
[Sách dịch] Sổ tay Bệnh Gan – Handbook of Liver Disease – (C) NXB Elsevier, 2022
Dịch và chú giải: Ths.Bs. Lê Đình Sáng
CHƯƠNG 25: BỆNH GAN Ở TRẺ EM
Pediatric Liver Disease
Chatmanee Lertudomphonwanit MD and William F. Balistreri MD
Handbook of Liver Disease, Chapter 25, 335-350
ĐIỂM CHÍNH
|
Hậu quả của sự chưa trưởng thành về mặt sinh lý của gan
- Nồng độ đường huyết sau sinh thấp:
Hạ đường huyết đáng kể không phổ biến ở trẻ đủ tháng có chế độ ăn đều đặn; tuy nhiên, có nguy cơ hạ đường huyết vì quá trình tân tạo đường và ly giải glycogen trưởng thành nhanh chóng sau khi sinh. Trẻ sinh non có nguy cơ hạ đường huyết cao nhất do dự trữ glycogen giảm và phản ứng tạo ceton của gan không đủ đối với tình trạng hạ đường huyết, điều này có thể tiếp tục kéo dài trong 8 tuần sau sinh. - Thay đổi trong chuyển hóa và thanh thải các hợp chất độc hại nội sinh và ngoại sinh tiềm tàng:
- Nồng độ cytochrome P-450 ở gan thấp ở trẻ sơ sinh. Tương tự, hoạt động của aminopyrine N-demethylase và aniline p-hydroxylase cũng thấp. Các quá trình của gan, chẳng hạn như thanh thải một số loại thuốc hoặc bilirubin phụ thuộc vào các hệ thống này, đều không hiệu quả. Do đó, nồng độ các hợp chất này trong huyết thanh có thể đạt đến mức độc hại.
- Nồng độ glutathione peroxidase và glutathione S-transferase (GST) thấp hơn ở trẻ sơ sinh, do đó gan của trẻ sơ sinh có khả năng dễ bị tổn thương do oxy hóa.
- Thay đổi về kích thước và thành phần của chu trình axit mật: Điều này có thể dẫn đến sự hòa tan micelle không hiệu quả hoặc tích tụ các axit mật không điển hình có hại, có thể làm trầm trọng thêm tình trạng ứ mật và tổn thương gan.
- Vàng da sinh lý:
- Lên đến một phần ba trẻ sơ sinh bị tăng bilirubin không liên hợp trong tuần đầu tiên của cuộc đời; tình trạng này tự khỏi mà không có biến chứng.
- Trẻ bú mẹ có nguy cơ bị vàng da cao hơn trẻ bú sữa công thức.
- Vàng da ở trẻ sinh non có thể xuất hiện sớm hơn, kéo dài hơn và nặng hơn so với trẻ đủ tháng.
- Vàng da sinh lý phản ánh sự chuyển đổi trong việc thanh thải và chuyển hóa bilirubin không liên hợp từ hệ thống của mẹ sang trẻ. Sinh bệnh học có thể do nhiều yếu tố:
- Tăng sản xuất bilirubin: Trẻ sơ sinh có khối lượng hồng cầu lớn, và các tế bào này có chu kỳ sống ngắn hơn hồng cầu của người lớn.
- Giảm liên hợp nội bào do biểu hiện thấp của men uridine diphosphate (UDP)-glucuronyl transferase ở gan.
- Tăng tái hấp thu bilirubin không liên hợp qua chu trình gan-ruột do hệ vi sinh vật đường ruột bị thay đổi và có nhiều beta-glucuronidase nội sinh hoặc ngoại sinh hơn.
- Thường không cần điều trị vàng da sinh lý. Không cần thiết phải ngưng cho con bú.
- Mặc dù vàng da bệnh lý không phổ biến, việc nhận biết và thực hiện các xét nghiệm sâu hơn là rất quan trọng. Các dấu hiệu cảnh báo của vàng da bệnh lý được trình bày trong Hộp 25.1.
Tăng Bilirubin máu
SINH LÝ BỆNH
Sự thay đổi ở bất kỳ bước nào trong quá trình chuyển hóa bilirubin đều có thể gây vàng da (Hình 25.1; các số trong hình tương ứng với các số [1 đến 8] trong phần trình bày sau).
- Tăng sản xuất bilirubin: Điều này có thể do sự gia tăng giải phóng heme từ hồng cầu, vì những lý do sau.
- Các bệnh tán huyết do bất đồng nhóm máu (ABO, Rh, và các nhóm máu phụ khác), khiếm khuyết men hồng cầu (glucose-6-phosphate dehydrogenase [G6PD]; hoặc pyruvate kinase [PK], hexokinase [HK]), và các khiếm khuyết cấu trúc/màng hồng cầu (bệnh hồng cầu hình cầu bẩm sinh, bệnh hồng cầu hình elip di truyền).
- Tái hấp thu máu đã tán huyết từ một khối máu tụ.
- Giảm hấp thu bilirubin vào tế bào gan:
- Điều này có thể do suy giáp hoặc các hormone thai kỳ có thể ức chế sự hấp thu bilirubin qua màng tế bào gan.
- Sự giảm lượng bilirubin gắn với protein huyết thanh cũng dẫn đến giảm hấp thu bởi tế bào gan. Việc giảm gắn kết bilirubin có thể là kết quả của giảm albumin máu, giảm protein máu toàn phần, hoặc sự dịch chuyển của bilirubin khỏi các protein này bởi một số loại thuốc.
- Bất thường trong việc gắn kết hoặc lưu trữ bilirubin nội bào trong tế bào gan: Đây là những rối loạn hiếm gặp và bao gồm sự thiếu hụt hoặc thay đổi của GST, protein gắn kết nội bào chính cho bilirubin. Không cần điều trị, vì không có bệnh suất hoặc tử suất liên quan.
- Liên hợp bilirubin không hiệu quả: Trong tế bào gan, bilirubin được liên hợp với axit glucuronic bởi (UDP)-glucuronyl transferase để tạo thành bilirubin monoglucuronide hoặc diglucuronide.
a. Hội chứng Gilbert
* Là hội chứng tăng bilirubin máu di truyền phổ biến nhất; gây ra bởi các đột biến trong vùng promoter của gen UGT1, dẫn đến giảm hoạt độ của (UDP)-glucuronyl transferase.
* Các đặc điểm lâm sàng chính là sự tăng nồng độ bilirubin không liên hợp trong huyết thanh một cách không liên tục và lành tính ở những người không có triệu chứng khác; điều này đặc biệt nổi bật trong các giai đoạn căng thẳng như bệnh do virus. Hội chứng Gilbert cũng có thể góp phần gây tăng bilirubin máu ở trẻ sơ sinh và dẫn đến nồng độ bilirubin trong huyết thanh cao hơn.
* Chẩn đoán dựa trên việc loại trừ tán huyết hoặc tổn thương tế bào gan; xét nghiệm di truyền có sẵn.
* Không cần điều trị; hậu quả lâu dài duy nhất là tăng nguy cơ sỏi mật.
b. Hội chứng Crigler-Najjar (các bệnh di truyền lặn trên nhiễm sắc thể thường do các loại đột biến khác nhau trong gen UGT1)
* Hội chứng Crigler-Najjar type I được đặc trưng bởi sự vắng mặt của men bilirubin (UDP)-glucuronyl transferase và dẫn đến tăng bilirubin máu nghiêm trọng (thường mg/dL). Điều này có liên quan đến các ảnh hưởng thần kinh thứ phát do vàng da nhân.
* Hội chứng Crigler-Najjar type II gây ra bởi sự giảm sút (UDP)-glucuronyl transferase và có kiểu hình nhẹ hơn (tổng bilirubin thường mg/dL).
* Điều trị bao gồm thay máu và chiếu đèn tích cực để giữ nồng độ bilirubin dưới ngưỡng gây vàng da nhân. Liệu pháp phenobarbital có thể được sử dụng trong hội chứng Crigler-Najjar type II, vốn thường có tiên lượng tốt hơn type I. Ghép gan là phương pháp điều trị dứt điểm cho Crigler-Najjar type I. - Thay đổi trong việc bài tiết bilirubin liên hợp từ tế bào gan:
Trong điều kiện bình thường, bilirubin diglucuronide được bài tiết chủ yếu vào tiểu quản mật bởi một protein vận chuyển khu trú tại protein liên quan đến đa kháng thuốc (MRP2) trên màng tiểu quản mật. Một phần bilirubin glucuronide được tiết vào máu xoang gan; sự tái hấp thu vào tế bào gan xảy ra thông qua các chất vận chuyển (gen/protein) ABCC3/MRP3 hoặc SLCO1B1/OATP1B1 và SLCO1B3/OATP1B3 (OATP là viết tắt của polypeptide vận chuyển anion hữu cơ).
Các bệnh gây ra sự thay đổi trong các bước này có thể dẫn đến tăng bilirubin máu (cả bilirubin không liên hợp và liên hợp) và bao gồm:
a. Hội chứng Dubin-Johnson:
* Một bệnh di truyền lặn trên nhiễm sắc thể thường gây ra bởi đột biến gen ABCC2 trên nhiễm sắc thể 10q24 dẫn đến thay đổi trong bài tiết bilirubin qua MRP2.
* Đặc trưng bởi nồng độ bilirubin liên hợp và không liên hợp trong huyết thanh tăng cao và các chỉ số sinh hóa gan khác bình thường; thường biểu hiện ở tuổi thanh niên.
* Tăng bilirubin máu có thể trở nên rõ rệt hơn trong thai kỳ hoặc khi sử dụng thuốc tránh thai đường uống.
* Chẩn đoán dựa trên nồng độ coproporphyrin toàn phần bình thường trong nước tiểu nhưng có sự gia tăng tỷ lệ coproporphyrin I. Mẫu sinh thiết gan, nếu được thực hiện, cho thấy một sắc tố giống melanin đặc trưng lắng đọng trong tế bào gan trên nền gan có mô học bình thường.
* Do tính chất lành tính của hội chứng này, không cần điều trị.
b. Hội chứng Rotor:
* Một bệnh di truyền lặn trên nhiễm sắc thể thường gây ra bởi các đột biến trong gen SLCO1B1 và SLCO1B3 trên nhiễm sắc thể 12 dẫn đến sự vắng mặt của OATP1B1 và OATP1B3 trên bề mặt đáy-bên của tế bào gan.
* Về mặt lâm sàng, không thể phân biệt được với hội chứng Dubin-Johnson.
* Xét nghiệm nước tiểu là quyết định: Có sự gia tăng nồng độ coproporphyrin toàn phần. * Giống như hội chứng Dubin-Johnson, hội chứng Rotor lành tính và không cần điều trị. - Bất thường chức năng bài tiết của tế bào gan hoặc đường mật trong gan (ứ mật trong gan [hoặc cả hai]); suy giảm chức năng của các protein vận chuyển màng tiểu quản mật hoặc tắc nghẽn dòng chảy mật ở mức độ đường mật trong gan.
Có thể là kết quả của một bất thường di truyền trong một protein vận chuyển của tế bào gan (từ a đến d, sau đây), chức năng bài tiết của tế bào gan (e, sau đây), hoặc đường mật trong gan (f, sau đây):
a. Ứ mật trong gan tiến triển có tính gia đình, type I (PFIC-1; thiếu hụt FIC1), trước đây được gọi là bệnh Byler, là một bệnh di truyền lặn trên nhiễm sắc thể thường gây ra bởi các khiếm khuyết trong gen ATP8B1 trên nhiễm sắc thể 18q21-22 mã hóa cho protein FIC1.
