
Sách dịch: CÁC NGUYÊN LÝ Y HỌC HÔ HẤP, ẤN BẢN THỨ 8
Dịch và chú giải Tiếng Việt: Ts.Bs. Lê Nhật Huy, Ths.Bs. Lê Đình Sáng
CHƯƠNG 11. Bệnh Phổi Mô Kẽ Lan Tỏa Không Rõ Nguyên Nhân
Diffuse parenchymal lung diseases of unknown etiology
Steven E. Weinberger MD, MACP, FRCP, Barbara A. Cockrill MD and Jess Mandel MD, MACP, FRCP
Principles of Pulmonary Medicine, 11, 148-164
NỘI DUNG CHƯƠNG
| Xơ Phổi Vô Căn
Các Bệnh Viêm Phổi Kẽ Vô Căn Khác Tổn Thương Mô Kẽ Phổi trong Bệnh Thấp Khớp Hệ Thống Bệnh Sarcoidosis Các Rối Loạn Khác Liên Quan Đến Mô Kẽ Phổi Bệnh Mô Bào Langerhans ở Phổi |
Khoảng 65% bệnh nhân mắc bệnh phổi mô kẽ lan tỏa là nạn nhân của một quá trình bệnh lý mà không xác định được tác nhân gây bệnh, mặc dù bệnh có thể có một tên gọi cụ thể. Nhóm này bao gồm xơ phổi vô căn (IPF), bệnh phổi mô kẽ liên quan đến bệnh thấp khớp hệ thống (ví dụ: lupus ban đỏ hệ thống, xơ cứng bì, viêm khớp dạng thấp), sarcoidosis, bệnh mô bào Langerhans ở phổi (PLCH), và một loạt các rối loạn khác. Nhiều khía cạnh chung của các vấn đề này đã được thảo luận trong Chương 9. Chương này tập trung vào các bệnh cụ thể và các đặc điểm riêng của chúng.
XƠ PHỔI VÔ CĂN
Mặc dù thuật ngữ xơ phổi vô căn thường được sử dụng không đặc hiệu để mô tả bệnh phổi kẽ dạng xơ hóa không xác định được chẩn đoán, hầu hết các bác sĩ lâm sàng và nhà nghiên cứu tin rằng IPF đại diện cho một thực thể bệnh riêng biệt. Chương này chấp nhận giả định đó và xem xơ phổi liên quan đến một bệnh thấp khớp hệ thống tiềm ẩn là một thực thể riêng biệt. Một tên gọi khác đã được sử dụng thay thế cho IPF là viêm phế nang xơ hóa tự phát. Thuật ngữ viêm phổi kẽ thông thường (UIP) không phải là một bệnh lâm sàng mà đề cập đến kiểu tổn thương bệnh học liên quan đến IPF, nhưng cũng có thể được thấy trong các bối cảnh lâm sàng khác ngoài IPF, chẳng hạn như khi bệnh phổi đi kèm với một bệnh thấp khớp hệ thống.
Như tên gọi của nó, IPF không có tác nhân khởi phát rõ ràng, mặc dù hầu hết các nghiên cứu đều cho thấy có mối liên quan với việc phơi nhiễm khói thuốc lá. Lý thuyết về cơ chế bệnh sinh của IPF đã thay đổi đáng kể trong 25 năm qua. Trong nhiều năm, quan điểm phổ biến cho rằng việc phơi nhiễm với một tác nhân không xác định (có thể là một kháng nguyên dẫn đến sự hình thành các phức hợp kháng nguyên-kháng thể) đã gây ra viêm phế nang, tình trạng này kéo dài do sự giải phóng các yếu tố hóa ứng động từ các tế bào viêm. Tình trạng viêm liên tục này được cho là nguyên nhân gây ra sự phát triển xơ hóa sau đó. Tuy nhiên, mô hình được chấp nhận rộng rãi hiện nay là các vi tổn thương tái phát đối với các tế bào biểu mô phế nang nhạy cảm dẫn đến một quá trình lành vết thương bất thường, cuối cùng gây ra xơ hóa. Theo lý thuyết hiện tại này, viêm phế nang không đóng vai trò quan trọng trong sự phát triển cuối cùng của xơ hóa, và tổn thương tế bào biểu mô phế nang là sự kiện khởi đầu chính. Mặc dù tổn thương các tế bào biểu mô phế nang type I thông thường sẽ được theo sau bởi một quá trình sửa chữa bao gồm sự tăng sinh của các tế bào type II và biệt hóa thành các tế bào type I, quá trình sửa chữa này bị suy giảm, ít nhất một phần là do sự phá vỡ màng đáy, vốn rất quan trọng cho quá trình tái biểu mô hóa. Một số khiếm khuyết trong chức năng tế bào type II đã được liên kết với cơ chế bệnh sinh của IPF, bao gồm các bất thường về tự thực bào, apoptosis và chức năng tế bào tiền thân. Đồng thời, các tế bào biểu mô phế nang biểu hiện nhiều loại cytokine và yếu tố tăng trưởng gây xơ, bao gồm yếu tố tăng trưởng có nguồn gốc từ tiểu cầu (PDGF) và yếu tố tăng trưởng biến đổi (TGF)-β1, làm tăng cường sự di chuyển và tăng sinh của nguyên bào sợi. Các ổ nguyên bào sợi phát triển tại các vị trí tổn thương phế nang và dường như chịu trách nhiệm cho sự lắng đọng chất nền ngoại bào gia tăng. Quá trình này được tóm tắt trong Hình 11.1.
Sự phát triển của IPF có liên quan đến các đột biến gen trong một số con đường sinh học được biết là có liên quan đến tổn thương phổi, sửa chữa phổi hoặc sản xuất mucin trong các đường thở ngoại vi. Các đột biến trong con đường đầu tiên ảnh hưởng đến các gen mã hóa protein surfactant A2 (SFP-A2) và C (SFP-C), có thể làm tăng tính nhạy cảm với tổn thương phổi mạn tính bằng cách gây ra căng thẳng lưới nội chất gia tăng trong các tế bào biểu mô phế nang type II. Các đột biến trong con đường thứ hai ảnh hưởng đến các gen mã hóa men sao chép ngược telomerase (TERT) và thành phần RNA của telomerase (TERC), hệ thống enzyme đa tiểu đơn vị sửa chữa các telomere bị rút ngắn. Các bất thường trong chức năng telomerase dường như làm suy yếu quá trình lành vết thương bằng cách giảm sự sao chép của các tế bào tiền thân. Cuối cùng, một biến thể dẫn đến sự biểu hiện quá mức của gen MUC5B, liên quan đến sản xuất mucin ở các đường thở ngoại vi, cũng có liên quan đến nguy cơ IPF gia tăng thông qua một hoặc nhiều cơ chế chưa được làm sáng tỏ. Mặc dù các đột biến khác nhau này không có ở nhiều bệnh nhân IPF, việc xác định các con đường liên quan này đã giúp hiểu rõ hơn về cơ chế bệnh sinh ở ít nhất một số bệnh nhân IPF, làm dấy lên hy vọng về các liệu pháp mới.
Xơ phổi vô căn được cho là đại diện cho một mô hình xơ hóa bị rối loạn điều hòa để đáp ứng với tổn thương biểu mô phế nang.
Về mặt lâm sàng, độ tuổi phổ biến nhất khi bệnh nhân IPF đến khám là từ 50 đến 70 tuổi. Bệnh thường khởi phát âm thầm, và các triệu chứng tương tự như các bệnh phổi kẽ khác; khó thở là triệu chứng nổi bật nhất. Ngoài dấu hiệu kinh điển là ran nổ hoặc ran ẩm khô thì hít vào khi khám thực thể, bệnh nhân thường có biểu hiện ngón tay dùi trống.
Dòng tế bào viêm đổ vào -> Kích thích khởi phát/ nguồn tổn thương biểu mô phế nang -> Tổn thương tế bào biểu mô -> Các chất trung gian cytokine (ví dụ, TGF-β1, và PDGF) -> Tăng sinh nguyên bào sợi (ổ nguyên bào sợi) -> Phá vỡ màng đáy và tái cấu trúc chất nền ngoại bào -> Bất thường “lành vết thương” với xơ hóa
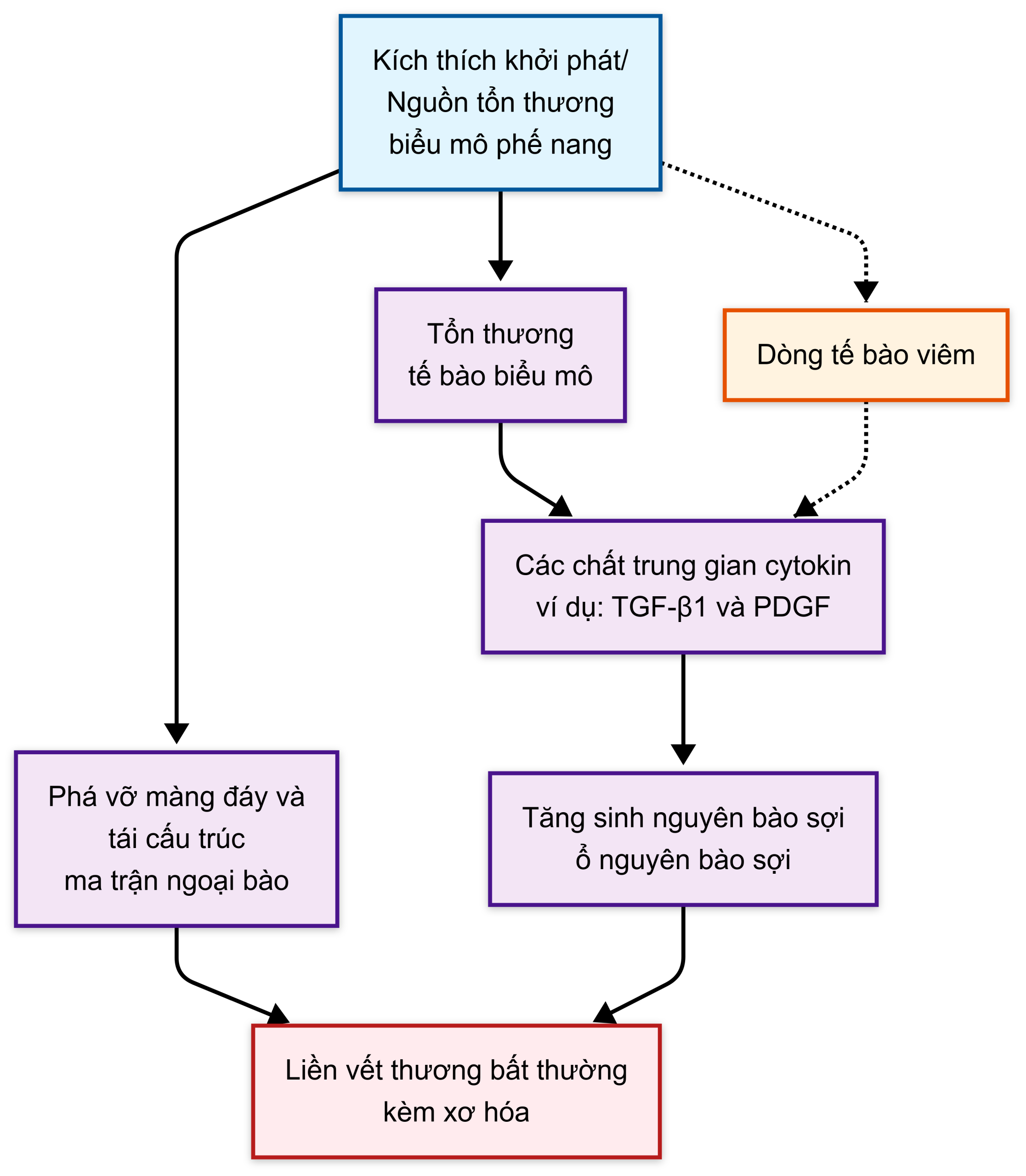
Hình 11.1 Chuỗi quá trình bệnh sinh được đề xuất trong xơ phổi vô căn. Các đường chấm chấm chỉ ra rằng mặc dù có sự đổ vào của các tế bào viêm, điều này không được cho là một thành phần chính của cơ chế bệnh sinh. PDGF, yếu tố tăng trưởng có nguồn gốc từ tiểu cầu; TGF-B1, yếu tố tăng trưởng biến đổi-β1.
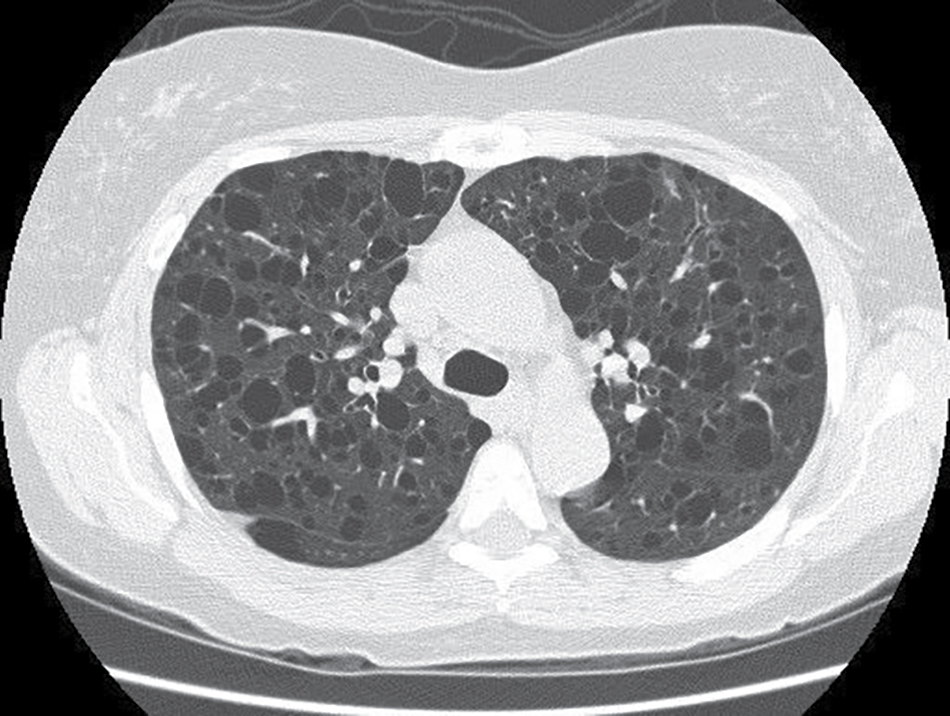
X-quang ngực cho thấy một dạng tổn thương kẽ (dạng lưới) thường ở hai bên và tương đối lan tỏa nhưng điển hình là nổi bật hơn ở đáy phổi, đặc biệt là ở các vùng dưới màng phổi ngoại vi (xem Hình 3.6). Cả rốn phổi to và tràn dịch màng phổi đều không phổ biến. Chụp cắt lớp vi tính độ phân giải cao (HRCT) thường có hình ảnh đặc trưng, cho thấy các tổn thương đậm độ kẽ dạng mảng, ở ngoại vi, dưới màng phổi, và kết hợp với các khoang dạng nang nhỏ (Hình 11.2). Dạng tổn thương nang nhỏ ở ngoại vi trên HRCT được gọi là hình ảnh tổ ong và cho thấy tình trạng xơ hóa không hồi phục. Nhiều bệnh nhân có các bất thường huyết thanh học, chẳng hạn như kết quả xét nghiệm kháng thể kháng nhân dương tính, thường thấy ở những bệnh nhân bị bệnh tự miễn hoặc bệnh thấp khớp hệ thống. Tuy nhiên, khi không có các đặc điểm lâm sàng gợi ý khác, những bất thường này được cho là không đặc hiệu và không chỉ ra một bệnh thấp khớp tiềm ẩn.
X-quang ngực trong IPF cho thấy tổn thương kẽ lan tỏa không có bệnh lý màng phổi hoặc rốn phổi to.
Chẩn đoán được xác định chắc chắn bằng sinh thiết phổi phẫu thuật. Tuy nhiên, nếu phim HRCT cho thấy một hình ảnh kinh điển bao gồm hình ảnh tổ ong và một số đặc điểm khác, chẩn đoán có thể được đưa ra với độ chắc chắn tương đối mà không cần sinh thiết phổi. Các phương pháp sinh thiết phổi qua nội soi phế quản truyền thống không cho mẫu đủ lớn để xác định chẩn đoán, mặc dù một kỹ thuật mới hơn gọi là sinh thiết lạnh qua phế quản, lấy được mẫu mô lớn hơn, có vẻ hữu ích hơn. Biểu hiện mô học của IPF là ở dạng UIP (xem Hình 9.3), và những bệnh nhân có kiểu bệnh học tương thích hơn với viêm phổi kẽ tróc vảy (DIP) hoặc NSIP (xem Các bệnh viêm phổi kẽ vô căn khác; cũng xem Chương 9) không nên được coi là mắc IPF. Không nên thấy u hạt trên mẫu sinh thiết IPF và nếu có, nó gợi ý một rối loạn khác.
Mặc dù IPF là một bệnh mạn tính, xơ hóa, một số bệnh nhân có thể xuất hiện đợt cấp của IPF. Vấn đề xen kẽ này được đặc trưng bởi sự gia tăng triệu chứng tương đối cấp tính, các phát hiện X-quang về hình ảnh kính mờ chồng lên bệnh lý có từ trước, và một hình ảnh bệnh học phù hợp với tổn thương phổi cấp tính giống như hội chứng suy hô hấp cấp (ARDS) (xem Chương 29). Các đợt cấp thường được điều trị bằng corticosteroid và kháng sinh (do khó loại trừ dứt điểm nhiễm trùng), mặc dù không rõ liệu chúng có mang lại lợi ích gì không, và tỷ lệ tử vong liên quan đến đợt cấp là tương đối cao.
