
[Sách dịch] Sổ tay Bệnh Gan – Handbook of Liver Disease – (C) NXB Elsevier, 2022
Dịch và chú giải: Ths.Bs. Lê Đình Sáng
CHƯƠNG 19: Bệnh Wilson và các Rối Loạn Liên Quan
Wilson Disease and Related Disorders
Michael L. Schilsky MD
Handbook of Liver Disease, Chapter 19, 253-268
NHỮNG ĐIỂM CHÍNH
|
Chuyển hóa Đồng (Hình 19.1 và 19.2)
- Khoảng 1 đến 2 mg đồng trong chế độ ăn được hấp thu hàng ngày bởi các tế bào biểu mô ruột non đoạn gần. Chất vận chuyển đồng CTR1 chịu trách nhiệm cho việc hấp thu đồng của tế bào biểu mô ruột, và protein bệnh Menkes ATP7A tham gia vào việc vận chuyển đồng từ tế bào biểu mô vào tuần hoàn.
- Đồng được vận chuyển vào tuần hoàn cửa sẽ gắn với albumin huyết thanh và các axit amin. Lượng đồng còn lại trong tế bào biểu mô chủ yếu gắn với peptide thải độc nội sinh metallothionein và sau đó được bài tiết khi các tế bào biểu mô ruột bong ra. Không có chu trình gan-ruột đáng kể của đồng.
- Chỉ một phần nhỏ đồng trong tuần hoàn (<50 µg/24 giờ) được bài tiết bình thường qua thận; phần lớn được tế bào gan hấp thu và lượng dư thừa được bài tiết vào mật.
- Trong tế bào gan, đồng được tạo phức và giải độc bởi metallothionein hoặc glutathione và được sử dụng làm đồng yếu tố cho các enzyme tế bào đặc hiệu, được tích hợp vào ceruloplasmin rồi bài tiết vào tuần hoàn, hoặc được bài tiết vào mật.
- Vị trí gắn đồng vào ceruloplasmin trong tế bào gan là bộ máy trans-Golgi. ATP7B được cho là chịu trách nhiệm vận chuyển đồng trong khoang này và sau đó gắn vào ceruloplasmin.
- Việc vận chuyển đồng trong bào tương đến các vị trí nội bào đặc hiệu được trung gian bởi các protein nhỏ gọi là chaperone đồng.
- Sự di chuyển của ATP7B từ mạng lưới trans-Golgi đến một khoang túi gần màng tiểu quản mật để đáp ứng với nồng độ đồng trong tế bào gan tăng lên tạo điều kiện cho việc bài tiết đồng qua tiểu quản mật và đường mật. Một con đường thứ cấp để bài tiết đồng qua mật của tế bào gan là thông qua vận chuyển phức hợp đồng-glutathione. Ngoài ra, một số đồng nội bào được vận chuyển ngược qua màng sinh chất đáy bên của tế bào gan vào tuần hoàn.

Hình 19.1 Sự hấp thu và bài tiết đồng (Cu). Đồng trong chế độ ăn (1 đến 2 mg/ngày) được vận chuyển vào tế bào biểu mô ruột, với sản phẩm gen Menkes điều hòa vận chuyển vào tuần hoàn cửa (25% đến 60%). Lượng đồng còn lại trong tế bào biểu mô được gắn với metallothionein và sau đó được bài tiết qua phân khi các tế bào biểu mô ruột bong ra. Một lượng nhỏ đồng được hấp thu được bài tiết qua nước tiểu, nhưng phần lớn được tế bào gan hấp thu, tổng hợp thành ceruloplasmin và dự trữ trong gan hoặc bài tiết qua mật.
Di truyền học
- Bệnh Wilson là một bệnh di truyền lặn trên nhiễm sắc thể thường với tỷ lệ mắc từ 1/20.000 đến 30.000 ở hầu hết các quần thể. Tần số gen của bệnh Wilson được ước tính từ 0.3% đến 0.7%, do đó tỷ lệ người mang gen dị hợp tử là hơn 1/150 đến 200 một chút.
- Năm 1985, gen bệnh Wilson được chứng minh có liên kết với enzyme hồng cầu, esterase D, một mối liên kết xác định vị trí trên nhiễm sắc thể 13. Năm 1993, ba nhóm nhà nghiên cứu khác nhau đã xác định gen bệnh Wilson là gen mã hóa cho một ATPase vận chuyển đồng được đặt tên là ATP7B. Gen này trải dài trên một vùng 80 kb của nhiễm sắc thể, mã hóa cho một bản phiên mã 7.5 kb được biểu hiện chủ yếu ở gan, với một lượng nhỏ hơn biểu hiện ở thận và nhau thai.
- ATP7B là một protein gồm 1466 axit amin, là thành viên của phân họ ATPase vận chuyển cation P-type được bảo tồn cao trong quá trình tiến hóa. ATP7B có độ tương đồng cao với sản phẩm của gen Menkes (ATP7A) và ATPase vận chuyển đồng (cop A) được tìm thấy ở các chủng Enterococcus hirae kháng đồng.
- Hơn 600 đột biến gây bệnh của gen Wilson đã được xác định cho đến nay. Hầu hết các đột biến là đột biến sai nghĩa (missense). Tương đối ít bệnh nhân là đồng hợp tử cho cùng một đột biến; tuy nhiên, hầu hết là dị hợp tử phức hợp (tức là mang các đột biến khác nhau trên mỗi alen).
- Mặc dù có sự đa dạng lâm sàng của bệnh Wilson, sự không đồng nhất của các alen tại locus ATP7B dường như không giải thích được sự thay đổi đáng kể về kiểu hình và lâm sàng quan sát được ở bệnh nhân.
- Mặc dù một alen ATP7B bình thường là đủ để ngăn ngừa bệnh lâm sàng, người mang gen dị hợp tử với một đột biến của gen Wilson có thể biểu hiện các bất thường cận lâm sàng trong chuyển hóa đồng như tăng nhẹ nồng độ đồng trong gan trên mức bình thường và, ở 20% trường hợp, giảm nồng độ ceruloplasmin trong tuần hoàn.

Hình 19.2 Chuyển hóa đồng trong tế bào gan. Đồng (Cu) được tế bào gan hấp thu, nơi nó tương tác với glutathione và metallothionein. Một phần đồng nội bào được tích hợp vào các metalloenzyme (ví dụ: superoxide dismutase, cytochrome oxidase), và một số được vận chuyển vào mạng lưới trans-Golgi bởi protein gen bệnh Wilson (ATP7B), nơi nó được tích hợp vào ceruloplasmin. Người ta giả định rằng đồng cũng được chuyển từ bộ máy trans-Golgi đến một khoang túi trong lysosome để sau đó bài tiết qua mật. Đồng gắn với glutathione cũng được bài tiết vào tiểu quản mật thông qua chất vận chuyển anion hữu cơ (CMOAT [hoặc MRP2]). Không chắc chắn liệu ATP7B có ở tại hoặc gần màng tiểu quản mật ở đỉnh hay không, nhưng đây là con đường quan trọng bị ảnh hưởng trong bệnh Wilson. CMOAT, chất vận chuyển anion hữu cơ đa đặc hiệu ở tiểu quản mật 1; MRP2, protein liên quan đến kháng đa thuốc 2.
Cơ chế bệnh sinh
- Việc duy trì cân bằng nội môi đồng bình thường phụ thuộc vào sự cân bằng giữa hấp thu ở đường tiêu hóa và bài tiết qua mật. Hấp thu đồng ở ruột ở bệnh nhân Wilson không khác so với người không mắc bệnh.
- Bài tiết đồng qua mật bị giảm trong bệnh Wilson do chức năng ATP7B bị khiếm khuyết hoặc không có, điều này có thể gây ra khiếm khuyết trong việc đưa đồng từ bào tương vào thành phần túi của con đường bài tiết vào mật.
- Giảm bài tiết đồng qua mật trong bệnh Wilson dẫn đến sự tích tụ đồng bệnh lý trong tế bào gan thông qua tổn thương oxy hóa do gốc tự do gây ra cho lipid, protein và axit nucleic; sự cạn kiệt các chất chống oxy hóa; và sự trùng hợp của đồng-metallothionein. Do đó, tổn thương do đồng dẫn đến hoại tử và chết theo chương trình của tế bào gan. Các bất thường về hình thái do tổn thương oxy hóa đã được xác định, đặc biệt là ở ty thể (ví dụ: phình to, giãn các mào và lắng đọng tinh thể).
- Sự tích tụ đồng trong gan và tổn thương tế bào gan dẫn đến tăng đồng không gắn ceruloplasmin trong tuần hoàn, là nguyên nhân gây tích tụ đồng ngoài gan. Độc tính của đồng đóng một vai trò chính trong cơ chế bệnh sinh của các biểu hiện ngoài gan của bệnh Wilson. Các cơ quan bị ảnh hưởng, đặc biệt là hệ thần kinh trung ương, luôn cho thấy nồng độ đồng tăng cao.
- Sự lắng đọng đồng bệnh lý trong não, chủ yếu ở nhân đuôi và nhân bèo của hạch nền, dẫn đến các biểu hiện thần kinh và tâm thần của bệnh. Sự lắng đọng quá mức của đồng trong màng Descemet của giác mạc làm phát sinh vòng Kayser-Fleischer (KF) và hiếm khi là đục thủy tinh thể hình hoa hướng dương.
- Sự thiếu hụt protein đồng huyết tương ceruloplasmin không có vai trò trong cơ chế bệnh sinh của bệnh Wilson. Nồng độ ceruloplasmin huyết thanh thấp ở bệnh nhân Wilson là kết quả của việc giảm tích hợp đồng vào peptide ceruloplasmin không có đồng (aceruloplasmin), chất này có thời gian bán hủy ngắn hơn so với ceruloplasmin có gắn đồng (holoceruloplasmin).
Đặc điểm Lâm sàng
- Bệnh nhân có thể không có triệu chứng, mặc dù hầu hết đều có biểu hiện ở gan hoặc thần kinh. Ít phổ biến hơn, bệnh nhân có các triệu chứng ở thận, xương khớp, tim mạch, mắt, nội tiết hoặc da.
- Các triệu chứng lâm sàng hiếm khi được quan sát thấy trước 3 đến 5 tuổi, và hầu hết bệnh nhân không được điều trị sẽ có triệu chứng trước 40 tuổi. Các triệu chứng gan thường xuất hiện ở thập kỷ thứ hai hoặc thứ ba của cuộc đời, và các triệu chứng thần kinh ở thập kỷ thứ ba và thứ tư.
- Trong một loạt nghiên cứu lớn, các biểu hiện lâm sàng ban đầu là ở gan trong 42% trường hợp, thần kinh 34%, tâm thần 10%, và huyết học 12%. Ít bệnh nhân biểu hiện bệnh Wilson sau 50 tuổi; anh chị em ruột được báo cáo lớn tuổi nhất biểu hiện bệnh ở 70 và 72 tuổi.
GAN
- Các biểu hiện ở gan có xu hướng xảy ra ở độ tuổi trẻ hơn (trung bình, 10 đến 12 tuổi) so với các biểu hiện thần kinh. Tốc độ tiến triển của bệnh gan thay đổi ở các bệnh nhân Wilson. Bệnh nhân không có triệu chứng thường có các bất thường trong xét nghiệm sinh hóa gan tương quan về mặt mô học với tình trạng gan nhiễm mỡ và viêm gan. Bệnh nhân trẻ tuổi có thể có các đặc điểm không thể phân biệt được với viêm gan siêu vi mạn tính hoặc viêm gan tự miễn.
- Tình trạng viêm đang diễn ra dẫn đến tiến triển của xơ hóa và cuối cùng là xơ gan với suy gan tiến triển và suy gan. Các biến chứng của tăng áp lực tĩnh mạch cửa trở nên rõ ràng khi xơ gan tiến triển.