* FIC1 là một P-type adenosine triphosphatase (ATPase) có chức năng vận chuyển aminophospholipid qua màng plasma tiểu quản mật của tế bào gan.
* Bệnh nhân đặc trưng có nồng độ gamma-glutamyltranspeptidase (GGTP) huyết thanh thấp trong bối cảnh ứ mật.
* Ngứa dữ dội được ghi nhận ở độ tuổi trung bình khởi phát là 3 tháng.
* Do gen ATP8B1 được phân bố ở nhiều mô khác nhau, các đặc điểm ngoài gan có thể bao gồm chậm tăng trưởng, tiêu chảy mạn tính, viêm tụy và mất thính lực thần kinh giác quan.
* Thiếu hụt FIC1 có diễn biến lâm sàng thay đổi với khả năng tiến triển thành xơ gan và bệnh gan giai đoạn cuối.
* Điều trị nhằm mục đích cải thiện tình trạng ngứa. Ghép gan chữa khỏi bệnh gan và thường được yêu cầu trong thập kỷ đầu tiên của cuộc đời; tuy nhiên, gan nhiễm mỡ và tiêu chảy mạn tính có thể trầm trọng hơn sau khi ghép gan.
* Một dạng đột biến ATP8B1 nhẹ hơn được gọi là ứ mật trong gan tái phát lành tính (BRIC) và được đặc trưng bởi các đợt vàng da và ngứa tái phát mà không tiến triển thành bệnh gan giai đoạn cuối.
b. Ứ mật trong gan tiến triển có tính gia đình, type II (PFIC-2; thiếu hụt BSEP), về mặt lâm sàng tương tự như PFIC-1 nhưng không có các đặc điểm ngoài gan. Cả hai đều liên quan đến nồng độ GGTP huyết thanh thấp. Bệnh là kết quả của các khiếm khuyết trong bơm xuất khẩu muối mật của tế bào gan (BSEP) do đột biến gen ABCB11 trên nhiễm sắc thể 2q24 dẫn đến tích tụ muối mật trong tế bào gan và cuối cùng ảnh hưởng đến chức năng tế bào gan.
* Thiếu hụt BSEP cũng có thể thay đổi từ kiểu hình nhẹ (BRIC) đến dạng nặng hơn cần ghép gan.
* Mẫu sinh thiết gan sẽ cho thấy viêm gan sơ sinh với sự biến đổi thành tế bào khổng lồ của tế bào gan.
* Bệnh nhân thiếu hụt BSEP, đặc biệt là những người có đột biến cắt cụt hai alen, có nguy cơ cao phát triển ung thư biểu mô tế bào gan (HCC). Do đó, việc tầm soát là cần thiết.
c. Ứ mật trong gan tiến triển có tính gia đình, type III (PFIC-3; thiếu hụt MDR3) tiến triển nhanh chóng đến xơ gan và suy gan.
* Không giống như các loại PFIC khác, hội chứng này được đặc trưng bởi nồng độ GGTP huyết thanh tăng cao.
* PFIC-3 gây ra bởi các đột biến trong gen ABCB4 trên nhiễm sắc thể 7q21-36, mã hóa một bơm xuất khẩu chủ động tham gia vào việc chuyển vị phosphatidylcholine qua màng tiểu quản mật của tế bào gan.
d. Thiếu hụt TJP2 gây ra bởi các đột biến cắt cụt protein trong gen TJP2, dẫn đến thất bại trong việc định vị protein và phá vỡ cấu trúc của liên kết chặt. Các đột biến này đã được phát hiện có liên quan đến kiểu hình ứ mật có GGTP thấp ở trẻ sơ sinh.
e. Tổn thương tế bào gan do nhiều nguyên nhân khác nhau, chẳng hạn như rối loạn chuyển hóa, nhiễm trùng huyết, nhiễm trùng đường tiết niệu, và độc tính của thuốc hoặc độc tố, cũng có thể biểu hiện bằng tăng bilirubin máu, đặc biệt là ứ mật, có lẽ thứ phát sau tổn thương tế bào gan hoặc dòng chảy mật bị thay đổi (hoặc cả hai).
f. Thiểu sản đường mật trong gan, được định nghĩa là tỷ lệ giảm của các ống mật liên tiểu thùy so với khoảng cửa (bình thường là 0.9 đến 1.8; thiểu sản là <0.5), có thể là không hội chứng hoặc có hội chứng, như trong hội chứng Alagille, có liên quan đến hẹp động mạch phổi ngoại biên, đốt sống hình cánh bướm, phôi độc tố màng sau và các đặc điểm khuôn mặt đặc trưng. - Bất thường cấu trúc của đường mật ngoài gan ngăn cản sự dẫn lưu mật từ tiểu quản mật vào ruột và có thể gây tích tụ mật và trào ngược bilirubin vào tuần hoàn hệ thống.
a. Teo đường mật là một bệnh tiến triển được đặc trưng bởi viêm và xơ hóa đường mật ngoài gan dẫn đến tắc nghẽn một phần hoặc hoàn toàn các ống mật ngoài gan.
* Teo đường mật thường biểu hiện dưới dạng ứ mật (tăng bilirubin liên hợp) với phân bạc màu trong khoảng từ 2 đến 6 tuần tuổi.
* Về kiểu hình, có ít nhất hai dạng:
(1) Đa số bệnh nhân (85%) biểu hiện dưới dạng teo đường mật đơn độc (còn được gọi là dạng sau sinh);
(2) một nhóm khác có liên quan đến các dị tật lớn có hoặc không có khiếm khuyết về bên. Các dị tật liên quan trong nhóm sau bao gồm dị tật lách (không lách, đa lách) và các hệ thống tim mạch, tiêu hóa (xoay ruột bất toàn, teo ruột), và niệu-sinh dục; <10% trường hợp có thể có giãn nang ống mật ngoài gan ngoài tình trạng tắc nghẽn do xơ hóa.
* Chẩn đoán dựa trên dữ liệu lâm sàng, sinh hóa và mô học. Mẫu sinh thiết gan cho thấy xơ hóa khoảng cửa và tăng sinh ống mật; nếu không thể loại trừ tắc nghẽn ống mật ngoài gan, nên thực hiện chụp đường mật trong mổ.
* Dị tật này ban đầu được điều trị bằng phẫu thuật tạo một cầu nối cửa-ruột Kasai, cho phép dẫn lưu mật trực tiếp từ gan vào ruột. Mặc dù thủ thuật này không chữa khỏi bệnh, nó có thể làm chậm quá trình tiến triển của bệnh.
* Bệnh gan giai đoạn cuối thứ phát sau teo đường mật là lý do phổ biến nhất cho việc ghép gan ở trẻ em.
b. Nang đường mật chủ, một sự giãn nang của đường mật, có thể chỉ ở ngoài gan hoặc bao gồm cả sự giãn nở của đường mật trong gan.
* Biểu hiện lâm sàng với đau bụng và vàng da, có hoặc không có khối u bụng sờ thấy, có thể xảy ra ở mọi lứa tuổi.
* Chẩn đoán có thể được thực hiện bằng siêu âm, chụp cắt lớp vi tính (CT), hoặc chụp mật tụy ngược dòng nội soi hoặc cộng hưởng từ (ERCP, MRCP).
* Điều trị là phẫu thuật cắt bỏ đoạn giãn, thay vì làm cầu nối hoặc dẫn lưu, do tần suất ác tính tăng lên trong biểu mô của nang. - Thay đổi trong chu trình gan-ruột có thể làm tăng tái hấp thu bilirubin từ ruột. Nguyên nhân có thể là tắc ruột, như trong teo ruột hoặc bệnh Hirschsprung, hoặc thay đổi hệ vi khuẩn đường ruột do sử dụng kháng sinh.
BIẾN CHỨNG
- Tăng bilirubin không liên hợp
- Vàng da nhân (bệnh não do bilirubin) có thể là kết quả của nồng độ bilirubin không liên hợp tăng cao. Các nhóm nguy cơ bao gồm trẻ sơ sinh và những người mắc hội chứng Crigler-Najjar, type I.
- Nồng độ bilirubin không liên hợp > 30 mg/dL có liên quan đến sự phát triển của bệnh não.
- Các yếu tố làm tăng nguy cơ vàng da nhân bao gồm giảm albumin máu và sự dịch chuyển bilirubin khỏi albumin bởi thuốc hoặc các anion hữu cơ.
- Ứ mật
- Suy dinh dưỡng thứ phát sau kém hấp thu chất béo đường ruột có thể dẫn đến chậm phát triển và thiếu hụt vitamin tan trong chất béo.
- Ngứa khó chữa.
- U vàng (Xanthomatosis) thứ phát sau những thay đổi trong chuyển hóa cholesterol.
- Ung thư biểu mô tế bào gan (HCC) có thể xảy ra trong nhiều bệnh liên quan đến ứ mật trong gan, chẳng hạn như thiếu hụt FIC1, BSEP và MDR3; việc tầm soát HCC là cần thiết.
ĐIỀU TRỊ
- Tăng bilirubin không liên hợp
- Thay máu thể tích kép làm giảm nguy cơ vàng da nhân ở trẻ sơ sinh bằng cách giảm nhanh nồng độ bilirubin huyết thanh.
- Chiếu đèn: Quang đồng phân hóa bilirubin thành một hợp chất phân cực hơn cho phép bài tiết bilirubin trong nước tiểu.
- Chuyển hóa bilirubin có thể được thực hiện bằng cách dùng phenobarbital, chất gây cảm ứng các men vi thể giúp tạo điều kiện cho quá trình chuyển hóa bilirubin.
- Ứ mật
- Điều trị tất cả các dạng ứ mật trong gan là điều trị triệu chứng, với sự cân nhắc đặc biệt đến việc quản lý suy dinh dưỡng và ngứa.
- Axit Ursodeoxycholic, một axit mật có tác dụng lợi mật, liều 15 mg/kg mỗi ngày chia nhiều lần, có thể được sử dụng để tăng cường dòng chảy của mật ở bệnh nhân ứ mật.
- Bổ sung vitamin tan trong chất béo cũng cần thiết vì sự hấp thu ở ruột kém nếu không có dòng chảy mật bình thường.
- Ghép gan có thể được yêu cầu trong một số trường hợp.
Suy Gan (xem Chương 2)
- Bệnh não rất khó đánh giá ở trẻ nhỏ và có thể không rõ ràng về mặt lâm sàng cho đến các giai đoạn sau của bệnh; nó không phải là một tiêu chí chẩn đoán bắt buộc đối với suy gan cấp ở trẻ em.
- Tiêu chí chẩn đoán suy gan cấp ở trẻ em: Không có bằng chứng đã biết về bệnh gan mạn tính, bằng chứng sinh hóa về tổn thương gan cấp, và bệnh đông máu không được điều chỉnh bằng vitamin K (thời gian prothrombin [PT] > 15 giây hoặc tỷ lệ chuẩn hóa quốc tế [INR] > 1.5 kèm theo bệnh não gan và PT ≥ 20 hoặc INR ≥ 2 không có bệnh não gan).
- Các nguyên nhân cụ thể rất khác nhau ở mỗi nhóm tuổi, và nguyên nhân có thể không được xác định trong 50% trường hợp. Ngộ độc acetaminophen là nguyên nhân có thể xác định phổ biến nhất ở trẻ lớn và thanh thiếu niên, trong khi các nguyên nhân nhiễm trùng (ví dụ, virus viêm gan A) phổ biến hơn ở các nước đang phát triển. Các bệnh chuyển hóa, bao gồm tyrosinemia type I (xem Chương 20) và viêm gan siêu vi (ví dụ, virus herpes simplex [xem Chương 6]) là những nguyên nhân có thể xác định phổ biến của suy gan cấp ở trẻ sơ sinh và trẻ nhỏ (Bảng 25.2).
- Nhận biết sớm suy gan cấp và các xét nghiệm kịp thời (Bảng 25.3) cùng với điều trị hỗ trợ và theo dõi tại đơn vị chăm sóc tích cực được khuyến nghị.
- Kết quả của suy gan ở trẻ em thay đổi tùy theo nguyên nhân. Vì ghép gan thường cứu sống được bệnh nhân, nên cần liên hệ sớm với một trung tâm ghép tạng trong quá trình diễn biến bệnh.
GALACTOSEMIA
Galactosemia do thiếu hụt galactose-1-phosphate uridyltransferase (GALT) thường biểu hiện trong vài ngày đầu đời.
- Thiếu hụt GALT dẫn đến tích tụ galactose-1-phosphate và galactitol.
- Biểu hiện ban đầu có thể là nhiễm trùng huyết do Escherichia coli ở trẻ sơ sinh.
- Hạ đường huyết có thể xảy ra khi trẻ đang bú sữa mẹ hoặc sữa công thức chứa lactose. Có thể tìm thấy chất khử trong nước tiểu.
- Chẩn đoán dựa trên sự vắng mặt hoạt độ GALT trong hồng cầu. Các chương trình sàng lọc sơ sinh cũng có sẵn ở nhiều tiểu bang tại Hoa Kỳ.
- Nếu rối loạn không được điều trị, trẻ sơ sinh bị ảnh hưởng sẽ tử vong do suy gan.
- Điều trị bằng cách loại bỏ lactose (và galactose) khỏi chế độ ăn, vì lactose được phân hủy thành glucose và galactose.
BỆNH GAN DO TY THỂ NGUYÊN PHÁT
Các khiếm khuyết ty thể thường biểu hiện dưới dạng suy gan sơ sinh, bệnh gan tiến triển với sự suy giảm đột ngột ở thời thơ ấu (thường liên quan đến các triệu chứng thần kinh-cơ), hoặc tiến triển thành bệnh gan xơ hóa mạn tính. Các đặc điểm điển hình có thể gợi ý bệnh gan do ty thể bao gồm tổn thương thần kinh, các đợt hạ đường huyết, tăng amoniac máu và nhiễm toan lactic.
- Khiếm khuyết oxy hóa axit béo như thiếu hụt medium-chain acyl-coenzyme A (CoA) dehydrogenase (MCAD) và thiếu hụt long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) biểu hiện bằng gan to, hạ đường huyết, và tăng nồng độ aminotransferase huyết thanh. Các khiếm khuyết này dẫn đến không có khả năng sử dụng chất béo, chất này sẽ tích tụ trong gan. Ở những bệnh nhân mất bù, tình trạng giảm ceton máu biểu hiện trong bối cảnh hạ đường huyết.
- Mất bù thường được khởi phát bởi các bệnh thông thường ở trẻ em như viêm tai giữa hoặc viêm dạ dày ruột cấp và được đặc trưng bởi tình trạng lơ mơ và hạ đường huyết nghiêm trọng. Các đợt này đáp ứng nhanh với việc bù dịch và glucose.
- Chẩn đoán được gợi ý bởi một hồ sơ axit hữu cơ trong nước tiểu bất thường. Tỷ lệ thể ceton trên axit dicarboxylic thấp, cho thấy không có khả năng chuyển hóa chất béo dự trữ. Nồng độ carnitine toàn phần trong huyết thanh thấp, trong khi phần acylcarnitine thường cao.
- Khiếm khuyết chuỗi hô hấp, như trong hội chứng Alpers (các rối loạn liên quan đến POLG), thường biểu hiện bằng bệnh thần kinh (co giật kháng trị và thoái triển tâm thần vận động) và suy gan tiến triển. Trong một số trường hợp, tổn thương gan có thể bị thúc đẩy bởi việc phơi nhiễm axit valproic. Ban đầu, thường khó phân biệt các rối loạn này với các tác dụng phụ của thuốc chống co giật. Sinh hóa gan điển hình cho thấy ALT và AST tăng nhẹ với chức năng tổng hợp bị suy giảm (hạ đường huyết, giảm albumin máu và bệnh đông máu).
- Hội chứng suy giảm DNA ty thể, giống với hội chứng Alpers, được đặc trưng bởi sự giảm số lượng bản sao DNA ty thể đặc hiệu cho mô. Bệnh nhân bị ảnh hưởng với dạng gan-não của rối loạn này thường biểu hiện trong vài tuần hoặc vài tháng đầu đời với suy gan tiến triển và các triệu chứng thần kinh bao gồm giảm trương lực cơ và co giật. Nhiễm toan lactic không ngừng và hạ đường huyết là những phát hiện xét nghiệm phổ biến.
BỆNH GAN ĐỒNG MIỄN DỊCH THAI KỲ
Bệnh gan đồng miễn dịch thai kỳ là kết quả của một tổn thương gan đồng miễn dịch trong tử cung và là nguyên nhân phổ biến nhất của suy gan sơ sinh. Nó được đặc trưng bởi sự kết hợp của bệnh gan nặng ở trẻ sơ sinh và nhiễm siderosis ngoài gan.
- Một trẻ sơ sinh bị ảnh hưởng thường biểu hiện ngay sau khi sinh với hạ đường huyết, bệnh đông máu rõ rệt, và vàng da (cả tăng bilirubin liên hợp và không liên hợp); nồng độ aminotransferase huyết thanh thấp một cách không tương xứng. Nồng độ alpha fetoprotein (AFP) trong huyết thanh đặc trưng cao (100,000 đến 600,000 ng/mL).
- Các xét nghiệm sắt cho thấy độ bão hòa sắt cao với nồng độ transferrin thấp. Ferritin huyết thanh tăng có thể được tìm thấy nhưng không đặc hiệu.
- Việc chứng minh nhiễm siderosis ngoài gan, bằng sinh thiết niêm mạc miệng hoặc MRI bụng, là cần thiết để chẩn đoán.
- Điều trị bằng thay máu thể tích kép và globulin miễn dịch tiêm tĩnh mạch đã được chứng minh là cải thiện kết quả.
- Nguy cơ tái phát trong các lần mang thai tiếp theo là cao; do đó, điều trị phòng ngừa bằng globulin miễn dịch tiêm tĩnh mạch, 1 g/kg trọng lượng cơ thể bắt đầu từ tuần 14 và 16 của thai kỳ sau đó hàng tuần từ tuần 18 cho đến cuối thai kỳ, được khuyến nghị cho phụ nữ có nguy cơ cao.
THIẾU HỤT CITRIN
Thiếu hụt citrin là một rối loạn di truyền lặn trên nhiễm sắc thể thường do sự thiếu hụt chất vận chuyển glutamate-aspartate trong màng ty thể gây ra bởi các đột biến trong gen SLC25A13. Bệnh phổ biến hơn ở Nhật Bản và các nước Đông Á. Hai dạng được công nhận:
- Ứ mật trong gan sơ sinh liên quan đến thiếu hụt citrin (NICCD) biểu hiện trong giai đoạn sơ sinh. Các triệu chứng khác nhau từ ứ mật trong gan sơ sinh thoáng qua đến suy gan cấp.
- Hạ đường huyết và phát hiện chất khử trong nước tiểu là phổ biến, như trong galactosemia. Các phát hiện xét nghiệm đặc trưng khác bao gồm nồng độ AFP cao và tỷ lệ axit amin chuỗi nhánh trên axit amin thơm trong huyết tương tăng.
- Điều trị bao gồm sữa công thức không chứa lactose với bổ sung triglyceride chuỗi trung bình ở trẻ sơ sinh. Rối loạn chức năng gan thường khỏi trong năm đầu đời. Trẻ em bị ảnh hưởng sau đó có thể phát triển sở thích ăn chế độ giàu protein và ác cảm với thực phẩm nhiều carbohydrate.
- Citrullinemia type 2 biểu hiện ở trẻ lớn hoặc người trưởng thành với sự khởi phát cấp tính của các vấn đề tâm thần kinh và tăng amoniac máu. Điều trị bao gồm bổ sung arginine và chế độ ăn giàu protein, ít carbohydrate.
HỘI CHỨNG REYE
Hội chứng Reye là một nguyên nhân hiếm gặp của suy gan tối cấp ở trẻ em.
- Nó thường biểu hiện sau một bệnh có sốt tiền triệu như nhiễm trùng đường hô hấp trên hoặc thủy đậu và có liên quan đến việc điều trị bằng aspirin.
- Nôn kéo dài xảy ra 5 đến 7 ngày sau khi khởi phát bệnh ban đầu, thường là khi bệnh khởi phát đang cải thiện. Sự tiến triển đến suy gan với suy giảm thần kinh liên quan, co giật và hôn mê có thể xảy ra nhanh chóng.
- Nồng độ aminotransferase huyết thanh thường cao hơn ba đến bốn lần giới hạn trên của mức bình thường. Nồng độ amoniac tăng rõ rệt, với sự kéo dài nhẹ đến trung bình của PT và nồng độ bilirubin huyết thanh bình thường. Hạ đường huyết cũng phổ biến.
- Mô học gan cho thấy các tế bào gan có sự tích tụ triglyceride dạng bọt. Kính hiển vi điện tử cho thấy sự thay đổi cấu trúc ty thể; những thay đổi tương tự được thấy trong ty thể ở não.
- Điều trị là hỗ trợ và tập trung vào việc kiểm soát áp lực nội sọ và nồng độ glucose. Sự sống còn phụ thuộc vào chẩn đoán sớm; bệnh nhân được điều trị trước khi có tổn thương thần kinh nghiêm trọng có cơ hội hồi phục hoàn toàn cao hơn.
Gan to
Gan có thể tăng kích thước do tăng sản hoặc phì đại tế bào, xơ hóa, sung huyết tĩnh mạch, thâm nhiễm mỡ hoặc tích tụ các chất thường không có trong gan, và thâm nhiễm khối u.
THÂM NHIỄM TẾ BÀO VIÊM VÀ TĂNG SINH TẾ BÀO KUPFFER
Bao gồm nhiều nguyên nhân khác nhau như viêm gan siêu vi và viêm gan tự miễn (xem Chương 3 đến 7).
XƠ HÓA
Bệnh gan xơ nang là kết quả của sự thiếu hụt phát triển phôi bình thường của đường mật. Một phổ bệnh được thấy, tùy thuộc vào kích thước của ống mật bị ảnh hưởng. Nhiều biến thể có liên quan đến các rối loạn nang của thận (xem thêm Chương 30).
- Xơ gan bẩm sinh được đặc trưng bởi xơ hóa gan và tăng áp lực tĩnh mạch cửa, và thường liên quan đến bệnh thận đa nang di truyền lặn trên nhiễm sắc thể thường (ARPKD). Bệnh nhân có biểu hiện gan nặng hơn thường xuất hiện ở độ tuổi lớn hơn hoặc trong giai đoạn thanh thiếu niên. Gan to và lách to, thứ phát sau tăng áp lực tĩnh mạch cửa, là những dấu hiệu thực thể phổ biến.
- Bệnh Caroli và hội chứng Caroli: Bệnh gan dạng nang liên quan đến các ống mật lớn trong gan. Do ứ đọng mật trong các nang, bệnh nhân bị ảnh hưởng có khuynh hướng bị viêm đường mật tái phát. Dị dạng tấm ống mật, như trong xơ gan bẩm sinh, cũng được tìm thấy trong hội chứng Caroli. Tăng áp lực tĩnh mạch cửa là một biểu hiện phổ biến và thường đi trước viêm đường mật.
SUNG HUYẾT TĨNH MẠCH
Rối loạn chức năng tim hoặc tắc nghẽn dòng chảy ra của gan (hội chứng Budd-Chiari) có thể dẫn đến sung huyết thụ động của gan, có thể biểu hiện bằng gan to cùng với cổ trướng và đau bụng (xem Chương 21 và 22).
TÍCH TỤ CÁC CHẤT CHUYỂN HÓA
- Mỡ
- Gan to do tích tụ mỡ được thấy trong nhiều rối loạn, phổ biến nhất là béo phì, thay đổi cân nặng nhanh, đái tháo đường, và suy dinh dưỡng. Béo phì cũng như tăng cân nhanh có thể dẫn đến gan to, liên quan đến nhiễm mỡ, viêm nhẹ, và tăng sinh tế bào Kupffer. Bệnh gan nhiễm mỡ không do rượu (NAFLD) được ước tính ảnh hưởng từ 3% đến 10% dân số nhi khoa nói chung và lên đến 40% đến 80% trong dân số nhi khoa béo phì (xem Chương 9).
- Sự tiến triển từ nhiễm mỡ sang viêm gan nhiễm mỡ đến xơ hóa tiến triển và xơ gan đã được ghi nhận ở trẻ em cũng như ở người lớn, ngay cả ở những trẻ có ALT huyết thanh bình thường hoặc tăng nhẹ (tiêu chuẩn dựa trên bằng chứng cho mức ALT bình thường được đề xuất ở trẻ em là ≤25 U/L đối với bé trai và ≤22 U/L đối với bé gái).
- Sự hiện diện của hội chứng chuyển hóa (béo phì nội tạng, tăng huyết áp, kháng insulin/đái tháo đường, và rối loạn lipid máu) dường như quan trọng hơn so với chỉ riêng béo phì trong sự phát triển của nhiễm mỡ và viêm gan nhiễm mỡ.
- Nguồn gốc dân tộc cũng là một yếu tố nguy cơ; người gốc Tây Ban Nha và người Mỹ bản địa có nguy cơ cao nhất, và người Mỹ gốc Phi có nguy cơ thấp nhất.
- Vì NAFLD hiếm gặp ở trẻ em từ 3 đến 10 tuổi, bệnh nhân trong nhóm tuổi này cần một cuộc khảo sát chẩn đoán chi tiết để loại trừ các nguyên nhân khác, đặc biệt là các bệnh gan chuyển hóa khác.
- Điều trị chủ yếu bao gồm giảm cân và kiểm soát tăng đường huyết và tăng lipid máu. Giảm cân không nên quá nhanh, vì có khả năng làm trầm trọng thêm tình trạng viêm gan; giảm 500 g/tuần đã được khuyến nghị ở trẻ em.
- Một thử nghiệm ngẫu nhiên, có đối chứng giả dược ở trẻ em đã cho thấy lợi ích về mặt mô học của việc điều trị bằng vitamin E ở những bệnh nhân bị NASH hoặc NASH ranh giới cũng như cải thiện nồng độ aminotransferase huyết thanh.
- Cholesterol Thiếu hụt lipase axit lysosome (LAL-D) là một rối loạn di truyền lặn trên nhiễm sắc thể thường được đặc trưng bởi sự giảm men lipase axit lysosome. Điều này dẫn đến giảm sự phân hủy cholesterol.
- LAL-D gây ra bởi đột biến gen LIPA trên nhiễm sắc thể 10q23.2-q23.3 dẫn đến các mức độ hoạt động khác nhau của lipase axit lysosome.
- Các đặc điểm gan phổ biến bao gồm gan to (do tích tụ este cholesteryl và triglyceride), tăng nồng độ aminotransferase huyết thanh, xơ hóa gan tiến triển và xơ gan.
a. Bệnh dự trữ este cholesteryl (CESD), một bệnh nhẹ hơn với hoạt độ enzyme còn lại, biểu hiện bằng gan to không giải thích được và rối loạn lipid máu (thường là tăng lipoprotein máu type IIb). Sự khởi phát của bệnh có thể được ghi nhận ở mọi lứa tuổi. Bệnh gan tiến triển với nhiễm mỡ, xơ hóa và xơ gan có thể xảy ra ở hơn một nửa số bệnh nhân.
b. Bệnh Wolman, một dạng LAL-D nghiêm trọng, là do thiếu hụt gần như hoàn toàn hoạt độ enzyme LAL.- Bệnh thường biểu hiện trong giai đoạn sơ sinh với nôn mửa dai dẳng, tiêu chảy, kém hấp thu (thứ phát sau sự tích tụ lipid trong biểu mô ruột), chậm phát triển và gan lách to. Vôi hóa tuyến thượng thận là một đặc điểm nổi bật.
- Suy giảm thần kinh và tử vong xảy ra vào khoảng 6 đến 12 tháng tuổi.
- Liệu pháp thay thế enzyme bằng lipase axit lysosome người tái tổ hợp đã được phê duyệt để điều trị cho trẻ em và người lớn mắc LAL-D. Việc sử dụng thuốc này qua đường tĩnh mạch đã được chứng minh là cải thiện kết quả sống còn ở trẻ sơ sinh mắc bệnh Wolman, với sự cải thiện nồng độ ALT và lipid huyết thanh và hàm lượng mỡ gan ở bệnh nhân CESD sau 20 tuần điều trị.
- Glycogen
a. Bệnh dự trữ glycogen (GSD) biểu hiện bằng gan to, thường không có lách to, thứ phát sau sự tích tụ glycogen trong tế bào gan (xem thêm Chương 20).
* Trong GSD type I, hoạt độ của glucose-6-phosphatase không có hoặc bất thường; do đó, quá trình tân tạo đường không thể diễn ra. Hạ đường huyết sâu phát triển sau những khoảng thời gian nhịn ăn ngắn với nhiễm toan lactic, tăng axit uric máu, giảm phosphat máu và tăng lipid máu. Điều trị bao gồm chế độ ăn nhiều tinh bột thường ở dạng bột ngô hoặc cho ăn liên tục để cung cấp nguồn glucose liên tục. Bệnh nhân có nguy cơ cao bị u tuyến gan.
* GSD type IV hiếm gặp và biểu hiện ở trẻ sơ sinh với gan lách to và cân nặng kém; nó gây ra bởi sự thiếu hụt enzyme khử nhánh. Giống như GSD I, GSD IV dẫn đến tân tạo đường bị lỗi và tích tụ glycogen. Bởi vì loại bệnh này có thể tiến triển thành xơ gan với suy gan, ghép gan được coi là một phương pháp điều trị hiệu quả; tuy nhiên, các đặc điểm tim và thần kinh đã được báo cáo trở nên rõ ràng sau khi ghép.
b. Hội chứng Mauriac, được tìm thấy ở những bệnh nhân đái tháo đường type 1, được đặc trưng bởi bộ ba đái tháo đường kiểm soát kém, chậm phát triển và gan to.
* Mô học gan cho thấy các tế bào gan to lan tỏa với sự tích tụ glycogen, như trong GSD.
* Các mức độ khác nhau của thâm nhiễm mỡ và xơ hóa có thể hiện diện. * Rối loạn này khỏi khi kiểm soát đường huyết được cải thiện. - Sphingolipid
a. Bệnh Gaucher là kết quả của sự thiếu hụt glucocerebrosidase di truyền lặn trên nhiễm sắc thể thường, là enzyme lysosome chịu trách nhiệm phân hủy sphingolipid (xem thêm Chương 20). Ba dạng được công nhận:
* Type I thường biểu hiện bằng gan lách to và là một dạng mạn tính không có tổn thương thần kinh của bệnh. Loại này là biến thể phổ biến nhất và chiếm 90% các trường hợp.
* Type II cũng biểu hiện bằng gan lách to nhưng có các đặc điểm thần kinh và thường gây tử vong trước 2 tuổi.
* Type III có liên quan đến gan lách to và sự khởi phát muộn hơn của các đặc điểm thần kinh.
b. Bệnh Niemann-Pick
* Bệnh Niemann-Pick type A và B là các bệnh di truyền lặn trên nhiễm sắc thể thường gây ra bởi các đột biến SMPD1 dẫn đến giảm hoạt độ sphingomyelinase, gây tích tụ sphingomyelin trong hệ thống võng nội mô ở nhiều cơ quan bao gồm cả gan. Đốm đỏ anh đào được nhìn thấy khi khám mắt. Mẫu sinh thiết gan được đặc trưng bởi các “tế bào bọt” chứa đầy lipid và sphingomyelin được lưu trữ trong đại thực bào.
* Bệnh Niemann-Pick type C, không giống như type A và B, là kết quả của các khiếm khuyết vận chuyển lipid gây ra sự tích tụ cholesterol không este hóa và sphingolipid trong các tế bào bị ảnh hưởng. Sự khởi phát của rối loạn chức năng thần kinh nhận thức và bệnh nội tạng là khác nhau nhưng phổ biến hơn ở thời thơ ấu so với giai đoạn sơ sinh và trưởng thành. - Alpha-1 antitrypsin bất thường
Thiếu hụt alpha-1 antitrypsin, đặc biệt là các kiểu hình PiZZ và PiSZ, có liên quan đến bệnh gan do sự tích tụ alpha-1 antitrypsin bất thường trong lưới nội chất. Bệnh nhân bị ảnh hưởng có thể biểu hiện ứ mật sơ sinh hoặc các đặc điểm của bệnh gan sau này ở giai đoạn sơ sinh hoặc thời thơ ấu. Ghép gan đã được thực hiện để điều trị bệnh gan liên quan đến rối loạn này (xem Chương 20). - Bệnh dự trữ đồng (xem thêm Chương 19)
Bệnh Wilson là một bệnh di truyền do quá tải đồng; gen bất thường nằm trên nhiễm sắc thể 13.- Tỷ lệ người mang gen là 1 trên 90; sự biểu hiện của bệnh dường như thay đổi. Sự bài tiết đồng bị khiếm khuyết dẫn đến tích tụ quá mức trong gan với sự tích tụ sau đó trong hệ thần kinh trung ương và các cơ quan khác.
- Bệnh gan biểu hiện trong thập kỷ thứ hai đến thứ tư của cuộc đời. Biểu hiện muộn hơn có xu hướng là thần kinh hoặc tâm thần.
- Chẩn đoán bằng nồng độ ceruloplasmin huyết thanh <20 mg/dL, đồng trong gan >250 μg/g trọng lượng khô, và bài tiết đồng qua nước tiểu >100 μg/ngày.
- Mẫu sinh thiết gan cho thấy nhiễm mỡ sớm trong quá trình bệnh; bệnh tiến triển đến viêm và xơ hóa với xơ gan. Nhuộm đồng của gan có thể hữu ích trong chẩn đoán nhưng không đặc hiệu, và sự vắng mặt của đồng có thể nhuộm không loại trừ bệnh Wilson.
- Nếu không điều trị, bệnh có thể tiến triển và gây tử vong do suy gan. Bệnh có thể kiểm soát được bằng liệu pháp thải đồng. D-penicillamine và trientine là các chất thải sắt làm tăng bài tiết đồng qua nước tiểu. Kẽm, chất ngăn chặn sự hấp thu đồng ở ruột, cũng đã được sử dụng.
- Một số bệnh nhân biểu hiện suy gan cấp; lựa chọn điều trị hiệu quả duy nhất trong tình huống này là ghép gan, đây là phương pháp chữa khỏi bệnh.
THÂM NHIỄM KHỐI U
Thâm nhiễm khối u của gan có thể góp phần gây gan to.
- Các khối u nguyên phát bao gồm rhabdomyosarcoma phôi thai của đường mật, u quái, hepatoblastoma, hemangioendothelioma, và HCC.
- Các khối u có thể thâm nhiễm gan thứ phát bao gồm neuroblastoma, u Wilms, và lymphoma.
- CT có thể xác định các khối u ban đầu là các bất thường khu trú thay vì thâm nhiễm lan tỏa. Chẩn đoán bằng sinh thiết.
- Điều trị phụ thuộc vào loại khối u.
Viêm gan siêu vi (xem Chương 3 đến 5)
- Viêm gan A và viêm gan B là những nguyên nhân phổ biến nhất của viêm gan siêu vi ở trẻ em.
- Mặc dù cả viêm gan A và viêm gan B đều có thể biểu hiện như một bệnh sốt cấp tính với vàng da và gan to, diễn biến có thể khác nhau.
- Ngoài ra, các biểu hiện và diễn biến khác với người lớn.
- Viêm gan A
- Virus viêm gan A lây truyền qua đường phân-miệng, và các đợt bùng phát thường có thể được truy nguyên từ các trung tâm giữ trẻ nơi vệ sinh có thể không tối ưu. Người lớn làm việc với trẻ em tại các trung tâm giữ trẻ có nguy cơ cao mắc bệnh này.
- Bệnh thường có triệu chứng nhẹ (75% đến 95%) ở trẻ em không có vàng da rõ rệt, trong khi người lớn thường có triệu chứng hơn (75% đến 95%).
- Vì bệnh tự giới hạn, không cần điều trị cụ thể, nhưng cần theo dõi để loại trừ tiến triển thành suy gan cấp.
- Tiêm phòng viêm gan A có hiệu quả cao trong việc ngăn ngừa bệnh lâm sàng (94%) cho trẻ em và người lớn có nguy cơ cao.
- Viêm gan B
- Lây truyền virus viêm gan B từ mẹ sang con trong giai đoạn chu sinh là một con đường lây nhiễm quan trọng ở các vùng lưu hành. Các triệu chứng cấp tính không phổ biến, đặc biệt ở trẻ sơ sinh và trẻ nhỏ. Trẻ lớn hơn biểu hiện một diễn biến lâm sàng tương tự như người lớn.
- Hiếm khi, một bệnh ngoài gan qua trung gian phức hợp miễn dịch liên quan, chẳng hạn như viêm cầu thận màng hoặc viêm da đầu chi dạng sẩn ở trẻ em (hội chứng Gianotti-Crosti), xảy ra.
- Trẻ sơ sinh có mẹ dương tính với kháng nguyên bề mặt viêm gan B (HBsAg) (đặc biệt là những người dương tính với kháng nguyên e của viêm gan B [HBeAg]) có nguy cơ cao bị viêm gan B mạn tính, thường ở trong giai đoạn dung nạp miễn dịch trong thời thơ ấu, và có nguy cơ phát triển HCC sau này trong cuộc đời.
- Đối với trẻ sơ sinh có mẹ dương tính với HBsAg, việc tiêm globulin miễn dịch viêm gan B trong vòng 4 đến 6 giờ sau khi sinh, sau đó là liều vắc-xin viêm gan B đầu tiên và hoàn thành loạt tiêm sau đó có thể ngăn ngừa bệnh ở trẻ sơ sinh.
- Liệu pháp kháng virus nên được xem xét cho những phụ nữ cũng dương tính với HBeAg và có nồng độ DNA virus viêm gan B trong huyết thanh cao.
- Viêm gan C
- Lây truyền từ mẹ sang con là một con đường phổ biến cho nhiễm trùng ở trẻ em, với tỷ lệ lây truyền chu sinh là 5%; lạm dụng ma túy qua đường tĩnh mạch là một con đường lây truyền phổ biến ở thanh thiếu niên.
- Viêm gan C cấp tính không thường được phát hiện ở trẻ em; bệnh tiến triển chậm trong giai đoạn thơ ấu, và bệnh gan tiến triển và các biến chứng nghiêm trọng là hiếm gặp.
- Tại Hoa Kỳ, điều trị được phê duyệt cho trẻ em bao gồm peginterferon-alfa2b kết hợp với ribavirin. Tỷ lệ đáp ứng thấp, và tác dụng phụ thường xuyên. Năm 2017, sự kết hợp của các thuốc kháng virus tác động trực tiếp đường uống ledipasvir và sofosbuvir đã được phê duyệt cho trẻ em từ 12 đến 17 tuổi bị nhiễm trùng do genotype 1, 4, 5 hoặc 6. Vì các thuốc này có hiệu quả cao và ít độc hơn các phác đồ dựa trên interferon, việc điều trị có thể được hoãn lại cho đến khi các thuốc mới này được phê duyệt cho trẻ em dưới 12 tuổi hoặc các phác đồ khác được phê duyệt cho trẻ em bị nhiễm genotype-2 hoặc genotype-3.
Các tình trạng hệ thống ảnh hưởng đến gan (xem Chương 24)
- Xơ nang (CF)
- CF là một bệnh về sự bài tiết clorua bị thay đổi, ảnh hưởng phổ biến nhất đến phổi và tuyến tụy. Nhiều bệnh nhân CF có liên quan đến xơ gan mật khu trú và đa tiểu thùy, với các biến chứng như tăng áp lực tĩnh mạch cửa.
- Sự hiện diện của bệnh gan dường như không phụ thuộc vào kiểu gen của CF, cũng không liên quan đến mức độ nghiêm trọng của bệnh phổi.
- Bệnh nhân biểu hiện bằng gan to, có thể bị quy nhầm là do căng phồng phổi.
- Bệnh nhân CF được biết là có tần suất cao bị bùn mật, sỏi mật, hẹp đường mật, túi mật nhỏ và ứ mật sơ sinh kéo dài.
- Điều trị bằng axit ursodeoxycholic đã được chứng minh là cải thiện các kết quả xét nghiệm bất thường liên quan đến bệnh gan ở CF; tuy nhiên, không rõ liệu liệu pháp dự phòng bằng axit ursodeoxycholic có lợi cho tất cả bệnh nhân CF hay không.
- Bệnh hồng cầu hình liềm
- Bệnh nhân mắc bệnh hồng cầu hình liềm thường có gan to, rõ ràng là thứ phát sau sự giãn xoang gan và tăng sinh tế bào Kupffer.
- Tần suất sỏi mật tăng thứ phát sau sự luân chuyển hemoglobin nhanh được thấy ở nhóm dân số này.
- Nuôi dưỡng hoàn toàn qua đường tĩnh mạch (TPN)
- Ở trẻ em, đặc biệt là trẻ sơ sinh, TPN kéo dài có thể liên quan đến ứ mật, có thể tiến triển thành xơ gan và suy gan.
- Sinh bệnh học chính xác của bệnh gan do TPN vẫn chưa được biết. Nó có thể do nhiều yếu tố, bao gồm các chất nền độc hại trong dung dịch TPN, thiếu hụt chất dinh dưỡng và vi chất dinh dưỡng, và các sản phẩm phụ độc hại của vi khuẩn (lipopolysaccharide gây viêm) đi qua hàng rào niêm mạc ruột bị teo.
- Trẻ sơ sinh có nguy cơ cao bị ứ mật do TPN vì gan chưa trưởng thành và nhu cầu năng lượng cao để đảm bảo tăng trưởng đầy đủ. Giới hạn trên 3.5 g/kg mỗi ngày lipid đã được khuyến nghị ở trẻ sơ sinh; tuy nhiên, tỷ lệ truyền thấp hơn đã được liên kết với tỷ lệ ứ mật giảm.
- Các yếu tố nguy cơ bao gồm sinh non, phẫu thuật bụng, viêm ruột hoại tử, và nhiễm trùng, đặc biệt là nhiễm trùng huyết liên quan đến catheter.
- Điều trị hiệu quả nhất là cho ăn qua đường ruột và ngừng TPN. Dầu cá hoặc nhũ tương lipid đa nguồn làm thành phần lipid có liên quan đến sự khỏi vàng da và có thể làm giảm bệnh gan mật.
- Bệnh Celiac
- Bệnh nhân mắc bệnh celiac có thể biểu hiện tăng nồng độ aminotransferase huyết thanh, thời gian prothrombin kéo dài, hoặc những thay đổi mô học gan không đặc hiệu ngay cả khi không có triệu chứng tiêu hóa.
- Bệnh Celiac cũng có liên quan đến viêm gan tự miễn, viêm đường mật xơ hóa nguyên phát (PSC), và viêm đường mật nguyên phát.
- Chế độ ăn không chứa gluten thường làm bình thường hóa cả các bất thường về xét nghiệm và mô học gan.
- PSC và bệnh viêm ruột (IBD) (xem Chương 17)
- Bệnh nhân IBD, đặc biệt là viêm loét đại tràng, có thể phát triển PSC. Một tỷ lệ đáng kể có các đặc điểm tự miễn (viêm đường mật xơ hóa tự miễn).
- Sự tiến triển của PSC không liên quan đến thời gian hoặc mức độ nghiêm trọng của IBD và có thể đi trước các triệu chứng đường ruột.
- Điều trị IBD bằng ức chế miễn dịch không làm giảm bớt các triệu chứng hoặc sự tiến triển của PSC.
- Các hội chứng mô bào ở trẻ em:
Sự hoạt hóa bất thường của hệ thống võng nội mô có thể dẫn đến bệnh gan.
a. Bệnh mô bào Langerhans (LCH)
* Tỷ lệ mắc LCH là bốn đến năm trường hợp trên 100,000, với độ tuổi trung bình tại thời điểm chẩn đoán là 30 tháng.
* Các tế bào Langerhans bị hoạt hóa bất thường có thể thâm nhiễm vào gan, do đó dẫn đến tăng nồng độ aminotransferase huyết thanh, giảm albumin máu, kéo dài thời gian prothrombin và gan to.
* Mô học gan thường cho thấy một thâm nhiễm viêm ở khoảng cửa bao gồm các tế bào lympho, bạch cầu trung tính và bạch cầu ái toan. LCH có thể rõ ràng nếu mô gan được nhuộm hóa mô miễn dịch cho protein S-100.
* Viêm đường mật xơ hóa là quá trình kinh điển được cho là do LCH. Bệnh nhân cần ghép gan có thể có nguy cơ cao bị thải ghép tế bào cấp tính và bệnh tăng sinh lympho sau ghép.
b. Bệnh thực bào máu lympho mô bào (HLH)
* Tỷ lệ mắc HLH là 1.2 trường hợp trên 100,000 mỗi năm, với độ tuổi trung bình tại thời điểm chẩn đoán là 2.9 tháng.
* Bệnh đa cơ quan này gây ra bởi sự hoạt hóa bất thường của các đại thực bào không ác tính.
* Biểu hiện lâm sàng thay đổi và bao gồm suy gan cấp ở trẻ sơ sinh. * Mô học gan cho thấy các thâm nhiễm khoảng cửa (với tế bào lympho) có kích thước khác nhau.
* Tiêu chí chẩn đoán bao gồm năm trong số tám đặc điểm sau: Sốt, lách to, giảm tế bào máu (≥2 dòng tế bào): Hemoglobin <9 g/dL; tiểu cầu <100×10⁹/L; bạch cầu trung tính <1.0×10⁹/L, tăng triglyceride máu (≥265 mg/dL) và/hoặc giảm fibrinogen máu (≤150 mg/dL), thực bào máu trong tủy xương hoặc lách hoặc hạch bạch huyết (không có bằng chứng ác tính), hoạt độ tế bào NK thấp hoặc không có, ferritin huyết thanh ≥500 μg/L, và nồng độ IL-2R alpha hòa tan ≥2400 U/mL.
* Điều trị bằng ghép tủy xương. - Loạn dưỡng cơ:
Không liên quan đến bệnh gan nhưng thường có nồng độ aspartate aminotransferase (AST) huyết thanh tăng cao khiến bác sĩ lâm sàng tin rằng gan có liên quan. Đánh giá sâu hơn cho thấy nồng độ creatine kinase và/hoặc aldolase tăng, do đó xác nhận rằng nguồn gốc của AST là từ cơ. - Lỗi bẩm sinh của quá trình glycosyl hóa:
Các hội chứng glycoprotein thiếu carbohydrate bao gồm một nhóm các rối loạn đa hệ thống với các khiếm khuyết trong việc lắp ráp oligosaccharide liên kết N.- Type Ia, loại phổ biến nhất và được mô tả rõ nhất, gây ra bởi các khiếm khuyết trong gen phosphomannomutase 2 (PMM2) và có tỷ lệ mắc là 1 trên 80,000.
- Trẻ sơ sinh có nguy cơ tử vong cao do bệnh đa hệ thống. Những trẻ sống sót qua giai đoạn sơ sinh thường bị chậm phát triển tâm thần vận động nghiêm trọng. Bệnh nhân có thể biểu hiện ở giai đoạn sơ sinh với các mức độ rối loạn chức năng gan khác nhau thứ phát sau nhiễm mỡ hoặc xơ hóa.
- Chẩn đoán được thực hiện bằng phương pháp điện di đẳng điện bất thường của transferrin huyết thanh.
- Điều trị bằng D-mannose có thể cải thiện các triệu chứng gan và tiêu hóa ở những bệnh nhân mắc type Ib (thiếu hụt phosphomannose isomerase, một rối loạn chủ yếu ở gan và ruột với tổn thương thần kinh nhẹ).
Bảng 25.1: Dấu hiệu gợi ý chẩn đoán bệnh gan chuyển hóa ở trẻ sơ sinh, trẻ nhỏ và trẻ em
| Bệnh sử | Khám thực thể | Kết quả xét nghiệm |
|---|---|---|
| Triệu chứng bị kích hoạt bởi bệnh tật hoặc nhịn ăn | Gan to rõ rệt, Đục thủy tinh thể | Hạ đường huyết (không có rối loạn chức năng gan nặng), Bệnh đông máu nặng (không có rối loạn chức năng gan nặng) |
| Mùi bất thường | Chậm phát triển, chậm phát triển tâm thần vận động | Tăng amoniac máu (không có rối loạn chức năng gan nặng) |
| Hôn nhân cận huyết trong gia đình | Giảm trương lực cơ, co giật | Nhiễm toan axit hữu cơ khó chữa, nhiễm toan lactic |
| Bệnh gan của mẹ trong thai kỳ | ||
| Tiền sử sẩy thai ở lần mang thai trước |
Chú thích: Bảng này cung cấp các manh mối lâm sàng và xét nghiệm để hướng tới chẩn đoán bệnh gan chuyển hóa ở các nhóm tuổi nhi khoa khác nhau.
Hộp 25.1: Các dấu hiệu cảnh báo vàng da bệnh lý ở trẻ sơ sinh
|
Chú thích: Việc nhận biết sớm các dấu hiệu này là rất quan trọng để can thiệp kịp thời và ngăn ngừa các biến chứng thần kinh.
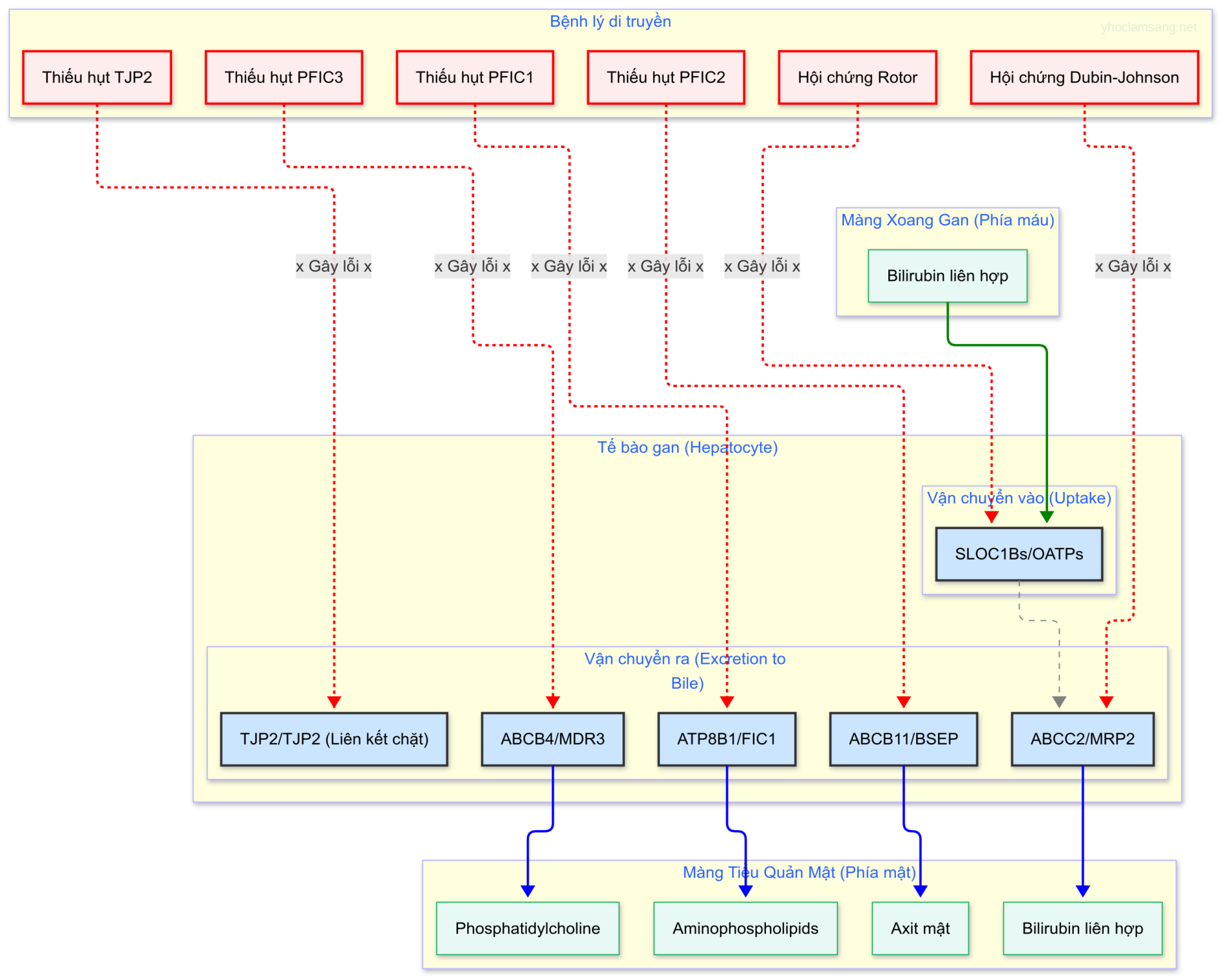
Hình 25.1: Các bước trong quá trình chuyển hóa bilirubin (Hình được vẽ lại trên cơ sở hình ảnh gốc)
Chú thích: (1) Sản xuất bilirubin; (2) hấp thu bilirubin vào tế bào gan; (3) gắn kết nội bào; (4) liên hợp; (5) bài tiết bilirubin liên hợp; (6) bài tiết thành phần mật qua các chất vận chuyển màng tế bào gan đến các ống mật trong gan; (7) đường mật ngoài gan; (8) chu trình gan-ruột. Các con số tương ứng với các bước được mô tả trong văn bản. Phần chèn cho thấy các gen và các protein tương ứng (với các sản phẩm vận chuyển của chúng được hiển thị bên ngoài tế bào gan trên) bị đột biến trong các rối loạn (hiển thị bên ngoài tế bào gan dưới) ảnh hưởng đến các bước từ 3 đến 6. Tên đầy đủ của các gen và protein được đưa ra trong văn bản.
Bảng 25.2: Nguyên nhân suy gan cấp ở trẻ sơ sinh và trẻ nhỏ
| Loại | Nguyên nhân |
|---|---|
| Nhiễm trùng | Herpesvirus, enterovirus, virus viêm gan B |
| Bệnh chuyển hóa | Galactosemia, tyrosinemia, không dung nạp fructose di truyền (sau khi giới thiệu fructose và/hoặc sucrose), thiếu hụt citrin, khiếm khuyết glycosyl hóa, bệnh Niemann-Pick type C, bệnh gan do ty thể |
| Thiếu máu cục bộ | Bệnh tim bẩm sinh, ngạt nặng |
| Rối loạn điều hòa miễn dịch | Bệnh thực bào máu lympho mô bào, bệnh gan đồng miễn dịch thai kỳ |
| Thuốc và độc tố | Axit valproic, acetaminophen |
| Khác | Hội chứng Reye, bệnh ác tính |
Chú thích: Bảng tóm tắt các nhóm nguyên nhân chính gây suy gan cấp ở nhóm tuổi sơ sinh và trẻ nhỏ.
Bảng 25.3: Xét nghiệm chẩn đoán và điều trị đặc hiệu trong suy gan cấp ở trẻ sơ sinh và trẻ nhỏ
| Bệnh | Xét nghiệm chẩn đoán | Điều trị đặc hiệu |
|---|---|---|
| Galactosemia | Galactose-1-phosphate uridyltransferase (GALT) hồng cầu | Sữa công thức không chứa lactose |
| Không dung nạp fructose di truyền | Định lượng enzyme fructose-1-phosphate aldolase (aldolase B) | Chế độ ăn không chứa fructose |
| Tyrosinemia type I (xem Chương 20) | Succinylacetone trong nước tiểu | NTBC (0.5-1 mg/kg/ngày), chế độ ăn loại bỏ |
| Bệnh gan đồng miễn dịch thai kỳ | Sinh thiết niêm mạc miệng hoặc MRI bụng (lắng đọng sắt ngoài gan) | Thay máu thể tích kép sau đó là globulin miễn dịch IV (1 g/kg) |
| Thiếu hụt Citrin | Axit amin huyết tương, xét nghiệm di truyền | Chế độ ăn ít carbohydrate (không lactose), giàu protein, và giàu chất béo, bổ sung MCT |
| Nhiễm Herpesvirus | Huyết thanh học virus và PCR | Acyclovir (60 mg/kg/ngày IV) |
| Bệnh thực bào máu lympho mô bào (HLH) | Tiêu chí chẩn đoán (xem văn bản) | Phác đồ điều trị HLH-2004: Etoposide, dexamethasone, cyclosporine, methotrexate nội tủy (nếu có tổn thương thần kinh) |
Chú thích: IV, Tiêm tĩnh mạch; MCT, triglyceride chuỗi trung bình; MRI, chụp cộng hưởng từ; NTBC, 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexenedione; PCR, phản ứng chuỗi polymerase.
TÀI LIỆU THAM KHẢO
- Beath SV, Kelly DA. Total parenteral nutrition-induced cholestasis: prevention and management. Clin Liver Dis. 2016;20:159-176.
- Feldman AG, Mack CL. Biliary atresia: clinical lessons learned. J Pediatr Gastroenterol Nutr. 2015;61:167-175.
- Fretzayas A, Moustaki M, Liapi O, et al. Gilbert syndrome. Eur J Pediatr. 2012;171:11-15.
- Grijalva J, Vakili K. Neonatal liver physiology. Semin Pediatr Surg. 2013;22:185-189.
- Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012;36:S26-S35.
- Memon N, Weinberger BI, Hegyi T, et al. Inherited disorders of bilirubin clearance. Pediatr Res. 2016;79:378-386.
- Mieli-Vergani G, Vergani D. Paediatric autoimmune liver disease. Arch Dis Child. 2013;98:1012-1017.
- Mieli-Vergani G, Vergani D. Sclerosing cholangitis in children and adolescents. Clin Liver Dis. 2016;20:99-111.
- Mitchel EB, Lavine JE. Review article: the management of paediatric nonalcoholic fatty liver disease. Aliment Pharmacol Ther. 2014;40:1155-1170.
- Molleston JP, Schwimmer JB, Yates KP, et al. Histological abnormalities in children with nonalcoholic fatty liver disease and normal or mildly elevated alanine aminotransferase levels. J Pediatr. 2014;164:707-713.
- Pan X, Kelly S, Melin-Aldana H, et al. Novel mechanism of fetal hepatocyte injury in congenital alloimmune hepatitis involves the terminal complement cascade. Hepatology. 2010;51:2061-2068.
- Santos JL, Choquette M, Bezerra JA. Cholestatic liver disease in children. Curr Gastroenterol Rep. 2010;12:30-39.
- Stender S, Frikke-Schmidt R, Nordestgaard BG, et al. Extreme bilirubin levels as a causal risk factor for symptomatic gallstone disease. JAMA Intern Med. 2013;173:1222-1228.
- Suchy FJ, Sokol RJ, Balistreri WF, eds. Liver Disease in Children. 4th ed. Cambridge: Cambridge University Press; 2014.
BẢNG CHÚ GIẢI THUẬT NGỮ Y HỌC ANH-VIỆT
| STT | Thuật ngữ tiếng Anh | Phiên âm IPA | Nghĩa Tiếng Việt |
|---|---|---|---|
| 1 | Pediatric Liver Disease | /ˌpiːdiˈætrɪk ˈlɪvər dɪˈziːz/ | Bệnh Gan Ở Trẻ Em |
| 2 | Perinatal period | /ˌpɛrɪˈneɪtəl ˈpɪəriəd/ | Giai đoạn chu sinh |
| 3 | Maturational changes | /ˌmætjʊˈreɪʃənl ˈtʃeɪndʒɪz/ | Những thay đổi trong quá trình trưởng thành |
| 4 | Hepatic metabolic processes | /hɪˈpætɪk ˌmɛtəˈbɒlɪk ˈprəʊsɛsɪz/ | Các quá trình chuyển hóa ở gan |
| 5 | Toxin exposures | /ˈtɒksɪn ɪkˈspəʊʒərz/ | Phơi nhiễm độc tố |
| 6 | Inherited liver diseases | /ɪnˈhɛrɪtɪd ˈlɪvər dɪˈziːzɪz/ | Bệnh gan di truyền |
| 7 | Metabolic liver diseases | /ˌmɛtəˈbɒlɪk ˈlɪvər dɪˈziːzɪz/ | Bệnh gan chuyển hóa |
| 8 | Etiologies | /ˌiːtiˈɒlədʒiz/ | Nguyên nhân gây bệnh |
| 9 | Hyperbilirubinemia | /ˌhaɪpərˌbɪlɪˌruːbɪˈniːmiə/ | Tăng bilirubin máu |
| 10 | Hepatomegaly | /ˌhɛpətəʊˈmɛɡəli/ | Gan to |
| 11 | Liver failure | /ˈlɪvər ˈfeɪljər/ | Suy gan |
| 12 | Acute hepatitis | /əˈkjuːt ˌhɛpəˈtaɪtɪs/ | Viêm gan cấp |
| 13 | Chronic hepatitis | /ˈkrɒnɪk ˌhɛpəˈtaɪtɪs/ | Viêm gan mạn tính |
| 14 | Portal hypertension | /ˈpɔːrtl ˌhaɪpərˈtɛnʃən/ | Tăng áp lực tĩnh mạch cửa |
| 15 | Systemic disease | /sɪˈstɛmɪk dɪˈziːz/ | Bệnh hệ thống |
| 16 | Metabolic derangements | /ˌmɛtəˈbɒlɪk dɪˈreɪndʒmənts/ | Rối loạn chuyển hóa |
| 17 | Hypoglycemia | /ˌhaɪpəʊɡlaɪˈsiːmiə/ | Hạ đường huyết |
| 18 | Coagulopathy | /kəʊˌæɡjʊˈlɒpəθi/ | Bệnh đông máu |
| 19 | Clotting factors | /ˈklɒtɪŋ ˈfæktərz/ | Các yếu tố đông máu |
| 20 | Intracranial hemorrhage | /ˌɪntrəˈkreɪniəl ˈhɛmərɪdʒ/ | Xuất huyết nội sọ |
| 21 | Endogenous toxin | /ɛnˈdɒdʒɪnəs ˈtɒksɪn/ | Độc tố nội sinh |
| 22 | Galactosemia | /ɡəˌlæktəʊˈsiːmiə/ | Bệnh galactose huyết |
| 23 | Fructosemia | /ˌfrʌktəʊˈsiːmiə/ | Bệnh fructose huyết |
| 24 | Hypersplenism | /ˌhaɪpərˈspliːnɪzəm/ | Cường lách |
| 25 | Gastrointestinal bleeding | /ˌɡæstrəʊɪnˈtɛstɪnl ˈbliːdɪŋ/ | Xuất huyết tiêu hóa |
| 26 | Physiologic immaturity | /ˌfɪziəˈlɒdʒɪk ˌɪməˈtjʊərɪti/ | Sự chưa trưởng thành về mặt sinh lý |
| 27 | Gluconeogenesis | /ˌɡluːkəʊˌniːəʊˈdʒɛnɪsɪs/ | Tân tạo đường |
| 28 | Glycogenolysis | /ˌɡlaɪkəʊdʒəˈnɒlɪsɪs/ | Ly giải glycogen |
| 29 | Premature infants | /ˌprɛməˈtjʊər ˈɪnfənts/ | Trẻ sinh non |
| 30 | Glycogen reserves | /ˈɡlaɪkədʒən rɪˈzɜːrvz/ | Dự trữ glycogen |
| 31 | Ketogenic response | /ˌkiːtəʊˈdʒɛnɪk rɪˈspɒns/ | Phản ứng tạo ceton |
| 32 | Cytochrome P-450 | /ˈsaɪtəkrəʊm piː fɔːrˈfɪfti/ | Cytochrome P-450 |
| 33 | Aminopyrine N-demethylase | /əˌmiːnəʊˈpaɪriːn ɛn diːˈmɛθɪleɪs/ | Aminopyrine N-demethylase |
| 34 | Aniline p-hydroxylase | /ˈænɪliːn piː haɪˈdrɒksɪleɪs/ | Aniline p-hydroxylase |
| 35 | Glutathione peroxidase | /ˌɡluːtəˈθaɪəʊn pəˈrɒksɪdeɪs/ | Glutathione peroxidase |
| 36 | Glutathione S-transferase (GST) | /ˌɡluːtəˈθaɪəʊn ɛs ˈtrænsfəreɪs/ | Glutathione S-transferase (GST) |
| 37 | Oxidant injury | /ˈɒksɪdənt ˈɪndʒəri/ | Tổn thương do oxy hóa |
| 38 | Bile acid pool | /baɪl ˈæsɪd puːl/ | Chu trình axit mật |
| 39 | Micelle solubilization | /ˈmaɪsɛl ˌsɒljʊbɪlaɪˈzeɪʃən/ | Sự hòa tan micelle |
| 40 | Atypical bile acids | /eɪˈtɪpɪkl baɪl ˈæsɪdz/ | Các axit mật không điển hình |
| 41 | Cholestasis | /ˌkɒlɪˈsteɪsɪs/ | Ứ mật |
| 42 | Physiologic jaundice | /ˌfɪziəˈlɒdʒɪk ˈdʒɔːndɪs/ | Vàng da sinh lý |
| 43 | Unconjugated hyperbilirubinemia | /ʌnˈkɒndʒʊɡeɪtɪd ˌhaɪpərˌbɪlɪˌruːbɪˈniːmiə/ | Tăng bilirubin không liên hợp |
| 44 | Formula-fed infants | /ˈfɔːrmjələ fɛd ˈɪnfənts/ | Trẻ bú sữa công thức |
| 45 | Pathogenesis | /ˌpæθəʊˈdʒɛnɪsɪs/ | Sinh bệnh học |
| 46 | Red cell mass | /rɛd sɛl mæs/ | Khối lượng hồng cầu |
| 47 | Half-life | /hɑːf laɪf/ | Chu kỳ sống, thời gian bán hủy |
| 48 | Uridine diphosphate (UDP)-glucuronyl transferase | /ˈjʊərɪdiːn daɪˈfɒsfeɪt ɡluːˌkjʊərəˈnɪl ˈtrænsfəreɪs/ | Men (UDP)-glucuronyl transferase |
| 49 | Enterohepatic circulation | /ˌɛntərəʊhɪˈpætɪk ˌsɜːrkjʊˈleɪʃən/ | Chu trình gan-ruột |
| 50 | Beta-glucuronidase | /ˈbiːtə ɡluːˌkjʊərəˈnɪdeɪs/ | Men Beta-glucuronidase |
| 51 | Pathologic jaundice | /ˌpæθəˈlɒdʒɪk ˈdʒɔːndɪs/ | Vàng da bệnh lý |
| 52 | Conjugated serum bilirubin | /ˈkɒndʒʊɡeɪtɪd ˈsɪərəm ˌbɪlɪˈruːbɪn/ | Bilirubin liên hợp trong huyết thanh |
| 53 | Bilirubin metabolism | /ˌbɪlɪˈruːbɪn məˈtæbəlɪzəm/ | Chuyển hóa bilirubin |
| 54 | Heme | /hiːm/ | Heme (nhân của hemoglobin) |
| 55 | Hemolytic diseases | /ˌhiːməˈlɪtɪk dɪˈziːzɪz/ | Các bệnh tán huyết |
| 56 | Blood group incompatibility | /blʌd ɡruːp ˌɪnkəmˌpætəˈbɪlɪti/ | Bất đồng nhóm máu |
| 57 | Red cell enzyme defects | /rɛd sɛl ˈɛnzaɪm ˈdiːfɛkts/ | Khiếm khuyết men hồng cầu |
| 58 | Congenital spherocytosis | /kənˈdʒɛnɪtl ˌsfɪərəʊsaɪˈtəʊsɪs/ | Bệnh hồng cầu hình cầu bẩm sinh |
| 59 | Hereditary elliptocytosis | /hɪˈrɛdɪtəri ɪˌlɪptəʊsaɪˈtəʊsɪs/ | Bệnh hồng cầu hình elip di truyền |
| 60 | Hematoma | /ˌhiːməˈtəʊmə/ | Khối máu tụ |
| 61 | Hypothyroidism | /ˌhaɪpəʊˈθaɪrɔɪdɪzəm/ | Suy giáp |
| 62 | Gestational hormones | /dʒɛˈsteɪʃənl ˈhɔːrməʊnz/ | Hormone thai kỳ |
| 63 | Hypoalbuminemia | /ˌhaɪpəʊˌælbjʊmɪˈniːmiə/ | Giảm albumin máu |
| 64 | Hypoproteinemia | /ˌhaɪpəʊˌprəʊtiːˈniːmiə/ | Giảm protein máu |
| 65 | Intracellular binding | /ˌɪntrəˈsɛljʊlər ˈbaɪndɪŋ/ | Gắn kết nội bào |
| 66 | Morbidity | /mɔːrˈbɪdɪti/ | Bệnh suất |
| 77 | Mortality | /mɔːrˈtælɪti/ | Tử suất |
| 68 | Conjugation of bilirubin | /ˌkɒndʒʊˈɡeɪʃən əv ˌbɪlɪˈruːbɪn/ | Sự liên hợp bilirubin |
| 69 | Gilbert syndrome | /ˈɡɪlbərt ˈsɪndrəʊm/ | Hội chứng Gilbert |
| 70 | Crigler-Najjar syndrome | /ˈkriːɡlər ˈnɑːdʒɑːr ˈsɪndrəʊm/ | Hội chứng Crigler-Najjar |
| 71 | Kernicterus | /kərˈnɪktərəs/ | Vàng da nhân |
| 72 | Exchange transfusion | /ɪksˈtʃeɪndʒ trænsˈfjuːʒən/ | Thay máu |
| 73 | Phototherapy | /ˌfəʊtəʊˈθɛrəpi/ | Chiếu đèn |
| 74 | Phenobarbital | /ˌfiːnəʊˈbɑːrbɪtɑːl/ | Phenobarbital |
| 75 | Liver transplantation | /ˈlɪvər ˌtrænsplænˈteɪʃən/ | Ghép gan |
| 76 | Canaliculus | /ˌkænəˈlɪkjʊləs/ | Tiểu quản mật |
| 77 | Multidrug resistance-associated protein (MRP2) | /ˈmʌltidrʌɡ rɪˈzɪstəns əˈsəʊʃieɪtɪd ˈprəʊtiːn/ | Protein liên quan đến đa kháng thuốc (MRP2) |
| 78 | Sinusoidal blood | /ˌsaɪnjəˈsɔɪdəl blʌd/ | Máu xoang gan |
| 79 | Organic anion-transporting polypeptide (OATP) | /ɔːrˈɡænɪk ˈænaɪən trænsˈpɔːrtɪŋ ˌpɒliˈpɛptaɪd/ | Polypeptide vận chuyển anion hữu cơ (OATP) |
| 80 | Dubin-Johnson syndrome | /ˈduːbɪn ˈdʒɒnsən ˈsɪndrəʊm/ | Hội chứng Dubin-Johnson |
| 81 | Basolateral surface | /ˌbeɪsəʊˈlætərəl ˈsɜːrfɪs/ | Bề mặt đáy-bên |
| 82 | Rotor syndrome | /ˈroʊtər ˈsɪndrəʊm/ | Hội chứng Rotor |
| 83 | Coproporphyrin | /ˌkɒprəʊˈpɔːrfɪrɪn/ | Coproporphyrin |
| 84 | Intrahepatic cholestasis | /ˌɪntrəhɪˈpætɪk ˌkɒlɪˈsteɪsɪs/ | Ứ mật trong gan |
| 85 | Progressive familial intrahepatic cholestasis (PFIC) | /prəˈɡrɛsɪv fəˈmɪliəl ˌɪntrəhɪˈpætɪk ˌkɒlɪˈsteɪsɪs/ | Ứ mật trong gan tiến triển có tính gia đình (PFIC) |
| 86 | Byler disease | /ˈbaɪlər dɪˈziːz/ | Bệnh Byler |
| 87 | Gamma-glutamyltranspeptidase (GGTP) | /ˈɡæmə ˌɡluːtəˈmɪlˌtrænsˈpɛptɪdeɪz/ | Gamma-glutamyltranspeptidase (GGTP) |
| 88 | Pruritus | /prʊəˈraɪtəs/ | Ngứa |
| 89 | Sensorineural hearing loss | /ˌsɛnsəriˈnjʊərəl ˈhɪərɪŋ lɒs/ | Mất thính lực thần kinh giác quan |
| 90 | Benign recurrent intrahepatic cholestasis (BRIC) | /bɪˈnaɪn rɪˈkʌrənt ˌɪntrəhɪˈpætɪk ˌkɒlɪˈsteɪsɪs/ | Ứ mật trong gan tái phát lành tính (BRIC) |
| 91 | Bile salt export pump (BSEP) | /baɪl sɔːlt ˈɛkspɔːrt pʌmp/ | Bơm xuất khẩu muối mật (BSEP) |
| 92 | Giant cell transformation | /ˈdʒaɪənt sɛl ˌtrænsfərˈmeɪʃən/ | Biến đổi thành tế bào khổng lồ |
| 93 | Hepatocellular carcinoma (HCC) | /ˌhɛpətəʊˈsɛljʊlər ˌkɑːrsɪˈnəʊmə/ | Ung thư biểu mô tế bào gan (HCC) |
| 94 | Phosphatidylcholine | /ˌfɒsfəˌtaɪdɪlˈkəʊliːn/ | Phosphatidylcholine |
| 95 | Tight junction | /taɪt ˈdʒʌŋkʃən/ | Liên kết chặt |
| 96 | Interlobular bile ducts | /ˌɪntərˈlɒbjʊlər baɪl dʌkts/ | Ống mật liên tiểu thùy |
| 97 | Portal tracts | /ˈpɔːrtl trækts/ | Khoảng cửa |
| 98 | Alagille syndrome | /ˈælədʒiːl ˈsɪndrəʊm/ | Hội chứng Alagille |
| 99 | Peripheral pulmonic stenosis | /pəˈrɪfərəl pʊlˈmɒnɪk stɪˈnəʊsɪs/ | Hẹp động mạch phổi ngoại biên |
| 100 | Butterfly vertebrae | /ˈbʌtərflaɪ ˈvɜːrtɪbreɪ/ | Đốt sống hình cánh bướm |
| 101 | Posterior embryotoxon | /pɒˈstɪəriər ɛmˈbraɪəʊˌtɒksən/ | Phôi độc tố màng sau |
| 102 | Biliary atresia | /ˈbɪliəri əˈtriːʒə/ | Teo đường mật |
| 103 | Acholic stools | /eɪˈkoʊlɪk stuːlz/ | Phân bạc màu |
| 104 | Laterality defects | /ˌlætəˈrælɪti ˈdiːfɛkts/ | Khiếm khuyết về bên |
| 105 | Asplenia | /eɪˈspliːniə/ | Không lách |
| 106 | Polysplenia | /ˌpɒliˈspliːniə/ | Đa lách |
| 107 | Intestinal malrotation | /ɪnˈtɛstɪnl ˌmælrəʊˈteɪʃən/ | Xoay ruột bất toàn |
| 108 | Intraoperative cholangiography | /ˌɪntrəˈɒpərətɪv ˌkoʊlænʤiˈɒɡrəfi/ | Chụp đường mật trong mổ |
| 109 | Kasai portoenterostomy | /kəˈsaɪ ˌpɔːrtəʊˌɛntəˈrɒstəmi/ | Phẫu thuật nối cửa-ruột Kasai |
| 110 | Choledochal cyst | /ˌkoʊlɪˈdɒkəl sɪst/ | Nang đường mật chủ |
| 111 | Malignancy | /məˈlɪɡnənsi/ | Bệnh ác tính |
| 112 | Hirschsprung disease | /ˈhɜːrʃsprʊŋ dɪˈziːz/ | Bệnh Hirschsprung |
| 113 | Bilirubin encephalopathy | /ˌbɪlɪˈruːbɪn ɛnˌsɛfəˈlɒpəθi/ | Bệnh não do bilirubin |
| 114 | Fat-soluble vitamin deficiencies | /fæt ˈsɒljʊbl ˈvaɪtəmɪn dɪˈfɪʃənsiz/ | Thiếu hụt vitamin tan trong chất béo |
| 115 | Intractable pruritus | /ɪnˈtræktəbl prʊəˈraɪtəs/ | Ngứa khó chữa |
| 116 | Xanthomatosis | /ˌzænθəʊməˈtəʊsɪs/ | U vàng |
| 117 | Double-volume exchange transfusion | /ˈdʌbl ˈvɒljuːm ɪksˈtʃeɪndʒ trænsˈfjuːʒən/ | Thay máu thể tích kép |
| 118 | Photoisomerization | /ˌfəʊtəʊˌaɪsəˌmɛraɪˈzeɪʃən/ | Quang đồng phân hóa |
| 119 | Ursodeoxycholic acid | /ˌɜːrsəʊˌdiːˌɒksɪˈkoʊlɪk ˈæsɪd/ | Axit Ursodeoxycholic |
| 120 | Pediatric acute liver failure | /ˌpiːdiˈætrɪk əˈkjuːt ˈlɪvər ˈfeɪljər/ | Suy gan cấp ở trẻ em |
| 121 | Prothrombin time (PT) | /proʊˈθrɒmbɪn taɪm/ | Thời gian prothrombin (PT) |
| 122 | International normalized ratio (INR) | /ˌɪntərˈnæʃənl ˈnɔːrməlaɪzd ˈreɪʃiəʊ/ | Tỷ lệ chuẩn hóa quốc tế (INR) |
| 123 | Hepatic encephalopathy | /hɪˈpætɪk ɛnˌsɛfəˈlɒpəθi/ | Bệnh não gan |
| 124 | Tyrosinemia | /ˌtaɪrəʊsɪˈniːmiə/ | