəˈriːə/ | Vô kinh thứ phát |
| 99 | Leukoencephalopathy | /ˌluːkoʊɛnˌsɛfəˈlɒpəθi/ | Bệnh não chất trắng |
| 100 | Vanishing white matter | /ˈvænɪʃɪŋ waɪt ˈmætər/ | (Bệnh) chất trắng biến mất |
| 101 | Ovarian insufficiency | /oʊˈvɛəriən ˌɪnsəˈfɪʃənsi/ | Suy buồng trứng |
| 102 | Autosomal dominant | /ˌɔːtəˈsoʊməl ˈdɒmɪnənt/ | Trội trên nhiễm sắc thể thường |
| 103 | Fragile X premutation | /ˈfrædʒaɪl ɛks ˌpriːmjuːˈteɪʃən/ | Tiền đột biến Fragile X |
| 104 | Trinucleotide expansion | /traɪˈnjuːkliəˌtaɪd ɪkˈspænʃən/ | Mở rộng lặp lại bộ ba nucleotide |
| 105 | Blepharophimosis | /ˌblɛfəroʊfaɪˈmoʊsɪs/ | Hẹp khe mi |
| 106 | Deletion/duplications | /dɪˈliːʃən/ /ˌdjuːplɪˈkeɪʃənz/ | Mất đoạn/nhân đoạn |
| 107 | Cardiomyopathy | /ˌkɑːrdioʊmaɪˈɒpəθi/ | Bệnh cơ tim |
| 108 | McKusick-Kaufman syndrome | /məˈkjuːsɪk-ˈkɔːfmən ˈsɪndroʊm/ | Hội chứng McKusick-Kaufman |
| 109 | Hypogenitalism | /ˌhaɪpoʊˈdʒɛnɪtlˌɪzəm/ | Thiểu sản sinh dục |
| 110 | Hydrometrocolpos | /ˌhaɪdroʊˌmiːtroʊˈkɒlpoʊs/ | Ứ dịch trong tử cung và âm đạo |
| 111 | Primary ovarian insufficiency | /ˈpraɪmeri oʊˈvɛəriən ˌɪnsəˈfɪʃənsi/ | Suy buồng trứng nguyên phát |
| 112 | Secondary ovarian insufficiency | /ˈsɛkənˌdɛri oʊˈvɛəriən ˌɪnsəˈfɪʃənsi/ | Suy buồng trứng thứ phát |
| 113 | Sex reversal | /sɛks rɪˈvɜːrsəl/ | Đảo ngược giới tính |
| 114 | Cerebellar atrophy | /ˌsɛrəˈbɛlər ˈætrəfi/ | Teo tiểu não |
| 115 | Mental impairment | /ˈmɛntəl ɪmˈpɛərmənt/ | Suy giảm trí tuệ |
| 116 | Pigmentary retinopathy | /ˈpɪɡməntəri ˌrɛtɪˈnɒpəθi/ | Bệnh võng mạc sắc tố |
| 117 | Virilizing | /ˈvɪrəlaɪzɪŋ/ | Nam hóa |
| 118 | Duplication syndrome | /ˌdjuːplɪˈkeɪʃən ˈsɪndroʊm/ | Hội chứng nhân đoạn |
| 119 | Waardenburg syndrome | /ˈvɑːrdənbɜːrɡ ˈsɪndroʊm/ | Hội chứng Waardenburg |
| 120 | Peripheral demyelinating neuropathy | /pəˈrɪfərəl diːˈmaɪəlɪneɪtɪŋ njʊəˈrɒpəθi/ | Bệnh thần kinh ngoại biên mất myelin |
| 121 | Mullerian aplasia | /mʌˈlɪəriən eɪˈpleɪʒə/ | Bất sản ống Müller |
| 122 | Hyperandrogenism | /ˌhaɪpərænˈdrɒdʒəˌnɪzəm/ | Tăng androgen |
| 123 | Phenotypic spectrum | /ˌfiːnoʊˈtɪpɪk ˈspɛktrəm/ | Phổ kiểu hình |
