
Sperling Nội tiết học Nhi khoa, Ấn bản thứ 5 – Biên dịch: Ths.Bs. Lê Đình Sáng
Sperling Pediatric Endocrinology, Fifth Edition
Tác giả: Sperling, Mark A., MD – Nhà xuất bản: Elsevier Inc.
PHẦN III. NỘI TIẾT HỌC TRẺ EM VÀ THANH THIẾU NIÊN
Chương 14: Vỏ thượng thận và các rối loạn liên quan
Walter L. Miller; Christa E. Flück; David T. Breault; Brian J. Feldman
The Adrenal Cortex and Its Disorders
Sperling Pediatric Endocrinology, 14, 425-490
Lịch sử, phôi thai học và giải phẫu học
Vỏ thượng thận sản xuất ba nhóm hormone steroid chính, điều hòa một loạt các quá trình sinh lý từ giai đoạn bào thai đến tuổi trưởng thành. Mineralocorticoid, chủ yếu là aldosterone, điều hòa sự giữ lại natri ở thận và do đó ảnh hưởng sâu sắc đến cân bằng điện giải, thể tích nội mạch và huyết áp. Glucocorticoid, chủ yếu là cortisol, được đặt tên theo hoạt tính huy động carbohydrate, nhưng chúng là những chất điều hòa sinh lý phổ biến, ảnh hưởng đến nhiều chức năng của cơ thể. Androgen tuyến thượng thận không có vai trò sinh lý nào được biết đến nhưng có vai trò trung gian trong một số đặc điểm sinh dục phụ ở phụ nữ (ví dụ, lông mu và lông nách), và sự sản xuất quá mức của chúng có thể dẫn đến nam hóa. Do đó, vỏ thượng thận rất được quan tâm vì những tác động lan tỏa từ các chất bài tiết của nó và vì các dẫn xuất của những steroid này được sử dụng rộng rãi làm dược phẩm. Các rối loạn của vỏ thượng thận, từng được cho là hiếm gặp, đang ngày càng được ghi nhận thường xuyên hơn. Các thể tăng sản thượng thận bẩm sinh (CAH) nặng ảnh hưởng đến gần 1 trên 10.000 người, và các thể rất nhẹ có thể ảnh hưởng đến 1 trên 100 người ở một số quần thể. Bệnh Cushing, từng được coi là rất hiếm trong nhi khoa, có thể ảnh hưởng đến trẻ em nhiều như người lớn. Cường aldosteron nguyên phát là một nguyên nhân phổ biến của tăng huyết áp, đặc biệt ở người lớn.
Lịch sử
Lịch sử nghiên cứu về tuyến thượng thận gần đây đã được tổng hợp lại. Tuyến thượng thận dường như được mô tả lần đầu tiên vào năm 1563 bởi nhà giải phẫu học người Ý Bartolomeo Eustaccio, người được biết đến nhiều hơn với mô tả về vòi Eustachi của tai. Sự quan tâm y học đến tuyến thượng thận như một thực thể không chỉ là một sự tò mò về giải phẫu bắt đầu vào giữa thế kỷ 19 với mô tả kinh điển của Addison về suy thượng thận và việc Brown-Sequard tạo ra các rối loạn tương tự trên động vật thực nghiệm bằng cách cắt bỏ tuyến thượng thận. Các dấu hiệu và triệu chứng của tình trạng dư thừa glucocorticoid do khối u tuyến thượng thận đã được biết đến rõ vào năm 1932, khi Cushing mô tả các khối u tuyến yên gây ra bệnh mà ngày nay được gọi là bệnh Cushing. Tác động của việc cắt bỏ tuyến thượng thận đối với chuyển hóa muối và nước đã được báo cáo vào năm 1927, Loeb đã chỉ ra rằng việc truyền nước muối giúp kéo dài sự sống của bệnh nhân Addison vào năm 1933, và đến cuối những năm 1930, Selye đã đề xuất các thuật ngữ glucocorticoid và mineralocorticoid để phân biệt hai nhóm tác động chính của các chiết xuất từ tuyến thượng thận.
Nhiều steroid tuyến thượng thận đã được phân lập một cách công phu và cấu trúc của chúng được xác định trong những năm 1930 trong các phòng thí nghiệm của Reichstein và Kendall. Nhiều trong số các steroid này đã được tổng hợp hóa học, cung cấp vật liệu tinh khiết cho mục đích thực nghiệm. Năm 1949, Kendall và Hench báo cáo rằng glucocorticoid làm giảm các triệu chứng của viêm khớp dạng thấp, điều này đã kích thích mạnh mẽ sự quan tâm đến việc tổng hợp các chất tương tự có hoạt tính dược lý mới của các steroid tự nhiên. Kendall, Reichstein và Hench đã cùng nhận giải Nobel Y học năm 1950. Cấu trúc của các steroid tuyến thượng thận khác nhau gợi ý về mối quan hệ tiền chất/sản phẩm, dẫn đến việc điều trị CAH bằng cortisone lần đầu tiên vào năm 1950 bởi cả Wilkins và Bartter. Điều này đã mở ra một kỷ nguyên sôi nổi của các nghiên cứu lâm sàng về các con đường sinh tổng hợp steroid trong một loạt các rối loạn di truyền của tuyến thượng thận và tuyến sinh dục. Mối liên hệ giữa cytochrome P450 với quá trình 21-hydroxyl hóa được xác định vào năm 1965, và một số enzyme sinh steroid sau đó đã được phân lập trong những năm 1970, nhưng phải đến khi các gen của hầu hết các enzyme này được nhân bản vào những năm 1980, người ta mới làm rõ được protein nào tham gia vào quá trình biến đổi steroid nào. Việc xác định các gen này (Bảng 14.1) sau đó đã dẫn đến sự hiểu biết về các tổn thương di truyền gây ra các rối loạn di truyền trong sinh tổng hợp steroid. Đồng thời, các nghiên cứu về tác động của hormone steroid đã dẫn đến việc phát hiện ra các thụ thể hormone steroid vào những năm 1960, nhưng phải đến khi chúng được nhân bản vào những năm 1980, sinh học của chúng mới bắt đầu được hiểu rõ.
Bảng 14.1. Đặc điểm Vật lý của các Gen Người Mã hóa cho các Enzyme Sinh steroid
| Enzyme | Gen | Kích thước Gen (kb) | Vị trí Nhiễm sắc thể | Số Exon | Kích thước mRNA (kb) |
|---|---|---|---|---|---|
| StAR | STAR | 8 | 8p11.2 | 8 | 1.6 |
| P450scc | CYP11A1 | >30 | 15q23-q24 | 9 | 2 |
| P450c11β | CYP11B1 | 9.5 | 8q21-22 | 9 | 4.2 |
| P450c11AS | CYP11B2 | 9.5 | 8q21-22 | 9 | 4.2 |
| P450c17 | CYP17A1 | 6.6 | 10q24.3 | 8 | 1.9 |
| P450c21 | CYP21A2 | 3.4 | 6p21.1 | 10 | 2 |
| P450aro | CYP19A1 | >130 | 15q21.1 | 10 | 1.5-4.5 |
| 3βHSD1 | HSD3B1 | 8 | 1p13.1 | 4 | 1.7 |
| 3βHSD2 | HSD3B2 | 8 | 1p13.1 | 4 | 1.7 |
| 11βHSD1 | HSD11B1 | >7 | 1q32-q41 | 6 | 1.6 |
| 11βHSD2 | HSD11B2 | 6.2 | 16q22 | 5 | 1.6 |
| 17βHSD1 | HSD17B1 | 3.3 | 17q11-q21 | 6 | 1.4, 2.4 |
| 17βHSD2 | HSD17B2 | 63 | 16q24.1-q24.2 | 5 | 1.5 |
| 17βHSD3 | HSD17B3 | >67 | 9q22 | 11 | 1.2 |
| 17βHSD6 (RoDH) | HSD17B6 | 24.5 | 12q13 | 5 | 1.6 |
| AKR1C1 | AKR1C1 | 14.3 | 10p14-p15 | 9 | 1.2 |
| AKR1C2 | AKR1C2 | 13.8 | 10p14-p15 | 9 | 1.3 |
| AKR1C3 | AKR1C3 | 13 | 10p14-p15 | 9 | 1.2 |
| AKR1C4 | AKR1C4 | 22.1 | 10p14-p15 | 9 | 1.2 |
| 5α-Reductase 1 | SRD5A1 | >36 | 5p15 | 5 | 2.4 |
| 5α-Reductase 2 | SRD5A2 | 56 | 2p23 | 5 | 2.4 |
| SULT2A1 | SULT2A1 | 17 | 19q13.3 | 6 | 2 |
| PAPSS2 | PAPSS2 | >85 | 10q24 | 13 | 3.9 |
| P450 Oxidoreductase | POR | >69 | 7q11.2 | 16 | 2.5 |
| Ferredoxin | FDX1 | 35 | 11q22 | 5 | 1.0, 1.4, 1.7, 3.2 |
| Ferredoxin Reductase | FDXR | >11 | 17q24-q25 | 12 | 2 |
| Cytochrome b5 | CYB5A | >32 | 18q23 | 5 | 0.9 |
| H6PDH | H6PD | 36.5 | 1p36 | 5 | >9 |
mRNA, RNA thông tin.
Phôi thai học
Các tế bào của vỏ thượng thận có nguồn gốc từ trung bì, trái ngược với các tế bào của tủy thượng thận có nguồn gốc từ ngoại bì thần kinh. Ở phôi người, các tế bào tiền thân thượng thận-sinh dục lần đầu tiên xuất hiện vào khoảng tuần thứ tư của thai kỳ dưới dạng một lớp dày lên của biểu mô khoang cơ thể (hoặc trung bì trung gian) giữa mào niệu-sinh dục và mạc treo lưng (Hình 14.1). Những tế bào tiền thân này tạo ra các tế bào sinh steroid của tuyến sinh dục và vỏ thượng thận. Các tế bào thượng thận và sinh dục sau đó tách ra, với các tế bào thượng thận di chuyển ra sau phúc mạc đến cực trên của trung thận và các tế bào sinh dục di chuyển xuống dưới. Giữa tuần thứ bảy và thứ tám của quá trình phát triển, mầm thượng thận bị xâm nhập bởi các tế bào giao cảm có nguồn gốc từ mào thần kinh, tạo ra tủy thượng thận. Đến cuối tuần thứ tám, tuyến thượng thận sơ khai đã được bao bọc và liên kết rõ ràng với cực trên của thận, lúc này nhỏ hơn nhiều so với tuyến thượng thận.
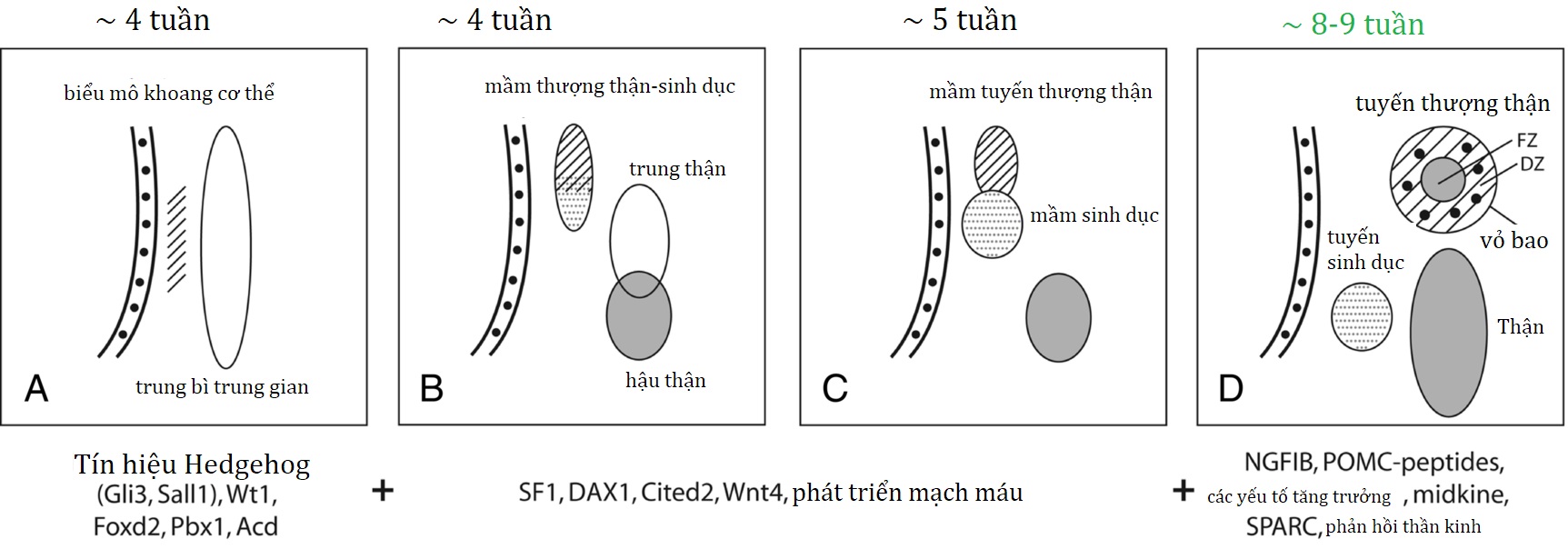
Hình 14.1: Tổng quan về sự phát triển tuyến thượng thận của người. A-C, Mầm tuyến thượng thận-sinh dục phát triển vào khoảng tuần thứ 4 của thai kỳ, sau đó mầm tuyến thượng thận trở thành một cấu trúc riêng biệt rồi di chuyển ra sau phúc mạc đến cực trên của trung thận. D, Đến tuần thứ 8 đến 9 của thai kỳ, tuyến thượng thận được bao bọc, chứa các tế bào ưa crôm (màu đen) và có các vùng bào thai (FZ) và vùng trưởng thành (DZ) riêng biệt. Một số phân tử tín hiệu, yếu tố phiên mã và yếu tố tăng trưởng liên quan đến sự phát triển của tuyến thượng thận được trình bày bên dưới, mặc dù thời gian chính xác và sự tương tác của nhiều yếu tố này hiện vẫn chưa được hiểu rõ.
Vỏ thượng thận của thai nhi bao gồm một vùng “xác định” bên ngoài, là nơi chính tổng hợp glucocorticoid và mineralocorticoid, và một vùng “bào thai” lớn hơn nhiều, tạo ra các tiền chất androgen (dehydroepiandrosterone [DHEA], dehydroepiandrosterone sulfate [DHEAS]) mà nhau thai chuyển đổi thành estriol. Một vùng “chuyển tiếp” được cho là tồn tại giữa các vùng này vào cuối quá trình phát triển của thai nhi, nhưng vai trò của nó chưa rõ ràng. Tuyến thượng thận của thai nhi rất lớn so với các cấu trúc khác và tiếp tục phát triển cho đến ba tháng cuối thai kỳ (Hình 14.2). Khi sinh ra, tuyến thượng thận nặng từ 8 đến 9 g, có kích thước tương đương với tuyến thượng thận của người trưởng thành và chiếm khoảng 0,4% tổng trọng lượng cơ thể. Sau khi sinh, các tế bào của vùng bào thai trải qua quá trình apoptosis, và vùng thượng thận của thai nhi nhanh chóng thoái triển và gần như biến mất hoàn toàn sau 6 đến 12 tháng tuổi. Sau đó, sự phát triển của tuyến thượng thận tương đối chậm, do đó tuyến thượng thận chỉ chiếm 0,01% trọng lượng cơ thể ở người trưởng thành.
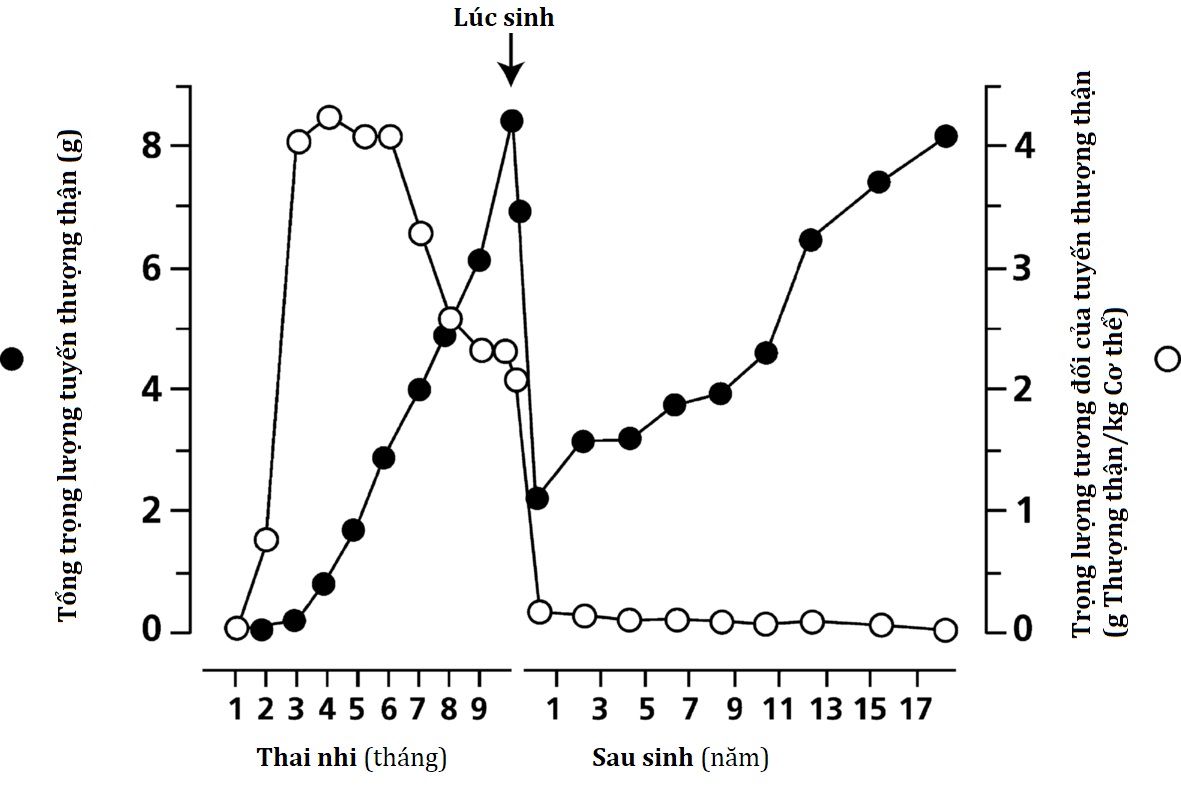
Hình 14.2: Tổng trọng lượng tuyến thượng thận (chấm tròn đen) và trọng lượng tương đối của tuyến thượng thận (vòng tròn trắng) từ ba tháng đầu thai kỳ cho đến khi trưởng thành sớm.
Các cơ chế phức tạp điều hòa sự phát triển của tuyến thượng thận vẫn còn chưa được hiểu rõ. Tuy nhiên, những hiểu biết quan trọng về các yếu tố chính đã được thu thập từ các nghiên cứu trên chuột biến đổi gen và từ các bệnh nhân mắc các rối loạn phát triển tuyến thượng thận. Ví dụ, các giai đoạn đầu của sự biệt hóa và phát triển tuyến thượng thận liên quan đến một số con đường tín hiệu (hedgehog/GLI3, WNT3/WNT4/WNT11, midkine), các yếu tố phiên mã (SALL1, FOXD2, PBX1, WT1, SF1 [NR5A1], DAX1 [NR0B1]), các yếu tố đồng điều hòa (CITED2), các protein chất nền (SPARC), và các chất điều hòa hoạt động telomerase (ACD). Sự phát triển sau đó của tuyến thượng thận ở thai nhi phụ thuộc nhiều vào tác động dinh dưỡng của hormone vỏ thượng thận (ACTH), thụ thể của nó (MC2R), và các con đường tín hiệu xuôi dòng, cũng như các con đường tín hiệu của yếu tố tăng trưởng, chẳng hạn như yếu tố tăng trưởng giống insulin-2 (IGF-2), yếu tố tăng trưởng nguyên bào sợi cơ bản (bFGF, hiện được gọi là FGF2), và yếu tố tăng trưởng biểu bì (EGF).
Giải phẫu học
Tuyến thượng thận, từng được gọi là tuyến trên thận, có tên gọi bắt nguồn từ vị trí giải phẫu của chúng, nằm trên cực trên của mỗi quả thận. Không giống như hầu hết các cơ quan khác, các động mạch và tĩnh mạch cung cấp cho tuyến thượng thận không chạy song song. Máu động mạch được cung cấp bởi một số động mạch nhỏ phát sinh từ động mạch thận và động mạch hoành, động mạch chủ, và đôi khi là động mạch buồng trứng và động mạch tinh hoàn trái. Các tĩnh mạch có cấu trúc thông thường hơn, với tĩnh mạch thượng thận trái đổ vào tĩnh mạch thận trái và tĩnh mạch thượng thận phải đổ trực tiếp vào tĩnh mạch chủ. Máu động mạch đi vào hệ tuần hoàn xoang của vỏ và đổ về phía tủy, do đó các tế bào ưa crom của tủy được tắm trong nồng độ hormone steroid rất cao. Nồng độ cortisol cao là cần thiết cho sự biểu hiện của phenylethanolamine-N-methyltransferase ở tủy, enzyme này chuyển đổi norepinephrine thành epinephrine, liên kết các phản ứng của vỏ và tủy thượng thận với stress.
Vỏ thượng thận bao gồm ba vùng có thể nhận biết về mặt mô học: lớp cầu (zona glomerulosa) ngay dưới bao, lớp bó (zona fasciculata) ở giữa, và lớp lưới (zona reticularis) nằm cạnh tủy. Lớp cầu, lớp bó và lớp lưới lần lượt chiếm khoảng 15%, 75% và 10% vỏ thượng thận của trẻ lớn và người trưởng thành. Các vùng này dường như khác biệt về chức năng cũng như mô học, nhưng có sự chồng chéo đáng kể, và dữ liệu hóa mô miễn dịch cho thấy các vùng này xen kẽ với nhau về mặt vật lý. Sau khi sinh, vùng bào thai lớn bắt đầu thoái triển và biến mất sau khoảng 3 đến 6 tháng tuổi. Vùng xác định đồng thời mở rộng, nhưng hai trong số các vùng của người trưởng thành, lớp cầu và lớp bó, chưa được biệt hóa hoàn toàn cho đến khoảng 3 tuổi, và lớp lưới có thể chưa được biệt hóa hoàn toàn cho đến khoảng 15 tuổi. Nguồn gốc của các vùng vỏ thượng thận riêng biệt và các cơ chế điều hòa sự tăng sinh của chúng vẫn chưa được hiểu rõ. Một mô hình cho thấy có một quần thể tế bào gốc chưa biệt hóa tồn tại giữa lớp cầu và lớp bó, đại diện cho một nhóm tế bào tiền thân chung có thể góp phần vào cả vùng trong và vùng ngoài. Ngược lại, lý thuyết “di chuyển hướng tâm” đề xuất rằng một quần thể tế bào tiền thân/tế bào gốc dưới bao đầu tiên biệt hóa thành các tế bào của lớp cầu, sau đó chuyển thành các tế bào lớp bó (và có lẽ sau đó là các tế bào lớp lưới), khi chúng di chuyển hướng tâm về phía tủy thượng thận, nơi chúng hoàn thành vòng đời và trải qua quá trình apoptosis.
Sinh tổng hợp hormone steroid
Các bước ban đầu: Hấp thu, dự trữ và vận chuyển Cholesterol
Hiện nay, chúng ta đã biết nhiều về sinh tổng hợp steroid và về các bước ban đầu trong quá trình vận chuyển cholesterol nội bào. Tuyến thượng thận của người có thể tự tổng hợp cholesterol từ acetate, nhưng phần lớn nguồn cung cấp cholesterol đến từ lipoprotein tỷ trọng thấp (LDL) trong huyết tương có nguồn gốc từ cholesterol trong chế độ ăn. Tuyến thượng thận của loài gặm nhấm lấy hầu hết cholesterol từ lipoprotein tỷ trọng cao thông qua một thụ thể được gọi là SR-B1, nhưng con đường này đóng vai trò nhỏ trong sinh tổng hợp steroid ở người. Nồng độ LDL đủ sẽ ức chế 3-hydroxy-3-methylglutaryl co-enzyme A (HMG-CoA) reductase, enzyme giới hạn tốc độ trong quá trình tổng hợp cholesterol. ACTH, chất kích thích sinh tổng hợp steroid tuyến thượng thận, cũng kích thích hoạt động của HMG-CoA reductase, các thụ thể LDL, và sự hấp thu cholesterol LDL. Các este cholesterol LDL được hấp thu bởi quá trình nhập bào qua trung gian thụ thể, sau đó được dự trữ trực tiếp hoặc chuyển đổi thành cholesterol tự do và được sử dụng để tổng hợp hormone steroid. Cholesterol có thể được este hóa bởi acyl-CoA:cholesterol transferase (ACAT), được lưu trữ trong các giọt lipid, và được tiếp cận bằng cách kích hoạt lipase nhạy cảm với hormone (HSL) và bởi các protein được gọi là NPC, có tên gọi bắt nguồn từ vai trò gây bệnh Niemann-Pick type C. ACTH kích thích HSL và ức chế ACAT, do đó làm tăng sự sẵn có của cholesterol tự do để tổng hợp hormone steroid.
Các Enzyme sinh steroid
Cytochrome P450
Hầu hết các enzyme sinh steroid là các enzyme cytochrome P450. “Cytochrome P450” chỉ một nhóm các enzyme oxy hóa, tất cả đều có khoảng 500 axit amin và chứa một nhóm heme duy nhất. “P450” có nghĩa là “sắc tố 450” vì tất cả đều hấp thụ ánh sáng ở bước sóng 450 nm ở trạng thái khử. Con người có 57 gen mã hóa các enzyme P450; bảy P450 ở người được nhắm đến ty thể và 50 được nhắm đến mạng lưới nội chất, đặc biệt là ở gan, nơi chúng chuyển hóa vô số độc tố, thuốc, các chất lạ và các chất ô nhiễm môi trường. Mỗi enzyme P450, bao gồm cả các P450 sinh steroid, có thể chuyển hóa nhiều cơ chất, xúc tác cho một loạt các phản ứng oxy hóa.
Năm P450 tham gia vào quá trình sinh tổng hợp steroid của tuyến thượng thận (Hình 14.3). P450scc (CYP11A1) ở ty thể là enzyme cắt chuỗi bên cholesterol, xúc tác cho một loạt các phản ứng trước đây được gọi là 20,22 desmolase. Hai isozyme của P450c11 ở ty thể, P450c11β (CYP11B1) và P450c11AS (CYP11B2), xúc tác cho các hoạt động 11β-hydroxylase, 18-hydroxylase và 18-methyl oxidase. P450c17 (CYP17A1), được tìm thấy trong mạng lưới nội chất, xúc tác cho cả hoạt động 17α-hydroxylase và 17,20 lyase, và P450c21 (CYP21A2) xúc tác cho quá trình 21-hydroxyl hóa cả glucocorticoid và mineralocorticoid. Trong tuyến sinh dục và các nơi khác, P450aro (CYP19A1) trong mạng lưới nội chất xúc tác cho quá trình thơm hóa androgen thành estrogen.
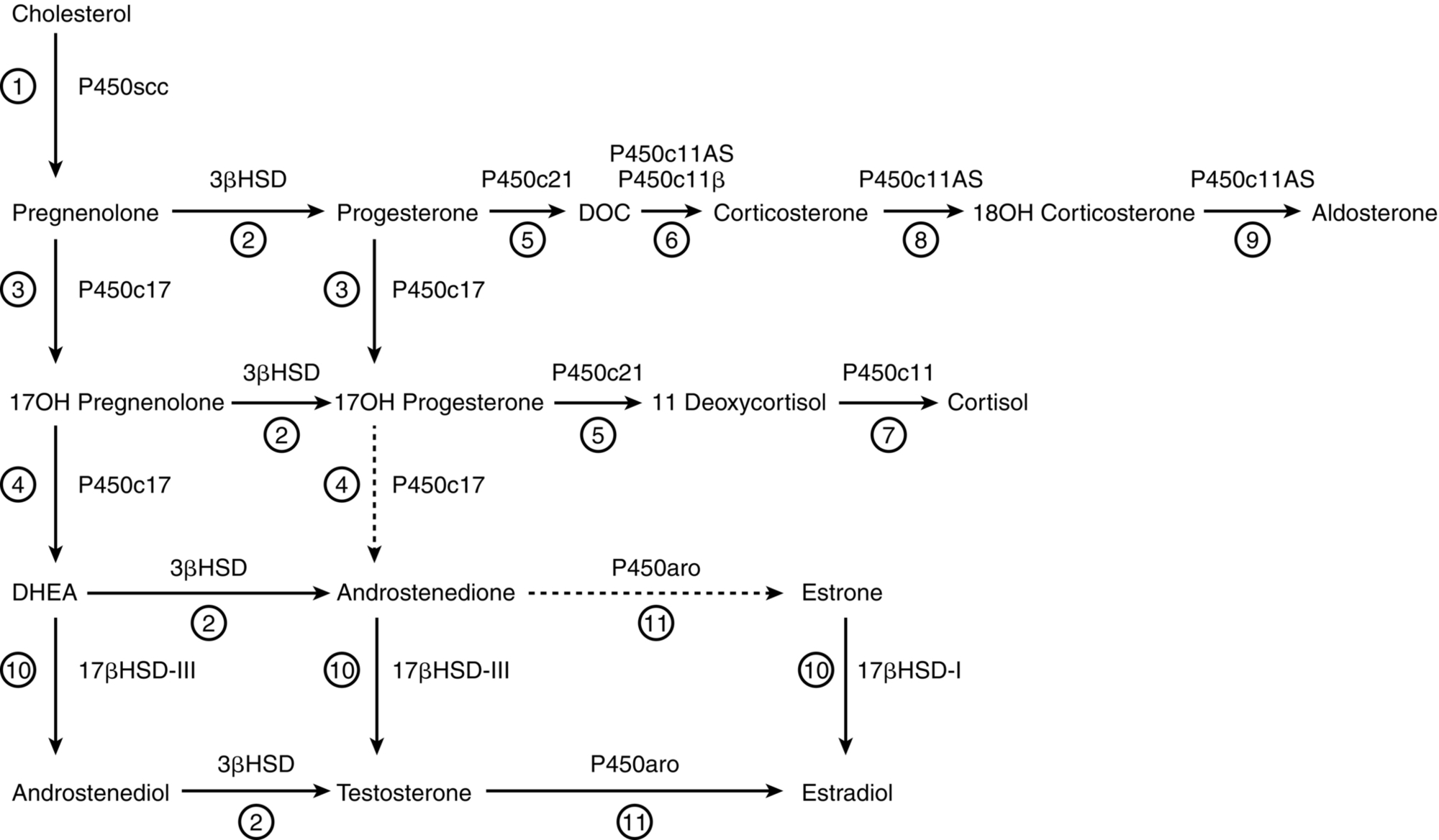
Hình 14.3: Các con đường chính của quá trình tổng hợp hormone steroid tuyến thượng thận ở người. Các steroid khác có số lượng và vai trò sinh lý thứ yếu cũng được sản xuất. Tên của các enzyme được hiển thị bên cạnh mỗi phản ứng, và tên truyền thống của các hoạt động enzyme tương ứng với các số được khoanh tròn. Phản ứng 1: Cytochrome P450scc ty thể xúc tác quá trình 20α-hydroxyl hóa, 22-hydroxyl hóa và cắt đứt liên kết carbon C20-22. Phản ứng 2: 3βHSD xúc tác các hoạt động 3β-hydroxysteroid dehydrogenase và isomerase, chuyển đổi các steroid Δ5 thành steroid Δ4. Phản ứng 3: P450c17 xúc tác quá trình 17α-hydroxyl hóa pregnenolone thành 17OH-pregnenolone và progesterone thành 17OH-progesterone. Phản ứng 4: Hoạt động 17,20 lyase của P450c17 chuyển đổi 17OH-pregnenolone thành dehydroepiandrosterone (DHEA); chỉ có một lượng không đáng kể 17OH-progesterone được chuyển đổi thành androstenedione Δ4 bởi P450c17 của người, mặc dù phản ứng này xảy ra ở các loài khác. Phản ứng 5: P450c21 xúc tác quá trình 21-hydroxyl hóa progesterone thành deoxycorticosterone (DOC) và 17OH-progesterone thành 11-deoxycortisol. Phản ứng 6: DOC được chuyển đổi thành corticosterone bởi hoạt động 11-hydroxylase của P450c11AS ở lớp cầu và bởi P450c11β ở lớp bó. Phản ứng 7: 11-Deoxycortisol trải qua quá trình 11β-hydroxyl hóa bởi P450c11β để tạo ra cortisol ở lớp bó. Phản ứng 8 và 9: Hoạt động 18-hydroxylase và 18-methyl oxidase của P450c11AS lần lượt chuyển đổi corticosterone thành 18OH-corticosterone và aldosterone, ở lớp cầu. Phản ứng 10 và 11 chủ yếu được tìm thấy trong tinh hoàn và buồng trứng. Phản ứng 10: 17βHSD-III chuyển đổi DHEA thành androstenediol và androstenedione thành testosterone, trong khi 17βHSD-I chuyển đổi estrone thành estradiol. Phản ứng 11: Testosterone có thể được chuyển đổi thành estradiol và androstenedione có thể được chuyển đổi thành estrone bởi P450aro.
Hydroxysteroid Dehydrogenase
Các hydroxysteroid dehydrogenase (HSD) có khối lượng phân tử khoảng 35 đến 45 kDa, không có nhóm heme và cần nicotinamide adenine dinucleotide (NAD+) hoặc nicotinamide adenine dinucleotide phosphate (NADP+) làm đồng yếu tố. Trong khi hầu hết các phản ứng sinh steroid do P450 xúc tác được thực hiện bởi một dạng P450 duy nhất, mỗi phản ứng do HSD xúc tác có thể được thực hiện bởi ít nhất hai isozyme. Các HSD bao gồm 3α- và 3β-hydroxysteroid dehydrogenase, hai 11β-hydroxysteroid dehydrogenase, và một loạt các 17β-hydroxysteroid dehydrogenase; các 5α-reductase không liên quan đến họ này. Dựa trên cấu trúc của chúng, các HSD được chia thành hai nhóm: họ dehydrogenase/reductase chuỗi ngắn (SDR), có đặc điểm cấu trúc là “nếp gấp Rossman”, và họ aldo-keto reductase (AKR), có đặc điểm là mô-típ thùng triosephosphate isomerase. Các enzyme SDR bao gồm 11β-HSD 1 và 2, và 17β-HSD 1, 2, 3, và 4; các enzyme AKR bao gồm 17β-HSD5, quan trọng trong việc kích hoạt các tiền chất androgen ngoại tuyến, và các 3α-hydroxysteroid dehydrogenase tham gia vào cái gọi là con đường cửa sau của quá trình tổng hợp androgen ở thai nhi (xem sau). Dựa trên hoạt động của chúng, việc phân loại chúng thành dehydrogenase hoặc reductase hữu ích hơn về mặt sinh lý. Các dehydrogenase sử dụng NAD+ làm đồng yếu tố để oxy hóa hydroxysteroid thành ketosteroid, và các reductase chủ yếu sử dụng NADPH để khử ketosteroid thành hydroxysteroid. Mặc dù các enzyme này thường có thể hoạt động hai chiều trong ống nghiệm, chúng có xu hướng chỉ hoạt động theo một hướng trong các tế bào nguyên vẹn, với hướng được xác định bởi (các) đồng yếu tố có sẵn.
P450scc
Việc chuyển đổi cholesterol thành pregnenolone trong ty thể là bước đầu tiên, giới hạn tốc độ và được điều hòa bởi hormone trong quá trình tổng hợp tất cả các hormone steroid. Quá trình này bao gồm 20α-hydroxyl hóa, 22-hydroxyl hóa cholesterol, và cắt chuỗi bên của nó để tạo ra pregnenolone và axit isocaproic. Bởi vì 20-hydroxycholesterol, 22-hydroxycholesterol, và 20,22-hydroxycholesterol có thể được phân lập từ tuyến thượng thận với số lượng đáng kể, trước đây người ta cho rằng có ba enzyme riêng biệt tham gia. Tuy nhiên, một protein duy nhất, được gọi là P450scc (‘scc’ là viết tắt của side-chain cleavage – cắt chuỗi bên của cholesterol) được mã hóa bởi gen CYP11A1 trên nhiễm sắc thể 15, xúc tác cho tất cả các bước giữa cholesterol và pregnenolone. Việc xóa gen cyp11a1 ở chuột hoặc thỏ đã loại bỏ hoàn toàn quá trình sinh tổng hợp steroid, cho thấy rằng tất cả quá trình sinh tổng hợp steroid đều được khởi đầu bởi một enzyme này.
Vận chuyển Điện tử đến P450scc: Ferredoxin Reductase và Ferredoxin
Các enzyme P450 ty thể (P450scc, P450c11β, P450c11AS, và các vitamin D 1α- và 24-hydroxylase) là các oxidase cuối cùng trong một hệ thống vận chuyển điện tử: NADPH tặng điện tử cho ferredoxin reductase (FDXR, còn được gọi là adrenodoxin reductase), một flavoprotein liên kết lỏng lẻo với màng trong ty thể. FDXR chuyển các điện tử đến ferredoxin (FDX, còn được gọi là adrenodoxin), một protein sắt/lưu huỳnh 14 kDa, sau đó chuyển các điện tử đến P450scc (Hình 14.4). FDX tạo thành một phức hợp 1:1 với FDXR, phân ly, sau đó tái tạo một phức hợp 1:1 tương tự với P450, do đó hoạt động như một cơ chế con thoi điện tử khuếch tán. FDXR và FDX được biểu hiện rộng rãi trong các mô của người, nhưng biểu hiện của FDXR cao hơn hai bậc độ lớn trong các mô sinh steroid. Bản phiên mã axit ribonucleic (RNA) chính từ gen FDXR được cắt nối luân phiên tạo ra hai loại RNA thông tin (mRNA) mã hóa các protein khác nhau sáu axit amin, nhưng chỉ có protein ngắn hơn mới hoạt động trong sinh tổng hợp steroid. FDXR cũng rất cần thiết trong việc hình thành các trung tâm sắt/lưu huỳnh được nhiều enzyme sử dụng. Có hai isozyme FDX ở người được mã hóa bởi các gen trên các nhiễm sắc thể khác nhau: FDX1 tương tác với các enzyme P450 ty thể; FDX2 được sử dụng để tổng hợp các cụm sắt-lưu huỳnh, nhưng không dùng cho sinh tổng hợp steroid. FDX1 được biểu hiện khắp nơi nhưng đặc biệt phong phú ở vỏ thượng thận. Các đột biến ở FDX ở người chưa được mô tả, nhưng một số đột biến FDXR làm suy giảm một phần sự hình thành các trung tâm sắt/lưu huỳnh có liên quan đến mất thính lực do bệnh lý thần kinh.
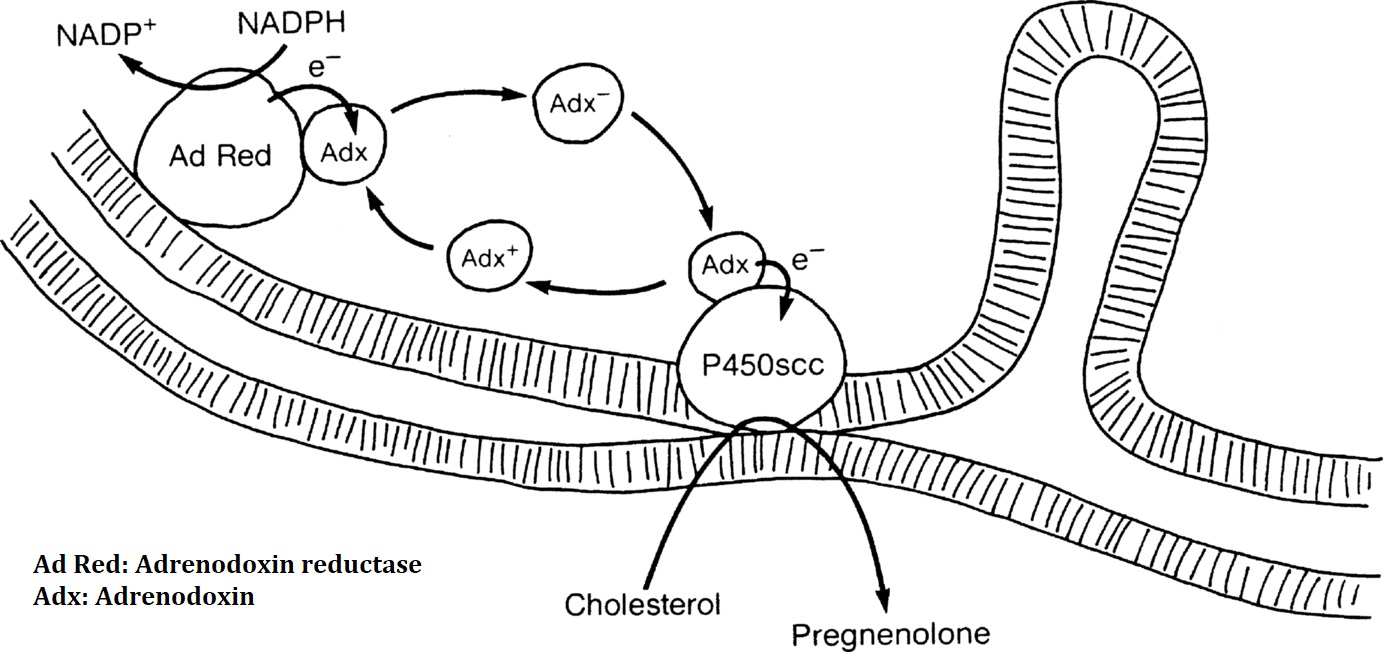
Hình 14.4: Vận chuyển điện tử đến các dạng cytochrome P450 ở ty thể. Adrenodoxin reductase (AdRed), một flavoprotein liên kết lỏng lẻo với màng trong ty thể, nhận các điện tử (e-) từ nicotinamide adenine dinucleotide phosphate hydrogen (NADPH), chuyển đổi nó thành NADP+. Các điện tử này được chuyển đến adrenodoxin (Adx), một protein sắt/lưu huỳnh trong dung dịch trong chất nền ty thể, hoạt động như một cơ chế con thoi vận chuyển điện tử khuếch tán tự do. Các điện tử từ adrenodoxin tích điện (Adx-) được chấp nhận bởi bất kỳ cytochrome P450 nào có sẵn, chẳng hạn như P450c11, hoặc P450scc được hiển thị ở đây. Adrenodoxin không tích điện (Adx+) sau đó có thể được liên kết lại với adrenodoxin reductase để nhận một cặp điện tử khác. Đối với P450scc, ba cặp điện tử phải được vận chuyển đến P450 để chuyển đổi cholesterol thành pregnenolone. Dòng cholesterol vào ty thể được tạo điều kiện bởi protein điều hòa cấp tính sinh steroid, không được hiển thị trong sơ đồ này.
Hấp thu Cholesterol vào Ty thể: Protein Điều hòa Cấp tính Sinh steroid, StAR
ACTH điều hòa khả năng sinh steroid (điều hòa mãn tính) bằng cách cảm ứng phiên mã các gen cho các enzyme sinh steroid, nhưng sự điều hòa cấp tính, nơi steroid được giải phóng trong vòng vài phút sau một kích thích, diễn ra ở mức độ tiếp cận của cholesterol với P450scc. Việc xử lý các tế bào sinh steroid hoặc chuột nguyên vẹn bằng các chất ức chế tổng hợp protein (ví dụ, cycloheximide) đã loại bỏ phản ứng sinh steroid cấp tính, cho thấy một protein tồn tại ngắn đã kích hoạt phản ứng này. Một cuộc tìm kiếm dài đã dẫn đến việc xác định và nhân bản protein điều hòa cấp tính sinh steroid, StAR. Vai trò trung tâm của StAR trong sinh tổng hợp steroid đã được chứng minh bằng việc phát hiện ra rằng các đột biến của StAR gây ra tăng sản thượng thận dạng mỡ bẩm sinh. Do đó, StAR là tác nhân kích hoạt cấp tính cần thiết cho dòng cholesterol nhanh chóng từ màng ngoài đến màng trong ty thể, cần thiết cho phản ứng cấp tính của aldosterone đối với angiotensin II, của cortisol đối với ACTH, và của steroid sinh dục đối với một xung hormone tạo hoàng thể (LH).
Một số quá trình sinh tổng hợp steroid của tuyến thượng thận độc lập với StAR; quá trình sinh tổng hợp steroid không phụ thuộc StAR này chiếm khoảng 14% tốc độ do StAR gây ra. Nhau thai sử dụng P450scc để bắt đầu sinh tổng hợp steroid nhưng không biểu hiện StAR. Cơ chế sinh tổng hợp steroid không phụ thuộc StAR chưa rõ ràng; nó có thể xảy ra mà không cần một protein kích hoạt, hoặc một protein khác có thể có hoạt tính giống StAR để thúc đẩy dòng cholesterol, nhưng không có động học nhanh như của StAR. Cơ chế hoạt động của StAR chưa rõ ràng, nhưng đã xác định được rằng StAR hoạt động trên màng ngoài ty thể (OMM), không cần phải đi vào ty thể để hoạt động, và trải qua những thay đổi cấu trúc trên OMM cần thiết cho hoạt động của StAR. Một số nghiên cứu cho thấy StAR hoạt động như một thành phần của một cỗ máy phân tử bao gồm StAR, TSPO (protein vận chuyển trước đây được gọi là thụ thể benzodiazepine ngoại vi), và các protein khác trên OMM, mặc dù các nghiên cứu với chuột knockout TSPO đặt câu hỏi về vai trò của nó. Cách thức chính xác mà cholesterol được nạp vào OMM và di chuyển từ OMM đến P450scc với sự hỗ trợ của StAR vẫn chưa rõ ràng, và vẫn đang được tích cực nghiên cứu.
3β-Hydroxysteroid Dehydrogenase/Δ5− > Δ4 Isomerase
Khi pregnenolone được sản xuất từ cholesterol, nó có thể trải qua quá trình 17α-hydroxyl hóa bởi P450c17 để tạo ra 17-hydroxypregnenolone (17-Preg), hoặc nó có thể được chuyển đổi thành progesterone. Hai isozyme 42 kDa của 3β-hydroxysteroid dehydrogenase (3βHSD), được mã hóa bởi các gen HSD3B1 và HSD3B2, có thể xúc tác cho cả việc chuyển đổi nhóm hydroxyl thành nhóm keto ở carbon 3 và sự đồng phân hóa của liên kết đôi từ vòng B (steroid Δ5) sang vòng A (steroid Δ4). Các isozyme này có 93,5% tương đồng về trình tự axit amin và rất giống nhau về mặt enzyme: cả hai đều có thể chuyển đổi pregnenolone thành progesterone, 17-Preg thành 17α-hydroxyprogesterone (17OHP), DHEA thành androstenedione, và androstenediol thành testosterone. Tuy nhiên, 3βHSD2, isozyme được biểu hiện ở tuyến thượng thận và tuyến sinh dục, có hằng số Michaelis-Menten (Km) cao khoảng 5,5 μM, cao hơn khoảng 10 lần so với 3βHSD1 được biểu hiện ở nhau thai, não và các mô “ngoại tuyến”. Km thấp của 3βHSD1 ngoại tuyến cho phép nó tác động lên nồng độ thấp của một số steroid được tìm thấy trong tuần hoàn. Dữ liệu siêu cấu trúc cho thấy 3βHSD có thể được tìm thấy trong ty thể, mạng lưới nội chất và tế bào chất. Không rõ liệu sự phân bố dưới tế bào này có khác nhau ở các loại tế bào sinh steroid khác nhau hay không, nhưng đây có thể là một điểm điều hòa mới cho hướng đi của quá trình sinh tổng hợp steroid.
P450c17
P450c17 (CYP17A1) xúc tác cho cả hoạt động 17α-hydroxylase và 17,20-lyase. Hoạt động 17α-hydroxylase của P450c17 có thể chuyển đổi pregnenolone thành 17-Preg và progesterone thành 17OHP. Hoạt động 17,20-lyase của P450c17 có thể chuyển đổi 17-Preg thành DHEA, nhưng rất ít 17OHP được chuyển đổi thành androstenedione vì P450c17 của người xúc tác phản ứng này chỉ ở mức khoảng 2% đến 3% tốc độ chuyển đổi 17-Preg thành DHEA. P450c17 là điểm phân nhánh chính trong quá trình tổng hợp hormone steroid: khi không có nó, như ở lớp cầu của tuyến thượng thận, hoạt động 17α-hydroxylase và 17,20-lyase không có, do đó pregnenolone được chuyển đổi thành mineralocorticoid; khi có hoạt động 17α-hydroxylase của nó ở lớp bó, chỉ có hoạt động 17α-hydroxylase hiện diện, do đó pregnenolone được chuyển đổi tuần tự thành cortisol; ở lớp lưới, nơi cả hai hoạt động đều có mặt, pregnenolone được chuyển đổi tuần tự thành các steroid sinh dục (xem Hình 14.3).
17α-hydroxylase và 17,20 lyase từng được cho là các enzyme riêng biệt. Tuyến thượng thận của trẻ em trước tuổi dậy thì tổng hợp đủ cortisol nhưng hầu như không có DHEA (tức là có hoạt động 17α-hydroxylase nhưng không có hoạt động 17,20 lyase) cho đến khi giai đoạn thượng thận hóa (adrenarche) bắt đầu sản xuất androgen tuyến thượng thận (tức là bật hoạt động 17,20 lyase). Hơn nữa, các bệnh nhân đã được mô tả là thiếu hoạt động 17,20 lyase nhưng vẫn giữ được hoạt động 17α-hydroxylase bình thường. Tuy nhiên, cả hai hoạt động 17α-hydroxylase và 17,20 lyase đều nằm trong cùng một vị trí hoạt động, và các tế bào được chuyển nạp với một vector biểu hiện P450c17 có được cả hai hoạt động 17α-hydroxylase và 17,20 lyase. P450c17 được mã hóa bởi gen CYP17A1 trên nhiễm sắc thể 10q24.3, và có liên quan về cấu trúc với gen CYP21A2 của P450c21 (21-hydroxylase). Do đó, sự phân biệt giữa 17α-hydroxylase và 17,20 lyase là về mặt chức năng chứ không phải di truyền hay cấu trúc. P450c17 của người xúc tác quá trình 17α-hydroxyl hóa pregnenolone và progesterone tốt như nhau, nhưng hoạt động 17,20 lyase của nó gần như chỉ sử dụng 17-Preg, chứ không phải 17OHP, điều này phù hợp với lượng lớn DHEA được tiết ra bởi cả tuyến thượng thận của thai nhi và người lớn. Yếu tố chính điều hòa phản ứng 17,20 lyase là sự vận chuyển điện tử từ NADPH thông qua P450 oxidoreductase (POR).
Vận chuyển Điện tử đến P450c17: P450 Oxidoreductase và Cytochrome b5
POR, một flavoprotein gắn màng không liên quan đến ferredoxin reductase ty thể, nhận điện tử từ NADPH và chuyển chúng đến 50 dạng cytochrome P450 ở vi thể, bao gồm P450c17, P450c21 và P450aro. Đối với phản ứng 17,20 lyase của P450c17, việc vận chuyển điện tử được tạo điều kiện bởi hoạt động của cytochrome b5, hoạt động như một yếu tố dị lập thể chứ không phải là một nhà tài trợ điện tử thay thế; hoạt động 17,20 lyase cũng được tăng lên bởi sự phosphoryl hóa serine/threonine của P450c17 bởi p38α, một protein kinase phụ thuộc cyclic adenosine monophosphate (cAMP) (Hình 14.5). Do đó, sự sẵn có của các điện tử quyết định liệu P450c17 chỉ thực hiện 17α-hydroxyl hóa hay còn thực hiện cả việc cắt liên kết 17,20: việc tăng tỷ lệ POR hoặc cytochrome b5 so với P450c17 làm tăng tỷ lệ hoạt động 17,20 lyase so với hoạt động 17α-hydroxylase. Do đó, sự điều hòa hoạt động 17,20 lyase, và do đó là sản xuất DHEA, phụ thuộc vào các yếu tố tạo điều kiện cho dòng điện tử đến P450c17: nồng độ POR cao, sự hiện diện của cytochrome b5, và sự phosphoryl hóa serine của P450c17.
P450c21
Sau khi tổng hợp progesterone và 17OHP, các steroid này được 21-hydroxyl hóa để tạo ra deoxycorticosterone (DOC) và 11-deoxycortisol, tương ứng (xem Hình 14.3). Bản chất của bước 21-hydroxyl hóa rất được quan tâm vì rối loạn 21-hydroxyl hóa gây ra hơn 90% tất cả các trường hợp CAH. Các phát hiện lâm sàng trong CAH rất phức tạp và có khả năng tàn phá. Giảm tổng hợp cortisol và aldosterone thường dẫn đến mất natri, giữ kali, nhiễm toan và hạ huyết áp, điều này sẽ dẫn đến trụy tim mạch và tử vong, thường trong vòng một tháng sau khi sinh nếu không được điều trị thích hợp. Giảm sinh tổng hợp steroid tuyến thượng thận trong tử cung dẫn đến sản xuất quá mức androgen tuyến thượng thận thông qua một số con đường (xem sau), dẫn đến nam hóa nặng ở thai nhi nữ. Quá trình 21-hydroxyl hóa của tuyến thượng thận được xúc tác bởi P450c21 được tìm thấy trong mạng lưới nội chất trơn, sử dụng POR để nhận điện tử từ NADPH. Các gen lặp lại CYP21A1P và CYP21A2 nằm trên nhiễm sắc thể 6p21, nhưng chỉ có gen CYP21A2 của người mã hóa P450c21. Vì gen này nằm ở giữa locus tương hợp mô chính, các rối loạn 21-hydroxyl hóa của tuyến thượng thận có liên quan chặt chẽ với các loại kháng nguyên bạch cầu người (HLA) cụ thể. Hoạt động 21-hydroxylase ngoại thượng thận được tìm thấy ở nhiều mô của người lớn và thai nhi, đặc biệt là ở gan, nhưng được xúc tác bởi các enzyme khác, đáng chú ý là CYP2C19 và CYP3A4, chủ yếu tham gia vào quá trình chuyển hóa thuốc. CYP2C19 và CYP3A4 có thể 21-hydroxyl hóa progesterone nhưng không phải 17OHP, và do đó có thể góp phần vào việc tổng hợp mineralocorticoid nhưng không phải glucocorticoid.
P450c11β và P450c11AS
Các enzyme P450c11β và P450c11AS có liên quan chặt chẽ với nhau xúc tác cho các bước cuối cùng trong quá trình tổng hợp glucocorticoid và mineralocorticoid. Hai isozyme này có 93% tương đồng về trình tự axit amin và được mã hóa bởi các gen lặp lại song song trên nhiễm sắc thể 8q21-22. Giống như P450scc, hai dạng P450c11 được tìm thấy trên màng trong ty thể, và sử dụng ferredoxin và ferredoxin reductase để nhận điện tử từ NADPH. Isozyme phong phú hơn nhiều trong hai loại là P450c11β (được mã hóa bởi CYP11B1), là enzyme 11β-hydroxylase cổ điển chuyển đổi 11-deoxycortisol thành cortisol và 11-deoxycorticosterone thành corticosterone. Isozyme ít phong phú hơn, P450c11AS (được mã hóa bởi CYP11B2), chỉ được tìm thấy ở lớp cầu, nơi nó có các hoạt động 11β-hydroxylase, 18-hydroxylase và 18-methyl oxidase (aldosterone synthase); do đó P450c11AS có thể xúc tác cho tất cả các phản ứng cần thiết để chuyển đổi DOC thành aldosterone. CYP11B1 được cảm ứng bởi ACTH thông qua cAMP và bị ức chế bởi glucocorticoid; CYP11B2 được cảm ứng bởi angiotensin II và K+. Bệnh nhân có đột biến ở CYP11B1 bị thiếu hụt 11β-hydroxylase cổ điển nhưng vẫn có thể sản xuất aldosterone, trong khi bệnh nhân có đột biến ở CYP11B2 có các dạng thiếu hụt aldosterone hiếm gặp (cái gọi là thiếu hụt corticosterone methyl oxidase), trong khi vẫn giữ được khả năng sản xuất cortisol.
17β-Hydroxysteroid Dehydrogenase
Androstenedione được chuyển đổi thành testosterone, DHEA được chuyển đổi thành androstenediol, và estrone được chuyển đổi thành estradiol bởi các 17β-hydroxysteroid dehydrogenase (17βHSD; HSD17B), đôi khi còn được gọi là 17-oxidoreductase hoặc 17-ketosteroid reductase. Thuật ngữ cho các enzyme này khác nhau, tùy thuộc vào hướng của phản ứng đang được xem xét. Y văn về 17βHSD có thể gây nhầm lẫn vì (1) có một số 17βHSD khác nhau; (2) một số là oxidase ưu tiên, trong khi những loại khác là reductase ưu tiên; (3) chúng khác nhau về sở thích cơ chất và vị trí biểu hiện; (4) danh pháp không nhất quán; và (5) một số protein được gọi là 17βHSD thực sự có rất ít hoạt động 17βHSD, và chủ yếu tham gia vào các phản ứng khác.
17βHSD loại 1 (17βHSD1, được mã hóa bởi HSD17B1), là một enzyme SDR khử, tạo estrogen 34 kDa được biểu hiện ở các tế bào hạt của buồng trứng (nơi nó sản xuất estradiol) và nhau thai (nơi nó sản xuất estriol). 17βHSD1 sử dụng NADPH làm đồng yếu tố, hoạt động như một dimer, và chỉ chấp nhận các cơ chất steroid có vòng A thơm, do đó hoạt động của nó chỉ giới hạn ở việc kích hoạt estrogen. Chưa có hội chứng thiếu hụt di truyền nào đối với 17βHSD1 được mô tả.
17βHSD2 là một oxidase ở vi thể sử dụng NAD+ để bất hoạt estradiol thành estrone và testosterone thành Δ4-androstenedione. 17βHSD2 được tìm thấy ở nhau thai, gan, ruột non, tuyến tiền liệt, nội mạc tử cung chế tiết và buồng trứng. Trong khi 17βHSD1 được tìm thấy ở các tế bào hợp bào nuôi của nhau thai, 17βHSD2 được biểu hiện ở các tế bào nội mô của các mạch máu trong nhung mao nhau thai, phù hợp với vai trò rõ ràng của nó trong việc bảo vệ tuần hoàn thai nhi khỏi sự vận chuyển qua nhau thai của estradiol hoặc testosterone từ mẹ. Chưa có tình trạng thiếu hụt nào đối với 17βHSD2 được báo cáo.
17βHSD3 ở vi thể là dạng 17βHSD androgenic chính, dường như chỉ được biểu hiện ở tinh hoàn. Enzyme này bị rối loạn trong hội chứng lưỡng tính giả nam kinh điển thường được gọi là thiếu hụt 17-ketosteroid reductase.
Một enzyme được gọi là 17βHSD4 ban đầu được xác định là một oxidase phụ thuộc NAD+ với các hoạt động tương tự như 17βHSD2, nhưng protein peroxisome này chủ yếu là một enoyl-CoA hydratase và 3-hydroxyacyl-CoA dehydrogenase. Sự thiếu hụt 17βHSD4 gây ra một dạng của hội chứng Zellweger, trong đó quá trình sinh tổng hợp axit mật bị xáo trộn nhưng quá trình sinh tổng hợp steroid thì không.
17βHSD5, ban đầu được nhân bản như một 3α-hydroxysteroid dehydrogenase, là một enzyme AKR (trong khi 17βHSD loại 1-4 là các enzyme SDR) được gọi là AKR1C3, xúc tác cho sự khử của Δ4-androstenedione thành testosterone. Hoạt động 17βHSD của 17βHSD5 khá không ổn định trong ống nghiệm, và enzyme này xúc tác các hoạt động khác nhau trong các điều kiện khác nhau. Lớp lưới của tuyến thượng thận biểu hiện enzyme này ở mức độ thấp, giải thích cho lượng testosterone nhỏ do tuyến thượng thận sản xuất.
17βHSD6, được mã hóa bởi gen HSD17B6 trên nhiễm sắc thể 12q13.3, còn được gọi là RoDH vì sự tương đồng của nó với các retinol dehydrogenase. 17βHSD6 được biểu hiện ở mức độ thấp nhưng có thể phát hiện được ở tinh hoàn của thai nhi, nơi nó dường như xúc tác cho các hoạt động 3αHSD oxy hóa trong cái gọi là “con đường cửa sau” để tổng hợp dihydrotestosterone (DHT).
Steroid Sulfotransferase và Sulfatase
Các steroid sulfat có thể được tổng hợp trực tiếp từ cholesterol sulfat hoặc có thể được hình thành bằng cách sulfat hóa các steroid bởi các enzyme sulfotransferase (SULT) trong bào tương. Các SULT sử dụng 3′-phosphoadenine-5′-phosphosulfate (PAPS) làm chất cho sulfat, được tổng hợp từ adenosine triphosphate (ATP) và sulfat bởi hai isozyme của PAPS synthase: PAPSS1, được biểu hiện khắp nơi, và PAPSS2, chủ yếu được biểu hiện ở sụn, tuyến thượng thận và gan. Ít nhất 44 đồng dạng riêng biệt của các enzyme này đã được xác định thuộc năm họ gen SULT; nhiều gen trong số này tạo ra các sản phẩm cắt nối luân phiên, giải thích cho số lượng lớn các enzyme. Các enzyme SULT sulfat hóa steroid bao gồm SULT1E (estrogen), SULT2A1 (steroid không thơm), và SULT2B1 (sterol). SULT2A1 là sulfotransferase chính được biểu hiện ở tuyến thượng thận, nơi nó sulfat hóa nhóm 3β hydroxyl của các steroid Δ5 (pregnenolone, 17OH-pregnenolone, DHEA, androsterone) nhưng không phải cholesterol. SULT2B1a cũng sẽ sulfat hóa pregnenolone nhưng không phải cholesterol, trong khi cholesterol là cơ chất chính cho SULT2B1b ở da, gan và các nơi khác. Không rõ liệu hầu hết các steroid sulfat chỉ đơn giản là các dạng steroid không hoạt động hay chúng có vai trò hormone cụ thể. Việc knockout gen sult1e1 ở chuột có liên quan đến nồng độ estrogen tăng cao, tăng biểu hiện yếu tố mô ở nhau thai và tăng kích hoạt tiểu cầu, dẫn đến huyết khối nhau thai và mất thai có thể được cải thiện bằng liệu pháp chống đông. Các đột biến làm mất chức năng của các enzyme SULT ở người chưa được mô tả, nhưng các đột biến PAPSS2 làm cạn kiệt PAPS ở tuyến thượng thận, làm tăng sản xuất DHEA không liên hợp. Người Mỹ gốc Phi có tỷ lệ đa hình cao ở SULT2A1, dường như ảnh hưởng đến tỷ lệ DHEA:DHEAS trong huyết tương, điều này có thể tương quan với nguy cơ ung thư tuyến tiền liệt và các loại ung thư khác.
Các steroid sulfat cũng có thể được thủy phân thành steroid gốc bởi steroid sulfatase. Việc xóa gen steroid sulfatase trên nhiễm sắc thể Xp22.3 gây ra bệnh vảy cá liên kết X. Việc nam giới có một bản sao duy nhất của gen này có lẽ giải thích tại sao nam giới có nồng độ DHEAS cao hơn nữ giới cùng tuổi. Ở tuyến thượng thận và nhau thai của thai nhi, sự thiếu hụt hoặc không có sulfatase làm giảm lượng DHEA tự do có sẵn để nhau thai chuyển đổi thành estrogen, dẫn đến nồng độ estriol thấp trong máu và nước tiểu của mẹ. Sự tích tụ của các steroid sulfat trong lớp sừng của da gây ra bệnh vảy cá.
Aromatase: P450aro
Estrogen được sản xuất bằng cách thơm hóa androgen, bao gồm cả androgen tuyến thượng thận, bằng một loạt các phản ứng phức tạp được xúc tác bởi aromatase, P450aro. Cytochrome P450 ở vi thể này được mã hóa bởi một gen duy nhất (CYP19A1) trên nhiễm sắc thể 15q21.1, sử dụng một số trình tự promoter, vị trí bắt đầu phiên mã và các exon đầu tiên được chọn lựa thay thế khác nhau để mã hóa P450aro giống hệt nhau ở các mô khác nhau dưới sự điều hòa hormone khác nhau. Biểu hiện của Aromatase ở các mô ngoại tuyến, đặc biệt là mô mỡ, có thể chuyển đổi androgen tuyến thượng thận thành estrogen. Aromatase ở các đầu xương của xương đang phát triển chuyển đổi testosterone thành estradiol; tầm vóc cao, sự trưởng thành đầu xương chậm và loãng xương ở nam giới bị thiếu hụt aromatase, và sự đảo ngược nhanh chóng của chúng khi thay thế estrogen cho thấy rằng estrogen, chứ không phải androgen, chịu trách nhiệm cho sự trưởng thành đầu xương ở nam giới.
Các bệnh nhân hiếm gặp bị thiếu hụt aromatase minh họa rằng hoạt động của aromatase ở nhau thai và hậu quả là quá trình tổng hợp estrogen của thai nhi-nhau thai không cần thiết cho sự phát triển bình thường của thai nhi. Tuy nhiên, khi không có hoạt động aromatase ở nhau thai, androgen và tiền chất androgen của tuyến thượng thận thai nhi sẽ đi vào tuần hoàn của mẹ, gây ra nam hóa ở mẹ. Hơn nữa, aromatase ở nhau thai chuyển đổi androgen của mẹ thành estrogen, bảo vệ thai nhi khỏi nguy cơ bị nam hóa (như ở phụ nữ mang thai bị thiếu hụt 21-hydroxylase [21OHD] điều trị không đủ). Trẻ em bị thiếu hụt aromatase phát triển bình thường và tiếp tục phát triển sau tuổi dậy thì, bởi vì chỉ có estrogen, chứ không phải androgen, làm trung gian cho sự cốt hóa đầu xương. Điều trị bệnh nhân thiếu hụt aromatase bằng estrogen sẽ làm cốt hóa đầu xương của họ và ngừng tăng trưởng. Những quan sát này là cơ sở cho việc sử dụng các chất ức chế aromatase để ức chế sự trưởng thành xương nhanh.
5α-Reductase
Testosterone được chuyển đổi thành androgen mạnh hơn, DHT, bởi 5α-reductase, một enzyme được tìm thấy trong các mô đích của testosterone. Có hai isozyme 30 kDa kỵ nước của 5α-reductase có 50% tương đồng. Enzyme loại I, được tìm thấy ở da đầu và các mô ngoại vi khác, được mã hóa bởi gen SRD5A1 trên nhiễm sắc thể 5; enzyme loại II, dạng chủ yếu được tìm thấy ở các mô sinh sản của nam giới, được mã hóa bởi gen SRD5A2 có cấu trúc liên quan trên nhiễm sắc thể 2p23. Hội chứng thiếu hụt 5α-reductase, một rối loạn biệt hóa giới tính nam, được gây ra bởi một loạt các đột biến trong gen SRD5A2. Ở thai nhi, gen SRD5A1 được biểu hiện ở tinh hoàn, sau đó được biểu hiện một thời gian ngắn ở da của trẻ sơ sinh, và sau đó không được biểu hiện cho đến khi hoạt động và protein của nó được tìm thấy trở lại sau tuổi dậy thì. Gen SRD5A2 được biểu hiện ở da bộ phận sinh dục của thai nhi, ở tuyến tiền liệt bình thường, và trong tăng sản và ung thư biểu mô tuyến tiền liệt. Do đó, enzyme loại I có thể chịu trách nhiệm cho sự nam hóa ở tuổi dậy thì được thấy ở những bệnh nhân bị thiếu hụt 5α-reductase cổ điển, và enzyme loại II có thể liên quan đến chứng hói đầu ở nam giới.
11β-Hydroxysteroid Dehydrogenase
Mặc dù một số steroid thường được phân loại là glucocorticoid hoặc mineralocorticoid, thụ thể “mineralocorticoid” (glucocorticoid loại II) có ái lực bằng nhau đối với cả aldosterone và cortisol. Tuy nhiên, cortisol không hoạt động như một mineralocorticoid trong cơ thể sống, mặc dù nồng độ cortisol có thể vượt quá nồng độ aldosterone từ 100 đến 1000 lần, bởi vì các mô đáp ứng với mineralocorticoid (như thận) chuyển đổi cortisol thành cortisone, một steroid không có hoạt tính chuyển hóa. Sự chuyển đổi qua lại giữa cortisol và cortisone được trung gian bởi hai isozyme gắn màng của 11β-hydroxysteroid dehydrogenase (11βHSD; HSD11B), mỗi loại có thể xúc tác cho cả hoạt động oxidase và reductase, tùy thuộc vào đồng yếu tố có sẵn (NADP+ hoặc NADPH). Tỷ lệ NADP+/NADPH được điều hòa bởi hexose-6-phosphate dehydrogenase (H6PDH).
Enzyme loại 1 (HSD11B1; 11βHSD1) được biểu hiện chủ yếu ở các mô đáp ứng với glucocorticoid, chẳng hạn như gan, mỡ, tinh hoàn và phổi. HSD11B1 có thể xúc tác cho cả quá trình oxy hóa cortisol thành cortisone sử dụng NADP+ làm đồng yếu tố (Km 1–2 μM), hoặc quá trình khử cortisone thành cortisol sử dụng NADPH làm đồng yếu tố (Km 0,1–0,3 μM); phản ứng được xúc tác phụ thuộc vào đồng yếu tố nào có sẵn, nhưng enzyme chỉ có thể hoạt động với nồng độ steroid cao (micromolar). HSD11B1 nằm ở phía lòng của mạng lưới nội chất, và do đó không tiếp xúc với tế bào chất, cho phép HSD11B1 nhận NADPH được cung cấp bởi H6PDH. Điều này liên kết HSD11B1 với con đường pentose monophosphate, cung cấp một liên kết cận tiết trực tiếp giữa sản xuất glucocorticoid tại chỗ và lưu trữ năng lượng dưới dạng mỡ. Prednisone và cortisone là các 11-ketosteroid không hoạt động, phải được khử thành các dẫn xuất 11β-hydroxy hoạt động của chúng bởi HSD11B1, chủ yếu ở gan. Bằng cách kích hoạt cortisone, HSD11B1 khuếch đại tác động của glucocorticoid trong các mô mà nó được biểu hiện, đặc biệt là ở gan và mỡ, có khả năng góp phần vào béo phì, kháng insulin, hội chứng chuyển hóa và có thể là bệnh gan nhiễm mỡ không do rượu; do đó, các nghiên cứu về các chất ức chế HSD11B1 đang được quan tâm đáng kể về mặt dược phẩm.
