
Sperling Nội tiết học Nhi khoa, Ấn bản thứ 5 – Biên dịch: Ths.Bs. Lê Đình Sáng
Sperling Pediatric Endocrinology, Fifth Edition
Tác giả: Sperling, Mark A., MD – Nhà xuất bản: Elsevier Inc.
PHẦN III. NỘI TIẾT HỌC TRẺ EM VÀ THANH THIẾU NIÊN
Chương 11. Rối Loạn Tăng Trưởng Ở Trẻ Em
Alexander A.L. Jorge; Adda Grimberg; Mehul T. Dattani; Jeffrey Baron
Disorders of Childhood Growth
Sperling Pediatric Endocrinology, 11, 299-356
Giới thiệu
Sự tăng trưởng của con người là một quá trình đáng kinh ngạc. Khởi đầu của nó gắn liền với các cơ chế vô cùng phức tạp biến một tế bào đơn lẻ thành một phôi thai phức tạp. Sau khi hình thành, thai nhi và sau đó là trẻ em tiếp tục phát triển trong khoảng 15 năm, đạt đến khối lượng cơ thể lớn hơn 10.000.000 lần so với hợp tử ban đầu. Tốc độ tăng trưởng ban đầu là rất lớn. Thai nhi phát triển chiều dài với tốc độ hơn 100 cm mỗi năm, nhưng khi sinh ra, tốc độ tăng trưởng chiều dài đã chậm lại còn 50 cm mỗi năm và đến giữa thời thơ ấu là 5 cm mỗi năm. Sự suy giảm tốc độ tăng trưởng này bị gián đoạn trong một thời gian ngắn bởi giai đoạn tăng vọt ở tuổi dậy thì nhưng sau đó lại tiếp tục, khiến sự tăng trưởng của cơ thể dần dần dừng lại khi trẻ bước vào tuổi trưởng thành. Trong giai đoạn cơ thể lớn lên, sự phát triển của các cơ quan khác nhau, chẳng hạn như thận, tim, phổi, gan và xương, diễn ra đồng bộ, được điều phối để duy trì tỷ lệ cơ thể.
Ở phần lớn trẻ em, các cơ chế chi phối sự tăng trưởng của cơ thể, vốn phần lớn còn là bí ẩn, diễn ra một cách đều đặn, tạo ra chiều cao trưởng thành thường từ 1,5 đến 2 mét. Tuy nhiên, ở một số trẻ, sự tăng trưởng của cơ thể lớn hơn hoặc nhỏ hơn so với mức bình thường, thường khiến gia đình lo lắng và tìm đến bác sĩ để được đánh giá.
Khi đó, nhân viên y tế được yêu cầu trả lời hai câu hỏi. Thứ nhất, tại sao sự tăng trưởng của trẻ lại nằm ngoài phạm vi bình thường và thứ hai, cần phải làm gì, nếu có. Câu hỏi đầu tiên, “tại sao,” rất quan trọng vì nhiều lý do. Nhiều trẻ em thấp hoặc cao vì chúng được di truyền từ cha mẹ một tập hợp lớn các đa hình bình thường khiến trẻ nằm ở một trong hai thái cực của phân bố chiều cao bình thường. Tình trạng tầm vóc thấp hoặc cao đa gen như vậy thường là lành tính. Tuy nhiên, một số trẻ thấp hoặc cao do bất thường một gen duy nhất có thể có những ảnh hưởng sức khỏe quan trọng, chẳng hạn như xu hướng dị tật tim hoặc các bệnh ác tính. Những trẻ khác bị thấp còi do một bệnh hệ thống mắc phải không rõ ràng, chẳng hạn như bệnh viêm ruột (IBD), viêm tuyến giáp tự miễn, hoặc khối u hệ thần kinh trung ương (CNS) gây rối loạn chức năng tuyến yên. Do đó, nhân viên y tế đánh giá sự tăng trưởng của trẻ em phải đối mặt với thách thức phân loại số ít trẻ có bệnh lý tiềm ẩn quan trọng khỏi số đông trẻ có tình trạng lành tính.
Câu hỏi thứ hai, “phải làm gì,” cũng có thể là một thách thức. Nếu việc đánh giá chẩn đoán phát hiện ra một rối loạn tiềm ẩn có thể điều trị được, chẳng hạn như bệnh celiac, thì hướng xử trí rất rõ ràng. Nhưng thường thì không tìm thấy nguyên nhân đơn giản có thể điều trị được. Khi đó, nhân viên y tế và gia đình phải cố gắng đánh giá xem tầm vóc thấp hoặc cao có đủ bất thường và đủ gây phiền muộn cho trẻ, hoặc có khả năng gây phiền muộn trong tương lai, để những lợi ích có thể có của liệu pháp y tế lớn hơn những nỗ lực, chi phí, sự khó chịu và những rủi ro tiềm tàng của việc can thiệp.
Để giải quyết hai câu hỏi trung tâm này, cần có sự hiểu biết sâu sắc về sự tăng trưởng bình thường ở trẻ em và nhiều rối loạn ảnh hưởng đến nó, sự quyết tâm tìm kiếm cẩn thận các nguyên nhân tiềm ẩn, cũng như sự sẵn lòng và khả năng lắng nghe một cách cảm thông những lo lắng của trẻ và gia đình. Trong chương này, chúng tôi đã cố gắng trình bày cho nhân viên y tế một cái nhìn tổng quan rộng rãi về kiến thức cần thiết cho nỗ lực này.
Tăng trưởng ở trẻ em
Cơ sở sinh học của sự tăng trưởng theo chiều dọc
Ở trẻ em, nhiều mô và cơ quan phát triển đồng thời. Tuy nhiên, hầu hết trẻ em đến khám bác sĩ nội tiết nhi khoa để đánh giá sự tăng trưởng là do chiều cao của trẻ bất thường. Chiều cao chủ yếu được quyết định bởi chiều dài của các xương dài ở chi dưới và chiều cao của các thân đốt sống. Chiều dài của các xương này lại được quyết định bởi hoạt động của các sụn tăng trưởng xương, là những lớp sụn mỏng nằm gần các đầu xương dài và các đốt sống.
Bên trong sụn tăng trưởng, các tế bào sụn được sắp xếp thành các cột song song với trục dài của xương (Hình 11.1A). Ở phần trên (gần đầu xương hơn) của các cột, các tế bào sụn tăng sinh (Hình 11.1B). Ở phần dưới của các cột, các tế bào sụn ngừng phân chia và thay vào đó là phì đại về kích thước (Hình 11.1B). Hai quá trình này, tăng sinh tế bào và phì đại tế bào, dẫn đến sự hình thành sụn (chondrogenesis). Nếu chỉ riêng quá trình này, nó sẽ làm cho sụn tăng trưởng dày lên dần. Tuy nhiên, đồng thời, ở phần dưới của sụn tăng trưởng, sụn mới hình thành được tu sửa thành xương. Kết quả cuối cùng là mô xương mới được tạo ra liên tục ở phần dưới của sụn tăng trưởng, làm cho toàn bộ xương dài ra và trẻ cao lên.
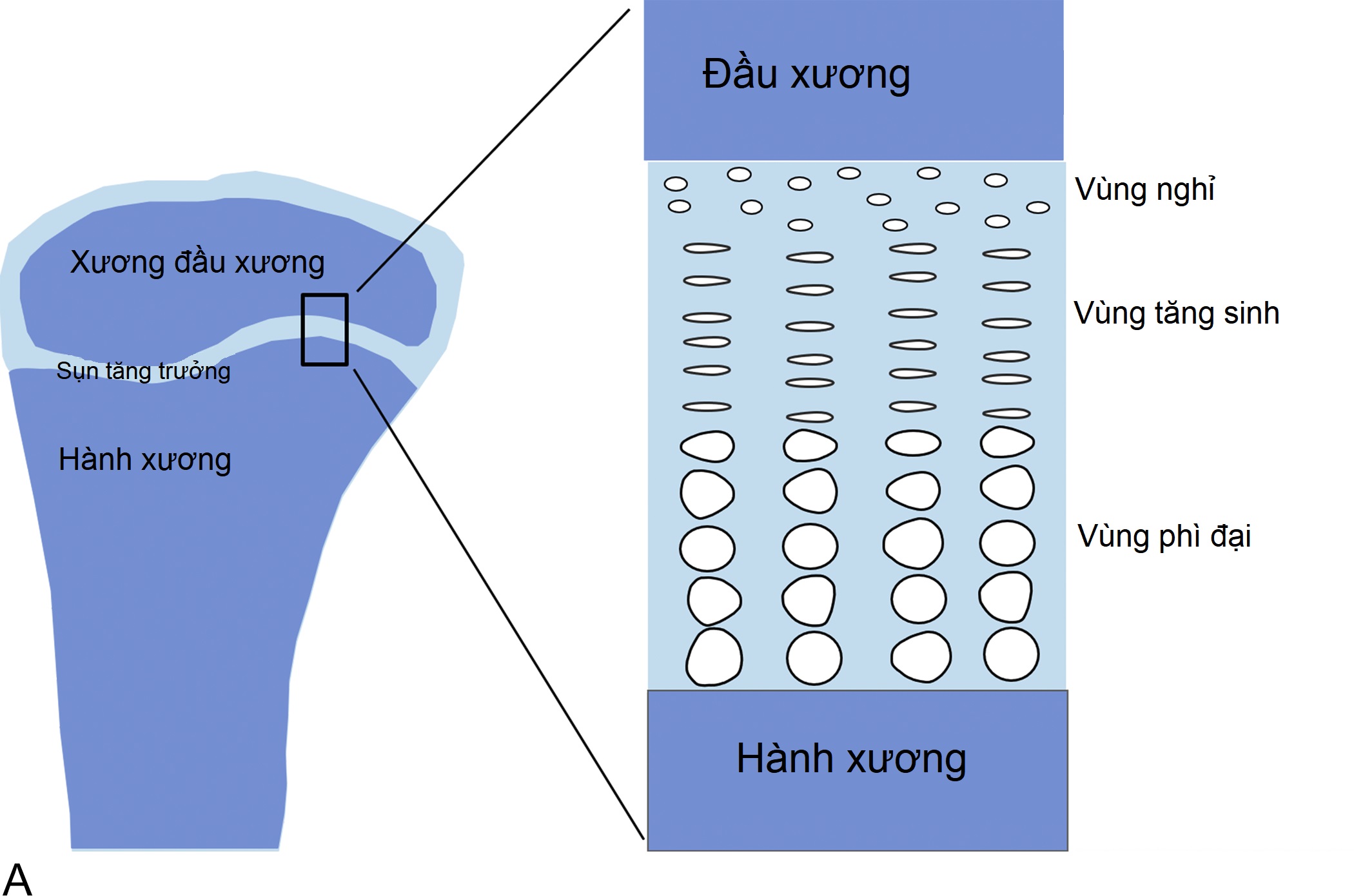
Hình 11.1A Cấu trúc sụn tăng trưởng. Ở trẻ em, sự tăng trưởng theo chiều dọc (tăng chiều cao) là kết quả của quá trình tạo sụn ở sụn tăng trưởng. Sụn tăng trưởng là một lớp sụn mỏng được tìm thấy gần các đầu xương dài và trong các đốt sống. Sụn tăng trưởng bao gồm ba vùng.
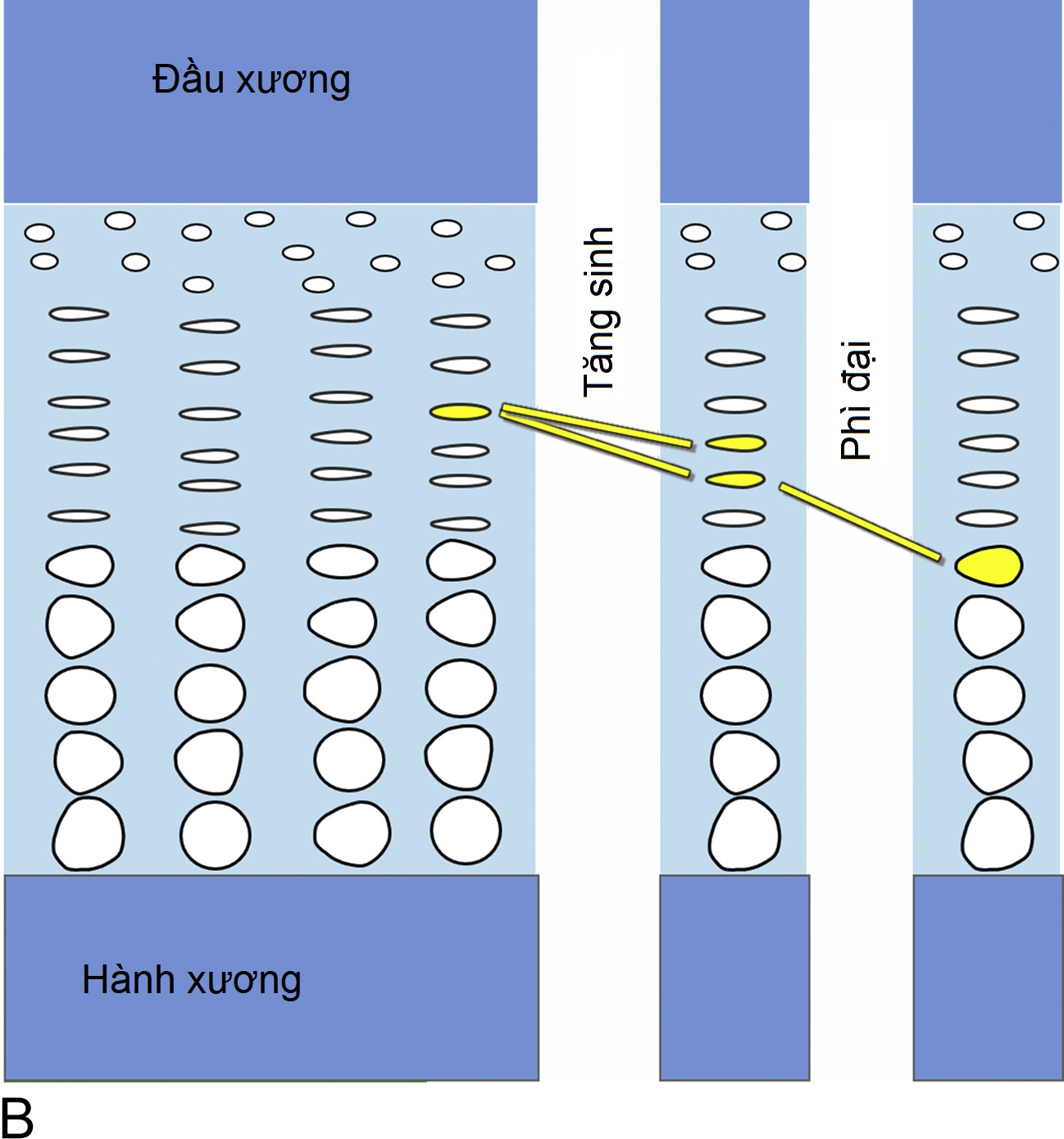
Hình 11.1B Chức năng của sụn tăng trưởng. Bên trong sụn tăng trưởng, các tế bào sụn trước tiên tăng sinh và sau đó phì đại. Kết quả là tạo ra nhiều sụn hơn (quá trình tạo sụn). Ở phần dưới của sụn tăng trưởng, sụn mới hình thành được tu sửa thành xương (không được hiển thị). Kết quả cuối cùng là xương mới được tạo ra ở phần dưới của sụn tăng trưởng, dần dần đẩy các đầu xương ra xa nhau, làm cho xương dài ra và trẻ cao lên.
Do đó, sự tăng trưởng theo chiều dọc (tăng chiều cao) ở trẻ em là kết quả của quá trình tạo sụn ở sụn tăng trưởng. Vì vậy, tầm vóc thấp là do giảm quá trình tạo sụn ở sụn tăng trưởng và tầm vóc cao là do tăng quá trình tạo sụn ở sụn tăng trưởng.
Điều hòa sự tăng trưởng theo chiều dọc
Bởi vì sự tăng trưởng theo chiều dọc là kết quả của quá trình tạo sụn ở sụn tăng trưởng, nên sự điều hòa tăng trưởng theo chiều dọc là kết quả của sự điều hòa các tế bào sụn ở sụn tăng trưởng. Các yếu tố kích thích sự tăng sinh và phì đại của tế bào sụn ở sụn tăng trưởng sẽ kích thích sự tăng trưởng theo chiều dọc, trong khi các yếu tố ức chế sự tăng sinh và phì đại của tế bào sụn ở sụn tăng trưởng sẽ ức chế sự tăng trưởng theo chiều dọc. Sự điều hòa chức năng của tế bào sụn ở sụn tăng trưởng này xảy ra ở nhiều cấp độ khác nhau; các tín hiệu nội tiết, cytokine gây viêm, lượng dinh dưỡng nạp vào, tín hiệu cận tiết/tự tiết, các tác động của chất nền ngoại bào và các hệ thống nội bào đều có thể điều chỉnh sự tăng sinh và phì đại của tế bào sụn và do đó ảnh hưởng đến tốc độ tăng trưởng theo chiều dọc. Do đó, những bất thường ở bất kỳ cấp độ nào trong số này đều có thể gây ra tầm vóc thấp hoặc cao.
Điều hòa nội tiết của sự tăng trưởng theo chiều dọc
Hormone Tăng Trưởng
Tuyến yên
Tuyến yên nằm trong hố yên (sella turcica), là hố tuyến yên của xương bướm, nằm ở trung tâm của nền sọ. Quan niệm về tuyến yên là một “tuyến chủ” kiểm soát các hoạt động nội tiết của cơ thể đã trở nên lỗi thời và được thay thế bằng sự công nhận tầm quan trọng của não bộ, đặc biệt là vùng dưới đồi, trong việc điều hòa sản xuất và bài tiết hormone. Tuy nhiên, tuyến yên vẫn là trung tâm trong sự hiểu biết của chúng ta về sự điều hòa tăng trưởng, chuyển hóa và cân bằng nội môi, phản ứng với căng thẳng, tiết sữa và sinh sản.
Về mặt phôi thai học, tuyến yên được hình thành từ hai nguồn riêng biệt, đó là túi Rathke, một túi thừa của khoang miệng nguyên thủy (ngoại bì miệng), tạo ra tuyến yên trước (adenohypophysis), và ngoại bì thần kinh của sàn não trước, tạo ra tuyến yên sau (neurohypophysis). Tuyến yên trước thường chiếm 80% trọng lượng của tuyến yên và bao gồm phần xa (còn được gọi là thùy trước), phần trung gian và phần phễu (còn được gọi là phần gần).
Phần lớn kiến thức của chúng ta về sự phát triển bình thường của trục dưới đồi-tuyến yên có được từ các mô hình động vật, đặc biệt là loài gặm nhấm. Ở chuột, sự dày lên của ngoại bì ở đường giữa của mào thần kinh trước, tạo thành mảng tuyến yên (hypophyseal placode), báo hiệu sự bắt đầu phát triển của tuyến yên ở ngày 7.5 sau khi giao phối (dpc). Sự hình thành túi Rathke sơ khai diễn ra sau đó vào ngày 9 dpc, với sự hình thành một túi hoàn chỉnh vào ngày 12 dpc và sau đó, tuyến yên trước bao gồm năm loại tế bào khác nhau tiết ra sáu loại hormone khác nhau (Hình 11.2).
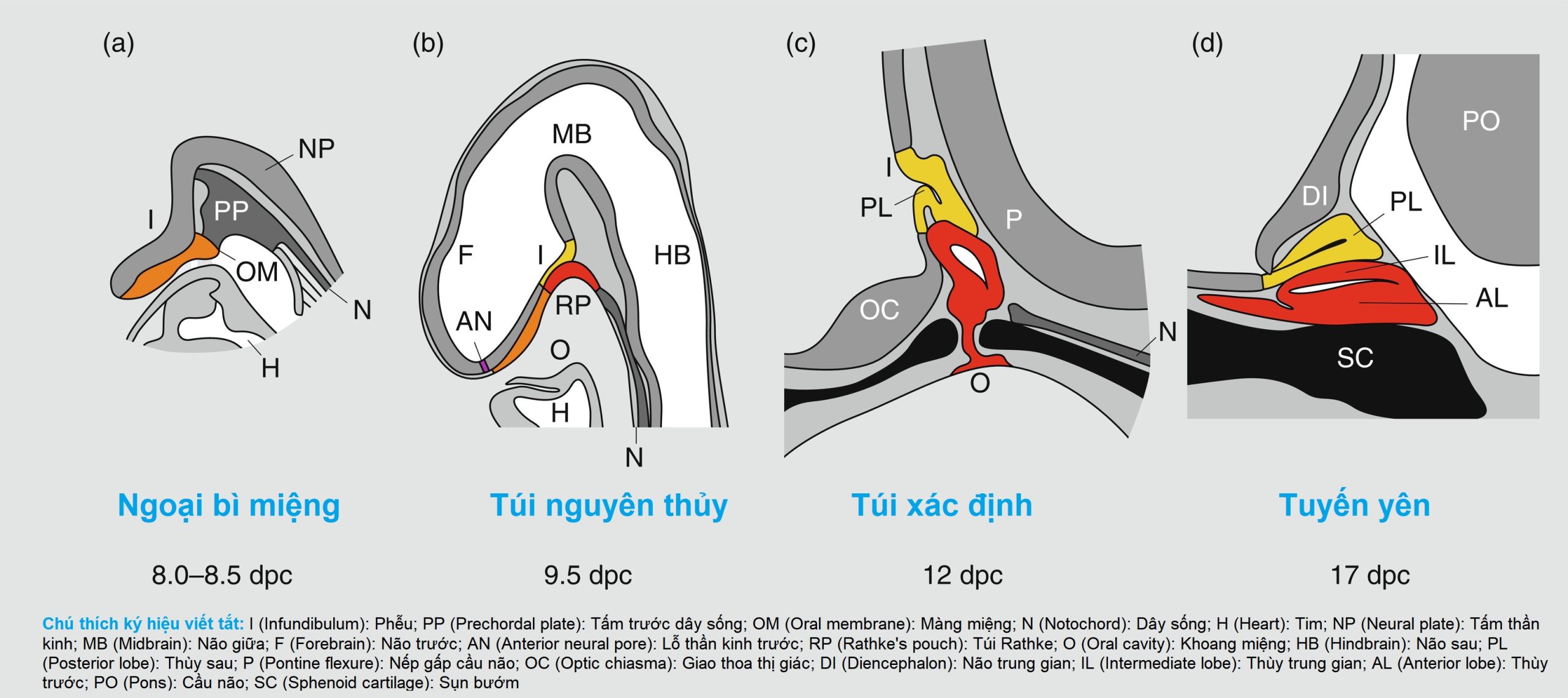
Hình 11.2 Sự phát triển của tuyến yên chuột trong mặt cắt dọc. Các giai đoạn phát triển được chỉ định bằng dpc. AL, Thùy trước; AN, Lỗ thần kinh trước; DI, Gian não; F, Não trước; H, Tim; HB, Não sau; I, Cuống tuyến yên; IL, Thùy trung gian; MB, Não giữa; N, Dây sống; NP, Tấm thần kinh; O, Khoang miệng; OC, Giao thoa thị giác; OM, Màng miệng; P, Nếp gấp cầu não; PL, Thùy sau; PO, Cầu não; PP, Tấm trước dây sống; RP, Túi Rathke; SC, Sụn bướm. (Chỉnh sửa từ Sheng, H. Z., & Westphal, H. (1999). Early steps in pituitary organogenesis. Trends Genet, 15:236–240. Với sự cho phép của Elsevier.)
Ở người, phần xa là phần lớn nhất của tuyến yên trước và chứa phần lớn các tế bào sản xuất hormone. Trái ngược với chuột, phần trung gian ở người còn sơ khai và bao gồm một số khoang nang được lót bởi một lớp biểu mô hình khối duy nhất vì nó phần lớn biến mất trong quá trình phát triển phôi thai. Phần xa và phần trung gian được ngăn cách bởi một khe, một cấu trúc di tích của túi Rathke mà từ đó nó phát triển. Cấu trúc này thường có thể phát triển thành một nang (nang khe Rathke). Phần phễu đại diện cho một phần mở rộng hướng lên của phần xa lên cuống tuyến yên và có thể chứa một số lượng hạn chế các tế bào sản xuất gonadotropin. Tuyến yên sau (neurohypophysis) bao gồm cuống phễu hoặc cuống tuyến yên, lồi giữa của củ xám, và mỏm phễu (thùy sau, thùy thần kinh). Tuyến yên sau chứa các đầu tận cùng sợi trục của các neuron tế bào lớn từ các nhân cận não thất và trên thị của vùng dưới đồi; chúng sản xuất oxytocin, cần thiết trong quá trình tiết sữa và sinh con, và vasopressin, cần thiết cho việc điều hòa thẩm thấu. Nó không có chức năng nào được biết đến trong việc điều hòa tăng trưởng và sẽ không được thảo luận thêm trong chương này.
Túi Rathke, nguồn gốc của tuyến yên trước, có thể được xác định ở phôi thai 3 mm trong tuần thứ ba của thai kỳ ở người. Túi Rathke sau đó bắt đầu phát triển, dẫn đến một túi hoàn chỉnh tách rời khỏi ngoại bì miệng vào cuối tuần thứ sáu của thai kỳ. Các tế bào sản xuất hormone tăng trưởng (GH) có thể được xác định vào tuần thứ 9 của thai kỳ. Vào khoảng thời gian này, các kết nối mạch máu giữa thùy trước của tuyến yên và vùng dưới đồi phát triển, mặc dù đã được chứng minh rằng việc sản xuất hormone của tuyến yên có thể xảy ra khi không có kết nối với vùng dưới đồi. Do đó, các tế bào hướng thân (somatotropes) thường có thể được chứng minh trong tuyến yên của trẻ sơ sinh vô não. Tuy nhiên, có vẻ như việc khởi đầu sự phát triển của tuyến yên trước phụ thuộc vào khả năng đáp ứng của ngoại bì miệng với các yếu tố gây cảm ứng từ gian não bụng. Hiếm khi, ống sọ hầu (đánh dấu sự di chuyển phôi thai của túi Rathke) vẫn còn thông và có thể chứa các cụm nhỏ tế bào tuyến yên trước—tạo ra một tuyến yên hầu có thể có khả năng tổng hợp hormone.
Sự tiếp giáp và tương tác duy trì giữa ngoại bì miệng và ngoại bì thần kinh là rất quan trọng cho sự phát triển bình thường của tuyến yên trước. Các thao tác thực nghiệm trên phôi của một số loài, cũng như các thí nghiệm cấy ghép túi Rathke ở loài gặm nhấm, đã cho thấy rằng các tín hiệu từ gian não không chỉ cần thiết cho sự cảm ứng và duy trì túi Rathke, mà còn cho sự phân vùng trong túi cho phép sự xuất hiện của các loại tế bào nội tiết khác nhau. Trong thai kỳ, các tế bào tiền thân đang tăng sinh tập trung xung quanh lòng túi, và chúng dường như tách ra khi thoát khỏi chu kỳ tế bào và biệt hóa. Trong giai đoạn cuối của thai kỳ ở chuột và giai đoạn sau sinh, các tế bào tiền thân của thùy trước tái nhập chu kỳ tế bào và mở rộng quần thể các tế bào chuyên biệt, sản xuất hormone. Khi sinh ra, tất cả các loại tế bào đều có mặt, và vị trí của chúng dường như được phân tầng, dựa trên loại tế bào. Các mô hình hiện tại về sự chuyên biệt hóa tế bào ở thùy trước cho thấy rằng các gradient đối lập của tín hiệu yếu tố tăng trưởng nguyên bào sợi (FGF) và protein hình thái xương (BMP) tạo khuôn mẫu cho các tế bào tiền thân trong túi Rathke trước khi chúng di chuyển đến thùy trước nơi chúng biệt hóa. Một số nghiên cứu đã tiết lộ rằng sự phát triển bình thường của tuyến yên phụ thuộc vào một chuỗi phức tạp các yếu tố phiên mã và các phân tử tín hiệu được biểu hiện theo không gian và thời gian.
Các phân tử tín hiệu liên quan đến sự phát triển của tuyến yên có thể là nội tại, bắt nguồn từ ngoại bì miệng, chẳng hạn như sonic hedgehog (Shh), hoặc ngoại lai từ ngoại bì thần kinh, chẳng hạn như Nkx2.1, FGFs (ví dụ: FGF8) và BMPs (ví dụ: BMP4) (Hình 11.3). Những phân tử này có thể kích hoạt hoặc ức chế các yếu tố phiên mã, chẳng hạn như Hesx1, Lhx3 và Lhx4. Chúng cũng có thể hoạt động như các chất tạo hình thái (morphogen) tạo ra môi trường thích hợp cho sự biệt hóa tế bào, do đó đóng một vai trò quan trọng trong số phận của tế bào. Các phân tử tín hiệu như vậy bao gồm các thành viên của họ Shh, FGFs, các yếu tố tăng trưởng biến đổi-β (TGFs)/BMPs, Wingless/Wnts, và các phân tử trong con đường Notch, chỉ là một vài ví dụ.
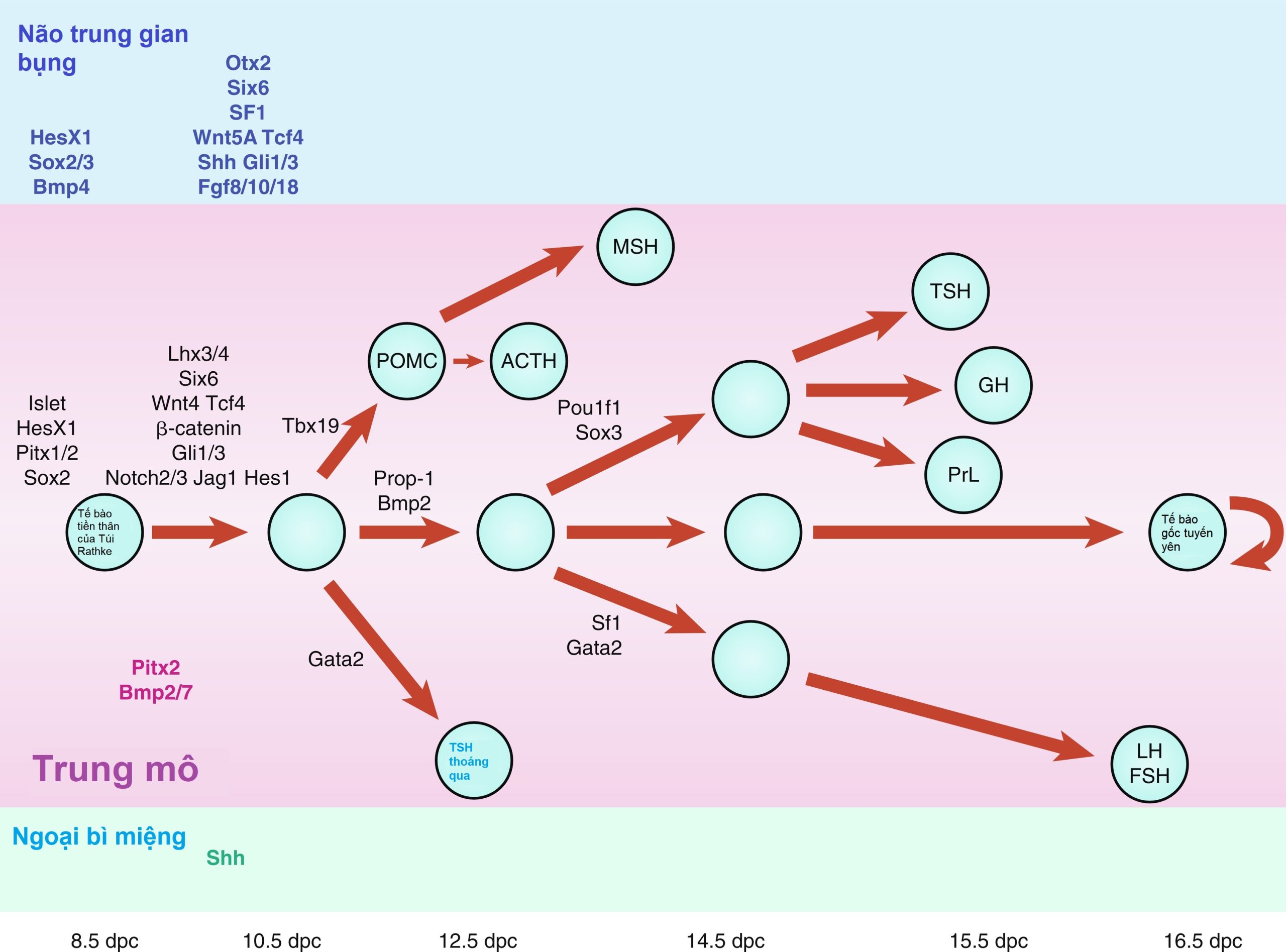
Hình 11.3 Sơ đồ biểu diễn chuỗi phát triển của các gen liên quan đến sự phát triển tuyến yên ở người với sự tham chiếu đặc biệt đến sự biệt hóa tế bào tuyến yên. (Từ Kelberman, D., Rizzoti, K., Lovell-Badge, R., và cộng sự. (2009). Genetic regulation of pituitary gland development in human and mouse. Endocr Rev, 30, 790–829.)
Gần đây, Davis và các đồng nghiệp đã thách thức giáo điều hiện tại về sự chuyên biệt hóa tế bào tuyến yên, đưa ra bằng chứng cho thấy rằng, ở chuột, mô hình chuyên biệt hóa tế bào dẫn đến vị trí phía trước của các tế bào hướng sinh dục (gonadotropes), vị trí phía sau cho các tế bào hướng thân (somatotropes) và một vị trí trung gian hơn cho các tế bào hướng vỏ thượng thận (corticotropes) và tế bào hướng tuyến giáp (thyrotropes), dường như không phải là kết quả của một trật tự thoát khỏi chu kỳ tế bào, như đã được đề xuất trước đây. Tất cả các loại tế bào của thùy trước dường như bắt đầu quá trình biệt hóa đồng thời (E11.5-E14.5), thay vì theo một cách rời rạc về mặt thời gian.
Cho đến nay, không có nhiều kiểu hình tuyến yên được báo cáo liên quan đến các đột biến trong các phân tử tín hiệu này. Quan trọng là, con đường tín hiệu Wnt gần đây đã được liên quan đến sự hình thành khối u tuyến yên. Ví dụ, có bằng chứng rõ ràng rằng con đường Wnt/β catenin có liên quan đến cơ chế bệnh sinh của u sọ hầu (craniopharyngioma), một khối u hiếm gặp ở vùng dưới đồi-tuyến yên.
Nhiều yếu tố phiên mã đặc hiệu cho tuyến yên có liên quan đến việc xác định các dòng tế bào tuyến yên và sự biểu hiện đặc hiệu của các hormone tuyến yên trước (xem Hình 11.3). Một số yếu tố phiên mã homeodomain đã được chứng minh là có liên quan đến sự phát triển và biệt hóa của tuyến yên trước ở người. Các khiếm khuyết trong một số yếu tố này hiện đã được liên kết với các sự kết hợp khác nhau của tình trạng thiếu hụt hormone tuyến yên (Bảng 11.1). Bởi vì các khiếm khuyết gen bổ sung đã được liên quan đến sự phát triển bất thường của trục dưới đồi-tuyến yên ở chuột, có vẻ như số lượng các khiếm khuyết di truyền đã biết ở người sẽ tăng lên.
Bảng 11.1 Các Khiếm Khuyết Di Truyền Trong Sự Phát Triển Tuyến Yên và Kiểu Hình của Chúng Chỉnh sửa và cập nhật từ Alatzoglou KS, Gregory LC, Dattani MT. Development of the pituitary gland. Compr Physiol. 2020;10(2):389–413.
| Gen | Các Thiếu Hụt Tuyến Yên | Kiểu Hình MRI | Kiểu Di Truyền | Các Đặc Điểm Kiểu Hình Khác |
|---|---|---|---|---|
| POU1F1 | GH, TSH, prolactin | Tuyến yên trước nhỏ hoặc kích thước bình thường | Lặn NST thường và Trội NST thường | |
| PROP1 | GH, TSH, LH, FSH, prolactin, thiếu hụt ACTH tiến triển | Tuyến yên trước nhỏ, bình thường hoặc to – có thể tiến triển theo thời gian | Lặn NST thường | |
| HESX1 | Từ thiếu hụt GH đơn độc đến suy toàn bộ tuyến yên với thiếu hụt TSH, LH, FSH, ACTH, và prolactin | Thiểu sản thần kinh thị, không có vách trong suốt, tuyến yên sau lạc chỗ, thiểu sản tuyến yên trước | Lặn NST thường và Trội NST thường | Chậm phát triển, bất thường thị giác, Loạn sản vách-thị (SOD) |
| LHX3 | GH, TSH, LH, FSH, prolactin với thiếu hụt ACTH muộn | Tuyến yên trước nhỏ, bình thường hoặc to | Lặn NST thường | Cổ ngắn với khả năng xoay hạn chế |
| LHX4 | Thiếu hụt GH, TSH, ACTH, và gonadotrophin | Tuyến yên trước nhỏ, tuyến yên sau lạc chỗ, bất thường tiểu não, thiểu sản thể chai | Trội NST thường (độ thấm biến đổi) | |
| SOX3 | Thiếu hụt GH, TSH, LH, FSH, ACTH. Phổ biến nhất là thiếu hụt GH đơn độc | Thiểu sản tuyến yên trước và cuống tuyến yên, tuyến yên sau lạc chỗ, bất thường thể chai bao gồm cả nang; ống sọ hầu tồn tại | Lặn liên kết NST X | Khó khăn trong học tập |
| SOX2 | Thiếu hụt LH, FSH, GH biến đổi | Thiểu sản tuyến yên trước, thiểu sản thần kinh thị, SOD, u mô thừa vùng dưới đồi | Trội NST thường (thường là de novo) | Tật mắt nhỏ, không có mắt, dương vật nhỏ, điếc thần kinh giác quan, khiếm khuyết đường tiêu hóa với teo thực quản |
| GLI2 | GH, TSH, và ACTH với thiếu hụt gonadotrophin biến đổi | Thiểu sản tuyến yên trước | Trội NST thường | Holoprosencephaly, sứt môi và hở hàm ếch, không có mắt, thừa ngón sau trục, hậu môn không thủng, khe thanh quản, bất sản thận |
| GLI3 | GH, TSH, LH, FSH, ACTH | Thiểu sản tuyến yên trước | Trội NST thường | Hội chứng Pallister-Hall, Thừa ngón sau trục, u mô thừa |
| OTX2 | Thiếu hụt GH, TSH, LH, FSH, và ACTH | Tuyến yên trước bình thường hoặc nhỏ, bất sản cuống tuyến yên, tuyến yên sau lạc chỗ, dị dạng Chiari I | Trội NST thường (thường là de novo) | Đầu nhỏ, không có mắt hai bên, chậm phát triển, hở hàm ếch |
| FGFR1 | Thiếu hụt GH, TSH, LH, FSH, và ACTH | Tuyến yên trước bình thường hoặc nhỏ, bất sản thể chai | Trội NST thường | Thông liên nhĩ (ASD) và thông liên thất (VSD), tật ngón ngắn, đầu ngắn, mấu thịt thừa trước tai, bất thường mắt, co giật |
| FGF8 | Thiếu hụt GH, TSH, ACTH, gonadotrophin, và ADH | Không có thể chai, thiểu sản thần kinh thị, holoprosencephaly | Trội NST thường hoặc Lặn NST thường | |
| PROKR2 | Thiếu hụt GH, TSH, ACTH và gonadotrophin | Thiểu sản thể chai, tuyến yên trước bình thường hoặc nhỏ | Trội NST thường | |
| ARNT2 | Thiếu hụt GH, TSH, ACTH, LH, FSH, ADH | Không có tuyến yên sau, cuống mỏng, thể chai mỏng, chậm myelin hóa | Lặn NST thường | Loạn sản khớp háng, thận ứ nước, trào ngược bàng quang-niệu quản, bàng quang thần kinh, đầu nhỏ, trán dô, mắt sâu, cằm lẹm |
| TCFL7 | GH | Không có tuyến yên sau, thiểu sản tuyến yên trước, thiểu sản thần kinh thị, bất sản một phần thể chai, mép trước mỏng | Trội NST thường | |
| IGSF1 | GH (thoáng qua/một phần), TSH, prolactin | Bình thường trong đa số các trường hợp. U dịch não tủy vùng trán-đỉnh, thiểu sản thể chai, và tổn thương cuống nhỏ đã được báo cáo | Lặn liên kết NST X | Tinh hoàn to, dậy thì muộn |
| RNPC3 | Thiếu hụt GH | Tuyến yên trước nhỏ với cuống bình thường và tuyến yên sau đúng chỗ | Lặn NST thường | U xơ nướu di truyền từ mẹ |
| KCNQ1 | Thiếu hụt GH, ACTH, TSH, và gonadotrophin | Tuyến yên trước bình thường/nhỏ | Lặn NST thường |
ACTH, Hormone vỏ thượng thận; AD, Trội NST thường; ADH, Hormone chống bài niệu; AP, Tuyến yên trước; AR, Lặn NST thường; ASD, Thông liên nhĩ; FSH, Hormone kích thích nang trứng; GH, Hormone tăng trưởng; LH, Hormone tạo hoàng thể; MRI, Chụp cộng hưởng từ; PP, Tuyến yên sau; SOD, Loạn sản vách-thị; TSH, Hormone kích thích tuyến giáp; VSD, Thông liên thất.
Ở người trưởng thành, tuyến yên có trọng lượng trung bình 600 mg, với khoảng dao động từ 400 đến 900 mg. Trọng lượng tuyến yên ở phụ nữ lớn hơn một chút so với nam giới, và thường tăng lên trong tuổi dậy thì và thai kỳ. Ở trẻ sơ sinh, trọng lượng tuyến yên trung bình khoảng 100 mg. Tuyến yên thường nằm trong hố yên ngay phía trên và được bao quanh một phần bởi xương bướm. Thể tích của hố yên cung cấp một thước đo tốt về kích thước tuyến yên, có thể bị giảm ở trẻ bị thiểu sản tuyến yên. Tuy nhiên, điều quan trọng cần nhận ra là có sự thay đổi đáng kể về kích thước tuyến yên một cách bình thường. Tuyến yên được bao phủ phía trên bởi cơ hoành yên (diaphragma sellae), và giao thoa thị giác nằm ngay trên cơ hoành. Sự gần gũi về mặt giải phẫu giữa giao thoa thị giác và tuyến yên rất quan trọng vì thiểu sản giao thoa thị giác có thể xảy ra cùng với rối loạn chức năng dưới đồi/tuyến yên, như trong tình trạng loạn sản vách-thị (SOD), và vì các khối u tuyến yên có thể tác động đến giao thoa thị giác, dẫn đến suy giảm thị lực. Bệnh nhân bị mù bẩm sinh hoặc rung giật nhãn cầu nên được đánh giá ban đầu và sau đó được theo dõi cẩn thận về tình trạng suy tuyến yên, vì tình trạng này có thể tiến triển. Ngoài ra, sự phát triển trên yên của một khối u tuyến yên ban đầu có thể biểu hiện bằng các phàn nàn về thị lực hoặc bằng chứng về sự suy giảm thị trường ngoại vi tiến triển, đặc biệt là bán manh thái dương hai bên.
Sự tồn tại của hệ thống tuần hoàn cửa trong tuyến yên là rất quan trọng đối với chức năng tuyến yên bình thường. Nguồn cung cấp máu của tuyến yên bắt nguồn từ các động mạch tuyến yên trên và dưới, là các nhánh của động mạch cảnh trong. Các nhánh trước và sau của động mạch tuyến yên trên có thể kết thúc trong cuống tuyến yên và phần gần của cuống tuyến yên. Các peptide vùng dưới đồi, được sản xuất trong các neuron kết thúc ở cuống tuyến yên, đi vào đám rối sơ cấp của tuần hoàn cửa tuyến yên và được vận chuyển bằng các tĩnh mạch cửa tuyến yên đến các mao mạch của tuyến yên trước. Hệ thống cửa này do đó cung cấp một phương tiện liên lạc giữa các neuron của vùng dưới đồi và các tế bào sản xuất hormone của tuyến yên trước. Nguồn cung cấp máu của tuyến yên sau là riêng biệt, bắt nguồn từ động mạch tuyến yên dưới. Sự điều hòa của thùy sau của tuyến yên không liên quan đến tuần hoàn cửa tuyến yên mà được trung gian thông qua các kết nối thần kinh trực tiếp.
Túi Rathke hoàn chỉnh bao gồm các tế bào tiền thân tăng sinh sẽ dần dần di chuyển về phía bụng, ra xa khỏi lòng túi khi chúng biệt hóa. Một vùng tăng sinh chứa các tế bào tiền thân được duy trì trong phôi ở khu vực quanh lòng túi và gần đây được phát hiện vẫn tồn tại ở người trưởng thành. Tuy nhiên, bản chất chính xác của các tế bào tiền thân trong tuyến yên vẫn chưa được biết rõ. Các thành viên của họ yếu tố phiên mã Sox có khả năng tham gia vào các bước sớm nhất của sự tăng sinh tế bào gốc tuyến yên và các chuyển đổi sớm nhất sang biệt hóa. Yếu tố phiên mã Prophet of Pit1 (PROP1) và con đường tín hiệu NOTCH sau đó có thể điều chỉnh quá trình chuyển sang biệt hóa. Người ta đã đề xuất rằng ổ của các tế bào tiền thân có thể là vùng rìa xung quanh lòng túi Rathke, giữa thùy trước và thùy trung gian của tuyến yên chuột, bởi vì các tế bào trong vùng này có khả năng tạo ra tất cả năm dòng tế bào hormone tuyến yên (xem Hình 11.3).
Tế bào gốc đã được chứng minh đóng một vai trò trong sự hình thành khối u ở một số mô, và vai trò của chúng trong tăng sản tuyến yên, u tuyến yên, và các khối u là một lĩnh vực quan trọng cho các nghiên cứu trong tương lai. Khả năng nuôi cấy và phát triển tế bào gốc tuyến yên ở trạng thái tiền biệt hóa cũng có thể hữu ích cho việc điều trị lâu dài các tình trạng thiếu hụt tuyến yên. Thật vậy, một nghiên cứu tiên phong đã dẫn đến sự tự hình thành hiệu quả của một tuyến yên trước ba chiều trong một khối nuôi cấy tổng hợp từ các tế bào gốc phôi (ES) chuột. Nhiều tế bào nội tiết khác nhau đã được tạo ra, và các tế bào này có khả năng đáp ứng với các hormone dinh dưỡng. Khi các khối tế bào có nguồn gốc từ ES này được cấy dưới bao thận ở những con chuột bị cắt bỏ tuyến yên, corticosterone đã được sản xuất. Do đó, các nghiên cứu này có thể phản ánh bước đầu tiên hướng tới việc điều trị bằng tế bào gốc.
Các tế bào tiết đã biệt hóa hoàn toàn không được phân bố ngẫu nhiên theo kiểu chắp vá trên toàn bộ tuyến yên. Thay vào đó, các tế bào này dường như tự tổ chức thành các mạng lưới cùng loại tế bào. Sự kết nối giữa các tế bào của mạng lưới này rất quan trọng để cung cấp các xung tiết hormone phối hợp đến các mô đích của chúng và để tạo điều kiện cho phản ứng sinh lý phối hợp với các kích thích.
Hóa học Hormone Tăng Trưởng
Hormone tăng trưởng (GH) ở người được sản xuất từ các tế bào hướng thân trong tuyến yên trước dưới dạng một protein chuỗi đơn, không glycosyl hóa, gồm 191 axit amin, 22 kDa, bao gồm một lõi gồm bốn chuỗi xoắn theo hướng song song/đối song song với hai liên kết disulfide giữa các cysteine 53–165 và 182–189. Tiền chất GH 217 axit amin được cắt để loại bỏ peptide tín hiệu trước khi bài tiết.
GH tương đồng với một số protein khác được sản xuất bởi tuyến yên hoặc nhau thai, bao gồm prolactin, chorionic somatomammotropin (CS, placental lactogen), và một biến thể GH 22 kDa hGH-V, chỉ được nhau thai tiết ra. Biến thể sau khác với GH tuyến yên ở 13 axit amin. Các gen của các protein này có lẽ đã có nguồn gốc từ một gen tổ tiên chung, mặc dù hiện nay các gen này nằm trên các nhiễm sắc thể khác nhau.
Gen GH1 nằm trên nhánh dài của nhiễm sắc thể 17 (17q22-24) trong một cụm năm gen tương đồng bao gồm một khoảng cách khoảng 65 kb (CSHP [pseudogene CS], CSH-1 [gen CS], GH-2 và CSH-2). Sự biểu hiện của GH1 được điều hòa bởi promoter gần có tính đa hình cao và một vùng kiểm soát locus (LCR) cách gen 15–32 kb về phía thượng nguồn, mang lại sự biểu hiện đặc hiệu cho tuyến yên và mức độ cao của GH. Thông thường, phần lớn GH (75%) do tuyến yên sản xuất là dạng 22-kDa trưởng thành. Việc cắt nối thay thế dẫn đến việc mất các axit amin từ 32 đến 46, tạo ra một dạng 20-kDa thường chiếm ít hơn 10% GH tuyến yên. Phần còn lại của GH tuyến yên bao gồm các dạng đã khử amid và N-acetyl hóa, cũng như các oligomer GH khác nhau. Một biến thể 17.5-kDa là kết quả của việc bỏ qua hoàn toàn exon 3 và thiếu các axit amin 32–71 có mức độ phong phú thấp hơn nhiều (1%–5%).
Sự bài tiết Hormone Tăng Trưởng
GH có thể được xác định trong huyết thanh thai nhi vào cuối tam cá nguyệt đầu tiên. Mô hình xung đặc trưng của sự bài tiết GH phần lớn phản ánh sự tương tác của nhiều yếu tố điều hòa, bao gồm hai peptide điều hòa của vùng dưới đồi: hormone giải phóng hormone tăng trưởng (GHRH) và somatostatin (yếu tố ức chế giải phóng somatotropin [SRIF]). GHRHR mã hóa một thụ thể kết hợp với protein G bao gồm bảy miền xuyên màng. Sự biểu hiện của GHRHR được điều hòa tăng bởi yếu tố phiên mã đặc hiệu tuyến yên 1 (POU1F1) và cần thiết cho sự tăng sinh của các tế bào hướng thân.
Sự điều hòa sản xuất GH bởi GHRH phần lớn được trung gian qua phiên mã và phụ thuộc vào sự kích thích adenylate cyclase và sự gia tăng nồng độ cyclic adenosine monophosphate (cAMP) nội bào. Các khối u đặc tiết GHRH là một nguyên nhân hiếm gặp của tình trạng thừa GH. GHRH được sử dụng trong chẩn đoán, đặc biệt để xác định tình trạng thiếu hụt GH ở người lớn, khi nó thường được sử dụng kết hợp với arginine. Tuy nhiên, nó không được sử dụng thông thường cho mục đích điều trị ở những bệnh nhân thiếu hụt GH.
Các tác động của peptide somatostatin 14 axit amin dường như điều chỉnh thời gian và biên độ của sự bài tiết GH theo xung, hơn là tổng hợp GH. Sự liên kết của somatostatin với thụ thể đặc hiệu của nó dẫn đến sự ức chế hoạt động của adenylate cyclase và giảm nồng độ canxi nội bào. Sự bài tiết GH theo xung được quan sát in vivo được cho là kết quả của sự giảm đồng thời giải phóng somatostatin từ vùng dưới đồi và tăng hoạt động của GHRH. Ngược lại, một đáy của sự bài tiết GH xảy ra khi giải phóng somatostatin tăng lên trong khi hoạt động của GHRH giảm. Tác động tổng hợp của hai hormone vùng dưới đồi này là điều chỉnh sự bài tiết GH, cũng như thời gian và biên độ của các đỉnh, dẫn đến sự bài tiết GH theo xung. Nhiều chất dẫn truyền thần kinh và neuropeptide tham gia vào việc điều chỉnh sự giải phóng các yếu tố vùng dưới đồi này, bao gồm serotonin, histamine, norepinephrine, dopamine, acetylcholine, axit gamma-aminobutyric (GABA), hormone giải phóng tuyến giáp, peptide ruột vận mạch, gastrin, neurotensin, chất P, calcitonin, neuropeptide Y, vasopressin, hormone giải phóng corticotropin, và galanin. Các yếu tố này có liên quan đến những thay đổi trong sự bài tiết GH được quan sát thấy trong nhiều trạng thái sinh lý khác nhau (chẳng hạn như căng thẳng, giấc ngủ, xuất huyết, nhịn ăn, hạ đường huyết và tập thể dục) và tạo cơ sở cho một số nghiệm pháp kích thích GH được sử dụng trong việc đánh giá khả năng/dự trữ tiết GH. Sự bài tiết GH cũng bị ảnh hưởng bởi nhiều loại hormone không phải peptide, bao gồm androgen, estrogen, thyroxine và glucocorticoid. Các cơ chế chính xác mà các hormone này điều chỉnh sự bài tiết GH rất phức tạp, có thể liên quan đến các tác động ở cấp độ dưới đồi và tuyến yên. Về mặt thực tế, suy giáp và thừa glucocorticoid đều có thể làm giảm sự bài tiết GH tự phát và do kích thích (và do đó nên được điều chỉnh trước khi xét nghiệm GH). Steroid sinh dục, khi bắt đầu dậy thì hoặc được dùng dưới dạng thuốc, dường như chịu trách nhiệm cho sự gia tăng bài tiết GH đặc trưng của tuổi dậy thì. Các hexapeptide tổng hợp có khả năng kích thích bài tiết GH đã được phát triển và được gọi là peptide giải phóng GH (GHRP). Các peptide này, sau này được công nhận là các chất tương tự của hormone ghrelin từ dạ dày, có khả năng kích thích trực tiếp giải phóng GH và tăng cường phản ứng GH với GHRH. Các tác nhân này có lợi thế tiềm năng là dùng đường uống, và ở bệnh nhân có tuyến yên nguyên vẹn, có thể có khả năng tăng cường đáng kể sự bài tiết GH. Khi các tác nhân này được dùng mãn tính cho bệnh nhân cao tuổi và một số trẻ em thiếu GH, biên độ của các xung GH đã tăng lên đáng kể. Các phối tử bắt chước ghrelin đã được sử dụng để xác định một thụ thể chung được gọi là thụ thể chất kích thích tiết GH (GHS-R) cho các chất giải phóng GH. GHS-R là một thụ thể kết hợp với protein G, khác biệt với thụ thể GHRH. Thụ thể này được biểu hiện mạnh ở vùng dưới đồi, nhưng các vị trí gắn kết đặc hiệu cho GHRP cũng đã được xác định ở các vùng khác của hệ thần kinh trung ương và các mô nội tiết và không nội tiết ngoại vi ở cả người và các sinh vật khác. Ghrelin là một peptide 28 axit amin đã được xác định là phối tử nội sinh cho GHS-R. Nó được biểu hiện chủ yếu ở dạ dày, nhưng một lượng nhỏ hơn cũng được sản xuất trong ruột, tuyến tụy, thận, hệ miễn dịch, nhau thai, tuyến yên, tinh hoàn, buồng trứng và vùng dưới đồi. Ghrelin là một sản phẩm gen độc đáo đòi hỏi phải được octanoyl hóa để có chức năng bình thường. Việc tiêm ghrelin vào tĩnh mạch, não thất và phúc mạc trong các mô hình động vật kích thích lượng thức ăn ăn vào và béo phì và làm tăng nồng độ GH trong huyết tương, và ở mức độ thấp hơn, nồng độ prolactin (PRL) và hormone vỏ thượng thận (ACTH). Ngoài ra, nó ảnh hưởng đến chức năng nội tiết của tuyến tụy và chuyển hóa glucose, chức năng tuyến sinh dục và hành vi. Nó cũng kiểm soát nhu động dạ dày và bài tiết axit và có tác dụng tim mạch và chống tăng sinh. Cả ghrelin và GHRP đều giải phóng GH một cách hiệp đồng với GHRH nhưng hiệu quả của các hợp chất này như các tác nhân thúc đẩy tăng trưởng là kém. Các biến thể trong thụ thể ghrelin đã được xác định là một nguyên nhân có thể gây ra tầm vóc thấp vô căn (ISS) và thiếu hụt GH. Tuy nhiên, lưu ý rằng các mô hình chuột bị xóa mục tiêu của thụ thể (ghsr −/−) có kiểu hình gần như bình thường. Một peptide thứ hai được mã hóa bởi cùng một gen với ghrelin đã được xác định và được đặt tên là obestatin. Gen này dường như điều chỉnh cân nặng nhưng không điều chỉnh sự bài tiết GH.
Ngoài các quá trình điều hòa phức tạp được mô tả trước đây, sự tổng hợp và bài tiết GH cũng được điều hòa bởi cơ chế phản hồi ngược của các polypeptide giống insulin (IGF). Các thụ thể IGF đã được xác định trong tuyến yên. Sự ức chế bài tiết GH bởi IGF-1 đã được chứng minh trong nhiều hệ thống. Ngoài ra, sự ức chế bài tiết GH tự phát đã được chứng minh ở người được điều trị bằng các mũi tiêm dưới da của IGF-1 tái tổ hợp. GH cũng điều chỉnh sự bài tiết của chính nó bằng cách tác động trực tiếp lên các thụ thể GH (GHR) ở vùng dưới đồi, thông qua một vòng phản hồi ngược ngắn.
Trong tuổi dậy thì, steroid sinh dục làm tăng biên độ xung GH, góp phần vào nồng độ IGF-1 trong huyết thanh rất cao đặc trưng của tuổi dậy thì. Sự bài tiết GH bắt đầu giảm vào cuối tuổi vị thành niên và tiếp tục giảm trong suốt cuộc đời trưởng thành. Thật vậy, tuổi dậy thì có thể được coi là một giai đoạn “to đầu chi” (acromegaly), trong khi quá trình lão hóa (với đặc điểm giảm bài tiết GH) đã được gọi là somatopause.
Ngoài lão hóa, một loạt các điều kiện sinh lý ảnh hưởng đến sự bài tiết GH. Chúng bao gồm giai đoạn của giấc ngủ, tình trạng dinh dưỡng, nhịn ăn cấp tính, tập thể dục, căng thẳng và steroid sinh dục. Ho và các cộng sự đã báo cáo rằng cả tuổi tác và giới tính đều không ảnh hưởng đến nồng độ GH tích hợp trong huyết thanh khi các tác động của estradiol được loại bỏ khỏi phân tích. Tác động của testosterone lên nồng độ IGF-1 trong huyết thanh có thể ít nhất một phần độc lập với GH vì những người có đột biến GHR vẫn có sự gia tăng IGF-1 trong huyết thanh trong tuổi dậy thì.
Bản chất xung của sự bài tiết GH có thể được chứng minh dễ dàng bằng cách lấy mẫu huyết thanh thường xuyên, đặc biệt khi kết hợp với các xét nghiệm nhạy cho GH. Các xét nghiệm như vậy cho thấy rằng, trong điều kiện bình thường, nồng độ GH trong huyết thanh nhỏ hơn 0,2 ng/mL giữa các đợt bùng phát bài tiết GH. Do đó, không thực tế để đánh giá sự bài tiết GH bằng một mẫu huyết thanh ngẫu nhiên duy nhất. Sự bài tiết GH tối đa xảy ra vào ban đêm, đặc biệt là khi bắt đầu giấc ngủ sóng chậm đầu tiên (giai đoạn III và IV). Giấc ngủ chuyển động mắt nhanh (REM), mặt khác, có liên quan đến sự bài tiết GH thấp.
Nam thanh niên bình thường thường trải qua trung bình 12 đợt bùng phát bài tiết GH mỗi 24 giờ. Béo phì được đặc trưng bởi sự giảm bài tiết GH, phản ánh bởi số lượng các đợt bùng phát bài tiết GH giảm. Nhịn ăn làm tăng số lượng và biên độ của các đợt bùng phát bài tiết GH, có lẽ phản ánh sự giảm bài tiết somatostatin. Tác động của bản chất bài tiết theo xung của GH đối với các hoạt động sinh học của nó vẫn chưa chắc chắn.
Thụ thể Hormone Tăng Trưởng/Protein Gắn Hormone Tăng Trưởng
Thụ thể GH (GHR) bao gồm một miền ngoại bào, gắn hormone, tiếp theo là một miền xuyên màng duy nhất và một miền tế bào chất. Hai dạng đồng phân GHR gen chỉ tồn tại ở người đã phát sinh từ sự tái tổ hợp tương đồng của tổ tiên. Chúng khác nhau ở việc giữ lại hoặc xóa exon 3. Exon 3 của GHR đã được chứng minh là bị xóa ở một số cá nhân bình thường. Đa hình delta-3 GHR này đã được một số, nhưng không phải tất cả, các nhà điều tra chứng minh là ảnh hưởng đến khả năng đáp ứng với GH và có liên quan đến kích thước khi sinh và sự tăng trưởng sau sinh. Cũng có những dạng đồng phân ngắn của GHR do cắt nối thay thế, dẫn đến một thụ thể bị cắt cụt với sự mất đi một phần lớn của miền tế bào chất.
GHR là một thành viên của họ cytokine tạo máu loại 1 và có tính tương đồng cao với thụ thể prolactin và chia sẻ sự tương đồng về trình tự với nhiều thụ thể cho interleukin (IL), cũng như các thụ thể cho erythropoietin (EPO), leptin, yếu tố kích thích cụm tế bào hạt-đại thực bào, và interferon.
Kiểm tra cấu trúc tinh thể của phức hợp GH-GHR cho thấy phức hợp này bao gồm một phân tử GH liên kết với hai phân tử GHR, cho thấy sự dimer hóa thụ thể do GH gây ra—điều cần thiết cho hoạt động của GH. Điều thú vị là, một phức hợp dung hợp được biến đổi gen của GH và GHR đã được chứng minh là có hiệu quả được cải thiện đáng kể và thời gian bán hủy dài hơn đáng kể so với GH đơn độc khi được thử nghiệm trên các mô hình động vật gặm nhấm.
GHR, giống như thành viên cùng nhóm gia đình EPO-R, được hình thành trước dưới dạng dimer và được vận chuyển ở trạng thái không liên kết với phối tử đến bề mặt tế bào. GH sau đó liên kết theo một cách tuần tự với dimer GHR, trong đó GHR đầu tiên liên kết với vị trí 1 mạnh hơn của phân tử GH, tiếp theo là GHR thứ hai liên kết với vị trí 2 yếu hơn. Sự liên kết của GH dẫn đến một sự thay đổi về cấu hình, theo đó sự quay của các GHR dẫn đến việc tái định vị các miền nội bào và của Janus Kinase 2 (JAK2) liên kết với Hộp 11-1, một tyrosine kinase chính liên kết với GHR. Kết quả là, JAK2 được tự phosphoryl hóa và kích hoạt, điều này lần lượt dẫn đến sự phosphoryl hóa chéo của các gốc tyrosine ở đầu xa của GHR. Điều này cho phép các phân tử miền SH2 (Src homology 2) gắn vào các vị trí này. Bản thân GHR dường như không có hoạt động kinase nội tại. Có khả năng sự đồng định vị của hai phân tử JAK2 bởi GHR đã được dimer hóa dẫn đến sự phosphoryl hóa chéo của một JAK2 bởi phân tử kia, dẫn đến sự kích hoạt JAK2. Các phân tử truyền tín hiệu và hoạt hóa phiên mã (Stat)5a và 5b chứa các miền SH2 và liên kết với các vị trí tyrosine đã được phosphoryl hóa này, và sau đó chúng lần lượt bị phosphoryl hóa. Các phân tử Stat5 đã được phosphoryl hóa (homo- và hetero-) dimer hóa và chuyển vị vào nhân, nơi chúng liên kết với axit deoxyribonucleic (DNA), dưới dạng dimer hoặc tetramer, và kích hoạt các gen đích. Quan trọng là, sự dimer hóa GHR bởi GH được khai thác bằng cách sử dụng Pegvisomant, một dạng GH biến đổi gen, chỉ liên kết với một thụ thể và do đó không thể dimer hóa thụ thể. Do đó, nó hoạt động như một chất đối kháng GH và có thể được sử dụng để điều trị bệnh khổng lồ tuyến yên và bệnh to đầu chi.
Cả Stat5a và Stat5b đều có thể được kích hoạt bởi GH, và chúng có cả chức năng chồng chéo và riêng biệt. Các mô hình chuột bất hoạt gen đã cho thấy rằng Stat5b có tầm quan trọng lớn hơn đối với việc kích thích tăng trưởng so với Stat5a.
Điều hòa tiêu cực của tín hiệu Hormone Tăng Trưởng
Sự kích hoạt tín hiệu JAK-STAT xảy ra nhanh chóng, trong vòng vài phút sau khi kích thích GH, nhưng chỉ là tạm thời do sự kiểm soát chặt chẽ của việc chấm dứt tín hiệu. Sự điều hòa tiêu cực của tín hiệu này xảy ra ở nhiều cấp độ: sự nội hóa GHR, các chất ức chế tín hiệu cytokine (SOCS), các protein tyrosine phosphatase (PTP), và các chất ức chế protein của các Stats đã được kích hoạt (PIAS).
Ở người, protein gắn GH (GHBP) lưu hành dường như bắt nguồn từ sự phân cắt protein của miền ngoại bào của GHR. GHBP có ái lực với GH giống hệt GHR. Nó liên kết với GH với độ đặc hiệu và ái lực cao, nhưng với dung lượng tương đối thấp. Nó làm tăng thời gian bán hủy của GH trong tuần hoàn và có thể có chức năng vận chuyển GH đến các mô đích và liên kết sau đó với thụ thể. GHBP, giống như GHR, có mặt ở nhiều mô, nhưng GHBP trong tuần hoàn chủ yếu có nguồn gốc từ gan. Mặc dù GHR và GHBP được điều hòa bởi và rất nhạy cảm với GH, và GHR và GHBP thường thay đổi song song, việc đo GHBP trong huyết tương đã không được chứng minh là phản ánh GHR và khả năng đáp ứng với GH. Tuy nhiên, việc đo nồng độ GHBP trong huyết thanh có thể hỗ trợ xác định những bệnh nhân không nhạy cảm với GH (GHI) do các bất thường di truyền của GHR. Bệnh nhân bị GHI do các bất thường không phải thụ thể, các bất thường của phần nội bào của GHR, hoặc không có khả năng dimer hóa của thụ thể có thể có nồng độ GHBP trong huyết thanh bình thường. Gần đây, một phức hợp của GH và phân tử GHBP đã được chứng minh là hiệu quả hơn GH đơn độc—cho thấy một vai trò sinh lý và có thể có vai trò điều trị cho GHBP.
Tác động của Hormone Tăng Trưởng
Theo giả thuyết somatomedin, các tác động đồng hóa của GH được trung gian thông qua các polypeptide IGF. Mặc dù giả thuyết này ít nhất một phần là đúng, dường như GH có khả năng kích thích nhiều tác động khác nhau độc lập với hoạt động của IGF. Thật vậy, các tác động của GH và IGF đôi khi trái ngược nhau, ví dụ như tác động “gây đái tháo đường” của GH và hoạt động hạ đường huyết của IGF. Green và các đồng nghiệp đã cố gắng giải quyết một số khác biệt này trong một mô hình “tác nhân kép” trong đó GH kích thích các tế bào tiền thân, chẳng hạn như tiền tế bào sụn, biệt hóa. Khi các tế bào đã biệt hóa hoặc các tế bào lân cận sau đó tiết ra IGF, các peptide này hoạt động như các chất gây phân bào và kích thích sự mở rộng dòng vô tính. Giả thuyết này dựa trên khả năng của các peptide IGF hoạt động không chỉ như các yếu tố nội tiết cổ điển được vận chuyển qua máu mà còn như các yếu tố tăng trưởng cận tiết hoặc tự tiết. GH cũng kích thích nhiều tác động chuyển hóa khác nhau, một số trong đó dường như xảy ra độc lập với việc sản xuất IGF, chẳng hạn như phân giải lipid, vận chuyển axit amin trong cơ hoành và tim, và sản xuất các protein gan đặc hiệu. Do đó, có nhiều vị trí tác động của GH, và không hoàn toàn rõ ràng tác động nào trong số này được trung gian qua hệ thống IGF và tác động nào có thể đại diện cho các tác động độc lập với IGF-1 của GH.
Yếu tố tăng trưởng giống Insulin-1
Yếu tố tăng trưởng giống Insulin-1: Bối cảnh lịch sử
Các IGF (hoặc somatomedin) tạo thành một họ peptide ít nhất một phần phụ thuộc vào GH và được cho là trung gian cho nhiều tác động đồng hóa và gây phân bào của GH. Mặc dù ban đầu chúng được xác định vào năm 1957 bởi khả năng kích thích sự kết hợp của [35S] sulfate vào sụn chuột, trong 45 năm sau đó, người ta đã xác định rằng chúng tham gia vào các hoạt động chuyển hóa đa dạng.
Cấu trúc và Sinh học phân tử của Yếu tố tăng trưởng giống Insulin
IGF-1, trước đây được gọi là somatomedin-C, là một polypeptide kiềm gồm 70 axit amin, trong khi IGF-2 là một polypeptide có tính axit nhẹ gồm 67 axit amin. Hai peptide này có liên quan về mặt cấu trúc, chia sẻ 45 trong số 73 vị trí axit amin có thể có. Chúng có khoảng 50% tương đồng axit amin với insulin. Giống như insulin, cả hai IGF đều có chuỗi A và B được nối với nhau bằng các liên kết disulfide và một vùng nối trung gian (peptide C). Sự tương đồng về cấu trúc này giải thích khả năng của cả hai IGF liên kết với thụ thể insulin và khả năng của insulin liên kết với thụ thể IGF loại 1. Mặt khác, những khác biệt về cấu trúc giải thích sự thất bại của insulin trong việc liên kết với các protein gắn IGF.
Nồng độ các Yếu tố tăng trưởng giống Insulin trong huyết thanh
Trong huyết thanh thai nhi người, nồng độ IGF-1 tương đối thấp và tương quan thuận với tuổi thai. Một số nhóm đã báo cáo mối tương quan giữa nồng độ IGF-1 trong huyết thanh dây rốn của thai nhi với cân nặng khi sinh, mặc dù những nhóm khác không báo cáo mối tương quan nào. Nồng độ IGF-1 trong huyết thanh trẻ sơ sinh người thường bằng 30% đến 50% nồng độ của người lớn. Có sự gia tăng chậm, dần dần của nồng độ trong huyết thanh trong thời thơ ấu, đạt đến nồng độ của người lớn khi bắt đầu trưởng thành về mặt sinh dục. Trong tuổi dậy thì, nồng độ IGF-1 tăng lên 2 đến 3 lần so với nồng độ ở người lớn. Do đó, nồng độ trong tuổi vị thành niên tương quan tốt hơn với giai đoạn Tanner (tuổi xương) hơn là với tuổi theo thời gian. Các bé gái bị loạn sản tuyến sinh dục không cho thấy sự gia tăng IGF-1 trong huyết thanh ở tuổi vị thành niên, điều này rõ ràng xác định mối liên hệ giữa sự gia tăng IGF-1 ở tuổi dậy thì với việc sản xuất steroid sinh dục. Estrogen kích thích bài tiết GH, làm tăng sản xuất IGF-1 ở gan. Tuy nhiên, điều đáng chú ý là những bệnh nhân bị GHI do đột biến GHR vẫn cho thấy sự gia tăng IGF-1 trong huyết thanh ở tuổi dậy thì, điều này có thể được trung gian bởi các tác động trực tiếp của androgen lên IGF-1.