| 124 | Genital ambiguity | /ˈdʒɛnɪtl ˌæmbɪˈɡjuːəti/ | Cơ quan sinh dục không điển hình |
| 125 | Meiotic arrest | /maɪˈɒtɪk əˈrɛst/ | Ngừng giảm phân |
| 126 | Germ cell degeneration | /dʒɜːrm sɛl dɪˌdʒɛnəˈreɪʃən/ | Thoái hóa tế bào mầm |
| 127 | Molecular etiologies | /məˈlɛkjələr ˌiːtiˈɒlədʒiz/ | Nguyên nhân phân tử |
| 128 | Secondary sexual characteristics | /ˈsɛkənˌdɛri ˈsɛkʃuəl ˌkærəktəˈrɪstɪks/ | Đặc điểm sinh dục thứ phát |
| 129 | Upstream | /ʌpˈstriːm/ | Ngược dòng (gen) |
| 130 | Overexpression | /ˌoʊvərɪkˈsprɛʃən/ | Biểu hiện quá mức |
| 131 | Congenital anomalies | /kənˈdʒɛnɪtl əˈnɒməliz/ | Bất thường bẩm sinh |
| 132 | Single exon gene | /ˈsɪŋɡəl ˈɛksɒn dʒiːn/ | Gen có một exon |
| 133 | Pituitary development | /pɪˈtjuːɪtəri dɪˈvɛləpmənt/ | Sự phát triển tuyến yên |
| 134 | Craniofacial development | /ˌkreɪnioʊˈfeɪʃəl dɪˈvɛləpmənt/ | Sự phát triển sọ mặt |
| 135 | Bilateral cryptorchidism | /baɪˈlætərəl krɪpˈtɔːrkɪˌdɪzəm/ | Tinh hoàn ẩn hai bên |
| 136 | Duplication | /ˌdjuːplɪˈkeɪʃən/ | Nhân đoạn |
| 137 | Preclinical transgenic XX mouse model | /ˌpriːˈklɪnɪkəl trænsˈdʒɛnɪk ɛks-ɛks maʊs ˈmɒdl/ | Mô hình chuột XX chuyển gen tiền lâm sàng |
| 138 | Surrogate | /ˈsɜːrəɡət/ | Thay thế |
| 139 | Hypoplasia | /ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản |
| 140 | Anterior pituitary | /ænˈtɪəriər pɪˈtjuːɪtəri/ | Thùy trước tuyến yên |
| 141 | Ectopic position | /ɛkˈtɒpɪk pəˈzɪʃən/ | Vị trí lạc chỗ |
| 142 | Posterior pituitary | /pɒˈstɪəriər pɪˈtjuːɪtəri/ | Thùy sau tuyến yên |
| 143 | AMH concentrations | /ˌeɪ ɛm ˈeɪtʃ ˌkɒnsənˈtreɪʃənz/ | Nồng độ AMH (Hormone kháng Müller) |
| 144 | Sertoli cell function | /sərˈtoʊli sɛl ˈfʌŋkʃən/ | Chức năng tế bào Sertoli |
| 145 | Sexually dimorphic | /ˈsɛkʃuəli daɪˈmɔːrfɪk/ | Dị hình giới tính |
| 146 | Anorchia | /æˈnɔːrkiə/ | Không có tinh hoàn |
| 147 | Cryptorchidism | /krɪpˈtɔːrkɪˌdɪzəm/ | Tinh hoàn ẩn |
| 148 | Testicular dysgenesis | /tɛˈstɪkjələr dɪsˈdʒɛnəsɪs/ | Loạn sản tinh hoàn |
| 149 | Virilized females | /ˈvɪrəlaɪzd ˈfiːmeɪlz/ | Nữ giới bị nam hóa |
| 150 | Inhibin B concentrations | /ɪnˈhɪbɪn biː ˌkɒnsənˈtreɪʃənz/ | Nồng độ Inhibin B |