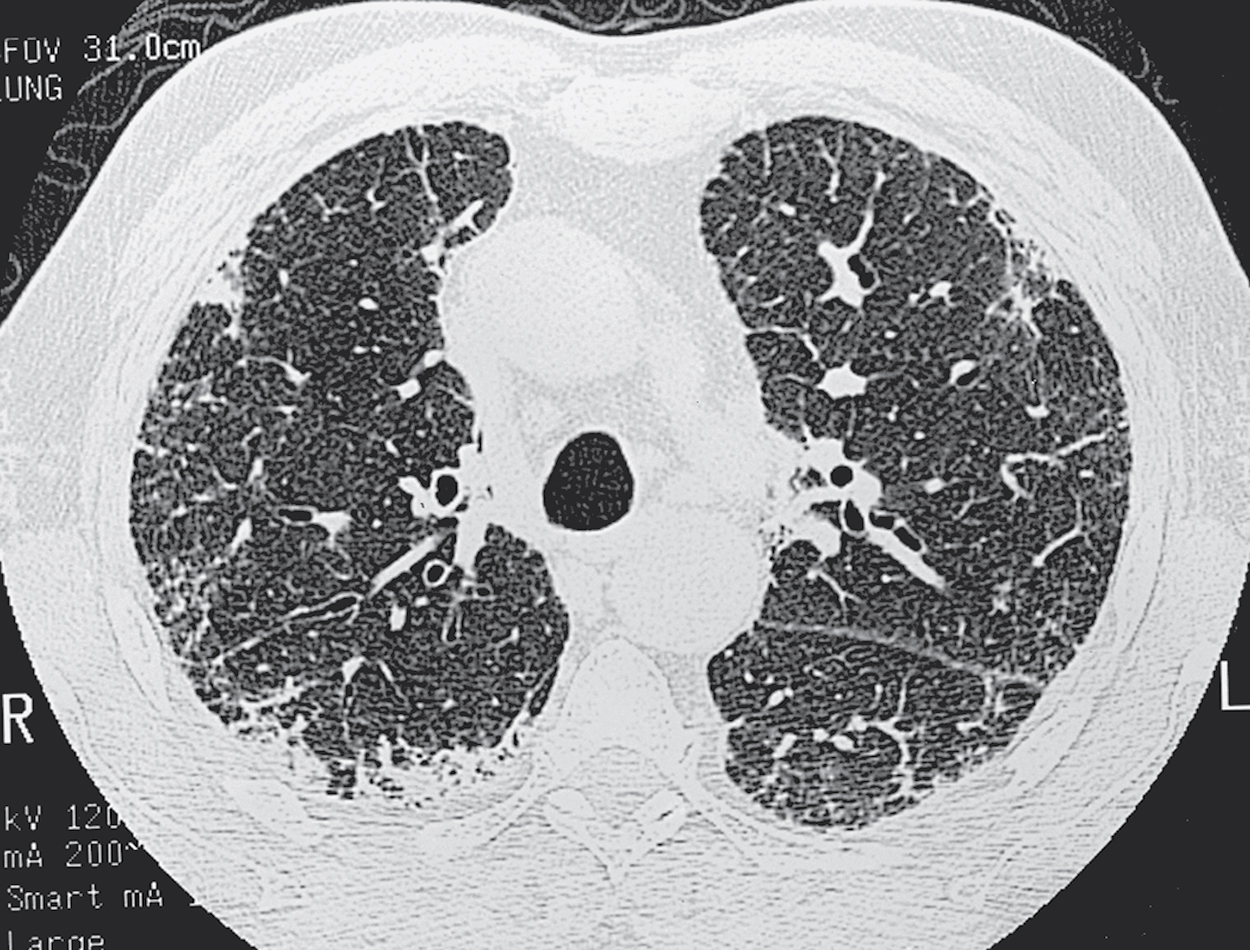
Hình 11.2 Phim chụp cắt lớp vi tính độ phân giải cao của xơ phổi vô căn cho thấy các đám mờ dạng lưới rải rác, đặc biệt ở các vùng dưới màng phổi.
Hiện tại, không có liệu pháp hiệu quả nào được chứng minh để ngăn chặn hoặc đảo ngược quá trình xơ hóa tiến triển của IPF. Mặc dù corticosteroid và các thuốc độc tế bào đã được sử dụng thường xuyên trong quá khứ cho IPF, các nghiên cứu sau đó không may đã chứng minh rằng những tác nhân này không hiệu quả và dường như có hại. Hiện nay người ta cho rằng nhóm nhỏ bệnh nhân trước đây được cho là đáp ứng với corticosteroid thực chất đã mắc một bệnh phổi mô kẽ lan tỏa đáp ứng với corticosteroid nhưng bị chẩn đoán nhầm là IPF. Điều trị IPF hiện nay tập trung vào các tác nhân ức chế xơ hóa hoặc can thiệp vào các chất trung gian liên quan đến quá trình xơ hóa. Hai loại thuốc hiện có đã được chứng minh là làm giảm (nhưng không ngăn chặn) sự tiến triển của bệnh, được đo bằng sự suy giảm chức năng phổi. Một trong số đó, pirfenidone, ức chế sự tổng hợp collagen và tăng sinh nguyên bào sợi qua trung gian TGF-β. Tác nhân còn lại, nintedanib, một chất ức chế tyrosine kinase, ngăn chặn các thụ thể và tín hiệu xuôi dòng của một số yếu tố tăng trưởng gây xơ, bao gồm PDGF, yếu tố tăng trưởng nguyên bào sợi và yếu tố tăng trưởng nội mô mạch máu. Ở một số bệnh nhân bị IPF nặng, đặc biệt là những người trẻ tuổi, ghép phổi là lựa chọn điều trị duy nhất thay thế cho suy hô hấp tiến triển và tử vong. Mặc dù tiên lượng chung thường xấu, với thời gian sống trung bình dưới 5 năm, một số bệnh nhân có diễn biến kéo dài hơn, với thời gian sống có thể vượt quá một thập kỷ.
Tiên lượng trong xơ phổi vô căn thường xấu. Các lựa chọn điều trị bao gồm các loại thuốc nhằm làm chậm sự tiến triển của bệnh (nintedanib, pirfenidone) hoặc, ở những bệnh nhân được chọn lọc, ghép phổi.
CÁC BỆNH VIÊM PHỔI KẼ VÔ CĂN KHÁC
Ngoài IPF, một số rối loạn khác cũng thuộc nhóm viêm phổi kẽ vô căn và trước đây thường bị nhầm lẫn với IPF. Mặc dù các rối loạn này không phổ biến, một số được mô tả ngắn gọn ở đây, chủ yếu để làm rõ các đặc điểm bệnh học của chúng khác với UIP như thế nào và các đặc điểm lâm sàng của chúng khác với IPF như thế nào. Chúng cũng được đề cập trong Chương 9 trong phần thảo luận về bệnh học của viêm phổi kẽ (xem Bảng 9.2).
Bệnh phổi kẽ liên quan đến viêm tiểu phế quản hô hấp (RB-ILD) và DIP thường được xem xét cùng nhau như một phần của một phổ bệnh do mối liên quan chặt chẽ của chúng với hút thuốc, mặc dù hiện nay chúng được phân loại là các bệnh viêm phổi kẽ liên quan đến hút thuốc riêng biệt. RB-ILD thường có các triệu chứng tương đối nhẹ, chỉ xảy ra ở người hút thuốc, và thường hồi phục khi cai thuốc lá. Phim HRCT ở RB-ILD thường cho thấy hình ảnh mờ dạng kính (hazy) kèm theo các nốt trung tâm tiểu thùy. Mô hình bệnh học tương ứng với hình ảnh này trên HRCT là sự tích tụ của các đại thực bào chứa sắc tố trong lòng các đường thở nhỏ kèm theo một số viêm quanh tiểu phế quản với các tế bào lympho và đại thực bào (xem Hình 9.5).
DIP gần như luôn luôn liên quan đến hút thuốc, mặc dù hiếm khi các trường hợp cũng đã được mô tả ở những người không hút thuốc. Bệnh thường có khởi phát bán cấp hơn là mạn tính. Các nghiên cứu hình ảnh với X-quang ngực và chụp HRCT thường cho thấy hình ảnh kính mờ. Trên sinh thiết phổi, có sự tích tụ đồng nhất của các đại thực bào trong phế nang, với ít hoặc không có xơ hóa. So với sự phân bố quanh tiểu phế quản ở RB-ILD, DIP cho thấy sự liên quan đến mô kẽ lan tỏa hơn. Tiên lượng tốt hơn so với IPF, và bệnh nhân thường có thể cải thiện sau khi cai thuốc lá và có thể đáp ứng với corticosteroid.
NSIP khác với UIP về hình ảnh X-quang, hình thái mô học, tiên lượng và đáp ứng điều trị. Tương tự như DIP, các nghiên cứu hình ảnh thường cho thấy hình ảnh kính mờ phản ánh tình trạng viêm hơn là xơ hóa. Sinh thiết phổi cho thấy một phản ứng viêm chiếm ưu thế trong thành phế nang, với tương đối ít xơ hóa (xem Hình 9.4). Mặc dù NSIP thường là vô căn, nó có thể đại diện cho hình ảnh mô học của bệnh phổi mô kẽ liên quan đến một trong các bệnh thấp khớp hệ thống hoặc độc tính phổi do thuốc. Tiên lượng của NSIP dường như phụ thuộc vào mức độ liên quan xơ hóa có mặt trên cả hình ảnh và bệnh học. Nếu tình trạng viêm chiếm ưu thế hơn là xơ hóa, tiên lượng tốt hơn đáng kể so với IPF, và bệnh nhân thường đáp ứng với điều trị bằng corticosteroid.
Viêm phổi tổ chức hóa tự phát (COP) là một rối loạn được đặc trưng bởi các nút mô liên kết trong các đường thở nhỏ kèm theo sự xâm nhập của tế bào đơn nhân vào nhu mô phổi xung quanh (xem Hình 9.6). Như đã lưu ý trong Chương 9, các thuật ngữ viêm phổi tổ chức hóa tự phát (COP) và viêm tiểu phế quản tắc nghẽn với viêm phổi tổ chức hóa (BOOP) thường được sử dụng thay thế cho nhau, nhưng thuật ngữ BOOP tốt nhất nên được dành cho hình ảnh bệnh học hơn là hội chứng lâm sàng. Mặc dù mô hình bệnh học của BOOP có thể liên quan đến bệnh thấp khớp hệ thống, hít phải khói độc hoặc nhiễm trùng, phần lớn các trường hợp không có nguyên nhân xác định và được coi là vô căn. Thuật ngữ COP là phù hợp nhất cho những bệnh nhân có “BOOP vô căn” – nghĩa là, các phát hiện mô học của BOOP mà không có nguyên nhân rõ ràng.
Trên X-quang ngực, viêm phổi tổ chức hóa tự phát (COP) thường giống như viêm phổi với một hoặc nhiều đám thâm nhiễm phế nang.
Giống như viêm phổi tăng bạch cầu ái toan mạn tính (xem phần sau), COP thường có biểu hiện bán cấp (kéo dài hàng tuần đến hàng tháng) với các triệu chứng toàn thân (toàn trạng) cũng như các triệu chứng hô hấp. Các nghiên cứu hình ảnh ngực cho thấy các đám thâm nhiễm dạng mảng, thường có dạng phế nang hơn là dạng kẽ, thường giống với viêm phổi mắc phải tại cộng đồng (Hình 11.3). Giống như viêm phổi tăng bạch cầu ái toan mạn tính, đáp ứng với corticosteroid thường ngoạn mục và xảy ra trong vài ngày đến vài tuần. Liệu pháp thường được duy trì trong nhiều tháng để ngăn ngừa tái phát.
Viêm phổi kẽ cấp tính (AIP) là một loại bệnh nhu mô phổi cấp tính hoặc tối cấp hơn được đặc trưng bởi bức tranh lâm sàng của ARDS (xem Chương 29) nhưng không có bất kỳ sự kiện khởi phát thông thường nào liên quan đến sự phát triển của ARDS. Các nghiên cứu hình ảnh của AIP thường cho thấy các đặc điểm của ARDS, bao gồm các vùng mờ dạng kính và lấp đầy phế nang (trái ngược với dạng tổn thương kẽ đơn thuần). Mô hình mô học là tổn thương phế nang lan tỏa, thường cho thấy một số tổ chức hóa và xơ hóa. Mặc dù tỷ lệ tử vong nói chung là cao, một tỷ lệ nhỏ bệnh nhân có diễn biến tốt, với sự giải quyết bệnh trên lâm sàng và không có di chứng lâu dài.
Một khía cạnh khó hiểu của danh pháp viêm phổi kẽ vô căn là mối quan hệ cơ bản giữa AIP, UIP (hoặc IPF), và một rối loạn được gọi là hội chứng Hamman-Rich. Hơn 80 năm trước, Hamman và Rich đã mô tả một số trường hợp bệnh nhu mô phổi mà sau đó được cho là đại diện cho những trường hợp IPF được mô tả đầu tiên, và trong nhiều năm, thuật ngữ hội chứng Hamman-Rich đã được sử dụng đồng nghĩa với IPF. Tuy nhiên, các trường hợp được mô tả bởi Hamman và Rich hiện được cho là các trường hợp AIP chứ không phải IPF, và phù hợp hơn khi hội chứng Hamman-Rich được coi là đồng nghĩa với AIP thay vì UIP hoặc IPF.
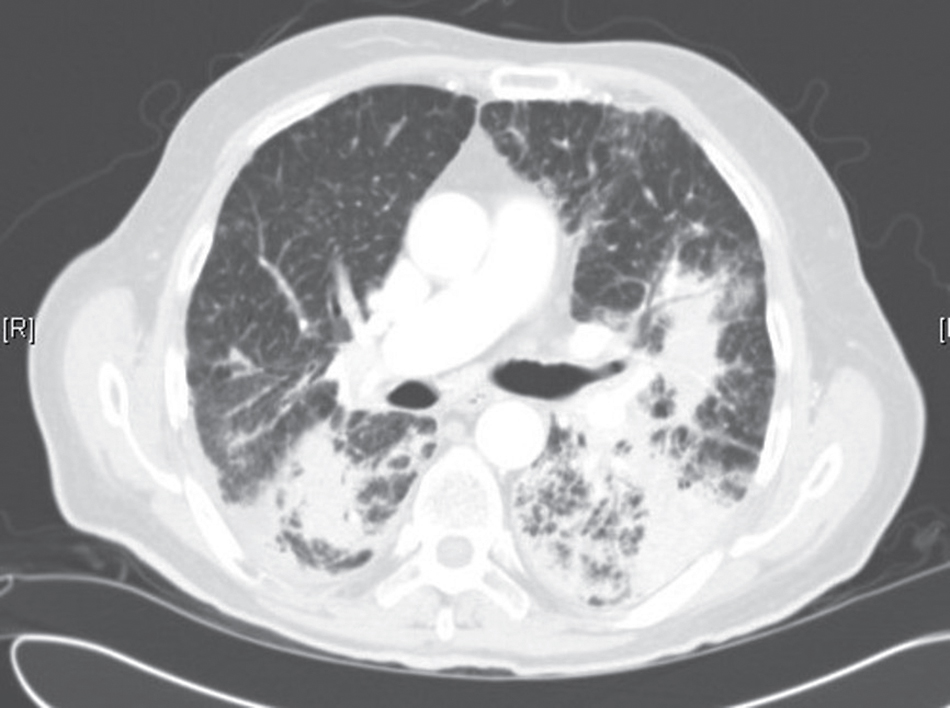
Hình 11.3 Phim chụp CT ngực cho thấy các đám mờ phế nang dạng mảng ở một bệnh nhân bị viêm phổi tổ chức hóa tự phát.
TỔN THƯƠNG MÔ KẼ PHỔI TRONG BỆNH THẤP KHỚP HỆ THỐNG
Các bệnh thấp khớp hệ thống, còn được gọi là bệnh mạch máu collagen hoặc bệnh mô liên kết, bao gồm viêm khớp dạng thấp, lupus ban đỏ hệ thống, xơ cứng bì hệ thống (scleroderma), viêm đa cơ-viêm da cơ, hội chứng Sjögren, và các hội chứng chồng lấp có các đặc điểm của nhiều hơn một trong những rối loạn này. Mặc dù chúng bao gồm một nhóm đa dạng, tất cả đều là các bệnh viêm đa hệ thống được trung gian miễn dịch và thường được coi là các rối loạn tự miễn. Các hệ cơ quan có khả năng bị ảnh hưởng thay đổi theo từng bệnh và được đề cập ngắn gọn trong phần thảo luận sau đây về từng thực thể. Mỗi bệnh đều phức tạp và đã là trọng tâm của nghiên cứu sâu rộng về nguyên nhân và cơ chế bệnh sinh. Tuy nhiên, vì không có bệnh nào trong số này chủ yếu ảnh hưởng đến phổi, chúng không được xem xét chi tiết ở đây. Thay vào đó, một cuộc thảo luận ngắn gọn sẽ lưu ý cách chúng ảnh hưởng đến hệ hô hấp, đặc biệt là liên quan đến sự phát triển của bệnh phổi mô kẽ. Một số bác sĩ lâm sàng bao gồm thêm các rối loạn khác dưới nhãn bệnh mô liên kết, nhưng cuộc thảo luận này được giới hạn ở những bệnh trong đoạn trước, mỗi bệnh đều có khả năng liên quan đến phổi.