HSD11B2 (11βHSD2) xúc tác quá trình oxy hóa cortisol thành cortisone sử dụng NADH, và có thể hoạt động với nồng độ steroid thấp (nanomolar) (Km 10–100 nM). HSD11B2 được biểu hiện ở các mô đáp ứng với mineralocorticoid, chẳng hạn như nephron xa và do đó phục vụ để “bảo vệ” thụ thể mineralocorticoid (MR) bằng cách bất hoạt cortisol thành cortisone, để chỉ những “mineralocorticoid thực sự”, chẳng hạn như aldosterone hoặc deoxycorticosterone, mới có thể gây ra tác dụng mineralocorticoid; do đó HSD11B2 ngăn cortisol tràn ngập các MR ở thận. HSD11B2 ở nhau thai cũng bất hoạt cortisol của mẹ thành cortisone (và prednisolone thành prednisone), và nhau thai có nhiều NADP+ ủng hộ hoạt động oxy hóa của HSD11B1, do đó trong nhau thai, cả hai enzyme đều bảo vệ thai nhi khỏi nồng độ cortisol cao của mẹ, nhưng không bảo vệ khỏi betamethasone hoặc dexamethasone do mẹ dùng, vì chúng không phải là cơ chất cho các enzyme HSD11B. Do đó, các isozyme HSD11B quyết định liệu một steroid có “qua được nhau thai” hay không.
3α-Hydroxysteroid Dehydrogenase
Các 3α-hydroxysteroid dehydrogenase (3αHSD) không quen thuộc với hầu hết các nhà nội tiết học, nhưng có tầm quan trọng lâm sàng vì sự phát hiện gần đây về “con đường cửa sau” của sinh tổng hợp steroid. Con đường đáng chú ý này (Hình 14.6), lần đầu tiên được phát hiện là cơ chế mà tinh hoàn của bào thai thú có túi đực tạo ra androgen, đóng một vai trò trung tâm trong sự biệt hóa giới tính nam ở người. Trong con đường này, 17OHP được chuyển đổi thành DHT mà không đi qua DHEA, androstenedione, hoặc testosterone, và do đó cung cấp một cơ chế để 17OHP góp phần vào sự nam hóa của thai nhi nữ bị 21OHD. Các 3αHSD khử là các enzyme AKR và các 3αHSD oxy hóa là các enzyme SDR. Có bốn 3αHSD khử, được gọi là AKR1C4-1. Các enzyme này rất giống nhau về cấu trúc, được mã hóa bởi một cụm gen rất giống nhau trên nhiễm sắc thể 10p14-15, và xúc tác cho một loạt các chuyển đổi steroid (đáng chú ý là khử 17-ketosteroid và khử 20α) và các phản ứng khác. AKR1C3, còn được gọi là 17βHSD5, chuyển đổi androstenedione thành testosterone ở tuyến thượng thận.
Con đường cửa sau được đặc trưng bởi cả hoạt động 3αHSD khử và oxy hóa; hoạt động khử dường như có thể được xúc tác bởi AKR1C2 hoặc AKR1C4. Các 3αHSD oxy hóa là các SDR tương tự như phân họ retinol dehydrogenase hoặc cis-retinol/androgen dehydrogenase (RoDH/CRAD). Thành viên hoạt động mạnh nhất trong số này là RoDH, còn được gọi là 17βHSD6, có lẽ xúc tác cho bước cuối cùng trong con đường cửa sau. Trong não, 3αHSD khử 5α-dihydroprogesterone thành allopregnanolone, là một chất hoạt hóa dị lập thể của các thụ thể gamma-aminobutyric acid (GABA)A. Các nghiên cứu sâu hơn về vai trò của con đường cửa sau sẽ là trung tâm của nội tiết học nhi khoa.
Sinh tổng hợp steroid ở tuyến thượng thận của thai nhi
Quá trình sinh tổng hợp steroid của vỏ thượng thận ở phôi bắt đầu vào khoảng 7 tuần sau khi thụ tinh. Các enzyme sinh steroid có thể được xác định bằng phương pháp hóa mô miễn dịch, chủ yếu ở vùng bào thai, vào ngày thứ 50 đến 52 sau khi thụ thai, và đến tuần thứ 8 sau khi thụ thai, tuyến thượng thận chứa cortisol và đáp ứng với ACTH trong các hệ thống nuôi cấy nguyên phát. Quá trình tổng hợp cortisol này được điều hòa bởi ACTH tuyến yên và liên quan đến sự biểu hiện tạm thời của 3βHSD2 ở tuyến thượng thận: sau tuần thứ chín sau khi thụ thai, biểu hiện của 3βHSD2 và quá trình tổng hợp cortisol suy yếu, và 3βHSD2 hầu như không thể phát hiện được ở tuần thứ 10 đến 11 và không có ở tuần thứ 14. Đồng thời, tuyến thượng thận của thai nhi cũng sản xuất 17βHSD5, có thể chuyển đổi androstenedione thành testosterone. Do đó, tuyến thượng thận của thai nhi tạo ra cortisol cùng thời điểm trong thai kỳ khi testosterone của tinh hoàn thai nhi đang nam hóa bộ phận sinh dục của thai nhi nam bình thường. Cortisol của tuyến thượng thận thai nhi này dường như ức chế ACTH, nếu không sẽ thúc đẩy quá trình tổng hợp androgen tuyến thượng thận và nam hóa thai nhi 46,XX.
Các thai nhi bị ảnh hưởng bởi các tổn thương di truyền trong quá trình sinh tổng hợp steroid tuyến thượng thận có thể sản xuất đủ androgen tuyến thượng thận để nam hóa một thai nhi nữ đến ngoại hình gần như nam giới, và quá trình nam hóa bộ phận sinh dục này hoàn tất vào khoảng tuần thứ 12 của thai kỳ. Vùng xác định của tuyến thượng thận thai nhi sản xuất hormone steroid theo các con đường trong Hình 14.3. Ngược lại, vùng bào thai lớn của tuyến thượng thận tương đối thiếu hoạt động 3βHSD2 sau 12 tuần. Hoạt động 17,20 lyase mạnh mẽ và hoạt động 3βHSD thấp ở tuyến thượng thận của thai nhi giải thích cho sự sản xuất dồi dào DHEA và sulfat của nó, DHEAS, sau đó được nhau thai chuyển đổi thành estrogen (Hình 14.7). Tuyến thượng thận của thai nhi cũng có hoạt động sulfotransferase đáng kể nhưng ít hoạt động steroid sulfatase, cũng ủng hộ việc chuyển đổi DHEA thành DHEAS. DHEAS kết quả không phải là cơ chất cho 3βHSD2 của tuyến thượng thận, và được tiết ra, 16α-hydroxyl hóa ở gan của thai nhi, và sau đó được tác động bởi 3βHSD1, 17βHSD1, và P450aro của nhau thai để sản xuất estriol; các steroid thoát khỏi quá trình 16α-hydroxyl hóa ở gan của thai nhi sẽ tạo ra estrone và estradiol. Estrogen của nhau thai ức chế hoạt động 3βHSD của tuyến thượng thận, cung cấp một hệ thống phản hồi để thúc đẩy sản xuất DHEAS. Các steroid tuyến thượng thận của thai nhi chiếm 50% estrone và estradiol và 90% estriol trong tuần hoàn của mẹ.
Mặc dù đơn vị thai nhi-nhau thai sản xuất một lượng lớn DHEA, DHEAS và estriol (và các steroid khác), chúng dường như không có vai trò thiết yếu. Một thai kỳ thành công đòi hỏi nhau thai tổng hợp progesterone, chất này ức chế sự co bóp của tử cung và ngăn ngừa sảy thai tự nhiên; tuy nhiên, các thai nhi bị rối loạn di truyền trong sinh tổng hợp steroid tuyến thượng thận và tuyến sinh dục phát triển bình thường, đến đủ tháng, và trải qua quá trình chuyển dạ và sinh nở bình thường. Việc sản xuất mineralocorticoid chỉ cần thiết sau khi sinh, estrogen không cần thiết, và androgen chỉ cần thiết cho sự biệt hóa giới tính nam. Dường như glucocorticoid của thai nhi người cần thiết vào khoảng 8 đến 12 tuần, nhưng không rõ liệu chúng có cần thiết sau đó hay không; nếu có, một lượng nhỏ cortisol của mẹ thoát khỏi sự bất hoạt của nhau thai là đủ. Một trẻ sơ sinh đã được mô tả với tình trạng kháng glucocorticoid sâu sắc, đồng hợp tử cho một đột biến dịch khung ở codon 772 trong miền gắn glucocorticoid của thụ thể glucocorticoid (GR). Mặc dù trẻ bị hạ đường huyết và tăng huyết áp nặng sau sinh, sự phát triển của phổi và các khía cạnh khác của sự phát triển thai nhi là bình thường, cho thấy tác động của glucocorticoid không cần thiết cho sự phát triển bình thường của thai nhi người.
Sự điều hòa sinh tổng hợp steroid và sự phát triển của tuyến thượng thận thai nhi chưa được hiểu đầy đủ, nhưng cả hai đều liên quan đến ACTH. ACTH kích thích hiệu quả quá trình sinh tổng hợp steroid bởi các tế bào tuyến thượng thận của thai nhi trong ống nghiệm, và ACTH dư thừa rõ ràng có liên quan đến sự phát triển của tuyến thượng thận và sản xuất quá mức androgen ở các thai nhi bị CAH. Điều trị thử nghiệm trước sinh cho các thai nhi như vậy bằng cách cho mẹ dùng liều dược lý dexamethasone vào tuần thứ 6 đến 10 của thai kỳ có thể làm giảm đáng kể sản xuất androgen tuyến thượng thận của thai nhi và do đó làm giảm sự nam hóa của thai nhi nữ, cho thấy trục hạ đồi-tuyến yên-thượng thận (HPA) hoạt động rất sớm trong đời sống thai nhi. Ngược lại, các thai nhi vô não thiếu ACTH tuyến yên có tuyến thượng thận chứa một bộ enzyme sinh steroid khá bình thường và giữ được khả năng sinh tổng hợp steroid. Do đó, sinh tổng hợp steroid của tuyến thượng thận thai nhi có thể được điều hòa bởi cả cơ chế phụ thuộc và không phụ thuộc ACTH.
Điều hòa sinh tổng hợp steroid
Trục Hạ đồi-Tuyến yên-Thượng thận
Vùng dưới đồi: Yếu tố giải phóng Corticotropin và Arginine Vasopressin
Sản phẩm steroid chính của tuyến thượng thận ở người là cortisol, chủ yếu được tiết ra để đáp ứng với ACTH (corticotropin) do tuyến yên sản xuất; sự tiết ACTH chủ yếu được kích thích bởi yếu tố giải phóng corticotropin (CRF) từ vùng dưới đồi. Lịch sử khám phá các thành phần của trục HPA và mối quan hệ qua lại của chúng đã được tổng hợp gần đây. CRF của vùng dưới đồi là một peptide gồm 41 axit amin được tổng hợp chủ yếu bởi các neuron trong nhân cạnh não thất. Các neuron vùng dưới đồi này cũng sản xuất decapeptide arginine vasopressin (AVP, còn được gọi là hormone chống bài niệu hoặc ADH). Cả CRF và AVP đều được tạo ra từ quá trình phân giải protein của các tiền chất lớn hơn, với tiền chất AVP chứa trình tự cho neurophysin, là protein gắn kết với AVP. CRF và AVP di chuyển qua các sợi trục đến lồi giữa, nơi giải phóng chúng vào tuần hoàn cửa tuyến yên, mặc dù hầu hết các sợi trục AVP kết thúc ở thùy sau tuyến yên. AVP được đồng tiết với CRF để đáp ứng với stress, và cả CRF và AVP đều kích thích tổng hợp và giải phóng ACTH, nhưng chúng dường như thực hiện điều này bằng các cơ chế khác nhau. CRF liên kết với một thụ thể cặp đôi protein G trên màng của các tế bào corticotrope của tuyến yên và kích hoạt adenylyl cyclase, làm tăng cAMP, từ đó kích hoạt con đường tín hiệu protein kinase A (PKA). PKA kích hoạt sự tiết ACTH bằng cách điều hòa phối hợp các dòng kali và canxi của tế bào, và tăng cường phiên mã gen proopiomelanocortin (POMC). AVP liên kết với thụ thể cặp đôi protein G của nó và kích hoạt phospholipase C, dẫn đến giải phóng Ca++ nội bào và kích hoạt protein kinase C (PKC). AVP dường như khuếch đại tác dụng của CRF đối với sự tiết ACTH mà không ảnh hưởng đến quá trình tổng hợp. Tuy nhiên, CRF là chất kích thích sinh lý quan trọng hơn đối với việc giải phóng ACTH, mặc dù liều AVP tối đa có thể gây ra phản ứng ACTH tối đa. Khi được dùng cùng nhau, CRF và AVP có tác dụng hiệp đồng, như mong đợi từ các cơ chế hoạt động độc lập của chúng.
Tuyến yên: Hormone Vỏ thượng thận và Proopiomelanocortin
ACTH tuyến yên là một peptide gồm 39 axit amin có nguồn gốc từ POMC, một protein gồm 241 axit amin. POMC trải qua một loạt các quá trình phân cắt protein, tạo ra một số peptide có hoạt tính sinh học (Hình 14.8). Glycopeptide đầu N (POMC 1-75) có thể kích thích sinh tổng hợp steroid và có thể hoạt động như một chất gây tăng sinh tuyến thượng thận. POMC 112-150 là ACTH 1-39, POMC 112-126 và POMC 191-207 lần lượt tạo thành α- và β-MSH (hormone kích thích tế bào hắc tố), và POMC 210-241 là β-endorphin. POMC cũng được sản xuất với số lượng nhỏ ở não, tinh hoàn, gan, thận và nhau thai, nhưng POMC ngoại tuyến yên này không đóng góp đáng kể vào ACTH lưu hành. Các khối u ác tính thường sẽ sản xuất “ACTH lạc chỗ” ở người lớn và hiếm khi ở trẻ em; ACTH này có nguồn gốc từ quá trình sinh tổng hợp lạc chỗ của cùng một tiền chất POMC. Chỉ cần 20 đến 24 axit amin đầu tiên của ACTH là đủ cho hoạt tính sinh học đầy đủ của nó, và ACTH 1-24 tổng hợp được sử dụng rộng rãi trong các xét nghiệm chẩn đoán chức năng tuyến thượng thận. Tuy nhiên, các dạng ACTH ngắn hơn này có thời gian bán hủy ngắn hơn so với ACTH 1-39 tự nhiên. Phiên mã gen POMC được kích thích bởi CRF và bị ức chế bởi glucocorticoid.
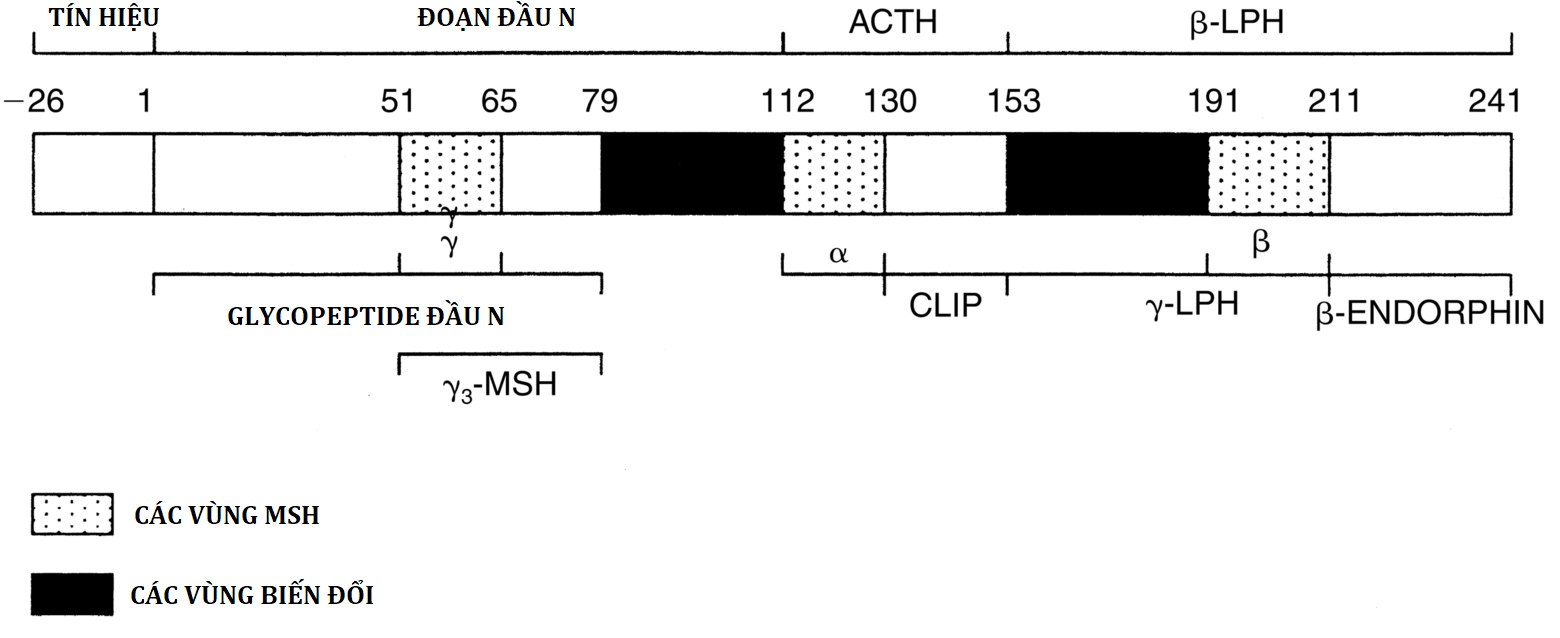
Hình 14.8: Cấu trúc của preproopiomelanocortin (POMC) ở người. Các con số chỉ vị trí axit amin, với số 1 được gán cho axit amin đầu tiên của POMC sau peptide tín hiệu 26 axit amin. Các vùng α-, β-, và γ-MSH, đặc trưng cho ba vùng “bất biến”, được chỉ định bằng các đường chéo; các vùng “biến đổi” là màu đen. Các số axit amin được hiển thị chỉ axit amin ở đầu N của mỗi vị trí cắt; vì các axit amin này bị loại bỏ, các con số không tương ứng chính xác với các số axit amin của các peptide như được sử dụng trong văn bản. ACTH, Hormone vỏ thượng thận; CLIP, peptide thùy trung gian giống corticotropin; LPH, hormone hướng mỡ: MSH, hormone kích thích tế bào hắc tố.
Tác dụng của Hormone Vỏ thượng thận
ACTH kích thích thụ thể melanocortin 2 (MCR2) cặp đôi protein G, nằm gần như độc quyền ở vỏ thượng thận. Việc kích hoạt MCR2 sẽ khởi động quá trình sản xuất cAMP, kích hoạt PKA xúc tác cho quá trình phosphoryl hóa nhiều protein liên quan đến sinh tổng hợp steroid, từ đó điều chỉnh hoạt động của chúng. ACTH gây ra cả tác dụng cấp tính và lâu dài. ACTH kích thích sinh tổng hợp các thụ thể LDL và sự hấp thu LDL, cung cấp phần lớn cholesterol được sử dụng cho sinh tổng hợp steroid, và kích thích phiên mã gen HMG-CoA reductase, bước giới hạn tốc độ trong sinh tổng hợp cholesterol, nhưng sinh tổng hợp cholesterol của tuyến thượng thận về mặt số lượng ít quan trọng hơn nhiều so với việc hấp thu cholesterol LDL. Cholesterol được lưu trữ trong các mô sinh steroid dưới dạng este cholesterol trong các giọt lipid. ACTH kích thích hoạt động của cholesterol esterase, đồng thời ức chế cholesterol ester synthetase, do đó làm tăng lượng cholesterol tự do nội bào, cơ chất cho P450scc. Cuối cùng, ACTH tạo điều kiện vận chuyển cholesterol vào ty thể, bằng cách kích thích tổng hợp và phosphoryl hóa StAR, do đó làm tăng dòng cholesterol tự do vào ty thể. Tất cả những tác động này được trung gian bởi cAMP và xảy ra trong vòng vài phút, tạo thành tác dụng “cấp tính” của ACTH đối với sinh tổng hợp steroid. Tuyến thượng thận chứa một lượng hormone steroid tương đối khiêm tốn; do đó việc giải phóng cortisol được tạo sẵn không đóng góp đáng kể vào phản ứng cấp tính với ACTH; các phản ứng cấp tính xảy ra bằng cách cung cấp nhanh chóng một lượng lớn cholesterol cho P450scc của ty thể.
Các tác dụng “mãn tính” lâu dài của ACTH được trung gian trực tiếp ở cấp độ các enzyme sinh steroid. ACTH thông qua cAMP kích thích sự tích lũy của các enzyme sinh steroid và mRNA của chúng bằng cách kích thích phiên mã các gen của chúng. ACTH cũng làm tăng lưu lượng máu đến tuyến thượng thận, tăng cường sự xâm nhập của oxy và nhiên liệu chuyển hóa và việc cung cấp các hormone mới được tiết ra vào tuần hoàn. Do đó, ACTH làm tăng cả sự hấp thu cơ chất cholesterol và sự chuyển đổi của nó thành các sản phẩm steroid. Sự kích thích sinh tổng hợp steroid này xảy ra ở mỗi bước trong con đường, chứ không chỉ ở bước giới hạn tốc độ, P450scc.
Vai trò của ACTH và các peptide khác có nguồn gốc từ POMC, trong việc kích thích sự phát triển của tuyến thượng thận trưởng thành, được hỗ trợ bởi các quan sát cho thấy việc thiếu POMC tuyến yên gây ra thiểu sản tuyến thượng thận nặng, và thừa ACTH mãn tính gây ra tăng sản thượng thận. Ở tuyến thượng thận của thai nhi, ACTH kích thích sản xuất tại chỗ IGF-2, FGF2 và EGF. Những yếu tố này, và có thể cả các yếu tố khác, phối hợp với nhau để làm trung gian cho sự phát triển của tuyến thượng thận thai nhi do ACTH gây ra.
Nhịp sinh học ngày đêm của Hormone Vỏ thượng thận và Cortisol
Nồng độ ACTH và cortisol trong huyết tương có xu hướng cao vào buổi sáng và thấp vào buổi tối. Nồng độ ACTH đỉnh thường được thấy vào lúc 4 đến 6 giờ sáng và nồng độ cortisol đỉnh theo sau vào khoảng 8 giờ sáng. Cả ACTH và cortisol đều được giải phóng theo từng đợt không liên tục sau mỗi 30 đến 120 phút trong ngày, nhưng tần suất và biên độ của các đợt này lớn hơn nhiều vào buổi sáng. Cơ sở của nhịp sinh học ngày đêm này rất phức tạp và chưa được hiểu rõ. Hàm lượng CRF của chính vùng dưới đồi cho thấy một nhịp sinh học ngày đêm với hàm lượng đỉnh vào khoảng 4 giờ sáng. Ít nhất bốn yếu tố dường như đóng một vai trò trong nhịp điệu của ACTH và cortisol: nhịp điệu nội tại của quá trình tổng hợp và bài tiết CRF bởi vùng dưới đồi; chu kỳ sáng/tối; chu kỳ ăn uống; và nhịp điệu vốn có trong tuyến thượng thận, có thể được trung gian bởi sự chi phối thần kinh của tuyến thượng thận. Các yếu tố này rõ ràng là phụ thuộc lẫn nhau và có liên quan. Nhịp điệu ăn uống có thể đóng một vai trò lớn như chu kỳ sáng/tối. Các thí nghiệm trên động vật cho thấy việc thay đổi thời gian cho ăn có thể vượt qua chu kỳ ACTH/cortisol được thiết lập bởi chu kỳ sáng/tối. Ở những người bình thường, cortisol được giải phóng trước bữa trưa và bữa tối, nhưng không phải vào những thời điểm này ở những người ăn liên tục trong ngày. Do đó, glucocorticoid, làm tăng đường huyết, dường như được giải phóng vào những lúc nhịn ăn và bị ức chế bởi việc ăn uống.
Như tất cả các bậc cha mẹ đều biết, trẻ sơ sinh không có nhịp sinh học ngày đêm về giấc ngủ hoặc ăn uống. Trẻ sơ sinh có được các nhịp điệu hành vi như vậy để đáp ứng với môi trường của chúng rất lâu trước khi chúng có được nhịp điệu của ACTH và cortisol. Nhịp sinh học ngày đêm của ACTH và cortisol bắt đầu được thiết lập ở 6 đến 12 tháng tuổi và thường không được thiết lập tốt cho đến sau 3 tuổi. Một khi nhịp điệu được thiết lập tốt ở trẻ lớn hoặc người lớn, nó chỉ thay đổi một cách khó khăn. Khi mọi người di chuyển đến các nơi khác nhau trên thế giới, nhịp điệu ACTH/cortisol của họ thường mất từ 15 đến 20 ngày để điều chỉnh một cách thích hợp. Việc tái đồng bộ hóa đồng hồ sinh học sau khi bị lệch múi giờ cần khoảng 1 đến 2 giờ/ngày, nhiều hơn đối với việc đi về phía đông và ít hơn đối với việc đi về phía tây.
Stress thể chất (như phẫu thuật lớn, chấn thương nặng, mất máu, sốt cao, hoặc bệnh nặng) có thể làm tăng bài tiết cả ACTH và cortisol, nhưng phẫu thuật nhỏ và các bệnh nhẹ (như nhiễm trùng đường hô hấp trên) có ít ảnh hưởng đến bài tiết ACTH và cortisol. Nhiễm trùng, sốt và các chất gây sốt có thể kích thích giải phóng các cytokine, chẳng hạn như interleukin (IL)-1 và IL-6, kích thích bài tiết hormone giải phóng corticotropin (CRH), và cũng kích thích IL-2 và yếu tố hoại tử khối u (TNF), kích thích giải phóng ACTH, tạo thêm kích thích cho bài tiết cortisol trong quá trình viêm; hơn nữa, IL-6 có thể trực tiếp kích thích tổng hợp và giải phóng cortisol ở tuyến thượng thận. Ngược lại, glucocorticoid ức chế sản xuất cytokine trong hệ miễn dịch, tạo thành một vòng phản hồi âm. Hầu hết các loại thuốc tác động tâm thần, chẳng hạn như thuốc chống co giật, chất dẫn truyền thần kinh và thuốc chống trầm cảm, không ảnh hưởng đến nhịp sinh học ngày đêm của ACTH và cortisol, mặc dù cyproheptadine (một chất đối kháng serotonin) có thể ức chế giải phóng ACTH.
Tuyến thượng thận: Phản hồi ngược của Glucocorticoid
Trục HPA là một ví dụ kinh điển của một hệ thống phản hồi nội tiết. ACTH làm tăng sản xuất cortisol, và cortisol làm giảm sản xuất ACTH. Cortisol và các glucocorticoid khác tác động ức chế phản hồi ngược lên cả CRF và ACTH (và AVP), chủ yếu thông qua GR. Giống như các giai đoạn cấp tính và mãn tính của tác động của ACTH lên tuyến thượng thận, có các giai đoạn cấp tính và mãn tính của sự ức chế phản hồi ngược của ACTH (và có lẽ là CRF). Giai đoạn cấp tính, xảy ra trong vòng vài phút, ức chế giải phóng ACTH (và CRF) từ các hạt bài tiết. Khi tiếp xúc kéo dài, glucocorticoid ức chế tổng hợp ACTH bằng cách ức chế trực tiếp phiên mã gen POMC (và AVP), điều này có thể dẫn đến suy thượng thận thứ phát. Một số bằng chứng cũng cho thấy rằng glucocorticoid có thể trực tiếp ức chế sinh tổng hợp steroid ở cấp độ của chính tế bào lớp bó, nhưng đây dường như là một thành phần nhỏ về mặt sinh lý trong việc điều hòa bài tiết cortisol.
Bài tiết Mineralocorticoid: Hệ Renin-Angiotensin
Renin là một enzyme serine protease được tổng hợp chủ yếu bởi các tế bào cạnh cầu thận của thận, nhưng nó cũng được sản xuất ở nhiều mô khác, bao gồm cả các tế bào lớp cầu của vỏ thượng thận. Vai trò của renin do tuyến thượng thận sản xuất chưa được xác định rõ; nó dường như duy trì mức độ cơ bản của P450c11AS, nhưng không rõ liệu angiotensin II có liên quan đến tác động này hay không. Renin được tổng hợp dưới dạng tiền chất gồm 406 axit amin, được phân cắt thành prorenin (386 axit amin) và cuối cùng thành protein gồm 340 axit amin được tìm thấy trong huyết tương. Huyết áp giảm, tư thế đứng thẳng, suy giảm natri, thuốc giãn mạch, kallikrein, opiat, và kích thích β-adrenergic đều thúc đẩy giải phóng renin. Renin tấn công angiotensinogen, cơ chất của renin, trong tuần hoàn một cách enzyme. Angiotensinogen là một protein được glycosyl hóa cao, và do đó có trọng lượng phân tử rất thay đổi từ 50.000 đến 100.000 dalton. Renin phân giải giải phóng 10 axit amin đầu N của angiotensinogen, được gọi là angiotensin I. Decapeptide này không có hoạt tính sinh học cho đến khi enzyme chuyển đổi, một enzyme được tìm thấy chủ yếu ở phổi và mạch máu, cắt bỏ hai axit amin đầu C của nó, để tạo ra một octapeptide, được gọi là angiotensin II. Angiotensin II liên kết với các thụ thể màng đặc hiệu nằm ở lớp cầu của vỏ thượng thận để kích thích sản xuất aldosterone. Enzyme chuyển angiotensin có thể bị ức chế bởi captopril và các tác nhân liên quan; ngoài ra các thụ thể angiotensin II có thể bị chặn bởi các tác nhân dược lý, chẳng hạn như candesartan để chẩn đoán và điều trị tăng huyết áp (do tăng renin).
Angiotensin II có hai tác dụng chính, cả hai đều làm tăng huyết áp. Nó trực tiếp kích thích co tiểu động mạch trong vòng vài giây và nó kích thích tổng hợp và bài tiết aldosterone trong vòng vài phút. Tăng kali huyết tương là một chất kích thích trực tiếp, rất mạnh mẽ đối với quá trình tổng hợp và giải phóng aldosterone. Aldosterone, được tiết ra bởi các tế bào lớp cầu của vỏ thượng thận, có hoạt tính mineralocorticoid lớn nhất trong tất cả các steroid tự nhiên. Aldosterone gây bài tiết kali qua thận và giữ lại natri, với hậu quả là tăng thể tích nội mạch và huyết áp. Sự gia tăng thể tích máu cung cấp tín hiệu phản hồi âm để điều hòa bài tiết renin và aldosterone. Angiotensin II liên kết với AT1, thụ thể cặp đôi protein G (Gq) của nó để kích thích sản xuất phosphatidylinositol, huy động Ca++ nội bào và ngoại bào, và kích hoạt tín hiệu thông qua PKC và hệ thống canxi-calmodulin. Các chất truyền tin thứ hai nội bào này kích thích phiên mã gen CYP11A1 cho P450scc độc lập với các tác động của ACTH và cAMP. Sự gia tăng kali huyết tương trực tiếp điều hòa sản xuất aldosterone bằng cách khử cực các tế bào lớp cầu, dẫn đến dòng Ca++ đi vào thông qua các kênh canxi phụ thuộc điện thế. Do đó, angiotensin II và nồng độ kali tăng cao hội tụ trên cùng một con đường truyền tin thứ hai nội bào để điều hòa tổng hợp và bài tiết aldosterone. Mặc dù hệ renin-angiotensin rõ ràng là cơ quan điều hòa chính của bài tiết mineralocorticoid, ACTH (và có thể các peptide khác có nguồn gốc từ POMC) cũng có thể thúc đẩy bài tiết aldosterone. Ion amoni, hạ natri máu, các chất đối kháng dopamin, và một số tác nhân khác cũng có thể kích thích bài tiết aldosterone, và yếu tố lợi niệu nhĩ là một chất ức chế sinh lý mạnh mẽ của bài tiết aldosterone.
Vai trò dinh dưỡng của angiotensin II và tăng kali máu được hỗ trợ bởi các nghiên cứu trên động vật cho thấy sự mở rộng năng động của lớp cầu để đáp ứng với tình trạng suy giảm thể tích mãn tính, phong tỏa MR, chế độ ăn ít natri, và/hoặc chế độ ăn nhiều kali. Ngược lại, tình trạng tăng thể tích mãn tính, như xảy ra với chế độ ăn nhiều natri và/hoặc hạ kali máu, ức chế sự phát triển của lớp cầu.
Bài tiết Androgen tuyến thượng thận và Điều hòa Giai đoạn thượng thận hóa (Adrenarche)
DHEA, DHEAS và androstenedione, gần như chỉ được tiết ra bởi lớp lưới của tuyến thượng thận, thường được gọi là androgen tuyến thượng thận vì chúng có thể được chuyển đổi ngoại vi thành testosterone. Tuy nhiên, các steroid này có rất ít hoặc không có khả năng liên kết và kích hoạt các thụ thể androgen, do đó chúng chỉ là tiền chất androgen, chứ không phải là androgen thực sự. Tuyến thượng thận của thai nhi tiết ra một lượng lớn DHEA và DHEAS, và các steroid này có nhiều ở trẻ sơ sinh, nhưng nồng độ của chúng giảm nhanh chóng khi vùng bào thai của tuyến thượng thận thoái triển sau khi sinh. Sau năm đầu đời, tuyến thượng thận của trẻ nhỏ tiết ra một lượng rất nhỏ DHEA, DHEAS và androstenedione cho đến khi bắt đầu giai đoạn thượng thận hóa, thường là khoảng 7 đến 8 tuổi, trước khi bắt đầu dậy thì khoảng 2 năm. Giai đoạn thượng thận hóa độc lập với dậy thì, tuyến sinh dục, hoặc gonadotropin, và cơ chế kích hoạt sự khởi đầu của giai đoạn thượng thận hóa vẫn chưa được biết. Sự bài tiết DHEA và DHEAS tiếp tục tăng trong và sau tuổi dậy thì và đạt giá trị tối đa ở tuổi trưởng thành trẻ, sau đó có sự giảm chậm, dần dần trong bài tiết các steroid này ở người già (“adrenopause”) (Hình 14.9). Nam giới có nồng độ DHEAS trong huyết thanh cao hơn phụ nữ, có lẽ vì nam giới có một bản sao duy nhất của gen steroid sulfatase liên kết X. Trong phần lớn cuộc đời trưởng thành, bài tiết DHEAS của tuyến thượng thận vượt quá bài tiết cortisol; ở phụ nữ trưởng thành, bài tiết tiền chất androgen và androgen của tuyến thượng thận tương đương với bài tiết từ buồng trứng. Mặc dù có sự gia tăng rất lớn trong bài tiết DHEA và DHEAS của tuyến thượng thận trong giai đoạn thượng thận hóa, nồng độ ACTH và cortisol lưu hành không thay đổi theo tuổi. Do đó, ACTH đóng vai trò cho phép trong giai đoạn thượng thận hóa nhưng không kích hoạt nó. Các cuộc tìm kiếm các hormone polypeptide giả định có thể kích thích đặc biệt lớp lưới đã không thành công. Giai đoạn thượng thận hóa là một hiện tượng độc đáo chỉ giới hạn ở một vài loài linh trưởng bậc cao, chẳng hạn như tinh tinh hoặc đười ươi, nhưng ý nghĩa của giai đoạn thượng thận hóa vẫn chưa được biết.
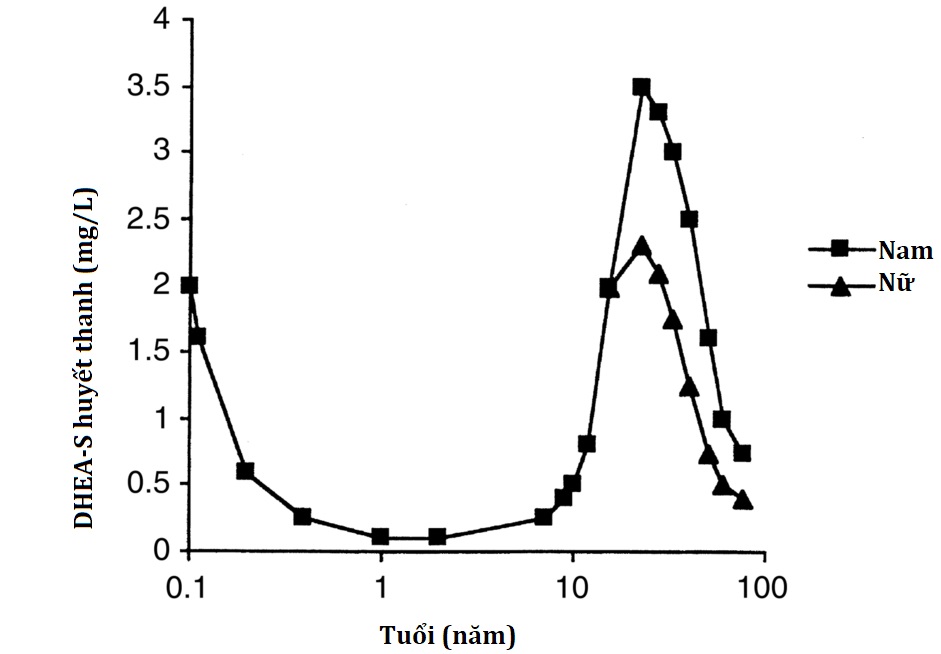
Hình 14.9: Nồng độ dehydroepiandrosterone sulfat theo tuổi. Lưu ý rằng trục x ở thang logarit.
Các nghiên cứu gần đây về giai đoạn thượng thận hóa đã tập trung vào vai trò của 3βHSD và P450c17. Sự phong phú của 3βHSD trong lớp lưới dường như giảm khi bắt đầu giai đoạn thượng thận hóa, và sự biểu hiện của cytochrome b5 ở tuyến thượng thận, chất thúc đẩy hoạt động 17,20 lyase của P450c17, gần như chỉ giới hạn ở lớp lưới; những yếu tố này ủng hộ mạnh mẽ việc sản xuất DHEA. Sự phosphoryl hóa của P450c17, dường như bởi p38α, cũng làm tăng hoạt động 17,20 lyase, nhưng vai trò tiềm năng của nó trong giai đoạn thượng thận hóa vẫn chưa được khám phá. Giai đoạn thượng thận hóa sớm và quá mức có thể liên quan đến kháng insulin và/hoặc thừa cân, và các bé gái có giai đoạn thượng thận hóa sớm quá mức dường như có nguy cơ cao hơn nhiều bị hội chứng buồng trứng đa nang khi trưởng thành (đặc trưng bởi tăng androgen, ít chu kỳ rụng trứng hơn, kháng insulin và tăng triglyceride máu). Bằng chứng gần đây cho thấy trẻ sơ sinh nhẹ cân so với tuổi thai có thể có nguy cơ cao hơn đối với hội chứng này. Do đó, các nghiên cứu về sinh lý, sinh hóa và các mối tương quan lâm sàng của giai đoạn thượng thận hóa cho thấy giai đoạn thượng thận hóa sớm có thể là một dấu hiệu sớm của một rối loạn chuyển hóa. Gợi ý rằng việc thay thế DHEA có thể cải thiện trí nhớ và cảm giác khỏe khoắn ở người già và trong suy thượng thận vẫn còn gây tranh cãi.
Trong khi từ lâu người ta cho rằng DHEA, DHEAS và androstenedione là “androgen tuyến thượng thận”, công trình gần đây đã chỉ ra rằng các androgen tuyến thượng thận chính là các steroid 11-oxygenated (Oxo), chủ yếu là 11-keto-testosterone (11-KT). Ngoài việc được biểu hiện ở lớp bó, P450c11β, enzyme 11-hydroxylase cổ điển chuyển đổi 11-deoxycortisol thành cortisol, cũng được biểu hiện ở lớp lưới, nơi nó chuyển đổi androstenedione và testosterone lần lượt thành 11OH-androstenedione và 11OH-testosterone. Các steroid 11-Oxo này có thể được chuyển đổi thành 11-keto androstenedione và 11-KT không thể thơm hóa bởi HSD11B2 (Hình 14.10). 11-KT cũng có thể bị khử 5α thành 11-keto dihydrotestosterone (11-KDHT); 11-KT và 11-KDHT là các androgen liên kết và kích hoạt thụ thể androgen với ái lực tương tự như testosterone và DHT; các androgen này được tìm thấy ở mức độ tương tự ở nam và nữ, cao hơn trong tĩnh mạch thượng thận so với tĩnh mạch ngoại vi, và tăng cao trong CAH và hội chứng buồng trứng đa nang. Do đó, 11-KT dường như là androgen hoạt tính sinh học lưu hành chủ yếu trong cả giai đoạn thượng thận hóa bình thường và sớm, cũng như trong CAH cổ điển. Các steroid 11-Oxo này tương quan với kích thước tuyến thượng thận và có thể là dấu ấn sinh học hữu ích cho các khối u lạc chỗ tuyến thượng thận ở bệnh nhân CAH.

Hình 14.10: Tổng hợp androgen tuyến thượng thận. Quan điểm truyền thống về tổng hợp androgen tuyến thượng thận là cholesterol → pregnenolone → 17OH-pregnenolone (17OH-Preg) → DHEA → androstenedione, như được hiển thị ở bên phải. Tuy nhiên, thay vì là androgen, dehydroepiandrosterone (DHEA), DHEA sulfat (S), và androstenedione hầu như không có khả năng liên kết và kích hoạt thụ thể androgen, và do đó chỉ đơn giản là các tiền chất C19 (19-carbon) của androgen. Một lượng nhỏ androstenedione có thể được chuyển đổi thành testosterone bởi 17β-hydroxysteroid dehydrogenase 5 (17βHSD5) (AKR1C3) của tuyến thượng thận, nhưng steroid androgen chính được tiết ra bởi tuyến thượng thận là 11-keto-testosterone (11Keto-T). Cả androstenedione và testosterone đều có thể được 11-hydroxyl hóa bởi P450c11β để tạo ra lần lượt 11OH-androstenedione (11OH-Δ4) và 11OH-testosterone (11OH-T). Các steroid này có thể bị oxy hóa bởi 11βHSD (cùng một enzyme oxy hóa cortisol thành cortisone) thành lần lượt 11-keto-androstenedione (11Keto-Δ4) và 11-keto-testosterone (11Keto-T). Mặc dù 11Keto-T là androgen chính trong tĩnh mạch thượng thận, nó có thể bị khử 5α bởi 5α-reductase type 2 ở các mô ngoại vi và có thể cả bởi 5α-reductase type 1 trong chính tuyến thượng thận, thành lần lượt 5α-dihydrotestosterone. Con đường chiếm ưu thế về số lượng là androstenedione → 11OH-Δ4 → 11Keto-Δ4 → 11-Keto-T, như được chỉ ra bởi các mũi tên đậm.
Các steroid trong huyết tương và sự thải trừ của chúng
Cấu trúc và Danh pháp
Tất cả các hormone steroid đều là dẫn xuất của pregnenolone (Hình 14.11). Pregnenolone và các dẫn xuất của nó chứa 21 nguyên tử carbon thường được gọi là steroid C21. Mỗi nguyên tử carbon được đánh số, cho biết vị trí xảy ra các phản ứng sinh steroid khác nhau (ví dụ: 21-hydroxyl hóa, 11-hydroxyl hóa). Hoạt tính 17,20 lyase của P450c17 cắt đứt liên kết giữa các nguyên tử carbon 17 và 20, tạo ra các steroid C19, bao gồm tất cả các androgen; P450aro chuyển đổi androgen C19 thành estrogen C18. Ngoại trừ estrogen, tất cả các hormone steroid đều có một liên kết đôi carbon-carbon không bão hòa duy nhất. Các steroid có liên kết đôi này giữa các nguyên tử carbon 4 và 5, bao gồm tất cả các steroid có hoạt tính sinh học chính, được gọi là steroid Δ4; các tiền chất của chúng có liên kết đôi giữa các nguyên tử carbon 5 và 6 được gọi là steroid Δ5. Hai isozyme của 3βHSD chuyển đổi steroid Δ5 thành Δ4.
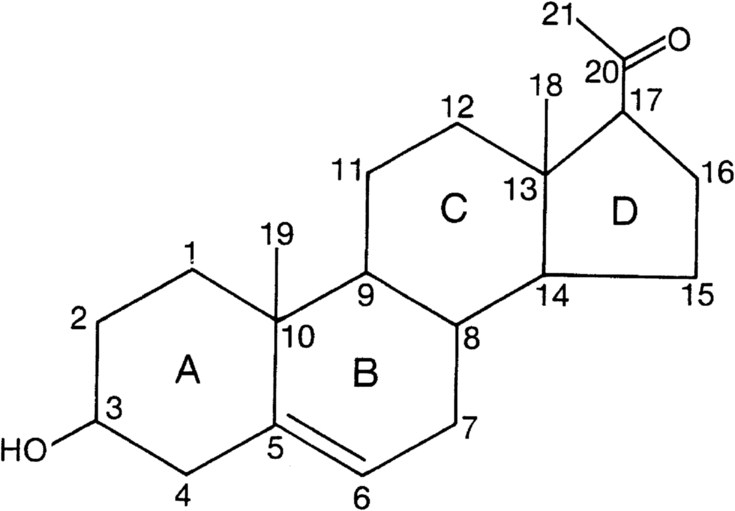
Hình 14.11: Cấu trúc của pregnenolone. Các nguyên tử carbon được chỉ định bằng số và các vòng được chỉ định bằng chữ cái theo quy ước tiêu chuẩn. Pregnenolone có nguồn gốc từ cholesterol, có một chuỗi bên 6-carbon gắn vào carbon số 21. Pregnenolone là một hợp chất “Δ5”, có một liên kết đôi giữa các carbon số 5 và 6; tác động của 3β-hydroxysteroid dehydrogenase/isomerase di chuyển liên kết đôi này từ vòng B đến các carbon số 4 và 5 trong vòng A, tạo thành các hợp chất Δ4. Tất cả các hormone steroid có hoạt tính sinh học chính đều là các hợp chất Δ4.
Có một thuật ngữ hóa học logic, hệ thống và rõ ràng để mô tả chính xác cấu trúc của tất cả các hormone steroid và tất cả các dẫn xuất có thể có của chúng. Tuy nhiên, thuật ngữ này cực kỳ rườm rà (ví dụ: cortisol là 11β,17α,21-trihydroxy-pregn-4-ene-3,20-dione, và dexamethasone là 9α-fluoro-11β,17α,21-trihydroxypregna-1,4-diene-3,20-dione). Do đó, chúng tôi chỉ sử dụng các “tên thông thường” tiêu chuẩn. Trước khi cấu trúc của các hormone steroid được xác định vào những năm 1930, Reichstein, Kendall và những người khác đã xác định chúng là các đốm trên sắc ký đồ giấy và ký hiệu chúng là A, B, C, v.v. Thật không may, một số người vẫn tiếp tục sử dụng thuật ngữ lỗi thời này hơn 80 năm sau, do đó corticosterone đôi khi được gọi là hợp chất B, cortisol là hợp chất F, và 11-deoxycortisol là hợp chất S. Thuật ngữ cổ xưa này làm lu mờ mối quan hệ tiền chất-sản phẩm của các steroid, gây nhầm lẫn cho sinh viên và không nên được sử dụng.
Các Steroid trong tuần hoàn
Mặc dù hơn 50 loại steroid khác nhau đã được phân lập từ mô vỏ thượng thận, các con đường chính của quá trình sinh tổng hợp steroid tuyến thượng thận chỉ bao gồm khoảng một chục steroid, trong đó chỉ một vài loại được tiết ra với số lượng đáng kể. Tốc độ bài tiết của DHEA và cortisol ở người trưởng thành mỗi loại khoảng 20 mg/24 giờ và bài tiết corticosterone, một glucocorticoid yếu, là khoảng 2 mg/24 giờ. Mặc dù glucocorticoid, như cortisol, và mineralocorticoid, như aldosterone, đều cần thiết cho sự sống và do đó có tầm quan trọng sinh lý, các sơ đồ như Hình 14.3 không chỉ ra rằng các steroid này không được tiết ra với lượng tương đương về mặt phân tử gam. Tốc độ bài tiết aldosterone ở người trưởng thành chỉ khoảng 0,1 mg/24 giờ. Sự khác biệt từ 100 đến 1000 lần về mặt phân tử gam trong tốc độ bài tiết của cortisol và aldosterone phải được ghi nhớ khi xem xét tác dụng của các protein gắn steroid trong huyết tương và khi hình dung các biểu hiện sinh lý của các khiếm khuyết không hoàn toàn trong sinh tổng hợp steroid.
Hầu hết các steroid trong tuần hoàn được gắn với các protein huyết tương, bao gồm globulin gắn corticosteroid (CBG), albumin và glycoprotein axit α1. CBG có ái lực rất cao với cortisol nhưng khả năng gắn kết tương đối thấp; albumin có ái lực thấp và khả năng gắn kết cao; và glycoprotein axit α1 ở mức trung gian cho cả hai biến số. Kết quả là khoảng 90% cortisol trong tuần hoàn được gắn với CBG và một lượng nhỏ hơn được gắn với các protein khác. Điều thú vị là, sự vắng mặt của CBG không gây ra một rối loạn sinh lý có thể phát hiện được. Do đó, các protein huyết tương này được cho là hoạt động như một kho dự trữ không thiết yếu cho các steroid, tạo điều kiện cho sự phân bố rộng rãi và đồng đều của cortisol khắp cơ thể. Hầu hết các glucocorticoid tổng hợp được sử dụng trong điều trị không gắn kết đáng kể với CBG và gắn kết kém với albumin, điều này một phần giải thích cho hiệu lực tăng lên của chúng, cũng liên quan đến ái lực gắn kết thụ thể tăng lên. Aldosterone không được gắn kết tốt bởi bất kỳ protein huyết tương nào; do đó những thay đổi nồng độ protein huyết tương không ảnh hưởng đến nồng độ aldosterone huyết tương, nhưng ảnh hưởng lớn đến nồng độ cortisol huyết tương. Estradiol và testosterone gắn kết mạnh với một protein huyết tương khác gọi là globulin gắn steroid sinh dục và cũng gắn kết yếu với albumin.
Bởi vì steroid là hormone, người ta thường cho rằng nồng độ steroid “tự do” (tức là không gắn kết) trong tuần hoàn quyết định hoạt tính sinh học. Tuy nhiên, các mô đích của nhiều hormone steroid chứa các enzyme làm biến đổi các steroid đó. Do đó, nhiều tác động của testosterone thực sự được gây ra bởi DHT được sản xuất bởi 5α-reductase tại chỗ. Tương tự, cortisol sẽ có những tác động khác nhau trên các mô khác nhau do sự hiện diện hay vắng mặt của hai isozyme 11βHSD, có thể bất hoạt cortisol thành cortisone hoặc tái kích hoạt cortisone trở lại thành cortisol. Quá trình chuyển hóa ngoại vi tương tự xảy ra thông qua 21-hydroxylase, P450aro, 3βHSD và 17βHSD “ngoại tuyến”. Do đó, các steroid trong tuần hoàn vừa là hormone cổ điển vừa là tiền chất cho các yếu tố tự tiết hoặc cận tiết tác động tại chỗ.
Chuyển hóa Steroid
Chỉ khoảng 1% cortisol và aldosterone trong huyết tương lưu hành được bài tiết dưới dạng không đổi trong nước tiểu; phần còn lại được chuyển hóa bởi gan. Các công nghệ như chất đánh dấu phóng xạ và khối phổ đã nâng cao hiểu biết của chúng ta về sinh lý tuyến thượng thận và thúc đẩy chăm sóc lâm sàng. Hiểu biết về các quá trình và con đường chuyển hóa steroid có thể tạo điều kiện chẩn đoán nhanh hơn và chính xác hơn cũng như tránh các tác dụng phụ không mong muốn từ thuốc. Sắc ký lỏng-khối phổ (LC-MS/MS) hiện có thể xác định và định lượng một cách đặc hiệu và đồng thời các chất chuyển hóa chính và phụ. Các enzyme chính chuyển hóa steroid được tóm tắt dưới đây.
Các Enzyme chính chuyển hóa Steroid
Gan là nơi chính chuyển hóa glucocorticoid. Cortisol bị khử, oxy hóa, hoặc hydroxyl hóa và sau đó được liên hợp với sulfat hoặc axit glucuronic, làm cho nó tan trong nước và được bài tiết qua nước tiểu. 3α-Hydroxysteroid dehydrogenase (ví dụ, AKR1C2 và AKR1C4) là nhóm enzyme chính khử cortisol ở nhóm 3-keto, và 5α- hoặc 5β-reductase (lần lượt là SRD5A1 và AKR1D1) khử liên kết đôi 4-5 trong vòng A. Các tetrahydrocortisol và tetrahydrocortisone tạo ra từ các phản ứng này có thể bị khử bởi các 20-hydroxysteroid dehydrogenase, tạo ra cortol và cortolone. Tùy thuộc vào sự sẵn có của đồng yếu tố, 11β-hydroxysteroid dehydrogenase loại 1 (HSD11B1) có thể có hoạt động dehydrogenase (bất hoạt) hoặc reductase (kích hoạt), nhưng ở gan, enzyme này thường hoạt động như một reductase để kích hoạt cortisone thành cortisol. Cortisol, và các chất chuyển hóa được tạo ra trong các phản ứng được mô tả trước đó, bị oxy hóa. Quá trình này loại bỏ chuỗi bên C20-C21, dẫn đến các steroid C19 với một nhóm 17-ketone, nhưng enzyme thúc đẩy phản ứng này vẫn chưa được xác định. CYP3A4 có thể 6β-hydroxyl hóa cortisol; khi nồng độ cortisol bình thường, tốc độ của phản ứng này rất thấp. Tuy nhiên, khi nồng độ cortisol tăng cao, tốc độ phản ứng tăng lên, điều này có thể làm cho việc đo lường 6β-hydroxycortisol trong nước tiểu trở thành một xét nghiệm bổ trợ hữu ích cho tình trạng dư thừa glucocorticoid. Cuối cùng, các uridine diphosphoglucuronosyl transferase có thể liên hợp axit glucuronic hoặc sulfat với các chất chuyển hóa C19 và C21, làm tăng khả năng hòa tan trong nước và tạo điều kiện cho chúng được bài tiết qua nước tiểu.
Tương tự như cortisol, aldosterone bị khử và liên hợp ở gan. 4,3-Ketosteroid-reductase (cũng có hoạt tính 5β-hydroxysteroid reductase) và một 3α-hydroxysteroid dehydrogenase là các enzyme chính dẫn đến việc chuyển đổi aldosterone thành 3α, 5β-tetrahydroaldosterone. Tetrahydroaldosterone được liên hợp với axit glucuronic ở vị trí 3-keto (làm tăng khả năng hòa tan trong nước) và là chất chuyển hóa chính của aldosterone được bài tiết qua nước tiểu.
DHEA là steroid tuyến thượng thận được sản xuất với số lượng lớn nhất. DHEA được chuyển đổi thành androstenedione và bị khử 5α thành androsterone, sau đó được chuyển đổi thành etiocholanolone bởi 5β-reductase. Cuối cùng, các 17β-hydroxysteroid dehydrogenase khử etiocholanolone thành các dẫn xuất -diol, có thể được liên hợp và bài tiết qua nước tiểu. Tuy nhiên, sự bài tiết DHEA và các chất chuyển hóa của nó qua phân cao hơn các steroid khác. Mặt khác, este sulfat của DHEA (DHEAS), có thể được bài tiết trực tiếp qua nước tiểu. Mặc dù các con đường này có vẻ hơi khó hiểu, như được minh họa sau đây, chúng có thể bị xáo trộn bởi các liệu pháp và tình trạng phổ biến, do đó chúng cần được xem xét trong các kế hoạch chẩn đoán và điều trị lâm sàng.
Ví dụ về các loại thuốc làm thay đổi chuyển hóa Steroid
Hormone tuyến giáp làm tăng tốc độ chuyển hóa cortisol bằng cách cảm ứng hoạt động của 5α- và 5β-reductase ở gan và ức chế các enzyme CYP3A. Quá trình ngược lại xảy ra ở những bệnh nhân bị suy giáp. Trong cả hai tình huống, nồng độ cortisol huyết thanh thường bình thường vì trục HPA vẫn còn nguyên vẹn. Tuy nhiên, trong các trường hợp suy tuyến yên, nên thận trọng thay thế cortisol trước khi bắt đầu thay thế hormone tuyến giáp, để tránh một cơn suy thượng thận cấp do thanh thải cortisol tăng nhanh.
Spironolactone làm tăng nồng độ aldosterone huyết thanh, một phần do ức chế quá trình 18-glucuronid hóa aldosterone.
Troglitazone cảm ứng hoạt động của CYP3A4, làm gián đoạn chuyển hóa cortisol. Mặc dù cả rosiglitazone và pioglitazone đều không ảnh hưởng đến CYP3A4, các loại thuốc thiazolidinedione nói chung ức chế P450c17 và HSD3B2 sinh steroid.
Các loại thuốc làm thay đổi enzyme P450. Kết quả của một thử nghiệm đối chứng ngẫu nhiên đã ủng hộ các báo cáo rằng việc tiêu thụ Hypericum perforatum (Cỏ St. John, một phương thuốc thảo dược phổ biến) làm giảm hiệu quả của thuốc tránh thai đường uống dựa trên estradiol. Cỏ St. John cảm ứng CYP3A4, CYP2C19 và CYP2C9, làm tăng tốc độ chuyển hóa của thuốc tránh thai đường uống, làm giảm hiệu quả của chúng. Ngược lại, thuốc chống nấm ketoconazole hoạt động chủ yếu bằng cách ức chế CYP51 (lanosterol demethylase), nhưng nó cũng ức chế một số enzyme P450 sinh steroid và đã được sử dụng như một liệu pháp bổ trợ trong ung thư tuyến tiền liệt và hội chứng Cushing.
Ví dụ về các Rối loạn làm thay đổi chuyển hóa Steroid
Stress: Đáng ngạc nhiên là, sự gia tăng nồng độ ACTH thường là một quá trình thoáng qua, ngay cả khi stress kéo dài. Nồng độ cortisol tăng cao liên tục trong thời gian stress mãn tính là do các cytokine kích thích và giảm thanh thải cortisol thông qua sự bất hoạt enzyme bởi các 5α- và 5β-reductase và bởi HSD11B2.
Hội chứng Cushing: Người ta cho rằng khi nồng độ glucocorticoid vẫn tăng cao liên tục, các con đường oxy hóa và khử chuyển hóa chúng trở nên bão hòa và CYP3A4 được cảm ứng. Điều này dẫn đến tăng nồng độ 6β-hydroxyl hóa (và đồng thời giảm nồng độ cortol, cortolone, tetrahydrocortisone và 5α-tetrahydrocortisol) được bài tiết, điều này có thể được xác nhận bằng phân tích nước tiểu. Sự gia tăng nồng độ glucocorticoid cũng kích thích hoạt động HSD11B2 ở gan.
Béo phì: Những người béo phì có nồng độ chất chuyển hóa cortisol trong nước tiểu tăng cao, ngay cả khi đã chuẩn hóa theo diện tích bề mặt cơ thể. Điều quan trọng là phải phân biệt quá trình này với hội chứng Cushing. Ở bệnh nhân béo phì, nồng độ cortisol huyết thanh bình thường vì tốc độ bài tiết cao hơn được bù đắp bởi tốc độ sản xuất cao hơn.
Kháng Insulin: Những người bị kháng insulin, bao gồm cả những người có tăng insulin máu, đã tăng mức độ hoạt động của SRD5A1 ở gan, làm tăng bài tiết qua nước tiểu các chất chuyển hóa khử 5α của cortisol.
Bệnh thận: Mặc dù chuyển hóa cortisol thường bình thường ở bệnh nhân mắc bệnh thận, việc thanh thải các glucuronid có thể bị giảm, gây tích tụ các hợp chất liên hợp không hoạt động trong tuần hoàn. Quá trình này có khả năng được sử dụng để theo dõi bệnh nhưng không được cho là góp phần vào bệnh lý.
Suy tim: Bệnh nhân suy tim sung huyết nặng và giảm tưới máu gan cũng bị suy giảm chuyển hóa aldosterone, dẫn đến nồng độ tetrahydroaldosterone glucuronid trong nước tiểu thấp hơn.
Đánh giá lâm sàng và xét nghiệm chức năng tuyến thượng thận
Đánh giá lâm sàng
Đánh giá lâm sàng sắc sảo nói chung có thể tiết lộ sự hiện diện của suy thượng thận nguyên phát hoặc tăng tiết trước khi thực hiện các xét nghiệm. Thomas Addison đã mô tả suy thượng thận vào năm 1849, rất lâu trước khi các xét nghiệm miễn dịch ra đời. Hầu như tất cả các bệnh nhân bị suy thượng thận mãn tính sẽ có yếu, mệt mỏi, chán ăn, sụt cân, hạ huyết áp và tăng sắc tố da. Bệnh nhân bị suy thượng thận cấp có thể có hạ huyết áp, sốc, yếu, thờ ơ, lú lẫn, chán ăn, buồn nôn, nôn, mất nước, đau bụng hoặc đau hông, tăng thân nhiệt và/hoặc hạ đường huyết. Thiếu hụt bài tiết androgen tuyến thượng thận sẽ ảnh hưởng đến việc đạt được các đặc điểm sinh dục phụ nam hóa (lông mu và lông nách, mụn trứng cá, mùi hôi nách) ở nữ thanh thiếu niên. Các dấu hiệu sớm của dư thừa glucocorticoid bao gồm tăng cảm giác thèm ăn, tăng cân, thay đổi tâm trạng và ngừng tăng trưởng mà không có sự chậm trễ tương ứng về tuổi xương. Dư thừa glucocorticoid mãn tính ở trẻ em dẫn đến bộ mặt Cushing điển hình và teo cơ, nhưng “bướu trâu” và sự phân bố mỡ trung tâm đặc trưng của bệnh Cushing ở người lớn chỉ được thấy ở bệnh không được chẩn đoán kéo dài ở trẻ em. Dư thừa mineralocorticoid chủ yếu được đặc trưng bởi tăng huyết áp, nhưng những bệnh nhân ăn chế độ ăn rất ít natri (ví dụ, trẻ sơ sinh) sẽ không bị tăng huyết áp, vì mineralocorticoid làm tăng huyết áp chủ yếu bằng cách giữ lại natri và do đó làm tăng thể tích nội mạch. Tăng tiết androgen tuyến thượng thận ở mức độ vừa phải được đặc trưng bởi các dấu hiệu nam hóa nhẹ, trong khi tăng tiết androgen tuyến thượng thận đáng kể được đặc trưng bởi tăng trưởng nhanh, với sự gia tăng không cân xứng về tuổi xương, tăng khối lượng cơ, mụn trứng cá, rậm lông và giọng nói trầm hơn. Một đặc điểm quan trọng của bất kỳ cuộc khám thực thể nào đối với một nam giới bị nam hóa là kiểm tra và đo lường cẩn thận tinh hoàn. Tinh hoàn to hai bên gợi ý dậy thì sớm thật sự (trung ương); tinh hoàn to một bên gợi ý khối u tinh hoàn; tinh hoàn trước tuổi dậy thì ở một nam giới bị nam hóa cho thấy một nguồn androgen ngoài tinh hoàn, chẳng hạn như tuyến thượng thận.
Các nghiên cứu hình ảnh có tiện ích hạn chế trong bệnh vỏ thượng thận. Chụp cắt lớp vi tính (CT) chỉ hiếm khi phát hiện được các khối u tuyến yên tăng tiết ACTH, và chụp cộng hưởng từ (MRI) sẽ phát hiện được ít hơn một nửa trong số này, ngay cả khi có tăng cường gadolinium. Kích thước nhỏ, hình dạng kỳ lạ và vị trí gần các cấu trúc khác cũng làm giảm việc sử dụng các kỹ thuật hình ảnh cho tuyến thượng thận. CT hiện là lựa chọn đầu tiên để chụp ảnh tuyến thượng thận; mặc dù có phơi nhiễm phóng xạ, nó có lợi thế về độ phân giải cao hơn so với MRI. Bệnh nhân mắc bệnh Cushing hoặc CAH sẽ có tuyến thượng thận to ra khiêm tốn, nhưng sự gia tăng kích thước như vậy không thể phát hiện được bằng các kỹ thuật hình ảnh với bất kỳ mức độ chắc chắn hữu ích nào. Các nghiên cứu hình ảnh có thể hỗ trợ trong việc chẩn đoán sự gia tăng kích thước rõ rệt của tuyến thượng thận trong tăng sản thượng thận dạng mỡ bẩm sinh, thiểu sản của chúng trong thiểu sản thượng thận bẩm sinh, hoặc trong các hội chứng không đáp ứng với ACTH di truyền, và với nhiều khối u ác tính; tuy nhiên, nhiều adenoma tuyến thượng thận quá nhỏ (< 2 mm) để có thể phát hiện được. Do đó, các nghiên cứu hình ảnh có thể xác định sự hiện diện của các khối u tuyến yên hoặc tuyến thượng thận, nhưng chúng không bao giờ có thể xác định sự vắng mặt của chúng. Các xét nghiệm chức năng bổ sung, với các dược phẩm phóng xạ nhắm vào vỏ và tủy thượng thận, có thể tăng cường tiện ích chẩn đoán của các nghiên cứu hình ảnh và được sử dụng kết hợp với xạ hình, chụp cắt lớp phát xạ positron (PET) và chụp cắt lớp phát xạ đơn photon (SPECT).
Lấy mẫu hormone qua ống thông hiếm khi được thực hiện ở trẻ em. Lấy mẫu xoang đá có thể hữu ích trong trường hợp nghi ngờ bệnh Cushing, khi các nghiên cứu hình ảnh không phát hiện được khối u tiết ACTH ở tuyến yên. Lấy mẫu tĩnh mạch thượng thận có thể phân biệt adenoma thượng thận tăng tiết một bên với hai bên (ví dụ, cường aldosteron, hội chứng Conn), nhưng những trường hợp này thường thấy ở người lớn.
Đánh giá xét nghiệm
Việc đánh giá chẩn đoán chức năng tuyến thượng thận về cơ bản là hóa học. Sự không đặc hiệu của nhiều dấu hiệu lâm sàng được mô tả ở trên và kết quả đáng thất vọng với các nghiên cứu hình ảnh nhắc nhở chúng ta rằng bất kỳ đánh giá đúng đắn nào về chức năng HPA phải dựa vào một loạt các thao tác sinh lý được thực hiện cẩn thận kết hợp với các xét nghiệm hormone. Sự phát triển của các xét nghiệm có độ đặc hiệu cao, độ nhạy tinh tế có thể được thực hiện trên một lượng máu nhỏ hiện cho phép kiểm tra trực tiếp hầu như mọi hormone liên quan đến chuyển hóa tuyến thượng thận. Ngoài ra, việc phân tích hồ sơ steroid và phân tích các mẫu huyết thanh và nước tiểu, sử dụng các phương pháp sắc ký, khối phổ, mà qua đó các chất chuyển hóa steroid thông thường và mới có thể được phát hiện đồng thời từ một mẫu duy nhất, đã phát triển thành một công cụ mạnh mẽ để xác định các dấu hiệu steroid đặc trưng của các rối loạn phức tạp.
Đo lường Steroid
Các kỹ thuật đo lường Steroid thường được đo bằng các phương pháp xét nghiệm miễn dịch (IA) hoặc khối phổ (MS). IA là kỹ thuật truyền thống được sử dụng trong hầu hết các phòng thí nghiệm thông thường: chúng dễ thực hiện, nhanh, rẻ và phổ biến. IA bị hạn chế bởi độ đặc hiệu của chúng, bị giới hạn bởi (1) sự không tương xứng về kích thước giữa các phân tử steroid nhỏ và đoạn gắn kết của globulin miễn dịch, (2) phản ứng chéo của kháng huyết thanh với các steroid liên quan, và (3) chất lượng của chính các kháng huyết thanh. Hơn nữa, các hiệu ứng nền gây ra bởi các phân tử không phải steroid phản ứng chéo, các hợp chất ưa béo khác, hoặc các protein gắn kết trong các mẫu chưa qua xử lý cũng có thể ảnh hưởng đến độ đặc hiệu của IA. Hiệu ứng này có thể được cải thiện bằng cách tinh chế mẫu bằng chiết pha lỏng hoặc rắn, sắc ký, hoặc kết tủa. Tuy nhiên, các IA trực tiếp trên các thiết bị tự động thông lượng cao đã đẩy IA đến giới hạn của chúng. Mặc dù độ tin cậy của IA gần đây đã bị đặt câu hỏi, đặc biệt là để đo các steroid sinh dục, các IA đã được xác nhận thực hiện đúng cách vẫn là một công cụ có giá trị cho các phép đo steroid lâm sàng.
MS có thể là phương pháp linh hoạt và chính xác nhất để đo lường định tính và định lượng các steroid. Các phương pháp sắc ký (khí hoặc lỏng) phục vụ như các kỹ thuật tách, và máy khối phổ phục vụ như máy dò; cùng nhau, các phương pháp này đạt được độ đặc hiệu và độ chọn lọc phân tích cao nhất. Trái ngược với IA, nơi một xét nghiệm đo một steroid duy nhất, một lần chạy trong một xét nghiệm dựa trên MS cung cấp một hồ sơ steroid của nhiều steroid. Tùy thuộc vào các phương pháp cụ thể được áp dụng, các thiết bị có thể được chạy ở chế độ quét không phân biệt, không nhắm mục tiêu, thường được sử dụng cho các nghiên cứu. Ngược lại, trong các ứng dụng lâm sàng, các thiết bị ghi lại các ion được chọn trước tương ứng với các chất phân tích steroid cụ thể. Phân tích định lượng các steroid đòi hỏi các tiêu chuẩn nội bộ được đánh dấu bằng các đồng vị ổn định; những tiêu chuẩn này hiện có sẵn trên thị trường cho hầu hết các steroid quan tâm, bao gồm cả các chất chuyển hóa steroid mới được xác định có nguồn gốc từ các con đường “cửa sau” và 11-Oxo. Nói chung, sắc ký khí kết hợp khối phổ (GC-MS) là phương pháp lý tưởng để tách các steroid và được sử dụng rộng rãi trong các phòng thí nghiệm nghiên cứu để mô tả đặc điểm các chất chuyển hóa steroid trong các rối loạn sinh tổng hợp steroid mới hoặc phức tạp. Tuy nhiên, GC-MS/MS đòi hỏi phải dẫn xuất hóa mẫu, phức tạp và tốn thời gian, và không phù hợp để phân tích thông lượng cao các mẫu lâm sàng. Ngược lại, LC-MS/MS dễ dàng hơn, nhanh hơn và có thể phân tích các steroid được sulfat hóa và glucuronid hóa. Do đó, LC-MS/MS được sử dụng như một phương pháp thông lượng cao để phân tích steroid và hiện đã trở thành phương pháp được lựa chọn. Tuy nhiên, tất cả các phương pháp dựa trên MS đều đòi hỏi kiến thức về phân tích và sinh hóa phức tạp và do đó chỉ được thiết lập trong các phòng thí nghiệm chuyên biệt. Việc giải thích một hồ sơ steroid của một mẫu huyết thanh hoặc nước tiểu không phải là chuyện đơn giản. Nồng độ hoặc tốc độ bài tiết của nhiều chất chuyển hóa được so sánh với các giá trị tham chiếu và/hoặc tỷ lệ tiền chất:sản phẩm được tính toán như là các đại diện cho hoạt động của enzyme. Chẩn đoán các rối loạn steroid phức tạp đòi hỏi phải nhận dạng mẫu từ các bộ dữ liệu định lượng của nhiều steroid. Điều này có thể được tạo điều kiện bởi các phương pháp toán học không thiên vị (“học máy”) và đã được sử dụng cho một số rối loạn steroid.