| 151 | Testosterone secretion | /tɛˈstɒstəˌroʊn sɪˈkriːʃən/ | Sự tiết testosterone |
| 152 | Palpation | /pælˈpeɪʃən/ | Sờ nắn |
| 153 | Ultrasound | /ˈʌltrəˌsaʊnd/ | Siêu âm |
| 154 | Hormone responses | /ˈhɔːrmoʊn rɪˈspɒnsɪz/ | Đáp ứng hormone |
| 155 | Carboxy-terminal | /kɑːrˌbɒksiˈtɜːrmɪnəl/ | Đầu tận cùng Carboxy |
| 156 | Biological activity | /ˌbaɪəˈlɒdʒɪkəl ækˈtɪvəti/ | Hoạt tính sinh học |
| 157 | Terminal sialic acid | /ˈtɜːrmɪnəl saɪˈælɪk ˈæsɪd/ | Axit sialic đầu cuối |
| 158 | Carbohydrate chain | /ˌkɑːrboʊˈhaɪdreɪt tʃeɪn/ | Chuỗi carbohydrate |
| 159 | Half-life | /ˈhæf laɪf/ | Thời gian bán hủy |
| 160 | hCG preparations | /ˌeɪtʃ siː ˈdʒiː ˌprɛpəˈreɪʃənz/ | Các chế phẩm hCG |
| 161 | Recombinant hCG | /riˈkɒmbɪnənt ˌeɪtʃ siː ˈdʒiː/ | hCG tái tổ hợp |
| 162 | Batch-to-batch consistency | /bætʃ-tə-bætʃ kənˈsɪstənsi/ | Tính nhất quán giữa các lô |
| 163 | Specific activity | /spəˈsɪfɪk ækˈtɪvəti/ | Hoạt tính đặc hiệu |
| 164 | Purity | /ˈpjʊərəti/ | Độ tinh khiết |
| 165 | Dosing frequency | /ˈdoʊsɪŋ ˈfriːkwənsi/ | Tần suất dùng thuốc |
| 166 | Leydig cell hypoplasia | /ˈlaɪdɪɡ sɛl ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản tế bào Leydig |
| 167 | Testosterone biosynthesis | /tɛˈstɒstəˌroʊn ˌbaɪoʊˈsɪnθəsɪs/ | Sinh tổng hợp testosterone |
| 168 | Androgen responsiveness | /ˈændrədʒən rɪˈspɒnsɪvnəs/ | Khả năng đáp ứng androgen |
| 169 | Subcutaneously | /ˌsʌbkjuːˈteɪniəsli/ | Dưới da |
| 170 | Blood sampling | /blʌd ˈsæmplɪŋ/ | Lấy mẫu máu |
| 171 | Intermittent injections | /ˌɪntərˈmɪtənt ɪnˈdʒɛkʃənz/ | Tiêm ngắt quãng |
| 172 | Penile growth | /ˈpiːnaɪl ɡroʊθ/ | Sự phát triển dương vật |
| 173 | Target tissue responsiveness | /ˈtɑːrɡɪt ˈtɪʃuː rɪˈspɒnsɪvnəs/ | Khả năng đáp ứng của mô đích |
| 174 | Androstenedione | /ˌændroʊˌstiːnˈdaɪoʊn/ | Androstenedione |
| 175 | Dihydrotestosterone (DHT) | /daɪˌhaɪdroʊtɛˈstɒstəˌroʊn/ | Dihydrotestosterone |
| 176 | Poststimulation hormone ratios | /poʊstˌstɪmjəˈleɪʃən ˈhɔːrmoʊn ˈreɪʃioʊz/ | Tỷ lệ hormone sau kích thích |