Bốn khẳng định sau đây đúng với mỗi rối loạn này. Thứ nhất, mặc dù bệnh nhân thường có bằng chứng về bệnh thấp khớp hệ thống tiềm ẩn trước khi các biểu hiện ở phổi phát triển, một số bệnh nhân có bệnh phổi là vấn đề khởi phát, đôi khi đi trước các biểu hiện khác của bệnh vài năm. Thứ hai, đánh giá chi tiết về mô học, sinh lý học hoặc tử thi của bệnh nhân mắc các bệnh này cho thấy rằng sự liên quan đến phổi phổ biến hơn nhiều trong các tình trạng tự miễn này so với nghi ngờ trên lâm sàng. Thứ ba, mô bệnh học của bệnh phổi kẽ liên quan đến các bệnh thấp khớp hệ thống thường là NSIP hoặc UIP và không thể phân biệt được với NSIP vô căn hoặc IPF, tương ứng. Tuy nhiên, đôi khi, mô bệnh học cho thấy viêm phổi tổ chức hóa không thể phân biệt được với COP. Thứ tư, bệnh phổi kẽ có thể phát triển với mỗi thực thể này ưu tiên ảnh hưởng đến các vùng phổi dưới hơn là các vùng phổi trên. Thực tế này thường rõ ràng khi kiểm tra X-quang ngực.
Các thay đổi mô học và sinh lý học cho thấy sự tham gia của phổi trong hầu hết các bệnh thấp khớp hệ thống là phổ biến, thường với mô hình mô học của viêm phổi kẽ không đặc hiệu hoặc viêm phổi kẽ thông thường.
Viêm khớp dạng thấp là một rối loạn với các biểu hiện chính bao gồm bệnh khớp viêm. Vị trí liên quan phổ biến nhất trong lồng ngực là màng phổi. Sự liên quan có dạng viêm màng phổi, tràn dịch màng phổi, hoặc cả hai. Nhu mô phổi có thể bị ảnh hưởng, với một hoặc nhiều nốt hoặc với sự phát triển của bệnh phổi kẽ. Hình ảnh bệnh học của UIP là phổ biến nhất, NSIP ít phổ biến hơn một chút, và một số bệnh nhân có mô hình viêm phổi tổ chức hóa giống COP. Ít phổ biến hơn, bệnh nhân viêm khớp dạng thấp phát triển các biến chứng đường thở dưới dạng viêm tiểu phế quản (một quá trình viêm liên quan đến các đường thở nhỏ) hoặc giãn phế quản. Vì bệnh nhân viêm khớp dạng thấp thường được điều trị bằng các loại thuốc có thể liên quan đến độc tính phổi, chẳng hạn như methotrexate hoặc rituximab, khả năng bệnh phổi do thuốc cũng phải được xem xét.
Trong viêm khớp dạng thấp và lupus, bệnh lý màng phổi phổ biến hơn bệnh phổi mô kẽ lan tỏa rõ ràng trên lâm sàng.
Lupus ban đỏ hệ thống là một bệnh đa hệ thống chủ yếu ảnh hưởng đến khớp và da nhưng thường có sự liên quan nghiêm trọng hơn đến một số hệ cơ quan, bao gồm thận, phổi, hệ thần kinh và tim. Biểu hiện thường gặp nhất của nó trong lồng ngực có dạng bệnh lý màng phổi, cụ thể là đau ngực kiểu màng phổi, tràn dịch màng phổi, hoặc cả hai. Nhu mô phổi có thể bị ảnh hưởng bởi một đợt viêm phổi cấp tính giống như AIP, với các đám thâm nhiễm thường liên quan đến cả khoang phế nang và thành phế nang. Một biến chứng phổi cấp tính khác trong lupus là sự phát triển của xuất huyết phế nang lan tỏa. Ít thường xuyên hơn, bệnh nhân lupus có thể phát triển bệnh phổi kẽ mạn tính có bệnh học NSIP hoặc UIP, mặc dù xơ hóa lan rộng tương đối không phổ biến trong lupus.
Xơ cứng bì hệ thống, hay scleroderma, là một bệnh có các biểu hiện rõ ràng nhất ở da và các mạch máu nhỏ. Các hệ cơ quan khác, bao gồm đường tiêu hóa, phổi, thận và tim, bị ảnh hưởng tương đối thường xuyên. Như với các bệnh thấp khớp hệ thống khác, sự liên quan đến nhu mô phổi với xơ cứng bì có thể là NSIP hoặc UIP. Xơ phổi biến chứng của xơ cứng bì dường như có liên quan chặt chẽ với sự hiện diện của một dấu ấn huyết thanh cụ thể, một tự kháng thể kháng topoisomerase I (antitopoisomerase I, còn gọi là Scl70). Một biểu hiện phổi tiềm tàng khác của xơ cứng bì là bệnh của các mạch máu nhỏ ở phổi, gây tăng áp động mạch phổi, được thảo luận trong Chương 14. Sự liên quan này dường như độc lập với quá trình xơ hóa ảnh hưởng đến thành phế nang. Vì bệnh nhân xơ cứng bì thường có bệnh thực quản dẫn đến trào ngược dạ dày-thực quản, việc hít sặc tái phát có thể đóng một vai trò trong sự phát triển hoặc tiến triển của bệnh phổi kẽ rõ ràng.
Ngoài xơ hóa kẽ, bệnh nhân xơ cứng bì có thể phát triển bệnh mạch máu phổi liên quan đến các mạch phổi nhỏ độc lập với quá trình kẽ.
Trong viêm đa cơ-viêm da cơ, cơ và da là những vị trí chính của quá trình viêm. Bệnh phổi kẽ của viêm đa cơ-viêm da cơ tương đối không thường xuyên và thường không có đặc điểm phân biệt đặc biệt nào. Bệnh nhân cũng có thể có các vấn đề về hô hấp do bệnh cơ, với sự yếu của cơ hoành hoặc các cơ hít vào khác. Sự tham gia của cơ vân ở thực quản đoạn gần có thể dẫn đến khó nuốt và các đợt viêm phổi hít tái phát.
Trong hội chứng Sjögren, sự xâm nhập của tế bào lympho ảnh hưởng đến các tuyến nước bọt và tuyến lệ và có liên quan đến khô miệng và khô mắt (viêm kết giác mạc khô). Khi bệnh nhân mắc hội chứng Sjögren có tổn thương nhu mô phổi, hình ảnh mô học phổ biến nhất là NSIP hoặc sự xâm nhập của tế bào lympho trong thành phế nang được gọi là viêm phổi kẽ tế bào lympho. Các biến chứng tế bào lympho khác của phổi có thể phát triển ở bệnh nhân mắc hội chứng Sjögren, cụ thể là một tổn thương dạng nốt cục bộ được gọi là u lympho giả hoặc một u lympho thực sự.
Cuối cùng, một số hội chứng chồng lấp, thường được gọi là bệnh mô liên kết không biệt hóa, có các đặc điểm của một số rối loạn này, đặc biệt là xơ cứng bì, lupus và viêm đa cơ. Bệnh nhân có thể phát triển bất kỳ biến chứng nào được ghi nhận với các rối loạn riêng lẻ kinh điển hơn, bao gồm bệnh phổi mô kẽ, bệnh màng phổi và bệnh mạch máu phổi.
BỆNH SARCOIDOSIS
Sarcoidosis là một rối loạn hệ thống trong đó các u hạt, điển hình là không hoại tử bã đậu, có thể được tìm thấy trong các mô hoặc hệ cơ quan bị ảnh hưởng. Một điều kiện quan trọng là các u hạt này xảy ra khi không có bất kỳ tác nhân ngoại sinh nào (truyền nhiễm hoặc môi trường) được biết là có liên quan đến viêm u hạt. Phổi là cơ quan bị ảnh hưởng thường xuyên nhất, với các biểu hiện tiềm tàng bao gồm bệnh phổi mô kẽ, hạch bạch huyết rốn phổi và trung thất to, hoặc cả hai.
Sarcoidosis là một bệnh u hạt hệ thống thường ảnh hưởng nhất đến phổi, các hạch bạch huyết rốn phổi và trung thất, hoặc cả hai.
Sarcoidosis là một rối loạn tương đối phổ biến, đặc biệt ảnh hưởng đến người trẻ tuổi từ 20 đến 40 tuổi. Bệnh phổ biến hơn một chút ở phụ nữ so với nam giới. Ở Hoa Kỳ, bệnh phổ biến hơn ở người da đen so với người da trắng. Tuy nhiên, sự thiên vị này không được thấy trên toàn thế giới vì bệnh đặc biệt phổ biến ở người da trắng ở Scandinavia. Trong tất cả các rối loạn không rõ nguyên nhân ảnh hưởng đến thành phế nang, sarcoidosis là phổ biến nhất.
Mặc dù kiến thức về các tế bào tham gia vào phản ứng viêm và u hạt trong sarcoidosis và việc xác định nhiều cytokine và chemokine dường như có liên quan đến cơ chế bệnh sinh của bệnh ngày càng tăng, nguyên nhân cơ bản của sarcoidosis vẫn còn bí ẩn như khi bệnh được mô tả lần đầu tiên hơn 125 năm trước. Người ta giả thuyết rằng sarcoidosis đại diện cho một phản ứng miễn dịch với một tác nhân ngoại sinh ở một cá nhân nhạy cảm về mặt di truyền. Nhiều kháng nguyên ngoại sinh và một số kháng nguyên bạch cầu người và các gen ứng cử viên khác đã được liên kết với tính nhạy cảm với sarcoidosis. Tuy nhiên, cả tác nhân ngoại sinh cụ thể lẫn tính nhạy cảm di truyền cụ thể đều chưa được chứng minh một cách nhất quán. Sự quan tâm đến các tác nhân kích thích ngoại sinh tiềm tàng đã tập trung vào các vi sinh vật như virus, mycobacteria và các vi khuẩn khác (ví dụ, Propionibacterium acnes), cũng như các loại bụi vô cơ như silica. Tuy nhiên, danh tính của một tác nhân gây ra sarcoidosis vẫn còn khó nắm bắt, và liệu một tác nhân hoặc nhóm tác nhân như vậy có tồn tại hay không vẫn chưa được biết. Hiện tại, có vẻ như sarcoidosis rất có thể đại diện cho một sự tương tác phức tạp giữa nhiều loại kháng nguyên hoặc hạt và ảnh hưởng của nhiều gen.
Ngược lại, có rất nhiều thông tin về các tế bào và chất trung gian dường như quan trọng trong phản ứng viêm và u hạt trong sarcoidosis (Hình 11.4). Các tế bào quan trọng là tế bào trình diện kháng nguyên và tế bào lympho T. Việc xử lý kháng nguyên (chưa được xác định) chịu trách nhiệm bởi các đại thực bào phế nang hoặc tế bào tua dẫn đến việc huy động các tế bào lympho T hỗ trợ (tế bào ) với hồ sơ . Sự biểu hiện tăng rõ rệt của interferon (IFN)-γ là đặc trưng, và các cytokine và chemokine gây viêm khác, chẳng hạn như interleukin (IL)-2, yếu tố hoại tử u (TNF)-α, và IL-12, dường như quan trọng trong việc huy động và kích hoạt các tế bào viêm, duy trì phản ứng viêm, và gây ra sự hình thành các u hạt. Các cytokine gây xơ, chẳng hạn như TGF-β, PDGF, và yếu tố tăng trưởng giống insulin (IGF)-1, sau đó có thể dẫn đến xơ hóa như một biến chứng của phản ứng viêm ban đầu.
Sự tích tụ các tế bào lympho tại các vị trí bệnh đang hoạt động dường như dẫn đến các hiện tượng miễn dịch thứ cấp được công nhận rõ trong sarcoidosis. Thứ nhất, có lẽ do sự tập trung các tế bào lympho hoạt hóa này trong các mô bị ảnh hưởng, có sự suy giảm tương đối của các tế bào trong máu ngoại vi. Sự suy giảm này dẫn đến sự suy giảm rõ ràng của miễn dịch qua trung gian tế bào, ít nhất là khi được đo bằng test quá mẫn muộn trên da (test da). Tuy nhiên, bệnh nhân sarcoidosis không quá nhạy cảm với các bệnh nhiễm trùng cơ hội đặc trưng ảnh hưởng đến vật chủ bị suy giảm miễn dịch có miễn dịch tế bào suy yếu. Thứ hai, các tế bào lympho T trong sarcoidosis kích hoạt không đặc hiệu các tế bào lympho B và hệ thống miễn dịch dịch thể, dẫn đến sản xuất nhiều loại globulin miễn dịch và phát hiện phổ biến về tăng gammaglobulin đa dòng.
Kích thích (ví dụ, KN) -> ? Khuynh hướng di truyền -> Đại thực bào -> Yếu tố gây xơ và yếu tố tăng trưởng -> Xơ hóa Huy động và kích hoạt tế bào do cytokine -> Tế bào lympho CD4+ -> Hình thành u hạt -> Xơ hóa -> Kích thích tế bào lympho B -> Tế bào lympho B hoạt hóa -> Sản xuất KT
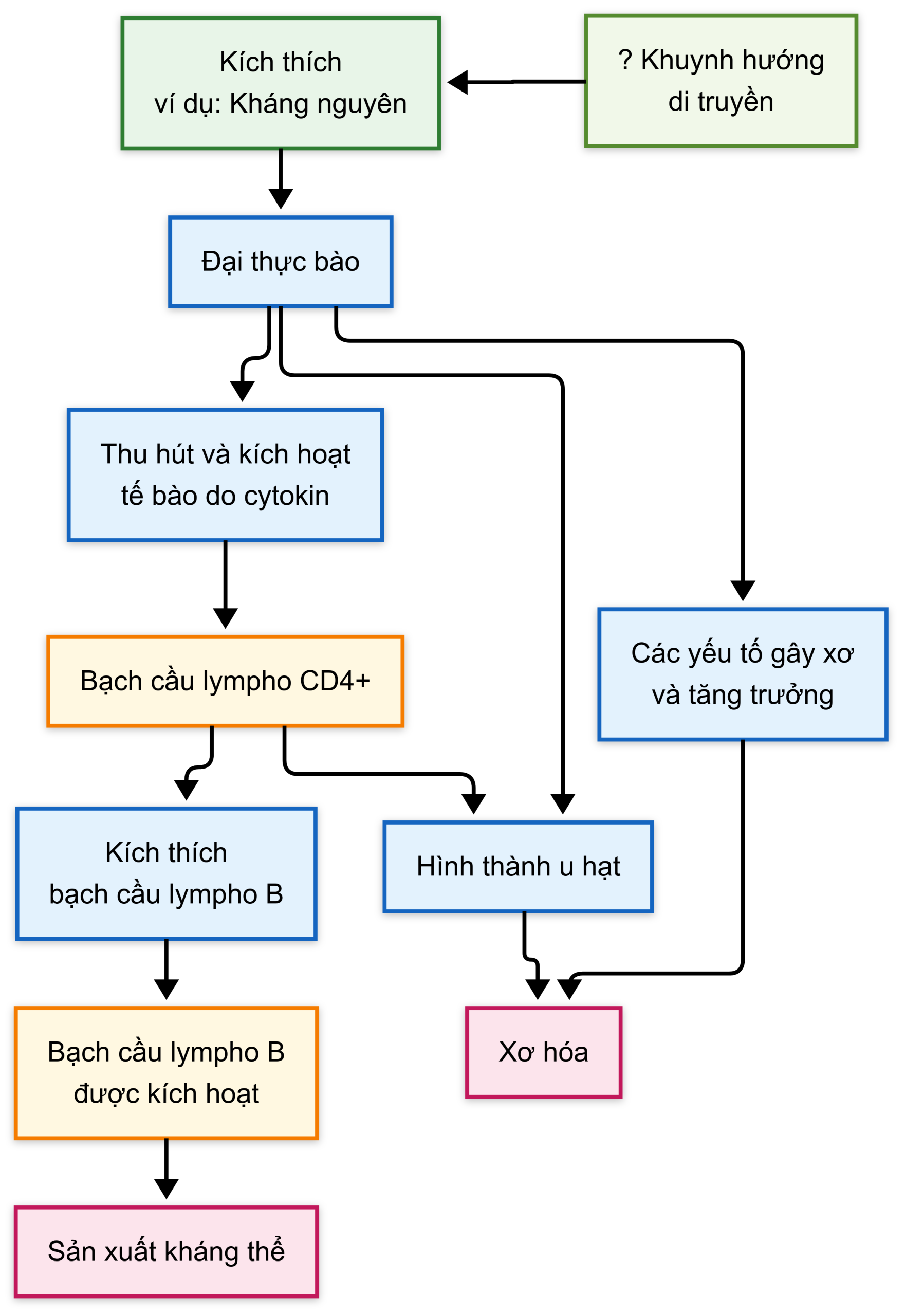
Hình 11.4 Chuỗi bệnh sinh được đề xuất đã được đơn giản hóa trong bệnh sarcoidosis. KT, kháng thể; KN, kháng nguyên.
Đặc điểm mô bệnh học đặc trưng của sarcoidosis là u hạt không hoại tử bã đậu (xem Hình 9.2). Những u hạt được hình thành tốt này đại diện cho một tập hợp các đại thực bào mô (còn gọi là tế bào mô bào dạng biểu mô), các tế bào khổng lồ đa nhân, và các tế bào lympho T, đặc biệt là ở phía ngoại vi hoặc ở rìa của u hạt. Trái ngược với mô bệnh học thấy trong bệnh lao hoặc bệnh histoplasmosis, trung tâm của một u hạt sarcoid không cho thấy bằng chứng hoại tử rõ ràng hoặc hoại tử bã đậu. Một tình trạng viêm phế nang thường đi kèm với các u hạt trong nhu mô phổi hoặc các hạch bạch huyết trong lồng ngực. Viêm phế nang chủ yếu bao gồm các tế bào lympho T hỗ trợ , được cho là có tầm quan trọng đặc biệt trong cơ chế bệnh sinh của bệnh.