- Suy gan cấp (ALF) phát triển ở một số ít bệnh nhân (xem thảo luận sau trong chương).
- Ung thư biểu mô tế bào gan, từng được coi là hiếm gặp, đã được báo cáo ở những bệnh nhân mắc bệnh Wilson.
- Bệnh nhân được điều trị có tiên lượng tốt ngay cả khi họ đã phát triển xơ gan. Điều trị thậm chí có thể cho phép thoái lui xơ hóa trong một số trường hợp. Việc ngưng điều trị dẫn đến tiến triển bệnh và phát triển suy gan cấp hoặc suy gan tiến triển.
Suy Gan Cấp
- Bệnh nhân có xu hướng trẻ, ở thập kỷ thứ hai của cuộc đời, và bệnh cảnh lâm sàng có thể không thể phân biệt được với hoại tử gan lan tỏa do virus. Bệnh cảnh lâm sàng tương tự này cũng có thể xuất hiện ở những bệnh nhân ngưng điều trị bệnh Wilson.
- Mặc dù nồng độ aminotransferase huyết thanh chỉ tăng nhẹ đến trung bình, nhưng có sự tăng rõ rệt bilirubin huyết thanh, nồng độ phosphatase kiềm huyết thanh thấp, và bằng chứng của thiếu máu tan máu Coombs âm tính. Nồng độ ceruloplasmin huyết thanh kém giá trị tiên đoán bệnh Wilson trong bối cảnh cấp tính; tuy nhiên, nồng độ đồng trong nước tiểu 24 giờ và nồng độ đồng trong tuần hoàn tăng rõ rệt.
- Các đặc điểm lâm sàng đặc trưng bao gồm rối loạn đông máu, tan máu không do miễn dịch, lách to, vòng KF, và diễn biến tối cấp; bệnh nhân hiếm khi sống sót quá vài ngày đến vài tuần trừ khi được thực hiện ghép gan (LT). Chỉ một số ít bệnh nhân có thể được cứu sống chỉ bằng điều trị nội khoa.
- Sinh thiết gan, nếu được thực hiện (thường qua đường tĩnh mạch cảnh do rối loạn đông máu), cho thấy nồng độ đồng trong gan tăng cao và thường là xơ hóa tiến triển hoặc xơ gan với tổn thương tế bào gan nặng. Có thể thấy rõ sự chết theo chương trình và hoại tử.
THẦN KINH
- Tổn thương thần kinh có xu hướng xảy ra ở thập kỷ thứ ba đến thứ tư của cuộc đời. Chẩn đoán bệnh Wilson thường bị trì hoãn từ 1 đến 2 năm ở những bệnh nhân có các đặc điểm thần kinh chiếm ưu thế.
- Các triệu chứng thần kinh sớm phổ biến là khó nói (dysarthria), vụng về, run, chảy nước dãi, rối loạn dáng đi, khuôn mặt vô cảm và chữ viết xấu đi.
- Cứng cơ với các đặc điểm Parkinson rõ rệt, co cứng gập và co cứng cơ ít gặp hơn và ở giai đoạn sau của bệnh. Múa vờn (athetosis – các cử động uốn éo không tự chủ) hoặc một rối loạn vận động nặng hơn có thể xuất hiện. Hiếm khi, có thể xảy ra co giật toàn thể.
- Rối loạn chức năng thần kinh tự chủ có thể xuất hiện, phổ biến nhất là liên quan đến các dấu hiệu thần kinh tiến triển khác.
- Khả năng nhận thức thường vẫn bình thường nhưng có thể bị suy giảm ở những bệnh nhân bị suy giảm thần kinh nặng.
- Các triệu chứng thần kinh có thể cải thiện rõ rệt với điều trị nội khoa hoặc sau khi ghép gan, mặc dù các di chứng còn lại là phổ biến, đặc biệt ở những người có triệu chứng kéo dài trước khi bắt đầu điều trị.
- Chụp cộng hưởng từ (MRI) cho thấy các tổn thương khu trú ở nhân bèo và cầu nhạt, và trong một số trường hợp có thêm các tổn thương ở cầu não và thân não.
TÂM THẦN
- Trong số tất cả các bệnh nhân mắc bệnh Wilson, một phần ba có thể có các triệu chứng tâm thần. Bệnh nhân có thể bị chẩn đoán nhầm là một bệnh tâm thần tiến triển, do đó làm trì hoãn chẩn đoán bệnh Wilson và tăng khả năng mắc bệnh gan đồng thời hoặc tiến triển.
- Các triệu chứng sớm ở thanh thiếu niên có thể chỉ giới hạn ở những thay đổi hành vi tinh tế và suy giảm thành tích học tập và công việc.
- Bệnh nhân sau này có thể có những thay đổi về nhân cách, tính khí thất thường, dễ xúc động, hành vi bốc đồng và chống đối xã hội, trầm cảm và tăng ham muốn tình dục. Rối loạn tâm thần thực sự có thể xảy ra.
- Các triệu chứng tâm thần có thể giải quyết bằng liệu pháp nội khoa hoặc sau khi ghép gan.
NHÃN KHOA
Vòng Kayser-Fleischer
- Các hạt đậm đặc electron giàu đồng và lưu huỳnh lắng đọng trong màng Descemet của giác mạc, tạo ra vòng KF. Vòng KF có màu nâu vàng hoặc màu xanh lục ở vùng rìa giác mạc, ban đầu rõ ở cực trên và dưới của giác mạc khi khám bằng đèn khe. Vòng cuối cùng trở thành chu vi, và chiều rộng của vòng tăng lên ở những bệnh nhân không được điều trị. Vòng KF giảm kích thước và có thể biến mất khi điều trị, thường là sau vài tháng đến vài năm.
- Sự hiện diện hay vắng mặt của vòng KF nên được xác nhận bởi một bác sĩ nhãn khoa có kinh nghiệm bằng cách sử dụng khám đèn khe.
- Vòng KF có mặt ở hầu hết các bệnh nhân Wilson có triệu chứng và gần như luôn có ở những người có biểu hiện thần kinh; chúng thường không có trong các trường hợp không triệu chứng và ở 40% đến 50% bệnh nhân bị bệnh gan.
- Sự xuất hiện trở lại sau khi thoái lui hoặc sự xuất hiện mới của vòng KF cho thấy sự không tuân thủ điều trị nội khoa.
- Vòng KF không phải là dấu hiệu đặc hiệu cho bệnh Wilson vì chúng cũng thỉnh thoảng được thấy ở những bệnh nhân bị ứ mật kéo dài do các nguyên nhân khác.
Đục thủy tinh thể hình hoa hướng dương
- Thường được quan sát thấy cùng với vòng KF, nhưng ít gặp hơn nói chung.
- Thị lực không bị suy giảm.
- Hết khi điều trị bệnh Wilson.
THẬN
- Các dấu hiệu bao gồm toan hóa ống thận gần hoặc các đặc điểm của hội chứng Fanconi ở những bệnh nhân có biểu hiện bệnh mạn tính. Tổn thương ống thận cấp có thể xảy ra trong suy gan cấp do bệnh Wilson do giải phóng lượng lớn đồng và các phức hợp đồng từ các tế bào gan bị tổn thương.
- Toan hóa ống thận xa cũng có thể xảy ra và có thể là nguyên nhân làm tăng tỷ lệ sỏi thận trong bệnh Wilson.
- Tiểu máu, chủ yếu là vi thể, có thể do sỏi thận hoặc bệnh cầu thận.
- Protein niệu đã được ghi nhận là một biểu hiện của bệnh Wilson, mặc dù hội chứng thận hư và hội chứng Goodpasture có nhiều khả năng là một tác dụng phụ của liệu pháp với D-penicillamine hoặc ít phổ biến hơn là trientine (xem thảo luận sau trong chương).
- Liệu pháp thải đồng thường dẫn đến cải thiện rõ rệt chức năng thận.
XƯƠNG KHỚP
- Hơn một nửa số bệnh nhân mắc bệnh Wilson có biểu hiện giảm mật độ xương (osteopenia) do nhuyễn xương (osteomalacia), loãng xương (osteoporosis), hoặc cả hai.
- Bệnh khớp có triệu chứng xảy ra ở 25% đến 50% bệnh nhân; bệnh khớp thoái hóa này giống như viêm xương khớp và liên quan đến cột sống và các khớp lớn.
- Viêm xương sụn bóc tách (osteochondritis dissecans), nhuyễn sụn xương bánh chè (chondromalacia patellae), và vôi hóa sụn khớp (chondrocalcinosis) cũng đã được mô tả.
KHÁC
- Một đợt tan máu nội mạch cấp tính có thể là biểu hiện khởi phát ở 15% bệnh nhân; nó thường thoáng qua và tự giới hạn nhưng có thể liên quan đến suy gan cấp do bệnh Wilson.
- Tần suất tổn thương tim đã bị đánh giá thấp trong quá khứ; các bất thường trên điện tâm đồ có mặt trong một phần ba số trường hợp. Rối loạn thần kinh tự chủ có thể gây loạn nhịp.
- Móng xanh (Azure lunulae – sự đổi màu xanh của liềm móng [gốc móng tay]) là một dấu hiệu không phổ biến nhưng đặc trưng.
- Dậy thì muộn, nữ hóa tuyến vú và vô kinh đã được ghi nhận. Chúng xảy ra thường xuyên nhất ở những bệnh nhân bị bệnh gan tiến triển và có thể do mất cân bằng nội tiết tố từ bệnh gan chứ không phải do chính bệnh Wilson.
Chẩn đoán
Nên xem xét bệnh Wilson ở những người có:
- Tăng aminotransferase huyết thanh không giải thích được, viêm gan mạn tính với nhiễm mỡ, viêm gan tự miễn đáp ứng kém, xơ gan và suy gan cấp.
- Các đặc điểm thần kinh không rõ nguồn gốc (hành vi bất thường, mất phối hợp, run, rối loạn vận động).
- Một rối loạn thần kinh hoặc tâm thần với các dấu hiệu của bệnh gan đồng thời.
- Vòng KF được phát hiện khi khám mắt định kỳ.
- Thiếu máu tan máu Coombs âm tính mắc phải không giải thích được.
- Anh, chị, em ruột hoặc cha mẹ được chẩn đoán mắc bệnh Wilson.
CÁC XÉT NGHIỆM
- Ceruloplasmin
- Nồng độ huyết thanh bình thường là 20 đến 40 mg/dL (tham khảo khoảng tham chiếu của phòng xét nghiệm địa phương, vì các biến thể nhỏ là phổ biến).
- 95% tất cả các bệnh nhân mắc bệnh Wilson mạn tính có nồng độ thấp hơn khoảng bình thường.
- Nồng độ bình thường được tìm thấy ở ít nhất 5% bệnh nhân mắc bệnh Wilson.
- Ceruloplasmin có thể tăng lên mức bình thường hoặc gần bình thường ở bệnh nhân Wilson có đáp ứng pha cấp với tổn thương gan và ở những bệnh nhân có nồng độ estrogen huyết thanh tăng thứ phát do mang thai hoặc dùng thuốc ngoại sinh.
- Nồng độ ceruloplasmin giảm không phải là dấu hiệu đặc hiệu cho bệnh Wilson. Các nguyên nhân không phải Wilson sau đây cũng nên được xem xét:
- Lên đến 20% người mang gen dị hợp tử không triệu chứng.
- Trẻ em dưới 6 tháng tuổi (nồng độ thấp sinh lý).
- Giảm chức năng tổng hợp do bệnh gan nặng.
- Hội chứng thận hư, bệnh ruột mất protein và kém hấp thu ở ruột.
- Bệnh di truyền không có ceruloplasmin (aceruloplasminemia), không liên quan đến bệnh Wilson và có thể liên quan đến quá tải sắt.