| 177 | 17β-hydroxysteroid dehydrogenase-3 deficiency | /… ˌhaɪdrɒksiˈstɪərɔɪd ˌdiːhaɪˈdrɒdʒəneɪs-θriː dɪˈfɪʃənsi/ | Thiếu hụt 17β-hydroxysteroid dehydrogenase-3 |
| 178 | 5α-reductase deficiency | /faɪv ˈælfə rɪˈdʌkteɪs dɪˈfɪʃənsi/ | Thiếu hụt 5α-reductase |
| 179 | Mutation testing | /mjuːˈteɪʃən ˈtɛstɪŋ/ | Xét nghiệm đột biến |
| 180 | Newborn screening (NBS) | /ˈnjuːbɔːrn ˈskriːnɪŋ/ | Sàng lọc sơ sinh |
| 181 | Classic CAH (Congenital Adrenal Hyperplasia) | /ˈklæsɪk siː eɪ eɪtʃ/ | Tăng sản thượng thận bẩm sinh thể cổ điển |
| 182 | Whole-blood | /hoʊl blʌd/ | Máu toàn phần |
| 183 | Dried filter paper blood spot | /draɪd ˈfɪltər ˈpeɪpər blʌd spɒt/ | Đốm máu khô trên giấy lọc |
| 184 | Immunoassay | /ˌɪmjənoʊˈæseɪ/ | Xét nghiệm miễn dịch |
| 185 | Dissociation-enhanced lanthanide fluoroimmunoassay | /dɪˌsoʊsiˈeɪʃən-ɪnˈhænst ˈlænθənaɪd ˌflʊəroʊˌɪmjənoʊˈæseɪ/ | Xét nghiệm miễn dịch huỳnh quang lanthanide tăng cường phân ly |
| 186 | Preterm infants | /ˈpriːtɜːrm ˈɪnfənts/ | Trẻ sinh non |
| 187 | Clinical decision-making | /ˈklɪnɪkəl dɪˈsɪʒən ˈmeɪkɪŋ/ | Ra quyết định lâm sàng |
| 188 | Glucocorticoid therapy | /ˌɡluːkoʊˈkɔːrtɪkɔɪd ˈθɛrəpi/ | Liệu pháp glucocorticoid |
| 189 | False positive results | /fɔːls ˈpɒzətɪv rɪˈzʌlts/ | Kết quả dương tính giả |
| 190 | Prematurity | /ˌpriːməˈtʃʊərəti/ | Sinh non |
| 191 | Cross-reacting steroids | /krɒs riˈæktɪŋ ˈstɪərɔɪdz/ | Các steroid phản ứng chéo |
| 192 | Heterozygosity | /ˌhɛtəroʊzaɪˈɡɒsəti/ | Tình trạng dị hợp tử |
| 193 | 21-hydroxylase deficiency | /ˈtwɛnti wʌn haɪˈdrɒksɪleɪs dɪˈfɪʃənsi/ | Thiếu hụt 21-hydroxylase |
| 194 | Stressed infant | /strɛst ˈɪnfənt/ | Trẻ bị stress |
| 195 | Late-onset CAH | /leɪt ˈɒnsɛt siː eɪ eɪtʃ/ | Tăng sản thượng thận bẩm sinh khởi phát muộn |
| 196 | False negative results | /fɔːls ˈnɛɡətɪv rɪˈzʌlts/ | Kết quả âm tính giả |
| 197 | Maternal treatment | /məˈtɜːrnl ˈtriːtmənt/ | Điều trị cho mẹ |
| 198 | Differential diagnosis | /ˌdɪfəˈrɛnʃəl ˌdaɪəɡˈnoʊsɪs/ | Chẩn đoán phân biệt |
| 199 | Cutoff levels | /ˈkʌtɒf ˈlɛvəlz/ | Mức ngưỡng |
| 200 | Classic salt-losing | /ˈklæsɪk sɔːlt ˈluːzɪŋ/ | (Thể) mất muối cổ điển |