Sau tuổi vị thành niên, hoặc ít nhất là sau 20 đến 30 tuổi, nồng độ IGF-1 trong huyết thanh cho thấy sự suy giảm dần dần và tiến triển theo tuổi. Người ta cho rằng sự suy giảm này có thể là nguyên nhân gây ra cân bằng nitơ âm, giảm khối lượng cơ bắp và loãng xương đặc trưng của quá trình lão hóa. Mặc dù giả thuyết gây tranh cãi này vẫn chưa được chứng minh vào thời điểm này, nó đã tạo ra sự quan tâm đáng kể đến việc sử dụng tiềm năng liệu pháp GH và/hoặc IGF-1 trong quá trình lão hóa bình thường.
Nồng độ IGF-2 ở trẻ sơ sinh người thường bằng 50% nồng độ của người lớn. Tuy nhiên, đến 1 tuổi, nồng độ của người lớn đã đạt được với rất ít, nếu có, sự suy giảm sau đó ngay cả đến thập kỷ thứ bảy hoặc thứ tám của cuộc đời. Điều này trái ngược với loài gặm nhấm, có sự biểu hiện IGF-2 giảm sớm sau khi sinh. Việc thiếu các mô hình động vật gặm nhấm đã cản trở sự hiểu biết của chúng ta về vai trò sinh lý của sự tồn tại của IGF-2 ở người.
Thụ thể của Yếu tố tăng trưởng giống Insulin
Các IGF liên kết (mặc dù thường với ái lực thấp) với các thụ thể insulin, do đó cung cấp một lời giải thích cho hoạt động giống insulin của chúng. Ngoài ra, các IGF liên kết với ít nhất hai loại thụ thể IGF.
Thụ thể IGF loại 1 có liên quan chặt chẽ với thụ thể insulin. Cả hai đều là các heterotetramer bao gồm hai tiểu đơn vị alpha giống hệt nhau xuyên màng và hai tiểu đơn vị beta giống hệt nhau trong tế bào chất. Các tiểu đơn vị alpha chứa các vị trí liên kết cho IGF-1 và được liên kết bởi các liên kết disulfide. Các tiểu đơn vị beta chứa một miền xuyên màng, một vị trí liên kết adenosine triphosphate (ATP) và một miền tyrosine kinase, tạo thành cơ chế truyền tín hiệu được cho là của thụ thể.
Mặc dù thụ thể IGF loại 1 thường được gọi là thụ thể IGF-1, thụ thể này có khả năng liên kết cả IGF-1 và IGF-2 với ái lực cao—và cả hai polypeptide IGF dường như đều có khả năng kích hoạt tyrosine kinase bằng cách liên kết với thụ thể này. Ái lực của thụ thể loại 1 đối với insulin thường thấp hơn 100 lần, do đó cung cấp một trong những cơ chế cho các tác động gây phân bào của insulin. IGF-1 hoạt động chủ yếu thông qua IGF1R nhưng có thể liên kết với ái lực thấp hơn với thụ thể insulin (IR) có tính tương đồng cao, và với các dị dimer IGF1R/IR. Ngược lại, insulin có khả năng truyền tín hiệu thông qua IGF1R. Dấu hiệu của tín hiệu thay thế như vậy có thể trở nên rõ ràng trong tín hiệu IGF-1 hoặc insulin bệnh lý.
Thụ thể IGF loại 1 trung gian cho các tác động của IGF trên nhiều loại tế bào, và các tác động này đa dạng và đặc hiệu cho từng mô. Nhìn chung, người ta tin rằng tất cả các tác động của việc kích hoạt thụ thể IGF đều được trung gian bởi sự kích hoạt tyrosine kinase và sự phosphoryl hóa các cơ chất, kích hoạt các con đường tế bào cụ thể, dẫn đến các hoạt động sinh học khác nhau. Trong số các tác động này có sự cảm ứng tăng trưởng tế bào thông qua việc kích hoạt bộ máy chu kỳ tế bào, duy trì sự sống sót của tế bào (ngăn chặn apoptosis) được trung gian bởi các tác động lên các thành viên họ Bcl, và sự cảm ứng biệt hóa tế bào, xảy ra bởi các cơ chế chưa được mô tả đầy đủ.
Các cơ chất được phosphoryl hóa bởi thụ thể IGF bao gồm các thành viên của họ cơ chất thụ thể insulin (đặc biệt là IRS-1 và IRS-2); việc loại bỏ các gen này dẫn đến tăng trưởng kém ở chuột (cũng như kháng insulin). Ngoài ra, một số phân tử tín hiệu khác đáp ứng với việc kích hoạt thụ thể IGF. Việc phong tỏa thụ thể loại I đã được đề xuất như một liệu pháp điều trị ung thư.
Các đột biến làm giảm tín hiệu IGF dẫn đến sự kéo dài tuổi thọ ở giun tròn, ruồi và chuột. Tuy nhiên, không rõ IGF-1/IR có liên quan gì đến tuổi thọ của con người. Dữ liệu gần đây đã cho thấy rằng các biến thể mất chức năng trong IGF1R có tỷ lệ cao hơn ở những phụ nữ sống trăm tuổi, cho thấy vai trò của con đường này trong việc điều chỉnh tuổi thọ của con người.
Tuy nhiên, thụ thể IGF loại 2 không có sự tương đồng về cấu trúc với thụ thể insulin hoặc thụ thể IGF loại 1. Thụ thể loại 2 không chứa một miền tyrosine kinase nội tại hoặc bất kỳ cơ chế truyền tín hiệu nào có thể nhận biết khác và đã được phát hiện là giống hệt với thụ thể mannose-6-phosphate độc lập với cation (CIM6P), một protein liên quan đến việc nhắm mục tiêu lysosome nội bào của nhiều loại hydrolase axit và các protein mannosyl hóa khác. Không giống như thụ thể IGF loại 1, liên kết cả hai polypeptide IGF với ái lực cao và insulin với ái lực thấp hơn 100 lần, thụ thể loại 2 chỉ liên kết IGF-2 với ái lực cao. IGF-1 liên kết với ái lực thấp hơn đáng kể, và insulin hoàn toàn không liên kết. Hầu hết các nghiên cứu đã chỉ ra rằng các tác động gây phân bào và chuyển hóa cổ điển của IGF-1 và IGF-2 được trung gian thông qua thụ thể IGF loại 1, với cơ chế truyền tín hiệu tyrosine kinase của nó.
Siêu họ Protein Gắn Yếu tố tăng trưởng giống Insulin
Mặc dù insulin và các IGF có sự tương đồng đáng kể về cấu trúc, và mặc dù có sự tương đồng về cấu trúc-chức năng của các thụ thể insulin và IGF loại 1, các IGF khác với insulin ở chỗ các IGF lưu hành trong huyết tương ở dạng phức hợp với một họ các protein gắn. Các protein mang này kéo dài thời gian bán hủy trong huyết thanh của các peptide IGF, vận chuyển các IGF đến các tế bào đích và điều chỉnh sự tương tác của các IGF với các thụ thể màng bề mặt của chúng. Sáu IGFBP riêng biệt đã được xác định. Các protein gắn IGF có ái lực thấp hơn bổ sung (được đặt tên là các protein liên quan đến IGFBP, IGFBPrP) đã được tìm thấy bằng các tìm kiếm in silico về sự tương đồng với các IGFBP đã biết; nhiều phân tử này đã được biết đến trước đây trong các bối cảnh khác, phục vụ các vai trò trong sự tăng trưởng bình thường hoặc tân sản.
Trong hầu hết các điều kiện, các IGFBP dường như ức chế tác động của IGF—có lẽ bằng cách cạnh tranh với các thụ thể IGF để giành các polypeptide IGF. Khái niệm này được hỗ trợ bởi quan sát rằng các chất tương tự IGF có ái lực giảm đối với các IGFBP thường dường như có hiệu lực sinh học tăng lên. Ngoài ra, IGFBP-3 dường như ức chế sự tăng trưởng của tế bào ngay cả khi không có IGF được thêm vào—cho thấy một vai trò ức chế trực tiếp của protein gắn. Tuy nhiên, trong các điều kiện cụ thể, một số IGFBP dường như có khả năng tăng cường tác động của IGF—có lẽ bằng cách tạo điều kiện thuận lợi cho việc cung cấp IGF đến các thụ thể đích.
Bằng chứng cho thấy IGFBP là các phân tử hoạt tính sinh học thiết yếu, ngoài việc gắn với IGF, còn có nhiều chức năng độc lập với IGF-1. Chúng bao gồm ức chế tăng trưởng ở một số loại tế bào, kích thích tăng trưởng ở các mô khác, gây ra apoptosis trực tiếp và điều chỉnh tác động của các yếu tố tăng trưởng không phải IGF khác. Các tác động này của IGFBP được trung gian bởi việc gắn vào các thụ thể của riêng chúng. Vì IGFBP-3 được điều hòa bởi GH, điều thú vị là in vivo, IGFBP-3 tăng cường tác động của IGF-1 khi được dùng cho chuột bị cắt bỏ tuyến yên (thay vì ức chế nó). Các cơ chế liên quan đến tác động này chưa được làm sáng tỏ nhưng có thể giải thích các tác động hạn chế của liệu pháp IGF-1 đối với sự tăng trưởng của bệnh nhân Laron.
Lượng tương đối của mỗi IGFBP thay đổi giữa các dịch sinh học. IGFBP-1 là IGFBP chính trong nước ối của người. IGFBP-2 chiếm ưu thế trong dịch não tủy và huyết tương tinh dịch. IGFBP-3 là IGFBP chính trong huyết thanh người bình thường và cho thấy sự phụ thuộc rõ ràng vào GH. Trong số các IGFBP, IGFBP-3 và IGFBP-5 là duy nhất ở chỗ chúng thường lưu hành trong huyết thanh người lớn như một phần của một phức hợp tam phân bao gồm IGFBP-3 hoặc IGFBP-5, một polypeptide IGF, và một tiểu đơn vị không bền với axit (ALS).
Phân tích các IGFBP còn phức tạp hơn bởi sự hiện diện của các protease IGFBP, có khả năng phân giải IGFBP ở các mức độ khác nhau. Ban đầu được báo cáo trong huyết thanh của phụ nữ mang thai, các protease cho IGFBP-3, -4 và -5 đã được chứng minh trong nhiều loại dịch sinh học. Sự phân giải protein của các IGFBP làm phức tạp việc xét nghiệm chúng bằng cả phương pháp Western ligand blotting và phương pháp xét nghiệm miễn dịch phóng xạ và phải được tính đến khi báo cáo nồng độ của các IGFBP khác nhau trong các dịch sinh học. Ý nghĩa sinh lý của sự phân giải protein hạn chế của các IGFBP vẫn cần được xác định, mặc dù bằng chứng cho thấy rằng hoạt động của protease dẫn đến giảm ái lực của IGFBP đối với các peptide IGF. Gần đây, các đột biến lặn trong metalloproteinase protein huyết tương A2 liên quan đến thai kỳ (PAPP-A2) có liên quan đến tầm vóc thấp. Các đột biến này dẫn đến tăng liên kết của IGF-1 trong phức hợp tam phân và giảm IGF-1 tự do.
Phá vỡ có mục tiêu các thành phần của hệ thống Yếu tố tăng trưởng giống Insulin
Vai trò quan trọng của hệ thống IGF trong sự tăng trưởng của thai nhi và sau sinh đã được chứng minh trong một loạt các nghiên cứu loại bỏ gen tinh vi ở chuột. Không giống như các con chuột bị loại bỏ GH và GHR, có kích thước gần như bình thường khi sinh ra, những con chuột không có Igf1 có cân nặng khi sinh bằng 60% so với bình thường. Sự tăng trưởng sau sinh cũng bất thường. Một kiểu hình tăng trưởng trước và sau sinh tương tự đã được quan sát thấy trong một trường hợp được báo cáo ở người về việc xóa gen Igf1. Những con chuột không có Igf2 cũng cho thấy sự tăng trưởng trước và sau sinh bị suy giảm. Gen Igf2 được in dấu ở người, do đó chỉ có gen của cha được biểu hiện. Do đó, các đột biến mất chức năng trên alen của cha gây ra sự suy giảm tăng trưởng trong khi các đột biến trên alen của mẹ thì không. Khi gen của thụ thể IGF loại 1 bị loại bỏ (những con chuột không có Igf1r), những con chuột này bị chậm tăng trưởng nghiêm trọng. Ở người, các đột biến IGF1R trội và lặn có liên quan đến chậm tăng trưởng trong tử cung (IUGR), suy giảm tăng trưởng sau sinh và khó khăn trong học tập (xem phần Rối loạn tăng trưởng ở trẻ em để biết chi tiết). Mối quan hệ giữa GH và IGF-1 trong việc kiểm soát sự tăng trưởng sau sinh đã được phân tích ở những con chuột đột biến thiếu GHR, IGF-1, hoặc cả hai. Điều này đã chứng minh rằng GH và IGF-1 thúc đẩy sự tăng trưởng sau sinh bằng các chức năng độc lập và chung.
Hormone tuyến giáp
Hormone tuyến giáp là cần thiết cho sự tăng trưởng theo chiều dọc bình thường. Trẻ sơ sinh bị suy giáp có chiều dài khi sinh bình thường, cho thấy sự tăng trưởng của thai nhi không bị ảnh hưởng nhiều bởi việc sản xuất hormone tuyến giáp của thai nhi. Ngược lại, suy giáp sau sinh nghiêm trọng, kéo dài có thể làm suy giảm sự tăng trưởng theo chiều dọc và làm chậm sự trưởng thành của xương. Các đột biến ở thụ thể hormone tuyến giáp-alpha hoặc -beta gây ra một số sự suy giảm trong tăng trưởng theo chiều dọc, cho thấy cả hai thụ thể này đều góp phần vào việc điều hòa tăng trưởng theo chiều dọc. Hormone tuyến giáp có cả tác động trực tiếp lên các tế bào sụn ở sụn tăng trưởng, đặc biệt là hỗ trợ sự phì đại của tế bào sụn, và một tác động gián tiếp, được trung gian bởi tác động kích thích lên sự bài tiết hormone tăng trưởng.
Glucocorticoid
Glucocorticoid, khi có mặt ở nồng độ vượt quá mức sinh lý, sẽ làm suy giảm sự tăng trưởng theo chiều dọc. Sự suy giảm tăng trưởng theo chiều dọc này một phần phát sinh từ tác động trực tiếp của glucocorticoid lên sự tăng sinh của tế bào sụn ở sụn tăng trưởng. Glucocorticoid cũng có thể ảnh hưởng gián tiếp đến quá trình tạo sụn ở sụn tăng trưởng bằng cách thay đổi sự bài tiết GH. Tuy nhiên, không phải tất cả các nghiên cứu đều cho thấy một tác động, và bất kỳ tác động nào cũng có thể là do tình trạng béo phì do glucocorticoid gây ra. Nồng độ IGF-1 lưu hành, một dấu hiệu và chất trung gian của hoạt động GH, dường như không bị giảm do thừa glucocorticoid, điều này phù hợp với ý tưởng rằng nồng độ GH giảm chủ yếu là do béo phì. Việc điều trị bằng GH không có khả năng bù đắp hoàn toàn cho sự suy giảm tăng trưởng theo chiều dọc do thừa glucocorticoid gây ra càng cho thấy rằng sự suy giảm tăng trưởng chủ yếu là do tác động trực tiếp của glucocorticoid lên các tế bào sụn ở sụn tăng trưởng.
Estrogen
Ở thanh thiếu niên, cả nam và nữ, estrogen kích thích sự tăng trưởng theo chiều dọc, góp phần vào giai đoạn tăng vọt ở tuổi dậy thì. Ở bé trai, estrogen kích thích giai đoạn tăng vọt ở tuổi vị thành niên được sản xuất bởi quá trình thơm hóa các androgen của tinh hoàn và tuyến thượng thận. Sự tăng tốc tăng trưởng được trung gian một phần bởi tác động kích thích của estrogen lên sự bài tiết GH. Ngoài ra, estrogen dường như có các tác động trực tiếp lên các tế bào sụn ở sụn tăng trưởng. Estrogen kích thích sự lão hóa của sụn tăng trưởng, chương trình phát triển trong sụn tăng trưởng chịu trách nhiệm cho sự chậm lại dần dần của sự tăng trưởng theo chiều dọc theo tuổi tác. Do đó, việc tiếp xúc với estrogen dẫn đến sự “lão hóa” nhanh hơn của sụn tăng trưởng, ngừng tăng trưởng sớm hơn và cốt hóa sụn đầu xương sớm hơn. Tác động này của estrogen giải thích cho sự cốt hóa sụn đầu xương sớm ở trẻ dậy thì sớm và sự cốt hóa sụn đầu xương muộn ở những người bị suy sinh dục, thiếu hụt aromatase và kháng estrogen.
Do đó, tác động của estrogen lên sự tăng trưởng theo chiều dọc là một “con dao hai lưỡi” ở chỗ nó có hai tác động đối lập lên chiều cao người lớn. Estrogen kích thích tốc độ tăng trưởng theo chiều dọc, điều này được kỳ vọng sẽ làm tăng chiều cao người lớn, nhưng cũng đẩy nhanh quá trình lão hóa của sụn tăng trưởng, điều này được kỳ vọng sẽ làm giảm chiều cao người lớn. Nhìn chung, tác động sau chiếm ưu thế. Kết quả là, dậy thì sớm không được điều trị có thể gây ra tầm vóc cao ban đầu ở trẻ em nhưng cuối cùng là tầm vóc thấp khi trưởng thành, trong khi suy sinh dục không được điều trị hoặc dậy thì muộn có thể gây ra tầm vóc thấp ở tuổi vị thành niên nhưng cuối cùng là tầm vóc cao khi trưởng thành. Những khái niệm này cũng cho thấy rằng các chất tương tự hormone giải phóng gonadotropin (GnRH) và các chất ức chế aromatase sẽ làm tăng chiều cao người lớn (xem phần Điều trị tầm vóc thấp, ở phần sau của chương này).
Androgen
Androgen cũng có thể đẩy nhanh sự tăng trưởng theo chiều dọc và do đó góp phần vào giai đoạn tăng vọt ở tuổi dậy thì. Androgen, được tiết ra bởi tuyến sinh dục hoặc tuyến thượng thận, có thể được thơm hóa thành estrogen ở các mô ngoại vi khác nhau, bao gồm cả mô mỡ và do đó ảnh hưởng đến sự tăng trưởng theo chiều dọc. Aromatase cũng được biểu hiện trong sụn tăng trưởng; do đó, sự chuyển đổi cục bộ thành estrogen có thể xảy ra. Ngoài vai trò của androgen như một tiền chất cho estrogen, bản thân androgen dường như cũng kích thích sự tăng trưởng theo chiều dọc. Ví dụ, dihydrotestosterone, không thể thơm hóa được, dường như đẩy nhanh sự tăng trưởng theo chiều dọc ở các bé trai vị thành niên. Các nghiên cứu trên động vật và trong ống nghiệm cho thấy rằng những tác động này được trung gian, ít nhất một phần, thông qua một tác động trực tiếp lên sụn tăng trưởng.
Insulin
Insulin điều hòa tích cực sự tăng trưởng của thai nhi người. Do đó, sự chậm tăng trưởng của thai nhi xảy ra ở những cá nhân bị giảm sản xuất insulin của thai nhi do đái tháo đường sơ sinh vĩnh viễn và những người bị giảm độ nhạy insulin do các khiếm khuyết thụ thể insulin hai alen nghiêm trọng. Ngược lại, sự tăng trưởng của thai nhi tăng lên trong các thai kỳ bị biến chứng bởi đái tháo đường thai kỳ. Tình trạng thai to này, chủ yếu liên quan đến cân nặng hơn là chiều dài, được cho là do nồng độ glucose cao kích thích nồng độ insulin của thai nhi cao.
Điều hòa dinh dưỡng của sự tăng trưởng theo chiều dọc
Việc hấp thu không đủ dinh dưỡng gây ra tình trạng không nhạy cảm với GH chức năng, với nồng độ IGF-1 lưu hành giảm và nồng độ GH tăng. Các cơ chế được đề xuất bao gồm nồng độ insulin giảm, gây ra sự điều hòa giảm biểu hiện của GHR và nồng độ FGF-21 tăng, gây ra sự giảm GHR ở gan và STAT5 đã được phosphoryl hóa (có liên quan đến sự truyền tín hiệu GH). Nồng độ FGF-21 tăng trong tình trạng suy dinh dưỡng có thể làm suy giảm độ nhạy với GH không chỉ ở gan mà còn ở sụn tăng trưởng. Ngoài ra, suy dinh dưỡng có thể làm giảm nồng độ hormone tuyến giáp, như một phần của hội chứng bệnh không do tuyến giáp, tăng nồng độ cortisol, và giảm steroid sinh dục (được trung gian bởi leptin giảm và do đó gonadotropin giảm), tất cả đều có thể góp phần vào sự suy giảm tăng trưởng theo chiều dọc.
Ngược lại, dinh dưỡng quá mức có thể kích thích sự tăng trưởng theo chiều dọc. Trẻ em béo phì có xu hướng cao với tuổi xương tăng. Do sự trưởng thành xương được đẩy nhanh và, ở các bé gái, dậy thì sớm hơn, có ít ảnh hưởng đến chiều cao người lớn. Các cơ chế được đề xuất mà béo phì ảnh hưởng đến sự tăng trưởng theo chiều dọc bao gồm nồng độ estrogen tăng do sự thơm hóa ngoại vi và tăng IGF-1 tự do, leptin, prolactin, và androgen tuyến thượng thận.
Điều hòa Cytokine của sự tăng trưởng theo chiều dọc
Sự suy giảm tăng trưởng theo chiều dọc thường xảy ra ở trẻ em bị các rối loạn viêm hệ thống, bao gồm viêm khớp tự phát thiếu niên (JIA), bệnh viêm ruột (IBD) và xơ nang. Các cơ chế cơ bản có khả năng phức tạp và liên quan đến nhiều chất trung gian, bao gồm suy dinh dưỡng và thừa glucocorticoid. Ngoài ra, có bằng chứng cho thấy mức độ tăng của các cytokine tiền viêm góp phần vào sự tăng trưởng kém. Các cytokine tiền viêm khác nhau, bao gồm yếu tố hoại tử khối u-α (TNF-α), IL-1β, và IL-6, tác động trực tiếp lên các tế bào sụn ở sụn tăng trưởng để ức chế sự phát triển của xương. Các cytokine tiền viêm cũng có thể tác động gián tiếp lên sụn tăng trưởng bằng cách làm giảm nồng độ IGF-1, steroid sinh dục và hormone tuyến giáp lưu hành. Các tác động của các cytokine này có thể được trung gian một phần bởi sự điều hòa tăng của một protein nội bào được gọi là SOCS, làm giảm tín hiệu GHR.
Điều hòa tự tiết/cận tiết của sự tăng trưởng theo chiều dọc
Nhiều yếu tố tự tiết/cận tiết khác nhau được tiết ra bởi các tế bào sụn ở sụn tăng trưởng và tác động cục bộ lên các tế bào sụn khác. Các yếu tố này phục vụ để điều phối các quá trình phức tạp của sự tăng sinh và biệt hóa trong sụn tăng trưởng. Do đó, các khiếm khuyết di truyền trong các yếu tố tăng trưởng cục bộ này, các thụ thể của chúng, hoặc các gen khác liên quan đến tín hiệu yếu tố tăng trưởng có thể làm suy giảm quá trình tạo sụn ở sụn tăng trưởng và do đó làm suy giảm sự tăng trưởng theo chiều dọc của trẻ. Tùy thuộc vào hệ thống cận tiết cụ thể liên quan và mức độ nghiêm trọng của bất thường di truyền, xương có thể vừa ngắn vừa bị dị dạng, biểu hiện lâm sàng như một bệnh loạn sản sụn, hoặc ngắn nhưng không bị dị dạng, biểu hiện lâm sàng như tầm vóc thấp đơn độc. Sau đây là một số ví dụ về các yếu tố tăng trưởng tự tiết/cận tiết điều hòa các tế bào sụn ở sụn tăng trưởng và có tầm quan trọng lâm sàng.
Peptide lợi niệu loại C
Peptide lợi niệu loại C (CNP) được đặt tên dựa trên sự tương đồng về cấu trúc của nó với các peptide lợi niệu tâm nhĩ và não. Tuy nhiên, CNP phục vụ các chức năng khác nhau. Nó tác động cục bộ trong sụn tăng trưởng để kích thích sự tăng sinh và phì đại của tế bào sụn. Do đó, các đột biến mất chức năng trong NPR2, thụ thể của CNP, gây ra tầm vóc thấp trong khi các đột biến tăng chức năng gây ra tầm vóc cao (xem phần Rối loạn tăng trưởng ở trẻ em để biết chi tiết). Các chất tương tự CNP đang trong các thử nghiệm lâm sàng để tăng cường sự phát triển của xương trong bệnh loạn sản sụn (achondroplasia – ACH).
Các Yếu tố tăng trưởng Nguyên bào sợi
Các FGF, tác động thông qua thụ thể FGF-3 (FGFR3), điều hòa tiêu cực quá trình tạo sụn ở sụn tăng trưởng. Kết quả là, các đột biến tăng chức năng trong FGFR3 làm suy giảm sự tăng trưởng theo chiều dọc. Tùy thuộc vào mức độ nghiêm trọng của đột biến, biểu hiện lâm sàng có thể bao gồm một số bệnh loạn sản xương với tầm vóc thấp nghiêm trọng. Bằng chứng gần đây cho thấy rằng các đột biến tăng chức năng nhẹ hơn có thể biểu hiện như tầm vóc thấp đơn độc mà không có bệnh loạn sản xương rõ ràng. Ngược lại, các đột biến mất chức năng gây ra tầm vóc cao.
Các Yếu tố tăng trưởng giống Insulin
IGF-1 và -2 tác động thông qua thụ thể IGF1R để kích thích sự tăng trưởng của nhiều mô. Trong sụn tăng trưởng, chúng tác động lên các tế bào sụn để kích thích sự tăng sinh và phì đại. IGF-1, được sản xuất chủ yếu ở gan, tác động theo kiểu nội tiết lên sụn tăng trưởng (xem phần Điều hòa nội tiết của sự tăng trưởng theo chiều dọc-Điều hòa nội tiết của sự tăng trưởng theo chiều dọc), nhưng IGF-1 cũng được cho là được sản xuất cục bộ trong sụn tăng trưởng, bởi các tế bào trong màng sụn và/hoặc các tế bào sụn, và tác động cục bộ lên các tế bào sụn như một yếu tố cận tiết. Ở loài gặm nhấm, cả hai tác động nội tiết và cận tiết đều quan trọng, nhưng tầm quan trọng tương đối ở người vẫn chưa rõ ràng. IGF-2 tác động chủ yếu như một yếu tố cận tiết và được biểu hiện ở mức độ cao trong các mô của thai nhi. Gen IGF2 được in dấu và do đó chỉ được biểu hiện bởi alen của cha. Ở người, các đột biến trong IGF1, IGF2, và IGFR1 đều có thể làm suy giảm cả sự tăng trưởng của thai nhi và sau sinh (xem phần Rối loạn tăng trưởng ở trẻ em để biết chi tiết) cho thấy cả IGF-1 và IGF-2 đều quan trọng cho sự tăng trưởng trước và sau khi sinh.
Protein liên quan đến Hormone cận giáp và Indian Hedgehog
Protein liên quan đến hormone cận giáp (PTHrP) được biểu hiện ở các tế bào sụn gần đầu xương dài trong phôi và ở vùng nghỉ của sụn tăng trưởng sau sinh. Nó khuếch tán xuống qua sụn tăng trưởng. PTHrP tác động thông qua PTH1R (cùng một thụ thể được PTH sử dụng) và một phần, thông qua Gs-alpha, để ngăn chặn các tế bào sụn ở vùng tăng sinh trải qua quá trình biệt hóa phì đại cho đến khi chúng đạt đến một khoảng cách đủ xa so với nguồn PTHrP. Indian hedgehog (IHH) được sản xuất bởi các tế bào sụn vừa mới bắt đầu biệt hóa phì đại. IHH kích thích sản xuất PTHrP, tạo thành một vòng phản hồi ngược âm tính. Các đột biến trong các gen mã hóa các thành phần của hệ thống này làm suy giảm sự phát triển của xương. Ví dụ, các đột biến mất chức năng trong PTH1R gây ra bệnh loạn sản sụn gây chết Blomstrand, và các đột biến tăng chức năng trong PTH1R gây ra bệnh loạn sản sụn hành xương Jansen. Tương tự, các đột biến mất chức năng trong IHH gây ra tật ngón ngắn loại A1 hoặc tầm vóc thấp với các bất thường xương không đặc hiệu, và các đột biến mất chức năng trong Gs-alpha gây ra bệnh loạn dưỡng xương di truyền Albright. Các đột biến trong các gen ảnh hưởng đến các bước tiếp theo trong con đường truyền tín hiệu PTHrP gây ra bệnh acrodysostosis.
Điều hòa sự tăng trưởng theo chiều dọc bởi chất nền sụn
Các tế bào sụn ở sụn tăng trưởng tiết ra một chất nền sụn có chức năng cấu trúc, cho phép sụn tăng trưởng chịu được các lực cơ học đáng kể cần thiết cho hệ xương. Chất nền sụn cũng cung cấp môi trường mà qua đó các yếu tố nội tiết và cận tiết phải di chuyển để đến được các tế bào sụn ở sụn tăng trưởng. Chất nền này là một hỗn hợp phức tạp của các phân tử, bao gồm nhiều loại collagen, ví dụ, collagen loại II và X, và các proteoglycan, chẳng hạn như aggrecan. Các đột biến trong nhiều gen mã hóa, ví dụ, COL2A1, COL10A1, và ACAN (aggrecan), gây ra các bệnh loạn sản sụn. Các đột biến dị hợp tử trong ACAN cũng có thể biểu hiện như tầm vóc thấp với tuổi xương cao (xem phần Rối loạn tăng trưởng ở trẻ em để biết chi tiết). Các đột biến trong các gen collagen loại 1, COL1A1 và COL1A2, gây ra bệnh xương thủy tinh. Bệnh nhân bị ảnh hưởng có xương giòn, phản ánh vai trò quan trọng của collagen loại I trong chất nền xương, nhưng cũng có thể có tầm vóc thấp.
Điều hòa sự tăng trưởng theo chiều dọc trong tế bào
Sự tăng sinh và biệt hóa phì đại của các tế bào sụn ở sụn tăng trưởng cũng được kiểm soát phức tạp bởi các yếu tố nội bào. Ví dụ, gen chứa hộp homeobox tầm vóc thấp (SHOX) mã hóa một yếu tố phiên mã cần thiết cho chức năng sụn tăng trưởng bình thường và do đó, các đột biến trong SHOX gây ra tầm vóc thấp. Một ví dụ khác về một hệ thống nội bào điều hòa chức năng của tế bào sụn ở sụn tăng trưởng là con đường RAS-mitogen-activated protein kinase (MAPK). Đây là một hệ thống truyền tín hiệu điều hòa sự tăng sinh và biệt hóa tế bào ở nhiều mô. Sự liên kết của phối tử với các thụ thể tyrosine kinase gây ra sự truyền tín hiệu liên quan đến sự phosphoryl hóa của một loạt các protein. Các đột biến làm tăng cường tín hiệu RAS-MAPK dẫn đến hội chứng Noonan (NS), bao gồm cả tầm vóc thấp.
Sự lão hóa của sụn tăng trưởng
Sự tăng trưởng theo chiều dọc cực kỳ nhanh chóng ở thai nhi người và chậm dần trong giai đoạn sơ sinh và thời thơ ấu. Sự chậm lại này bị gián đoạn trong một thời gian ngắn bởi giai đoạn tăng vọt ở tuổi dậy thì nhưng sau đó sự chậm lại lại tiếp tục, khiến sự tăng trưởng cuối cùng cũng ngừng lại. Sự suy giảm tăng trưởng theo chiều dọc là do một chương trình phát triển nội tại của sụn tăng trưởng được gọi là sự lão hóa của sụn tăng trưởng. Chương trình phát triển này bao gồm sự suy giảm chức năng trong sự tăng sinh của tế bào sụn và cả sự teo nhỏ dần của sụn tăng trưởng với sự giảm số lượng tế bào sụn và giảm chiều cao của sụn tăng trưởng. Sự lão hóa của sụn tăng trưởng tiến triển cho đến khi sự tăng sinh của tế bào sụn về cơ bản ngừng lại. Tại thời điểm đó, sụn tăng trưởng không còn gây ra sự kéo dài xương, và sụn hiện đã trơ được tu sửa thành xương, một sự kiện được gọi là cốt hóa sụn đầu xương. Do đó, cốt hóa sụn đầu xương không phải là nguyên nhân, mà là kết quả của việc ngừng tăng trưởng. Tuổi xương (thường được đánh giá bằng X-quang bàn tay và cổ tay không thuận) đóng vai trò như một dấu hiệu X-quang cho sự lão hóa của sụn tăng trưởng. Do đó, tuổi xương cung cấp một ước tính về mức độ lão hóa đã tiến triển ở một đứa trẻ và do đó, bao nhiêu tiềm năng tăng trưởng của trẻ đã được sử dụng và bao nhiêu còn lại. Vì lý do này, tuổi xương hữu ích cho việc dự đoán chiều cao người lớn.
Các tình trạng ức chế tăng trưởng, chẳng hạn như suy dinh dưỡng, suy giáp và thiếu hụt GH, thường làm chậm không chỉ tốc độ tăng trưởng mà còn cả tốc độ lão hóa của sụn tăng trưởng. Sự chậm lão hóa của sụn tăng trưởng này được phản ánh bởi sự chậm trễ của tuổi xương. Từ quan điểm tiến hóa, sự chậm lão hóa của sụn tăng trưởng này có khả năng phục vụ một chức năng quan trọng ở trẻ bị suy dinh dưỡng, cho phép sự tăng trưởng chậm lại để bảo tồn chất dinh dưỡng, đồng thời bảo tồn phần lớn tiềm năng tăng trưởng cho những thời điểm tốt hơn.
Ngược lại, estrogen đẩy nhanh quá trình lão hóa của sụn tăng trưởng, giải thích cho tuổi xương cao và mất chiều cao người lớn ở trẻ em dậy thì sớm và giải thích cho tuổi xương chậm và tăng chiều cao người lớn của thanh thiếu niên bị suy sinh dục, thiếu hụt aromatase và kháng estrogen. Ví dụ, ở một người đàn ông có đột biến hai alen trong thụ thể estrogen-alpha, sự tăng trưởng theo chiều dọc tiếp tục với tốc độ chậm cho đến tuổi trưởng thành, dẫn đến tầm vóc cao, và cốt hóa sụn đầu xương xảy ra ở tuổi 35. Tác động này của estrogen lên sự lão hóa của sụn tăng trưởng cũng giải thích việc sử dụng tiềm năng trong điều trị của các chất tương tự GnRH và các chất ức chế aromatase để tăng chiều cao người lớn.
Tăng trưởng bắt kịp
Tăng trưởng bắt kịp xảy ra ở trẻ em sau một thời gian bị ức chế tăng trưởng. Sự tăng trưởng theo chiều dọc có thể bị ức chế bởi nhiều tình trạng khác nhau, bao gồm suy dinh dưỡng, suy giáp, thiếu hụt hormone tăng trưởng và thừa glucocorticoid. Nếu tình trạng này kéo dài đủ lâu, điểm độ lệch chuẩn (SDS) chiều cao theo tuổi của trẻ sẽ giảm (phần trăm trên đường cong chiều cao giảm). Thường thì tình trạng ức chế tăng trưởng cuối cùng sẽ được giải quyết, ví dụ, nếu thức ăn trở nên sẵn có hơn, liệu pháp thay thế hormone tuyến giáp hoặc hormone tăng trưởng được bắt đầu, hoặc có thể ngừng sử dụng glucocorticoid dược lý. Trong tình huống này, tốc độ tăng trưởng theo chiều dọc của trẻ thường không chỉ tăng lên bằng với tốc độ tăng trưởng bình thường theo tuổi. Thay vào đó, tốc độ tăng trưởng của trẻ vượt quá mức bình thường, khiến SDS chiều cao được cải thiện. Sự tăng trưởng bắt kịp này thường không hoàn toàn ở chỗ SDS chiều cao của trẻ không trở lại hoàn toàn giá trị trước khi bị ức chế tăng trưởng, và chiều cao người lớn vẫn có thể bị ảnh hưởng phần nào.
Trong một thời gian, người ta cho rằng tăng trưởng bắt kịp là kết quả của một cơ chế trong hệ thần kinh trung ương đánh giá kích thước cơ thể thực tế của trẻ, so sánh kích thước cơ thể thực tế này với điểm đặt phù hợp với lứa tuổi, và sau đó điều chỉnh tốc độ tăng trưởng cho phù hợp. Sau một thời gian bị ức chế tăng trưởng, cơ chế giả định này sẽ cảm nhận được sự thiếu hụt chiều cao và tăng tốc độ tăng trưởng để gây ra tăng trưởng bắt kịp. Tuy nhiên, bằng chứng gần đây hơn cho thấy rằng cơ chế chính chịu trách nhiệm cho sự tăng trưởng bắt kịp không nằm ở hệ thần kinh trung ương mà ở sụn tăng trưởng và liên quan đến sự lão hóa chậm của sụn tăng trưởng. Các tình trạng ức chế tăng trưởng làm chậm quá trình lão hóa của sụn tăng trưởng. Khi tình trạng ức chế tăng trưởng được giải quyết, sụn tăng trưởng ít lão hóa hơn bình thường và do đó phát triển nhanh hơn bình thường và trong một khoảng thời gian dài hơn, gây ra sự tăng trưởng bắt kịp. Ở một số tình trạng, tốc độ tăng trưởng bắt kịp của con người phù hợp với giả thuyết này, nhưng ở những tình trạng khác, tốc độ tăng trưởng bắt kịp ban đầu cao cho thấy khả năng có các cơ chế đóng góp khác.
Biến thiên bình thường về tầm vóc
Các mô hình tăng trưởng thời thơ ấu và chiều cao người lớn là kết quả của sự tương tác giữa các yếu tố di truyền và môi trường. Kể từ nghiên cứu nổi tiếng của Francis Galton vào năm 1885, người ta đã biết rằng chiều cao của một đứa trẻ có mối tương quan mạnh mẽ với chiều cao của cha mẹ. Các nghiên cứu quy mô lớn sau này tập trung vào sự biến thiên chiều cao giữa các cặp song sinh cho thấy rằng các yếu tố môi trường có tầm quan trọng lớn hơn trong thời thơ ấu, trong khi các yếu tố di truyền có ảnh hưởng quyết định hơn đến chiều cao của thanh thiếu niên và người lớn. Gần đây, các gen cụ thể kiểm soát sự biến thiên chiều cao bình thường đã bắt đầu được làm sáng tỏ bởi các nghiên cứu liên kết toàn bộ bộ gen (GWAS), tìm kiếm các mối liên kết thống kê giữa chiều cao người lớn và các đa hình đơn nucleotide. Các nghiên cứu này đã xác định được các biến thể alen phổ biến (thường có tần số alen nhỏ > 5%) mà riêng lẻ chỉ có tác động kiểu hình nhỏ, nhưng cùng nhau quyết định phần lớn sự biến thiên của chiều cao. Phương pháp này đã được sử dụng để xác định các biến thể ở hơn 700 locus có ảnh hưởng đáng kể đến chiều cao. Mỗi biến thể này chịu trách nhiệm cho khoảng 1 mm biến thiên chiều cao trên mỗi alen và kết hợp lại, giải thích khoảng 25% tính di truyền của chiều cao người lớn. Ngoài ra, 83 biến thể mã hóa có tần số thấp hơn (tần số alen nhỏ ≤ 1%) đã được xác định là chịu trách nhiệm cho tới 2 cm chiều cao trên mỗi alen.
Đánh giá lâm sàng về sự tăng trưởng
Đo lường
Đo lường chính xác là nền tảng quan trọng của tất cả các đánh giá tăng trưởng. Mặc dù nó thường bị coi là đơn giản (xét cho cùng, có bao nhiêu phụ huynh theo dõi sự phát triển của con mình trên một bức tường trong nhà?), hai yếu tố làm cho nó thực sự trở nên thách thức. Thứ nhất, vì tăng trưởng được định nghĩa là sự thay đổi về chiều dài hoặc chiều cao trong một khoảng thời gian nhất định, hai phép đo ở mỗi đầu của khoảng thời gian đó phải được thực hiện theo kỹ thuật giống hệt nhau; bất kỳ sai lệch nào của người đo này hay người đo khác có thể tạo ra sự tăng tốc hoặc giảm tốc vi mô giả tạo trong sự tăng trưởng được tính toán, trở thành những sai số khuếch đại khi vận tốc được tính theo năm để phân tích (Hộp 11.1). Thứ hai, cố hữu trong kỹ thuật giống hệt nhau là định vị đúng mỗi lần, điều này có thể là thách thức với một trẻ sơ sinh hoặc trẻ em hay ngọ nguậy, lo lắng, buồn chán, không có khả năng phát triển để hiểu hoặc thực hiện các hướng dẫn, hoặc gặp khó khăn về thể chất trong việc đạt được và duy trì tư thế đúng (ví dụ, do giảm trương lực cơ, tăng trương lực cơ, co cứng, các vấn đề về thăng bằng, bất đối xứng chiều dài chân, dị tật, chấn thương hoặc bệnh tật). Làm phức tạp thêm khó khăn trong việc duy trì kỹ thuật giống hệt nhau mỗi lần là cả vấn đề nhân sự (cùng một đứa trẻ thường được đo bởi các nhân viên khác nhau trong các lần khám nối tiếp vì thời gian đã trôi qua hoặc các địa điểm chăm sóc khác nhau) và vấn đề thiết bị (phải được hiệu chuẩn thường xuyên và sử dụng hợp lý). Một nguồn sai sót cuối cùng có thể xảy ra ở giai đoạn ghi chép, khi phép đo được nhập vào hồ sơ y tế, chẳng hạn như hoán vị các chữ số, lỗi đánh máy khác, hoặc vô tình ghi lại phép đo bằng inch hoặc centimet khi nó đã được đo bằng đơn vị kia.
| Hộp 11.1 Vận tốc tăng trưởng hàng năm
|

Chiều cao đứng nên được đo bằng thước đo chiều cao gắn tường (stadiometer). Trẻ nên đứng thẳng dựa vào tường hoặc tấm lưng, cởi giày, tháo bất kỳ phụ kiện tóc gây cản trở nào, và đầu ở mặt phẳng Frankfurt (bờ dưới của hốc mắt và bờ trên của ống tai ngoài đều nằm trên cùng một mặt phẳng ngang), tức là, nhìn thẳng về phía trước. Tấm đầu, được hạ xuống trên đỉnh đầu để đọc số đo, phải được cố định chắc chắn sao cho nó vuông góc với tường hoặc tấm lưng. Các thanh ngang lật lên (cánh tay lỏng lẻo) trên cân đo trọng lượng, thường được sử dụng để đo chiều cao, có thể gây ra sai số lớn do tư thế chùng xuống của trẻ và sự thay đổi đáng kể về góc của thanh ngang.
Chiều dài nằm được đo tương tự cho trẻ sơ sinh và trẻ mới biết đi bằng cách sử dụng một bảng đo chiều dài nằm, bao gồm một nền tảng chắc chắn, một tấm đầu cố định và một tấm chân có thể di chuyển để đọc số đo. Cần có hai người để đo chiều dài chính xác: một người giữ đầu ở mặt phẳng Frankfurt, nhìn thẳng lên, và người thứ hai giữ thẳng đầu gối và đưa bảng đo đến chân, chân cũng phải được giữ vuông góc ở mắt cá chân. Nhiều bác sĩ, khi đo trẻ sơ sinh một mình, đặt bệnh nhân lên tờ giấy lót bàn khám, đánh dấu bằng bút trên giấy các vị trí của đầu và chân, nhấc bệnh nhân ra, và sau đó sử dụng thước dây để đo khoảng cách giữa hai vạch bút. Kỹ thuật phổ biến này rất không chính xác do định vị sai trẻ sơ sinh, sự di chuyển và nhàu nát của giấy, và không thể có được các vạch bút vuông góc.
Mặc dù có các quy trình được thiết lập tốt, được chấp nhận chung để đo lường trẻ em, các nghiên cứu đã chỉ ra rằng nó thường được thực hiện không đúng cách. Quan sát 55 cơ sở chăm sóc ban đầu của Hoa Kỳ (44 nhi khoa, 11 thực hành gia đình) cho thấy các phép đo được coi là chính xác (được định nghĩa là ≤ 0,5 cm) chỉ trong 30% trường hợp, và khác với các phép đo của cùng một trẻ em bởi các nhà điều tra nghiên cứu trung bình 1,3 cm. Bởi vì sai số trung bình vượt quá sự khác biệt giữa vận tốc tăng trưởng bình thường (5 cm/năm) và dưới mức bình thường (4 cm/năm), sai số đo lường có khả năng góp phần vào cả việc điều tra không phù hợp và/hoặc giới thiệu đến chuyên gia chuyên sâu đối với trẻ em tăng trưởng bình thường và bỏ sót việc phát hiện các vấn đề tăng trưởng thực sự ở những trẻ khác. Tuy nhiên, điều này có thể dễ dàng được khắc phục thông qua đào tạo. Việc phân ngẫu nhiên 55 cơ sở này vào các nhóm can thiệp giáo dục và nhóm đối chứng cho thấy tỷ lệ đo lường chính xác tăng liên tục trong nhóm can thiệp sau 3 tháng (55% so với 37% ở nhóm đối chứng) và 6 tháng (70% so với 34% ở nhóm đối chứng). Mặc dù nhiều cơ sở chăm sóc ban đầu bận rộn chỉ đo bệnh nhân một lần, các phép đo ba lần được thực hiện (với giá trị trung bình được ghi lại) như một phương tiện để cải thiện độ chính xác khi sự chính xác là chìa khóa (ví dụ, đối với trẻ em đang được đánh giá và điều trị tăng trưởng tại các phòng khám nội tiết hoặc tham gia vào các nghiên cứu).
Với thiết bị phù hợp, nhân viên có kinh nghiệm và một đứa trẻ hợp tác, phần trăm chiều cao đứng có thể được đánh giá với độ chính xác đáng kể. Ngược lại, ngay cả với kỹ thuật đo tốt, sai số trong đánh giá vận tốc tăng trưởng ngắn hạn là một vấn đề. Sai số tương đối (sai số tuyệt đối chia cho số đo) lớn hơn nhiều đối với vận tốc tăng trưởng so với chiều cao chủ yếu là do mẫu số cho vận tốc tăng trưởng (sự khác biệt nhỏ giữa hai phép đo liên tiếp) nhỏ hơn rất nhiều so với mẫu số cho chiều cao. Ví dụ, đối với một đứa trẻ 5 tuổi có chiều cao quan sát được ở phần trăm thứ 3, khoảng tin cậy 95% kéo dài từ phần trăm thứ 2 đến thứ 4 nhưng khoảng tin cậy cho vận tốc chiều cao 12 tháng kéo dài từ phần trăm thứ 8 đến 52. Do đó, ngay cả với vận tốc tăng trưởng 1 năm, trẻ em có thể dễ dàng bị phân loại sai là có vận tốc tăng trưởng bình thường so với bất thường. Cố gắng đo vận tốc tăng trưởng trong các khoảng thời gian dưới một năm đầy đủ dẫn đến một sai số đo lường tương đối thậm chí còn lớn hơn. Vận tốc tăng trưởng, được đo trong một năm, được phát hiện là có ít giá trị chẩn đoán để xác định trẻ em bị thiếu hụt GH vô căn đơn độc hoặc hội chứng Turner. Vận tốc chiều cao phải được đo trong khoảng thời gian 3 năm để đạt được giá trị chấp nhận được. Do đó, việc quá phụ thuộc vào vận tốc tăng trưởng được tính toán có thể gây hiểu lầm. Tuy nhiên, điều quan trọng đối với bác sĩ lâm sàng là xác định những trẻ có chiều cao và/hoặc tăng cân kém, điều này có thể cho thấy một rối loạn tiềm ẩn quan trọng. Như một giải pháp thay thế cho các vận tốc tăng trưởng được tính toán, sự sụt giảm phần trăm chiều cao và/hoặc cân nặng trên biểu đồ tăng trưởng tiêu chuẩn có thể cung cấp những manh mối có giá trị về tốc độ tăng trưởng giảm.
Biểu đồ tăng trưởng
Bởi vì sự tăng trưởng bình thường tuân theo một mô hình có thể dự đoán được, các biểu đồ tăng trưởng được xây dựng mô tả mô hình này dưới dạng các đường cong phần trăm tuần tự cho thấy sự phân bố của các số đo cơ thể được chọn (thường là chiều dài hoặc chiều cao, cân nặng, và cân nặng theo chiều dài hoặc chỉ số khối cơ thể [BMI]) ở trẻ em của một quốc gia nhất định. Tăng trưởng nhanh nhất trong giai đoạn bào thai và giảm dần trong giai đoạn sơ sinh, với chiều dài tăng khoảng 25 cm trong năm đầu đời và 10 đến 15 cm trong năm thứ hai. Trong giai đoạn sơ sinh, trẻ em thường xuyên vượt qua các kênh phần trăm chiều dài (sự tái định kênh sinh lý) khi chúng chuyển từ các yếu tố quyết định tăng trưởng trước khi sinh sang sau khi sinh; sức khỏe và dinh dưỡng của mẹ và thai kỳ có tác động lớn đến yếu tố trước, trong khi yếu tố sau thường phù hợp với tiềm năng chiều cao di truyền của trẻ. Sau sinh nhật thứ hai, tốc độ tăng trưởng theo chiều dọc tiếp tục giảm, nhưng dần dần hơn, và việc dịch chuyển các kênh phần trăm là bất thường. Với sự khởi đầu của tuổi dậy thì, sự tăng trưởng tầm vóc tăng tốc, dẫn đến giai đoạn tăng vọt ở tuổi dậy thì, sau đó sự suy giảm tốc độ tăng trưởng theo chiều dọc lại tiếp tục, khiến việc tăng chiều cao dần dần dừng lại. Tuy nhiên, việc đạt được thành phần cơ thể người lớn vẫn tiếp tục trong giai đoạn chuyển tiếp sau đó.
Quần thể được chọn để tham khảo là quan trọng khi đưa ra các phán đoán về chiều cao và sự tăng trưởng của một cá nhân. Nhiều biểu đồ tăng trưởng đã được xây dựng cho trẻ em Hoa Kỳ trong những thập kỷ qua, khác nhau về thành phần chủng tộc và kinh tế xã hội của các quần thể tham khảo mà chúng dựa vào. Các biểu đồ tăng trưởng cho trẻ em của các quốc gia khác cũng có sẵn, phản ánh sự phân bố chiều cao khác nhau của các quần thể địa phương. Các biểu đồ tăng trưởng gần đây nhất và hiện được khuyến nghị của Hoa Kỳ, được công bố vào năm 2000 bởi Trung tâm Kiểm soát và Phòng ngừa Dịch bệnh (CDC; có sẵn tại https://www.cdc.gov/growthcharts/cdc_charts.htm), dựa trên một số nguồn dữ liệu cắt ngang và đã giới thiệu các biểu đồ BMI nhi khoa của Hoa Kỳ. Bởi vì các biểu đồ của CDC và các biểu đồ quốc gia khác được sử dụng để đánh giá lâm sàng mặc dù là các biểu đồ tham khảo (tức là, mô tả quan sát cách trẻ em trong các quần thể phát triển), Tổ chức Y tế Thế giới (WHO) đã đặt ra mục tiêu tạo ra các biểu đồ tiêu chuẩn tăng trưởng (tức là, mô tả cách trẻ em khỏe mạnh nên phát triển). Các biểu đồ tăng trưởng năm 2006 của WHO (có sẵn tại: https://www.cdc.gov/growthcharts/who_charts.htm) dựa trên các phép đo theo chiều dọc từ sơ sinh đến 2 tuổi của 882 trẻ em từ sáu quốc gia khác nhau, đại diện cho sự đa dạng chủng tộc nhưng phát triển trong điều kiện “lý tưởng”, và dữ liệu cắt ngang trên các trẻ em khác sau 2 tuổi. Do đó, CDC khuyến nghị sử dụng các biểu đồ tăng trưởng năm 2006 của WHO dưới 2 tuổi và các biểu đồ tăng trưởng năm 2000 của CDC sau 2 tuổi để đánh giá sự tăng trưởng của trẻ em Hoa Kỳ. Một điểm khác biệt quan trọng là các biểu đồ của WHO dựa trên trẻ sơ sinh bú mẹ so với trẻ sơ sinh chủ yếu bú sữa công thức của các biểu đồ CDC, dẫn đến các đường cong cân nặng có hình dạng khác nhau có thể làm thay đổi tỷ lệ suy dinh dưỡng được nhận thấy tùy thuộc vào phương thức cho ăn của trẻ sơ sinh được đánh giá lâm sàng.
Hai loại biểu đồ tăng trưởng đặc biệt cũng hữu ích. Biểu đồ vận tốc vẽ các mức tăng hàng năm thay vì chiều cao (khoảng cách) và được điều chỉnh theo nhịp độ, cho phép sự thay đổi về thời gian và cường độ của giai đoạn tăng vọt ở tuổi vị thành niên. Các biểu đồ này có thể hữu ích để đánh giá các mô hình tăng trưởng của trẻ em có nhịp độ trưởng thành chậm hơn hoặc nhanh hơn (“phát triển sớm và muộn”), những biến thể của chúng bị mất đi một cách hiệu quả trong các biểu đồ chiều cao cắt ngang tính trung bình sự tăng trưởng giữa các bạn đồng lứa ở các giai đoạn phát triển khác nhau. Một hạn chế của các biểu đồ tăng trưởng theo chiều dọc như vậy là dữ liệu của chúng được lấy từ các mẫu quần thể tương đối nhỏ, không đại diện được ghi nhận từ lâu. Một hạn chế thứ hai là sai số tương đối lớn trong các phép đo vận tốc tăng trưởng, đã được thảo luận trước đó, tạo ra cả sự không chắc chắn trong dữ liệu của từng đứa trẻ và cả trong dữ liệu tham khảo mà các biểu đồ vận tốc tăng trưởng dựa vào. Do đó, dựa trên các nghiên cứu về sai số đo lường, Voss và các đồng nghiệp đã kết luận, “Mặc dù các biểu đồ vận tốc hấp dẫn về mặt khái niệm, chúng dường như không có khả năng phân biệt hơn các biểu đồ chiều cao trong thực tế, và có thể gây hiểu lầm về mặt lâm sàng trừ khi được giải thích một cách hết sức cẩn thận.” Ngoài ra, các biểu đồ tăng trưởng cho thấy chiều cao, thay vì vận tốc tăng trưởng, nhưng dựa trên dữ liệu theo chiều dọc, có thể hữu ích cho trẻ em có nhịp độ trưởng thành chậm hoặc nhanh.
Loại biểu đồ tăng trưởng đặc biệt thứ hai bao gồm các biểu đồ cụ thể cho trẻ em mắc các hội chứng di truyền cụ thể. Các biểu đồ này được sử dụng về cơ bản giống như các biểu đồ cho trẻ em bình thường. Sự sai lệch của sự tăng trưởng so với đường cong tăng trưởng liên quan đến bệnh thích hợp cho thấy khả năng có một vấn đề tiềm ẩn thứ hai, chẳng hạn như suy giáp mắc phải ở bệnh nhân mắc hội chứng Down hoặc Turner.
Tỷ lệ cơ thể
Bộ xương không phát triển một cách cân đối. Tỷ lệ phân đoạn cơ thể trên so với dưới bắt đầu ở mức 1,7 khi sinh, cho thấy thân mình dài hơn đáng kể so với chi dưới. Tuy nhiên, sau khi sinh, chi dưới phát triển nhanh hơn thân mình, và tỷ lệ này giảm xuống khoảng 1,0 vào lúc 10 tuổi. Việc ngừng tăng trưởng theo chiều dọc sớm, chẳng hạn như xảy ra ở tuổi dậy thì sớm, dẫn đến tỷ lệ trẻ con dai dẳng (chi ngắn so với thân mình), trong khi tăng trưởng kéo dài, chẳng hạn như xảy ra ở tình trạng suy sinh dục, tạo ra một dáng người chân dài. Tỷ lệ chi so với thân mình có thể được đánh giá bằng ba kỹ thuật khác nhau, tất cả đều kết hợp đo chiều cao đứng để so sánh. Đối với tỷ lệ phân đoạn trên-dưới, phân đoạn cơ thể dưới được đo từ bờ trên của khớp mu đến sàn nhà trong khi bệnh nhân đang đứng thẳng. Chiều dài phân đoạn dưới này được trừ đi khỏi chiều cao đứng để xác định chiều dài phân đoạn trên và sau đó chia cho chiều dài phân đoạn dưới để xác định tỷ lệ, có thể được so sánh với dữ liệu tham khảo. Kỹ thuật thứ hai đo sải tay (giữa các đầu ngón tay giữa với hai cánh tay duỗi thẳng hoàn toàn vuông góc với thân mình, một lần nữa với trẻ đứng thẳng dựa vào tường) để so sánh với chiều cao đứng. Thông thường sải tay nhỏ hơn chiều cao đứng trước 8 tuổi, bằng chiều cao ở độ tuổi 8 đến 12, và lớn hơn chiều cao trên 12 tuổi. Kỹ thuật thứ ba đo chiều cao ngồi, được chia cho chiều cao đứng để mang lại chỉ số chiều cao ngồi. Chỉ số chiều cao ngồi nhạy cảm trong việc phát hiện sự tăng trưởng không cân đối của thân mình (các đốt sống) so với chi dưới và có thể được sử dụng để sàng lọc các nguyên nhân cụ thể gây ra tầm vóc thấp không cân đối, chẳng hạn như thiếu hụt SHOX và loạn sản sụn. Dữ liệu tham khảo xuất sắc có sẵn từ NHANES III (Bảng 11.2).
Bảng 11.2 Giá trị trung bình, độ lệch chuẩn và các phân vị của chỉ số chiều cao ngồi (Chiều cao ngồi/Tầm vóc × 100) theo tuổi cho nam và nữ từ 2 đến 90 tuổi (Từ Frisancho, A.R., Anthropometric Standards: An Interactive Nutritional Reference of Body Size and Body Composition for Children and Adults, The University of Michigan Press, Ann Arbor, MI: 2008. Với sự cho phép.)
| Nhóm Tuổi (năm) | Tuổi Trung Bình (năm) | Phân vị | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | M | SD | 5 | 10 | 15 | 25 | 50 | 75 | 85 | 90 | ||
| Nam | ||||||||||||
| 2.0–2.9 | 2.46 | 619 | 56.9 | 2.01 | 53.7 | 54.4 | 54.9 | 55.6 | 56.9 | 58.3 | 59.0 | 59.6 |
| 3.0–3.9 | 3.45 | 508 | 55.5 | 1.8 | 52.6 | 53.2 | 53.6 | 54.3 | 55.6 | 56.8 | 57.5 | 58.0 |
| 4.0–4.9 | 4.47 | 553 | 54.4 | 1.8 | 51.6 | 52.2 | 52.6 | 53.2 | 54.4 | 55.6 | 56.3 | 56.7 |
| 5.0–5.9 | 5.43 | 496 | 53.5 | 1.7 | 50.8 | 51.4 | 51.8 | 52.4 | 53.5 | 54.6 | 55.3 | 55.7 |
| 6.0–6.9 | 6.45 | 262 | 52.7 | 1.6 | 50.2 | 50.7 | 51.1 | 51.7 | 52.7 | 53.8 | 54.4 | 54.8 |
| 7.0–7.9 | 7.47 | 273 | 52.1 | 0.0 | 49.7 | 50.2 | 50.6 | 51.1 | 52.2 | 53.2 | 53.8 | 54.2 |
| 8.0–8.9 | 8.46 | 267 | 51.7 | 0.1 | 49.3 | 49.8 | 50.2 | 50.7 | 51.7 | 52.8 | 53.3 | 53.7 |
| 9.0–9.9 | 9.50 | 281 | 51.4 | 0.1 | 49.0 | 49.5 | 49.9 | 50.4 | 51.4 | 52.4 | 53.0 | 53.4 |
| 10.0–10.9 | 10.45 | 288 | 51.2 | 1.5 | 48.8 | 49.3 | 49.7 | 50.2 | 51.2 | 52.2 | 52.8 | 53.1 |
| 11.0–11.9 | 11.44 | 275 | 51.0 | 1.4 | 48.7 | 49.2 | 49.6 | 50.1 | 51.1 | 52.1 | 52.6 | 53.0 |
| 12.0–12.9 | 12.47 | 204 | 51.0 | 1.4 | 48.7 | 49.2 | 49.5 | 50.0 | 51.0 | 52.0 | 52.6 | 52.9 |
| 13.0–13.9 | 13.47 | 192 | 51.0 | 1.4 | 48.7 | 49.2 | 49.5 | 50.0 | 51.0 | 52.0 | 52.6 | 52.9 |
| 14.0–14.9 | 14.49 | 186 | 51.1 | 1.5 | 48.7 | 49.2 | 49.6 | 50.1 | 51.1 | 52.1 | 52.6 | 53.0 |
| 15.0–15.9 | 15.45 | 182 | 51.2 | 1.5 | 48.8 | 49.3 | 49.6 | 50.2 | 51.1 | 52.2 | 52.7 | 53.1 |
| 16.0–16.9 | 16.45 | 196 | 51.3 | 1.5 | 48.9 | 49.4 | 49.7 | 50.3 | 51.3 | 52.3 | 52.8 | 53.2 |
| 17.0–17.9 | 17.45 | 191 | 51.4 | 1.5 | 49.0 | 49.5 | 49.8 | 50.4 | 51.4 | 52.4 | 53.0 | 53.4 |
| 18.0–18.9 | 18.45 | 172 | 51.6 | 1.5 | 49.1 | 49.6 | 50.0 | 50.5 | 51.5 | 52.6 | 53.1 | 53.5 |
| 19.0–19.9 | 19.43 | 160 | 51.7 | 1.5 | 49.2 | 49.7 | 50.1 | 50.6 | 51.7 | 52.7 | 53.3 | 53.7 |
| 20.0–29.9 | 24.96 | 1620 | 52.3 | 1.5 | 49.8 | 50.3 | 50.7 | 51.2 | 52.2 | 53.3 | 53.8 | 54.2 |
| 30.0–39.9 | 34.72 | 1452 | 52.4 | 1.6 | 49.9 | 50.4 | 50.8 | 51.3 | 52.4 | 53.4 | 54.0 | 54.4 |
| 40.0–49.9 | 44.35 | 1195 | 52.2 | 1.5 | 49.9 | 50.4 | 50.8 | 51.3 | 52.3 | 53.4 | 53.9 | 54.3 |
| 50.0–59.9 | 54.89 | 835 | 52.1 | 1.5 | 49.8 | 50.3 | 50.7 | 51.2 | 52.2 | 53.3 | 53.8 | 54.2 |
| 60.0–69.9 | 64.83 | 1129 | 51.9 | 1.5 | 49.7 | 50.2 | 50.6 | 51.1 | 52.1 | 53.1 | 53.6 | 54.0 |
| 70.0–79.9 | 74.16 | 798 | 51.7 | 1.4 | 49.5 | 50.0 | 50.4 | 50.9 | 51.8 | 52.8 | 53.3 | 53.7 |
| 80.0–90.9 | 84.09 | 566 | 51.5 | 1.4 | 49.3 | 49.8 | 50.1 | 50.6 | 51.5 | 52.5 | 53.0 | 53.3 |
| Nữ | ||||||||||||
| 2.0–2.9 | 2.45 | 584 | 56.9 | 1.9 | 52.9 | 53.5 | 54.0 | 54.7 | 55.9 | 57.2 | 57.9 | 58.4 |
| 3.0–3.9 | 3.46 | 586 | 55.3 | 1.8 | 52.1 | 52.7 | 53.2 | 53.8 | 55.0 | 56.2 | 56.9 | 57.4 |
| 4.0–4.9 | 4.43 | 528 | 54.1 | 1.6 | 51.5 | 52.1 | 52.5 | 53.1 | 54.3 | 55.4 | 56.1 | 56.5 |
| 5.0–5.9 | 5.46 | 543 | 53.2 | 1.6 | 50.9 | 51.5 | 51.9 | 52.5 | 53.6 | 54.7 | 55.4 | 55.8 |
| 6.0–6.9 | 6.47 | 273 | 52.5 | 1.6 | 50.4 | 51.0 | 51.4 | 52.0 | 53.1 | 54.2 | 54.8 | 55.2 |
| 7.0–7.9 | 7.44 | 268 | 52.1 | 1.4 | 50.1 | 50.7 | 51.0 | 51.6 | 52.7 | 53.7 | 54.3 | 54.7 |
| 8.0–8.9 | 8.47 | 247 | 51.8 | 1.5 | 49.8 | 50.4 | 50.7 | 51.3 | 52.3 | 53.4 | 54.0 | 54.3 |
| 9.0–9.9 | 9.43 | 273 | 51.7 | 1.5 | 49.6 | 50.1 | 50.5 | 51.0 | 52.1 | 53.1 | 53.7 | 54.1 |
| 10.0–10.9 | 10.43 | 260 | 51.6 | 1.5 | 49.5 | 50.0 | 50.3 | 50.9 | 51.9 | 52.9 | 53.5 | 53.9 |
| 11.0–11.9 | 11.46 | 288 | 51.6 | 1.4 | 49.4 | 49.9 | 50.2 | 50.8 | 51.8 | 52.8 | 53.3 | 53.7 |
| 12.0–12.9 | 12.46 | 221 | 51.7 | 1.5 | 49.3 | 49.9 | 50.2 | 50.7 | 51.7 | 52.7 | 53.3 | 53.6 |
| 13.0–13.9 | 13.45 | 227 | 51.8 | 1.5 | 49.3 | 49.8 | 50.2 | 50.7 | 51.7 | 52.7 | 53.2 | 53.6 |
| 14.0–14.9 | 14.47 | 216 | 52.0 | 1.5 | 49.4 | 49.9 | 50.2 | 50.7 | 51.7 | 52.7 | 53.3 | 53.6 |
| 15.0–15.9 | 15.47 | 189 | 52.1 | 1.5 | 49.4 | 50.0 | 50.3 | 50.8 | 51.8 | 52.8 | 53.3 | 53.7 |
| 16.0–16.9 | 16.46 | 224 | 52.2 | 1.5 | 49.5 | 50.0 | 50.4 | 50.9 | 51.9 | 52.9 | 53.4 | 53.8 |
| 17.0–17.9 | 17.45 | 213 | 52.4 | 1.4 | 49.6 | 50.1 | 50.5 | 51.0 | 52.0 | 53.0 | 53.5 | 53.9 |
| 18.0–18.9 | 18.43 | 186 | 52.5 | 1.4 | 49.8 | 50.3 | 50.6 | 51.1 | 52.1 | 53.1 | 53.6 | 54.0 |
| 19.0–19.9 | 19.48 | 191 | 52.6 | 1.3 | 49.9 | 50.4 | 50.8 | 51.3 | 52.3 | 53.2 | 53.8 | 54.2 |
| 20.0–29.9 | 24.91 | 1842 | 52.9 | 1.5 | 50.2 | 50.8 | 51.1 | 51.7 | 52.7 | 53.8 | 54.3 | 54.7 |
| 30.0–39.9 | 34.85 | 1837 | 52.8 | 1.5 | 50.5 | 51.0 | 51.4 | 51.9 | 52.9 | 53.9 | 54.5 | 54.9 |
| 40.0–49.9 | 44.28 | 1328 | 52.8 | 1.5 | 50.5 | 51.1 | 51.4 | 51.9 | 52.9 | 53.9 | 54.5 | 54.9 |
| 50.0–59.9 | 54.83 | 979 | 52.8 | 1.5 | 50.4 | 50.9 | 51.3 | 51.8 | 52.8 | 53.8 | 54.3 | 54.7 |
| 60.0–69.9 | 64.82 | 1110 | 52.3 | 1.5 | 50.1 | 50.6 | 50.9 | 51.4 | 52.4 | 53.4 | 54.0 | 54.3 |
| 70.0–79.9 | 74.46 | 893 | 52.0 | 1.5 | 49.5 | 50.0 | 50.4 | 50.9 | 51.9 | 52.9 | 53.5 | 53.9 |
| 80.0–90.9 | 84.45 | 599 | 51.1 | 1.5 | 48.8 | 49.3 | 49.7 | 50.2 | 51.2 | 52.2 | 52.8 | 53.2 |
M, Trung bình; SD, độ lệch chuẩn.
Sự mất cân đối có thể xảy ra không chỉ giữa các chi và thân mình mà còn ở các đoạn chi cụ thể, phân loại bệnh nhân vào các nhóm chẩn đoán cụ thể. Rhizomelia đề cập đến sự ngắn không cân đối của các chi gần (xương cánh tay và xương đùi), mezomelia đến sự ngắn không cân đối của phần giữa của các chi (xương quay/xương trụ và xương chày/xương mác), và acromelia đến sự ngắn không cân đối của các chi xa (bàn tay và bàn chân). Do đó, mọi trẻ em đến khám vì vấn đề tăng trưởng đều nên được đánh giá về sự mất cân đối vì thông tin này giúp thu hẹp chẩn đoán phân biệt, bao gồm cả khả năng mắc các bệnh loạn sản xương.
Sự trưởng thành của xương
Sự thay đổi bình thường về tốc độ trưởng thành giữa trẻ em và thanh thiếu niên ảnh hưởng đến tiên lượng tăng trưởng, vì trẻ có nhiều thời gian để phát triển hơn có thể đạt được chiều cao người lớn cao hơn. Một phim X-quang trước-sau (AP) của bàn tay và cổ tay không thuận được sử dụng để xác định tuổi xương của trẻ như một sự xác định định lượng về sự trưởng thành của cơ thể. Tuổi xương đánh giá mức độ mà bộ xương đã cốt hóa. Trong phôi người, bộ xương đầu tiên hình thành từ sụn, và sau đó sụn được tu sửa thành xương. Quá trình này tiếp tục trong suốt thời thơ ấu. Ở các xương dài, sự cốt hóa này bắt đầu như một trung tâm cốt hóa chính ở giữa thân xương và sau đó lan ra ngoài. Sau đó, các trung tâm cốt hóa thứ cấp hình thành ở các đầu xương và cũng lan ra ngoài. Tuy nhiên, các trung tâm cốt hóa chính và thứ cấp vẫn tách biệt trong thời thơ ấu bởi một lớp sụn mỏng, được gọi là sụn tăng trưởng, chịu trách nhiệm cho sự kéo dài xương và do đó tăng chiều cao. Ở tuổi vị thành niên, khi sự tăng trưởng dừng lại, chính sụn tăng trưởng cuối cùng cũng được cốt hóa, một sự kiện được gọi là cốt hóa sụn đầu xương, chỉ để lại sụn ở bề mặt khớp. Bởi vì xương cản quang hơn sụn hoặc các mô mềm, một phim X-quang của bàn tay và cổ tay trái cho thấy mức độ cốt hóa đã tiến triển ở một đứa trẻ cụ thể. Hình ảnh sau đó được so sánh với các tiêu chuẩn đã được công bố (Greulich và Pyle hoặc Tanner-Whitehouse [TW]). Có các bộ tiêu chuẩn riêng cho bé trai và bé gái. Nếu, ví dụ, hình ảnh tuổi xương của một bệnh nhân giống nhất với tiêu chuẩn cho một đứa trẻ 9 tuổi, thì bệnh nhân được cho là có tuổi xương 9 tuổi. Gần đây, một phương pháp tự động để xác định tuổi xương đã được phát triển.