Nồng độ Cortisol và các Steroid khác trong Huyết tương/Huyết thanh
Cortisol huyết tương có thể được đo bằng các IA, chẳng hạn như xét nghiệm miễn dịch phóng xạ (RIA), xét nghiệm đo miễn dịch phóng xạ (IRMA), và xét nghiệm điện hóa phát quang (ECLIA), phương pháp sau là phương pháp ưa thích trong các phòng thí nghiệm tự động thông lượng cao. Ngoài ra, cortisol cũng có thể được xác định bằng LC-MS/MS trong một môi trường được tiêu chuẩn hóa. Điều cần thiết là phải biết phòng thí nghiệm của mình đang sử dụng phương pháp nào và phương pháp đó đang đo chính xác những gì. Tất cả các xét nghiệm miễn dịch đều có một mức độ phản ứng chéo nhất định với các steroid khác. Hầu hết các IA cortisol sẽ phát hiện cả cortisol và cortisone; ngược lại, chúng dễ dàng được phân biệt bằng các kỹ thuật khối phổ. Huyết tương của trẻ sơ sinh chủ yếu chứa cortisone thay vì cortisol, do đó việc so sánh dữ liệu của trẻ sơ sinh thu được bằng sắc ký lỏng hiệu năng cao (HPLC) hoặc LC-MS/MS với các tiêu chuẩn đã công bố thu được bằng IA có thể gợi ý sai về suy thượng thận. Bảng 14.2 và Bảng 14.3 tóm tắt nồng độ huyết tương bình thường cho một loạt các steroid được đo bằng LC-MS/MS (https://www.endocrinesciences.com/). Với ngoại lệ đáng chú ý là DHEAS, hầu hết các steroid tuyến thượng thận đều biểu hiện sự biến thiên theo ngày đêm, dựa trên nhịp điệu ngày đêm của ACTH. Bởi vì stress do bệnh tật hoặc nhập viện có thể làm tăng bài tiết steroid tuyến thượng thận và vì nhịp điệu ngày đêm có thể không được thiết lập tốt ở trẻ em dưới 3 tuổi, tốt nhất là nên lấy hai hoặc nhiều mẫu để đo lường bất kỳ steroid nào.
Bảng 14.2. Nồng độ Steroid sinh dục theo Tuổi
| Tuổi | Prog | 17OHP | DHEA | DHEA-S | Δ4A | E1 | T | E2 | DHT | |
|---|---|---|---|---|---|---|---|---|---|---|
| Nữ | Nam | Nữ | Nam | |||||||
| Đơn vị | ng/dL | ng/dL | ng/dL | μg/dL | ng/dL | pg/mL | ng/dL | ng/dL | pg/mL | |
| TRẺ EM | ||||||||||
| Trẻ non tháng | ||||||||||
| 26-28 tuần | Ngày 4 sau sinh | 124-841 | 82-1484 | 63-935 | 59-125 | 5-16 | 123-882 | 2-13 | 10-53 | |
| 31-35 tuần | Ngày 4 sau sinh | 26-568 | 56-1853 | 37-198 | 50-449 | 5-22 | 122-710 | 2-13 | 10-53 | |
| Trẻ đủ tháng | Ngày 3 sau sinh | <78 | 41-1292 | <10-279 | 75-400 | 20-64 | 88-356 | <2-15 | 5-60 | |
| Trẻ nhũ nhi | ||||||||||
| 1 tuần đến 6 tháng | 13-106 | <948 | <10-37 | 20-50 | <10 | <15 | <15 | <112 | <15 | |
| 6-12 tháng | 13-106 | <136 | <2.5-10 | <10-17 | <10 | <15 | <15 | <49 | <15 | |
| Dậy thì mini | 2-3 tháng | 40-200 (Nam) | 5-50 | 60-400 | 12-85 | |||||
| TRẺ EM | 1-5 tuổi | <91 | <68 | <2.5-10 | <10-17 | <15 | <15 | <2.5-10 | <3 | <3 |
| 6-7 tuổi | <111 | <57 | ||||||||
| NAM | 8-10 tuổi | <186 | <193 | |||||||
| Tuổi dậy thì | 1-16 tuổi | <10-15 | <491 | |||||||
| Tanner I | <91 | <186 | 5-11 | <10-17 | 13-83 | <5-17 | <2.5-10 | <3 | ||
| Tanner II | <116 | 42-109 | <10-33 | <202 | 18-150 | 5-16 | 10-25 | 3-17 | ||
| Tanner III | 10-138 | <319 | 48-200 | 17-72 | 100-320 | 15-25 | 5-25 | 8-33 | ||
| Tanner IV | 29-180 | 39-481 | 200-620 | 33-192 | 102-385 | 10-36 | 15-45 | 15-115 | 22-52 | |
| Người lớn | Tanner V | 24-175 | 40-491 | 350-970 | 120-370 | 20-45 | 264-916 | 10-36 | 10-50 | 24-65 |
| 20-49 tuổi | 27-199 | 31-701 | 44-186 | 30-85 | 16-523 | |||||
| >50 tuổi | <10-11 | <298 | 21-402 | |||||||
| NỮ | ||||||||||
| Tuổi dậy thì | Tanner I | <10-26 | <83 | <186 | 19-144 | 4-29 | <10-17 | 5-20 | <2.5-10 | <3 |
| Tanner II | 11-98 | <10-255 | <202 | 34-129 | <10-72 | 10-24 | 7-28 | 10-33 | 5-12 | |
| Tanner III | 11-115 | <10-852 | <319 | 50-170 | 32-226 | 15-35 | 15-43 | 7-60 | 7-19 | |
| Tanner IV | 18-230 | <10-1076 | 39-481 | 58-260 | 47-208 | 16-77 | 21-85 | 13-32 | 4-13 | |
| Tanner V | <10-1294 | 20-265 | 40-491 | 50-224 | 44-248 | 34-170 | 29-105 | 20-38 | 3-18 | |
| Người lớn | 31-701 | 28-230 | 17-372 | 10-55 | 4-22 | |||||
| Pha nang trứng | <10-1563 | 15-70 | 30-100 | 30-100 | ||||||
| Pha hoàng thể | 35-290 <10-2555 | Đỉnh (N17-23) 350-3750 | 70-300 | 9-160 | ||||||
| Mãn kinh | <10 | <298 | <40 | <10-93 | <215 | <15 | 7-40 |
Các giá trị steroid mới tính bằng ng/dL chỉ có ở người lớn: 11-Hydroxy-testosterone, Nam 5.2-43.4, Nữ <39.8; 11-Keto-testosterone, Nam 9.5-70.8, Nữ 5-60.6; 11OH Δ4A, Nam 36.4-313, Nữ 19.2-333; androsterone, Nam <28, Nữ <23.
Tất cả các steroid được đo bằng sắc ký lỏng hiệu năng cao khối phổ (HPLC-MS/MS).
Tất cả các giá trị là khoảng dao động tính bằng ng/dL, μg/dL, hoặc pg/mL như đã chỉ định; để chuyển đổi các giá trị này sang nmol/L, nhân các giá trị đã cho với hệ số sau: androstenedione, 0.035; DHEA, 0.035; DHT, 0.034; E1, 0.0037; E2, 0.0037; Prog, 0.032; 17OHP, 0.03; DHEA-S, 27.17; T toàn phần, 0.035; DHT, 0.034; androsterone, 0.034.
Δ4A, Androstenedione; 17OHP, 17-Hydroxyprogesterone; N, ngày; DHEA, dehydroepiandrosterone; DHEA-S, DHEA sulfat; DHT, dihydrotestosterone; E1, estrone; E2, estradiol; Nữ; Nam; th, tháng; PROG, progesterone; T, testosterone; t, tuổi.
(Từ LabCorp (Laboratory Corporation of America)/Endocrine Sciences, Calabasas Hills, California, 2019.)
Bảng 14.3. Nồng độ Glucocorticoid và Mineralocorticoid
| Tuổi | Cortisol | Corticosterone | DOC | 11-Deoxycortisol | 21-Deoxycortisol | 18OH Corticosterone | Aldosterone | Hoạt độ Renin huyết tương | |
|---|---|---|---|---|---|---|---|---|---|
| Đơn vị | μg/dL | ng/dL | ng/dL | ng/dL | ng/dL | ng/dL | ng/dL | ng/mL/h | |
| TRẺ EM | |||||||||
| Trẻ non tháng | 11000-167000 | ||||||||
| 26-28 tuần | 1-11 | 235-1108 | 20-105 | 110-1376 | 10-670 | 5-635 | |||
| 31-35 tuần | 2.5-9.1 | 150-1700 | 28-78 | 48-579 | 57-410 | 19-141 | 2000-35000 | ||
| Trẻ sơ sinh đủ tháng | <78 | ||||||||
| Ngày 3 sau sinh | 1.7-14 | 70-850 | 13-147 | 31-546 | 7-184 | ||||
| Ngày 7 sau sinh | 2-11 | 70-850 | 5-175 | ||||||
| Trẻ nhũ nhi | 1-12 tháng | 2.8-23 | 80-1500 | 7-49 | <10-156 | 5-220 | 5-90 | 2000-37000 | |
| Trẻ em | |||||||||
| 8:00 sáng | 1-15 tuổi | 3-21 | 135-1860 | 2-34 | 12-158 | <10 | |||
| 4:00 chiều | 1-15 tuổi | 70-620 | |||||||
| 1-2 tuổi | 18-155 | 7-54 | 1700-11200 | ||||||
| 3-9 tuổi | 6-85 | 5-80 | 500-6500 | ||||||
| 10-14 tuổi | 10-72 | 4-485 | 500-3300 | ||||||
| NGƯỜI LỚN | |||||||||
| 8:00 sáng | 8-19 | 130-820 | 2-19 | 12-158 | <10 | 9-58 | 167-5380 | ||
| 4:00 chiều | 4-11 | 60-220 | |||||||
| Nằm/đứng | 4-21/5-46 | <31 |
Tất cả các giá trị được đưa ra dưới dạng khoảng và được đo bằng sắc ký lỏng hiệu năng cao khối phổ (HPLC-MS/MS).
Để chuyển đổi các giá trị đã cho sang đơn vị SI nmol/L, nhân với các hệ số sau: cortisol, 27.59; corticosterone, 0.029; 18OH corticosterone, 0.028; aldosterone, 0.028; DOC, 0.03; 11DOC, 0.029; 21-Deoxycorticol, 0.029.
Giá trị ở: tư thế nằm và đứng.
N3, Ngày 3; DOC, deoxycorticosterone; th, tháng; KXĐ, không xác định; t, tuổi.
(Từ LabCorp (Laboratory Corporation of America)/Endocrine Sciences, Calabasas Hills, California 2019.)
Dữ liệu tồn tại cho nồng độ của một số lượng lớn các hormone steroid trong suốt giai đoạn sơ sinh, thời thơ ấu và thanh thiếu niên bình thường (xem Bảng 14.2 và 14.3). Không phải tất cả các phòng thí nghiệm nội tiết đều thực hiện tất cả các xét nghiệm này, và, tùy thuộc vào quy trình xét nghiệm được sử dụng, các phòng thí nghiệm khác nhau có thể có các giá trị “bình thường” khác nhau. Hầu hết các bệnh viện trung ương và các phòng thí nghiệm thương mại chủ yếu được thiết kế để phục vụ bệnh nhân người lớn, thay vì bệnh nhi. Do đó, điều quan trọng là phải biết liệu các xét nghiệm có sẵn có đủ nhạy với một lượng máu nhỏ để hữu ích trong việc đo lường các giá trị ở trẻ em hay không. Điều này đặc biệt đúng đối với việc đo lường các steroid sinh dục (và gonadotropin), có thể biểu hiện sự gia tăng bệnh lý ở trẻ em và vẫn nằm dưới giới hạn phát hiện của hầu hết các xét nghiệm “người lớn”. Những nỗ lực để đạt được sự tiêu chuẩn hóa các phương pháp phân tích và các giá trị định chuẩn là những chủ đề tích cực trong lĩnh vực này.
Bài tiết Steroid qua nước tiểu
Đo lường sự bài tiết qua nước tiểu trong 24 giờ của các chất chuyển hóa steroid, một trong những quy trình lâu đời nhất để đánh giá chức năng tuyến thượng thận, gần đây đã được hồi sinh bởi các phương pháp phân tích khối phổ mới. Việc kiểm tra tổng lượng bài tiết steroid qua nước tiểu trong 24 giờ loại bỏ sự dao động theo ngày đêm của nồng độ steroid trong huyết thanh, và các biến thể do các đợt bùng phát của ACTH và stress thoáng qua (chẳng hạn như một chuyến đi đến phòng khám hoặc lấy máu tĩnh mạch khó khăn). Việc thu thập một mẫu nước tiểu 24 giờ hoàn chỉnh có thể khó khăn ở trẻ sơ sinh hoặc trẻ nhỏ, do đó nước tiểu cũng nên được xét nghiệm creatinine để theo dõi tính đầy đủ của việc thu thập. Do bản chất theo ngày đêm và từng đợt của sự bài tiết steroid, không bao giờ nên suy ra tốc độ bài tiết 24 giờ từ các lần thu thập một phần. Điều này rất quan trọng khi đánh giá tốc độ bài tiết tuyệt đối của một steroid duy nhất, chẳng hạn như cortisol tự do. Ngược lại, một mẫu nước tiểu tại chỗ (tốt nhất là lần đi tiểu đầu tiên vào buổi sáng, cô đặc nhất và được sản xuất dưới sự kích thích ACTH cao nhất), trong đó nhiều steroid được bài tiết được định lượng liên quan đến creatinine, có thể đủ cho chẩn đoán định tính kiểu mẫu của một rối loạn steroid cụ thể.
Các quy trình hiện đại để phân tích steroid trong nước tiểu sử dụng sắc ký để tách các steroid, sau đó là một phương pháp phát hiện MS, thường là GC-MS/MS, cho phép các xét nghiệm steroid trong nước tiểu rất nhạy và đặc hiệu. Tuy nhiên, mỗi steroid được chuyển hóa thành nhiều dạng trước khi được bài tiết qua nước tiểu, và sự chuyển hóa này có thể thay đổi theo tuổi và giới tính ở các quần thể nhi khoa, do đó các phân tích rất phức tạp và đòi hỏi chuyên môn chuyên biệt chưa được phổ biến rộng rãi. Các phép đo cortisol tự do trong nước tiểu (UFC) rất đáng tin cậy để chẩn đoán hội chứng Cushing ở người lớn. Sự bài tiết UFC và tổng các chất chuyển hóa cortisol có tương quan chặt chẽ với tuổi, diện tích bề mặt cơ thể và tình trạng béo phì, nhưng thường là 11 ± 5 μg/m2/ngày. Các giá trị thay đổi đáng kể giữa các phòng thí nghiệm tham chiếu khác nhau, phản ánh sự khác biệt trong công nghệ xét nghiệm, do đó điều cần thiết là sử dụng một phòng thí nghiệm có dữ liệu tốt cho trẻ em bình thường. Việc đo creatinine trong nước tiểu vẫn quan trọng để theo dõi tính đầy đủ của việc thu thập.
Renin huyết tương
Renin có thể được định lượng bằng hoạt độ enzyme của nó hoặc bằng cách đo trực tiếp nồng độ của nó. Hoạt độ renin huyết tương (PRA) đơn giản là một xét nghiệm về lượng angiotensin I được tạo ra trên mỗi mililit huyết thanh, mỗi giờ ở 37° C. Trong huyết thanh bình thường, nồng độ của cả renin và angiotensinogen (cơ chất của renin) đều bị giới hạn. Một xét nghiệm khác, về mặt kỹ thuật khó khăn hơn, đo trực tiếp hàm lượng renin huyết tương (PRC) bằng xét nghiệm miễn dịch.
PRA nhạy cảm với lượng natri ăn vào, tư thế, liệu pháp lợi tiểu, hoạt động và các steroid sinh dục. Bởi vì các giá trị PRA có thể thay đổi rộng rãi, tốt nhất là nên đo PRA hai lần, một lần vào buổi sáng, sau khi nằm ngửa qua đêm, và sau đó một lần nữa sau khi duy trì tư thế thẳng đứng trong 4 giờ. Một mẫu nước tiểu 24 giờ đồng thời để xác định tổng lượng natri bài tiết thường cần thiết để giải thích kết quả PRA. Lượng natri trong chế độ ăn và nước tiểu giảm, thể tích nội mạch giảm, thuốc lợi tiểu và estrogen sẽ làm tăng PRA. Tải natri, tăng aldosteron máu và tăng thể tích nội mạch làm giảm PRA.
Các phép đo renin được sử dụng trong đánh giá tăng huyết áp và trong quản lý CAH. Đánh giá hệ renin-angiotensin là cần thiết ở trẻ em bị CAH thể nam hóa đơn thuần (SV) không có bằng chứng lâm sàng về mất muối qua nước tiểu (hạ natri máu, tăng kali máu, nhiễm toan, hạ huyết áp, sốc), nhưng vẫn có thể có PRA tăng, đặc biệt là khi lượng natri trong chế độ ăn bị hạn chế. Đây là một dấu hiệu lâm sàng cho thấy dạng 21OHD này đơn giản là một dạng nhẹ hơn của dạng mất muối phổ biến hơn. Điều trị CAH thể SV bằng đủ mineralocorticoid, để ức chế PRA vào phạm vi bình thường, sẽ làm giảm nhu cầu glucocorticoid của trẻ, do đó tối đa hóa chiều cao cuối cùng của người trưởng thành và giảm phơi nhiễm với glucocorticoid dư thừa. Trẻ em bị CAH cần được theo dõi liệu pháp thay thế mineralocorticoid một cách thường xuyên bằng cách đo PRA. Việc đo angiotensin II cũng có thể thực hiện được ở một số phòng thí nghiệm nghiên cứu, nhưng hầu hết các kháng thể đối với angiotensin II đều phản ứng chéo mạnh với angiotensin I. Do đó, PRA vẫn là cách thông thường để đánh giá hệ renin-angiotensin-aldosterone.
ACTH huyết tương và các Peptide Proopiomelanocortin khác
Xét nghiệm miễn dịch chính xác ACTH huyết tương có sẵn ở hầu hết các trung tâm, nhưng việc đo lường nó vẫn khó khăn và biến đổi hơn so với các xét nghiệm cho hầu hết các hormone tuyến yên khác. Việc xử lý các mẫu phải được thực hiện cẩn thận; các mẫu phải được lấy vào một ống tiêm nhựa chứa heparin hoặc axit ethylenediamine tetraacetic (EDTA) và nhanh chóng vận chuyển trong các ống nhựa trên đá, vì ACTH bám vào thủy tinh và nhanh chóng bị bất hoạt. Do đó, nồng độ ACTH huyết tương tăng cao có thể rất hữu ích, nhưng hầu hết các xét nghiệm không thể phát hiện các giá trị thấp hoặc bình thường thấp, và các giá trị như vậy có thể là giả mạo nếu các mẫu được xử lý kém. Ở người lớn và trẻ lớn có nhịp sinh học ngày đêm của ACTH đã được thiết lập tốt, các giá trị lúc 8 giờ sáng bình thường hiếm khi vượt quá 50 pg/mL, trong khi các giá trị lúc 8 giờ tối thường không thể phát hiện được. Bệnh nhân mắc bệnh Cushing thường có các giá trị buổi sáng bình thường, nhưng chẩn đoán có thể được gợi ý bởi các giá trị buổi chiều và buổi tối tăng cao liên tục; bệnh nhân mắc hội chứng ACTH lạc chỗ có thể có các giá trị lên tới 1000 pg/mL.
Tốc độ bài tiết
Tốc độ bài tiết của cortisol và aldosterone (hoặc các steroid khác) có thể được đo bằng cách sử dụng một liều nhỏ cortisol hoặc aldosterone được đánh dấu bằng tritium và đo hoạt độ riêng của một hoặc nhiều chất chuyển hóa đã biết trong một mẫu nước tiểu 24 giờ. Quy trình này cho phép đo lường một số steroid nhất định, chẳng hạn như aldosterone, trước khi có các xét nghiệm miễn dịch đặc hiệu. Các quy trình này cũng đã cung cấp nhiều thông tin về tốc độ sản xuất bình thường của các steroid khác nhau. Dựa trên quy trình này, hầu hết các chuyên gia đã đồng ý rằng trẻ em và người lớn tiết ra khoảng 6 đến 9 mg cortisol trên mỗi mét vuông diện tích bề mặt cơ thể mỗi ngày.
Nghiệm pháp ức chế Dexamethasone
Việc sử dụng dexamethasone, một glucocorticoid tổng hợp mạnh, sẽ ức chế bài tiết ACTH tuyến yên và cortisol tuyến thượng thận; nghiệm pháp ức chế dexamethasone hữu ích để phân biệt xem tình trạng dư thừa glucocorticoid là do bệnh lý tuyến yên hay bệnh lý tuyến thượng thận gây ra. Vì dexamethasone cũng ức chế bài tiết androgen tuyến thượng thận, nghiệm pháp này hữu ích để phân biệt giữa các nguồn steroid sinh dục từ tuyến thượng thận và tuyến sinh dục. Một nghiệm pháp ức chế dexamethasone chính thức hoàn chỉnh đòi hỏi phải đo các giá trị cơ bản và các giá trị thu được để đáp ứng với cả dexamethasone liều thấp và liều cao. Điều này được mô tả trong phần về đánh giá hội chứng Cushing. Các biến thể của nghiệm pháp này thường được sử dụng, đáng chú ý là liều duy nhất 1,0 mg ở người lớn hoặc 0,3 mg/m2 ở trẻ em. Đây là một quy trình sàng lọc ngoại trú hữu ích để phân biệt hội chứng Cushing với béo phì ngoại sinh. Nó có thể hữu ích cho cùng mục đích ở thanh thiếu niên và trẻ lớn, nhưng mặt khác lại có tiện ích hạn chế trong nhi khoa. Một nghiệm pháp ức chế dexamethasone liều cao qua đêm có lẽ đáng tin cậy hơn so với nghiệm pháp liều cao tiêu chuẩn 2 ngày trong việc phân biệt người lớn mắc bệnh Cushing với những người mắc hội chứng ACTH lạc chỗ. Tuy nhiên, tiện ích của nghiệm pháp này ở bệnh nhân nhi chưa được xác định.
Các nghiệm pháp kích thích
Kích thích trực tiếp tuyến thượng thận bằng ACTH là một cách nhanh chóng, an toàn, dễ dàng để đánh giá chức năng vỏ thượng thận. Nghiệm pháp ACTH ban đầu bao gồm truyền 0,5 đơn vị/kg ACTH(1-39) trong 4 đến 6 giờ. Điều này sẽ kích thích tối đa sự bài tiết cortisol của tuyến thượng thận, và do đó phân biệt hiệu quả suy thượng thận nguyên phát (bệnh Addison), trong đó tuyến thượng thận không có khả năng đáp ứng, với suy thượng thận thứ phát do suy tuyến yên. Trong suy thượng thận thứ phát, một số khả năng sinh steroid vẫn còn, do đó một số cortisol được sản xuất để đáp ứng với ACTH; do đó sự bài tiết cortisol thấp hơn bình thường nhưng lớn hơn các giá trị không đáng kể thấy trong suy thượng thận nguyên phát. Nghiệm pháp ACTH truyền tĩnh mạch 4 đến 6 giờ đã được thay thế bằng nghiệm pháp 60 phút, trong đó một liều bolus duy nhất ACTH(1-24) được tiêm tĩnh mạch và cortisol và có thể các steroid khác được đo ở thời điểm 0 và 60 phút. Các đáp ứng bình thường đối với nghiệm pháp 60 phút được trình bày trong Bảng 14.4. ACTH(1-24) tổng hợp (cosyntropin) được ưu tiên sử dụng, vì nó có tác dụng nhanh hơn và thời gian bán hủy ngắn hơn so với ACTH(1-39). Liều thông thường là 15 μg/kg ở trẻ em đến 2 tuổi, và 0,25 mg cho trẻ em trên 2 tuổi và người lớn. Tất cả các liều này đều là liều dược lý. Một nghiệm pháp liều rất thấp (1 μg) có thể hữu ích trong việc đánh giá sự phục hồi của tuyến thượng thận sau khi bị ức chế bởi glucocorticoid. Dữ liệu mới hơn cho thấy rằng các đáp ứng steroid tối đa có thể đạt được chỉ sau 30 phút, nhưng các tiêu chuẩn tốt nhất hiện có là cho nghiệm pháp 60 phút. Một trong những ứng dụng rộng rãi nhất của các nghiệm pháp ACTH tĩnh mạch trong nhi khoa là chẩn đoán CAH. Kích thích tuyến thượng thận bằng ACTH làm tăng sinh tổng hợp steroid, dẫn đến tích tụ các steroid ở phía trước enzyme bị rối loạn. Ví dụ, xem xét Hình 14.3 cho thấy hoạt động suy giảm của P450c21 (21-hydroxylase) sẽ dẫn đến sự tích tụ progesterone và 17OHP. Tuy nhiên, progesterone không tích tụ với số lượng đáng kể, vì nó được chuyển đổi thành 17OHP. Trong thực hành thường quy, việc đo lường đáp ứng của 17OHP đối với một thử thách 60 phút với ACTH tĩnh mạch, là phương tiện duy nhất mạnh mẽ và đáng tin cậy nhất để chẩn đoán 21OHD; xét nghiệm di truyền có thể cung cấp một sự xác nhận hữu ích. So sánh các giá trị 17OHP cơ bản và sau kích thích ACTH của bệnh nhân với các giá trị từ số lượng lớn các bệnh nhân được nghiên cứu kỹ lưỡng thường cho phép phân biệt người bình thường, người dị hợp tử, bệnh nhân CAH không cổ điển và bệnh nhân CAH cổ điển, mặc dù không thể tránh khỏi có sự chồng chéo giữa các nhóm (Hình 14.12). Đo lường testosterone hoặc Δ4-androstenedione để đáp ứng với ACTH có thể phân biệt người bình thường với bệnh nhân CAH cổ điển, nhưng người dị hợp tử và bệnh nhân CAH thể ẩn có các giá trị chồng chéo cả với người bình thường và CAH cổ điển.
Hình 14.12: Các giá trị 17α-Hydroxyprogesterone (tính bằng ng/100 mL) trước và sau khi kích thích bằng hormone vỏ thượng thận ở người bình thường, bệnh nhân bị tăng sản thượng thận bẩm sinh, và những người dị hợp tử.
Bảng 14.4. Đáp ứng của các Steroid thượng thận trong huyết tương với Nghiệm pháp ACTH tiêm tĩnh mạch 60 phút
| 1-6 Tháng | 2-6 Tuổi | Trước dậy thì | Nam trưởng thành | Nữ trưởng thành | ||
|---|---|---|---|---|---|---|
| Đơn vị | Nền/Kích thích | Nền/Kích thích | Nền/Kích thích | Nền/Kích thích | Nền/Kích thích | |
| Cortisol | μg/dL | 3-22 / 27-50 | 6-19 / 20-33 | 5-16 / 20-31 | 7-15 / 19-31 | 7-21 / 17-39 |
| 17OHP | ng/dL | 13-173 / 85-250 | 7-114 / 50-269 | 7-100 / 85-280 | 35-150 / 45-258 | 22-140 / 65-250 |
| 17OH Pregnenolone | ng/dL | 52-828 / 633-3286 | 10-103 / 45-347 | 10-186 / 70-656 | 20-187 / 240-1000 | 48-320 / 290-1382 |
| 11-Deoxycortisol | ng/dL | 10-200 / 101-392 | 7-210 / 95-323 | 14-136 / 95-254 | 20-65 / 73-214 | 15-158 / 65-237 |
| DOC | ng/dL | 7-48 / 40-158 | 4-49 / 26-139 | 4-17 / 22-120 | 3-13 / 14-38 | 3-19 / 12-90 |
| Pregnenolone | ng/dL | 10-150 / 100-359 | 17-50 / 34-99 | 15-63 / 39-130 | 10-85 / 20-200 | 46-150 / 70-220 |
| Δ4A | ng/dL | <10-48 / <10-87 | <10 / <10-35 | <10 / <10-69 | 50-210 / 78-285 | 61-222 / 98-295 |
Các giá trị nền được đánh giá trong khoảng 8-9 giờ sáng.
Tất cả các giá trị được đo bằng sắc ký lỏng hiệu năng cao khối phổ (HPLC-MS/MS) và được đưa ra dưới dạng khoảng tính bằng ng/dL, ngoại trừ cortisol tính bằng μg/dL. Hệ số chuyển đổi sang Đơn vị SI được đưa ra trong Bảng 14.2 và 14.3.
Δ4A, Androstenedione; 17OHP, 17-hydroxyprogesterone; ACTH, hormone vỏ thượng thận (corticotropin); DOC, deoxycorticosterone.
(Từ LabCorp (Laboratory Corporation of America)/Endocrine Sciences, Calabasas Hills, California.)
Các nghiệm pháp ACTH kéo dài hơn đến 3 ngày cũng đã được sử dụng để đánh giá chức năng tuyến thượng thận. Điều quan trọng cần nhớ là ACTH có cả tác dụng cấp tính và mãn tính. Do đó, các nghiệm pháp ngắn chỉ đo lường các tác dụng cấp tính của ACTH, tức là sự kích thích tối đa của bộ máy sinh steroid đã có từ trước. Ngược lại, một nghiệm pháp 3 ngày sẽ kiểm tra các tác dụng mãn tính hơn của ACTH để kích thích tăng khả năng sinh tổng hợp steroid bằng cách tăng tổng hợp bộ máy sinh steroid. Rất ít tình huống cần chỉ định một nghiệm pháp ACTH tiêm bắp 3 ngày, mặc dù đôi khi nó hữu ích trong việc chẩn đoán các hội chứng hiếm gặp về không đáp ứng di truyền với ACTH.
Hạ đường huyết do insulin gây ra là một nghiệm pháp hiệu quả nhưng có khả năng rủi ro, và do đó hiếm khi được sử dụng. Insulin (0,1 U/kg/IV) được tiêm và máu được lấy ở các thời điểm 0, 30, 45 và 60 phút. Hạ đường huyết do insulin gây ra sẽ kích thích giải phóng các hormone “phản điều hòa” có tác dụng làm tăng nồng độ glucose huyết tương: ACTH và cortisol, hormone tăng trưởng, epinephrine và glucagon. Do nguy cơ co giật vốn có do hạ đường huyết, một bác sĩ có kinh nghiệm phải có mặt (không chỉ đơn thuần là “sẵn sàng”) trong suốt quá trình thực hiện nghiệm pháp. Đường huyết phải giảm xuống một nửa giá trị ban đầu hoặc xuống 45 mg/dL để đạt được một nghiệm pháp đầy đủ, và nên kết thúc nghiệm pháp sau khi đạt được mức này. Hầu hết bệnh nhân sẽ cảm thấy đói, cáu kỉnh, toát mồ hôi và nhịp tim nhanh; khi những triệu chứng này được theo sau bởi buồn ngủ hoặc ngủ, mức đường huyết có khả năng dưới giới hạn chấp nhận được. Nếu điều này xảy ra, nên lấy một mẫu máu và tiêm tĩnh mạch 2 mL/kg glucose 20% đến 25%, tối đa là 100 mL.
Nghiệm pháp Metyrapone
Metyrapone ngăn chặn hoạt động của P450c11β và, ở một mức độ thấp hơn nhiều, P450scc. Do đó, nó là một phương tiện hóa học để gây ra sự thiếu hụt tạm thời hoạt động của 11-hydroxylase, dẫn đến giảm bài tiết cortisol và sau đó là tăng bài tiết ACTH. Nghiệm pháp metyrapone được thực hiện để đánh giá khả năng của tuyến yên sản xuất ACTH để đáp ứng với một kích thích sinh lý. Nghiệm pháp này hữu ích trong việc đánh giá trục hạ đồi-tuyến yên khi có tổn thương hệ thần kinh trung ương, sau phẫu thuật thần kinh, hoặc ức chế lâu dài bằng liệu pháp glucocorticoid. Bệnh nhân có tiền sử bệnh hạ đồi, tuyến yên hoặc tuyến thượng thận, hoặc những người đã ngưng liệu pháp glucocorticoid nên được đánh giá lại bằng nghiệm pháp metyrapone. Một đáp ứng bình thường cho thấy sự phục hồi của trục HPA và dự đoán rằng bệnh nhân sẽ đáp ứng bình thường với stress phẫu thuật.
Metyrapone thường được dùng đường uống với liều 300 mg/m2 mỗi 4 giờ cho tổng cộng sáu liều (24 giờ). Không giống như nhiều loại thuốc khác, việc tiếp tục tăng liều ở những bệnh nhân lớn tuổi hoặc thừa cân là phù hợp, nhưng tổng liều không được vượt quá 3,0 g. Máu nên được lấy để xét nghiệm cortisol, 11-deoxycortisol và ACTH, trước và sau nghiệm pháp. Trong một đáp ứng bình thường với metyrapone, cortisol giảm, ACTH tăng, và 11-deoxycortisol (cơ chất cho P450c11β) tăng lên rất nhiều, đến khoảng 5 μg/dL. Người lớn và trẻ lớn có thể được kiểm tra bằng cách dùng một liều duy nhất 30 mg/kg đường uống vào nửa đêm, uống cùng với thức ăn để giảm kích ứng đường tiêu hóa. Các mẫu máu được lấy vào lúc 8 giờ sáng các buổi sáng trước và sau khi dùng thuốc.
Các tổn thương di truyền trong sinh tổng hợp steroid
Các rối loạn di truyền lặn trên nhiễm sắc thể thường phá vỡ mỗi bước được trình bày trong Hình 14.3; hầu hết các rối loạn này làm giảm tổng hợp cortisol. Để đáp ứng với tình trạng giảm cortisol máu, tuyến yên sản xuất lượng POMC và ACTH tăng lên, thúc đẩy tăng sinh tổng hợp steroid; ACTH và các peptide đầu N của POMC cũng kích thích phì đại và tăng sản tuyến thượng thận. Do đó, thuật ngữ “tăng sản thượng thận bẩm sinh” (CAH) đề cập đến một nhóm các bệnh theo truyền thống được nhóm lại với nhau trên cơ sở phát hiện nổi bật nhất khi tử thiết.
Về lý thuyết, các bệnh CAH rất dễ hiểu: một tổn thương di truyền ở một enzyme sinh steroid cản trở quá trình sinh tổng hợp steroid bình thường. Các dấu hiệu và triệu chứng của bệnh bắt nguồn từ sự thiếu hụt sản phẩm steroid cuối cùng và tác động của các tiền chất steroid tích tụ, ở phía trước bước bị tắc nghẽn. Do đó, việc tham khảo các con đường trong Hình 14.3 và kiến thức về tác dụng sinh học của mỗi steroid sẽ cho phép người ta suy ra các biểu hiện của bệnh.
Trong thực tế, các bệnh CAH có thể gây nhầm lẫn về mặt lâm sàng và khoa học. Mỗi enzyme sinh steroid có nhiều hoạt tính và nhiều mô ngoại thượng thận, đặc biệt là gan, biểu hiện các enzyme khác có hoạt tính sinh steroid, do đó việc loại bỏ hoàn toàn một enzyme tuyến thượng thận có thể không dẫn đến việc loại bỏ hoàn toàn các sản phẩm steroid của nó khỏi tuần hoàn. Hơn nữa, có những “thiếu hụt một phần” ở mỗi enzyme, nơi một số hoạt tính enzyme vẫn còn, thường gây ra bệnh “không cổ điển” nhẹ hơn với khởi phát muộn hơn. Các đặc điểm lâm sàng, xét nghiệm và điều trị chính của mỗi dạng CAH được tóm tắt trong Bảng 14.5.
Bảng 14.5. Các phát hiện lâm sàng và xét nghiệm trong các bệnh Tăng sản thượng thận bẩm sinh
| Thiếu hụt Enzyme | Biểu hiện | Phát hiện Xét nghiệm | Các biện pháp điều trị |
|---|---|---|---|
| CAH dạng mỡ (StAR hoặc P450scc) | Cơn mất muối, 46,XY DSD | Nồng độ tất cả các hormone steroid thấp/không có, Đáp ứng giảm/không có với ACTH, Đáp ứng giảm/không có với hCG ở 46,XY DSD, ↑ ACTH và PRA | Thay thế glucocorticoid và mineralocorticoid, bổ sung muối, Thay thế estrogen ở tuổi ≥12 tuổi, Cắt bỏ tuyến sinh dục ở 46,XY DSD và bổ sung muối |
| 3β-HSD | Cơn mất muối, 46,XX và 46,XY DSD | ↑ steroid Δ5 trước và sau ACTH, ↑ tỷ lệ steroid Δ5/Δ4 trong huyết thanh, Ức chế các steroid thượng thận tăng cao sau khi dùng glucocorticoid, ↑ ACTH và PRA | Thay thế glucocorticoid và mineralocorticoid, Bổ sung muối, Phẫu thuật chỉnh sửa bộ phận sinh dục, Thay thế hormone sinh dục khi cần thiết |
| P450c21 | Các thể cổ điển: Cơn mất muối, 46,XX DSD, Nam hóa trước và sau sinh. Thể không cổ điển: Thượng thận hóa sớm, kinh nguyệt không đều, rậm lông, mụn trứng cá, vô sinh | ↑ 17OHP trước và sau ACTH, ↑ androgen huyết thanh và 17KS nước tiểu, Ức chế các steroid thượng thận tăng cao sau khi dùng R glucocorticoid | Thay thế glucocorticoid và mineralocorticoid, Bổ sung muối, Phẫu thuật sửa chữa ở 46,XX DSD |
| P450c11β | 46,XX DSD, Nam hóa sau sinh ở nam và nữ, Tăng huyết áp, Hạ kali máu | ↑ ACTH và PRA, ↑ 11-deoxycortisol và DOC trước và sau ACTH, ↑ androgen huyết thanh và 17KS nước tiểu, Ức chế các steroid tăng cao sau khi dùng glucocorticoid | Dùng glucocorticoid, Phẫu thuật sửa chữa ở 46,XX DSD |
| P450c11AS | Chậm phát triển, Yếu, Mất muối | Hạ natri máu, tăng kali máu, ↓ Corticosterone, ↓ Aldosterone, ↑ PRA | Thay thế mineralocorticoid, Bổ sung muối |
| P450c17 | 46,XY DSD, Thiểu năng sinh dục, Tăng huyết áp | ↑ DOC, 18-OHDOC, corticosterone, 18-hydroxycorticosterone, Các steroid 17α-hydroxyl hóa thấp và đáp ứng kém với ACTH, Đáp ứng kém với hCG ở 46,XY DSD, Ức chế các steroid thượng thận tăng cao sau khi dùng glucocorticoid, ↑ ACTH và PRA, Hạ kali máu | Dùng glucocorticoid, Phẫu thuật chỉnh sửa bộ phận sinh dục và thay thế steroid sinh dục ở 46,XY DSD phù hợp với giới tính nuôi dưỡng, Thay thế estrogen ở nữ ở >12 tuổi, Thay thế testosterone nếu được nuôi dưỡng là nam (hiếm) |
| POR | 46,XX và 46,XY DSD, Hội chứng Antley-Bixler, Vô sinh ở người lớn | ↑ ACTH, Prog, 17OHP, ↓ DHEA, Andro, T, Điện giải bình thường | Thay thế glucocorticoid và steroid sinh dục, Phẫu thuật chỉnh sửa các dị tật xương |
17OHP, 17-Hydroxyprogesterone; 17KS, 17-ketosteroid; 18-OHDOC, 18-hydroxy deoxycorticosterone; ACTH, hormone vỏ thượng thận (corticotropin); DOC, deoxycorticosterone; DSD, rối loạn phát triển giới tính; hCG, gonadotropin màng đệm người; PRA, hoạt độ renin huyết tương.
Rối loạn tổng hợp và vận chuyển Cholesterol
Ngoài các bước enzyme được trình bày trong Hình 14.3, các rối loạn ở thượng nguồn trong quá trình tổng hợp và vận chuyển cholesterol nội bào có thể làm suy giảm sinh tổng hợp steroid. Thai nhi giai đoạn đầu cần tự tổng hợp cholesterol của mình, với ít hơn được cung cấp qua nhau thai, do đó các rối loạn sinh tổng hợp cholesterol gây ra các hội chứng dị tật bẩm sinh được xác định rõ: hội chứng Smith-Lemli-Opitz (SLOS, thiếu hụt 7-dehydrocholesterol reductase), lathosterolosis (thiếu hụt lathosterol Δ5-desaturase), desmosterolosis (thiếu hụt sterol Δ24-reductase), hội chứng CHILD (rối loạn phức hợp demethyl hóa C-4), và hội chứng Conradi-Hunermann (thiếu hụt sterol isomerase liên kết X). Hậu quả là suy giảm sinh tổng hợp steroid của thai nhi có thể dẫn đến rối loạn phát triển giới tính (DSD) 46,XY, nhưng suy thượng thận không được báo cáo.
Bệnh Wolman và Bệnh ứ đọng Ester Cholesteryl
Hầu hết cholesterol tế bào có nguồn gốc từ việc hấp thu các lipoprotein lưu hành chứa các este cholesterol; các este này được phân cắt thành cholesterol tự do bởi lipase axit lysosome (cholesterol esterase), được mã hóa bởi gen LIPA trên nhiễm sắc thể 10q23.31. Các đột biến của LIPA gây ra bệnh Wolman (WD, bệnh u vàng nguyên phát) và biến thể nhẹ hơn của nó, bệnh ứ đọng este cholesteryl (CESD). Trong WD, các este cholesteryl và triglyceride tích tụ trong gan, lách, các hạch bạch huyết và các mô khác. Ở tuyến thượng thận, không có đủ cholesterol tự do để sinh tổng hợp steroid, gây ra suy thượng thận. Bệnh ít nghiêm trọng hơn so với CAH dạng mỡ về mặt sinh tổng hợp steroid, và bệnh nhân có thể sống sót trong vài tháng sau khi sinh. Tuy nhiên, bệnh ảnh hưởng đến tất cả các tế bào, không chỉ các tế bào sinh steroid, vì tất cả các tế bào phải lưu trữ và sử dụng cholesterol; do đó, rối loạn này không ngừng tiến triển và gây tử vong. Nôn, tiêu chảy mỡ, chậm phát triển, gan lách to, vàng da và thiếu máu là những phát hiện thường gặp khi trình bày, đôi khi bắt đầu trong tuần đầu tiên của cuộc đời, dẫn đến chậm phát triển và suy dinh dưỡng do kém hấp thu. Vôi hóa dưới bao tuyến thượng thận hai bên đặc trưng phác họa tuyến thượng thận có thể được nhìn thấy trên siêu âm trước hoặc sau sinh. Chẩn đoán được xác định bằng chọc hút tủy xương, cho thấy các tế bào bọt chứa các không bào lysosome lớn, căng phồng este cholesterol, và được xác nhận bằng cách tìm thấy hoạt tính cholesterol esterase không có trong nguyên bào sợi, bạch cầu, tế bào tủy xương, hoặc tế bào ối được nuôi cấy (để chẩn đoán trước sinh). Điều trị bằng ghép tủy xương dường như cải thiện diễn biến của bệnh trong khoảng một nửa số trường hợp, nhưng cơ chế của tác dụng này chưa rõ ràng; liệu pháp thay thế enzyme có thể cải thiện sự tiến triển của bệnh. Các phương pháp điều trị hỗ trợ bao gồm thay thế corticosteroid, bổ sung vitamin và khoáng chất. Một cuộc khảo sát cho thấy WD chiếm khoảng 3% các trường hợp suy thượng thận ở trẻ em.
CESD là một dạng WD nhẹ hơn, khởi phát muộn hơn do các đột biến LIPA vẫn giữ được một phần hoạt tính. CESD thường biểu hiện ở giai đoạn cuối thời thơ ấu và thanh thiếu niên với tăng cholesterol máu, xơ vữa động mạch và gan lách to (thâm nhiễm gan với các đại thực bào chứa este cholesteryl); xơ gan có thể dẫn đến giãn tĩnh mạch thực quản. Có thể có khởi phát sớm với xơ gan nặng hoặc khởi phát muộn hơn với bệnh gan tiến triển chậm hơn. Một số bệnh nhân sống đến tuổi trưởng thành.
Bệnh Niemann-Pick Type C
Quá trình xử lý các este cholesterol trong endosome đòi hỏi hoạt động của NPC1 và NPC2, hai protein được đặt tên theo bệnh NPC. NPC là một rối loạn thần kinh hiếm gặp về vận chuyển cholesterol đặc trưng bởi sự tích tụ cholesterol và glycosphingolipid trong endosome, dẫn đến thoái hóa thần kinh tiến triển, thâm nhiễm tế bào đệm mạnh và tử vong. NPC thường biểu hiện ở 2 đến 4 tuổi, với suy giảm trí tuệ nhẹ, mất khả năng nói, liệt nhìn dọc trên nhân, thất điều, loạn trương lực cơ, sa sút trí tuệ, co giật và thiếu hụt ngoại tháp. Một dạng tiến triển nhanh ở trẻ sơ sinh biểu hiện với rối loạn chức năng gan nặng, chậm phát triển tâm thần vận động, liệt nhìn dọc trên nhân, thất điều, co cứng và sa sút trí tuệ; gan lách to và xơ gan ở trẻ em có thể gây tử vong. Tuy nhiên, mặc dù vai trò dường như là trung tâm của các protein NPC trong vận chuyển cholesterol nội bào, suy thượng thận không được báo cáo trong NPC. Do đó, mặc dù có bằng chứng rõ ràng về rối loạn sinh tổng hợp steroid trong não của bệnh nhân NPC, tuyến thượng thận dường như thoát khỏi, cho thấy nó có thể sử dụng các con đường khác để vận chuyển cholesterol nội bào.
Tăng sản thượng thận bẩm sinh dạng mỡ
CAH dạng mỡ cổ điển, rối loạn di truyền nghiêm trọng nhất của sinh tổng hợp steroid, thường biểu hiện ở trẻ sơ sinh với nồng độ thấp hoặc không có của tất cả các steroid, ACTH cơ bản và renin huyết tương cao, không có đáp ứng steroid với điều trị dài hạn bằng liều cao ACTH hoặc gonadotropin màng đệm người (hCG), và tuyến thượng thận to ra rõ rệt chứa đầy cholesterol và các este cholesterol; những phát hiện này cho thấy một tổn thương trong việc chuyển đổi cholesterol thành pregnenolone. Ban đầu người ta cho rằng tổn thương nằm ở một enzyme, và trước khi vai trò của P450scc được hiểu rõ, CAH dạng mỡ đã bị gọi sai là “thiếu hụt 20,22-desmolase.” Tuy nhiên, gen của P450scc là bình thường ở những bệnh nhân này, cũng như mRNA cho adrenodoxin reductase và adrenodoxin; các rối loạn hiếm gặp ở P450scc về mặt lâm sàng không thể phân biệt được với CAH dạng mỡ (xem sau). Hơn nữa, sinh tổng hợp steroid của nhau thai vẫn tồn tại trong CAH dạng mỡ, cho phép thai kỳ đủ tháng bình thường. Những phát hiện này cho thấy tổn thương có thể do một yếu tố liên quan đến việc vận chuyển cholesterol vào ty thể. Một cuộc tìm kiếm dài đã dẫn đến việc phát hiện ra StAR, và vai trò gây bệnh của nó trong CAH dạng mỡ đã xác lập chức năng thiết yếu của StAR. StAR tạo điều kiện cho dòng cholesterol đi vào từ OMM đến màng trong ty thể, nơi nó có thể được chuyển đổi thành pregnenolone bởi P450scc. P450scc là enzyme sinh steroid chậm nhất, nhưng bước giới hạn tốc độ thực sự trong sinh tổng hợp steroid là sự nhập khẩu cholesterol vào ty thể, được tạo điều kiện bởi StAR.
CAH dạng mỡ là thí nghiệm knockout gen StAR của tự nhiên, tiết lộ sinh lý phức tạp của protein StAR. StAR thúc đẩy sinh tổng hợp steroid bằng cách tăng sự di chuyển của cholesterol vào ty thể, nhưng khi không có StAR, các tế bào sinh steroid tạo ra steroid ở khoảng 14% mức độ do StAR gây ra. Việc phát hiện ra sinh tổng hợp steroid độc lập với StAR đã dẫn đến mô hình hai tác động của CAH dạng mỡ (Hình 14.13). Tác động thứ nhất là sự mất mát của chính StAR, dẫn đến mất hầu hết, nhưng không phải tất cả, quá trình sinh tổng hợp steroid, và sự gia tăng bù trừ của ACTH và LH. Các hormone này làm tăng cAMP tế bào, làm tăng sinh tổng hợp các thụ thể LDL, dẫn đến sự hấp thu cholesterol LDL và tổng hợp cholesterol de novo. Khi không có StAR, lượng cholesterol nội bào tăng lên này tích tụ như trong một bệnh lưu trữ, gây ra tác động thứ hai, đó là tổn thương ty thể và tế bào do cholesterol, este cholesterol tích tụ và các sản phẩm tự oxy hóa của chúng.
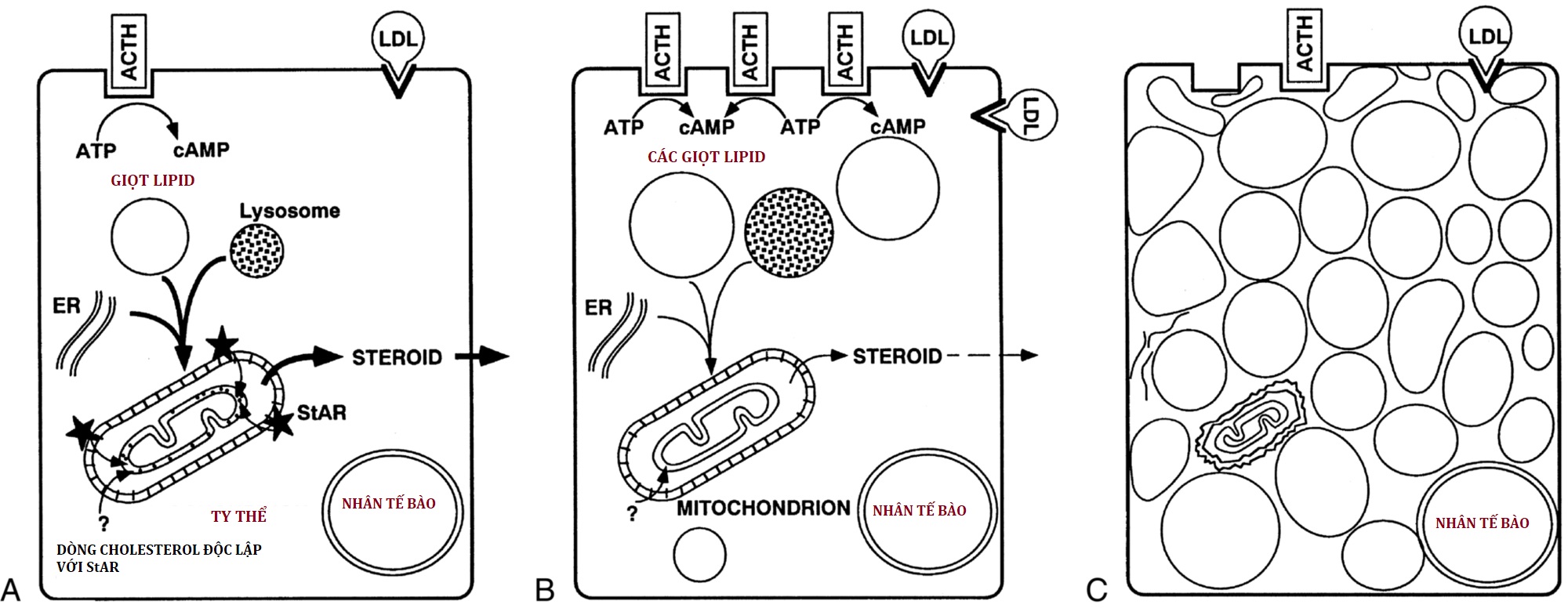
Hình 14.13: Mô hình hai tác động của tăng sản thượng thận bẩm sinh dạng mỡ (CAH). A, Trong một tế bào thượng thận bình thường, cholesterol chủ yếu có nguồn gốc từ lipoprotein mật độ thấp (LDL) bằng cách nội bào qua trung gian thụ thể và được xử lý trong lysosome trước khi vào kho dự trữ của tế bào, nhưng cholesterol cũng có thể được tổng hợp de novo từ acetyl CoA. Cholesterol từ cả hai nguồn được lưu trữ dưới dạng este cholesterol trong các giọt lipid. Cholesterol đến ty thể bằng các quá trình chưa được xác định rõ, sau đó đi từ màng ngoài đến màng trong ty thể bằng cả cơ chế phụ thuộc và độc lập với protein điều hòa cấp tính sinh steroid (StAR). B, Trong giai đoạn đầu của CAH dạng mỡ, sự vắng mặt của StAR làm giảm dòng cholesterol và quá trình sinh steroid, nhưng một số quá trình sinh steroid vẫn tồn tại thông qua con đường độc lập với StAR. Sự giảm tiết cortisol dẫn đến tăng hormone vỏ thượng thận, kích thích thêm việc hấp thu và tổng hợp cholesterol; cholesterol này tích tụ trong các giọt lipid. C, Các giọt lipid tích tụ làm hỏng tế bào, cả thông qua sự phá vỡ vật lý của cấu trúc tế bào và bởi tác động hóa học của các sản phẩm tự oxy hóa, cuối cùng phá hủy tất cả khả năng sinh steroid. Trong buồng trứng, các tế bào nang vẫn không bị kích thích và không bị tổn thương cho đến khi chúng được huy động vào đầu mỗi chu kỳ. Sau đó, chúng có thể sản xuất một lượng nhỏ estradiol, như trong bảng B, dẫn đến nữ hóa và các chu kỳ không rụng trứng ở những người nữ bị ảnh hưởng.
Mô hình hai tác động giải thích những phát hiện lâm sàng bất thường trong CAH dạng mỡ. Trong tinh hoàn của thai nhi, nơi thường tạo ra một lượng lớn testosterone trong đời sống thai nhi, các tế bào Leydig bị phá hủy sớm trong thai kỳ, loại bỏ sinh tổng hợp testosterone, do đó một thai nhi 46,XY bị ảnh hưởng không trải qua quá trình nam hóa bình thường, và được sinh ra với bộ phận sinh dục ngoài của nữ và một túi âm đạo bịt kín. Tuy nhiên, các dẫn xuất ống Wolff được phát triển tốt, cho thấy sự hiện diện của một số quá trình tổng hợp testosterone sớm trong đời sống thai nhi, như dự đoán của mô hình hai tác động. Các tế bào Sertoli không bị tổn thương sản xuất hormone ức chế Müllerian, do đó thai nhi 46,XY có kiểu hình nữ không có cổ tử cung, tử cung hoặc ống dẫn trứng. Vùng bào thai hoạt động sinh steroid của tuyến thượng thận cũng bị ảnh hưởng tương tự, loại bỏ hầu hết sinh tổng hợp DHEA và do đó loại bỏ sản xuất estriol của thai nhi-nhau thai, do đó nồng độ estriol của mẹ và thai nhi ở giữa thai kỳ rất thấp. Vùng xác định của tuyến thượng thận thai nhi, nơi biệt hóa thành lớp cầu và lớp bó, thường sản xuất rất ít aldosterone, và vì chuyển hóa muối và nước của thai nhi được duy trì bởi nhau thai, sự kích thích lớp cầu bởi angiotensin II thường không bắt đầu cho đến khi sinh. Phù hợp với điều này, nhiều trẻ sơ sinh bị CAH dạng mỡ không bị khủng hoảng mất muối cho đến sau vài tuần tuổi, khi sự kích thích mãn tính sau đó dẫn đến tổn thương tế bào.
Mô hình hai tác động cũng giải thích sự nữ hóa tự phát của các cá thể nữ 46,XX bị ảnh hưởng được điều trị trong giai đoạn sơ sinh và đến tuổi vị thành niên. Buồng trứng của thai nhi tạo ra rất ít hoặc không có steroid và không chứa các enzyme sinh steroid sau ba tháng đầu thai kỳ; do đó, buồng trứng phần lớn không bị tổn thương cho đến khi nó được kích thích bởi các gonadotropin vào thời điểm dậy thì, khi đó nó tạo ra một số estrogen bằng sinh tổng hợp steroid độc lập với StAR. Sự kích thích liên tục dẫn đến tích tụ cholesterol và tổn thương tế bào, do đó sinh tổng hợp progesterone trong phần sau của chu kỳ bị suy giảm. Bởi vì sự kích thích gonadotropin chỉ chiêu mộ các nang trứng riêng lẻ và không thúc đẩy sinh tổng hợp steroid trong toàn bộ buồng trứng, hầu hết các nang trứng vẫn không bị tổn thương và có sẵn cho các chu kỳ trong tương lai. Tính chu kỳ được xác định bởi trục hạ đồi-tuyến yên, và vẫn bình thường. Với mỗi chu kỳ mới, một nang trứng mới được chiêu mộ và nhiều estradiol hơn được sản xuất bằng sinh tổng hợp steroid độc lập với StAR. Mặc dù tổng thể sinh tổng hợp steroid của buồng trứng bị suy giảm, đủ estrogen được sản xuất để gây ra sự phát triển vú, nữ hóa chung, chảy máu do ngưng estrogen hàng tháng và chảy máu âm đạo theo chu kỳ. Tuy nhiên, tổng hợp progesterone trong nửa sau của chu kỳ bị xáo trộn bởi các este cholesterol tích tụ do đó các chu kỳ không có rụng trứng và có thể phát triển các nang buồng trứng. Các phép đo estradiol, progesterone và gonadotropin trong suốt chu kỳ ở những phụ nữ trưởng thành bị CAH dạng mỡ đã xác nhận mô hình này, cũng như các thí nghiệm với chuột knockout StAR. Do đó, việc kiểm tra bệnh nhân bị CAH dạng mỡ đã làm sáng tỏ sinh lý của protein StAR trong mỗi mô sinh steroid.
Các nghiên cứu trên hơn 100 bệnh nhân bị CAH dạng mỡ đã xác định được hơn 40 đột biến StAR. CAH dạng mỡ là dạng CAH phổ biến thứ hai ở Nhật Bản và Hàn Quốc, nơi đột biến Q258X chiếm ưu thế, với tần suất người mang gen khoảng 1 trên 300, do đó cứ 250.000 đến 300.000 trẻ sơ sinh ở các quốc gia này thì có một trẻ bị ảnh hưởng. CAH dạng mỡ cũng phổ biến ở các quần thể Ả Rập, với R182L được tìm thấy ở người Palestine, R188C ở miền đông Ả Rập Saudi, và R182H và c.201-202delCT ở các quần thể Ả Rập khác; ở Thụy Sĩ, L260P là một đột biến tái phát.
Biểu hiện lâm sàng điển hình trong CAH dạng mỡ là một trẻ sơ sinh có bộ phận sinh dục ngoài trông bình thường của nữ, bị chậm phát triển và mất muối trong những tuần đầu đời. Các biểu hiện lâm sàng khác bao gồm hội chứng đột tử ở trẻ sơ sinh (SIDS) và khởi phát muộn của mất muối vào khoảng 1 tuổi. CAH dạng mỡ không cổ điển do các đột biến giữ lại khoảng 20% đến 25% hoạt tính StAR bình thường đã được tìm thấy ở trẻ 46,XY có bộ phận sinh dục ngoài của nam trông bình thường, bị suy thượng thận có triệu chứng ở vài tuổi. Người lớn bị CAH dạng mỡ không cổ điển được ghi nhận ngày càng thường xuyên và thường bị nhầm lẫn là bị “thiếu hụt glucocorticoid gia đình,” một thuật ngữ chung đề cập đến các rối loạn hoạt động của ACTH; nhiều bệnh nhân trong số này có suy giảm bài tiết mineralocorticoid nhẹ, với điện giải huyết thanh bình thường và renin huyết tương tăng cao, và một số có thể bị suy sinh dục tăng gonadotropin nhẹ. Hầu hết các bệnh nhân này mang cùng một đột biến R188C được mô tả lần đầu tiên ở trẻ em. Do đó, phổ biểu hiện lâm sàng của tăng sản thượng thận dạng mỡ bẩm sinh rộng hơn đáng kể so với nhận thức ban đầu.
Điều trị CAH dạng mỡ rất đơn giản nếu chẩn đoán được thực hiện. Thay thế sinh lý bằng glucocorticoid, mineralocorticoid và muối, trong giai đoạn sơ sinh, sẽ cho phép sống sót đến tuổi trưởng thành. Liều glucocorticoid tương tự như trong bệnh Addison và thấp hơn trong các bệnh tăng sản thượng thận nam hóa vì không cần phải ức chế quá mức sản xuất androgen tuyến thượng thận dư thừa, do đó sự tăng trưởng ở những bệnh nhân này sẽ bình thường. Liều mineralocorticoid không tăng theo tuổi hoặc kích thước, vì trẻ em ngày càng nhạy cảm hơn với mineralocorticoid theo tuổi; bổ sung muối được giảm dần sau khoảng 1 năm. Trẻ sơ sinh 46,XY bị ảnh hưởng nặng có bộ phận sinh dục ngoài của nữ bình thường và có thể được khuyên cắt bỏ tinh hoàn sau này trong cuộc đời và được nuôi dưỡng như nữ, với liệu pháp thay thế hormone sinh dục, bắt đầu ở tuổi dậy thì. Các cá thể nữ 46,XX bị ảnh hưởng thường có nữ hóa dậy thì tự phát, nhưng các chu kỳ không rụng trứng và vô kinh thứ phát sớm, sau đó cũng cần liệu pháp thay thế hormone sinh dục. Thai kỳ đã được gây ra trong hai báo cáo.
Các rối loạn giống Tăng sản thượng thận bẩm sinh dạng mỡ: Thiếu hụt P450scc và Thiếu hụt Yếu tố sinh steroid 1
Các đột biến ở các gen khác có thể tạo ra một kiểu hình lâm sàng về cơ bản không thể phân biệt được với kiểu hình do đột biến StAR gây ra, nhưng các rối loạn này không nên được gọi là CAH dạng mỡ. Bắt đầu từ năm 2001, một số bệnh nhân đã được mô tả có đột biến ở P450scc. Các phát hiện lâm sàng và hormone của họ không thể phân biệt được với những người có đột biến StAR, do đó việc giải trình tự gen là cách duy nhất để phân biệt các rối loạn này; tuy nhiên, cho đến nay không có bệnh nhân nào bị đột biến P450scc có tăng sản thượng thận thường thấy trong CAH dạng mỡ. Có vẻ hợp lý rằng việc loại bỏ tất cả hoạt tính P450scc sẽ không tương thích với thai kỳ đủ tháng, vì nhau thai, một mô của thai nhi, phải sản xuất progesterone trong nửa sau của thai kỳ để ức chế các cơn co tử cung của mẹ, do đó ngăn ngừa sẩy thai. Tuy nhiên, khoảng 40 trường hợp như vậy đã được báo cáo cho đến năm 2018, nhiều trường hợp từ đông nam Thổ Nhĩ Kỳ. Các thai nhi bị đột biến P450scc có lẽ đến đủ tháng khi có sự duy trì kéo dài của thể vàng thai kỳ của mẹ, nơi thường thoái triển trong ba tháng giữa thai kỳ, nhưng điều này chưa được điều tra trực tiếp. Thiếu hụt P450scc không cổ điển về mặt lâm sàng và hormone không thể phân biệt được với CAH dạng mỡ không cổ điển đã được báo cáo ở những bệnh nhân có đột biến P450scc giữ lại 10% đến 20% hoạt tính của loại hoang dã.
Hơn 50 bệnh nhân cũng đã được mô tả mang đột biến trong gen của yếu tố sinh steroid 1 (SF1), một yếu tố phiên mã cần thiết cho sự biểu hiện của các gen cho các enzyme sinh steroid ở tuyến thượng thận và tuyến sinh dục, nhưng không phải ở nhau thai. Có sự biến đổi kiểu hình rộng rãi ở những bệnh nhân thiếu SF1; một số là 46,XY với kiểu hình nữ và suy thượng thận, do đó giống CAH dạng mỡ, nhưng trong hầu hết các trường hợp, kiểu hình tuyến sinh dục chiếm ưu thế và có rất ít hoặc không có suy giảm sinh tổng hợp steroid tuyến thượng thận. Các đột biến SF1 có thể được tìm thấy ở khoảng 10% bệnh nhân 46,XY bị rối loạn phát triển giới tính. Các tế bào Leydig có thể có tích tụ lipid và thoái hóa tiến triển, tương tự như các phát hiện trong CAH dạng mỡ.
Thiếu hụt 3β-Hydroxysteroid Dehydrogenase
Thiếu hụt 3βHSD là một nguyên nhân hiếm gặp, có khả năng gây tử vong của thiếu hụt glucocorticoid, mineralocorticoid và steroid sinh dục. Có hai gen chức năng của người cho 3βHSD: gen loại 1 (HSD3B1) được biểu hiện ở nhau thai và các mô ngoại vi, và gen loại 2 (HSD3B2) được biểu hiện ở tuyến thượng thận và tuyến sinh dục. Tất cả các đột biến gây ra thiếu hụt 3βHSD đều được tìm thấy trong gen loại 2; các đột biến chưa bao giờ được tìm thấy ở 3βHSD1, có lẽ vì điều này sẽ ngăn cản sinh tổng hợp progesterone của nhau thai, dẫn đến sẩy thai tự nhiên trong ba tháng đầu thai kỳ. Thiếu hụt 3βHSD gây ra DSD ở cả hai giới: các cá thể nữ về mặt di truyền có phì đại âm vật và nam hóa nhẹ vì tuyến thượng thận của thai nhi sản xuất quá mức một lượng lớn DHEA, một phần nhỏ trong số đó được chuyển đổi thành testosterone bởi 3βHSD1; các cá thể nam về mặt di truyền tổng hợp một số androgen bằng cách chuyển đổi ngoại vi DHEA của tuyến thượng thận và tinh hoàn, nhưng nồng độ không đủ để phát triển hoàn toàn bộ phận sinh dục nam.
Người ta có thể mong đợi rằng trẻ sơ sinh bị thiếu hụt 3βHSD2 sẽ có nồng độ 17OHP thấp, nhưng một số trẻ sơ sinh bị thiếu hụt 3βHSD có nồng độ 17OHP huyết thanh rất cao, gần bằng nồng độ thấy ở những bệnh nhân bị 21OHD cổ điển, và có kết quả sàng lọc sơ sinh dương tính với 21OHD. Trong khi 3βHSD2 có Km cao khoảng 5,5 μM, Km của 3βHSD1 chỉ khoảng 0,2 μM, cho phép 3βHSD1 của gan tác động lên nồng độ 17-Preg lưu hành cao ở những bệnh nhân bị thiếu hụt 3βHSD2, tạo ra 17OHP. 17OHP này không được tuyến thượng thận hấp thu hiệu quả để chuyển đổi thành cortisol sau đó vì nồng độ lưu hành thấp hơn Km của P450c17 (~ 0,8 μM 17OHP, hoặc ~ 40.000 ng/dL). Tỷ lệ của các hợp chất Δ5 so với Δ4 vẫn cao, phù hợp với sự thiếu hụt 3βHSD của tuyến thượng thận và tuyến sinh dục. Do đó, xét nghiệm chẩn đoán chính trong thiếu hụt 3βHSD là tiêm ACTH tĩnh mạch, với việc đo ba hợp chất Δ5 (Preg, 17-Preg, DHEA), và các hợp chất Δ4 tương ứng (progesterone, 17OHP, androstenedione). Không giống như trường hợp của 21OHD, nơi những người mang gen dị hợp tử có thể được chẩn đoán bằng đáp ứng của 17OHP với ACTH, các đáp ứng steroid với ACTH không thể được sử dụng để xác định những người mang gen thiếu hụt 3βHSD.
Các khiếm khuyết “nhẹ” hoặc “một phần” của hoạt tính 3βHSD tuyến thượng thận đã được báo cáo trên cơ sở tỷ lệ steroid Δ5 so với steroid Δ4, sau một nghiệm pháp ACTH vượt quá 2 hoặc 3 độ lệch chuẩn (SD) so với giá trị trung bình; những bệnh nhân này thường là các bé gái bị thượng thận hóa sớm, hoặc phụ nữ trẻ có tiền sử thượng thận hóa sớm và phàn nàn về rậm lông, nam hóa và thiểu kinh. Tuy nhiên, những bệnh nhân này không bị thiếu hụt 3βHSD vì gen HSD3B2 của họ bình thường. Bệnh nhân có đột biến 3βHSD2 nhẹ có tỷ lệ steroid Δ5 so với Δ4 vượt quá 8 SD so với giá trị trung bình. Do đó, tỷ lệ steroid Δ5 so với Δ4 không đáng tin cậy và không thể được sử dụng để chẩn đoán thiếu hụt 3βHSD; chẩn đoán đòi hỏi một nghiệm pháp ACTH với sự gia tăng các steroid Δ5 (thường là sự gia tăng 17-Preg lên > 3000 ng/dL). Cơ sở của tỷ lệ steroid Δ5 so với Δ4 tăng nhẹ, ở những cá thể rậm lông có gen HSD3B bình thường, vẫn chưa được biết. Ở phụ nữ trưởng thành, tình trạng rậm lông có thể được cải thiện và kinh nguyệt đều đặn có thể được phục hồi bằng cách ức chế ACTH với 0,25 mg dexamethasone uống mỗi ngày, nhưng việc điều trị như vậy bị chống chỉ định ở các bé gái chưa đạt đến chiều cao trưởng thành cuối cùng.
Thiếu hụt 17α-Hydroxylase/17,20-Lyase
P450c17 xúc tác cho cả hoạt động 17α-hydroxylase và 17,20-lyase. Thiếu hụt 17-hydroxylase đã được hiểu rõ về mặt lâm sàng và di truyền, và phổ biến ở Brazil và Trung Quốc. Hoạt tính 17α-hydroxylase bị thiếu hụt dẫn đến giảm tổng hợp cortisol, sản xuất quá mức ACTH, và kích thích các bước trước P450c17. Những bệnh nhân này có thể có các triệu chứng nhẹ của thiếu hụt glucocorticoid, nhưng điều này không đe dọa tính mạng, vì sự thiếu hụt cortisol được bù đắp bằng việc sản xuất quá mức corticosterone, chất có hoạt tính glucocorticoid. Điều này tương tự như tình hình ở loài gặm nhấm, tuyến thượng thận của chúng thiếu P450c17 và do đó sản xuất corticosterone làm glucocorticoid của chúng. Bệnh nhân bị ảnh hưởng cũng thường sản xuất quá mức DOC ở lớp bó, gây giữ natri, tăng huyết áp và hạ kali máu, đồng thời ức chế hoạt độ renin huyết tương và bài tiết aldosterone từ lớp cầu, mặc dù sự ức chế aldosterone khá thay đổi. Khi thiếu hụt P450c17 được điều trị bằng glucocorticoid, bài tiết DOC bị ức chế và hoạt độ renin huyết tương và nồng độ aldosterone tăng lên mức bình thường.