Đặc điểm bệnh học đặc trưng của sarcoidosis là u hạt không hoại tử bã đậu. Có thể xảy ra viêm phế nang bao gồm chủ yếu các tế bào đơn nhân.
Bệnh nhân sarcoidosis thường được xác định do các bất thường phát hiện trên X-quang ngực tình cờ hoặc do các triệu chứng hô hấp, chủ yếu là khó thở hoặc ho khan. Không giống như dấu hiệu ran nổ phổ biến và sớm ở bệnh nhân IPF, ran nổ thường không có trong sarcoidosis, ngay cả khi có các nghiên cứu hình ảnh bất thường đáng kể. Phổi là vị trí bị ảnh hưởng phổ biến nhất, nhưng nhiều cơ quan khác có thể bị ảnh hưởng bởi các u hạt không hoại tử bã đậu. Tổn thương mắt (ví dụ, viêm màng bồ đào trước [viêm ở tiền phòng của mắt]) và tổn thương da (ví dụ, sẩn hoặc mảng da) là các biểu hiện ngoài lồng ngực đặc biệt phổ biến của sarcoidosis, nhưng các phát hiện về tim, thần kinh, huyết học, gan, nội tiết và hạch bạch huyết ngoại vi cũng có thể được thấy.
Mặc dù các triệu chứng thường khởi phát âm thầm, một số bệnh nhân sarcoidosis có biểu hiện cấp tính hơn được gọi là hội chứng Löfgren, trong đó phát hiện X-quang ngực là hạch rốn phổi hai bên to kèm theo hồng ban nút (các nốt đỏ đau, thường ở mặt trước của cẳng chân) và khởi phát cấp tính sốt và đau khớp chi dưới. Vì những lý do chưa rõ, những bệnh nhân có hội chứng Löfgren thường có tiên lượng rất tốt, với tỷ lệ thuyên giảm tự phát lớn hơn 80%.
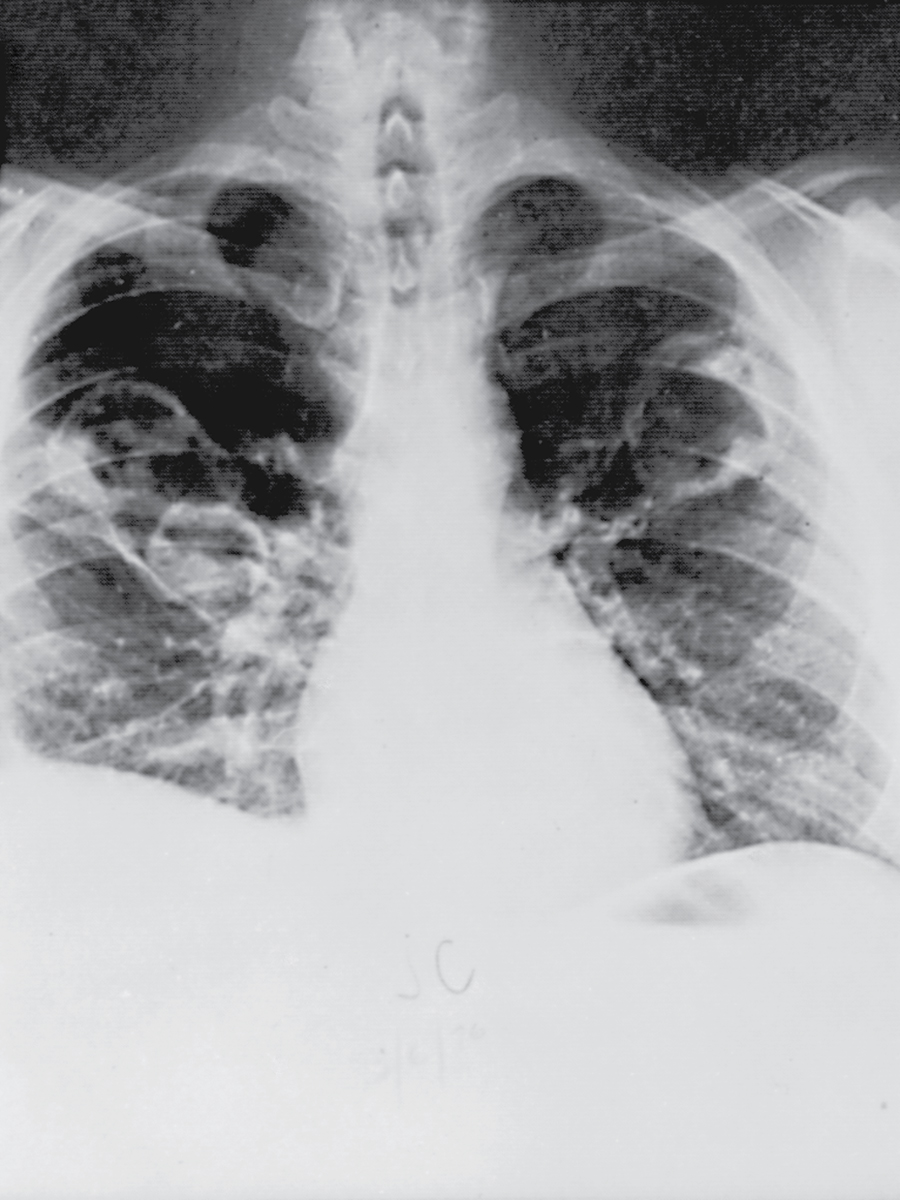
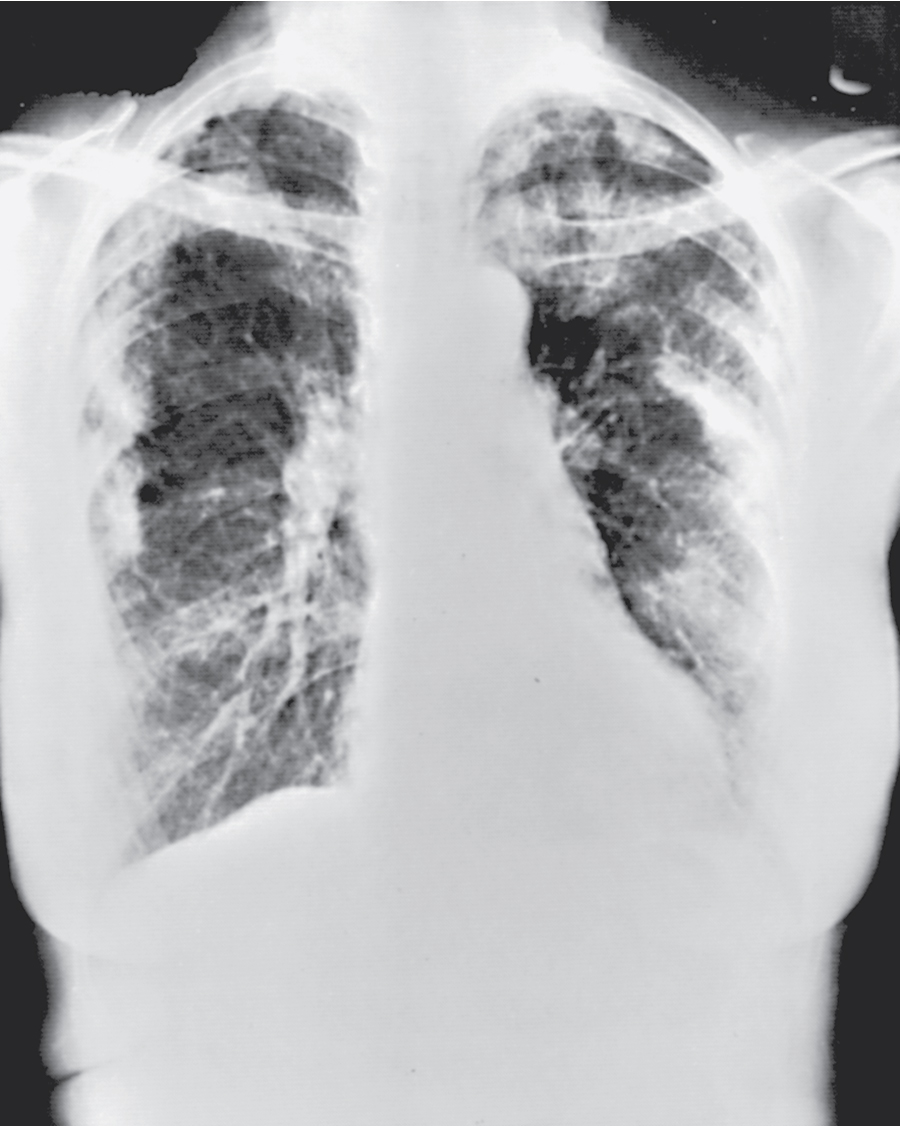
X-quang ngực trong sarcoidosis thường cho thấy một trong các dạng sau: (1) hạch bạch huyết to, phổ biến nhất là hạch rốn phổi hai bên to có hoặc không có hạch cạnh khí quản to (Hình 11.5); (2) bệnh phổi mô kẽ (dưới dạng bệnh kẽ, nốt, hoặc thâm nhiễm phế nang) (Hình 11.6); hoặc (3) cả hạch to và bệnh mô kẽ. Chụp HRCT nhạy hơn X-quang ngực thông thường trong việc phát hiện bệnh phổi mô kẽ. Nó có thể cho thấy một mô hình đặc biệt đặc trưng của các nốt nhỏ phân bố ưu tiên dọc theo các bó mạch-phế quản (Hình 11.7). Ngoài ra, HRCT thường cho thấy hạch trung thất to mà không thể nhận thấy trên X-quang ngực thông thường.
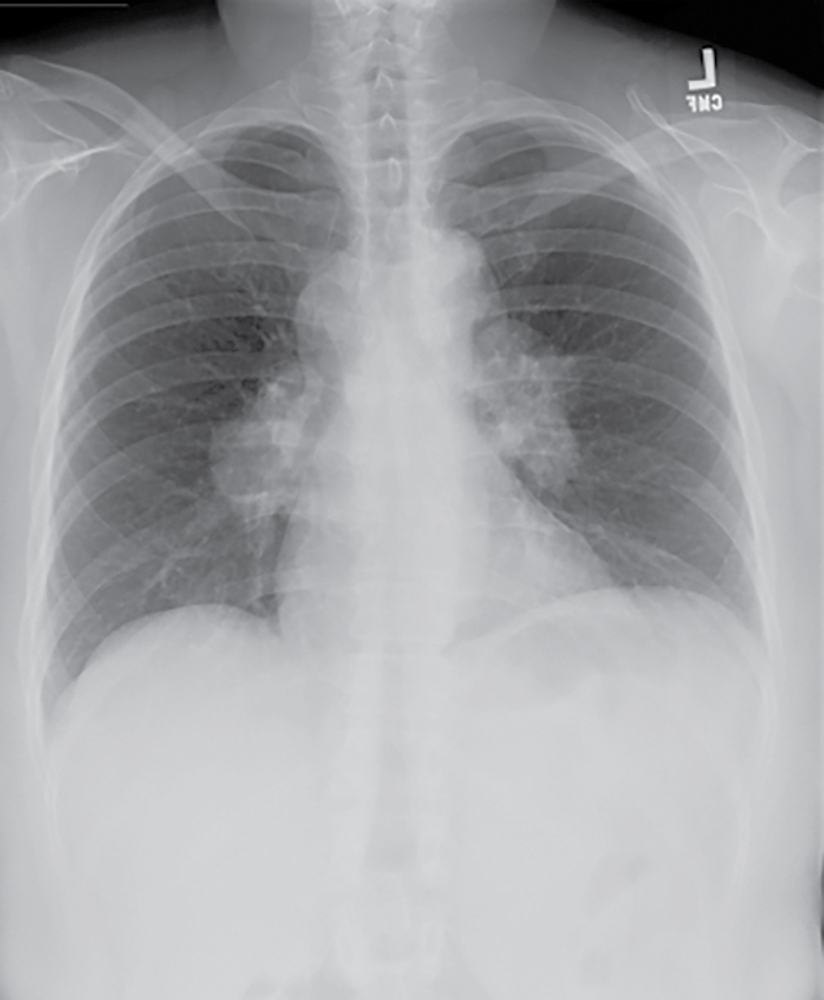
Hình 11.5 X-quang ngực thẳng sau-trước (PA) của bệnh sarcoidosis giai đoạn I cho thấy hạch rốn phổi hai bên và hạch cạnh khí quản to mà không có tổn thương nhu mô phổi rõ ràng.
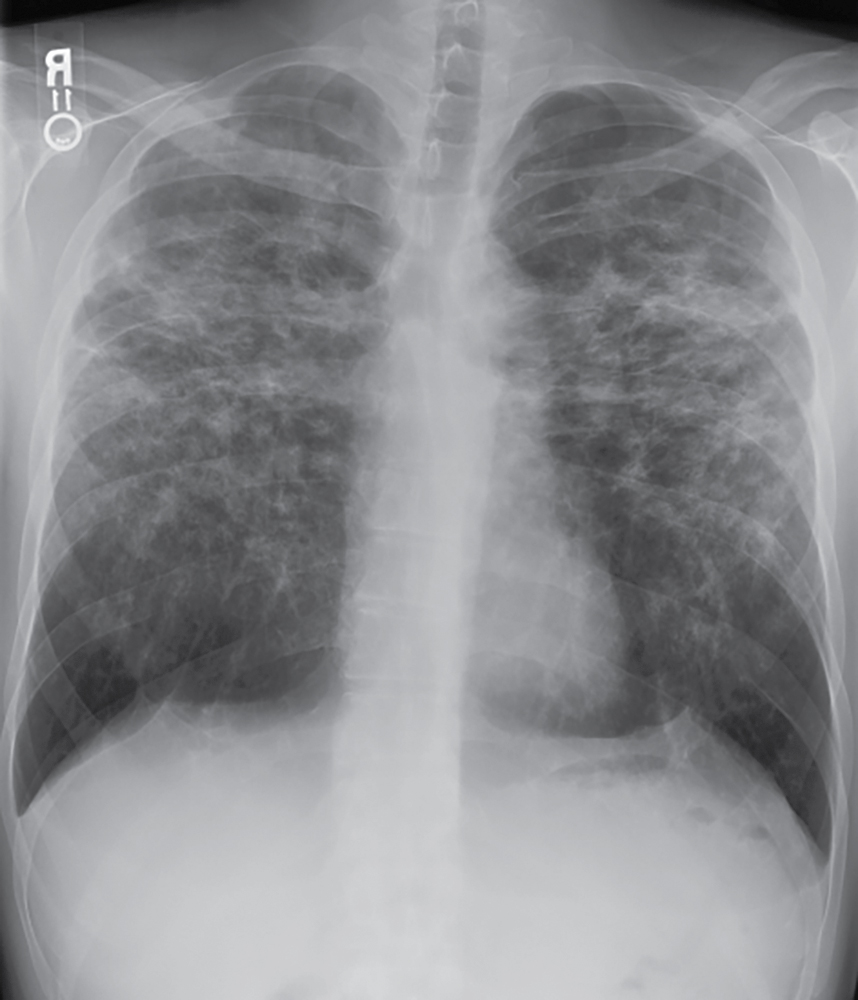
Hình 11.6 X-quang ngực thẳng sau-trước (PA) của bệnh sarcoidosis giai đoạn III. Có các đám thâm nhiễm kẽ hai bên, nổi bật nhất ở các vùng phổi trên. Không có hạch rốn phổi hoặc trung thất rõ ràng.
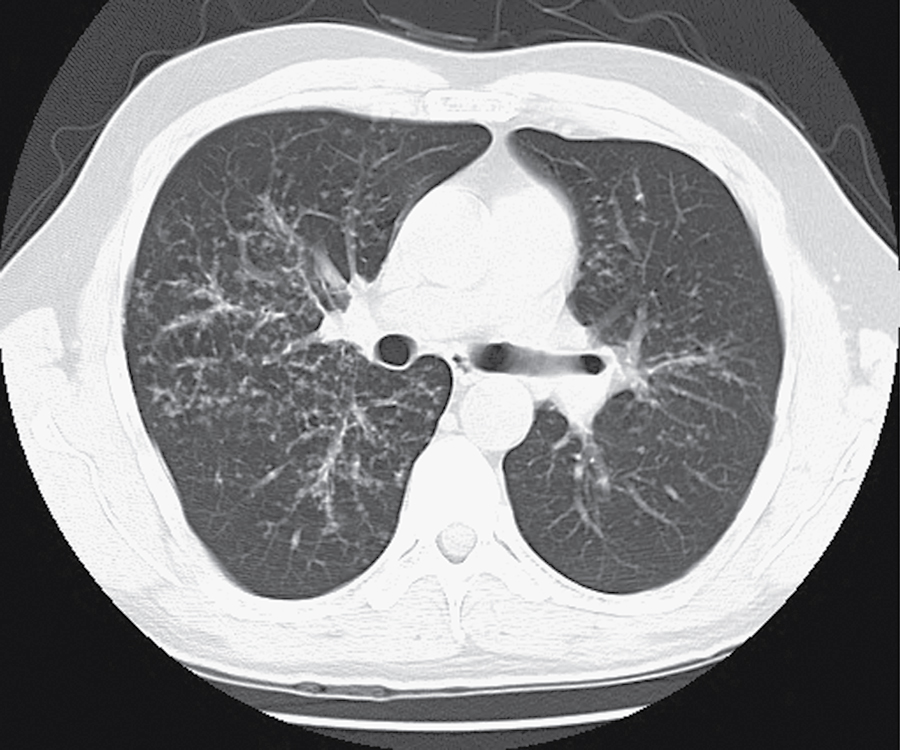
Hình 11.7 Phim chụp cắt lớp vi tính ngực cho thấy hình ảnh nốt nhỏ li ti ở một bệnh nhân bị sarcoidosis.