Cũng giống như các biện pháp đo lường nhân trắc học khác, tuổi xương có các phạm vi bình thường, dựa trên tuổi và giới tính. Các phạm vi bình thường xác định sự chênh lệch bình thường giữa tuổi xương và tuổi theo thời gian của một đứa trẻ. Sự không thống nhất giữa các trung tâm xương, với các mức độ trưởng thành khác nhau giữa các nhóm xương, có thể là một biến thể bình thường. Bởi vì việc xác định tuổi xương là một biện pháp chủ quan, các bác sĩ nội tiết nhi khoa nên tự đọc phim thay vì chỉ dựa vào báo cáo của các bác sĩ X-quang. Điều này sẽ loại bỏ sự thay đổi giữa những người đọc giữa các nghiên cứu lặp đi lặp lại, do đó cải thiện tính hợp lệ của việc theo dõi sự tiến triển của tuổi xương theo chiều dọc ở một đứa trẻ.
Tuổi xương phản ánh nhịp độ phát triển thể chất của trẻ, đặc biệt phản ánh sự tiến triển của quá trình lão hóa sụn tăng trưởng, và cho biết tiềm năng tăng trưởng còn lại. Phim chụp đầu gối có thể được sử dụng thay thế ở lứa tuổi rất nhỏ khi phim chụp bàn tay/cổ tay chưa cung cấp thông tin do thiếu sự cốt hóa phù hợp với sự phát triển. Tuổi xương cũng liên quan chặt chẽ hơn tuổi theo thời gian với thời điểm dậy thì trung ương trong một số tình huống, ví dụ, các bé trai bị tăng sản tuyến thượng thận bẩm sinh, dậy thì sớm giới hạn ở nam giới có tính gia đình, và tầm vóc thấp vô căn. Tuy nhiên, đáng ngạc nhiên là tuổi xương không dự đoán khởi phát dậy thì tốt hơn tuổi theo thời gian ở trẻ em bình thường.
Ngay cả trước khi xác định tuổi xương, một bác sĩ lâm sàng có thể phỏng đoán tốc độ trưởng thành của một đứa trẻ bằng cách kiểm tra răng của chúng; mức độ trưởng thành của răng có xu hướng tương quan với sự trưởng thành của xương (tức là, mọc răng chậm có thể ngụ ý tuổi xương chậm).
Sự trưởng thành của xương, giống như sự phát triển của xương, được điều hòa cao bởi các yếu tố nội tiết, dinh dưỡng và di truyền. Nhìn chung, các yếu tố điều hòa tích cực sự tăng trưởng theo chiều dọc, chẳng hạn như hormone tuyến giáp, GH, estrogen và lượng dinh dưỡng, cũng điều hòa tích cực sự trưởng thành của xương, trong khi các yếu tố điều hòa tiêu cực sự tăng trưởng, chẳng hạn như glucocorticoid ở nồng độ cao, cũng điều hòa tiêu cực sự trưởng thành của xương.
Dự đoán chiều cao người lớn
Dự đoán chiều cao người lớn được sử dụng để cố gắng xác định những trẻ nào đang phát triển theo hướng đạt chiều cao người lớn bình thường. Phạm vi “bình thường” cho chiều cao người lớn có thể được xác định bằng các phân vị ngoài cùng được phân định trên biểu đồ tăng trưởng của quốc gia, nhận ra rằng các phân vị được chọn cho mục đích này (ví dụ, thứ 5 và 95 hoặc thứ 3 và 97) được chọn một cách tùy tiện.
Bởi vì chiều cao là một đặc điểm có tính di truyền cao, một mục tiêu cụ thể hơn có thể được thiết lập từ chiều cao của cha mẹ ruột của đứa trẻ. Điều quan trọng là phải đo chiều cao của cha mẹ thay vì dựa vào các báo cáo tự khai, thường không chính xác. Chiều cao của hai cha mẹ có thể được kết hợp bằng cách sử dụng các công thức cho chiều cao trung bình của cha mẹ được điều chỉnh theo giới tính (Hộp 11.2), điều chỉnh cho sự khác biệt 13 cm (5 inch) giữa chiều cao trung bình của nam và nữ trưởng thành, và cung cấp chiều cao trung bình của cha mẹ ± 2 SD (1 SD sẽ là khoảng 5 cm, hoặc 2 inch) như là phạm vi chiều cao người lớn “bình thường” cho con cái của họ. Tuy nhiên, chiều cao trung bình của cha mẹ được điều chỉnh giả định rằng chiều cao của đứa trẻ được di truyền từ cha mẹ theo kiểu đa gen, với nhiều đa hình được di truyền góp phần vào chiều cao của đứa trẻ. Nó không áp dụng cho các tình trạng đơn gen. Nếu, ví dụ, một trong hai cha mẹ thấp do đột biến dị hợp tử trong SHOX và người cha mẹ kia có chiều cao bình thường, con cái sẽ có xu hướng thấp, giống như người cha mẹ bị ảnh hưởng, hoặc có tầm vóc bình thường, giống như người cha mẹ không bị ảnh hưởng, chứ không phải ở giữa. Ngay cả đối với di truyền đa gen, dữ liệu thực nghiệm cho thấy rằng con của những bậc cha mẹ thấp có xu hướng không thấp bằng cha mẹ của chúng và những đứa trẻ cao có xu hướng không cao bằng cha mẹ của chúng. Ví dụ, khi chiều cao trung bình của cha mẹ là –2 SD, chiều cao của con cái có xu hướng gần với –1 SD.
| Hộp 11.2. Công thức tính chiều cao trung bình của cha mẹ được điều chỉnh theo giới tính
|

Phương pháp đơn giản nhất để dự đoán chiều cao người lớn là giả định rằng phân vị chiều cao của trẻ sẽ không đổi cho đến khi trưởng thành. Tuy nhiên, phương pháp này không chính xác và có thể thất bại nếu trẻ có tuổi xương cao hoặc chậm, một tình trạng ảnh hưởng đến sự tăng trưởng, hoặc thời điểm dậy thì sớm hoặc muộn.
Có ba phương pháp phổ biến để dự đoán chiều cao người lớn có tính đến sự trưởng thành của xương (tức là tuổi xương), và tất cả đều đặc hiệu cho từng giới tính. Phương pháp được sử dụng phổ biến nhất trong ba phương pháp, các bảng dự đoán Bayley-Pinneau từ năm 1952, sử dụng dữ liệu theo chiều dọc có được từ những trẻ em Hoa Kỳ khỏe mạnh sống ở Cleveland để ước tính phần tăng trưởng còn lại ở mỗi tuổi xương (theo phương pháp Greulich-Pyle). Chiều cao người lớn được dự đoán bằng cách kết hợp tuổi xương của trẻ với chiều cao được đo tại thời điểm nghiên cứu X-quang. Các bảng dự đoán và sai số dự đoán được cung cấp trong Tập bản đồ Greulich và Pyle. Tuổi xương Greulich và Pyle (đọc thủ công hoặc bằng hệ thống tự động) cũng có thể được sử dụng trong một mô hình dự đoán toán học phức tạp hơn (hiện có sẵn trực tuyến), dường như có độ chính xác cao hơn một chút so với các bảng Greulich và Pyle ở trẻ em có tầm vóc thấp vô căn. Phương pháp TW, bắt nguồn từ trẻ em Anh, sử dụng các tiêu chuẩn TW để đánh giá tuổi xương, trong đó mỗi xương ở bàn tay và cổ tay được đánh giá riêng lẻ. Ngoài tuổi xương và chiều cao đo được, phương pháp này còn tính đến tuổi theo thời gian, chiều cao của cha mẹ, và ở các bé gái, sự xuất hiện của kinh nguyệt. Phương pháp thứ ba để dự đoán chiều cao người lớn là phương pháp Roche-Wainer-Thissen (RWT). Phương pháp này tính đến cân nặng hoặc tình trạng dinh dưỡng của trẻ và sử dụng chiều dài nằm thay vì chiều cao đứng (được tính là lớn hơn 1,25 cm so với chiều cao đứng). Năm biến số dự đoán trong phương pháp RWT là chiều dài nằm, cân nặng, tuổi xương, tuổi theo thời gian và chiều cao của cha mẹ. Một phương pháp dự đoán chiều cao bổ sung gần đây đã được phát triển sử dụng các phương pháp toán học phức tạp hơn và có thể cho thấy độ chính xác tốt hơn các phương pháp trước đó. Nó hiện có sẵn trực tuyến.
Thật không may, ngay cả khi sử dụng tuổi xương, các dự đoán chiều cao cũng không chính xác cao. Sai số dự đoán là đáng kể nhưng giảm dần theo tuổi tác. Ví dụ, sử dụng các bảng Bailey-Pinneau cho các bé gái, dự đoán có thể sai lệch đối với một đứa trẻ 9 tuổi khoảng 7,4 cm hoặc 3 inch (2 SD của sai số đo lường), và khoảng 3 cm hoặc 1,2 inch đối với một đứa trẻ 13 tuổi. Hơn nữa, các phương pháp dự đoán này và các ước tính sai số liên quan dựa trên dữ liệu từ trẻ em khỏe mạnh. Có thể có các sai số hệ thống trong bối cảnh bệnh tật hoặc trong tầm vóc thấp vô căn. Ví dụ, các phương pháp này dự đoán quá cao chiều cao người lớn trong dậy thì sớm không được điều trị, hội chứng Turner và tầm vóc thấp nguyên phát. Các nguồn không chính xác trong dự đoán chiều cao người lớn bao gồm sự không chính xác của chính việc ước tính tuổi xương, không có khả năng dự đoán khởi phát và nhịp độ dậy thì, và khó khăn trong việc tính đến các tác động của bệnh tật. Một sự khác biệt nhỏ trong việc xác định tuổi xương có thể dẫn đến một sự khác biệt lớn trong dự đoán chiều cao người lớn, đặc biệt là trong giai đoạn tăng vọt ở tuổi dậy thì. Một nghiên cứu mô phỏng trên máy tính, so sánh ba phương pháp dự đoán chiều cao người lớn—Bayley-Pinneau, RWT và Khamis-Roche (tính toán chiều cao người lớn không cần tuổi xương từ chiều cao, cân nặng của trẻ và chiều cao trung bình của cha mẹ, sử dụng các hệ số đặc hiệu theo giới tính và tuổi)—cho thấy sự đồng thuận tổng thể kém giữa ba phương pháp (κ = 0,21 đối với nam và âm đối với nữ) trong việc dự đoán chiều cao người lớn dưới phân vị thứ 1,2 (ngưỡng cho chỉ định được Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ [FDA] phê duyệt điều trị bằng hormone tăng trưởng cho tầm vóc thấp vô căn). Điều quan trọng đối với cả bác sĩ lâm sàng và gia đình bệnh nhân là phải hiểu những hạn chế đáng kể của việc dự đoán chiều cao.
Di truyền tầm vóc
Phần lớn sự biến thiên bình thường về chiều cao giữa các cá nhân là do ảnh hưởng đa gen. Nhiều biến thể di truyền, cả phổ biến và hiếm gặp, mỗi biến thể có ảnh hưởng nhỏ hoặc trung bình đến sự tăng trưởng theo chiều dọc, kết hợp lại để xác định chiều cao của cá nhân. Tuy nhiên, tầm vóc thấp hoặc cao, đặc biệt khi nghiêm trọng hoặc liên quan đến các bất thường khác, cũng có thể chủ yếu là đơn gen, tức là, do một biến thể duy nhất có ảnh hưởng mạnh đến sự tăng trưởng theo chiều dọc. Các nguyên nhân đơn gen này có thể được xác định vì chúng tuân theo các kiểu di truyền Mendel cổ điển, thường là trội trên nhiễm sắc thể thường hoặc lặn trên nhiễm sắc thể thường và ít thường xuyên hơn là liên kết với nhiễm sắc thể X. Về mặt lâm sàng, kiểu di truyền được điều tra bằng cách khai thác bệnh sử gia đình cẩn thận về chiều cao của các thành viên trong gia đình mở rộng, có thể được hình dung bằng một phả hệ được viết ra. Để thảo luận chi tiết hơn, xem phần Phương pháp tiếp cận chẩn đoán bệnh nhân có tầm vóc thấp ở phần sau của chương này. Khi phả hệ cho thấy một kiểu di truyền đa gen, bác sĩ lâm sàng có thể tính toán chiều cao trung bình của cha mẹ được điều chỉnh, cung cấp một ước tính về chiều cao người lớn có khả năng của đứa trẻ, dựa trên chiều cao của hai cha mẹ. Chủ đề này được thảo luận chi tiết hơn trong một phần trước của chương này: Đánh giá lâm sàng về sự tăng trưởng—Dự đoán chiều cao người lớn.
Rối loạn tăng trưởng ở trẻ em
Nhiều rối loạn ảnh hưởng đến sự tăng trưởng ở trẻ em, đặc biệt là làm giảm sự tăng trưởng gây ra tầm vóc thấp. Nhiều nguyên nhân chính được thảo luận sau đây. Tuy nhiên, danh sách này không đầy đủ. Một danh sách mở rộng, có tổ chức cũng có thể được tìm thấy trong Phân loại Quốc tế về Chẩn đoán Nội tiết Nhi khoa (ICPED, http://www.icped.org/).
Tầm vóc thấp
Tầm vóc thấp là kết quả của sự giảm tạo sụn ở sụn tăng trưởng. Sự giảm tạo sụn có thể do các yếu tố nội tại của sụn tăng trưởng (suy giảm tăng trưởng nguyên phát) hoặc có thể là thứ phát do các yếu tố ở nơi khác trong cơ thể ảnh hưởng thứ phát đến các tế bào sụn ở sụn tăng trưởng (suy giảm tăng trưởng thứ phát) (Hình 11.4).
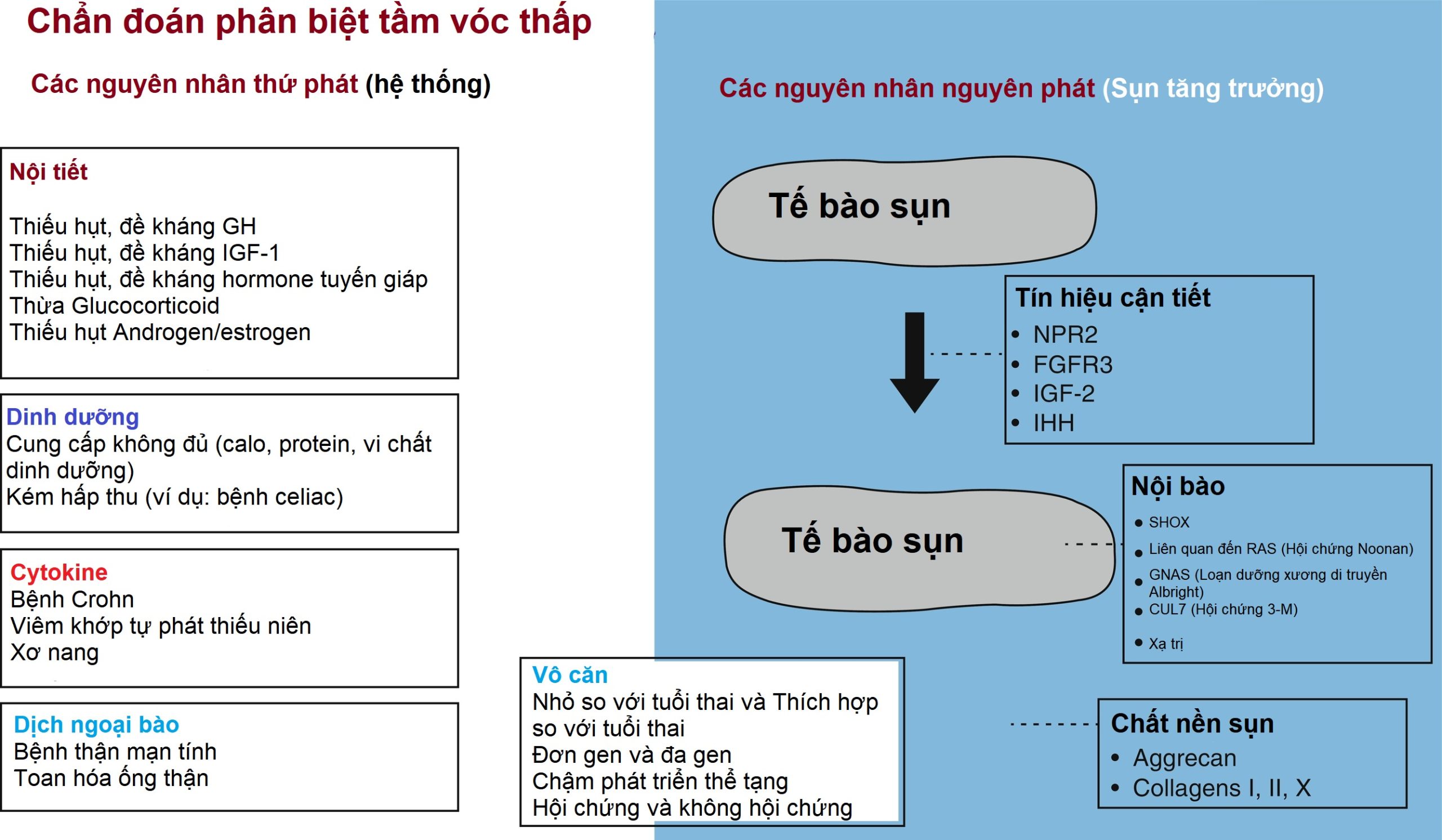
Hình 11.4 Chẩn đoán phân biệt tầm vóc thấp. Tầm vóc thấp là do giảm quá trình tạo sụn ở sụn tăng trưởng, có thể là kết quả của một bất thường tại chỗ, bên trong chính sụn tăng trưởng, hoặc một bất thường toàn thân, sau đó ảnh hưởng thứ phát đến các tế bào sụn ở sụn tăng trưởng. Các nguyên nhân tại chỗ bao gồm các khiếm khuyết trong tín hiệu tự tiết/cận tiết, chất nền sụn, và các hệ thống điều hòa nội bào. Các nguyên nhân toàn thân bao gồm các bất thường về nội tiết, dinh dưỡng, cytokine, và dịch ngoại bào. Tầm vóc thấp vô căn có khả năng rất không đồng nhất về nguyên nhân và bao gồm cả các cơ chế toàn thân và tại chỗ. Đối với mỗi loại, sơ đồ cho thấy một số ví dụ về các rối loạn cụ thể và các gen liên quan nhưng không trình bày một danh sách đầy đủ các nguyên nhân đã biết.
Suy giảm sụn tăng trưởng nguyên phát có thể liên quan đến những bất thường trong tín hiệu cận tiết, chất nền sụn, hoặc các yếu tố nội bào. Ở một số tình trạng, khiếm khuyết trong quá trình tạo sụn ở sụn tăng trưởng không chỉ làm xương ngắn mà còn bị dị dạng, trong trường hợp đó tình trạng này được gọi là loạn sản xương hoặc, chính xác hơn là, loạn sản sụn, ví dụ, ACH. Thường thì, các khiếm khuyết di truyền ảnh hưởng đến sụn tăng trưởng cũng ảnh hưởng đến sự phát triển của các mô khác, dẫn đến một hội chứng bao gồm không chỉ tầm vóc thấp mà còn các bất thường bẩm sinh khác và rối loạn chức năng cơ quan, ví dụ, NS hoặc giả suy cận giáp loại 1A.
Suy giảm tăng trưởng thứ phát có thể liên quan đến thiếu hụt dinh dưỡng, bất thường nội tiết, các rối loạn viêm, và các bất thường dịch ngoại bào. Thường thì các rối loạn này là do một khiếm khuyết ở một số hệ cơ quan khác ngoài bộ xương, chẳng hạn như thận, tuyến yên, tuyến giáp, hoặc hệ miễn dịch. Các rối loạn này xảy ra ở nơi khác trong cơ thể sau đó dẫn đến nồng độ bất thường của một số phân tử cần thiết cho chức năng bình thường của tế bào sụn ở sụn tăng trưởng. Các phân tử này có thể bao gồm các hormone steroid hoặc polypeptide, các cytokine tiền viêm, hoặc các phân tử nhỏ, chẳng hạn như phosphate hoặc ion hydro.
Cả suy giảm tăng trưởng nguyên phát và thứ phát đều có thể là đơn gen, thiểu gen, đa gen, hoặc không do di truyền. Một số tình trạng ảnh hưởng đến sự tăng trưởng của thai nhi, và do đó trẻ sinh ra nhỏ so với tuổi thai. Các tình trạng khác chỉ ảnh hưởng đến sự tăng trưởng sau sinh để trẻ sinh ra có kích thước phù hợp với tuổi thai, và tầm vóc thấp phát triển sau đó. Tuy nhiên, các tình trạng khác ảnh hưởng đến cả sự tăng trưởng trước và sau sinh.
Nguyên nhân của tầm vóc thấp
Suy giảm tăng trưởng thứ phát
Thiếu hụt dinh dưỡng
Dinh dưỡng đầy đủ là cần thiết để hỗ trợ sự tăng trưởng tầm vóc tối ưu, và thường có một độ trễ giữa sự giảm/tăng tốc cân nặng và sự giảm/tăng tốc chiều cao. Suy dinh dưỡng có thể là kết quả của việc ăn uống không đủ, kém hấp thu hoặc một số quá trình tiêu hóa khác, hoặc sự kết hợp của cả hai.
Suy dinh dưỡng là nguyên nhân phổ biến nhất của sự tăng trưởng kém trên toàn cầu, với việc ăn uống không đủ (tổng lượng calo và/hoặc suy dinh dưỡng protein-calo) thường liên quan đến mất an ninh lương thực và nghèo đói. Suy dinh dưỡng cũng phổ biến ở các cộng đồng có kinh tế xã hội cao hơn, khi nhu cầu năng lượng (ví dụ, tăng do bệnh tật, thể thao hoặc các hoạt động) không được đáp ứng bằng lượng ăn vào (ví dụ, do bữa ăn không có cấu trúc, kén ăn, sợ béo phì). Giảm lượng chất dinh dưỡng ăn vào có thể là kết quả của các dị tật hầu họng (ví dụ, chuỗi Pierre Robin, sứt môi/hở hàm ếch), chức năng vận động miệng bất thường (ví dụ, chậm phát triển lan tỏa), cũng như mất cảm giác thèm ăn do một số loại thuốc (ví dụ, chất kích thích để điều trị rối loạn tăng động giảm chú ý hoặc các tác nhân hóa trị). Các rối loạn ăn uống (ví dụ, chán ăn tâm thần) liên quan đến các hành vi ăn uống bị xáo trộn liên quan đến hình ảnh cơ thể bị thay đổi hoặc tránh thức ăn do ám ảnh hoặc các khía cạnh cảm quan của thực phẩm. Kém hấp thu cũng có thể gây ra suy dinh dưỡng, và các rối loạn như bệnh celiac, IBD, hoặc xơ nang có thể biểu hiện như suy giảm tăng trưởng đơn độc. Tuổi xương và dậy thì thường bị trì hoãn.
Ngoài suy dinh dưỡng, thiếu hụt một số vi chất dinh dưỡng có thể làm suy giảm sự tăng trưởng tầm vóc. Sự xuất hiện thường xuyên của các thiếu hụt đồng thời làm phức tạp việc nghiên cứu và tài liệu về bất kỳ vi chất dinh dưỡng đơn lẻ nào. Mặc dù thiếu sắt thường được tìm thấy ở trẻ em bị còi cọc do dinh dưỡng và nên được điều chỉnh, nhiều phân tích tổng hợp và đánh giá hệ thống của các thử nghiệm can thiệp ngẫu nhiên có đối chứng về việc bổ sung sắt cho thấy rằng liệu pháp sắt không có tác dụng đáng kể đối với sự tăng trưởng ở trẻ em đủ sắt. Ngược lại, sự chậm phát triển ở người (và suy sinh dục ở nam giới) do thiếu kẽm lần đầu tiên được báo cáo bởi Prasad và các đồng nghiệp vào năm 1963, và một số, nhưng không phải tất cả, các đánh giá hệ thống cho thấy sự gia tăng chiều cao và/hoặc cân nặng từ việc bổ sung kẽm ở trẻ em. Ion kẽm cần thiết cho hoạt động của hơn 300 enzyme (metalloenzyme kẽm), bao gồm DNA polymerase, RNA polymerase và thymidine kinase, rất quan trọng cho quá trình tổng hợp axit nucleic và protein và phân chia tế bào. Xương chứa một lượng lớn kẽm, nơi nó đóng một vai trò quan trọng trong sự phát triển của xương, và thiếu kẽm làm giảm sản xuất IGF-1, khả năng đáp ứng của tế bào với IGF-1, và phản ứng tăng trưởng đối với việc điều trị GH ở trẻ em thiếu GH.
Đánh giá cẩn thận sự tiến triển của cân nặng và chiều cao sẽ xác định những trẻ không tăng cân và phát triển phù hợp. Việc ghi lại bệnh sử ăn uống chi tiết là điều cần thiết để thiết lập chẩn đoán này, chẳng hạn như nhật ký ăn uống 3 ngày để phân tích bất kỳ sự thiếu hụt chất dinh dưỡng đa lượng hoặc vi lượng nào. Điều trị, tất nhiên, là bổ sung dinh dưỡng, có thể bao gồm giáo dục bệnh nhân-gia đình, thay đổi hành vi, bổ sung dinh dưỡng và trong những trường hợp nghiêm trọng, cho ăn qua ống thông, cũng như điều trị bất kỳ vấn đề tiêu hóa tiềm ẩn nào. Tuy nhiên, điều quan trọng cần lưu ý là một số tình trạng gây ra tầm vóc thấp có liên quan đến BMI thấp (hoặc có BMI thấp là một thành phần trong kiểu hình của chúng), chẳng hạn như hội chứng Silver-Russell (SRS), NS, và hội chứng Bloom.
Thiếu hụt nội tiết
Thiếu hụt/Không nhạy cảm với Hormone tăng trưởng Các rối loạn của trục GH/IGF bao gồm một nhóm lớn và không đồng nhất các tình trạng có các kiểu hình riêng biệt (Bảng 11.3). Các đánh giá lâm sàng và nội tiết tố thường là nền tảng của chẩn đoán các rối loạn này.
Bảng 11.3 Kiểu hình do các khiếm khuyết di truyền của trục Hormone tăng trưởng-Yếu tố tăng trưởng giống Insulin
| Đo lường Hormone | ||||||
|---|---|---|---|---|---|---|
| Gen | Di truyền | Mô hình tăng trưởng | GH | IGF-1 | IGFBP-3 | |
| Thiếu hụt GH | nhiều | Lặn/Trội NST thường | mức độ nặng/nhẹ khác nhau/sau sinh | ↓ đến ↓↓↓ | ↓ đến ↓↓↓ | ↓ đến ↓↓↓ |
| GH không hoạt tính sinh học | GH1 | Lặn/Trội NST thường | mức độ nặng/nhẹ khác nhau/sau sinh | ↑↑ | ↓↓ | ↓↓ |
| Không nhạy cảm với GH (GHI) | GHR | Lặn > Trội NST thường | nặng/sau sinh | Bth đến ↑↑ | ↓ đến ↓↓↓ | ↓ đến ↓↓↓ |
| GHI liên quan đến rối loạn chức năng miễn dịch | STAT5b | Lặn > Trội NST thường | nặng/sau sinh | ↑↑ | ↓↓ | ↓↓ |
| Khiếm khuyết phức hợp tam phân | IGFALS | Lặn NST thường | nhẹ/sau sinh | Bth đến ↑ | ↓↓ | ↓↓↓ |
| PAPPA2 | Lặn NST thường | mức độ nặng/nhẹ khác nhau | ↑↑ | ↑↑↑ | ↑↑↑ | |
| Khiếm khuyết IGF1 | IGF1 | Lặn/Trội NST thường | nặng/trước và sau sinh | ↑↑ | ↓↓↓↓ | Bth hoặc ↑ |
| Khiếm khuyết IGF2 | IGF2 | Trội NST thường (di truyền từ cha) | nặng/trước và sau sinh | Bth hoặc ↑ | Bth hoặc ↑ | Bth hoặc ↑ |
| IGF-1 không hoạt tính sinh học | IGF1 | Lặn NST thường | nặng/trước và sau sinh | ↑↑ | ↑↑↑↑ | Bth |
| Không nhạy cảm với IGF | IGF1R | Trội > Lặn NST thường | mức độ nặng/nhẹ khác nhau/trước và sau sinh | Bth đến ↑↑ | Bth đến ↑↑ | Bth đến ↑↑ |
Các đặc điểm được liệt kê là điển hình của các rối loạn liên quan nhưng có thể không có ở một số trẻ mắc các tình trạng này. ↑, Tăng; ↓, giảm; AD, trội NST thường; ADp, trội NST thường với sự truyền từ cha; AR, lặn NST thường; GH, hormone tăng trưởng; IGF, yếu tố tăng trưởng giống insulin; IGFBP, protein gắn yếu tố tăng trưởng giống insulin; Nl, bình thường; PRL, prolactin.
Thiếu hụt Hormone tăng trưởng Thiếu hụt GH là một rối loạn hiếm gặp với tỷ lệ hiện mắc khoảng 1 trên 4000 trong thời thơ ấu. Thiếu hụt GH có thể là đơn độc (IGHD) hoặc kết hợp với các thiếu hụt hormone khác, khi đó nó được gọi là thiếu hụt hormone tuyến yên kết hợp hoặc đa hormone (CPHD/MPHD). Tình trạng này có thể là bẩm sinh hoặc mắc phải (Bảng 11.4). Chẩn đoán và điều trị sớm là rất quan trọng để đảm bảo kết quả chiều cao cuối cùng tối ưu. Chẩn đoán thiếu hụt GH dựa trên sự kết hợp của đo lường nhân trắc học, loại trừ các bệnh lý khác dẫn đến tầm vóc thấp, điều tra sinh hóa trục GH-IGF-1, và hình ảnh của khu vực dưới đồi-tuyến yên. Gần đây hơn, một chẩn đoán phân tử có thể được tìm kiếm ở những người bị thiếu hụt GH “vô căn” hoặc MPHD (xem Bảng 11.1); tuy nhiên, phần lớn bệnh nhân bị thiếu hụt IGH hoặc CPHD vẫn chưa được giải thích về mặt nguyên nhân. Trước khi đánh giá trục GH-IGF-1, các chẩn đoán khác, chẳng hạn như tầm vóc thấp gia đình, suy giáp, hội chứng Turner, bệnh celiac, bệnh mãn tính, chẳng hạn như bệnh Crohn hoặc suy thận, và các bệnh loạn sản xương nên được loại trừ. Các hướng dẫn đồng thuận về chẩn đoán thiếu hụt GH ở trẻ em đã được công bố vào năm 2000, và đề nghị điều tra trong các tình huống sau:
- Tầm vóc thấp nghiêm trọng, được định nghĩa là chiều cao thấp hơn 3 SD so với mức trung bình.
- Chiều cao thấp hơn 1,5 SD so với chiều cao trung bình của cha mẹ.
- Chiều cao thấp hơn 2 SD so với mức trung bình và vận tốc chiều cao trong 1 năm thấp hơn 1 SD so với mức trung bình cho tuổi theo thời gian, hoặc giảm SD chiều cao hơn 0,5 trong 1 năm ở trẻ em trên 2 tuổi.
- Trong trường hợp không có tầm vóc thấp, vận tốc chiều cao thấp hơn 2 SD so với mức trung bình trong 1 năm hoặc hơn –1,5 SD kéo dài trong 2 năm, có thể xảy ra ở tình trạng thiếu hụt GH biểu hiện ở trẻ sơ sinh hoặc trong thiếu hụt GH mắc phải thực thể.
- Các dấu hiệu chỉ ra một tổn thương nội sọ.
- Các dấu hiệu của MPHD.
- Các triệu chứng và dấu hiệu sơ sinh của thiếu hụt GH.
Bảng 11.4. Các nguyên nhân bẩm sinh và mắc phải của thiếu hụt Hormone tăng trưởng
| Nguyên nhân | Chi tiết |
|---|---|
| 1. Bẩm sinh | |
| Di truyền | Xem Bảng 11.2 |
| Liên quan đến khiếm khuyết cấu trúc não | – Bất sản thể chai – Loạn sản vách–thị – Holoprosencephaly – Thoát vị não – Não úng thủy |
| Liên quan đến khiếm khuyết đường giữa khuôn mặt | – Sứt môi/hở hàm ếch – Một răng cửa giữa duy nhất |
| Hội chứng liên quan | – Hội chứng Cornelia de Lange – Hội chứng CHARGE |
| 2. Mắc phải | |
| Khối u | – U trong và quanh tuyến yên (u sọ hầu, u tế bào mầm) – Khối u di căn |
| Tổn thương khác | – Nang – Xạ trị – Hóa trị – Chấn thương não |
| Viêm/Nhiễm trùng | – Sarcoidosis – Bệnh mô bào Langerhans – Viêm màng não do lao |
| Khác | – Thâm nhiễm – Nhồi máu tuyến yên – Thiếu thốn tâm lý xã hội |
Biểu hiện trong giai đoạn sơ sinh có thể bao gồm hạ đường huyết, vàng da kết hợp kéo dài (thường liên quan đến thiếu hụt ACTH), và dương vật nhỏ. Kích thước khi sinh thường nằm trong phạm vi bình thường, mặc dù có thể có sự giảm khoảng 10%, thường xảy ra vào cuối thai kỳ. Vận tốc tăng trưởng giảm trong năm đầu đời ở trẻ em bị thiếu hụt GH nghiêm trọng với sự suy giảm tăng trưởng biểu hiện vào lúc 6 tháng tuổi, nhưng kiểu hình tiến triển sau 1 tuổi ở những trẻ bị thiếu hụt GH nhẹ hơn. Các biểu hiện sớm nhất là giảm vận tốc chiều cao, sau đó là giảm SDS chiều cao được điều chỉnh theo SDS chiều cao trung bình của cha mẹ. SDS chiều cao của trẻ cuối cùng sẽ giảm xuống dưới –2 SD, thời gian này phụ thuộc vào mức độ nghiêm trọng và thời gian của tình trạng thiếu hụt GH. Một đứa trẻ bị thiếu hụt GH nghiêm trọng thường có thiểu sản giữa mặt, giảm trương lực cơ, giọng nói cao, vẻ ngoài chưa trưởng thành, mọc răng chậm, tóc mỏng thưa, móng tay mọc chậm và béo phì thân mình. Thiếu hụt GH cũng liên quan đến các tác động lên nhận thức. Ở những con chuột bị thiếu hụt GH, khả năng học tập và ghi nhớ không gian giảm đã được báo cáo trong khi ở những đứa trẻ không được điều trị bị thiếu hụt GH, chỉ số IQ, khả năng hiểu bằng lời nói và tốc độ xử lý bị giảm. Ở trẻ em và người lớn, các khiếm khuyết về thần kinh nhận thức trong tình trạng thiếu hụt GH được cải thiện bằng liệu pháp GH. Với việc điều trị bằng hormone tăng trưởng người tái tổ hợp (rhGH) kịp thời, triển vọng về chiều cao cuối cùng là rất tốt. Các thiếu hụt hormone khác có thể cần điều trị; đặc biệt những người có bất thường cấu trúc của vùng dưới đồi-tuyến yên có nhiều khả năng tiến triển và phát triển các thiếu hụt hormone khác.
Nguyên nhân gây thiếu hụt hormone tăng trưởng Các rối loạn bẩm sinh của sự phát triển vùng dưới đồi
Rối loạn chức năng dưới đồi-tuyến yên có thể phát sinh từ các dị tật bẩm sinh của não hoặc vùng dưới đồi hoặc của tuyến yên. Vô não dẫn đến một tuyến yên nhỏ hoặc hình thành bất thường và thường lạc chỗ. Holoprosencephaly, kết quả từ sự phát triển đường giữa bất thường của não phôi, cũng thường liên quan đến suy dưới đồi. Phổ lâm sàng của holoprosencephaly có thể từ cyclopia đến hai mắt xa nhau, đi kèm với không có rãnh môi trên hoặc vách ngăn mũi và các khe hở đường giữa của vòm miệng hoặc môi. Trong những tình huống này, đặc điểm nội tiết kinh điển là đái tháo nhạt (DI), thường đi kèm với thiếu hụt GH, hormone kích thích tuyến giáp (TSH) và ACTH.
Tranh cãi vẫn tiếp tục về việc liệu tỷ lệ thiếu hụt GH có tăng lên trong các trường hợp sứt môi và/hoặc hở hàm ếch đơn giản hay không. Rõ ràng, trẻ em bị sứt môi đang phát triển bất thường cần được đánh giá thêm, và cả thiếu hụt GH và CPHD đều thường xuyên hơn ở nhóm này. SOD là một dị tật hiếm gặp, bẩm sinh, không đồng nhất với tỷ lệ hiện mắc từ 6,3 đến 10,9 trên 100.000. Tình trạng này được xác định bởi sự hiện diện của bất kỳ hai trong ba đặc điểm: khiếm khuyết đường giữa của não trước, chẳng hạn như không có vách trong suốt và/hoặc thể chai; thiểu sản thần kinh thị (ONH); và suy tuyến yên do sự phát triển bất thường của vùng dưới đồi-tuyến yên. De Morsier, vào năm 1956, đã mô tả các phát hiện sau khi khám nghiệm tử thi về ONH và bất sản vách trong suốt và đặt ra thuật ngữ SOD, còn được gọi là hội chứng De Morsier. Khoảng 30% bệnh nhân SOD biểu hiện bộ ba lâm sàng hoàn chỉnh, 62% bệnh nhân có một mức độ nào đó của suy tuyến yên, và 60% không có vách trong suốt. Tình trạng này có tỷ lệ hiện mắc như nhau ở nam và nữ. ONH có thể là một bên hoặc hai bên và có thể là đặc điểm biểu hiện đầu tiên, với sự khởi phát sau đó của rối loạn chức năng nội tiết. ONH hai bên phổ biến hơn (88% so với 12% trường hợp một bên). Ngoài ra, dường như có ít mối tương quan giữa kích thước của dây thần kinh thị giác và chức năng thị giác của nó. Các bất thường về thần kinh X-quang có mặt ở 75% đến 80% bệnh nhân bị ONH. Thiểu sản tuyến yên có thể biểu hiện như các thiếu hụt nội tiết khác nhau từ thiếu hụt GH đơn độc đến suy toàn bộ tuyến yên. Đã có một số gợi ý rằng các bất thường của vách trong suốt và trục dưới đồi-tuyến yên trên hình ảnh thần kinh có thể dự đoán mức độ nghiêm trọng của rối loạn chức năng nội tiết. Giảm tốc độ tăng trưởng do thiếu hụt GH là đặc điểm phổ biến nhất, với hạ đường huyết và tiểu nhiều và khát nhiều là ít phổ biến hơn. Có thể xảy ra dậy thì sớm hoặc không phát triển trong tuổi dậy thì. Giải phẫu thần kinh hoặc chức năng dưới đồi bất thường và DI có thể là một đặc điểm. Bệnh nội tiết tiến triển với sự mất dần chức năng nội tiết theo thời gian. Bệnh nội tiết phổ biến nhất là thiếu hụt GH, sau đó là thiếu hụt TSH và ACTH. Sự bài tiết Gonadotropin có thể được giữ lại khi có các thiếu hụt hormone tuyến yên khác. Thiếu hụt thần kinh là phổ biến, nhưng không phải luôn luôn như vậy, và các thiếu hụt từ chậm phát triển toàn bộ đến các thiếu hụt khu trú, chẳng hạn như động kinh hoặc liệt nửa người. Các bất thường giải phẫu thần kinh khác bao gồm khoang vách trong suốt, thiểu sản tiểu não, schizencephaly, và bất sản vòm não. Mối liên quan giữa SOD và các dị tật bẩm sinh khác, chẳng hạn như các bất thường về ngón tay, không phải là hiếm.
Nguyên nhân của SOD phần lớn vẫn chưa được biết cho đến gần đây. Cả yếu tố di truyền và môi trường đều có liên quan đến nguyên nhân của tình trạng này. Các đột biến trong các gen liên quan đến sự phát triển của não trước, mắt và tuyến yên chiếm một số lượng nhỏ bệnh nhân bị SOD, và các gen này bao gồm HESX1, SOX2, OTX2, và TCF7L1. Các tác nhân môi trường, chẳng hạn như nhiễm virus, các thay đổi mạch máu hoặc thoái hóa, và tiếp xúc với rượu hoặc ma túy cũng đã được liên quan đến nguyên nhân của SOD. Tình trạng này biểu hiện phổ biến hơn ở trẻ em sinh ra từ những bà mẹ trẻ hơn và tập trung ở các khu vực địa lý có tần suất mang thai ở tuổi vị thành niên cao. Tuy nhiên, tần suất chung của các đột biến di truyền trong SOD thấp, cho thấy rằng các đột biến trong các gen đã biết hoặc chưa biết khác có thể góp phần vào rối loạn phức tạp này.
Gần đây, con đường tín hiệu Sonic Hedgehog (SHH) đã được liên quan đến các rối loạn phức tạp hơn của sự phát triển tuyến yên. Các đột biến trong SHH có liên quan đến holoprosencephaly. Các yếu tố phiên mã Gli tham gia vào sự truyền tín hiệu hedgehog, và các đột biến mất chức năng dị hợp tử trong GLI2 đã được xác định ở những bệnh nhân bị holoprosencephaly. Độ thấm của kiểu hình có thể thay đổi. Ở một số cá nhân bị ảnh hưởng có đột biến GLI2, chức năng tuyến yên bất thường, đi kèm với các bất thường sọ mặt khác nhau. Các đặc điểm khác bao gồm thừa ngón sau trục, một lỗ mũi duy nhất, một răng cửa giữa duy nhất và bất sản một phần thể chai. Gần đây hơn, các đột biến trong GLI2 đã được liên kết với suy tuyến yên mà không có bất kỳ khiếm khuyết đường giữa nào.
SOX2 và SOX3 là các thành viên của họ yếu tố phiên mã SOX (hộp HMG liên quan đến SRY) và là các dấu hiệu sớm của các tế bào tiền thân; sự biểu hiện của chúng bị điều hòa giảm khi các tế bào biệt hóa. Ban đầu, các đột biến SOX2 đã được liên kết với không có mắt hai bên, tật mắt nhỏ nghiêm trọng, khó khăn trong học tập, teo thực quản và các bất thường về bộ phận sinh dục. Tuy nhiên, việc xác định đặc điểm kiểu hình sâu hơn đã tiết lộ sự hiện diện của thiểu sản tuyến yên trước, suy sinh dục do thiếu gonadotropin, và thiếu hụt GH thay đổi, thường đi kèm với các kiểu hình như bất thường hồi hải mã, bất sản thể chai, teo thực quản, u mô thừa vùng dưới đồi, và mất thính lực thần kinh giác quan. Mặc dù, ở phần lớn bệnh nhân, chụp cộng hưởng từ (MRI) cho thấy một tuyến yên trước nhỏ, trong một số trường hợp, tuyến yên bị to ra và vẫn như vậy trong nhiều năm.
Sự phát triển bình thường của trục dưới đồi-tuyến yên phụ thuộc rất nhiều vào liều lượng của SOX3; quá liều hoặc thiếu liều có thể dẫn đến suy tuyến yên hoặc thiểu sản cuống tuyến yên. Ở người, sự nhân đôi hoặc đột biến SOX3 có liên quan đến các kiểu hình khác nhau, bao gồm cả IGHD hoặc suy toàn bộ tuyến yên. Có sự chậm phát triển thay đổi, và MRI thường cho thấy một tuyến yên trước nhỏ, một tuyến yên sau lạc chỗ/không xuống, và loạn sản thể chai. Việc xóa SOX3 gần đây đã được liên kết với một ống sọ hầu tồn tại liên quan đến suy tuyến yên.
Chấn thương não và/hoặc vùng dưới đồi
Chấn thương sọ não (TBI) đã được công nhận là một nguyên nhân gây suy tuyến yên mắc phải trong một số nghiên cứu ở người lớn. Dữ liệu về bệnh nhân nhi vẫn còn rải rác, nhưng ngày càng có nhận thức rằng suy tuyến yên sau TBI bị chẩn đoán thiếu với những tác động tiêu cực có thể có đối với sự tăng trưởng và phát triển. Mặc dù tuyến yên được bảo vệ trong hố yên, mạng lưới mạch máu phong phú của vùng dưới đồi và tuyến yên và cấu trúc của cuống tuyến yên làm cho nó dễ bị tổn thương bởi các tác động của chấn thương sọ não. Vùng dưới đồi và tuyến yên có một nguồn cung cấp mạch máu phức tạp bao gồm các mạch tuyến yên dài và một mạng lưới mao mạch cửa phong phú bao quanh tuyến yên và cuống tuyến yên. Sinh lý bệnh của suy tuyến yên liên quan đến TBI không được xác định rõ ràng, nhưng người ta cho rằng đó là kết quả của chấn thương trực tiếp hoặc của tổn thương mạch máu dẫn đến thiếu máu cục bộ và nhồi máu. Điều này được hỗ trợ bởi các phát hiện giải phẫu của các cuộc khám nghiệm tử thi sau chấn thương đầu, bao gồm hoại tử thùy trước, xơ hóa tuyến yên, xuất huyết, nhồi máu, hoặc hoại tử cuống tuyến yên.
Đáng chú ý là lớp ngoại vi của các tế bào tuyến yên trước, dưới bao, nhận máu động mạch từ bao chứ không phải từ hệ thống tĩnh mạch cửa, và điều này có thể giải thích tại sao các tế bào này và những tế bào trong một khu vực nhỏ liền kề với thùy sau là những tế bào duy nhất còn sống sót trong các trường hợp hoại tử thùy trước đơn thuần. Các tế bào hướng thân nằm ở các cánh của tuyến yên, nguồn cung cấp mạch máu của chúng đến từ các mạch cửa, và chúng dễ bị gián đoạn nguồn cung cấp máu sau chấn thương đầu. Mặt khác, các tế bào tiết ACTH và TSH nằm ở phần giữa của tuyến yên và nhận nguồn cung cấp máu từ các mạch cửa và động mạch tuyến yên trước. Điều này có thể giải thích tại sao thiếu hụt GH là thiếu hụt phổ biến nhất được thấy sau TBI. Các thiếu hụt hormone có thể được xác định trong những ngày đến vài tuần đầu sau chấn thương (giai đoạn cấp tính) hoặc có thể phát triển theo thời gian (tác động muộn). Vì có sự chồng chéo giữa các triệu chứng và dấu hiệu của suy tuyến yên và các di chứng thần kinh-tâm lý của TBI, có thể các thiếu hụt tiến triển muộn hoặc một phần có thể không được chẩn đoán trong nhiều năm. Do đó, không có gì ngạc nhiên khi, trong các nghiên cứu khác nhau, thời gian để chẩn đoán dao động từ 1 đến 40 năm. Dường như phần lớn bệnh nhân cho thấy một mức độ rối loạn chức năng hormone tuyến yên trong những ngày đầu sau TBI (53%–76%); tuy nhiên, có sự thay đổi lớn trong các phản ứng hormone được báo cáo, phản ánh sự khác biệt trong việc lựa chọn bệnh nhân và thời gian xét nghiệm. Tất cả các hormone tuyến yên trước đều có thể bị ảnh hưởng. Các thiếu hụt hormone tuyến yên biểu hiện trong giai đoạn cấp tính thường là tạm thời, nhưng chúng có thể tồn tại hoặc xuất hiện và tiến triển theo thời gian. Ở người lớn, tỷ lệ suy tuyến yên vĩnh viễn dao động từ 23% đến 69% tùy thuộc vào nghiên cứu. Trục GH bị ảnh hưởng thường xuyên nhất (10%–33%), sau đó là các trục sinh dục (8%–23%), tuyến thượng thận (5%–23%) và tuyến giáp (2%–22%). Tỷ lệ hiện mắc của DI vĩnh viễn thay đổi từ 0% đến 6%.
Cho đến gần đây, chỉ có các báo cáo rải rác về suy tuyến yên sau TBI ở trẻ em, nhưng các nghiên cứu tiến cứu được thiết kế để xác định đặc điểm tốt hơn của rối loạn này trong dân số nhi khoa và vị thành niên đang được tiến hành. Tỷ lệ suy tuyến yên được báo cáo dao động từ 10% đến 60% và, mặc dù tỷ lệ này ở trẻ em thấp hơn so với người lớn, nhưng nó không phải là hiếm. Nói chung, kết quả lâu dài của TBI dường như thuận lợi hơn ở trẻ em. Bệnh nhân bị suy tuyến yên sau chấn thương đầu có thể không có các dấu hiệu và triệu chứng lâm sàng gợi ý về rối loạn này và chẩn đoán kịp thời đòi hỏi mức độ nghi ngờ cao. Một hướng dẫn đồng thuận về việc sàng lọc bệnh nhân sau TBI cho thấy rằng tất cả các bệnh nhân đã bị TBI, bất kể mức độ nghiêm trọng của nó, nên được đánh giá nội tiết cơ bản 3 và 12 tháng sau sự kiện hoặc xuất viện từ đơn vị điều trị tích cực. Đối với trẻ em và thanh thiếu niên, một thuật toán để đánh giá và theo dõi nội tiết cũng đã được đề xuất. Tuy nhiên, một nghiên cứu gần đây đã báo cáo rằng suy tuyến yên vĩnh viễn là không phổ biến sau chấn thương sọ não nghiêm trọng ở trẻ nhỏ khi các tiêu chí lâm sàng nghiêm ngặt cho suy tuyến yên được sử dụng. Việc xét nghiệm chức năng tuyến yên định kỳ, như được đề xuất cho người lớn, có thể không cần thiết ở trẻ em và có thể dẫn đến một số lượng lớn các kết quả xét nghiệm bất thường.
Chấn thương chu sinh đối với não, vùng dưới đồi, cuống tuyến yên, hoặc tuyến yên trước có thể dẫn đến thiếu hụt GH đơn độc hoặc thiếu hụt nhiều hormone của tuyến yên trước, cũng như lạm dụng trẻ em. Nhiều loạt bài đã được công bố về các bệnh nhân bị thiếu hụt GH cho thấy tỷ lệ chấn thương khi sinh tăng lên, chẳng hạn như sinh ngôi mông, sử dụng kẹp forceps rộng rãi, chuyển dạ kéo dài, hoặc sinh đột ngột. Tranh cãi vẫn tiếp tục về việc liệu thiếu hụt GH là hậu quả của một cuộc sinh khó hay chỉ đơn thuần phản ánh những hậu quả chu sinh của tình trạng suy tuyến yên của thai nhi.
Viêm và thâm nhiễm não và/hoặc vùng dưới đồi
Viêm não (viêm màng não hoặc viêm não do nhiễm vi khuẩn, virus hoặc nấm) có thể dẫn đến suy dưới đồi/tuyến yên, với hầu hết các trường hợp được báo cáo dưới dạng các báo cáo ca đơn lẻ. Mặc dù có sự thiên vị trong việc lựa chọn các trường hợp, tỷ lệ thiếu hụt nội tiết có khả năng phụ thuộc vào độc lực của sinh vật gây nhiễm, mức độ nghiêm trọng và vị trí của bệnh, và tình trạng miễn dịch của vật chủ. Trong một nghiên cứu trên 19 bệnh nhân người lớn được điều tra từ 10 đến 56 tháng sau khi bị nhiễm trùng hệ thần kinh trung ương, 21% bị thiếu hụt ACTH và 11% bị thiếu hụt gonadotropin, trong khi không có báo cáo về thiếu hụt GH hoặc DI. Suy tuyến yên đã được báo cáo sau khi nhiễm nhiều loại tác nhân khác nhau, bao gồm liên cầu khuẩn nhóm B, Haemophilus influenza, và Mycobacterium tuberculosis. Vùng dưới đồi và/hoặc tuyến yên cũng có thể bị liên quan trong bệnh sarcoidosis. Sarcoidosis là một bệnh u hạt đa hệ thống không rõ nguyên nhân, ảnh hưởng lâm sàng đến hệ thần kinh trung ương trong 5% đến 10% trường hợp. Các tác động của sarcoidosis lên trục dưới đồi-tuyến yên là kết quả của sự thâm nhiễm bởi mô u hạt. Trên MRI, tổn thương có thể thâm nhiễm vào vùng dưới đồi và tuyến yên và gây dày cuống tuyến yên, và nó thường ngấm thuốc cản quang gadolinium. Bất thường nội tiết được báo cáo thường xuyên nhất là DI, được báo cáo ở 25% đến 50% bệnh nhân bị sarcoidosis thần kinh. Tiếp theo là tăng prolactin máu mặc dù rối loạn chức năng tuyến yên trước với suy sinh dục cũng đã được báo cáo. Sarcoidosis của hệ thần kinh có tiên lượng xấu, nhưng các đợt thuyên giảm lâu dài đã được báo cáo với liệu pháp methylprednisolone liều cao tiêm tĩnh mạch. Các khiếm khuyết nội tiết kéo dài dưới 1 năm có thể đáp ứng với điều trị bằng steroid, nhưng các thiếu hụt kéo dài hơn thường tồn tại.
Viêm tuyến yên là một tình trạng viêm của tuyến yên có thể là nguyên phát hoặc thứ phát và có thể là kết quả của nhiễm trùng, bệnh hệ thống, hoặc kích thích từ các tổn thương lân cận. Quá trình viêm này bắt chước, về mặt lâm sàng và X-quang, các khối u của khu vực tuyến yên. Có ba loại mô học của viêm tuyến yên nguyên phát: lympho bào, u hạt, và xanthomatous. Viêm tuyến yên lympho bào là loại phổ biến nhất; nó liên quan đến tuyến yên trước và có thể thâm nhiễm vào cuống tuyến yên và thùy sau. Viêm tuyến yên lympho bào xảy ra chủ yếu ở phụ nữ trẻ và có liên quan đến thai kỳ hoặc sự hiện diện của các bệnh tự miễn, bao gồm viêm tuyến giáp Hashimoto, bệnh Graves, đái tháo đường loại I, và lupus ban đỏ hệ thống. Có những báo cáo ca hiếm gặp về viêm tuyến yên ở trẻ em và thanh thiếu niên, và trong hầu hết các trường hợp, chẩn đoán chỉ được thực hiện sau khi sinh thiết và kiểm tra mô học, mặc dù trong thực hành lâm sàng, điều này hiếm khi được thực hiện. Trong nhiều trường hợp, viêm tuyến yên biểu hiện bằng DI và suy tuyến yên và có trước chẩn đoán một khối u nội sọ, chẳng hạn như u tế bào mầm. Trong các trường hợp khác, nó có thể biểu hiện bằng DI và suy sinh dục do thiếu gonadotropin. Sau khi chẩn đoán được xác lập, việc quản lý thường là bảo tồn, trừ khi có các dấu hiệu tăng áp lực nội sọ hoặc chèn ép dây thần kinh thị giác.
Bệnh mô bào Langerhans (LCH) được đặc trưng bởi sự tăng sinh dòng vô tính và tích tụ các tế bào đuôi gai bất thường có thể ảnh hưởng đến một vị trí duy nhất hoặc nhiều hệ thống gây ra rối loạn chức năng đa cơ quan. Ở trẻ em, tuổi trung bình của chẩn đoán dao động từ 1,8 đến 3,4 tuổi. LCH thâm nhiễm khu vực dưới đồi-tuyến yên ở 15% đến 35% bệnh nhân với sự phát triển sau đó của ít nhất một thiếu hụt hormone tuyến yên. Trong một nghiên cứu quốc gia đa trung tâm của Pháp trên 589 bệnh nhân nhi bị LCH, 145 bệnh nhân (25%) bị rối loạn chức năng tuyến yên. Ở 60 bệnh nhân, sự liên quan của tuyến yên đã có mặt tại thời điểm chẩn đoán và ở 20 người trong số họ, đó là biểu hiện đầu tiên của bệnh. Bệnh nhân có nguy cơ cao bị liên quan đến tuyến yên dường như là những người mắc bệnh đa hệ thống liên quan đến xương sọ và mặt, xương chũm, các xoang, và màng nhầy (tức là nướu, tai, mũi, và vùng họng). Hơn nữa, so với những bệnh nhân không bị liên quan đến tuyến yên, những bệnh nhân bị liên quan đến tuyến yên có tỷ lệ tái phát cao hơn (10% ở 5 năm so với 4,8% ở 5 năm), và tỷ lệ mắc LCH thoái hóa thần kinh cao hơn.
DI là hậu quả vĩnh viễn được báo cáo thường xuyên nhất của LCH và là bệnh nội tiết phổ biến nhất; gần như tất cả các bệnh nhân bị liên quan đến tuyến yên đều có DI, thường biểu hiện sớm trong quá trình của bệnh, trong vòng 3 đến 5 năm đầu tiên, và đôi khi có thể có trước chẩn đoán LCH. Trẻ em bị LCH và DI cũng có thể bị thiếu hụt hormone tuyến yên trước, với hầu hết các thiếu hụt phát triển trong 6 năm sau khi chẩn đoán DI. Bệnh nội tiết phổ biến thứ hai là thiếu hụt GH, xảy ra ở 14% tổng số bệnh nhân bị LCH và ở hơn 40% bệnh nhân bị liên quan đến tuyến yên. Trong đại đa số bệnh nhân, thiếu hụt GH có liên quan đến DI, với khoảng thời gian trung bình từ 2,9 đến 3,5 năm giữa chẩn đoán DI và sự phát triển của thiếu hụt GH. Thiếu hụt GH đơn độc, hoặc sự kết hợp của thiếu hụt GH với các thiếu hụt hormone tuyến yên trước khác, xảy ra ít phổ biến hơn.
Các phát hiện MRI tuyến yên ở bệnh nhân bị LCH bao gồm dày cuống tuyến yên, gợi ý quá trình thâm nhiễm, các thay đổi ngấm thuốc trong tuyến yên và vùng dưới đồi, và không có tín hiệu sáng của tuyến yên sau trên hình ảnh T1, do mất các hạt tiết hormone chống bài niệu giàu phospholipid. Dấu hiệu sau là một đặc điểm không thay đổi của những bệnh nhân phát triển DI. Mặc dù, tại thời điểm chẩn đoán DI, 75% cho thấy một cuống tuyến yên dày, chỉ có 24% có cuống dày dai dẳng sau 5 năm. Những thay đổi này có thể thay đổi và không tương quan với điều trị hoặc với sự phục hồi lâm sàng vì DI tồn tại trong tất cả các trường hợp. Vai trò của MRI trong việc dự đoán sự phát triển của các thiếu hụt hormone trước là không chắc chắn. Đã có báo cáo rằng những bệnh nhân sẽ bị thiếu hụt GH có nhiều khả năng có một tuyến yên trước nhỏ hơn, trong khi kích thước của cuống và tuyến yên sau không khác biệt đáng kể.
Theo dõi lâu dài các bệnh nhân bị LCH đã cho thấy rằng các thiếu hụt hormone đã được xác lập không thể được đảo ngược bằng điều trị.
Các khối u của não và/hoặc vùng dưới đồi
Các khối u của hệ thần kinh trung ương là một nguyên nhân chính gây suy dưới đồi. Điều này đặc biệt đúng đối với các khối u đường giữa của não, chẳng hạn như u tế bào mầm, u màng não, u thần kinh đệm, nang keo của não thất ba, u màng não thất, và u thần kinh đệm của dây thần kinh thị giác. Mặc dù di căn từ các ung thư ngoài sọ hiếm khi được tìm thấy ở trẻ em, suy dưới đồi có thể là kết quả của sự lan rộng tại chỗ của ung thư biểu mô vòm họng hoặc bệnh Hodgkin của vòm họng. U tuyến yên chiếm ít hơn 3% các khối u trên lều ở trẻ em và khoảng 3,5% đến 6% tất cả các khối u tuyến yên ở trẻ em được điều trị bằng phẫu thuật. Trong hầu hết các trường hợp, chúng hoạt động về mặt nội tiết, phát sinh từ bất kỳ trong năm loại tế bào của tuyến yên trước, và có thể sản xuất prolactin (prolactinoma, 52%), ACTH dẫn đến bệnh Cushing (corticotropinoma 33,3%), GH (somatotropinoma 8%), hoặc hiếm hơn là TSH (thyrotropinoma). Các u tuyến yên không hoạt động hiếm gặp ở trẻ em (2,7%) so với người lớn, nơi chúng chiếm gần 20% các u tuyến yên. Mặc dù phần lớn các u tuyến yên ở trẻ em là prolactinoma biểu hiện ở tuổi vị thành niên, corticotropinoma là các khối u phổ biến nhất ở trẻ em trước tuổi dậy thì. Các u tuyến yên xảy ra đơn độc hoặc có thể là một phần của một hội chứng di truyền, chẳng hạn như đa u tuyến nội tiết loại 1 (MEN1), hội chứng McCune-Albright (MAS), hoặc phức hợp Carney. Cơ chế bệnh sinh của chúng không rõ ràng nhưng ngày càng có nhiều bằng chứng cho thấy sự rối loạn điều hòa trong tín hiệu thụ thể hormone, những thay đổi trong các phân tử điều hòa chu kỳ tế bào hoặc quan trọng đối với sự kết dính với chất nền ngoại bào, cũng như những thay đổi trong các yếu tố tăng trưởng, có thể có liên quan. Biểu hiện lâm sàng của chúng là kết quả của sự tăng tiết hoặc thiếu hụt hormone tuyến yên, sự gián đoạn của sự tăng trưởng và trưởng thành sinh dục, và các hiệu ứng chèn ép. Trên MRI, các u tuyến yên cho thấy sự ngấm thuốc gadolinium chậm và xuất hiện dưới dạng các tổn thương giảm ngấm thuốc có thể làm lệch cuống tuyến yên.
U thần kinh đệm thị giác
Các khối u của đường thị giác chiếm 4% đến 6% tất cả các khối u nội sọ ở trẻ em và trong số này phổ biến nhất là u thần kinh đệm thị giác (65%). Hầu hết các u thần kinh đệm thị giác là các tổn thương độ thấp với tiên lượng thuận lợi nếu được điều trị tối ưu. Các u thần kinh đệm giới hạn ở dây thần kinh thị giác có khuynh hướng ở nữ giới (60%–70%) và có liên quan đến bệnh u xơ thần kinh loại 1 (NF-1) trong hơn một nửa số trường hợp, trong khi 38% là lẻ tẻ. Trẻ em bị u thần kinh đệm lẻ tẻ có nhiều khả năng biểu hiện tăng áp lực nội sọ, giảm hoạt động thị giác, và các biến chứng nội tiết được ghi nhận phổ biến hơn. Các triệu chứng thường gặp nhất khi trình bày là các khiếm khuyết thị giác (giảm thị lực, teo dây thần kinh thị giác, lác, rung giật nhãn cầu, lồi mắt), mất điều hòa, và dậy thì sớm. Do mối quan hệ giải phẫu gần gũi của chúng với vùng dưới đồi và tuyến yên, sự rối loạn điều hòa của trục dưới đồi-tuyến yên là phổ biến và là do chính khối u hoặc thứ phát sau điều trị. Trưởng thành sinh dục sớm là một triệu chứng trình bày thường xuyên trong khi khiếm khuyết phổ biến nhất sau xạ trị sọ não là thiếu hụt GH. Các u thần kinh đệm thị giác liên quan đến NF-1 cũng có thể gây ra thừa GH và tăng trưởng quá mức.
Các tổn thương dạng nang
Các tổn thương dạng nang ở vùng tuyến yên bao gồm nang khe Rathke, nang màng nhện, và u tuyến dạng nang và u sọ hầu. Nang khe Rathke là những di tích dạng nang lành tính của túi Rathke. Chúng thường nhỏ (< 5 mm), không có triệu chứng, và được tìm thấy trong gần 20% các cuộc khám nghiệm tử thi định kỳ. Chúng bao gồm các tế bào biểu mô hình trụ hoặc hình khối biệt hóa tốt và thành phần của nang thay đổi. Nang khe Rathke có thể phát triển dần dần và trở nên có triệu chứng, đặc biệt nếu chúng lan rộng trên yên. Các triệu chứng bao gồm đau đầu, khiếm khuyết thị giác, và rối loạn chức năng tuyến yên từ tăng prolactin đến thiếu hụt hormone tuyến yên. Chẩn đoán phân biệt với các tổn thương dạng nang khác trong khu vực không phải lúc nào cũng dễ dàng, vì trên MRI, dịch nang cho thấy cường độ tín hiệu thay đổi và do đó nang có thể xuất hiện dưới dạng giảm hoặc tăng đậm độ. Gần 50% nang khe Rathke cho thấy sự ngấm thuốc viền. Sự tái phát của nang sau khi điều trị là hiếm (2 trong số 14 bệnh nhân trong một loạt ca) và khuyến cáo rằng việc điều trị nên bao gồm cả dẫn lưu dịch và loại bỏ thành nang để tránh tái phát.
Nang màng nhện bao gồm một tập hợp dịch giống dịch não tủy (CSF) được bao quanh bởi một bức tường làm bằng các cấu trúc màng nhện. Chúng chủ yếu nằm trên yên, chỉ có những trường hợp hiếm gặp là trong yên. Các nang trên yên thường được chẩn đoán sau các triệu chứng không phải nội tiết, chẳng hạn như thiếu hụt thần kinh, đầu to, và các triệu chứng thị giác. Do sự gần gũi của tổn thương với khu vực dưới đồi-tuyến yên, nang màng nhện có thể gây ra dậy thì sớm trung ương, vô kinh, và tăng prolactin máu, ngoài ra còn có thiếu hụt thyrotropin, ACTH, hoặc GH.
U sọ hầu có thể dẫn đến rối loạn chức năng vùng dưới đồi (sẽ được thảo luận sau).
Xạ trị não và/hoặc vùng dưới đồi
Xạ trị sọ não là một nguyên nhân quan trọng gây rối loạn chức năng dưới đồi/tuyến yên. Xạ trị có thể làm suy giảm trực tiếp chức năng của vùng dưới đồi và tuyến yên, và không phải lúc nào cũng dễ dàng phân biệt được tổn thương ở mỗi cấp độ này. Ngoài ra, chức năng của tuyến giáp và tuyến sinh dục cũng có thể bị ảnh hưởng trực tiếp nếu áp dụng xạ trị toàn thân hoặc xạ trị sọ-tủy sống. Mức độ suy giảm tuyến yên có liên quan đến liều bức xạ nhận được. Liều thấp thường dẫn đến thiếu hụt GH đơn độc. Với liều cao hơn, quan sát thấy thiếu hụt nhiều hormone tuyến yên. Hai đến 5 năm sau xạ trị, 100% trẻ em nhận được hơn 3000 cGy trong 3 tuần vào trục dưới đồi-tuyến yên cho thấy các phản ứng GH dưới mức bình thường đối với các nghiệm pháp kích thích bằng insulin.
Mức độ thiếu hụt tuyến yên quan sát được tăng lên theo thời gian kể từ khi xạ trị. Trẻ em có kết quả xét nghiệm bình thường sau 1 năm xạ trị vẫn có thể phát triển các thiếu hụt tuyến yên vào những thời điểm sau đó. Ngay cả khi nồng độ GH trong huyết thanh sau các nghiệm pháp kích thích là bình thường, các phép đo sự bài tiết GH tự phát có thể cho thấy một sự suy giảm. Chỉ cần 1800 cGy đã được chứng minh là ảnh hưởng đến sự bài tiết GH tự phát ở trẻ em tuổi dậy thì.
Sự giảm bài tiết GH có thể còn phức tạp hơn bởi tác động của xạ trị lên sự phát triển của cột sống, có thể dẫn đến tầm vóc thấp và mất cân đối xương. Đáng ngạc nhiên là, xạ trị sọ não cũng có thể dẫn đến dậy thì sớm—với hậu quả là cốt hóa sụn đầu xương xảy ra ở độ tuổi sớm hơn lý tưởng từ quan điểm tối đa hóa sự tăng trưởng. Các chất tương tự GnRH có thể được sử dụng để trì hoãn sự tiến triển của tuổi dậy thì. Bác sĩ nội tiết theo dõi trẻ đã được xạ trị sọ-tủy sống phải xem xét ba yếu tố này: suy tuyến yên tiến triển, giảm tiềm năng tăng trưởng cột sống, và dậy thì sớm và cốt hóa sụn đầu xương sớm. Cần phải cẩn thận để tối đa hóa tiềm năng tăng trưởng của bệnh nhân mà không gây ra sự mất cân đối xương và không trì hoãn tuổi dậy thì quá mức hoặc cho phép sự tăng trưởng kết thúc quá sớm.
Các bất thường di truyền dẫn đến thiếu hụt hormone tuyến yên kết hợp
Cơ sở phân tử của SOD và các tình trạng đường giữa khác đã được thảo luận trước đây.
Các đột biến lặn trong gen LHX3 đã được xác định ở một số gia đình có quan hệ huyết thống, ngoài ra còn có một bệnh nhân duy nhất được phát hiện là dị hợp tử phức hợp cho hai đột biến sai nghĩa trong gen. Những bệnh nhân này thường có nhiều thiếu hụt hormone tuyến yên trước, bao gồm cả thiếu hụt ACTH, mà ban đầu được cho là không bị ảnh hưởng. Hình thái tuyến yên thay đổi giữa các bệnh nhân có đột biến LHX3: hầu hết bệnh nhân có tuyến yên trước thiểu sản với tuyến yên sau và các cấu trúc đường giữa bình thường, ngược lại, một tuyến yên trước to ra cũng đã được báo cáo ở một bệnh nhân mà không rõ ràng trong một lần quét MRI trước đó được thực hiện 10 năm trước. Ngoài ra, một bệnh nhân có tổn thương giảm tín hiệu ở tuyến yên trước phù hợp với một u tuyến nhỏ cũng đã được mô tả. Phần lớn bệnh nhân có đột biến LHX3 được báo cáo cho đến nay cũng đã biểu hiện một cột sống cổ ngắn, cứng với khả năng xoay cổ và cử động thân mình hạn chế. Một lần nữa, các kiểu hình xương có thể thay đổi, và một bệnh nhân duy nhất có khả năng xoay cổ bình thường và không có các đặc điểm hội chứng khác đã được báo cáo. Ngoài ra, hầu hết bệnh nhân có đột biến LHX3 biểu hiện điếc thần kinh giác quan, mức độ nghiêm trọng của nó cũng rất thay đổi và có thể từ sâu đến rất nhẹ và có thể bị bỏ sót trong một số trường hợp. Một vai trò trực tiếp có thể được liên quan ở đây vì LHX3 được biểu hiện ở các vùng cụ thể của tai trong theo một mô hình được bảo tồn cao giữa người và chuột, và nó có khả năng có vai trò trong sự phát triển của tế bào lông ốc tai.