Sự vắng mặt của hoạt tính 17α-hydroxylase và 17,20-lyase trong thiếu hụt P450c17 hoàn toàn ngăn cản việc tổng hợp các steroid sinh dục của tuyến thượng thận và tuyến sinh dục. Kết quả là, các cá thể nữ bị ảnh hưởng có kiểu hình bình thường, nhưng không trải qua giai đoạn thượng thận hóa và dậy thì, và các cá thể nam về mặt di truyền có sự phát triển không có hoặc không hoàn toàn của bộ phận sinh dục ngoài (lưỡng tính giả nam; DSD 46,XY). Biểu hiện cổ điển là của một nữ thiếu niên bị nhi hóa sinh dục và tăng huyết áp. Chẩn đoán được thực hiện bằng cách tìm thấy các steroid C21 và C19 được 17-hydroxyl hóa trong huyết tương thấp hoặc không có, đáp ứng kém với kích thích bằng ACTH, và DOC, corticosterone, và 18OH-corticosterone tăng cao, đáp ứng quá mức với ACTH và có thể bị ức chế bằng glucocorticoid. Liệu pháp thay thế glucocorticoid sinh lý sẽ ức chế tăng huyết áp do mineralocorticoid; liệu pháp thay thế steroid sinh dục phù hợp với giới tính nuôi dưỡng được bắt đầu ở tuổi vị thành niên.
Hơn 100 đột biến riêng biệt đã được tìm thấy trong gen CYP17A1 trên nhiễm sắc thể 10q24.3, gây ra thiếu hụt 17α-hydroxylase. Bốn đột biến xuất hiện tái phát: một sự nhân đôi của bốn nucleotide gây ra dịch khung được tìm thấy ở con cháu của người Friesland Hà Lan; 7-489 phổ biến ở châu Á; một đột biến dịch khung phổ biến ở miền Bắc Trung Quốc và Hàn Quốc; và các đột biến phổ biến W406R và R362C, được tìm thấy ở người Brazil gốc Tây Ban Nha và Bồ Đào Nha, tương ứng. Hầu hết các đột biến P450c17 được báo cáo làm mất hoàn toàn hoạt tính, trong khi một số khác ảnh hưởng một phần đến cả hai hoạt tính, thường là tương đương nhau.
Thiếu hụt 17,20 Lyase: P450c17, Cytochrome b5 và các yếu tố khác
Các báo cáo ban đầu về thiếu hụt 17,20-lyase chọn lọc ban đầu đã dẫn đến kết luận không chính xác rằng 17α-hydroxylase và 17,20-lyase là các enzyme riêng biệt. Thiếu hụt 17,20 lyase riêng lẻ có thể do đột biến ở một số gen, nhưng tất cả đều liên quan đến việc vận chuyển điện tử đến P450c17 từ POR. Các trường hợp thiếu hụt 17,20 lyase riêng lẻ được chứng minh về mặt di truyền đầu tiên là hai bệnh nhân có bộ phận sinh dục không rõ ràng, bài tiết bình thường 17OHCS (17-hydroxycorticosteroid), và sản xuất C19 steroid giảm rõ rệt. Những bệnh nhân này đồng hợp tử cho các đột biến P450c17 R347H hoặc R358Q; cả hai đột biến đều thay đổi sự phân bố điện tích bề mặt ở vị trí gắn kết đối tác redox của P450c17, và các thí nghiệm enzyme và sinh học tế bào cho thấy sự mất hoạt tính lyase là do suy giảm vận chuyển điện tử. Cũng như việc thay đổi sự phân bố điện tích ở vị trí gắn kết đối tác redox của P450c17 từ dương sang âm với các đột biến R347H hoặc R358Q gây ra hoạt tính 17,20 lyase riêng lẻ, việc thay đổi điện tích tĩnh điện từ âm sang dương với đột biến G539R trong miền cho điện tử của POR cũng được tìm thấy ở những bệnh nhân bị thiếu hụt 17,20 lyase riêng lẻ. Do đó, các đột biến ở vị trí gắn kết đối tác redox của P450c17 hoặc ở miền cho điện tử của POR có thể gây ra thiếu hụt 17,20 lyase riêng lẻ, cho thấy thiếu hụt 17,20 lyase riêng lẻ là một rối loạn vận chuyển điện tử đến P450c17.
Ngoài các đột biến hiếm gặp, đặc hiệu ở P450c17 và POR, các đột biến ở cytochrome b5, một hemoprotein nhỏ kích thích hoạt tính 17,20 lyase khoảng 10 lần, cũng sẽ gây ra thiếu hụt 17,20 lyase. Sự biểu hiện của b5 ở tuyến thượng thận là đặc hiệu cho lớp lưới và trùng với thời điểm bắt đầu giai đoạn thượng thận hóa. Báo cáo đầu tiên về thiếu hụt b5 là ở một nam giới bị DSD và methemoglobin huyết không được đánh giá về hormone. Methemoglobin huyết là một hậu quả được mong đợi của thiếu hụt b5 vì sự khử methemoglobin là vai trò sinh lý chính của b5, và nguyên nhân thông thường của methemoglobin huyết là thiếu hụt cytochrome b5 reductase. Nồng độ methemoglobin tăng cao, không có methemoglobin huyết lâm sàng, có thể phổ biến hơn. Hoạt tính 17,20-lyase cũng có thể được tăng cường bởi sự phosphoryl hóa serine/threonine của P450c17, được xúc tác bởi p38α (MAPK14), nhưng sự gián đoạn của hệ thống này chưa (chưa) được báo cáo là gây ra thiếu hụt 17,20 lyase lâm sàng. Điều thú vị là, những bệnh nhân đầu tiên được báo cáo là bị thiếu hụt 17,20 lyase sau đó đã được phát hiện có đột biến ở AKR1C2 và AKR1C4, hai enzyme xúc tác cho quá trình khử 3α trong con đường cửa sau của tổng hợp androgen. Con đường này tham gia vào sự nam hóa thấy trong 21OHD, nhưng không có vai trò trong hoạt tính 17,20 lyase. Những nguyên nhân di truyền đa dạng này minh họa rằng thiếu hụt 17,20 lyase là một hội chứng chứ không phải là một bệnh cụ thể.
Thiếu hụt P450 Oxidoreductase
Thiếu hụt POR là một dạng CAH mới được công nhận. POR là protein 2-flavin vận chuyển điện tử từ NADPH đến tất cả 50 dạng cytochrome P450 ở vi thể, bao gồm P450c17, P450c21 và P450aro, cũng như các enzyme P450 chuyển hóa thuốc của gan (xem Hình 14.5). Vì POR tham gia vào nhiều chức năng, đột biến của nó có thể được dự kiến sẽ tạo ra một kiểu hình rất nặng, và chuột thiếu POR chết trong quá trình phát triển của thai nhi, nhưng nhiều bệnh nhân bị thiếu hụt POR đã được báo cáo từ năm 2004. Một loạt các đột biến POR đã được mô tả, ảnh hưởng đến các enzyme P450 khác nhau ở các mức độ khác nhau, dường như giải thích cho sự biến đổi lớn trong các phát hiện lâm sàng và hormone trong thiếu hụt POR. Các steroid trong huyết thanh và nước tiểu cho thấy các khiếm khuyết ở P450c17, với các khiếm khuyết ở P450c21 và P450aro biến đổi nhiều hơn; các phát hiện lâm sàng thay đổi từ trẻ sơ sinh bị ảnh hưởng nặng với bộ phận sinh dục không rõ ràng, thiếu hụt cortisol, và hội chứng dị tật xương Antley-Bixler (ABS) đến phụ nữ bị ảnh hưởng nhẹ dường như có một dạng hội chứng buồng trứng đa nang, hoặc nam giới bị ảnh hưởng nhẹ, với suy sinh dục. ABS được đặc trưng bởi dính khớp sọ, đầu ngắn, dính khớp trụ-quay hoặc trụ-cánh tay, xương đùi cong, ngón nhện, thiểu sản mặt giữa, lồi mắt, và hẹp lỗ mũi sau. Khi ABS được thấy kết hợp với các steroid bất thường và bộ phận sinh dục không rõ ràng ở cả hai giới, nguyên nhân là một đột biến lặn trên nhiễm sắc thể thường ở POR; Ngược lại, khi ABS được thấy mà không có tổn thương trong sinh tổng hợp steroid hoặc phát triển bộ phận sinh dục, nguyên nhân là một đột biến trội trên nhiễm sắc thể thường, tăng chức năng ở thụ thể FGF 2. Do đó, thuật ngữ “hội chứng Antley-Bixler” nên được dành riêng cho việc mô tả kiểu hình của các dị tật xương, và không nên được đánh đồng với thiếu hụt POR, có thể có hoặc không liên quan đến ABS. Kiểu hình xương ABS có lẽ là kết quả của hoạt động suy giảm của CYP26B1, enzyme vi thể phụ thuộc POR phân hủy axit retinoic. Các nghiên cứu về hai gia đình có đột biến CYP26B1, và tái tạo các đột biến như vậy ở chuột biến đổi gen và cá ngựa vằn cung cấp bằng chứng mạnh mẽ rằng axit retinoic phải được phân hủy tại chỗ ở các vị trí phôi thai thường hình thành các khớp xương và đường khớp sọ; sự can thiệp vào hoạt động này trong thiếu hụt POR dường như là cơ chế chính giải thích cho kiểu hình xương. Các cơ chế khác, bao gồm tín hiệu khiếm khuyết bởi các protein hedgehog thứ phát sau một khiếm khuyết liên quan đến POR trong tổng hợp cholesterol, cũng có thể đóng một vai trò.
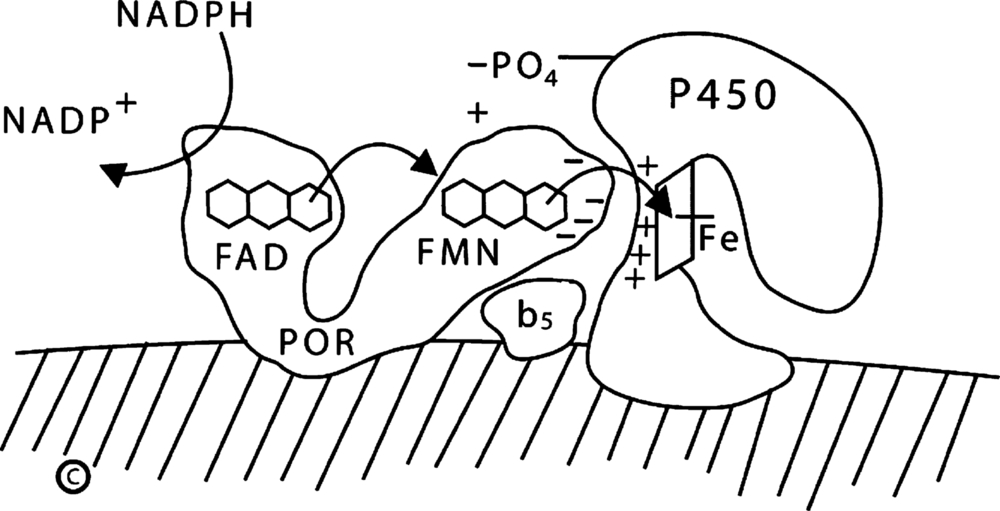
Hình 14.5: Vận chuyển điện tử đến các dạng cytochrome P450 ở microsome. Nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) tương tác với P450 oxidoreductase (POR), liên kết với lưới nội chất, và nhường một cặp điện tử (e-), được nhận bởi phần flavin adenine dinucleotide (FAD). Việc nhận điện tử gây ra một sự thay đổi cấu dạng, cho phép các vòng isoalloxazine của các phần FAD và flavin mononucleotide (FMN) đến gần nhau, do đó các điện tử đi từ FAD đến FMN. Sau một sự thay đổi cấu dạng khác đưa protein trở lại hướng ban đầu, miền FMN của POR tương tác với vị trí liên kết đối tác redox của P450. Các điện tử từ miền FMN của POR đến nhóm heme để làm trung gian cho quá trình xúc tác. Sự tương tác của POR và P450 được điều phối bởi các gốc axit mang điện tích âm trên bề mặt của miền FMN của POR, và các gốc bazơ mang điện tích dương trong vị trí liên kết đối tác redox lõm của P450. Vị trí hoạt động chứa steroid nằm ở phía sắt của vòng heme (Fe) đối diện với vị trí liên kết đối tác redox. Trong trường hợp của P450c17 ở người, sự tương tác này được tạo điều kiện bởi tác động dị lập thể của cytochrome b5, và bởi sự phosphoryl hóa serine của P450c17.
Ở một số bệnh nhân thiếu POR, sự suy giảm hoạt động của P450c21 có thể tạo ra mức 17OHP được phát hiện bằng sàng lọc sơ sinh cho 21OHD. Bệnh nhân thiếu POR thường có điện giải và chức năng mineralocorticoid bình thường, mức cortisol gần như bình thường đáp ứng kém với kích thích bằng ACTH, nồng độ 17OHP cao đáp ứng thay đổi với ACTH, và mức C19 tiền chất của steroid sinh dục thấp. Một đặc điểm đáng chú ý của thiếu hụt POR là có sự không rõ ràng về bộ phận sinh dục ở cả hai giới; nữ có thể bị nam hóa và nam có thể kém phát triển, mặc dù có sự thay đổi đáng kể giữa các cá nhân. Bởi vì hoạt tính 17,20 lyase của P450c17 đặc biệt nhạy cảm với các nhiễu loạn trong vận chuyển điện tử, các khiếm khuyết trong sinh tổng hợp steroid của tinh hoàn thai nhi dẫn đến bộ phận sinh dục ngoài phát triển không hoàn toàn ở nam 46,XY là kết quả được dự đoán. Ngược lại, sự nam hóa một phần thấy ở các cá thể nữ 46,XX dường như là kết quả của hai nguyên nhân. Thứ nhất, aromatase của nhau thai (P450aro) cần POR. Phụ nữ mang thai mang thai nhi có đột biến POR R457H (nhưng không phải POR A287P) có thể bị nam hóa trong thai kỳ, tương tự như những phụ nữ mang thai nhi bị thiếu hụt P450aro. Thai nhi thường thải một lượng lớn steroid C19 của tuyến thượng thận bằng cách bài tiết chúng qua nhau thai, nơi thơm hóa chúng thành các estrogen mẹ của thai kỳ (xem Hình 14.7). Một khiếm khuyết trong hoạt động aromatase của nhau thai này, do đột biến của POR hoặc chính P450aro, sẽ cho phép một lượng lớn steroid C19 của thai nhi đi vào và nam hóa người mẹ. Điều này được chứng minh bằng các giá trị estriol thấp thấy ở những phụ nữ mang thai nhi có một số đột biến POR nhất định. Thứ hai, phân tích các steroid trong nước tiểu từ các bệnh nhân thiếu POR cho thấy rằng con đường cửa sau thay thế của sản xuất androgen (xem Hình 14.6) cũng góp phần vào sự nam hóa trước sinh của các cá thể nữ bị ảnh hưởng.
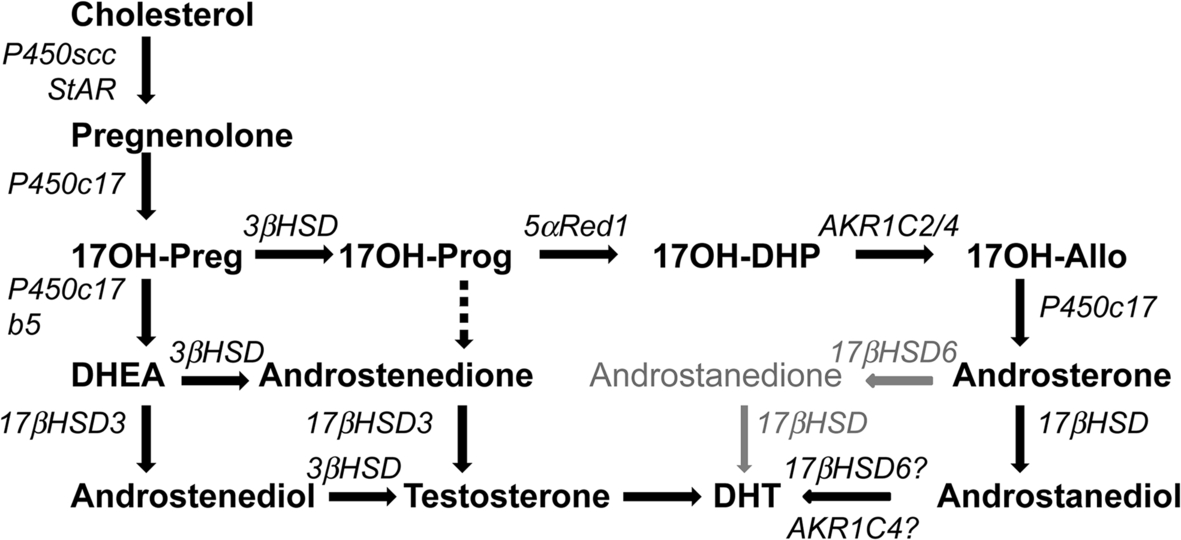
Hình 14.6: Các con đường tạo androgen trong tăng sản thượng thận bẩm sinh (CAH). Khi không có hoạt tính P450c21, tuyến thượng thận có thể sản xuất androgen theo ba con đường. Thứ nhất, con đường từ cholesterol đến dehydroepiandrosterone (DHEA) vẫn còn nguyên vẹn trong thiếu hụt 21-hydroxylase, và việc tăng sản xuất DHEA sẽ dẫn đến một phần DHEA được chuyển đổi thành androstenedione và sau đó là testosterone. Thứ hai, mặc dù một lượng tối thiểu 17α-hydroxyprogesterone (17OHP) được chuyển đổi thành androstenedione trong tuyến thượng thận bình thường, lượng lớn 17OHP được sản xuất trong CAH cho phép một phần 17OHP được chuyển đổi thành androstenedione và sau đó là testosterone. Thứ ba, cái gọi là “con đường cửa sau” phụ thuộc vào sự khử 5α và 3α của 17OHP thành 17OH-allopregnenolone. Steroid này dễ dàng được chuyển đổi thành androstanediol, sau đó có thể được oxy hóa thành dihydrotestosterone (DHT) bởi một enzyme 3-hydroxysteroid dehydrogenase (HSD), AKR1C2. Các phân tích khối phổ của các steroid trong nước tiểu người chỉ ra rằng con đường này là một yếu tố đóng góp chính trong CAH.
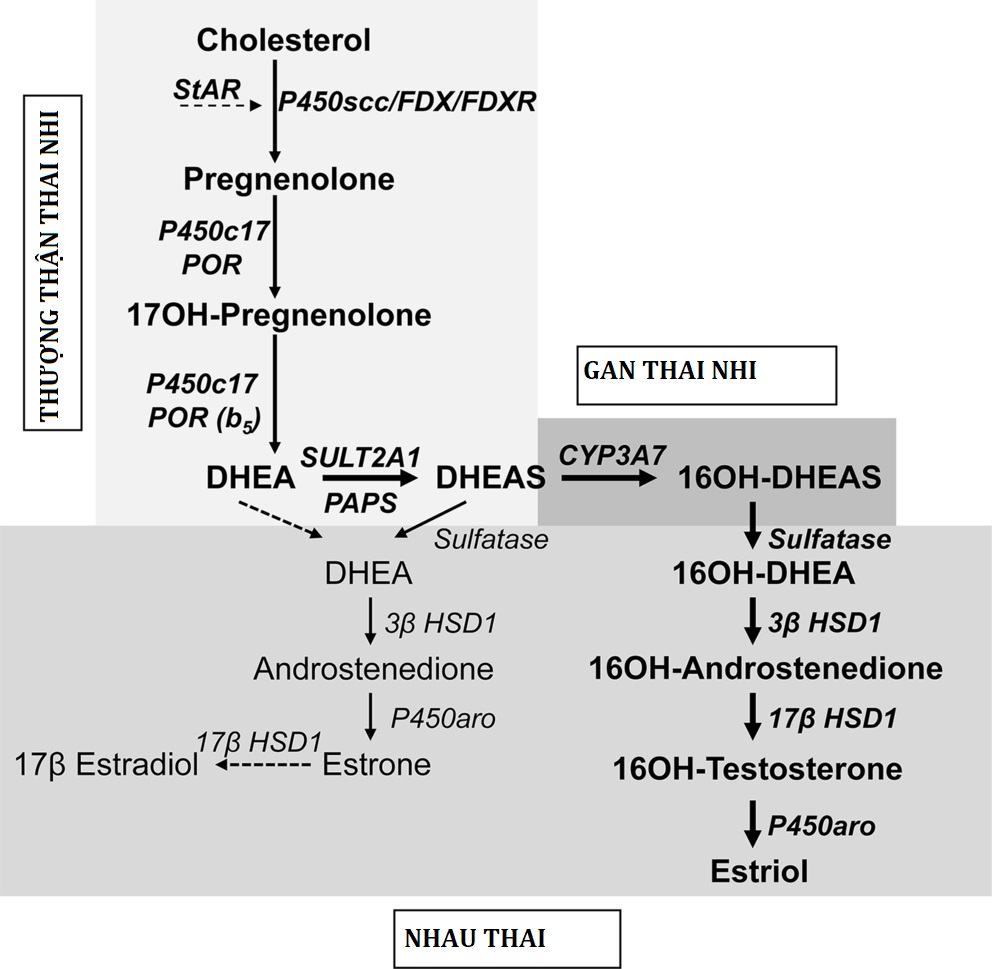
Hình 14.7: Tổng hợp steroid bởi hệ thống thai-nhau thai. Tuyến thượng thận của thai nhi có hoạt tính 3β-hydroxysteroid dehydrogenase (3βHSD) ở mức tối thiểu, do đó con đường từ cholesterol đến dehydroepiandrosterone (DHEA) chiếm ưu thế. Hầu hết DHEA được chuyển đổi thành DHEAS bởi sulfotransferase SULT2A1 và sau đó được 16α-hydroxyl hóa bởi CYP3A7 trong gan của thai nhi. 16α-hydroxy DHEAS đến nhau thai, nơi tác động tuần tự của steroid sulfatase, 3βHSD1, 17βHSD1 và aromatase (P450aro) tạo ra estriol, sản phẩm steroid chính của nhau thai. Một lượng nhỏ DHEA đến nhau thai mà không bị 16α-hydroxyl hóa, nơi nó có thể được chuyển đổi thành estrone bởi 3βHSD1 và P450aro; một lượng nhỏ estrone cũng có thể được chuyển đổi thành estradiol. Khoảng 80% estrogen của nhau thai là estriol, 15% là estrone, và chỉ 5% là estradiol.
Bởi vì POR cần thiết cho các enzyme cytochrome P450 chuyển hóa thuốc của gan, hợp lý khi mong đợi sự suy giảm chuyển hóa thuốc ở những bệnh nhân thiếu POR. Mặc dù chuột biến đổi gen có khiếm khuyết POR đặc hiệu ở gan chuyển hóa thuốc kém và tích tụ lipid gan, các vấn đề tương tự vẫn chưa được mô tả ở những bệnh nhân thiếu POR. Nhiều nghiên cứu về các enzyme chuyển hóa thuốc trong ống nghiệm cho thấy sự suy giảm lớn bởi các đột biến POR; tuy nhiên, chỉ có một nghiên cứu đã tìm thấy một tác động như vậy ở một bệnh nhân thiếu POR. Còn nhiều điều cần tìm hiểu về thiếu hụt POR.
Tỷ lệ mắc thiếu hụt POR thay đổi giữa các nhóm dân tộc. Hai đột biến đặc biệt phổ biến: A287P, đột biến chiếm ưu thế ở bệnh nhân gốc châu Âu, và R457H, đột biến chiếm ưu thế ở bệnh nhân gốc Nhật Bản. Kiểu hình lâm sàng ở nữ bị thiếu hụt POR phụ thuộc vào đột biến POR của họ. POR A287P phá vỡ hoạt động của P450c17 nhưng không phải P450c21, và R457H phá vỡ hoạt động của P450aro nhưng A287P thì không. Hoạt động P450aro của nhau thai bị khiếm khuyết có thể cho phép các tiền chất androgen của thai nhi nam hóa người mẹ, do đó một phụ nữ mang thai nhi có POR R457H (nhưng không phải A287P) có thể bị nam hóa trong thai kỳ; những thai kỳ như vậy được đặc trưng bởi nồng độ estriol của mẹ thấp. Biến thể đa hình A503V, ảnh hưởng nhẹ đến nhiều hoạt động P450, được tìm thấy ở khoảng 28% các alen POR, thay đổi từ khoảng 19% ở người Mỹ gốc Phi đến khoảng 37% ở người Mỹ gốc Hoa.
Điều trị thiếu hụt POR đòi hỏi sự quản lý đa chuyên khoa về dính khớp sọ, các vấn đề chỉnh hình, và DSD, bao gồm liệu pháp thay thế hormone sinh dục, bắt đầu ở tuổi dậy thì ở cả hai giới. Một số bệnh nhân có thể được hưởng lợi từ liệu pháp thay thế glucocorticoid liều thấp, đặc biệt là trong các bệnh nặng; điều này nên được xác định riêng lẻ bằng cách đánh giá đáp ứng của cortisol với ACTH.
Thiếu hụt 21-Hydroxylase
21OHD, do đột biến trong gen CYP21A2 mã hóa P450c21 của tuyến thượng thận, là một trong những lỗi chuyển hóa bẩm sinh phổ biến nhất, và chiếm hơn 90% tất cả các bệnh nhân bị CAH. Do chẩn đoán và điều trị được cải thiện ở trẻ sơ sinh, bệnh nhân bị 21OHD nặng hiện thường xuyên sống đến tuổi trưởng thành, do đó việc quản lý 21OHD liên quan đến các bác sĩ làm việc với tất cả các nhóm tuổi.
Sinh lý bệnh
Trong 21OHD nặng, có sự bất lực trong việc chuyển đổi progesterone thành DOC, dẫn đến thiếu hụt aldosterone gây ra hạ natri máu nặng, tăng kali máu và nhiễm toan. Hạ huyết áp, sốc và trụy tim mạch liên quan có thể dẫn đến tử vong ở trẻ sơ sinh không được điều trị. Vì việc kiểm soát chất lỏng và điện giải ở thai nhi được duy trì bởi nhau thai và thận của người mẹ, khủng hoảng mất muối này chỉ phát triển sau khi sinh, thường là trong tuần thứ hai của cuộc đời. Việc không thể chuyển đổi 17OHP thành 11-deoxycortisol dẫn đến thiếu hụt cortisol, làm suy giảm chuyển hóa carbohydrate sau sinh và làm trầm trọng thêm trụy tim mạch, vì một tác động cho phép của cortisol là cần thiết cho tác động tăng huyết áp đầy đủ của catecholamine. Nồng độ cortisol cao trong dòng mao mạch vỏ thượng thận tắm tủy là cần thiết cho sự phiên mã của phenylethanolamine N-methyltransferase ở tủy, chất chuyển đổi norepinephrine thành epinephrine, do đó trẻ em bị 21OHD có nồng độ epinephrine thấp, có thể làm trầm trọng thêm tình trạng hạ đường huyết liên quan đến thiếu hụt cortisol.
Mặc dù vai trò của cortisol trong sinh lý thai nhi chưa được xác định rõ, thiếu hụt cortisol được biểu hiện trước sinh. Cortisol của thai nhi thấp kích thích bài tiết ACTH, kích thích tăng sản thượng thận và phiên mã các gen cho tất cả các enzyme sinh steroid, đặc biệt là P450scc, enzyme giới hạn tốc độ trong sinh tổng hợp steroid. Sự gia tăng phiên mã này làm tăng sản xuất và hoạt động của enzyme, với hậu quả là sự tích tụ của các steroid không được 21-hydroxyl hóa. Có bốn con đường mà các steroid này được chuyển hướng đến androgen (xem Hình 14.6). Thứ nhất, 17-Preg có thể được chuyển đổi thành DHEA bởi hoạt tính 17,20 lyase của P450c17; nếu không bị bất hoạt bởi sulfat hóa thành DHEAS, DHEA có thể được chuyển đổi thành androstenedione bởi 3βHSD2. Androstenedione sau đó có thể được chuyển đổi thành testosterone trong tuyến thượng thận của thai nhi hoặc trong các mô đích bởi 17βHSD5 (xem Hình 14.3). Thứ hai, mặc dù 17OHP thường không phải là một cơ chất hiệu quả cho hoạt tính 17,20 lyase của P450c17, mức 17OHP rất cao đặc trưng của 21OHD sẽ “ép” một số chuyển đổi thành androstenedione bằng tác động khối lượng. Thứ ba, như được mô tả trong Mục III (Bài tiết Androgen tuyến thượng thận và Điều hòa Giai đoạn thượng thận hóa), androstenedione có thể được chuyển đổi thành 11-keto-testosterone, androgen tuyến thượng thận chính, ở lớp lưới. Thứ tư, trong con đường cửa sau thay thế đến DHT, 17OHP bị khử 5α- và 3α- thành 17OH-allopregnenolone, chất này dễ dàng được chuyển đổi bởi P450c17 thành androsterone và từ đó bởi 17βHSD thành androstanediol và một 3αHSD oxy hóa thành DHT, do đó androgen mạnh nhất này được sản xuất mà không có DHEA, androstenedione, hoặc testosterone làm chất trung gian. Công trình gần đây đã xác định rằng con đường này tồn tại ở thai nhi người và nó góp phần đáng kể vào việc sản xuất androgen ở trẻ em bị 21OHD. Cũng như được mô tả trước đó (xem Sinh tổng hợp steroid ở tuyến thượng thận của thai nhi), tuyến thượng thận của thai nhi biểu hiện tạm thời 3βHSD2 vào khoảng 7 đến 12 tuần, cho phép tổng hợp cortisol, từ đó ức chế ACTH và do đó ức chế sản xuất DHEA và các steroid C-19 khác của tuyến thượng thận thai nhi. Do đó, tổng hợp androgen của tuyến thượng thận thai nhi thường bị ức chế trong thời gian bộ phận sinh dục ngoài có thể bị nam hóa. Trong 21OHD, việc sản xuất cortisol tạm thời này là không thể, càng thúc đẩy sự nam hóa của thai nhi nữ.
Sớm trong thai kỳ, tinh hoàn của thai nhi sản xuất testosterone bằng con đường androgen thông thường và DHT bằng con đường cửa sau, cả hai đều cần thiết để biệt hóa các cấu trúc tiền thân phôi đa năng thành bộ phận sinh dục ngoài của nam. Ở thai nhi nam bị 21OHD, các androgen bổ sung được sản xuất ở tuyến thượng thận không có tác động kiểu hình. Ngược lại, buồng trứng của thai nhi thường sản xuất rất ít steroid sinh dục hoặc các yếu tố khác cần thiết cho sự biệt hóa của bộ phận sinh dục ngoài của nữ. Các androgen được sản xuất không phù hợp bởi tuyến thượng thận của thai nhi nữ bị 21OHD gây ra các mức độ khác nhau của sự nam hóa bộ phận sinh dục ngoài, có thể từ phì đại âm vật nhẹ, có hoặc không có sự hợp nhất phía sau của các nếp gấp môi-bìu, đến sự hợp nhất môi-bìu hoàn toàn bao gồm một niệu đạo đi qua âm vật bị phì đại (Hình 14.14). Những trẻ sơ sinh này có buồng trứng, ống dẫn trứng và tử cung bình thường, nhưng có bộ phận sinh dục ngoài “không rõ ràng” hoặc có thể bị nam hóa đủ để trông giống như nam giới, dẫn đến sai sót trong việc xác định giới tính khi sinh. 21OHD là dạng phổ biến nhất của DSD 46,XX.
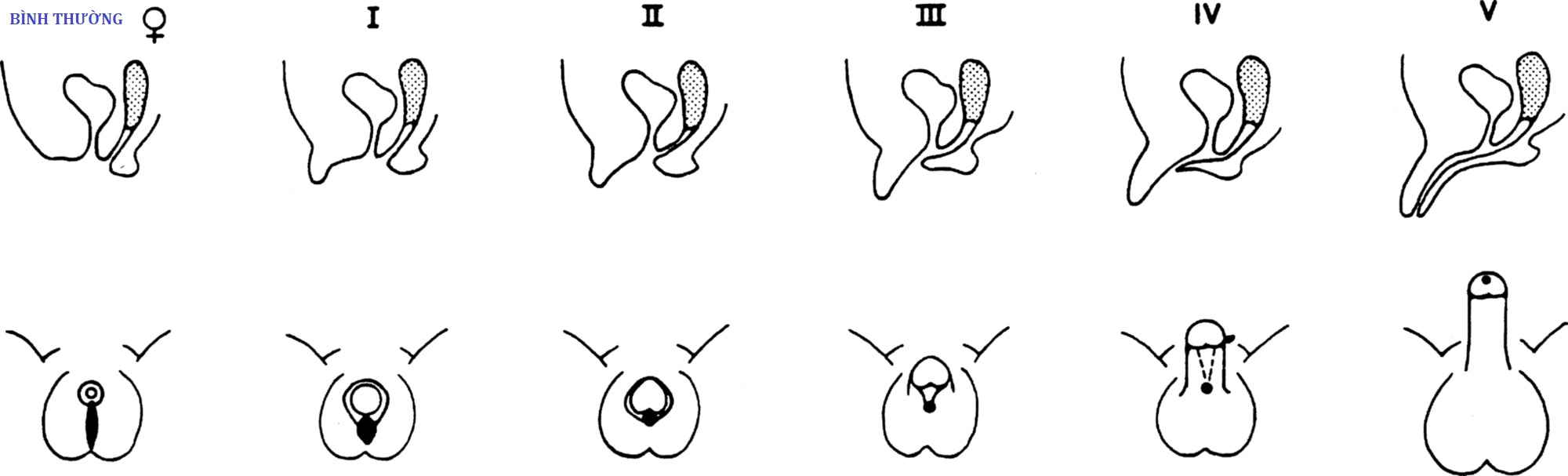
Hình 14.14: Nam hóa bộ phận sinh dục ngoài. Một phổ liên tục được hiển thị từ nữ bình thường đến nam bình thường ở cả mặt cắt dọc giữa (trên) và hình ảnh đáy chậu (dưới), sử dụng hệ thống phân độ của Prader. Rối loạn bộ phận sinh dục ngoài có thể xảy ra do nam hóa một người nữ bình thường, như trong tăng sản thượng thận bẩm sinh, hoặc do lỗi trong quá trình tổng hợp testosterone ở nam giới. Ở nữ giới bị tăng sản thượng thận bẩm sinh do thiếu hụt 21-hydroxylase, mức độ nam hóa có tương quan kém với sự hiện diện hay vắng mặt của các dấu hiệu lâm sàng mất muối.
Chẩn đoán 21OHD được gợi ý bởi DSD ở nữ, một đợt mất muối ở cả hai giới, hoặc tăng trưởng nhanh và nam hóa ở nam hoặc nữ trong giai đoạn sơ sinh. 17OHP huyết tương tăng rõ rệt (> 2000 ng/dL hoặc 60 nmol/L, và thường vượt quá 40.000 ng/dL hoặc 1200 nmol/L sau 24 giờ tuổi, ở một trẻ sơ sinh đủ tháng khỏe mạnh), và đáp ứng quá mức với kích thích bằng ACTH (xem Hình 14.12). Bác sĩ lâm sàng cũng phải đo 11-deoxycortisol, DHEA và androstenedione, cả để phân biệt giữa các dạng CAH và vì các khối u tuyến thượng thận và tinh hoàn cũng có thể sản xuất 17OHP. ACTH cũng sẽ gây ra sự gia tăng đáng kể 21-deoxycortisol huyết thanh trong tất cả các dạng 21OHD, nhưng không phải ở người bình thường, cung cấp một xét nghiệm hữu ích khi steroid này có thể được đo. Các giá trị 17OHP sơ sinh cao tăng thêm sau ACTH cũng có thể được thấy trong thiếu hụt 3βHSD2 (do hoạt động của 3βHSD1 của gan) và trong thiếu hụt P450c11β (do ức chế sản phẩm cuối của P450c21). 17OHP thường cao trong máu cuống rốn, nhưng giảm xuống mức sơ sinh bình thường sau khoảng 24 giờ (Hình 14.15), do đó việc đánh giá nồng độ 17OHP không nên được thực hiện trong ngày đầu tiên của cuộc đời. Trẻ sinh non và trẻ đủ tháng bị stress nặng (ví dụ, bị bệnh tim hoặc phổi) có thể có nồng độ 17OHP tăng cao liên tục với 21-hydroxylase bình thường. Các chương trình sàng lọc sơ sinh cho CAH, dựa trên các phép đo 17OHP, được sử dụng trên khắp thế giới công nghiệp hóa. Các công nghệ được sử dụng và các giá trị “cắt” được sử dụng khác nhau trong các hệ thống chăm sóc sức khỏe khác nhau. Khi xét nghiệm được thực hiện trên trẻ đủ tháng hơn 24 giờ sau khi sinh, sàng lọc là đáng tin cậy. Các nhà nội tiết học và sơ sinh học phải làm quen với các xét nghiệm địa phương và các giá trị được tìm thấy ở trẻ sinh cực non, có thể được đọc là dương tính giả cho CAH đòi hỏi phải xét nghiệm lại. Ngoài ra, sàng lọc sơ sinh âm tính không loại trừ 21OHD nhẹ hơn gây ra 21OHD không cổ điển ngoài giai đoạn sơ sinh.
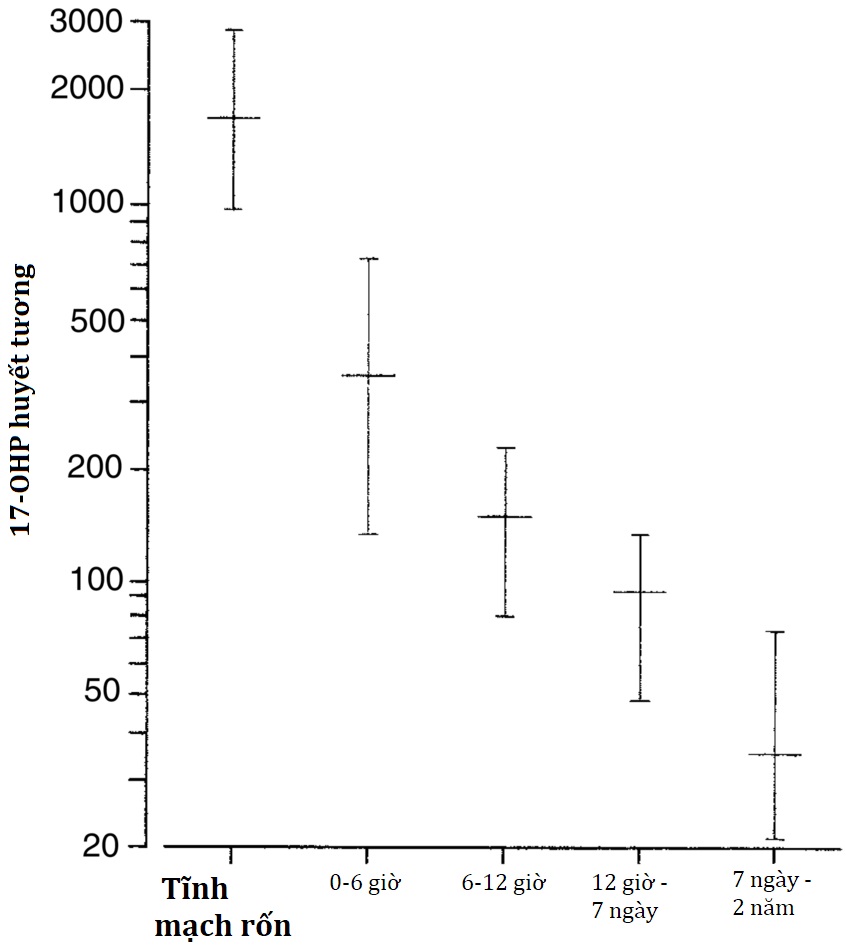
Hình 14.15: Giá trị trung bình và khoảng dao động của 17α-Hydroxyprogesterone ở trẻ sơ sinh bình thường (dữ liệu tính bằng ng/100 mL). Lưu ý rằng các giá trị có thể rất cao và khá thay đổi trong 24 giờ đầu đời.
Các dạng lâm sàng của Thiếu hụt 21-Hydroxylase
Có một phổ rộng các biểu hiện lâm sàng của 21OHD, tùy thuộc vào đột biến gen cụ thể. Các dạng khác nhau này của 21OHD không phải là các bệnh khác nhau, vì có một phổ liên tục các biểu hiện, từ dạng “mất muối” nặng đến các dạng không có biểu hiện lâm sàng có thể là các biến thể bình thường. Do đó, các dạng bệnh điển hình được thảo luận sau đây chủ yếu là một sự tiện lợi về mặt lâm sàng.
Thiếu hụt 21-hydroxylase thể mất muối 21OHD thể mất muối (SW) là do thiếu hụt gần như hoàn toàn hoạt tính của P450c21, loại bỏ hiệu quả cả việc tổng hợp glucocorticoid và mineralocorticoid. Các cá thể nữ bị 21OHD thể SW thường được chẩn đoán khi sinh do DSD của họ. Sau khi hồi sức thích hợp cho trụy tim mạch, nhiễm toan và các rối loạn điện giải, các mineralocorticoid và glucocorticoid có thể được thay thế bằng đường uống và DSD có thể được điều chỉnh bằng các thủ thuật phẫu thuật tạo hình. Việc quản lý thay thế steroid rất khó khăn do nhu cầu thay đổi nhanh chóng của một trẻ sơ sinh hoặc trẻ em đang phát triển (xem phần “Điều trị”). Liều thuốc phải được điều chỉnh thường xuyên, và có sự thay đổi cá nhân về những gì cấu thành sự thay thế “sinh lý”. Vì việc dùng thiếu liều glucocorticoid có thể đe dọa tính mạng, đặc biệt là trong khi bị bệnh, hầu hết các bác sĩ nhi khoa đã có xu hướng “phạm sai lầm về phía an toàn,” do đó những trẻ này thường nhận được liều glucocorticoid lớn không phù hợp. Không thể bù đắp cho sự tăng trưởng bị mất trong 2 năm đầu đời, khi sự tăng trưởng nhanh nhất, do đó những trẻ này thường có chiều cao thấp hơn so với dự đoán từ tiềm năng di truyền của chúng. Phụ nữ trưởng thành có thể bị rối loạn chức năng tình dục, kết hôn với tần suất thấp, ngại hình thành các mối quan hệ thân mật hơn và giảm khả năng sinh sản. Androgen trước sinh dường như ảnh hưởng đến hành vi, nhưng không ảnh hưởng đến bản dạng giới. Các cá thể nam bị 21OHD thể SW thường không được chẩn đoán khi sinh và được chú ý y tế thông qua sàng lọc sơ sinh hoặc trong cuộc khủng hoảng mất muối xảy ra vào ngày thứ 5 đến 15 của cuộc đời; trẻ sơ sinh không được điều trị thường tử vong. Đàn ông trưởng thành dường như có các mối quan hệ dị tính ít ổn định hơn và giảm khả năng sinh sản, đặc biệt là khi điều trị không đủ, điều này thúc đẩy sự xuất hiện của các khối u lạc chỗ tuyến thượng thận ở tinh hoàn. Các đột biến CYP21A2 phổ biến nhất gây ra khoảng 50% các trường hợp 21OHD thể SW là các xóa gen lớn, chuyển đổi gen lớn và một sự thay thế C thành G cách vị trí chấp nhận nối ghép 13 base trong intron 2.
Thiếu hụt 21-hydroxylase thể nam hóa đơn thuần Các cá thể nữ bị nam hóa có nồng độ 17OHP tăng cao, nhưng không bị khủng hoảng mất muối, từ lâu đã được công nhận là mắc dạng SV của 21OHD. Trừ khi được phát hiện bằng sàng lọc sơ sinh, các cá thể nam bị 21OHD thể SV có thể thoát khỏi chẩn đoán cho đến 3 đến 7 tuổi, khi họ được chú ý y tế do sự phát triển sớm của lông mu, lông nách và lông mặt, mụn trứng cá và sự phát triển của dương vật. Những dấu hiệu này có thể bị nhầm lẫn với dậy thì sớm, nhưng tinh hoàn vẫn có kích thước trước tuổi dậy thì vì không có sự kích thích của gonadotropin, trong khi các bé trai bị dậy thì sớm trung ương thật sự có tinh hoàn kích thước dậy thì (≥ 4 mL). Những trẻ này phát triển nhanh và cao so với tuổi khi được chẩn đoán, nhưng sự trưởng thành của đầu xương (tuổi xương) của chúng tiến triển nhanh không tương xứng, do đó chiều cao trưởng thành cuối cùng bị ảnh hưởng. Trẻ em bị 21OHD không được điều trị hoặc điều trị kém có thể không trải qua dậy thì bình thường, và các bé trai có thể có tinh hoàn nhỏ và không có tinh trùng do phản hồi của testosterone do tuyến thượng thận sản xuất lên các gonadotropin của tuyến yên. Khi điều trị được bắt đầu ở vài tuổi, việc ức chế bài tiết testosterone của tuyến thượng thận có thể loại bỏ sự ức chế trương lực của vùng dưới đồi, đôi khi dẫn đến dậy thì sớm trung ương thật sự, đòi hỏi phải điều trị bằng một chất chủ vận hormone giải phóng gonadotropin (GnRH). Nồng độ ACTH cao có thể kích thích sự phát triển của các khối u lạc chỗ tuyến thượng thận ở tinh hoàn (TART). Do tỷ lệ mắc TART cao (~ 25%), tinh hoàn của nam giới sau tuổi dậy thì bị 21OHD nên được kiểm tra 1 đến 2 năm một lần bằng siêu âm. Bởi vì tuyến thượng thận thường sản xuất cortisol nhiều hơn aldosterone từ 100 đến 1000 lần, các khiếm khuyết nhẹ (đột biến thay thế axit amin) ở P450c21 ít có khả năng ảnh hưởng đến bài tiết mineralocorticoid hơn là bài tiết cortisol. Điều này được phản ánh về mặt sinh lý bởi hoạt độ renin huyết tương tăng thấy ở những bệnh nhân bị 21OHD thể SV, sau khi hạn chế muối vừa phải. Đột biến phổ biến nhất gây ra 21OHD thể SV là I172N, giữ lại khoảng 5% hoạt tính bình thường.
Thiếu hụt 21-hydroxylase không cổ điển Các dạng 21OHD rất nhẹ, không cổ điển (NC) có thể được biểu hiện bằng rậm lông, nam hóa, mụn trứng cá, kinh nguyệt không đều và giảm khả năng sinh sản ở phụ nữ trưởng thành từ nhẹ đến trung bình (đôi khi được gọi là 21OHD khởi phát muộn), hoặc có thể không có biểu hiện kiểu hình nào khác ngoài sự gia tăng đáp ứng của 17OHP huyết tương với nghiệm pháp ACTH tĩnh mạch (cái gọi là 21OHD thể ẩn). Những cá thể này cũng có thể có bằng chứng hormone về sự suy giảm nhẹ trong bài tiết mineralocorticoid, được chứng minh bằng sự gia tăng lớn hơn bất thường của PRA khi lượng natri trong chế độ ăn bị hạn chế. Các phân tích kiểu gen CYP21A2 đã xác định được tỷ lệ người mang gen dị hợp tử là 15% (khoảng tin cậy 95% [CI], 10,4–20,7) trong số 200 người Do Thái Ashkenazi và tỷ lệ người mang gen là 9,5% (khoảng tin cậy 95%, 5,8–14,4) trong số 200 người da trắng không phải Do Thái; sự khác biệt này không có ý nghĩa (P =,13), cho tần suất chung khoảng 1 trên 200. Đột biến CYP21A2 phổ biến nhất gây ra 21OHD thể NC là V281L, giữ lại khoảng 20% hoạt tính bình thường.
Có một số sự không nhất quán trong việc phân loại bệnh nhân bị 21OHD vì ba “dạng” không phải là các bệnh riêng biệt mà đại diện cho các hình ảnh điển hình trong một phổ bệnh liên tục, gây ra bởi một phổ các tổn thương di truyền. Bởi vì một số alen CYP21A2 đột biến phổ biến trong dân số chung, hầu hết các bệnh nhân là dị hợp tử phức hợp, mang một đột biến khác nhau trên alen được thừa hưởng từ mỗi cha mẹ. Cuối cùng, các yếu tố khác ngoài các đột biến cụ thể được tìm thấy trong CYP21A2 sẽ ảnh hưởng đến kiểu hình lâm sàng, bao gồm sự hiện diện của các 21-hydroxylase ngoại thượng thận và các biến thể trong chuyển hóa và độ nhạy của androgen. Do đó, sự không tương hợp giữa kiểu gen và kiểu hình là điều được mong đợi.
Tỷ lệ mắc Thiếu hụt 21-Hydroxylase
Sàng lọc chu sinh cho 17OHP huyết thanh tăng cao đã cho thấy tỷ lệ mắc 21OHD cổ điển thể SW và SV là khoảng 1 trên 14.000, cho tỷ lệ người mang gen dị hợp tử là 1 trên 60. Sàng lọc 1,9 triệu trẻ sơ sinh ở Texas cho tỷ lệ mắc chung là 1 trên 16.000, bao gồm tỷ lệ mắc 1 trên 15.600 người da trắng, 1 trên 14.500 người gốc Tây Ban Nha (chủ yếu là người Mỹ gốc Mexico có tổ tiên là người Mỹ bản địa), và 1 trên 42.300 người Mỹ gốc Phi. Bởi vì khoảng 20% vốn gen của người Mỹ gốc Phi có nguồn gốc châu Âu, tỷ lệ mắc được tính toán ở những cá thể hoàn toàn có tổ tiên châu Phi cận Sahara là khoảng 1 trên 250.000; cơ sở cho sự khác biệt dân tộc rõ ràng này vẫn chưa được biết. 21OHD thể NC phổ biến hơn nhiều, với tỷ lệ mắc cao nhất ở các dân tộc Địa Trung Hải. Tỷ lệ mắc cao của 21OHD thể NC, tính không gây tử vong của nó, và tác động không đáng kể của nó đối với khả năng sinh sản cho thấy nó là một biến thể đa hình chứ không phải là một bệnh theo nghĩa cổ điển. Tuy nhiên, bệnh nhân bị 21OHD thể NC có thể tìm kiếm sự giúp đỡ cho các phàn nàn về nam hóa, rối loạn kinh nguyệt và khả năng sinh sản.
Di truyền học của Thiếu hụt 21-Hydroxylase
Các Gen CYP21 Gen CYP21A2 chức năng và gen giả CYP21A1P không chức năng gần đó bao gồm 10 exon, dài ~3.4kb, được nhân đôi song song với các gen C4A và C4B mã hóa thành phần thứ tư của bổ thể huyết thanh, và nằm trong vùng loại III của phức hợp tương hợp mô chính (MHC) của người (Hình 14.16). CYP21A1P được phiên mã, nhưng các RNA kết quả không mã hóa protein. Locus MHC trên nhiễm sắc thể 6p21 được đặc trưng bởi một tỷ lệ tái tổ hợp di truyền rất cao; tỷ lệ trao đổi DNA giữa các gen cao này dẫn đến các gen CYP21A2 và CYP21A1P chỉ khác nhau 87 hoặc 88 base. Các gen cyp21 của chuột và bò cũng được nhân đôi tương tự trong các locus MHC, nhưng các ranh giới nhân đôi gen khác nhau, và một số động vật có vú chỉ có một gen cyp21 duy nhất, cho thấy sự nhân đôi gen xảy ra sau sự hình thành loài của động vật có vú.
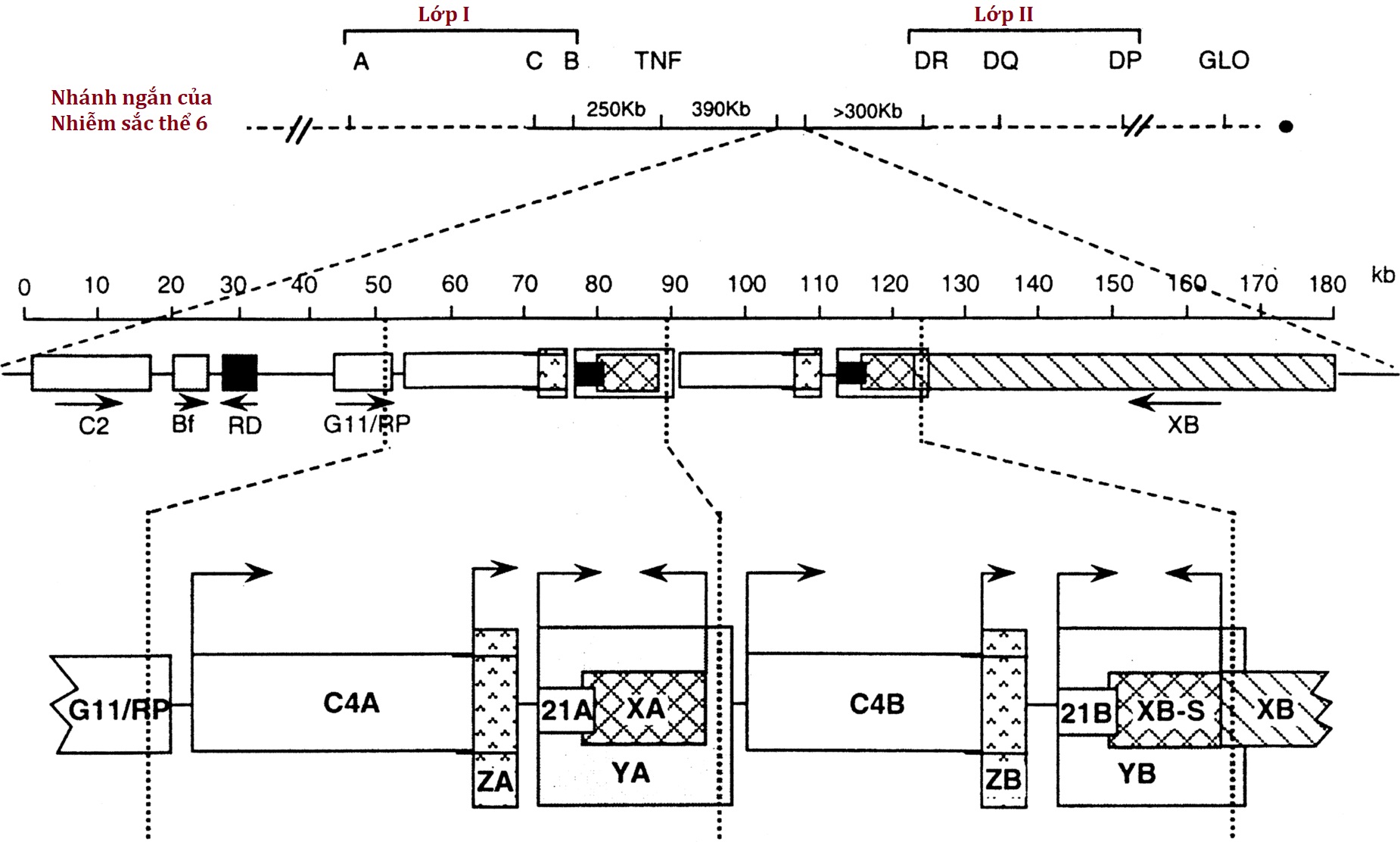
Hình 14.16: Bản đồ di truyền của locus kháng nguyên bạch cầu người (HLA) chứa các gen P450c21. Dòng trên cùng cho thấy vùng p21.1 của nhiễm sắc thể 6, với telomere ở bên trái và tâm động ở bên phải. Hầu hết các gen HLA được tìm thấy ở các vùng lớp I và lớp II; vùng lớp III chứa các gen P450c21 nằm giữa hai vùng này. Dòng thứ hai cho thấy thang đo (tính bằng kilobase) cho sơ đồ ngay sau đó, cho thấy (từ trái sang phải) các gen cho yếu tố bổ thể C2, yếu tố properdin Bf, và các gen RD và G11/RP STK19 có chức năng chưa rõ; các mũi tên chỉ hướng phiên mã. Dòng dưới cùng cho thấy locus 21-hydroxylase trên một thang đo mở rộng, bao gồm các gen C4A và C4B cho thành phần thứ tư của bổ thể, gen CYP21A1P không hoạt động (21A) và gen CYP21A2 hoạt động (21B) mã hóa cho P450c21. XA, YA, và YB là các bản phiên mã đặc hiệu của tuyến thượng thận không có khung đọc mở. Gen XB mã hóa protein ma trận ngoại bào Tenascin-X; XB-S mã hóa một dạng Tenascin-X bị cắt ngắn, đặc hiệu của tuyến thượng thận mà chức năng của nó chưa được biết. ZA và ZB là các bản phiên mã đặc hiệu của tuyến thượng thận phát sinh trong các gen C4 và có các khung đọc mở, nhưng không biết liệu chúng có được dịch mã thành protein hay không; tuy nhiên, các yếu tố promoter của các bản phiên mã này là các thành phần thiết yếu của các promoter CYP21A và CYP21B. Các mũi tên chỉ hướng phiên mã. Các đường chấm dọc chỉ định ranh giới của sự kiện nhân đôi di truyền dẫn đến sự hiện diện của các vùng A và B.
Liên kết với Kháng nguyên Bạch cầu Người Bởi vì các gen CYP21 nằm trong locus MHC, việc định type HLA đã được sử dụng để chẩn đoán trước sinh và để xác định các thành viên gia đình dị hợp tử, nhưng cách tiếp cận này hiện đã được thay thế bằng phân tích di truyền trực tiếp. Các mối liên quan thống kê (mất cân bằng liên kết) đã được thiết lập rõ ràng giữa 21OHD và một số loại HLA cụ thể. 21OHD thể SW có liên quan đến HLA-B60 và HLA-40 ở một số quần thể, và HLA Bw47 có liên quan mạnh mẽ đến 21OHD thể SW. HLA-Bw51 có liên quan đến 21OHD thể SV ở một số quần thể, và khoảng 40% các haplotype cho 21OHD thể NC mang HLA-B14. HLA-B14 thường liên quan đến sự nhân đôi của gen C4B, nhưng tất cả các alen HLA-B đều có thể được tìm thấy liên kết với 21OHD. Các cá nhân có HLA giống hệt nhau trong một gia đình có thể có các đặc điểm lâm sàng khác nhau của 21OHD, mặc dù có sự giống nhau về HLA, có thể đại diện cho 21-hydroxyl hóa ngoại thượng thận, các đột biến de novo, hoặc nhiều sự kiện trao đổi chéo di truyền.
Các Gen khác trong Locus CYP21 Các gen C4A và C4B sản xuất các đồng dạng rất giống nhau của thành phần bổ thể C4; protein C4B có hoạt tính tan máu lớn hơn, mặc dù có hơn 99% tương đồng về trình tự với C4A. Gen C4A dài 22 kb, nhưng có các dạng dài (22 kb) và ngắn (16 kb) của C4B do một biến thể trong một intron. Các đầu 3′ của các gen C4 chỉ cách vị trí bắt đầu phiên mã của các gen CYP21 2466bp về phía thượng nguồn. Các trình tự promoter cần thiết cho sự phiên mã của CYP21A2 nằm trong intron 35 của C4B. Một số gen khác cũng nằm trong khoảng 100 kb của CYP21A2, bao gồm các gen cho yếu tố bổ thể C2 và yếu tố properdin Bf (xem Hình 14.16). Nằm ngay sau gen Bf, trên sợi DNA đối diện với CYP21A2, là gen STK19 (trước đây được gọi là G11), mã hóa một kinase serine/threonine hạt nhân.
Tenascin-X và hội chứng tăng sản thượng thận bẩm sinh-X (CAH-X) Tenascin-X (TNX), được mã hóa bởi gen TNXB, là một protein chất nền ngoại bào có tác dụng chủ yếu là chống dính, nhưng mở rộng ra ngoài kiến trúc tế bào. Các gen TNXA và TNXB được nhân đôi cùng với các gen C4 và CYP21, và nằm trên sợi DNA đối diện với các gen C4 và CYP21. Các exon cuối cùng của TNXA và TNXB nằm trong vùng không được dịch 3′ của exon 10 trong CYP21A1P và CYP21A2, tương ứng. TNXA đã bị cắt ngắn trong quá trình nhân đôi của đơn vị di truyền tổ tiên C4-CYP21-TNX; nó được phiên mã trong tuyến thượng thận, nhưng không có chức năng nào được biết đến. Protein TNX được mã hóa bởi TNXB là một protein chất nền ngoại bào lớn được biểu hiện ở hầu hết các mô, đặc biệt là mô liên kết. Gen TNXB trải dài khoảng 65 kb và bao gồm 43 exon mã hóa một mRNA dài 12 kb. TNXB cũng mã hóa một dạng TNX bị cắt ngắn 74 kDa (XB-Short; XB-S), giống hệt với 673 axit amin cuối C của TNX, phát sinh từ một promoter giữa các gen, và được biểu hiện duy nhất trong tuyến thượng thận. XB-S liên kết với kinesin động cơ phân bào Eg5, nhưng chức năng chính xác của nó vẫn chưa rõ ràng. Việc xác định một bệnh nhân 21OHD bị “hội chứng gen liền kề”, bao gồm việc xóa cả gen CYP21A2 và TNXB, đã chứng minh rằng thiếu hụt TNX dẫn đến hội chứng Ehlers-Danlos (EDS). Hầu hết các dạng EDS là do đột biến trội trên nhiễm sắc thể thường ở các gen collagen; các dạng lặn là do đột biến ở các gen cho các enzyme biến đổi collagen, bao gồm TNX, chất liên kết và ổn định các sợi collagen. Thiếu hụt TNX đồng hợp tử gây ra một dạng EDS lặn nghiêm trọng, khác biệt về mặt lâm sàng, có hoặc không có 21OHD đi kèm; bán thiếu hụt TNX gây ra dạng EDS “tăng động nhẹ”, biểu hiện bằng tăng động khớp, đau, trật khớp nhiều lần, và ít phổ biến hơn là đau khớp mãn tính, thoát vị, và các khiếm khuyết tim. Có đến 10% bệnh nhân bị 21OHD thể SW có xóa gen CYP21A2 trên một alen kéo dài vào gen TNX, do đó họ cũng bị bán thiếu hụt TNX và có một dạng 21OHD riêng biệt được gọi là CAH-X.
Các tổn thương Gen CYP21A2 gây Thiếu hụt 21-Hydroxylase 21OHD có thể do xóa gen CYP21A2, chuyển đổi gen, và các đột biến điểm biểu kiến. Các đột biến điểm biểu kiến phổ biến nhất thực sự là các sự kiện chuyển đổi gen nhỏ, do đó chuyển đổi gen chiếm khoảng 85% các tổn thương trong 21OHD. Mỗi người có hai alen CYP21A2, một alen do mỗi cha mẹ đóng góp. Hầu hết các bệnh nhân bị 21OHD là dị hợp tử phức hợp, có các tổn thương khác nhau trên hai alen của họ. Xóa gen và chuyển đổi lớn loại bỏ phiên mã gen; đồng hợp tử cho những tổn thương này gây ra 21OHD thể mất muối. Một số chuyển đổi vi mô, chẳng hạn như những chuyển đổi tạo ra sự kết thúc dịch mã sớm, cũng gây ra 21OHD thể mất muối. Các dạng nhẹ hơn, chẳng hạn như 21OHD thể SV và NC, có liên quan đến việc thay thế axit amin trong protein P450c21 do các sự kiện chuyển đổi vi mô gen. Bệnh nhân có các dạng 21OHD này thường là dị hợp tử phức hợp mang một alen bị rối loạn nặng và một alen bị rối loạn nhẹ, do đó các biểu hiện lâm sàng dựa trên bản chất của alen bị rối loạn nhẹ. Các gen CYP21A2 và CYP21A1P chỉ khác nhau 87 hoặc 88 nucleotide. Hai đặc điểm bất thường và liên quan của locus 21-hydroxylase làm phức tạp việc phân tích nó. Thứ nhất, việc xóa gen trong locus này là bất thường nhất ở chỗ chúng kéo dài 30 kb từ một trong một số điểm ở giữa CYP21A1P đến điểm tương đồng chính xác trong CYP21A2. Thứ hai, chuyển đổi gen cực kỳ phổ biến.
Chuyển đổi và Vi chuyển đổi Gen gây Thiếu hụt 21-Hydroxylase thể mất muối Nếu một đoạn của gen A thay thế đoạn tương ứng của gen B liên quan, cấu trúc của gen nhận B được cho là “chuyển đổi” thành cấu trúc của gen cho A. Dấu hiệu của chuyển đổi gen là số lượng các gen liên quan chặt chẽ vẫn không đổi, trong khi sự đa dạng của chúng giảm đi. Hai loại chuyển đổi gen thường gây ra 21OHD: chuyển đổi gen lớn có thể bị nhầm lẫn với xóa gen, và chuyển đổi vi mô nhỏ giống như đột biến điểm. Tần suất của chuyển đổi gen lớn so với xóa gen trước đây gây tranh cãi, chủ yếu vì các nghiên cứu ban đầu sử dụng các nhóm bệnh nhân tương đối nhỏ từ các địa điểm hoặc nhóm dân tộc đơn lẻ. Một tập hợp của nhiều nghiên cứu cho thấy khoảng 19% các alen đột biến có xóa gen, 8% có chuyển đổi gen lớn, 67% có chuyển đổi vi mô, và 6% có các tổn thương khác (Hình 14.17). Do sai lệch trong việc xác định, những con số như vậy nhấn mạnh đến các bệnh nhân bị ảnh hưởng nặng hơn. Khoảng 75% các gen CYP21A2 bị đột biến vẫn còn nguyên vẹn về mặt tổng thể và dường như mang các đột biến điểm, nhưng hầu hết các đột biến điểm biểu kiến cũng được tìm thấy trong gen giả CYP21A1P, cho thấy rằng chúng là kết quả của các sự kiện chuyển đổi gen nhỏ (Bảng 14.6). Ba thay đổi trong gen giả (xóa 8 bp, exon 3; chèn T, exon 7; Gly 318 Stop, exon 8) làm cho sản phẩm RNA của nó không chức năng, do khung đọc bị thay đổi và/hoặc codon dừng sớm; những thay đổi này đã được tìm thấy trong các alen CYP21A2 gây ra 21OHD thể SW. Ba thay đổi base cụm lại gần nhau trong CYP21A1P làm thay đổi trình tự axit amin bình thường Ile-Val-Glu-Met ở các codon 236 đến 239 trong exon 6 thành Asn-Glu-Glu-Lys trong một số ít gen gây ra 21OHD thể SW.
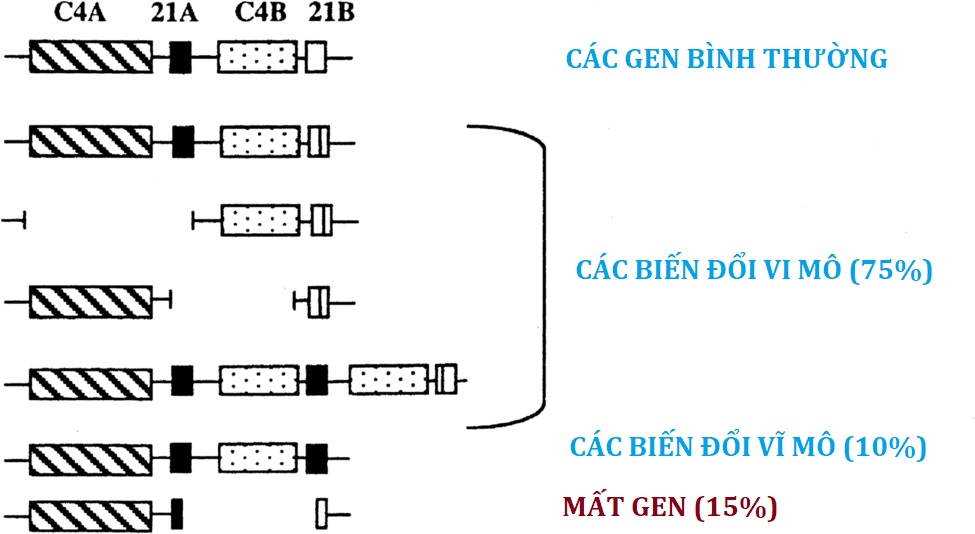
Hình 14.17: Các loại sắp xếp lại di truyền gây ra thiếu hụt 21-hydroxylase. Sự xóa hoặc nhân đôi các gen C4A và C4B có thể xảy ra có hoặc không có các tổn thương liên quan trong gen P450c21B. Lưu ý rằng tất cả các “đột biến điểm” trong P450c21B thực ra là “biến đổi vi mô”. Nhiều tác giả kết hợp các nhóm “xóa gen” và “biến đổi vĩ mô” vì chúng khó phân biệt bằng phương pháp Southern blotting, vì cả hai đều dẫn đến mất gen P450c21B, nhưng các kiểu gen rõ ràng là khác biệt, như được hiển thị.
Bảng 14.6. Các biến đổi vi mô trong Gen CYP21A2 gây ra Thiếu hụt 21-Hydroxylase
| Đột biến | Vị trí | Kiểu hình liên quan | Hoạt tính |
|---|---|---|---|
| Pro 30→Leu | Exon 1 | NC/SV | 30%-60% |
| A→G | Intron 2 | SV/SW | Tối thiểu |
| Xóa 8 bp | Exon 3 | SW | 0 |
| Ile 172→Asn | Exon 4 | SV | 3%-7% |
| Ile 236→Asp | Exon 6 | SW | 0 |
| Val 237→Glu | |||
| Met 239→Lys | |||
| Val 281→Leu | Exon 7 | NC | 18% ± 9% |
| Gly 292→Ser | Exon 7 | SW | 0 |
| Chèn T @ 306 | Exon 7 | SW | 0 |
| Gly 318→Stop | Exon 8 | SW | 0 |
| Arg 339→His | Exon 8 | NC | 20%-50% |
| Arg 356→Trp | Exon 8 | SV/SW | <2% |
| Pro 453→Ser | Exon 10 | NC | 20%-50% |
| GGC @ 484 | Exon 10 | SW | 0 |
Tổn thương phổ biến nhất trong 21OHD cổ điển là sự thay đổi A → G trong intron thứ hai, cách vị trí chấp nhận nối ghép 3′ bình thường của nó 13 base về phía thượng nguồn; đây là một vi chuyển đổi được tìm thấy ở hơn 25% các alen 21OHD bị ảnh hưởng nặng. Đột biến trong intron này gây ra sự nối ghép bất thường của RNA được mã hóa, làm mất hoạt tính. Tuy nhiên, một phần nhỏ của mRNA này có thể được nối ghép chính xác ở một số bệnh nhân do đó biểu hiện kiểu hình có thể thay đổi; hầu hết các bệnh nhân như vậy bị 21OHD thể SW, nhưng một số bị 21OHD thể SV. Vi chuyển đổi intron 2 này thường liên quan đến đa hình Ser/Thr ở codon 268, không làm thay đổi hoạt tính enzyme. Vi chuyển đổi R356W, được tìm thấy ở khoảng 10% các alen bị ảnh hưởng nặng, có thể giữ lại một chút hoạt tính và đã được tìm thấy ở cả 21OHD thể SW và SV. Một số lượng lớn các đột biến hiếm gặp đã được mô tả ở các cá nhân đơn lẻ.