- Đồng huyết thanh không gắn ceruloplasmin
- Ở những bệnh nhân không bị ảnh hưởng, đồng trong ceruloplasmin chiếm khoảng 90% tổng lượng đồng trong huyết thanh. Ở những bệnh nhân Wilson không được điều trị, tổng lượng đồng huyết thanh thường giảm do nồng độ ceruloplasmin trong tuần hoàn giảm (ngoại trừ những bệnh nhân bị suy gan cấp do bệnh Wilson, trong đó một lượng lớn đồng có mặt trong tuần hoàn).
- Ước tính nồng độ đồng “tự do” (không gắn ceruloplasmin) trong huyết thanh có thể được tính bằng cách trừ đi lượng đồng ceruloplasmin (0.047 µmol đồng/mg ceruloplasmin) khỏi tổng nồng độ đồng huyết thanh. Một công thức gần đúng là: [tổng đồng huyết thanh (µg/dL) – (3.15 × ceruloplasmin [mg/dL])].
- Bệnh nhân mắc bệnh Wilson có tỷ lệ đồng gắn với albumin huyết thanh, axit amin, hoặc các peptide khác (đồng không gắn ceruloplasmin) tăng so với người không bị ảnh hưởng (trong đó khoảng 10% tổng lượng đồng trong tuần hoàn là đồng không gắn ceruloplasmin). Ở những bệnh nhân Wilson không được điều trị, đồng không gắn ceruloplasmin vượt quá 25 µg/dL; ở những bệnh nhân suy gan do bệnh Wilson, nồng độ tăng rõ rệt và có thể vượt quá 200 µg/dL.
- Nồng độ đồng huyết thanh không gắn ceruloplasmin rất hữu ích để theo dõi sự đầy đủ của liệu pháp thải đồng trong quá trình điều trị duy trì. Tỷ lệ đồng không gắn ceruloplasmin giảm ở những bệnh nhân được điều trị, và nồng độ thường là 5 đến 15 µg/dL.
- Các xét nghiệm mới hơn về “đồng trao đổi” đo trực tiếp đồng không gắn ceruloplasmin từ các mẫu huyết tương và trong tương lai có thể hữu ích để theo dõi điều trị.
- Bài tiết đồng qua nước tiểu
- Bài tiết đồng qua nước tiểu bình thường là <40 µg/24 giờ.
- Hầu hết bệnh nhân Wilson có triệu chứng có bài tiết đồng qua nước tiểu lớn hơn 100 µg/24 giờ, và bệnh nhân bị suy gan cấp thường có nồng độ vượt quá 1000 µg/24 giờ.
- Bệnh nhân Wilson không triệu chứng có thể có bài tiết đồng qua nước tiểu bình thường, và 16% đến 23% bệnh nhân Wilson có biểu hiện bệnh gan bài tiết <100 µg/24 giờ. Do đó, giá trị tiên đoán của xét nghiệm này một cách riêng lẻ là hạn chế.
- Nồng độ tăng có thể thấy trong các rối loạn gan khác, chẳng hạn như xơ gan mật nguyên phát và viêm gan mạn tính, và trong protein niệu nặng do mất ceruloplasmin trong nước tiểu.
- Xét nghiệm này hữu ích trong việc xác nhận chẩn đoán bệnh Wilson và trong việc theo dõi sự tuân thủ và đáp ứng với liệu pháp thải đồng.
- Nghiệm pháp kích thích D-penicillamine, được dùng với liều 0.5 g trước và 12 giờ sau đó trong quá trình thu thập nước tiểu 24 giờ, đã được chứng minh là làm tăng bài tiết đồng qua nước tiểu, nhưng nó không phân biệt một cách đáng tin cậy bệnh nhân Wilson với người mang gen dị hợp tử và những người mắc các bệnh gan khác và do đó có tiện ích hạn chế ở người lớn. Việc hạ ngưỡng bài tiết đồng nước tiểu cơ bản (không có D-penicillamine) xuống 40 µg từ 100 µg làm tăng độ nhạy của xét nghiệm để chẩn đoán bệnh Wilson.
- Sinh thiết gan
- Những thay đổi mô học trên kính hiển vi quang học thường không đặc hiệu trong bệnh Wilson. Các đặc điểm sớm có thể bao gồm thể vùi glycogen trong nhân của các tế bào gan quanh khoảng cửa (nhân glycogen hóa) và thâm nhiễm mỡ mức độ trung bình có thể là cả nhiễm mỡ hạt nhỏ (microsteatotic) và hạt lớn (macrosteatotic). Ở giai đoạn này, quan sát thấy những thay đổi siêu cấu trúc của ty thể.
- Trong các trường hợp tiến triển hơn, có hiện tượng xơ hóa hoặc xơ gan.
- Trong viêm gan cấp tính nặng và viêm gan mạn tính, có hoại tử bán lan tỏa với thể hyalin Mallory (thể Mallory-Denk) và xơ gan hoặc xơ hóa tiến triển. Bằng chứng về chết theo chương trình cũng như hoại tử có thể thấy rõ trong các mẫu bệnh phẩm này.
- Nhuộm hóa mô học các mẫu sinh thiết gan để tìm đồng bằng rhodanine hoặc axit rubeanic có giá trị hạn chế trừ khi kết quả dương tính, bởi vì trong giai đoạn đầu của sự tích tụ đồng trong tế bào gan, kim loại được phân bố lan tỏa trong bào tương và không nhuộm màu hóa mô học bằng các phương pháp này. Nhuộm bạc sulfide Timm có thể phát hiện protein gắn đồng trong bào tương, nhưng xét nghiệm này không được thực hiện thường quy.
- Nồng độ đồng trong gan >250 µg/g gan khô (bình thường, 15 đến 55 µg/g) kèm theo ceruloplasmin huyết thanh thấp sẽ xác định chẩn đoán bệnh Wilson, với hai lưu ý:
- Kim sinh thiết và vật chứa mẫu phải không có đồng. Một kim dùng một lần làm bằng thép là chấp nhận được. Ở các kim tái sử dụng cũ hơn có thể làm bằng đồng thau, chẳng hạn như kim Klatskin hoặc Menghini, khuyến cáo là rửa kim trong 0.1 M axit ethylenediaminetetraacetic (EDTA) và rửa lại bằng nước khử khoáng trước khi sử dụng. Sử dụng các hộp nhựa vô trùng tránh được nhu cầu rửa EDTA các hộp đựng mẫu bằng thủy tinh có thể bị nhiễm kim loại.
- Việc tìm thấy nồng độ đồng trong gan bình thường sẽ loại trừ chẩn đoán, nhưng chỉ riêng nồng độ tăng cũng có thể được tìm thấy trong các bệnh gan khác.
- Các rối loạn ứ mật (ví dụ, xơ gan mật nguyên phát, viêm đường mật xơ cứng nguyên phát, ứ mật trong gan ở trẻ em, teo đường mật).
- Ngộ độc đồng gan không do Wilson (ví dụ, xơ gan trẻ em Ấn Độ, xơ gan trẻ sơ sinh Tyrolean lưu hành, ngộ độc đồng vô căn).
- Gắn đồng phóng xạ dùng đường uống vào ceruloplasmin
- Hoạt độ phóng xạ huyết thanh (chủ yếu là ceruloplasmin chứa đồng phóng xạ) được đo sau khi uống đồng được đánh dấu phóng xạ (
 Cu hoặc 67Cu) tại các thời điểm 1, 2, 4, và 48 giờ. Bình thường, người ta thấy sự xuất hiện nhanh chóng của đồng được đánh dấu phóng xạ trong huyết thanh, sau đó là sự biến mất của nó theo thời gian. Đỉnh sớm này không có ở bệnh nhân mắc bệnh Wilson.
Cu hoặc 67Cu) tại các thời điểm 1, 2, 4, và 48 giờ. Bình thường, người ta thấy sự xuất hiện nhanh chóng của đồng được đánh dấu phóng xạ trong huyết thanh, sau đó là sự biến mất của nó theo thời gian. Đỉnh sớm này không có ở bệnh nhân mắc bệnh Wilson. - Xét nghiệm này chủ yếu được sử dụng cho các bệnh nhân nghi ngờ mắc bệnh Wilson có nồng độ ceruloplasmin huyết thanh bình thường; tuy nhiên, do khó khăn trong việc lấy đồng phóng xạ và sự sẵn có ngày càng tăng của xét nghiệm di truyền phân tử, xét nghiệm này hiện nay ít được sử dụng.
- Hoạt độ phóng xạ huyết thanh (chủ yếu là ceruloplasmin chứa đồng phóng xạ) được đo sau khi uống đồng được đánh dấu phóng xạ (
- Chẩn đoán di truyền
- Trong các nghiên cứu gia đình, phân tích haplotype có sẵn để chẩn đoán ở anh chị em ruột của bệnh nhân đã được xác định, với tỷ lệ sai sót <1% đến 2% (sai sót có thể xảy ra với tái tổ hợp kép).
- Việc sử dụng phân tích đột biến trực tiếp của ATP7B trong chẩn đoán bệnh Wilson đã được cải thiện rõ rệt với những tiến bộ trong công nghệ giải trình tự và phân tích DNA. Xét nghiệm này hiện đã có sẵn rộng rãi tại các phòng xét nghiệm thương mại; tuy nhiên, vẫn còn những hạn chế đối với xét nghiệm.
- Chi phí phân tích vẫn còn tương đối đắt.
- Bệnh Wilson do nhiều đột biến đặc hiệu gây ra, hiện đã lên tới hơn 600. Có nhiều đa hình khác với tác động chưa rõ lên chức năng protein.
- Sàng lọc gia đình cho anh chị em ruột có thể được thực hiện với chi phí thấp hơn khi chẩn đoán bệnh Wilson đã được xác định ở một thành viên trong gia đình bằng cách sử dụng các đột biến đã biết của bệnh nhân chỉ điểm làm tham chiếu.
- Đánh giá sinh hóa vẫn có thể được thực hiện khi chẩn đoán bệnh Wilson được xác định bằng phân tích haplotype hoặc bằng phân tích đột biến để giúp mô tả kiểu hình của bệnh nhân.
HỆ THỐNG TÍNH ĐIỂM
- Một hệ thống tính điểm (tiêu chuẩn Leipzig) đã được Ferenci và các đồng nghiệp phát triển để hỗ trợ các bác sĩ lâm sàng trong việc xác định khi nào nên xem xét bệnh Wilson và giúp xác định chẩn đoán.
- Hệ thống này sử dụng một điểm số có trọng số của sự kết hợp các dấu hiệu và triệu chứng bệnh, các giá trị xét nghiệm, và cuối cùng là phân tích đột biến (Bảng 19.1). Điểm số <3 cho thấy nên xem xét một chẩn đoán khác; điểm số 3 cho thấy nên tiếp tục các xét nghiệm chẩn đoán thêm; và điểm số ≥4 xác định chẩn đoán bệnh Wilson.
- Hệ thống tính điểm này đã được đưa vào các hướng dẫn được công bố bởi Hiệp hội Nghiên cứu Gan Châu Âu (EASL) vào năm 2012.
- Một chỉ số dự đoán tử vong (thang điểm Nazer) đã được phát triển (Bảng 19.2).
TIẾP CẬN CHẨN ĐOÁN
Xem Hình 19.3.