Các đột biến dị hợp tử trong gen LHX4 cũng đã được báo cáo, với tất cả các bệnh nhân đều biểu hiện thiếu hụt GH và tầm vóc thấp liên quan khi khám, một lần nữa với các thiếu hụt nội tiết bổ sung và các bất thường ngoài tuyến yên khác nhau. Ban đầu, một đột biến intron dị hợp tử làm mất đi sự cắt nối bình thường của LHX4 đã được báo cáo bởi Machinis và các đồng nghiệp trong một gia đình ba thế hệ phân ly theo kiểu trội và có độ thấm hoàn toàn. Các bệnh nhân khởi phát có tầm vóc thấp và được phát hiện là thiếu hụt GH, TSH và ACTH, với thiểu sản tuyến yên trước, tuyến yên sau không xuống, và không có cuống tuyến yên trên MRI. Các thành viên khác trong gia đình bị ảnh hưởng có tầm vóc thấp liên quan đến IGHD và một tuyến yên sau bình thường. Các biểu hiện bổ sung bao gồm một hố yên hình thành kém và các hạnh nhân tiểu não nhọn. Một số đột biến đã được mô tả sau đó và rõ ràng là thay đổi về độ thấm và kiểu hình. Gần đây, một đột biến đồng hợp tử mới trong LHX4 có liên quan đến một kiểu hình lặn nghiêm trọng dẫn đến một dạng suy tuyến yên gây chết, cho thấy rằng độ thấm thay đổi được quan sát thấy ở phần lớn các đột biến dị hợp tử có thể chỉ ra sự hiện diện của một bất thường di truyền thứ hai chưa được phát hiện cho đến nay.
Gen mã hóa PROP1 tham gia vào việc xác định và biệt hóa sớm của nhiều dòng tế bào tuyến yên trước. Các bất thường của PROP1 dẫn đến CPHD, được đặc trưng bởi các mức độ thiếu hụt khác nhau của GH, prolactin, TSH, hormone kích thích nang trứng (FSH), hormone tạo hoàng thể (LH), và đôi khi là ACTH. Các đột biến đầu tiên trong PROP1 đã được xác định ở những bệnh nhân người bị suy tuyến yên được đặc trưng bởi thiếu hụt GH, TSH, và prolactin (PRL) ngoài ra còn giảm gonadotropin và không thể bước vào tuổi dậy thì tự phát. Sau đó, các đột biến PROP1 đã được xác định ở các bệnh nhân từ hơn 21 quốc gia khác nhau, cho thấy các đột biến PROP1 là nguyên nhân di truyền phổ biến nhất của CPHD chiếm khoảng 50% các trường hợp gia đình, mặc dù tỷ lệ mắc ở các trường hợp lẻ tẻ thấp hơn nhiều. Các cá nhân bị ảnh hưởng biểu hiện di truyền lặn, và phần lớn các đột biến được xác định liên quan đến miền homeodomain liên kết DNA. Các đột biến trong PROP1 được xác định cho đến nay bao gồm các đột biến vô nghĩa, sai nghĩa, dịch khung, intron, và xóa đoạn. Phần lớn các đột biến được dự đoán sẽ dẫn đến mất hoàn toàn chức năng bằng cách xóa bỏ liên kết DNA và kích hoạt phiên mã, mặc dù một số đột biến sai nghĩa vẫn giữ được một phần hoạt động. Cho đến nay, đột biến phổ biến nhất, chiếm 50% đến 72% tất cả các đột biến PROP1 gia đình ở nhiều gia đình không liên quan, là một đoạn xóa 2 bp trong exon 2 dẫn đến dịch khung (thường được gọi là p.S109X). Đột biến này có khả năng đại diện cho một điểm nóng đột biến trong gen, thay vì một đột biến sáng lập chung, và kết hợp với đột biến c.150delA, chiếm hơn 90% tất cả các đột biến PROP1 đã biết.
Đồng hợp tử cho các đột biến trong PROP1 thường liên quan đến sự thiếu hụt GH, TSH, PRL, và gonadotropin, mặc dù thời gian khởi phát và mức độ nghiêm trọng của sự thiếu hụt hormone thay đổi. Hầu hết các bệnh nhân có thiếu hụt GH khởi phát sớm và chậm phát triển; tuy nhiên, sự tăng trưởng bình thường trong thời thơ ấu đã được báo cáo ở một bệnh nhân đạt được chiều cao cuối cùng bình thường mà không cần điều trị thay thế GH. Bệnh nhân có thể có thiếu hụt gonadotropin với sự tiến triển của các thiếu hụt hormone khác sau này trong cuộc đời. Thiếu hụt TSH cũng rất thay đổi và đã được báo cáo là triệu chứng khởi phát ban đầu trong một số trường hợp, trong khi các bệnh nhân khác cho thấy khởi phát muộn. Tần suất thiếu hụt ACTH tăng theo tuổi. Hầu hết các bệnh nhân biểu hiện nồng độ ACTH và cortisol bình thường trong những năm đầu đời; tuy nhiên, những bệnh nhân trẻ đến 6 tuổi đã được mô tả với thiếu hụt cortisol, nhấn mạnh sự cần thiết phải đánh giá lâm sàng hoàn chỉnh và liên tục đối với các bệnh nhân có đột biến PROP1. Phổ thiếu hụt gonadotropin ở người cực kỳ thay đổi ở những bệnh nhân có đột biến PROP1, từ suy sinh dục sớm với dương vật nhỏ và tinh hoàn không xuống và hoàn toàn không phát triển dậy thì đến khởi phát dậy thì tự phát, mặc dù thường bị trì hoãn, với sự thiếu hụt gonadotropin sau đó, đòi hỏi điều trị thay thế hormone.
Hình thái tuyến yên ở bệnh nhân có đột biến PROP1 là không thể đoán trước; hầu hết các trường hợp cho thấy một tuyến yên trước thiểu sản hoặc có kích thước bình thường trên hình ảnh, với một cuống tuyến yên và thùy sau bình thường, mặc dù một số báo cáo đã ghi nhận một tuyến yên trước to ra. Các phân tích theo chiều dọc về kích thước tuyến yên trước theo thời gian đã cho thấy rằng một số bệnh nhân có tuyến yên trước to ra khi quét ban đầu ở thời thơ ấu cho thấy sự thoái triển và teo nhỏ tự phát, do đó MRI ở những bệnh nhân lớn tuổi thường cho thấy thiểu sản tuyến yên trước, mặc dù kích thước của tuyến yên có thể tăng và giảm trong thời gian này. Sự to ra của tuyến yên bao gồm một tổn thương khối nằm giữa thùy trước và sau, có thể bắt nguồn từ thùy trung gian hoặc di tích túi Rathke trong khe, mặc dù cơ chế cơ bản cho khối u vẫn chưa được biết.
Bản chất tiến triển của sự thiếu hụt hormone ở những bệnh nhân có đột biến PROP1 cho thấy sự suy giảm tiến triển của trục tuyến yên trước, vì vậy những bệnh nhân như vậy cần được theo dõi thường xuyên để phát hiện sự phát triển của các thiếu hụt hormone có thể không rõ ràng khi khám ban đầu. Bản chất rất thay đổi của kiểu hình liên quan đến các đột biến PROP1, ngay cả giữa các anh chị em trong cùng một gia đình mang các đột biến giống hệt nhau, một lần nữa cho thấy các yếu tố điều chỉnh di truyền chưa được xác định đóng một vai trò trong mức độ nghiêm trọng và sự khởi phát của cơ chế bệnh sinh.
POU1F1 là gen tương đồng ở người của gen Pit1 ở chuột và mã hóa một yếu tố phiên mã tham gia vào việc kích hoạt các gen GH và PRL, điều hòa promoter TSH-b, và sự chuyên biệt hóa, tăng sinh và tồn tại của các dòng tế bào tương ứng. Các đột biến trong POU1F1 (PIT1) (được xem xét trong) thường liên quan đến thiếu hụt GH, PRL và TSH với thiểu sản tuyến yên trước thay đổi. Thiếu hụt GH và PRL thường hoàn toàn và xuất hiện sớm trong đời, trong khi thiếu hụt TSH có thể rất thay đổi. Phần lớn các trường hợp có thiếu hụt TSH sớm; tuy nhiên, trong một số trường hợp, suy giáp xảy ra muộn hơn trong thời thơ ấu. MRI của bệnh nhân có đột biến POU1F1 cho thấy một tuyến yên trước nhỏ hoặc có kích thước bình thường với một tuyến yên sau và cuống tuyến yên bình thường, nhưng không có bất thường ngoài tuyến yên. Hơn 28 đột biến khác nhau trong POU1F1 đã được mô tả cho đến nay; phần lớn trong số này cho thấy di truyền lặn, bao gồm cả việc xóa toàn bộ gen, cũng như các đột biến vị trí cắt nối, trong khi một số ít là các đột biến trội. Trong số này, sự thay thế axit amin p.R271W là thường gặp nhất, đã được xác định ở một số bệnh nhân không liên quan từ nhiều nguồn gốc dân tộc khác nhau. Sự thay thế p.R271W dẫn đến việc sản xuất một protein vẫn có khả năng liên kết với DNA nhưng hoạt động như một chất ức chế phiên mã trội âm.
Các nghiên cứu về POU1F1 trong các nhóm bệnh nhân CPHD cho thấy rằng tỷ lệ đột biến trong các trường hợp CPHD lẻ tẻ khá thấp (khoảng 3%–6%), trong khi tỷ lệ mắc ở những bệnh nhân gia đình bị suy tuyến yên lớn hơn nhiều (25%). Nhìn chung, các nghiên cứu sàng lọc cho thấy rằng các bất thường của POU1F1 là một nguyên nhân ít phổ biến hơn của CPHD so với các bất thường của gen PROP1.
Sự thiếu hụt một nửa của gen homeobox RIEG dẫn đến hội chứng Rieger, một rối loạn trội trên nhiễm sắc thể thường liên quan đến sự phát triển bất thường của buồng trước của mắt, răng và rốn—đôi khi có liên quan đến thiếu hụt GH.
Các bất thường di truyền của sản xuất và/hoặc bài tiết GH dẫn đến thiếu hụt GH đơn độc
Đã có báo cáo rằng có tới 30% bệnh nhân bị thiếu hụt IGH có cha mẹ hoặc anh chị em bị ảnh hưởng. Ngoài thiếu hụt hormone tuyến yên kết hợp, chẳng hạn như do các bất thường của PROP1 và POU1F1 được mô tả trước đây, bốn dạng thiếu hụt IGH theo Mendel đã được báo cáo.
Thiếu hụt GH đơn độc GHDIA là kết quả của việc xóa (đã báo cáo các đoạn xóa 6.7-, 7.0-, 7.6-, và 45-kb) hoặc các đột biến của gen GH1 làm chặn hoàn toàn quá trình tổng hợp hoặc bài tiết GH. Sự di truyền của thiếu hụt GH đơn độc GHDIA là lặn trên nhiễm sắc thể thường, và bệnh nhân có thiếu hụt GH bẩm sinh sâu sắc. Bởi vì GH chưa bao giờ được bệnh nhân sản xuất, ngay cả trong cuộc sống thai nhi, bệnh nhân không dung nạp miễn dịch với GH và thường phát triển kháng thể kháng GH khi được điều trị bằng GH có nguồn gốc từ tuyến yên hoặc có nguồn gốc từ DNA tái tổ hợp, mặc dù sự phát triển của các kháng thể làm suy giảm tăng trưởng dường như ít thường xuyên hơn với các chế phẩm GH tổng hợp mới hơn.
Khi các kháng thể ngăn cản bệnh nhân đáp ứng với GH, nên xem xét liệu pháp IGF-1 tái tổ hợp (rh). Dạng thiếu hụt GH lặn trên nhiễm sắc thể thường ít nghiêm trọng hơn (thiếu hụt GH đơn độc GHDIB) có khả năng cũng là kết quả của các đột biến hoặc sắp xếp lại của GH1, có lẽ dẫn đến một phân tử GH vẫn giữ được một số chức năng nhưng có lẽ không ổn định. Ngoài ra, các đột biến trong GHRHR cũng dẫn đến GHD1B. Tuy nhiên, cho đến nay, hầu hết các bệnh nhân được cho là bị thiếu hụt GH đơn độc GHDIB đã không chứng minh được sự thay đổi của GH1 và GHRHR và nguyên nhân gây ra sự thiếu hụt GH của họ vẫn chưa rõ ràng. Những bệnh nhân này thường đáp ứng tốt với liệu pháp GH, và sự phát triển của các kháng thể kháng GH có ý nghĩa lâm sàng là không phổ biến.
Thiếu hụt GH đơn độc GHDII được di truyền theo kiểu trội trên nhiễm sắc thể thường. Những bệnh nhân như vậy thường có các bất thường của GH1 hoạt động theo kiểu trội âm. Nguyên nhân phổ biến nhất của rối loạn này dường như là các đột biến vị trí cắt nối và intron làm bất hoạt vị trí cho cắt nối 5′ của intron 3, dẫn đến việc bỏ qua exon 3 với việc tạo ra dạng đồng phân 17,5 kDa của GH có tác dụng trội âm lên sự bài tiết của phân tử 22 kDa. Chuột chuyển gen biểu hiện quá mức dạng đồng phân 17,5 kDa có một khiếm khuyết trong sự trưởng thành của các túi tiết chứa GH và biểu hiện thiểu sản tuyến yên trước với sự phát triển của các thiếu hụt hormone khác. Dạng đồng phân 17,5 kDa bị giữ lại trong lưới nội chất, bị gấp sai, và làm gián đoạn con đường bài tiết. Các đột biến tương tự được xác định ở người cũng có liên quan đến GHDII và một bệnh nội tiết tiến triển.
Các đột biến sai nghĩa ảnh hưởng đến sự bài tiết và/hoặc hoạt động của GH cũng có thể gây ra thiếu hụt IDH trội trên nhiễm sắc thể thường. Bệnh nhân có đột biến p.Arg183His bị suy giảm giải phóng GH vì, ở cấp độ tế bào, các túi tiết chứa protein Arg183His đột biến không được xuất bào hiệu quả như những túi chứa hormone kiểu hoang dã. Các đột biến khác (ví dụ, p.Pro89leu) có thể gây ra những rối loạn sâu sắc và sớm hơn trong con đường bài tiết bằng cách thay đổi hướng của các chuỗi xoắn GH và ảnh hưởng đến việc gấp đúng của phân tử.
Bệnh nhân bị thiếu hụt IGH trội trên nhiễm sắc thể thường cho thấy sự thay đổi đáng kể về mức độ nghiêm trọng của thiếu hụt GH. Họ có nồng độ GH trong huyết thanh thấp nhưng có thể phát hiện được, thiếu hụt chiều cao thay đổi, và có thể cho thấy thiểu sản tuyến yên trước trên MRI (38%–50%). Dữ liệu về các phả hệ có đột biến Arg183His hoặc e3 + 1G > A nhấn mạnh thực tế rằng những bệnh nhân có cùng một đột biến có thể thay đổi đáng kể về chiều cao (≤–4 SDS đến bình thường) và thậm chí đạt được chiều cao người lớn bình thường mà không cần điều trị. Bệnh nhân có đột biến vị trí cắt nối được cho là bị ảnh hưởng nghiêm trọng hơn những người có đột biến sai nghĩa; tuy nhiên, những bệnh nhân có đột biến vị trí cắt nối IvS3 + 1 hoặc IvS3 + 2 hoặc đột biến p.Pro89leu có thể phát triển các thiếu hụt hormone tuyến yên bổ sung, bao gồm thiếu hụt ACTH, prolactin, TSH, hoặc gonadotropin. Kiểu hình tiến triển này là không thể đoán trước và đòi hỏi sự cần thiết phải theo dõi suốt đời đối với những cá nhân bị ảnh hưởng. Một quan sát hấp dẫn khác là, ngay cả ở những bệnh nhân có nguyên nhân di truyền (ví dụ, p.Glu32Ala), thiếu hụt GH dường như đảo ngược khi họ được kiểm tra lại vào cuối giai đoạn tăng trưởng, trong giai đoạn chuyển tiếp trước khi chuyển từ dịch vụ nhi khoa sang người lớn. Tuy nhiên, hiệu ứng này là tạm thời, và được quan sát thấy ở những bệnh nhân được xét nghiệm tại thời điểm chuyển tiếp, và những người không nên được cho xuất viện khỏi việc theo dõi.
Chưa có trường hợp thuyết phục nào về các đột biến của gen GHRH ở người được xác định. Mặt khác, các bất thường của gen cho GHRHR đã được tìm thấy ở một số phả hệ. GHRHR là một thụ thể có bảy miền xuyên màng. Sự biểu hiện của nó đòi hỏi sự có mặt của POU1F1. Các đột biến đồng hợp tử hoặc dị hợp tử phức hợp đã được báo cáo trong GHRHR (sai nghĩa, vô nghĩa, vị trí cắt nối, xóa đoạn, hoặc các đột biến điều hòa) dẫn đến thiếu hụt IGH loại IB. Các đột biến GHRHR được xác định ở khoảng 10% bệnh nhân bị thiếu hụt IGH lặn gia đình, và ở khoảng 3% trong một nhóm bệnh nhân được chọn bị thiếu hụt IGH. Các đột biến có nhiều khả năng được xác định hơn ở các phả hệ có quan hệ huyết thống hoặc các bệnh nhân từ các nguồn gốc dân tộc nhất định, chẳng hạn như từ tiểu lục địa Ấn Độ và Brazil.
Trẻ em có đột biến trong GHRHR bị thiếu hụt GH nghiêm trọng và tầm vóc thấp, nhưng thiểu sản giữa mặt, hạ đường huyết sơ sinh, và dương vật nhỏ, như được tìm thấy ở những người có đột biến GH1 lặn, là hiếm gặp. Tuyến yên trước hiếm khi có thể bình thường; ở phần lớn nó bị thiểu sản. Tuyến yên sau và cuống tuyến yên bình thường. Các đột biến mất chức năng một phần có thể liên quan đến một kiểu hình thiếu hụt GH tương đối nhẹ. Nhìn chung, các đột biến trong các gen điều hòa sự bài tiết GH hiếm khi được xác định, cho thấy rằng các gen mới vẫn cần được xác định.
Thiếu hụt GH đơn độc GHDIII được di truyền theo kiểu liên kết X. Rất ít được biết về nguyên nhân của tình trạng này, mặc dù các đột biến hiếm gặp trong SOX3 có thể liên quan đến IGHD có hoặc không có khó khăn trong học tập.
Các bất thường bẩm sinh của tuyến yên
Một số báo cáo đã mô tả mối liên quan của thiếu hụt GH “vô căn” với một tuyến yên sau lạc chỗ. Các phát hiện MRI đã được mô tả trong một số loạt bệnh nhân bị thiếu hụt GH vô căn. Abrahams và các đồng nghiệp đã nghiên cứu 35 bệnh nhân bị thiếu hụt GH vô căn và thấy rằng những người có phát hiện MRI có thể được chia thành hai nhóm: 43% có một tuyến yên sau lạc chỗ (tuyến yên sau nằm gần lồi giữa), không có cuống tuyến yên, và không có điểm sáng bình thường của tuyến yên sau, và 43% có một tuyến yên trước nhỏ là một phát hiện đơn độc hoặc kết hợp với một tuyến yên sau lạc chỗ. Nói chung, một tuyến yên sau lạc chỗ được tìm thấy ở 87% các trường hợp có thiếu hụt nhiều hormone tuyến yên nhưng chỉ ở 10% các trường hợp có thiếu hụt IGH.
Tuy nhiên, trong các nghiên cứu khác, các phát hiện MRI có độ phân giải cao của một hoặc nhiều trong số những điều sau đây đã được đề xuất là các chỉ số nhạy cảm và/hoặc đặc hiệu của suy tuyến yên: một tuyến yên trước nhỏ, cuống tuyến yên bị suy yếu, và tuyến yên sau lạc chỗ. Trong một nghiên cứu, các bất thường tuyến yên được tìm thấy ở 80% bệnh nhân bị thiếu hụt IGH và 93% bệnh nhân bị MPHD. Ở những bệnh nhân có nồng độ GH đỉnh thấp hơn 3 ng/mL, 90% có các phát hiện MRI bất thường—so với 39% những người có nồng độ GH từ 3 ng/mL trở lên. Trong một nghiên cứu khác, cuống bị bất thường ở 90% bệnh nhân bị IGHD và không có ở 96% bệnh nhân bị MPHD. Do đó, các bất thường MRI là phổ biến ở trẻ em bị thiếu hụt IGH và MPHD và có liên quan chặt chẽ với mức độ nghiêm trọng của thiếu hụt GH. Bệnh nhân có các bất thường cấu trúc sẽ cần theo dõi suốt đời ở tuổi trưởng thành do nguy cơ phát triển các thiếu hụt hormone tuyến yên khác. Tuy nhiên, những bệnh nhân có tuyến yên sau lạc chỗ cũng có thể cho thấy sự đảo ngược của tình trạng thiếu hụt GH của họ khi kiểm tra lại tại thời điểm chuyển từ dịch vụ nhi khoa sang người lớn hoặc ở tuổi trưởng thành.
Các khối u liên quan đến tuyến yên
Nhiều khối u ảnh hưởng đến chức năng vùng dưới đồi cũng tác động trực tiếp đến sự bài tiết GH của tuyến yên. Đặc biệt, u sọ hầu là một nguyên nhân chính gây suy tuyến yên. Những khối u này phát sinh từ những di tích của túi Rathke, túi thừa của nóc khoang miệng phôi mà thường tạo ra tuyến yên trước. Một số người coi khối u này là một dị tật bẩm sinh vì nó được cho là đã có mặt từ khi sinh ra, phát triển dần dần trong những năm và thập kỷ sau đó. Khối u phát sinh từ các phần còn lại của các tế bào vảy ở chỗ nối của tuyến yên trước và tuyến yên sau. Khi nó lớn lên, nó tạo thành một nang chứa các tế bào bị thoái hóa và nó có thể bị vôi hóa nhưng không trải qua quá trình thoái hóa ác tính. Ở trẻ em, chúng chiếm 5% đến 15% các khối u nội sọ và là khối u tân sản phổ biến nhất của khu vực dưới đồi-tuyến yên, chiếm tới 80% các khối u ở vị trí này. Tỷ lệ mắc bệnh của chúng ở Hoa Kỳ là 1,3 trên một triệu mỗi năm và gần 28% ảnh hưởng đến trẻ em dưới 14 tuổi. Có một đỉnh mắc bệnh hai pha: đỉnh đầu tiên xảy ra ở trẻ em từ 5 đến 14 tuổi và đỉnh thứ hai ở người lớn trên 50 tuổi. Tuy nhiên, chúng có thể được chẩn đoán ở mọi lứa tuổi và thậm chí đã được báo cáo trong giai đoạn sơ sinh.
Khi xuất hiện, hầu hết các u sọ hầu có vị trí kết hợp trong và trên yên (74,2%) và gần một nửa có liên quan đến vùng dưới đồi (51,6%). Một tỷ lệ nhỏ hơn chỉ nằm hoàn toàn trên yên (22,6%) hoặc giới hạn trong hố yên (6%–3%). Gần một phần ba xâm lấn vào sàn của não thất ba và có thể gây ra não úng thủy tắc nghẽn. Ở bệnh nhân nhi, u sọ hầu chủ yếu là dạng nang (56,7%), đa nang (16,7%), chủ yếu là dạng đặc (13,3%), hoàn toàn đặc (10%), hoặc hoàn toàn dạng nang (3,3%). Dịch nang nhớt và giàu cholesterol và tỷ lệ vôi hóa cao hơn nhiều ở trẻ em (83,3%) so với người lớn.
Có hai loại mô học chính: loại phổ biến nhất là loại adamantinomatous và bao gồm các tế bào tân sản biểu mô giống như những tế bào được tìm thấy trong các tổn thương của hàm. Loại nhú hiếm hơn nhiều và gần như chỉ được tìm thấy ở người lớn. Mặc dù u sọ hầu về mặt mô học là lành tính, chúng có thể lan rộng từ vị trí ban đầu, phát triển các nhú, và xâm lấn các mô quan trọng xung quanh, bao gồm cả vùng dưới đồi và giao thoa thị giác. Sự gắn kết này làm cho việc cắt bỏ hoàn toàn của chúng trở nên khó khăn, nếu không muốn nói là không thể, và góp phần vào sự tái phát của khối u và tỷ lệ bệnh tật.
U sọ hầu thường là các khối u lẻ tẻ. Tuy nhiên, đã có những báo cáo ca hiếm gặp về các thành viên trong gia đình bị ảnh hưởng cho thấy di truyền lặn. Sự hoạt hóa quá mức của con đường tín hiệu Wnt dường như đóng một vai trò trong sự phát triển của u sọ hầu; các đột biến hoạt hóa trong beta-catenin, một thành phần hạ nguồn của con đường tín hiệu Wnt, đã được xác định trong các u sọ hầu adamantinomatous.
Các dấu hiệu và triệu chứng lâm sàng của u sọ hầu có thể phát sinh ở mọi lứa tuổi, từ sơ sinh đến tuổi trưởng thành, nhưng điển hình nhất là ở giữa thời thơ ấu. Biểu hiện phổ biến nhất là với các triệu chứng tăng áp lực nội sọ (lên đến 75%), chẳng hạn như đau đầu, nôn mửa, hoặc các bất thường vận nhãn. Suy giảm thị lực là phổ biến. Khiếm khuyết thị trường có thể là kết quả của sự chèn ép giao thoa thị giác, và có thể quan sát thấy phù gai thị hoặc teo dây thần kinh thị giác. Ảo giác thị giác và khứu giác đã được báo cáo, cũng như co giật và sa sút trí tuệ.
Ước tính có 70% đến 80% trẻ em có bằng chứng về thiếu hụt nội tiết khi khám và suy giảm tăng trưởng được quan sát thấy ở 32% đến 52% trẻ em trước khi chẩn đoán. Nồng độ IGF-1 thấp đã được báo cáo ở 80% trẻ em tại thời điểm chẩn đoán. Thiếu hụt GH là thiếu hụt hormone phổ biến nhất, được ghi nhận ở 75% đến 100% những người được xét nghiệm trước khi điều trị. Tiếp theo là thiếu hụt ACTH (20%–70%) và TSH (3%–30%). Sự chèn ép của cuống tuyến yên hoặc tổn thương các neuron dopaminergic của vùng dưới đồi dẫn đến nồng độ PRL tăng cao, được quan sát thấy ở 8% đến 20% trẻ em khi chẩn đoán. Tỷ lệ mắc DI khi khám thay đổi từ 10% đến 29% bệnh nhân, tùy thuộc vào nghiên cứu. Đặc biệt ở tuổi vị thành niên, u sọ hầu có thể biểu hiện bằng dậy thì muộn hoặc ngừng dậy thì. Các biểu hiện hiếm gặp bao gồm dậy thì sớm và hội chứng bài tiết hormone chống bài niệu không phù hợp.
Phim chụp sọ nghiêng thường cho thấy sự to ra hoặc biến dạng của hố yên, thường đi kèm với vôi hóa trên yên. Tuy nhiên, một số trẻ em bị u sọ hầu sẽ có phim chụp thường quy bình thường, và các kỹ thuật X-quang thay thế được khuyến nghị. Chụp cắt lớp vi tính (CT) là một kỹ thuật nhạy để xác định một lượng nhỏ vôi hóa hoặc các bất thường dạng nang. MRI có lẽ là kỹ thuật nhạy nhất. U sọ hầu xuất hiện dưới dạng các tổn thương khối ở khu vực yên và/hoặc trên yên có thể lan đến vùng dưới đồi và xâm lấn não thất ba. U sọ hầu adamantinomatous chủ yếu là dạng nang và phần nang của tổn thương xuất hiện tăng tín hiệu trên hình ảnh T1 và T2. Phần đặc của khối u cho thấy các vùng có cường độ tín hiệu cao và thấp đại diện cho các vùng vôi hóa và lắng đọng hemosiderin. Trong phần lớn các trường hợp (58%–76%), kích thước của u sọ hầu, được ước tính bằng MRI hoặc CT, đã được báo cáo là 2 đến 4 cm, trong khi nó nhỏ hơn 2 cm trong 4% đến 28% trường hợp và hơn 4 cm trong 14% đến 20%.
Việc quản lý u sọ hầu rất phức tạp, gây tranh cãi, và tốt nhất là được thực hiện bằng một phương pháp tiếp cận đa ngành. Mục tiêu của việc điều trị là làm giảm các dấu hiệu và triệu chứng cấp tính của sự chèn ép (tăng áp lực nội sọ, đe dọa suy giảm thị lực); bảo tồn chức năng vùng dưới đồi, do đó làm giảm tỷ lệ bệnh tật và tử vong sau này; và cung cấp sự kiểm soát lâu dài và ngăn ngừa tái phát. Điều rõ ràng từ các nghiên cứu khác nhau là mức độ cắt bỏ phẫu thuật có lẽ là yếu tố quan trọng nhất ảnh hưởng đến sự tái phát của u sọ hầu. Ở những bệnh nhân chỉ được phẫu thuật, tỷ lệ sống không tái phát trong 10 năm là 83% sau khi cắt bỏ toàn bộ, 50,5% sau khi cắt bỏ gần toàn bộ và 15,6% sau khi cắt bỏ một phần. Sự tái phát của khối u thường xảy ra trong 5 năm đầu và tương đối hiếm sau đó. Tuy nhiên, ngay cả sau khi cắt bỏ hoàn toàn được xác nhận bằng X-quang, sự tái phát xảy ra ở 15% đến 25% bệnh nhân. Tuy nhiên, trong các loạt ca cũ hơn, khi bệnh nhân được điều trị bằng các phẫu thuật cắt bỏ triệt để và lặp đi lặp lại, tỷ lệ tử vong cao (25%–50%) và bệnh tật vùng dưới đồi, thị giác và nhận thức xảy ra ở đại đa số (75%), đặc biệt là ở các u sọ hầu có sự lan rộng trên yên hoặc sau giao thoa.
Các hướng dẫn về quản lý đa ngành đối với trẻ em bị u sọ hầu gần đây đã được công bố. Hiện nay người ta đề nghị rằng bệnh nhân có thể được phân loại thành hai nhóm nguy cơ về quản lý và tiên lượng. Nhóm nguy cơ tốt bao gồm trẻ lớn hơn có các khối u nhỏ (2–4 cm) và không có hội chứng dưới đồi hoặc não úng thủy. Trẻ nhỏ hơn, có các khối u lớn hơn (> 2–4 cm) và hội chứng dưới đồi hoặc não úng thủy thuộc nhóm nguy cơ xấu. Phẫu thuật cắt bỏ triệt để hoàn toàn, có hoặc không có xạ trị bổ trợ, được đề nghị cho nhóm nguy cơ tốt, trong khi phẫu thuật hạn chế và xạ trị ngay lập tức hoặc trì hoãn là lựa chọn điều trị cho nhóm nguy cơ xấu. Sự sẵn có gần đây của liệu pháp chùm proton là một sự bổ sung có giá trị cho kho vũ khí điều trị.
Thiếu hụt hormone tăng trưởng tuyến yên vô căn
Như đã thảo luận trước đây, nhiều quá trình bệnh ảnh hưởng đến sự điều hòa của vùng dưới đồi đối với sự bài tiết GH cũng tác động đến chức năng tuyến yên. Với những hạn chế chẩn đoán hiện tại, không phải lúc nào cũng có thể phân biệt hoàn toàn giữa rối loạn chức năng vùng dưới đồi và tuyến yên. Hơn nữa, có khả năng nhiều trường hợp được gọi là thiếu hụt GH vô căn sẽ được phát hiện là có cơ sở phân tử cho rối loạn này. Thật vậy, trong 5 năm qua, chúng ta đã chứng kiến một sự bùng nổ thông tin liên quan đến các gen tham gia một cách quan trọng vào sự biệt hóa và chức năng của các tế bào hướng thân của tuyến yên.
Tuy nhiên, tại thời điểm này, một nguyên nhân rõ ràng cho sự thiếu hụt GH của tuyến yên thường không được xác định—do đó có thuật ngữ vô căn. Tỷ lệ thiếu hụt GH của tuyến yên là 1:60.000 trẻ sinh sống đã được báo cáo từ Vương quốc Anh. Một cuộc khảo sát gần đây hơn trên 48.000 học sinh Scotland đã chỉ ra một tỷ lệ cao tới 1:4000, trong khi ước tính tốt nhất hiện có trong dân số Hoa Kỳ là ít nhất 1:3480.
Tuy nhiên, có khả năng là thiếu hụt GH ở trẻ em là một tình trạng được chẩn đoán quá mức. Đặc biệt, chẩn đoán thiếu hụt IGH vô căn khởi phát muộn nên luôn được đặt câu hỏi. Mặc dù người ta có thể lập luận rằng các tổn thương phá hủy hoặc viêm của vùng dưới đồi hoặc tuyến yên có thể chỉ ảnh hưởng đến sự bài tiết GH, rằng thiếu hụt IGH do một đột biến/xóa nhẹ của gen GHRHR hoặc gen GH1 có thể xuất hiện muộn, hoặc CPHD có thể biểu hiện lần đầu với những gì dường như là thiếu hụt IGH, những trường hợp như vậy dường như là hiếm. Trong trường hợp không có các bất thường giải phẫu rõ ràng trên các nghiên cứu hình ảnh và/hoặc bằng chứng sinh hóa của CPHD, chẩn đoán thiếu hụt GH đơn độc vô căn mắc phải đòi hỏi tài liệu cẩn thận và kỹ lưỡng. Thật vậy, dữ liệu gần đây cho thấy rằng việc kiểm tra lại những bệnh nhân như vậy có thể liên quan đến sự đảo ngược sớm của chẩn đoán. Việc chẩn đoán quá mức thiếu hụt GH có khả năng phát sinh từ độ đặc hiệu kém của xét nghiệm GH kích thích, đặc biệt là ở trẻ em trước tuổi dậy thì.
Hormone tăng trưởng không hoạt tính sinh học
Một số ít bệnh nhân có khiếm khuyết trong gen GH1 tạo ra một phân tử GH không hoạt tính sinh học nhưng có phản ứng miễn dịch đã được báo cáo. Những bệnh nhân này có một kiểu hình chung và được đặc trưng bởi tầm vóc thấp nghiêm trọng (thường là SDS chiều cao <–3) và nồng độ IGF-1 và IGFBP-3 rất thấp, nhưng nồng độ GH bình thường hoặc tăng cao (cơ bản hoặc sau một nghiệm pháp kích thích). Bệnh nhân có thể có các đặc điểm lâm sàng nhẹ, chẳng hạn như trán dô và mũi yên ngựa, là những đặc điểm điển hình của GHI. Trái với những gì được mong đợi ở một bệnh nhân bị GHI, những bệnh nhân này cho thấy một số cải thiện về tốc độ tăng trưởng và mức IGF-1 khi điều trị bằng GH ngoại sinh. Các đột biến sai nghĩa trong GH1, được di truyền theo kiểu trội hoặc lặn trên nhiễm sắc thể thường, tạo ra một GH đột biến có khả năng liên kết với thụ thể, nhưng không thể kích hoạt đúng sự truyền tín hiệu bởi GHR.
Không nhạy cảm với hormone tăng trưởng
GHI là một rối loạn di truyền được định nghĩa là không có khả năng đáp ứng với GH nội sinh và ngoại sinh với các tác động tăng trưởng và chuyển hóa thích hợp. Dạng cổ điển của GHI, còn được gọi là hội chứng Laron, được đặc trưng bởi tầm vóc thấp nghiêm trọng biểu hiện sau sinh và có liên quan đến các đặc điểm dị dạng, chẳng hạn như mất cân đối sọ mặt với khuôn mặt tương đối nhỏ, sống mũi lõm, giọng nói cao, béo phì thân mình, và dương vật nhỏ, một kiểu hình tương tự như được thấy ở những bệnh nhân bị thiếu hụt GH bẩm sinh hoàn toàn (Hình 11.5). Thông thường, bệnh nhân GHI cho thấy nồng độ IGF-1, IGFBP-3, và ALS rất thấp, nhưng nồng độ GH cơ bản và được kích thích tăng cao. Trong hầu hết các trường hợp, GHI là do một đột biến lặn trên nhiễm sắc thể thường có độ thấm hoàn toàn trong GHR. Phần lớn các đột biến này nằm ở các exon mã hóa miền ngoại bào của GHR và làm gián đoạn miền liên kết phối tử và/hoặc làm suy giảm sự vận chuyển của thụ thể đến màng plasma. Vì những lý do này, bệnh nhân bị GHI cổ điển thường cho thấy mức GHBP thấp hoặc không thể phát hiện được vì GHBP được tạo ra bởi sự phân cắt protein của miền GHR ngoại bào. Tuy nhiên, các đột biến ở những nơi khác trong gen GHR có thể dẫn đến nồng độ GHBP bình thường hoặc tăng cao.
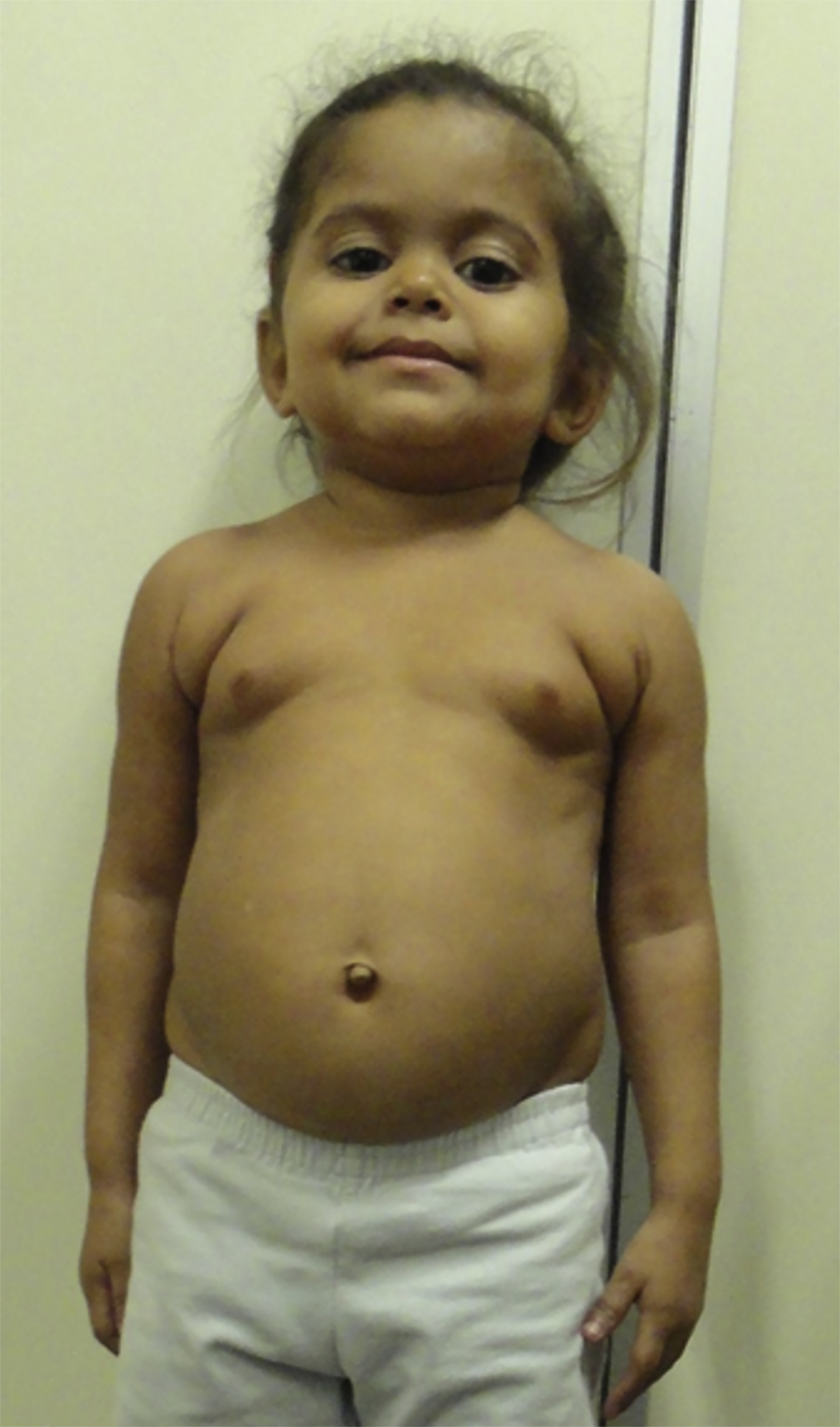
Hình 11.5 Kiểu hình của một bệnh nhân bị thiếu hụt hoặc không nhạy cảm với hormone tăng trưởng nghiêm trọng: trán dô, sống mũi lõm, thiểu sản giữa mặt, và béo phì thân mình.
Sau khi xác định đặc điểm của các dạng GHI hoàn chỉnh, một số trường hợp có kiểu hình GHI nhẹ hơn hoặc không điển hình khác đã được mô tả. Phổ lâm sàng của GHI một phần có thể thay đổi giữa các bệnh nhân có các phát hiện xét nghiệm điển hình của GHI nhưng thiếu các đặc điểm khuôn mặt và dị dạng cổ điển liên quan đến hội chứng Laron, và những bệnh nhân ban đầu được phân loại là có ISS. Thông thường, bệnh nhân GHI một phần là dị hợp tử cho các đột biến GHR trội âm hoặc có một khiếm khuyết hai alen liên quan đến các đột biến GHR giảm hình thái. Một số hoạt động GHR còn lại được giữ lại trong cả hai tình huống, gây ra nhiều kiểu hình một phần khác nhau. Ngoài ra, GHBP thường có thể phát hiện được, và thậm chí tăng cao, ở một số bệnh nhân bị GHI một phần.
Thông thường, chẩn đoán bệnh nhân bị GHI nghiêm trọng không khó, và các nghiên cứu phân tử bổ sung rất ít thông tin khác vào đánh giá lâm sàng. Ngược lại, sự vắng mặt của một kiểu hình điển hình đặt ra một thách thức cho việc thiết lập chẩn đoán GHI một phần; do đó, việc sử dụng các nghiên cứu di truyền phân tích GHR là một công cụ chẩn đoán phân tử có giá trị.
Đáng chú ý là các nghiên cứu dược di truyền học GH đã cho thấy rằng một đa hình GHR duy nhất, sự hiện diện hay vắng mặt của GHR-exon 3, có liên quan đến một sự điều chỉnh nhỏ nhưng có ý nghĩa của phản ứng tăng trưởng trong quá trình điều trị bằng hormone tăng trưởng người tái tổ hợp. Ngoài ra, một nghiên cứu liên kết toàn bộ bộ gen đã chứng minh rằng một biến thể vô nghĩa GHR, gây ra GHI nghiêm trọng ở trạng thái đồng hợp tử, dẫn đến giảm 4,2 cm chiều cao ở những người dị hợp tử, cho thấy rằng các biến thể GHR cũng đóng một vai trò quan trọng trong sự biến thiên chiều cao trong một mô hình đa gen.
Không nhạy cảm với Hormone tăng trưởng liên quan đến rối loạn chức năng miễn dịch
Có một nhóm bệnh nhân riêng biệt bị GHI biểu hiện một rối loạn chức năng miễn dịch liên quan, chủ yếu được đặc trưng bởi viêm phổi kẽ lympho bào, chàm, và bệnh tự miễn. Những bệnh nhân này mang các đột biến hai alen trong gen STAT5B. Bệnh nhân có khiếm khuyết STAT5B hai alen có một hồ sơ nội tiết tố tương tự như bệnh nhân có các khiếm khuyết đồng hợp tử trong GHR. STAT5B là một phân tử quan trọng tham gia vào sự truyền tín hiệu GHR, trung gian cho các hoạt động thúc đẩy tăng trưởng của GHR. Sự rối loạn điều hòa miễn dịch do các đột biến STAT5B có thể được giải thích bởi thực tế là các phân tử khác sử dụng con đường tín hiệu STAT5B, bao gồm một số IL. Khiếm khuyết trong hoạt động của IL-2 đặc biệt quan trọng vì IL này tham gia vào việc kích hoạt các tế bào lympho T và trong sự phát triển của các tế bào lympho T điều hòa. Nồng độ PRL tăng cao là một đặc điểm riêng biệt của các đột biến STAT5B hai alen, bởi vì tín hiệu STAT5B cần thiết cho phản hồi ngược âm của sản xuất PRL. Những bệnh nhân như vậy cũng cho thấy giảm bạch cầu lympho vừa phải, với sự giảm rõ rệt của các tế bào tiêu diệt tự nhiên và tế bào T, có một sự suy giảm đáng chú ý trong các tế bào T-điều hòa, góp phần vào khả năng miễn dịch của niêm mạc và sự bảo vệ của vật chủ chống lại các mầm bệnh nội bào. Các cá nhân mang các đột biến STAT5B mất chức năng dị hợp tử có chiều cao giảm (khoảng 3,9 cm) và mức IGF-1 và IGFBP-3 thấp hơn so với các đối chứng không mang gen. Mặc dù những cá nhân này thường nằm trong phạm vi chiều cao bình thường, phát hiện này ủng hộ giả thuyết rằng sự dị hợp tử của các biến thể gây bệnh hiếm gặp góp phần vào tính di truyền của chiều cao bình thường.
Gần đây, các bệnh nhân mang các biến thể STAT5B dị hợp tử trội âm đã được báo cáo là bị suy giảm tăng trưởng sau sinh, chàm, và tăng immunoglobin E nhưng không bị bệnh miễn dịch nghiêm trọng, mở rộng phạm vi của các biểu hiện lâm sàng và di truyền liên quan đến các kiểu gen STAT5B bất thường. Hơn nữa, có một số báo cáo về các bệnh nhân bị GHI một phần và rối loạn chức năng miễn dịch mang các biến thể tăng chức năng STAT3 có liên quan đến hoạt động phiên mã STAT5B giảm.
Thiếu hụt/Kháng Yếu tố tăng trưởng giống Insulin-1 Thiếu hụt tiểu đơn vị không bền với axit
Các khiếm khuyết đồng hợp tử trong gen mã hóa tiểu đơn vị không bền với axit của yếu tố tăng trưởng giống insulin (IGFALS) gây ra thiếu hụt ALS nghiêm trọng và không hình thành phức hợp tam phân (IGFs/ALS/IGFBP-3). Bệnh nhân bị thiếu hụt ALS được đặc trưng bởi tầm vóc thấp từ nhẹ đến trung bình, nồng độ ALS trong huyết thanh không thể phát hiện được hoặc rất thấp, và nồng độ IGF-1 và IGFBP-3 trong huyết thanh rất thấp, với IGFBP-3 bị ảnh hưởng nhiều hơn IGF-1. Một số bệnh nhân này cũng có sự giảm kích thước khi sinh thay đổi, dậy thì muộn, các mức độ không nhạy cảm với insulin khác nhau, và/hoặc mật độ khoáng xương thấp. Người ta cho rằng sự hạn chế tăng trưởng ở những bệnh nhân này là nhẹ, mặc dù có sự giảm rõ rệt nồng độ IGF-1 và IGFBP-3, do sự bảo tồn tương đối của nồng độ IGF-1 tự do lưu hành và/hoặc sự biểu hiện của IGF-1 được sản xuất tại chỗ. Các khiếm khuyết IGFALS là các đặc điểm lặn trên nhiễm sắc thể thường và bệnh nhân có thể là đồng hợp tử hoặc dị hợp tử phức hợp cho các đột biến ALS. Những người mang gen dị hợp tử có sự giảm nhẹ về chiều cao (khoảng 1 SDS) so với những người thân không mang gen của họ.
Khiếm khuyết trong sự phân cắt protein của IGFBP (Gen PAPPA2)
PAPP-A2 là một protease có độ đặc hiệu cao cho việc phân cắt IGFBP-3 và -5. Việc phân cắt các IGFBP này giải phóng IGF-1 khỏi phức hợp tam phân (IGFBP-3 và ALS) và cho phép IGF-1 tự do hoạt động trên các mô đích. Cho đến nay, chỉ có hai gia đình được báo cáo có các đột biến đồng hợp tử trong gen PAPPA2. Các thành viên bị ảnh hưởng của các gia đình này có một lượng IGF-1 tăng lên bị liên kết trong các phức hợp tam phân và giảm IGF-1 tự do hoặc có hoạt tính sinh học. Những bệnh nhân này được đặc trưng lâm sàng bởi sự suy giảm tăng trưởng tiến triển, đầu nhỏ, và xương mỏng. Họ cũng có sự tăng rõ rệt nồng độ IGF-1, IGF-2, IGFBP-3, IGFBP-5, và ALS toàn phần, nhưng IGF-1 tự do thấp. GH cũng tăng cao ở những bệnh nhân này, có lẽ do phản hồi ngược âm giảm gây ra bởi sự giảm IGF-1 tự do.
Thiếu hụt Yếu tố tăng trưởng giống Insulin nguyên phát Khiếm khuyết IGF1
Một số ít bệnh nhân có các biến thể gây bệnh trong IGF1 đã được mô tả cho đến nay, khoảng một nửa trong số họ mang các biến thể đồng hợp tử và một kiểu di truyền lặn trên nhiễm sắc thể thường. Những bệnh nhân này được đặc trưng bởi sự suy giảm tăng trưởng nghiêm trọng trong tử cung và sau sinh, đầu nhỏ, điếc thần kinh giác quan, các thiếu hụt trí tuệ thay đổi, và giảm khối lượng xương cột sống. Bệnh nhân cho thấy nồng độ IGF-1 không thể phát hiện được và nồng độ GH cơ bản và được kích thích tăng cao nhưng có nồng độ IGFBP-3 và ALS bình thường. Kháng insulin cũng đã được quan sát thấy ở những bệnh nhân này (thứ phát sau thừa GH), cũng như rối loạn chức năng sinh dục một phần và béo phì. Bệnh nhân đồng hợp tử cho các đột biến IGF1 ít gây rối loạn hơn hoặc mang các đột biến IGF1 dị hợp tử có tầm vóc thấp đáng kể, có hoặc không có đầu nhỏ hoặc thiểu năng trí tuệ, và nồng độ IGF-1 thấp hoặc thấp-bình thường. Biểu hiện lâm sàng nhẹ hơn này, đặc biệt ở những bệnh nhân có các biến thể IGF1 dị hợp tử, làm cho việc chẩn đoán trở nên khó khăn nếu không có các cuộc điều tra di truyền phân tử.
Yếu tố tăng trưởng giống Insulin-1 không hoạt tính sinh học
Có một báo cáo mô tả một bệnh nhân đồng hợp tử cho một đột biến IGF1 dẫn đến sự hình thành một polypeptide có phản ứng miễn dịch nhưng không hoạt tính sinh học. Phân tử IGF-1 bất thường này là sản phẩm của một biến thể sai nghĩa trong gen IGF1 và có ái lực giảm 90 lần đối với thụ thể của nó. Trong nghiên cứu này, bệnh nhân bị ảnh hưởng cho thấy sự suy giảm tăng trưởng nghiêm trọng trước và sau sinh, đầu nhỏ, điếc thần kinh giác quan, thiểu năng trí tuệ, và loãng xương, một kiểu hình rất giống với kiểu hình được thấy ở những bệnh nhân bị thiếu hụt IGF-1 nghiêm trọng. Nồng độ GH rất cao để đáp ứng với hạ đường huyết do insulin gây ra và IGF-1 cũng tăng rõ rệt.
Không nhạy cảm với Yếu tố tăng trưởng giống Insulin
Các khiếm khuyết IGF1R đã được xác định ở những bệnh nhân sinh ra nhỏ so với tuổi thai (SGA) không cho thấy sự tăng trưởng bắt kịp. Bệnh nhân có các đột biến IGF1R hai alen (đồng hợp tử hoặc dị hợp tử phức hợp) có một kiểu hình lâm sàng đồng nhất tương tự như kiểu hình của bệnh nhân có các đột biến IGF1 đồng hợp tử. Trái ngược với sự hiếm gặp của các đột biến IGF1 và IGF1R hai alen, các biến thể IGF1R dị hợp tử tương đối thường xuyên và biểu hiện sự thay đổi lâm sàng đáng kể. Mức độ hạn chế tăng trưởng có thể thay đổi cũng như các mức độ chậm phát triển tâm thần vận động với sự vắng mặt hoặc hiện diện của đầu nhỏ. Bệnh nhân có đột biến IGF1R cũng cho thấy tuổi xương chậm và nồng độ IGF-1, IGFBP-3, và GH dao động từ bình thường đến cao.
Cũng có các báo cáo về các bệnh nhân có bất thường ở nhiễm sắc thể 15, trong đó gen IGF1R bị xóa. Các kiểu hình được biểu hiện bởi những bệnh nhân này phụ thuộc vào loại và mức độ của sự bất thường. Theo quy luật, bệnh nhân có các khiếm khuyết IGF1R hai alen hoặc các đột biến vô nghĩa dị hợp tử có một biểu hiện lâm sàng nghiêm trọng hơn so với những bệnh nhân dị hợp tử cho các đột biến sai nghĩa, phản ánh một phổ liên quan đến hoạt động còn lại của IGF-1. Có tới 2% trẻ em sinh ra SGA không có sự tăng trưởng bắt kịp có thể có các khiếm khuyết dị hợp tử trong IGF1R. Sự thiếu hụt một nửa IGF1R đã được quan sát thấy ngay cả ở những bệnh nhân sinh ra có kích thước phù hợp với tuổi thai, và thường được phân loại là tầm vóc thấp vô căn. Do sự thay đổi lâm sàng đáng kể, việc chẩn đoán không nhạy cảm với IGF chỉ dựa trên các phát hiện lâm sàng và xét nghiệm là không thể, củng cố tầm quan trọng của chẩn đoán di truyền phân tử cho những bệnh nhân này.
Suy giáp và kháng Hormone tuyến giáp Suy giáp
Nhiều đặc điểm lâm sàng đặc trưng của phù niêm ở người lớn không có ở bệnh nhân nhi bị suy giáp mắc phải. Mặc dù người ta tin rộng rãi rằng biểu hiện phổ biến và nổi bật nhất của suy giáp mắc phải mãn tính là suy giảm tăng trưởng, một số nghiên cứu đã chỉ ra rằng suy giảm tăng trưởng thường là một biểu hiện tương đối không phổ biến của suy giáp, vì chẩn đoán thường được thực hiện tương đối sớm, trước khi suy giảm tăng trưởng đáng kể bắt đầu. Suy giáp có thể biểu hiện bằng tầm vóc thấp, tăng cân quá mức, và một vận tốc tăng trưởng dưới mức tối ưu. Các đặc điểm kinh điển, chẳng hạn như táo bón, da khô, rụng tóc, phản xạ gân sâu chậm và mặt thô có thể không có hoặc nhẹ. Chậm phát triển sau sinh cũng có thể được quan sát thấy ở trẻ sơ sinh bị suy giáp bẩm sinh, nhưng sự phát triển của các chương trình sàng lọc sơ sinh cho bệnh suy giáp thường dẫn đến chẩn đoán và điều trị kịp thời cho những bệnh nhân như vậy ở thế giới phương Tây. Trong suy giáp mắc phải, sự chậm phát triển có thể mất vài năm để trở nên rõ ràng về mặt lâm sàng. Tuy nhiên, một khi đã có mặt, sự suy giảm tăng trưởng thường nghiêm trọng và tiến triển.
Mặc dù suy giáp mãn tính thường được đặc trưng bởi dậy thì muộn, dậy thì sớm và thậm chí có kinh sớm có thể xảy ra ở trẻ bị suy giáp—một thực thể được gọi là hội chứng VanWyk-Grumbach. Tuy nhiên, ở nam giới, tinh hoàn thường to ra với sự nam hóa tương đối tối thiểu có thể do sự bài tiết FSH chiếm ưu thế. Ở một số phụ nữ bị suy giáp nguyên phát nghiêm trọng, có thể biểu hiện các nang buồng trứng lớn tái phát. Tuổi xương thường bị trì hoãn rõ rệt.
Việc xác nhận chẩn đoán suy giáp nguyên phát thường đơn giản. Nồng độ thyroxine tự do (FT4) trong huyết thanh giảm, và nồng độ TSH tăng cao. Sự hiện diện của các kháng thể kháng tuyến giáp phù hợp với chẩn đoán viêm tuyến giáp Hashimoto, nguyên nhân phổ biến nhất của suy giáp mắc phải ở thế giới phương Tây. Suy giáp thứ phát và tam phát đơn độc, do thiếu hụt TSH và hormone giải phóng tuyến giáp (tương ứng), là những nguyên nhân rất hiếm gặp của suy giáp mắc phải.
Liệu pháp thay thế bằng levothyroxine có liên quan đến một giai đoạn tăng trưởng bắt kịp nhanh chóng. Tuy nhiên, mặc dù có phản ứng đáng mừng này, sự tăng trưởng nhanh chóng thường không dẫn đến việc phục hồi hoàn toàn tiềm năng tăng trưởng—phần lớn là do sự gia tăng nhanh chóng của tuổi xương trong 18 tháng đầu điều trị, thường với sự tiến triển nhanh chóng qua tuổi dậy thì. Trong nghiên cứu của Rivkees và các cộng sự, những đứa trẻ bị chậm phát triển nghiêm trọng do suy giáp kéo dài, được điều trị ở tuổi theo thời gian trung bình ban đầu là 11 tuổi, có chiều cao người lớn thấp hơn khoảng 2 SD so với mức trung bình theo giới tính. Chiều cao cuối cùng này thấp hơn đáng kể so với chiều cao trung bình của cha mẹ hoặc chiều cao người lớn được dự đoán ban đầu dựa trên dữ liệu của Bayley và Pinneau. Sự thiếu hụt về tầm vóc người lớn tương quan đáng kể với thời gian suy giáp trước khi bắt đầu điều trị.
Kháng Hormone tuyến giáp RTHbeta
Kháng hormone tuyến giáp qua trung gian TRβ (RTH) ở người có tỷ lệ mắc khoảng 1 trên 40.000 và phát sinh do các đột biến điểm trong gen THRB, mã hóa thụ thể hormone tuyến giáp beta (TRβ). Các cá nhân bị RTHβ biểu hiện một hồ sơ sinh hóa đặc trưng (FT4 tăng, triiodothyronine tự do [FT3] với TSH không bị ức chế) do sự trơ trong hệ thống tín hiệu phản hồi ngược âm phụ thuộc TRβ, điều hòa trục dưới đồi-tuyến yên-tuyến giáp. Điều này dẫn đến việc trục cân bằng xung quanh một “điểm đặt” mới cao hơn với nồng độ FT4 và FT3 tăng cao.
Các biểu hiện lâm sàng của RTHβ rất thay đổi và phần lớn các cá nhân tương đối không có triệu chứng. Các đặc điểm lâm sàng liên quan có thể là do sự biểu hiện khác nhau của các dạng đồng phân TR ở các cơ quan khác nhau sao cho các mô chủ yếu biểu hiện TRβ (gan, thận, vùng dưới đồi, tuyến yên) biểu hiện sự kháng với hormone tuyến giáp, trong khi các mô chủ yếu biểu hiện TRα (cơ xương và tim, xương, não), vẫn nhạy cảm với nồng độ hormone tuyến giáp lưu hành tăng cao và do đó có thể biểu hiện nhiễm độc giáp tương đối. Do đó, các đối tượng RTH có nhịp tim tăng so với các đối chứng bình giáp, điều này có thể phản ánh sự chiếm ưu thế của TRα trong cơ tim, và tăng chi tiêu năng lượng khi nghỉ ngơi có lẽ được trung gian bởi sự chiếm ưu thế của TRα trong cơ xương. Trẻ em bị RTHβ có thể có chậm phát triển hoặc không phát triển và rối loạn tăng động giảm chú ý cũng có thể có mặt. Bướu cổ cũng phổ biến.
Hiếm khi, các đột biến điểm hoặc xóa đoạn THRB đồng hợp tử đã được mô tả liên quan đến một kiểu hình nghiêm trọng hơn bao gồm suy giảm trí tuệ rõ rệt, mất thính lực, các đặc điểm dị dạng (mặt giống chim, ngực ức gà, xương vai có cánh), điếc-câm, và mù màu.
RTHalpha
RTH xảy ra là kết quả của các đột biến mất chức năng trội âm, dị hợp tử trong gen THRA, mã hóa thụ thể hormone tuyến giáp alpha (TRα). Nó biểu hiện với suy giáp đặc hiệu cho mô ở các mô chủ yếu biểu hiện dạng đồng phân TRα và các xét nghiệm chức năng tuyến giáp gần như bình thường vì TRα là dạng đồng phân chủ yếu điều hòa phản hồi ngược âm của hormone tuyến giáp ở vùng dưới đồi và tuyến yên. Bệnh nhân có thể biểu hiện khuôn mặt rộng đặc trưng với hai mắt xa nhau, mũi tẹt, lưỡi nổi bật, và môi dày và đầu to, có lẽ do sự đóng thóp chậm. Một số lượng lớn các mấu thịt thừa đã được ghi nhận trong nhiều trường hợp.
Chậm phát triển thường nổi bật, phù hợp với vai trò quan trọng của TRα đối với sự trưởng thành xương bình thường và thường chủ yếu ở đoạn dưới. Các đặc điểm X-quang của loạn sản xương ở trẻ em bị ảnh hưởng có thể bao gồm xương Wormian (cốt hóa nội màng, rối loạn), loạn sản sụn đầu xương (cốt hóa nội sụn, rối loạn), và mọc răng chậm. Táo bón là phổ biến và có thể nghiêm trọng.
TRα là dạng đồng phân TR chủ yếu trong não và các cá nhân bị ảnh hưởng thường biểu hiện các thiếu hụt thần kinh nhận thức, bao gồm các mốc phát triển chậm (vận động, lời nói) ở thời thơ ấu, phối hợp vận động kém và khởi đầu cử động chậm, biểu hiện như chứng khó phối hợp động tác hoặc dáng đi rộng, và nói chậm. Chỉ số IQ giảm ở các mức độ khác nhau.
Các bất thường sinh hóa phổ biến nhất bao gồm nồng độ T4 thấp/thấp-bình thường và T3 cao/cao-bình thường, tỷ lệ T4/T3 dưới mức bình thường, và T3 đảo ngược giảm ở các mức độ khác nhau, điều này có thể phản ánh sự thay đổi chuyển hóa của hormone tuyến giáp.
Liệu pháp Levothyroxine đã có lợi trong một số trường hợp ở trẻ em, cải thiện sự tăng trưởng, giảm táo bón, và cải thiện sự phát triển vận động và sức khỏe. Nồng độ TSH dễ dàng bị ức chế khi điều trị với sự tăng FT3 lên các mức siêu sinh lý, điều này làm dấy lên khả năng rằng việc tiếp xúc với TH dư thừa mãn tính ở bệnh nhân RTH được điều trị bằng thyroxine có thể dẫn đến các độc tính không mong muốn ở các mô bình thường chứa TRα, chẳng hạn như gan hoặc xương.
Thừa Glucocorticoid (Hội chứng Cushing)
Thừa glucocorticoid có ảnh hưởng sâu sắc đến sự phát triển của xương cho dù nguyên nhân của hội chứng Cushing là tăng tiết ACTH, một khối u tuyến thượng thận nguyên phát, hay liệu pháp glucocorticoid. Glucocorticoid dường như làm chậm sự tăng trưởng chủ yếu thông qua một tác động trực tiếp lên sụn tăng trưởng. Sự bài tiết GH thường bình thường và nồng độ trong huyết thanh của các peptide IGF và IGFBP thường không bị ảnh hưởng. Khái niệm này cũng giải thích quan sát rằng việc điều trị bằng GH không thể khắc phục hoàn toàn các tác động ức chế tăng trưởng của glucocorticoid dư thừa.
Nếu tình trạng thừa glucocorticoid được giải quyết, sự tăng trưởng bắt kịp xảy ra nhưng thường không hoàn toàn đến mức chiều cao người lớn có thể bị giảm. Thời gian càng dài và cường độ của tình trạng thừa glucocorticoid càng lớn, bệnh nhân càng ít có khả năng trải qua sự tăng trưởng bắt kịp hoàn toàn. Do đó, điều quan trọng là phải hạn chế tiếp xúc với glucocorticoid dư thừa ở mức tối đa mà tình trạng cơ bản đang được điều trị cho phép. Các dấu hiệu đặc trưng của hội chứng Cushing (chẳng hạn như béo phì thân mình, giảm khối lượng cơ, các vết rạn da, dễ bầm tím, da mỏng, tăng huyết áp, và loãng xương) đã được biết rõ. Các khối u tuyến thượng thận tiết ra một lượng lớn glucocorticoid thường cũng sản xuất androgen dư thừa, có thể che giấu các tác động ức chế tăng trưởng của glucocorticoid, nhưng cũng làm tăng tốc độ trưởng thành của xương.
Trẻ em bị hội chứng Cushing có thể thiếu nhiều dấu hiệu và triệu chứng lâm sàng liên quan đến rối loạn này ở người lớn và có thể biểu hiện chủ yếu bằng sự tăng tốc tăng cân đi kèm với sự giảm tốc tăng trưởng theo chiều dọc. Do đó, một biểu đồ tăng trưởng cho thấy các phân vị cân nặng tăng và các phân vị chiều cao giảm cho thấy hội chứng Cushing. Ngược lại, béo phì ngoại sinh có liên quan đến sự phát triển xương bình thường hoặc thậm chí tăng tốc.
Tiếp xúc với Steroid sinh dục
Cả việc tiếp xúc sớm và quá mức với steroid sinh dục, cũng như dậy thì muộn đều có thể ảnh hưởng đến chiều cao của trẻ.
Tiếp xúc sớm/Quá mức với Steroid sinh dục
Các nguyên nhân bao gồm trưởng thành sinh dục sớm do các nguyên nhân phụ thuộc và không phụ thuộc gonadotropin, tăng androgen tuyến thượng thận do khởi phát tuyến thượng thận, các khối u tuyến thượng thận nam hóa hoặc tăng sản tuyến thượng thận bẩm sinh (CAH), các khối u buồng trứng và tinh hoàn, và các khối u sản xuất hormone gonadotropin màng đệm người. Việc sản xuất quá mức steroid sinh dục sẽ dẫn đến sự gia tăng tốc độ tăng trưởng với tầm vóc cao ban đầu, sau đó là sự cốt hóa sớm của các sụn đầu xương và cuối cùng là tầm vóc thấp. Ví dụ, điều trị không đủ hoặc chẩn đoán muộn CAH dẫn đến tăng trưởng nhanh với tầm vóc cao ban đầu, với sự ngừng tăng trưởng và cốt hóa sụn đầu xương sớm và tầm vóc thấp ở tuổi trưởng thành. Estrogen đẩy nhanh sự tăng trưởng theo chiều dọc một phần bằng cách kích thích bài tiết GH, nhưng estrogen cũng tác động lên sụn tăng trưởng để đẩy nhanh quá trình lão hóa của sụn tăng trưởng và do đó làm giảm tiềm năng tăng trưởng còn lại. Androgen dường như có một tác động kích thích, trực tiếp lên sự tăng trưởng tại sụn tăng trưởng.
Dậy thì muộn
Dậy thì muộn do chậm phát triển và dậy thì sinh lý là một trong những nguyên nhân phổ biến nhất của tầm vóc thấp trong những năm quanh tuổi dậy thì. Đây là một biến thể của dậy thì bình thường dường như phổ biến hơn ở nam giới và được đặc trưng bởi sự trưởng thành chậm của trục dưới đồi-tuyến yên-sinh dục (HPG) với hậu quả là thiếu sự bài tiết steroid sinh dục của tuyến sinh dục và sự phát triển sinh dục. Tầm vóc thấp với sự bài tiết GH giảm, tuổi xương chậm, và nồng độ IGF-1 thấp có thể liên quan đến vận tốc tăng trưởng giảm, vì sự bài tiết GH phụ thuộc nhiều vào việc sản xuất steroid sinh dục trong tuổi vị thành niên. Tình trạng này là lành tính và tự giới hạn nhưng có thể liên quan đến sự đau khổ tâm lý đáng kể và lòng tự trọng kém.
Các Rối Loạn Viêm
Các rối loạn viêm mãn tính, chẳng hạn như JIA, IBD, và xơ nang, có thể làm suy giảm sự tăng trưởng theo chiều dọc gây ra tầm vóc thấp. Các cơ chế cơ bản có lẽ phức tạp; sự suy giảm tăng trưởng có thể được trung gian bởi các cytokine tiền viêm, bao gồm TNF-α, IL1-beta, và IL-6, suy dinh dưỡng, và liệu pháp glucocorticoid và có thể liên quan đến cả các tác động trực tiếp lên các tế bào sụn ở sụn tăng trưởng và các tác động gián tiếp được trung gian bởi các bất thường nội tiết (xem các phần trước về Điều hòa tăng trưởng theo chiều dọc, Điều hòa Cytokine của sự tăng trưởng theo chiều dọc, Điều hòa dinh dưỡng của sự tăng trưởng theo chiều dọc, và Glucocorticoid).
JIA là một nhóm rối loạn không đồng nhất, như tên gọi của nó, chia sẻ các đặc điểm chung của viêm khớp dai dẳng, khởi phát ở thời thơ ấu, và một nguyên nhân chưa được hiểu rõ. Các loại JIA khác nhau về biểu hiện lâm sàng, bao gồm số lượng khớp bị ảnh hưởng, các biểu hiện viêm hệ thống, và sự hiện diện của bệnh vẩy nến. Các phương pháp điều trị bao gồm các thuốc chống viêm không steroid, glucocorticoid nội khớp và toàn thân, methotrexate, và nhiều liệu pháp kháng cytokine khác nhau. Suy giảm tăng trưởng theo chiều dọc phổ biến hơn ở JIA hệ thống và đa khớp nhưng cũng có thể xảy ra ở JIA ít khớp. Tầm vóc thấp không phổ biến khi khám lâm sàng ban đầu nhưng trở nên thường xuyên hơn trong những năm tiếp theo. Chiều cao người lớn có thể bị ảnh hưởng xấu. Giảm IGF-1 lưu hành được báo cáo ở những trẻ phát triển chậm bị JIA. Giảm GH cũng đã được báo cáo, nhưng việc giải thích những phát hiện này ít đơn giản hơn và có thể bị phức tạp bởi việc điều trị bằng glucocorticoid và độ đặc hiệu của xét nghiệm kém. Các thử nghiệm ngẫu nhiên nhỏ cho thấy rằng điều trị bằng GH làm tăng tốc độ tăng trưởng và có thể làm tăng chiều cao người lớn. Các nghiên cứu có đối chứng, dài hạn, lớn hơn ở các phân nhóm JIA cụ thể là cần thiết để xác định hiệu quả và sự an toàn. Hy vọng rằng những tiến bộ gần đây trong điều trị chống viêm sẽ cải thiện các kết quả lâu dài, bao gồm cả tầm vóc.