| 201 | Simple virilizing forms | /ˈsɪmpəl ˈvɪrəlaɪzɪŋ fɔːrmz/ | Các dạng nam hóa đơn thuần |
| 202 | Specificity | /ˌspɛsɪˈfɪsəti/ | Độ đặc hiệu |
| 203 | Organic extraction | /ɔːrˈɡænɪk ɪkˈstrækʃən/ | Chiết xuất hữu cơ |
| 204 | Chromatography | /ˌkroʊməˈtɒɡrəfi/ | Sắc ký |
| 205 | Liquid chromatography-tandem mass spectrometry (LC/MS/MS) | /ˈlɪkwɪd ˌkroʊməˈtɒɡrəfi-ˈtændəm mæs spɛkˈtrɒmɪtri/ | Sắc ký lỏng-khối phổ song song |
| 206 | Steroidogenesis | /ˌstɪərɔɪdoʊˈdʒɛnəsɪs/ | Sinh tổng hợp steroid |
| 207 | Sensitivity | /ˌsɛnsɪˈtɪvəti/ | Độ nhạy |
| 208 | Gestational-age based cut-points | /dʒɛˈsteɪʃənəl eɪdʒ beɪst kʌt pɔɪnts/ | Các điểm cắt dựa trên tuổi thai |
| 209 | 21-deoxycortisol concentrations | /ˈtwɛnti wʌn diːˌɒksiˈkɔːrtɪsɒl ˌkɒnsənˈtreɪʃənz/ | Nồng độ 21-deoxycortisol |
| 210 | Gas chromatography-mass spectrometry | /ɡæs ˌkroʊməˈtɒɡrəfi mæs spɛkˈtrɒmɪtri/ | Sắc ký khí-khối phổ |
| 211 | Hormone ratios | /ˈhɔːrmoʊn ˈreɪʃioʊz/ | Tỷ lệ hormone |
| 212 | 11-deoxycortisol | /ɪˈlɛvən diːˌɒksiˈkɔːrtɪsɒl/ | 11-deoxycortisol |
| 213 | Endoscopic studies | /ˌɛndoʊˈskɒpɪk ˈstʌdiz/ | Các nghiên cứu nội soi |
| 214 | Vaginal-urethral confluence | /ˈvædʒɪnəl jʊəˈriːθrəl ˈkɒnfluəns/ | Hợp lưu âm đạo-niệu đạo |
| 215 | Bladder neck | /ˈblædər nɛk/ | Cổ bàng quang |
| 216 | Urogenital sinus | /ˌjuːroʊˈdʒɛnɪtl ˈsaɪnəs/ | Xoang niệu-sinh dục |
| 217 | Cystoscope | /ˈsɪstoʊskoʊp/ | Ống soi bàng quang |
| 218 | Catheter placement | /ˈkæθətər ˈpleɪsmənt/ | Đặt ống thông |
| 219 | Retrograde contrast studies | /ˈrɛtroʊɡreɪd ˈkɒntræst ˈstʌdiz/ | Các nghiên cứu cản quang ngược dòng |
| 220 | Urethra | /jʊəˈriːθrə/ | Niệu đạo |
| 221 | Vagina | /vəˈdʒaɪnə/ | Âm đạo |
| 222 | Uterus | /ˈjuːtərəs/ | Tử cung |
| 223 | Cervix | /ˈsɜːrvɪks/ | Cổ tử cung |
| 224 | Uterine cavity | /ˈjuːtəraɪn ˈkævəti/ | Khoang tử cung |
| 225 | Reconstructive surgery | /ˌriːkənˈstrʌktɪv ˈsɜːrdʒəri/ | Phẫu thuật tạo hình |
| 226 | Medical complications | /ˈmɛdɪkəl ˌkɒmplɪˈkeɪʃənz/ | Biến chứng y tế |
| 227 | Laparoscopy | /ˌlæpəˈrɒskəpi/ | Nội soi ổ bụng |
| 228 | Internal genital structures | /ɪnˈtɜːrnl ˈdʒɛnɪtl ˈstrʌktʃərz/ | Các cấu trúc sinh dục trong |