X-quang ngực trong sarcoidosis cho thấy các hạch rốn phổi to đối xứng, bệnh phổi kẽ, hoặc cả hai. Phim CT thường cho thấy một mô hình đặc trưng của các nốt nhỏ quanh bó mạch-phế quản kèm theo hạch rốn phổi và trung thất to.
Diễn biến của các phát hiện X-quang trong sarcoidosis khá thay đổi. Theo thời gian, cả hạch to và bệnh phổi kẽ có thể thoái lui tự phát. Ở thái cực khác, bệnh kẽ có thể tiến triển đến tình trạng sẹo lan rộng và bệnh phổi giai đoạn cuối, lúc đó bệnh nhân bị suy hô hấp nặng.
Bệnh nhân thường biểu hiện các bất thường hệ thống miễn dịch. Về mặt lâm sàng, những bệnh nhân này có thể bị mất phản ứng, không đáp ứng với các xét nghiệm da đòi hỏi phản ứng quá mẫn muộn còn nguyên vẹn. Họ cũng có thể bị tăng gammaglobulin máu, đây là bằng chứng của một hệ thống miễn dịch dịch thể hoạt động quá mức. Chuyển hóa canxi có thể bất thường trong sarcoidosis, do sự hình thành gia tăng dạng hoạt động của vitamin D (1,25-dihydroxy-D3) bởi các đại thực bào hoạt hóa trong các u hạt. Lượng vitamin D dạng hoạt động tăng lên dẫn đến tăng cường hấp thu canxi từ đường tiêu hóa, có khả năng gây ra tăng canxi niệu hoặc, ít thường xuyên hơn, tăng canxi máu.
Chẩn đoán sarcoidosis có thể được xác định bằng nhiều cách. Khi chẩn đoán lâm sàng gợi ý mạnh mẽ đến sarcoidosis, việc xác nhận bằng mô học đôi khi không cần thiết. Một ví dụ về một biểu hiện như vậy là bệnh nhân có bức tranh kinh điển của hội chứng Löfgren. Ngược lại, khi bệnh nhân có các triệu chứng hoặc phát hiện khác hoặc khi có thắc mắc về chẩn đoán, việc lấy mẫu mô thường được thực hiện để tìm kiếm các u hạt không hoại tử bã đậu và loại trừ các nguyên nhân khác. Phổi hoặc một hạch bạch huyết trong trung thất thường là nguồn mô phù hợp nhất, giả sử không có vị trí sinh thiết dễ tiếp cận như tổn thương da hoặc hạch bạch huyết ngoại vi to. Các mẫu mô phổi thường được lấy bằng sinh thiết xuyên phế quản qua nội soi phế quản ống mềm. Điều thú vị là, ngay cả khi X-quang ngực chỉ cho thấy hạch rốn phổi to mà không có bệnh phổi mô kẽ rõ ràng, thành phế nang và các đường thở nhỏ thường rải rác các u hạt có thể được nhìn thấy trên sinh thiết phổi xuyên phế quản. Với việc sử dụng ngày càng nhiều siêu âm nội soi phế quản trong quá trình nội soi, chọc hút bằng kim qua thành đường thở vào một hạch bạch huyết trung thất hoặc rốn phổi liền kề đã trở thành một lựa chọn thường xuyên để lấy vật liệu tế bào nhằm xác định tình trạng viêm u hạt. Các cách xâm lấn hơn khác để lấy mô bao gồm thực hiện sinh thiết hạch bạch huyết ở trung thất (qua nội soi trung thất) hoặc sinh thiết phổi qua nội soi lồng ngực. Các vị trí sinh thiết tiềm năng bên ngoài lồng ngực phụ thuộc vào sự hiện diện của bệnh rõ ràng ở những vị trí đó. Chúng có thể bao gồm da, hạch bạch huyết ngoại vi, kết mạc, tuyến nước bọt phụ và gan.
Trong sarcoidosis, sinh thiết phổi xuyên phế quản qua nội soi phế quản ống mềm thường cho thấy các u hạt trong nhu mô phổi, ngay cả khi X-quang ngực không cho thấy bệnh phổi kẽ.
Nồng độ men chuyển angiotensin (ACE) trong huyết thanh tăng đã được tìm thấy ở một tỷ lệ lớn bệnh nhân sarcoidosis. Enzyme này, thường được tổng hợp bởi các tế bào nội mô mạch máu, dường như được sản xuất trong các u hạt của sarcoidosis. Tuy nhiên, vì nó không đặc hiệu cho sarcoidosis và thường ở mức bình thường khi bệnh tương đối không hoạt động, nồng độ ACE không được coi là đáng tin cậy trong cả việc chẩn đoán sarcoidosis lẫn đánh giá đáp ứng điều trị của nó.
Diễn biến tự nhiên của sarcoidosis khá thay đổi. Ở một số bệnh nhân, tất cả các biểu hiện lâm sàng và X-quang đều khỏi trong vòng 1 đến 2 năm. Các bệnh nhân khác có những thay đổi X-quang dai dẳng, có hoặc không có các triệu chứng kéo dài. Nhìn chung, gần hai phần ba bệnh nhân có sự thuyên giảm tự phát. Một thiểu số bệnh nhân (10% -30%) cho thấy sự tiến triển liên tục của các bất thường X-quang, có hoặc không có thêm bệnh ngoài lồng ngực, và có thể có các triệu chứng hô hấp gây suy nhược. Các yếu tố lâm sàng liên quan đến tiên lượng xấu hơn bao gồm tuổi khởi phát lớn hơn 40 tuổi, viêm màng bồ đào mạn tính, tăng canxi máu mạn tính, một số biến thể di truyền, xơ hóa nhu mô phổi tiến triển và sự hiện diện của lupus pernio, một tổn thương da ảnh hưởng đến mặt. Ở Hoa Kỳ, nhiều nghiên cứu cho thấy chủng tộc Da đen và tình trạng thu nhập thấp là các yếu tố tiên lượng bất lợi; mức độ mà các yếu tố kinh tế xã hội giải thích cho mối liên quan với tiên lượng là một lĩnh vực được quan tâm hiện nay.
Các xét nghiệm chức năng phổi hữu ích nhất để định lượng sự suy giảm chức năng. Phế dung kế, thể tích phổi và khả năng khuếch tán đều được đo lường. Có lẽ đáng ngạc nhiên, các bất thường trên xét nghiệm chức năng phổi không nhất thiết tương quan tốt với mức độ nghiêm trọng của các phát hiện trên X-quang ngực hoặc chụp HRCT. Mặc dù kiểu hạn chế là phổ biến nhất, một số bệnh nhân phát triển tắc nghẽn đường thở đơn độc hoặc đồng thời, thường là do các u hạt liên quan đến đường thở hoặc do sự biến dạng của đường thở ở những bệnh nhân bị xơ hóa lan rộng.
Quyết định điều trị ban đầu mà bác sĩ lâm sàng phải đối mặt là có nên bắt đầu trị liệu cho bệnh nhân sarcoidosis hay không. Nhiều bệnh nhân không cần điều trị, đặc biệt khi bệnh không gây ra các triệu chứng đáng kể hoặc tổn thương chức năng cơ quan đáng kể. Việc bệnh có thể cải thiện hoặc tự khỏi cũng làm phức tạp các quyết định về việc bắt đầu điều trị. Khi điều trị được chỉ định do các triệu chứng và tổn thương mô đáng kể ảnh hưởng đến chức năng cơ quan, điều trị hàng đầu thường là corticosteroid toàn thân. Vì điều trị lâu dài bằng glucocorticoid toàn thân có liên quan đến các tác dụng phụ đáng kể, việc xem xét sớm các tác nhân tiết kiệm steroid được khuyến nghị nếu bệnh nhân cần điều trị kéo dài. Các loại thuốc không steroid thường được sử dụng bao gồm methotrexate, azathioprine, mycophenolate mofetil và leflunomide. Ở những bệnh nhân mắc bệnh kháng trị, các kháng thể đơn dòng nhắm vào TNF-α, đặc biệt là infliximab và adalimumab, có thể được sử dụng.
Diễn biến tự nhiên hay thay đổi của sarcoidosis thường làm cho các quyết định về điều trị trở nên khó khăn.
CÁC RỐI LOẠN KHÁC LIÊN QUAN ĐẾN MÔ KẼ PHỔI
Không thể trình bày ở đây một mô tả đầy đủ về tất cả các bệnh còn lại không rõ nguyên nhân ảnh hưởng đến nhu mô phổi. Thay vào đó, một mô tả ngắn gọn về một số bệnh bổ sung sẽ giúp người đọc làm quen với các đặc điểm chính của chúng. Chúng bao gồm (1) PLCH, (2) bệnh cơ trơn bạch huyết mạch (LAM), (3) hội chứng Goodpasture, (4) bệnh u hạt kèm viêm đa mạch (GPA), (5) viêm phổi tăng bạch cầu ái toan mạn tính, và (6) bệnh tích protein phế nang (PAP). Đối với mỗi rối loạn tương đối không phổ biến này, một số đặc điểm bệnh học, lâm sàng hoặc X-quang nhất định phân biệt chúng với các bệnh phổi mô kẽ lan tỏa được mô tả trước đó trong chương này. Tuy nhiên, đặc điểm xác định cho mỗi rối loạn này là một hình ảnh bệnh học tương đối đặc hiệu liên quan đến các thành phần khác nhau của nhu mô phổi.
Bệnh Mô Bào Langerhans ở Phổi
PLCH, trước đây được gọi là u hạt ái toan của phổi hoặc bệnh mô bào X ở phổi, được cho là một phần của một phổ các rối loạn liên quan đến sự xâm nhập của tế bào mô bào vào một hoặc nhiều hệ cơ quan. Mặc dù sự liên quan đa hệ thống trong bệnh mô bào Langerhans hoặc bệnh mô bào X thường thấy ở các rối loạn thời thơ ấu được gọi là bệnh Letterer-Siwe hoặc bệnh Hand-Schüller-Christian (không được thảo luận ở đây), sự liên quan đơn độc hoặc chủ yếu đến phổi trong PLCH xảy ra chủ yếu ở người trẻ đến trung niên.
Bệnh mô bào Langerhans ở phổi (trước đây được gọi là u hạt ái toan của phổi hoặc bệnh mô bào X ở phổi) được đưa vào chẩn đoán phân biệt của bệnh kẽ không giải thích được, đặc biệt ở người trẻ hoặc trung niên.
Tế bào mô bào chịu trách nhiệm là một tế bào tua có nguồn gốc từ dòng monocyte/đại thực bào được gọi là tế bào Langerhans. Gần đây, các đột biến trong con đường protein kinase hoạt hóa bởi mitogen (MAPK) và nguồn gốc đơn dòng đã được xác định trong các tế bào tua này, dẫn đến việc phân loại PLCH là một khối u tủy viêm. Một đặc điểm siêu cấu trúc thú vị của các tế bào này là sự hiện diện của các cấu trúc hình que trong bào tương được gọi là thể X (do đó có tên là bệnh mô bào X) hoặc hạt Birbeck, có thể được nhìn thấy bằng kính hiển vi điện tử. Những tế bào này cũng đáng chú ý vì nhuộm hóa mô miễn dịch dương tính với protein S-100. Kiểm tra bằng kính hiển vi quang học của phổi, ngoài việc chứng minh sự hiện diện của các tế bào mô bào này, còn cho thấy sự xâm nhập của bạch cầu ái toan, tế bào lympho, đại thực bào và tương bào. Quá trình này ban đầu liên quan đến phổi theo sự phân bố quanh tiểu phế quản và sau đó trở nên lan tỏa hơn. Bệnh xảy ra gần như độc quyền ở những người hút thuốc hiện tại và trước đây, và dường như các tế bào tua mang đột biến MAPK tích tụ trong phổi để đáp ứng với khói thuốc lá. Điều này dẫn đến sự kích hoạt và di chuyển của các tế bào miễn dịch và sự hình thành các nốt quanh tiểu phế quản và sự phá hủy mô.
Bệnh nhân thường có biểu hiện lâm sàng với khó thở, ho, hoặc cả hai. Trên X-quang ngực, PLCH thường có dạng bệnh lý nốt hoặc nốt-lưới, có xu hướng nổi bật hơn ở các vùng phổi trên. Phim HRCT cho thấy các nang nhỏ ngoài những thay đổi dạng nốt hoặc nốt-lưới (Hình 11.8). Các nang đôi khi vỡ, dẫn đến tràn khí màng phổi tự phát, có thể là biểu hiện khởi phát của bệnh. Trong một số trường hợp, sự tiến triển dẫn đến một mô hình bệnh nang lan rộng và hình ảnh tổ ong. Không giống như mô hình hạn chế điển hình trong hầu hết các bệnh phổi mô kẽ lan tỏa, xét nghiệm chức năng phổi trong PLCH có thể cho thấy các thay đổi hạn chế, tắc nghẽn, hoặc cả hai. Sự hiện diện của các nang chứa khí thường dẫn đến thể tích phổi bình thường hoặc lớn trên X-quang ngực, mặc dù có bệnh kẽ.
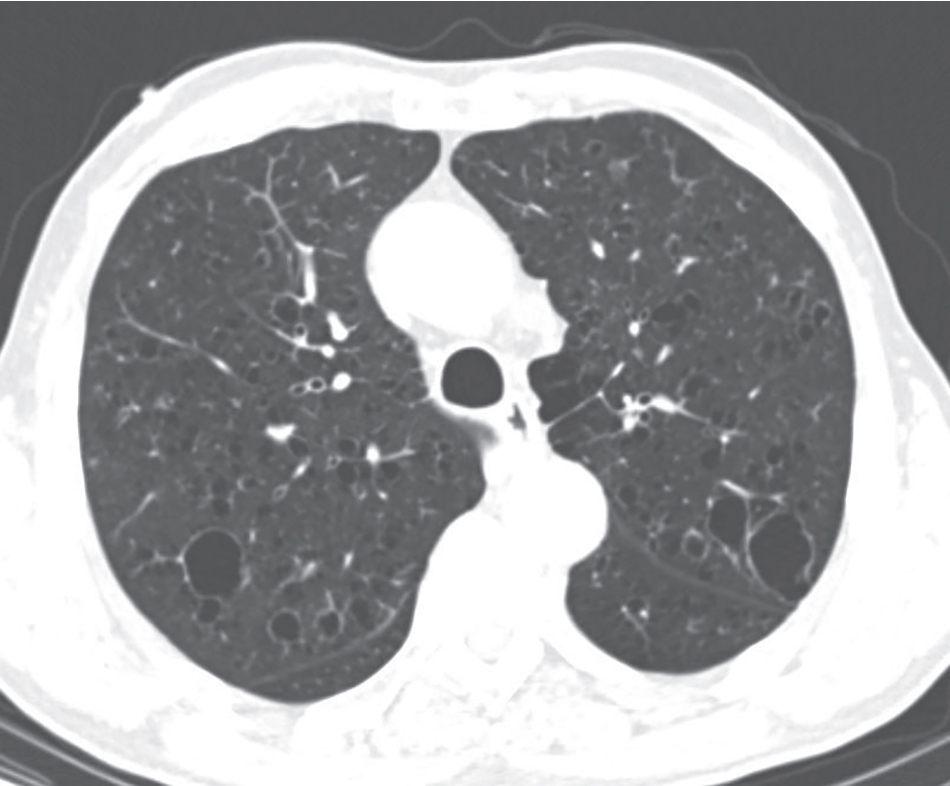
Hình 11.8 Phim chụp CT ngực cho thấy nhiều nang có kích thước khác nhau ở một bệnh nhân bị bệnh mô bào Langerhans ở phổi. (Tư liệu của Bác sĩ Seth Kligerman.)
Diễn biến tự nhiên của bệnh là thay đổi. Ở một số bệnh nhân, bệnh tự giới hạn, và các thay đổi về X-quang và chức năng có thể ổn định theo thời gian, đặc biệt là khi cai thuốc lá. Ở những bệnh nhân khác, bệnh lan rộng và suy giảm chức năng đáng kể theo sau. Không có phương pháp điều trị hiệu quả rõ ràng nào, mặc dù corticosteroid đôi khi được thử nếu chỉ cai thuốc lá không hiệu quả.
Bệnh Cơ Trơn Bạch Huyết Mạch
LAM là một bệnh phổi hiếm gặp, hiện được coi là một khối u, được đặc trưng bởi sự tăng sinh của các tế bào cơ trơn không điển hình xung quanh các mạch bạch huyết, mạch máu và đường thở, kèm theo nhiều nang nhỏ khắp nhu mô phổi. LAM xảy ra gần như độc quyền ở phụ nữ trong độ tuổi sinh đẻ. Đặc điểm nhân khẩu học này, cũng như việc các tế bào LAM biểu hiện các thụ thể cho estrogen và progesterone, cho thấy rằng các ảnh hưởng nội tiết tố đóng một vai trò trong sự phát triển của bệnh. Ngoài việc xảy ra lẻ tẻ, LAM cũng phát triển ở 30% đến 40% bệnh nhân nữ mắc bệnh di truyền phức hợp xơ cứng củ (TSC). Một khía cạnh thú vị là quá trình bệnh lý thấy trong phổi ở LAM về cơ bản giống hệt với quá trình thấy trong nhiều hệ cơ quan trong TSC, cho thấy một cơ chế bệnh sinh chung. Các đột biến dòng mầm ở hai gen, TSC1 và TSC2, có liên quan đến TSC, trong khi ở LAM, các tế bào cơ trơn bất thường có đột biến ở gen TSC2.