IBD bao gồm viêm loét đại tràng, trong đó tình trạng viêm được giới hạn ở lớp niêm mạc của đại tràng, và bệnh Crohn, trong đó tình trạng viêm là xuyên thành và có thể liên quan đến bất kỳ phần nào của đường tiêu hóa. Suy giảm tăng trưởng đáng kể phổ biến hơn ở bệnh Crohn. Một số trẻ em có thể đến khám y tế với sự tăng trưởng kém là biểu hiện chủ yếu, thay vì các triệu chứng rõ rệt của bệnh đường tiêu hóa. Các xét nghiệm sàng lọc, chẳng hạn như hemoglobin, số lượng tiểu cầu, albumin, protein phản ứng C (CRP), và tốc độ lắng hồng cầu thường, nhưng không phải luôn luôn, bất thường khi khám. Ảnh hưởng đến chiều cao người lớn là nhẹ ở hầu hết các bệnh nhân. Các cơ chế cơ bản của sự tăng trưởng giảm có thể liên quan đến thiếu hụt dinh dưỡng, viêm, và liệu pháp glucocorticoid. Nồng độ IGF-1 lưu hành có thể bị giảm. Giảm GH cũng đã được báo cáo, nhưng việc giải thích bị phức tạp bởi độ đặc hiệu của xét nghiệm kém. Các thử nghiệm ngẫu nhiên ngắn hạn cho thấy rằng điều trị bằng GH có thể làm tăng tốc độ tăng trưởng theo chiều dọc. Các thử nghiệm dài hạn là cần thiết để xác định sự an toàn và hiệu quả.
Các Rối Loạn Liên Quan Đến Dịch Ngoại Bào Bất Thường và/hoặc Suy Giảm Các Hệ Cơ Quan Cụ Thể
Các bất thường ở nhiều hệ cơ quan, chẳng hạn như thận, gan, tim và phổi có thể làm suy giảm sự tăng trưởng ở trẻ em. Nhiều rối loạn này ảnh hưởng đến thành phần của dịch ngoại bào, một dung dịch nước chứa nhiều chất tan khác nhau bao gồm các ion vô cơ, chất dinh dưỡng, protein, khí hòa tan và chất thải chuyển hóa. Các bất thường trong dịch ngoại bào có thể làm chậm sự tăng trưởng theo chiều dọc cả thông qua các tác động trực tiếp lên quá trình tạo sụn ở sụn tăng trưởng và các tác động gián tiếp liên quan đến các yếu tố dinh dưỡng và nội tiết.
Ở trẻ em bị bệnh thận mãn tính, tầm vóc thấp là phổ biến, và chiều cao người lớn thường bị ảnh hưởng. Tầm vóc thấp phổ biến hơn với suy thận nghiêm trọng hơn và với tuổi khởi phát sớm hơn. Nguyên nhân được cho là đa yếu tố. Việc ăn uống kém do chán ăn thường góp phần vào sự suy giảm tăng trưởng, cũng như việc sử dụng glucocorticoid. Có bằng chứng cho thấy các cơ chế có thể bao gồm không nhạy cảm với GH và giảm khả dụng sinh học của IGF do nồng độ IGFBP tăng. Ở trẻ em bị bệnh thận mãn tính, các phương pháp tiếp cận để giải quyết sự suy giảm tăng trưởng bao gồm cải thiện lượng dinh dưỡng, tối ưu hóa lọc máu, và, khi có thể, giảm thiểu việc sử dụng glucocorticoid. Điều trị bằng GH tái tổ hợp làm tăng tốc độ tăng trưởng ở trẻ em bị suy thận mãn tính và được sử dụng trong điều trị ở những bệnh nhân được chọn (xem phần Điều trị tầm vóc thấp).
Tầm vóc thấp cũng xảy ra ở trẻ em bị nhiễm toan ống thận. Trong các rối loạn này, nhiễm toan chuyển hóa tăng clo máu là kết quả của sự suy giảm bài tiết axit của thận. Suy giảm tăng trưởng theo chiều dọc được thấy ở cả nhiễm toan ống thận đầu xa và đầu gần.
Giảm tăng trưởng theo chiều dọc cũng được tìm thấy ở trẻ em bị tăng phosphate máu, bao gồm cả những trẻ bị còi xương giảm phosphate máu liên kết X, liên quan đến nồng độ FGF-23 tăng và cả còi xương giảm phosphate máu di truyền với tăng canxi niệu, do các chất đồng vận chuyển natri-phosphate của thận bị khiếm khuyết. Trong các rối loạn này, các sụn tăng trưởng cho thấy các bất thường điển hình của bệnh còi xương. Có bằng chứng cho thấy nồng độ phosphate thấp trong dịch ngoại bào trực tiếp làm suy giảm sự biệt hóa của tế bào sụn ở sụn tăng trưởng, góp phần vào sự tăng trưởng theo chiều dọc bất thường.
Bệnh tim bẩm sinh thường làm suy giảm sự tăng trưởng ở trẻ em. Suy giảm tăng trưởng xảy ra ở cả các rối loạn tím và không tím. Suy dinh dưỡng, liên quan đến cả chán ăn và nhu cầu dinh dưỡng tăng, được cho là một yếu tố đóng góp chính.
Thiếu Thốn Tâm Lý Xã Hội
Suy giảm tăng trưởng do thiếu thốn tâm lý xã hội là một rối loạn tăng trưởng liên quan đến sự thiếu thốn tình cảm và/hoặc một môi trường bệnh lý làm gián đoạn sự gắn bó bình thường giữa cha mẹ và con cái. Nó có thể ảnh hưởng đến trẻ em ở mọi lứa tuổi; chậm phát triển thường thấy ở trẻ sơ sinh, trong khi trẻ mới biết đi và trẻ lớn hơn thường bị trầm cảm và thường biểu hiện các hành vi kỳ quái xung quanh thức ăn và việc ăn uống (chẳng hạn như uống nước từ bồn cầu và chậu rửa bát, ăn từ thùng rác và đĩa của vật nuôi, và ăn uống vô độ). Chậm phát triển tầm vóc và dậy thì, các đường ngừng tăng trưởng ở xương dài, và sự giãn rộng tạm thời của các đường khớp sọ đã được báo cáo. Mặc dù suy dinh dưỡng thường có liên quan, có một số bằng chứng cho thấy rằng sự thiếu thốn tâm lý xã hội có thể gây ra còi cọc mặc dù có đủ nguồn cung cấp chất dinh dưỡng. Ví dụ, Widdowson đã mô tả hai trại trẻ mồ côi ở Đức do những người phụ nữ có tính cách đối lập điều hành. Mặc dù có lượng ăn vào tương tự, trẻ em dưới sự chăm sóc của người phụ nữ khó chịu, hung hăng, không nuôi dưỡng đã không phát triển tốt, trong khi những trẻ dưới sự chăm sóc của người phụ nữ có những đặc điểm tính cách đối lập đã phát triển tốt.
Sự ức chế bài tiết GH, không liên quan đến giấc ngủ bị xáo trộn, đã được báo cáo trong một số trường hợp, nhưng phát hiện này bị gây nhiễu bởi độ đặc hiệu kém của xét nghiệm GH. Dấu hiệu của tình trạng này là khả năng đảo ngược của sự còi cọc và các bất thường bài tiết GH (khi được tìm thấy) khi được đưa ra khỏi môi trường thù địch và đặt vào một môi trường nuôi dưỡng. Nồng độ huyết thanh thấp hơn của một số dấu hiệu thần kinh nội tiết (melatonin, serotonin, β-endorphin, và hormone vỏ thượng thận) đã được tìm thấy ở trẻ em bị thiếu thốn xã hội, đặc biệt là những trẻ bị suy giảm tăng trưởng, so với nhóm đối chứng hoặc trẻ em bị chậm phát triển.
Việc sớm đưa trẻ em bị thiếu thốn xã hội vào một môi trường nuôi dưỡng là rất quan trọng không chỉ để phục hồi chiều cao và tăng cân, mà còn để cải thiện nhận thức, và sự bắt kịp về chiều cao đóng vai trò như một yếu tố dự báo tích cực về sự phục hồi nhận thức. Một nghiên cứu trên trẻ em Romania bị thiếu thốn xã hội được phân ngẫu nhiên vào các gia đình nuôi dưỡng so với việc chăm sóc tại cơ sở liên tục, tuổi từ 5 đến 32 tháng tại thời điểm ban đầu, cho thấy sự gia tăng trung bình 12,6 điểm (SD, 4,7 điểm) trong chỉ số IQ lời nói cho mỗi sự gia tăng của 1 SD trong điểm Z-score chiều cao giữa thời điểm ban đầu và 42 tháng. Ngay cả khi phục hồi tăng trưởng, trẻ em đã phải chịu đựng sự thiếu thốn sớm vẫn có thể phải đối mặt với các bất thường dai dẳng của hệ thống dưới đồi-tuyến yên-thượng thận; dậy thì sớm; tăng nguy cơ mắc hội chứng chuyển hóa ở tuổi trưởng thành; và các di chứng về nhận thức, hành vi và cảm xúc.
Suy giảm tăng trưởng nguyên phát
Các Rối Loạn Liên Quan Đến Các Yếu Tố Tự Tiết/Cận Tiết
Các tế bào sụn ở sụn tăng trưởng tiết ra nhiều yếu tố hoạt động cục bộ theo kiểu cận tiết trên các tế bào sụn gần đó và theo kiểu tự tiết trên tế bào tiết ra. Các yếu tố này, nhiều trong số đó là các polypeptide, phục vụ để phối hợp sự tăng sinh và biệt hóa của các tế bào ở các vùng khác nhau của sụn tăng trưởng (xem phần Điều hòa tự tiết/cận tiết của sự tăng trưởng theo chiều dọc trước đó). Do đó, các đột biến trong các gen tham gia vào sự giao tiếp giữa các tế bào cục bộ này có thể làm suy giảm quá trình tạo sụn ở sụn tăng trưởng, gây ra tầm vóc thấp, hoặc đôi khi tăng cường quá trình tạo sụn ở sụn tăng trưởng, gây ra tầm vóc cao. Nếu quá trình tạo sụn ở sụn tăng trưởng bất thường ảnh hưởng không chỉ đến chiều dài xương mà còn cả hình dạng xương, sẽ xảy ra dị dạng, và tình trạng này được gọi là loạn sản sụn. Thảo luận sau đây cung cấp một số ví dụ về các rối loạn tăng trưởng liên quan đến các khiếm khuyết tín hiệu cận tiết, đặc biệt tập trung vào những rối loạn có thể biểu hiện như tầm vóc thấp đơn độc mà không có một bệnh loạn sản sụn rõ ràng.
Các Rối Loạn Liên Quan Đến Yếu Tố Tăng Trưởng Giống Insulin-2
IGF2 là một gen được in dấu được biểu hiện từ cha trước khi sinh và, ở một số mô, sau khi sinh. Sự giảm methyl hóa DNA trong vùng của gen IGF2 dẫn đến giảm biểu hiện IGF2 từ cha và biểu hiện lâm sàng như hội chứng Silver-Russell (SRS) (xem phần Rối loạn In Dấu—hội chứng Silver-Russell để biết chi tiết), ảnh hưởng đến cả sự tăng trưởng trước và sau sinh ở người. Gần đây, một số bệnh nhân có các biến thể gen IGF2 gây bệnh liên quan đến hạn chế tăng trưởng trong tử cung và sau sinh đã được mô tả. Thông thường, chỉ những người được di truyền biến thể qua đường truyền từ cha mới bị ảnh hưởng, một phát hiện phù hợp với sự in dấu của gen IGF2. Những bệnh nhân này cũng có các dấu hiệu lâm sàng tương thích với SRS, chẳng hạn như cân nặng khi sinh thấp với chu vi vòng đầu bình thường, mặt hình tam giác, và cằm nhỏ. Một số bệnh nhân có chậm phát triển tâm thần vận động. Về mặt nội tiết, bệnh nhân có nồng độ IGF-2 thấp, nhưng nồng độ IGF-1, IGFBP-3, và GH từ bình thường đến cao. Người ta cũng đã đề xuất rằng các biến thể gây bệnh dị hợp tử trong các gen HMGA2 và PLAG1 có thể gây ra các kiểu hình tương tự. PLAG1 kích hoạt IGF2 thông qua một promoter và HMGA2 là một chất điều hòa thượng nguồn của PLAG1. Cả hai gen đều có liên quan đến các bất thường tăng trưởng trong các mô hình chuột và trong các phân tích tổng hợp GWAS về chiều cao ở người.
Các Rối Loạn Liên Quan Đến Tín Hiệu Peptide Lợi Niệu Loại C
Peptide lợi niệu loại C (CNP) tác động lên thụ thể của nó, NPR2 (còn được gọi là NPRB và GC-B) để kích thích quá trình tạo sụn ở sụn tăng trưởng. Do đó, các đột biến trong phối tử hoặc thụ thể đều có thể làm suy giảm sự tăng trưởng theo chiều dọc. Các đột biến mất chức năng hai alen (đồng hợp tử hoặc dị hợp tử phức hợp) trong NPR2 gây ra một bệnh loạn sản sụn với tầm vóc thấp, được gọi là loạn sản đầu chi-giữa chi, loại Maroteaux. Các đột biến mất chức năng một alen (dị hợp tử) trong NPR2 có thể biểu hiện như tầm vóc thấp đơn độc, thường với tỷ lệ chiều cao ngồi so với đứng tăng. Tương tự, các đột biến dị hợp tử trong NPPC, mã hóa CNP, gây ra tầm vóc thấp trội trên nhiễm sắc thể thường với xu hướng bàn tay nhỏ.
Ngược lại, các đột biến tăng chức năng trong NPR2 có thể gây ra tầm vóc cao có thể đi kèm với vẹo cột sống, tật dính ngón, và đặc biệt là các ngón chân cái dài. Sự biểu hiện quá mức của CNP dường như gây ra một kiểu hình tương tự.
Các Rối Loạn Liên Quan Đến Tín Hiệu Yếu Tố Tăng Trưởng Nguyên Bào Sợi
Các FGF, tác động thông qua FGFR3, điều hòa tiêu cực quá trình tạo sụn ở sụn tăng trưởng. Do đó, các đột biến tăng chức năng trong FGFR3 làm suy giảm sự tăng trưởng theo chiều dọc. Mức độ nghiêm trọng của kiểu hình thay đổi tùy thuộc vào đột biến, bao gồm (theo thứ tự mức độ nghiêm trọng giảm dần) loạn sản tử vong, ACH, và loạn sản sụn, tất cả đều bao gồm tầm vóc thấp ảnh hưởng không cân đối đến các chi so với thân mình.
ACH là bệnh loạn sản sụn phổ biến nhất. Phần lớn bệnh nhân bị ACH có cùng một đột biến điểm trong FGFR3 do tỷ lệ đột biến cao ở cặp bazơ đó. Nó được di truyền theo kiểu trội trên nhiễm sắc thể thường, nhưng, do tỷ lệ đột biến cao, hầu hết các trường hợp là lẻ tẻ, phát sinh từ các đột biến de novo. ACH dẫn đến các chi ngắn gốc chi, đầu to, và thiểu sản giữa mặt. Suy giảm sự phát triển của lỗ chẩm có thể dẫn đến chèn ép tủy cổ, có thể gây tử vong.
Loạn sản sụn có một kiểu hình nhẹ hơn ACH và có thể biểu hiện bằng tầm vóc thấp, tật ngón ngắn, đầu to, và hạn chế duỗi khuỷu tay. Chẩn đoán bao gồm các đặc điểm lâm sàng, các đặc điểm xương X-quang, và xét nghiệm di truyền phân tử. Một số bệnh nhân có các đặc điểm lâm sàng và X-quang của loạn sản sụn có thể không có các đột biến trong FGFR3, cho thấy sự không đồng nhất về locus.
Các đột biến tăng chức năng nhẹ hơn đã được báo cáo là biểu hiện như tầm vóc thấp cân đối.
Ngược lại, các đột biến mất chức năng một alen gây ra tầm vóc cao với tật co ngón, tật dính ngón, vẹo cột sống, và mất thính lực, được gọi là hội chứng CATSHL.
Các Rối Loạn Liên Quan Đến Tín Hiệu Protein Liên Quan Đến Hormone Cận Giáp và Indian Hedgehog
PTHrP và IHH tạo thành một vòng phản hồi ngược âm tại chỗ trong sụn tăng trưởng để kiểm soát sự tăng sinh và đặc biệt là sự phì đại của tế bào sụn. PTHrP tác động thông qua PTH1R, cùng một thụ thể mà PTH tác động thông qua. Điều thú vị là cả các đột biến mất chức năng và tăng chức năng trong PTH1R đều làm suy giảm quá trình tạo sụn ở sụn tăng trưởng, gây ra các bệnh loạn sản xương—loạn sản sụn gây chết Blomstrand và loạn sản sụn hành xương Jansen, tương ứng.
PTH1R là một thụ thể có bảy miền xuyên màng, truyền tín hiệu một phần thông qua Gs-alpha. Các đột biến dị hợp tử trong GNAS, gen mã hóa Gs-alpha, gây ra bệnh loạn dưỡng xương di truyền Albright (AHO), được đặc trưng bởi tầm vóc thấp, vôi hóa dưới da, và tật ngón ngắn thay đổi (đặc biệt với các đốt ngón tay xa và các xương bàn tay thứ năm, thứ tư và thứ ba bị rút ngắn). Tầm vóc thấp phát triển theo tuổi do sự chậm lại và ngừng tăng trưởng theo chiều dọc sớm, đi kèm với tuổi xương X-quang cao và sự đóng sụn đầu xương sớm. AHO xảy ra với sự di truyền đột biến từ cha hoặc mẹ. Khi các đột biến dị hợp tử trong GNAS được di truyền trên alen của mẹ, các phát hiện AHO có thể đi kèm với sự kháng với PTH, gonadotropin, TSH, và GHRH, cũng như béo phì như một phần của giả suy cận giáp loại 1A (PHP1a). Các đặc điểm bổ sung này xảy ra với sự di truyền từ mẹ vì GNAS được in dấu, do đó nó thường được biểu hiện ưu tiên bởi alen của mẹ ở một số mô. Vai trò của việc điều trị bằng GH trong PHP1a đang được điều tra. Khi đột biến được di truyền từ cha, bệnh nhân chỉ biểu hiện AHO, một tình trạng được gọi là giả-giả suy cận giáp (PPHP). Các đột biến trong protein kinase phụ thuộc cyclic adenosine monophosphate loại I tiểu đơn vị điều hòa alpha (PRKAR1A), nằm ở hạ nguồn trong tín hiệu Gs-alpha, gây ra bệnh acrodysostosis có các đặc điểm tương tự như AHO và PHP1a.
Các đột biến dị hợp tử trong IHH gây ra tầm vóc thấp trội trên nhiễm sắc thể thường như một phần của tật ngón ngắn loại A1 hoặc có thể gây ra tầm vóc thấp với các bất thường xương không đặc hiệu. Tình trạng sau được đặc trưng bởi tầm vóc thấp không cân đối nhẹ thường với sự rút ngắn của đốt ngón tay giữa của ngón thứ năm nhưng không có các đặc điểm kinh điển của tật ngón ngắn loại A1.
Các Rối Loạn Liên Quan Đến Chất Nền Ngoại Bào Sụn
Các tế bào sụn ở sụn tăng trưởng tiết ra một chất nền sụn, bao gồm nhiều protein khác nhau, bao gồm nhiều loại collagen, và các proteoglycan. Các đột biến trong các gen mã hóa các protein và proteoglycan này có thể làm suy giảm quá trình tạo sụn ở sụn tăng trưởng và do đó làm suy giảm sự tăng trưởng theo chiều dọc. Biểu hiện lâm sàng có thể là một bệnh loạn sản sụn. Ví dụ, COL10A1 mã hóa collagen X, là một thành phần chính của chất nền sụn tăng trưởng, và các đột biến dị hợp tử trong COL10A1 gây ra loạn sản sụn hành xương, loại Schmid. Tương tự, các đột biến dị hợp tử trong protein ma trận oligomeric sụn có thể biểu hiện như giả loạn sản sụn hoặc loạn sản sụn đầu xương đa phát-1 nhẹ hơn, cả hai đều gây ra tầm vóc thấp.
Các đột biến trong các gen mã hóa các thành phần của chất nền ngoại bào sụn cũng có thể có một kiểu hình nhẹ hơn, biểu hiện, không phải là một bệnh loạn sản sụn, mà thay vào đó là tầm vóc thấp mà không có một bệnh loạn sản sụn rõ ràng. Ví dụ, ACAN mã hóa aggrecan, một proteoglycan là một thành phần quan trọng của chất nền sụn. Các đột biến đồng hợp tử có thể gây ra một bệnh loạn sản sụn nghiêm trọng với tầm vóc cực thấp được gọi là loạn sản cột sống-đầu xương-hành xương, loại aggrecan. Các đột biến dị hợp tử có thể gây ra một bệnh loạn sản xương nhẹ hơn hoặc có thể biểu hiện lâm sàng như tầm vóc thấp đơn độc mà không có các dấu hiệu rõ ràng của một bệnh loạn sản sụn. Tình trạng này có thể được di truyền như một đặc điểm trội trên nhiễm sắc thể thường. Thông thường, tuổi xương cao, và sự ngừng tăng trưởng xảy ra ở độ tuổi sớm. Các thành viên gia đình bị ảnh hưởng có thể phát triển viêm xương khớp khởi phát sớm và bệnh đĩa đệm liên đốt sống do sự bất thường của chất nền sụn.
Các Rối Loạn Liên Quan Đến Các Yếu Tố Nội Bào và/hoặc Các Quá Trình Tế Bào Cơ Bản
Các khiếm khuyết di truyền trong một số con đường nội bào hoặc các quá trình tế bào cơ bản dẫn đến một nhóm lớn và không đồng nhất các hội chứng tầm vóc thấp. Các hội chứng này thường cho thấy các kiểu hình riêng biệt phản ánh nhiều cơ chế suy giảm tăng trưởng, một số trong đó chưa được làm sáng tỏ hoàn toàn. Một cuộc thảo luận về các rối loạn đại diện trong danh mục này, được chọn vì tần suất hoặc tầm quan trọng của chúng như các mô hình bệnh tật, được bao gồm trong văn bản này, nhưng hãy nhớ rằng đây là một nhóm lớn các rối loạn bao gồm hàng trăm tình trạng riêng biệt.
Khiếm khuyết trong SHOX
Gen SHOX nằm bên trong vùng giả nhiễm sắc thể thường 1 (PAR1) của các nhiễm sắc thể giới tính X và Y. Các gen nằm trong PAR1 thoát khỏi sự bất hoạt X; do đó hai bản sao của SHOX được biểu hiện ở cả nam và nữ. SHOX mã hóa một yếu tố phiên mã cần thiết cho sự phát triển chi bình thường và chức năng sụn tăng trưởng, đặc biệt là cho cẳng tay và cẳng chân. Đơn nhiễm sắc thể X, ở những bệnh nhân bị hội chứng Turner, dẫn đến sự thiếu hụt một nửa SHOX và giải thích một phần tầm vóc thấp và một số đặc điểm xương được quan sát thấy ở những bệnh nhân này (khuỷu tay vẹo ngoài và sự cong và ngắn của cẳng tay và cẳng chân) (xem Chương 17 về hội chứng Turner để biết chi tiết). Hơn nữa, đột biến dị hợp tử trong các vùng mã hóa hoặc điều hòa của SHOX gây ra sự thiếu hụt một nửa SHOX và có liên quan đến một phổ các kiểu hình tầm vóc thấp, bao gồm loạn sản sụn Leri-Weill (LWD) và tầm vóc thấp không có bất kỳ đặc điểm xương cụ thể nào. LWD là một bệnh loạn sản xương di truyền trội được đặc trưng bởi tầm vóc thấp không cân đối, sự rút ngắn chi giữa, và biến dạng Madelung (Hình 11.6). Ngoài ra, bệnh nhân LWD thường có vòm miệng cao, khuỷu tay vẹo ngoài, chân vòng kiềng, phì đại cơ, và xu hướng có BMI tăng. Các khiếm khuyết SHOX dị hợp tử được xác định ở 70% đến 90% bệnh nhân bị LWD và ở 2% đến 15% trẻ em được phân loại là tầm vóc thấp vô căn. Các khiếm khuyết SHOX đồng hợp tử có liên quan đến loạn sản giữa chi Langer, là một dạng lùn chi ngắn hiếm gặp và nghiêm trọng. Tỷ lệ hiện mắc ước tính của thiếu hụt SHOX là 1:1000, và vì lý do này, nó được coi là một trong những nguyên nhân đơn gen chính của tầm vóc thấp.
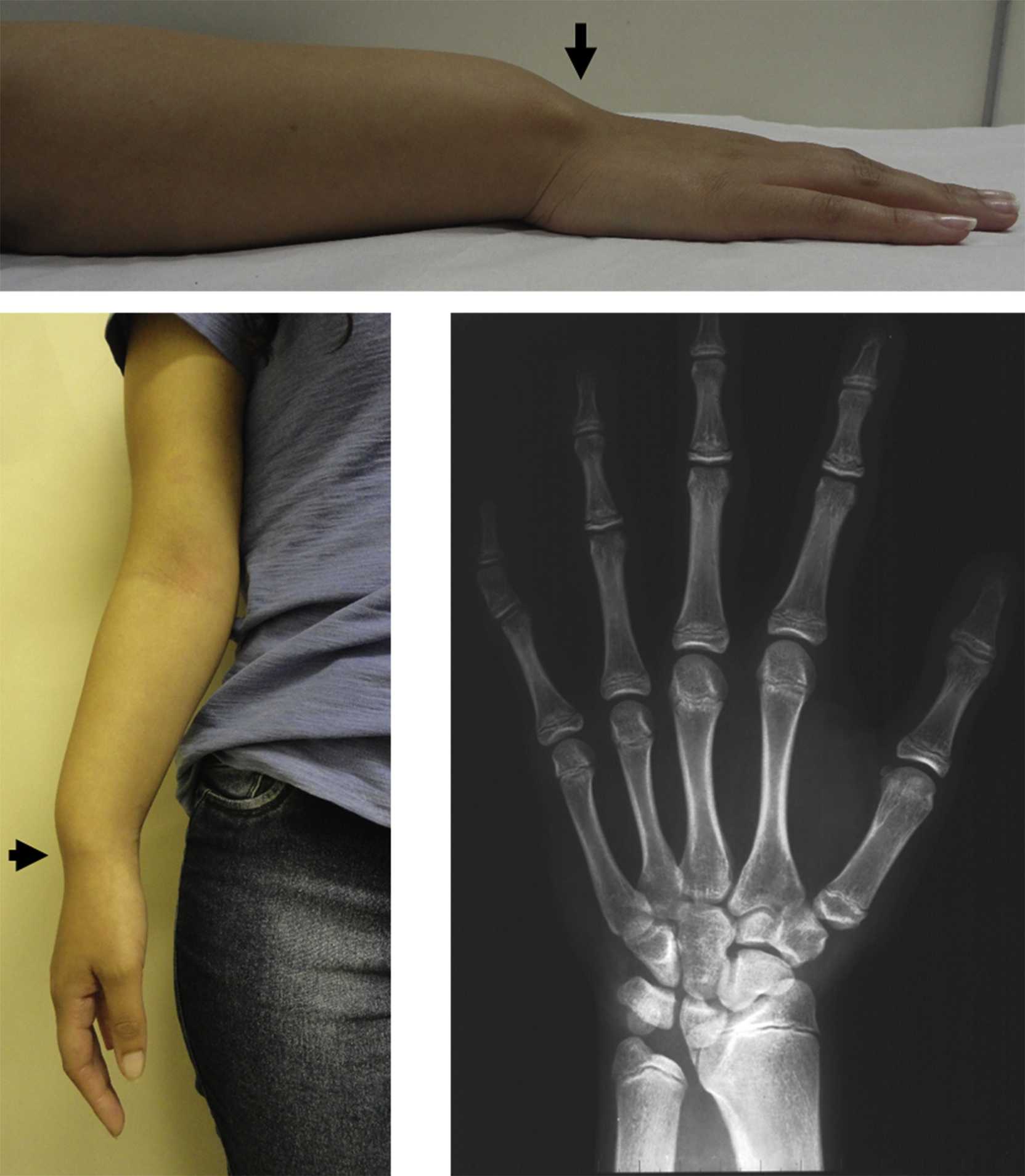
Hình 11.6 Biến dạng Madelung được quan sát thấy ở những bệnh nhân bị loạn sản sụn Leri-Weill liên quan đến sự thiếu hụt một nửa SHOX: sự hiện diện của trật khớp sau của khớp quay trụ dưới. Hình ảnh X-quang bàn tay và cổ tay với sự tam giác hóa của sụn đầu xương quay dưới; hàng xương cổ tay gần hình chữ “V” (hoặc hình chóp) và sự ngừng tăng trưởng sụn đầu xương sớm ở phần trong của xương quay dưới, dẫn đến sự cong của xương quay dưới.
Vị trí nhiễm sắc thể của SHOX, trong một vùng có số lượng lớn các trình tự lặp lại, có xu hướng bị xóa, chịu trách nhiệm cho 80% các trường hợp thiếu hụt một nửa SHOX. Hầu hết 20% còn lại là do các đột biến nucleotide đơn. Điều tra di truyền phân tử về một khiếm khuyết SHOX được chỉ định ở những bệnh nhân và gia đình bị LWD và ở trẻ em được phân loại là ISS vì chẩn đoán phân tử của các khiếm khuyết SHOX cho phép xác định nguyên nhân của tầm vóc thấp. Các chẩn đoán như vậy có ý nghĩa điều trị vì điều trị bằng hGH có hiệu quả trong việc cải thiện chiều cao người lớn. Cả hai kiểu hình, LWD và ISS, đều có thể được quan sát thấy ở các thành viên bị ảnh hưởng trong cùng một gia đình; vì lý do này, sự hiện diện của một người thân bị LWD trong một gia đình có tầm vóc thấp trội trên nhiễm sắc thể thường làm tăng khả năng có khiếm khuyết SHOX. Ngoài ra, sự hiện diện của tầm vóc thấp không cân đối, được chứng minh bằng chỉ số chiều cao ngồi (chiều cao ngồi/tổng chiều cao) SDS trên 2, mà không có bằng chứng về loạn sản xương, là một công cụ đơn giản và hữu ích để lựa chọn trẻ em ISS cho sàng lọc di truyền phân tử SHOX. Bởi vì độ nhạy của chỉ số chiều cao ngồi ở người lớn lớn hơn ở trẻ em, cũng hữu ích khi đo chỉ số chiều cao ngồi của cha mẹ thấp của một đứa trẻ thấp.
Hội chứng Noonan và các bệnh liên quan đến RAS (RASopathies)
Hội chứng Noonan (NS) là một rối loạn di truyền tương đối phổ biến, với tỷ lệ hiện mắc ước tính từ 1 trên 1000 đến 1 trên 2500 trẻ sinh sống với tỷ lệ nam và nữ ngang nhau. Nó có kiểu di truyền trội trên nhiễm sắc thể thường, với độ thấm gần như hoàn toàn và tính không đồng nhất lâm sàng rõ rệt. Khoảng một phần ba bệnh nhân được chẩn đoán mắc NS có cha hoặc mẹ bị ảnh hưởng, phần còn lại của bệnh nhân mang các đột biến de novo. NS được đặc trưng bởi các đặc điểm dị dạng khuôn mặt đặc biệt (Hình 11.7), các khuyết tật tim bẩm sinh (phổ biến nhất là hẹp van động mạch phổi, bệnh cơ tim phì đại, và các khuyết tật vách ngăn), các bất thường thành ngực (ngực ức gà và/hoặc ngực lõm), và chậm phát triển. Bệnh nhân mắc NS cũng có thể cho thấy sự chậm phát triển từ nhẹ đến trung bình, khó khăn trong học tập, gù vẹo cột sống, và phù bạch huyết, thường xuyên dễ bị bầm tím, và xu hướng chảy máu. Các cá nhân mắc NS có nguy cơ phát triển các bệnh ác tính tăng nhẹ (chủ yếu là bệnh bạch cầu tủy bào đơn nhân vị thành niên, các rối loạn tăng sinh tủy khác, và các khối u não) trong thời thơ ấu. Tinh hoàn ẩn một bên hoặc hai bên thường được quan sát thấy ở các bé trai bị ảnh hưởng. Mặc dù cân nặng và chiều dài khi sinh thường nằm trong giới hạn bình thường, hầu hết trẻ sơ sinh mắc NS đều gặp khó khăn trong việc ăn uống có thể dẫn đến chậm phát triển. Chậm phát triển xảy ra sau khi sinh và đi kèm với nồng độ IGF-1 giảm, nhưng thường là sự bài tiết GH bình thường, mặc dù GHD đã được mô tả ở một số bệnh nhân. Các bé trai và bé gái mắc NS thường có khởi phát dậy thì muộn, dẫn đến một giai đoạn tăng trưởng vị thành niên kéo dài. Chiều cao trung bình của người lớn đối với bệnh nhân mắc NS là khoảng -2 SDS, và gần đây người ta đã nhận ra rằng bệnh nhân mắc NS cho thấy một kiểu hình cơ thể gầy và chống lại việc tăng cân trong thời thơ ấu và cuộc sống trưởng thành.
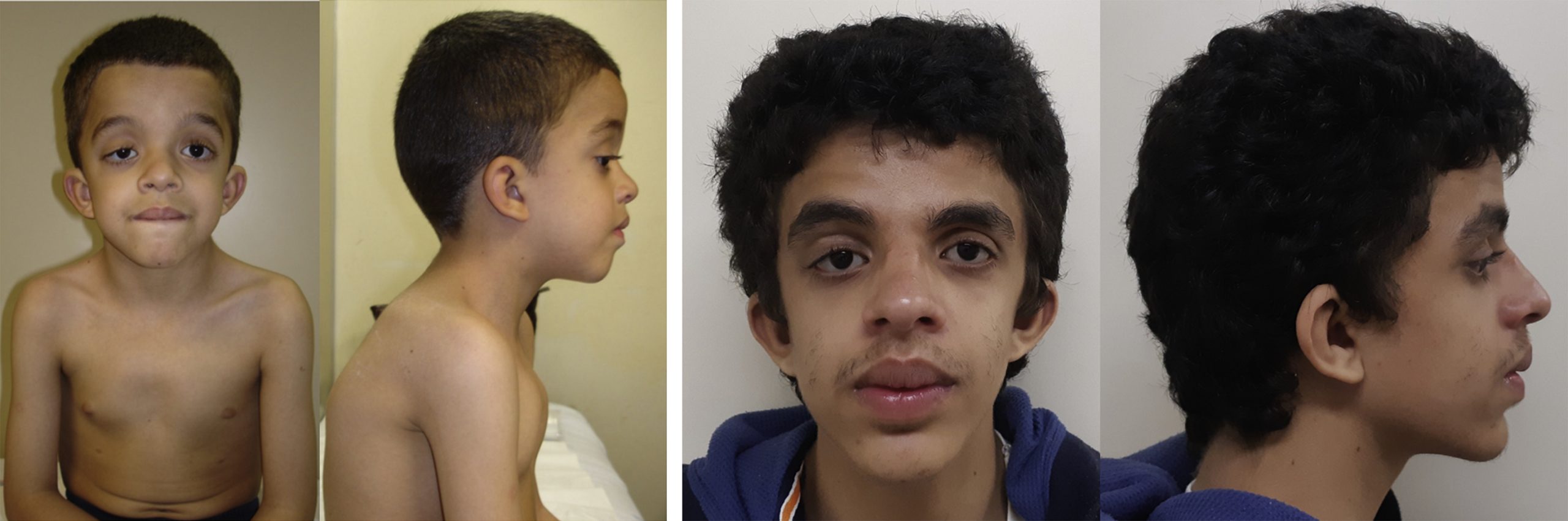
Hình 11.7 Các đặc điểm khuôn mặt của một bệnh nhân mắc hội chứng Noonan. Cùng một bệnh nhân được hiển thị, đầu tiên ở tuổi 8 (trái) và 17 (phải). Các đặc điểm điển hình của hội chứng Noonan được quan sát thấy: hai mắt xa nhau nhẹ, sụp mí, khe mi mắt xếch xuống, tai đóng thấp xoay ra sau, và ngực lõm.
Các đột biến trong hơn 15 gen hoạt động trong con đường RAS/MAPK gây ra NS, cũng như các hội chứng hiếm gặp khác có các đặc điểm chồng chéo với NS, bao gồm hội chứng tim-mặt-da, hội chứng Costello, NS với nhiều nốt ruồi (trước đây được gọi là hội chứng LEOPARD), và hội chứng giống Noonan với tóc anagen lỏng lẻo. Các hội chứng này đại diện cho một nhóm các tình trạng di truyền được gọi chung là RASopathies. Các khiếm khuyết phân tử liên quan đến NS và các RASopathies khác dẫn đến sự hoạt hóa quá mức của con đường RAS/MAPK, chịu trách nhiệm cho các tác động đa dạng được quan sát thấy trong các rối loạn này.
Chẩn đoán NS chủ yếu nên dựa trên các phát hiện lâm sàng. Các đặc điểm khuôn mặt cổ điển hoặc dị tật tim điển hình thường gây ra nghi ngờ về NS, đặc biệt khi kết hợp với chậm phát triển và/hoặc tầm vóc thấp. Chẩn đoán phân tử của NS có thể được thiết lập bằng một bảng xét nghiệm đa gen hoặc giải trình tự toàn bộ exome (WES). Xét nghiệm gen đơn lẻ nối tiếp có thể được xem xét nếu xét nghiệm bảng không khả thi và nên bắt đầu bằng việc giải trình tự protein-tyrosine phosphatase nonreceptor-type 11 (PTPN11) vì 40% bệnh nhân NS mang các biến thể gây bệnh dị hợp tử trong gen này. Các biến thể gây bệnh PTPN11 có nhiều khả năng được tìm thấy hơn khi có hẹp van động mạch phổi, và các đột biến ở các codon 61, 71, 72, và 76 có liên quan đến bệnh bạch cầu tủy bào đơn nhân vị thành niên. Đáng chú ý là một số nghiên cứu điều tra trẻ em có tầm vóc thấp vô căn bằng cách giải trình tự nhắm mục tiêu nhiều gen đã xác định được những bệnh nhân dị hợp tử cho các biến thể gây bệnh trong PTPN11, phản ánh phần cuối nhẹ nhất của phổ lâm sàng của NS.
Các Rối Loạn bất hoạt GNAS
Tiểu đơn vị alpha S của protein liên kết guanine-nucleotide (protein G) (Gs-alpha) là một protein quan trọng trung gian cho tín hiệu nội bào của nhiều thụ thể hormone bằng cách kích hoạt adenylyl cyclase. Gs-alpha là một trong những bản phiên mã được tạo ra từ locus GNAS, một locus phức tạp, được in dấu, sản xuất nhiều bản phiên mã bằng cách sử dụng các promoter thay thế và cắt nối thay thế. Gs-alpha cho thấy sự biểu hiện hai alen ở hầu hết các tế bào (ví dụ, tế bào sụn), nhưng ở một số tế bào (ví dụ, tế bào hướng thân của tuyến yên, các tế bào ống lượn gần của thận, các tế bào biểu mô tuyến giáp, các tế bào sinh dục), nó được biểu hiện ưu tiên từ alen của mẹ.
Sự bất hoạt dị hợp tử của GNAS chịu trách nhiệm cho hai kiểu hình chính: PHP và PPHP. Bệnh nhân bị PHP cho thấy sự kháng với PTH ở các tế bào ống lượn gần của thận và hậu quả là hạ canxi máu và tăng phosphate máu, mặc dù nồng độ PTH cao. Sự kháng với các hormone khác có thể được quan sát thấy, chủ yếu là TSH, gonadotropin, và GHRH. Ngoài ra, bệnh nhân bị PHP cũng có thể có béo phì khởi phát sớm và suy giảm nhận thức. PHP có thể do các đột biến bất hoạt trong các vùng mã hóa của GNAS trên alen của mẹ (PHP1a) hoặc đột biến ngoại di truyền (epimutation) (ví dụ, các mô hình methyl hóa bất thường), làm giảm sự biểu hiện của Gs-alpha của mẹ (PHP1b). Tầm vóc thấp với sự liên quan đến xương có mặt ở những bệnh nhân bị PHP1a và PPHP, do sự giảm hoạt động của PTHrP thông qua thụ thể PTH (PTH1R) do thiếu hụt Gs-alpha. Bệnh nhân bị PPHP mang các đột biến trên alen GNAS của cha và do đó không cho thấy sự kháng PTH ở các tế bào ống thận nơi alen của mẹ được biểu hiện. Kiểu hình xương được quan sát thấy ở bệnh nhân bị PHP1a và PPHP được gọi là AHO. AHO được đặc trưng bởi khuôn mặt tròn, tầm vóc thấp, đóng sớm các sụn tăng trưởng, tật ngón ngắn, và cốt hóa lạc chỗ.
Ở hầu hết trẻ em bị PHP1a và PPHP, phân vị chiều cao giảm dần theo tuổi, và tuổi xương thường cao. Do đó, hầu hết bệnh nhân bị PHP1a và PPHP đều có tầm vóc thấp khi đến tuổi trưởng thành. Trong cả hai tình trạng, sự kháng với PTHrP ở sụn tăng trưởng có khả năng đóng một vai trò quan trọng, và ở PHP1a, sự kháng GHRH cũng có thể góp phần. Ngược lại, hầu hết bệnh nhân bị PHP1b đều có tầm vóc bình thường.
Khuyến cáo rằng chẩn đoán phân tử của AHO nên bao gồm giải trình tự DNA, sau đó là phân tích biến thể số lượng bản sao (CNV) của locus GNAS cho bệnh nhân bị PHP và PPHP. Phân tích methyl hóa của locus GNAS được khuyến nghị cho bệnh nhân PHP có kết quả xét nghiệm di truyền âm tính.
Hội chứng 3-M
Hội chứng 3-M là một rối loạn lặn trên nhiễm sắc thể thường được đặc trưng bởi sự chậm phát triển trước và sau sinh, chu vi vòng đầu bình thường, dị dạng khuôn mặt (mặt tròn với trán rộng, thiểu sản giữa mặt, lông mày rậm, mũi ngắn với lỗ mũi hếch, miệng nổi bật, và cằm nhọn), các thay đổi xương nhẹ (cổ ngắn, ưỡn cột sống quá mức, thân đốt sống cao, và gót chân nổi bật), và trí thông minh bình thường. Suy sinh dục và lỗ tiểu lệch thấp đã được báo cáo ở một số bệnh nhân nam. Cho đến nay, các đột biến mất chức năng hai alen trong ba gen (CUL7, OBSL1, hoặc CCDC8) đã được liên quan đến hội chứng 3-M, nhưng dự kiến rằng các đột biến của các gen khác trong cùng một con đường có thể liên quan ở những bệnh nhân không có khiếm khuyết phân tử được xác định. CUL7 là một ubiquitin ligase và tham gia vào quá trình phân hủy một số protein, chẳng hạn như p53, điều hòa chu kỳ tế bào, tín hiệu IGF-1, và sự tăng sinh của tế bào sụn. Hội chứng 3-M là một phần của chẩn đoán phân biệt của SRS (xem phần Rối loạn In Dấu—Hội chứng Silver-Russell để biết chi tiết), và nên được xem xét ở những bệnh nhân có các phát hiện xương điển hình và/hoặc có cha mẹ là họ hàng gần. Chẩn đoán phân tử của hội chứng 3-M tốt nhất nên được thiết lập bằng bảng xét nghiệm đa gen hoặc WES do tính không đồng nhất về mặt di truyền.
Các hội chứng sửa chữa tổn thương DNA
Là một nhóm, các hội chứng sửa chữa tổn thương DNA được đặc trưng bởi sự bất ổn định di truyền, suy giảm tăng trưởng, thiểu năng trí tuệ, tăng nguy cơ ung thư, và các đặc điểm của sự lão hóa nhanh. Đây là những tình trạng hiếm gặp, nhưng hội chứng Bloom và thiếu máu Fanconi là hai chẩn đoán phân biệt quan trọng đối với trẻ em sinh ra SGA không cho thấy sự tăng trưởng bắt kịp. Cả hai đều là các rối loạn lặn trên nhiễm sắc thể thường liên quan đến sự khởi phát chậm phát triển và đầu nhỏ trước khi sinh. Hội chứng Bloom thường liên quan đến ban đỏ giãn mao mạch ở giữa mặt theo hình cánh bướm, với sự nhạy cảm với ánh sáng và các dấu hiệu của suy giảm miễn dịch (tức là, tăng nhạy cảm với viêm phổi, giãn phế quản, và bệnh phổi mãn tính). Thiếu máu Fanconi nên bị nghi ngờ khi bệnh nhân có dị tật ngón tay cái (thiểu sản hoặc bất sản), và các dấu hiệu của suy tủy xương (thiếu máu, giảm bạch cầu trung tính, và/hoặc giảm tiểu cầu). Ngoài suy giảm tăng trưởng và BMI thấp, cả hai tình trạng này cũng liên quan đến các bất thường nội tiết khác. Suy sinh dục do tăng gonadotropin thường được quan sát thấy trong cả hai tình trạng. Ngoài ra, bệnh nhân bị thiếu máu Fanconi cũng có nguy cơ mắc bệnh đái tháo đường, rối loạn lipid máu, và rối loạn chức năng tuyến giáp tăng rõ rệt. Chẩn đoán cả hội chứng Bloom và thiếu máu Fanconi có thể được thiết lập thông qua một xét nghiệm tế bào di truyền chuyên biệt cho các bất thường nhiễm sắc thể hoặc bằng xét nghiệm di truyền phân tử cho các biến thể gây bệnh hai alen trong một gen liên quan.
Các dạng lùn nguyên thủy
Lùn nguyên thủy là thuật ngữ cho một nhóm các rối loạn được đặc trưng bởi sự suy giảm tăng trưởng toàn cầu, cực kỳ nghiêm trọng trước và sau sinh, đầu nhỏ rõ rệt, và các mức độ thiểu năng trí tuệ khác nhau. Nó bao gồm các bệnh di truyền lặn hiếm gặp, chẳng hạn như hội chứng Seckel, lùn nguyên thủy loạn sản xương đầu nhỏ (MOPD), và hội chứng Meier-Gorlin. Mỗi hội chứng này đều có các đặc điểm lâm sàng cụ thể và do các khiếm khuyết trong các gen cụ thể gây ra. Các khiếm khuyết di truyền liên quan đến các hội chứng này làm tổn hại đến sự phân chia tế bào và bao gồm các bất thường trong các gen tham gia vào các quá trình tế bào cơ bản. Ví dụ, hội chứng Seckel loại I là do các khiếm khuyết trong gen ATR, một chất điều hòa chính của một chuỗi tín hiệu phản ứng tổn thương DNA, là một chất điều hòa thiết yếu của sự toàn vẹn bộ gen. MOPD loại II là do các khiếm khuyết trong gen pericentrin (PCNT), mã hóa một protein trung tâm của trung thể thiết yếu cho sự hình thành thoi vô sắc trong quá trình nguyên phân.
Các Rối Loạn In Dấu
Các rối loạn in dấu là một nhóm các rối loạn bẩm sinh do các thay đổi phân tử ảnh hưởng đến các vùng và gen nhiễm sắc thể được in dấu. In dấu là một hiện tượng ngoại di truyền trong đó các gen được biểu hiện theo một cách thức đặc hiệu theo nguồn gốc của cha mẹ—hoặc được biểu hiện từ alen của mẹ hoặc của cha, chứ không phải từ cả hai. In dấu bộ gen liên quan đến sự methyl hóa DNA hoặc histone không làm thay đổi mã di truyền. Mặc dù các gen được in dấu có thể trải qua các đột biến ảnh hưởng đến chức năng protein (ví dụ, xem khiếm khuyết IGF2), ngoài ra, sự biểu hiện của chúng có thể bị ảnh hưởng bởi các thay đổi ngoại di truyền, bao gồm các thay đổi trong sự methyl hóa DNA (đột biến ngoại di truyền hoặc epimutation) hoặc sự thừa hưởng cả hai alen từ cùng một cha hoặc mẹ (lưỡng bội đơn thân). SRS, hội chứng Temple, và hội chứng Prader-Willi (PWS) là những ví dụ về các rối loạn tăng trưởng liên quan đến các gen được in dấu và có thể do các đột biến ngoại di truyền gây ra.
Hội chứng Silver-Russell
SRS là một nguyên nhân được công nhận rõ ràng của sự chậm phát triển trước và sau sinh. Bệnh nhân điển hình được sinh ra SGA với chu vi vòng đầu được bảo tồn (đầu to tương đối) và không trải qua sự tăng trưởng bắt kịp tự phát trong cuộc sống sau sinh (SDS chiều cao <–2 sau năm thứ hai của cuộc đời). Ngoài ra, bệnh nhân bị SRS thường có trán dô, khó khăn trong việc ăn uống với chỉ số khối cơ thể thấp, và bất đối xứng cơ thể (Hình 11.8). Các đặc điểm phổ biến khác được quan sát thấy ở bệnh nhân bị SRS là mặt hình tam giác, cằm nhỏ, răng chen chúc hoặc không đều, tật ngón út cong, khối lượng cơ thấp, và đổ mồ hôi nhiều; nam giới cũng có thể biểu hiện lỗ tiểu lệch thấp. Hạ đường huyết cũng có thể là một đặc điểm và cần cho ăn qua đêm.
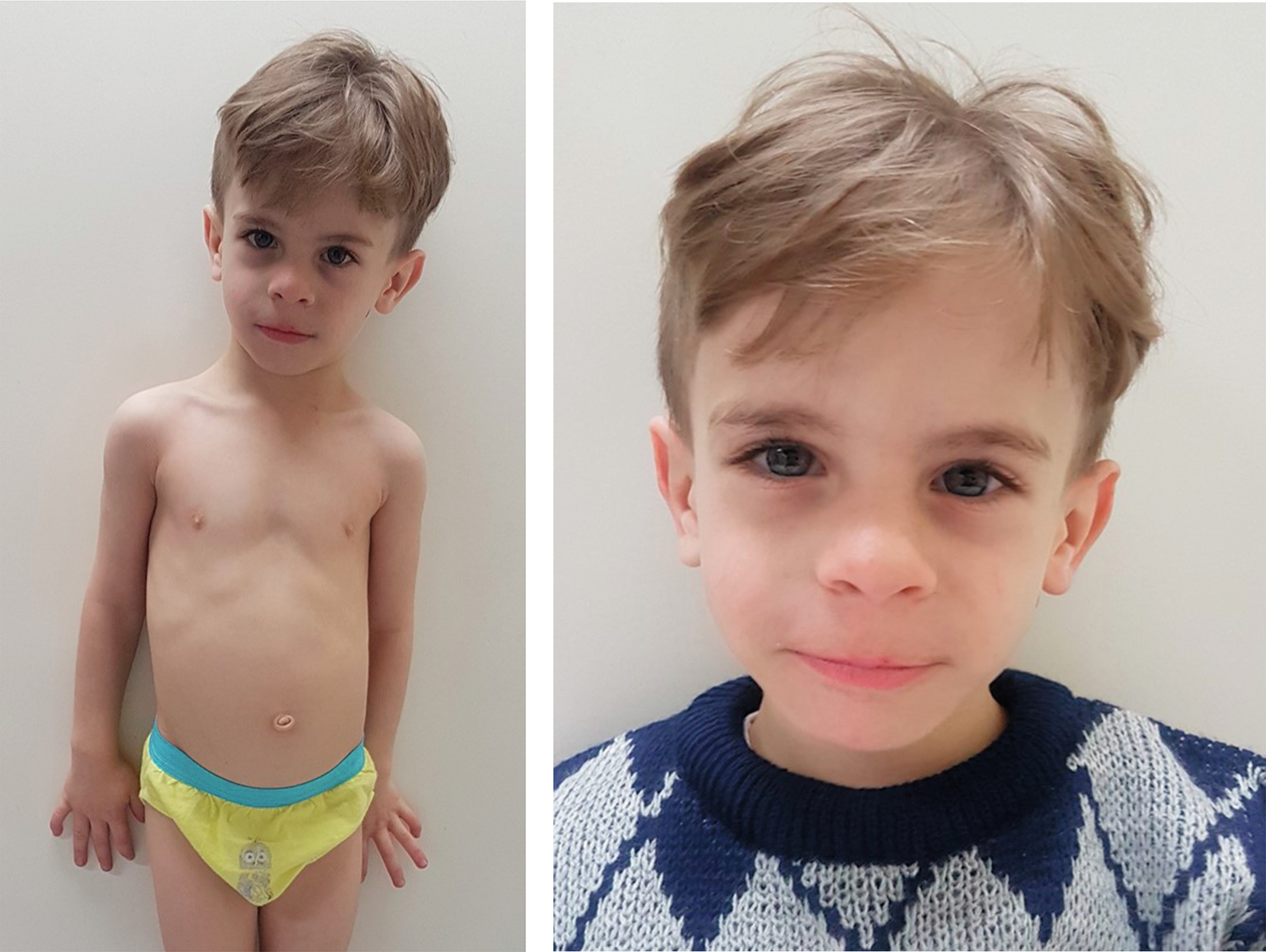
Hình 11.8 Kiểu hình của một bệnh nhân mắc hội chứng Silver-Russell: chỉ số khối cơ thể thấp, mặt hình tam giác, và trán rộng với đầu to tương đối.
SRS thường do các bất thường in dấu liên quan đến vùng nhiễm sắc thể 11p15.5. Locus này chứa gen IGF2, được biểu hiện độc quyền từ alen của cha trong cuộc sống thai nhi ở hầu hết các mô phôi. Sự giảm methyl hóa của vùng được methyl hóa khác biệt H19/IGF2 được xác định ở 40% đến 60% bệnh nhân bị SRS, trong khi chỉ có 1% bệnh nhân mang CNV trong vùng này. Điều thú vị là, bệnh nhân có đột biến ngoại di truyền 11p15.5 thường có các rối loạn in dấu đa locus, mặc dù ý nghĩa lâm sàng của hiện tượng này vẫn chưa rõ ràng. Lưỡng bội đơn thân của mẹ liên quan đến vùng này [UPD(11p)m], mặc dù có thể, nhưng hiếm khi được mô tả. Tất cả các khiếm khuyết phân tử này dẫn đến sự giảm biểu hiện IGF2 của cha, góp phần vào sự hạn chế tăng trưởng. Ít hơn 10% bệnh nhân bị SRS mang UPD của mẹ của một locus được in dấu trên nhiễm sắc thể 7 [UPD(7q)m] chứa hai gen được in dấu: một gen được biểu hiện từ mẹ, GRB10 (7p12.1), và một gen khác được biểu hiện từ cha MEST (7q32).
Có sự chồng chéo rộng rãi trong các kiểu hình lâm sàng liên quan đến các kiểu gen (ngoại) liên quan đến SRS. Bất đối xứng cơ thể, tật ngón út cong, các dị tật bẩm sinh, và suy giảm tăng trưởng trước khi sinh rõ rệt thường được quan sát thấy hơn ở những bệnh nhân bị giảm methyl hóa 11p15.5, trong khi các vấn đề về thần kinh nhận thức thường xuyên hơn ở những bệnh nhân bị UPD(7q)m. Các xét nghiệm phân tử hữu ích để xác nhận chẩn đoán lâm sàng và thường được đánh giá bằng khuếch đại phản ứng chuỗi polymerase qua trung gian ghép nối đa mồi đặc hiệu methyl hóa (MS-MLPA), có hiệu quả về chi phí và cho phép phân tích song song số lượng bản sao và methyl hóa DNA. Phân tích karyotype phân tử (mảng đa hình đơn nucleotide [SNP] hoặc lai so sánh bộ gen [CGH]) nên được thực hiện ở những bệnh nhân mà chẩn đoán SRS được xem xét nhưng mUPD7 và giảm methyl hóa 11p15 đã được loại trừ.
Hội chứng Prader-Willi
PWS là một rối loạn in dấu được đặc trưng bởi giảm trương lực cơ sơ sinh, chậm phát triển trong giai đoạn đầu của trẻ sơ sinh, chậm phát triển toàn bộ, tầm vóc thấp, và béo phì khởi phát nhanh trong thời thơ ấu với khối lượng mỡ cơ thể cao và khối lượng cơ thấp. Bệnh nhân thường cũng có một mức độ nào đó của thiểu năng trí tuệ, các rối loạn hành vi, và bàn tay và bàn chân nhỏ. Bệnh nhân bị PWS có tỷ lệ mắc ngưng thở khi ngủ và đột tử tăng lên. Ngoài ra, PWS có liên quan đến rối loạn chức năng vùng dưới đồi gây suy sinh dục do thiếu gonadotropin, giảm bài tiết GH, và tăng nguy cơ suy thượng thận trung ương.
PWS là do thiếu sự biểu hiện của các gen được di truyền từ cha trong vùng nhiễm sắc thể 15q11.2-q13. Một số gen trong locus này góp phần vào kiểu hình PWS được biểu hiện độc quyền từ alen của cha. Phần lớn bệnh nhân (70%) có một đoạn xóa của alen của cha, trong khi một phần tư có lưỡng bội đơn thân của mẹ [UPD(15q)m]. Các khiếm khuyết in dấu làm im lặng các gen của cha trong locus PWS cũng đã được quan sát thấy ở bệnh nhân PWS nhưng ít phổ biến hơn. Mặc dù một phân tích methyl hóa DNA bằng MS-MLPA phù hợp với PWS là đủ để chẩn đoán, việc xác định cơ chế di truyền cơ bản là cần thiết để tư vấn di truyền phù hợp.
Bằng chứng cho thấy thiếu hụt GH có thể góp phần vào tầm vóc thấp và các bất thường về thành phần cơ thể trong PWS đã thúc đẩy các thử nghiệm lâm sàng về liệu pháp GH cho rối loạn này. Các thử nghiệm này cho thấy rằng việc điều trị bằng GH cho trẻ em bị PWS làm tăng sự tăng trưởng theo chiều dọc, khối lượng cơ nạc, chức năng thể chất, và giảm khối lượng mỡ. Kết quả là, việc điều trị bằng GH cho PWS đã được FDA chấp thuận. Tuy nhiên, chiều cao cuối cùng có thể bị ảnh hưởng do sự xuất hiện tương đối thường xuyên của khởi phát tuyến thượng thận sớm. Bởi vì trẻ em bị PWS có nguy cơ cao bị ngưng thở khi ngủ và đột tử và khả năng GH có thể làm trầm trọng thêm nguy cơ này, việc đánh giá lâm sàng và đa ký giấc ngủ cho rối loạn thở liên quan đến giấc ngủ được khuyến nghị.
Hội chứng Temple
Hội chứng Temple là một rối loạn in dấu được xem xét trong chẩn đoán phân biệt của SRS và PWS do có sự chồng chéo về kiểu hình. Nó chủ yếu do lưỡng bội đơn thân của mẹ của nhiễm sắc thể 14 [UPD(14)m] gây ra nhưng cũng có thể do đột biến ngoại di truyền và/hoặc xóa vi đoạn ảnh hưởng đến một vùng in dấu của cha trên nhiễm sắc thể 14q32.2. Bệnh nhân điển hình mắc hội chứng Temple được sinh ra SGA với đầu to tương đối và không trải qua sự tăng trưởng bắt kịp trong thời thơ ấu. Những cá nhân này thường cho thấy giảm trương lực cơ và khó khăn trong việc ăn uống trong giai đoạn sơ sinh nhưng phát triển béo phì thân mình sau này trong thời thơ ấu. Các đặc điểm khác chồng chéo với SRS hoặc PWS bao gồm trán dô, trán nổi bật, bàn tay và bàn chân nhỏ, và thiểu năng trí tuệ nhẹ. Dậy thì sớm và các bất thường chuyển hóa, chẳng hạn như thừa cân/béo phì, không dung nạp glucose/đái tháo đường loại 2 khởi phát sớm, và tăng lipid máu, là những đặc điểm phân biệt quan trọng của bệnh nhân mắc hội chứng Temple. Các mối liên quan chuyển hóa này được cho là do sự thiếu hụt một nửa của DLK1, một gen được biểu hiện từ cha nằm ở 14q32.2, hoạt động như một chất điều hòa âm của sự biệt hóa tế bào mỡ.
Các bất thường nhiễm sắc thể và biến thể số lượng bản sao
Các bất thường nhiễm sắc thể tạo thành một nguyên nhân đã được xác lập rõ ràng của các rối loạn tăng trưởng được xác định lần đầu tiên sau sự phát triển của các kỹ thuật tế bào di truyền cổ điển, chẳng hạn như karyotyping. Karyotyping cho phép xác định cơ sở phân tử của hội chứng Turner và Down lần lượt là đơn nhiễm sắc thể X hoàn toàn hoặc một phần và tam nhiễm sắc thể 21. Kiến thức có được từ những nghiên cứu tiên phong này chịu trách nhiệm cho việc đưa xét nghiệm di truyền vào việc đánh giá thường quy trẻ em bị rối loạn tăng trưởng. Người ta chấp nhận rằng karyotyping nên được yêu cầu ở bất kỳ bé gái nào bị suy giảm tăng trưởng không giải thích được. Các đặc điểm lâm sàng của hội chứng Turner được mô tả trong Chương 17.
Sau sự phát triển của lai tại chỗ huỳnh quang và các kỹ thuật dựa trên mảng, việc xác định một số lượng ngày càng tăng các sắp xếp lại cấu trúc nhỏ, không cân bằng nằm dưới giới hạn phân giải của kính hiển vi quang học đã trở nên khả thi. Các đoạn xóa và nhân đôi dưới kính hiển vi này liên quan đến một gen duy nhất hoặc có thể liên quan đến một tập hợp các gen ở gần nhau và do đó có thể gây ra các hội chứng gen liền kề. Kiểu hình của bệnh nhân được xác định bởi sự kết hợp của các kiểu hình liên quan đến mỗi gen có mặt trong đoạn nhiễm sắc thể không cân bằng. Có rất nhiều ví dụ về các hội chứng gen liền kề liên quan đến các rối loạn tăng trưởng, chẳng hạn như hội chứng Williams-Beuren, do 7q11.23del gây ra, và hội chứng DiGeorge, do 22q11.2del gây ra. Tỷ lệ hiện mắc của CNV gây bệnh ở trẻ em có tầm vóc thấp không rõ nguyên nhân được báo cáo dao động từ 10,4% đến 15,5%, thường xuyên hơn ở những bệnh nhân bị chậm phát triển, thiểu năng trí tuệ, hoặc các dị tật lớn bổ sung. Tuy nhiên, cả trong các nghiên cứu và trong thực hành lâm sàng, thường khó xác định liệu một CNV cụ thể là gây bệnh hay lành tính và trùng hợp ngẫu nhiên.
Suy giảm tăng trưởng vô căn
Thường thì trẻ em đến khám bác sĩ nội tiết với tầm vóc thấp khởi phát sau sinh, và không thể xác định được nguyên nhân. Trong những tình trạng vô căn này, có những manh mối cho thấy rằng các nguyên nhân cơ bản chưa được biết có khả năng cực kỳ đa dạng. Một số tầm vóc thấp vô căn được di truyền theo một mô hình cho thấy sự di truyền đa gen, trong khi những trường hợp khác cho thấy một mô hình lặn đơn gen hoặc trội đơn gen. Một số tầm vóc thấp là cân đối, ảnh hưởng đến các xương khác nhau trong cơ thể một cách tương tự, trong khi một số là không cân đối, thường với sự phát triển của các chi bị ảnh hưởng nhiều hơn sự phát triển của cột sống. Ở một số trẻ, đứa trẻ có vẻ hoàn toàn khỏe mạnh và bình thường trong khi những đứa trẻ khác có các đặc điểm dị dạng, chậm phát triển, hoặc các vấn đề sức khỏe khác, cho thấy rằng một sự thay đổi di truyền cơ bản có thể ảnh hưởng nhiều hơn là chỉ các tế bào sụn ở sụn tăng trưởng. Ở một số trẻ bị tầm vóc thấp vô căn, cân nặng bị ảnh hưởng nhiều hơn chiều cao, cho thấy rằng sự giảm tăng trưởng theo chiều dọc là thứ phát sau sự giảm dinh dưỡng, có lẽ liên quan đến giảm cảm giác thèm ăn. Ở một số trẻ bị tầm vóc thấp không rõ nguyên nhân, chiều dài cơ thể đã ở dưới mức bình thường khi sinh, cho thấy rằng sự tăng trưởng theo chiều dọc đã bị suy giảm ở thai nhi. Ở những trẻ khác, kích thước khi sinh là bình thường, và tầm vóc thấp phát triển vào một thời điểm nào đó trong thời thơ ấu, cho thấy rằng sự suy giảm tăng trưởng theo chiều dọc chỉ biểu hiện trong cuộc sống sau sinh.
Nguyên nhân có thể là gì? Chúng ta có thể suy đoán rằng nguyên nhân của sự giảm tạo sụn ở sụn tăng trưởng ở những trẻ này có thể thuộc vào cùng các loại như các nguyên nhân đã biết của tầm vóc thấp. Một số có thể là do nội tiết, ví dụ, các tác động của GH hoặc IGF-1 quá tinh vi để có thể được phát hiện bằng các xét nghiệm hiện tại của chúng ta. Những nguyên nhân khác có thể liên quan đến các hệ thống tín hiệu cận tiết/tự tiết tại chỗ, chất nền sụn, hoặc các cơ chế điều hòa nội bào. Một số có thể là các dạng nhẹ của các bệnh loạn sản xương hoặc các hội chứng dị dạng đã biết, nhưng với các biểu hiện quá tinh vi để có thể nhận biết trên lâm sàng. Những nguyên nhân khác có khả năng là do các gen có ảnh hưởng đến quá trình tạo sụn ở sụn tăng trưởng hiện chưa được biết.
Theo các sơ đồ phân loại truyền thống, tầm vóc thấp khởi phát sau sinh và tầm vóc thấp khởi phát trước khi sinh được thảo luận riêng trong các phần sau.
Tầm vóc thấp vô căn khởi phát sau sinh, bao gồm chậm phát triển sinh lý
Ở nhiều trẻ em có tầm vóc thấp khởi phát sau sinh, không thể xác định được nguyên nhân mặc dù đã khai thác bệnh sử cẩn thận, khám thực thể, và các xét nghiệm. Ở một số trẻ này, cả cha mẹ, anh chị em, và các thành viên khác trong gia đình đều thấp, cho thấy sự di truyền đa gen. Có lẽ, những đứa trẻ này đã được di truyền nhiều biến thể trình tự mà mỗi biến thể đều có một ảnh hưởng tiêu cực, nhẹ đến sự tăng trưởng theo chiều dọc. Một số biến thể này có khả năng là các đa hình phổ biến đã được xác định bởi các GWAS gần đây.
Ở những trẻ khác, tầm vóc thấp dường như được di truyền chủ yếu từ một cha hoặc mẹ, và một bệnh sử gia đình cẩn thận cho thấy sự di truyền trội trên nhiễm sắc thể thường. Ở những đứa trẻ này, tầm vóc thấp chủ yếu phát sinh từ một biến thể di truyền duy nhất. Một số nguyên nhân trội, chẳng hạn như các đột biến trong SHOX hoặc NPR2 đã được biết đến, nhưng có khả năng là có nhiều nguyên nhân đơn gen vẫn chưa được khám phá.
Một số trẻ bị tầm vóc thấp cho thấy sự chậm trễ rõ rệt trong sự trưởng thành thể chất, với tuổi xương chậm, khởi phát dậy thì muộn, và sự tiếp tục tăng trưởng theo chiều dọc đến một độ tuổi muộn hơn so với những đứa trẻ khác. Giáo sư James Tanner, một người tiên phong trong lĩnh vực tăng trưởng trẻ em, đã sử dụng thuật ngữ âm nhạc tempo để mô tả nhịp độ trưởng thành tổng thể này, thay đổi giữa các đứa trẻ khác nhau. Thuật ngữ chậm phát triển và dậy thì sinh lý được sử dụng để mô tả những đứa trẻ có nhịp độ chậm. Thuật ngữ này có thể gộp hai tình trạng—chậm phát triển sinh lý có khởi phát lâm sàng ở tuổi dậy thì và chậm phát triển sinh lý có khởi phát lâm sàng ở thời thơ ấu.
Một số trẻ có sự chậm trễ trong khởi phát dậy thì trong đó nguyên nhân chính dường như là sự chậm trễ trong việc kích hoạt HPG. Thường thì có các thành viên khác trong gia đình có sự chậm trễ tương tự trong việc kích hoạt dậy thì và đôi khi có các thành viên trong gia đình bị suy sinh dục do thiếu gonadotropin. Những đứa trẻ này thường phát triển bình thường trong những năm trước tuổi dậy thì. Sau độ tuổi khởi phát dậy thì bình thường, sự chậm trễ tạm thời trong việc bài tiết steroid sinh dục ở những đứa trẻ này làm cho phân vị tăng trưởng theo chiều dọc của chúng giảm xuống. Sự giảm phân vị này một phần là do những đứa trẻ khác trải qua một giai đoạn tăng vọt ở tuổi dậy thì, làm cho các đường cong phân vị bình thường kéo ra xa khỏi đứa trẻ bị chậm. Tuy nhiên, ngay cả khi người ta bỏ qua sự lệch lên của phạm vi bình thường, tốc độ tăng trưởng theo chiều dọc của đứa trẻ bị dậy thì muộn có xu hướng giảm so với tốc độ trước tuổi dậy thì trước đó của nó, có lẽ do sự lão hóa sụn tăng trưởng tiến triển mà không có tác động tăng tốc đối nghịch của steroid sinh dục. Ở những đứa trẻ này, một khi dậy thì bắt đầu ở độ tuổi muộn, một giai đoạn tăng vọt ở tuổi dậy thì sẽ xảy ra. Mặc dù cường độ của giai đoạn tăng vọt ở tuổi dậy thì thường giảm so với một đứa trẻ có khởi phát dậy thì sớm hơn, thời gian tăng trưởng kéo dài tổng thể cho phép có một tầm vóc người lớn bình thường hoặc thậm chí cao.
Ở những trẻ khác bị chậm phát triển sinh lý, sự chậm trễ dường như có khởi phát trước tuổi dậy thì. Những đứa trẻ này thấp trong suốt thời thơ ấu và có tuổi xương chậm ngay cả trước tuổi dậy thì. Một số trẻ này có thể thấp trong thời thơ ấu nhưng, do sự ngừng tăng trưởng muộn ở tuổi vị thành niên, sẽ có chiều cao người lớn bình thường. Ở những trẻ khác có dạng chậm phát triển sinh lý này, tầm vóc người lớn có thể hơi thấp. Ở những đứa trẻ này, một tổn thương chính ảnh hưởng đến trục HPG không thể giải thích cho tình trạng này. Một yếu tố đóng góp có thể có để giải thích sự tăng trưởng giảm, sự chậm trễ tuổi xương, và sự chậm trễ dậy thì là giảm cảm giác thèm ăn.