| 229 | MRI (Magnetic Resonance Imaging) | /ˌɛm ɑːr ˈaɪ/ | Chụp cộng hưởng từ |
| 230 | Anatomic relationships | /ˌænəˈtɒmɪk rɪˈleɪʃənʃɪps/ | Các mối quan hệ giải phẫu |
| 231 | DSD team | /diː ɛs diː tiːm/ | Đội ngũ (chuyên gia) DSD |
| 232 | Pediatric endocrinologist | /ˌpiːdiˈætrɪk ˌɛndoʊkrɪˈnɒlədʒɪst/ | Bác sĩ nội tiết nhi |
| 233 | Pediatric surgeon | /ˌpiːdiˈætrɪk ˈsɜːrdʒən/ | Bác sĩ phẫu thuật nhi |
| 234 | Pediatric urologist | /ˌpiːdiˈætrɪk jʊəˈrɒlədʒɪst/ | Bác sĩ tiết niệu nhi |
| 235 | Urogenital reconstructive surgery | /ˌjuːroʊˈdʒɛnɪtl ˌriːkənˈstrʌktɪv ˈsɜːrdʒəri/ | Phẫu thuật tạo hình niệu-sinh dục |
| 236 | Behavioral health professional | /bɪˈheɪvjərəl hɛlθ prəˈfɛʃənl/ | Chuyên gia sức khỏe hành vi |
| 237 | Psychologist | /saɪˈkɒlədʒɪst/ | Nhà tâm lý học |
| 238 | Social worker | /ˈsoʊʃəl ˈwɜːrkər/ | Nhân viên xã hội |
| 239 | Psychiatrist | /saɪˈkaɪətrɪst/ | Bác sĩ tâm thần |
| 240 | Neonatologist | /ˌniːoʊneɪˈtɒlədʒɪst/ | Bác sĩ sơ sinh |
| 241 | Ethics consultant | /ˈɛθɪks kənˈsʌltənt/ | Nhà tư vấn đạo đức |
| 242 | Stigmatization | /ˌstɪɡmətaɪˈzeɪʃən/ | Sự kỳ thị |
| 243 | Gender identity | /ˈdʒɛndər aɪˈdɛntəti/ | Bản dạng giới |
| 244 | Fertility potential | /fərˈtɪləti pəˈtɛnʃəl/ | Tiềm năng sinh sản |
| 245 | Gonadal tumor risk | /ɡoʊˈnædəl ˈtuːmər rɪsk/ | Nguy cơ u tuyến sinh dục |
| 246 | Self-esteem | /sɛlf ɪˈstiːm/ | Lòng tự trọng |
| 247 | Coordination of care | /koʊˌɔːrdɪˈneɪʃən əv kɛər/ | Điều phối chăm sóc |
| 248 | Tertiary care facility | /ˈtɜːrʃiˌɛri kɛər fəˈsɪləti/ | Cơ sở chăm sóc tuyến ba |
| 249 | Family traditions and customs | /ˈfæməli trəˈdɪʃənz ænd ˈkʌstəmz/ | Truyền thống và phong tục gia đình |
| 250 | Informed discussion | /ɪnˈfɔːrmd dɪˈskʌʃən/ | Thảo luận có đầy đủ thông tin |
| 251 | Pathophysiology | /ˌpæθoʊˌfɪziˈɒlədʒi/ | Sinh lý bệnh |
| 252 | Internal reproductive system anatomy | /ɪnˈtɜːrnl ˌriːprəˈdʌktɪv ˈsɪstəm əˈnætəmi/ | Giải phẫu hệ thống sinh sản bên trong |
| 253 | Spontaneous pubertal development | /spɒnˈteɪniəs pjuːˈbɜːrtl dɪˈvɛləpmənt/ | Sự phát triển dậy thì tự nhiên |
| 254 | Sexual activity | /ˈsɛkʃuəl ækˈtɪvəti/ | Hoạt động tình dục |
| 255 | Orgasm | /ˈɔːrɡæzəm/ | Cực khoái |
| 256 | Fertility preservation | /fərˈtɪləti ˌprɛzərˈveɪʃən/ | Bảo tồn khả năng sinh sản |
| 257 | Natal sex | /ˈneɪtl sɛks/ | Giới tính khai sinh |
| 258 | Psychosexual development | /ˌsaɪkoʊˈsɛkʃuəl dɪˈvɛləpmənt/ | Phát triển tâm lý tình dục |
| 259 | Gender role | /ˈdʒɛndər roʊl/ | Vai trò giới |
| 260 | Sexual orientation | /ˈsɛkʃuəl ˌɔːriənˈteɪʃən/ | Xu hướng tính dục |
| 261 | Prenatal hormonal exposure | /ˌpriːˈneɪtl hɔːrˈmoʊnəl ɪkˈspoʊʒər/ | Phơi nhiễm hormone trước sinh |
| 262 | Nonbinary spectrum | /nɒnˈbaɪnəri ˈspɛktrəm/ | Phổ phi nhị nguyên |
| 263 | Complete Androgen Insensitivity Syndrome (CAIS) | /kəmˈpliːt ˈændrədʒən ɪnˌsɛnsɪˈtɪvəti ˈsɪndroʊm/ | Hội chứng không nhạy cảm androgen hoàn toàn |
| 264 | Partial Androgen Insensitivity Syndrome (PAIS) | /ˈpɑːrʃəl ˈændrədʒən ɪnˌsɛnsɪˈtɪvəti ˈsɪndroʊm/ | Hội chứng không nhạy cảm androgen một phần |
| 265 | CNS (Central Nervous System) development | /siː ɛn ɛs dɪˈvɛləpmənt/ | Sự phát triển hệ thần kinh trung ương |
| 266 | Exogenous testosterone | /ɛkˈsɒdʒənəs tɛˈstɒstəˌroʊn/ | Testosterone ngoại sinh |
| 267 | Intramuscular injections | /ˌɪntrəˈmʌskjələr ɪnˈdʒɛkʃənz/ | Tiêm bắp |
| 268 | Sex assignment | /sɛks əˈsaɪnmənt/ | Chỉ định giới tính |
| 269 | Self-reassign | /sɛlf ˌriːəˈsaɪn/ | Tự tái chỉ định (giới tính) |
| 270 | Genital surgery | /ˈdʒɛnɪtl ˈsɜːrdʒəri/ | Phẫu thuật bộ phận sinh dục |
| 271 | Delayed diagnosis | /dɪˈleɪd ˌdaɪəɡˈnoʊsɪs/ | Chẩn đoán chậm trễ |
| 272 | Homosexual orientation | /ˌhoʊmoʊˈsɛkʃuəl ˌɔːriənˈteɪʃən/ | Xu hướng đồng tính luyến ái |
| 273 | Sexual imagery | /ˈsɛkʃuəl ˈɪmədʒəri/ | Hình ảnh tình dục |
| 274 | Sexual attraction | /ˈsɛkʃuəl əˈtrækʃən/ | Sức hấp dẫn tình dục |
| 275 | Psychological adjustment | /ˌsaɪkəˈlɒdʒɪkəl əˈdʒʌstmənt/ | Sự điều chỉnh tâm lý |
| 276 | Unaffected siblings | /ˌʌnəˈfɛktɪd ˈsɪblɪŋz/ | Anh chị em không bị ảnh hưởng |
| 277 | Cognitive behavior | /ˈkɒɡnətɪv bɪˈheɪvjər/ | Hành vi nhận thức |
| 278 | Self-concept | /sɛlf ˈkɒnsɛpt/ | Quan niệm bản thân |