Bệnh cơ trơn bạch huyết mạch được đặc trưng bởi sự tăng sinh của các tế bào cơ trơn không điển hình trong phổi.
Các sản phẩm bình thường của TSC1 và TSC2 là các protein tạo thành một phức hợp hoạt động như một chất ức chế mạnh mẽ sự phát triển và tăng sinh tế bào thông qua con đường đích cơ học của rapamycin (mTOR). Do đó, các protein bất thường dẫn đến mất hoạt động ức chế này, dẫn đến sự tăng trưởng không kiểm soát. Bệnh nhân LAM dường như đã phát triển một đột biến mắc phải trong các tế bào cơ trơn ở phổi, trong khi bệnh nhân TSC dường như có một lỗi di truyền bẩm sinh. Hơn nữa, sản phẩm gen của TSC2 cũng tương tác trực tiếp với các thụ thể estrogen nội bào để gây ức chế sự phát triển tế bào. Có lẽ, một đột biến trong gen TSC2 dẫn đến mất chức năng này, giải thích cho một số ảnh hưởng nội tiết tố trong LAM. Các tế bào LAM cũng biểu hiện một yếu tố tăng trưởng mạch bạch huyết gọi là yếu tố tăng trưởng nội mô mạch máu D, và việc tìm thấy nồng độ tăng của yếu tố tăng trưởng này có thể hữu ích trong việc xác định chẩn đoán.
Các biểu hiện lâm sàng của LAM là kết quả của sự hiện diện của các nang và sự liên quan của bệnh đến các mạch bạch huyết, mạch máu và đường thở. Quá trình bệnh lý tổng thể trong nhu mô phổi có thể dẫn đến khó thở và ho. Sự liên quan đến mạch máu có thể dẫn đến ho ra máu, tắc nghẽn mạch bạch huyết có thể tạo ra tràn dịch màng phổi dưỡng chấp (màu trắng sữa), và sự liên quan đến đường thở có thể gây tắc nghẽn luồng khí. Vỡ các nang dưới màng phổi có thể dẫn đến phát triển tràn khí màng phổi tự phát. LAM cũng thường đi kèm với các khối u lành tính của thận được gọi là u cơ mỡ mạch máu.
X-quang ngực thường cho thấy một mô hình dạng lưới, và có thể thấy các thay đổi dạng nang. Chụp HRCT vượt trội hơn nhiều so với X-quang ngực thông thường trong việc chứng minh bệnh nang trên khắp nhu mô phổi (Hình 11.9). Cơ chế hình thành nang được cho là sự kết hợp của hiện tượng van một chiều do tắc nghẽn đường thở nhỏ bởi sự tăng sinh cơ trơn bất thường và sự phá hủy mô do sự sản xuất metalloproteinase bởi các tế bào LAM. Giống như PLCH, kết quả xét nghiệm chức năng phổi không điển hình cho hầu hết các bệnh nhu mô lan tỏa, vì bệnh nhân có thể biểu hiện bệnh tắc nghẽn, bệnh hạn chế, hoặc cả hai. Tương tự, thể tích phổi trên X-quang ngực có vẻ bình thường hoặc tăng thay vì giảm do bẫy khí trong các vùng nang.
Hình 11.9 Phim chụp CT ngực cho thấy vô số nang ở hai bên ở một bệnh nhân bị bệnh cơ trơn bạch huyết mạch. (Tư liệu của Bác sĩ Seth Kligerman.)
Điều trị bằng thuốc nhắm vào việc tăng cường sự ức chế tăng sinh tế bào cơ trơn qua trung gian con đường mTOR, vốn bị mất do đột biến gen TSC2. Sirolimus là một chất ức chế sự phát triển và tăng sinh tế bào thông qua cùng một con đường như các sản phẩm gen TSC2. Sirolimus (còn gọi là rapamycin) ngăn chặn tín hiệu mTOR và do đó phục hồi một số chức năng của sản phẩm gen TSC2 bất thường. Sirolimus đã được chứng minh là có hiệu quả trong việc ổn định chức năng phổi và cải thiện các triệu chứng và chất lượng cuộc sống ở bệnh nhân LAM. Trước đây, bệnh nhân thường được điều trị bằng các biện pháp điều hòa nội tiết tố để ngăn chặn tác dụng của estrogen đối với sự tăng trưởng cơ trơn bất thường; tuy nhiên, điều trị kháng estrogen thường quy không còn được khuyến cáo.
Hội Chứng Goodpasture
Hội chứng Goodpasture là một bệnh đã trở nên nổi tiếng không phải vì tỷ lệ mắc bệnh, vốn cực kỳ thấp, mà vì các đặc điểm bệnh sinh và miễn dịch thú vị của nó. Hai hệ cơ quan có liên quan trong hội chứng này: phổi và thận. Ở phổi, bệnh nhân có các đợt xuất huyết phổi, và xơ phổi có thể phát triển, có lẽ là hậu quả của các đợt chảy máu tái phát. Ở thận, bệnh nhân có viêm cầu thận được đặc trưng bởi sự lắng đọng kháng thể dạng đường thẳng dọc theo màng đáy cầu thận (GBM). Các nghiên cứu trên máu ngoại vi đã chứng minh rằng bệnh nhân có các kháng thể lưu hành chống lại một thành phần của collagen type IV trong chính GBM của họ, thường được viết tắt là kháng thể kháng GBM. Những kháng thể này phản ứng chéo với các kháng nguyên trong màng đáy của thành phế nang, gây ra tổn thương chịu trách nhiệm cho các biểu hiện lâm sàng của bệnh ở cả hai hệ cơ quan.
Trong hội chứng Goodpasture, các tự kháng thể nhắm vào màng đáy cầu thận có thể phản ứng chéo với màng đáy của thành phế nang.
Tại sao những tự kháng thể thực sự này phát triển ở bệnh nhân mắc hội chứng Goodpasture vẫn chưa rõ. Ở một số bệnh nhân, bệnh khởi phát dường như sau khi nhiễm cúm hoặc phơi nhiễm với một hydrocarbon độc hại. Có lẽ, tổn thương màng đáy và giải phóng các yếu tố quyết định kháng nguyên chưa từng tiếp xúc trước đây có liên quan, hoặc sự hình thành ngẫu nhiên của kháng thể (chống lại một kháng nguyên không liên quan) có thể phản ứng chéo với màng đáy phế nang và cầu thận. Bệnh có liên quan đến một số kháng nguyên bạch cầu người nhất định, cho thấy một sự nhạy cảm di truyền tiềm ẩn. Không giống như nhiều bệnh liên quan đến tự kháng thể, kháng thể kháng GBM rõ ràng có vai trò gây bệnh. Liệu pháp cho hội chứng Goodpasture dựa trên việc giảm gánh nặng kháng thể kháng GBM được trình diện cho phổi và thận. Lọc huyết tương có khả năng loại bỏ trực tiếp các kháng thể kháng GBM khỏi tuần hoàn. Liệu pháp ức chế miễn dịch (ví dụ, glucocorticoid cộng với cyclophosphamide), nhằm giảm sự hình thành kháng thể kháng GBM, thường được dùng kết hợp với lọc huyết tương.
Bệnh U Hạt Kèm Viêm Đa Mạch
Một nhóm các rối loạn được gọi là viêm mạch u hạt có thể ảnh hưởng đến thành phế nang như một phần của một bệnh tổng quát hơn. Bệnh được biết đến nhiều nhất trong số các rối loạn này là GPA (trước đây gọi là bệnh u hạt Wegener), một bệnh đặc trưng chủ yếu nhưng không độc quyền bởi sự liên quan của đường hô hấp trên, phổi và thận. Quá trình bệnh lý ở phổi và đường hô hấp trên bao gồm viêm mạch u hạt hoại tử các mạch máu nhỏ, trong khi viêm cầu thận khu trú có mặt ở thận. Trên X-quang ngực, bệnh nhân thường có một hoặc nhiều nốt (thường lớn) hoặc các đám thâm nhiễm, thường có kèm theo hang hóa của (các) tổn thương (Hình 11.10). Xuất huyết phổi là một biểu hiện tiềm tàng khác của sự liên quan đến đường hô hấp. Không giống như hầu hết các rối loạn khác của nhu mô phổi được thảo luận trong Chương 10 và chương này, bệnh phổi mô kẽ lan tỏa không phổ biến trong thực thể này.
Bệnh u hạt kèm viêm đa mạch được đặc trưng về mặt bệnh học bởi viêm mạch u hạt của phổi và đường hô hấp trên và bởi viêm cầu thận. Hệ quả lâm sàng là bệnh phổi, đường hô hấp trên và bệnh thận.
Bệnh nhân mắc GPA thường có các kháng thể trong huyết thanh nhắm vào proteinase 3, một serine protease có trong các hạt azurophil được tìm thấy trong bào tương của bạch cầu trung tính. Những kháng thể này có thể được phát hiện bằng các kỹ thuật huỳnh quang miễn dịch, cho thấy một mô hình nhuộm bào tương thô, lan tỏa khi huyết thanh của bệnh nhân được ủ với các bạch cầu trung tính bình thường. Sự hiện diện của kháng thể kháng bào tương bạch cầu trung tính (ANCA), đặc biệt là với mô hình nhuộm bào tương (c-ANCA), là một thành phần quan trọng trong đánh giá chẩn đoán GPA, mặc dù một số bệnh nhân có thể có kết quả âm tính, đặc biệt là với bệnh giới hạn. Nồng độ kháng thể tương quan với hoạt động của bệnh, và những kháng thể này có khả năng đóng một vai trò nào đó trong cơ chế bệnh sinh của bệnh. Tuy nhiên, các yếu tố khác có lẽ cũng có liên quan.
Mặc dù GPA từng được coi là một bệnh hung hãn và gây tử vong, tiên lượng của nó đã cải thiện đáng kể kể từ khi các tác nhân độc tế bào, đặc biệt là cyclophosphamide, được sử dụng trong điều trị. Prednisone cũng thường được thêm vào trong giai đoạn điều trị ban đầu. Gần đây hơn, rituximab, một kháng thể đơn dòng nhắm vào kháng nguyên CD20 được tìm thấy chủ yếu trên các tế bào lympho B, đã đóng một vai trò quan trọng trong việc điều trị bệnh này. Mặc dù thời gian sống trung bình nếu không điều trị là 5 tháng, bệnh nhân thường đạt được sự thuyên giảm hoàn toàn và lâu dài khi bắt đầu điều trị thích hợp. Hầu hết bệnh nhân cần điều trị duy trì, và rituximab được sử dụng phổ biến nhất do có hồ sơ tác dụng phụ tốt hơn so với cyclophosphamide.
Viêm Phổi Tăng Bạch Cầu Ái Toan Mạn Tính
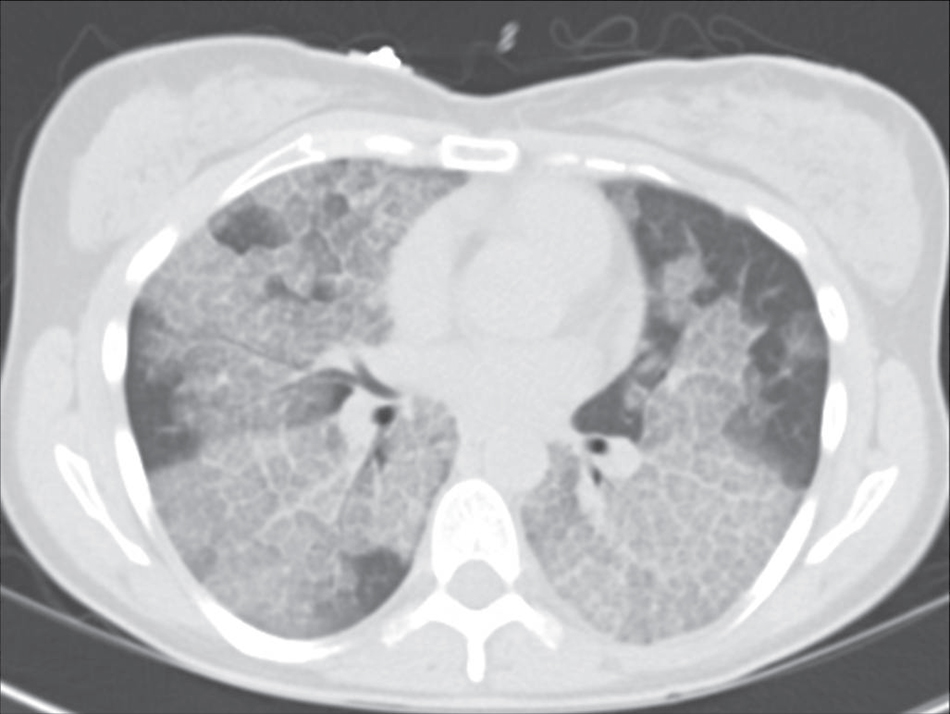
Viêm phổi tăng bạch cầu ái toan mạn tính là một rối loạn trong đó mô kẽ phổi và các khoang phế nang bị xâm nhập chủ yếu bởi bạch cầu ái toan và, ở mức độ thấp hơn, bởi các đại thực bào. Biểu hiện lâm sàng thường xảy ra trong vài tuần đến vài tháng, với các triệu chứng toàn thân như sốt và sụt cân đi kèm với khó thở và ho khan. Các manh mối gợi ý chẩn đoán này thường được tìm thấy trên X-quang ngực và công thức bạch cầu thông thường. X-quang thường cho thấy các đám thâm nhiễm phổi có sự phân bố ở ngoại vi và một mô hình gợi ý lấp đầy phế nang hơn là bệnh kẽ (Hình 11.11). Bởi vì mô hình X-quang điển hình của phù phổi do suy tim sung huyết có các đám thâm nhiễm trung tâm phổi với sự chừa lại vùng ngoại vi phổi, mô hình ngoại vi nổi bật thường thấy trong viêm phổi tăng bạch cầu ái toan mạn tính đã được mô tả là “phim âm bản của phù phổi.” Phần lớn bệnh nhân cũng có số lượng bạch cầu ái toan tăng trong máu ngoại vi, mặc dù phát hiện này không phải lúc nào cũng có mặt và do đó không quan trọng cho chẩn đoán. Rửa phế quản phế nang thường cho thấy một tỷ lệ cao bạch cầu ái toan, phản ánh quá trình bệnh lý trong nhu mô phổi.
Hình 11.10 X-quang ngực cho thấy nhiều nốt phổi có hang ở một bệnh nhân bị bệnh u hạt kèm viêm đa mạch.
Hình 11.11 X-quang ngực cho thấy mô hình thâm nhiễm phổi ngoại vi đặc trưng của viêm phổi tăng bạch cầu ái toan mạn tính.
Viêm phổi tăng bạch cầu ái toan mạn tính thường được gợi ý trên X-quang ngực bởi một mô hình thâm nhiễm phổi ngoại vi.
Điều trị rất khả quan cho cả bệnh nhân và bác sĩ vì viêm phổi tăng bạch cầu ái toan mạn tính đặc trưng bởi đáp ứng ngoạn mục với liệu pháp corticosteroid. Cải thiện lâm sàng và giải quyết trên X-quang thường xảy ra trong vài ngày đến vài tuần, mặc dù liệu pháp thường phải được kéo dài trong nhiều tháng để ngăn ngừa tái phát.
Bệnh Tích Protein Phế Nang
PAP là một bệnh nhu mô phổi trong đó quá trình bệnh lý chính ảnh hưởng đến các khoang phế nang, không phải thành phế nang. Các khoang phế nang chứa đầy một vật liệu phospholipid giàu protein đại diện cho các thành phần của surfactant phổi. Sự tích tụ các thành phần surfactant là do giảm thoái biến hoặc rối loạn chức năng surfactant. PAP được phân loại là tự miễn, thứ phát (thường liên quan đến các bệnh ác tính huyết học), hoặc bẩm sinh/di truyền. PAP tự miễn (trước đây được gọi là PAP nguyên phát hoặc vô căn) là loại phổ biến nhất trong ba loại và được thảo luận ở đây.
Trong PAP tự miễn, cơ chế cơ bản là sản xuất tự kháng thể kháng yếu tố kích thích cụm bạch cầu hạt-đại thực bào (GM-CSF). GM-CSF, hoạt động thông qua các yếu tố phiên mã đặc hiệu của đại thực bào phế nang, ảnh hưởng đến một số chức năng thiết yếu của đại thực bào, bao gồm điều hòa sự thoái biến surfactant, chuyển hóa lipid nội bào và thực bào. Do đó, việc ức chế hoạt động của GM-CSF thông qua tự kháng thể dẫn đến chức năng đại thực bào bất thường và giảm thanh thải surfactant khỏi các khoang phế nang. Cơ chế bệnh đã được phát hiện một cách tình cờ khi người ta ghi nhận rằng những con chuột bị loại bỏ gen GM-CSF (trong đó cả hai alen cho GM-CSF đều bị vô hiệu hóa) luôn phát triển một quá trình bệnh lý ở phổi với bệnh học về cơ bản giống hệt như thấy ở PAP ở người.