Tầm vóc thấp không rõ nguyên nhân và khởi phát trước khi sinh
Ở một số trẻ bị tầm vóc thấp, kích thước cơ thể đã bị giảm khi sinh, tức là, đứa trẻ được sinh ra SGA. Các định nghĩa khác nhau về SGA đã được sử dụng. Tuy nhiên, một tuyên bố đồng thuận định nghĩa SGA bằng cân nặng và/hoặc chiều dài nhỏ hơn –2 SDS. Tiền sử được sinh ra SGA cho thấy một quá trình làm giảm sự tăng trưởng của thai nhi do các bất thường của thai nhi, nhau thai, hoặc của mẹ. Các nguyên nhân từ thai nhi bao gồm các rối loạn di truyền (chẳng hạn như các khiếm khuyết gen đơn lẻ, các khiếm khuyết in dấu, các đoạn xóa/nhân đôi, và lệch bội—xem các thảo luận trước đó trong chương), nhiễm trùng thai nhi (chẳng hạn như cytomegalovirus và toxoplasmosis), và đa thai. Các nguyên nhân từ nhau thai bao gồm thể khảm nhau thai giới hạn, nhau bong non, nhau tiền đạo, và các tình trạng khác liên quan đến suy nhau thai-tử cung. Các nguyên nhân từ mẹ bao gồm tiền sản giật; tăng huyết áp; đái tháo đường trước khi mang thai; và các rối loạn hệ thống của mẹ ở thận, tim, phổi, huyết học, và thấp khớp, cũng như việc mẹ sử dụng rượu hoặc thuốc lá. Tuy nhiên, ở nhiều trẻ sơ sinh sinh ra SGA, nguyên nhân cơ bản vẫn chưa được biết.
Nếu trẻ SGA tiếp tục tụt lại xa hơn so với phạm vi bình thường sau sinh, thì nguyên nhân cơ bản có khả năng liên quan đến một quá trình cơ bản ảnh hưởng đến cả sự tăng trưởng của thai nhi và sau sinh, ví dụ, một số bệnh loạn sản sụn. Các rối loạn di truyền gây ra sự suy giảm tăng trưởng rõ rệt cả ở thai nhi và sau sinh được gọi là các dạng lùn nguyên thủy. Thông thường, sự suy giảm tăng trưởng ảnh hưởng đến nhiều mô, bao gồm cả hệ thần kinh trung ương, dẫn đến kích thước đầu tương đối nhỏ. Thường thì, ngoài sự tăng trưởng giảm, còn có các dị tật bẩm sinh. Loại này không đồng nhất và bao gồm các hội chứng di truyền được công nhận, chẳng hạn như hội chứng Seckel, lùn nguyên thủy loạn sản xương đầu nhỏ loại I và II, và hội chứng Meier-Gorlin. Nhiều khiếm khuyết di truyền đã được xác định, liên quan đến các chức năng tế bào cơ bản, ví dụ, sao chép DNA, sửa chữa DNA, cắt nối RNA, và hình thành trung thể. SRS được đặc trưng bởi sự giảm tăng trưởng của thai nhi và sau sinh, có thể ảnh hưởng đến kích thước cơ thể một cách không đối xứng. Một khuôn mặt hình tam giác, trán dô, và các dị tật nhỏ đặc hiệu thường được tìm thấy (xem Suy giảm tăng trưởng nguyên phát—Các Rối Loạn In Dấu trước đó).
Tuy nhiên, ở hầu hết trẻ em sinh ra SGA, tốc độ tăng trưởng sau khi sinh vượt quá mức bình thường, gây ra sự tăng trưởng bắt kịp. Phần lớn sự tăng trưởng bắt kịp này xảy ra trong năm đầu đời và khoảng 90% trẻ SGA đủ tháng đạt được tầm vóc lớn hơn –2 SDS vào lúc 2 tuổi. Tuy nhiên, chiều cao người lớn của những cá nhân sinh ra SGA bị giảm khoảng 1 SDS so với dân số bình thường. Sự tăng trưởng bắt kịp có xu hướng dần dần và ít hoàn toàn hơn ở trẻ SGA sinh non. Đối với trẻ em sinh ra SGA có chiều cao không bắt kịp vào phạm vi bình thường, việc điều trị bằng GH tái tổ hợp làm tăng sự tăng trưởng theo chiều dọc và được chấp thuận cho việc sử dụng này ở cả Hoa Kỳ và Châu Âu.
Có bằng chứng cho thấy những cá nhân sinh ra SGA có xu hướng kháng insulin và béo phì nhiều hơn những người sinh ra với kích thước bình thường. Các tác động dường như lớn hơn ở những người là SGA nhưng tăng cân nhanh chóng trong 3 tháng đầu đời. Cũng có bằng chứng cho thấy các bé gái sinh ra SGA có xu hướng có kinh sớm hơn, mặc dù thường vẫn trong phạm vi bình thường, và cũng có nhiều khả năng có khởi phát tuyến thượng thận sớm, mặc dù không phải tất cả các nghiên cứu đều ủng hộ những kết luận này.
Phương pháp tiếp cận chẩn đoán bệnh nhân có tầm vóc thấp
Mặc dù bản thân tầm vóc thấp không phải là một bệnh, nhưng suy giảm tăng trưởng có thể là một dấu hiệu nhạy cảm của một vấn đề sức khỏe tiềm ẩn. Việc đánh giá chẩn đoán trước tiên nên cố gắng phân biệt giữa các biến thể khỏe mạnh và các mô hình tăng trưởng bệnh lý và, đối với loại sau, xác định nguyên nhân cơ bản và điều trị chúng một cách phù hợp.
Diễn biến theo thời gian
Sự khởi phát của sự chững lại tăng trưởng có thể là một manh mối rất hữu ích để thu hẹp chẩn đoán phân biệt. Để xác định diễn biến theo thời gian một cách chính xác, bác sĩ lâm sàng phải có sẵn hồ sơ tăng trưởng của trẻ, cả về chiều dài/chiều cao và cân nặng, tốt nhất là bắt đầu từ khi sinh. Bởi vì những hồ sơ này có giá trị lớn đối với việc đánh giá chẩn đoán, cha mẹ nên được khuyến khích để có được một bộ hoàn chỉnh. Nếu dữ liệu chưa được vẽ trên các biểu đồ tăng trưởng chiều dài/chiều cao và cân nặng phù hợp, bác sĩ lâm sàng nên làm như vậy. Bác sĩ lâm sàng sau đó đánh giá trực quan mô hình tăng trưởng tổng thể để xác định khi nào tốc độ tăng trưởng dưới mức bình thường, tức là, khi chiều cao hoặc cân nặng của trẻ đang vượt qua các phân vị xuống dưới. Thường thì đường cong tăng trưởng sẽ cho thấy một số sự bất thường do sai số đo lường, và bác sĩ lâm sàng có kinh nghiệm học cách làm mịn dữ liệu trong mắt mình.
Nếu chiều cao và/hoặc cân nặng của trẻ dưới mức bình thường khi sinh (SGA), điều này ngụ ý rằng tốc độ tăng trưởng dưới mức bình thường trong tử cung và do đó một tình trạng cản trở sự tăng trưởng đã có mặt trong cuộc sống thai nhi. Nếu các đường cong tăng trưởng sau đó cho thấy sự phục hồi sau khi sinh, mô hình tổng thể cho thấy một tình trạng đã có mặt trong tử cung nhưng sau đó đã được giải quyết, chẳng hạn như một rối loạn của mẹ hoặc của nhau thai. Ngược lại, nếu trẻ SGA tiếp tục cho thấy sự tăng trưởng chậm sau sinh, mô hình cho thấy một vấn đề tăng trưởng nội tại, chẳng hạn như một hội chứng di truyền hoặc loạn sản xương. Tương tự, các khiếm khuyết trong hoạt động của IGF-1, bao gồm các khiếm khuyết gen IGF-1, các khiếm khuyết gen IGF1R, cũng như các khiếm khuyết gen IGF-2 và SRS, gây ra suy giảm tăng trưởng kết hợp trước và sau sinh. Nếu đứa trẻ được sinh ra với kích thước bình thường nhưng sau đó phát triển chậm trong giai đoạn đầu của cuộc sống sau sinh, bác sĩ lâm sàng nên nghi ngờ một rối loạn bẩm sinh ảnh hưởng đến sự tăng trưởng sau sinh nhưng không phải của thai nhi, bao gồm thiếu hụt GH bẩm sinh hoặc hội chứng không nhạy cảm với GH. Sự khởi phát của sự chững lại tăng trưởng trong thời thơ ấu cho thấy một vấn đề mắc phải, ví dụ, IBD, một khối u gây ra rối loạn chức năng tuyến yên, hoặc một loại thuốc cản trở sự tăng trưởng. Bác sĩ lâm sàng tinh ý đôi khi có thể tìm thấy một sự tương ứng giữa sự khởi phát của sự tăng trưởng chậm trên biểu đồ tăng trưởng và một sự kiện nào đó trong bệnh sử của đứa trẻ, chẳng hạn như bắt đầu một loại thuốc ức chế tăng trưởng hoặc sự khởi phát của một căn bệnh. Một sự tương ứng về thời gian như vậy cho thấy một mối quan hệ nhân quả. Sự khởi phát của sự chậm tăng trưởng trong tuổi vị thành niên cũng có thể là kết quả của dậy thì muộn.
Kiểu di truyền
Một bệnh sử gia đình cẩn thận, lý tưởng nhất là bao gồm chiều cao của cha mẹ, anh chị em, ông bà, cô, chú, và anh chị em họ, có thể cung cấp nhiều thông tin. Theo truyền thống, bệnh sử gia đình được sử dụng để phân loại tầm vóc thấp là có tính gia đình hoặc không có tính gia đình. Tuy nhiên, sự phân loại này gây nhầm lẫn về nguyên nhân di truyền cơ bản. Tầm vóc thấp gia đình gộp chung tầm vóc thấp đơn gen (chủ yếu do một đột biến trong một gen duy nhất, hoặc trội hoặc lặn) và tầm vóc thấp thiểu/đa gen. Tầm vóc thấp không có tính gia đình, mặc dù có nhãn hiệu như vậy, vẫn có thể là do di truyền (xem sau).
Do đó, sẽ hữu ích hơn khi sử dụng phả hệ để giúp phân biệt các nguyên nhân đơn gen (chủ yếu là trội trên nhiễm sắc thể thường và lặn trên nhiễm sắc thể thường) của tầm vóc thấp với các nguyên nhân thiểu/đa gen. Trong một số trường hợp, tầm vóc thấp dường như được di truyền từ một người cha hoặc mẹ bị ảnh hưởng và một người ông hoặc bà bị ảnh hưởng, cho thấy một khiếm khuyết di truyền trội, ví dụ thiếu hụt SHOX hoặc một đột biến NPR2 dị hợp tử. Ít thường xuyên hơn, tầm vóc thấp xảy ra ở hai anh chị em bị ảnh hưởng có cha mẹ không bị ảnh hưởng, cho thấy một rối loạn lặn, chẳng hạn như hội chứng không nhạy cảm với GH hoặc hội chứng 3-M. Trong các trường hợp khác, đứa trẻ thấp một cách bất thường so với tất cả các thành viên khác trong gia đình. Tầm vóc thấp lẻ tẻ này có thể do một vấn đề mắc phải hoặc do một khiếm khuyết di truyền là lặn trên nhiễm sắc thể thường, trội de novo, hoặc (nếu bệnh nhân là nam) lặn liên kết X. Nếu cha mẹ có tầm vóc trung bình thực sự hoặc thậm chí cao, tình trạng này ít có khả năng là đa gen. Ở các gia đình khác, nhiều thành viên trong gia đình thấp, theo một mô hình không thể được giải thích bằng một sự di truyền đơn gen, cho thấy một sự di truyền thiểu gen hoặc đa gen. Các tình trạng này thường lành tính.
Trong thực tế, phả hệ có thể không phân biệt rõ ràng giữa một sự di truyền đơn gen và đa gen, một phần do sự giao phối chọn lọc, tức là, xu hướng của những cá nhân thấp kết hôn với những cá nhân thấp khác. Kết quả là, các gia đình có thể có các thành phần của cả tầm vóc thấp đơn gen và thiểu/đa gen. Khi có sự chênh lệch lớn về chiều cao của cha mẹ, một đứa trẻ có thể giống với kênh chiều cao của một trong hai cha mẹ thay vì mức trung bình được tính toán, và với sự phân ly ngẫu nhiên của các gen ở mỗi lần thụ thai, không thể dự đoán trước được đứa trẻ sẽ giống với cha hay mẹ.
Bệnh sử và khám thực thể cẩn thận
Với chẩn đoán phân biệt rất rộng, một bệnh sử cẩn thận và toàn diện là cực kỳ quan trọng. Bệnh sử đa thế hệ về chiều cao người lớn và thời điểm dậy thì, cũng như các bệnh di truyền trong gia đình, là cần thiết để tạo tiền đề. Bệnh sử gia đình nên xác định các thành viên trong gia đình bị dậy thì muộn, điều này sẽ gợi ý đến chậm phát triển và dậy thì sinh lý, cũng như những người bị dậy thì sớm, cũng có thể là một yếu tố di truyền ảnh hưởng đến mô hình tăng trưởng của trẻ. Ngoài ra, bác sĩ lâm sàng nên hỏi về bệnh sử gia đình của các bệnh lý có thành phần di truyền và có thể ảnh hưởng đến sự tăng trưởng, chẳng hạn như suy giáp.
Bệnh sử trước khi sinh và khi sinh, đặc biệt là cân nặng và chiều dài khi sinh và tuổi thai, giúp xác định các tình trạng ảnh hưởng đến sự tăng trưởng của thai nhi. Việc hỏi bệnh chi tiết nên được sử dụng để sàng lọc các triệu chứng tinh vi của các bệnh hệ thống, ví dụ, bệnh đường tiêu hóa, các khối u hệ thần kinh trung ương, thiếu hụt GH hoặc các bệnh tuyến yên khác, suy giáp, hội chứng Cushing, và các rối loạn ăn uống. Cần xác định các loại thuốc đã dùng. Bệnh sử vàng da hoặc hạ đường huyết sơ sinh, các dị tật hệ thần kinh trung ương (đặc biệt là SOD và holoprosencephaly), bệnh sử chấn thương đầu hoặc tổn thương thần kinh, và bệnh sử tân sản, đặc biệt khi được điều trị bằng xạ trị sọ não, nên làm tăng nghi ngờ về khả năng thiếu hụt GH. Đối với thanh thiếu niên, tuổi có kinh, tuổi dậy thì, tuổi có kinh nguyệt, và thay đổi giọng nói, nếu phù hợp, là cần thiết để đặt mô hình tăng trưởng trong bối cảnh thời điểm dậy thì. Bệnh sử xã hội và dinh dưỡng đôi khi giúp đưa ra chẩn đoán.
Khám thực thể đặc biệt phục vụ cho việc sàng lọc các hội chứng dị dạng và các bệnh hệ thống. Bác sĩ lâm sàng nên tìm kiếm cẩn thận các đặc điểm dị dạng có thể chỉ ra một hội chứng, chẳng hạn như hội chứng Silver-Russell, Turner, Noonan, Down, Prader-Willi, hoặc Williams, hoặc thiếu hụt một nửa SHOX. Khám thực thể toàn diện nên bao gồm soi đáy mắt, kiểm tra thị trường bằng phương pháp đối đầu, và sờ nắn tuyến giáp. Thiếu hụt GH có thể liên quan đến các khiếm khuyết đường giữa (đặc biệt là răng cửa giữa của hàm trên), rung giật nhãn cầu (dấu hiệu khởi phát của SOD), và phù gai thị, và ở các bé trai, dương vật nhỏ, thiểu sản giữa mặt, béo phì bụng, và giọng nói cao. Tương tự, nên tìm kiếm các dấu hiệu của suy giáp.
Đánh giá tuổi răng có thể cung cấp cái nhìn sâu sắc về sự trưởng thành xương của trẻ. Có sự thay đổi lớn về thời gian mọc răng sữa và răng vĩnh viễn, có thể bị ảnh hưởng bởi các yếu tố tại chỗ và môi trường, chẳng hạn như kích thước hàm, vị trí của các răng chưa mọc, và mất răng sữa sớm. Trẻ em bị thiếu hụt GH hoặc suy giáp không được điều trị thường có mọc răng chậm đáng kể hoặc răng bất thường (thiếu răng, thường là các răng cửa hàm trên). Sự chậm trễ nhẹ trong tiến triển của răng có thể xảy ra trong chậm phát triển sinh lý. Sự chậm mọc răng thường liên quan đến sự đóng thóp chậm và tuổi xương chậm.
Đo lường nhân trắc học
Các phép đo nhân trắc học chính xác nên được thực hiện (xem phần Đo lường trước đó trong chương này) bao gồm chiều cao, cân nặng, chu vi vòng đầu, sải tay, và chiều cao ngồi hoặc phân đoạn dưới. Tỷ lệ cơ thể bất thường có thể là một manh mối có giá trị cho việc chẩn đoán một bệnh loạn sản sụn. Nhiều bệnh loạn sản sụn ảnh hưởng đến các sụn tăng trưởng của các chi nhiều hơn là của các đốt sống, dẫn đến một chỉ số chiều cao ngồi tăng, tỷ lệ phân đoạn trên-dưới tăng, và sải tay ngắn so với chiều cao. Ngược lại, một thân mình ngắn không cân đối có thể phản ánh các tình trạng ảnh hưởng xấu đến các đốt sống, chẳng hạn như vẹo cột sống, xạ trị cột sống, hoặc một số bệnh loạn sản sụn nhất định. Khi nghi ngờ có tầm vóc thấp không cân đối, nên thực hiện thêm các phép đo các đoạn chi khác nhau để phân biệt các tình trạng rhizomelic, mesomelic, và acromelic. Chẩn đoán một bệnh loạn sản sụn cụ thể thường có thể được thực hiện bằng cách giải thích chuyên môn của các phim X-quang xương được bổ sung bằng các xét nghiệm sinh hóa và di truyền cụ thể nhắm vào các đột biến gây bệnh cụ thể.
Sự tăng cân tương đối so với sự tăng chiều cao là quan trọng để xem xét ở mọi trẻ em đang được đánh giá tăng trưởng, ngay cả những trẻ không có bất kỳ triệu chứng tiêu hóa nào, để loại trừ một nguyên nhân dinh dưỡng của sự chững lại tăng trưởng. BMI (Hộp 11.3) được tính cho trẻ em trên 2 tuổi, và cân nặng theo chiều dài có thể được sử dụng từ sơ sinh đến 5 tuổi; cả hai đều được vẽ trên các biểu đồ tăng trưởng tương ứng của chúng (có sẵn tại https://www.cdc.gov/growthcharts/cdc_charts.htm). Mặc dù BMI hoặc cân nặng theo chiều dài “dưới các đường cong” là một dấu hiệu rõ ràng của suy dinh dưỡng nghiêm trọng, sự chững lại tăng trưởng có thể là kết quả của các trường hợp suy dinh dưỡng nhẹ hơn vẽ trong các phạm vi “bình thường”. Sự sụt giảm qua các phân vị chính của BMI hoặc cân nặng theo chiều dài và/hoặc sự sụt giảm phân vị cân nặng theo tuổi trước khi sụt giảm phân vị chiều cao theo tuổi nên cảnh báo về một vấn đề dinh dưỡng tiềm ẩn có thể có. Ngược lại, khi chiều cao bị ảnh hưởng tương tự như cân nặng hoặc nhiều hơn cân nặng (dẫn đến BMI bình thường đến tăng nhẹ), người ta nên nghi ngờ các rối loạn trong đó các sụn tăng trưởng bị ảnh hưởng nhiều hơn sự phát triển của các mô khác, ví dụ, thiếu hụt GH, suy giáp, và các bệnh loạn sản sụn. Khi phân vị chiều cao giảm nhưng phân vị cân nặng tăng, nên xem xét tình trạng thừa glucocorticoid.
| Hộp 11.3 Chỉ số khối cơ thể
|

Suy dinh dưỡng trước khi sinh có thể dẫn đến một kích thước khi sinh được đặc trưng bởi SGA hoặc IUGR. Trẻ sơ sinh tiếp xúc với suy dinh dưỡng thai nhi sớm có cân nặng khi sinh thấp và nhỏ đối xứng (IUGR cân đối). Suy dinh dưỡng vào cuối thai kỳ dẫn đến một đứa trẻ bị hạn chế tăng trưởng không đối xứng có chu vi vòng đầu được bảo tồn bởi một sự thích ứng sinh lý (hiện tượng bảo vệ não).
Tuổi xương
Mặc dù tuổi xương hữu ích trong việc đánh giá tiềm năng tăng trưởng của bệnh nhân, nó chỉ cung cấp thông tin hạn chế về nguyên nhân cơ bản của tầm vóc thấp. Tuổi xương có thể bị trì hoãn do chậm phát triển sinh lý, nhiều bệnh hệ thống hoặc nội tiết làm suy giảm sự tăng trưởng, điều trị glucocorticoid mãn tính, hoặc suy dinh dưỡng. Tuổi xương phù hợp với tuổi theo thời gian có thể xảy ra trong tầm vóc thấp gia đình đa gen, một số nguyên nhân đơn gen của tầm vóc thấp đơn độc, một số nguyên nhân hội chứng, và một số bệnh loạn sản sụn. Tuổi xương cao, hiếm khi xảy ra ở trẻ em có tầm vóc thấp, chỉ ra các nguyên nhân di truyền cụ thể, chẳng hạn như các đột biến ACAN dị hợp tử, AHO, và thiếu hụt SHOX.
Các xét nghiệm sàng lọc
Không phải tất cả trẻ em có tầm vóc thấp đều cần một cuộc điều tra xét nghiệm sàng lọc. Ví dụ, ở một đứa trẻ đã phát triển đều đặn ngay dưới và song song với giới hạn dưới của tầm vóc bình thường, có vẻ khỏe mạnh trong bệnh sử và khám thực thể, và có một phả hệ cho thấy tầm vóc thấp đa gen, hiệu suất của việc điều tra xét nghiệm sẽ rất thấp. Một ví dụ thứ hai sẽ là một đứa trẻ có một tình trạng, chẳng hạn như một hội chứng di truyền, giải thích rõ ràng tầm vóc thấp. Tuy nhiên, khi mô hình tăng trưởng, bệnh sử, khám thực thể, hoặc phả hệ đáng lo ngại về một nguyên nhân bệnh lý tiềm ẩn (xem các phần về Diễn biến theo thời gian, Kiểu di truyền, Bệnh sử và khám thực thể cẩn thận, và Đo lường nhân trắc học), thì một cuộc điều tra xét nghiệm có thể được bảo đảm.
Các xét nghiệm sàng lọc được khuyến nghị có thể bao gồm công thức máu toàn phần, tốc độ lắng hồng cầu, creatinine, glucose, điện giải, bicarbonate, các xét nghiệm chức năng gan, canxi, phosphate, albumin, sàng lọc bệnh celiac, FT4, TSH, IGF-1 và, ở trẻ nhỏ hơn, IGFBP-3. Nên thực hiện karyotype hoặc microarray ở các bé gái có tầm vóc thấp không giải thích được để loại trừ chẩn đoán hội chứng Turner và ở các bé trai thấp có các bất thường bộ phận sinh dục liên quan. Nên chụp X-quang bàn tay và cổ tay trái để đánh giá tuổi xương bởi một chuyên gia để ước tính tiềm năng tăng trưởng còn lại. Các dị tật trên phim X-quang này đôi khi có thể chỉ ra một bệnh loạn sản xương. Tuổi xương cao ở một đứa trẻ có tầm vóc thấp cho thấy các nguyên nhân di truyền cụ thể, chẳng hạn như các đột biến ACAN. Nên xem xét một cuộc khảo sát xương ở những trẻ bị nghi ngờ có bệnh loạn sản xương, chẳng hạn như những trẻ có sự mất cân đối hoặc những trẻ có tiền sử gia đình về tầm vóc thấp không cân đối.
Có bằng chứng cho thấy rằng các phương pháp sàng lọc xét nghiệm như vậy có hiệu suất thấp trong một quần thể trẻ em dưới phân vị thứ ba về chiều cao, bệnh sử âm tính, tổng quan hệ thống, và khám thực thể bình thường, một lần nữa nhấn mạnh rằng các xét nghiệm này có thể được bỏ qua một cách hợp lý ở một số trẻ, như đã thảo luận trước đó.
Đo nồng độ Yếu tố tăng trưởng giống Insulin-1 trong các rối loạn tăng trưởng
Nồng độ IGF-1 trong huyết thanh bị ảnh hưởng lớn bởi tuổi theo thời gian, mức độ trưởng thành sinh dục, và tình trạng dinh dưỡng. Do đó, việc sử dụng các giá trị chuẩn dựa trên cả tuổi và giai đoạn dậy thì là rất quan trọng. Ví dụ, việc sử dụng một phạm vi bình thường cho trẻ em Tanner I là có vấn đề vì nồng độ IGF-1 bình thường tăng đáng kể theo tuổi ngay cả trước tuổi dậy thì. Ngược lại, việc sử dụng phạm vi bình thường cho các bé trai 14 tuổi làm dữ liệu chuẩn cho một bệnh nhân trước tuổi dậy thì ở độ tuổi đó cũng có vấn đề vì bệnh nhân sẽ có nồng độ IGF-1 thấp hơn do steroid sinh dục thấp hơn. Do đó, một phương pháp tiếp cận thực tế là sử dụng dữ liệu chuẩn đặc hiệu theo tuổi cho trẻ em ở độ tuổi trước dậy thì và sử dụng dữ liệu chuẩn đặc hiệu theo giai đoạn dậy thì cho trẻ em ở độ tuổi dậy thì.
Nồng độ IGF-1 trong huyết thanh thường được sử dụng như một xét nghiệm sàng lọc cho thiếu hụt GH. Việc sử dụng này dựa trên quan sát sinh lý đã được xác lập rõ ràng rằng GH kích thích nồng độ IGF-1 lưu hành. Do đó, nồng độ IGF-1, trung bình, thấp hơn ở trẻ em bị thiếu hụt GH so với trẻ em bình thường hoặc trẻ em có tầm vóc thấp không do thiếu hụt GH. Tuy nhiên, có sự chồng chéo rộng rãi giữa nồng độ IGF-1 trong huyết thanh ở trẻ em thiếu hụt GH và không thiếu hụt cả vì trẻ em thiếu hụt GH có thể có IGF-1 trong huyết thanh trong phần dưới của phạm vi bình thường và vì trẻ em không thiếu hụt GH có tầm vóc thấp có thể có IGF-1 trong huyết thanh dưới phạm vi bình thường. Do đó, IGF-1 trong huyết thanh dường như cho thấy cả độ nhạy và độ đặc hiệu không hoàn toàn cho thiếu hụt GH. Tuy nhiên, các nghiên cứu trong lĩnh vực này rất phức tạp để giải thích vì chúng ta không có một xét nghiệm tiêu chuẩn vàng cho thiếu hụt GH, mà thay vào đó dựa vào các nghiệm pháp GH kích thích, cũng có vấn đề (xem phần Các xét nghiệm và hình ảnh học theo dõi sau). Về mặt thực tế, nồng độ IGF-1 trong huyết thanh có thể giúp định hướng đánh giá về phía hoặc ra khỏi thiếu hụt GH nhưng nên được giải thích một cách thận trọng. Nồng độ IGF-1 thấp cũng có thể được thấy trong các tình trạng khác, ví dụ, suy dinh dưỡng, không nhạy cảm với GH, các đột biến IGF1, và thiếu hụt ALS. Nồng độ IGF-1 cao có thể xảy ra trong một số nguyên nhân của tầm vóc thấp, ví dụ, các đột biến IGF1R, trong đó nồng độ IGF-1 có xu hướng cao hơn mức trung bình theo tuổi trừ khi bệnh nhân bị suy dinh dưỡng, và các đột biến PAPPA2.
Bởi vì GH không chỉ kích thích IGF-1 mà còn cả sản xuất IGFBP-3, IGFBP-3 trong huyết thanh cũng có thể được sử dụng tương tự như một xét nghiệm sàng lọc cho thiếu hụt GH. Blum và các cộng sự đã đề nghị rằng việc xác định nồng độ IGFBP-3 trong huyết thanh bằng phương pháp xét nghiệm miễn dịch phóng xạ có thể đặc hiệu hơn (nhưng ít nhạy hơn) so với các xét nghiệm IGF-1 trong chẩn đoán thiếu hụt GH và có thể hữu ích ở trẻ nhỏ mà nồng độ IGF-1 bình thường thấp và do đó khó phân biệt với thiếu hụt GH.
Các xét nghiệm và hình ảnh học theo dõi
Sau khi các xét nghiệm sàng lọc (lấy máu ngẫu nhiên) thu hẹp chẩn đoán phân biệt, có thể cần đến các xét nghiệm chức năng theo dõi. Các xét nghiệm chức năng được thực hiện phổ biến nhất là các nghiệm pháp GH kích thích để chẩn đoán thiếu hụt GH. Do sự bài tiết theo xung của nó trong giấc ngủ sâu, các mức GH ngẫu nhiên thường không cung cấp thông tin ngoại trừ trong giai đoạn sơ sinh, trước khi sự đồng bộ hóa giấc ngủ phát triển. Việc đo nồng độ IGF-1 và IGFBP-3 lưu hành có thể hữu ích nhưng thường không mang tính quyết định, như đã thảo luận trước đó. Do đó, một nhóm các nghiệm pháp kích thích đã được phát triển trong đó nồng độ GH được đo nối tiếp qua một ống thông tĩnh mạch đặt trong sau khi dùng các tác nhân dược lý được xác định là chất kích thích tiết GH (Bảng 11.5). Các xét nghiệm này nên được thực hiện khi đói và suy giáp, nếu có, nên được điều chỉnh trước khi xét nghiệm. Do tần suất cao của các kết quả “thất bại” giả trên bất kỳ nghiệm pháp kích thích nào, một tiêu chuẩn thông thường đã được phát triển rằng chẩn đoán thiếu hụt GH đòi hỏi nồng độ GH đỉnh không đủ trên hai nghiệm pháp GH kích thích khác nhau (có thể được thực hiện nối tiếp trong cùng một ngày). Ngưỡng GH chẩn đoán được chấp nhận chung là 10 mcg/L.
Bảng 11.5 Các Nghiệm Pháp Kích Thích Hormone Tăng Trưởng Được Sử Dụng Để Chẩn Đoán Thiếu Hụt Hormone Tăng Trưởng Từ Ranke MB. Diagnosis of GH Deficiency and GH Stimulation Tests. Trong: Ranke MB, ed. Diagnostics of Endocrine Function in Children and Adolescents. Tái bản lần 3. Basel, Thụy Sĩ: Karger; 2007:107–128. Bảng 3.
| Kích thích | Liều lượng và quy trình | Lấy mẫu GH, phút |
|---|---|---|
| Tập thể dục | Leo cầu thang cưỡng bức, Xe đạp ergometer, 2 W/kg BW (10 phút) | 0, 15–20 sau khi bắt đầu |
| Arginine HCl | 0.5 g/kg BW (tối đa, 30 g) (truyền IV 10% arginine HCl trong 0.9% NaCl với tốc độ không đổi trong 30 phút) | − 30, 0, 15, 30, 45, 60, 90, 120 |
| Ornithine HCl | 12g/m2 BSA (truyền IV 6.25% ornithine HCl trong 0.9% NaCl với tốc độ không đổi trong 30 phút) | − 30, 0, 15, 30, 45, 60, 90, 120 |
| Insulin (regular) | 0.1–0.05 IU/kg BW IV | − 30, 0, 15, 30, 45, 60, 90, 120 |
| Arginine HCl-insulin | Xem nghiệm pháp arginine (insulin được tiêm [xem ở trên] 60 phút sau khi dùng arginine) | − 30, 0, 15, 30, 45, 60, 80, 90, 105, 120, 150 |
| Clonidine (2-[2,6-dichlorphenyl]amino-2-imidazoline) | 0.15 mg/m2 BSA uống | − 30, 0, 30, 60, 90, 120, 150 |
| Glucagon | 0.03 mg/kg BW (IM hoặc SC) (có thể kết hợp với propranolol, 0.75 mg/kg BW uống 2 giờ trước glucagon) | 0, 60, 90, 120, 150, 180 |
| L-Dopa (L-3,4-dihydroxyphenylalanine) | < 15 kg BW: 125 mg, < 35 kg BW: 250 mg, > 35 kg BW: 500 mg (uống) | − 30, 0, 30, 60, 90, 120 |
| GHRH (1–40), (1–44), (1–29)NH2 | 1 g/kg BW tiêm tĩnh mạch bolus | − 30, 0, 15, 30, 45, 60, 90, 120 |
BSA, Diện tích bề mặt cơ thể; BW, trọng lượng cơ thể; GHRH, hormone giải phóng GH. IM, tiêm bắp; IV, tiêm tĩnh mạch; SC, tiêm dưới da.
Thật không may, ngay cả sự kết hợp của hai nghiệm pháp GH kích thích, như thường được thực hiện, cũng có độ đặc hiệu kém. Marin và các đồng nghiệp đã thực hiện xét nghiệm GH kích thích (tập thể dục và arginine-insulin) trên trẻ em bình thường và thấy rằng phần lớn trẻ em khỏe mạnh trước tuổi dậy thì có phản ứng GH đỉnh dưới 10 mcg/L trên cả ba xét nghiệm. Giới hạn dưới của phạm vi bình thường là khoảng 2 mcg/L. Những dữ liệu này cho thấy rằng xét nghiệm kích thích GH dẫn đến một số lượng rất lớn các chẩn đoán dương tính giả. Độ đặc hiệu của xét nghiệm được cải thiện với giai đoạn dậy thì Tanner tăng lên. Cũng có bằng chứng cho thấy phản ứng GH và do đó độ đặc hiệu của xét nghiệm giảm theo BMI, ngay cả trong phạm vi BMI bình thường và trẻ em béo phì có nồng độ GH đặc biệt thấp. Chẩn đoán quá mức thiếu hụt GH có những hậu quả bất lợi tiềm tàng, bao gồm các tác động tâm lý của việc gán nhãn sai cho một đứa trẻ là mắc bệnh, việc kích hoạt các xét nghiệm bổ sung, chẳng hạn như MRI tuyến yên và các xét nghiệm chức năng tuyến yên, sự thất bại trong việc theo đuổi các xét nghiệm khác để tìm ra nguyên nhân thực sự của suy giảm tăng trưởng, và lấy đi khả năng của gia đình để đưa ra quyết định sáng suốt về việc có nên sử dụng điều trị bằng GH ở một đứa trẻ có tầm vóc thấp không do thiếu hụt GH hay không. Mối quan tâm sau có thể được giải quyết một phần bằng cách thông báo cho gia đình về tỷ lệ cao của các xét nghiệm kích thích dương tính giả. Độ đặc hiệu kém của các nghiệm pháp GH kích thích ở trẻ em cũng có thể giải thích tại sao thiếu hụt GH ở trẻ em đơn độc thường không được xác nhận khi bệnh nhân được kiểm tra lại khi còn trẻ.
Việc mồi steroid sinh dục trước khi xét nghiệm GH kích thích làm tăng khả năng của các chất kích thích tiết để gây ra một phản ứng GH và cải thiện độ đặc hiệu của xét nghiệm. Trong nghiên cứu của Marin và các đồng nghiệp được trích dẫn trước đó, khi trẻ em trước tuổi dậy thì được mồi estrogen trước khi xét nghiệm GH kích thích và khi một ngưỡng chẩn đoán là 7 mcg/L được sử dụng, thì nghiệm pháp GH kích thích cho thấy độ đặc hiệu chấp nhận được. Các nghiên cứu tương tự cho thấy rằng việc mồi estrogen cải thiện độ đặc hiệu trong khi vẫn giữ được độ nhạy đầy đủ, nhưng độ nhạy của xét nghiệm rất khó đánh giá, đặc biệt đối với thiếu hụt GH một phần, vì chúng ta thiếu một tiêu chuẩn vàng cho chẩn đoán để các xét nghiệm khác có thể được so sánh. Trong các nghiên cứu ủng hộ việc mồi estrogen, việc mồi đã được sử dụng ngay cả ở trẻ nhỏ; mặc dù nồng độ estrogen cao là không sinh lý ở độ tuổi trẻ, estrogen đã phục vụ như một chất kích thích dược lý bổ trợ hữu ích. Mặc dù có những dữ liệu này, xét nghiệm kích thích GH thường được thực hiện mà không cần mồi steroid sinh dục.
Do những hạn chế đáng kể của các nghiệm pháp GH kích thích, Hướng dẫn của Hội Nội tiết Nhi khoa về điều trị bằng GH đã khuyến nghị “chống lại việc dựa vào kết quả nghiệm pháp GH kích thích như là tiêu chí chẩn đoán duy nhất của thiếu hụt GH,” mà thay vào đó đề nghị rằng kết quả xét nghiệm nên được giải thích trong bối cảnh mô hình tăng trưởng của bệnh nhân, các yếu tố nguy cơ thiếu hụt GH, và các kết quả xét nghiệm chẩn đoán khác, chẳng hạn như nồng độ IGF-1 và IGFBP-3, tuổi xương, và hình ảnh học tuyến yên (xem sau).
Logic tương tự đã được sử dụng trong việc tạo ra nghiệm pháp tạo IGF-1 để chẩn đoán thiếu hụt IGF-1 nguyên phát (PIGFD). Trong trường hợp này, GH được sử dụng như chất kích thích tiết, và phản ứng IGF-1 trong huyết thanh được đo để xác định những bệnh nhân có nồng độ IGF-1 ngẫu nhiên thấp là kết quả của sự không nhạy cảm với GH. Những bệnh nhân như vậy được dự kiến sẽ thất bại tương tự trong việc tăng sản xuất IGF-1 (và do đó, sự tăng trưởng) để đáp ứng với liệu pháp GH và có thể được phục vụ tốt hơn bằng cách điều trị bằng IGF-1 tái tổ hợp thay thế. Tuy nhiên, xét nghiệm này cũng đầy những hạn chế; mặc dù nhiều quy trình xét nghiệm đã được đề xuất, không có quy trình xét nghiệm tạo IGF-1 tiêu chuẩn nào với một ngưỡng IGF-1 có độ nhạy và độ đặc hiệu cao đã được thiết lập.
MRI tuyến yên, có và không có thuốc cản quang, là tiêu chuẩn chăm sóc cho tất cả các bệnh nhân được chẩn đoán thiếu hụt GH. Mục tiêu là để sàng lọc các dị tật tuyến yên và các khối u tân sản. Tuy nhiên, phần lớn các trường hợp thiếu hụt IGH vẫn là vô căn, một phần vì hình ảnh học chỉ có thể ghi lại các khiếm khuyết cấu trúc thô, trong khi thiếu hụt GH có thể phát sinh từ các khiếm khuyết tế bào và phân tử không thể được hình dung bằng MRI. Ngoài ra, tỷ lệ chẩn đoán dương tính giả cao của thiếu hụt GH có thể góp phần vào hiệu suất thấp của các phát hiện trên hình ảnh học. Ở những bệnh nhân bị thiếu hụt GH, nên tìm kiếm các thiếu hụt hormone tuyến yên bổ sung.
Xét nghiệm di truyền
Các xét nghiệm di truyền đang dần được đưa vào thực hành lâm sàng để điều tra các bệnh nhân bị rối loạn tăng trưởng. Xét nghiệm di truyền hiện tại chủ yếu chỉ có thể xác định được nguyên nhân ở trẻ em bị rối loạn tăng trưởng đơn gen, không phải đa gen. Bệnh nhân có tầm vóc thấp nghiêm trọng (SDS chiều cao <–3), từ cha mẹ có quan hệ huyết thống hoặc các gia đình có tầm vóc thấp trội trên nhiễm sắc thể thường rõ ràng, có bằng chứng về loạn sản xương hoặc có kiểu hình phức tạp (tình trạng hội chứng) có nhiều khả năng mắc bệnh di truyền đơn gen. Ngay cả ở trẻ em bị rối loạn tăng trưởng đơn gen, xét nghiệm di truyền thường không khám phá ra được nguyên nhân. Sự thất bại thường xuyên này một phần là do nguyên nhân di truyền của các rối loạn tăng trưởng rất không đồng nhất và nhiều gen liên quan vẫn chưa được khám phá. Ngoài ra, xét nghiệm di truyền có thể cho thấy một biến thể trong một gen được biết là chịu trách nhiệm cho rối loạn tăng trưởng, nhưng có thể không có đủ bằng chứng để xác định xem biến thể cụ thể đó có gây bệnh hay không.
Có ba kịch bản chính trong đó nên yêu cầu xét nghiệm di truyền cho trẻ em có tầm vóc thấp (Hình 11.9). Đầu tiên là khi có nghi ngờ lâm sàng về một tình trạng di truyền cụ thể ở một bệnh nhân. Trong tình huống này, các xét nghiệm di truyền phục vụ để xác nhận một chẩn đoán nghi ngờ. Một xác nhận phân tử của chẩn đoán có tác động rõ ràng hơn đến tư vấn di truyền và chăm sóc bệnh nhân khi kiểu hình không cho phép một chẩn đoán rõ ràng. Ví dụ, đối với một bệnh nhân 2 tuổi có kiểu hình điển hình của ACH, xét nghiệm di truyền có thể được hoãn lại. Ngược lại, đối với một bệnh nhân ở cùng độ tuổi bị nghi ngờ mắc NF-1, nhưng không đáp ứng các tiêu chí chẩn đoán do tuổi còn trẻ, một xét nghiệm di truyền dương tính có thể chứng minh thông tin có giá trị, với những hậu quả ngay lập tức trong việc đánh giá theo dõi. Ở những bệnh nhân có tình trạng có thể nhận biết được chủ yếu do các đột biến trong một gen duy nhất, việc giải trình tự gen ứng cử viên duy nhất đó là khả thi. Tuy nhiên, trong các tình trạng có tính không đồng nhất về mặt di truyền hoặc liên quan đến các gen lớn, việc sử dụng bảng giải trình tự nhắm mục tiêu hoặc giải trình tự exome có thể được ưu tiên hơn.
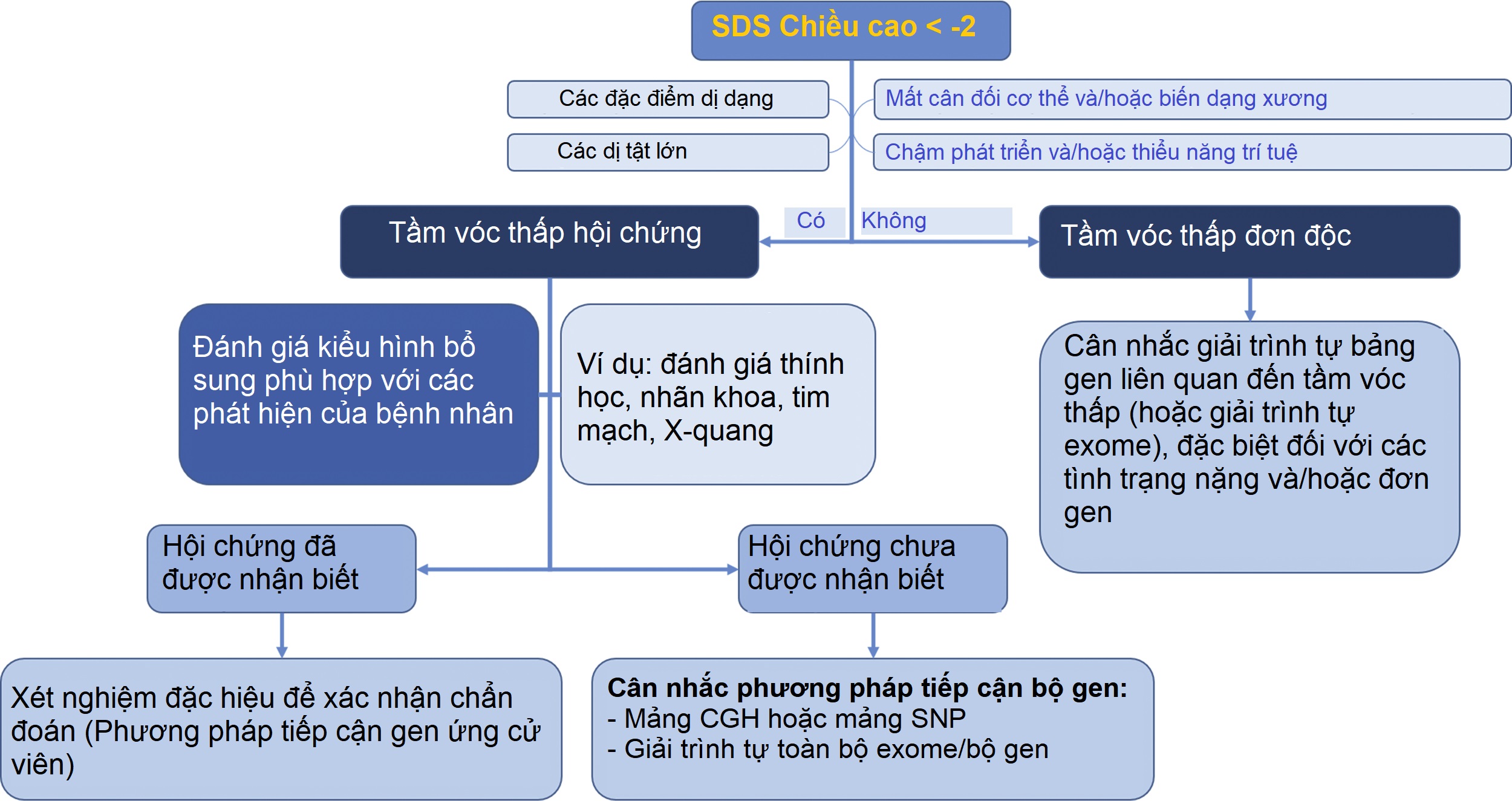
Hình 11.9 Tổng quan về việc điều tra di truyền phân tử ở trẻ em có tầm vóc thấp.
Kịch bản thứ hai liên quan đến các bệnh nhân có kiểu hình hội chứng không phù hợp với bất kỳ tình trạng nào đã được công nhận. Đối với những bệnh nhân này, điều tra di truyền phân tử bằng phương pháp tiếp cận bộ gen, chẳng hạn như bằng karyotyping phân tử (mảng SNP hoặc mảng-CGH) và/hoặc WES có thể xác định một biến thể di truyền gây bệnh trong một số lượng đáng kể các trường hợp.
Kịch bản thứ ba áp dụng cho các bệnh nhân có tầm vóc thấp đơn độc, nói cách khác, là những trẻ em khỏe mạnh có SDS chiều cao <–2 và có kết quả điều tra xét nghiệm và X-quang bình thường. Chắc chắn, kịch bản này bao gồm phần lớn trẻ em được đánh giá về suy giảm tăng trưởng. Nhiều trẻ em này có cha mẹ có tầm vóc thấp, ủng hộ sự hiện diện của một yếu tố di truyền trong chiều cao của chúng. Từ lâu người ta đã cho rằng những đứa trẻ như vậy có một nguyên nhân đa gen, nhưng các nghiên cứu gần đây đã chứng minh một số nguyên nhân đơn gen ở những bệnh nhân ban đầu được phân loại là ISS (Bảng 11.6). Một tỷ lệ trẻ em này nằm ở phần cuối nhẹ của phổ kiểu hình của một rối loạn hội chứng đã biết liên quan đến tầm vóc thấp, chẳng hạn như NS. Hơn nữa, nhiều nghiên cứu đã liên kết các gen cụ thể chủ yếu liên quan đến sự điều hòa sụn tăng trưởng, chẳng hạn như SHOX, ACAN, NPR2, NPPC, và IHH, dẫn đến các kiểu hình tầm vóc thấp đơn độc. Bệnh nhân có tầm vóc thấp do các khiếm khuyết trong các gen này cũng có thể cho thấy các phát hiện X-quang không đặc hiệu (Hình 11.10), chẳng hạn như tuổi xương cao hoặc các bất thường ở xương bàn tay hoặc xương ngón tay. Mỗi gen này chiếm một phần nhỏ các trường hợp tầm vóc thấp (≤ 1%–2%), nhưng có thể cao hơn đáng kể trong các trường hợp tầm vóc thấp gia đình. Ở trẻ em bị ISS, sự vắng mặt của các phát hiện lâm sàng cụ thể làm cho việc nhận biết trở nên khó khăn nếu không có một nghiên cứu di truyền phân tử và làm cho phương pháp tiếp cận gen ứng cử viên trở nên không thực tế. Vì những lý do này, một phương pháp xét nghiệm đa gen sử dụng giải trình tự thế hệ tiếp theo được ưu tiên hơn để điều tra ISS. Việc lựa chọn sử dụng WES hay phân tích bảng gen phụ thuộc vào sự sẵn có của các cơ sở và đánh giá chi phí-lợi ích. Ngay cả khi sử dụng WES, phân tích nên ưu tiên các gen đã được liên kết với kiểu hình tầm vóc thấp đơn độc và các gen liên quan đến các tình trạng hội chứng biểu hiện sự thay đổi kiểu hình được ghi nhận rõ ràng (xem Bảng 11.6). Mặc dù một chẩn đoán kết luận không thể được đưa ra cho hầu hết các bệnh nhân bị ISS sau khi điều tra di truyền rộng rãi, một kết quả dương tính sẽ có một tác động đáng kể đối với bệnh nhân và gia đình của họ. Dự kiến rằng sàng lọc di truyền phân tử bằng cách sử dụng một bảng gen hoặc phân tích exome sẽ ngày càng trở nên có giá trị trong việc đánh giá chẩn đoán trẻ em có tầm vóc thấp đơn độc vì, theo thời gian, các gen gây bệnh bổ sung và các biến thể gây bệnh bổ sung trong các gen đã biết sẽ được khám phá và vì chi phí, phạm vi bao phủ, và độ chính xác của việc giải trình tự sẽ được cải thiện.
Bảng 11.6 Các gen liên quan đến kiểu hình tầm vóc thấp đơn độc, thường được phân loại ban đầu là tầm vóc thấp vô căn
| Gen | Kiểu di truyền | Tần suất | Bằng chứng liên kết | Quan sát | Tham khảo |
|---|---|---|---|---|---|
| Các gen liên quan đến trục GH-IGF | |||||
| GH1 | Trội NST thường | Không có | Hạn chế | 386 | |
| GHSR | Trội/Lặn NST thường | Không có | Hạn chế | Có thể liên quan đến thiếu hụt GH trong cùng một gia đình | 13 |
| GHR | Trội NST thường | Không có | Hạn chế | Kiểu hình không nhạy cảm với GH nhẹ (Mức IGF-1 và GHBP thấp) | 387 |
| STAT5B | Trội NST thường | Không có | Hạn chế | Kiểu hình không nhạy cảm với GH nhẹ với bệnh chàm | 206 |
| IGFALS | Lặn NST thường | Không có | Mạnh | Thiếu hụt IGF-1 và IGFBP-3 nghiêm trọng không tương xứng với thiếu hụt chiều cao nhẹ | 388 |
| IGF1 | Trội NST thường | Không có | Hạn chế | 210 | |
| IGF1R | Trội NST thường | Không có | Trung bình | Phần lớn sinh ra SGA và nồng độ IGF-1 tăng cao hoặc là cơ bản hoặc trong quá trình điều trị rhGH | 389 |
| PAPPA2 | Lặn NST thường | Không có | Hạn chế | Nồng độ IGF-1 và IGFBP-3 tăng cao | 28 |
| Các gen liên quan đến sự phát triển sụn tăng trưởng | |||||
| SHOX | Trội NST thường | 1%–16% | Mạnh | Mất cân đối cơ thể nhẹ, đôi khi với các thành viên gia đình có LWD điển hình | 390 |
| ACAN | Trội NST thường | 1.4%–2.1% | Mạnh | Tuổi xương cao | 391 |
| NPPC | Trội NST thường | Không có | Hạn chế | Tật ngón ngắn nhẹ | 262 |
| NPR2 | Trội NST thường | 1.2%–3.4% | Mạnh | Xương bàn tay ngắn ở một số bệnh nhân | 392 |
| FGFR3 | Trội NST thường | Không có | Hạn chế | Tỷ lệ cơ thể bình thường trái ngược với kiểu hình loạn sản sụn | 55 |
| IHH | Trội NST thường | 1.6% | Trung bình | Rút ngắn các đốt ngón tay giữa của ngón thứ 2 và thứ 5 ở một số bệnh nhân | 61 |
| Gen liên quan đến con đường RAS-MAPK | |||||
| PTPN11 | Trội NST thường | 1.5% | Hạn chế | Các dạng nhẹ không được nhận biết trên lâm sàng của hội chứng Noonan | 279 |
AD, Trội nhiễm sắc thể thường; AR, Lặn nhiễm sắc thể thường; GH, hormone tăng trưởng; GHBP, protein gắn GH; IGF, yếu tố tăng trưởng giống insulin; IGFBP, protein gắn yếu tố tăng trưởng giống insulin; IHH, Indian hedgehog; LWD, loạn sản sụn Leri-Weill, NA, không có sẵn; rhGH, GH người tái tổ hợp; SGA, nhỏ so với tuổi thai.
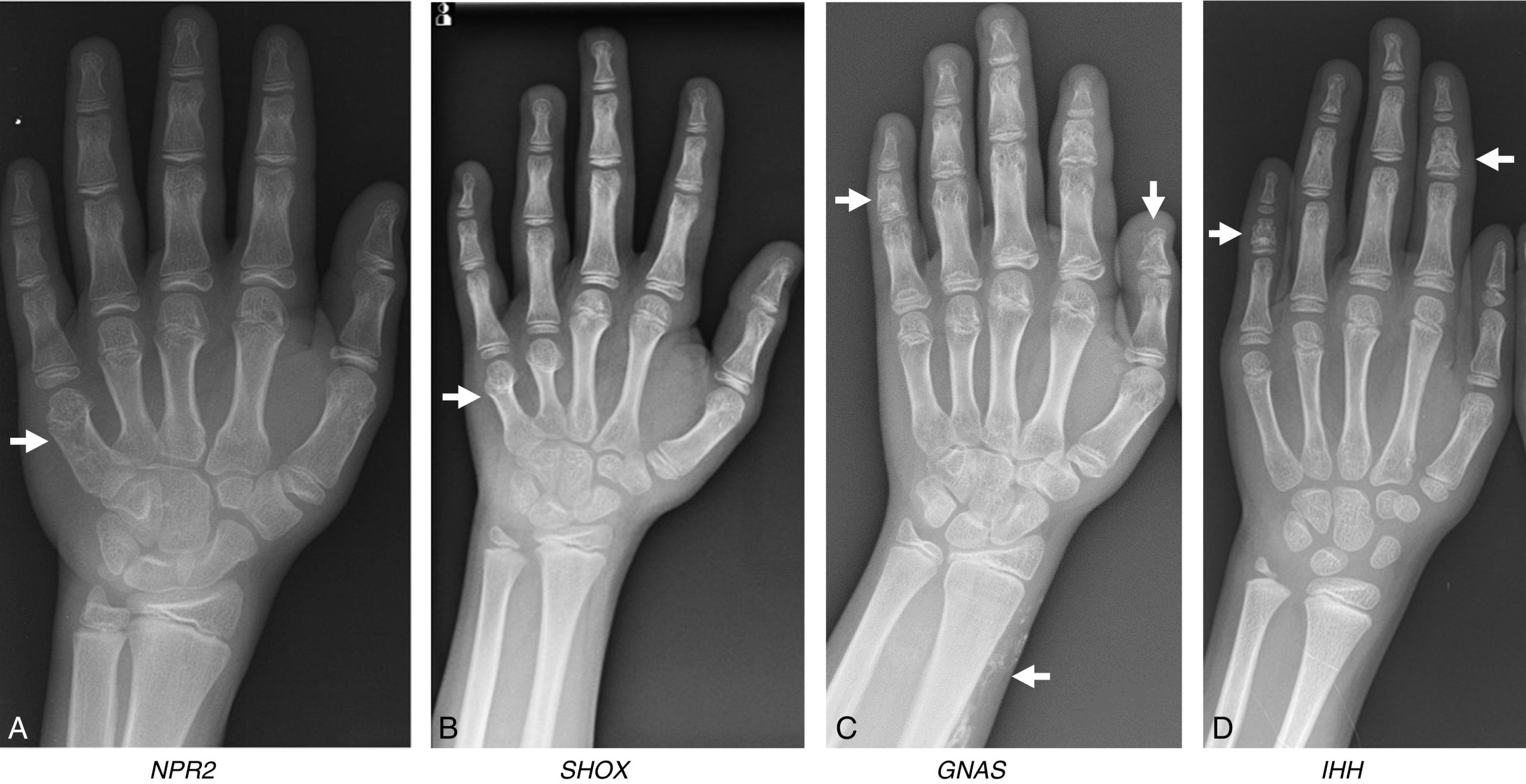
Hình 11.10 X-quang bàn tay của trẻ em có tầm vóc thấp với các bất thường xương không đặc hiệu liên quan đến các biến thể dị hợp tử trong các gen liên quan đến sự phát triển sụn tăng trưởng. A, Xương bàn tay ngắn được quan sát thấy ở một số bệnh nhân có các biến thể thụ thể peptide lợi niệu 2 (NPR2); B, xương bàn tay thứ tư và thứ năm ngắn ở một bệnh nhân bị thiếu hụt một nửa hộp homeobox tầm vóc thấp (SHOX); C, rút ngắn các đốt ngón tay giữa và đốt ngón tay xa của ngón cái với sự hiện diện của cốt hóa dưới da ở một bệnh nhân bị giả-giả suy cận giáp do khiếm khuyết protein liên kết nucleotide guanine (GNAS) được di truyền từ người cha; và D, rút ngắn đốt ngón tay giữa của ngón thứ hai và thứ năm với các sụn đầu xương hình nón liên quan đến các khiếm khuyết gen Indian hedgehog (IHH).
Điều trị Tầm vóc thấp
Khi một nguyên nhân có thể điều trị được của tầm vóc thấp được phát hiện, ví dụ, bệnh celiac hoặc suy giáp, thường có rất ít khó khăn trong việc quyết định một kế hoạch điều trị vấn đề cơ bản. Tuy nhiên, thường thì nguyên nhân vẫn chưa rõ ràng (tầm vóc thấp vô căn) hoặc nguyên nhân đã được biết nhưng một phương pháp điều trị cụ thể để khắc phục nguyên nhân cơ bản thì không được biết. Trong những trường hợp này, bác sĩ lâm sàng và gia đình có thể xem xét một phương pháp điều trị không đặc hiệu để cố gắng bù đắp cho nguyên nhân cơ bản của sự tăng trưởng giảm. Mặc dù có nhiều phương pháp điều trị tiềm năng có thể được sử dụng, nhưng sự cân nhắc phổ biến nhất là điều trị bằng GH.
Hormone tăng trưởng
Việc điều trị bằng hGH cho một bệnh nhân bị thiếu hụt GH lần đầu tiên được báo cáo bởi Tiến sĩ Maurice Raben vào năm 1958. Ông đã tinh chế hGH từ các chiết xuất tuyến yên từ các tử thi hiến tặng bằng cách sử dụng axit axetic băng nóng, giữ lại GH nhưng phá hủy TSH, LH và FSH. Ngay sau đó, Wilhelmi đã phát triển một phương pháp chiết xuất khác; ông thu thập các tuyến yên trong nước cất, đông lạnh chúng, và thực hiện chiết xuất cột cho ra hGH, cũng như các hormone tuyến yên trước khác. Chẳng mấy chốc, các cơ quan tuyến yên quốc gia đã được thành lập ở các quốc gia khác nhau để thu thập, chiết xuất và phân phối hGH từ tuyến yên để sử dụng lâm sàng. Cần khoảng 400 tuyến yên mỗi năm để điều trị cho một đứa trẻ bị suy tuyến yên, vì vậy việc điều trị bị giới hạn ở những trường hợp nghiêm trọng nhất và các chế độ liều lượng được quyết định bởi sự sẵn có hạn chế của hGH. Các chương trình tuyến yên đã bị chấm dứt bắt đầu từ năm 1985 khi bệnh Creutzfeld-Jacob, một bệnh não xốp tiến triển và gây tử vong do prion trung gian, được chẩn đoán ở những bệnh nhân trước đây đã nhận được hGH bị ô nhiễm được chiết xuất bằng phương pháp Wilhelmi.
Thật tình cờ, năm 1985 cũng chứng kiến sự ra đời của việc sản xuất GH bằng công nghệ DNA tái tổ hợp để sử dụng lâm sàng; rhGH đã thay thế hGH từ tuyến yên và vẫn là nguồn duy nhất của việc điều trị bằng GH. Bởi vì nguồn cung rhGH đột nhiên không giới hạn—các công ty dược phẩm có thể sản xuất và bán bao nhiêu tùy theo thị trường sẵn sàng chịu đựng—sự ra đời của rhGH không chỉ làm tăng sự an toàn của việc điều trị bằng GH mà còn mở ra khả năng điều trị nhiều bệnh nhân hơn với các chỉ định khác nhau ở liều cao hơn và trong thời gian dài hơn. Việc sử dụng rhGH đã mở rộng từ việc thay thế thiếu hụt GH (ở những bệnh nhân bị thiếu hụt GH và PWS) đến việc tăng cường sự tăng trưởng ở những bệnh nhân đủ GH nhưng có các khiếm khuyết khác làm suy giảm sự tăng trưởng của họ (Hộp 11.4). Ví dụ, việc điều trị bằng GH cho ISS đã được FDA chấp thuận vào năm 2003 cho những cá nhân có chiều cao dưới –2,25 SD (phân vị 1,2), có vận tốc tăng trưởng không được kỳ vọng sẽ cho phép đạt được chiều cao người lớn trong phạm vi bình thường và có đánh giá chẩn đoán đã loại trừ các nguyên nhân khác của suy giảm tăng trưởng cần được theo dõi hoặc điều trị bằng các phương tiện khác.
| Hộp 11.4. Các chỉ định điều trị bằng Hormone tăng trưởng được Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ chấp thuận
AIDS, hội chứng suy giảm miễn dịch mắc phải; GH, hormone tăng trưởng; HIV, virus gây suy giảm miễn dịch ở người. CÁC CHỈ ĐỊNH của Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ với các năm được phê duyệt Chỉ định cho trẻ em
|
Việc điều trị bằng rhGH hiện tại bao gồm các mũi tiêm dưới da hàng đêm—dưới da vì bản chất polypeptide của rhGH không cho phép dùng đường uống, và hàng đêm, vì hầu hết sự bài tiết GH nội sinh xảy ra vào ban đêm. Hầu hết các nhà sản xuất rhGH cũng tạo ra các thiết bị bút để tạo điều kiện thuận lợi cho việc tiêm của các gia đình. Tuy nhiên, việc tuân thủ với nhiều năm tiêm hàng đêm vẫn còn nhiều thách thức, và nhiều công ty đã tìm cách phát triển các chế phẩm rhGH tác dụng kéo dài có thể hỗ trợ việc tuân thủ lâu dài thông qua việc giảm tần suất tiêm; các chế phẩm như vậy vẫn đang được nghiên cứu.
Lợi ích
Đối với trẻ em bị thiếu hụt GH, điều trị bằng rhGH mang lại cả lợi ích về chiều cao và sức khỏe. Sự tăng tốc của sự tăng trưởng tầm vóc là rõ ràng trong vài tháng đầu tiên bắt đầu điều trị bằng rhGH, với vận tốc chiều cao lớn nhất được thấy trong năm đầu tiên của điều trị, ít hơn một chút trong năm thứ 2, và ít hơn một chút nữa nhưng vẫn dai dẳng trong các năm từ 3 đến cuối điều trị. Sự gia tăng vận tốc chiều cao chuyển thành sự gia tăng điểm Z-score chiều cao. Mặc dù trong lịch sử, những cá nhân bị thiếu hụt GH đơn độc không được điều trị có điểm Z-score chiều cao người lớn trung bình là –4,7 (phạm vi, –3,9 đến –6), những người được điều trị bằng rhGH thường đạt được chiều cao người lớn trong phạm vi bình thường (tức là, thấp hơn 2 SD so với mức trung bình của dân số và chiều cao trung bình của cha mẹ họ). Bệnh nhân có cha mẹ cao hơn có xu hướng có phản ứng với điều trị tốt hơn những người có cha mẹ thấp hơn, cũng như những bệnh nhân vẫn còn ở giai đoạn trước dậy thì khi bắt đầu điều trị so với những người đã ở tuổi dậy thì.
Ngoài lợi ích về chiều cao, bệnh nhân bị thiếu hụt GH còn nhận được lợi ích sức khỏe từ việc điều trị bằng rhGH do tác dụng của GH đối với chuyển hóa lipid, protein và glucose. Nhiều nghiên cứu đã chỉ ra rằng việc điều trị bằng rhGH cải thiện mật độ khoáng xương giảm, khối lượng cơ nạc giảm, và khối lượng mỡ tăng được thấy ở những đứa trẻ không được điều trị bị thiếu hụt GH. Mặc dù các nghiên cứu ngắn hạn cho thấy tác động tích cực của việc điều trị bằng rhGH đối với khối lượng tâm thất trái, dữ liệu không nhất quán về hiệu suất tim được đo bằng siêu âm tim (phân suất co rút và phân suất tống máu tâm thất trái), chức năng mạch máu (độ dày lớp nội trung mạc ở các động mạch cảnh chung), hồ sơ lipid (mặc dù việc điều trị bằng rhGH làm giảm tổng số và mức cholesterol lipoprotein mật độ thấp, dữ liệu không nhất quán về việc liệu trẻ em không được điều trị bị thiếu hụt GH có hồ sơ bình thường hay bất thường), và các chỉ số gây xơ vữa.
Đối với trẻ em được điều trị bằng rhGH không bị thiếu hụt GH (tất cả các chỉ định ngoại trừ thiếu hụt GH và hội chứng Prader-Willi), lợi ích chỉ giới hạn ở chiều cao và ngay cả ở đó, thường ít ấn tượng hơn so với trẻ em bị thiếu hụt GH. Trong một thử nghiệm ngẫu nhiên có đối chứng ở trẻ em bị ISS, khoảng 6 năm điều trị bằng GH đã làm tăng chiều cao người lớn khoảng 6 cm. Các phản ứng điều trị rất khác nhau, có lẽ vì ISS là một phân loại không đồng nhất; một số bệnh nhân có thể không có sự gia tăng Z-score chiều cao có thể đo lường được. Hơn nữa, không rõ liệu sự gia tăng chiều cao do điều trị bằng GH có lợi ích tâm lý quan trọng hay không. Do đó, điều quan trọng là phải tư vấn cho bệnh nhân và gia đình của họ một cách phù hợp trước khi bắt đầu điều trị bằng rhGH, để cho phép họ đưa ra quyết định sáng suốt và quản lý kỳ vọng.
Các tác dụng phụ tiềm tàng
Các tác dụng phụ tiềm tàng của việc điều trị bằng rhGH đã được xem xét rộng rãi gần đây bởi Hội Nội tiết Nhi khoa và Hội Nghiên cứu Hormone Tăng trưởng, và kết quả được tóm tắt sau đây. Phần lớn dữ liệu về sự an toàn bắt nguồn từ các sổ đăng ký giám sát sau khi đưa ra thị trường của rhGH, ban đầu được FDA bắt buộc đối với tất cả các nhà sản xuất rhGH cho các sản phẩm tương ứng của họ và sau đó được chính các nhà sản xuất tiếp tục. Mặc dù các sổ đăng ký này có đủ sức mạnh nhờ quy mô của chúng (phạm vi quốc tế và nhiều thập kỷ thu thập dữ liệu) để xác định các tác dụng phụ thảm khốc hoặc thường xuyên, vẫn có một số hạn chế. Chúng bao gồm: (1) sự phụ thuộc vào việc xác định tự nguyện cho cả sự xuất hiện và sự liên quan đến việc điều trị bằng rhGH có thể dẫn đến việc thu thập dữ liệu không đầy đủ, (2) việc giám sát trong quá trình điều trị có thể bỏ lỡ bất kỳ tác dụng phụ nào chỉ biểu hiện sau khi điều trị đã kết thúc, (3) những thay đổi theo thời gian về liều rhGH và/hoặc các đặc điểm của bệnh nhân có thể làm thay đổi nguy cơ tác dụng phụ, và (4) thiếu một quần thể đối chứng hợp lệ không cho phép so sánh để xác định các tác động thực tế của việc điều trị bằng rhGH đối với nguy cơ.
Các tác dụng phụ khi điều trị bằng rhGH xảy ra với tần suất thấp (< 3% trẻ em được điều trị) nhưng có khả năng nghiêm trọng và nên được xem xét với các gia đình bệnh nhân trước khi họ quyết định bắt đầu điều trị. Tăng áp lực nội sọ xảy ra với tỷ lệ hiện mắc tổng thể ước tính là 28 trên 100.000 năm điều trị, thường là trong quá trình bắt đầu điều trị bằng rhGH hoặc tăng liều; nó sẽ hết khi ngừng rhGH, sau đó rhGH có thể được bắt đầu lại với liều một nửa và tăng cẩn thận hơn. Trượt chỏm xương đùi (SCFE) xảy ra với tỷ lệ hiện mắc tổng thể ước tính là 73 trên 100.000 năm điều trị, với thời gian khởi phát trung bình từ 0,4 đến 2,5 năm sau khi bắt đầu điều trị bằng rhGH, và đòi hỏi phải đóng đinh phẫu thuật chỏm xương đùi để khắc phục vị trí lệch của nó. Sự tiến triển của vẹo cột sống có thể xảy ra, đặc biệt là ở các quần thể bệnh nhân có nguy cơ cơ bản cao hơn (PWS và hội chứng Turner), và dường như là kết quả của sự tăng trưởng nhanh chóng hơn là do rhGH.
Một số “tác dụng phụ” rõ ràng của việc điều trị bằng rhGH thực ra phản ánh các hoạt động sinh lý của GH. Các tác động sinh lý của GH đối với chuyển hóa glucocorticoid (tức là, làm giảm sự chuyển đổi qua trung gian 11β-hydroxysteroid dehydrogenase 1 của gan từ cortisone không hoạt động thành cortisol hoạt động và làm tăng quá trình dị hóa cortisol qua trung gian cytochrome P450 3A4) có thể làm lộ ra tình trạng suy thượng thận tiềm ẩn ở những người mà sự thiếu hụt GH của họ có thể liên quan đến MPHD. Tương tự như vậy, bằng cách tăng quá trình khử iod ngoại vi của T4 thành T3, rhGH có thể làm giảm nồng độ T4 tự do trong huyết thanh, thường được sử dụng để chẩn đoán suy giáp trung ương. GH làm giảm sinh lý sự nhạy cảm với insulin, và do đó việc điều trị bằng rhGH có thể gây ra tình trạng không dung nạp glucose và biểu hiện tăng đường huyết ở trẻ em có sự bài tiết hoặc nhạy cảm với insulin bị tổn hại, và dẫn đến tăng nhu cầu insulin ở trẻ em bị đái tháo đường đã được thay thế bằng insulin. Tuy nhiên, các tác động của rhGH đối với sự nhạy cảm với insulin chỉ xảy ra trong quá trình điều trị, với sự trở lại mức ban đầu khi chấm dứt điều trị bằng rhGH.
Nguy cơ tân sản với việc điều trị bằng rhGH đã là một mối quan tâm lâu dài, một phần dựa trên bằng chứng gián tiếp. Ví dụ, IGF-1 thúc đẩy sự tăng trưởng và di căn của các tế bào ác tính khác nhau trong các mô hình thí nghiệm, và nồng độ IGF-1 lưu hành cao hơn có liên quan về mặt dịch tễ học với nguy cơ ung thư vú trước mãn kinh, ung thư ruột kết, và có thể là ung thư tuyến tiền liệt. Các nghiên cứu dịch tễ học đã cho thấy nguy cơ gia tăng của một số bệnh ác tính nhất định ở những bệnh nhân bị bệnh to đầu chi, trong khi những cá nhân bị hội chứng Laron (không nhạy cảm với GH) dường như được bảo vệ khỏi việc phát triển ung thư.