Các đột biến gây Thiếu hụt 21-Hydroxylase thể nam hóa đơn thuần và không cổ điển Vi chuyển đổi I172N là nguyên nhân phổ biến nhất của 21OHD thể SV. Khi dư lượng này được thay đổi thành Asn, Leu, Gln, hoặc His và protein P450c21 đột biến kết quả được xét nghiệm trong ống nghiệm, nó giữ lại khoảng 3% đến 7% hoạt tính 21-hydroxylase bình thường. Vi chuyển đổi intron 2 đôi khi được thấy trong 21OHD thể SV. Vi chuyển đổi P30L thường liên quan đến 21OHD thể NC, nhưng được tìm thấy ở một số bệnh nhân bị 21OHD thể SV. Đột biến phổ biến nhất gây ra 21OHD thể NC là V281L, một vi chuyển đổi liên kết với HLA-B14 và HLA-DR1, nhưng cũng được tìm thấy ở những bệnh nhân có các loại HLA khác. Vi chuyển đổi P30L được tìm thấy ở khoảng 15% đến 20% các alen 21OHD thể NC, và các đột biến R339H và P453S đã được liên kết với 21OHD thể NC; đột biến P453S là đa hình ở khoảng 20% các gen giả CYP21A1P, và do đó cũng đại diện cho một sự kiện vi chuyển đổi.
Chẩn đoán Thiếu hụt 21-Hydroxylase
Chẩn đoán trước sinh Chẩn đoán 21OHD có thể được tiếp cận trước sinh bằng cách đo các steroid do thai nhi sản xuất, chẳng hạn như 17OHP hoặc Δ4-androstenedione trong nước ối, nhưng các steroid này có thể không tăng trên phạm vi rộng của mức bình thường ở các thai nhi bị 21OHD thể SV hoặc NC. Việc định type HLA của các tế bào ối của thai nhi có thể cung cấp thông tin nếu có phân tích liên kết trước đó của một trường hợp chỉ điểm bị ảnh hưởng và cha mẹ, nhưng một số alen HLA được biểu hiện yếu trong các tế bào ối được nuôi cấy, do đó quy trình này hiếm khi được sử dụng. Giải trình tự DNA hiện được sử dụng rộng rãi, nhưng di truyền học phức tạp của locus CYP21 có nghĩa là quy trình này chỉ đáng tin cậy khi tổn thương di truyền ở một anh chị em bị ảnh hưởng trước đó hoặc cha mẹ đã được biết.
Sàng lọc sơ sinh Các chương trình sàng lọc sơ sinh trên khắp thế giới công nghiệp hóa hiện kết hợp các phép đo 17OHP để phát hiện 21OHD. Bằng cách giảm thời gian chẩn đoán, sàng lọc đã làm giảm đáng kể tỷ lệ tử vong, bệnh tật do hạ natri máu, thời gian nằm viện, khuyết tật học tập và sai sót trong việc xác định giới tính. Tuy nhiên, các chương trình sàng lọc ở các khu vực pháp lý khác nhau sử dụng các xét nghiệm 17OHP, ngưỡng và quy trình theo dõi khác nhau, do đó bác sĩ phải làm quen với các quy trình địa phương. Các hướng dẫn hiện hành khuyến nghị một xét nghiệm miễn dịch 17OHP được tiêu chuẩn hóa từ các mẫu máu khô được sử dụng cho các sàng lọc sơ sinh khác. Bởi vì 17OHP tăng ở trẻ sinh non và trẻ ốm, và ở trẻ khỏe mạnh trong 2 ngày đầu, sàng lọc thứ cấp bằng sắc ký lỏng kết hợp khối phổ song song (LC-MS/MS) được khuyến nghị cho tất cả các kết quả tăng cao. Kết quả sàng lọc ban đầu được cải thiện khi trẻ nhỏ được phân tầng theo tuổi thai thay vì cân nặng khi sinh. Bởi vì các xét nghiệm miễn dịch có thể cho kết quả dương tính giả, nếu không có LC-MS/MS, xét nghiệm theo dõi nên được thực hiện bằng nghiệm pháp ACTH tĩnh mạch. Khoảng 40% các mẫu ban đầu được báo cáo là dương tính sẽ có mức 17OHP bình thường khi được đo bằng LC-MS/MS. Việc xác định kiểu gen không được khuyến nghị cho các chương trình sàng lọc do chi phí, tỷ lệ lỗi cao, các đột biến không được phát hiện, và không có khả năng phân biệt các đột biến ở dạng cis so với trans mà không cần xác định kiểu gen của ít nhất một phụ huynh. Việc đo thêm các steroid và tỷ lệ của chúng có thể cải thiện độ đặc hiệu của sàng lọc bằng LC-MS/MS: tỷ lệ tổng của 17OHP cộng với 21-deoxycortisol chia cho mức cortisol đã xác định được tất cả trẻ em bị ảnh hưởng mà không có dương tính giả.
Chẩn đoán sau sinh Khi không có sàng lọc sơ sinh, bộ phận sinh dục không rõ ràng và/hoặc một cuộc khủng hoảng mất muối thường sẽ cảnh báo các bác sĩ nhi khoa về hầu hết các trường hợp 21OHD nặng. Các cuộc khủng hoảng mất muối thường xảy ra trong tuần thứ hai của cuộc đời, biểu hiện bằng nôn, tiêu chảy, mất nước, tăng kali máu và hạ natri máu. Đôi khi, những trẻ sơ sinh như vậy được cho là mắc các hội chứng do virus hoặc tắc nghẽn đường tiêu hóa; việc không chẩn đoán được như vậy có thể dẫn đến tử vong của trẻ. Các bé trai bị 21OHD thể SV có thể thoát khỏi chẩn đoán cho đến khi 3 đến 7 tuổi, khi chúng biểu hiện với dậy thì đồng giới sớm, tuổi xương tiến triển, và đặc trưng là tinh hoàn trước tuổi dậy thì. Các nữ thiếu niên và trưởng thành bị 21OHD thể NC có thể tham khảo ý kiến bác sĩ nội khoa, sản phụ khoa, hoặc da liễu về nam hóa, rậm lông, kinh nguyệt không đều, vô sinh, hoặc mụn trứng cá.
Thao tác chẩn đoán chính là đo 17OHP và các steroid khác để đáp ứng với ACTH tổng hợp tiêm tĩnh mạch. Các liều thông thường là 15 μg/kg ở trẻ sơ sinh, 0,125 mg ở trẻ em đến 2 tuổi, và 0,25 mg ở trẻ lớn và người lớn. 17OHP và cortisol nên được đo ở thời điểm 0 và 60 phút. Các đáp ứng của từng bệnh nhân phải được so sánh với dữ liệu phù hợp với tuổi và giới tính từ trẻ em bình thường. Các đáp ứng bình thường được trình bày trong Bảng 14.4 và Hình 14.12. Cả mức 17OHP cơ bản và sau kích thích đều tăng rõ rệt ở bệnh nhân bị 21OHD thể SW và SV. Mức cơ bản thường lớn hơn 2000 ng/dL và tăng lên hơn 5000 đến 10.000 ng/dL sau ACTH. Bệnh nhân bị 21OHD thể NC thường có mức cơ bản bình thường hoặc tăng nhẹ, nhưng có đáp ứng trên mức bình thường với kích thích ACTH, tức là 1500 đến hơn 10.000 ng/dL. Đáp ứng cortisol với ACTH không có hoặc dưới mức bình thường ở bệnh nhân bị 21OHD thể SW hoặc SV và bình thường ở bệnh nhân bị 21OHD thể NC. Mức ACTH huyết tương cơ bản có thể tăng rõ rệt ở các dạng nặng và có thể bình thường ở bệnh nhân có các dạng nhẹ hơn không bị suy thượng thận rõ ràng.
Các xét nghiệm phụ trợ khác được liệt kê trong Bảng 14.5. Sự bài tiết 17-ketosteroid trong nước tiểu thường tăng, nhưng xét nghiệm này không còn được sử dụng chung và hữu ích hơn để theo dõi liệu pháp ức chế hơn là để chẩn đoán ban đầu. Khi các steroid trong nước tiểu được đo, phải lấy một mẫu 24 giờ hoàn chỉnh, và cần phải đo đồng thời sự bài tiết creatinine để theo dõi tính đầy đủ của việc thu thập. Các mẫu nước tiểu dưới 24 giờ không chính xác về mặt định lượng do sự thay đổi theo ngày đêm trong bài tiết steroid. Phân tích hồ sơ steroid trong nước tiểu bằng các phương pháp khối phổ (thường là GC/MS-MS) thường cho thấy các chất chuyển hóa của 17OHP tăng cao: pregnanetriol, 17-OH-pregnanolone và pregnanetriolone.
Renin huyết tương, được đo bằng hoạt độ renin hoặc tổng hàm lượng, và đáp ứng của nó với việc hạn chế muối có thể đặc biệt hữu ích. Hầu hết các bệnh nhân bị 21OHD thể SV có renin huyết tương cao, tăng thêm khi hạn chế natri, xác nhận rằng những bệnh nhân này bị thiếu hụt mineralocorticoid một phần và chỉ có thể duy trì natri huyết thanh bình thường bằng cách kích thích quá mức lớp cầu.
Điều trị
Việc quản lý 21OHD vẫn còn khó khăn. Điều trị quá mức bằng glucocorticoid gây chậm tăng trưởng, ngay cả khi mức độ điều trị quá mức không đủ để tạo ra các dấu hiệu của hội chứng Cushing. Điều trị không đủ dẫn đến việc tiếp tục sản xuất quá mức androgen tuyến thượng thận, làm tăng tốc độ trưởng thành và đóng sớm của đầu xương, một lần nữa dẫn đến sự tăng trưởng bị ảnh hưởng và các biểu hiện khác của dư thừa androgen.
Liều glucocorticoid nên dựa trên tốc độ bài tiết cortisol bình thường dự kiến, khoảng 6 đến 8 mg/m2 mỗi ngày. Tuy nhiên, việc ức chế hiệu quả ACTH và sản xuất androgen tuyến thượng thận đòi hỏi liều cao hơn một chút khoảng 10 đến 15 mg/m2 mỗi ngày. Bệnh nhân mới được chẩn đoán, đặc biệt là trẻ sơ sinh, cần liều ban đầu cao hơn để ức chế trục CRH-ACTH-thượng thận hoạt động quá mức của họ. Loại glucocorticoid được sử dụng là quan trọng. Hầu hết các bảng tương đương liều glucocorticoid dựa trên sự tương đương của chúng trong các xét nghiệm chống viêm; tuy nhiên, sự tương đương về ức chế tăng trưởng không song song với sự tương đương chống viêm: các steroid tổng hợp tác dụng dài, chẳng hạn như dexamethasone, có tác dụng ức chế tăng trưởng lớn hơn không tương xứng và do đó phải tránh sử dụng trong điều trị trẻ em và thanh thiếu niên đang phát triển (Bảng 14.7). Hầu hết các chuyên gia ủng hộ việc sử dụng hydrocortisone đường uống chia ba liều hàng ngày ở trẻ em đang phát triển. Tuy nhiên, người lớn và thanh thiếu niên lớn tuổi đã đóng đầu xương có thể được quản lý bằng prednisone.
Liệu pháp mineralocorticoid trong 21OHD đưa thể tích huyết tương trở lại bình thường và loại bỏ động lực giảm thể tích đối với bài tiết ACTH. Do đó, liệu pháp mineralocorticoid thường cho phép sử dụng liều glucocorticoid thấp hơn ở những bệnh nhân bị 21OHD thể SV, tối ưu hóa sự tăng trưởng ở trẻ em và giảm tăng cân không mong muốn ở người lớn. Chế phẩm mineralocorticoid đường uống duy nhất thường có sẵn là fludrocortisone (9α-fluorocortisol). Khi đường uống không khả dụng ở những bệnh nhân bị bệnh nặng, việc thay thế mineralocorticoid được thực hiện bằng hydrocortisone tiêm tĩnh mạch cộng với natri clorua. Khoảng 20 mg hydrocortisone có tác dụng mineralocorticoid tương đương khoảng 0,1 mg 9α-fluorocortisol (xem Bảng 14.7). Liều mineralocorticoid không dựa trên khối lượng cơ thể hoặc diện tích bề mặt: trẻ sơ sinh khá không nhạy cảm với mineralocorticoid như được phản ánh bởi nồng độ aldosterone huyết thanh cao của chúng (xem Hình 14.17), và thường cần liều lớn hơn so với người lớn (0,15–0,30 mg/ngày, tùy thuộc vào việc bổ sung natri). Ở trẻ lớn hơn, liều thay thế của 9α-fluorocortisol là 0,05 đến 0,15 mg mỗi ngày. Mineralocorticoid không hiệu quả trừ khi có đủ natri được cung cấp cho các ống thận, do đó cần bổ sung thêm muối, thường là 1 đến 2 g NaCl/ngày ở trẻ sơ sinh. Một số bệnh nhân trưởng thành bị 21OHD thể SW có thể ngừng thay thế mineralocorticoid và bổ sung muối; điều này có lẽ phản ánh sự nhạy cảm với mineralocorticoid tăng lên ở người lớn, việc họ tự do tiếp cận thực phẩm mặn, và sự cảm ứng của các enzyme 21-hydroxyl hóa ngoại thượng thận ở gan.
Bảng 14.7. Hiệu lực của các Steroid điều trị khác nhau (Đặt tương đối so với hiệu lực của Cortisol)
| Steroid | Tác dụng Glucocorticoid chống viêm | Tác dụng Glucocorticoid kìm hãm tăng trưởng | Tác dụng Mineralocorticoid giữ muối | Thời gian bán hủy trong huyết tương (phút) | Thời gian bán hủy sinh học (giờ) |
|---|---|---|---|---|---|
| Cortisol (hydrocortisone) | 1.0 | 1.0 | 1.0 | 80-120 | 8 |
| Cortisone acetate (uống) | 0.8 | 0.8 | 0.8 | 80-120 | 8 |
| Cortisone acetate (IM) | 0.8 | 1.3 | 0.8 | >18 | |
| Prednisone | 4 | 5 | 0.25 | >200 | 16-36 |
| Prednisolone | 4 | 0.25 | 120-300 | 16-36 | |
| Methyl prednisolone | 5 | 7.5 | 0.4 | ||
| Betamethasone | 25 | 0 | 130-330 | ||
| Triamcinolone | 5 | 0 | |||
| Dexamethasone | 30 | 80 | 0 | 150-300 | 36-54 |
| 9α-fluorocortisone | 15 | >200 | |||
| DOC acetate | 0 | 20 | |||
| Aldosterone | 0.3 | 200-1000 |
DOC, Deoxycorticosterone; IM, tiêm bắp.
Quản lý lâu dài đòi hỏi phải theo dõi lâm sàng và xét nghiệm cẩn thận. Sự tăng trưởng nên được theo dõi sau mỗi 3 đến 4 tháng ở trẻ em; tuổi xương nên được đánh giá hàng năm. Mỗi lần khám nên đi kèm với việc đo huyết áp, renin huyết tương và androstenedione, DHEA, DHEAS, và testosterone trong huyết thanh. 17OHP huyết tương thường được đo nhưng có thể khó giải thích do sự biến đổi của nó theo thời gian dùng liều glucocorticoid, sự biến đổi theo ngày đêm của nó, và sự đáp ứng quá mức của nó với stress (ví dụ, các chuyến đi đến phòng khám).
Điều trị thử nghiệm trước sinh cho Thiếu hụt 21-Hydroxylase
Bởi vì việc điều trị 21OHD tập trung vào việc ức chế trục HPA bằng glucocorticoid, việc ức chế trục HPA của thai nhi đã được đề xuất bằng cách cho mẹ dùng glucocorticoid qua nhau thai. Thai nhi nữ bị CAH bắt đầu bị nam hóa vào khoảng 6 đến 8 tuần thai, thời điểm mà tinh hoàn của thai nhi nam bình thường sản xuất testosterone, gây ra sự hợp nhất của các nếp gấp môi-bìu, sự phát triển của củ sinh dục thành dương vật, và sự hình thành của niệu đạo dương vật. Tuyến thượng thận của thai nhi nữ bị 21OHD có thể sản xuất nồng độ androgen gần bằng nồng độ của một nam giới bình thường, dẫn đến các mức độ khác nhau của sự nam hóa bộ phận sinh dục ngoài; việc ức chế sinh tổng hợp steroid của tuyến thượng thận thai nhi ở một thai nhi nữ bị 21OHD về mặt lý thuyết có thể cải thiện sự nam hóa. Dexamethasone đã được sử dụng cho mục đích này, nhưng việc điều trị như vậy vẫn còn mang tính thử nghiệm và gây nhiều tranh cãi. Cách tiếp cận này đã được đề xuất khi cha mẹ được biết là dị hợp tử do trước đó đã có một đứa con bị ảnh hưởng. Tuy nhiên, ngay cả trong những thai kỳ như vậy, chỉ có một trong bốn thai nhi sẽ bị 21OHD, và vì việc điều trị trước sinh không cần thiết cho thai nhi nam bị 21OHD, chỉ có một trong tám thai kỳ của cha mẹ dị hợp tử sẽ mang thai nhi nữ bị ảnh hưởng có khả năng được hưởng lợi từ việc điều trị trước sinh. Việc điều trị phải được bắt đầu vào khoảng 6 tuần sau khi thụ thai (8 tuần vô kinh); việc xác định giới tính của thai nhi bằng DNA của thai nhi trong máu của mẹ có thể được thực hiện vào khoảng thời gian này, cho phép không điều trị hoặc ngừng sớm điều trị dexamethasone cho thai nhi nam. Tuy nhiên, ở thai nhi nữ, sinh thiết gai rau để chẩn đoán DNA không thể được thực hiện cho đến 12 đến 13 tuần, và thời gian trả kết quả trong các phòng thí nghiệm DNA chậm nếu các đột biến do cả hai cha mẹ mang chưa được xác định trước đó. Do đó, bảy trong tám thai kỳ có thể được điều trị một cách không cần thiết để điều trị cho một thai nhi nữ bị ảnh hưởng. Một số trung tâm tiên tiến có thể tránh điều trị cho thai nhi nam bằng cách xác định DNA nhiễm sắc thể Y trong tuần hoàn của mẹ vào tuần thứ 6; tuy nhiên, điều này chỉ làm giảm, chứ không loại bỏ, số lượng thai nhi bị phơi nhiễm dexamethasone một cách không cần thiết, và khả năng áp dụng chung của nó vẫn chưa được xác định.
Đạo đức của việc điều trị trước sinh như vậy vẫn còn gây nhiều tranh cãi. Cơ sở lý luận là dexamethasone, không được chuyển hóa bởi 11βHSD2 của nhau thai, sẽ đi qua nhau thai, ức chế ACTH của thai nhi, và do đó ức chế sinh tổng hợp steroid của tuyến thượng thận. Tuy nhiên, không rõ chính xác khi nào vùng dưới đồi của thai nhi bắt đầu sản xuất CRH, khi nào tuyến yên của thai nhi bắt đầu sản xuất ACTH, liệu tất cả sản xuất ACTH của thai nhi có được điều hòa bởi CRH hay không, hoặc liệu các hormone này có thể bị ức chế bởi dexamethasone ở thai nhi giai đoạn đầu hay không. Mặc dù có bằng chứng cho thấy liều dược lý của glucocorticoid không gây hại cho phụ nữ mang thai, có rất ít dữ liệu cho thai nhi. Phụ nữ mang thai mắc các bệnh như hội chứng thận hư và lupus ban đỏ hệ thống thường được điều trị bằng prednisone, không đến được thai nhi vì nó bị bất hoạt bởi 11βHSD của nhau thai. Việc điều trị một thai nhi bị 21OHD đòi hỏi phải sử dụng các steroid có chứa fluor thoát khỏi sự chuyển hóa bởi 11βHSD, và có rất ít dữ liệu về việc sử dụng lâu dài các tác nhân như vậy trong suốt thai kỳ.
Giao thức thông thường sử dụng liều dexamethasone là 20 μg/kg trọng lượng cơ thể của mẹ (tối đa 1,5 mg/ngày); đối với một phụ nữ 60 kg, đây là 1,2 mg, gấp khoảng 6 lần liều thay thế sinh lý. Tuy nhiên, thai nhi thường phát triển trong sự hiện diện của nồng độ cortisol rất thấp chỉ khoảng 20 đến 60 nmol/L (0,7–2,0 μg/dL), chỉ bằng khoảng 10% mức tương ứng của mẹ. Do đó, các liều được sử dụng trong điều trị trước sinh dường như đạt được nồng độ glucocorticoid hiệu quả gấp khoảng 60 lần sinh lý cho thai nhi. Lợi ích tiềm năng của việc điều trị trước sinh là giảm hoặc loại bỏ sự nam hóa của bộ phận sinh dục ngoài và não, giảm nguy cơ nhầm lẫn giới tính và nhu cầu phẫu thuật. Các nghiên cứu trên động vật cho thấy dexamethasone được dùng trước sinh làm tăng nguy cơ hở hàm ếch; làm suy giảm sự phát triển của não, thận; và tế bào đảo tụy; giảm cân nặng khi sinh; và tăng nguy cơ tăng huyết áp. Các nghiên cứu trên người đã bị hạn chế về phương pháp luận hoặc quy mô, nhưng các nghiên cứu ở Thụy Điển cho thấy rằng trẻ em được điều trị trước sinh không bị 21OHD có trí nhớ làm việc kém hơn và tự nhận thức về năng lực học tập kém hơn, và tăng lo âu xã hội tự đánh giá; cũng có những ảnh hưởng nhẹ đến tính xã hội và hành vi vai trò giới ở các bé trai, dẫn đến các nhà điều tra Thụy Điển ngừng nghiên cứu thêm về phương pháp điều trị này. Các phương pháp tiếp cận thay thế, chẳng hạn như chẩn đoán di truyền tiền cấy phôi, cũng có một số rủi ro. Do đó, các rủi ro của việc điều trị trước sinh dường như lớn hơn lợi ích, và tất cả các hiệp hội y khoa đã xem xét vấn đề này tính đến năm 2018 đều khuyến nghị rằng việc điều trị như vậy chỉ nên được thực hiện với sự đồng ý hoàn toàn của cha mẹ sau khi được thông báo đầy đủ theo các giao thức nghiên cứu được Hội đồng Đánh giá Thể chế (IRB) phê duyệt, tại các trung tâm có đủ bệnh nhân như vậy để đạt được kết quả có ý nghĩa.
Điều trị thử nghiệm sau sinh cho Thiếu hụt 21-Hydroxylase
Sự tăng trưởng bị ảnh hưởng ở hầu hết trẻ em bị 21OHD. Việc sử dụng glucocorticoid tác dụng ngắn và bổ sung mineralocorticoid có giúp ích, nhưng chiều cao trưởng thành thường thấp hơn 1 SD hoặc hơn so với chiều cao dự đoán. Sự mất chiều cao trong 21OHD một phần là do tác động của các steroid sinh dục lên việc đóng đầu xương và một phần là do sự kháng lại tác động của hormone tăng trưởng do glucocorticoid gây ra; những thời điểm quan trọng nhất khi chiều cao bị mất là trong 2 năm đầu đời và trong giai đoạn tăng trưởng vượt bậc của tuổi dậy thì. Do đó, một số nghiên cứu đã đề cập đến việc tối ưu hóa chiều cao cuối cùng của trẻ em bị CAH.
Thuốc kháng androgen và Thuốc ức chế Aromatase Các loại thuốc này đã được thử nghiệm với sự hiện diện của mineralocorticoid liều thấp và liều glucocorticoid thấp hơn. Bởi vì estrogen là hormone chính trong việc thúc đẩy sự hợp nhất đầu xương, việc ức chế chuyển đổi androgen thành estrogen bằng một chất ức chế aromatase (testolactone) thúc đẩy sự tăng trưởng, trong khi chất kháng androgen (flutamide) cải thiện sự nam hóa. Cách tiếp cận này cho phép sử dụng liều thay thế sinh lý của hydrocortisone (8 mg/m2/ngày), thay vì liều trên sinh lý thông thường là 12 đến 15 mg/m2/ngày, ủng hộ sự tăng trưởng bình thường. Tương tự, các thử nghiệm đang được tiến hành với abiraterone để ức chế P450c17, do đó cản trở việc tổng hợp tất cả các steroid sinh dục. Các loại thuốc này đắt tiền và không được chấp thuận cho mục đích sử dụng này, tính an toàn và hiệu quả lâu dài chưa được xác định, và các chất kháng androgen có khả năng gây độc cho gan. Do đó, giống như tất cả các liệu pháp thử nghiệm, các phương pháp tiếp cận này chỉ nên được theo đuổi trong các thử nghiệm tiền cứu, có kiểm soát được Hội đồng Đánh giá Thể chế phê duyệt.
Liệu pháp Hormone tăng trưởng và Chất chủ vận Hormone giải phóng Gonadotropin Các loại thuốc thúc đẩy tăng trưởng đã được đề xuất ở trẻ em gần tuổi dậy thì vì liệu pháp hormone tăng trưởng dược lý có thể khắc phục một phần tác động của liều glucocorticoid cao hơn, và chất chủ vận GnRH sẽ làm chậm quá trình dậy thì, cho phép có nhiều thời gian hơn để phát triển. Các nghiên cứu sơ bộ nhỏ với các tác nhân này rất hứa hẹn, nhưng cả hai tác nhân đều đắt tiền và đều không được chấp thuận cho mục đích sử dụng này; cần có các thử nghiệm tiền cứu có kiểm soát.
Cắt bỏ tuyến thượng thận Bởi vì tuyến thượng thận mang các đột biến CYP21A2 nặng không thể sản xuất aldosterone hoặc cortisol, người ta đã lập luận rằng các tuyến bị ảnh hưởng này gây hại nhiều hơn là lợi, và nên được cắt bỏ. Trong số 18 bệnh nhân được cắt bỏ tuyến thượng thận, năm người bị cơn suy thượng thận cấp khi điều trị không tối ưu và hai người bị hạ đường huyết trong các bệnh xen kẽ. Do nguy cơ cao về bệnh tật và tử vong sau phẫu thuật, cách tiếp cận này không được khuyến nghị.
Các tổn thương ở Isozyme của P450c11
Thiếu hụt 11β-Hydroxylase
Thiếu hụt 11β-Hydroxylase (11OHD) là dạng CAH tăng androgen. Có hai isozyme của 11-hydroxylase. P450c11β, được mã hóa bởi gen CYP11B1, được biểu hiện ở lớp bó và lớp cầu, nơi nó xúc tác cho quá trình 11β-hydroxyl hóa của 11-deoxycortisol thành cortisol và của DOC thành corticosterone; nó cũng có một số hoạt tính 18-hydroxylase, nhưng không có hoạt tính 18-methyl oxidase. P450c11AS (aldosterone synthase) được mã hóa bởi gen CYP11B2, chỉ được tìm thấy ở lớp cầu và xúc tác cho 11β-hydroxyl hóa, 18-hydroxyl hóa, và 18-oxy hóa; do đó nó là enzyme duy nhất cần thiết để chuyển đổi DOC thành aldosterone. Các gen CYP11B1 và CYP11B2 giống nhau 93% và liên kết chặt chẽ trên nhiễm sắc thể 8q22, nhưng, không giống như các gen CYP21A1P và CYP21A2 trong locus HLA, sự tái tổ hợp di truyền của chúng rất hiếm. Thiếu hụt nặng P450c11β làm giảm bài tiết cortisol, gây ra CAH và nam hóa các cá thể nữ bị ảnh hưởng. Khiếm khuyết trong con đường đến cortisol dẫn đến tích tụ 11-deoxycortisol và khiếm khuyết trong con đường 17-deoxy trong quá trình tổng hợp corticosterone ở lớp bó có thể dẫn đến sản xuất quá mức DOC; nồng độ 11-deoxycortisol tăng cao đáp ứng quá mức với ACTH xác lập chẩn đoán. Bởi vì DOC là một mineralocorticoid, những bệnh nhân này có thể giữ natri. Mặc dù DOC kém hiệu lực hơn aldosterone, nó được tiết ra ở mức độ cao trong 11OHD, do đó muối được giữ lại và natri huyết thanh vẫn bình thường. Sản xuất quá mức DOC thường dẫn đến tăng huyết áp; kết quả là, 11OHD thường được gọi là dạng CAH tăng huyết áp khi được phát hiện ở trẻ lớn. Tuy nhiên, trẻ sơ sinh thường biểu hiện mất muối nhẹ, thoáng qua, do sự kháng mineralocorticoid bình thường ở trẻ sơ sinh (Hình 14.18); điều này có thể dẫn đến chẩn đoán và điều trị không chính xác. Do đó, có thể có sự tương quan kém giữa nồng độ DOC, kali huyết thanh và huyết áp hoặc giữa mức độ nam hóa ở các cá thể nữ bị ảnh hưởng và các biểu hiện điện giải và tim mạch. Trẻ sơ sinh cũng có thể có nồng độ 17OHP tăng cao, có lẽ là một hiện tượng “dự phòng” của nồng độ 11-deoxycortisol cao ức chế P450c21, do đó 11OHD có thể được phát hiện trong sàng lọc sơ sinh cho 21OHD. Chẩn đoán được xác lập bằng cách chứng minh nồng độ DOC và 11-deoxycortisol cơ bản tăng cao, đáp ứng quá mức với ACTH; hoạt độ renin huyết tương bình thường hoặc bị ức chế cũng là một dấu hiệu của bệnh này.
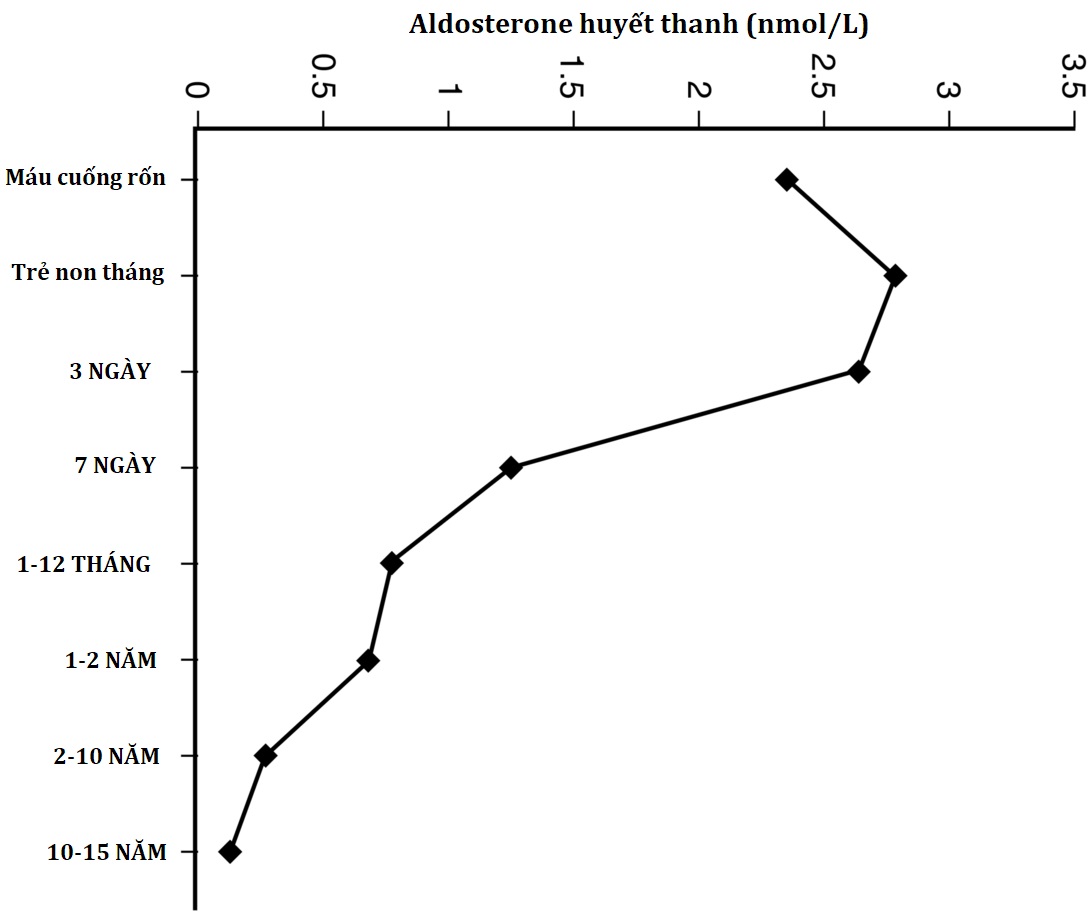
Hình 14.18: Nồng độ aldosterone theo tuổi.
11OHD dường như gây ra khoảng 5% các trường hợp CAH ở những người gốc châu Âu, nhưng phổ biến hơn ở cả các quần thể Hồi giáo và Do Thái Trung Đông, và chiếm tới 13,5% các trường hợp CAH ở Thổ Nhĩ Kỳ, nơi 13 đột biến CYP11B1 khác nhau đã được tìm thấy. Hơn 100 đột biến CYP11B1 đã được báo cáo. Các đột biến CYP11B1 phổ biến bao gồm R448H ở người Do Thái Ma-rốc, và Q356X và G379V ở người Ả Rập Tunisia. Một dạng 11OHD nhẹ hơn, không cổ điển, tương tự như 21OHD không cổ điển, đã được báo cáo ở những phụ nữ không có triệu chứng khác bị rậm lông, nam hóa và kinh nguyệt không đều, và ở nam giới bị dậy thì sớm. Tuy nhiên, 11OHD không cổ điển thực sự là hiếm; chỉ có hai trong số năm phụ nữ tăng androgen máu có giá trị 11-deoxycortisol cao hơn 3 lần so với phân vị thứ 95, để đáp ứng với kích thích bằng ACTH, có đột biến P450c11β, tất cả đều giữ lại 15% đến 37% hoạt tính bình thường. Xét nghiệm ACTH lặp lại ở hai trong số ba phụ nữ không có đột biến cho thấy giá trị 11-deoxycortisol thấp hơn nhiều (nhưng vẫn tăng). Do đó, cũng như trong trường hợp thiếu hụt 3βHSD không cổ điển, một đáp ứng steroid bất thường với ACTH không đủ để chẩn đoán một tổn thương di truyền. Điều trị 11OHD bao gồm thay thế glucocorticoid với liều tương tự như liều được sử dụng trong 21OHD, nhưng không thay thế mineralocorticoid.
Thiếu hụt Corticosterone Methyl Oxidase
P450c11AS được biểu hiện độc quyền ở lớp cầu, nơi nó xúc tác cho các hoạt động 11β-hydroxylase, 18-hydroxylase và 18 methyl oxidase. Các rối loạn của P450c11AS gây ra cái gọi là thiếu hụt corticosterone methyl oxidase (CMO), trong đó sinh tổng hợp aldosterone bị suy giảm, trong khi lớp bó tiếp tục sản xuất corticosterone và DOC. Sự vắng mặt của sinh tổng hợp aldosterone thường sẽ dẫn đến một cuộc khủng hoảng mất muối ở trẻ sơ sinh, tại thời điểm đó tốc độ bài tiết DOC bình thường không đủ để đáp ứng nhu cầu mineralocorticoid của trẻ sơ sinh (tương tự như trẻ sơ sinh bị 11OHD). Những trẻ sơ sinh này thường biểu hiện với hạ natri máu, tăng kali máu và nhiễm toan chuyển hóa, nhưng hội chứng mất muối thường ít nghiêm trọng hơn so với bệnh nhân bị 21OHD hoặc CAH dạng mỡ do sự bài tiết liên tục của DOC. Những bệnh nhân này có thể tự phục hồi và phát triển đến tuổi trưởng thành mà không cần điều trị, có lẽ phản ánh sự nhạy cảm ngày càng tăng đối với tác động của mineralocorticoid khi phát triển, được thể hiện bằng sự giảm aldosterone huyết thanh theo tuổi (xem Hình 14.18). Phù hợp với điều này, hoạt độ renin huyết tương tăng rõ rệt ở trẻ em bị ảnh hưởng nhưng có thể bình thường ở người lớn bị ảnh hưởng.
Thiếu hụt CMOI là kết quả của sự mất hoàn toàn hoạt tính của P450c11AS do đó không có hoạt tính 18-hydroxylase hoặc 18 methyl oxidase nào tồn tại, loại bỏ sinh tổng hợp 18OH-corticosterone và aldosterone, trong khi vẫn duy trì sinh tổng hợp corticosterone bởi P450c11β. Do đó, chẩn đoán cho thiếu hụt CMOI thường dựa trên tỷ lệ corticosterone so với 18OH-corticosterone tăng. Thiếu hụt CMOII là kết quả của các đột biến thay thế axit amin trong P450c11AS làm mất chọn lọc hoạt tính 18-methyl oxidase, trong khi vẫn duy trì hoạt tính 18-hydroxylase: 18OH-corticosterone huyết thanh cao và aldosterone thấp. Chẩn đoán thiếu hụt CMOII đòi hỏi 18OH-corticosterone tăng và nồng độ aldosterone rất thấp. Thiếu hụt CMOII phổ biến ở người Do Thái Sephardic gốc Iran, nơi tất cả các cá nhân bị ảnh hưởng dường như đồng hợp tử cho hai đột biến khác nhau, R181W và V385A. Các thành viên gia đình đồng hợp tử chỉ cho một trong những đột biến này không bị ảnh hưởng về mặt lâm sàng; cả hai đột biến đều cần thiết để gây bệnh. Các phát hiện lâm sàng và hormone trong CMOI và CMOII chồng chéo lên nhau, và các hội chứng này nên được coi là các mức độ nghiêm trọng khác nhau trên một phổ lâm sàng liên tục, tương tự như phổ lâm sàng chồng chéo của 21OHD.
Tăng Aldosteron có thể ức chế bằng Glucocorticoid
Mặc dù các chuyển đổi gen CYP11B rất hiếm, một sự nhân đôi gen bất thường gây ra tăng aldosteron có thể ức chế bằng glucocorticoid. Một sự kiện tái tổ hợp tương đồng tạo ra một gen CYP11B thứ ba hợp nhất DNA sườn 5′ của gen CYP11B1 cho P450c11β vào gen CYP11B2 cho P450c11AS, do đó đặt sự điều hòa của P450c11AS dưới sự kiểm soát của ACTH thay vì hệ renin-angiotensin, do đó những bệnh nhân này tạo ra P450c11AS để đáp ứng với sinh lý lẽ ra phải kích thích P450c11β. P450c11AS dư thừa gây ra tăng aldosteron và tăng huyết áp; điều này sau đó có thể bị ức chế bởi sự ức chế ACTH bằng glucocorticoid, điều thường ức chế P450c11β, do đó có tên là “tăng huyết áp có thể chữa bằng glucocorticoid”; rối loạn này dường như chiếm khoảng 2% các trường hợp tăng huyết áp.
Các tổn thương ở Isozyme của 11β-Hydroxysteroid Dehydrogenase
Cortisone và prednisone là các tiền hormone không hoạt động phải được khử thành cortisol hoặc prednisolone để liên kết và kích hoạt GR. Sự chuyển đổi qua lại của các keto- và hydroxysteroid này được xúc tác bởi hai isozyme của 11β-hydroxysteroid dehydrogenase, 11βHSD1 (được mã hóa bởi HSD11B1) và 11βHSD2 (được mã hóa bởi HSD11B2). Cả hai enzyme đều có thể đảo ngược trong ống nghiệm, do đó cả hai đều có thể hoạt động như một oxidase hoặc một reductase, tùy thuộc vào sự sẵn có của các đồng yếu tố, nhưng trong các tình huống sinh lý, 11βHSD1 thường hoạt động để kích hoạt cortisone thành cortisol, trong khi 11βHSD2 đảo ngược sự kích hoạt này. 11βHSD1 chủ yếu được biểu hiện ở gan và mỡ, và 11βHSD2 được biểu hiện ở các mô đáp ứng với mineralocorticoid, nơi nó bất hoạt cortisol, cho phép nồng độ aldosterone thấp kích hoạt các MR (cũng liên kết với cortisol); 11βHSD2 không hoạt động đối với aldosterone, DOC và fludrocortisol. Sự quan tâm đến các enzyme này vượt xa các trạng thái thiếu hụt của chúng, vì chúng đóng vai trò trung tâm trong chuyển hóa; điều này đã kích thích sự quan tâm đến việc sử dụng các chất ức chế 11βHSD1 để điều trị hội chứng chuyển hóa, nhưng các tác nhân như vậy chưa có sẵn trên lâm sàng.
Các tổn thương ở 11βHSD1—Thiếu hụt (Biểu kiến) Cortisone Reductase
Hoạt tính 11βHSD1 bị khiếm khuyết, được chẩn đoán bằng tỷ lệ giảm của các chất chuyển hóa cortisol trong nước tiểu so với các chất chuyển hóa của cortisone, làm suy giảm phản hồi cortisol ở trục hạ đồi/tuyến yên, làm tăng bài tiết ACTH và do đó làm tăng bài tiết steroid C19 của tuyến thượng thận, dẫn đến tăng androgen, dậy thì sớm và buồng trứng đa nang. Những bệnh nhân này có thể có các đột biến dị hợp tử (K187N, R137C) trong gen HSD11B1 mã hóa 11βHSD1; vì 11βHSD1 thường hoạt động như một dimer, sự hiện diện của một số protein đột biến có thể gây ra các hiệu ứng trội âm. Ngoài ra, một tổn thương di truyền có thể được tìm thấy trong gen H6PDH, mã hóa H6PDH, enzyme tạo ra NADPH được 11βHSD1 sử dụng trong lòng mạng lưới nội chất. Mặc dù ban đầu người ta cho rằng các đột biến ở cả 11βHSD1 và H6PDH tương tác để gây ra bệnh này, các đột biến của H6PDH ở bệnh nhân dường như biểu hiện với một kiểu hình nặng hơn và đủ để gây ra rối loạn này, và chỉ một vài trường hợp là do đột biến ở HSD11B1.
Các tổn thương ở 11βHSD2—Dư thừa Mineralocorticoid biểu kiến
Bệnh nhân bị dư thừa mineralocorticoid biểu kiến (AME) có tăng huyết áp tăng thể tích, giữ muối và kiềm hóa hạ kali máu—bức tranh cổ điển của cường aldosteron—nhưng với hoạt độ renin huyết tương bị ức chế và không có mineralocorticoid huyết thanh đo được do các đột biến lặn của 11βHSD2. Khoảng 30 đột biến khác nhau ở 11βHSD2 đã được mô tả ở khoảng 60 bệnh nhân bị AME, và những người mang gen dị hợp tử có thể có nguy cơ tăng huyết áp cao hơn. Các đặc điểm điển hình của trẻ em bị AME bao gồm chậm phát triển, dậy thì muộn, khát nhiều, tiểu nhiều, yếu cơ và tăng huyết áp. Tăng huyết áp nghiêm trọng, thường gây tổn thương cơ quan đích ở tuổi sớm. Chẩn đoán được thực hiện từ tỷ lệ cao của các chất chuyển hóa cortisol so với cortisone trong nước tiểu. Điều trị bao gồm đối kháng MR bằng spironolactone, điều chỉnh hạ kali máu, chế độ ăn ít muối và thuốc lợi tiểu, nhưng chỉ thành công một phần, và 10% bệnh nhân tử vong do tai biến mạch máu não.
Thiếu hụt PAPSS2
Tuyến thượng thận, tuyến sinh dục và não tạo ra DHEA, nhưng chỉ có tuyến thượng thận sulfat hóa hiệu quả DHEA thành DHEAS, được xúc tác bởi SULT2A1; DHEAS có thể được khử sulfat trở lại thành DHEA bởi steroid sulfatase. SULT2A1 sử dụng PAPS làm chất cho sulfat, chất này lại được tổng hợp bởi PAPSS2 (xem Mục II). Sự sulfat hóa bởi SULT2A1 chiếm ưu thế, đặc biệt là ở lớp lưới; hoạt động của steroid sulfatase đóng một vai trò tương đối nhỏ trong tuyến thượng thận, và sự thiếu hụt của nó gây ra bệnh vảy cá liên kết X. Thiếu hụt SULT2A1 đã bị nghi ngờ ở một bé gái bị biến dạng đốt sống nhẹ, dậy thì sớm, tuổi xương tiến triển và rậm lông; DHEA, androstenedione và testosterone của cô bé tăng cao nhưng DHEAS không có. Gen SULT2A1 của bệnh nhân này bình thường, nhưng có sự dị hợp tử phức hợp cho các đột biến PAPSS2. Thiếu hụt PAPSS2 làm cạn kiệt PAPS của tuyến thượng thận, làm tăng sản xuất DHEA không liên hợp, chất này bị tác động bởi 3βHSD1 của gan, tạo ra androgen. Thiếu hụt PAPSS2 đã được báo cáo trong một gia đình Pakistan bị một khiếm khuyết xương hiếm gặp được gọi là “loạn sản đầu xương-đốt sống (SEMD)/brachyolmia loại 4,”, nhưng các nghiên cứu nội tiết chưa được thực hiện ở những cá nhân đó; những bệnh nhân này và các bệnh nhân tương tự khác đã mất hoàn toàn hoạt tính của PAPSS2.
Suy thượng thận
Nhiều tình trạng sẽ gây ra suy thượng thận, bao gồm CAH, suy tuyến yên với thiếu hụt ACTH, và các rối loạn thượng thận nguyên phát. Suy thượng thận nguyên phát thường được gọi là bệnh Addison, nhưng đây là một thuật ngữ mơ hồ bao gồm nhiều rối loạn. Cho đến Chiến tranh thế giới thứ hai, hầu hết bệnh nhân mắc “bệnh Addison” đều bị lao tuyến thượng thận, nhưng hơn 80% bệnh nhân người lớn đương thời bị viêm thượng thận tự miễn, do đó thuật ngữ bệnh Addison hiện được sử dụng rộng rãi để chỉ một nguyên nhân tự miễn hoặc vô căn.
Phổ các rối loạn thượng thận biểu hiện ở trẻ sơ sinh, trẻ em và thanh thiếu niên khác với phổ biểu hiện ở người lớn (Bảng 14.8). CAH và bệnh thượng thận tự miễn chiếm tỷ lệ lớn nhất trong các trường hợp, nhưng một số nguyên nhân di truyền về phát triển và chuyển hóa gây suy thượng thận cũng khá phổ biến. Chẩn đoán một số rối loạn này rất quan trọng để đánh giá các đặc điểm liên quan tiềm ẩn, bắt đầu quản lý lâu dài và thực hiện tư vấn di truyền. Các rối loạn thượng thận thường được chia thành các nguyên nhân mạn tính và cấp tính, nhưng nhiều biểu hiện cấp tính phản ánh một quá trình mạn tính hoặc phát triển tiềm ẩn chưa được chẩn đoán (Bảng 14.9). Các biểu hiện cấp tính có thể được khởi phát bởi bệnh xen kẽ, chấn thương, hoặc phẫu thuật, với lượng dịch và natri đưa vào kém. Một cách khác để nhóm các rối loạn thượng thận bao gồm các dạng hội chứng và không hội chứng, với các dạng hội chứng có liên quan đến các dấu hiệu và triệu chứng bổ sung của các hệ cơ quan khác.
Bảng 14.8. Các nguyên nhân gây Suy thượng thận
| SUY THƯỢNG THẬN NGUYÊN PHÁT |
|---|
| Tăng sản thượng thận bẩm sinh |
| Các rối loạn tự miễn |
| Viêm thượng thận tự miễn |
| Hội chứng đa tuyến nội tiết tự miễn |
| Thiểu sản thượng thận bẩm sinh |
| Thiểu sản thượng thận liên kết X |
| Các nguyên nhân khác (SF1, hội chứng IMAGE) |
| Các hội chứng kháng ACTH |
| Thiếu hụt glucocorticoid gia đình, type 1 và 2 |
| Hội chứng Triple A (Allgrove) |
| Các rối loạn chuyển hóa |
| Loạn dưỡng chất trắng thượng thận |
| Các rối loạn sinh tổng hợp peroxisome (ví dụ, Zellweger) |
| Chuyển hóa cholesterol (Smith-Lemli-Opitz, Wolman) |
| Bệnh ty thể (Kearn-Sayres, xóa ty thể) |
| Các rối loạn nhiễm trùng |
| Nhiễm trùng huyết |
| Lao |
| Nhiễm nấm |
| Nhiễm virus |
| Các nguyên nhân thâm nhiễm/phá hủy |
| Xuất huyết |
| Bệnh amyloidosis, sarcoidosis, di căn |
| Các thuốc ức chế sinh tổng hợp steroid |
| SUY THƯỢNG THẬN THỨ PHÁT |
| Các khối u vùng dưới đồi, xạ trị hoặc phẫu thuật |
| Suy tuyến yên |
| Thiếu hụt ACTH đơn độc |
| Các khiếm khuyết trong tổng hợp và xử lý POMC |
| Ngừng liệu pháp glucocorticoid |
ACTH, Hormone vỏ thượng thận; IMAGE, chậm tăng trưởng trong tử cung, loạn sản hành xương, thiểu sản thượng thận, dị tật sinh dục-tiết niệu; POMC, proopiomelanocortin; SF1, yếu tố sinh steroid 1.
Suy thượng thận nguyên phát cấp tính
Cơn suy thượng thận cấp xảy ra phổ biến nhất ở trẻ bị suy thượng thận mạn tính chưa được chẩn đoán phải chịu thêm một stress nặng, chẳng hạn như bệnh nặng, chấn thương, hoặc phẫu thuật. Các triệu chứng và dấu hiệu chính khi nhập viện bao gồm đau bụng, sốt, hạ đường huyết kèm co giật, yếu, thờ ơ, buồn nôn, nôn, chán ăn, hạ natri máu, hạ clo máu, nhiễm toan, tăng kali máu, hạ huyết áp, sốc, trụy tim mạch và tử vong. Điều trị bao gồm hồi sức dịch và điện giải, liều glucocorticoid dồi dào, thay thế glucocorticoid và mineralocorticoid mạn tính, và điều trị bệnh khởi phát.
Xuất huyết thượng thận ồ ạt, với sốc do mất máu, có thể xảy ra ở trẻ sơ sinh lớn có cuộc đẻ sang chấn. Một khối u ở hông thường có thể sờ thấy và có thể được phân biệt với huyết khối tĩnh mạch thận bằng tiểu máu vi thể thay vì đại thể; chẩn đoán sau đó được xác nhận bằng CT hoặc siêu âm. Xuất huyết thượng thận ồ ạt thường liên quan đến nhiễm não mô cầu (hội chứng Waterhouse-Friederichsen). Viêm màng não thường, nhưng không phải luôn luôn, có mặt. Ban xuất huyết chấm đặc trưng của nhiễm não mô cầu có thể tiến triển nhanh chóng thành các mảng xuất huyết lớn; huyết áp giảm và nhịp thở trở nên khó khăn, thường dẫn đến hôn mê và tử vong nhanh chóng. Can thiệp ngay lập tức bằng dịch truyền tĩnh mạch, kháng sinh và glucocorticoid không phải lúc nào cũng thành công. Một cơn suy thượng thận tương tự cũng có thể xảy ra hiếm khi với nhiễm trùng huyết do Streptococcus, Pneumococcus, Pseudomonas, bạch hầu, và các chủng Staphylococcus aureus nhạy cảm và kháng methicillin. Xuất huyết thượng thận cũng đã được báo cáo với hội chứng kháng phospholipid và ở những bệnh nhân đang điều trị chống đông.
Suy thượng thận nguyên phát mạn tính
Các rối loạn tự miễn
Viêm thượng thận tự miễn thường thấy nhất ở người lớn từ 25 đến 45 tuổi, khoảng 60% đến 70% trong số đó là phụ nữ, với tỷ lệ hiện mắc ở người lớn khoảng 1 trên 25.000. Sự phá hủy tự miễn của các mô nội tiết khác thường liên quan đến viêm thượng thận tự miễn. Suy thượng thận mạn tính được gợi ý bởi tăng cân kém hoặc sụt cân, yếu, mệt mỏi, chán ăn, hạ huyết áp, hạ natri máu, hạ clo máu, tăng kali máu, bệnh tật thường xuyên, buồn nôn, và các phàn nàn về đường tiêu hóa mơ hồ (xem Bảng 14.9), phản ánh sự thiếu hụt mạn tính của cả glucocorticoid và mineralocorticoid. Giai đoạn đầu của viêm thượng thận tự miễn, người ta có thể thấy các dấu hiệu thiếu hụt glucocorticoid (yếu, mệt mỏi, sụt cân, hạ đường huyết, chán ăn) mà không có dấu hiệu thiếu hụt mineralocorticoid (hạ natri máu, tăng kali máu, nhiễm toan, nhịp tim nhanh, hạ huyết áp, điện thế thấp trên điện tâm đồ, tim nhỏ trên X-quang ngực), hoặc bằng chứng về thiếu hụt mineralocorticoid mà không có thiếu hụt glucocorticoid. Do đó, một biểu hiện lâm sàng ban đầu chỉ ảnh hưởng đến một loại steroid thượng thận không có nghĩa là nó sẽ không bị ảnh hưởng trong dài hạn. Các triệu chứng được liệt kê trong Bảng 14.9 có thể được thấy trong suy thượng thận mạn tính nguyên phát hoặc thứ phát. Trong suy thượng thận mạn tính nguyên phát, nồng độ cortisol huyết tương thấp kích thích sự tăng tiết ACTH và các peptide POMC khác, bao gồm các dạng khác nhau của MSH; do đó, suy thượng thận mạn tính nguyên phát cũng được đặc trưng bởi tăng sắc tố da và niêm mạc, trong khi suy thượng thận thứ phát thì không. Sự tăng sắc tố như vậy nổi bật nhất ở vùng da tiếp xúc với ánh nắng và ở các bề mặt gấp, chẳng hạn như đầu gối, khuỷu tay và khớp ngón tay. Chẩn đoán được gợi ý bởi các dấu hiệu và triệu chứng đã đề cập ở trên, được xác minh bằng nồng độ cortisol buổi sáng thấp với ACTH cao, và được xác nhận bằng đáp ứng tối thiểu của cortisol với nghiệm pháp ACTH tĩnh mạch 60 phút. Hạ natri máu, tăng kali máu, aldosterone thấp, và PRA tăng gợi ý một sự xáo trộn trong sản xuất mineralocorticoid. Các phát hiện liên quan có thể bao gồm sự xuất hiện của một quả tim nhỏ trên X-quang ngực, thiếu máu, tăng ure huyết, tăng bạch cầu ái toan, tăng lympho bào, và hạ đường huyết khi đói. Điều trị suy thượng thận nguyên phát mạn tính bao gồm liệu pháp thay thế glucocorticoid và mineralocorticoid sinh lý.
Bảng 14.9. Các Dấu hiệu và Triệu chứng của Suy thượng thận
| CÁC ĐẶC ĐIỂM CHUNG CỦA SUY THƯỢNG THẬN CẤP VÀ MẠN TÍNH |
|---|
| Chán ăn |
| Thờ ơ và lú lẫn |
| Mất nước |
| Mệt mỏi |
| Tăng kali máu |
| Hạ đường huyết |
| Hạ natri máu |
| Giảm thể tích máu và nhịp tim nhanh |
| Buồn nôn và nôn |
| Hạ huyết áp tư thế |
| Vàng da sơ sinh kéo dài |
| Thèm muối |
| Yếu |
| CÁC ĐẶC ĐIỂM CỦA SUY THƯỢNG THẬN CẤP (CƠN SUY THƯỢNG THẬN CẤP) |
| Đau bụng |
| Sốt |
| CÁC ĐẶC ĐIỂM CỦA SUY THƯỢNG THẬN MẠN TÍNH (BỆNH ADDISON) |
| Giảm lông mu và lông nách |
| Tiêu chảy |
| Tăng sắc tố da |
| Điện tâm đồ điện thế thấp |
| Tim nhỏ trên X-quang |
| Sụt cân |
Viêm thượng thận tự miễn có liên quan chặt chẽ với các haplotype HLA cụ thể và với các đa hình trong gen của kháng nguyên 4 liên quan đến tế bào lympho T gây độc tế bào (CTLA-4), có thể liên quan rộng rãi đến tính nhạy cảm với bệnh tự miễn. Chẩn đoán suy thượng thận mạn tính tự miễn chủ yếu dựa vào việc tìm thấy các kháng thể lưu hành chống lại các tế bào thượng thận hoặc các thành phần tế bào thượng thận. Trong nhiều trường hợp, các kháng nguyên thượng thận là các enzyme cytochrome P450 sinh steroid, đặc biệt là P450scc, P450c17 và P450c21. Không rõ làm thế nào các enzyme này tiếp cận các tế bào miễn dịch để gây ra phản ứng kháng thể, nhưng các nghiên cứu tử thiết cho thấy sự thâm nhiễm của vỏ thượng thận. Do đó, có khả năng miễn dịch qua trung gian tế bào chịu trách nhiệm cho sự phá hủy các tế bào vỏ thượng thận, dẫn đến sự giải phóng thứ phát các thành phần tế bào (bao gồm cả các enzyme P450) vào tuần hoàn, với sự phát triển sau đó của các kháng thể “dấu ấn” thứ phát chống lại các P450 này. Khoảng một nửa số bệnh nhân người lớn bị viêm thượng thận lympho bào cũng sẽ mắc bệnh tự miễn của một mô nội tiết khác với hiệu giá kháng thể cao chống lại các thành phần cụ thể của mô bị ảnh hưởng. Phát hiện này đã dẫn đến việc xác định các hội chứng đa tuyến nội tiết tự miễn (APS) cụ thể, một số trong đó phổ biến hơn ở trẻ em.
Hội chứng Đa tuyến nội tiết Tự miễn type 1 (APS1)
Còn được gọi là bệnh đa tuyến nội tiết-nấm candida-loạn sản ngoại bì tự miễn (APECED), APS1 được đặc trưng bởi nhiễm nấm candida da niêm mạc mạn tính, bệnh Addison tự miễn và suy cận giáp. Ít nhất hai trong số các đặc điểm này phải có mặt để đưa ra chẩn đoán, và tuổi khởi phát của chúng có thể rất thay đổi. Nói chung, nhiễm nấm candida da niêm mạc mạn tính xuất hiện ở trẻ nhỏ và ảnh hưởng đến miệng và móng. Suy cận giáp mắc phải có thể biểu hiện bằng hạ canxi máu lâm sàng trong giai đoạn giữa hoặc cuối thời thơ ấu, mặc dù trong một số trường hợp, hạ canxi máu có thể bị che giấu bởi suy thượng thận không được điều trị. Rối loạn thượng thận thường biểu hiện ở trẻ em hoặc thanh thiếu niên; bệnh thượng thận tự miễn có thể là một đặc điểm biểu hiện ở khoảng 5% các trường hợp và được tìm thấy ở hơn 60%, chủ yếu ở nam giới (~2nam:1nữ). Các đặc điểm tự miễn bổ sung của tình trạng này bao gồm rụng tóc và bạch biến; viêm dạ dày, tiêu chảy mạn tính và kém hấp thu, có hoặc không có thiếu máu ác tính; suy sinh dục tăng gonadotropin, đặc biệt ở phụ nữ; và ít phổ biến hơn là viêm gan, viêm tuyến giáp, viêm thận kẽ, viêm cơ, thiểu sản men răng, mất lách mắc phải, viêm giác-kết mạc và đái tháo đường type 1. Viêm giác-kết mạc là một đặc điểm liên quan quan trọng cần được theo dõi và điều trị cẩn thận để ngăn ngừa mù lòa. Ung thư biểu mô tế bào vảy ở miệng hoặc thực quản xảy ra ở 10% cá nhân khi trưởng thành. APS1 hiếm gặp ở hầu hết các quần thể, nhưng phổ biến ở những người có tổ tiên Phần Lan (1:25.000), Sardinia (1:14.000) và Do Thái Iran (1:9.000). APS1 là do các đột biến di truyền lặn ở một yếu tố phiên mã 58 kDa được gọi là yếu tố điều hòa tự miễn (AIRE). Hơn 100 đột biến khác nhau trong gen AIRE đã được mô tả, mặc dù sự thay đổi R257X đồng hợp tử hoặc dị hợp tử phức hợp đặc biệt phổ biến ở dân số Phần Lan, trong khi một đoạn xóa 13 bp (p.C322del 13) phổ biến ở những người từ Na Uy. Gen AIRE được biểu hiện trong các tế bào biểu mô tuyến ức và các tế bào đuôi gai ngoại vi hiếm gặp làm trung gian cho sự trình diện lạc chỗ của các peptide bị hạn chế ở mô và thúc đẩy sự dung nạp bản thân của các tế bào thymocyte tự phản ứng. Ngoài ra, AIRE dường như gây ra các tế bào T điều hòa cụ thể trong tuyến ức có khả năng ức chế tự miễn. Việc xóa AIRE ở chuột dẫn đến sự biểu hiện lạc chỗ của các kháng nguyên mô ngoại vi trong các tế bào biểu mô tủy tuyến ức, dẫn đến sự phát triển của một rối loạn tự miễn tương tự như APS1/APECED. Nói chung, các bệnh đa tuyến nội tiết tự miễn được điều trị bằng liệu pháp thay thế hormone. Các cuộc hẹn tái khám được khuyến nghị hàng năm và nên sàng lọc các thành viên trong gia đình. APS1 cũng được thảo luận chi tiết trong Chương 22.
Hội chứng Đa tuyến nội tiết Tự miễn type 2 (APS2)
Còn được gọi là hội chứng Schmidt, APS2 đề cập đến sự kết hợp tương đối phổ biến của viêm thượng thận tự miễn với viêm tuyến giáp và/hoặc đái tháo đường type 1. APS2 phổ biến hơn ở nữ giới (tỷ lệ 3:1), có liên quan đến HLA, và thường thấy ở người lớn trẻ hoặc trung niên, nhưng có thể biểu hiện ở hầu hết mọi lứa tuổi. Suy buồng trứng nguyên phát (tăng gonadotropin) được thấy ở một phần tư phụ nữ sau tuổi dậy thì bị APS2, nhưng suy tinh hoàn nguyên phát thì hiếm. Thiếu máu ác tính, viêm gan, bạch biến và rụng tóc cũng có thể được thấy, nhưng suy cận giáp và nhiễm nấm candida da niêm mạc điển hình của APS1 không được thấy trong APS2. APS2 có liên quan đến các dấu ấn HLA giống như viêm thượng thận tự miễn vô căn, có thể đơn giản là một dạng của APS2. Nhưng tính di truyền của APS2 rất phức tạp. APS2 và hội chứng rối loạn điều hòa miễn dịch, đa tuyến nội tiết và bệnh ruột liên kết X (IPEX) sau đó được thảo luận chi tiết trong Chương 22.
Rối loạn điều hòa miễn dịch, Đa tuyến nội tiết và Bệnh ruột liên kết X (IPEX)
IPEX là một hội chứng hiếm gặp do các đột biến trong gen FOXP3; hơn 70 đột biến đã được mô tả. IPEX bao gồm đái tháo đường type 1 khởi phát sớm, bệnh ruột tự miễn biểu hiện bằng tiêu chảy và kém hấp thu, và viêm da. Có thể thấy tăng bạch cầu ái toan và nồng độ immunoglobulin (Ig)E tăng cao. Bệnh thận, viêm tuyến giáp tự miễn, rụng tóc, cũng như viêm gan tự miễn, và viêm tụy ngoại tiết, thiếu máu tan máu và giảm tiểu cầu có thể là các biểu hiện muộn. Bệnh chủ yếu gây tử vong ở trẻ sơ sinh do nhiễm trùng, và ghép tủy xương đồng loại có thể là phương pháp chữa trị duy nhất.
Bệnh tự miễn liên quan đến Liệu pháp miễn dịch
Liệu pháp miễn dịch đã rất thành công với nhiều bệnh ung thư, nhưng liệu pháp miễn dịch có thể liên quan đến độc tính miễn dịch và tự miễn. Các biểu hiện tự miễn do các chất ức chế điểm kiểm soát miễn dịch (ICI) gây ra phụ thuộc vào (các) con đường được nhắm mục tiêu. ICI phá vỡ việc duy trì sự dung nạp miễn dịch đối với các kháng nguyên bản thân. Các biến cố bất lợi đã được báo cáo đối với nhiều hệ cơ quan, bao gồm đường tiêu hóa, da và tất cả các tuyến nội tiết. Các biểu hiện nội tiết thường được quan sát là suy và cường giáp, viêm tuyến yên, đái tháo đường tự miễn và suy thượng thận nguyên phát. Việc sử dụng kháng thể đơn dòng, ipilimumab, ngăn chặn CTLA-4, có liên quan đến viêm tuyến yên, trong khi liệu pháp kháng thể protein-1 gây chết tế bào theo chương trình thường liên quan đến suy giáp. Viêm tuyến yên liên quan đến ICI thường dẫn đến suy thượng thận thứ phát đe dọa tính mạng, đòi hỏi phải thay thế glucocorticoid kịp thời, và nên được xem xét hoặc điều trị trước khi bắt đầu bất kỳ liệu pháp nào cho bệnh suy giáp đồng thời. Suy thượng thận nguyên phát hiếm gặp với điều trị ICI đơn tác nhân, nhưng nguy cơ có thể vượt quá 4% với các liệu pháp kết hợp.
Thiểu sản thượng thận bẩm sinh
Thiểu sản thượng thận bẩm sinh, (AHC) còn được gọi là thiểu sản thượng thận bẩm sinh hoặc loạn sản thượng thận, là một rối loạn phát triển tuyến thượng thận dẫn đến suy thượng thận nguyên phát. Tình trạng này có thể xảy ra với nhiều kiểu di truyền khác nhau, và với một loạt các đặc điểm liên quan hoặc hội chứng.
Thiểu sản thượng thận bẩm sinh liên kết X
Dạng AHC phổ biến nhất này là do các đột biến của gen NR0B1 mã hóa DAX1 trên nhiễm sắc thể Xp21. Trong dạng thiểu sản thượng thận nguyên phát phổ biến nhất này, vùng xác định của tuyến thượng thận thai nhi không phát triển, và vùng bào thai bị không bào hóa và tế bào khổng lồ. Khoảng một nửa số bé trai bị AHC biểu hiện mất muối và suy glucocorticoid ở giai đoạn đầu của trẻ sơ sinh; phần còn lại biểu hiện âm thầm hơn với suy thượng thận mạn tính trong suốt thời thơ ấu. Suy sinh dục do thiếu gonadotropin và phát triển dậy thì không hoàn toàn là các đặc điểm liên quan, mặc dù dậy thì sớm, với sự ngừng dậy thì sau đó, đã được báo cáo trong các trường hợp hiếm. Một khiếm khuyết tiềm ẩn trong quá trình sinh tinh cũng có thể có mặt.
DAX1 là một yếu tố phiên mã hạt nhân liên quan đến sự phát triển của tuyến thượng thận và tinh hoàn, cũng như được biểu hiện trong các tế bào gonadotrope của tuyến yên. Khoảng hai phần ba số bé trai bị AHC có đột biến điểm, một phần ba còn lại có xóa gen NR0B1 hoặc là đơn lẻ hoặc là một phần của hội chứng xóa gen liền kề liên quan đến một locus chậm phát triển tâm thần liên kết X ở đầu mút (IL1RAPL1) và/hoặc các gen ở tâm động cho thiếu hụt glycerol kinase (GKD) và đôi khi là ornithine transcarbamylase (OTC) và loạn dưỡng cơ Duchenne (DMD). Một dạng AHC khởi phát ở người lớn do đột biến điểm cũng đã được mô tả.
Các bé trai bị AHC đáp ứng tốt với liệu pháp thay thế glucocorticoid và mineralocorticoid. Cần điều trị bằng testosterone để gây ra các đặc điểm sinh dục phụ ở tuổi vị thành niên; khả năng sinh sản tự nhiên rất hiếm và các nỗ lực gây ra quá trình sinh tinh bằng gonadotropin hiếm khi thành công. Các cá thể nữ mang gen không bị ảnh hưởng, nhưng một nửa số con trai của họ sẽ bị ảnh hưởng. Theo dõi chặt chẽ và tư vấn di truyền có thể giúp ngăn ngừa các cơn suy thượng thận đe dọa tính mạng ở các thành viên khác trong gia đình hoặc các lần mang thai trong tương lai. Do đó, tiền sử gia đình về suy thượng thận, tử vong không giải thích được, hoặc các bất thường về dậy thì ở các họ hàng nam của một bé trai bị suy thượng thận nên gợi ý AHC; thực sự, một tỷ lệ đáng kể các bé trai bị thiểu sản thượng thận (lẻ tẻ) có đột biến DAX1.
Các dạng Thiểu sản thượng thận trên nhiễm sắc thể thường
Ngoài AHC liên kết X, các dạng thiểu sản thượng thận lặn trên nhiễm sắc thể thường đã được báo cáo và đang được phân tích di truyền. Các đột biến ở SF1 (NR5A1), đã được thảo luận trước đó như một rối loạn sinh tổng hợp steroid giống như CAH dạng mỡ, đôi khi được phân loại là một dạng của thiểu sản thượng thận. Tuy nhiên, các đột biến SF1 ảnh hưởng đến tuyến sinh dục nghiêm trọng hơn tuyến thượng thận, và chưa được báo cáo ở các cá thể nam có kiểu hình bị thiểu sản thượng thận. Suy thượng thận nguyên phát cũng có liên quan đến hội chứng Pena-Shoekir loại I (DOK7, RAPSN), trisomy 13 giả, hội chứng Meckel (MKS1), hội chứng hydrolethalus (HYLS1), hội chứng Galloway-Mowat (WDR73), hội chứng Pallister-Hall (GLI3), và với các khiếm khuyết ở WNT3.
Hội chứng Triple A (Allgrove)
Rối loạn này bao gồm (1) suy thượng thận (glucocorticoid) kháng ACTH (adrenal), (2) co thắt tâm vị (achalasia), và (3) không có nước mắt (alacrima). Không có nước mắt là triệu chứng sớm nhất và nhất quán nhất, và co thắt tâm vị và suy thượng thận phát triển trong 2 thập kỷ đầu tiên, mặc dù co thắt tâm vị thường được ghi nhận trước. Suy mineralocorticoid được báo cáo ở khoảng 15% các trường hợp, và có tới 60% bệnh nhân phát triển các triệu chứng thần kinh tiến triển, chẳng hạn như suy giảm trí tuệ, điếc thần kinh giác quan, bệnh thần kinh ngoại biên và sọ não, teo dây thần kinh thị giác, bệnh Parkinson, và rối loạn chức năng tự chủ. Khoảng 80% bệnh nhân bị ảnh hưởng có các đột biến lặn trên nhiễm sắc thể thường ở AAAS, mã hóa một protein lặp lại WD được gọi là ALADIN; cơ sở của bệnh ở những bệnh nhân không có đột biến AAAS vẫn chưa được giải quyết hoàn toàn. ALADIN đồng định vị ở phía tế bào chất của lỗ nhân, nơi nó tương tác và tham gia vào quá trình chuyển vị hạt nhân của protein chuỗi nặng ferritin, FTH1, do đó làm cho các tế bào ngày càng nhạy cảm với tổn thương oxy hóa. Thiếu hụt ALADIN làm suy giảm tiềm năng oxy hóa khử của các tế bào thượng thận và ức chế sinh tổng hợp steroid. ALADIN cũng tham gia vào việc điều hòa các bộ điều khiển phân bào, cần thiết cho sự hình thành của thoi vô sắc. Các phát hiện lâm sàng của AAAS có thể khá thay đổi ngay cả trong cùng một gia đình. Suy thượng thận hiếm khi là đặc điểm biểu hiện. Do đó, một tiền sử gia đình chi tiết về co thắt tâm vị, không có nước mắt, hoặc các rối loạn thần kinh là quan trọng khi đánh giá một bệnh nhân bị suy thượng thận nguyên phát và có thể hướng dẫn trực tiếp đến rối loạn tiềm ẩn chính xác.