Sự hấp thu surfactant bị khiếm khuyết bởi các đại thực bào phế nang, do lượng hoặc tác dụng của GM-CSF giảm, là cơ sở của cơ chế bệnh sinh của bệnh tích protein phế nang.
Bệnh nhân bị bệnh tích protein phế nang chủ yếu có biểu hiện khó thở và ho. X-quang ngực đáng chú ý với các đám thâm nhiễm phế nang hai bên. HRCT thường cho thấy một hình ảnh đặc biệt nhưng không hoàn toàn đặc trưng được gọi là mô hình “lát đá không đều” (crazy paving) (được tạo ra bởi sự dày lên của các vách liên tiểu thùy kèm theo sự lấp đầy phế nang dạng kính mờ) gợi ý chẩn đoán (Hình 11.12). Bệnh nhân dễ bị một số loại nhiễm trùng hô hấp chồng chất không phổ biến ở vật chủ bình thường, đặc biệt là với sinh vật Nocardia. Sự nhạy cảm với các mầm bệnh bất thường dường như là do chức năng đại thực bào bất thường cũng như các bất thường trong chức năng bạch cầu trung tính cũng do GM-CSF trung gian.
Hình 11.12 Phim chụp CT ngực ở một bệnh nhân bị bệnh tích protein phế nang cho thấy mô hình “lát đá không đều” đặc trưng đại diện cho các vách liên tiểu thùy dày lên chồng lên trên đám mờ dạng kính.
Đối với những bệnh nhân mắc bệnh từ trung bình đến nặng, phương pháp điều trị chính của PAP là rửa toàn bộ phổi, bao gồm việc rửa sạch vật liệu lấp đầy các khoang phế nang trong khi bệnh nhân đang được gây mê toàn thân. Việc sử dụng GM-CSF tái tổ hợp dạng hít hoặc tiêm dưới da có thể được sử dụng như một liệu pháp thay thế nhưng vẫn đang được nghiên cứu. Tiên lượng của bệnh tương đối tốt, mặc dù bệnh nhân có thể cần các phương pháp điều trị bổ sung bằng cách rửa toàn bộ phổi. Các tác dụng lâu dài của GM-CSF ngoại sinh trong bệnh này vẫn chưa được biết.
TÀI LIỆU THAM KHẢO ĐỀ XUẤT
- Albert R.K., Schwartz D.A.: Revealing the secrets of idiopathic pulmonary fibrosis. New England Journal of Medicine 2019; 380: pp. 94-96.
- Collard H.R., Ryerson C.J., Corte T.J., Jenkins G., Kondoh Y., Lederer D.J., et. al.: Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. American Journal of Respiratory and Critical Care Medicine 2016; 194: pp. 265-275.
- Flaherty K.R., Wells A.U., Cottin V., Devaraj A., Walsh S.L.F., Inoue Y., et. al.: Nintedanib in progressive fibrosing interstitial lung diseases. New England Journal of Medicine 2019; 381: pp. 1718-1727.
- Katzen J., Beers M.F.: Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. Journal of Clinical Investigation 2020; 130: pp. 5088-5099.
- King T.E.Jr., Bradford W.Z., Castro-Bernardini S., Fagan E.A., Glaspole I., Glassberg M.K., et. al.: A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. New England Journal of Medicine 2014; 370: pp. 2083-2092.
- Lederer D.J., Martinez F.J.: Idiopathic pulmonary fibrosis. New England Journal of Medicine 2018; 378: pp. 1811-1823.
- Rackow A.R., Nagel D.J., McCarthy C., Judge J., Lacy S., Freeberg M.A.T., et. al.: The self-fulfilling prophecy of pulmonary fibrosis: A selective inspection of pathological signaling loops. European Respiratory Journal 2020; 56: pp. 2000075.
- Raghu G., Remy-Jardin M., Myers J.L., Richeldi L., Ryerson C.J., Lederer D.J., et. al.: Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. American Journal of Respiratory and Critical Care Medicine 2018; 198: pp. e44-e68.
- Thomson C.C., Duggal A., Bice T., Lederer D.J., Wilson K.C., Raghu G.: 2018 Clinical practice guideline summary for clinicians: Diagnosis of idiopathic pulmonary fibrosis. Annals of the American Thoracic Society 2019; 16: pp. 285-290.
- Whitsett J.A., Kalin T.V., Xu Y., Kalinichenko V.V.: Building and regenerating the lung cell by cell. Physiological Reviews 2019; 99: pp. 513-554.
- Wijsenbeek M., Cottin V.: Spectrum of fibrotic lung diseases. New England Journal of Medicine 2020; 383: pp. 958-968.
- American Thoracic Society, European Respiratory Society.: American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. American Journal of Respiratory and Critical Care Medicine 2002; 165: pp. 277-304.
- King T.E.,Jr., Lee J.S.: Cryptogenic organizing pneumonia. New England Journal of Medicine 2022; 386: pp. 1058-1069.
- Kumar A., Cherian S.V., Vassallo R., Yi E.S., Ryu J.H.: Current concepts in pathogenesis, diagnosis, and management of smoking-related interstitial lung diseases. Chest 2018; 154: pp. 394-408.
- Raghu G., Meyer K.C.: Cryptogenic organising pneumonia: Current understanding of an enigmatic lung disease. European Respiratory Review 2021; 30: pp. 210094.
- Travis W.D., Costabel U., Hansell D.M., King T.E.Jr., Lynch D.A., Nicholson A.G., et. al.: An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. American Journal of Respiratory and Critical Care Medicine 2013; 188: pp. 733-748.
- Doyle T.J., Dellaripa P.F.: Lung manifestations in the rheumatic diseases. Chest 2017; 152: pp. 1283-1295.
- Fischer A., du Bois R.: Interstitial lung disease in connective tissue disorders. Lancet 2012; 380: pp. 689-698.
- Hannah J.R., D’Cruz D.P.: Pulmonary complications of systemic lupus erythematosus. Seminars in Respiratory and Critical Care Medicine 2019; 40: pp. 227-234.
- Kadura S., Raghu G.: Rheumatoid arthritis-interstitial lung disease: Manifestations and current concepts in pathogenesis and management. European Respiratory Review 2021; 30: pp. 210011.
- Khanna D., Tashkin D.P., Denton C.P., Renzoni E.A., Desai S.R., Varga J.: Etiology, risk factors, and biomarkers in systemic sclerosis with interstitial lung disease. American Journal of Respiratory and Critical Care Medicine 2020; 201: pp. 650-660.
- Lee A.S., Scofield R.H., Hammitt K.M., Gupta N., Thomas D.E., Moua T., et. al.: Consensus guidelines for evaluation and management of pulmonary disease in Sjögren’s. Chest 2021; 159: pp. 683-698.
- Morisset J., Johnson C., Rich E., Collard H.R., Lee J.S.: Management of myositis-related interstitial lung disease. Chest 2016; 150: pp. 1118-1128.
- Perelas A., Silver R.M., Arrossi A.V., Highland K.B.: Systemic sclerosis-associated interstitial lung disease. Lancet Respiratory Medicine 2020; 8: pp. 304-320.
- Tsuchiya Y., Fischer A., Solomon J.J., Lynch D.A.: Connective tissue disease-related thoracic disease. Clinics in Chest Medicine 2015; 36: pp. 283-297.
- Baughman R.P., Valeyre D., Korsten P., Mathioudakis A.G., Wuyts W.A., Wells A., et. al.: ERS clinical practice guidelines on treatment of sarcoidosis. European Respiratory Journal 2021; 58: pp. 2004079.
- Belperio J.A., Shaikh F., Abtin F.G., Fishbein M.C., Weigt S.S., Saggar R., et. al.: Diagnosis and treatment of pulmonary sarcoidosis. A review. JAMA 2022; 327: pp. 856-867.
- Celada L.J., Hawkins C., Drake W.P.: The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clinics in Chest Medicine 2015; 36: pp. 561-568.
- Crouser E.D., Maier L.A., Wilson K.C., Bonham C.A., Morgenthau A.S., Patterson K.C., et. al.: Diagnosis and detection of sarcoidosis. An official American Thoracic Society clinical practice guideline. American Journal of Respiratory and Critical Care Medicine 2020; 201: pp. e26-e51.
- Drent M., Crouser E.D., Grunewald J.: Challenges of sarcoidosis and its management. New England Journal of Medicine 2021; 385: pp. 1018-1032.
- Fingerlin T.E., Hamzeh N., Maier L.A.: Genetics of sarcoidosis. Clinics in Chest Medicine 2015; 36: pp. 569-584.
- Patterson K.C., Chen E.S.: The pathogenesis of pulmonary sarcoidosis and implications for treatment. Chest 2018; 153: pp. 1432-1442.
- Rahaghi F.F., Baughman R.P., Saketkoo L.A., Sweiss N.J., Barney J.B., Birring S.S., et. al.: Delphi consensus recommendations for a treatment algorithm in pulmonary sarcoidosis. European Respiratory Review 2020; 29: pp. 190146.
- Singha A., Liao S.Y., Herman D.D., Crouser E.D., Maier L.A., Baughman R.P., et. al.: Summary for clinicians: Clinical practice guideline for the diagnosis and detection of sarcoidosis. Annals of the American Thoracic Society 2020; 17: pp. 1510-1515.
- Thillai M., Atkins C.P., Crawshaw A., Hart S.P., Ho L.P., Kouranos V., et. al.: BTS clinical statement on pulmonary sarcoidosis. Thorax 2021; 76: pp. 4-20.
- Zissel G., Müller-Quernheim J.: Cellular players in the immunopathogenesis of sarcoidosis. Clinics in Chest Medicine 2015; 36: pp. 549-560.
- Allen C.E., Merad M., McClain K.L.: Langerhans-cell histiocytosis. New England Journal of Medicine 2018; 379: pp. 856-868.
- Bernheim A., McLoud T.: A review of clinical and imaging findings in eosinophilic lung diseases. AJR 2017; 208: pp. 1002-1010.
- Cottin V.: Eosinophilic lung diseases. Clinics in Chest Medicine 2016; 37: pp. 535-556.
- Feemster L.C., Lyons P.G., Chatterjee R.S., Kidambi P., McCormack F.X., Moss J., et. al.: Summary for clinicians: Lymphangioleiomyomatosis diagnosis and management clinical practice guideline. Annals of the American Thoracic Society 2017; 14: pp. 1073-1075.
- Gupta N., Finlay G.A., Kotloff R.M., Strange C., Wilson K.C., Young L.R., et. al.: Lymphangioleiomyomatosis diagnosis and management: High-resolution chest computed tomography, transbronchial lung biopsy, and pleural disease management. An official American Thoracic Society/Japanese Respiratory Society clinical practice guideline. American Journal of Respiratory and Critical Care Medicine 2017; 196: pp. 1337-1348.
- Kadura S., Raghu G.: Antineutrophil cytoplasmic antibody-associated interstitial lung disease: A review. European Respiratory Review 2021; 30: pp. 210123.
- Kumar A., Abdelmalak B., Inoue Y., Culver D.A.: Pulmonary alveolar proteinosis in adults: Pathophysiology and clinical approach. Lancet Respiratory Medicine 2018; 6: pp. 554-565.
- McCarthy C., Carey B.C., Trapnell B.C.: Autoimmune pulmonary alveolar proteinosis. American Journal of Respiratory and Critical Care Medicine 2022; 205: pp. 1016-1035.
- McCarthy C., Gupta N., Johnson S.R., Yu J.J., McCormack F.X.: Lymphangioleiomyomatosis: Pathogenesis, clinical features, diagnosis, and management. Lancet Respiratory Medicine 2021; 9: pp. 1313-1327.
- McCormack F.X., Gupta N., Finlay G.R., Young L.R., Taveira-DaSilva A.M., Glasgow C.G., et. al.: Official American Thoracic Society/Japanese Respiratory Society clinical practice guidelines: Lymphangioleiomyomatosis diagnosis and management. American Journal of Respiratory and Critical Care Medicine 2016; 194: pp. 748-761.
- Radzikowska E.: Update on pulmonary Langerhans cell histiocytosis. Frontiers in Medicine 2021; 7: pp. 582581.
- Shaw B., Borchers M., Zander D., Gupta N.: Pulmonary Langerhans cell histiocytosis. Seminars in Respiratory and Critical Care Medicine 2020; 41: pp. 269-279.
- Shin J.I., Geetha D., Szpirt W.M., Windpessl M., Kronbichler A.: Anti-glomerular basement membrane disease (Goodpasture disease): From pathogenesis to plasma exchange to IdeS. Therapeutic Apheresis and Dialysis 2022; 26: pp. 24-31.
- Trapnell B.C., Inoue Y., Bonella F., Morgan C., Jouneau S., Bendstrup E., et. al.: Inhaled molgramostim therapy in autoimmune pulmonary alveolar proteinosis. New England Journal of Medicine 2020; 383: pp. 1635-1644.
- Walsh M., Merkel P.A., Peh C.A., Szpirt W.M., Puéchal X., Fujimoto S., et. al.: Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. New England Journal of Medicine 2020; 382: pp. 622-631.
BẢNG CHÚ GIẢI THUẬT NGỮ Y HỌC ANH-VIỆT
Chương 11: Các bệnh phổi kẽ lan tỏa nguyên nhân chưa rõ căn nguyên
| STT | Thuật ngữ tiếng Anh | Phát âm | Nghĩa tiếng Việt |
|---|---|---|---|
| 1 | Diffuse parenchymal lung disease | /dɪˈfjuːs pəˈreŋkɪməl lʌŋ dɪˈziːz/ | Bệnh nhu mô phổi lan tỏa |
| 2 | Idiopathic pulmonary fibrosis (IPF) | /ˌɪdiəˈpæθɪk ˈpʌlmənəri faɪˈbroʊsɪs/ | Bệnh xơ phổi vô căn |
| 3 | Systemic rheumatic disease | /sɪˈstemɪk ruˈmætɪk dɪˈziːz/ | Bệnh thấp khớp hệ thống |
| 4 | Sarcoidosis | /ˌsɑːrkɔɪˈdoʊsɪs/ | Bệnh sarcoidosis |
| 5 | Pulmonary Langerhans cell histiocytosis (PLCH) | /ˈpʌlmənəri ˈlæŋərhænz sel ˌhɪstioʊsaɪˈtoʊsɪs/ | Bệnh tế bào Langerhans phổi |
| 6 | Cryptogenic fibrosing alveolitis | /krɪpˈtoʊdʒenɪk ˈfaɪbroʊsɪŋ ˌælvioʊˈlaɪtɪs/ | Viêm phế nang xơ hóa không rõ nguyên nhân |
| 7 | Usual interstitial pneumonia (UIP) | /ˈjuːʒuəl ˌɪntərˈstɪʃəl nʊˈmoʊniə/ | Viêm phổi kẽ thể thông thường |
| 8 | Pathogenesis | /ˌpæθoʊˈdʒenəsɪs/ | Cơ chế bệnh sinh |
| 9 | Alveolar epithelial cells | /ælˈviːələr ˌepɪˈθiːliəl selz/ | Tế bào biểu mô phế nang |
| 10 | Microinjuries | /ˈmaɪkroʊˌɪndʒəriz/ | Vi tổn thương |
| 11 | Wound healing | /wuːnd ˈhiːlɪŋ/ | Liền vết thương |
| 12 | Fibrosis | /faɪˈbroʊsɪs/ | Xơ hóa |
| 13 | Type I pneumocytes | /taɪp wʌn ˈnuːməsaɪts/ | Tế bào biểu mô phế nang loại I |
| 14 | Type II pneumocytes | /taɪp tuː ˈnuːməsaɪts/ | Tế bào biểu mô phế nang loại II |
| 15 | Basement membrane | /ˈbeɪsmənt ˈmembreɪn/ | Màng đáy |
| 16 | Re-epithelialization | /riː ˌepɪˌθiːliəlaɪˈzeɪʃən/ | Tái tạo biểu mô |
| 17 | Autophagy | /ɔːˈtɒfədʒi/ | Tự thực bào |
| 18 | Apoptosis | /ˌæpəpˈtoʊsɪs/ | Chết tế bào có chương trình |
| 19 | Progenitor cells | /proʊˈdʒenɪtər selz/ | Tế bào tiền thân |
| 20 | Profibrotic cytokines | /proʊfaɪˈbrɒtɪk ˈsaɪtoʊkaɪnz/ | Cytokin gây xơ hóa |
| 21 | Platelet-derived growth factor (PDGF) | /ˈpleɪtlət dɪˈraɪvd ɡroʊθ ˈfæktər/ | Yếu tố tăng trưởng nguồn gốc tiểu cầu |
| 22 | Transforming growth factor-β1 (TGF-β1) | /trænsˈfɔːrmɪŋ ɡroʊθ ˈfæktər/ | Yếu tố tăng trưởng biến đổi β1 |
| 23 | Fibroblast migration | /ˈfaɪbroʊblæst maɪˈɡreɪʃən/ | Di chuyển nguyên bào sợi |
| 24 | Fibroblastic foci | /ˌfaɪbroʊˈblæstɪk ˈfoʊsaɪ/ | Ổ nguyên bào sợi |
| 25 | Extracellular matrix | /ˌekstrəˈseljələr ˈmeɪtrɪks/ | Ma trận ngoại bào |
| 26 | Gene mutations | /dʒiːn mjuːˈteɪʃənz/ | Đột biến gen |
| 27 | Surfactant proteins | /ˈsɜːrfəktənt ˈproʊtiːnz/ | Protein chất hoạt động bề mặt |
| 28 | Endoplasmic reticulum stress | /ˌendoʊˈplæzmɪk rɪˈtɪkjələm stres/ | Stress lưới nội chất |
| 29 | Telomerase reverse transcriptase (TERT) | /ˈteloʊməreɪs rɪˈvɜːrs trænˈskrɪpteɪs/ | Enzym telomerase phiên mã ngược |
| 30 | Telomerase RNA component (TERC) | /ˈteloʊməreɪs ˌɑːrˌenˈeɪ kəmˈpoʊnənt/ | Thành phần RNA telomerase |
| 31 | Telomeres | /ˈteloʊmɪrz/ | Telomere (đầu mút nhiễm sắc thể) |
| 32 | MUC5B gene | /mjuːk faɪv biː dʒiːn/ | Gen MUC5B |
| 33 | Mucin production | /ˈmjuːsɪn prəˈdʌkʃən/ | Sản xuất mucin |
| 34 | Peripheral airways | /pəˈrɪfərəl ˈerweɪz/ | Đường thở ngoại vi |
| 35 | Dyspnea | /dɪspˈniːə/ | Khó thở |
| 36 | Inspiratory crackles | /ɪnˈspaɪrətɔːri ˈkrækəlz/ | Ran ẩm khô khi hít vào |
| 37 | Digital clubbing | /ˈdɪdʒɪtəl ˈklʌbɪŋ/ | Vồ ngón tay chân |
| 38 | Reticular pattern | /rɪˈtɪkjələr ˈpætərn/ | Hình ảnh dạng lưới |
| 39 | Bilateral | /ˌbaɪˈlætərəl/ | Hai bên |
| 40 | Subpleural regions | /sʌbˈplʊrəl ˈriːdʒənz/ | Vùng dưới màng phổi |
| 41 | Hilar enlargement | /ˈhaɪlər ɪnˈlɑːrdʒmənt/ | Phì đại rốn phổi |
| 42 | Pleural effusion | /ˈplʊrəl ɪˈfjuːʒən/ | Tràn dịch màng phổi |
| 43 | High-resolution computed tomography (HRCT) | /haɪ ˌrezəˈluːʃən kəmˈpjuːtəd təˈmɒɡrəfi/ | Chụp cắt lớp vi tính độ phân giải cao |
| 44 | Honeycombing | /ˈhʌnikəʊmɪŋ/ | Dạng tổ ong |
| 45 | Ground-glass opacity | /ɡraʊnd ɡlæs oʊˈpæsəti/ | Mờ kính |
| 46 | Antinuclear antibodies | /ˌæntiˈnuːkliər ˈæntɪˌbɒdiz/ | Kháng thể kháng nhân |
| 47 | Autoimmune disease | /ˌɔːtoʊɪˈmjuːn dɪˈziːz/ | Bệnh tự miễn |
| 48 | Surgical lung biopsy | /ˈsɜːrdʒɪkəl lʌŋ ˈbaɪɒpsi/ | Sinh thiết phổi phẫu thuật |
| 49 | Transbronchial lung biopsy | /trænsˈbrɒŋkiəl lʌŋ ˈbaɪɒpsi/ | Sinh thiết phổi xuyên phế quản |
| 50 | Transbronchial cryobiopsy | /trænsˈbrɒŋkiəl ˈkraɪoʊbaɪɒpsi/ | Sinh thiết đông lạnh xuyên phế quản |
| 51 | Desquamative interstitial pneumonia (DIP) | /dɪˈskwæmətɪv ˌɪntərˈstɪʃəl nʊˈmoʊniə/ | Viêm phổi kẽ bong vảy |
| 52 | Nonspecific interstitial pneumonia (NSIP) | /ˌnɒnspəˈsɪfɪk ˌɪntərˈstɪʃəl nʊˈmoʊniə/ | Viêm phổi kẽ không đặc hiệu |
| 53 | Granulomas | /ˌɡrænjəˈloʊməz/ | U hạt |
| 54 | Acute exacerbation | /əˈkjuːt ɪɡˌzæsərˈbeɪʃən/ | Đợt cấp |
| 55 | Acute respiratory distress syndrome (ARDS) | /əˈkjuːt ˈrespərətɔːri dɪˈstres ˈsɪndroʊm/ | Hội chứng suy hô hấp cấp |
| 56 | Corticosteroids | /ˌkɔːrtɪkoʊˈsterɔɪdz/ | Corticosteroid |
| 57 | Cytotoxic agents | /ˌsaɪtoʊˈtɒksɪk ˈeɪdʒənts/ | Thuốc độc tế bào |
| 58 | Pirfenidone | /pɪrˈfenɪdoʊn/ | Pirfenidone |
| 59 | Nintedanib | /nɪnˈtedənɪb/ | Nintedanib |
| 60 | Tyrosine kinase inhibitor | /ˈtaɪrəsiːn ˈkaɪneɪs ɪnˈhɪbɪtər/ | Chất ức chế tyrosine kinase |
| 61 | Fibroblast growth factor | /ˈfaɪbroʊblæst ɡroʊθ ˈfæktər/ | Yếu tố tăng trưởng nguyên bào sợi |
| 62 | Vascular endothelial growth factor | /ˈvæskjələr ˌendoʊˈθiːliəl ɡroʊθ ˈfæktər/ | Yếu tố tăng trưởng nội mô mạch máu |
| 63 | Lung transplantation | /lʌŋ ˌtrænsplænˈteɪʃən/ | Cấy ghép phổi |
| 64 | Respiratory bronchiolitis-interstitial lung disease (RB-ILD) | /ˈrespərətɔːri ˌbrɒŋkioʊˈlaɪtɪs ˌɪntərˈstɪʃəl lʌŋ dɪˈziːz/ | Bệnh lý đường hô hấp-phổi kẽ |
| 65 | Smoking-related interstitial pneumonias | /ˈsmoʊkɪŋ rɪˈleɪtɪd ˌɪntərˈstɪʃəl nʊˈmoʊniəz/ | Viêm phổi kẽ liên quan hút thuốc |
| 66 | Centrilobular nodules | /ˌsentroʊˈlɒbjələr ˈnɒdjuːlz/ | Nốt trung tâm tiểu thùy |
| 67 | Pigmented macrophages | /ˈpɪɡməntɪd ˈmækroʊfeɪdʒəz/ | Đại thực bào có sắc tố |
| 68 | Peribronchiolar inflammation | /ˌperibrɒŋˈkaɪələr ˌɪnfləˈmeɪʃən/ | Viêm quanh tiểu phế quản |
| 69 | Intra-alveolar macrophages | /ˌɪntrə ælˈviːələr ˈmækroʊfeɪdʒəz/ | Đại thực bào trong phế nang |
| 70 | Cryptogenic organizing pneumonia (COP) | /krɪpˈtoʊdʒenɪk ˈɔːrɡənaɪzɪŋ nʊˈmoʊniə/ | Viêm phổi tổ chức hóa không rõ nguyên nhân |
| 71 | Bronchiolitis obliterans organizing pneumonia (BOOP) | /ˌbrɒŋkioʊˈlaɪtɪs əˈblɪtərænz ˈɔːrɡənaɪzɪŋ nʊˈmoʊniə/ | Viêm tiểu phế quản tắc kèm viêm phổi tổ chức hóa |
| 72 | Connective tissue plugs | /kəˈnektɪv ˈtɪʃuː plʌɡz/ | Nút mô liên kết |
| 73 | Mononuclear cell infiltration | /ˌmɒnoʊˈnuːkliər sel ˌɪnfɪlˈtreɪʃən/ | Thâm nhiễm tế bào đơn nhân |
| 74 | Community-acquired pneumonia | /kəˈmjuːnəti əˈkwaɪərd nʊˈmoʊniə/ | Viêm phổi mắc phải ngoài cộng đồng |
| 75 | Acute interstitial pneumonia (AIP) | /əˈkjuːt ˌɪntərˈstɪʃəl nʊˈmoʊniə/ | Viêm phổi kẽ cấp |
| 76 | Fulminant | /ˈfʊlmɪnənt/ | Bùng phát |
| 77 | Diffuse alveolar damage | /dɪˈfjuːs ælˈviːələr ˈdæmɪdʒ/ | Tổn thương phế nang lan tỏa |
| 78 | Hamman-Rich syndrome | /ˈhæmən rɪtʃ ˈsɪndroʊm/ | Hội chứng Hamman-Rich |
| 79 | Collagen vascular diseases | /ˈkɒlədʒən ˈvæskjələr dɪˈziːzəz/ | Bệnh mạch máu collagen |
| 80 | Connective tissue diseases | /kəˈnektɪv ˈtɪʃuː dɪˈziːzəz/ | Bệnh mô liên kết |
| 81 | Rheumatoid arthritis | /ˈruːmətɔɪd ɑːrˈθraɪtɪs/ | Viêm khớp dạng thấp |
| 82 | Systemic lupus erythematosus | /sɪˈstemɪk ˈluːpəs ˌerɪθiːməˈtoʊsəs/ | Lupus ban đỏ hệ thống |
| 83 | Systemic sclerosis (scleroderma) | /sɪˈstemɪk skləˈroʊsɪs/ | Xơ cứng bì hệ thống |
| 84 | Polymyositis-dermatomyositis | /ˌpɒlimaɪəˈsaɪtɪs ˌdɜːrmætoʊmaɪəˈsaɪtɪs/ | Viêm đa cơ-viêm da cơ |
| 85 | Sjögren syndrome | /ˈʃoʊɡrənz ˈsɪndroʊm/ | Hội chứng Sjögren |
| 86 | Overlap syndromes | /ˈoʊvərlæp ˈsɪndroʊmz/ | Hội chứng chồng chéo |
| 87 | Multisystem inflammatory diseases | /ˌmʌltiˈsɪstəm ɪnˈflæmətɔːri dɪˈziːzəz/ | Bệnh viêm đa hệ thống |
| 88 | Immunologically mediated | /ˌɪmjənoʊˈlɒdʒɪkəli ˈmiːdieɪtɪd/ | Có trung gian miễn dịch |
| 89 | Pleuritic chest pain | /plʊˈrɪtɪk tʃest peɪn/ | Đau ngực màng phổi |
| 90 | Acute pneumonitis | /əˈkjuːt ˌnuːməˈnaɪtɪs/ | Viêm phổi cấp |
| 91 | Diffuse alveolar hemorrhage | /dɪˈfjuːs ælˈviːələr ˈheməridʒ/ | Xuất huyết phế nang lan tỏa |
| 92 | Antitopoisomerase I antibody | /ˌæntitoʊpɔɪˈsɒməreɪs wʌn ˈæntɪˌbɒdi/ | Kháng thể chống topoisomerase I |
| 93 | Pulmonary arterial hypertension | /ˈpʌlmənəri ɑːrˈtɪriəl ˌhaɪpərˈtenʃən/ | Tăng áp phổi động mạch |
| 94 | Gastroesophageal reflux | /ˌɡæstroʊɪˈsɒfəɡiːəl ˈriːflʌks/ | Trào ngược dạ dày-thực quản |
| 95 | Recurrent aspiration | /rɪˈkɜːrənt ˌæspəˈreɪʃən/ | Hít sặc tái diễn |
| 96 | Bronchiectasis | /ˌbrɒŋkiˈektəsɪs/ | Giãn phế quản |
| 97 | Methotrexate | /ˌmeθoʊˈtrekseit/ | Methotrexate |
| 98 | Rituximab | /rɪˈtʌksɪmæb/ | Rituximab |
| 99 | Drug-induced lung disease | /drʌɡ ɪnˈduːst lʌŋ dɪˈziːz/ | Bệnh phổi do thuốc |
| 100 | Striated muscle | /ˈstraɪeɪtɪd ˈmʌsəl/ | Cơ vân |
| 101 | Diaphragmatic weakness | /ˌdaɪəfræɡˈmætɪk ˈwiːknəs/ | Yếu cơ hoành |
| 102 | Inspiratory muscles | /ɪnˈspaɪrətɔːri ˈmʌsəlz/ | Cơ hít vào |
| 103 | Aspiration pneumonia | /ˌæspəˈreɪʃən nʊˈmoʊniə/ | Viêm phổi hít sặc |
| 104 | Lymphocytic infiltration | /ˌlɪmfoʊˈsɪtɪk ˌɪnfɪlˈtreɪʃən/ | Thâm nhiễm bạch cầu lympho |
| 105 | Salivary glands | /ˈsæləveri ɡlændz/ | Tuyến nước bọt |
| 106 | Lacrimal glands | /ˈlækrəməl ɡlændz/ | Tuyến lệ |
| 107 | Keratoconjunctivitis sicca | /ˌkerətoʊkəndʒʌŋkˈtaɪvətɪs ˈsɪkə/ | Viêm kết giác mạc khô |
| 108 | Lymphocytic interstitial pneumonia | /ˌlɪmfoʊˈsɪtɪk ˌɪntərˈstɪʃəl nʊˈmoʊniə/ | Viêm phổi kẽ lympho |
| 109 | Pseudolymphoma | /ˌsuːdoʊlɪmˈfoʊmə/ | Giả u lympho |
| 110 | Lymphoma | /lɪmˈfoʊmə/ | U lympho |
| 111 | Undifferentiated connective tissue disease | /ˌʌndɪfəˈrenʃieɪtɪd kəˈnektɪv ˈtɪʃuː dɪˈziːz/ | Bệnh mô liên kết không biệt định |
| 112 | Granulomatous inflammation | /ˌɡrænjəˈlɒmətəs ˌɪnfləˈmeɪʃən/ | Viêm u hạt |
| 113 | Noncaseating granulomas | /ˌnɒnˈkeɪsieɪtɪŋ ˌɡrænjəˈloʊməz/ | U hạt không hoại tử |
| 114 | Infectious agents | /ɪnˈfekʃəs ˈeɪdʒənts/ | Tác nhân nhiễm trùng |
| 115 | Environmental agents | /ɪnˌvaɪrənˈmentəl ˈeɪdʒənts/ | Tác nhân môi trường |
| 116 | Immunologic response | /ˌɪmjənoʊˈlɒdʒɪk rɪˈspɒns/ | Đáp ứng miễn dịch |
| 117 | Exogenous agent | /ɪɡˈzɒdʒənəs ˈeɪdʒənt/ | Tác nhân ngoại sinh |
| 118 | Genetic susceptibility | /dʒəˈnetɪk səˌseptəˈbɪləti/ | Khuynh hướng di truyền |
| 119 | Human leukocyte antigens | /ˈhjuːmən ˈluːkəsaɪt ˈæntɪdʒənz/ | Kháng nguyên bạch cầu người |
| 120 | Candidate genes | /ˈkændɪdət dʒiːnz/ | Gen ứng viên |
| 121 | Microorganisms | /ˌmaɪkroʊˈɔːrɡənɪzəmz/ | Vi sinh vật |
| 122 | Mycobacteria | /ˌmaɪkoʊbækˈtɪriə/ | Vi khuẩn lao |
| 123 | Propionibacterium acnes | /ˌproʊpioʊnibækˈtɪriəm ˈækniːz/ | Vi khuẩn Propionibacterium acnes |
| 124 | Inorganic dusts | /ˌɪnɔːrˈɡænɪk dʌsts/ | Bụi vô cơ |
| 125 | Silica | /ˈsɪlɪkə/ | Silica |
| 126 | Antigen-presenting cells | /ˈæntɪdʒən prɪˈzentɪŋ selz/ | Tế bào trình diện kháng nguyên |
| 127 | T lymphocytes | /tiː ˈlɪmfəsaɪts/ | Bạch cầu lympho T |
| 128 | Alveolar macrophages | /ælˈviːələr ˈmækroʊfeɪdʒəz/ | Đại thực bào phế nang |
| 129 | Dendritic cells | /denˈdrɪtɪk selz/ | Tế bào dendritic |
| 130 | Helper T lymphocytes (CD4+ cells) | /ˈhelpər tiː ˈlɪmfəsaɪts/ | Bạch cầu lympho T helper |
| 131 | TH1 profile | /tiː eɪtʃ wʌn ˈproʊfaɪl/ | Đặc điểm TH1 |
| 132 | Interferon-γ (IFN-γ) | /ˌɪntərˈfɪrɒn ˈɡæmə/ | Interferon-γ |
| 133 | Proinflammatory cytokines | /ˌproʊɪnˈflæmətɔːri ˈsaɪtoʊkaɪnz/ | Cytokin gây viêm |
| 134 | Chemokines | /ˈkiːmoʊkaɪnz/ | Chemokin |
| 135 | Interleukin-2 (IL-2) | /ˌɪntərˈluːkɪn tuː/ | Interleukin-2 |
| 136 | Tumor necrosis factor-α (TNF-α) | /ˈtuːmər nəˈkroʊsɪs ˈfæktər ˈælfə/ | Yếu tố hoại tử khối u-α |
| 137 | Interleukin-12 (IL-12) | /ˌɪntərˈluːkɪn ˈtwelv/ | Interleukin-12 |
| 138 | Insulin-like growth factor-1 (IGF-1) | /ˈɪnsəlɪn laɪk ɡroʊθ ˈfæktər wʌn/ | Yếu tố tăng trưởng tương tự insulin-1 |
| 139 | CD4+ lymphocytes | /siː diː fɔːr ˈpɒzətɪv ˈlɪmfəsaɪts/ | Bạch cầu lympho CD4+ |
| 140 | Peripheral blood | /pəˈrɪfərəl blʌd/ | Máu ngoại vi |
| 141 | Cell-mediated immunity | /sel ˈmiːdieɪtɪd ɪˈmjuːnəti/ | Miễn dịch qua trung gian tế bào |
| 142 | Delayed hypersensitivity | /dɪˈleɪd ˌhaɪpərsensəˈtɪvəti/ | Quá mẫn chậm |
| 143 | Skin testing | /skɪn ˈtestɪŋ/ | Xét nghiệm da |