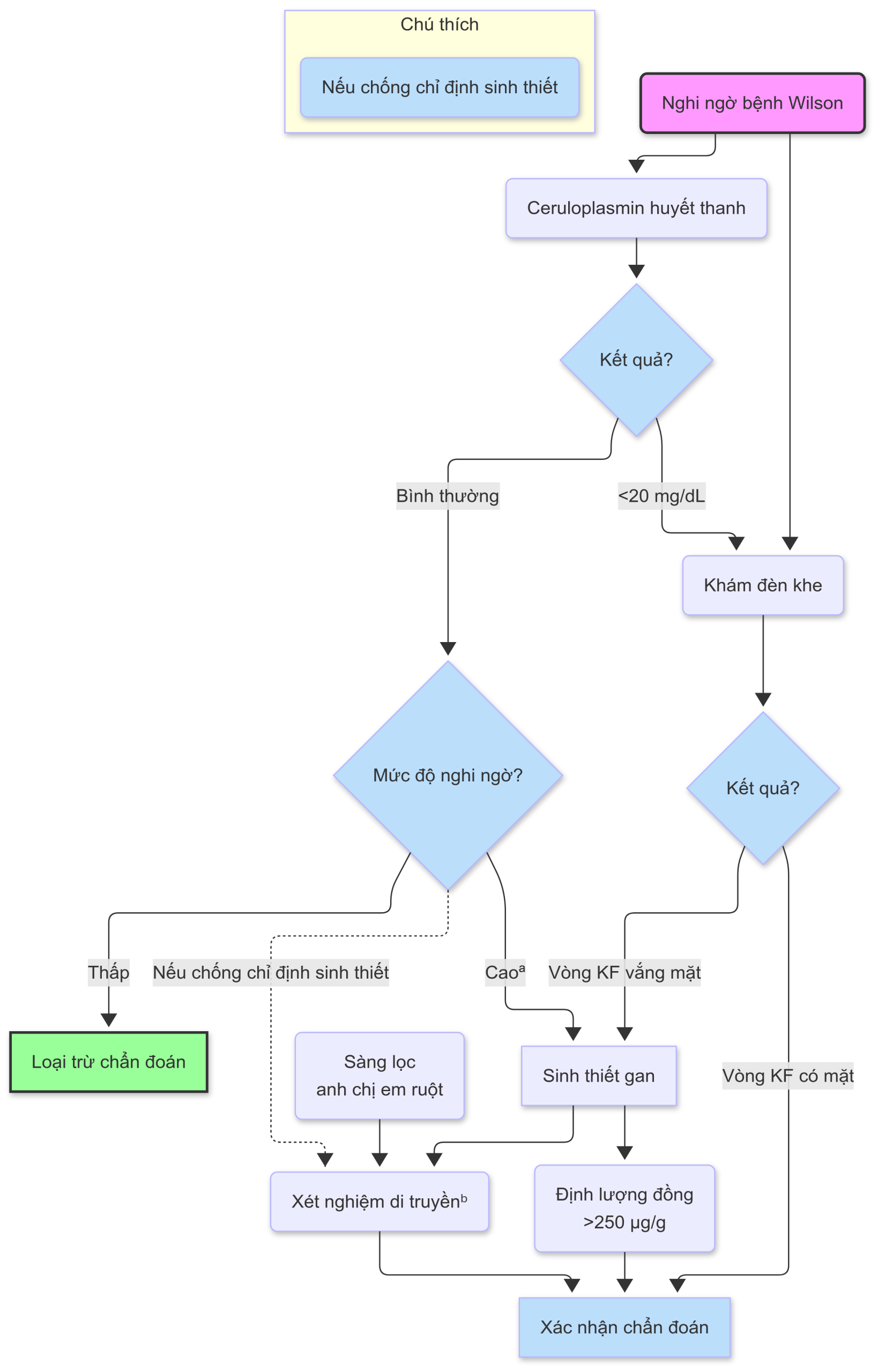
Hình 19.3 Lưu đồ chẩn đoán bệnh Wilson (WD). Trước đây, xét nghiệm đồng phóng xạ đã được sử dụng, với việc thiếu sự tích hợp của đồng phóng xạ dùng đường uống vào ceruloplasmin hỗ trợ chẩn đoán WD; xét nghiệm này hiện nay hiếm khi được sử dụng. aVí dụ: Sự hiện diện của vòng Kayser-Fleischer (KF), bài tiết đồng qua nước tiểu cao, và các triệu chứng tâm thần kinh với tổn thương gan. bXét nghiệm di truyền được thực hiện cho anh chị em ruột khi chẩn đoán đã được xác định ở một thành viên trong gia đình bằng cách sử dụng DNA của bệnh nhân chỉ điểm làm tham chiếu. Các thành viên khác trong gia đình có thể được sàng lọc bằng các nghiên cứu lâm sàng và sinh hóa.
Điều trị
CHẾ ĐỘ ĂN
- Khuyến nghị một chế độ ăn ít đồng. Nên tránh các loại thực phẩm có hàm lượng đồng cao (ví dụ: gan, sô cô la, các loại hạt, nấm, các loại đậu và động vật có vỏ).
- Danh sách các loại thực phẩm và hàm lượng đồng cũng như các chất dinh dưỡng khác có sẵn trên trang web của Bộ Nông nghiệp Hoa Kỳ.
- Sử dụng nước khử ion hoặc nước cất nếu hàm lượng đồng trong nước uống tại nhà >0.2 ppm.
THUỐC
- D-Penicillamine
- D-penicillamine là một dẫn xuất axit amin được xác định trong nước tiểu của bệnh nhân dùng penicillin.
- Cơ chế tác dụng của nó bao gồm thải sắt đồng, giải độc, và có thể là cảm ứng tổng hợp metallothionein tế bào, giúp tăng cường tỷ lệ Cu-metallothionein không độc.
- Liều khởi đầu: Khoảng 20 mg/kg chia liều hàng ngày bắt đầu bằng một phần nhỏ của liều này và tăng dần lên đủ 20 mg/kg trong khoảng 2 đến 4 tuần, với liều duy trì tiêu chuẩn là 10 đến 15 mg/kg hàng ngày; thuốc được hấp thu tốt nhất nếu uống lúc đói.
- Nên dùng liều nhỏ pyridoxine (25 mg/ngày) hàng ngày, vì tác dụng kháng pyridoxine yếu của D-penicillamine.
- Tác dụng phụ phát triển ở khoảng 20% bệnh nhân trong tháng đầu tiên điều trị, phổ biến nhất là phản ứng quá mẫn, bao gồm sốt, khó chịu, phát ban, và thỉnh thoảng là hạch to. Hầu hết bệnh nhân có thể được giải mẫn cảm đối với các triệu chứng này bằng cách sử dụng lại thuốc một cách từ từ. Ức chế tủy xương hoặc protein niệu đáng kể (>1 g/24 giờ) hoặc xấu đi thường đòi hỏi phải ngưng thuốc.
- Thuốc cũng có thể gây ra sự xấu đi đáng kể của các đặc điểm thần kinh hoặc khởi phát các đặc điểm tự miễn như nhược cơ, viêm đa cơ, hoặc lupus ban đỏ hệ thống. Nếu các tác dụng phụ này xảy ra, nên ngưng D-penicillamine và bắt đầu liệu pháp thay thế thích hợp. Các tác dụng phụ về da bao gồm pemphigus, acanthosis nigricans, và elastosis perforans serpiginosa.
- Trientine
- Trientine được giới thiệu vào năm 1969 như một tác nhân thải sắt thay thế cho D-penicillamine.
- Cơ chế tác dụng của nó bao gồm thải sắt đồng và giải độc.
- Liều hàng ngày (tương tự như penicillamine): Liều điều trị ban đầu khoảng 20 mg/kg chia liều hàng ngày bắt đầu bằng một phần nhỏ của liều này và tăng dần lên đủ 20 mg/kg trong khoảng 2 đến 4 tuần, với liều duy trì tiêu chuẩn là 10 đến 15 mg/kg hàng ngày chia liều; hấp thu tốt nhất nếu uống lúc đói.
- Dữ liệu thử nghiệm mới cho thấy thuốc này có thể được dùng một liều duy nhất hàng ngày, do đó cải thiện sự tuân thủ và giảm sự bất tiện liên quan đến việc tránh dùng thuốc cùng với bữa ăn.
- Thiếu máu nguyên hồng cầu sắt (sideroblastic anemia) là tác dụng phụ chính duy nhất của tác nhân này và xảy ra khi điều trị quá liều; các tác dụng phụ khác được báo cáo không thường xuyên bao gồm phát ban da, rối loạn tiêu hóa và viêm đại tràng, và hiếm khi là tiêu cơ vân. Hầu hết các tác dụng phụ của D-penicillamine, ngoại trừ elastosis perforans serpiginosa, sẽ giảm khi bệnh nhân được chuyển sang trientine.
- Do hồ sơ an toàn tốt hơn so với D-penicillamine, thuốc này đã được khuyến nghị là liệu pháp hàng đầu cho những bệnh nhân mắc bệnh Wilson bắt đầu bằng liệu pháp thải sắt; tuy nhiên, nó rất đắt.
- Kẽm
- Muối kẽm dùng đường uống chia liều có thể được sử dụng để điều trị bệnh Wilson. Kẽm hoạt động bằng cách cảm ứng tổng hợp metallothionein ở biểu mô ruột, do đó làm giảm hấp thu đồng ở ruột. Đồng trong tế bào ruột được bài tiết khi các tế bào biểu mô ruột bong ra, xảy ra sau mỗi 24 đến 48 giờ, do đó tạo ra sự cân bằng đồng âm tính theo thời gian.
- Kẽm tương đối an toàn; tác dụng phụ bao gồm khó chịu ở đường tiêu hóa và tăng amylase và lipase mà không có bằng chứng lâm sàng hoặc hình ảnh của viêm tụy.
- Liều: 150 mg kẽm acetate hàng ngày ở người lớn, chia thành ba liều giữa các bữa ăn; liều trẻ em: 75 mg chia thành ba liều giữa các bữa ăn.
- Vai trò của kẽm trong điều trị chủ yếu là ở bệnh Wilson tiền triệu chứng và là liệu pháp duy trì ở bệnh nhân.
- Có thể được sử dụng trong thai kỳ mà không cần giảm liều.
- Liệu pháp đơn trị bằng kẽm không được khuyến nghị là liệu pháp ban đầu cho bệnh nhân có triệu chứng, nhưng kết quả tốt đã được chứng minh ở những bệnh nhân có bệnh thần kinh chiếm ưu thế, ngay cả khi kẽm được sử dụng như liệu pháp ban đầu. Kết quả ít thỏa đáng hơn khi kẽm được sử dụng như một tác nhân duy nhất để duy trì bệnh nhân chủ yếu có bệnh gan.
- Ammonium tetrathiomolybdate (TM)
- Việc sử dụng TM vẫn còn trong giai đoạn thử nghiệm ở Hoa Kỳ. Các thử nghiệm điều trị cho bệnh nhân sử dụng một dạng ổn định của thuốc đang được tiến hành.
- Sau khi hấp thu, TM tạo thành các phức hợp không độc với đồng huyết thanh và albumin và ngăn chặn sự hấp thu đồng của mô.
- Vì ái lực của nó với đồng cao hơn so với metallothionein, TM có thể loại bỏ đồng gắn với metallothionein, do đó có khả năng làm cho nó trở thành một chất thải sắt mạnh hơn D-penicillamine và trientine.
- Các thử nghiệm lâm sàng cũ hơn sử dụng một dạng TM kém ổn định hơn cho thấy nó có thể hiệu quả hơn trong việc ngăn ngừa sự suy giảm thần kinh so với penicillamine hoặc trientine trong quá trình điều trị ban đầu cho bệnh nhân có bệnh thần kinh.
- Các tác dụng phụ tiềm ẩn của TM dường như liên quan đến liều và có thể hồi phục; chúng bao gồm ức chế tủy xương và các mức xét nghiệm sinh hóa gan bất thường.
PHÁC ĐỒ ƯU TIÊN
- Điều trị ban đầu
- Cần xác định đồng nước tiểu 24 giờ, đồng huyết thanh, ceruloplasmin, công thức máu với tiểu cầu, tỷ lệ chuẩn hóa quốc tế (INR), xét nghiệm sinh hóa gan và phân tích nước tiểu cơ bản.