Hội chứng IMAGe
Thiểu sản thượng thận nguyên phát và cơn suy thượng thận cấp sơ sinh cũng là một phần của hội chứng IMAGe (intrauterine growth retardation – chậm tăng trưởng trong tử cung, metaphyseal dysplasia – loạn sản hành xương, adrenal hypoplasia – thiểu sản thượng thận, genitourinary anomalies – dị tật sinh dục-tiết niệu). Một gia đình lớn, nhiều thế hệ, với nhiều cá nhân được mô tả đặc điểm rõ ràng, đã được nghiên cứu bằng phân tích liên kết, xác định một đột biến sai nghĩa trội trong gen CDKN1C là nguyên nhân của rối loạn này. Ngoài ra, bốn bệnh nhân khác, không có quan hệ họ hàng, có bốn đột biến sai nghĩa khác nhau ở CDKN1C. Gen CDKN1C mã hóa chất ức chế khối u Wilms p57KIP2, chất ức chế một số kinase phụ thuộc cyclin. Gen này nằm trong vùng được in dấu của nhiễm sắc thể 11p15.5, do đó chỉ có alen của mẹ được biểu hiện. Các đột biến khác nhau trong cùng một gen này gây ra hội chứng tăng trưởng quá mức Beckwith-Wiedemann. Các đột biến Beckwith ức chế chu kỳ tế bào, trong khi các đột biến IMAGe thì không, và, khi được biểu hiện ở drosophila, các đột biến Beckwick không có tác động đến sự phát triển của mắt, nhưng các đột biến IMAGe gây ra sự tăng trưởng hạn chế. Do đó, các đột biến trong cùng một gen gây ra cả hội chứng thiểu sản thượng thận IMAGe và hội chứng tăng sản thượng thận Beckwith-Wiedemann. Một đột biến sai nghĩa CDKN1C cũng đã được mô tả ở một bệnh nhân bị hội chứng Silver Russell có chung đặc điểm hạn chế tăng trưởng nhưng không có các đặc điểm khác với hội chứng IMAGe.
Hội chứng MIRAGE
Hội chứng MIRAGE được đặc trưng bởi myelodysplasia – loạn sản tủy, infections – nhiễm trùng, restricted growth – hạn chế tăng trưởng, adrenal hypoplasia – thiểu sản thượng thận, genital anomalies – dị tật sinh dục, và enteropathy – bệnh ruột, và có liên quan đến tiên lượng xấu. Các đột biến dị hợp tử SAMD9 (trên nhiễm sắc thể 7q) đã được xác định ở các cá nhân bị ảnh hưởng, một số trong đó sống sót lâu hơn, nhưng đã phát triển hội chứng loạn sản tủy (MDS). Những đột biến này gây ra tăng chức năng của chất ức chế tăng trưởng SAMD9. Ở một số bệnh nhân, sự mất dần SAMD9 bị đột biến bởi các thay đổi thích ứng soma trong tủy xương đã dẫn đến monosomy 7 và do đó đã cứu vãn tác động tiêu cực lên sự tăng trưởng dẫn đến sự sống sót kéo dài; nhưng với cái giá là MDS.
Đột biến MCM4
Trong quần thể Người du mục Ailen, gen MCM4 mã hóa thành phần phức hợp duy trì nhiễm sắc thể nhỏ 4 cũng đã được tìm thấy ở những bệnh nhân biểu hiện suy thượng thận nguyên phát. Những bệnh nhân này được tìm thấy trong một quần thể bị cô lập về mặt di truyền biểu hiện với thiếu hụt cortisol đơn độc, suy giảm tăng trưởng, tăng gãy nhiễm sắc thể, và thiếu hụt tế bào diệt tự nhiên. Họ có thể dễ bị nhiễm trùng hơn và có nguy cơ cao bị các khối u tân sinh, nhưng cần theo dõi lâu hơn và hiểu rõ hơn về cơ chế bệnh sinh tiềm ẩn.
Các hội chứng kháng Hormone Vỏ thượng thận
Không đáp ứng di truyền với ACTH, còn được gọi là thiếu hụt glucocorticoid gia đình (FGD), có thể biểu hiện dưới dạng một cơn suy thượng thận cấp, được thúc đẩy bởi một bệnh xen kẽ ở trẻ sơ sinh, hoặc với các dấu hiệu và triệu chứng của suy thượng thận mạn tính ở trẻ em. Một số nguyên nhân di truyền lặn khác nhau của FGD đã được xác định cho đến nay; chúng bao gồm MC2R, MRAP1, StAR, CYP11A1, NNT, MCM4, và TXNRD2. Không giống như các cá nhân bị viêm thượng thận tự miễn, thiểu sản thượng thận, hoặc các dạng phá hủy mô thượng thận khác, bệnh nhân không đáp ứng di truyền với ACTH thường tiếp tục sản xuất mineralocorticoid, vì việc sản xuất aldosterone bởi lớp cầu của tuyến thượng thận được điều hòa chủ yếu bởi hệ renin-angiotensin. Do đó, bức tranh biểu hiện bao gồm chậm phát triển, thờ ơ, xanh xao, tăng sắc tố da, và hạ đường huyết, thường liên quan đến co giật. Các trường hợp hiếm cũng có thể kéo theo các bất thường về điện giải hoặc tăng hoạt độ renin huyết tương, dẫn đến chẩn đoán sai là một dạng suy thượng thận khác.
Đột biến MC2R: Thiếu hụt Glucocorticoid Gia đình Type 1
Thụ thể ACTH là một thành viên 7-xoắn màng, cặp đôi protein G của họ các thụ thể melanocortin được gọi là MC2R. Các đột biến MC2R dường như là nguyên nhân phổ biến nhất của kháng ACTH (25%), với vài chục trường hợp được báo cáo, nhưng sự phân bố thống kê của các trường hợp này không rõ ràng, vì nhiều tiêu chí lâm sàng khác nhau đã được áp dụng. Hầu hết các đột biến MC2R là các đột biến sai nghĩa, dẫn đến sự lưu giữ của thụ thể trong mạng lưới nội chất. Điều thú vị là, một đột biến hoạt động cấu thành (F278C) của protein MC2R đã được mô tả ở một bệnh nhân bị hội chứng Cushing. Tuy nhiên, tất cả các bệnh nhân khác mang đột biến MC2R đều biểu hiện với thiếu hụt glucocorticoid đơn độc và tăng sắc tố da, thường là sớm sau khi sinh cho đến 2 tuổi; hạ đường huyết là phổ biến và nồng độ ACTH tăng rõ rệt, giải thích cho sự tăng sắc tố da. Sản xuất mineralocorticoid và steroid sinh dục thường không bị ảnh hưởng. Tuy nhiên, không có giai đoạn thượng thận hóa do DHEAS thấp được quan sát thấy ở nhiều người và xác lập một vai trò cho ACTH trong giai đoạn thượng thận hóa. Tầm vóc cao, chu vi vòng đầu tăng, và tuổi xương tiến triển đã được báo cáo trong một số trường hợp; đây có thể là các tác động thứ cấp của nồng độ ACTH tăng rõ rệt ảnh hưởng đến MC1R trong đĩa tăng trưởng hoặc kích thích tổng hợp estradiol. Ngoài ra, trục hormone tăng trưởng bị kích thích trước khi điều trị cũng có thể chịu trách nhiệm. Điều trị bằng liều thay thế glucocorticoid thường ngăn ngừa các cơn suy thượng thận cấp, nhưng có thể không ức chế hoàn toàn nồng độ ACTH tăng cao; tuy nhiên, nên tránh sử dụng liều steroid trên mức sinh lý để ức chế ACTH.
Đột biến MRAP: Thiếu hụt Glucocorticoid Gia đình Type 2
Protein phụ kiện MC2R, MRAP1, là một protein xuyên màng nhỏ tạo thành các homodimer đối song bất thường và phục vụ hai chức năng: nó tạo điều kiện cho việc vận chuyển MC2R từ mạng lưới nội chất đến màng tế bào, và nó tương tác với MC2R trên bề mặt tế bào để tạo điều kiện cho hoạt động của thụ thể. Cả protein MRAP1 và protein MRAP2 liên quan đều phục vụ các chức năng tương tự với các thành viên khác của họ MCR. Trong khi MRAP1 hợp tác với MC2R, MRAP2, được biểu hiện nhiều ở vùng dưới đồi, hợp tác với MC4R và các thụ thể prokineticin (PKR), và quan trọng trong cân bằng nội môi năng lượng và do đó là béo phì nặng. Các đột biến MRAP1 là một nguyên nhân phổ biến của FGD chiếm khoảng 10% các trường hợp, nhưng các đột biến MRAP1 về mặt lâm sàng không thể phân biệt được với các đột biến MC2R. Ngược lại, các đột biến MRAP2 ở người đã được mô tả trong bệnh béo phì nặng không hội chứng.
Đột biến Nicotinamide Nucleotide Transhydrogenase
Nicotinamide nucleotide transhydrogenase (NNT) là một protein của màng trong ty thể tạo ra NADPH từ năng lượng của gradient proton ty thể. Các enzyme sinh steroid của ty thể (ví dụ, CYP11A1 hoặc CYP11B2/1) cần NADPH cho hoạt động của chúng. Ngoài ra, NNT và NADPH quan trọng đối với sự cân bằng của các loại oxy phản ứng (ROS) của tế bào. Được mô tả lần đầu tiên vào năm 2012, các đột biến ở NNT hiện đã được tìm thấy ở khoảng 10% bệnh nhân FGD. Tương tự như thiếu hụt MC2R và MRAP1, chúng gây ra thiếu hụt cortisol đơn độc và chỉ có thể được phân biệt bằng xét nghiệm di truyền.
Các dạng khác của Thiếu hụt Glucocorticoid Gia đình
FGD do các đột biến ở các gen và protein khác phá vỡ hệ thống stress oxy hóa trong ty thể bao gồm TXNRD2, GPX1, và PRDX3. Hơn nữa, các đột biến nhẹ ở StAR gây ra CAH dạng mỡ không cổ điển hoặc ở CYP11A1 có thể bị nhầm lẫn với FGD. Nhìn chung, giải trình tự thông lượng cao của các bệnh nhân và gia đình biểu hiện với một kiểu hình FGD đã tiết lộ các đột biến gây bệnh ở khoảng hai phần ba số bệnh nhân, trong khi phần còn lại vẫn chưa có chẩn đoán di truyền.
Các rối loạn chuyển hóa
Các rối loạn chuyển hóa cũng có thể gây ra suy thượng thận nguyên phát mạn tính, bao gồm loạn dưỡng chất trắng thượng thận (bệnh Schilder), các rối loạn sinh tổng hợp peroxisome (ví dụ, phổ hội chứng Zellweger), các rối loạn tổng hợp và chuyển hóa cholesterol (ví dụ, bệnh Wolman/CESD (được thảo luận trong Mục VI), SLOS), và các rối loạn ty thể (ví dụ, hội chứng Kearns-Sayre).
Loạn dưỡng chất trắng thượng thận
Loạn dưỡng chất trắng thượng thận (ALD) là một bệnh peroxisome và là nguyên nhân chuyển hóa phổ biến nhất của suy thượng thận. Hầu hết các trường hợp là do các đột biến trong protein màng peroxisome ALDP được mã hóa bởi gen ABCD1 trên nhiễm sắc thể Xq28; ALDP thuộc siêu họ các chất vận chuyển cassette liên kết ATP. Có nhiều dạng lâm sàng của ALD, bao gồm dạng não ở trẻ em (CCALD) với sự hủy myelin não, dạng não ở thanh thiếu niên, dạng não ở người lớn, bệnh tủy-thần kinh thượng thận, với bệnh lý sợi trục của các bó kim tự tháp và cảm giác thân thể, và bệnh thần kinh ngoại biên, dạng trám-cầu-tiểu não, và một dạng chỉ biểu hiện với bệnh Addison. CCALD là kiểu hình phổ biến nhất. Tỷ lệ hiện mắc của tình trạng này thường được báo cáo là từ 1:20.000 đến 100.000, mặc dù tần suất chung có thể cao tới 1:17.000. Một dạng lặn trên nhiễm sắc thể thường hiếm gặp của tình trạng này cũng tồn tại, thường biểu hiện ở trẻ sơ sinh (xem sau).
ALDP nhập các dẫn xuất acyl-CoA đã được hoạt hóa của các axit béo chuỗi rất dài (VLCFA) vào peroxisome nơi chúng được rút ngắn bằng quá trình β-oxy hóa. Do đó, ALD được đặc trưng bởi tỷ lệ C26 so với C22 VLCFA cao trong huyết tương và các mô, cho phép chẩn đoán các bệnh nhân riêng lẻ và các thai nhi bị ảnh hưởng. Những người mang gen thường có thể được phát hiện bằng sàng lọc VLCFA, mặc dù phân tích di truyền có thể cần thiết trong một số trường hợp. Các triệu chứng của CCALD liên kết X thường phát triển ở giữa thời thơ ấu, bệnh tủy-thần kinh thượng thận biểu hiện ở tuổi trưởng thành. Cùng một đột biến ALDP có thể gây ra cả ALD và bệnh tủy-thần kinh thượng thận, do đó có khả năng các locus di truyền khác cũng có liên quan. Các phát hiện sớm nhất trong CCALD có liên quan đến loạn dưỡng chất trắng hệ thần kinh trung ương và bao gồm thay đổi hành vi, kết quả học tập kém, nói khó, và trí nhớ kém tiến triển đến sa sút trí tuệ nặng. Các triệu chứng suy thượng thận thường xuất hiện sau các triệu chứng của bệnh chất trắng, nhưng suy thượng thận có thể là phát hiện ban đầu ở tới 20% trẻ em hoặc thanh niên. Ngược lại, bệnh tủy-thần kinh thượng thận thường bắt đầu bằng suy thượng thận ở trẻ em và thanh thiếu niên, và các dấu hiệu bệnh thần kinh theo sau 10 đến 15 năm sau đó. Khoảng 1% đến 3% các cá thể nữ mang gen ALD liên kết X có thể phát triển các triệu chứng thần kinh hoặc rối loạn chức năng thượng thận. Bởi vì sàng lọc là chính xác và chẩn đoán có ý nghĩa lâu dài, VLCFA nên được phân tích ở tất cả các bé trai biểu hiện suy thượng thận mà chẩn đoán không rõ ràng. Thực tế, một số quốc gia, như Hoa Kỳ, gần đây đã bắt đầu đưa ALD vào chương trình sàng lọc sơ sinh của họ để không bỏ lỡ cơ hội cho việc ghép tế bào gốc tạo máu đồng loại để điều trị CCALD (https://adrenoleukodystrophy.info/clinical-diagnosis/newborn-screening). Tỷ lệ hiện mắc suốt đời của suy thượng thận ở nam giới bị ALD là khoảng 80%, nguy cơ cao nhất trong 10 năm đầu tiên (gần 50%) và giảm xuống 5% sau 40 tuổi. Khi khởi phát giảm cortisol máu, sản xuất mineralocorticoid thường vẫn còn nguyên vẹn và do đó không cần điều trị thay thế cho đến khi có biểu hiện thiếu hụt.
Liệu pháp ăn kiêng với cái gọi là dầu Lorenzo (hỗn hợp 4:1 của glyceryl-trioleate và glyceryl-trierucate) cải thiện nồng độ VLCFA, nhưng nó không hiệu quả trong việc điều trị bệnh não đã được xác định, mặc dù vai trò trong việc ngăn ngừa sự khởi phát của bệnh não chưa được đánh giá. Các lựa chọn điều trị khác bao gồm ghép tế bào gốc tạo máu, thành công nhất đối với bệnh não giai đoạn sớm với tỷ lệ sống sót sau 5 đến 8 năm được báo cáo là 56%. Điều trị bằng Lovastatin làm giảm C24:0 và C26:0 huyết tương, nhưng không còn được khuyến nghị vì nó không có tác dụng đối với C26:0 trong tế bào hoặc đối với VLCFA.
Các rối loạn Sinh tổng hợp Peroxisome
Các rối loạn sinh tổng hợp peroxisome (PBD) là một nhóm các tình trạng lặn trên nhiễm sắc thể thường do các đột biến ở các protein PEX. Hội chứng Zellweger, loạn dưỡng chất trắng thượng thận sơ sinh, và bệnh Refsum ở trẻ sơ sinh tạo thành “phổ Zellweger” và khác biệt về mặt lâm sàng với loạn sản sụn chấm đầu xương do các đột biến gen PEX7. Các rối loạn này được đặc trưng bởi chậm phát triển, giảm trương lực cơ, điếc thần kinh giác quan, teo dây thần kinh thị giác, và phát triển khuôn mặt dị dạng. Bệnh nhân thường phát triển co giật, đặc biệt là những người bị loạn dưỡng chất trắng thượng thận sơ sinh. Chẩn đoán có thể được xác nhận bằng sự hiện diện của C26:1, C26:0 tăng, và tỷ lệ C26:C22 và C24:C22 VLCFA tăng. Hầu hết trẻ em mắc các dạng nặng của hội chứng Zellweger và ALD sơ sinh không sống qua 2 tuổi, mặc dù các biến thể khác của phổ Zellweger có liên quan đến sự sống sót lâu hơn.
Hội chứng Smith-Lemli-Opitz
SLOS là một khiếm khuyết lặn trên nhiễm sắc thể thường trong sinh tổng hợp cholesterol do các bất thường trong gen sterol Δ-7-reductase, DHCR7. Tỷ lệ mắc SLOS được ước tính từ 1:13.000 đến 1:80.000. Hơn 190 đột biến đã được mô tả, nhưng có hai đột biến chiếm ưu thế: đột biến c.964G > C chủ yếu được tìm thấy ở Bắc Mỹ và Tây Âu, trong khi đột biến W151X thường được tìm thấy ở Trung và Đông Âu, cho thấy hiệu ứng người sáng lập cho hai đột biến này. Các đặc điểm lâm sàng của SLOS bao gồm đầu nhỏ, chậm phát triển, ngoại hình khuôn mặt điển hình (mũi ngắn với sống mũi rộng và lỗ mũi hếch, rãnh nhân trung dài, cằm lẹm nhỏ, sụp mí, tai đóng thấp, xoay ra sau, hở hàm ếch và/hoặc vòm miệng cao), ngón tay cái gần, và dính ngón thứ hai và thứ ba (> 97%). Các bất thường về tim, thận, phổi và đường tiêu hóa cũng phổ biến. Các dị tật sinh dục bao gồm lỗ tiểu lệch thấp, tinh hoàn ẩn, và thậm chí bộ phận sinh dục không rõ ràng ở nam giới. Suy thượng thận có mặt trong một số trường hợp, đặc biệt là trong thời gian stress hoặc khi các nguồn cholesterol có nguồn gốc từ LDL không đủ (ví dụ, thiếu hụt chế độ ăn/suy giảm muối mật). Phổ lâm sàng của SLOS cực kỳ rộng, có khả năng phản ánh sự khác biệt trong sinh tổng hợp cholesterol còn lại trong tử cung và/hoặc sự khác biệt trong việc cung cấp cholesterol qua nhau thai hỗ trợ sự phát triển của thai nhi. SLOS có thể bị nghi ngờ trong tử cung bởi estriol của mẹ thấp và các phát hiện siêu âm. Phân tích sinh hóa sau sinh về hoạt tính sterol Δ-7-reductase, kết hợp với phân tích di truyền, có thể xác nhận chẩn đoán. Ngoài ra, LC-MS/MS là một phương pháp tốt để phát hiện sterol có thể được sử dụng để xác nhận chẩn đoán SLOS. Cách tiếp cận này có thể được áp dụng cho sàng lọc sơ sinh cho SLOS thông qua các mẫu máu khô trong tương lai. Tuổi thọ thay đổi theo mức độ nghiêm trọng của bệnh. Bổ sung cholesterol trong chế độ ăn, từ 20 đến 300 mg/kg/ngày, đã trở thành một can thiệp điều trị được thiết lập. Các tác dụng có lợi bao gồm cung cấp cholesterol cho các mô bên ngoài hệ thần kinh trung ương (CNS) và điều hòa giảm HMG-CoA reductase, có lẽ ức chế tổng hợp 7-DHC. Mặc dù điều này có thể dẫn đến sự cải thiện của kiểu hình ngoài CNS, một tác dụng có lợi đối với não là không thể vì cholesterol huyết tương không đi qua hàng rào máu não. Việc bổ sung simvastatin vào việc bổ sung cholesterol không thực sự cải thiện kết quả.
Các rối loạn ty thể
Rối loạn chức năng ty thể có thể liên quan đến rối loạn chức năng thượng thận nguyên phát. Các đặc điểm lâm sàng điển hình bao gồm nhiễm toan lactic, đục thủy tinh thể, điếc thần kinh giác quan, và bệnh cơ/liệt mắt. Suy thượng thận hiếm gặp ở trẻ em, nhưng suy vỏ thượng thận cận lâm sàng có thể được tìm thấy ở người lớn mắc bệnh đa hệ thống, và là một yếu tố tiên lượng xấu. Hội chứng Kearns-Sayre là kết quả của việc xóa DNA ty thể quy mô lớn và có thể liên quan đến các bệnh nội tiết bổ sung, chẳng hạn như suy giáp, suy sinh dục, đái tháo đường, suy giảm tăng trưởng và suy cận giáp.
Các rối loạn chuyển hóa khác cũng đã được báo cáo là gây ra suy thượng thận, bao gồm bệnh Niemann-Pick type B (không nên nhầm lẫn với bệnh type C, được thảo luận trong Mục VI), một bệnh lưu trữ lipid lysosome do các đột biến trong gen sphingomyelin phosphodiesterase 1 (SMPD1), biểu hiện bằng gan lách to và bệnh phổi.
Sphingosine-1-Phosphate Lyase
Thiếu hụt Sphingosine-1-phosphate lyase (SGPL1) là một dạng suy thượng thận lặn trên nhiễm sắc thể thường có hội chứng mới được mô tả, biểu hiện kết hợp với hội chứng thận hư kháng steroid. Các đặc điểm liên quan khác là bệnh vảy cá, suy giáp, tinh hoàn ẩn, các triệu chứng thần kinh, dị tật xương và suy giảm miễn dịch. SGPL1 là enzyme chịu trách nhiệm cho bước cuối cùng trong quá trình chuyển hóa sphingolipid. Các đột biến ở các thành phần thượng nguồn của con đường này dẫn đến các bệnh sphingolipidosis, chẳng hạn như các bệnh Gaucher, Niemann-Pick, Fabry, Krabbe, Sandhoff và Tay-Sachs, và GM1-gangliosidosis, là các rối loạn lưu trữ lysosome dẫn đến sự tích tụ sphingolipid trong các cơ quan. Các bệnh sphingolipidosis thường là các rối loạn đa hệ thống, tiến triển, một số trong đó có kiểu hình thận, nhưng các kiểu hình thượng thận chưa được mô tả. Với sự thiếu hụt SGPL1, sphingosine-1-phospate và các tiền chất khác tích tụ, dẫn đến loạn sản thượng thận và gián đoạn sinh tổng hợp steroid. Kiểu hình lâm sàng với các đột biến SGPL1 rộng hơn dự kiến, có thể do sự khác biệt giữa các đột biến di truyền và hoạt tính enzyme còn lại.
Các nguyên nhân khác
Xuất huyết và nhiễm trùng, được thảo luận trước đó là nguyên nhân của suy thượng thận nguyên phát cấp tính, có thể chừa lại một số mô thượng thận, để lại chức năng thượng thận bị tổn hại nghiêm trọng, thay vì hoàn toàn không có. Kết quả, cũng như với viêm thượng thận tự miễn, là một rối loạn mạn tính với sự khởi phát âm thầm của một loạt các phát hiện không đặc hiệu được mô tả ở trên. Bệnh lao, nhiễm nấm (ví dụ, histoplasmosis, coccidiomycosis), nhiễm virus (ví dụ, virus gây suy giảm miễn dịch ở người [HIV], cytomegalovirus), di căn, bệnh amyloidosis và bệnh sarcoidosis có thể gây ra một bức tranh lâm sàng tương tự. Sinh tổng hợp Cortisol cũng có thể bị ức chế bởi các loại thuốc, chẳng hạn như aminoglutethimide, etomidate, suramin, ketoconazole, hoặc các phương pháp điều trị chống HIV; những loại thuốc này có thể gây suy thượng thận, và phải được sử dụng thận trọng.
Suy thượng thận thứ phát
ACTH cần thiết cho sự phát triển của tế bào thượng thận và cho sự phiên mã của các gen cho các yếu tố sinh steroid, do đó bất kỳ sự suy giảm nào trong tổng hợp, giải phóng, hoặc hoạt động của ACTH đều có thể gây ra suy thượng thận thứ phát. Các ví dụ bao gồm các khiếm khuyết vùng dưới đồi, suy tuyến yên, các rối loạn tổng hợp và xử lý POMC, và sự ức chế trục hạ đồi-tuyến yên, sau khi điều trị bằng steroid ngoại sinh.
Hầu hết các dạng suy thượng thận thứ phát ảnh hưởng đến tổng hợp glucocorticoid và androgen hơn là giải phóng mineralocorticoid, vì angiotensin II là động lực chính cho lớp cầu. Tuy nhiên, đánh giá lâm sàng và sinh hóa có thể phức tạp, vì glucocorticoid cần thiết cho việc thanh thải nước tự do của thận và có thể có sự thiếu hụt vasopressin (AVP, ADH) đồng thời. Trên thực tế, việc điều trị suy thượng thận thứ phát có thể làm lộ ra một sự thiếu hụt ADH trước đây không rõ ràng và do đó gây ra bệnh đái tháo nhạt lâm sàng, do đó phải chú ý chặt chẽ đến cân bằng dịch và điện giải, khi bắt đầu thay thế steroid. Ngược lại, suy giáp do thiếu hụt hormone kích thích tuyến giáp (TSH) sẽ dẫn đến chuyển hóa chậm của lượng nhỏ cortisol được sản xuất, và do đó bảo vệ bệnh nhân khỏi các triệu chứng suy thượng thận. Điều trị suy giáp bằng thyroxine sẽ làm tăng tốc độ chuyển hóa của lượng nhỏ cortisol này, do đó làm lộ ra suy thượng thận do thiếu hụt ACTH và, đôi khi, có thể gây ra một cơn suy thượng thận cấp. Do đó, cần phải đánh giá cẩn thận trục tuyến yên-thượng thận trong suy tuyến yên với suy giáp thứ phát. Nhiều bác sĩ lâm sàng sẽ chọn “che chở” một bệnh nhân bằng liều nhỏ glucocorticoid (một phần tư đến một nửa liều thay thế sinh lý) trong quá trình điều trị ban đầu cho bệnh suy giáp thứ phát như vậy. Cuối cùng, điều quan trọng là phải nhận ra rằng sự thiếu hụt kết hợp của hormone tăng trưởng và ACTH sẽ khiến bệnh nhân có nguy cơ cao bị hạ đường huyết, vì cả hai hormone đều có tác dụng làm tăng glucose huyết tương. Tác dụng này đặc biệt quan trọng ở trẻ sơ sinh và trẻ nhỏ, khi trẻ em dễ bị hạ đường huyết trong các giai đoạn nhịn ăn kéo dài.
Nguyên nhân từ Vùng dưới đồi
Các nguyên nhân từ vùng dưới đồi gây thiếu hụt ACTH bao gồm các khối u và xạ trị. Các khối u, chẳng hạn như u sọ hầu, có liên quan đến thiếu hụt ACTH ở khoảng 25% bệnh nhân. Tần suất có thể cao hơn ở các khối u, chẳng hạn như u mầm và u tế bào hình sao. Suy thượng thận hiếm khi là phàn nàn khi nhập viện nhưng có thể góp phần vào bức tranh lâm sàng. Sau phẫu thuật và/hoặc xạ trị, đại đa số bệnh nhân có khối u vùng dưới đồi sẽ bị thiếu hụt ACTH như một phần của tổn thương hạ đồi-tuyến yên do phẫu thuật hoặc bức xạ gây ra. Do đó, tất cả các bệnh nhân như vậy nên được bao phủ bằng glucocorticoid trong quá trình điều trị, bất kể tình trạng của trục HPA tại thời điểm khối u được xác định. Bởi vì việc điều trị suy thượng thận thứ phát có thể gây ra bệnh đái tháo nhạt, việc chú ý chặt chẽ đến cân bằng nước là cần thiết. Thiếu hụt ACTH cũng có thể xảy ra sau khi chiếu xạ toàn bộ não cho các khối u não và các khối u ác tính trung ương khác. Điều này có thể liên quan đến cả cơ chế vùng dưới đồi và tuyến yên. Tần suất thiếu hụt ACTH trong những trường hợp như vậy thấp hơn nhiều so với sau khi điều trị các khối u vùng dưới đồi nhưng chỉ có thể biểu hiện sau vài năm điều trị.
Thiếu hụt Hormone Vỏ thượng thận
ACTH có thể bị thiếu hụt như một phần của thiếu hụt nhiều hormone tuyến yên (MPHD, suy toàn bộ tuyến yên) hoặc như thiếu hụt ACTH đơn độc. MPHD có thể là kết quả của phẫu thuật tuyến yên hoặc xạ trị, từ một quá trình thâm nhiễm (ví dụ, bệnh mô bào Langerhans), hoặc từ một dạng rối loạn chức năng vùng dưới đồi chưa được hiểu rõ. Trong hầu hết các trường hợp, sự bài tiết hormone tăng trưởng bị mất trước, sau đó theo thứ tự là các gonadotropin, TSH, và ACTH, do đó cần phải cảnh giác và đánh giá liên tục những bệnh nhân này trong nhiều năm. MPHD cũng có thể là kết quả của các rối loạn phát triển hạ đồi-tuyến yên liên quan đến các yếu tố phiên mã, chẳng hạn như HESX1, LHX4 và SOX3. Các đặc điểm liên quan, chẳng hạn như thiểu sản dây thần kinh thị giác (HESX1) và các bất thường tiểu não (LHX4), có thể giúp tập trung chẩn đoán. Suy giảm bài tiết ACTH cũng đã được mô tả ở những cá nhân có đột biến PROP1, một trong những nguyên nhân di truyền thường gặp nhất của MPHD. Phát hiện này có thể xảy ra nhiều năm sau khi biểu hiện với thiếu hụt hormone tăng trưởng, TSH và gonadotropin, nhấn mạnh tầm quan trọng của việc theo dõi lâu dài bệnh nhân bị rối loạn tuyến yên.
Bệnh nhân bị MPHD thường có một dạng suy thượng thận tương đối nhẹ. Bài tiết mineralocorticoid là bình thường, trong khi bài tiết cortisol giảm nhưng không hoàn toàn không có. Tuy nhiên, dự trữ thượng thận bị tổn hại nghiêm trọng do sự kích thích dưới mức mạn tính của sinh tổng hợp enzyme sinh steroid. Bởi vì một số quá trình tổng hợp cortisol vẫn tiếp tục, chẩn đoán có thể không rõ ràng trừ khi thực hiện nghiệm pháp CRF hoặc metyrapone để đánh giá khả năng sản xuất ACTH của tuyến yên và nghiệm pháp ACTH tĩnh mạch để đánh giá dự trữ thượng thận. Điều này có thể đặc biệt đúng khi thiếu hụt TSH là một thành phần của suy tuyến yên, như đã nêu ở trên; điều trị suy giáp thứ phát bằng thyroxine sẽ làm tăng tốc độ chuyển hóa của lượng nhỏ cortisol này, do đó làm lộ ra suy thượng thận do thiếu hụt ACTH và, đôi khi, có thể gây ra một cơn suy thượng thận cấp.
Thiếu hụt ACTH đơn độc là một tình trạng hiếm gặp có thể do các đột biến di truyền lặn trong gen TPIT. TPIT mã hóa một yếu tố T-box (TBX19) điều hòa phiên mã của promoter POMC trong các tế bào corticotrope. Những bệnh nhân này thường biểu hiện với thiếu hụt ACTH nặng, khởi phát sớm. Hạ đường huyết và vàng da kéo dài thường có mặt, và có thể dẫn đến tử vong sơ sinh. Vì khiếm khuyết này chỉ giới hạn ở việc tổng hợp POMC trong các tế bào corticotrope, các đặc điểm bổ sung của thiếu hụt POMC toàn thể (được nêu sau) không có mặt. Các đột biến TPIT không được tìm thấy ở khoảng một nửa số bệnh nhân bị thiếu hụt ACTH đơn độc, cho thấy rằng các nguyên nhân khác vẫn chưa được tìm ra. Giảm cortisol máu do thiếu hụt ACTH đơn độc cũng đã được mô tả với các khiếm khuyết hồi hải mã (trí nhớ) và các bất thường về tóc (rụng tóc) như một phần của “hội chứng Triple H,” một mối liên quan tự miễn có thể có.
Các rối loạn của Proopiomelanocortin
Các khiếm khuyết trong tổng hợp và xử lý POMC cũng có thể gây ra sản xuất và hoạt động ACTH bất thường. Ở những bệnh nhân có đột biến POMC, nồng độ ACTH và cortisol huyết tương thường cực kỳ thấp, nhưng tùy thuộc vào khiếm khuyết di truyền, nồng độ ACTH huyết tương cũng có thể tăng cao phản ánh việc sản xuất một protein có phản ứng miễn dịch nhưng không có hoạt tính sinh học. Các đột biến hoặc xóa gen POMC di truyền lặn sẽ ảnh hưởng đến nhiều peptide POMC, bao gồm MSH và β-endorphin. Do đó, tóc đỏ, da nhợt nhạt và béo phì là các đặc điểm liên quan của dạng suy thượng thận thứ phát này. Những dấu hiệu lâm sàng này có thể tinh vi hơn ở những cá nhân có tóc đen tự nhiên và da sẫm màu, hoặc tóc đỏ có thể sẫm lại ở tuổi trưởng thành. Các đột biến ở prohormone convertase 1 (PC1, PCSK1), cần thiết cho việc xử lý POMC thành ACTH, gây ra sự phân cắt và xử lý bất thường của một số hệ thống hormone, bao gồm cả việc tạo ra ACTH có hoạt tính sinh học từ POMC. Bệnh nhân mắc rối loạn lặn hiếm gặp này có thể bị giảm cortisol máu cùng với chuyển hóa glucose bất thường, béo phì, suy sinh dục do thiếu gonadotropin, và tiêu chảy kém hấp thu kéo dài. Những bệnh nhân này cần liệu pháp thay thế glucocorticoid cho suy thượng thận và tình trạng béo phì nặng và chứng ăn nhiều của họ có thể được điều trị thành công bằng setmelanotide, một chất chủ vận MC4R.
Liệu pháp Steroid dài hạn
Liệu pháp glucocorticoid dài hạn có thể ức chế phiên mã gen POMC và sự tổng hợp và lưu trữ của ACTH. Hơn nữa, liệu pháp dài hạn dường như làm giảm sự tổng hợp và lưu trữ của CRF và làm giảm số lượng thụ thể cho CRF trong tuyến yên. Do đó, sự phục hồi của trục HPA sau liệu pháp glucocorticoid dài hạn đòi hỏi sự phục hồi của nhiều thành phần trong một chuỗi tuần tự, và do đó thường đòi hỏi thời gian đáng kể (xem phần về Liệu pháp Glucocorticoid và Ngừng thuốc). Bệnh nhân được ngừng thành công liệu pháp glucocorticoid hoặc được điều trị thành công bệnh Cushing có thể biểu hiện sự bình thường hóa khá nhanh của nồng độ cortisol huyết tương, trong khi tiếp tục có dự trữ thượng thận giảm trong hơn 12 tháng. Steroid dạng hít, thuốc xịt mũi, và thậm chí cả thuốc nhỏ mắt steroid có thể gây ức chế trục thượng thận, do đó cần phải cảnh giác sau khi ngừng thuốc hoặc vào thời điểm có thêm stress (ví dụ, phẫu thuật, bệnh xen kẽ). Điều trị bằng cortisone và prednisone trong thai kỳ sẽ chỉ dẫn đến sự ức chế tối thiểu của tuyến thượng thận thai nhi, do tác dụng bảo vệ của 11βHSD của nhau thai; điều trị bằng dexamethasone trong thai kỳ có thể ảnh hưởng đến sinh tổng hợp steroid của tuyến thượng thận thai nhi.
Tăng tiết hormon tuyến thượng thận
Hội chứng Cushing
Thuật ngữ “hội chứng Cushing” mô tả bất kỳ dạng nào của tình trạng dư thừa glucocorticoid; “bệnh Cushing” chỉ định tình trạng tăng cortisol máu do tuyến yên sản xuất quá mức ACTH. Rối loạn liên quan gây ra bởi ACTH có nguồn gốc không phải từ tuyến yên được gọi là “hội chứng ACTH lạc chỗ.” Thuật ngữ “hội chứng Cushing” đôi khi được sử dụng để chỉ riêng tình trạng tăng tiết cortisol từ các khối u tuyến thượng thận, nhưng điều này không rõ ràng và nên tránh. Các nguyên nhân khác của hội chứng Cushing bao gồm adenoma tuyến thượng thận, carcinoma tuyến thượng thận và tăng sản thượng thận đa nốt. Tất cả những điều đã đề cập ở trên đều khác biệt với hội chứng Cushing do thầy thuốc, là một tập hợp lâm sàng tương tự do việc sử dụng mạn tính một lượng ACTH hoặc glucocorticoid trên mức sinh lý.
Các phát hiện lâm sàng
Các đặc điểm thể chất của hội chứng Cushing là kết quả của tình trạng tăng cortisol máu mạn tính có hoặc không có tăng androgen máu. Béo phì trung tâm, “mặt tròn như mặt trăng,” rậm lông, và đỏ bừng mặt được thấy ở hơn 80% người lớn mắc hội chứng Cushing. Các vết rạn da, tăng huyết áp, yếu cơ, đau lưng, phân bố mỡ “bướu trâu,” rối loạn tâm lý, mụn trứng cá, và dễ bầm tím thường được mô tả (35%–80%). Tuy nhiên, đây là những dấu hiệu và đặc điểm của hội chứng Cushing giai đoạn muộn. Khi có sẵn các bức ảnh hàng năm của những bệnh nhân như vậy, thường rõ ràng rằng những đặc điểm này có thể mất 5 năm hoặc lâu hơn để phát triển. Do đó, “diện mạo Cushingoid” cổ điển thường sẽ không phải là hình ảnh ban đầu được thấy ở trẻ mắc hội chứng Cushing. Các chỉ số sớm nhất, đáng tin cậy nhất của tình trạng tăng cortisol máu ở trẻ em là tăng cân và ngừng tăng trưởng (Bảng 14.10). Dữ liệu tích lũy từ ba nghiên cứu lớn về bệnh Cushing ở trẻ em đã xác định được tăng cân khi nhập viện ở 91/97 (94%) trường hợp và suy giảm tăng trưởng ở 82/95 (86%) trường hợp. Do đó, bất kỳ trẻ thừa cân nào ngừng phát triển nên được đánh giá về hội chứng Cushing. Glucocorticoid ức chế tăng trưởng bằng cách tăng bài tiết somatostatin ở vùng dưới đồi, ức chế bài tiết hormone tăng trưởng và sản xuất IGF-1, và bằng cách tác động trực tiếp lên các đầu xương để ức chế quá trình sulfat hóa sụn, ức chế khoáng hóa, và ức chế sự tăng sinh tế bào. Ngược lại, trẻ em bị béo phì do chế độ ăn đơn thuần thường phát triển nhanh hơn và cao hơn so với tuổi (có lẽ do tăng insulin máu thứ phát mạn tính). Béo phì của bệnh Cushing ở trẻ em ban đầu là toàn thể thay vì trung tâm, và bướu trâu là bằng chứng của bệnh kéo dài. Rối loạn tâm lý, đặc biệt là hành vi nỗ lực quá mức mang tính cưỡng chế, được thấy ở khoảng 40% trẻ em và thanh thiếu niên mắc bệnh Cushing, và khác biệt rõ rệt so với trầm cảm thường thấy ở người lớn. Sự bất ổn về cảm xúc đã được mô tả ở khoảng 30% các trường hợp. Một khía cạnh chưa được đánh giá đúng mức của Cushing ở trẻ em là mức độ mất xương và giảm khoáng hóa đáng kể ở những bệnh nhân này. Do đó, bệnh Cushing thường được chẩn đoán ở người trẻ tuổi vì chẩn đoán đã bị bỏ sót, chứ không phải không có, trong giai đoạn thanh thiếu niên. Hiếm khi, hội chứng Cushing do carcinoma tuyến thượng thận hoặc hội chứng ACTH lạc chỗ có thể gây ra một diễn biến bùng phát nhanh chóng.
Bảng 14.10. Các phát hiện ở 39 trẻ mắc Bệnh Cushing
| Dấu hiệu/Triệu chứng | Số bệnh nhân | % |
|---|---|---|
| Tăng cân | 36/39 | 92 |
| Chậm tăng trưởng | 31/37 | 84 |
| Giảm mật độ xương | 14/19 | 74 |
| Mệt mỏi | 26/39 | 67 |
| Tăng huyết áp | 22/35 | 63 |
| Dậy thì muộn hoặc ngừng dậy thì | 21/35 | 60 |
| Da đỏ | 18/39 | 46 |
| Mụn trứng cá | 18/39 | 46 |
| Rậm lông | 18/39 | 46 |
| Hành vi cưỡng chế | 17/39 | 44 |
| Vết rạn da | 14/39 | 36 |
| Dễ bầm tím | 11/39 | 28 |
| Bướu trâu | 11/39 | 28 |
| Đau đầu | 10/39 | 26 |
| Tuổi xương chậm | 2/23 | 13 |
| Tiểu đêm | 3/39 | 8 |
(Từ Devoe, D. J., Miller, W. L., Conte, F. A., và cộng sự. (1997). Long-term outcome in children and adolescents after transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab, 82, 3196-3202.)
Bệnh Cushing
Mặc dù thường được mô tả rất chi tiết và minh họa bằng những bức ảnh ấn tượng trong các sách giáo khoa nội tiết, bệnh Cushing khá hiếm ở người lớn. Hơn nữa, khoảng 25% bệnh nhân được giới thiệu đến các trung tâm lớn vì bệnh Cushing là trẻ em, và nhiều bệnh nhân được khám lần đầu khi trưởng thành thực sự đã trải qua sự khởi phát các triệu chứng từ thời thơ ấu hoặc thanh thiếu niên. Ví dụ, bệnh nhân ban đầu của Harvey Cushing là một phụ nữ trẻ 23 tuổi có các đặc điểm lâm sàng cho thấy bệnh đã kéo dài, và chỉ cao 144 cm, gợi ý bệnh khởi phát vào khoảng 11 tuổi. Ở người lớn và trẻ em trên 7 tuổi, nguyên nhân phổ biến nhất của hội chứng Cushing là bệnh Cushing thật sự (tăng sản thượng thận do tăng tiết ACTH của tuyến yên). Các bé trai bị ảnh hưởng thường xuyên hơn các bé gái trong giai đoạn trước tuổi dậy thì, mặc dù tỷ lệ giới tính bằng nhau trong giai đoạn thanh thiếu niên và phụ nữ có tỷ lệ mắc bệnh Cushing cao hơn ở tuổi trưởng thành. Ở trẻ sơ sinh và trẻ em dưới 7 tuổi, các khối u tuyến thượng thận chiếm ưu thế. Trong số 60 trẻ sơ sinh dưới 1 tuổi mắc hội chứng Cushing, 48 trẻ có khối u tuyến thượng thận (Bảng 14.11).
Ở người lớn, hơn 90% bệnh nhân mắc bệnh Cushing có các microadenoma tuyến yên có thể xác định được. Các khối u này thường có đường kính từ 2 đến 10 mm, không có vỏ bọc, có ranh giới không rõ ràng, và thường có thể phát hiện được bằng MRI tuyến yên có cản quang. Các khối u này thường chỉ có thể xác định được bằng những khác biệt nhỏ về hình dạng và cấu trúc so với mô xung quanh, do đó tần suất chữa khỏi bằng phẫu thuật có tương quan với kỹ năng kỹ thuật của bác sĩ phẫu thuật. Ở trẻ em và thanh thiếu niên, khoảng 80% đến 85% những người mắc bệnh Cushing có microadenoma có thể xác định được bằng phẫu thuật. Mặc dù việc loại bỏ khối u thường có vẻ chữa khỏi, 20% những bệnh nhân được “chữa khỏi” như vậy bị tái phát và lại biểu hiện bệnh Cushing trong vòng khoảng 5 năm, do đó tỷ lệ chữa khỏi thực tế là 65% đến 75%.
Tỷ lệ chữa khỏi cao của phẫu thuật cắt bỏ microadenoma qua xương bướm trong bệnh Cushing cho thấy phần lớn những bệnh nhân như vậy mắc bệnh nguyên phát của chính tuyến yên, thay vì tăng năng tuyến yên thứ phát do sự kích thích quá mức của tuyến yên bởi CRF hoặc các tác nhân khác. Các nghiên cứu theo dõi cẩn thận những bệnh nhân này đã xác nhận điều này. Ở hầu hết các bệnh nhân sau phẫu thuật, nhịp sinh học ngày đêm của ACTH và cortisol trở lại bình thường, ACTH và cortisol đáp ứng thích hợp với hạ đường huyết, cortisol dễ dàng bị ức chế bởi liều thấp dexamethasone, và các hệ thống hạ đồi-tuyến yên khác trở lại bình thường.
Tuy nhiên, một số bệnh nhân mắc bệnh Cushing không có microadenoma có thể xác định được, và một số bệnh nhân được “chữa khỏi” lại tái phát. Điều này cho thấy rằng nhóm bệnh nhân nhỏ hơn này có thể có một rối loạn hạ đồi nguyên phát, hoặc một số mô tuyến yên chịu trách nhiệm cho việc tăng tiết ACTH đã không được cắt bỏ. Nghiên cứu lâm sàng hiện tại cho thấy rằng bệnh Cushing thường do một adenoma tuyến yên nguyên phát gây ra, nhưng đôi khi nó có thể do rối loạn chức năng vùng dưới đồi. Vi phẫu có thể chữa khỏi trong trường hợp đầu tiên, nhưng không phải trong trường hợp thứ hai. Thật không may, không có thủ thuật chẩn đoán nào để phân biệt hai khả năng này; do đó, thăm dò qua xương bướm vẫn là phương pháp điều trị ban đầu được ưu tiên cho bệnh nhân mắc bệnh Cushing.
Giải trình tự toàn bộ exome của các microadenoma tiết ACTH đã xác định được các đột biến tăng chức năng tái phát trong gen mã hóa ubiquitin-specific protease 8 (USP8), một deubiquitinase điều hòa sự luân chuyển của các thụ thể EGF và các protein màng khác. Cho đến nay, các đột biến soma này chủ yếu được tìm thấy ở bệnh nhân nữ trưởng thành. Điều này và các tiến bộ tương tự có thể dẫn đến các liệu pháp y tế có mục tiêu hơn.
Bệnh Cushing: Điều trị
Phẫu thuật qua xương bướm cung cấp phương pháp tiếp cận ban đầu tốt nhất để chữa khỏi nhanh chóng và hoàn toàn cho hầu hết các bệnh nhân, nhưng các phương pháp tiếp cận thay thế có thể cần thiết ở trẻ nhỏ hơn nếu xoang bướm chưa được thông khí. Các hậu quả ngắn hạn của phẫu thuật qua xương bướm bao gồm đái tháo nhạt thoáng qua và rò dịch não tủy qua mũi. Suy toàn bộ tuyến yên dai dẳng rất hiếm, nhưng ảnh hưởng của tăng cortisol máu đối với sự bài tiết hormone tăng trưởng có thể kéo dài trong 1 đến 2 năm sau khi điều trị, và thiếu hụt hormone tăng trưởng có thể xảy ra ngay cả ở những trẻ không được xạ trị. Chiều cao cuối cùng có thể bị giảm từ 1,5 đến 2,0 SD do tăng cortisol máu kéo dài; điều trị bằng hormone tăng trưởng có thể cải thiện sự mất mát tăng trưởng này ở những bệnh nhân bị suy giảm hormone tăng trưởng; Việc quản lý tình trạng giảm cortisol máu trong giai đoạn sau phẫu thuật là quan trọng, vì trục HPA sẽ bị ức chế. Cần theo dõi cẩn thận sự phục hồi của trục trong vài tháng, cho đến khi khả năng đáp ứng với stress trở lại.
Việc quản lý bệnh Cushing không đáp ứng hoặc tái phát là một thách thức. Phẫu thuật qua xương bướm lặp lại thường là phương pháp tiếp cận đầu tiên, đặc biệt nếu có bằng chứng về một tổn thương riêng biệt hoặc tăng tiết ACTH một bên. Các phương pháp tiếp cận hàng thứ hai bao gồm cắt bỏ tuyến yên bán phần, xạ trị Gamma Knife, chất chủ vận dopamine (cabergoline), chất tương tự somatostatin (pasireotide), cắt bỏ tuyến thượng thận, và các loại thuốc nhắm vào tuyến thượng thận (mitotane), sinh tổng hợp steroid (ketoconazole, metyrapone, etomidate), hoặc các thụ thể GR (mifepristone). Tất cả đều có những nhược điểm đáng kể, đặc biệt là ở trẻ em. Cắt bỏ tuyến yên bán phần có khả năng loại bỏ sự bài tiết của tuyến yên đối với hormone tăng trưởng, TSH và gonadotropin, gây ra suy giảm tăng trưởng, suy giáp và không tiến triển trong tuổi dậy thì. Mặc dù suy giáp dễ dàng được điều trị bằng thay thế thyroxine đường uống, thiếu hụt hormone tăng trưởng đòi hỏi liệu pháp thay thế rất tốn kém. Thay thế steroid sinh dục có thể được sử dụng để đạt được các đặc điểm sinh dục phụ ở tuổi dậy thì; tuy nhiên, cần thay thế gonadotropin để đạt được khả năng sinh sản. Xạ trị tuyến yên đã được ca ngợi để tránh nhiều vấn đề này và có hiệu quả trong điều trị bệnh Cushing, nhưng thiếu hụt hormone tăng trưởng xảy ra trong hầu hết các trường hợp và các bệnh nội tiết bổ sung có thể xảy ra theo thời gian. Khoảng thời gian từ xạ trị đến khi chữa khỏi có thể hơn 1 năm, trong thời gian đó cần phải ngăn chặn điều trị tăng cortisol máu để ngăn ngừa các tác động liên tục của bệnh Cushing đối với sự tăng trưởng, cân nặng và khoáng hóa xương. Hơn nữa, liều bức xạ lớn làm tăng nguy cơ viêm động mạch não, bệnh não chất trắng, bệnh bạch cầu, các khối u tế bào đệm, và các khối u xương liên quan đến hộp sọ; xạ trị lập thể có thể làm giảm các tác động tiềm ẩn này, nhưng có rất ít dữ liệu ở trẻ em.
Cắt bỏ tuyến thượng thận hai bên qua nội soi là liệu pháp dứt điểm cho những bệnh nhân bị bệnh Cushing dai dẳng; xạ trị tuyến yên có thể được sử dụng khi khả năng sinh sản trong tương lai không phải là vấn đề. Ngoài những ảnh hưởng rõ ràng của việc loại bỏ sản xuất bình thường của glucocorticoid và mineralocorticoid, việc cắt bỏ tuyến thượng thận cũng loại bỏ sự ức chế phản hồi sinh lý của tuyến yên. Ở một số người lớn có adenoma tuyến yên còn sót lại, điều này dẫn đến sự phát triển của các macroadenoma tuyến yên, sản xuất một lượng rất lớn ACTH. Chúng có thể mở rộng và chèn ép vào các dây thần kinh thị giác và có thể sản xuất đủ POMC để tạo ra đủ MSH gây sạm da sâu (hội chứng Nelson), nhưng điều này hiếm khi thấy ở trẻ em. Có tương đối ít kinh nghiệm ở trẻ em với ketoconazole và các loại thuốc khác ức chế sinh tổng hợp steroid, nhưng chúng có thể cung cấp một hình thức điều trị hữu ích cho các bệnh nhân được lựa chọn hoặc để kiểm soát tăng cortisol máu trong ngắn hạn. Metyrapone không hữu ích cho điều trị dài hạn; mitotane, một tác nhân tiêu hủy tuyến thượng thận, có thể được sử dụng để thực hiện một “cuộc cắt bỏ tuyến thượng thận bằng hóa chất,” nhưng các tác dụng phụ của nó như buồn nôn, chán ăn và nôn mửa rất nghiêm trọng. Etomidate có thể hữu ích trong tình huống cấp tính đối với bệnh Cushing nặng hoặc đe dọa tính mạng trước khi phẫu thuật. Mifepristone đã được báo cáo là điều trị thành công các triệu chứng liên quan đến tăng cortisol máu trong hội chứng Cushing ở cả bệnh nhân người lớn và trẻ em.
Các nguyên nhân nội sinh khác của Hội chứng Cushing
Các khối u tuyến thượng thận
Carcinoma tuyến thượng thận là nguyên nhân điển hình của hội chứng Cushing ở trẻ sơ sinh và trẻ nhỏ (xem Bảng 14.11); báo cáo đầu tiên về carcinoma vỏ thượng thận ở trẻ em là của một bé gái 6 tuổi qua đời vào năm 1688. Chúng có xu hướng xảy ra thường xuyên hơn ở các bé gái, vì những lý do chưa rõ. Adenoma tuyến thượng thận hầu như luôn tiết ra cortisol với sự bài tiết tối thiểu của mineralocorticoid hoặc steroid sinh dục. Ngược lại, carcinoma tuyến thượng thận có xu hướng tiết ra cả cortisol và androgen, và thường liên quan đến sự nam hóa tiến triển. Adenoma hoặc carcinoma tuyến thượng thận có thể liên quan đến phì đại một bên cơ thể, đôi khi là một phần của hội chứng Beckwith-Wiedemann, với sự sản xuất quá mức IGF-2. Các mối liên quan di truyền bổ sung bao gồm các đột biến dòng mầm hoặc mất tính dị hợp tử của gen ức chế khối u p53, đôi khi là một phần của hội chứng Li-Fraumeni, cũng như các đột biến trong con đường Wnt/β-catenin kinh điển. Giải trình tự DNA của các adenoma tuyến thượng thận gây ra hội chứng Cushing độc lập với ACTH ở người lớn cũng đã cho thấy các đột biến tăng chức năng lẻ tẻ ở PRKACA, gen mã hóa tiểu đơn vị xúc tác của kinase protein phụ thuộc cAMP (PKA), cũng như ở các phosphodiesterase thường bất hoạt cAMP, PDE11A và PDE8B.
CT và MRI hữu ích trong chẩn đoán các khối u tuyến thượng thận, và phân tích steroid có thể cung cấp thông tin tại thời điểm biểu hiện và để theo dõi khả năng tái phát. Điều trị cho cả adenoma và carcinoma là phẫu thuật, và cần phải cắt bỏ hoàn toàn để chữa khỏi. Trong một số trường hợp, việc phân biệt mô học giữa adenoma và carcinoma tuyến thượng thận rất khó khăn, nhưng tiên lượng xấu hơn có liên quan đến kích thước khối u tăng, xâm lấn bao và/hoặc mạch máu, các hạch bạch huyết sau phúc mạc, di căn, hoặc không bình thường hóa được các giá trị hormone sau phẫu thuật. Mặc dù một số ít bệnh nhân có bệnh còn sót lại hoặc di căn đã đáp ứng tốt với liệu pháp bổ trợ bằng mitotane hoặc các chiến lược hóa trị liệu khác, tiên lượng chung vẫn còn kém. Để cải thiện kết quả cho các bệnh hiếm gặp như vậy, bệnh nhân nên được ghi danh vào các thử nghiệm lâm sàng và các sổ đăng ký quốc tế.
Tăng sản thượng thận đa nốt độc lập với Adrenocorticotropic Hormone
Nhóm các bệnh tăng sản thượng thận thường hiếm gặp và lành tính này bao gồm tăng sản vỏ thượng thận dạng nốt lớn độc lập với ACTH (còn được gọi là bệnh vỏ thượng thận dạng nốt lớn ồ ạt) và các bệnh tăng sản dạng nốt nhỏ, chủ yếu là bệnh vỏ thượng thận dạng nốt sắc tố nguyên phát (PPNAD). Theo truyền thống, các rối loạn dạng nốt lớn có liên quan đến các nốt lớn hơn 1 cm và các rối loạn dạng nốt nhỏ bao gồm các nốt nhỏ hơn 1 cm; tuy nhiên, trong kỷ nguyên gen, ngày càng được công nhận rằng các chỉ định như vậy có thể là tùy ý.
Ở trẻ em, tăng sản dạng nốt lớn hai bên chủ yếu được thấy với hội chứng McCune-Albright do các đột biến soma trong gen polypeptide alpha-kích thích, protein liên kết nucleotide guanine (GNAS) 1. Bệnh nhân thường biểu hiện với bộ ba loạn sản xơ đa xương, các đốm da café-au-lait, với bờ không đều, và dậy thì sớm độc lập với gonadotropin. Tuy nhiên, vì rối loạn này là do các đột biến tế bào soma, chứ không phải các đột biến dòng mầm, các biểu hiện của nó không đồng nhất về mặt lâm sàng và có thể bao gồm các rối loạn nội tiết khác, chẳng hạn như nhiễm độc giáp, cường cận giáp, bệnh khổng lồ do tuyến yên, và tăng prolactin máu. Hội chứng Cushing hiếm gặp trong hội chứng McCune-Albright và thường biểu hiện trước 6 tháng tuổi. Hầu hết các trường hợp tăng sản vỏ thượng thận dạng nốt lớn dẫn đến hội chứng Cushing là lẻ tẻ, đơn độc, và xảy ra ở tuổi trung niên. Nguyên nhân của các tổn thương như vậy có liên quan đến sự hình thành lạc chỗ của các thụ thể hormone, chẳng hạn như vasopressin, serotonin, và LH/hCG, cũng như đến việc sản xuất ACTH tại chỗ từ chính vỏ thượng thận. Một nguyên nhân khác là các đột biến dòng mầm ở các gen ức chế khối u, bao gồm gen chứa lặp lại armadillo 5 (ARMC5), tân sinh đa tuyến nội tiết loại 1 (MEN1), polyp tuyến đại tràng (APC), và fumarate hydratase (FH).
Nhóm các bệnh tăng sản thượng thận lành tính dạng nốt nhỏ bao gồm PPNAD và bệnh vỏ thượng thận dạng nốt nhỏ đơn độc. PPNAD, được đặc trưng bởi sự bài tiết của cả cortisol và androgen tuyến thượng thận, là một thực thể hiếm gặp thấy ở trẻ sơ sinh, trẻ em và thanh niên, với nữ giới bị ảnh hưởng thường xuyên hơn. Nó thường được thấy như một phần của “phức hợp Carney,” là một dạng của tân sinh đa tuyến nội tiết (MEN), bao gồm các đốm sắc tố và nốt ruồi xanh trên mặt, môi, và kết mạc, và một loạt các khối u, bao gồm schwannoma và u nhầy nhĩ, và đôi khi là các adenoma tuyến yên tiết hormone tăng trưởng, các khối u tế bào Leydig, các khối u tế bào Sertoli vôi hóa (có thể tiết ra estrogen), và carcinoma tủy của tuyến giáp. Các đặc điểm điển hình của hội chứng Cushing thường được thấy ở quần thể trẻ em. Tuyến thượng thận không thực sự tăng sản mà bao gồm các nốt sắc tố rời rạc được bao quanh bởi mô teo, cho phép xác định chúng bằng MRI hoặc CT. Bởi vì tình trạng tăng cortisol máu kháng lại sự ức chế bằng liều cao dexamethasone và vì cả glucocorticoid và steroid sinh dục đều được sản xuất, PPNAD về mặt lâm sàng khó phân biệt với hội chứng ACTH lạc chỗ, nhưng các xét nghiệm ACTH huyết tương thường có giá trị chẩn đoán. Cắt bỏ tuyến thượng thận hoàn toàn thường được chỉ định, mặc dù một số thành công đã được báo cáo với việc cắt bỏ bán phần.
Phức hợp Carney, giống như các rối loạn MEN khác, thường là trội trên nhiễm sắc thể thường. Mất tính dị hợp tử và các đột biến ở một tiểu đơn vị điều hòa của kinase protein A (PRKAR1A) đã được tìm thấy ở 73% bệnh nhân mắc phức hợp Carney, hoặc như các sự kiện dòng mầm hoặc soma lẻ tẻ trong các khối u tuyến thượng thận đơn độc (PPNAD đơn độc). Nói chung, bệnh nhân mắc Phức hợp Carney mang đột biến ở PRKAR1A biểu hiện ở độ tuổi trẻ hơn và có nhiều u nhầy, schwannoma, khối u tuyến giáp, và khối u tuyến sinh dục hơn so với bệnh nhân không có đột biến ở PRKAR1A. Bệnh nhân bị PPNAD đơn độc thường mang các đột biến PRKAR1A c.709-7del6 hoặc c.1A > G/p.M1V, điều này quan trọng đối với tư vấn và sàng lọc di truyền.
Bệnh nhân bị tăng sản thượng thận lành tính dạng nốt nhỏ không có Phức hợp Carney và đột biến PRKAR1A có thể mang các đột biến ở các protein tín hiệu cAMP khác. Các đột biến trong gen mã hóa phosphodiesterase 11A4 (PDE11A) đã được báo cáo ở những cá nhân bị bệnh vỏ thượng thận dạng nốt nhỏ đơn độc và PPNAD và các đột biến ở phosphodiesterase 8B (PDE8B) trong bệnh vỏ thượng thận dạng nốt nhỏ đơn độc. Cả PDE11A và PDE8B đều xúc tác cho quá trình thủy phân cAMP và cyclic guanosine monophosphate và được biểu hiện ở một số mô nội tiết, bao gồm cả tuyến thượng thận. Như đã lưu ý trước đó, các đột biến lẻ tẻ ở PRKAR1A, PDE11A, và PDE8B cũng có liên quan đến các adenoma tuyến thượng thận đơn độc gây ra hội chứng Cushing độc lập với ACTH. Do đó, các bất thường trong các con đường tín hiệu đóng một vai trò quan trọng trong tăng sản và hình thành khối u tuyến thượng thận.
Hội chứng Adrenocorticotropic Hormone Lạc chỗ
Hội chứng ACTH lạc chỗ thường thấy nhất ở người lớn bị carcinoma tế bào yến mạch của phổi, các khối u carcinoid, carcinoma tế bào đảo tụy và u tuyến ức, nhưng nó cũng có thể được thấy ở trẻ em. POMC và ACTH được sản xuất lạc chỗ có nguồn gốc từ cùng một gen sản xuất POMC của tuyến yên nhưng không nhạy cảm với phản hồi glucocorticoid trong các tế bào ác tính. Hiện tượng này cho phép phân biệt giữa ACTH của tuyến yên và lạc chỗ bằng khả năng ức chế của ACTH tuyến yên bởi liều cao dexamethasone. Mặc dù hội chứng ACTH lạc chỗ hiếm gặp ở trẻ em, nó đã được mô tả ở trẻ sơ sinh dưới 1 tuổi. Các khối u liên quan bao gồm neuroblastoma, pheochromocytoma, carcinoma tế bào đảo tụy và các khối u thần kinh nội tiết của tuyến ức. Hội chứng ACTH lạc chỗ thường liên quan đến nồng độ ACTH cao gấp 10 lần so với nồng độ thấy trong bệnh Cushing. Tuy nhiên, cả người lớn và trẻ em mắc rối loạn này có thể có ít hoặc không có bằng chứng lâm sàng về tăng cortisol máu, có lẽ do sự khởi phát nhanh chóng điển hình của bệnh và do sự dị hóa chung liên quan đến bệnh ác tính. Không giống như bệnh nhân mắc bệnh Cushing, người lớn và trẻ em mắc hội chứng ACTH lạc chỗ thường có kiềm hóa hạ kali máu hạ renin, có lẽ vì nồng độ ACTH cực kỳ cao kích thích sản xuất DOC bởi lớp bó của tuyến thượng thận, và/hoặc kích thích sản xuất aldosterone bởi lớp cầu của tuyến thượng thận. Các khối u nội sọ, không phải tuyến yên hiếm gặp có thể sản xuất ACTH lạc chỗ nhưng bị nhầm lẫn với bệnh Cushing của tuyến yên do vị trí của chúng.
Hội chứng DICER1 và Bệnh Cushing ở trẻ sơ sinh
Các đột biến ở DICER1, gen mã hóa ribonuclease DICER1 tham gia vào việc tạo ra các RNA can thiệp nhỏ (siRNA) và microRNA, lần đầu tiên được phát hiện trong u nguyên bào màng phổi-phổi (PPB), và sau đó được báo cáo trong u nguyên bào tuyến yên, và các khối u phôi khác. Hội chứng khối u và loạn sản gia đình PPB [OMIM #601200] là một hội chứng khuynh hướng khối u đa hiệu. U nguyên bào tuyến yên đã được mô tả trong hơn một chục trường hợp trẻ sơ sinh mắc bệnh Cushing, và có thể là lặn trên nhiễm sắc thể thường hoặc liên quan đến một đột biến dòng mầm di truyền cộng với một đột biến soma, cả hai đều ở DICER1; trong số ít trường hợp được báo cáo, tiên lượng đã kém.
Tiếp cận Trẻ bị Hội chứng Cushing Nội sinh
Hội chứng Cushing ở trẻ em thường được gợi ý bởi tăng cân, ngừng tăng trưởng, thay đổi tâm trạng, và thay đổi diện mạo khuôn mặt (da đỏ, mụn trứng cá, rậm lông). Chẩn đoán ở trẻ em có thể tinh vi và khó khăn khi nó được tìm kiếm ở một thời điểm tương đối sớm trong lịch sử tự nhiên của bệnh. Để sàng lọc ban đầu, ba cuộc điều tra xét nghiệm được khuyến nghị: (1) hồ sơ ACTH và cortisol ngày đêm từ máu (hồ sơ cortisol có thể được thực hiện trên nước bọt), (2) đo UFC 24 giờ, và (3) nghiệm pháp ức chế dexamethasone 1 mg qua đêm. Sự gia tăng tuyệt đối trên “giới hạn trên của bình thường” đối với nồng độ ACTH và cortisol huyết tương thường không có. Thay vì tìm thấy nồng độ cortisol buổi sáng lớn hơn 20 μg/dL hoặc ACTH lớn hơn 50 pg/mL, điển hình hơn là tìm thấy sự gia tăng nhẹ, thường không rõ ràng trong các giá trị buổi chiều và buổi tối. Sự mất nhịp điệu ngày đêm này, được chứng minh bằng việc tiếp tục bài tiết ACTH và cortisol trong suốt buổi chiều, buổi tối và ban đêm, thường là chỉ số xét nghiệm đáng tin cậy sớm nhất của bệnh Cushing. Một phép đo cortisol huyết tương duy nhất được lấy vào nửa đêm từ một ống thông tĩnh mạch lưu, trong khi bệnh nhân vẫn ngủ, nên nhỏ hơn 2 μg/dL ở những người bình thường, và lớn hơn 2 μg/dL trong bệnh Cushing. Tương tự, các giá trị cortisol tự do vào ban đêm, được đo trong nước bọt, thường nên nhỏ hơn khoảng 1 ng/mL (2,8 nmol/L) lúc 11 giờ tối. Việc giải thích các mẫu nước bọt vào ban đêm có thể bị ảnh hưởng bởi các yếu tố từ rối loạn giấc ngủ đến bệnh nướu răng. Do đó, các mẫu nên được thu thập trong điều kiện tối ưu (ví dụ, sau khi súc miệng nhưng trước khi đánh răng) để tránh kết quả dương tính giả. Các mẫu nước bọt ổn định ở nhiệt độ phòng trong vài ngày nhưng có thể được đông lạnh nếu cần lưu trữ lâu dài hơn.
Các giá trị của ACTH và cortisol thường cực kỳ cao trong hội chứng ACTH lạc chỗ, trong khi cortisol tăng, nhưng ACTH bị ức chế trong các khối u tuyến thượng thận và trong tăng sản thượng thận đa nốt (Bảng 14.12). Trong tất cả các dạng hội chứng Cushing, việc theo dõi UFC 24 giờ hỗ trợ quyết định xem có cần điều tra thêm hay không. Một giá trị UFC 24 giờ bình thường thường nhỏ hơn 70 μg/m2/ngày (với các xét nghiệm miễn dịch phóng xạ). Nên lặp lại việc thu thập nhiều lần, và điều quan trọng là sử dụng các phạm vi bình thường được điều chỉnh theo kích thước, cũng như tuổi, vì trẻ em bị béo phì đơn thuần có tốc độ bài tiết cortisol cao hơn. Một xét nghiệm cơ bản khác là nghiệm pháp ức chế dexamethasone liều thấp (1 mg) qua đêm với ngưỡng cắt cho cortisol thấp hơn 1,8 μg/dL (50 nmol/L). Nếu cả UFC 24 giờ và nghiệm pháp ức chế dexamethasone liều thấp qua đêm đều bình thường, chẩn đoán hội chứng Cushing thường được loại trừ. Một ngoại lệ là những bệnh nhân có tăng tiết cortisol không liên tục hoặc định kỳ cần theo dõi lâu hơn và xét nghiệm lặp lại để chẩn đoán hội chứng Cushing.
Bảng 14.11. Nguyên nhân Hội chứng Cushing ở trẻ nhũ nhi
| Nam | Nữ | |
|---|---|---|
| CÁC KHỐI U THƯỢNG THẬN (n = 48) | ||
| Carcinoma | 5 | 20 |
| Adenoma | 4 | 16 |
| Không xác định | 2 | 1 |
| Hội chứng ACTH lạc chỗ | 1 | 1 |
| Tăng sản thượng thận dạng nốt | 1 | 4 |
| Tăng sản thượng thận không xác định | 2 | 2 |
| Khối u sản xuất ACTH | 1 | 0 |
| TỔNG CỘNG | 16 | 44 |
ACTH, Hormone vỏ thượng thận.
(Từ Miller, W. L., Townsend, J. J., Grumbach, M. M., & Kaplan, S. L. (1979). An infant with Cushing’s disease due to an adrenocorticotropin-producing pituitary adenoma. J Clin Endocrinol Metab, 48, 1017-1025.)