| 279 | Gender dysphoria | /ˈdʒɛndər dɪsˈfɔːriə/ | Bồn chồn giới tính |
| 280 | Transgender individuals | /trænsˈdʒɛndər ˌɪndɪˈvɪdʒuəlz/ | Người chuyển giới |
| 281 | Multidisciplinary team approach | /ˌmʌltiˈdɪsɪplɪˌnɛri tiːm əˈproʊtʃ/ | Phương pháp tiếp cận nhóm đa ngành |
| 282 | Mineralocorticoid biosynthesis | /ˌmɪnərəloʊˈkɔːrtɪkɔɪd ˌbaɪoʊˈsɪnθəsɪs/ | Sinh tổng hợp mineralocorticoid |
| 283 | Hormone replacement therapy | /ˈhɔːrmoʊn rɪˈpleɪsmənt ˈθɛrəpi/ | Liệu pháp thay thế hormone |
| 284 | Sex steroid hormone | /sɛks ˈstɪərɔɪd ˈhɔːrmoʊn/ | Hormone steroid sinh dục |
| 285 | Induction of puberty | /ɪnˈdʌkʃən əv ˈpjuːbərti/ | Khởi phát dậy thì |
| 286 | Skeletal maturation | /ˈskɛlɪtl ˌmætʃəˈreɪʃən/ | Sự trưởng thành xương |
| 287 | Conjugated estrogens | /ˈkɒndʒəɡeɪtɪd ˈɛstrədʒənz/ | Estrogen liên hợp |
| 288 | Transdermal estrogen preparations | /trænsˈdɜːrməl ˈɛstrədʒən ˌprɛpəˈreɪʃənz/ | Các chế phẩm estrogen qua da |
| 289 | Matrix transdermal patches | /ˈmeɪtrɪks trænsˈdɜːrməl ˈpætʃɪz/ | Miếng dán qua da dạng ma trận |
| 290 | Progestational agent | /ˌproʊdʒɛˈsteɪʃənl ˈeɪdʒənt/ | Tác nhân progestational |
| 291 | Withdrawal bleeding | /wɪðˈdrɔːəl ˈbliːdɪŋ/ | Chảy máu kinh nguyệt (do ngưng thuốc) |
| 292 | Cyclic estrogen-progesterone therapy | /ˈsaɪklɪk ˈɛstrədʒən-proʊˈdʒɛstəˌroʊn ˈθɛrəpi/ | Liệu pháp estrogen-progesterone theo chu kỳ |
| 293 | Menstrual flow | /ˈmɛnstruəl floʊ/ | Dòng chảy kinh nguyệt |
| 294 | Calcium bone loss | /ˈkælsiəm boʊn lɒs/ | Mất canxi trong xương |
| 295 | Low-dosage estrogen birth control pills | /loʊ ˈdoʊsɪdʒ ˈɛstrədʒən bɜːrθ kənˈtroʊl pɪlz/ | Thuốc tránh thai estrogen liều thấp |
| 296 | Oral progestin | /ˈɔːrəl proʊˈdʒɛstɪn/ | Progestin đường uống |
| 297 | Estrogen-progestin transdermal patches | /ˈɛstrədʒən-proʊˈdʒɛstɪn trænsˈdɜːrməl ˈpætʃɪz/ | Miếng dán qua da estrogen-progestin |
| 298 | Libido | /lɪˈbiːdoʊ/ | Ham muốn tình dục |
| 299 | Gonadotropins | /ɡoʊˌnædoʊˈtroʊpɪnz/ | Gonadotropin (hormone hướng sinh dục) |
| 300 | Human menopausal gonadotropin | /ˈhjuːmən ˌmɛnoʊˈpɔːzəl ɡoʊˌnædoʊˈtroʊpɪn/ | Gonadotropin mãn kinh người |