- D-penicillamine hoặc trientine nên được bắt đầu với liều chia nhỏ ở mức 25% đến 50% liều mục tiêu ban đầu là 20 mg/kg trong 2 đến 4 tuần với sự theo dõi.
- Bệnh nhân không dung nạp D-penicillamine có thể được điều trị bằng trientine hoặc kẽm.
- Bằng chứng về sự cải thiện chức năng tổng hợp của gan hoặc các triệu chứng thần kinh và tâm thần thường bắt đầu trong vòng 6 đến 12 tháng điều trị không gián đoạn.
- Những bệnh nhân suy gan nặng không đáp ứng với liệu pháp dược lý, thường được định nghĩa là 3 tháng điều trị, nếu có thể, với thang điểm Nazer đã sửa đổi (dựa trên bilirubin huyết thanh, aspartate aminotransferase huyết thanh, tỷ lệ chuẩn hóa quốc tế, số lượng bạch cầu và albumin huyết thanh; phạm vi từ 0 đến 20) từ 10 trở lên, nên được xem xét để ghép gan (LT) (xem Bảng 19.2 và thảo luận sau trong chương).
- Điều trị duy trì
- Khi các triệu chứng và dấu hiệu lâm sàng đã ổn định, bài tiết đồng qua nước tiểu đang giảm so với giá trị ban đầu, và đồng không gắn ceruloplasmin giảm xuống <15 µg/dL, liều của các tác nhân thải sắt nên được giảm xuống mức duy trì.
- Sự sụt giảm đồng không gắn ceruloplasmin xuống <5 µg/dL có thể cho thấy sự suy kiệt đồng nghiêm trọng, có thể dẫn đến ức chế tủy xương với thiếu máu và giảm tiểu cầu và thậm chí dẫn đến giảm hoạt động ferroxidase với sự tích tụ sắt độc hại trong gan; trong tình huống này, tác nhân thải sắt hoặc kẽm nên được tạm dừng hoặc giảm liều.
- Điều trị cho bệnh nhân Wilson không triệu chứng, được chẩn đoán bằng cách sàng lọc các thành viên trong gia đình, có thể được bắt đầu sớm nhất là từ 2 đến 3 tuổi.
- Điều trị suốt đời không gián đoạn là cần thiết ở tất cả các bệnh nhân mắc bệnh Wilson; việc ngừng điều trị có thể dẫn đến suy giảm gan và thần kinh nhanh chóng và không thể hồi phục.
- Mang thai
- Nên tiếp tục điều trị trong suốt thai kỳ.
- Tính gây quái thai đã được báo cáo trong các nghiên cứu trên động vật với liều dược lý của D-penicillamine và trientine; tuy nhiên, các trường hợp mang thai thành công đã được báo cáo khi dùng các loại thuốc này. Kẽm cũng an toàn trong thai kỳ.
- Để giảm nguy cơ gây quái thai, ở những bệnh nhân được duy trì tốt bằng liệu pháp thải sắt, liều D-penicillamine hoặc trientine nên được giảm 50% trong thời gian dự kiến thụ thai và trong suốt thai kỳ. Bệnh nhân nên được theo dõi thường xuyên hơn, khoảng ba tháng một lần hoặc trong mỗi tam cá nguyệt nếu có thai, cho đến sau khi sinh.
- Liệu pháp duy trì bằng kẽm không cần điều chỉnh liều trong thai kỳ.
- Bệnh thần kinh
- Khoảng 20% bệnh nhân có biểu hiện bệnh thần kinh có triệu chứng thần kinh xấu đi trong quá trình điều trị ban đầu với D-penicillamine hoặc trientine; điều này rất có thể là do sự huy động và tái phân phối đồng trong não trong quá trình điều trị ban đầu. Việc tăng liều thuốc từ từ có thể làm giảm tần suất của biến chứng này.
- Có thể cần giảm liều hoặc ngưng điều trị thải sắt nếu tình trạng thần kinh xấu đi. Bệnh nhân có thể được cho dùng kẽm trong thời gian này.
- Ngoài ra, liệu pháp kết hợp với kẽm và liều thấp hơn của tác nhân thải sắt được dùng cách nhau về mặt thời gian có thể là một phương pháp điều trị thay thế cho những bệnh nhân này nhưng chưa được thử nghiệm trong các thử nghiệm lâm sàng.
- Các thử nghiệm lâm sàng đang đánh giá việc sử dụng TM cho những bệnh nhân có biểu hiện ban đầu là bệnh thần kinh. Một thử nghiệm đã cho thấy lợi ích của TM so với trientine trong việc ngăn ngừa sự xấu đi ban đầu của bệnh thần kinh trong quá trình bắt đầu điều trị.
- Các báo cáo ca bệnh về việc sử dụng liệu pháp kết hợp với một tác nhân thải sắt (D-penicillamine hoặc trientine) được dùng cách nhau về mặt thời gian với kẽm được cho dùng chia liều cho thấy kết quả đáng khích lệ ở những bệnh nhân có triệu chứng và ở những người bị bệnh gan nặng. Ở một số bệnh nhân có điểm Child-Turcotte-Pugh cao (xem Chương 32), sự cải thiện về lâm sàng và sinh hóa đã loại bỏ nhu cầu ghép gan.
GHÉP GAN (xem thêm Chương 33)
- Ghép gan (LT) là phương pháp chữa khỏi bệnh Wilson vì khiếm khuyết của bệnh nằm trong các tế bào gan. Sự đảo ngược hoàn toàn khiếm khuyết chuyển hóa đồng được quan sát thấy, cũng như sự cải thiện các biểu hiện ở gan và thần kinh ở hầu hết các bệnh nhân.
- Phần lớn người lớn được ghép gan vì bệnh Wilson bị suy gan mạn tính. Suy gan cấp là một chỉ định phổ biến hơn ở trẻ em. Trong một phân tích về ghép gan trong cơ sở dữ liệu của Hoa Kỳ về cấy ghép từ Mạng lưới Chia sẻ Nội tạng Thống nhất (UNOS) từ năm 1997 đến 2008, 400 người lớn và 170 trẻ em đã được ghép gan vì bệnh Wilson. Tỷ lệ sống sót của bệnh nhân và mảnh ghép đối với bệnh nhân người lớn và trẻ em được ghép gan vì bệnh Wilson là tuyệt vời. Tỷ lệ sống sót sau 1 và 5 năm, tương ứng, là 90% và 89% đối với trẻ em, so với 88% và 86% đối với người lớn.
- Bệnh Wilson ở bệnh nhân suy gan cấp có thể được xác định bằng cách sử dụng các xét nghiệm sinh hóa đơn giản và công thức máu ngay cả trước khi có kết quả xét nghiệm đồng trong máu, nước tiểu và mô gan. Bệnh nhân suy gan cấp do bệnh Wilson có giá trị phosphatase kiềm tương đối thấp và giảm dần khi suy gan tiến triển. Bởi vì những bệnh nhân này bị tan máu và bilirubin tăng nhanh chóng, tỷ lệ phosphatase kiềm so với bilirubin huyết thanh thường <4, và tỷ lệ AST so với ALT >2. Nếu bệnh nhân có hai thông số sinh hóa này và nồng độ hemoglobin thấp do tan máu, độ nhạy và độ đặc hiệu cho chẩn đoán bệnh Wilson là nguyên nhân gây suy gan cấp gần như 100%.
- Bệnh nhân suy gan cấp do bệnh Wilson nên được chuyển ngay để ghép gan. Các biện pháp để ổn định bệnh nhân được hướng đến việc hạ thấp nồng độ đồng tăng rõ rệt trong tuần hoàn. Điều này đã được thực hiện bằng một số phương tiện, bao gồm thay máu, trao đổi huyết tương kết hợp với lọc máu, lọc máu bằng albumin, và hệ thống tái tuần hoàn hấp phụ phân tử (MARS) (xem Chương 2). Chỉ một số ít bệnh nhân Wilson bị suy gan cấp sống sót mà không cần ghép gan, và các biện pháp trước đó chỉ nhằm ổn định bệnh nhân trong khi chờ đợi một cơ quan mới nhưng thường không đủ để hỗ trợ cho gan tái tạo.
- Các nghiên cứu về lịch sử tự nhiên của bệnh nhân Wilson gan trong thời đại trước ghép gan đã dẫn đến sự phát triển của thang điểm Nazer (xem Bảng 19.2 và thảo luận trước đó trong chương). Thang điểm này sau đó đã được sửa đổi để bao gồm các thông số khác về suy gan và viêm hệ thống. Những bệnh nhân có điểm trên 10 không sống sót nếu không có ghép gan, trong khi những người có điểm thấp hơn đã cải thiện bằng liệu pháp nội khoa. Mặc dù thang điểm tiên lượng không hoàn hảo trong việc dự đoán kết quả điều trị cho tất cả các bệnh nhân, nhưng nó là một hướng dẫn hữu ích.
- Trong trường hợp không có bệnh gan nặng, ghép gan cho các biểu hiện thần kinh kháng trị vẫn nên được coi là thử nghiệm; tuy nhiên, một số báo cáo đã ghi nhận sự cải thiện ở những bệnh nhân này sau khi ghép gan. Những bệnh nhân bị tổn thương thần kinh nặng không phải lúc nào cũng cải thiện sau khi ghép gan, và kết quả sau ghép kém hơn đã được báo cáo về tỷ lệ sống sót và các biến chứng do sử dụng các chất ức chế calcineurin để ngăn ngừa thải ghép.
CÁC LIỆU PHÁP TRONG TƯƠNG LAI
- Việc xác định gen ATP7B và cơ chế của bệnh sẽ làm cho các liệu pháp dựa trên phân tử, bao gồm sửa chữa gen và liệu pháp gen, trở nên khả thi.
- Một báo cáo về việc chuyển gen ATP7B qua trung gian virus liên quan đến adeno với sự biểu hiện lâu dài của protein trong một mô hình động vật của bệnh Wilson là đáng khích lệ và sẽ dẫn đến các thử nghiệm lâm sàng trong tương lai.
- Một liệu pháp tiềm năng khác là cấy ghép tế bào gan, đã được áp dụng trong mô hình động vật của bệnh Wilson, chuột Long Evans Cinnamon (LEC), và một mô hình chuột thiếu ATP7B, trong đó đã đạt được sự điều chỉnh chuyển hóa hoàn toàn. Hạn chế của phương pháp này đối với các thử nghiệm trên người là nhu cầu đạt được sự tái sinh đủ tế bào với các tế bào bình thường được cấy ghép và nhu cầu ức chế miễn dịch để ngăn ngừa sự thải loại các tế bào được cấy ghép.
Các Rối loạn Liên quan đến Đồng khác
XƠ GAN TRẺ EM ẤN ĐỘ (INDIAN CHILDHOOD CIRRHOSIS)
- Việc hấp thụ quá nhiều đồng từ môi trường, do sử dụng các bình chứa bằng đồng và đồng thau, là nguyên nhân có khả năng gây ra quá tải đồng trong rối loạn này, mặc dù một khuynh hướng di truyền dường như cùng tồn tại ở một số bệnh nhân.
- Xơ gan tiến triển nhanh chóng biểu hiện ở độ tuổi từ 6 tháng đến 5 tuổi; rối loạn này thường chỉ giới hạn ở tiểu lục địa Ấn Độ.
- Nồng độ đồng trong gan, nước tiểu và huyết thanh tăng rõ rệt được ghi nhận.
- Thực thể này từng là một nguyên nhân phổ biến của bệnh gan mạn tính ở Ấn Độ, nhưng hiện nay hiếm khi được thấy do giáo dục sức khỏe và tránh sử dụng các bình chứa bằng đồng thau.
NGỘ ĐỘC ĐỒNG VÔ CĂN (IDIOPATHIC COPPER TOXICOSIS)
- Đây là một rối loạn hiếm gặp, với các trường hợp lẻ tẻ xảy ra trên toàn thế giới.
- Xơ gan nặng, tiến triển xảy ra trong trường hợp không có bệnh thần kinh, với khởi phát lâm sàng thường vào 2 tuổi.
- Nồng độ ceruloplasmin huyết thanh bình thường; các mẫu sinh thiết gan cho thấy xơ gan với thể Mallory-Denk và nồng độ đồng trong gan >400 µg/g trọng lượng khô.
- Rối loạn này có thể do một khiếm khuyết di truyền chưa được xác định hoặc phơi nhiễm với đồng từ môi trường quá mức (ví dụ, nước suối bị ô nhiễm trong bệnh xơ gan trẻ sơ sinh Tyrolean lưu hành).
BỆNH MENKES (MENKES DISEASE)
- Bệnh Menkes là một rối loạn thoái hóa thần kinh di truyền lặn liên kết với nhiễm sắc thể X, thường biểu hiện ở <3 tháng tuổi và gây tử vong trong hầu hết các trường hợp vào 3 đến 6 tuổi.
- Nó được gây ra bởi các đột biến trong gen ATP7A, mã hóa cho một ATPase p-type, tương đồng với ATP7B, gây ra suy giảm vận chuyển đồng qua nhau thai, ruột và hàng rào máu não và do đó dẫn đến tình trạng thiếu đồng nghiêm trọng (với hoạt động thiếu hụt của các cuproenzyme thiết yếu).
- Trẻ sơ sinh bị ảnh hưởng có thể phát triển giảm trương lực cơ, co giật và chậm phát triển.
- Chức năng suy giảm của enzyme phụ thuộc đồng lysyl oxidase dẫn đến giảm liên kết chéo collagen và “tóc xoắn” (kinky hair) đặc trưng. Các đặc điểm khác là giảm sắc tố, loãng xương và động mạch ngoằn ngoèo.
- Sàng lọc sơ sinh không được thực hiện thường quy. Về mặt thực nghiệm, tỷ lệ dopamine/norepinephrine cao cũng như axit dihydroxyphenylacetic/dihydroxyphenylglycol có thể xác định bệnh nhân trước khi các triệu chứng nặng khởi phát. Cuối cùng, các dấu ấn huyết thanh về đồng và ceruloplasmin thấp là dấu hiệu của tình trạng thiếu đồng tương đối. Có thể thực hiện xét nghiệm di truyền phân tử cho các đột biến ATP7A, nhưng có sự đa dạng di truyền và không phải tất cả các đột biến đều được mô tả rõ.
- Điều trị bằng cách tiêm đồng histidine hàng ngày, nếu được bắt đầu sớm trong quá trình bệnh, có thể cải thiện một phần kết quả. Các thử nghiệm liệu pháp gen trên các mô hình động vật đang được tiến hành.
BẢNG BIỂU
Bảng 19.1: Tiêu chuẩn Leipzig để Chẩn đoán Bệnh Wilson
| Dấu hiệu và Triệu chứng Lâm sàng Điển hình | Điểm | Các Xét nghiệm khác | Điểm |
|---|---|---|---|
| Vòng Kayser-Fleischer: | Đồng trong gan (khi không có ứ mật): | ||
| Có mặt | 2 | >5 lần giới hạn trên của mức bình thường (ULN) (>4 µmol/g) | 2 |
| Vắng mặt | 0 | 0.8-4 µmol/g | 1 |
| Bình thường (<0.8 µmol/g). Các hạt dương tính với Rhodanine | 1 | ||
| Triệu chứng thần kinh: | Đồng trong nước tiểu (khi không có viêm gan cấp): | ||
| Nặng | 2 | Bình thường | 0 |
| Nhẹ | 1 | 1-2 lần ULN | 1 |
| Vắng mặt | 0 | >2 lần ULN. Bình thường, nhưng >5 lần ULN sau khi dùng D-penicillamine | 2 |
| Ceruloplasmin huyết thanh: | Phân tích đột biến: | ||
| Bình thường (>0.2 g/L) | 0 | Phát hiện đột biến trên cả hai nhiễm sắc thể | 4 |
| 0.1-0.2 g/L | 1 | Phát hiện đột biến trên 1 nhiễm sắc thể | 1 |
| <0.1 g/L | 2 | Không phát hiện đột biến | 0 |
| Thiếu máu tan máu không qua trung gian miễn dịch: | |||
| Có mặt | 1 | ||
| Vắng mặt | 0 | ||
| Tổng điểm | Diễn giải | ||
| ≥4 | Chẩn đoán xác định | ||
| 3 | Chẩn đoán có thể, cần thêm xét nghiệm | ||
| <2 | Chẩn đoán khó có khả năng | ||
| Chú thích: a nếu không có định lượng đồng trong gan, b hoặc các bất thường điển hình trên chụp cộng hưởng từ não. ULN, Giới hạn trên của mức bình thường. |
Bảng 19.2: Thang điểm Nazer sửa đổi để Dự đoán Tỷ lệ Tử vong trong Bệnh Wilson
| Điểm | Bilirubin (µmol/L) | INR | AST (U/L) | WBC (10⁹/L) | Albumin (g/L) |
|---|---|---|---|---|---|
| 0 | 0-100 | 0-1.29 | 0-100 | 0-6.7 | >45 |
| 1 | 101-150 | 1.3-1.6 | 101-150 | 6.8-8.3 | 34-44 |
| 2 | 151-200 | 1.7-1.9 | 151-300 | 8.4-10.3 | 25-33 |
| 3 | 201-300 | 2.0-2.4 | 301-400 | 10.4-15.3 | 21-24 |
| 4 | >300 | >2.5 | >400 | >15.4 | <20 |
| Chú thích: Bệnh nhân có tổng điểm ≥10 nên được xem xét để ghép gan. AST, Aspartate aminotransferase; INR, tỷ lệ chuẩn hóa quốc tế; WBC, số lượng bạch cầu. |
TÀI LIỆU THAM KHẢO
- Ala A, Aliu E, Schilsky ML. Prospective pilot study of a single daily dosage of trientine for the treatment of Wilson disease. Dig Dis Sci. 2015;60:1433-1439.
- Beinhardt S, Leiss W, Stättermayer AF, et al. Long-term outcomes of patients with Wilson disease in a large Austrian cohort. Clin Gastroenterol Hepatol. 2014;12:683-689.
- Brewer GJ, Askari F, Lorincz MT, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol. 2006;63:521-527.
- Ferenci P, Członkowska A, Stremmel W, et al. EASL clinical practice guidelines: Wilson’s disease. European Association for Study of Liver. J Hepatol. 2012;56:671-685.
- Kaler SG. Translational research investigations on ATP7A: an important human copper ATPase. Ann NY Acad Sci. 2014;1314:64-68.
- Koppikar S, Dhawan A. Evaluation of the scoring system for the diagnosis of Wilson’s disease in children. Liver Int. 2005;25:680-681.
- Korman JD, Volenberg I, Balko J, et al. Screening for Wilson disease in acute liver failure by serum testing: a comparison of currently used tests. Hepatology. 2008;48:1167-1174.
- Murillo O, Luqui DM, Gazquez C, et al. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy. J Hepatol. 2016;64:419-426.
- Roberts E, Schilsky ML. A practice guideline on Wilson disease. Hepatology. 2008;47:2089-2111.
- Schilsky ML. Liver transplantation for Wilson disease. Ann NY Acad Sci. 2014;1315:45-49.
- Zimbrean PC, Schilsky ML. The spectrum of psychiatric symptoms in Wilson’s disease: treatment and prognostic considerations. Am J Psychiatry. 2015;172:1068-1072.
BẢNG CHÚ GIẢI THUẬT NGỮ Y HỌC ANH-VIỆT
| STT | Thuật ngữ tiếng Anh | Phiên âm IPA | Nghĩa Tiếng Việt |
|---|---|---|---|
| 1 | Wilson disease (WD) | /ˈwɪlsən dɪˌziːz/ | Bệnh Wilson (WD) |
| 2 | autosomal recessive disorder | /ˌɔːtəʊˈsəʊməl rɪˈsɛsɪv dɪsˈɔːdə/ | Rối loạn di truyền lặn trên nhiễm sắc thể thường |
| 3 | copper metabolism | /ˈkɒpə mɛˈtæbəlɪzəm/ | Chuyển hóa đồng |
| 4 | ATP7B gene | /eɪ-tiː-piː ˈsɛvən biː dʒiːn/ | Gen ATP7B |
| 5 | copper-transporting P-type adenosine triphosphatase (ATPase) | /ˈkɒpə trænsˈpɔːtɪŋ piː-taɪp əˈdɛnəʊsiːn traɪˈfɒsfəteɪz/ | P-type adenosine triphosphatase (ATPase) vận chuyển đồng |
| 6 | trans-Golgi network | /træns-ˈɡɒldʒi ˈnɛtwɜːk/ | Mạng lưới trans-Golgi |
| 7 | hepatocytes | /hɛˈpætəʊsaɪts/ | Tế bào gan |
| 8 | biliary excretion | /ˈbɪliəri ɛksˈkriːʃən/ | Bài tiết qua mật |
| 9 | hepatic copper | /hɪˈpætɪk ˈkɒpə/ | Đồng ở gan |
| 10 | secondary organ injury | /ˈsɛkəndəri ˈɔːɡən ˈɪndʒəri/ | Tổn thương cơ quan thứ phát |
| 11 | ceruloplasmin | /səˌruːləʊˈplæzmɪn/ | Ceruloplasmin |
| 12 | phenotypic marker | /ˌfiːnəʊˈtɪpɪk ˈmɑːkə/ | Dấu ấn kiểu hình |
| 13 | symptomatic WD | /ˌsɪmptəˈmætɪk dʌbəljuː diː/ | Bệnh Wilson có triệu chứng |
| 14 | neuropsychiatric features | /ˌnjʊərəʊˌsaɪkiˈætrɪk ˈfiːtʃəz/ | Đặc điểm tâm thần kinh |
| 15 | hepatic disease | /hɪˈpætɪk dɪˈziːz/ | Bệnh gan |
| 16 | asymptomatic | /ˌeɪsɪmptəˈmætɪk/ | Không triệu chứng |
| 17 | chronic hepatitis | /ˈkrɒnɪk ˌhɛpəˈtaɪtɪs/ | Viêm gan mạn tính |
| 18 | cirrhosis | /sɪˈrəʊsɪs/ | Xơ gan |
| 19 | acute liver failure (ALF) | /əˈkjuːt ˈlɪvə ˈfeɪljə/ | Suy gan cấp (ALF) |
| 20 | biochemical testing | /ˌbaɪəʊˈkɛmɪkəl ˈtɛstɪŋ/ | Xét nghiệm sinh hóa |
| 21 | molecular testing | /məˈlɛkjʊlə ˈtɛstɪŋ/ | Xét nghiệm phân tử |
| 22 | family screening | /ˈfæməli ˈskriːnɪŋ/ | Sàng lọc gia đình |
| 23 | first-degree relatives | /fɜːst dɪˈɡriː ˈrɛlətɪvz/ | Người thân thế hệ thứ nhất |
| 24 | index patient | /ˈɪndɛks ˈpeɪʃənt/ | Bệnh nhân chỉ điểm |
| 25 | copper chelation therapy | /ˈkɒpə kiːˈleɪʃən ˈθɛrəpi/ | Liệu pháp thải đồng |
| 26 | D-penicillamine | /diː-ˌpɛnɪˈsɪləmiːn/ | D-penicillamine |
| 27 | trientine | /ˈtraɪəntiːn/ | Trientine |
| 28 | maintenance therapy | /ˈmeɪntənəns ˈθɛrəpi/ | Liệu pháp duy trì |
| 29 | zinc salts | /zɪŋk sɔːlts/ | Muối kẽm |
| 30 | liver transplantation (LT) | /ˈlɪvə ˌtrænsplɑːnˈteɪʃən/ | Ghép gan (LT) |
| 31 | decompensated cirrhosis | /ˌdiːkɒmpɛnˈseɪtɪd sɪˈrəʊsɪs/ | Xơ gan mất bù |
| 32 | dietary copper | /ˈdaɪətəri ˈkɒpə/ | Đồng trong chế độ ăn |
| 33 | proximal small intestinal epithelial cells | /ˈprɒksɪməl smɔːl ɪnˈtɛstɪnl ˌɛpɪˈθiːliəl sɛlz/ | Tế bào biểu mô ruột non đoạn gần |
| 34 | copper transporter CTR1 | /ˈkɒpə trænsˈpɔːtə siː-tiː-ɑː wʌn/ | Chất vận chuyển đồng CTR1 |
| 35 | Menkes disease protein ATP7A | /ˈmɛnkɪz dɪˈziːz ˈprəʊtiːn eɪ-tiː-piː ˈsɛvən eɪ/ | Protein bệnh Menkes ATP7A |
| 36 | portal circulation | /ˈpɔːtl ˌsɜːkjʊˈleɪʃən/ | Tuần hoàn cửa |
| 37 | serum albumin | /ˈsɪərəm ˈælbjʊmɪn/ | Albumin huyết thanh |
| 38 | amino acids | /əˈmiːnəʊ ˈæsɪdz/ | Axit amin |
| 39 | intraepithelial copper | /ˌɪntrəˌɛpɪˈθiːliəl ˈkɒpə/ | Đồng trong tế bào biểu mô |
| 40 | endogenous chelating peptide | /ɛnˈdɒdʒɪnəs ˈkiːleɪtɪŋ ˈpɛptaɪd/ | Peptide thải độc nội sinh |
| 41 | metallothionein | /mɛˌtæləʊˈθaɪəʊniːn/ | Metallothionein |
| 42 | enterohepatic circulation | /ˌɛntərəʊhɪˈpætɪk ˌsɜːkjʊˈleɪʃən/ | Chu trình gan-ruột |
| 43 | kidney | /ˈkɪdni/ | Thận |
| 44 | bile | /baɪl/ | Mật |
| 45 | glutathione | /ˌɡluːtəˈθaɪəʊn/ | Glutathione |
| 46 | cofactor | /ˌkəʊˈfæktə/ | Đồng yếu tố |
| 47 | cellular enzymes | /ˈsɛljʊlər ˈɛnzaɪmz/ | Enzyme tế bào |
| 48 | Golgi apparatus | /ˈɡɒldʒi ˌæpəˈreɪtəs/ | Bộ máy Golgi |
| 49 | copper chaperones | /ˈkɒpə ˈʃæpərəʊnz/ | Chaperone đồng |
| 50 | canalicular membrane | /ˌkænəˈlɪkjʊlə ˈmɛmbreɪn/ | Màng tiểu quản mật |
| 51 | canalicular and biliary copper excretion | /ˌkænəˈlɪkjʊlər ænd ˈbɪliəri ˈkɒpər ɛksˈkriːʃən/ | Bài tiết đồng qua tiểu quản mật và đường mật |
| 52 | copper-glutathione | /ˈkɒpə ˌɡluːtəˈθaɪəʊn/ | Đồng-glutathione |
| 53 | basolateral plasma membrane | /ˌbeɪsəʊˈlætərəl ˈplæzmə ˈmɛmbreɪn/ | Màng sinh chất đáy bên |
| 54 | chromosome 13 | /ˈkrəʊməsəʊm θɜːˈtiːn/ | Nhiễm sắc thể 13 |
| 55 | red cell enzyme, esterase D | /rɛd sɛl ˈɛnzaɪm, ˈɛstəreɪz diː/ | Enzyme hồng cầu, esterase D |
| 56 | transcript | /ˈtrænskrɪpt/ | Bản phiên mã |
| 57 | placenta | /pləˈsɛntə/ | Nhau thai |
| 58 | cation-transporting P-type ATPase | /ˈkætaɪən trænsˈpɔːtɪŋ piː-taɪp eɪ-tiː-piː-eɪz/ | ATPase vận chuyển cation P-type |
| 59 | Enterococcus hirae | /ˌɛntərəʊˈkɒkəs ˈhaɪəriː/ | Enterococcus hirae |
| 60 | missense mutations | /ˈmɪsɛns mjuːˈteɪʃənz/ | Đột biến sai nghĩa |
| 61 | homozygous | /ˌhəʊməʊˈzaɪɡəs/ | Đồng hợp tử |
| 62 | compound heterozygotes | /ˈkɒmpaʊnd ˌhɛtərəʊˈzaɪɡəʊts/ | Dị hợp tử phức hợp |
| 63 | allele | /əˈliːl/ | Alen |
| 64 | clinical diversity | /ˈklɪnɪkəl daɪˈvɜːsəti/ | Đa dạng lâm sàng |
| 65 | allelic heterogeneity | /əˈliːlɪk ˌhɛtərəʊdʒəˈniːəti/ | Không đồng nhất của các alen |
| 66 | subclinical abnormalities | /ˌsʌbˈklɪnɪkəl ˌæbnɔːˈmælətiz/ | Bất thường cận lâm sàng |
| 67 | heterozygotes | /ˌhɛtərəʊˈzaɪɡəʊts/ | Người mang gen dị hợp tử |
| 68 | pathogenesis | /ˌpæθəʊˈdʒɛnəsɪs/ | Cơ chế bệnh sinh |
| 69 | copper homeostasis | /ˈkɒpə ˌhəʊmiəʊˈsteɪsɪs/ | Cân bằng nội môi đồng |
| 70 | gastrointestinal absorption | /ˌɡæstrəʊɪnˈtɛstɪnl əbˈsɔːpʃən/ | Hấp thu ở đường tiêu hóa |
| 71 | vesicular component | /vɪˈsɪkjʊlə kəmˈpəʊnənt/ | Thành phần túi |
| 72 | free radical-induced oxidative injury | /friː ˈrædɪkəl ɪnˈdjuːst ˈɒksɪdətɪv ˈɪndʒəri/ | Tổn thương oxy hóa do gốc tự do gây ra |
| 73 | lipids | /ˈlɪpɪdz/ | Lipid |
| 74 | proteins | /ˈprəʊtiːnz/ | Protein |
| 75 | nucleic acids | /njuːˈkliːɪk ˈæsɪdz/ | Axit nucleic |
| 76 | antioxidants | /ˌæntiˈɒksɪdənts/ | Chất chống oxy hóa |
| 77 | polymerization | /pəˌlɪmərаɪˈzeɪʃən/ | Sự trùng hợp |
| 78 | hepatocellular necrosis | /hɛˌpætəʊˈsɛljʊlə nɛˈkrəʊsɪs/ | Hoại tử tế bào gan |
| 79 | apoptosis | /ˌæpəpˈtəʊsɪs/ | Chết theo chương trình |
| 80 | mitochondria | /ˌmaɪtəʊˈkɒndrɪə/ | Ty thể |
| 81 | cristae | /ˈkrɪstiː/ | Mào (ty thể) |
| 82 | crystalline deposits | /ˈkrɪstəlaɪn dɪˈpɒzɪts/ | Lắng đọng tinh thể |
| 83 | non-ceruloplasmin-bound copper | /nɒn-səˌruːləʊˈplæzmɪn baʊnd ˈkɒpə/ | Đồng không gắn ceruloplasmin |
| 84 | extrahepatic copper accumulation | /ˌɛkstrəhɪˈpætɪk ˈkɒpər əˌkjuːmjʊˈleɪʃən/ | Tích tụ đồng ngoài gan |
| 85 | copper toxicity | /ˈkɒpə tɒkˈsɪsəti/ | Độc tính của đồng |
| 86 | central nervous system | /ˈsɛntrəl ˈnɜːvəs ˈsɪstəm/ | Hệ thần kinh trung ương |
| 87 | caudate nucleus | /ˈkɔːdeɪt ˈnjuːklɪəs/ | Nhân đuôi |
| 88 | putamen | /pjuːˈteɪmən/ | Nhân bèo |
| 89 | basal ganglia | /ˈbeɪzəl ˈɡæŋɡlɪə/ | Hạch nền |
| 90 | Descemet membrane | /dəˈseɪmeɪ ˈmɛmbreɪn/ | Màng Descemet |
| 91 | cornea | /ˈkɔːnɪə/ | Giác mạc |
| 92 | Kayser-Fleischer (KF) rings | /ˈkaɪzə ˈflaɪʃə rɪŋz/ | Vòng Kayser-Fleischer (KF) |
| 93 | sunflower cataracts | /ˈsʌnˌflaʊə ˈkætərækts/ | Đục thủy tinh thể hình hoa hướng dương |
| 94 | aceruloplasmin | /əˌsɪərʊləʊˈplæzmɪn/ | Aceruloplasmin (ceruloplasmin không có đồng) |
| 95 | holoceruloplasmin | /ˌhəʊləʊsəˌruːləʊˈplæzmɪn/ | Holoceruloplasmin (ceruloplasmin có gắn đồng) |
| 96 | renal symptoms | /ˈriːnl ˈsɪmptəmz/ | Triệu chứng ở thận |
| 97 | skeletal symptoms | /ˈskɛlɪtl ˈsɪmptəmz/ | Triệu chứng ở xương khớp |
| 98 | cardiac symptoms | /ˈkɑːdɪæk ˈsɪmptəmz/ | Triệu chứng ở tim |
| 99 | ophthalmologic symptoms | /ˌɒfθælˈmɒlədʒɪk ˈsɪmptəmz/ | Triệu chứng ở mắt |
| 100 | endocrinologic symptoms | /ˌɛndəʊˌkrɪnəˈlɒdʒɪk ˈsɪmptəmz/ | Triệu chứng nội tiết |
| 101 | dermatologic symptoms | /ˌdɜːmətəˈlɒdʒɪk ˈsɪmptəmz/ | Triệu chứng ở da |
| 102 | hematologic manifestations | /ˌhiːmətəˈlɒdʒɪk ˌmænɪfɛsˈteɪʃənz/ | Biểu hiện huyết học |
| 103 | hepatic steatosis | /hɪˈpætɪk stiːəˈtəʊsɪs/ | Gan nhiễm mỡ |
| 104 | autoimmune hepatitis | /ˌɔːtəʊɪˈmjuːn ˌhɛpəˈtaɪtɪs/ | Viêm gan tự miễn |
| 105 | fibrosis | /faɪˈbrəʊsɪs/ | Xơ hóa |
| 106 | portal hypertension | /ˈpɔːtl ˌhaɪpəˈtɛnʃən/ | Tăng áp lực tĩnh mạch cửa |
| 107 | hepatocellular carcinoma | /hɛˌpætəʊˈsɛljʊlər ˌkɑːsɪˈnəʊmə/ | Ung thư biểu mô tế bào gan |
| 108 | viral-induced massive hepatic necrosis | /ˈvaɪrəl ɪnˈdjuːst ˈmæsɪv hɪˈpætɪk nɛˈkrəʊsɪs/ | Hoại tử gan lan tỏa do virus |
| 109 | serum aminotransferase | /ˈsɪərəm əˌmiːnəʊˈtrænsfəreɪz/ | Aminotransferase huyết thanh |
| 110 | serum bilirubin | /ˈsɪərəm ˌbɪlɪˈruːbɪn/ | Bilirubin huyết thanh |
| 111 | serum alkaline phosphatase | /ˈsɪərəm ˈælkəlaɪn ˈfɒsfəteɪz/ | Phosphatase kiềm huyết thanh |
| 112 | Coombs-negative hemolytic anemia | /kuːmz-ˈnɛɡətɪv ˌhiːməˈlɪtɪk əˈniːmɪə/ | Thiếu máu tan máu Coombs âm tính |
| 113 | coagulopathy | /kəʊˌæɡjʊˈlɒpəθi/ | Rối loạn đông máu |
| 114 | nonimmune hemolysis | /nɒnɪˈmjuːn hiːˈmɒlɪsɪs/ | Tan máu không do miễn dịch |
| 115 | splenomegaly | /ˌspliːnəʊˈmɛɡəli/ | Lách to |
| 116 | fulminant course | /ˈfʊlmɪnənt kɔːs/ | Diễn biến tối cấp |
| 117 | transjugular route | /trænsˈdʒʌɡjʊlə ruːt/ | Đường tĩnh mạch cảnh |
| 118 | dysarthria | /dɪsˈɑːθrɪə/ | Khó nói |
| 119 | drooling | /ˈdruːlɪŋ/ | Chảy nước dãi |
| 120 | gait disturbance | /ɡeɪt dɪsˈtɜːbəns/ | Rối loạn dáng đi |
| 121 | masklike facies | /ˈmɑːsklaɪk ˈfeɪʃiːz/ | Khuôn mặt vô cảm |
| 122 | rigidity | /rɪˈdʒɪdəti/ | Cứng cơ |
| 123 | Parkinsonian features | /ˌpɑːkɪnˈsəʊniən ˈfiːtʃəz/ | Đặc điểm Parkinson |
| 124 | flexion contractures | /ˈflɛkʃən kənˈtræktʃəz/ | Co cứng gập |
| 125 | spasticity | /spæsˈtɪsəti/ | Co cứng cơ |
| 126 | athetosis | /ˌæθɪˈtəʊsɪs/ | Múa vờn |
| 127 | generalized seizures | /ˈdʒɛnərəlaɪzd ˈsiːʒəz/ | Co giật toàn thể |
| 128 | autonomic dysfunction | /ˌɔːtəˈnɒmɪk dɪsˈfʌŋkʃən/ | Rối loạn chức năng thần kinh tự chủ |
| 129 | cognitive ability | /ˈkɒɡnɪtɪv əˈbɪləti/ | Khả năng nhận thức |
| 130 | magnetic resonance imaging (MRI) | /mæɡˈnɛtɪk ˈrɛzənəns ˈɪmɪdʒɪŋ/ | Chụp cộng hưởng từ (MRI) |
| 131 | globus pallidus | /ˈɡləʊbəs ˈpælɪdəs/ | Cầu nhạt |
| 132 | pons | /pɒnz/ | Cầu não |
| 133 | brainstem | /ˈbreɪnstɛm/ | Thân não |
| 134 | psychiatric illness | /ˌsaɪkiˈætrɪk ˈɪlnɪs/ | Bệnh tâm thần |
| 135 | behavioral changes | /bɪˈheɪvjərəl ˈtʃeɪndʒɪz/ | Thay đổi hành vi |
| 136 | personality changes | /ˌpɜːsəˈnæləti ˈtʃeɪndʒɪz/ | Thay đổi nhân cách |
| 137 | lability of mood | /ləˈbɪləti əv muːd/ | Tính khí thất thường |
| 138 | impulsive behavior | /ɪmˈpʌlsɪv bɪˈheɪvjə/ | Hành vi bốc đồng |
| 139 | antisocial behavior | /ˌæntiˈsəʊʃəl bɪˈheɪvjə/ | Hành vi chống đối xã hội |
| 140 | depression | /dɪˈprɛʃən/ | Trầm cảm |
| 141 | sexual preoccupation | /ˈsɛkʃʊəl priːˌɒkjʊˈpeɪʃən/ | Tăng ham muốn tình dục |
| 142 | psychosis | /saɪˈkəʊsɪs/ | Rối loạn tâm thần |
| 143 | electron-dense granules | /ɪˈlɛktrɒn-dɛns ˈɡrænjuːlz/ | Hạt đậm đặc electron |
| 144 | slit-lamp examination | /slɪt-læmp ɪɡˌzæmɪˈneɪʃən/ | Khám đèn khe |
| 145 | nonadherence | /ˌnɒnədˈhɪərəns/ | Không tuân thủ |
| 146 | cholestasis | /ˌkɒlɪˈsteɪsɪs/ | Ứ mật |
| 147 | proximal renal tubular acidosis | /ˈprɒksɪməl ˈriːnl ˈtjuːbjʊlər ˌæsɪˈdəʊsɪs/ | Toan hóa ống thận gần |
| 148 | Fanconi syndrome | /ˈfænkəʊni ˈsɪndrəʊm/ | Hội chứng Fanconi |
| 149 | acute tubular injury | /əˈkjuːt ˈtjuːbjʊlər ˈɪndʒəri/ | Tổn thương ống thận cấp |
| 150 | distal renal tubular acidosis | /ˈdɪstəl ˈriːnl ˈtjuːbjʊlər ˌæsɪˈdəʊsɪs/ | Toan hóa ống thận xa |
| 151 | renal calculi | /ˈriːnl ˈkælkjʊlaɪ/ | Sỏi thận |
| 152 | hematuria | /ˌhiːməˈtjʊərɪə/ | Tiểu máu |
| 153 | nephrolithiasis | /ˌnɛfrəʊlɪˈθaɪəsɪs/ | Sỏi thận |
| 154 | glomerular disease | /ɡləʊˈmɛrʊlə dɪˈziːz/ | Bệnh cầu thận |
| 155 | proteinuria | /ˌprəʊtɪˈnjʊərɪə/ | Protein niệu |
| 156 | nephrotic syndrome | /nɛˈfrɒtɪk ˈsɪndrəʊm/ | Hội chứng thận hư |
| 157 | Goodpasture syndrome | /ˈɡʊdˌpɑːstʃə ˈsɪndrəʊm/ | Hội chứng Goodpasture |
| 158 | osteopenia | /ˌɒstiəʊˈpiːnɪə/ | Giảm mật độ xương |
| 159 | osteomalacia | /ˌɒstiəʊməˈleɪʃɪə/ | Nhuyễn xương |
| 160 | osteoporosis | /ˌɒstiəʊpəˈrəʊsɪs/ | Loãng xương |
| 161 | symptomatic arthropathy | /ˌsɪmptəˈmætɪk ɑːˈθrɒpəθi/ | Bệnh khớp có triệu chứng |
| 162 | osteoarthritis | /ˌɒstiəʊɑːˈθraɪtɪs/ | Viêm xương khớp |
| 163 | osteochondritis dissecans | /ˌɒstiəʊkɒnˈdraɪtɪs ˈdɪsɪkænz/ | Viêm xương sụn bóc tách |
| 164 | chondromalacia patellae | /ˌkɒndrəʊməˈleɪʃɪə pəˈtɛliː/ | Nhuyễn sụn xương bánh chè |
| 165 | chondrocalcinosis | /ˌkɒndrəʊˌkælsɪˈnəʊsɪs/ | Vôi hóa sụn khớp |
| 166 | acute intravascular hemolysis | /əˈkjuːt ˌɪntrəˈvæskjʊlə hiːˈmɒlɪsɪs/ | Tan máu nội mạch cấp tính |
| 167 | electrocardiographic abnormalities | /ɪˌlɛktrəʊˌkɑːdɪəˈɡræfɪk ˌæbnɔːˈmælətiz/ | Bất thường trên điện tâm đồ |
| 168 | dysautonomia | /ˌdɪsɔːtəˈnəʊmɪə/ | Rối loạn thần kinh tự chủ |
| 169 | dysrhythmia | /dɪsˈrɪðmɪə/ | Loạn nhịp |
| 170 | azure lunulae | /ˈæʒə ˈluːnjuːliː/ | Móng xanh |
| 171 | delayed puberty | /dɪˈleɪd ˈpjuːbəti/ | Dậy thì muộn |
| 172 | gynecomastia | /ˌɡaɪnɪkəʊˈmæstɪə/ | Nữ hóa tuyến vú |
| 173 | amenorrhea | /ˌeɪmɛnəˈriːə/ | Vô kinh |
| 174 | hormonal imbalance | /hɔːˈməʊnl ɪmˈbæləns/ | Mất cân bằng nội tiết tố |
| 175 | dyskinesia | /ˌdɪskɪˈniːʒə/ | Rối loạn vận động |
| 176 | protein-losing enteropathy | /ˈprəʊtiːn-luːzɪŋ ˌɛntəˈrɒpəθi/ | Bệnh ruột mất protein |
| 177 | hereditary aceruloplasminemia | /hɪˈrɛdɪtəri əˌsɪərʊləʊˌplæzˈmiːnɪə/ | Bệnh di truyền không có ceruloplasmin |
| 178 | iron overload | /ˈaɪən ˈəʊvələʊd/ | Quá tải sắt |
| 179 | primary biliary cholangitis | /ˈpraɪməri ˈbɪliəri ˌkɒlænˈdʒaɪtɪs/ | Viêm đường mật tiên phát |
| 180 | light microscopy | /laɪt maɪˈkrɒskəpi/ | Kính hiển vi quang học |
| 181 | glycogen inclusions | /ˈɡlaɪkədʒən ɪnˈkluːʒənz/ | Thể vùi glycogen |
| 182 | Mallory hyaline | /ˈmæləri ˈhaɪəlɪn/ | Thể hyalin Mallory |
| 183 | Mallory-Denk bodies | /ˈmæləri-dɛŋk ˈbɒdiz/ | Thể Mallory-Denk |
| 184 | rhodanine | /ˈrəʊdəniːn/ | Rhodanine |
| 185 | rubeanic acid | /ruːˈbiːənɪk ˈæsɪd/ | Axit rubeanic |
| 186 | Timm sulfide staining | /tɪm ˈsʌlfaɪd ˈsteɪnɪŋ/ | Nhuộm bạc sulfide Timm |
| 187 | radiocopper | /ˌreɪdiəʊˈkɒpə/ | Đồng phóng xạ |
| 188 | haplotype analysis | /ˈhæpləʊtaɪp əˈnæləsɪs/ | Phân tích haplotype |
| 189 | double recombination | /ˈdʌbl ˌriːkɒmbɪˈneɪʃən/ | Tái tổ hợp kép |
| 190 | mutational analysis | /mjuːˈteɪʃənl əˈnæləsɪs/ | Phân tích đột biến |
| 191 | DNA sequencing | /diː-ɛn-eɪ ˈsiːkwənsɪŋ/ | Giải trình tự DNA |
| 192 | polymorphisms | /ˌpɒliˈmɔːfɪzəmz/ | Đa hình |
| 193 | deionized water | /diːˈaɪənaɪzd ˈwɔːtə/ | Nước khử ion |
| 194 | pyridoxine | /ˌpɪrɪˈdɒksiːn/ | Pyridoxine |
| 195 | hypersensitivity reaction | /ˌhaɪpəˌsɛnsɪˈtɪvəti riˈækʃən/ | Phản ứng quá mẫn |
| 196 | lymphadenopathy | /lɪmˌfædɪˈnɒpəθi/ | Hạch to |
| 197 | bone marrow suppression | /bəʊn ˈmærəʊ səˈprɛʃən/ | Ức chế tủy xương |
| 198 | myasthenia | /ˌmaɪəsˈθiːnɪə/ | Nhược cơ |
| 199 | polymyositis | /ˌpɒliˌmaɪəˈsaɪtɪs/ | Viêm đa cơ |
| 200 | systemic lupus erythematosus | /sɪsˈtɛmɪk ˈluːpəs ˌɛrɪθiːməˈtəʊsəs/ | Lupus ban đỏ hệ thống |