Các nghiên cứu giám sát nói chung đã không tìm thấy nguy cơ gia tăng đối với các khối u ác tính mới ở trẻ em khỏe mạnh không có tiền sử ung thư và không có khuynh hướng di truyền với ung thư. Đối với những bệnh nhân có khuynh hướng khối u đã biết do các tình trạng di truyền hoặc y tế khác, dữ liệu không đủ để xác định liệu việc điều trị bằng rhGH có làm tăng thêm nguy cơ đó hay không. Các hướng dẫn được công bố thường dựa trên dữ liệu hồi cứu vì các thử nghiệm ngẫu nhiên có đối chứng chưa được tiến hành. Theo các hướng dẫn gần đây, việc điều trị bằng rhGH chống chỉ định ở những bệnh nhân có bệnh ác tính đang hoạt động, nhưng những người sống sót sau ung thư ở trẻ em đã hoàn thành liệu pháp ung thư và không có dấu hiệu của bệnh tân sản đang hoạt động có thể là ứng cử viên cho việc điều trị bằng rhGH. Việc điều trị bằng rhGH đã không được phát hiện là làm tăng nguy cơ tái phát nội tại của họ. Mặc dù việc điều trị bằng rhGH có liên quan đến một sự gia tăng nhỏ ban đầu trong nguy cơ chung đối với các khối u tân sản nguyên phát sau đó, các nghiên cứu sau đó cho thấy nguy cơ liên quan đến xạ trị hơn là việc điều trị bằng rhGH. Các nghiên cứu này về nguy cơ ung thư là không có đối chứng và hồi cứu, và do đó các nguy cơ vẫn chưa chắc chắn. Mặc dù có ít dữ liệu để hướng dẫn khoảng thời gian thích hợp giữa việc hoàn thành liệu pháp ung thư và bắt đầu điều trị bằng rhGH, một khoảng thời gian chờ đợi tiêu chuẩn là 12 tháng để thiết lập “liệu pháp thành công” của tổn thương nguyên phát đã được khuyến nghị nhưng có thể được thay đổi tùy thuộc vào hoàn cảnh của từng bệnh nhân. Mặc dù về mặt mô học không phải là ác tính, u sọ hầu có tỷ lệ tái phát hoặc tiến triển cao sau khi cắt bỏ và đôi khi được điều trị bằng xạ trị, do đó làm tăng nguy cơ của bệnh nhân để phát triển các khối u tân sản nguyên phát sau đó. U sọ hầu thường liên quan đến thiếu hụt GH, và không có dữ liệu nào cho thấy việc điều trị chúng khác với các khối u ác tính về thời gian quan sát trước khi bắt đầu điều trị bằng rhGH.
Các nguy cơ lâu dài sau điều trị vẫn còn ít được biết đến vì: (1) rhGH chỉ có sẵn từ năm 1985, và do đó những bệnh nhân được điều trị lâu đời nhất hiện đang ở độ tuổi 40 hoặc đầu 50; (2) các sổ đăng ký giám sát sau khi đưa ra thị trường mất dấu vết của bệnh nhân một khi họ ngừng điều trị với nhãn hiệu rhGH cụ thể đó; (3) các nghiên cứu được thiết kế để trả lời các câu hỏi như vậy đòi hỏi quy mô và thời gian theo dõi đủ; và (4) thiếu các nhóm đối chứng phù hợp. Nghiên cứu đa trung tâm đang diễn ra “Sự an toàn và phù hợp của các phương pháp điều trị bằng hormone tăng trưởng ở Châu Âu” (SAGhE) được tạo ra để đánh giá tỷ lệ tử vong và bệnh tật ở người lớn ở tám quốc gia châu Âu (Bỉ, Pháp, Đức, Ý, Hà Lan, Thụy Điển, Thụy Sĩ và Vương quốc Anh [Anh, xứ Wales và Scotland]) đã được điều trị trong thời thơ ấu bằng rhGH (không phải hGH từ tuyến yên) cho các chỉ định được phê duyệt. Nghiên cứu đoàn hệ độc lập với ngành công nghiệp này đã so sánh 24.232 bệnh nhân đã được điều trị bằng rhGH trước đây, sau trung bình 17,1 năm theo dõi, với các tỷ lệ dự kiến từ dữ liệu dân số quốc gia và địa phương. Dữ liệu mâu thuẫn ban đầu từ các phân tích phân nhóm sơ bộ đã được xem xét trong các bài báo của xã hội nói trên. Hai phân tích SAGhE bổ sung đã được báo cáo sau đó. Đối với các bệnh nhân trong toàn bộ tập đoàn đã nhận được điều trị bằng rhGH cho “suy giảm tăng trưởng đơn độc” (tức là, thiếu hụt GH đơn độc, ISS, hoặc SGA), nguy cơ ung thư không tăng nói chung cũng như theo vị trí cụ thể. Tuy nhiên, đối với những bệnh nhân không bị “suy giảm tăng trưởng đơn độc”, các phân tích cho thấy tỷ lệ mắc ung thư xương và bàng quang tăng lên và, với thời gian theo dõi dài hơn, u lympho Hodgkin ở những người không bị ung thư trước đó, và nguy cơ tử vong do ung thư tăng lên liên quan đến việc tăng liều rhGH hàng ngày cho những bệnh nhân được điều trị sau khi bị ung thư trước đó. Phân tích của một đoàn hệ SAGhE năm quốc gia gồm 10.403 bệnh nhân cho thấy nguy cơ u màng não tăng lên liên quan đến xạ trị trước đó cho một bệnh ác tính tiềm ẩn, nhưng không liên quan đến việc điều trị bằng rhGH. Các nghiên cứu này có những hạn chế cố hữu, bao gồm thiếu một nhóm đối chứng phù hợp và thời gian theo dõi không đủ để đánh giá đầy đủ nguy cơ của các bệnh ác tính phổ biến (ví dụ, ung thư vú và ung thư ruột kết) có liên quan thống kê với nồng độ IGF-1 và xảy ra chủ yếu ở tuổi trưởng thành sau này. Cần có nghiên cứu sâu hơn.
Ngoài những cân nhắc nói trên, áp dụng cho tất cả các bệnh nhân nhi được điều trị bằng rhGH, còn có các nguy cơ bổ sung liên quan đến các quần thể đặc biệt. Bệnh nhân bị PWS có nguy cơ tiềm ẩn đối với cả ngưng thở khi ngủ trung ương và tắc nghẽn (OSA), đôi khi dẫn đến đột tử. Cả GH và IGF-1 đều có thể kích thích sự phát triển của amidan và vòm họng và do đó có thể làm trầm trọng thêm OSA. Do đó, đa ký giấc ngủ trước khi bắt đầu điều trị bằng rhGH và theo dõi trong quá trình điều trị bằng rhGH được khuyến nghị cho bệnh nhân bị PWS. Đối với những người phát triển OSA, việc điều trị bằng rhGH nên được gián đoạn cho đến khi tình trạng hô hấp của họ có thể được ổn định bằng phẫu thuật (cắt amidan và/hoặc cắt vòm họng, khi thích hợp) hoặc áp lực đường thở dương liên tục. Một quần thể đặc biệt khác bao gồm các bệnh nhân bị bệnh nặng trong môi trường chăm sóc đặc biệt. Tỷ lệ tử vong tăng đã được quan sát thấy với việc điều trị bằng rhGH trong hai thử nghiệm lâm sàng có đối chứng giả dược ở những bệnh nhân người lớn không bị thiếu hụt GH bị bệnh nặng cấp tính do các biến chứng sau phẫu thuật tim hở, phẫu thuật bụng hoặc đa chấn thương do tai nạn, hoặc những người bị suy hô hấp cấp tính. Sự an toàn của việc tiếp tục điều trị bằng rhGH ở bệnh nhân nhi nhận được liều thay thế cho các chỉ định được phê duyệt mà đồng thời phát triển các bệnh này chưa được thiết lập, và do đó các lợi ích tiềm năng của việc tiếp tục điều trị bằng rhGH nên được cân nhắc với các nguy cơ tiềm tàng.
Theo dõi trong quá trình điều trị
Trong quá trình điều trị, bệnh nhân phải được theo dõi ít nhất 6 tháng một lần với các phép đo nhân trắc học, phân loại Tanner, và đo nồng độ IGF-1. Nồng độ IGF-1 cung cấp thông tin về sự tuân thủ điều trị và khả năng đáp ứng cá nhân đối với những thay đổi liều rhGH. Mặc dù thiếu bằng chứng trực tiếp, việc duy trì nồng độ IGF-1 trong các phạm vi bình thường theo tuổi và giới tính dường như là thận trọng. Việc hỏi bệnh sử về đau đầu mới hoặc nặng hơn, rối loạn thị giác, hoặc sự hiện diện của phù gai thị khi soi đáy mắt nên thúc đẩy việc đánh giá thêm về tăng áp lực nội sọ, và bệnh sử đau hông hoặc rối loạn dáng đi nên gợi ý một cuộc đánh giá về SCFE. Nên thực hiện các cuộc khám cột sống nối tiếp để sàng lọc vẹo cột sống. Đánh giá sự đầy đủ của cortisol và hormone tuyến giáp ở những bệnh nhân mà sự thiếu hụt GH của họ có thể liên quan đến MPHD và sàng lọc hemoglobin A1c ở những bệnh nhân có nguy cơ bị rối loạn chuyển hóa carbohydrate là cần thiết.
Việc điều trị bằng rhGH ở liều nhi khoa không nên tiếp tục sau khi cốt hóa sụn đầu xương, để tránh gây ra những thay đổi giống bệnh to đầu chi (ví dụ, sự to ra không cân đối của bàn tay, bàn chân, hàm và lưỡi). Khi vận tốc tăng trưởng giảm xuống 2 đến 2,5 cm/năm, nên chụp X-quang tuổi xương để xác nhận sự cốt hóa sụn đầu xương. Tuy nhiên, sau một thời gian gián đoạn ngắn trong điều trị, việc đánh giá lại về thiếu hụt GH ở người lớn có thể dẫn đến việc tiếp tục điều trị thiếu hụt GH ở người lớn (với liều thấp hơn được thiết kế cho các lợi ích về chuyển hóa và thành phần cơ thể được nêu trước đó). Tuy nhiên, khi kiểm tra lại, phần lớn bệnh nhân bị thiếu hụt GH ở trẻ em đơn độc, vô căn không đáp ứng các tiêu chí cho thiếu hụt GH ở người lớn và không cần điều trị thêm.
Quá trình ra quyết định chung
Đối với trẻ em có tầm vóc thấp không do thiếu hụt GH, quyết định điều trị bằng GH là khó khăn, một phần vì vẫn còn rất nhiều lỗ hổng trong sự hiểu biết của chúng ta về lợi ích và rủi ro. Việc điều trị ISS bằng GH đặc biệt gây tranh cãi. Như được khuyến nghị bởi các hướng dẫn mới nhất về điều trị bằng GH của Hội Nội tiết Nhi khoa, “Tại Hoa Kỳ, đối với trẻ em đáp ứng các tiêu chí của FDA, chúng tôi đề nghị một phương pháp tiếp cận ra quyết định chung để theo đuổi việc điều trị bằng GH cho một đứa trẻ bị ISS. Quyết định có thể được đưa ra theo từng trường hợp sau khi đánh giá các gánh nặng về thể chất và tâm lý, và thảo luận về các rủi ro và lợi ích. Chúng tôi khuyến nghị chống lại việc sử dụng GH thường quy ở mọi trẻ em có SDS chiều cao (HtSDS) ≤–2,25.”
Một số yếu tố làm nền tảng cho phương pháp tiếp cận này. Nhiều gia đình bệnh nhân tìm kiếm sự đánh giá và điều trị tầm vóc thấp vì những lo ngại về sự thích ứng tâm lý xã hội, cả hiện tại ở thời thơ ấu và được dự kiến trong tương lai. Tuy nhiên, bằng chứng còn hạn chế để ủng hộ một trong hai quan niệm, rằng chiều cao có liên quan đến sức khỏe tâm lý và chất lượng cuộc sống hoặc việc điều trị bằng GH cải thiện điều này. Hơn nữa, mặc dù việc điều trị bằng GH đã được chứng minh là làm tăng chiều cao trung bình của các nhóm bệnh nhân được điều trị bị ISS, các phản ứng cá nhân rất khác nhau, bao gồm một số người không tăng Z-score chiều cao của họ. Với cùng một chiều cao có ý nghĩa khác nhau đối với những người khác nhau, khả năng đáp ứng thay đổi với điều trị, và các nhận thức khác nhau về gánh nặng điều trị, việc cân nhắc rủi ro-lợi ích của việc điều trị bằng GH là một vấn đề rất cá nhân.
Một số dấu hiệu của sự thành công xã hội, bao gồm cả nghề nghiệp và bạn đời, đã được liên kết với sự cao lớn ở nam giới. Do đó, áp lực xã hội đối với sự cao lớn dường như ảnh hưởng đến nam giới nhiều hơn nữ giới và điều này dẫn đến sự thiên vị, có ý thức hoặc vô thức, giữa các bậc cha mẹ tìm kiếm sự chăm sóc y tế cho những đứa trẻ thấp, giữa các bác sĩ chăm sóc ban đầu quyết định có nên theo đuổi các xét nghiệm chẩn đoán và/hoặc giới thiệu đến chuyên gia chuyên sâu hay không, và giữa các bác sĩ nội tiết kê đơn điều trị bằng GH. Kết quả cuối cùng là các bé trai đông hơn các bé gái trong các sổ đăng ký GH nhi khoa của Hoa Kỳ 2:1 cho tất cả các chỉ định và 3:1 cho ISS (chỉ định bị ảnh hưởng nhiều nhất bởi xã hội) mặc dù tỷ lệ hiện mắc của chiều cao dưới –2,25 SD (ngưỡng được FDA chấp thuận cho việc điều trị ISS bằng GH) không khác nhau theo giới tính trong một quần thể chăm sóc ban đầu nhi khoa lớn, khu vực. Tương tự như vậy, sự chênh lệch về chủng tộc/sắc tộc cũng đã được quan sát thấy trong các sổ đăng ký GH nhi khoa của Hoa Kỳ. Các bác sĩ lâm sàng nên lưu ý đến những xu hướng này, để các mô hình tăng trưởng bất thường được nhận biết và đánh giá một cách bình đẳng cho tất cả trẻ em như một dấu hiệu tiềm tàng của một vấn đề sức khỏe tiềm ẩn.
Yếu tố tăng trưởng giống Insulin-1
rhIGF-1 có sẵn để điều trị cho những bệnh nhân bị suy giảm tăng trưởng do nồng độ IGF-1 giảm mà sẽ không đáp ứng với điều trị bằng GH. Nó được sử dụng để điều trị thiếu hụt IGF-1 nguyên phát nghiêm trọng do hội chứng không nhạy cảm với GH (hội chứng Laron, do đột biến trong GHR), đột biến gen STAT5b, và các đột biến gen IGF1. Ngoài ra, rhIGF-1 được sử dụng để điều trị việc xóa gen GH1 khi bệnh nhân đã phát triển các kháng thể trung hòa làm cho việc điều trị bằng rhGH tiếp theo trở nên không hiệu quả. Đối với PIGFD nghiêm trọng không giải thích được, nơi mức độ đáp ứng với GH không rõ ràng, một thử nghiệm điều trị bằng rhGH được khuyến nghị trước khi chuyển sang điều trị bằng rhIGF-1. Việc điều trị bằng rhGH cho bệnh nhân bị thiếu hụt GH gây ra một phản ứng tăng trưởng lớn hơn so với việc điều trị bằng rhIGF-1 cho bệnh nhân bị PIGFD, có lẽ do các hoạt động độc lập với IGF-1 của GH tại sụn tăng trưởng, mà loại sau không thể thay thế được. Hơn nữa, việc điều trị bằng rhGH gây ra IGFBP-3, cũng như IGF-1, làm cho nó sinh lý hơn so với việc điều trị bằng rhIGF-1, chỉ cần tiêm một lần mỗi ngày (so với tiêm hai lần mỗi ngày của rhIGF-1), và không gây hạ đường huyết như loại sau (xem sau).
Nguyên nhân phổ biến nhất của nồng độ IGF-1 thấp ở trẻ em và thanh thiếu niên bị còi cọc là suy dinh dưỡng. Một bệnh sử ăn uống cẩn thận và kiểm tra các đường cong cân nặng và BMI là quan trọng để sàng lọc khả năng này. Đối với trẻ em bị nghi ngờ suy dinh dưỡng, can thiệp dinh dưỡng là phương pháp điều trị thích hợp hơn là liệu pháp hormone. Việc điều trị bằng rhIGF-1 cũng không phải là một phương pháp điều trị tiêu chuẩn cho trẻ em có nồng độ IGF-1 thấp do bệnh hệ thống mãn tính hoặc trẻ em có nồng độ IGF-1 thấp nhẹ liên quan đến ISS hoặc chậm phát triển sinh lý.
Khi được chỉ định, việc điều trị bằng rhIGF-1 được thực hiện dưới dạng tiêm dưới da hai lần mỗi ngày với liều 80 đến 120 mcg/kg (cũng được tiêm ở ngoài Hoa Kỳ dưới dạng tiêm một lần mỗi ngày 150–180 mcg/kg). Hạ đường huyết xảy ra ở 42% bệnh nhân trong quá trình điều trị bằng rhIGF-1, và mặc dù hầu hết các đợt đều nhẹ hoặc trung bình, các phản ứng hạ đường huyết nghiêm trọng (mất ý thức, co giật) có thể xảy ra. Do đó, để đảm bảo an toàn, các gia đình bệnh nhân cần được giáo dục về nguy cơ này, rhIGF-1 nên được tiêm 20 phút sau bữa ăn hoặc bữa ăn nhẹ có chứa carbohydrate và không tiêm nếu bệnh nhân không định ăn, và tăng cường cảnh giác trong các bệnh xen kẽ, bao gồm cả việc sử dụng máy đo đường huyết tại nhà. Việc điều trị bằng rhIGF-1 cũng có thể dẫn đến phì đại amidan và vòm họng, các phản ứng quá mẫn và dị ứng, và các phản ứng với thành phần cồn benzyl của dung môi. Các tác dụng phụ tiềm tàng khác của việc điều trị bằng rhIGF-1 tương tự như các tác dụng phụ của việc điều trị bằng rhGH.
Steroid sinh dục
Điều trị bằng estrogen làm tăng tốc độ tăng trưởng theo chiều dọc, gây ra một giai đoạn tăng vọt tạm thời, nhưng cũng làm tăng tốc độ lão hóa của sụn tăng trưởng, gây ra sự kết thúc sớm hơn của sự tăng trưởng theo chiều dọc. Nói chung, tác động sau chiếm ưu thế, do đó chiều cao người lớn bị giảm. Điều trị bằng các androgen có thể thơm hóa có thể có các tác động tương tự vì các androgen được chuyển đổi thành estrogen bởi enzyme aromatase. Tuy nhiên, điều trị bằng các androgen không thể thơm hóa, không thể được chuyển đổi thành estrogen, có thể làm tăng tốc độ tăng trưởng theo chiều dọc với ít sự tăng tốc hơn của sự lão hóa sụn tăng trưởng. Trong số các androgen không thể thơm hóa, oxandrolone được nghiên cứu nhiều nhất. Điều trị bằng Oxandrolone đã được chứng minh là làm tăng chiều cao người lớn một cách khiêm tốn ở các bé gái bị hội chứng Turner (xem Chương 17 về hội chứng Turner để biết chi tiết). Oxandrolone làm tăng tốc độ tăng trưởng theo chiều dọc ở các bé trai bị chậm phát triển sinh lý; tuy nhiên, không có các thử nghiệm ngẫu nhiên có đối chứng, dài hạn, có đủ sức mạnh đã đánh giá tác động lên chiều cao người lớn.
Các chất chủ vận Hormone giải phóng Gonadotropin
Điều trị bằng chất chủ vận GnRH tạo ra một sự ức chế hiệu quả cao, có thể đảo ngược của trục tuyến yên-sinh dục. Ở một đứa trẻ đang dậy thì, việc sản xuất steroid sinh dục bị giảm đi rất nhiều, làm chậm cả sự tăng trưởng theo chiều dọc và sự lão hóa của sụn tăng trưởng. Ít nhất trong một số tình huống, tác động sau chiếm ưu thế, dẫn đến chiều cao người lớn lớn hơn. Mặc dù không có các thử nghiệm có đối chứng tốt, dài hạn nào, người ta thường tin rằng việc điều trị bằng chất chủ vận GnRH ở trẻ em bị dậy thì sớm trung ương làm giảm sự suy giảm chiều cao người lớn; (xem Chương 16 và 18 về Dậy thì và các rối loạn của nó ở nữ và nam, tương ứng, để biết chi tiết). Ở thanh thiếu niên thấp có dậy thì đúng thời điểm, điều trị bằng GnRH trong 3,5 năm, trong một thử nghiệm ngẫu nhiên có đối chứng, ban đầu làm chậm sự tăng trưởng nhưng cũng làm chậm sự tiến triển của tuổi xương và cho phép sự tăng trưởng theo chiều dọc tiếp tục lâu hơn; tác động ròng là sự gia tăng chiều cao người lớn khoảng 4 cm. Các tác dụng phụ tiềm tàng bao gồm mật độ khoáng xương giảm, cũng như các hậu quả tâm lý của việc dậy thì muộn và tầm vóc thấp trong tuổi vị thành niên. Các lựa chọn cung cấp bao gồm tiêm bắp thường xuyên hoặc một thiết bị cấy ghép phải được tháo ra hoặc thay thế khoảng hàng năm. Việc điều trị này không được FDA chấp thuận cho trẻ em có tầm vóc thấp và dậy thì đúng thời điểm và được sử dụng ít thường xuyên hơn so với điều trị bằng GH.
Các chất ức chế Aromatase
Như đã lưu ý trước đó, estrogen tạm thời làm tăng tốc độ tăng trưởng theo chiều dọc, nhưng cũng làm tăng tốc độ lão hóa của sụn tăng trưởng, gây ra sự kết thúc sớm hơn của sự tăng trưởng theo chiều dọc, và tác động ròng thường là sự giảm chiều cao người lớn. Ở những người đàn ông bị thiếu hụt estrogen do các đột biến hai alen trong gen aromatase, sự giảm tốc và ngừng tăng trưởng theo chiều dọc bình thường ở giữa đến cuối tuổi vị thành niên bị trì hoãn, cho phép sự tăng trưởng theo chiều dọc chậm nhưng dai dẳng cho đến khi trưởng thành dẫn đến tầm vóc cao. Các tác động tương tự đã được báo cáo trong tình trạng kháng estrogen do đột biến hai alen trong gen thụ thể estrogen-alpha. Những quan sát này cho thấy rằng việc phong tỏa dược lý sản xuất estrogen bằng các chất ức chế aromatase trong tuổi vị thành niên của nam giới có thể trì hoãn sự trưởng thành của xương và kéo dài sự tăng trưởng theo chiều dọc, dẫn đến chiều cao người lớn lớn hơn. Về nguyên tắc, việc điều trị này có thể có một lợi thế so với việc điều trị bằng chất chủ vận GnRH bằng cách cho phép các tác động bình thường của androgen trong tuổi vị thành niên của nam giới.
Các thử nghiệm ngẫu nhiên có đối chứng của chất ức chế aromatase anastrozole hoặc letrozole mạnh hơn đã được tiến hành ở các bé trai có tầm vóc thấp và ở các bé trai bị chậm phát triển sinh lý. Tuy nhiên, các nghiên cứu này chưa được cung cấp đủ sức mạnh hoặc chưa được phân tích để đánh giá dứt khoát tác động lên chiều cao người lớn. Ngoài ra, các câu hỏi về sự an toàn vẫn còn, một số trong đó được gợi ý bởi kiểu hình của những người đàn ông thiếu hụt aromatase, bao gồm các tác động bất lợi có thể có đối với quá trình tạo tinh, mật độ khoáng xương, lipid, sự nhạy cảm với insulin, các chức năng nhận thức, và sự phát triển của tuyến tiền liệt. Ngoài ra, nồng độ testosterone có thể tăng lên các mức siêu sinh lý và các biến dạng đốt sống đã được báo cáo. Do các câu hỏi chưa được trả lời về hiệu quả và sự an toàn, các chất ức chế aromatase không được FDA chấp thuận để điều trị tầm vóc thấp và các hướng dẫn hiện tại của Hội Nội tiết Nhi khoa Hoa Kỳ kêu gọi thận trọng trong việc sử dụng các tác nhân này ngoài các thử nghiệm lâm sàng có đối chứng.
Các chất chủ vận Peptide Lợi Niệu Loại C cho bệnh Loạn Sản Sụn
ACH là bệnh loạn sản xương ở người phổ biến nhất và do một đột biến tăng chức năng trong FGFR3 dẫn đến tín hiệu quá mức trong con đường MAPK. Tín hiệu MAPK hoạt động quá mức dẫn đến sự tăng sinh và biệt hóa cuối cùng của các tế bào sụn bị suy giảm và sự tổng hợp chất nền ngoại bào trong sụn tăng trưởng bị suy giảm. Sự liên kết của CNP với thụ thể của nó, NPR-B, tạo ra guanosine monophosphate vòng kích hoạt cGKII, ức chế sự kích hoạt của con đường MAPK. Các mô hình động vật của ACH đã chứng minh rằng sự biểu hiện quá mức của CNP cứu vãn kiểu hình tăng trưởng xương bị suy giảm do sự kích hoạt FGFR3 gây ra. Bằng chứng này cho thấy rằng CNP có thể là một liệu pháp tiềm năng cho ACH. Một số thử nghiệm lâm sàng đang được tiến hành để đánh giá hiệu quả và sự an toàn của các chất tương tự CNP ở trẻ em bị ACH (www.clinicaltrials.gov).
Điều trị phẫu thuật
Dựa trên sự tạo xương kéo giãn (tức là, phẫu thuật cắt xương sau đó là sự gián đoạn nhịp nhàng dần dần của sự hình thành mô sẹo xương bằng cách sử dụng một thiết bị cố định bên ngoài, chẳng hạn như khung Ilizarov), phẫu thuật kéo dài chân ban đầu được phát triển để điều chỉnh sự chênh lệch chiều dài hoặc các biến dạng. Phẫu thuật kéo dài chân hai bên đã được đề xuất như một phương pháp điều trị cho một số dạng tầm vóc thấp nghiêm trọng, đặc biệt là những dạng có sự rút ngắn của các chi, ví dụ, ACH. Kỹ thuật này có thể được áp dụng để kéo dài xương chày và xương đùi một mình hoặc kết hợp. Quy trình này có khả năng thúc đẩy một sự gia tăng chiều cao tổng thể đáng kể (20 cm), nhưng có tần suất biến chứng và di chứng cao. Phẫu thuật có thể được thực hiện ở thời thơ ấu, nhưng không được khuyến nghị cho đến khi đứa trẻ đủ lớn để tham gia vào quyết định điều trị. Mặc dù kéo dài chân có khả năng mang lại sự thích ứng tốt hơn cho những bệnh nhân có tầm vóc thấp nghiêm trọng, nó có tính xâm lấn và liên quan đến nguy cơ cao của các biến chứng ngắn hạn và dài hạn và gây tranh cãi giữa những cá nhân bị loạn sản xương (https://www.lpaonline.org/). Vì những lý do này, quy trình nên được thực hiện tại các trung tâm chuyên biệt và chỉ trên những bệnh nhân được chọn có tầm vóc thấp nghiêm trọng đã nhận được thông tin đầy đủ về quy trình, rủi ro và hậu quả.
Tầm vóc cao
Một đứa trẻ có thể có chiều cao trên phạm vi bình thường vì nhiều lý do. Thường thì, cha mẹ cũng cao và tầm vóc của đứa trẻ dường như được di truyền như một đặc điểm thiểu gen hoặc đa gen. Trong những trường hợp này, tầm vóc cao được coi là một biến thể bình thường. Béo phì là một nguyên nhân phổ biến khác cho tầm vóc trên phạm vi bình thường. Tuy nhiên, cũng có một số lượng lớn các nguyên nhân không phổ biến của tầm vóc cao, hầu hết trong số chúng là di truyền. Một số nguyên nhân không phổ biến này, chẳng hạn như hội chứng Klinefelter, hội chứng Beckwith-Wiedemann (BWS), hội chứng Marfan, và thừa GH có những ý nghĩa sức khỏe quan trọng. Do đó, ở đứa trẻ có tầm vóc cao, một cuộc đánh giá lâm sàng cẩn thận là cần thiết.
Tầm vóc cao là kết quả của sự tạo sụn quá mức ở sụn tăng trưởng. Sự tạo sụn tăng lên này có thể phát sinh từ một nguyên nhân chính trong chính các tế bào sụn ở sụn tăng trưởng hoặc có thể phát sinh từ một nguyên nhân thứ cấp, xảy ra ở nơi khác trong cơ thể và ảnh hưởng đến sụn tăng trưởng thông qua một yếu tố lưu hành, thường là nội tiết.
Nguyên nhân của Tầm vóc cao
Tăng trưởng quá mức thứ phát
Nguyên nhân nội tiết của Tầm vóc cao
Thừa Hormone tăng trưởng/Yếu tố tăng trưởng giống Insulin-1
Ở trẻ em có tốc độ tăng trưởng tăng nhưng không có các dấu hiệu dậy thì, nên xem xét thừa GH hoặc cường giáp. Khi tình trạng trước xảy ra trước khi cốt hóa sụn đầu xương, kết quả là tăng trưởng nhanh, các thay đổi chuyển hóa tương tự như những thay đổi được quan sát thấy ở bệnh nhân bị bệnh to đầu chi, và nồng độ IGF-1 trong huyết thanh tăng cao. Nồng độ GH có thể tăng cao trên một hồ sơ GH qua đêm, không phát hiện được các điểm đáy. Hơn nữa, nồng độ GH trong huyết thanh không bị ức chế bởi việc dùng glucose đường uống trong một nghiệm pháp dung nạp glucose đường uống. MRI có thể cho thấy một khối u tuyến yên dưới dạng một u tuyến nhỏ hoặc lớn. Những tiến bộ gần đây đã cho thấy một nguyên nhân di truyền ở một tỷ lệ đáng kể các bệnh nhân này. Các đột biến somatic GNAS1, PIK3CA, và USP8 có thể được xác định, hoặc khối u có thể xảy ra như một thành phần của các hội chứng, chẳng hạn như MAS, phức hợp Carney, MEN 1 và Loại 4, và các hội chứng liên quan đến DICER và succinate dehydrogenase (SDH). Các đột biến trong protein tương tác với arylhydrocarbon (AIP) có liên quan đến các u tuyến yên gia đình (FIPA), và gần đây hơn, các vi nhân đôi liên quan đến nhiễm sắc thể Xq26.3 có liên quan đến bệnh khổng lồ tuyến yên do các khối u tuyến yên như một phần của hội chứng to đầu chi liên kết X (X-LAG).
U tuyến yên gia đình
Tình trạng này được đặc trưng bởi hai hoặc nhiều trường hợp u tuyến yên trong một gia đình mà không có các khối u liên quan khác. Bệnh to đầu chi/khổng lồ có liên quan đến 54% các khối u, u tuyến prolactin với 27%, các u tuyến yên không hoạt động ở 17%; u tuyến prolactin và bệnh khổng lồ/to đầu chi có thể xảy ra trong cùng một phả hệ. Các đột biến AIP (một gen ức chế khối u) có mặt ở 15% đến 30% các gia đình FIPA, và ở 20% các u tuyến yên ở trẻ em lẻ tẻ (PA). Độ thấm của PA ở những người mang đột biến AIP dương tính là khoảng 12% đến 30%, cho thấy vai trò của các yếu tố điều chỉnh bệnh khác. Tuổi trung bình khi chẩn đoán là 18 đến 24 tuổi; gần như tất cả các trường hợp dương tính với đột biến AIP đều được chẩn đoán trước 40 tuổi. Các PA dương tính với đột biến AIP có nhiều khả năng là các u tuyến lớn (90%) và hung hăng, cần điều trị đa mô thức.
Hội chứng X-LAG
Điều này có liên quan đến các vi nhân đôi của Xq26.3 bao gồm thụ thể GPR101 mồ côi được ghép cặp với protein G, được biểu hiện quá mức đáng kể trong tuyến yên của những bệnh nhân này. Các đột biến có thể là de novo lẻ tẻ, cũng như các vi nhân đôi Xq26.3 dòng mầm gia đình, và các đột biến có thể là thể khảm. GPR101 cũng được biểu hiện cao ở vùng dưới đồi và nhân accumbens. Bệnh nhân bị ảnh hưởng thường có bệnh khổng lồ khởi phát sớm, thường là trước 5 tuổi, và có thể có cảm giác thèm ăn tăng, bệnh gai đen, các đặc điểm khuôn mặt thô, và đổ mồ hôi nhiều. Có sự chiếm ưu thế của nữ giới, và tình trạng này có liên quan đến các u tuyến lớn và tăng sản tế bào hướng thân. Tăng prolactin máu đã được mô tả ở 85% các trường hợp. Các đột biến AIP chiếm 30% bệnh khổng lồ tuyến yên, trong khi X-LAG chiếm 10% các trường hợp.
Thừa Steroid sinh dục
Tiếp xúc sớm với steroid sinh dục gây ra sự tăng tốc tăng trưởng ở trẻ em. Sự tiếp xúc sớm này có thể là kết quả của dậy thì sớm trung ương; các dạng nam hóa của tăng sản tuyến thượng thận bẩm sinh; các nang buồng trứng; hoặc các khối u tuyến thượng thận, tinh hoàn, hoặc buồng trứng; hoặc steroid sinh dục ngoại sinh và các nguyên nhân khác. Nồng độ estrogen tăng cao làm tăng tốc độ trưởng thành của xương, bao gồm cả sự lão hóa của sụn tăng trưởng, dẫn đến tuổi xương cao, ngừng tăng trưởng sớm, và cốt hóa sụn đầu xương sớm. Do sự lão hóa sụn tăng trưởng tăng tốc và ngừng tăng trưởng sớm này, bệnh nhân thường cho thấy tầm vóc tăng ở thời thơ ấu nhưng tầm vóc giảm ở tuổi trưởng thành. Một trong những mục tiêu của việc điều trị dậy thì sớm, cho dù là trung ương hay ngoại vi, là để bảo tồn chiều cao người lớn. Trong dậy thì sớm trung ương được điều trị bằng một chất tương tự GnRH, chiều cao người lớn đạt được có xu hướng lớn hơn chiều cao người lớn được dự đoán trước khi điều trị, khiêm tốn ít hơn chiều cao được dự đoán vào cuối điều trị, và thường khiêm tốn ít hơn chiều cao trung bình của cha mẹ, đặc biệt nếu việc điều trị không được bắt đầu ngay sau khi khởi phát dậy thì.
Tăng trưởng kéo dài do sự trưởng thành xương bị trì hoãn
Điều này bao gồm một nhóm bệnh không đồng nhất, bao gồm suy sinh dục, thiếu hụt aromatase, và kháng estrogen. Trái ngược hoàn toàn với dậy thì sớm, những bệnh nhân này không có giai đoạn tăng vọt vì không có, hoặc kháng với, steroid sinh dục. Tốc độ tăng trưởng chậm nhưng, sự thiếu hụt trong hoạt động của estrogen cũng làm chậm sự lão hóa của sụn tăng trưởng, cho phép những bệnh nhân này tiếp tục phát triển đến tuổi trưởng thành, phát triển tầm vóc cao với các tỷ lệ hoạn quan (tăng sải tay và tỷ lệ phân đoạn trên-dưới) chỉ sau này trong cuộc đời. Do đó, những bệnh nhân không được điều trị bị suy sinh dục có thể cho thấy tầm vóc bình thường ở thời thơ ấu, tầm vóc hơi thấp ở đầu tuổi vị thành niên, và tầm vóc cao ở tuổi trưởng thành.
Thiếu hụt Aromatase
Thiếu hụt Aromatase là một bệnh hiếm gặp có di truyền lặn trên nhiễm sắc thể thường. Ở nam giới, chẩn đoán được đưa ra muộn hơn ở tuổi trưởng thành, khi có tầm vóc cao, đóng sụn đầu xương không hoàn toàn, các tỷ lệ xương của hoạn quan, loãng xương, và béo phì. Nồng độ estrogen thấp, trong khi FSH, LH, và testosterone tăng nhẹ. Các đặc điểm hấp dẫn nhất là sự hiện diện của viêm gan nhiễm mỡ, kháng insulin với bệnh gai đen, và nồng độ triglyceride cao. Việc dùng estrogen liều thấp cho phép hoàn thành sự trưởng thành của xương sau khi đóng hoàn toàn các sụn đầu xương và sau đó dẫn đến sự gia tăng mật độ xương. Hơn 30 đột biến (đột biến điểm, xóa và chèn) đã được tìm thấy trong gen CYP19A1, mã hóa enzyme P450 aromatase được biểu hiện ở một số mô, chẳng hạn như tuyến sinh dục, não, hợp bào lá nuôi nhau thai, vú, và mô mỡ, và xúc tác quá trình sinh tổng hợp estrogen từ androgen.
Thiếu hụt Thụ thể Estrogen α
Các trường hợp hiếm gặp của thiếu hụt thụ thể estrogen α (ERα) đã được báo cáo với sự tương đồng kiểu hình đáng kể với thiếu hụt aromatase với tầm vóc cao và các tỷ lệ cơ thể của hoạn quan. Các bệnh nhân cho thấy sự tăng trưởng theo chiều dọc tiếp tục đến tuổi trưởng thành do sự trưởng thành xương bị trì hoãn, loãng xương, không phát triển vú với nồng độ estrogen trong huyết thanh tăng rõ rệt, và buồng trứng đa nang và vô kinh ở nữ giới. Cho đến nay, những bệnh nhân kháng estrogen hiếm gặp đã có các đột biến sai nghĩa đồng hợp tử trong gen ERα.
Nhiễm độc giáp
Trong cường giáp, sự gia tăng tốc độ tăng trưởng có liên quan đến tuổi xương cao, mặc dù tầm vóc và tuổi xương thường vẫn trong phạm vi bình thường. Chẩn đoán được đưa ra bằng cách đo nồng độ T4 tự do và TSH. Nguyên nhân phổ biến nhất là bệnh Graves.
Hội chứng McCune-Albright
MAS là một nguyên nhân hiếm gặp của dậy thì sớm không phụ thuộc gonadotropin, thừa GH, và cường giáp. Đây là một rối loạn di truyền do một đột biến hoạt hóa trong gen GNAS, mã hóa tiểu đơn vị alpha của protein G kích thích. Đột biến somatic gây bệnh phát sinh trong quá trình phát triển phôi và được phân phối theo một mô hình khảm. Nó được đặc trưng bởi bộ ba loạn sản xơ xương đa ổ, dậy thì sớm, và các đốm da màu cà phê sữa. Hơn nữa, các bệnh nội tiết liên quan khác đã được công nhận, bao gồm cường giáp, thừa GH, lãng phí phosphate qua trung gian FGF23, và tăng cortisol máu. Các biểu hiện da là phổ biến và thường có mặt tại hoặc ngay sau khi sinh. Các đốm cà phê sữa thường có bờ không đều mang lại cho chúng một vẻ ngoài “bờ biển Maine”, và thường cho thấy một sự liên quan với đường giữa của cơ thể. Trong MAS, loạn sản xơ xương thường xảy ra ở một số vị trí (đa ổ), và thường biểu hiện bằng gãy xương, biến dạng, và/hoặc đau xương. X-quang cho thấy các tổn thương lan rộng đặc trưng với vẻ ngoài “kính mờ”.
Các nguyên nhân nội tiết khác
Thiếu hụt glucocorticoid gia đình (FGD) là một tình trạng được đặc trưng bởi sự kháng với ACTH. Ngoài các đột biến trong thụ thể melanocortin 2, một số nguyên nhân khác đã được xác định. FGD cũng liên quan đến tầm vóc cao.
Trẻ sơ sinh bị hạ đường huyết do tăng insulin bẩm sinh hoặc những trẻ sinh ra từ những bà mẹ bị đái tháo đường có xu hướng to hơn so với tuổi thai mặc dù chúng không có xu hướng cao về lâu dài.
Béo phì dinh dưỡng
Béo phì đã trở thành nguyên nhân phổ biến nhất của tầm vóc cao. Biểu đồ tăng trưởng là “xét nghiệm” chẩn đoán quan trọng nhất trong việc đánh giá béo phì; các nguyên nhân nội tiết của việc tăng cân và béo phì quá mức (thừa glucocorticoid—cả hội chứng Cushing do thuốc và nội sinh, suy giáp, và thiếu hụt GH) đều có xu hướng làm giảm sự tăng trưởng tầm vóc, trong khi béo phì dinh dưỡng (tức là, “ngoại sinh”) có liên quan đến sự tăng trưởng tầm vóc mạnh mẽ. Béo phì cũng làm tăng tốc độ trưởng thành của xương, bằng chứng là tuổi xương, và có liên quan đến tỷ lệ khởi phát tuyến thượng thận sớm tăng lên và, ở các bé gái, dậy thì sớm. Do đó, mặc dù các phân vị chiều cao của trẻ em béo phì thường vượt quá các phân vị chiều cao trung bình của cha mẹ chúng, những đứa trẻ này thường ngừng phát triển sớm hơn sao cho chiều cao người lớn kết thúc ở một phân vị thấp hơn so với phân vị được theo dõi trong thời thơ ấu, và do đó béo phì dường như có ít ảnh hưởng đến chiều cao người lớn.
Béo phì có nhiều tác động lên trục GH/IGF-1. Sự bài tiết GH tự phát bị giảm cả về tần số và biên độ xung, và sự bài tiết biên độ thấp hơn cũng có thể rõ ràng trên xét nghiệm GH kích thích. Mặc dù có sự giảm GH, nồng độ IGF-1 lưu hành tương tự hoặc, trong một số nghiên cứu, cao hơn ở những người béo phì so với những người không béo phì. Một số nghiên cứu cho thấy rằng lượng IGF-1 có sẵn sinh học tăng lên do sự giảm IGFBP-1 và IGFBP-2 lưu hành (IGFBP-3 không thay đổi). Nồng độ GHBP lưu hành cũng tăng.
Có lẽ ví dụ cực đoan nhất là một hiện tượng được gọi là tăng trưởng không có GH, theo đó trẻ em béo phì có thể duy trì vận tốc tăng trưởng bình thường mặc dù không có GH rõ ràng, chẳng hạn như do cắt bỏ phẫu thuật u sọ hầu. Các cơ chế liên quan để duy trì sự tăng trưởng tầm vóc của chúng bao gồm insulin, leptin, hormone sinh dục, và một adipokine thúc đẩy tăng trưởng, chẳng hạn như yếu tố tăng trưởng và biệt hóa 5.
Tăng trưởng quá mức nguyên phát
Ở một số trẻ, sự tạo sụn quá mức ở sụn tăng trưởng chịu trách nhiệm cho tầm vóc cao phát sinh từ một bất thường chính trong chính các tế bào sụn ở sụn tăng trưởng. Các nguyên nhân chính này bao gồm các khiếm khuyết gen đơn lẻ ảnh hưởng đến tín hiệu tự tiết/cận tiết trong sụn tăng trưởng, chất nền ngoại bào sụn, hoặc các con đường điều hòa nội bào. Ở những đứa trẻ khác, nguyên nhân cơ bản không phải là một khiếm khuyết gen đơn lẻ mà là một bất thường di truyền khác, chẳng hạn như một bất thường nhiễm sắc thể, một CNV di truyền (xóa hoặc nhân đôi), hoặc rối loạn in dấu.
Các Rối Loạn Liên Quan Đến Các Yếu Tố Tự Tiết/Cận Tiết
Yếu tố tăng trưởng nguyên bào sợi-3 (FGFR3) điều hòa tiêu cực quá trình tạo sụn ở sụn tăng trưởng. Do đó, như đã thảo luận trước đó trong chương, các đột biến tăng chức năng trong FGFR3 làm suy giảm quá trình tạo sụn ở sụn tăng trưởng và có thể biểu hiện lâm sàng như một bệnh loạn sản sụn với tầm vóc thấp, chẳng hạn như loạn sản sụn hoặc ACH. Các đột biến khác trong FGFR3, có khả năng liên quan đến sự mất chức năng, dường như gây ra sự tạo sụn quá mức ở sụn tăng trưởng, biểu hiện lâm sàng như tầm vóc cao với các bất thường xương.
CNP tác động thông qua NPR2 để kích thích quá trình tạo sụn ở sụn tăng trưởng (xem thảo luận trước đó). Do đó, các đột biến mất chức năng trong NPR2 làm chậm quá trình tạo sụn gây ra tầm vóc thấp. Ngược lại, các đột biến tăng chức năng trong NPR2 gây ra tầm vóc cao, liên quan đến vẹo cột sống và tật ngón to của ngón chân cái.
Hội chứng Simpson-Golabi-Behmel loại 1 (SGBS1) được đặc trưng bởi sự phát triển quá mức trước và sau sinh, các đặc điểm khuôn mặt đặc biệt, thiểu năng trí tuệ, và nhiều dị tật bẩm sinh khác nhau. Nó liên kết với nhiễm sắc thể X và có thể do các đột biến trong GPC3 hoặc 4 gây ra. Các glypican được mã hóa bởi các gen này là các proteoglycan heparan sulfate bề mặt tế bào. Có bằng chứng cho thấy rằng các glypican tham gia vào tín hiệu BMP, FGF, hedgehog (HH), và wingless-integrated (WNT).
Các Rối Loạn Liên Quan Đến Chất Nền/Dịch Ngoại Bào Sụn
Fibrillin-1 (FBN1) là một thành phần chính của các vi sợi, được tìm thấy ở ngoại bào trong các mô liên kết. Một số đột biến dị hợp tử trong FBN1 gây ra hội chứng Marfan, được di truyền theo kiểu trội trên nhiễm sắc thể thường và có nhiều biểu hiện ở xương, mắt, và tim mạch. Các bất thường xương bao gồm tầm vóc cao với các chi và ngón tay dài không cân đối, cũng như ngực lõm hoặc ngực ức gà và vẹo cột sống. Các phát hiện ở mắt bao gồm cận thị và lệch thủy tinh thể. Các biểu hiện tim mạch bao gồm sự giãn rộng của gốc động mạch chủ với nguy cơ cao bị bóc tách động mạch chủ. Các đột biến khác trong FBN1 có thể gây ra các bệnh loạn sản đầu chi, được đặc trưng bởi tầm vóc thấp với sự rút ngắn không cân đối của các chi xa.
Homocystin niệu do thiếu hụt cystathionine β-synthase cho thấy một số điểm tương đồng với hội chứng Marfan. Nó có thể ảnh hưởng đến bộ xương, gây ra tầm vóc cao với các chi dài không cân đối, vẹo cột sống, và ngực lõm. Các biểu hiện khác bao gồm cận thị và lệch thủy tinh thể, huyết khối tắc mạch, và chậm phát triển. Có bằng chứng cho thấy rằng sự tăng trưởng theo chiều dọc quá mức là do nồng độ homocysteine ngoại bào cao.
Các Rối Loạn Liên Quan Đến Các Yếu Tố Nội Bào
Hội chứng Klinefelter là một rối loạn nhiễm sắc thể tương đối phổ biến thường liên quan đến karyotype 47,XXY, đôi khi có thể khảm. Các đặc điểm kinh điển bao gồm một người nam có tầm vóc cao với các chi dưới tương đối dài, tinh hoàn nhỏ thường với vô tinh trùng, và nữ hóa tuyến vú (xem Chương 18 về Dậy thì và các rối loạn của nó ở Nam giới để biết chi tiết). Tầm vóc cao và chiều dài cẳng chân tăng được cho là do số lượng bản sao SHOX tăng lên, một yếu tố phiên mã dường như quan trọng đối với chức năng sụn tăng trưởng. SHOX nằm trong một vùng giả nhiễm sắc thể thường trên các nhiễm sắc thể X và Y và thoát khỏi sự bất hoạt X. Do đó, nam và nữ bình thường có hai bản sao hoạt động của SHOX, trong khi các bé gái bị hội chứng Turner chỉ có một bản sao, gây ra tầm vóc thấp, và các bé trai bị hội chứng Klinefelter có ba bản sao hoạt động, gây ra tầm vóc cao.
Các đột biến trong các gen ảnh hưởng đến các sửa đổi ngoại di truyền có thể gây ra sự phát triển quá mức. Hội chứng Sotos được đặc trưng bởi sự phát triển quá mức, thường là khởi phát trước khi sinh. Sự phát triển quá mức ảnh hưởng đến các chi nhiều hơn cột sống và đi kèm với đầu to và tuổi xương cao. Ngoài ra, có các đặc điểm khuôn mặt đặc trưng và chậm phát triển thay đổi. Nó có thể do các đột biến dị hợp tử trong NSD1, mã hóa một histone methyltransferase và do đó ảnh hưởng đến sự kiểm soát ngoại di truyền của biểu hiện gen. Hội chứng Malan, còn được gọi là hội chứng Sotos 2, là một rối loạn tương tự được đặc trưng bởi sự phát triển quá mức, đầu to, các đặc điểm khuôn mặt đặc trưng, và chậm phát triển và do các đột biến trong gen yếu tố hạt nhân I X (NFIX) gây ra. Các đột biến trong NFIX cũng có thể gây ra hội chứng Marshall-Smith, có thể bao gồm tầm vóc cao với tuổi xương cao, thiểu năng trí tuệ, và các đặc điểm khuôn mặt đặc biệt.
Hội chứng Weaver có các đặc điểm tương tự như hội chứng Sotos, bao gồm tầm vóc cao, tuổi xương cao, các đặc điểm khuôn mặt đặc trưng, và thiểu năng trí tuệ thay đổi. Nó có thể do các đột biến trong EZH2, EED, hoặc SUZ12, tham gia vào phức hợp protein PRC2 và có liên quan đến sự methyl hóa histone, một sửa đổi ngoại di truyền. Một hội chứng phát triển quá mức tương tự là do các đột biến dị hợp tử trong DNMT3a, mã hóa một DNA methyltransferase ảnh hưởng đến sự methyl hóa DNA.
BWS biểu hiện như sự phát triển quá mức, thường là cả trước và sau sinh, có thể ảnh hưởng không cân đối đến các mô khác nhau dẫn đến lưỡi to và phì đại nửa người. Bệnh nhân có nguy cơ cao bị các khiếm khuyết thành bụng khi sinh, tăng insulin máu, và các khối u phôi. BWS có thể do các bất thường di truyền hoặc ngoại di truyền trong một cụm các gen được in dấu ở vùng 11p15.5–11p15.4. Thường thì, có sự methyl hóa DNA bất thường trong vùng này và sự biểu hiện bất thường của các gen được in dấu, CDKN1C (một chất ức chế chu kỳ tế bào) và IGF2 (một yếu tố tăng trưởng cận tiết).
Hội chứng Perlman là một hội chứng phát triển quá mức tương tự như BWS nhưng được di truyền theo kiểu lặn trên nhiễm sắc thể thường. Nó do các đột biến trong DIS3L2, được cho là có liên quan đến sự phân hủy RNA thông tin.
Tăng trưởng quá mức vô căn
Phần lớn trẻ em cao khỏe mạnh không có nguyên nhân rõ ràng của sự tăng trưởng quá mức sẽ được phân loại là tầm vóc cao vô căn, bao gồm tầm vóc cao gia đình và sự tiến triển sinh lý trong tăng trưởng và tuổi vị thành niên. Cả hai đều là chẩn đoán loại trừ và là các chẩn đoán phổ biến nhất ở những người có tầm vóc cao. Trẻ em có tầm vóc cao gia đình thường duy trì sự tăng trưởng ở cùng một phân vị cao và đạt được chiều cao người lớn trong phạm vi mục tiêu của chúng. Sự tiến triển sinh lý trong tăng trưởng được đặc trưng bởi một mô hình phát triển ngược lại với sự chậm phát triển sinh lý. Nó có thể liên quan, mặc dù không phải độc quyền, với béo phì. Những bệnh nhân này cho thấy sự tăng trưởng nhanh với tuổi xương cao và dậy thì tương đối sớm, thường dẫn đến chiều cao người lớn bình thường. Trong nhóm trẻ em có tầm vóc cao đơn độc này, rất hiếm khi xác định được một nguyên nhân đơn gen cho sự phát triển quá mức. Mô hình tăng trưởng và chiều cao của những đứa trẻ này có lẽ được giải thích bởi sự kết hợp của một số biến thể phổ biến trong một tác động đa gen điển hình. Tuy nhiên, khả năng dự đoán tầm vóc cao bằng cách sử dụng các đa hình được xác định trong GWAS chiều cao là hạn chế.
Phương pháp tiếp cận chẩn đoán bệnh nhân có Tầm vóc cao
Mặc dù chúng ta có thể định nghĩa tầm vóc cao là một SDS chiều cao trên 2, trong trường hợp không có các triệu chứng và dấu hiệu khác, phần lớn trẻ em và thanh thiếu niên cao là khỏe mạnh và có tầm vóc cao gia đình hoặc sự tiến triển sinh lý trong tăng trưởng. Ngoài ra, tầm vóc cao được xã hội chấp nhận rộng rãi hơn tầm vóc thấp và các tình trạng thứ cấp dẫn đến sự phát triển quá mức là không thường xuyên. Vì những lý do này, các tiêu chí giới thiệu hạn chế hơn thường được sử dụng để cải thiện việc xác định các tình trạng phát triển quá mức bệnh lý và do đó giảm các xét nghiệm không cần thiết. Bệnh nhân có tầm vóc cao nghiêm trọng hơn, những người có chiều cao cao hơn đáng kể so với chiều cao trung bình của cha mẹ hoặc vượt qua các phân vị lên trên trong thời thơ ấu và/hoặc dậy thì có nhiều khả năng có tầm vóc cao do một tình trạng bệnh lý cụ thể và cần được điều tra lâm sàng. Hơn nữa, sự hiện diện của thiểu năng trí tuệ, đầu to, hoặc các đặc điểm dị dạng có thể chỉ ra một tình trạng tầm vóc cao hội chứng (nói chung có cơ sở di truyền). Ở trẻ em có tầm vóc cao và các đặc điểm bổ sung này, nên xem xét một cuộc đánh giá chi tiết hơn về các hội chứng di truyền.
Các chẩn đoán phân biệt chính của trẻ em có tầm vóc cao là tiếp xúc sớm với steroid sinh dục, các lệch bội nhiễm sắc thể giới tính thừa (ví dụ, hội chứng Klinefelter), thừa hormone tăng trưởng (tức là, bệnh khổng lồ tuyến yên), BWS, hội chứng Marfan, hội chứng Sotos, hội chứng Weaver, và các tình trạng di truyền hiếm gặp khác (xem phần Nguyên nhân của Tầm vóc cao trước đó). Mặc dù có sự phức tạp và đa dạng của các nguyên nhân tiềm tàng cho tầm vóc cao, một số chẩn đoán có thể được thu được từ một bệnh sử được ghi nhận cẩn thận và một cuộc khám thực thể toàn diện (Bảng 11.7). Tùy thuộc vào các phát hiện lâm sàng cụ thể từ bệnh sử và khám thực thể, các cuộc điều tra đặc biệt thường được yêu cầu để xác nhận ấn tượng lâm sàng ban đầu (Bảng 11.8).
Bảng 11.7 Các phát hiện chính trong Bệnh sử và Khám thực thể của trẻ em có Tầm vóc cao
| Phát hiện chính | Rối loạn |
|---|---|
| Tiền sử gia đình gợi ý di truyền lặn trên NST thường và/hoặc có quan hệ huyết thống | Homocystin niệu, hội chứng CATSHL |
| Tiền sử gia đình gợi ý di truyền trội trên NST thường của tầm vóc cao | Tầm vóc cao gia đình, hội chứng Marfan, loạn sản sụn đầu xương, loại Miura (tăng tín hiệu bởi CNP/NPR-B) |
| Chậm phát triển, thiểu năng trí tuệ và các vấn đề hành vi | Hội chứng Sotos, Weaver, homocystin niệu, nhiễm sắc thể X dễ gãy, đột biến DNMT3A, và các hội chứng khác |
| Cân nặng và chiều dài khi sinh tăng SDS theo tuổi thai | Hội chứng Beckwith-Wiedemann, Sotos, Weaver, nhân đôi IGF1R, và các hội chứng khác |
| SDS chu vi vòng đầu | Đầu to được quan sát thấy ở các hội chứng Sotos, Weaver, đột biến DNMT3A, và nhiễm sắc thể X dễ gãy; Đầu nhỏ được quan sát thấy ở hội chứng CATSHL |
| Sự hiện diện của các đặc điểm sinh dục thứ phát sớm | Tiếp xúc sớm với steroid sinh dục (dậy thì sớm, các rối loạn nam hóa) |
| Các dấu hiệu của suy sinh dục (tinh hoàn nhỏ, vô kinh, kém phát triển vú và lông cơ thể) | Một số nguyên nhân khác nhau; ở nam giới, chủ yếu là hội chứng Klinefelter |
| Dáng người Marfanoid (sải tay vượt quá chiều cao, tật dính ngón và tăng độ lỏng lẻo) | Hội chứng Marfan, Sotos, homocystin niệu, loạn sản sụn đầu xương, loại Miura |
| Tật ngón to của ngón chân cái | Loạn sản sụn đầu xương, loại Miura (tăng tín hiệu bởi CNP/NPR-B) |
| Mất cân đối cơ thể (sải tay vượt quá chiều cao và/hoặc chiều cao ngồi: tổng chiều cao SDS <–2) | Hội chứng Klinefelter |
| Các đặc điểm khuôn mặt dị dạng | Một số dạng hội chứng của các tình trạng phát triển quá mức, mỗi dạng có các đặc điểm riêng |
| Gù vẹo cột sống nghiêm trọng | Hội chứng CATSHL, loạn sản sụn đầu xương, loại Miura |
Các dấu hiệu và triệu chứng được liệt kê hữu ích cho chẩn đoán khi có mặt nhưng có thể không có ở một số trẻ mắc các rối loạn liên quan. CATSHL, Hội chứng co ngón, tầm vóc cao, và mất thính lực (đột biến FGFR3 lặn); CNP, peptide lợi niệu loại C; NPR-B, thụ thể peptide lợi niệu B; SDS, điểm độ lệch chuẩn.
Bảng 11.8 Các xét nghiệm chẩn đoán bổ sung có thể hữu ích trong quá trình điều tra một bệnh nhân có Tầm vóc cao
| Xét nghiệm | Mục tiêu |
|---|---|
| X-quang cổ tay và bàn tay để xác định tuổi xương | Tuổi xương cao thường được quan sát thấy ở những bệnh nhân mắc hội chứng Sotos, hội chứng Weaver, hội chứng Beckwith-Wiedemann, tiếp xúc sớm với steroid sinh dục, sự tiến triển sinh lý của sự tăng trưởng, và béo phì. Tuổi xương chậm được quan sát thấy trong suy sinh dục, thiếu hụt aromatase, và kháng estrogen |
| IGF-1 huyết thanh, GH ngẫu nhiên và OGTT để ức chế GH | Sàng lọc thừa GH |
| TSH huyết thanh, T4 tự do | Sàng lọc cường giáp |
| Mức LH, FSH, Testosterone/estradiol | Để đánh giá sự tiếp xúc sớm với steroid sinh dục khi có các dấu hiệu thực thể hoặc suy sinh dục |
| Homocysteine huyết thanh | Sàng lọc homocystin niệu chủ yếu ở những bệnh nhân có dáng người Marfanoid, cha mẹ có quan hệ huyết thống và/hoặc thiểu năng trí tuệ |
| Karyotype | Đánh giá các quang sai nhiễm sắc thể (tức là, hội chứng Klinefelter, 47 XYY, 47 XXX) hoặc CAIS |
| Siêu âm tim | Đánh giá sự giãn nở của gốc động mạch chủ (tức là, hội chứng Marfan) |
| Đánh giá nhãn khoa | Đánh giá lệch thủy tinh thể (tức là, hội chứng Marfan và homocystin niệu) |
| Trình tự FBN1 | Để xác nhận hoặc loại trừ chẩn đoán hội chứng Marfan ở những bệnh nhân có các phát hiện lâm sàng gợi ý |
| Karyotype phân tử (mảng SNP hoặc CGH) | Để xác định biến thể số lượng bản sao dưới kính hiển vi (xóa hoặc nhân đôi), chủ yếu ở những bệnh nhân bị chậm phát triển/thiểu năng trí tuệ không giải thích được, các rối loạn phổ tự kỷ, và/hoặc nhiều dị tật bẩm sinh |
| Trình tự thông lượng cao (trình tự toàn bộ exome hoặc bảng gen) | Để điều tra đồng thời sự hiện diện của các biến thể gây bệnh trong một số gen liên quan đến tầm vóc cao, đặc biệt là ở những bệnh nhân có nghi ngờ cao về các nguyên nhân di truyền |
CAIS, Không nhạy cảm với androgen hoàn toàn; CGH, lai so sánh bộ gen; FSH, hormone kích thích nang trứng; GH, hormone tăng trưởng; IGF-1, yếu tố tăng trưởng giống insulin-1; LH, hormone tạo hoàng thể; OGTT, nghiệm pháp dung nạp glucose đường uống; SNP, đa hình đơn nucleotide; T4, thyroxine, TSH, hormone kích thích tuyến giáp.
Việc xác định các mô hình tăng trưởng là một phần quan trọng của bệnh sử (Hình 11.11). Trẻ em cao vượt qua các phân vị lên trên nên được điều tra về sự tiếp xúc sớm với steroid sinh dục, thừa GH, và cường giáp. Mặt khác, những đứa trẻ cao trên 2 SD và vẫn ở cùng một phân vị và tương thích với chiều cao mục tiêu của phân vị của chúng cho thấy tầm vóc cao gia đình, trong trường hợp không có các phát hiện dương tính khác. Các hội chứng Sotos và Weaver nên được xem xét đặc biệt ở những trẻ sinh ra to so với tuổi thai vẫn còn có SDS chiều cao trên 2 và có liên quan đến đầu to và/hoặc suy giảm nhận thức. BWS nên được điều tra ở trẻ em có sự phát triển quá mức trước và sau sinh liên quan đến hạ đường huyết sơ sinh, lưỡi to, thoát vị rốn, thoát vị thành bụng, và/hoặc tiền sử có khối u phôi (ví dụ, u Wilms, u nguyên bào gan, u nguyên bào thần kinh, u cơ vân). Tầm vóc cao không cân đối (chân dài, sải tay vượt quá chiều cao, và/hoặc chiều cao ngồi: tổng chiều cao SDS <–2), tinh hoàn nhỏ, nữ hóa tuyến vú, khó khăn trong học tập, và/hoặc nồng độ gonadotropin tăng cho thấy hội chứng Klinefelter. Trẻ em cao có tiền sử cá nhân hoặc gia đình bị lệch thủy tinh thể (bán trật thủy tinh thể) hoặc giãn nở gốc động mạch chủ nên được điều tra về hội chứng Marfan. Cuối cùng, có những đứa trẻ có chiều cao trong phạm vi tham chiếu trong thời thơ ấu, nhưng có sự tăng trưởng kéo dài sau tuổi vị thành niên dẫn đến SDS chiều cao trên 2 ở tuổi trưởng thành, một mô hình có thể là kết quả của sự suy giảm sản xuất hoặc hoạt động của estrogen.
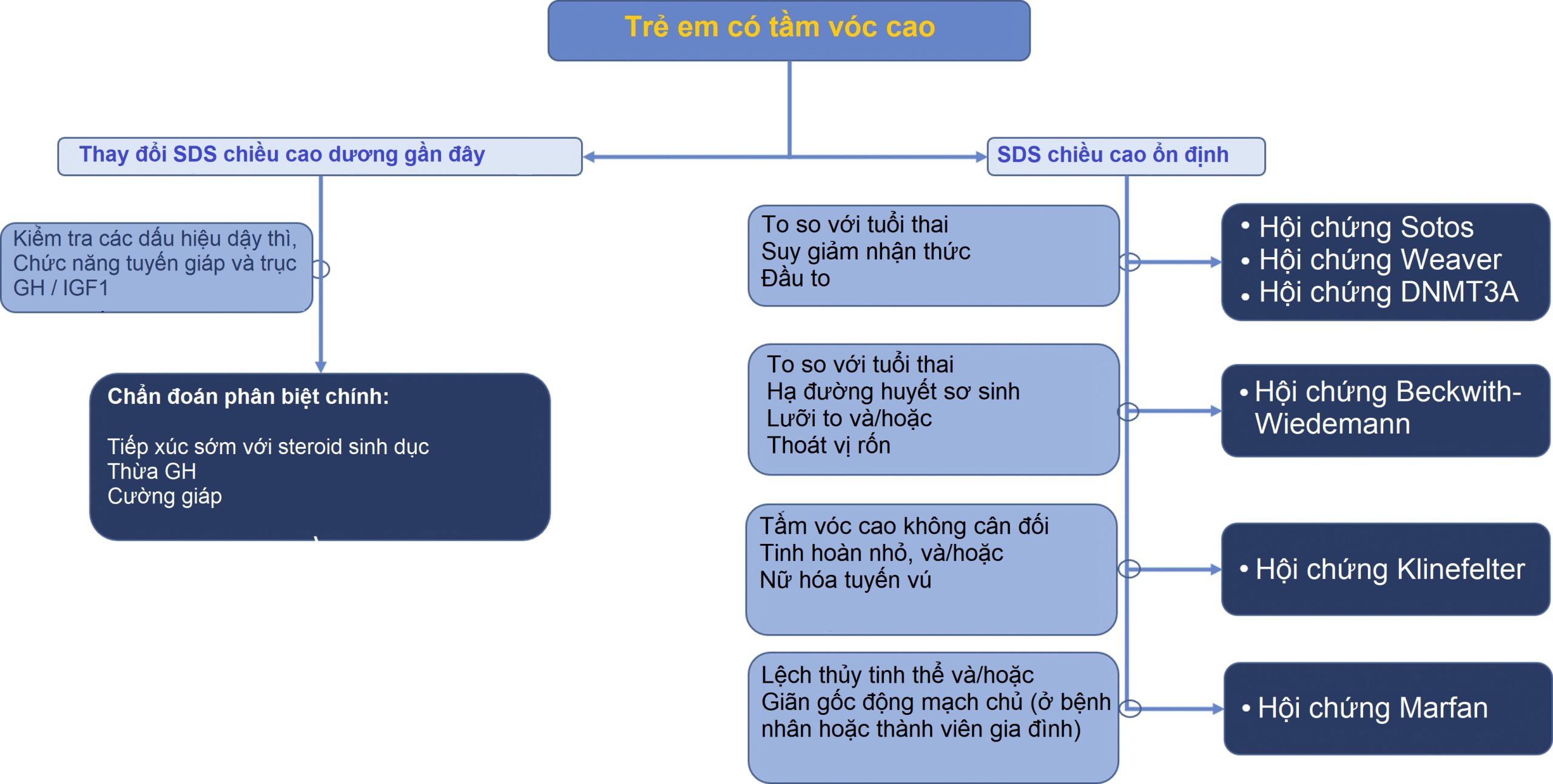
Hình 11.11 Tổng quan về việc điều tra trẻ em có tầm vóc cao.
Các xét nghiệm di truyền rất quan trọng để xác nhận các giả thuyết chẩn đoán và cũng để điều tra các bệnh nhân bị phát triển quá mức hội chứng không rõ nguyên nhân. Do sự không đồng nhất của các gen có thể gây ra tầm vóc cao bệnh lý, kích thước lớn của một số gen và sự hiếm gặp của các tình trạng này, một chiến lược đa gen sử dụng giải trình tự thế hệ tiếp theo (giải trình tự exome hoặc giải trình tự bảng gen nhắm mục tiêu) có thể hiệu quả hơn so với giải trình tự nhắm mục tiêu của một gen ứng cử viên duy nhất. Hơn nữa, karyotyping phân tử (mảng CGH hoặc SNP) cũng là một công cụ hữu ích để điều tra các bệnh nhân bị phát triển quá mức liên quan đến chậm phát triển, thiểu năng trí tuệ, hoặc các rối loạn phổ tự kỷ.
Điều trị Tầm vóc cao
Điều trị Thừa Hormone Tăng trưởng
Việc điều trị bệnh khổng lồ tuyến yên có thể phức tạp, và bao gồm liệu pháp y tế sử dụng các chất tương tự somatostatin, dẫn đến chữa khỏi về mặt sinh hóa ở 70% với sự co lại của khối u ở 75%. Cabergoline cũng có thể được thử và đặc biệt hiệu quả ở những bệnh nhân có các khối u tiết cả GH và prolactin. Chất đối kháng GH, Pegvisomant, cũng có thể được sử dụng mặc dù nó không gây co lại khối u. Cắt bỏ khối u qua xương bướm có liên quan đến sự chữa khỏi về mặt sinh hóa ở 75% đến 95%. Xạ trị được sử dụng trong các trường hợp kháng thuốc, nhưng có thể liên quan đến các biến chứng bao gồm suy tuyến yên, suy giảm thị lực, và có thể là các khối u thứ cấp.
Điều trị các nguyên nhân nội tiết khác của Tầm vóc cao
Khi tầm vóc cao là do thừa steroid sinh dục hoặc hormone tuyến giáp, sự bất thường nội tiết cơ bản thường có thể điều trị được. Các phương pháp điều trị cho dậy thì sớm, tăng sản tuyến thượng thận bẩm sinh, và cường giáp được thảo luận ở nơi khác.
Điều trị Tầm vóc cao không do nội tiết
Nói chung, tầm vóc cao không có nguyên nhân bệnh lý cụ thể không cần điều trị. Tuy nhiên, tầm vóc cao ở một đứa trẻ có thể gây lo lắng cho cha mẹ, những người sau đó có thể yêu cầu điều trị để ngừng sự tiến triển của sự tăng trưởng. Chưa có hướng dẫn dựa trên bằng chứng nào được tạo ra về việc lựa chọn các ứng cử viên tiềm năng để giảm chiều cao người lớn. Các quyết định điều trị thường bị ảnh hưởng bởi chiều cao người lớn được dự đoán, dựa trên một phim X-quang của bàn tay và cổ tay không thuận. Thông thường, việc điều trị chỉ được xem xét cho thanh thiếu niên có chiều cao người lớn được dự đoán cao hơn 2,5 SD so với mức trung bình của dân số. Thật không may, các phương pháp hiện có để dự đoán chiều cao người lớn, bao gồm phương pháp Bayley-Pinneau và các phương pháp TW Mark 1 và 2, có độ chính xác hạn chế và, tùy thuộc vào tuổi xương, có xu hướng đánh giá quá cao chiều cao người lớn.
Các loại điều trị khác nhau, hoặc nội tiết tố hoặc phẫu thuật, đã được sử dụng để giảm sự tăng trưởng ở trẻ em có tầm vóc cao.