Bảng 14.12. Các giá trị chẩn đoán trong các Nguyên nhân khác nhau của Hội chứng Cushing
| Xét nghiệm | Giá trị Bình thường | Carcinoma Thượng thận | Adenoma Thượng thận | Tăng sản Thượng thận dạng nốt | Bệnh Cushing | Hội chứng ACTH lạc chỗ |
|---|---|---|---|---|---|---|
| Nồng độ Cortisol huyết tương | ||||||
| Sáng | >14 | ↑ | ↑ | ↑ | ± | ↑↑ |
| Chiều | <8 | ↑ | ↑↑ | |||
| Nồng độ ACTH huyết tương | ||||||
| Sáng | <100 | ↓ | ↓ | ↓ | N hoặc ↑ | ↑↑ |
| Chiều | <50 | ↓ | ↓ | ↓ | ↑ | ↑↑ |
| Nghiệm pháp ức chế Dex liều thấp | ||||||
| Cortisol | <3 | Không Δ | Không Δ | Không Δ | Không ức chế | Không Δ |
| ACTH | <30 | Không Δ | Không Δ | Không Δ | Không Δ | |
| 17OHCS | <2 | Không Δ | Không Δ | Không Δ | ||
| Nghiệm pháp ức chế Dex liều cao | ||||||
| Cortisol | ↓↓ | Không Δ | Không Δ | b | ↓ | Không Δ |
| ACTH | Không Δ | Không Δ | b | ↓ | Không Δ | |
| 17OHCS | ↓↓ | Không Δ | b | ↓ | Không Δ | |
| Nghiệm pháp ACTH IV | ||||||
| Cortisol | >20 | Không Δ | ±↑ | ↑ | ± | Không Δ |
| Nghiệm pháp Metyrapone | ||||||
| Cortisol | ↓ | ±↓ | Không Δ | ↓↓ | ± | |
| 11-Deoxycortisol | ↑ | Không Δ | ↑ | ↑↑ | ± | |
| ACTH | ↑ | Không Δ | ↑↑ | Không Δ | ||
| 17OHCS | ↑ | Không Δ | ± | ↑↑ | Không Δ | |
| Bài tiết nước tiểu 24 giờ (Nền) | ||||||
| 17OHCS | ↑↑ | ↑ | ↑ | ↑ | ||
| 17KS | ↑↑ | N | ↑ | |||
| Nồng độ DHEA hoặc DHEA-S huyết tương | ↑↑ | ↓ | ±↑ | ↑ | ↑ |
a: Đáp ứng không hoàn toàn, tức là ↓. b: Thường không có Δ. Nồng độ Cortisol tính bằng μg/dL. Nồng độ ACTH tính bằng pg/mL. 17OHCS tính bằng mg/24h. Sáng thường chỉ 8 giờ sáng; Chiều chỉ 4 giờ chiều.
17OHCS, 17-hydroxycorticosteroid; 17KS, 17-ketosteroid; ACTH, hormone vỏ thượng thận; Dex, dexamethasone; DHEA, dehydroepiandrosterone; DHEA-S, DHEA sulfat; IV, tiêm tĩnh mạch.
Sau khi chẩn đoán hội chứng Cushing được xác định, có thể cần các cuộc điều tra sâu hơn để tìm ra nguồn gốc của bệnh. Các nghiệm pháp ức chế dexamethasone liều thấp và liều cao có thể hữu ích khi được thực hiện cẩn thận. Để đạt được kết quả đáng tin cậy ở bệnh nhân nhi, trẻ em nên được nhập viện, tốt nhất là tại một khoa nghiên cứu lâm sàng nhi khoa. Dữ liệu cơ bản (kiểm soát) trong hai ngày nên được thu thập. Dexamethasone liều thấp (20 μg/kg/ngày, tối đa 2,0 mg) nên được cho, chia thành các liều bằng nhau cho mỗi 6 giờ trong 2 ngày, sau đó là dexamethasone liều cao (80 μg/kg/ngày) được cho theo cách tương tự. Các giá trị 8 giờ sáng và 8 giờ tối (hoặc nửa đêm) cho ACTH và cortisol và các mẫu nước tiểu 24 giờ cho 17OHCS, 17-ketosteroid (17KS), cortisol tự do, và creatinine (để theo dõi tính đầy đủ của việc thu thập) nên được lấy vào mỗi trong 6 ngày của nghiệm pháp. Các phép đo UFC hoặc 17OHCS có lẽ đáng tin cậy như nhau nếu phòng thí nghiệm đã thiết lập các tiêu chuẩn nhi khoa tốt. Do các biến thể gây ra bởi sự bài tiết ACTH theo từng đợt, các giá trị máu lúc 8 giờ sáng và 8 giờ tối nên được lấy ba lần vào lúc 8:00, 8:15 và 8:30.
Ở những bệnh nhân bị béo phì ngoại sinh hoặc các rối loạn không phải Cushing khác, cortisol, ACTH và các steroid trong nước tiểu sẽ bị ức chế dễ dàng bởi dexamethasone liều thấp. Cortisol huyết tương nên nhỏ hơn 1,8 μg/dL, ACTH nhỏ hơn 20 pg/mL, và 17OHS trong nước tiểu 24 giờ nhỏ hơn 1 mg/g creatinine. Bệnh nhân bị adenoma tuyến thượng thận, carcinoma tuyến thượng thận, hoặc hội chứng ACTH lạc chỗ sẽ có các giá trị tương đối không nhạy cảm với cả dexamethasone liều thấp và liều cao, mặc dù một số bệnh nhân bị tăng sản thượng thận đa nốt có thể đáp ứng với sự ức chế liều cao bằng sự gia tăng nghịch lý của cortisol sau dexamethasone, như đã được báo cáo với phức hợp Carney. Bệnh nhân mắc bệnh Cushing cổ điển đáp ứng bằng sự ức chế ACTH, cortisol, và các steroid trong nước tiểu trong quá trình điều trị liều cao, nhưng không phải trong quá trình điều trị liều thấp. Tuy nhiên, một số trẻ em, đặc biệt là những trẻ ở giai đoạn đầu của bệnh, có thể biểu hiện sự ức chế một phần để đáp ứng với dexamethasone liều thấp. Do đó, nếu liều thấp được cho vượt quá 20 μg/kg/ngày hoặc nếu các xét nghiệm được sử dụng không đủ nhạy để phân biệt sự ức chế một phần với hoàn toàn, có thể dẫn đến các xét nghiệm âm tính giả.
Lấy mẫu xoang đá được sử dụng rộng rãi ở người lớn mắc bệnh Cushing, để phân biệt bệnh Cushing của tuyến yên với hội chứng ACTH lạc chỗ. Lòng mạch nhỏ hơn ở trẻ em làm tăng nguy cơ của thủ thuật này, nhưng lấy mẫu tĩnh mạch đá dưới đã được sử dụng với một số thành công ở thanh thiếu niên trong một nỗ lực để xác định vị trí các adenoma tuyến yên trước khi phẫu thuật. Các phương pháp tiếp cận như vậy chỉ nên được thực hiện tại các trung tâm chuyên môn cao; lấy mẫu tĩnh mạch cảnh có thể cung cấp một phương pháp tiếp cận thay thế, mặc dù dữ liệu sâu rộng ở quần thể nhi khoa không có sẵn. Nói chung, chẩn đoán bệnh Cushing ở trẻ em khó khăn hơn đáng kể so với người lớn. Ngoài các xét nghiệm, các nghiên cứu hình ảnh có thể giúp xác định chẩn đoán chính xác của hội chứng Cushing. Một phim CT hoặc MRI tuyến thượng thận có thể hình dung một khối u vỏ thượng thận hoặc một bệnh tăng sản thượng thận dạng nốt lớn/nốt nhỏ, trong khi siêu âm tuyến thượng thận thường thất bại và do đó không nên được tin cậy như một công cụ chẩn đoán cho các rối loạn tuyến thượng thận. MRI cũng là phương pháp ưa thích để hình dung vùng dưới đồi và tuyến yên trước để tìm các tổn thương.
Cường Aldosteron Nguyên phát
Aldosterone thường được sản xuất ở các tế bào lớp cầu để đáp ứng với thể tích nội mạch cạn kiệt thông qua hệ renin-angiotensin và/hoặc kali huyết tương cao. Trong cường aldosteron nguyên phát, tuyến thượng thận sản xuất aldosterone một cách cấu thành khi không có angiotensin II hoặc tăng kali máu. Cường aldosteron nguyên phát là dạng phổ biến nhất của tăng huyết áp thứ phát, ảnh hưởng đến khoảng 10% tất cả các bệnh nhân tăng huyết áp. Ngoài các kết cục tim mạch bất lợi liên quan đến tăng huyết áp, dư thừa aldosterone là một yếu tố nguy cơ độc lập cho bệnh tim mạch và tổn thương thận, có khả năng do tác động trực tiếp của aldosterone lên các mô khác ngoài thận.
Các phát hiện lâm sàng
Cường aldosteron nguyên phát cổ điển được đặc trưng là biểu hiện với tăng huyết áp, đa niệu, kiềm hóa hạ kali máu, và hoạt độ renin huyết tương thấp do sản xuất aldosterone tự chủ. Các adenoma sản xuất aldosterone được mô tả rõ ở người lớn nhưng hiếm gặp ở trẻ em. Các trường hợp gia đình của cường aldosteron nguyên phát cũng đã được mô tả, bao gồm cường aldosteron có thể chữa bằng glucocorticoid (GRA) (xem Mục VI). Cường aldosteron nguyên phát tồn tại trên một phổ liên tục về mức độ nghiêm trọng của bệnh, từ nhẹ đến nặng, cho thấy các ước tính hiện tại về tỷ lệ hiện mắc của bệnh có thể không đại diện đầy đủ cho gánh nặng thực sự của bệnh. Giai đoạn đầu của bệnh, cường aldosteron nguyên phát có thể chỉ đơn giản biểu hiện với huyết áp tăng mà không có hạ kali máu tương ứng hoặc hoạt độ renin thấp.
Chẩn đoán phân biệt
Kể từ mô tả ban đầu về cường aldosteron nguyên phát, một số nguyên nhân riêng biệt đã được mô tả, bao gồm (1) các adenoma sản xuất aldosterone một bên (APA, hội chứng Conn), chiếm 30% đến 40% các trường hợp; (2) cường aldosteron vô căn hai bên, chiếm 60% đến 70% các trường hợp; và các dạng hiếm hơn, chẳng hạn như (3) cường aldosteron gia đình (FH), và (4) các bệnh tăng sản thượng thận dạng nốt nguyên phát, như đã mô tả trước đó.
Các Adenoma Sản xuất Aldosterone (hội chứng Conn)
Các đột biến soma tái phát gây ra APA, phổ biến nhất là trong gen mã hóa kênh K+ KCNJ5 (còn được gọi là Kir3.4) đã được xác định gần đây. Các nghiên cứu đa trung tâm đã xác định được hai đột biến soma ở KCNJ5 (G151R hoặc L168R) ở 38% tất cả các APA. Các đột biến như vậy phổ biến hơn ở nữ giới và ở độ tuổi trẻ hơn, và biểu hiện với nồng độ aldosterone trước phẫu thuật cao hơn. Các đột biến KCNJ5 này ảnh hưởng đến bộ lọc chọn lọc ion của kênh K+, gây ra sự gia tăng dòng Na+ và sự khử cực vĩnh viễn của màng tế bào lớp cầu. Sự khử cực màng dẫn đến sự kích hoạt của các kênh Ca2+ phụ thuộc điện thế, làm tăng Ca2+ nội bào, do đó cung cấp tín hiệu bình thường cho việc sản xuất aldosterone và sự tăng sinh của tế bào lớp cầu. Gần đây, các nghiên cứu giải trình tự exome đã xác định được các đột biến soma tái phát bổ sung phát sinh độc lập với các đột biến ở KCNJ5, bao gồm ở ATP1A1, ATP2B3, CACNA1D, và CTNNB1. ATP1A1 mã hóa tiểu đơn vị alpha của Na+/K+ ATPase và chiếm ~5% các APA. ATP2B3 mã hóa một Ca2+ ATPase và chiếm khoảng 2% các APA. CACNA1D mã hóa một kênh canxi phụ thuộc điện thế và có thể chiếm 11% các APA. Mỗi đột biến liên quan đến kênh này đã được liên kết về mặt chức năng với sự khử cực của các tế bào lớp cầu và tăng tiết aldosterone. Tuy nhiên, vẫn chưa rõ liệu chúng có đủ để thúc đẩy phản ứng tăng sản dẫn đến APA hay không. CTNNB1 là gen mã hóa β-catenin, một thành viên kinh điển của con đường tín hiệu Wnt. Ngoài APA, các đột biến kích hoạt ở exon 3 của CTNNB1 có liên quan đến cả adenoma tuyến thượng thận không tiết và carcinoma vỏ thượng thận.
Cường Aldosteron Vô căn Hai bên
Cường aldosteron vô căn hai bên chiếm 60% đến 70% các trường hợp, mặc dù đôi khi nó có thể biểu hiện dưới dạng bệnh một bên. Cường aldosteron vô căn hai bên thường được coi là một bệnh của người lớn, nhưng nó có thể biểu hiện ban đầu sớm hơn trong cuộc đời. Mặc dù có tỷ lệ hiện mắc cao, sinh lý bệnh tiềm ẩn vẫn chưa được hiểu rõ. Mặc dù bệnh nhân bị cường aldosteron vô căn hai bên có độ nhạy cảm tăng với angiotensin II, vẫn chưa biết tại sao các tổn thương như vậy không đáp ứng với các chất đối kháng thụ thể angiotensin II. Các đột biến soma ở KCNJ5 có liên quan đến APA; tuy nhiên, các đột biến dòng mầm ở KCNJ5 có liên quan đến tăng sản thượng thận hai bên (như trong các trường hợp FH Loại III, sau này).
Cường Aldosteron Gia đình
Bốn phân nhóm của FH trội trên nhiễm sắc thể thường đã được mô tả. FH loại I, được đặc trưng bởi tăng huyết áp nặng biểu hiện trong 2 thập kỷ đầu đời là cường aldosteron có thể ức chế bằng glucocorticoid (được mô tả trong Mục VI). FH loại II, do các đột biến ở CLCN2, gần giống nhất với các dạng cường aldosteron nguyên phát lẻ tẻ, mặc dù trái ngược với dạng gia đình, chưa có đột biến di truyền nào được liên kết với tình trạng lẻ tẻ này. FH loại III có liên quan đến các đột biến dòng mầm ở KCNJ5 và cổ điển biểu hiện là tăng huyết áp nặng và hạ kali máu sớm trong đời. Gần đây, FH loại IV có liên quan đến các đột biến dòng mầm ở CACNA1H (mã hóa tiểu đơn vị α của một kênh canxi loại T, Cav3.2), biểu hiện với cường aldosteron nguyên phát trong thập kỷ đầu đời. Hiếm khi, các đột biến dòng mầm de novo ở CACNA1H cũng đã được mô tả ở trẻ em biểu hiện với tập hợp cường aldosteron nguyên phát, bại não, động kinh, và các bất thường thần kinh.
Nguồn gốc Tế bào của Cường Aldosteron Nguyên phát
Phân tích hóa mô miễn dịch của các tuyến thượng thận bình thường để tìm biểu hiện của CYP11B2 (aldosterone synthase) đã cho thấy sự tồn tại của các cụm tế bào sản xuất aldosterone (APCC) phát sinh dưới bao tuyến thượng thận và kéo dài vào lớp bó. Các APCC ban đầu được mô tả liền kề với các APA, cho thấy chúng có thể đại diện cho một tổn thương tiền thân. Phân tích gen của 23 APCC và mô thượng thận liền kề bình thường đã cho thấy các đột biến trước đây có liên quan đến các tổn thương sản xuất aldosterone ở 35% các trường hợp, với CACNA1D và ATP1A1 bị ảnh hưởng phổ biến nhất; các đột biến ở KCNJ5 không được tìm thấy trong các tổn thương APCC. Trong số các tuyến thượng thận từ các đối tượng Nhật Bản có huyết áp bình thường được tử thiết, tỷ lệ có ít nhất một APCC là khoảng 30%. Trong số các APCC được tìm thấy trong các tuyến thượng thận bình thường từ những người hiến thận, 12% được tìm thấy ở những người hiến dưới 19 tuổi, và 40% ở những người hiến dưới 35 tuổi, cho thấy cường aldosteron nguyên phát có thể thường bắt nguồn từ tuổi thanh thiếu niên và tuổi trưởng thành trẻ.
Tiếp cận Trẻ bị Cường Aldosteron
Nhiệm vụ chẩn đoán là phân biệt cường aldosteron nguyên phát với cường aldosteron thứ phát sinh lý xảy ra để đáp ứng với một rối loạn sinh lý khác. Bất kỳ sự mất natri, giữ kali, hoặc giảm thể tích máu nào cũng sẽ dẫn đến cường aldosteron thứ phát tăng renin máu. Nhiễm toan ống thận, điều trị bằng thuốc lợi tiểu, viêm thận mất muối, hoặc giảm thể tích máu do bệnh thận hư, cổ trướng, hoặc mất máu là những bối cảnh điển hình cho cường aldosteron thứ phát sinh lý. Mặc dù tập hợp tăng huyết áp, kiềm hóa hạ kali máu, và hoạt độ renin huyết tương thấp có thể chỉ ra cường aldosteron nguyên phát, những bệnh nhân bị nghi ngờ mắc tình trạng này nhưng không đáp ứng các tiêu chí này có thể được hưởng lợi từ việc đánh giá cẩn thận trong một môi trường nghiên cứu lâm sàng, nơi có thể đánh giá chính thức tác động của việc nạp natri và các can thiệp khác đối với nồng độ aldosterone và hoạt độ renin.
Các khối u tuyến thượng thận nam hóa và nữ hóa
Hầu hết các khối u tuyến thượng thận nam hóa là các carcinoma tuyến thượng thận sản xuất một loạt các androgen và glucocorticoid hỗn hợp; các adenoma tuyến thượng thận nam hóa và nữ hóa khá hiếm. Các khối u nam hóa ở các bé trai sẽ có biểu hiện tương tự như của CAH thể SV. Sẽ có sự phát triển dương vật, cương cứng, lông mu và lông nách, mụn trứng cá, tăng khối lượng cơ, giọng nói trầm hơn, và da bìu mỏng đi, nhưng kích thước tinh hoàn sẽ là trước tuổi dậy thì. Nồng độ testosterone tăng cao ở các bé trai làm thay đổi hành vi, với sự cáu kỉnh, ồn ào, hiếu động, và chơi thô bạo tăng lên, nhưng không có bằng chứng về ham muốn tình dục. Chẩn đoán dựa trên tăng androgen máu không thể ức chế bằng glucocorticoid. Điều trị là phẫu thuật; tất cả các khối u như vậy nên được xử lý như thể chúng là ác tính, với sự cẩn thận không cắt vào bao và gieo rắc các tế bào lên phúc mạc. Việc phân biệt mô học giữa adenoma và carcinoma tuyến thượng thận rất khó khăn, đặc biệt là ở bệnh nhân nhi.
Các khối u tuyến thượng thận nữ hóa cực kỳ hiếm ở cả hai giới. P450aro, enzyme thơm hóa các tiền chất androgen thành estrogen, thường không được tìm thấy trong tuyến thượng thận nhưng được tìm thấy ở các mô ngoại vi, chẳng hạn như mỡ, và cũng được tìm thấy trong một số carcinoma tuyến thượng thận. Không rõ liệu hầu hết các khối u tuyến thượng thận nữ hóa có biểu hiện sản xuất lạc chỗ enzyme này hay không, liệu một số enzyme khác có làm trung gian cho quá trình thơm hóa trong khối u hay không, hay liệu đây có thực sự là các khối u sản xuất androgen, nam hóa xảy ra trong một bối cảnh có sự thơm hóa ngoại vi hiệu quả bất thường của androgen tuyến thượng thận. Các khối u nữ hóa tuyến thượng thận (hoặc ngoài tuyến thượng thận) có thể được phân biệt với dậy thì sớm thật sự (trung ương) ở các bé gái bằng sự vắng mặt của nồng độ gonadotropin lưu hành tăng và bằng đáp ứng trước tuổi dậy thì của LH với một thử thách GnRH tĩnh mạch (yếu tố giải phóng hormone hoàng thể hóa [LRF], GnRH). Ở các bé trai, các khối u như vậy sẽ gây ra nữ hóa tuyến vú, giống như nữ hóa tuyến vú lành tính thường đi kèm với tuổi dậy thì. Tuy nhiên, cũng như với các khối u tuyến thượng thận nam hóa, kích thước tinh hoàn, và đáp ứng gonadotropin với xét nghiệm LRF sẽ là trước tuổi dậy thì. Chẩn đoán một khối u nữ hóa ở một bé trai tuổi dậy thì có thể cực kỳ khó khăn, nhưng nó thường được gợi ý bởi sự ngừng tiến triển dậy thì và có thể được chứng minh bằng sự tồn tại của estrogen huyết tương lưu hành sau khi dùng testosterone.
Các rối loạn khác
Kháng Glucocorticoid Gia đình
Kháng glucocorticoid gia đình là một rối loạn hiếm gặp do các đột biến ở đồng dạng α của GR. Giảm hoạt động của glucocorticoid dẫn đến tăng tiết ACTH rõ rệt, ngoài việc kích thích sản xuất cortisol, còn kích thích sản xuất các steroid thượng thận khác. Do đó, những bệnh nhân này có thể biểu hiện mệt mỏi, tăng huyết áp, và kiềm hóa hạ kali máu, gợi ý một hội chứng dư thừa mineralocorticoid, và cũng có các triệu chứng tăng androgen máu. Ngược lại, họ thường thiếu các đặc điểm Cushingoid với tăng cortisol máu sinh hóa. Nhịp sinh học ngày đêm của trục HPA được duy trì và quan sát thấy sự kháng lại sự ức chế dexamethasone. Các bệnh nhân đã được mô tả là đồng hợp tử cho các đột biến sai nghĩa, dị hợp tử cho một đoạn xóa gen, hoặc đồng hợp tử cho một đột biến hoàn toàn vô hiệu. Các đột biến điểm dị hợp tử với hoạt động trội âm không hoàn toàn hoặc nhiều tác động lên hoạt động của GRα cũng đã được mô tả. Các đột biến điểm có thể can thiệp vào sự điều hòa phiên mã phụ thuộc GRα thông qua sự thay đổi liên kết DNA, suy giảm liên kết phối tử, trì hoãn định vị hạt nhân, tập hợp hạt nhân bất thường, và gián đoạn tương tác với các đồng hoạt hóa, tùy thuộc vào vị trí của đột biến. Do đó, kháng glucocorticoid gia đình thường là một hội chứng kháng một phần đối với hoạt động của glucocorticoid. Điều trị bao gồm các liều dexamethasone trên mức sinh lý sẽ gây ra một đáp ứng sinh lý với thụ thể hoạt động kém, thường bắt đầu với 0,25 đến 0,5 mg/ngày trước khi đi ngủ và điều chỉnh để ức chế ACTH buổi sáng và do đó là androgen, huyết áp, và nồng độ kali huyết thanh. Liều dexamethasone có thể được giảm đến mức tối thiểu theo nhu cầu cá nhân. Tăng huyết áp có thể cần điều trị bổ sung bằng các chất đối kháng aldosterone.
Giả cường Aldosteron
Giả cường aldosteron (PHA) là một rối loạn mất muối ở trẻ sơ sinh được đặc trưng bởi hạ natri máu, tăng kali máu, và hoạt độ renin huyết tương tăng khi có nồng độ aldosterone tăng cao, phản ánh sự kháng aldosterone. Dạng PHA hệ thống, lặn trên nhiễm sắc thể thường, phổ biến hơn, nghiêm trọng hơn (“giả cường aldosteron Loại II”) là do các đột biến bất hoạt ở bất kỳ tiểu đơn vị nào trong ba tiểu đơn vị (α, β, γ) của kênh natri nhạy cảm với amiloride, ENaC (được mã hóa bởi các gen SCNN1A, B và G). Tình trạng này thường liên quan đến sung huyết đường hô hấp dưới, ho và khò khè (nhưng không phải nhiễm trùng phổi), và sổ mũi trong, vì các đột biến ENaC làm tăng thể tích dịch đường thở. PHA loại II tồn tại đến tuổi trưởng thành, đòi hỏi liệu pháp thay thế muối tích cực suốt đời. Các đột biến tăng chức năng do cắt ngắn đầu C của β-ENaC gây ra hội chứng Liddle, một dạng tăng huyết áp giữ muối trội trên nhiễm sắc thể thường.
Giả cường aldosteron loại 1 thận trội trên nhiễm sắc thể thường (PHA loại I) là do các đột biến bất hoạt ở MR (được mã hóa bởi gen NR3C2). Hơn 50 đột biến khác nhau đã được tìm thấy ở thụ thể này, can thiệp vào liên kết mineralocorticoid và phiên mã gen. PHA loại I nhẹ hơn các dạng PHA lặn do các đột biến ENaC và thuyên giảm theo tuổi, nhưng cần liệu pháp thay thế natri ở trẻ sơ sinh và trẻ em. Hiếm khi, các đột biến điểm ở MR đã được tìm thấy liên quan đến một dạng tăng huyết áp nặng trội trên nhiễm sắc thể thường, bắt đầu ở tuổi vị thành niên và xấu đi khi mang thai. Trong những trường hợp này, những thay đổi trong cấu trúc của miền liên kết phối tử của MR dẫn đến sự kích hoạt cấu thành nhẹ, cũng như cho phép liên kết và kích hoạt thụ thể bởi progesterone.
Một dạng PHA mắc phải, thoáng qua hiếm khi được thấy ở trẻ sơ sinh bị bệnh đường tiết niệu tắc nghẽn, đặc biệt là ngay sau khi phẫu thuật giải phóng tắc nghẽn, hoặc bị nhiễm trùng đường tiết niệu. Tổn thương là ở ống thận, giống như nhiễm toan ống thận, loại 4, do đó điều trị bằng mineralocorticoid thường không hiệu quả; thay thế muối thường là đủ, trong khi tổn thương thận tự khỏi. Nguyên nhân của sự kháng mineralocorticoid trong những trường hợp như vậy chưa được hiểu rõ.
Liệu pháp glucocorticoid và ngừng thuốc
Glucocorticoid được gọi như vậy vì các tác dụng chính của chúng là làm tăng nồng độ glucose huyết tương. Điều này xảy ra bởi các tác dụng đối kháng insulin đa dạng và sự cảm ứng phiên mã của các gen mã hóa các enzyme của con đường đường phân Embden-Meyerhof và các enzyme gan khác chuyển hướng các axit amin, chẳng hạn như alanine, đến việc sản xuất glucose. Kể từ khi được đưa vào y học lâm sàng vào đầu những năm 1950, glucocorticoid đã được sử dụng để cố gắng điều trị một số lượng đáng kể các bệnh. Hiện tại, việc sử dụng hợp lý của chúng được chia thành hai loại chính: thay thế trong suy thượng thận và sử dụng dược lý. Loại thứ hai phần lớn liên quan đến các đặc tính chống viêm và ức chế miễn dịch của glucocorticoid nhưng cũng bao gồm các tác dụng của chúng gây ngừng tăng trưởng và gây ra apoptosis ở các bạch cầu bình thường và bệnh bạch cầu, làm giảm nồng độ canxi huyết tương, và giảm tăng áp lực nội sọ.
Hầu như tất cả các tác dụng này đều qua trung gian của các thụ thể GR, được tìm thấy ở hầu hết các tế bào. Có ba biến thể chính của GR (α, β và γ) được tạo ra thông qua các kiểu nối ghép thay thế từ cùng một bản phiên mã mRNA từ gen GR. GRα là phong phú nhất, tương quan với mức độ của protein GR, và được cho là dạng chính làm trung gian cho hoạt động của glucocorticoid. Tuy nhiên, bằng chứng mới nổi cho thấy các GR khác có thể có vai trò quan trọng hơn so với đánh giá ban đầu. Ví dụ, GRβ và GRγ có hoạt tính trội âm so với GRα, và có thể góp phần vào sự phát triển của tình trạng kháng steroid có thể xảy ra trong quá trình điều trị. Hơn nữa, mặc dù mối quan hệ hormone-thụ thể ban đầu có vẻ đơn giản, các glucocorticoid liên kết với thụ thể lại có các hoạt động đa dạng đáng ngạc nhiên có thể đặc hiệu cho các mô cụ thể. Sự đa dạng này được trung gian bởi một loạt các cơ chế, bao gồm sự khác biệt về hình dạng thụ thể ở các mô khác nhau, dẫn đến các hoạt động phiên mã riêng biệt. Điều này có thể được thấy bằng cách so sánh các hồ sơ biểu hiện mRNA trên các mô và loại tế bào khác nhau để đáp ứng với cùng một tín hiệu glucocorticoid. Những khác biệt phân tử này ở GR trên các bối cảnh tế bào khác nhau là cơ chế cho các phản ứng mô khác nhau đối với glucocorticoid, chẳng hạn như thúc đẩy sự mở rộng mô mỡ nội tạng, đồng thời ức chế sự phát triển của cơ và xương. Các đặc điểm khác của hội chứng Cushing cũng có thể là do sự khác biệt đặc hiệu mô trong hoạt động phiên mã của các gen nhạy cảm với glucocorticoid cụ thể. Mặc dù làm phức tạp sự hiểu biết của chúng ta về hoạt động của glucocorticoid, sự đa dạng này có thể cho phép phát triển các loại thuốc chọn lọc hơn cho các đồng dạng GR nhất định, do đó duy trì hiệu quả điều trị, đồng thời giảm thiểu các tác dụng phụ bất lợi, tương tự như sự phát triển của các chất điều biến thụ thể estrogen chọn lọc (SERMS).
Tuy nhiên, sự khác biệt duy nhất giữa các chế phẩm glucocorticoid hiện có là tỷ lệ hoạt tính glucocorticoid so với mineralocorticoid, khả năng liên kết với các protein liên kết khác nhau, hiệu lực mol, và thời gian bán hủy sinh học của chúng. Một ngoại lệ quan trọng đối với mô hình này là việc sử dụng glucocorticoid tổng hợp mạnh dexamethasone để giảm tăng áp lực nội sọ và phù não. Kinh nghiệm phẫu thuật thần kinh cho thấy rằng các liều tối ưu cho tác dụng này cao hơn từ 10 đến 100 lần so với các liều sẽ bão hòa hoàn toàn tất cả các thụ thể có sẵn, cho thấy rằng tác dụng này của dexamethasone có thể đại diện cho hoạt động không qua trung gian của GR. Tuy nhiên, hoạt động glucocorticoid độc lập với thụ thể ở mức sinh lý của glucocorticoid vẫn chưa được xác định.
Trong dân số bình thường, có một sự biến đổi lớn giữa các cá nhân về độ nhạy cảm với glucocorticoid, ít nhất một phần được giải thích bởi các biến thể di truyền của gen GR. Một số đa hình nucleotide đơn đã được liên kết với những thay đổi về độ nhạy cảm với glucocorticoid. Đa hình A3669G (rs6198), có mặt ở khoảng 35% dân số bình thường, có liên quan đến sự mất nhạy cảm với glucocorticoid và một hệ miễn dịch hoạt động mạnh hơn với nguy cơ cao mắc bệnh tự miễn. Đa hình ER22/23EK (rs6189 và rs6190) có mặt ở khoảng 7% dân số và có liên quan đến kháng glucocorticoid nhẹ và một hồ sơ chuyển hóa lành mạnh hơn. Ngược lại, các đa hình N363S (rs1695) và Bcl1 (rs41423247) có mặt ở khoảng 8% và 45% dân số, tương ứng, có liên quan đến tăng nhạy cảm với glucocorticoid nhẹ, tạo ra một hồ sơ chuyển hóa kém may mắn hơn (nhiều mỡ cơ thể hơn, ít khối lượng nạc hơn, tăng cholesterol, và kháng insulin, v.v.). Một số sự thay đổi về độ nhạy cảm với glucocorticoid được giải thích bởi các phân nhóm GR phát sinh từ các sửa đổi sau dịch mã, chẳng hạn như phosphoryl hóa, ubiquitin hóa, sumoylation, và acetyl hóa. Các quá trình này góp phần vào phạm vi rộng của khả năng đáp ứng glucocorticoid để điều hòa đặc hiệu các gen phụ thuộc glucocorticoid. Do đó, độ nhạy cảm với glucocorticoid của cá nhân đóng một vai trò trong việc điều chỉnh nguy cơ mắc các bệnh (tim mạch, tự miễn, chuyển hóa) và cũng có thể ảnh hưởng đến khả năng đáp ứng với điều trị bằng glucocorticoid.
Cơ chế Tác động của Glucocorticoid
GR là thụ thể hormone hạt nhân đầu tiên được nhân bản, tiết lộ một loạt các miền chức năng trong protein hóa ra là nguyên mẫu cho họ thụ thể hạt nhân. Miền liên kết phối tử (LBD) nằm ở đầu C của protein, cũng tương tác với các protein đồng hoạt hóa. LBD của GR rất tương đồng với LBD của MR. Sự khác biệt cấu trúc tinh tế giữa MR và GR tạo ra tính đặc hiệu cho việc liên kết aldosterone với MR, nhưng cortisol có thể liên kết với cả hai thụ thể. Các mô nhạy cảm, như thận, biểu hiện HSD11B2, chất bất hoạt cortisol thành cortisone, bảo vệ các MR khỏi bị kích hoạt bởi cortisol. Miền liên kết DNA (DBD), được bảo tồn giữa các thụ thể hạt nhân, nằm ở trung tâm trong protein GR. DBD tạo điều kiện cho sự dimer hóa thụ thể và chứa hai họa tiết ngón tay kẽm có thể liên kết với rãnh lớn của DNA. Miền đầu N của GR là ít được bảo tồn nhất và cho phép tương tác với các đồng điều hòa.
Các GR không liên kết tồn tại chủ yếu trong tế bào chất; khi nồng độ glucocorticoid nội bào tăng lên, nhiều GR hơn bị liên kết bởi hormone và di chuyển vào nhân. Thay vì là một quá trình thụ động, sự liên kết của glucocorticoid gây ra những thay đổi về hình dạng của GR làm lộ ra các họa tiết định vị hạt nhân, thúc đẩy sự vận chuyển được tạo điều kiện của phức hợp hormone-thụ thể vào nhân. Một khi ở trong nhân, GR đã được liên kết phối tử hoạt động bằng cách (1) liên kết với các trình tự xác định của DNA (các yếu tố đáp ứng glucocorticoid; GRE), (2) tương tác với các đồng yếu tố và đồng điều hòa, và (3) sự tham gia của phức hợp glucocorticoid/GR với bộ máy phiên mã. Tổng hợp của các hành động này điều hòa sự biểu hiện của các gen đích của GR. Sự tiếp cận của GR với các GRE trong DNA cũng bị ảnh hưởng bởi cấu trúc chromatin, góp phần thêm vào hoạt động đặc hiệu mô của glucocorticoid. Các GR có thể liên kết với các GRE dưới dạng một monomer hoặc dimer GR (“đơn giản”) hoặc liên kết với một yếu tố phiên mã khác (“hỗn hợp”), hoặc GR có thể “neo” vào một GRE bằng cách liên kết với một protein khác đã liên kết với GRE. Do đó, GR không cần phải liên kết trực tiếp với DNA để điều hòa các gen đích. Các GRE có thể được tìm thấy ở vị trí “gần promoter” cổ điển trong các gen đích và cũng ở các khoảng cách đáng kể so với gen đích, sau đó chúng cuộn lại gần hơn với gen được điều hòa. Điều này có thể làm phức tạp việc gán các GRE cho các gen đích GR cụ thể, nhưng thêm vào nhiều cấp độ mà glucocorticoid có thể điều hòa sự biểu hiện gen. Tương tự, các sửa đổi sau dịch mã của GR tinh chỉnh thêm hoạt động của glucocorticoid trên các gen đích. Cùng với nhau, những biến số này cho phép hoạt động glucocorticoid đặc hiệu cho mô và tế bào, cũng như khả năng điều chỉnh tinh vi các mức phiên mã của các gen đích để đáp ứng với các nhu cầu sinh lý năng động.
Liệu pháp thay thế
Liệu pháp thay thế glucocorticoid phức tạp do các tác dụng phụ không mong muốn ngay cả với mức độ điều trị quá mức hoặc thiếu hụt nhỏ. Điều trị quá mức có thể gây ra các dấu hiệu và triệu chứng của hội chứng Cushing; và ngay cả điều trị quá mức tối thiểu cũng có thể làm suy giảm sự tăng trưởng. Điều trị thiếu hụt sẽ gây ra các dấu hiệu và triệu chứng của suy thượng thận (xem Bảng 14.9) chỉ khi mức độ thiếu hụt (liều lượng và thời gian) là đáng kể. Tuy nhiên, điều trị thiếu hụt có thể làm suy giảm khả năng đáp ứng với stress của cá nhân. Liệu pháp thay thế glucocorticoid được sử dụng phổ biến nhất trong CAH do 21OHD; tuy nhiên, trong bối cảnh này, điều trị thiếu hụt sẽ dẫn đến sản xuất quá mức androgen tuyến thượng thận, điều này sẽ thúc đẩy sự nam hóa và làm nhanh quá trình trưởng thành và đóng sớm của đầu xương, do đó ảnh hưởng đến chiều cao trưởng thành cuối cùng. Do đó, khi xây dựng một chương trình liệu pháp thay thế thượng thận cho trẻ đang phát triển, nên sử dụng liều thấp nhất có thể để đạt được mục tiêu điều trị.
Để tối ưu hóa liệu pháp thay thế glucocorticoid ở trẻ em, các bác sĩ giỏi đã điều chỉnh liệu pháp của họ để giống với tốc độ bài tiết cortisol nội sinh. Một số nghiên cứu chỉ ra rằng tốc độ bài tiết cortisol là 6 đến 8 mg/m2/ngày ở trẻ em và người lớn, nhưng không có dữ liệu cho trẻ sơ sinh và trẻ em dưới 5 tuổi. Phạm vi các giá trị bình thường thay đổi đáng kể, cho thấy rằng liệu pháp phải được điều chỉnh và cá nhân hóa cho từng bệnh nhân để đạt được kết quả tối ưu. Việc quản lý sự cân bằng mong manh này giữa điều trị quá mức và thiếu hụt ở trẻ, đòi hỏi liệu pháp thay thế, do đó bị làm phức tạp bởi sự thay đổi đáng kể trong tốc độ bài tiết cortisol “bình thường” giữa các trẻ khác nhau có cùng kích thước và khả năng hầu hết các hướng dẫn điều trị đều có xu hướng điều trị quá mức. Tuy nhiên, một số yếu tố bổ sung phải được xem xét khi điều chỉnh một chế độ thay thế glucocorticoid cụ thể cho một trẻ.
Dạng suy thượng thận cụ thể đang được điều trị ảnh hưởng đáng kể đến liệu pháp. Khi điều trị viêm thượng thận tự miễn hoặc bất kỳ dạng nào khác của “bệnh Addison,” nên thận trọng tránh điều trị quá mức. Điều này sẽ loại bỏ khả năng chậm phát triển do thầy thuốc gây ra do glucocorticoid và thường cho phép tuyến yên tiếp tục sản xuất nồng độ ACTH bình thường đến hơi cao. ACTH này sẽ tiếp tục kích thích bộ máy sinh steroid thượng thận chức năng còn lại và cũng cung cấp một phương tiện khá thuận tiện để theo dõi tác dụng của liệu pháp. Ngược lại, khi điều trị CAH nam hóa nặng, tuyến thượng thận nên được ức chế hoàn toàn hơn, vì về cơ bản tất cả quá trình sinh steroid thượng thận sẽ dẫn đến việc sản xuất các androgen không mong muốn, với sự nam hóa và tốc độ tiến triển của sự trưởng thành xương nhanh hơn tốc độ tiến triển của chiều cao. Tuy nhiên, điều trị quá mức cũng sẽ ảnh hưởng đến sự tăng trưởng và có thể có những tác động bất lợi lâu dài, bao gồm mật độ xương, thành phần cơ thể, và chuyển hóa.
Sự hiện diện hay vắng mặt của thiếu hụt mineralocorticoid đi kèm là một biến số quan trọng. Trẻ em bị thiếu hụt mineralocorticoid ở mức độ nhẹ, chẳng hạn như những trẻ bị CAH thể SV, có thể tiếp tục có giá trị ACTH hơi cao, cho thấy sự thay thế glucocorticoid không đủ kết hợp với PRA tăng. Ở một số trẻ, ACTH tăng để đáp ứng với tình trạng giảm thể tích máu mạn tính, bị tổn hại, cố gắng kích thích tuyến thượng thận sản xuất nhiều mineralocorticoid hơn. Ở những trẻ như vậy, không có các dấu hiệu và triệu chứng rõ ràng của thiếu hụt mineralocorticoid, liệu pháp thay thế mineralocorticoid đảm bảo thể tích máu bình thường, cho phép giảm lượng thay thế glucocorticoid cần thiết để ức chế ACTH huyết tương và 17KS trong nước tiểu. Việc giảm liệu pháp glucocorticoid này làm giảm khả năng chiều cao trưởng thành sẽ bị ảnh hưởng và giảm thiểu các tác động bất lợi tiềm tàng khác của dư thừa glucocorticoid.
Công thức cụ thể của glucocorticoid được sử dụng cũng rất quan trọng. Các glucocorticoid tác dụng dài, cực mạnh, chẳng hạn như dexamethasone hoặc prednisone, có thể được sử dụng trong điều trị người lớn nhưng hiếm khi thích hợp cho liệu pháp thay thế ở trẻ em. Vì trẻ em liên tục phát triển và thay đổi cân nặng và diện tích bề mặt cơ thể, cần phải điều chỉnh liều thường xuyên; những thay đổi nhỏ, tăng dần dễ thực hiện hơn với các glucocorticoid tương đối yếu hơn. Việc sử dụng các steroid tác dụng ngắn cho phép bệnh nhân có hoạt tính glucocorticoid ban đêm thấp về mặt sinh lý, tạo điều kiện cho nghỉ ngơi, tăng trưởng, và kích thích tuyến yên, cũng như bắt chước các dao động ngày đêm tự nhiên của nồng độ cortisol. Vì nồng độ ACTH và cortisol cao vào buổi sáng và thấp vào buổi tối, việc cố gắng sao chép nhịp điệu ngày đêm này trong liệu pháp thay thế là hấp dẫn về mặt trí tuệ và logic. Các dao động ngày đêm như vậy trong bài tiết glucocorticoid được thấy ở hầu hết các loài, bao gồm cả những loài không có thời gian nhịn ăn kéo dài, cho thấy rằng các quá trình sinh lý khác bị ảnh hưởng bởi nhịp điệu này. Một đồng hồ phân tử ngày đêm được nhúng trong một số con đường tín hiệu glucocorticoid quan trọng. Tuy nhiên, dữ liệu lâm sàng không đủ để khuyến nghị sự thay đổi liều steroid theo ngày đêm, mặc dù hầu hết các bác sĩ lâm sàng chọn bắt chước nhịp điệu ngày đêm bằng cách cho liều cao hơn vào buổi sáng.
Liệu pháp Dược lý
Liều dược lý của glucocorticoid được sử dụng trong nhiều tình huống lâm sàng, bao gồm ức chế miễn dịch trong ghép tạng, hóa trị liệu khối u, điều trị các hội chứng “tự miễn” collagen, mạch máu và thận hư, viêm ruột vùng, và viêm loét đại tràng. Hen suyễn, giả u não, viêm da, một số bệnh nhiễm trùng, viêm dây thần kinh, và một số bệnh thiếu máu cũng thường được điều trị bằng glucocorticoid. Việc lựa chọn chế phẩm glucocorticoid để sử dụng được hướng dẫn bởi các thông số dược lý được mô tả ở trên và trong Bảng 14.7, và theo thông lệ (ví dụ, việc sử dụng betamethasone thay vì dexamethasone để gây trưởng thành phổi thai nhi trong các trường hợp sinh non sắp xảy ra). Có sự thay đổi đáng kể trong các hoạt tính glucocorticoid và mineralocorticoid tương đối của mỗi steroid, tùy thuộc vào xét nghiệm được sử dụng, do đó Bảng 14.7 là sự tích hợp của nhiều nghiên cứu, và chỉ có thể được xem như một hướng dẫn sơ bộ. Hơn nữa, các liều tương đương giữa các glucocorticoid khác nhau có thể gây hiểu lầm. Hầu như tất cả các sổ tay điều trị đều công bố các bảng tương đương cho các chế phẩm dược phẩm được sử dụng phổ biến nhất của glucocorticoid; một bộ tương đương tương tự được trình bày trong Bảng 14.7. Bởi vì hầu hết các chế phẩm glucocorticoid được dùng cho mục đích dược lý hơn là liệu pháp thay thế, và vì chỉ định phổ biến nhất cho các liều dược lý của glucocorticoid là cho các đặc tính chống viêm của chúng, hầu như tất cả các bảng tương đương glucocorticoid đều dựa trên các tương đương chống viêm, ức chế miễn dịch. Tuy nhiên, sự khác biệt về thời gian bán hủy trong huyết tương và khả năng liên kết với các protein huyết tương dẫn đến các tương đương sinh học khác nhau khi người ta đánh giá, ví dụ, các tương đương chống viêm so với ức chế tăng trưởng. Do đó, dexamethasone được báo cáo rộng rãi là mạnh hơn cortisol khoảng 30 lần khi các khả năng chống viêm của nó được đo, nhưng hoạt tính ức chế tăng trưởng của dexamethasone cao hơn cortisol khoảng 80 lần (xem Bảng 14.7). Các biến số được thảo luận ở trên giải thích tại sao có rất ít sự đồng thuận trong các khuyến nghị để thiết kế một chế độ điều trị bằng glucocorticoid. Tuy nhiên, sự hiểu biết về những biến số này sẽ cho phép theo dõi bệnh nhân một cách thích hợp và khuyến khích bác sĩ thay đổi phương pháp điều trị theo các phản ứng và nhu cầu của từng trẻ.
Liều dược lý của glucocorticoid được dùng trong hơn 1 hoặc 2 tuần sẽ gây ra các dấu hiệu và triệu chứng của hội chứng Cushing do thầy thuốc. Chúng tương tự như các phát hiện do glucocorticoid gây ra trong bệnh Cushing, nhưng có thể nghiêm trọng hơn (nhưng thiếu các tác dụng của androgen tuyến thượng thận) do liều cao liên quan (Bảng 14.13). Tần suất của các tác dụng phụ này đã kích thích sự quan tâm rộng rãi đến việc phát triển các chất tương tự GR chọn lọc hơn, được thảo luận ở trên, nhưng chưa có chất nào được đưa vào sử dụng lâm sàng. Cho đến khi có các phương pháp tiếp cận có mục tiêu hơn, liệu pháp cách ngày thường có thể được thử để giảm độc tính của liệu pháp glucocorticoid dược lý, đặc biệt là sự ức chế trục HPA và sự ức chế tăng trưởng. Tiền đề cơ bản của liệu pháp cách ngày là tình trạng bệnh có thể được ức chế bằng liệu pháp không liên tục, trong khi có sự phục hồi đáng kể của trục HPA trong ngày “nghỉ”. Do đó, liệu pháp cách ngày đòi hỏi phải sử dụng một glucocorticoid tác dụng tương đối ngắn chỉ dùng một lần vào buổi sáng của mỗi ngày điều trị để đảm bảo rằng ngày nghỉ thực sự là nghỉ. Các glucocorticoid tác dụng dài, chẳng hạn như dexamethasone, không nên được sử dụng cho liệu pháp cách ngày; kết quả tốt nhất với prednisone hoặc methyl prednisolone đường uống.
Bảng 14.13. Các biến chứng của Liệu pháp Glucocorticoid liều cao
| Liệu pháp Ngắn hạn | Liệu pháp Dài hạn |
|---|---|
| Viêm dạ dày | Loét dạ dày |
| Ngừng tăng trưởng | Tầm vóc thấp |
| Tăng cảm giác thèm ăn | Tăng calci niệu |
| Tăng đường niệu | Loãng xương, gãy xương |
| Ức chế miễn dịch | Trượt chỏm xương đùi |
| Che lấp các triệu chứng nhiễm trùng, đặc biệt là sốt và viêm | Hoại tử xương do thiếu máu cục bộ |
| Rối loạn tâm thần do nhiễm độc | Chậm liền vết thương |
| Dị hóa | |
| Đục thủy tinh thể | |
| Dễ bầm tím (mỏng manh mao mạch) | |
| Ức chế thượng thận/tuyến yên | |
| Rối loạn tâm thần do nhiễm độc |
Các Chế phẩm Glucocorticoid thường được sử dụng
Nhiều dẫn xuất hóa học và các biến thể của các steroid tự nhiên có sẵn trên thị trường với một loạt các dạng bào chế, tá dược, và nồng độ. Việc lựa chọn sản phẩm thích hợp có thể được đơn giản hóa bằng cách chỉ xem xét các steroid được sử dụng rộng rãi nhất được liệt kê trong Bảng 14.7.
Có bốn yếu tố liên quan trong việc lựa chọn loại thuốc để sử dụng. Thứ nhất, hiệu lực glucocorticoid của các loại thuốc khác nhau thường được tính toán và mô tả theo hiệu lực chống viêm, nhưng điều này có thể không phải là yếu tố xem xét thích hợp trong nhiều trường hợp. Thứ hai, tác dụng ức chế tăng trưởng của một chế phẩm glucocorticoid có thể khác biệt đáng kể so với tác dụng chống viêm của nó. Điều này là do sự khác biệt về thời gian bán hủy, chuyển hóa và liên kết protein, và ái lực với thụ thể (hiệu lực). Thứ ba, hoạt tính mineralocorticoid của các chế phẩm glucocorticoid khác nhau thay đổi rộng rãi. Glucocorticoid có thể liên kết với cả GR và MR. Hoạt tính mineralocorticoid có liên quan mật thiết đến hoạt động của 11βHSD2, chất chuyển hóa glucocorticoid, nhưng không phải mineralocorticoid, thành các dạng không thể liên kết với thụ thể. Do đó, hiệu lực mineralocorticoid tương đối của các steroid khác nhau được xác định bởi cả ái lực của chúng đối với MR và sự kháng lại hoạt động của 11βHSD2. Sự hiểu biết rằng một số glucocorticoid thường được sử dụng, chẳng hạn như cortisol, cortisone, prednisolone, và prednisone, có hoạt tính mineralocorticoid đáng kể là đặc biệt quan trọng khi các liều lớn glucocorticoid được sử dụng làm “liều stress” ở một bệnh nhân đang điều trị thay thế. Các liều stress như vậy của chế phẩm glucocorticoid có thể cung cấp đủ hoạt tính mineralocorticoid để đáp ứng nhu cầu sinh lý; do đó không cần bổ sung mineralocorticoid và việc sử dụng kéo dài các liều rất cao của các chế phẩm này có khả năng dẫn đến các dấu hiệu dư thừa mineralocorticoid. Thứ tư, thời gian bán hủy trong huyết tương và thời gian bán hủy sinh học của các chế phẩm khác nhau có thể không tương đồng và sẽ thay đổi rộng rãi tùy thuộc vào sự liên kết với các protein huyết tương, chuyển hóa ở gan, và sự kích hoạt ở gan. Thời gian bán hủy trong huyết tương của cortisol (hydrocortisone) tiêm tĩnh mạch là thay đổi (trung bình 75–100 phút) và hơn 90% hydrocortisone dùng đường uống được hấp thu trong vòng một giờ. Cortisone và prednisone không có hoạt tính sinh học (và thậm chí có tác dụng đối kháng steroid nhẹ), cho đến khi chúng được chuyển hóa bởi 11βHSD1 của gan thành các dạng hoạt động của chúng, cortisol, và prednisolone. Do đó, hiệu lực glucocorticoid của các chế phẩm này bị ảnh hưởng bởi chức năng gan. Cortisone và prednisone được thanh thải nhanh hơn ở những bệnh nhân dùng các loại thuốc, chẳng hạn như phenobarbital hoặc phenytoin, gây cảm ứng các enzyme gan và được thanh thải chậm hơn ở những bệnh nhân bị suy gan.
Ngoài những cân nhắc về hóa học này trong việc lựa chọn glucocorticoid, đường dùng cũng rất quan trọng. Glucocorticoid có sẵn để sử dụng đường uống, tiêm bắp, tiêm tĩnh mạch, tiêm nội tủy, tiêm nội khớp, dạng hít, và dùng tại chỗ; các chế phẩm dùng tại chỗ bao gồm những chế phẩm được thiết kế để sử dụng trên da, niêm mạc, và kết mạc. Mỗi chế phẩm được thiết kế để cung cấp nồng độ steroid tối đa đến mô mong muốn, trong khi cung cấp ít steroid toàn thân hơn. Tuy nhiên, tất cả các chế phẩm như vậy đều được hấp thu ở các mức độ khác nhau, do đó các chế phẩm dạng hít được sử dụng rộng rãi để điều trị hen suyễn có thể, với liều đủ, gây chậm phát triển và các dấu hiệu khác của hội chứng Cushing và, trong các trường hợp hiếm, dẫn đến suy thượng thận khi ngừng đột ngột. Nói chung, và trái ngược với nhiều loại thuốc khác, các steroid dùng đường uống được hấp thu nhanh chóng, nhưng không hoàn toàn, trong khi các steroid tiêm bắp được hấp thu chậm, nhưng hoàn toàn. Do đó, nếu tốc độ bài tiết cortisol là 8 mg/m2 diện tích bề mặt cơ thể, liều thay thế tiêm bắp hoặc tiêm tĩnh mạch của cortisol (hydrocortisone) sẽ là 8 mg/m2. Tuy nhiên, các liều tương đương được liệt kê trong Bảng 14.7 và các bảng tương tự chỉ là các ước tính chung, và liều phải được điều chỉnh theo đáp ứng lâm sàng.
ACTH cũng có thể được sử dụng cho liệu pháp glucocorticoid bằng tác dụng của nó để kích thích sinh tổng hợp steroid thượng thận nội sinh. Mặc dù ACTH tiêm tĩnh mạch và tiêm bắp cực kỳ hữu ích trong các xét nghiệm chẩn đoán chức năng thượng thận, việc sử dụng ACTH như một tác nhân điều trị không còn được ưa chuộng, chủ yếu vì nó sẽ kích thích tổng hợp mineralocorticoid và androgen tuyến thượng thận, cũng như glucocorticoid. Hơn nữa, sự cần thiết phải dùng ACTH qua đường tiêm làm giảm thêm tính hữu dụng của nó. ACTH(1-39) tiêm bắp dưới dạng gel được khuyến nghị và là phương pháp điều trị được lựa chọn cho co giật ở trẻ sơ sinh và có thể cả cho các dạng động kinh khác ở trẻ sơ sinh kháng với các thuốc chống co giật thông thường. Liệu tác dụng này có qua trung gian của chính ACTH, bởi các steroid thượng thận do ACTH gây ra, hay bởi sự tổng hợp đáp ứng với ACTH của các “neurosteroid” mới trong não vẫn chưa được xác định. Khi các liều dược lý của ACTH được sử dụng điều trị, như trong co giật ở trẻ sơ sinh, bệnh nhân nên được cho chế độ ăn ít natri để cải thiện tình trạng tăng huyết áp do steroid. Mặc dù nồng độ ACTH tăng cao rõ rệt, như trong hội chứng ACTH lạc chỗ, sẽ gây ức chế tuyến yên, điều trị bằng tiêm ACTH hàng ngày dẫn đến ức chế hạ đồi-tuyến yên ít hơn so với điều trị bằng các liều tương đương của glucocorticoid đường uống, có lẽ vì tác dụng lên tuyến thượng thận là thoáng qua. Ngoài ra, ức chế thượng thận rõ ràng không xảy ra trong liệu pháp ACTH. Bởi vì tác dụng của ACTH đối với sinh tổng hợp steroid thượng thận rất thay đổi, việc xác định liều tương đương cho ACTH và các chế phẩm steroid đường uống thậm chí còn khó khăn hơn so với giữa các steroid khác nhau, như đã thảo luận ở trên. Một hướng dẫn rất sơ bộ từ các nghiên cứu ở người lớn là 40 đơn vị gel ACTH(1-39) tương đương với khoảng 100 mg cortisol.
Ngừng Liệu pháp Glucocorticoid
Ngừng liệu pháp glucocorticoid có thể khó khăn và có thể dẫn đến các triệu chứng thiếu hụt glucocorticoid. Khi liệu pháp glucocorticoid đã được sử dụng trong 10 ngày hoặc ít hơn, liệu pháp có thể được ngừng đột ngột, ngay cả khi đã sử dụng liều cao. Mặc dù chỉ cần một hoặc hai liều glucocorticoid để ức chế trục HPA, trục này phục hồi rất nhanh sau khi ức chế ngắn hạn. Khi liệu pháp đã kéo dài trong 2 tuần hoặc lâu hơn, sự phục hồi chức năng HPA chậm hơn, và cần giảm liều glucocorticoid. Việc ngừng đột ngột liệu pháp ở những bệnh nhân như vậy sẽ dẫn đến các triệu chứng thiếu hụt glucocorticoid, cái gọi là hội chứng cai steroid. Phức hợp triệu chứng này không bao gồm mất muối, vì chức năng lớp cầu của tuyến thượng thận, được điều hòa chủ yếu bởi hệ renin-angiotensin, vẫn bình thường. Tuy nhiên, huyết áp có thể giảm đột ngột, vì glucocorticoid cần thiết cho hoạt động của catecholamine trong việc duy trì trương lực mạch máu. Các triệu chứng nổi bật nhất của hội chứng cai steroid bao gồm khó chịu, chán ăn, đau đầu, thờ ơ, buồn nôn, và sốt. Khi giảm các liều dược lý của glucocorticoid, có vẻ hợp lý khi giảm liều đột ngột xuống các liều thay thế “sinh lý”. Tuy nhiên, điều này hiếm khi thành công và đôi khi gây tai hại. Ngay cả khi được cho liều thay thế sinh lý, những bệnh nhân đã nhận các liều dược lý của glucocorticoid sẽ trải qua hội chứng cai steroid. Liệu pháp glucocorticoid dược lý dài hạn ức chế tổng hợp GR, do đó nồng độ sinh lý của glucocorticoid sẽ gây ra các phản ứng tế bào dưới mức sinh lý, dẫn đến hội chứng cai steroid. Do đó, cần phải giảm liều dần dần ngay từ đầu. Thời gian điều trị bằng glucocorticoid là một yếu tố quan trọng trong việc thiết kế một chương trình ngừng glucocorticoid. Liệu pháp trong vài tháng sẽ ức chế hoàn toàn trục HPA nhưng sẽ không gây teo tuyến thượng thận. Liệu pháp kéo dài nhiều năm có thể dẫn đến teo gần như toàn bộ lớp bó/lớp lưới của tuyến thượng thận, và do đó có thể cần một chế độ ngừng thuốc kéo dài hàng tháng.
Các quy trình để giảm liều steroid là theo kinh nghiệm. Thành công của chúng được xác định bởi thời gian và phương thức điều trị và bởi các phản ứng của từng bệnh nhân. Bệnh nhân đã điều trị cách ngày có thể được ngừng thuốc dễ dàng hơn so với những người điều trị hàng ngày, đặc biệt là điều trị hàng ngày bằng một glucocorticoid tác dụng dài, chẳng hạn như dexamethasone. Ở những bệnh nhân điều trị lâu dài, thường khuyến nghị giảm 25% liều điều trị trước đó hàng tuần. Nếu bệnh nhân đã điều trị hàng ngày tương đương với 100 mg cortisol trong nhiều tháng, có thể cần một phác đồ giảm liều trong 8 đến 10 tuần. Một phác đồ 75% liều của tuần trước do đó sẽ là 75 mg/ngày trong tuần đầu tiên, 56 mg/ngày trong tuần thứ hai, sau đó là 42, 31,5, 24, 18, 13,5, 10, 7,5, 5,5 mg/ngày, sau đó ngừng điều trị. Một chế độ thực tế hơn dựa trên kích thước của các viên thuốc có sẵn sẽ là 75, 50, 37,5, 25, 17,5, 12,5, 10, 7,5, và 5 mg/ngày. Hầu hết các bệnh nhân có thể được giảm liều nhanh hơn, nhưng tất cả các bệnh nhân cần được theo dõi chặt chẽ. Khi việc ngừng thuốc được thực hiện bằng các steroid khác ngoài cortisol, việc đo các giá trị cortisol buổi sáng có thể là một biện pháp hỗ trợ hữu ích. Các giá trị cortisol buổi sáng từ 10 μg/dL trở lên cho thấy rằng liều có thể được giảm một cách an toàn.
Ngay cả sau khi ngừng thành công liệu pháp, trục HPA vẫn chưa hoàn toàn bình thường. Giống như ở bệnh nhân được điều trị thành công bệnh Cushing, trục HPA có thể không có khả năng đáp ứng với stress nặng trong 6 đến 12 tháng, sau khi ngừng thành công liệu pháp glucocorticoid liều cao, dài hạn. Do đó, việc đánh giá vùng dưới đồi và tuyến yên bằng nghiệm pháp CRF hoặc metyrapone (khi có sẵn), và đánh giá khả năng đáp ứng của tuyến thượng thận với kích thích tuyến yên bằng nghiệm pháp ACTH liều thấp tiêm tĩnh mạch, nên được thực hiện vào cuối chương trình ngừng thuốc, và 6 tháng sau đó. Kết quả của các xét nghiệm này sẽ cho biết liệu có cần “bao phủ bằng steroid” trong stress phẫu thuật cấp tính hoặc bệnh tật hay không.
Liều Stress của Glucocorticoid
Tốc độ bài tiết cortisol tăng đáng kể trong quá trình stress sinh lý, chẳng hạn như chấn thương, phẫu thuật, hoặc bệnh nặng. Bệnh nhân đang điều trị thay thế glucocorticoid hoặc những người gần đây đã ngừng liệu pháp dược lý cần được bao phủ bằng “liều stress” của steroid trong những tình huống như vậy. Tuy nhiên, các chỉ định cụ thể cho việc bao phủ này và liều lượng thích hợp gây tranh cãi và khó xác định; hầu hết các bác sĩ thực hành thích phạm sai lầm về phía “an toàn” của việc quá liều steroid. Đây là chiến thuật an toàn nhất trong ngắn hạn nhưng có thể có ảnh hưởng đáng kể đến sự tăng trưởng trong một khoảng thời gian nhiều năm và, nếu được sử dụng thường xuyên, có thể kéo dài thời gian cần thiết để trục HPA phục hồi.
Thường được cho là cần các liều gấp 3 lần liều thay thế sinh lý cho “stress của phẫu thuật,” nhưng stress này thay đổi rất nhiều. Trong quá khứ, nhiều “stress phẫu thuật” liên quan đến đau, mất dịch, giảm thể tích máu, và sốt, nhưng những vấn đề này được quản lý tốt hơn trong các dịch vụ nhi khoa đương đại. Các kỹ thuật gây mê hiện đại; các loại thuốc gây mê, giảm đau, và giãn cơ tốt hơn; và nhận thức tăng lên về các nhu cầu đặc biệt của trẻ em trong việc quản lý dịch và điện giải trong phẫu thuật đã làm giảm đáng kể stress phẫu thuật. Cortisol huyết thanh không vượt quá 10 ug/dL (276 nmol/L) ở trẻ em có tuyến thượng thận nguyên vẹn đang được gây mê và phẫu thuật nhỏ. Khi được sử dụng, liều stress nên được cho với liều không đổi cách nhau 6 giờ. Mặc dù vẫn còn phù hợp và cần thiết để cho khoảng 3 lần yêu cầu sinh lý trong các giai đoạn stress như vậy, có lẽ không cần thiết phải cho liều cao hơn. Tương tự, không cần thiết phải tăng gấp ba lần chế độ thay thế sinh lý của một trẻ trong các đợt cảm lạnh đơn giản, nhiễm trùng đường hô hấp trên, viêm tai giữa, hoặc sau khi tiêm chủng.
Việc chuẩn bị bệnh nhân suy thượng thận đang điều trị thay thế cho phẫu thuật đòi hỏi sự phối hợp với đội ngũ gây mê. Cách tiếp cận tốt nhất là đặt một liều stress 25 mg hydrocortisone trên mỗi mét vuông vào dịch truyền tĩnh mạch, để glucocorticoid được cung cấp liên tục trong quá trình phẫu thuật, thay vì được cho một liều bolus duy nhất vào đầu thủ thuật. Dường như stress lớn nhất là vào thời điểm hồi tỉnh sau gây mê, thay vì vào thời điểm khởi mê. Liệu pháp thông thường với liều gấp 2 đến 3 lần yêu cầu sinh lý sau đó có thể được bắt đầu lại vào ngày sau thủ thuật phẫu thuật.
Thay thế Mineralocorticoid
Liệu pháp thay thế bằng mineralocorticoid được chỉ định trong CAH mất muối và trong các hội chứng suy thượng thận ảnh hưởng đến lớp cầu. Chỉ có một loại mineralocorticoid, 9α-fluorocortisol (Fluorinef) đường uống, hiện có sẵn. Không có chế phẩm mineralocorticoid đường tiêm nên phải sử dụng hydrocortisone cộng với muối.
Do sự nhạy cảm với mineralocorticoid tăng theo tuổi (xem Hình 14.18), liều mineralocorticoid tương tự ở trẻ em và người lớn. Trẻ sơ sinh khá không nhạy cảm với mineralocorticoid và thường cần liều lớn hơn đáng kể so với người lớn. Liều thay thế ở người lớn của 9α-fluorocortisol thường là 0,05 đến 0,10 mg mỗi ngày, nhưng trẻ sơ sinh bị CAH có thể cần tới 0,4 mg. Natri phải có sẵn cho các nephron để mineralocorticoid thúc đẩy tái hấp thu natri, do đó trẻ sơ sinh bị CAH mất muối phải được điều trị bằng cả mineralocorticoid và natri clorua. Tương tự, mineralocorticoid sẽ gây tăng huyết áp chỉ bằng cách giữ lại natri.
Cortisol có hoạt tính mineralocorticoid: khoảng 20 mg cortisol tiêm tĩnh mạch có tác dụng mineralocorticoid tương đương với 0,1 mg 9α-fluorocortisol. Do đó, các liều stress của cortisol cung cấp đủ hoạt tính mineralocorticoid, và việc thay thế mineralocorticoid có thể bị gián đoạn. Không cần thêm 9α-fluorocortisol cho đến khi các liều stress trên mức sinh lý của cortisol được giảm xuống. Bởi vì 9α-fluorocortisol chỉ có thể được dùng bằng đường uống và vì điều này có thể không thể thực hiện được trong giai đoạn sau phẫu thuật, loại thuốc thích hợp để thay thế glucocorticoid là cortisol, có hoạt tính mineralocorticoid, thay vì các steroid tổng hợp, chẳng hạn như prednisone hoặc dexamethasone, có ít hoạt tính mineralocorticoid.
Lời kết
Bởi vì vỏ thượng thận chủ yếu liên quan đến việc tổng hợp steroid, hầu hết các rối loạn của nó phản ánh các tổn thương di truyền trong sự phát triển và sinh tổng hợp steroid của tuyến thượng thận. Sản xuất quá mức và thiếu hụt các steroid, có các tác dụng sinh lý đa dạng và phức tạp, dẫn đến các kiểu hình và biểu hiện lâm sàng phức tạp. Những rối loạn di truyền, nguyên phát này thường tự biểu hiện ở trẻ sơ sinh và trẻ nhỏ. Ngược lại, các rối loạn thứ phát, chẳng hạn như bệnh Cushing (thường là một rối loạn của tuyến yên) và bệnh Addison (thường là một rối loạn của miễn dịch tế bào), có thể được thấy ở mọi lứa tuổi. Do đó, bác sĩ nội tiết nhi khoa phải có một sự hiểu biết chi tiết về sinh học tế bào, di truyền học, và hóa sinh của sinh tổng hợp hormone steroid. Những cải tiến về tốc độ, độ chính xác, và tính kinh tế của việc giải trình tự DNA hiện cho phép chẩn đoán di truyền trực tiếp nhiều bệnh di truyền. Tuy nhiên, không có khả năng các phép đo hormone steroid sẽ trở nên lỗi thời trong tương lai gần, và sự hiểu biết về sinh lý steroid sẽ luôn cần thiết để hiểu các biểu hiện lâm sàng, xây dựng các chẩn đoán phân biệt, chọn các gen để nghiên cứu, và theo dõi liệu pháp. Do đó, di truyền học sẽ tiếp tục nâng cao sinh lý học lâm sàng, nhưng sẽ không thay thế nó.