Một phương pháp tiếp cận cho một dự đoán chiều cao quá mức đã được nghiên cứu là gây ra dậy thì sớm, do đó dẫn đến sự ngừng tăng trưởng sớm và cốt hóa sụn đầu xương sớm, bằng cách sử dụng testosterone ở nam giới và estrogen ở nữ giới. Tuy nhiên, việc sử dụng steroid sinh dục này đã giảm trong 20 năm qua, một phần do các tác dụng phụ của điều trị. Trong ngắn hạn, các bé trai được điều trị bằng androgen có thể có đau cơ, mụn trứng cá, nữ hóa tuyến vú, và tăng cân, trong khi các bé gái được điều trị bằng liều cao estrogen có thể bị tăng cân, chuột rút ban đêm, tiết sữa, nang buồng trứng, và khuynh hướng huyết khối. Cũng có bằng chứng cho thấy những phụ nữ cao được điều trị bằng estrogen liều cao có nguy cơ giảm khả năng sinh sản. Một số người đã ủng hộ việc gây ra dậy thì bằng cách sử dụng các liều steroid sinh dục sinh lý hơn trước khi đứa trẻ cao tiến vào tuổi dậy thì như một phương tiện để giảm chiều cao người lớn cuối cùng. Vẫn còn một lượng đáng kể sự không chắc chắn về hiệu quả của steroid sinh dục ngoại sinh trong việc hạn chế chiều cao cuối cùng, cho dù được sử dụng như một phần của chiến lược liều cao hay thấp hơn.
Truyền octreotide về đêm, một chất tương tự somatostatin, đã được báo cáo là làm giảm sự bài tiết GH và dự đoán chiều cao ở trẻ em cao. Tuy nhiên, một nghiên cứu gần đây hơn cho thấy rằng việc điều trị lâu dài bằng một chất tương tự somatostatin không làm giảm chiều cao cuối cùng một cách đủ để biện minh cho việc điều trị tầm vóc cao.
Thủ thuật phẫu thuật phổ biến nhất để giảm sự tăng trưởng là cốt hóa sụn đầu xương qua da hai bên của xương đùi xa và xương chày và xương mác gần. Cốt hóa sụn đầu xương là một can thiệp phẫu thuật để giảm chiều cao cuối cùng khoảng 5 cm, thường được thực hiện ở tuổi xương vượt quá 12,5 tuổi ở các bé gái có tầm vóc 170 cm và 14 tuổi ở các bé trai có tầm vóc hơn 185 cm.
Bảng chú giải thuật ngữ Y học Anh – Việt, Chương 11
| STT | Thuật ngữ tiếng Anh | Phiên âm IPA | Nghĩa Tiếng Việt |
|---|---|---|---|
| 1 | Endocrinology | /ˌɛndoʊkrɪˈnɒlədʒi/ | Nội tiết học |
| 2 | Growth | /ɡroʊθ/ | Sự tăng trưởng, sự phát triển |
| 3 | Embryo | /ˈɛmbrioʊ/ | Phôi thai |
| 4 | Fetus | /ˈfiːtəs/ | Thai nhi |
| 5 | Zygote | /ˈzaɪɡoʊt/ | Hợp tử |
| 6 | Linear growth | /ˈlɪniər ɡroʊθ/ | Tăng trưởng theo chiều dọc |
| 7 | Pubertal growth spurt | /pjuːˈbɜːrtəl ɡroʊθ spɜːrt/ | Giai đoạn tăng vọt tuổi dậy thì |
| 8 | Adulthood | /əˈdʌlthʊd/ | Tuổi trưởng thành |
| 9 | Polymorphisms | /ˌpɒliˈmɔːrfɪzəmz/ | Tính đa hình |
| 10 | Stature | /ˈstætʃər/ | Tầm vóc |
| 11 | Malformations | /ˌmælfɔːrˈmeɪʃənz/ | Dị tật |
| 12 | Malignancies | /məˈlɪɡnənsiz/ | Bệnh ác tính |
| 13 | Inflammatory bowel disease (IBD) | /ɪnˈflæmətɔːri ˈbaʊəl dɪˈziːz/ | Bệnh viêm ruột |
| 14 | Autoimmune thyroiditis | /ˌɔːtoʊɪˈmjuːn ˌθaɪrɔɪˈdaɪtɪs/ | Viêm tuyến giáp tự miễn |
| 15 | Central nervous system (CNS) | /ˈsɛntrəl ˈnɜːrvəs ˈsɪstəm/ | Hệ thần kinh trung ương |
| 16 | Pituitary dysfunction | /pɪˈtjuːɪtəri dɪsˈfʌŋkʃən/ | Rối loạn chức năng tuyến yên |
| 17 | Pathology | /pəˈθɒlədʒi/ | Bệnh học, bệnh lý |
| 18 | Celiac disease | /ˈsiːliæk dɪˈziːz/ | Bệnh Celiac (không dung nạp gluten) |
| 19 | Long bones | /lɔːŋ boʊnz/ | Xương dài |
| 20 | Vertebral bodies | /ˈvɜːrtəbrəl ˈbɒdiz/ | Thân đốt sống |
| 21 | Skeletal growth plates | /ˈskɛlɪtl ɡroʊθ pleɪts/ | Sụn tăng trưởng xương |
| 22 | Cartilage | /ˈkɑːrtəlɪdʒ/ | Sụn |
| 23 | Vertebrae | /ˈvɜːrtəbriː/ | Đốt sống |
| 24 | Chondrocytes | /ˈkɒndroʊsaɪts/ | Tế bào sụn |
| 25 | Proliferate | /prəˈlɪfəreɪt/ | Tăng sinh |
| 26 | Hypertrophy | /haɪˈpɜːrtrəfi/ | Phì đại |
| 27 | Chondrogenesis | /ˌkɒndroʊˈdʒɛnəsɪs/ | Sự tạo sụn |
| 28 | Remodeled | /ˌriːˈmɒdld/ | Tái cấu trúc |
| 29 | Endocrine signals | /ˈɛndəkrɪn ˈsɪɡnəlz/ | Tín hiệu nội tiết |
| 30 | Inflammatory cytokines | /ɪnˈflæmətɔːri ˈsaɪtəkaɪnz/ | Cytokine gây viêm |
| 31 | Nutritional intake | /njuːˈtrɪʃənəl ˈɪnteɪk/ | Lượng dinh dưỡng nạp vào |
| 32 | Paracrine/autocrine signals | /ˈpærəkrɪn/ /ˈɔːtəkrɪn ˈsɪɡnəlz/ | Tín hiệu cận tiết/tự tiết |
| 33 | Extracellular matrix | /ˌɛkstrəˈsɛljələr ˈmeɪtrɪks/ | Chất nền ngoại bào |
| 34 | Intracellular systems | /ˌɪntrəˈsɛljələr ˈsɪstəmz/ | Hệ thống nội bào |
| 35 | Pituitary gland | /pɪˈtjuːɪtəri ɡlænd/ | Tuyến yên |
| 36 | Sella turcica | /ˈsɛlə ˈtɜːrsɪkə/ | Hố yên |
| 37 | Hypophyseal fossa | /ˌhaɪpəˈfɪziəl ˈfɒsə/ | Hố tuyến yên |
| 38 | Sphenoid bone | /ˈsfiːnɔɪd boʊn/ | Xương bướm |
| 39 | Hypothalamus | /ˌhaɪpoʊˈθæləməs/ | Vùng dưới đồi |
| 40 | Hormonal production | /hɔːrˈmoʊnəl prəˈdʌkʃən/ | Sản xuất hormone |
| 41 | Metabolism | /məˈtæbəlɪzəm/ | Chuyển hóa |
| 42 | Homeostasis | /ˌhoʊmiəˈsteɪsɪs/ | Cân bằng nội môi |
| 43 | Lactation | /lækˈteɪʃən/ | Sự tiết sữa |
| 44 | Reproduction | /ˌriːprəˈdʌkʃən/ | Sinh sản |
| 45 | Rathke’s pouch | /ˈræθkiːz paʊtʃ/ | Túi Rathke |
| 46 | Stomodeal ectoderm | /ˌstoʊmoʊˈdiːəl ˈɛktədɜːrm/ | Ngoại bì miệng |
| 47 | Adenohypophysis | /əˌdiːnoʊhaɪˈpɒfəsɪs/ | Tuyến yên trước |
| 48 | Neural ectoderm | /ˈnʊərəl ˈɛktədɜːrm/ | Ngoại bì thần kinh |
| 49 | Neurohypophysis | /ˌnʊəroʊhaɪˈpɒfəsɪs/ | Tuyến yên sau |
| 50 | Pars distalis | /pɑːrz dɪsˈtɑːlɪs/ | Phần xa |
| 51 | Pars intermedia | /pɑːrz ˌɪntərˈmiːdiə/ | Phần trung gian |
| 52 | Pars tuberalis | /pɑːrz ˌtjuːbəˈrɑːlɪs/ | Phần củ |
| 53 | Hypothalamopituitary development | /ˌhaɪpoʊθəˌlæmoʊpɪˈtjuːɪtəri dɪˈvɛləpmənt/ | Sự phát triển của trục dưới đồi-tuyến yên |
| 54 | Rodent models | /ˈroʊdənt ˈmɒdəlz/ | Mô hình động vật gặm nhấm |
| 55 | Hypophyseal placode | /haɪpəˈfɪziəl ˈplækoʊd/ | Tấm tuyến yên |
| 56 | Diencephalon | /ˌdaɪənˈsɛfəlɒn/ | Não trung gian |
| 57 | Infundibulum | /ˌɪnfʌnˈdɪbjələm/ | Phễu |
| 58 | Notochord | /ˈnoʊtəkɔːrd/ | Dây sống |
| 59 | Prechordal plate | /ˌpriːˈkɔːrdəl pleɪt/ | Tấm trước dây sống |
| 60 | Sphenoid cartilage | /ˈsfiːnɔɪd ˈkɑːrtəlɪdʒ/ | Sụn bướm |
| 61 | Gestation | /dʒɛˈsteɪʃən/ | Thai kỳ |
| 62 | Progenitor cells | /proʊˈdʒɛnɪtər sɛlz/ | Tế bào tiền thân |
| 63 | Fibroblast growth factor (FGF) | /ˈfaɪbroʊblæst ɡroʊθ ˈfæktər/ | Yếu tố tăng trưởng nguyên bào sợi |
| 64 | Bone morphogenic protein (BMP) | /boʊn ˌmɔːrfoʊdʒəˈnɛtɪk ˈproʊtiːn/ | Protein tạo hình xương |
| 65 | Sonic hedgehog (Shh) | /ˈsɒnɪk ˈhɛdʒhɒɡ/ | (Gen) Sonic hedgehog |
| 66 | Transcription factors | /trænˈskrɪpʃən ˈfæktərz/ | Yếu tố phiên mã |
| 67 | Morphogens | /ˈmɔːrfədʒənz/ | Chất tạo hình thái |
| 68 | Cell differentiation | /sɛl ˌdɪfərɛnʃiˈeɪʃən/ | Biệt hóa tế bào |
| 69 | Transforming growth factors-β (TGFs) | /trænsˈfɔːrmɪŋ ɡroʊθ ˈfæktərz ˈbeɪtə/ | Yếu tố tăng trưởng biến đổi-β |
| 70 | Wnt signaling pathway | /wɪnt ˈsɪɡnəlɪŋ ˈpæθweɪ/ | Con đường tín hiệu Wnt |
| 71 | Pituitary tumorigenesis | /pɪˈtjuːɪtəri ˌtjuːmərɪdʒəˈnɛsɪs/ | Sự sinh u tuyến yên |
| 72 | Craniopharyngioma | /ˌkreɪnioʊfəˌrɪndʒiˈoʊmə/ | U sọ hầu |
| 73 | Pituitary cell lineages | /pɪˈtjuːɪtəri sɛl ˈlɪniɪdʒɪz/ | Dòng tế bào tuyến yên |
| 74 | Homeodomain | /ˈhoʊmioʊdoʊˌmeɪn/ | Miền homeo |
| 75 | Pituitary hormone deficiencies | /pɪˈtjuːɪtəri ˈhɔːrmoʊn dɪˈfɪʃənsiz/ | Thiếu hụt hormone tuyến yên |
| 76 | Panhypopituitarism | /ˌpænhypoʊpɪˈtjuːɪtərɪzəm/ | Suy toàn bộ tuyến yên |
| 77 | Optic nerve hypoplasia | /ˈɒptɪk nɜːrv ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản thần kinh thị |
| 78 | Septum pellucidum | /ˈsɛptəm pəˈluːsɪdəm/ | Vách trong suốt |
| 79 | Ectopic posterior pituitary | /ɛkˈtɒpɪk pɒˈstɪəriər pɪˈtjuːɪtəri/ | Tuyến yên sau lạc chỗ |
| 80 | Corpus callosum | /ˈkɔːrpəs kəˈloʊsəm/ | Thể chai |
| 81 | Cerebellar abnormalities | /ˌsɛrəˈbɛlər ˌæbnɔːrˈmælɪtiz/ | Bất thường tiểu não |
| 82 | Holoprosencephaly | /ˌhɒloʊˌprɒsənˈsɛfəli/ | Bất thường não trước không phân chia |
| 83 | Anophthalmia | /ˌænɒfˈθælmiə/ | Không có mắt |
| 84 | Postaxial polydactyly | /poʊstˈæksiəl ˌpɒliˈdæktɪli/ | Thừa ngón sau trục |
| 85 | Pallister-Hall syndrome | /ˈpælɪstər hɔːl ˈsɪndroʊm/ | Hội chứng Pallister-Hall |
| 86 | Hamartoma | /ˌhæmɑːrˈtoʊmə/ | U mô thừa |
| 87 | Microcephaly | /ˌmaɪkroʊˈsɛfəli/ | Đầu nhỏ |
| 88 | Chiari I malformation | /ˈkiɑːri wʌn ˌmælfɔːrˈmeɪʃən/ | Dị tật Chiari I |
| 89 | Brachydactyly | /ˌbrækɪˈdæktɪli/ | Tật ngón ngắn |
| 90 | Vesicoureteric reflux | /ˌvɛsɪkoʊˌjʊərɪˈtɛrɪk ˈriːflʌks/ | Trào ngược bàng quang-niệu quản |
| 91 | Hydronephrosis | /ˌhaɪdroʊnɛˈfroʊsɪs/ | Ứ nước thận |
| 92 | Gingival fibromatosis | /ˈdʒɪndʒɪvəl ˌfaɪbroʊməˈtoʊsɪs/ | U xơ nướu |
| 93 | Macroorchidism | /ˌmækroʊˈɔːrkɪdɪzəm/ | Tinh hoàn to |
| 94 | Adrenocorticotropic hormone (ACTH) | /əˌdriːnoʊˌkɔːrtɪkoʊˈtrɒpɪk ˈhɔːrmoʊn/ | Hormone vỏ thượng thận |
| 95 | Antidiuretic hormone (ADH) | /ˌæntidaɪjʊəˈrɛtɪk ˈhɔːrmoʊn/ | Hormone chống bài niệu |
| 96 | Follicle-stimulating hormone (FSH) | /ˈfɒlɪkəl ˈstɪmjəleɪtɪŋ ˈhɔːrmoʊn/ | Hormone kích thích nang trứng |
| 97 | Luteinizing hormone (LH) | /ˈluːtiənaɪzɪŋ ˈhɔːrmoʊn/ | Hormone tạo hoàng thể |
| 98 | Thyroid-stimulating hormone (TSH) | /ˈθaɪrɔɪd ˈstɪmjəleɪtɪŋ ˈhɔːrmoʊn/ | Hormone kích thích tuyến giáp |
| 99 | Septooptic dysplasia (SOD) | /ˌsɛptoʊˈɒptɪk dɪsˈpleɪʒə/ | Loạn sản vách-thị |
| 100 | Diaphragma sellae | /ˌdaɪəˈfræɡmə ˈsɛliː/ | Hoành yên |
| 101 | Optic chiasm | /ˈɒptɪk ˈkaɪæzəm/ | Giao thoa thị giác |
| 102 | Nystagmus | /nɪˈstæɡməs/ | Rung giật nhãn cầu |
| 103 | Bitemporal hemianopia | /baɪˈtɛmpərəl ˌhɛmiəˈnoʊpiə/ | Bán manh thái dương hai bên |
| 104 | Portal circulatory system | /ˈpɔːrtəl ˌsɜːrkjələˈtɔːri ˈsɪstəm/ | Hệ tuần hoàn cửa |
| 105 | Hypophyseal arteries | /haɪpəˈfɪziəl ˈɑːrtəriz/ | Động mạch tuyến yên |
| 106 | Hypophyseal portal veins | /haɪpəˈfɪziəl ˈpɔːrtəl veɪnz/ | Tĩnh mạch cửa tuyến yên |
| 107 | Somatotrope cells | /ˌsoʊmətəˈtroʊp sɛlz/ | Tế bào hướng thân |
| 108 | Nonglycosylated | /ˌnɒnɡlaɪˈkɒsəleɪtɪd/ | Không glycosyl hóa |
| 109 | Disulfide bonds | /daɪˈsʌlfaɪd bɒndz/ | Liên kết disulfide |
| 110 | Signal peptide | /ˈsɪɡnəl ˈpɛptaɪd/ | Peptide tín hiệu |
| 111 | Prolactin | /proʊˈlæktɪn/ | Prolactin |
| 112 | Chorionic somatomammotropin (CS) | /ˌkɔːriˈɒnɪk soʊˌmætəˌmæməˈtroʊpɪn/ | Somatomammotropin màng đệm |
| 113 | Placental lactogen | /pləˈsɛntəl ˈlæktədʒən/ | Lactogen nhau thai |
| 114 | Alternative splicing | /ɔːlˈtɜːrnətɪv ˈsplaɪsɪŋ/ | Cắt nối thay thế |
| 115 | Oligomers | /ˈɒlɪɡəmərz/ | Oligomer |
| 116 | Fetal serum | /ˈfiːtəl ˈsɪərəm/ | Huyết thanh thai nhi |
| 117 | Pulsatile pattern | /ˈpʌlsətaɪl ˈpætərn/ | Kiểu tiết xung |
| 118 | GH-releasing hormone (GHRH) | /dʒiː eɪtʃ rɪˈliːsɪŋ ˈhɔːrmoʊn/ | Hormone giải phóng GH |
| 119 | Somatostatin | /ˌsoʊmətəˈstætɪn/ | Somatostatin |
| 120 | G-protein–coupled receptor | /dʒiː ˈproʊtiːn ˈkʌpəld rɪˈsɛptər/ | Thụ thể kết cặp protein G |
| 121 | Somatotrophs | /ˌsoʊmətəˈtroʊfs/ | Tế bào hướng thân (tế bào tiết GH) |
| 122 | Adenylate cyclase | /əˈdɛnəleɪt ˈsaɪkleɪz/ | Adenylate cyclase |
| 123 | Cyclic adenosine monophosphate (cAMP) | /ˈsaɪklɪk əˈdɛnəsiːn ˌmɒnoʊˈfɒsfeɪt/ | Adenosine monophosphate vòng |
| 124 | Neurotransmitters | /ˌnʊəroʊtrænzˈmɪtərz/ | Chất dẫn truyền thần kinh |
| 125 | Neuropeptides | /ˌnʊəroʊˈpɛptaɪdz/ | Neuropeptide |
| 126 | Serotonin | /ˌsɛrəˈtoʊnɪn/ | Serotonin |
| 127 | Histamine | /ˈhɪstəmiːn/ | Histamine |
| 128 | Norepinephrine | /ˌnɔːrɛpɪˈnɛfrɪn/ | Norepinephrine |
| 129 | Dopamine | /ˈdoʊpəmiːn/ | Dopamine |
| 130 | Acetylcholine | /əˌsiːtəlˈkoʊliːn/ | Acetylcholine |
| 131 | Gamma-aminobutyric acid (GABA) | /ˈɡæmə əˌmiːnoʊbjuːˈtɪrɪk ˈæsɪd/ | Axit gamma-aminobutyric |
| 132 | Vasoactive intestinal peptide | /ˌveɪsoʊˈæktɪv ɪnˈtɛstɪnəl ˈpɛptaɪd/ | Peptide vận mạch ruột |
| 133 | Gastrin | /ˈɡæstrɪn/ | Gastrin |
| 134 | Neurotensin | /ˌnʊəroʊˈtɛnsɪn/ | Neurotensin |
| 135 | Substance P | /ˈsʌbstəns piː/ | Chất P |
| 136 | Calcitonin | /ˌkælsɪˈtoʊnɪn/ | Calcitonin |
| 137 | Neuropeptide Y | /ˌnʊəroʊˈpɛptaɪd waɪ/ | Neuropeptide Y |
| 138 | Vasopressin | /ˌveɪsoʊˈprɛsɪn/ | Vasopressin |
| 139 | Corticotrophin-releasing hormone | /ˌkɔːrtɪkoʊˈtrɒfɪn rɪˈliːsɪŋ ˈhɔːrmoʊn/ | Hormone giải phóng corticotropin |
| 140 | Galanin | /ˈɡælənin/ | Galanin |
| 141 | Hemorrhage | /ˈhɛmərɪdʒ/ | Xuất huyết |
| 142 | Fasting | /ˈfæstɪŋ/ | Nhịn ăn |
| 143 | Hypoglycemia | /ˌhaɪpoʊɡlaɪˈsiːmiə/ | Hạ đường huyết |
| 144 | Androgens | /ˈændrədʒənz/ | Androgen |
| 145 | Estrogens | /ˈɛstrədʒənz/ | Estrogen |
| 146 | Thyroxine | /θaɪˈrɒksiːn/ | Thyroxine |
| 147 | Glucocorticoids | /ˌɡluːkoʊˈkɔːrtɪkɔɪdz/ | Glucocorticoid |
| 148 | Hypothyroidism | /ˌhaɪpoʊˈθaɪrɔɪdɪzəm/ | Suy giáp |
| 149 | Hexapeptides | /ˌhɛksəˈpɛptaɪdz/ | Hexapeptide |
| 150 | Ghrelin | /ˈɡrɛlɪn/ | Ghrelin |
| 151 | Ghrelin-mimetic ligands | /ˈɡrɛlɪn mɪˈmɛtɪk ˈlaɪɡəndz/ | Phối tử bắt chước ghrelin |
| 152 | GH secretagogue receptor (GHS-R) | /dʒiː eɪtʃ səˈkriːtəɡɒɡ rɪˈsɛptər/ | Thụ thể chất kích thích tiết GH |
| 153 | Octanoylation | /ɒkˌtænoʊɪˈleɪʃən/ | Octanoyl hóa |
| 154 | Intravenous | /ˌɪntrəˈviːnəs/ | Nội tĩnh mạch |
| 155 | Intracerebroventricular | /ˌɪntrəsəˌriːbroʊvɛnˈtrɪkjələr/ | Nội não thất |
| 156 | Intraperitoneal | /ˌɪntrəˌpɛrɪtəˈniːəl/ | Nội phúc mạc |
| 157 | Obesity | /oʊˈbiːsəti/ | Béo phì |
| 158 | Endocrine pancreatic function | /ˈɛndəkrɪn ˌpæŋkriˈætɪk ˈfʌŋkʃən/ | Chức năng nội tiết tuyến tụy |
| 159 | Gonadal function | /ɡoʊˈnædəl ˈfʌŋkʃən/ | Chức năng tuyến sinh dục |
| 160 | Gastric motility | /ˈɡæstrɪk moʊˈtɪləti/ | Nhu động dạ dày |
| 161 | Acid secretion | /ˈæsɪd sɪˈkriːʃən/ | Tiết axit |
| 162 | Cardiovascular | /ˌkɑːrdioʊˈvæskjələr/ | Tim mạch |
| 163 | Antiproliferative effects | /ˌæntiproʊˈlɪfərətɪv ɪˈfɛkts/ | Tác dụng chống tăng sinh |
| 164 | Idiopathic short stature (ISS) | /ˌɪdioʊˈpæθɪk ʃɔːrt ˈstætʃər/ | Tầm vóc thấp vô căn |
| 165 | Obestatin | /oʊˈbɛstətɪn/ | Obestatin |
| 166 | Insulin-like growth factor (IGF) | /ˈɪnsəlɪn laɪk ɡroʊθ ˈfæktər/ | Yếu tố tăng trưởng giống insulin |
| 167 | Polypeptides | /ˌpɒliˈpɛptaɪdz/ | Polypeptide |
| 168 | Somatopause | /ˌsoʊmətəˈpɔːz/ | Mãn dục tố nam (liên quan đến GH) |
| 169 | Estradiol | /ˌɛstrəˈdaɪɒl/ | Estradiol |
| 170 | Testosterone | /tɛˈstɒstəroʊn/ | Testosterone |
| 171 | Slow-wave sleep | /sloʊ weɪv sliːp/ | Giấc ngủ sóng chậm |
| 172 | Rapid-eye-movement (REM) sleep | /ˈræpɪd aɪ ˈmuːvmənt sliːp/ | Giấc ngủ REM |
| 173 | Growth Hormone Receptor (GHR) | /ɡroʊθ ˈhɔːrmoʊn rɪˈsɛptər/ | Thụ thể hormone tăng trưởng |
| 174 | Growth Hormone-Binding Protein (GHBP) | /ɡroʊθ ˈhɔːrmoʊn ˈbaɪndɪŋ ˈproʊtiːn/ | Protein gắn hormone tăng trưởng |
| 175 | Extracellular domain | /ˌɛkstrəˈsɛljələr doʊˈmeɪn/ | Miền ngoại bào |
| 176 | Membrane-spanning domain | /ˈmɛmbreɪn ˈspænɪŋ doʊˈmeɪn/ | Miền xuyên màng |
| 177 | Cytoplasmic domain | /ˌsaɪtoʊˈplæzmɪk doʊˈmeɪn/ | Miền tế bào chất |
| 178 | Hematopoietic cytokine family | /hɪˌmætoʊpɔɪˈɛtɪk ˈsaɪtəkaɪn ˈfæməli/ | Họ cytokine tạo máu |
| 179 | Prolactin receptor | /proʊˈlæktɪn rɪˈsɛptər/ | Thụ thể prolactin |
| 180 | Interleukins (ILs) | /ˌɪntərˈluːkɪnz/ | Interleukin |
| 181 | Erythropoietin (EPO) | /ɪˌrɪθroʊˈpɔɪətɪn/ | Erythropoietin |
| 182 | Leptin | /ˈlɛptɪn/ | Leptin |
| 183 | Granulocyte-macrophage colony-stimulating factor | /ˈɡrænjəloʊsaɪt ˈmækroʊfeɪdʒ ˈkɒləni ˈstɪmjəleɪtɪŋ ˈfæktər/ | Yếu tố kích thích dòng bạch cầu hạt-đại thực bào |
| 184 | Interferon | /ˌɪntərˈfɪərɒn/ | Interferon |
| 185 | Receptor dimerization | /rɪˈsɛptər ˌdaɪmərəˈzeɪʃən/ | Sự nhị phân hóa thụ thể |
| 186 | Janus Kinase 2 (JAK2) | /ˈdʒænəs ˈkaɪneɪs tuː/ | Janus Kinase 2 |
| 187 | Tyrosine kinase | /ˈtaɪrəsiːn ˈkaɪneɪs/ | Tyrosine kinase |
| 188 | Autophosphorylated | /ˌɔːtoʊˌfɒsfəˈraɪleɪtɪd/ | Tự phosphoryl hóa |
| 189 | Signal transducer and activator of transcription (Stat) | /ˈsɪɡnəl trænˈsduːsər ænd ˈæktɪveɪtər əv trænˈskrɪpʃən/ | Chất truyền tín hiệu và hoạt hóa phiên mã |
| 190 | Deoxyribonucleic acid (DNA) | /diːˌɒksiˌraɪboʊnjuːˌkliːɪk ˈæsɪd/ | Axit deoxyribonucleic |
| 191 | Pegvisomant | /pɛɡˈvɪsoʊmænt/ | Pegvisomant |
| 192 | Pituitary gigantism | /pɪˈtjuːɪtəri ˈdʒaɪɡəntɪzəm/ | Bệnh khổng lồ tuyến yên |
| 193 | Acromegaly | /ˌækroʊˈmɛɡəli/ | Bệnh to đầu chi |
| 194 | Suppressors of cytokine signaling (SOCS) | /səˈprɛsərz əv ˈsaɪtəkaɪn ˈsɪɡnəlɪŋ/ | Chất ức chế tín hiệu cytokine |
| 195 | Protein tyrosine phosphatases (PTPs) | /ˈproʊtiːn ˈtaɪrəsiːn ˈfɒsfəteɪsɪz/ | Protein tyrosine phosphatase |
| 196 | Protein inhibitors of activated Stats (PIAS) | /ˈproʊtiːn ɪnˈhɪbɪtərz əv ˈæktɪveɪtɪd stæts/ | Chất ức chế protein của Stat hoạt hóa |
| 197 | Proteolytic cleavage | /ˌproʊtiəˈlɪtɪk ˈkliːvɪdʒ/ | Phân cắt protein |
| 198 | Growth hormone insensitivity (GHI) | /ɡroʊθ ˈhɔːrmoʊn ɪnˌsɛnsɪˈtɪvəti/ | Không nhạy cảm với hormone tăng trưởng |
| 199 | Somatomedin hypothesis | /ˌsoʊmətəˈmiːdɪn haɪˈpɒθəsɪs/ | Giả thuyết somatomedin |
| 200 | Anabolic actions | /ˌænəˈbɒlɪk ˈækʃənz/ | Tác dụng đồng hóa |
| 201 | Mitogens | /ˈmaɪtədʒənz/ | Chất gây phân bào |
| 202 | Diabetogenic | /ˌdaɪəˌbiːtoʊˈdʒɛnɪk/ | Gây đái tháo đường |
| 203 | Prechondrocytes | /ˌpriːˈkɒndroʊsaɪts/ | Tế bào tiền sụn |
| 204 | Lipolysis | /laɪˈpɒləsɪs/ | Tiêu mỡ |
| 205 | Diaphragm | /ˈdaɪəfræm/ | Cơ hoành |
| 206 | Somatomedins | /ˌsoʊmətəˈmiːdɪnz/ | Somatomedin |
| 207 | Mitogenic actions | /ˌmaɪtəˈdʒɛnɪk ˈækʃənz/ | Tác dụng gây phân bào |
| 208 | Somatomedin-C | /ˌsoʊmətəˈmiːdɪn siː/ | Somatomedin-C |
| 209 | Gonadal dysgenesis | /ɡoʊˈnædəl dɪsˈdʒɛnəsɪs/ | Loạn sản tuyến sinh dục |
| 210 | Insulin receptor (IR) | /ˈɪnsəlɪn rɪˈsɛptər/ | Thụ thể insulin |
| 211 | Heterotetramers | /ˌhɛtəroʊˈtɛtrəmərz/ | Dị tứ phân |
| 212 | Alpha subunits | /ˈælfə ˈsʌbˌjuːnɪts/ | Tiểu đơn vị alpha |
| 213 | Beta subunits | /ˈbeɪtə ˈsʌbˌjuːnɪts/ | Tiểu đơn vị beta |
| 214 | Adenosine triphosphate (ATP) | /əˈdɛnəsiːn traɪˈfɒsfeɪt/ | Adenosine triphosphate |
| 215 | Phosphorylation | /ˌfɒsfərɪˈleɪʃən/ | Phosphoryl hóa |
| 216 | Apoptosis | /ˌæpəˈtoʊsɪs/ | Chết tế bào theo chương trình |
| 217 | Insulin receptor substrate family | /ˈɪnsəlɪn rɪˈsɛptər ˈsʌbstreɪt ˈfæməli/ | Họ cơ chất của thụ thể insulin |
| 218 | Nematodes | /ˈnɛmətoʊdz/ | Giun tròn |
| 219 | Centenarians | /ˌsɛntəˈnɛəriənz/ | Người trăm tuổi |
| 220 | Cation-independent mannose-6-phosphate (CIM6P) receptor | /ˈkætaɪən ɪndɪˈpɛndənt ˈmænoʊs sɪks ˈfɒsfeɪt rɪˈsɛptər/ | Thụ thể mannose-6-phosphate không phụ thuộc cation |
| 221 | Lysosomal targeting | /ˌlaɪsəˈsoʊməl ˈtɑːrɡətɪŋ/ | Nhắm đích lysosome |
| 222 | Acid hydrolases | /ˈæsɪd ˈhaɪdrəleɪsɪz/ | Hydrolase axit |
| 223 | Mannosylated proteins | /məˈnɒsəleɪtɪd ˈproʊtiːnz/ | Protein được mannosyl hóa |
| 224 | IGF-binding protein (IGFBP) | /aɪ dʒiː ɛf ˈbaɪndɪŋ ˈproʊtiːn/ | Protein gắn IGF |
| 225 | Ternary complex | /ˈtɜːrnəri ˈkɒmplɛks/ | Phức hợp tam phân |
| 226 | Acid-labile subunit (ALS) | /ˈæsɪd ˈleɪbaɪl ˈsʌbˌjuːnɪt/ | Tiểu đơn vị không bền với axit |
| 227 | Proteases | /ˈproʊtieɪsɪz/ | Protease |
| 228 | Western ligand blotting | /ˈwɛstərn ˈlaɪɡənd ˈblɒtɪŋ/ | Thấm màng Western ligand |
| 229 | Radioimmunoassay | /ˌreɪdioʊˌɪmjʊnoʊˈæseɪ/ | Xét nghiệm miễn dịch phóng xạ |
| 230 | Metalloproteinase | /məˌtæloʊˈproʊtieɪneɪs/ | Metalloproteinase |
| 231 | Intrauterine growth retardation (IUGR) | /ˌɪntrəˈjuːtəraɪn ɡroʊθ ˌriːtɑːrˈdeɪʃən/ | Chậm tăng trưởng trong tử cung |
| 232 | Thyroid hormone receptor | /ˈθaɪrɔɪd ˈhɔːrmoʊn rɪˈsɛptər/ | Thụ thể hormone tuyến giáp |
| 233 | Skeletal maturation | /ˈskɛlɪtl ˌmætjʊəˈreɪʃən/ | Sự trưởng thành của xương |
| 234 | Growth plate senescence | /ɡroʊθ pleɪt səˈnɛsəns/ | Sự lão hóa của sụn tăng trưởng |
| 235 | Epiphyseal fusion | /ˌɛpɪˈfɪziəl ˈfjuːʒən/ | Cốt hóa sụn đầu xương |
| 236 | Aromatase deficiency | /əˈroʊməteɪs dɪˈfɪʃənsi/ | Thiếu hụt aromatase |
| 237 | Estrogen resistance | /ˈɛstrədʒən rɪˈzɪstəns/ | Đề kháng estrogen |
| 238 | Precocious puberty | /prɪˈkoʊʃəs ˈpjuːbərti/ | Dậy thì sớm |
| 239 | Hypogonadism | /ˌhaɪpoʊˈɡoʊnædɪzəm/ | Suy sinh dục |
| 240 | Gonadotropin-releasing hormone (GnRH) analogues | /ɡoʊˌnædəˈtroʊpɪn rɪˈliːsɪŋ ˈhɔːrmoʊn ˈænəlɒɡz/ | Chất tương tự hormone giải phóng gonadotropin |
| 241 | Aromatase inhibitors | /əˈroʊməteɪs ɪnˈhɪbɪtərz/ | Chất ức chế aromatase |
| 242 | Aromatized | /əˈroʊmətaɪzd/ | Được thơm hóa |
| 243 | Dihydrotestosterone | /daɪˌhaɪdroʊtɛˈstɒstəroʊn/ | Dihydrotestosterone |
| 244 | Neonatal diabetes mellitus | /ˌniːoʊˈneɪtəl ˌdaɪəˈbiːtiːz ˈmɛlɪtəs/ | Đái tháo đường sơ sinh |
| 245 | Biallelic | /baɪəˈliːlɪk/ | Hai alen |
| 246 | Gestational diabetes | /dʒɛˈsteɪʃənəl ˌdaɪəˈbiːtiːz/ | Đái tháo đường thai kỳ |
| 247 | Macrosomia | /ˌmækroʊˈsoʊmiə/ | Thai to |
| 248 | Nonthyroidal illness syndrome | /nɒnˌθaɪˈrɔɪdəl ˈɪlnəs ˈsɪndroʊm/ | Hội chứng bệnh không do tuyến giáp |
| 249 | Juvenile idiopathic arthritis (JIA) | /ˈdʒuːvənaɪl ˌɪdioʊˈpæθɪk ɑːrˈθraɪtɪs/ | Viêm khớp tự phát thiếu niên |
| 250 | Cystic fibrosis | /ˈsɪstɪk faɪˈbroʊsɪs/ | Xơ nang |
| 251 | Tumor necrosis factor-α (TNF-α) | /ˈtjuːmər nɛˈkroʊsɪs ˈfæktər ˈælfə/ | Yếu tố hoại tử khối u-α |
| 252 | Chondrodysplasia | /ˌkɒndroʊdɪsˈpleɪʒə/ | Loạn sản sụn |
| 253 | C-type natriuretic peptide (CNP) | /siː taɪp ˌneɪtriəˈrɛtɪk ˈpɛptaɪd/ | Peptide lợi niệu loại C |
| 254 | Achondroplasia (ACH) | /əˌkɒndroʊˈpleɪʒə/ | Loạn sản sụn |
| 255 | Hypochondroplasia | /ˌhaɪpoʊˌkɒndroʊˈpleɪʒə/ | Loạn sản sụn nhẹ |
| 256 | Perichondrium | /ˌpɛrɪˈkɒndriəm/ | Màng sụn |
| 257 | Parathyroid hormone-related protein (PTHrP) | /ˌpærəˈθaɪrɔɪd ˈhɔːrmoʊn rɪˈleɪtɪd ˈproʊtiːn/ | Protein liên quan đến hormone cận giáp |
| 258 | Indian hedgehog (IHH) | /ˈɪndiən ˈhɛdʒhɒɡ/ | (Gen) Indian hedgehog |
| 259 | Blomstrand lethal chondrodysplasia | /ˈblɒmstrænd ˈliːθəl ˌkɒndroʊdɪsˈpleɪʒə/ | Loạn sản sụn gây chết Blomstrand |
| 260 | Jansen metaphyseal chondrodysplasia | /ˈjænsən ˌmɛtəˈfɪziəl ˌkɒndroʊdɪsˈpleɪʒə/ | Loạn sản sụn hành xương Jansen |
| 261 | Brachydactyly type A1 | /ˌbrækɪˈdæktɪli taɪp eɪ wʌn/ | Tật ngón ngắn loại A1 |
| 262 | Albright hereditary osteodystrophy (AHO) | /ˈɔːlbraɪt həˈrɛdɪtəri ˌɒstioʊˈdɪstrəfi/ | Loạn dưỡng xương di truyền Albright |
| 263 | Acrodysostosis | /ˌækroʊdɪsɒˈstoʊsɪs/ | Loạn sản xương đầu chi |
| 264 | Proteoglycans | /ˌproʊtioʊˈɡlaɪkənz/ | Proteoglycan |
| 265 | Aggrecan | /ˈæɡrɪkæn/ | Aggrecan |
| 266 | Osteogenesis imperfecta | /ˌɒstioʊˈdʒɛnəsɪs ɪmpərˈfɛktə/ | Bệnh tạo xương bất toàn |
| 267 | Short stature homeobox-containing gene (SHOX) | /ʃɔːrt ˈstætʃər ˈhoʊmioʊbɒks kənˈteɪnɪŋ dʒiːn/ | Gen chứa hộp homeo tầm vóc thấp |
| 268 | RAS-mitogen-activated protein kinase (MAPK) pathway | /ræs ˈmaɪtədʒən ˈæktɪveɪtɪd ˈproʊtiːn ˈkaɪneɪs ˈpæθweɪ/ | Con đường RAS-mitogen-activated protein kinase |
| 269 | Noonan syndrome (NS) | /ˈnuːnən ˈsɪndroʊm/ | Hội chứng Noonan |
| 270 | Bone age | /boʊn eɪdʒ/ | Tuổi xương |
| 271 | Catch-up growth | /kætʃ ʌp ɡroʊθ/ | Tăng trưởng bắt kịp |
| 272 | Genome-wide association studies (GWAS) | /ˈdʒiːnoʊm waɪd əˌsoʊsiˈeɪʃən ˈstʌdiz/ | Nghiên cứu liên kết toàn bộ gen |
| 273 | Single nucleotide polymorphisms (SNPs) | /ˈsɪŋɡəl ˈnjuːkliətaɪd ˌpɒliˈmɔːrfɪzəmz/ | Đa hình đơn nucleotide |
| 274 | Allelic variants | /əˈliːlɪk ˈvɛəriənts/ | Biến thể alen |
| 275 | Stadiometer | /ˌsteɪdiˈɒmɪtər/ | Thước đo chiều cao đứng |
| 276 | Frankfurt plane | /ˈfræŋkfɜːrt pleɪn/ | Mặt phẳng Frankfurt |
| 277 | Recumbent length | /rɪˈkʌmbənt lɛŋθ/ | Chiều dài nằm |
| 278 | Growth charts | /ɡroʊθ tʃɑːrts/ | Biểu đồ tăng trưởng |
| 279 | Centile curves | /ˈsɛntaɪl kɜːrvz/ | Đường cong phân vị |
| 280 | Body mass index (BMI) | /ˈbɒdi mæs ˈɪndɛks/ | Chỉ số khối cơ thể |
| 281 | Physiological rechanneling | /ˌfɪziəˈlɒdʒɪkəl riːˈtʃænəlɪŋ/ | Tái phân kênh sinh lý |
| 282 | Tanner stage | /ˈtænər steɪdʒ/ | Giai đoạn Tanner |
| 283 | Velocity charts | /vəˈlɒsəti tʃɑːrts/ | Biểu đồ vận tốc |
| 284 | Upper-to-lower body segment ratio | /ˈʌpər tə ˈloʊər ˈbɒdi ˈsɛɡmənt ˈreɪʃioʊ/ | Tỷ lệ đoạn thân trên/dưới |
| 285 | Pubic symphysis | /ˈpjuːbɪk ˈsɪmfəsɪs/ | Khớp mu |
| 286 | Arm span | /ɑːrm spæn/ | Sải tay |
| 287 | Sitting height index | /ˈsɪtɪŋ haɪt ˈɪndɛks/ | Chỉ số chiều cao ngồi |
| 288 | Rhizomelia | /ˌraɪzoʊˈmiːliə/ | Ngắn gốc chi |
| 289 | Mezomelia | /ˌmɛzoʊˈmiːliə/ | Ngắn đoạn giữa chi |
| 290 | Acromelia | /ˌækroʊˈmiːliə/ | Ngắn đoạn xa chi |
| 291 | Anteroposterior (AP) radiograph | /ænˌtɪəroʊpɒˈstɪəriər ˈreɪdioʊɡræf/ | X-quang trước-sau |
| 292 | Ossification | /ˌɒsɪfɪˈkeɪʃən/ | Sự cốt hóa |
| 293 | Ossification center | /ˌɒsɪfɪˈkeɪʃən ˈsɛntər/ | Trung tâm cốt hóa |
| 294 | Epiphyses | /ɪˈpɪfəsiːz/ | Đầu xương |
| 295 | Articular surface | /ɑːrˈtɪkjələr ˈsɜːrfɪs/ | Bề mặt khớp |
| 296 | Radiopaque | /ˌreɪdioʊˈpeɪk/ | Cản quang |
| 297 | Midparental height | /mɪd pəˈrɛntəl haɪt/ | Chiều cao trung bình của cha mẹ |
| 298 | Menarche | /məˈnɑːrki/ | Hành kinh |
| 299 | Bayley-Pinneau prediction tables | /ˈbeɪli pɪˈnoʊ prɪˈdɪkʃən ˈteɪbəlz/ | Bảng dự đoán Bayley-Pinneau |
| 300 | Tanner-Whitehouse (TW) method | /ˈtænər ˈwaɪthaʊs ˈmɛθəd/ | Phương pháp Tanner-Whitehouse |
| 301 | Roche-Wainer-Thissen (RWT) method | /roʊʃ ˈweɪnər ˈθɪsən ˈmɛθəd/ | Phương pháp Roche-Wainer-Thissen |
| 302 | Khamis-Roche method | /ˈkɑːmɪs roʊʃ ˈmɛθəd/ | Phương pháp Khamis-Roche |
| 303 | Mendelian inheritance | /mɛnˈdiːliən ɪnˈhɛrɪtəns/ | Di truyền Mendel |
| 304 | Autosomal dominant | /ˌɔːtəˈsoʊməl ˈdɒmɪnənt/ | Trội trên nhiễm sắc thể thường |
| 305 | Autosomal recessive | /ˌɔːtəˈsoʊməl rɪˈsɛsɪv/ | Lặn trên nhiễm sắc thể thường |
| 306 | X-linked | /ɛks lɪŋkt/ | Liên kết X |
| 307 | Pedigree | /ˈpɛdɪɡriː/ | Phả hệ |
| 308 | International Classification of Pediatric Endocrine Diagnoses (ICPED) | /ˌɪntərˈnæʃənəl ˌklæsɪfɪˈkeɪʃən əv ˌpiːdiˈætrɪk ˈɛndəkrɪn ˌdaɪəɡˈnoʊsiːz/ | Phân loại quốc tế về chẩn đoán nội tiết nhi khoa |
| 309 | Primary growth impairment | /ˈpraɪməri ɡroʊθ ɪmˈpɛərmənt/ | Suy giảm tăng trưởng nguyên phát |
| 310 | Secondary growth impairment | /ˈsɛkəndəri ɡroʊθ ɪmˈpɛərmənt/ | Suy giảm tăng trưởng thứ phát |
| 311 | Skeletal dysplasia | /ˈskɛlɪtl dɪsˈpleɪʒə/ | Loạn sản xương |
| 312 | Pseudohypoparathyroidism 1A | /ˌsuːdoʊˌhaɪpoʊˌpærəˈθaɪrɔɪdɪzəm wʌn eɪ/ | Giả suy cận giáp loại 1A |
| 313 | Small for gestational age (SGA) | /smɔːl fər dʒɛˈsteɪʃənəl eɪdʒ/ | Nhỏ so với tuổi thai |
| 314 | Protein-calorie malnutrition | /ˈproʊtiːn ˈkæləri ˌmælnjuːˈtrɪʃən/ | Suy dinh dưỡng protein-năng lượng |
| 315 | Pierre Robin sequence | /pjɛər ˈroʊbɪn ˈsiːkwəns/ | Chuỗi Pierre Robin |
| 316 | Cleft lip/palate | /klɛft lɪp/ˈpælət/ | Sứt môi/hở hàm ếch |
| 317 | Pervasive developmental delay | /pərˈveɪsɪv dɪˌvɛləpˈmɛntəl dɪˈleɪ/ | Chậm phát triển lan tỏa |
| 318 | Anorexia nervosa | /ˌænəˈrɛksiə nɜːrˈvoʊsə/ | Chán ăn tâm thần |
| 319 | Silver-Russell syndrome (SRS) | /ˈsɪlvər ˈrʌsəl ˈsɪndroʊm/ | Hội chứng Silver-Russell |
| 320 | Bloom syndrome | /bluːm ˈsɪndroʊm/ | Hội chứng Bloom |
| 321 | Growth hormone deficiency (GHD) | /ɡroʊθ ˈhɔːrmoʊn dɪˈfɪʃənsi/ | Thiếu hụt hormone tăng trưởng |
| 322 | Combined/multiple pituitary hormone deficiency (CPHD/MPHD) | /kəmˈbaɪnd/ˈmʌltɪpəl pɪˈtjuːɪtəri ˈhɔːrmoʊn dɪˈfɪʃənsi/ | Thiếu hụt nhiều hormone tuyến yên |
| 323 | Hypothalamopituitary area | /ˌhaɪpoʊθəˌlæmoʊpɪˈtjuːɪtəri ˈɛəriə/ | Vùng dưới đồi-tuyến yên |
| 324 | Auxology | /ɔːkˈsɒlədʒi/ | Nhân trắc học tăng trưởng |
| 325 | Turner syndrome | /ˈtɜːrnər ˈsɪndroʊm/ | Hội chứng Turner |
| 326 | Crohn disease | /kroʊn dɪˈziːz/ | Bệnh Crohn |
| 327 | Renal failure | /ˈriːnəl ˈfeɪljər/ | Suy thận |
| 328 | Standard deviation score (SDS) | /ˈstændərd ˌdiːviˈeɪʃən skɔːr/ | Điểm độ lệch chuẩn |
| 329 | Intracranial lesion | /ˌɪntrəˈkreɪniəl ˈliːʒən/ | Tổn thương nội sọ |
| 330 | Hyperbilirubinemia | /ˌhaɪpərbɪlɪˌruːbɪˈniːmiə/ | Tăng bilirubin máu |
| 331 | Micropenis | /ˌmaɪkroʊˈpiːnɪs/ | Dương vật nhỏ |
| 332 | Midface hypoplasia | /mɪdˈfeɪs ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản giữa mặt |
| 333 | Hypotonia | /ˌhaɪpoʊˈtoʊniə/ | Giảm trương lực cơ |
| 334 | Truncal adiposity | /ˈtrʌŋkəl ˌædɪˈpɒsəti/ | Béo thân mình |
| 335 | Recombinant human GH (rhGH) | /rɪˈkɒmbɪnənt ˈhjuːmən dʒiː eɪtʃ/ | GH người tái tổ hợp |
| 336 | Anencephaly | /ˌænənˈsɛfəli/ | Thai vô sọ |
| 337 | Cyclopia | /saɪˈkloʊpiə/ | Mắt một mí |
| 338 | Hypertelorism | /ˌhaɪpərˈtiːlərɪzəm/ | Hai mắt xa nhau |
| 339 | Philtrum | /ˈfɪltrəm/ | Nhân trung |
| 340 | Diabetes insipidus (DI) | /ˌdaɪəˈbiːtiːz ɪnˈsɪpɪdəs/ | Đái tháo nhạt |
| 341 | De Morsier syndrome | /də mɔːrˈsjeɪ ˈsɪndroʊm/ | Hội chứng De Morsier |
| 342 | Polyuria | /ˌpɒliˈjʊəriə/ | Đa niệu |
| 343 | Polydipsia | /ˌpɒliˈdɪpsiə/ | Uống nhiều |
| 344 | Sexual precocity | /ˈsɛkʃuəl prɪˈkɒsəti/ | Dậy thì sớm |
| 345 | Endocrinopathy | /ˌɛndoʊkrɪˈnɒpəθi/ | Bệnh nội tiết |
| 346 | Global retardation | /ˈɡloʊbəl ˌriːtɑːrˈdeɪʃən/ | Chậm phát triển toàn bộ |
| 347 | Epilepsy | /ˈɛpɪlɛpsi/ | Động kinh |
| 348 | Hemiparesis | /ˌhɛmɪpəˈriːsɪs/ | Liệt nửa người |
| 349 | Cavum septum pellucidum | /ˈkeɪvəm ˈsɛptəm pəˈluːsɪdəm/ | Khoang vách trong suốt |
| 350 | Schizencephaly | /ˌskɪzənˈsɛfəli/ | Não nứt |
| 351 | Aplasia of the fornix | /əˈpleɪʒə əv ðə ˈfɔːrnɪks/ | Bất sản vòm não |
| 352 | Microphthalmia | /ˌmaɪkrɒfˈθælmiə/ | Mắt nhỏ |
| 353 | Esophageal atresia | /ɪˌsɒfəˈdʒiːəl əˈtriːʒə/ | Teo thực quản |
| 354 | Hippocampal abnormalities | /ˌhɪpoʊˈkæmpəl ˌæbnɔːrˈmælɪtiz/ | Bất thường hồi hải mã |
| 355 | Infundibular hypoplasia | /ˌɪnfʌnˈdɪbjələr ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản phễu |
| 356 | Craniopharyngeal canal | /ˌkreɪnioʊfəˈrɪndʒiəl kəˈnæl/ | Ống sọ hầu |
| 357 | Traumatic brain injury (TBI) | /trɔːˈmætɪk breɪn ˈɪndʒəri/ | Chấn thương sọ não |
| 358 | Ischemia | /ɪˈskiːmiə/ | Thiếu máu cục bộ |
| 359 | Infarction | /ɪnˈfɑːrkʃən/ | Nhồi máu |
| 360 | Anterior lobe necrosis | /ænˈtɪəriər loʊb nɛˈkroʊsɪs/ | Hoại tử thùy trước |
| 361 | Pituitary fibrosis | /pɪˈtjuːɪtəri faɪˈbroʊsɪs/ | Xơ hóa tuyến yên |
| 362 | Perinatal trauma | /ˌpɛrɪˈneɪtəl ˈtrɔːmə/ | Chấn thương chu sinh |
| 363 | Breech deliveries | /briːtʃ dɪˈlɪvəriz/ | Sinh ngôi mông |
| 364 | Meningitis | /ˌmɛnɪnˈdʒaɪtɪs/ | Viêm màng não |
| 365 | Encephalitis | /ɪnˌsɛfəˈlaɪtɪs/ | Viêm não |
| 366 | Streptococcus | /ˌstrɛptəˈkɒkəs/ | Liên cầu khuẩn |
| 367 | Haemophilus influenzae | /hiːˌmɒfɪləs ɪnfluˈɛnziː/ | Haemophilus influenzae |
| 368 | Mycobacterium tuberculosis | /ˌmaɪkoʊbækˈtɪəriəm ˌtjuːbərkjʊˈloʊsɪs/ | Mycobacterium tuberculosis |
| 369 | Sarcoidosis | /ˌsɑːrkɔɪˈdoʊsɪs/ | Bệnh sarcoidosis |
| 370 | Granulomatous disease | /ˌɡrænjəˈloʊmətəs dɪˈziːz/ | Bệnh u hạt |
| 371 | Gadolinium | /ˌɡædəˈlɪniəm/ | Gadolinium |
| 372 | Hyperprolactinemia | /ˌhaɪpərproʊˌlæktɪˈniːmiə/ | Tăng prolactin máu |
| 373 | Neurosarcoidosis | /ˌnʊəroʊˌsɑːrkɔɪˈdoʊsɪs/ | Bệnh sarcoidosis thần kinh |
| 374 | Methylprednisolone | /ˌmɛθəlprɛdˈnɪsəloʊn/ | Methylprednisolone |
| 375 | Hypophysitis | /haɪˌpɒfəˈsaɪtɪs/ | Viêm tuyến yên |
| 376 | Lymphocytic hypophysitis | /ˌlɪmfoʊˈsɪtɪk haɪˌpɒfəˈsaɪtɪs/ | Viêm tuyến yên lympho bào |
| 377 | Hashimoto thyroiditis | /ˌhæʃɪˈmoʊtoʊ ˌθaɪrɔɪˈdaɪtɪs/ | Viêm tuyến giáp Hashimoto |
| 378 | Graves disease | /ɡreɪvz dɪˈziːz/ | Bệnh Graves |
| 379 | Systemic lupus erythematosus | /sɪˈstɛmɪk ˈluːpəs ˌɛrɪθəˈmatoʊsəs/ | Lupus ban đỏ hệ thống |
| 380 | Langerhans cell histiocytosis (LCH) | /ˈlæŋərhænz sɛl ˌhɪstioʊsaɪˈtoʊsɪs/ | Bệnh mô bào Langerhans |
| 381 | Neurodegenerative | /ˌnʊəroʊdɪˈdʒɛnərətɪv/ | Thoái hóa thần kinh |
| 382 | Germinomas | /ˌdʒɜːrmɪˈnoʊməz/ | U tế bào mầm |
| 383 | Meningiomas | /məˌnɪndʒiˈoʊməz/ | U màng não |
| 384 | Gliomas | /ɡliˈoʊməz/ | U tế bào đệm |
| 385 | Colloid cysts | /ˈkɒlɔɪd sɪsts/ | Nang keo |
| 386 | Ependymomas | /ɪˌpɛndɪˈmoʊməz/ | U màng não thất |
| 387 | Craniopharyngeal carcinoma | /ˌkreɪnioʊfəˈrɪndʒiəl ˌkɑːrsɪˈnoʊmə/ | Ung thư biểu mô sọ hầu |
| 388 | Hodgkin disease | /ˈhɒdʒkɪn dɪˈziːz/ | Bệnh Hodgkin |
| 389 | Nasopharynx | /ˌneɪzoʊˈfærɪŋks/ | Vòm họng |
| 390 | Pituitary adenomas | /pɪˈtjuːɪtəri ˌædəˈnoʊməz/ | U tuyến tuyến yên |
| 391 | Prolactinomas | /proʊˌlæktɪˈnoʊməz/ | U tuyến prolactin |
| 392 | Cushing disease | /ˈkʊʃɪŋ dɪˈziːz/ | Bệnh Cushing |
| 393 | Corticotropinomas | /ˌkɔːrtɪkoʊtrəpɪˈnoʊməz/ | U tuyến corticotropin |
| 394 | Somatotropinomas | /soʊˌmætətrəpɪˈnoʊməz/ | U tuyến somatotropin |
| 395 | Thyrotropinomas | /θaɪroʊtrəpɪˈnoʊməz/ | U tuyến thyrotropin |
| 396 | Multiple endocrine neoplasia type 1 (MEN1) | /ˈmʌltɪpəl ˈɛndəkrɪn ˌniːəˈpleɪʒə taɪp wʌn/ | Đa u tuyến nội tiết loại 1 |
| 397 | McCune-Albright syndrome (MAS) | /məˈkjuːn ˈɔːlbraɪt ˈsɪndroʊm/ | Hội chứng McCune-Albright |
| 398 | Carney Complex | /ˈkɑːrni ˈkɒmplɛks/ | Phức hợp Carney |
| 399 | Optic gliomas | /ˈɒptɪk ɡliˈoʊməz/ | U tế bào đệm thần kinh thị |
| 400 | Neurofibromatosis type 1 (NF-1) | /ˌnʊəroʊˌfaɪbroʊməˈtoʊsɪs taɪp wʌn/ | Bệnh u xơ thần kinh loại 1 |
| 401 | Intracranial pressure | /ˌɪntrəˈkreɪniəl ˈprɛʃər/ | Áp lực nội sọ |
| 402 | Optic atrophy | /ˈɒptɪk ˈætrəfi/ | Teo dây thần kinh thị |
| 403 | Strabismus | /strəˈbɪzməs/ | Lác mắt |
| 404 | Proptosis | /prɒpˈtoʊsɪs/ | Lồi mắt |
| 405 | Ataxia | /əˈtæksiə/ | Mất điều hòa |
| 406 | Rathke’s cleft cysts | /ˈræθkiːz klɛft sɪsts/ | Nang khe Rathke |
| 407 | Arachnoidal cysts | /əˈræknɔɪdəl sɪsts/ | Nang màng nhện |
| 408 | Cystic adenomas | /ˈsɪstɪk ˌædəˈnoʊməz/ | U tuyến dạng nang |
| 409 | Amenorrhea | /əˌmɛnəˈriːə/ | Vô kinh |
| 410 | Adamantinomatous | /ˌædəmæntɪˈnoʊmətəs/ | Dạng adamantinoma |
| 411 | Papillary | /ˈpæpɪləri/ | Dạng nhú |
| 412 | Oculomotor abnormalities | /ˌɒkjəloʊˈmoʊtər ˌæbnɔːrˈmælɪtiz/ | Bất thường vận nhãn |
| 413 | Papilledema | /ˌpæpɪlɪˈdiːmə/ | Phù gai thị |
| 414 | Visual hallucinations | /ˈvɪʒuəl həˌluːsɪˈneɪʃənz/ | Ảo giác thị giác |
| 415 | Olfactory hallucinations | /ɒlˈfæktəri həˌluːsɪˈneɪʃənz/ | Ảo giác khứu giác |
| 416 | Dementia | /dɪˈmɛnʃə/ | Sa sút trí tuệ |
| 417 | Pubertal arrest | /pjuːˈbɜːrtəl əˈrɛst/ | Ngừng dậy thì |
| 418 | Syndrome of inappropriate antidiuretic hormone secretion | /ˈsɪndroʊm əv ɪnəˈproʊpriət ˌæntidaɪjʊəˈrɛtɪk ˈhɔːrmoʊn sɪˈkriːʃən/ | Hội chứng tiết hormone chống bài niệu không phù hợp |
| 419 | Computed tomography (CT) | /kəmˈpjuːtɪd təˈmɒɡrəfi/ | Chụp cắt lớp vi tính |
| 420 | Hemosiderin deposits | /ˌhiːmoʊˈsɪdərɪn dɪˈpɒzɪts/ | Lắng đọng hemosiderin |
| 421 | Obstructive hydrocephalus | /əbˈstrʌktɪv ˌhaɪdroʊˈsɛfələs/ | Não úng thủy tắc nghẽn |
| 422 | Adjuvant radiotherapy | /ˈædʒəvənt ˌreɪdioʊˈθɛrəpi/ | Xạ trị bổ trợ |
| 423 | Proton beam therapy | /ˈproʊtɒn biːm ˈθɛrəpi/ | Liệu pháp chùm proton |
| 424 | Laron syndrome | /ˈlærən ˈsɪndroʊm/ | Hội chứng Laron |
| 425 | Craniofacial disproportion | /ˌkreɪnioʊˈfeɪʃəl ˌdɪsprəˈpɔːrʃən/ | Mất cân đối sọ mặt |
| 426 | Depressed nasal bridge | /dɪˈprɛst ˈneɪzəl brɪdʒ/ | Sống mũi lõm |
| 427 | Lymphocytic interstitial pneumonia | /ˌlɪmfoʊˈsɪtɪk ˌɪntərˈstɪʃəl njuːˈmoʊniə/ | Viêm phổi kẽ lympho bào |
| 428 | Eczema | /ˈɛksəmə/ | Chàm |
| 429 | T lymphocytes | /tiː ˈlɪmfoʊsaɪts/ | Tế bào lympho T |
| 430 | Mucosal immunity | /mjuːˈkoʊsəl ɪˈmjuːnəti/ | Miễn dịch niêm mạc |
| 431 | Immunoglobulin E | /ˌɪmjʊnoʊˈɡlɒbjʊlɪn iː/ | Immunoglobulin E |
| 432 | Bone mineral density | /boʊn ˈmɪnərəl ˈdɛnsəti/ | Mật độ khoáng xương |
| 433 | Sensorineural deafness | /ˌsɛnsəriˈnʊərəl ˈdɛfnəs/ | Điếc thần kinh giác quan |
| 434 | Osteoporosis | /ˌɒstioʊpəˈroʊsɪs/ | Loãng xương |
| 435 | Myxedema | /ˌmɪksɪˈdiːmə/ | Phù niêm |
| 436 | VanWyk-Grumbach syndrome | /væn waɪk ˈɡrʌmbæk ˈsɪndroʊm/ | Hội chứng VanWyk-Grumbach |
| 437 | Virilization | /ˌvɪrɪlaɪˈzeɪʃən/ | Nam hóa |
| 438 | Deep tendon reflexes | /diːp ˈtɛndən ˈriːflɛksɪz/ | Phản xạ gân sâu |
| 439 | Goiter | /ˈɡɔɪtər/ | Bướu cổ |
| 440 | Thyrotoxicosis | /ˌθaɪroʊˌtɒksɪˈkoʊsɪs/ | Nhiễm độc giáp |
| 441 | Dysmorphic features | /dɪsˈmɔːrfɪk ˈfiːtʃərz/ | Đặc điểm dị dạng |
| 442 | Pigeon breast | /ˈpɪdʒən brɛst/ | Ngực ức gà |
| 443 | Winged scapulae | /wɪŋd ˈskæpjʊliː/ | Xương vai có cánh |
| 444 | Deaf-mutism | /dɛf ˈmjuːtɪzəm/ | Câm điếc |
| 445 | Color blindness | /ˈkʌlər ˈblaɪndnəs/ | Mù màu |
| 446 | Hypertelorism | /ˌhaɪpərˈtiːlərɪzəm/ | Hai mắt xa nhau |
| 447 | Fontanelle | /ˌfɒntəˈnɛl/ | Thóp |
| 448 | Wormian bones | /ˈwɜːrmiən boʊnz/ | Xương Wormian |
| 449 | Epiphyseal dysgenesis | /ˌɛpɪˈfɪziəl dɪsˈdʒɛnəsɪs/ | Loạn sản sụn đầu xương |
| 450 | Dyspraxia | /dɪsˈpræksiə/ | Chứng khó phối hợp động tác |
| 451 | Striae | /ˈstraɪiː/ | Vết rạn da |
| 452 | Adrenarche | /ˌædrɪˈnɑːrki/ | Khởi phát tuyến thượng thận |
| 453 | Congenital adrenal hyperplasia (CAH) | /kənˈdʒɛnɪtəl əˈdriːnəl ˌhaɪpərˈpleɪʒə/ | Tăng sản tuyến thượng thận bẩm sinh |
| 454 | Thelarche | /θiːˈlɑːrki/ | Khởi phát tuyến vú |
| 455 | Pubarche | /pjuːˈbɑːrki/ | Khởi phát lông mu |
| 456 | Pectus carinatum | /ˈpɛktəs ˌkærɪˈneɪtəm/ | Ngực ức gà |
| 457 | Pectus excavatum | /ˈpɛktəs ˌɛkskəˈveɪtəm/ | Ngực lõm |
| 458 | Kyphoscoliosis | /ˌkaɪfoʊˌskoʊliˈoʊsɪs/ | Gù vẹo cột sống |
| 459 | Lymphedema | /ˌlɪmfɪˈdiːmə/ | Phù bạch huyết |
| 460 | Cryptorchidism | /krɪpˈtɔːrkɪdɪzəm/ | Tinh hoàn ẩn |
| 461 | Myelomonocytic leukemia | /ˌmaɪəloʊˌmɒnoʊˈsɪtɪk luːˈkiːmiə/ | Bệnh bạch cầu tủy bào đơn nhân |
| 462 | Myeloproliferative disorders | /ˌmaɪəloʊprəˈlɪfərətɪv dɪsˈɔːrdərz/ | Rối loạn tăng sinh tủy |
| 463 | RASopathies | /ræsˈɒpəθiz/ | Các bệnh liên quan đến RAS |
| 464 | Cardiofaciocutaneous syndrome | /ˌkɑːrdioʊˌfeɪʃioʊkjuːˈteɪniəs ˈsɪndroʊm/ | Hội chứng tim-mặt-da |
| 465 | Costello syndrome | /kɒˈstɛloʊ ˈsɪndroʊm/ | Hội chứng Costello |
| 466 | Lentigines | /lɛnˈtɪdʒɪniːz/ | Nốt ruồi (đốm tàn nhang) |
| 467 | Anagen | /ˈænədʒən/ | Giai đoạn tăng trưởng (của tóc) |
| 468 | Pleiotropic effects | /ˌplaɪəˈtrɒpɪk ɪˈfɛkts/ | Tác động đa hiệu |
| 469 | Pulmonary stenosis | /ˈpʊlmənəri stɛˈnoʊsɪs/ | Hẹp van động mạch phổi |
| 470 | Guanine-nucleotide binding protein (G protein) | /ˈɡwɑːniːn ˈnjuːkliətaɪd ˈbaɪndɪŋ ˈproʊtiːn/ | Protein G (protein liên kết nucleotide guanine) |
| 471 | Adenylyl cyclase | /əˈdɛnəlɪl ˈsaɪkleɪz/ | Adenylyl cyclase |
| 472 | Epimutation | /ˌɛpɪmjuːˈteɪʃən/ | Đột biến ngoại di truyền |
| 473 | Hypocalcemia | /ˌhaɪpoʊkælˈsiːmiə/ | Hạ canxi máu |
| 474 | Hyperphosphatemia | /ˌhaɪpərfɒsfəˈtiːmiə/ | Tăng phosphate máu |
| 475 | Pseudopseudohypoparathyroidism (PPHP) | /ˈsuːdoʊˈsuːdoʊˌhaɪpoʊˌpærəˈθaɪrɔɪdɪzəm/ | Giả-giả suy cận giáp |
| 476 | Brachydactyly | /ˌbrækɪˈdæktɪli/ | Tật ngón ngắn |
| 477 | Ectopic ossifications | /ɛkˈtɒpɪk ˌɒsɪfɪˈkeɪʃənz/ | Cốt hóa lạc chỗ |
| 478 | Dysmorphism | /dɪsˈmɔːrfɪzəm/ | Dị dạng |
| 479 | Hyperlordosis | /ˌhaɪpərlɔːrˈdoʊsɪs/ | Ưỡn cột sống quá mức |
| 480 | Hypospadias | /ˌhaɪpoʊˈspeɪdiəs/ | Lỗ tiểu lệch thấp |
| 481 | Ubiquitin ligase | /juːˈbɪkwɪtɪn ˈlaɪɡeɪs/ | Ubiquitin ligase |
| 482 | Fanconi anemia | /fæŋˈkoʊni əˈniːmiə/ | Thiếu máu Fanconi |
| 483 | Telangiectatic erythema | /təˌlændʒiɛkˈtætɪk ˌɛrɪˈθiːmə/ | Ban đỏ giãn mao mạch |
| 484 | Photosensitivity | /ˌfoʊtoʊˌsɛnsɪˈtɪvəti/ | Nhạy cảm với ánh sáng |
| 485 | Bronchiectasis | /ˌbrɒŋkiˈɛktəsɪs/ | Giãn phế quản |
| 486 | Aplasia | /əˈpleɪʒə/ | Bất sản |
| 487 | Neutropenia | /ˌnjuːtrəˈpiːniə/ | Giảm bạch cầu trung tính |
| 488 | Thrombocytopenia | /ˌθrɒmboʊˌsaɪtoʊˈpiːniə/ | Giảm tiểu cầu |
| 489 | Hypergonadotropic hypogonadism | /ˌhaɪpərɡoʊˌnædəˈtrɒpɪk ˌhaɪpoʊˈɡoʊnædɪzəm/ | Suy sinh dục do tăng gonadotropin |
| 490 | Dyslipidemia | /ˌdɪslɪpɪˈdiːmiə/ | Rối loạn lipid máu |
| 491 | Primordial dwarfisms | /praɪˈmɔːrdiəl ˈdwɔːrfɪzəmz/ | Các dạng lùn nguyên thủy |
| 492 | Seckel syndrome | /ˈsɛkəl ˈsɪndroʊm/ | Hội chứng Seckel |
| 493 | Microcephalic osteodysplastic primordial dwarfism (MOPD) | /ˌmaɪkroʊsəˈfælɪk ˌɒstioʊdɪsˈplæstɪk praɪˈmɔːrdiəl ˈdwɔːrfɪzəm/ | Lùn nguyên thủy loạn sản xương đầu nhỏ |
| 494 | Meier-Gorlin syndrome | /ˈmaɪər ˈɡɔːrlɪn ˈsɪndroʊm/ | Hội chứng Meier-Gorlin |
| 495 | Centrosome | /ˈsɛntrəsoʊm/ | Trung thể |
| 496 | Mitotic spindle | /maɪˈtɒtɪk ˈspɪndəl/ | Thoi vô sắc |
| 497 | Imprinting disorders | /ˈɪmprɪntɪŋ dɪsˈɔːrdərz/ | Rối loạn in dấu |
| 498 | Epigenetic | /ˌɛpɪdʒəˈnɛtɪk/ | Ngoại di truyền |
| 499 | Uniparental disomy | /ˌjuːnɪpəˈrɛntəl ˈdaɪsoʊmi/ | Lưỡng bội đơn thân |
| 500 | Prader-Willi syndrome (PWS) | /ˈprɑːdər ˈwɪli ˈsɪndroʊm/ | Hội chứng Prader-Willi |