Bảng Chú giải Thuật ngữ – Chương 14
| STT | Thuật ngữ tiếng Anh | Phiên âm IPA | Nghĩa Tiếng Việt |
|---|---|---|---|
| 1 | Adrenal cortex | /əˈdriːnəl ˈkɔːrtɛks/ | Vỏ thượng thận |
| 2 | Steroid hormones | /ˈstɪərɔɪd ˈhɔːrmoʊnz/ | Hormone steroid |
| 3 | Mineralocorticoids | /ˌmɪnərəloʊˈkɔːrtɪkɔɪdz/ | Mineralocorticoid |
| 4 | Aldosterone | /ælˈdɒstəroʊn/ | Aldosterone |
| 5 | Electrolyte balance | /ɪˈlɛktrəlaɪt ˈbæləns/ | Cân bằng điện giải |
| 6 | Intravascular volume | /ˌɪntrəˈvæskjələr ˈvɒljuːm/ | Thể tích nội mạch |
| 7 | Blood pressure | /blʌd ˈprɛʃər/ | Huyết áp |
| 8 | Glucocorticoids | /ˌɡluːkoʊˈkɔːrtɪkɔɪdz/ | Glucocorticoid |
| 9 | Cortisol | /ˈkɔːrtɪsɒl/ | Cortisol |
| 10 | Carbohydrate-mobilizing | /ˌkɑːrboʊˈhaɪdreɪt ˈmoʊbəlaɪzɪŋ/ | Huy động carbohydrate |
| 11 | Adrenal androgens | /əˈdriːnəl ˈændrədʒənz/ | Androgen tuyến thượng thận |
| 12 | Virilism | /ˈvɪrəlɪzəm/ | Nam hóa |
| 13 | Congenital adrenal hyperplasias (CAHs) | /kənˈdʒɛnɪtəl əˈdriːnəl ˌhaɪpərˈpleɪʒəz/ | Tăng sản thượng thận bẩm sinh |
| 14 | Cushing disease | /ˈkʊʃɪŋ dɪˈziːz/ | Bệnh Cushing |
| 15 | Primary aldosteronism | /ˈpraɪmeri ˌældəˈstɛrəˌnɪzəm/ | Cường aldosteron nguyên phát |
| 16 | Hypertension | /ˌhaɪpərˈtɛnʃən/ | Tăng huyết áp |
| 17 | Adrenal glands | /əˈdriːnəl ɡlændz/ | Tuyến thượng thận |
| 18 | Adrenal insufficiency | /əˈdriːnəl ˌɪnsəˈfɪʃənsi/ | Suy thượng thận |
| 19 | Adrenalectomy | /əˌdriːnəˈlɛktəmi/ | Cắt tuyến thượng thận |
| 20 | Steroidogenesis | /ˌstɪərɔɪdoʊˈdʒɛnəsɪs/ | Sinh tổng hợp steroid |
| 21 | Cytochrome P450 | /ˈsaɪtəˌkroʊm piː fɔːr ˈfɪfti/ | Cytochrome P450 |
| 22 | 21-hydroxylation | /ˌtwɛntiˈwʌn haɪˌdrɒksɪˈleɪʃən/ | 21-hydroxyl hóa |
| 23 | Steroidogenic enzymes | /ˌstɪərɔɪdoʊˈdʒɛnɪk ˈɛnzaɪmz/ | Enzyme sinh steroid |
| 24 | Steroid hormone receptors | /ˈstɪərɔɪd ˈhɔːrmoʊn rɪˈsɛptərz/ | Thụ thể hormone steroid |
| 25 | Embryology | /ˌɛmbriˈɒlədʒi/ | Phôi thai học |
| 26 | Mesodermal origin | /ˌmɛsoʊˈdɜːrməl ˈɔːrɪdʒɪn/ | Nguồn gốc trung bì |
| 27 | Adrenal medulla | /əˈdriːnəl məˈdʌlə/ | Tủy thượng thận |
| 28 | Neuroectoderm | /ˌnʊəroʊˈɛktədɜːrm/ | Ngoại bì thần kinh |
| 29 | Adrenogonadal progenitor cells | /əˌdriːnoʊɡəˈnædəl prəˈdʒɛnɪtər sɛlz/ | Tế bào tiền thân thượng thận-sinh dục |
| 30 | Coelomic epithelium | /siːˈloʊmɪk ˌɛpɪˈθiːliəm/ | Biểu mô khoang cơ thể |
| 31 | Urogenital ridge | /ˌjʊəroʊˈdʒɛnɪtəl rɪdʒ/ | Gờ niệu-sinh dục |
| 32 | Dorsal mesentery | /ˈdɔːrsəl ˈmɛsənˌtɛri/ | Mạc treo lưng |
| 33 | Steroidogenic cells | /ˌstɪərɔɪdoʊˈdʒɛnɪk sɛlz/ | Tế bào sinh steroid |
| 34 | Gonads | /ˈɡoʊnædz/ | Tuyến sinh dục |
| 35 | Retroperitoneally | /ˌrɛtroʊˌpɛrɪtəˈniːəli/ | Sau phúc mạc |
| 36 | Mesonephros | /ˌmɛsoʊˈnɛfrəs/ | Trung thận |
| 37 | Adrenal primordium | /əˈdriːnəl praɪˈmɔːrdiəm/ | Mầm tuyến thượng thận |
| 38 | Sympathetic cells | /ˌsɪmpəˈθɛtɪk sɛlz/ | Tế bào giao cảm |
| 39 | Neural crest | /ˈnʊərəl krɛst/ | Mào thần kinh |
| 40 | Fetal adrenal cortex | /ˈfiːtəl əˈdriːnəl ˈkɔːrtɛks/ | Vỏ thượng thận thai nhi |
| 41 | Definitive zone | /dɪˈfɪnɪtɪv zoʊn/ | Vùng trưởng thành |
| 42 | Fetal zone | /ˈfiːtəl zoʊn/ | Vùng bào thai |
| 43 | Dehydroepiandrosterone (DHEA) | /diːˌhaɪdroʊˌɛpiænˈdrɒstəroʊn/ | Dehydroepiandrosterone (DHEA) |
| 44 | Dehydroepiandrosterone sulfate (DHEAS) | /diːˌhaɪdroʊˌɛpiænˈdrɒstəroʊn ˈsʌlfeɪt/ | Dehydroepiandrosterone sulfat (DHEAS) |
| 45 | Estriol | /ˈɛstriˌɒl/ | Estriol |
| 46 | Transitional zone | /trænˈzɪʃənəl zoʊn/ | Vùng chuyển tiếp |
| 47 | Apoptosis | /ˌæpəpˈtoʊsɪs/ | Chết tế bào theo chương trình |
| 48 | Postnatal life | /ˌpoʊstˈneɪtəl laɪf/ | Đời sống sau sinh |
| 49 | Adrenal differentiation | /əˈdriːnəl ˌdɪfəˌrɛnʃiˈeɪʃən/ | Biệt hóa tuyến thượng thận |
| 50 | Signaling pathways | /ˈsɪɡnəlɪŋ ˈpæθweɪz/ | Các con đường tín hiệu |
| 51 | Transcription factors | /trænˈskrɪpʃən ˈfæktərz/ | Các yếu tố phiên mã |
| 52 | Trophic effects | /ˈtroʊfɪk ɪˈfɛkts/ | Các tác dụng dinh dưỡng |
| 53 | Adrenocorticotropic hormone (ACTH) | /əˌdriːnoʊˌkɔːrtɪkoʊˈtroʊpɪk ˈhɔːrmoʊn/ | Hormone vỏ thượng thận (ACTH) |
| 54 | Insulin-like growth factor-2 (IGF-2) | /ˈɪnsəlɪn laɪk ɡroʊθ ˈfæktər tuː/ | Yếu tố tăng trưởng giống insulin-2 (IGF-2) |
| 55 | Basic fibroblast growth factor (bFGF) | /ˈbeɪsɪk ˌfaɪbroʊˈblæst ɡroʊθ ˈfæktər/ | Yếu tố tăng trưởng nguyên bào sợi cơ bản (bFGF) |
| 56 | Epidermal growth factor (EGF) | /ˌɛpɪˈdɜːrməl ɡroʊθ ˈfæktər/ | Yếu tố tăng trưởng biểu bì (EGF) |
| 57 | Anatomy | /əˈnætəmi/ | Giải phẫu học |
| 58 | Suprarenal glands | /ˌsuːprəˈriːnəl ɡlændz/ | Tuyến trên thận |
| 59 | Sinusoidal circulation | /ˌsaɪnəˈsɔɪdəl ˌsɜːrkjəˈleɪʃən/ | Tuần hoàn xoang |
| 60 | Chromaffin cells | /ˈkroʊməfɪn sɛlz/ | Tế bào ưa crôm |
| 61 | Phenylethanolamine-N-methyltransferase | /ˌfiːnəlˌɛθəˈnoʊləmiːn ɛn ˈmɛθəlˈtrænsfəreɪs/ | Phenylethanolamine-N-methyltransferase |
| 62 | Norepinephrine | /ˌnɔːrˌɛpɪˈnɛfrɪn/ | Norepinephrine |
| 63 | Epinephrine | /ˌɛpɪˈnɛfrɪn/ | Epinephrine |
| 64 | Zona glomerulosa | /ˈzoʊnə ɡləˌmɛrjəˈloʊsə/ | Lớp cầu |
| 65 | Capsule | /ˈkæpsuːl/ | Vỏ bao |
| 66 | Zona fasciculata | /ˈzoʊnə fəˌsɪkjəˈlɑːtə/ | Lớp bó |
| 67 | Zona reticularis | /ˈzoʊnə rɪˌtɪkjəˈlɛərɪs/ | Lớp lưới |
| 68 | Centripetal migration | /sɛnˈtrɪpɪtəl maɪˈɡreɪʃən/ | Di chuyển hướng tâm |
| 69 | Cholesterol | /kəˈlɛstərɒl/ | Cholesterol |
| 70 | Low-density lipoproteins (LDLs) | /loʊ ˈdɛnsəti ˌlaɪpoʊˈproʊtiːnz/ | Lipoprotein tỷ trọng thấp (LDL) |
| 71 | 3-hydroxy-3-methylglutaryl co-enzyme A (HMG-CoA) reductase | /θriː haɪˈdrɒksi θriː ˈmɛθəlˌɡluːtəˈrɪl koʊˈɛnzaɪm eɪ rɪˈdʌkteɪs/ | 3-hydroxy-3-methylglutaryl co-enzyme A (HMG-CoA) reductase |
| 72 | Receptor-mediated endocytosis | /rɪˈsɛptər ˈmiːdiˌeɪtɪd ˌɛndoʊsaɪˈtoʊsɪs/ | Nội bào qua trung gian thụ thể |
| 73 | Acyl-CoA:cholesterol transferase (ACAT) | /ˌeɪsəl koʊˈeɪ kəˈlɛstərɒl ˈtrænsfəreɪs/ | Acyl-CoA:cholesterol transferase (ACAT) |
| 74 | Lipid droplets | /ˈlɪpɪd ˈdrɒpləts/ | Giọt lipid |
| 75 | Hormone-sensitive lipase (HSL) | /ˈhɔːrmoʊn ˈsɛnsɪtɪv ˈlaɪpeɪs/ | Lipase nhạy cảm với hormone |
| 76 | Niemann-Pick type C disease | /ˈniːmən pɪk taɪp siː dɪˈziːz/ | Bệnh Niemann-Pick type C |
| 77 | Heme group | /hiːm ɡruːp/ | Nhóm heme |
| 78 | Mitochondria | /ˌmaɪtəˈkɒndriə/ | Ty thể |
| 79 | Endoplasmic reticulum | /ˌɛndoʊˈplæzmɪk rɪˈtɪkjələm/ | Lưới nội chất |
| 80 | Xenobiotics | /ˌziːnoʊbaɪˈɒtɪks/ | Chất lạ |
| 81 | Cholesterol side-chain cleavage enzyme | /kəˈlɛstərɒl saɪd tʃeɪn ˈkliːvɪdʒ ˈɛnzaɪm/ | Enzyme cắt chuỗi bên cholesterol |
| 82 | P450c11β (CYP11B1) | P450c11β (CYP11B1) | |
| 83 | P450c11AS (CYP11B2) | P450c11AS (CYP11B2) | |
| 84 | 11β-hydroxylase | /ɪˈlɛvən ˈbeɪtə haɪˈdrɒksɪleɪs/ | 11β-hydroxylase |
| 85 | 18-hydroxylase | /ˌeɪˈtiːn haɪˈdrɒksɪleɪs/ | 18-hydroxylase |
| 86 | 18-methyl oxidase | /ˌeɪˈtiːn ˈmɛθəl ˈɒksɪdeɪs/ | 18-methyl oxidase |
| 87 | P450c17 (CYP17A1) | P450c17 (CYP17A1) | |
| 88 | 17α-hydroxylase | /ˌsɛvənˈtiːn ˈælfə haɪˈdrɒksɪleɪs/ | 17α-hydroxylase |
| 89 | 17,20 lyase | /ˌsɛvənˈtiːn ˈtwɛnti ˈlaɪeɪs/ | 17,20 lyase |
| 90 | P450c21 (CYP21A2) | P450c21 (CYP21A2) | |
| 91 | P450aro (CYP19A1) | P450aro (CYP19A1) | |
| 92 | Aromatization | /əˌroʊmətəˈzeɪʃən/ | Aromat hóa |
| 93 | Androgens | /ˈændrədʒənz/ | Androgen |
| 94 | Estrogens | /ˈɛstrədʒənz/ | Estrogen |
| 95 | Hydroxysteroid dehydrogenases (HSDs) | /haɪˌdrɒksiˈstɪərɔɪd ˌdiːhaɪˈdrɒdʒəneɪsɪz/ | Hydroxysteroid dehydrogenase (HSD) |
| 96 | Nicotinamide adenine dinucleotide (NAD+) | /ˌnɪkəˈtɪnəmaɪd ˈædəniːn daɪˈnjuːkliətaɪd/ | Nicotinamide adenine dinucleotide (NAD+) |
| 97 | Nicotinamide adenine dinucleotide phosphate (NADP+) | /ˌnɪkəˈtɪnəmaɪd ˈædəniːn daɪˈnjuːkliətaɪd ˈfɒsfeɪt/ | Nicotinamide adenine dinucleotide phosphate (NADP+) |
| 98 | 5α-reductases | /faɪv ˈælfə rɪˈdʌkteɪsɪz/ | 5α-reductase |
| 99 | Short-chain dehydrogenase/reductase (SDR) | /ʃɔːrt tʃeɪn ˌdiːhaɪˈdrɒdʒəneɪs rɪˈdʌkteɪs/ | Dehydrogenase/reductase chuỗi ngắn (SDR) |
| 100 | Aldo-keto reductase (AKR) | /ˈældoʊ ˈkiːtoʊ rɪˈdʌkteɪs/ | Aldo-keto reductase (AKR) |
| 101 | Pregnenolone | /prɛɡˈnɛnəloʊn/ | Pregnenolone |
| 102 | Isocaproic acid | /ˌaɪsoʊkəˈproʊɪk ˈæsɪd/ | Axit isocaproic |
| 103 | Ferredoxin reductase (FDXR) | /ˌfɛrɪˈdɒksɪn rɪˈdʌkteɪs/ | Ferredoxin reductase (FDXR) |
| 104 | Ferredoxin (FDX) | /ˌfɛrɪˈdɒksɪn/ | Ferredoxin (FDX) |
| 105 | Flavoprotein | /ˌfleɪvoʊˈproʊtiːn/ | Flavoprotein |
| 106 | Steroidogenic acute regulatory protein (StAR) | /ˌstɪərɔɪdoʊˈdʒɛnɪk əˈkjuːt ˈrɛɡjələˌtɔːri ˈproʊtiːn/ | Protein điều hòa cấp tính sinh steroid (StAR) |
| 107 | Congenital lipoid adrenal hyperplasia | /kənˈdʒɛnɪtəl ˈlɪpɔɪd əˈdriːnəl ˌhaɪpərˈpleɪʒə/ | Tăng sản thượng thận bẩm sinh dạng mỡ |
| 108 | Luteinizing hormone (LH) | /ˈluːtiəˌnaɪzɪŋ ˈhɔːrmoʊn/ | Hormone tạo hoàng thể (LH) |
| 109 | Outer mitochondrial membrane (OMM) | /ˈaʊtər ˌmaɪtəˈkɒndriəl ˈmɛmbreɪn/ | Màng ngoài ty thể |
| 110 | 17-hydroxypregnenolone (17-Preg) | /ˌsɛvənˈtiːn haɪˈdrɒksi prɛɡˈnɛnəloʊn/ | 17-hydroxypregnenolone (17-Preg) |
| 111 | Progesterone | /proʊˈdʒɛstəroʊn/ | Progesterone |
| 112 | 3β-hydroxysteroid dehydrogenase (3βHSD) | /θriː ˈbeɪtə haɪˌdrɒksiˈstɪərɔɪd ˌdiːhaɪˈdrɒdʒəneɪs/ | 3β-hydroxysteroid dehydrogenase (3βHSD) |
| 113 | 17α-hydroxyprogesterone (17OHP) | /ˌsɛvənˈtiːn ˈælfə haɪˌdrɒksi proʊˈdʒɛstəroʊn/ | 17α-hydroxyprogesterone (17OHP) |
| 114 | Androstenedione | /ˌændroʊstiːnˈdaɪoʊn/ | Androstenedione |
| 115 | Androstenediol | /ˌændroʊstiːnˈdaɪɒl/ | Androstenediol |
| 116 | Testosterone | /tɛˈstɒstəroʊn/ | Testosterone |
| 117 | Michaelis-Menten constant (Km) | /ˈmaɪkəliːs ˈmɛntən ˈkɒnstənt/ | Hằng số Michaelis-Menten (Km) |
| 118 | P450 oxidoreductase (POR) | /piː fɔːr ˈfɪfti ˌɒksɪdoʊrɪˈdʌkteɪs/ | P450 oxidoreductase (POR) |
| 119 | Cytochrome b5 | /ˈsaɪtəˌkroʊm biː faɪv/ | Cytochrome b5 |
| 120 | Allosteric factor | /ˌæloʊˈstɛrɪk ˈfæktər/ | Yếu tố dị lập thể |
| 121 | Serine/threonine phosphorylation | /ˈsɛriːn ˈθriːəniːn ˌfɒsfəˌraɪˈleɪʃən/ | Phosphoryl hóa serine/threonine |
| 122 | Deoxycorticosterone (DOC) | /diːˌɒksiˌkɔːrtɪkoʊˈstɪəroʊn/ | Deoxycorticosterone (DOC) |
| 123 | 11-deoxycortisol | /ɪˈlɛvən diːˈɒksiˈkɔːrtɪsɒl/ | 11-deoxycortisol |
| 124 | Major histocompatibility locus | /ˈmeɪdʒər ˌhɪstoʊkəmˌpætəˈbɪləti ˈloʊkəs/ | Locus tương hợp mô chính |
| 125 | Human leukocyte antigen (HLA) | /ˈhjuːmən ˈluːkəsaɪt ˈæntɪdʒən/ | Kháng nguyên bạch cầu người (HLA) |
| 126 | Corticosterone | /ˌkɔːrtɪkoʊˈstɪəroʊn/ | Corticosterone |
| 127 | Aldosterone synthase | /ælˈdɒstəroʊn ˈsɪnθeɪs/ | Aldosterone synthase |
| 128 | Corticosterone methyl oxidase deficiency | /ˌkɔːrtɪkoʊˈstɪəroʊn ˈmɛθəl ˈɒksɪdeɪs dɪˈfɪʃənsi/ | Thiếu hụt corticosterone methyl oxidase |
| 129 | 17-oxidoreductases | /ˌsɛvənˈtiːn ˌɒksɪdoʊrɪˈdʌkteɪsɪz/ | 17-oxidoreductase |
| 130 | 17-ketosteroid reductases | /ˌsɛvənˈtiːn ˌkiːtoʊˈstɪərɔɪd rɪˈdʌkteɪsɪz/ | 17-ketosteroid reductase |
| 131 | Estrone | /ˈɛstroʊn/ | Estrone |
| 132 | Estradiol | /ˌɛstrəˈdaɪɒl/ | Estradiol |
| 133 | Ovarian granulosa cells | /oʊˈvɛəriən ˌɡrænjəˈloʊsə sɛlz/ | Tế bào hạt của buồng trứng |
| 134 | Placental syncytiotrophoblast cells | /pləˈsɛntəl sɪnˌsɪʃioʊˈtroʊfoʊblæst sɛlz/ | Tế bào hợp bào lá nuôi của nhau thai |
| 135 | Male pseudohermaphroditism | /meɪl ˌsuːdoʊhɜːrˈmæfrədaɪˌtɪzəm/ | Lưỡng tính giả nam |
| 136 | Zellweger syndrome | /ˈzɛlwɛɡər ˈsɪndroʊm/ | Hội chứng Zellweger |
| 137 | Dihydrotestosterone (DHT) | /daɪˌhaɪdroʊtɛˈstɒstəroʊn/ | Dihydrotestosterone (DHT) |
| 138 | Steroid sulfotransferase (SULT) | /ˈstɪərɔɪd ˌsʌlfoʊˈtrænsfəreɪs/ | Steroid sulfotransferase (SULT) |
| 139 | 3’-phosphoadenine-5’-phosphosulfate (PAPS) | /θriː praɪm ˌfɒsfoʊˈædəniːn faɪv praɪm ˌfɒsfoʊˈsʌlfeɪt/ | 3’-phosphoadenine-5’-phosphosulfate (PAPS) |
| 140 | Steroid sulfatase | /ˈstɪərɔɪd ˈsʌlfəteɪs/ | Steroid sulfatase |
| 141 | X-linked ichthyosis | /ɛks lɪŋkt ˌɪkθiˈoʊsɪs/ | Bệnh vảy cá liên kết X |
| 142 | Epiphyses | /ɪˈpɪfəsiːz/ | Đầu xương |
| 143 | Osteopenia | /ˌɒstioʊˈpiːniə/ | Giảm mật độ xương |
| 144 | Epiphyseal fusion | /ˌɛpɪˈfɪziəl ˈfjuːʒən/ | Cốt hóa đầu xương |
| 145 | Aromatase inhibitors | /əˈroʊməteɪs ɪnˈhɪbɪtərz/ | Thuốc ức chế aromatase |
| 146 | 5α-reductase deficiency | /faɪv ˈælfə rɪˈdʌkteɪs dɪˈfɪʃənsi/ | Thiếu hụt 5α-reductase |
| 147 | Male pattern baldness | /meɪl ˈpætərn ˈbɔːldnəs/ | Hói đầu kiểu nam |
| 148 | Cortisone | /ˈkɔːrtɪsoʊn/ | Cortisone |
| 149 | Hexose-6-phosphate dehydrogenase (H6PDH) | /ˈhɛksoʊs sɪks ˈfɒsfeɪt ˌdiːhaɪˈdrɒdʒəneɪs/ | Hexose-6-phosphate dehydrogenase (H6PDH) |
| 150 | Prednisone | /ˈprɛdnɪsoʊn/ | Prednisone |
| 151 | Metabolic syndrome | /ˌmɛtəˈbɒlɪk ˈsɪndroʊm/ | Hội chứng chuyển hóa |
| 152 | Nonalcoholic fatty liver disease | /nɒnˌælkəˈhɒlɪk ˈfæti ˈlɪvər dɪˈziːz/ | Bệnh gan nhiễm mỡ không do rượu |
| 153 | Distal nephron | /ˈdɪstəl ˈnɛfrɒn/ | Ống lượn xa |
| 154 | Prednisolone | /prɛdˈnɪsəloʊn/ | Prednisolone |
| 155 | Backdoor pathway | /ˈbækdɔːr ˈpæθweɪ/ | Con đường cửa sau |
| 156 | Allopregnanolone | /ˌæloʊprɛɡˈnænəloʊn/ | Allopregnanolone |
| 157 | Gamma-aminobutyric acid (GABA)A receptors | /ˈɡæmə əˈmiːnoʊbjuːˈtɪrɪk ˈæsɪd eɪ rɪˈsɛptərz/ | Thụ thể gamma-aminobutyric acid (GABA)A |
| 158 | Feto-placental unit | /ˈfiːtoʊ pləˈsɛntəl ˈjuːnɪt/ | Đơn vị thai-nhau thai |
| 159 | Parturition | /ˌpɑːrtjʊˈrɪʃən/ | Sự sinh đẻ |
| 160 | Glucocorticoid resistance | /ˌɡluːkoʊˈkɔːrtɪkɔɪd rɪˈzɪstəns/ | Kháng glucocorticoid |
| 161 | Hypothalamic-pituitary-adrenal (HPA) axis | /ˌhaɪpoʊθəˈlæmɪk pɪˈtjuːɪtəri əˈdriːnəl ˈæksɪs/ | Trục hạ đồi-tuyến yên-thượng thận (HPA) |
| 162 | Hypothalamus | /ˌhaɪpoʊˈθæləməs/ | Vùng dưới đồi |
| 163 | Corticotropin-releasing factor (CRF) | /ˌkɔːrtɪkoʊˈtroʊpɪn rɪˈliːsɪŋ ˈfæktər/ | Yếu tố giải phóng corticotropin (CRF) |
| 164 | Arginine vasopressin (AVP) | /ˈɑːrdʒɪniːn ˌveɪsoʊˈprɛsɪn/ | Arginine vasopressin (AVP) |
| 165 | Antidiuretic hormone (ADH) | /ˌæntiˌdaɪjʊˈrɛtɪk ˈhɔːrmoʊn/ | Hormone chống bài niệu (ADH) |
| 166 | Paraventricular nucleus | /ˌpærəvɛnˈtrɪkjələr ˈnjuːkliəs/ | Nhân cạnh não thất |
| 167 | Neurophysin | /ˌnʊəroʊˈfaɪsɪn/ | Neurophysin |
| 168 | Median eminence | /ˈmiːdiən ˈɛmɪnəns/ | Lồi giữa |
| 169 | Pituitary portal circulation | /pɪˈtjuːɪtəri ˈpɔːrtəl ˌsɜːrkjəˈleɪʃən/ | Tuần hoàn cửa tuyến yên |
| 170 | Posterior pituitary | /pɒˈstɪəriər pɪˈtjuːɪtəri/ | Thùy sau tuyến yên |
| 171 | Pituitary corticotropes | /pɪˈtjuːɪtəri ˈkɔːrtɪkoʊtroʊps/ | Tế bào hướng vỏ thượng thận của tuyến yên |
| 172 | Adenylyl cyclase | /əˌdɛnəˈlɪl ˈsaɪkleɪs/ | Adenylyl cyclase |
| 173 | Cyclic adenosine monophosphate (cAMP) | /ˈsaɪklɪk əˈdɛnəsiːn ˌmɒnoʊˈfɒsfeɪt/ | Cyclic adenosine monophosphate (cAMP) |
| 174 | Protein kinase A (PKA) | /ˈproʊtiːn ˈkaɪneɪs eɪ/ | Protein kinase A (PKA) |
| 175 | Proopiomelanocortin (POMC) | /proʊˌoʊpioʊməˌlænoʊˈkɔːrtɪn/ | Proopiomelanocortin (POMC) |
| 176 | Phospholipase C | /ˌfɒsfoʊˈlaɪpeɪs siː/ | Phospholipase C |
| 177 | Protein kinase C (PKC) | /ˈproʊtiːn ˈkaɪneɪs siː/ | Protein kinase C (PKC) |
| 178 | Pituitary | /pɪˈtjuːɪtəri/ | Tuyến yên |
| 179 | N-terminal glycopeptide | /ɛn ˈtɜːrmɪnəl ˌɡlaɪkoʊˈpɛptaɪd/ | Glycopeptide đầu N |
| 180 | Adrenal mitogen | /əˈdriːnəl ˈmaɪtədʒən/ | Chất gây phân bào tuyến thượng thận |
| 181 | α-MSH (melanocyte stimulating hormone) | /ˈælfə ɛm ɛs eɪtʃ/ | α-MSH (hormone kích thích tế bào hắc tố) |
| 182 | β-MSH | /ˈbeɪtə ɛm ɛs eɪtʃ/ | β-MSH |
| 183 | β-endorphin | /ˈbeɪtə ɛnˈdɔːrfɪn/ | β-endorphin |
| 184 | Ectopic ACTH | /ɛkˈtɒpɪk eɪ siː tiː eɪtʃ/ | ACTH lạc chỗ |
| 185 | Melanocortin 2 receptor (MC2R) | /məˌlænoʊˈkɔːrtɪn tuː rɪˈsɛptər/ | Thụ thể melanocortin 2 (MC2R) |
| 186 | Cholesterol esterase | /kəˈlɛstərɒl ˈɛstəreɪs/ | Cholesterol esterase |
| 187 | Cholesterol ester synthetase | /kəˈlɛstərɒl ˈɛstər ˈsɪnθəteɪs/ | Cholesterol ester synthetase |
| 188 | Diurnal rhythms | /daɪˈɜːrnəl ˈrɪðəmz/ | Nhịp ngày đêm |
| 189 | Circadian clock | /sərˈkeɪdiən klɒk/ | Đồng hồ sinh học |
| 190 | Jet lag | /dʒɛt læɡ/ | Lệch múi giờ |
| 191 | Cytokines | /ˈsaɪtəkaɪnz/ | Cytokine |
| 192 | Interleukin (IL)-1 | /ˌɪntərˈluːkɪn wʌn/ | Interleukin (IL)-1 |
| 193 | Interleukin (IL)-6 | /ˌɪntərˈluːkɪn sɪks/ | Interleukin (IL)-6 |
| 194 | Tumor necrosis factor (TNF) | /ˈtuːmər nɪˈkroʊsɪs ˈfæktər/ | Yếu tố hoại tử khối u (TNF) |
| 195 | Glucocorticoid feedback | /ˌɡluːkoʊˈkɔːrtɪkɔɪd ˈfiːdbæk/ | Phản hồi ngược glucocorticoid |
| 196 | Renin-angiotensin system | /ˈriːnɪn ˌændʒioʊˈtɛnsɪn ˈsɪstəm/ | Hệ renin-angiotensin |
| 197 | Renin | /ˈriːnɪn/ | Renin |
| 198 | Juxtaglomerular cells | /ˌdʒʌkstəɡləˈmɛrʊlər sɛlz/ | Tế bào cạnh cầu thận |
| 199 | Angiotensinogen | /ˌændʒioʊtɛnˈsɪnədʒən/ | Angiotensinogen |
| 200 | Angiotensin I | /ˌændʒioʊˈtɛnsɪn wʌn/ | Angiotensin I |
| 201 | Angiotensin II | /ˌændʒioʊˈtɛnsɪn tuː/ | Angiotensin II |
| 202 | Angiotensin-converting enzyme | /ˌændʒioʊˈtɛnsɪn kənˈvɜːrtɪŋ ˈɛnzaɪm/ | Enzyme chuyển angiotensin |
| 203 | Captopril | /ˈkæptoʊprɪl/ | Captopril |
| 204 | Candesartan | /ˌkændɪˈsɑːrtən/ | Candesartan |
| 205 | Vasoconstriction | /ˌveɪzoʊkənˈstrɪkʃən/ | Co mạch |
| 206 | Hyperkalemia | /ˌhaɪpərkəˈliːmiə/ | Tăng kali máu |
| 207 | Phosphatidylinositol | /ˌfɒsfəˌtaɪdəlɪˈnoʊsɪtɒl/ | Phosphatidylinositol |
| 208 | Calcium-calmodulin system | /ˈkælsiəm kælˈmɒdʒəlɪn ˈsɪstəm/ | Hệ calcium-calmodulin |
| 209 | Atrial natriuretic factor | /ˈeɪtriəl ˌnætriˌjʊəˈrɛtɪk ˈfæktər/ | Yếu tố bài niệu nhĩ |
| 210 | Adrenarche | /ˌædrɪˈnɑːrki/ | Thượng thận hóa |
| 211 | Adrenopause | /əˈdriːnoʊpɔːz/ | Mãn thượng thận |
| 212 | Insulin resistance | /ˈɪnsəlɪn rɪˈzɪstəns/ | Kháng insulin |
| 213 | Polycystic ovary syndrome | /ˌpɒliˈsɪstɪk ˈoʊvəri ˈsɪndroʊm/ | Hội chứng buồng trứng đa nang |
| 214 | Hyperandrogenism | /ˌhaɪpərˈændrədʒəˌnɪzəm/ | Cường androgen |
| 215 | Hypertriglyceridemia | /ˌhaɪpərˌtraɪɡlɪsəraɪˈdiːmiə/ | Tăng triglycerid máu |
| 216 | 11-oxygenated (11-Oxo) steroids | /ɪˈlɛvən ˌɒksɪdʒəˈneɪtɪd ˈstɪərɔɪdz/ | Steroid 11-oxygen hóa (11-Oxo) |
| 217 | 11-keto-testosterone (11-KT) | /ɪˈlɛvən ˈkiːtoʊ tɛˈstɒstəroʊn/ | 11-keto-testosterone (11-KT) |
| 218 | 11-keto dihydrotestosterone (11-KDHT) | /ɪˈlɛvən ˈkiːtoʊ daɪˌhaɪdroʊtɛˈstɒstəroʊn/ | 11-keto dihydrotestosterone (11-KDHT) |
| 219 | Testicular adrenal rest tumors (TARTs) | /tɛˈstɪkjələr əˈdriːnəl rɛst ˈtuːmərz/ | U tế bào lạc chỗ vỏ thượng thận tại tinh hoàn (TART) |
| 220 | Pregnenolone | /prɛɡˈnɛnəloʊn/ | Pregnenolone |
| 221 | C21 steroids | /siː ˌtwɛntiˈwʌn ˈstɪərɔɪdz/ | Steroid C21 |
| 222 | C19 steroids | /siː ˌnaɪnˈtiːn ˈstɪərɔɪdz/ | Steroid C19 |
| 223 | C18 estrogens | /siː ˌeɪˈtiːn ˈɛstrədʒənz/ | Estrogen C18 |
| 224 | Δ4 steroids | /ˈdɛltə fɔːr ˈstɪərɔɪdz/ | Steroid Δ4 |
| 225 | Δ5 steroids | /ˈdɛltə faɪv ˈstɪərɔɪdz/ | Steroid Δ5 |
| 226 | Dexamethasone | /ˌdɛksəˈmɛθəsoʊn/ | Dexamethasone |
| 227 | Corticosteroid-binding globulin (CBG) | /ˌkɔːrtɪkoʊˈstɪərɔɪd ˈbaɪndɪŋ ˈɡlɒbjəlɪn/ | Globulin gắn corticosteroid (CBG) |
| 228 | Albumin | /ælˈbjuːmɪn/ | Albumin |
| 229 | α1 acid glycoprotein | /ˈælfə wʌn ˈæsɪd ˌɡlaɪkoʊˈproʊtiːn/ | α1 acid glycoprotein |
| 230 | Sex steroid-binding globulin | /sɛks ˈstɪərɔɪd ˈbaɪndɪŋ ˈɡlɒbjəlɪn/ | Globulin gắn steroid sinh dục |
| 231 | Autocrine factors | /ˈɔːtoʊkrɪn ˈfæktərz/ | Các yếu tố tự tiết |
| 232 | Paracrine factors | /ˈpærəkrɪn ˈfæktərz/ | Các yếu tố cận tiết |
| 233 | Steroid catabolism | /ˈstɪərɔɪd kəˈtæbəlɪzəm/ | Chuyển hóa steroid |
| 234 | Labeled tracers | /ˈleɪbəld ˈtreɪsərz/ | Chất đánh dấu |
| 235 | Mass spectroscopy | /mæs spɛkˈtrɒskəpi/ | Quang phổ khối |
| 236 | Liquid chromatography-mass spectrometry (LC-MS/MS) | /ˈlɪkwɪd ˌkroʊməˈtɒɡrəfi mæs spɛkˈtrɒmɪtri/ | Sắc ký lỏng-khối phổ (LC-MS/MS) |
| 237 | Glucuronic acid | /ˌɡluːkjəˈrɒnɪk ˈæsɪd/ | Axit glucuronic |
| 238 | Tetrahydrocortisols | /ˌtɛtrəˌhaɪdroʊˈkɔːrtɪsɒlz/ | Tetrahydrocortisol |
| 239 | Tetrahydrocortisones | /ˌtɛtrəˌhaɪdroʊˈkɔːrtɪsoʊnz/ | Tetrahydrocortisone |
| 240 | Cortols | /ˈkɔːrtɒlz/ | Cortol |
| 241 | Cortolones | /ˈkɔːrtəloʊnz/ | Cortolone |
| 242 | 6β-hydroxycortisol | /sɪks ˈbeɪtə haɪˌdrɒksiˈkɔːrtɪsɒl/ | 6β-hydroxycortisol |
| 243 | Uridine diphosphoglucuronosyl transferases | /ˈjʊərɪdiːn daɪˌfɒsfoʊˌɡluːkjəˈrɒnəsɪl ˈtrænsfəreɪsɪz/ | Uridine diphosphoglucuronosyl transferase |
| 244 | 3α, 5β-tetrahydroaldosterone | /θriː ˈælfə faɪv ˈbeɪtə ˌtɛtrəˌhaɪdroʊælˈdɒstəroʊn/ | 3α, 5β-tetrahydroaldosterone |
| 245 | Androsterone | /ænˈdrɒstəroʊn/ | Androsterone |
| 246 | Etiocholanolone | /ˌiːtioʊkoʊˈlænəloʊn/ | Etiocholanolone |
| 247 | Thyroid hormone | /ˈθaɪrɔɪd ˈhɔːrmoʊn/ | Hormone tuyến giáp |
| 248 | Hypothyroidism | /ˌhaɪpoʊˈθaɪrɔɪdɪzəm/ | Suy giáp |
| 249 | Hypopituitarism | /ˌhaɪpoʊpɪˈtjuːɪtəˌrɪzəm/ | Suy tuyến yên |
| 250 | Adrenal crisis | /əˈdriːnəl ˈkraɪsɪs/ | Cơn suy thượng thận cấp |
| 251 | Spironolactone | /ˌspaɪrənoʊˈlæktoʊn/ | Spironolactone |
| 252 | Troglitazone | /troʊˈɡlɪtəzoʊn/ | Troglitazone |
| 253 | Rosiglitazone | /ˌroʊsɪˈɡlɪtəzoʊn/ | Rosiglitazone |
| 254 | Pioglitazone | /ˌpaɪoʊˈɡlɪtəzoʊn/ | Pioglitazone |
| 255 | Thiazolidinedione | /ˌθaɪəˌzoʊlɪdiːnˈdaɪoʊn/ | Thiazolidinedione |
| 256 | Hypericum perforatum (St. John’s Wort) | /haɪˈpɛrɪkəm pərˌfɔːˈreɪtəm/ | Cây Ban Âu (St. John’s Wort) |
| 257 | Oral contraception | /ˈɔːrəl ˌkɒntrəˈsɛpʃən/ | Thuốc tránh thai đường uống |
| 258 | Ketoconazole | /ˌkiːtoʊˈkɒnəzoʊl/ | Ketoconazole |
| 259 | Prostate cancer | /ˈprɒsteɪt ˈkænsər/ | Ung thư tuyến tiền liệt |
| 260 | Cushing syndrome | /ˈkʊʃɪŋ ˈsɪndroʊm/ | Hội chứng Cushing |
| 261 | Obesity | /oʊˈbiːsəti/ | Béo phì |
| 262 | Renal disease | /ˈriːnəl dɪˈziːz/ | Bệnh thận |
| 263 | Heart failure | /hɑːrt ˈfeɪljər/ | Suy tim |
| 264 | Hypotension | /ˌhaɪpoʊˈtɛnʃən/ | Hạ huyết áp |
| 265 | Hyperpigmentation | /ˌhaɪpərˌpɪɡmɛnˈteɪʃən/ | Tăng sắc tố da |
| 266 | Shock | /ʃɒk/ | Sốc |
| 267 | Hypoglycemia | /ˌhaɪpoʊɡlaɪˈsiːmiə/ | Hạ đường huyết |
| 268 | Hyperthermia | /ˌhaɪpərˈθɜːrmiə/ | Tăng thân nhiệt |
| 269 | Hirsutism | /ˈhɜːrsʊtɪzəm/ | Rậm lông |
| 270 | Bone age | /boʊn eɪdʒ/ | Tuổi xương |
| 271 | Muscle wasting | /ˈmʌsəl ˈweɪstɪŋ/ | Teo cơ |
| 272 | Buffalo hump | /ˈbʌfəloʊ hʌmp/ | Bướu trâu |
| 273 | Precocious puberty | /prɪˈkoʊʃəs ˈpjuːbərti/ | Dậy thì sớm |
| 274 | Testicular tumor | /tɛˈstɪkjələr ˈtuːmər/ | U tinh hoàn |
| 275 | Computed tomography (CT) | /kəmˈpjuːtɪd təˈmɒɡrəfi/ | Chụp cắt lớp vi tính (CT) |
| 276 | Magnetic resonance imaging (MRI) | /mæɡˈnɛtɪk ˈrɛzənəns ˈɪmədʒɪŋ/ | Chụp cộng hưởng từ (MRI) |
| 277 | Gadolinium enhancement | /ˌɡædəˈlɪniəm ɪnˈhænsmənt/ | Tăng cường gadolinium |
| 278 | Adrenal hypoplasia congenita | /əˈdriːnəl ˌhaɪpoʊˈpleɪʒə kənˈdʒɛnɪtə/ | Thiểu sản thượng thận bẩm sinh |
| 279 | ACTH unresponsiveness syndromes | /eɪ siː tiː eɪtʃ ˌʌnrɪˈspɒnsɪvnəs ˈsɪndroʊmz/ | Hội chứng không đáp ứng với ACTH |
| 280 | Adrenal adenomas | /əˈdriːnəl ˌædɪˈnoʊməz/ | U tuyến thượng thận |
| 281 | Radiopharmaceuticals | /ˌreɪdioʊˌfɑːrməˈsuːtɪkəlz/ | Dược chất phóng xạ |
| 282 | Scintigraphy | /sɪnˈtɪɡrəfi/ | Xạ hình |
| 283 | Positron emission tomography (PET) | /ˈpɒzɪtrɒn ɪˈmɪʃən təˈmɒɡrəfi/ | Chụp cắt lớp phát xạ positron (PET) |
| 284 | Single-photon emission computed tomography (SPECT) | /ˈsɪŋɡəl ˈfoʊtɒn ɪˈmɪʃən kəmˈpjuːtɪd təˈmɒɡrəfi/ | Chụp cắt lớp vi tính phát xạ đơn photon (SPECT) |
| 285 | Petrosal sinus sampling | /pɪˈtroʊsəl ˈsaɪnəs ˈsæmplɪŋ/ | Lấy mẫu xoang đá |
| 286 | Immunoassays (IAs) | /ˌɪmjənoʊˈæseɪz/ | Xét nghiệm miễn dịch (IA) |
| 287 | Mass spectrometry (MS) | /mæs spɛkˈtrɒmɪtri/ | Khối phổ (MS) |
| 288 | Radioimmunoassay (RIA) | /ˌreɪdioʊˌɪmjənoʊˈæseɪ/ | Xét nghiệm miễn dịch phóng xạ (RIA) |
| 289 | Immunoradiometric assay (IRMA) | /ˌɪmjənoʊˌreɪdiəˈmɛtrɪk ˈæseɪ/ | Xét nghiệm miễn dịch phóng xạ đo (IRMA) |
| 290 | Electrochemiluminescence assay (ECLIA) | /ɪˌlɛktroʊˌkiːmɪluːmɪˈnɛsəns ˈæseɪ/ | Xét nghiệm điện hóa phát quang (ECLIA) |
| 291 | High-performance liquid chromatography (HPLC) | /haɪ pərˈfɔːrməns ˈlɪkwɪd ˌkroʊməˈtɒɡrəfi/ | Sắc ký lỏng hiệu năng cao (HPLC) |
| 292 | Gas chromatography (GC) | /ɡæs ˌkroʊməˈtɒɡrəfi/ | Sắc ký khí (GC) |
| 293 | Urinary free cortisol (UFC) | /ˈjʊərɪˌnɛri friː ˈkɔːrtɪsɒl/ | Cortisol tự do trong nước tiểu (UFC) |
| 294 | Creatinine | /kriˈætɪniːn/ | Creatinin |
| 295 | Plasma renin activity (PRA) | /ˈplæzmə ˈriːnɪn ækˈtɪvəti/ | Hoạt độ renin huyết tương (PRA) |
| 296 | Plasma renin content (PRC) | /ˈplæzmə ˈriːnɪn ˈkɒntɛnt/ | Hàm lượng renin huyết tương (PRC) |
| 297 | Simple virilizing (SV) CAH | /ˈsɪmpəl ˈvɪrəlaɪzɪŋ siː eɪ eɪtʃ/ | CAH thể nam hóa đơn thuần (SV) |
| 298 | Ethylenediamine tetraacetic acid (EDTA) | /ˈɛθəliːnˌdaɪəˈmiːn ˌtɛtrəəˈsiːtɪk ˈæsɪd/ | Axit ethylenediamine tetraacetic (EDTA) |
| 299 | Secretory rates | /ˈsiːkrəˌtɔːri reɪts/ | Tốc độ bài tiết |
| 300 | Dexamethasone suppression test | /ˌdɛksəˈmɛθəsoʊn səˈprɛʃən tɛst/ | Nghiệm pháp ức chế bằng dexamethasone |
| 301 | Cosyntropin | /ˌkoʊsɪnˈtroʊpɪn/ | Cosyntropin |
| 302 | Heterozygotes | /ˌhɛtəroʊˈzaɪɡoʊts/ | Dị hợp tử |
| 303 | Nonclassic CAH | /nɒnˈklæsɪk siː eɪ eɪtʃ/ | CAH không cổ điển |
| 304 | Cryptic CAH | /ˈkrɪptɪk siː eɪ eɪtʃ/ | CAH thể ẩn |
| 305 | Insulin-induced hypoglycemia | /ˈɪnsəlɪn ɪnˈdjuːst ˌhaɪpoʊɡlaɪˈsiːmiə/ | Hạ đường huyết do insulin gây ra |
| 306 | Counterregulatory hormones | /ˌkaʊntərˈrɛɡjələˌtɔːri ˈhɔːrmoʊnz/ | Các hormone điều hòa ngược |
| 307 | Glucagon | /ˈɡluːkəɡɒn/ | Glucagon |
| 308 | Metyrapone test | /məˈtɪərəpoʊn tɛst/ | Nghiệm pháp metyrapone |
| 309 | Smith-Lemli-Opitz syndrome (SLOS) | /smɪθ ˈlɛmli ˈɒpɪts ˈsɪndroʊm/ | Hội chứng Smith-Lemli-Opitz (SLOS) |
| 310 | Lathosterolosis | /læθəˈstɛrəˌloʊsɪs/ | Bệnh lathosterolosis |
| 311 | Desmosterolosis | /ˌdɛsmoʊstəˈroʊləsɪs/ | Bệnh desmosterolosis |
| 312 | CHILD syndrome | /tʃaɪld ˈsɪndroʊm/ | Hội chứng CHILD |
| 313 | Conradi-Hunermann syndrome | /kənˈrɑːdi ˈhʊnərmən ˈsɪndroʊm/ | Hội chứng Conradi-Hunermann |
| 314 | Disorder of sex development (DSD) | /dɪsˈɔːrdər ʌv sɛks dɪˈvɛləpmənt/ | Rối loạn phát triển giới tính (DSD) |
| 315 | Wolman disease | /ˈwoʊlmən dɪˈziːz/ | Bệnh Wolman |
| 316 | Cholesteryl ester storage disease (CESD) | /kəˌlɛstəˈrɪl ˈɛstər ˈstɔːrɪdʒ dɪˈziːz/ | Bệnh dự trữ este cholesteryl (CESD) |
| 317 | Lysosomal acid lipase | /ˌlaɪsəˈsoʊməl ˈæsɪd ˈlaɪpeɪs/ | Lipase axit lysosome |
| 318 | Xanthomatosis | /ˌzænθoʊməˈtoʊsɪs/ | Bệnh u vàng |
| 319 | Steatorrhea | /ˌstiːətəˈriːə/ | Tiêu chảy phân mỡ |
| 320 | Hepatosplenomegaly | /ˌhɛpətoʊˌspliːnoʊˈmɛɡəli/ | Gan lách to |
| 321 | Jaundice | /ˈdʒɔːndɪs/ | Vàng da |
| 322 | Anemia | /əˈniːmiə/ | Thiếu máu |
| 323 | Adrenal calcification | /əˈdriːnəl ˌkælsɪfɪˈkeɪʃən/ | Vôi hóa tuyến thượng thận |
| 324 | Foam cells | /foʊm sɛlz/ | Tế bào bọt |
| 325 | Fibroblasts | /ˈfaɪbroʊblæsts/ | Nguyên bào sợi |
| 326 | Leukocytes | /ˈluːkəsaɪts/ | Bạch cầu |
| 327 | Amniocytes | /ˈæmnioʊsaɪts/ | Tế bào ối |
| 328 | Bone marrow transplantation | /boʊn ˈmæroʊ ˌtrænsplænˈteɪʃən/ | Ghép tủy xương |
| 329 | Enzyme replacement therapy | /ˈɛnzaɪm rɪˈpleɪsmənt ˈθɛrəpi/ | Liệu pháp thay thế enzyme |
| 330 | Cirrhosis | /sɪˈroʊsɪs/ | Xơ gan |
| 331 | Glycosphingolipids | /ˌɡlaɪkoʊˌsfɪŋɡoʊˈlɪpɪdz/ | Glycosphingolipid |
| 332 | Neurodegeneration | /ˌnʊəroʊdɪˌdʒɛnəˈreɪʃən/ | Thoái hóa thần kinh |
| 333 | Glial infiltration | /ˈɡliːəl ˌɪnfɪlˈtreɪʃən/ | Thâm nhiễm tế bào thần kinh đệm |
| 334 | Supranuclear vertical gaze paresis | /ˌsuːprəˈnjuːkliər ˈvɜːrtɪkəl ɡeɪz pəˈriːsɪs/ | Liệt nhìn dọc trên nhân |
| 335 | Ataxia | /əˈtæksiə/ | Thất điều |
| 336 | Dystonia | /dɪsˈtoʊniə/ | Loạn trương lực cơ |
| 337 | Dementia | /dɪˈmɛnʃə/ | Sa sút trí tuệ |
| 338 | Seizures | /ˈsiːʒərz/ | Co giật |
| 339 | Extrapyramidal deficits | /ˌɛkstrəpɪˈræmɪdəl ˈdɛfɪsɪts/ | Khiếm khuyết ngoại tháp |
| 340 | Leydig cells | /ˈlaɪdɪɡ sɛlz/ | Tế bào Leydig |
| 341 | Wolffian duct | /ˈwʊlfiən dʌkt/ | Ống Wolff |
| 342 | Sertoli cells | /sərˈtoʊli sɛlz/ | Tế bào Sertoli |
| 343 | Müllerian inhibitory hormone | /mjuːˈlɪəriən ɪnˈhɪbɪtəri ˈhɔːrmoʊn/ | Hormone ức chế Müller |
| 344 | Fallopian tubes | /fəˈloʊpiən tjuːbz/ | Vòi trứng |
| 345 | Anovulatory cycles | /ˌænˌɒvjəˈleɪtəri ˈsaɪkəlz/ | Chu kỳ không rụng trứng |
| 346 | Ovarian cysts | /oʊˈvɛəriən sɪsts/ | U nang buồng trứng |
| 347 | Sudden infant death syndrome (SIDS) | /ˈsʌdən ˈɪnfənt dɛθ ˈsɪndroʊm/ | Hội chứng đột tử ở trẻ sơ sinh (SIDS) |
| 348 | Nonclassic lipoid CAH | /nɒnˈklæsɪk ˈlɪpɔɪd siː eɪ eɪtʃ/ | CAH dạng mỡ không cổ điển |
| 349 | Hypergonadotropic hypogonadism | /ˌhaɪpərɡəˌnædoʊˈtroʊpɪk ˌhaɪpoʊˈɡoʊnæˌdɪzəm/ | Suy sinh dục do tăng gonadotropin |
| 350 | Orchiectomy | /ˌɔːrkiˈɛktəmi/ | Cắt bỏ tinh hoàn |
| 351 | Steroidogenic factor 1 (SF1) | /ˌstɪərɔɪdoʊˈdʒɛnɪk ˈfæktər wʌn/ | Yếu tố sinh steroid 1 (SF1) |
| 352 | Clitoromegaly | /ˌklaɪtərəˈmɛɡəli/ | Phì đại âm vật |
| 353 | 17-hydroxycorticosteroids (17OHCS) | /ˌsɛvənˈtiːn haɪˌdrɒksiˌkɔːrtɪkoʊˈstɪərɔɪdz/ | 17-hydroxycorticosteroid (17OHCS) |
| 354 | Methemoglobinemia | /ˌmɛθˌhiːməˌɡloʊbɪˈniːmiə/ | Methemoglobin máu |
| 355 | Antley-Bixler syndrome (ABS) | /ˈæntli ˈbɪkslər ˈsɪndroʊm/ | Hội chứng Antley-Bixler (ABS) |
| 356 | Craniosynostosis | /ˌkreɪnioʊˌsɪnəˈstoʊsɪs/ | Hẹp sọ |
| 357 | Brachycephaly | /ˌbrækiˈsɛfəli/ | Tật đầu ngắn |
| 358 | Radioulnar synostosis | /ˌreɪdioʊˈʌlnər ˌsɪnəˈstoʊsɪs/ | Dính xương quay-trụ |
| 359 | Radiohumeral synostosis | /ˌreɪdioʊˈhjuːmərəl ˌsɪnəˈstoʊsɪs/ | Dính xương quay-cánh tay |
| 360 | Bowed femora | /baʊd ˈfiːmərə/ | Xương đùi cong |
| 361 | Arachnodactyly | /əˌræknəˈdæktɪli/ | Tật ngón nhện |
| 362 | Midface hypoplasia | /ˈmɪdfeɪs ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản tầng giữa mặt |
| 363 | Proptosis | /prɒpˈtoʊsɪs/ | Lồi mắt |
| 364 | Choanal stenosis | /koʊˈeɪnəl stɪˈnoʊsɪs/ | Hẹp lỗ mũi sau |
| 365 | FGF receptor 2 | /ɛf dʒiː ɛf rɪˈsɛptər tuː/ | Thụ thể FGF 2 |
| 366 | Retinoic acid | /ˌrɛtɪˈnoʊɪk ˈæsɪd/ | Axit retinoic |
| 367 | 21-hydroxylase deficiency (21OHD) | /ˌtwɛntiˈwʌn haɪˈdrɒksɪleɪs dɪˈfɪʃənsi/ | Thiếu hụt 21-hydroxylase (21OHD) |
| 368 | Hyponatremia | /ˌhaɪpoʊnəˈtriːmiə/ | Hạ natri máu |
| 369 | Acidosis | /ˌæsɪˈdoʊsɪs/ | Nhiễm toan |
| 370 | Cardiovascular collapse | /ˌkɑːrdioʊˈvæskjələr kəˈlæps/ | Trụy tim mạch |
| 371 | Hypochloremia | /ˌhaɪpoʊklɔːˈriːmiə/ | Hạ clo máu |
| 372 | Meningococcemia | /məˌnɪŋɡoʊkɒkˈsiːmiə/ | Nhiễm não mô cầu huyết |
| 373 | Waterhouse-Friederichsen syndrome | /ˈwɔːtərhaʊs ˈfriːdərɪksən ˈsɪndroʊm/ | Hội chứng Waterhouse-Friederichsen |
| 374 | Petechial rash | /pɪˈtiːkiəl ræʃ/ | Ban xuất huyết dạng chấm |
| 375 | Ecchymoses | /ˌɛkɪˈmoʊsiːz/ | Mảng bầm |
| 376 | Septicemia | /ˌsɛptɪˈsiːmiə/ | Nhiễm trùng huyết |
| 377 | Antiphospholipid syndrome | /ˌæntɪˌfɒsfəˈlɪpɪd ˈsɪndroʊm/ | Hội chứng kháng phospholipid |
| 378 | Autoimmune adrenalitis | /ˌɔːtoʊɪˈmjuːn əˌdriːnəˈlaɪtɪs/ | Viêm thượng thận tự miễn |
| 379 | Electrocardiogram | /ɪˌlɛktroʊˈkɑːrdioʊɡræm/ | Điện tâm đồ |
| 380 | Azotemia | /ˌæzoʊˈtiːmiə/ | Nitơ huyết |
| 381 | Eosinophilia | /iːəˌsɪnəˈfɪliə/ | Tăng bạch cầu ái toan |
| 382 | Lymphocytosis | /ˌlɪmfoʊsaɪˈtoʊsɪs/ | Tăng bạch cầu lympho |
| 383 | Autoimmune polyendocrine syndromes (APSs) | /ˌɔːtoʊɪˈmjuːn ˌpɒliˈɛndoʊkrɪn ˈsɪndroʊmz/ | Hội chứng đa tuyến nội tiết tự miễn (APS) |
| 384 | Autoimmune polyendocrinopathy-candidiasis-ectodermal dysplasia (APECED) | /ˌɔːtoʊɪˈmjuːn ˌpɒliˌɛndoʊkrɪˈnɒpəθi ˌkændɪˈdaɪəsɪs ˌɛktoʊˈdɜːrməl dɪsˈpleɪʒə/ | Bệnh đa tuyến nội tiết-nhiễm nấm candida-loạn sản ngoại bì tự miễn (APECED) |
| 385 | Mucocutaneous candidiasis | /ˌmjuːkoʊkjuːˈteɪniəs ˌkændɪˈdaɪəsɪs/ | Bệnh nấm candida da niêm mạc |
| 386 | Hypoparathyroidism | /ˌhaɪpoʊˌpærəˈθaɪrɔɪdɪzəm/ | Suy cận giáp |
| 387 | Hypocalcemia | /ˌhaɪpoʊkælˈsiːmiə/ | Hạ canxi máu |
| 388 | Alopecia | /ˌæləˈpiːʃə/ | Rụng tóc |
| 389 | Vitiligo | /ˌvɪtɪˈlaɪɡoʊ/ | Bệnh bạch biến |
| 390 | Gastritis | /ɡæsˈtraɪtɪs/ | Viêm dạ dày |
| 391 | Pernicious anemia | /pərˈnɪʃəs əˈniːmiə/ | Thiếu máu ác tính |
| 392 | Hepatitis | /ˌhɛpəˈtaɪtɪs/ | Viêm gan |
| 393 | Thyroiditis | /ˌθaɪrɔɪˈdaɪtɪs/ | Viêm tuyến giáp |
| 394 | Interstitial nephritis | /ˌɪntərˈstɪʃəl nɪˈfraɪtɪs/ | Viêm thận kẽ |
| 395 | Myositis | /ˌmaɪəˈsaɪtɪs/ | Viêm cơ |
| 396 | Dental enamel hypoplasia | /ˈdɛntəl ɪˈnæməl ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản men răng |
| 397 | Asplenia | /eɪˈspliːniə/ | Vô lách |
| 398 | Keratoconjunctivitis | /ˌkɛrətoʊkənˌdʒʌŋktɪˈvaɪtɪs/ | Viêm giác-kết mạc |
| 399 | Type 1 diabetes mellitus | /taɪp wʌn ˌdaɪəˈbiːtiːz ˈmɛlɪtəs/ | Đái tháo đường type 1 |
| 400 | Squamous cell carcinoma | /ˈskweɪməs sɛl ˌkɑːrsɪˈnoʊmə/ | Ung thư biểu mô tế bào vảy |
| 401 | Autoimmune regulator (AIRE) | /ˌɔːtoʊɪˈmjuːn ˈrɛɡjəˌleɪtər/ | Chất điều hòa tự miễn (AIRE) |
| 402 | Schmidt’s syndrome | /ʃmɪts ˈsɪndroʊm/ | Hội chứng Schmidt |
| 403 | Immunodysregulation, polyendocrinopathy, and enteropathy (IPEX) | /ˌɪmjənoʊˌdɪsrɛɡjəˈleɪʃən ˌpɒliˌɛndoʊkrɪˈnɒpəθi ənd ˌɛntəˈrɒpəθi/ | Rối loạn điều hòa miễn dịch, bệnh đa tuyến nội tiết và bệnh ruột (IPEX) |
| 404 | Dermatitis | /ˌdɜːrməˈtaɪtɪs/ | Viêm da |
| 405 | Immunoglobulin (Ig)E | /ˌɪmjənoʊˈɡlɒbjəlɪn iː/ | Immunoglobulin (Ig)E |
| 406 | Exocrine pancreatitis | /ˈɛksəkrɪn ˌpæŋkriəˈtaɪtɪs/ | Viêm tụy ngoại tiết |
| 407 | Hemolytic anemia | /ˌhiːməˈlɪtɪk əˈniːmiə/ | Thiếu máu tan máu |
| 408 | Thrombocytopenia | /ˌθrɒmboʊˌsaɪtoʊˈpiːniə/ | Giảm tiểu cầu |
| 409 | Immune checkpoint inhibitors (ICIs) | /ɪˈmjuːn ˈtʃɛkpɔɪnt ɪnˈhɪbɪtərz/ | Thuốc ức chế điểm kiểm soát miễn dịch (ICI) |
| 410 | Hypophysitis | /ˌhaɪpoʊfɪˈsaɪtɪs/ | Viêm tuyến yên |
| 411 | Adrenal hypoplasia congenita (AHC) | /əˈdriːnəl ˌhaɪpoʊˈpleɪʒə kənˈdʒɛnɪtə/ | Thiểu sản thượng thận bẩm sinh (AHC) |
| 412 | Adrenal dysgenesis | /əˈdriːnəl dɪsˈdʒɛnəsɪs/ | Loạn sản tuyến thượng thận |
| 413 | Cytomegalic | /ˌsaɪtoʊmɛˈɡælɪk/ | Tế bào khổng lồ |
| 414 | Glycerol kinase deficiency (GKD) | /ˈɡlɪsərɒl ˈkaɪneɪs dɪˈfɪʃənsi/ | Thiếu hụt glycerol kinase (GKD) |
| 415 | Ornithine transcarbamylase (OTC) | /ˈɔːrnɪθiːn ˌtrænskɑːrbəˈmɪleɪs/ | Ornithine transcarbamylase (OTC) |
| 416 | Duchenne muscular dystrophy (DMD) | /duːˈʃɛn ˈmʌskjələr ˈdɪstrəfi/ | Loạn dưỡng cơ Duchenne (DMD) |
| 417 | Pena-Shoekir syndrome | /ˈpeɪnjə ˈʃoʊkɪr ˈsɪndroʊm/ | Hội chứng Pena-Shoekir |
| 418 | Pseudotrisomy 13 | /ˌsuːdoʊˈtraɪsoʊmi θɜːrˈtiːn/ | Giả trisomy 13 |
| 419 | Meckel syndrome | /ˈmɛkəl ˈsɪndroʊm/ | Hội chứng Meckel |
| 420 | Hydrolethalus syndrome | /ˌhaɪdroʊlɪˈθeɪləs ˈsɪndroʊm/ | Hội chứng Hydrolethalus |
| 421 | Galloway-Mowat syndrome | /ˈɡæləweɪ ˈmoʊæt ˈsɪndroʊm/ | Hội chứng Galloway-Mowat |
| 422 | Pallister-Hall syndrome | /ˈpælɪstər hɔːl ˈsɪndroʊm/ | Hội chứng Pallister-Hall |
| 423 | Triple A (Allgrove) syndrome | /ˈtrɪpəl eɪ (ˈɔːlɡroʊv) ˈsɪndroʊm/ | Hội chứng Triple A (Allgrove) |
| 424 | Achalasia | /ˌækəˈleɪʒə/ | Co thắt tâm vị |
| 425 | Alacrima | /əˈlækrɪmə/ | Vô lệ |
| 426 | Sensorineural deafness | /ˌsɛnsəriˈnʊərəl ˈdɛfnəs/ | Điếc thần kinh giác quan |
| 427 | Optic atrophy | /ˈɒptɪk ˈætrəfi/ | Teo dây thần kinh thị giác |
| 428 | Parkinsonism | /ˈpɑːrkɪnsəˌnɪzəm/ | Hội chứng Parkinson |
| 429 | Autonomic dysfunction | /ˌɔːtəˈnɒmɪk dɪsˈfʌŋkʃən/ | Rối loạn chức năng tự chủ |
| 430 | IMAGe syndrome | /ˈɪmədʒ ˈsɪndroʊm/ | Hội chứng IMAGe |
| 431 | Intrauterine growth retardation | /ˌɪntrəˈjuːtərɪn ɡroʊθ ˌriːtɑːrˈdeɪʃən/ | Chậm tăng trưởng trong tử cung |
| 432 | Metaphyseal dysplasia | /ˌmɛtəˈfɪziəl dɪsˈpleɪʒə/ | Loạn sản hành xương |
| 433 | Genitourinary anomalies | /ˌdʒɛnɪtoʊˈjʊərɪˌnɛri əˈnɒməliz/ | Dị tật sinh dục-tiết niệu |
| 434 | Beckwith-Wiedemann syndrome | /ˈbɛkwɪθ ˈviːdəmən ˈsɪndroʊm/ | Hội chứng Beckwith-Wiedemann |
| 435 | Silver Russell syndrome | /ˈsɪlvər ˈrʌsəl ˈsɪndroʊm/ | Hội chứng Silver Russell |
| 436 | MIRAGE syndrome | /mɪˈrɑːʒ ˈsɪndroʊm/ | Hội chứng MIRAGE |
| 437 | Myelodysplasia | /ˌmaɪəloʊdɪsˈpleɪʒə/ | Loạn sản tủy |
| 438 | Enteropathy | /ˌɛntəˈrɒpəθi/ | Bệnh lý ruột |
| 439 | Myelodysplastic syndrome (MDS) | /ˌmaɪəloʊdɪsˈplæstɪk ˈsɪndroʊm/ | Hội chứng loạn sản tủy (MDS) |
| 440 | Monosomy 7 | /ˌmɒnoʊˈsoʊmi ˈsɛvən/ | Monosomy 7 |
| 441 | Natural killer cell deficiency | /ˈnætʃərəl ˈkɪlər sɛl dɪˈfɪʃənsi/ | Thiếu hụt tế bào diệt tự nhiên |
| 442 | Neoplasias | /ˌniːoʊˈpleɪʒəz/ | U tân sinh |
| 443 | Familial glucocorticoid deficiency (FGD) | /fəˈmɪliəl ˌɡluːkoʊˈkɔːrtɪkɔɪd dɪˈfɪʃənsi/ | Thiếu hụt glucocorticoid gia đình (FGD) |
| 444 | Nicotinamide nucleotide transhydrogenase (NNT) | /ˌnɪkəˈtɪnəmaɪd ˈnjuːkliətaɪd ˌtrænshaɪˈdrɒdʒəneɪs/ | Nicotinamide nucleotide transhydrogenase (NNT) |
| 445 | Reactive oxygen species (ROS) | /riˈæktɪv ˈɒksɪdʒən ˈspiːʃiːz/ | Các loại oxy phản ứng (ROS) |
| 446 | Adrenoleukodystrophy (ALD) | /əˌdriːnoʊˌluːkoʊˈdɪstrəfi/ | Loạn dưỡng chất trắng thượng thận (ALD) |
| 447 | Peroxisomal disease | /pəˌrɒksɪˈsoʊməl dɪˈziːz/ | Bệnh peroxisome |
| 448 | Demyelination | /diːˌmaɪəlɪˈneɪʃən/ | Mất myelin |
| 449 | Adrenomyeloneuropathy | /əˌdriːnoʊˌmaɪəloʊnʊəˈrɒpəθi/ | Bệnh lý tủy-thần kinh thượng thận |
| 450 | Axonopathy | /ˌæksoʊˈnɒpəθi/ | Bệnh lý sợi trục |
| 451 | Very long chain fatty acids (VLCFA) | /ˈvɛri lɔːŋ tʃeɪn ˈfæti ˈæsɪdz/ | Axit béo chuỗi rất dài (VLCFA) |
| 452 | Hematopoietic stem cell transplantation | /ˌhiːmətoʊpɔɪˈɛtɪk stɛm sɛl ˌtrænsplænˈteɪʃən/ | Ghép tế bào gốc tạo máu |
| 453 | Lovastatin | /ˌloʊvəˈstætɪn/ | Lovastatin |
| 454 | Peroxisome biogenesis disorders (PBDs) | /pəˌrɒksɪˈsoʊm baɪoʊˈdʒɛnəsɪs dɪsˈɔːrdərz/ | Rối loạn sinh tổng hợp peroxisome (PBD) |
| 455 | Infantile Refsum disease | /ˈɪnfəntaɪl ˈrɛfsəm dɪˈziːz/ | Bệnh Refsum ở trẻ sơ sinh |
| 456 | Rhizomelic chondrodysplasia punctata | /ˌraɪzoʊˈmiːlɪk ˌkɒndroʊdɪsˈpleɪʒə pʌŋkˈteɪtə/ | Loạn sản sụn chấm đầu xương |
| 457 | Hypotonia | /ˌhaɪpoʊˈtoʊniə/ | Giảm trương lực cơ |
| 458 | Neurosensory deafness | /ˌnʊəroʊˈsɛnsəri ˈdɛfnəs/ | Điếc thần kinh giác quan |
| 459 | Optic atrophy | /ˈɒptɪk ˈætrəfi/ | Teo thị thần kinh |
| 460 | Simvastatin | /ˌsɪmvəˈstætɪn/ | Simvastatin |
| 461 | Kearns-Sayre syndrome | /kɜːrnz seɪr ˈsɪndroʊm/ | Hội chứng Kearns-Sayre |
| 462 | Lactic acidosis | /ˈlæktɪk ˌæsɪˈdoʊsɪs/ | Nhiễm toan lactic |
| 463 | Cataracts | /ˈkætərækts/ | Đục thủy tinh thể |
| 464 | Myopathy/ophthalmoplegia | /maɪˈɒpəθi ˌɒfθælmoʊˈpliːdʒə/ | Bệnh cơ/liệt mắt |
| 465 | Sphingosine-1-phosphate lyase (SGPL1) | /ˈsfɪŋɡəsiːn wʌn ˈfɒsfeɪt ˈlaɪeɪs/ | Sphingosine-1-phosphate lyase (SGPL1) |
| 466 | Steroid-resistant nephrotic syndrome | /ˈstɪərɔɪd rɪˈzɪstənt nɪˈfrɒtɪk ˈsɪndroʊm/ | Hội chứng thận hư kháng steroid |
| 467 | Ichthyosis | /ˌɪkθiˈoʊsɪs/ | Bệnh vảy cá |
| 468 | Cryptorchidism | /krɪpˈtɔːrkɪdɪzəm/ | Tinh hoàn ẩn |
| 469 | Sphingolipidoses | /ˌsfɪŋɡoʊˌlɪpɪˈdoʊsiːz/ | Bệnh dự trữ sphingolipid |
| 470 | Tuberculosis | /tjuːˌbɜːrkjəˈloʊsɪs/ | Bệnh lao |
| 471 | Fungal infections | /ˈfʌŋɡəl ɪnˈfɛkʃənz/ | Nhiễm nấm |
| 472 | Human immunodeficiency virus (HIV) | /ˈhjuːmən ˌɪmjənoʊdɪˈfɪʃənsi ˈvaɪrəs/ | Virus gây suy giảm miễn dịch ở người (HIV) |
| 473 | Cytomegalovirus | /ˌsaɪtoʊˌmɛɡəloʊˈvaɪrəs/ | Cytomegalovirus |
| 474 | Metastases | /məˈtæstəsiːz/ | Di căn |
| 475 | Amyloidosis | /ˌæmɪlɔɪˈdoʊsɪs/ | Bệnh amyloidosis |
| 476 | Sarcoidosis | /ˌsɑːrkɔɪˈdoʊsɪs/ | Bệnh sarcoidosis |
| 477 | Aminoglutethimide | /əˌmiːnoʊˌɡluːtəˈθaɪmaɪd/ | Aminoglutethimide |
| 478 | Etomidate | /ɪˈtɒmɪdeɪt/ | Etomidate |
| 479 | Suramin | /ˈsʊərəmɪn/ | Suramin |
| 480 | Panhypopituitarism | /ˌpænˌhaɪpoʊpɪˈtjuːɪtəˌrɪzəm/ | Suy toàn bộ tuyến yên |
| 481 | Diabetes insipidus | /ˌdaɪəˈbiːtiːz ɪnˈsɪpɪdəs/ | Đái tháo nhạt |
| 482 | Thyroxine | /θaɪˈrɒksiːn/ | Thyroxine |
| 483 | Craniopharyngioma | /ˌkreɪnioʊfəˌrɪndʒiˈoʊmə/ | U sọ hầu |
| 484 | Germinoma | /ˌdʒɜːrmɪˈnoʊmə/ | U tế bào mầm |
| 485 | Astrocytoma | /ˌæstroʊsaɪˈtoʊmə/ | U tế bào hình sao |
| 486 | Langerhans cell histiocytosis | /ˈlæŋərhænz sɛl ˌhɪstioʊsaɪˈtoʊsɪs/ | Bệnh mô bào Langerhans |
| 487 | Optic nerve hypoplasia | /ˈɒptɪk nɜːrv ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản thần kinh thị |
| 488 | Cerebellar abnormalities | /ˌsɛrəˈbɛlər ˌæbnɔːrˈmælətiz/ | Bất thường tiểu não |
| 489 | Red hair | /rɛd hɛər/ | Tóc đỏ |
| 490 | Pale skin | /peɪl skɪn/ | Da nhợt nhạt |
| 491 | Hyperphagia | /ˌhaɪpərˈfeɪdʒə/ | Chứng ăn nhiều |
| 492 | Setmelanotide | /sɛtˈmɛlənəˌtaɪd/ | Setmelanotide |
| 493 | Ectopic ACTH syndrome | /ɛkˈtɒpɪk eɪ siː tiː eɪtʃ ˈsɪndroʊm/ | Hội chứng ACTH lạc chỗ |
| 494 | Moon facies | /muːn ˈfeɪʃiːiːz/ | Mặt tròn như mặt trăng |
| 495 | Facial flushing | /ˈfeɪʃəl ˈflʌʃɪŋ/ | Đỏ bừng mặt |
| 496 | Striae | /ˈstraɪiː/ | Vết rạn da |
| 497 | Somatostatin | /ˌsoʊmətəˈstætɪn/ | Somatostatin |
| 498 | Transsphenoidal surgery | /ˌtrænsfiːˈnɔɪdəl ˈsɜːrdʒəri/ | Phẫu thuật qua xương bướm |
| 499 | Cerebrospinal fluid rhinorrhea | /ˌsɛrəbroʊˈspaɪnəl ˈfluːɪd ˌraɪnəˈriːə/ | Chảy dịch não tủy qua mũi |
| 500 | Gamma Knife irradiation | /ˈɡæmə naɪf ɪˌreɪdiˈeɪʃən/ | Xạ trị bằng dao Gamma |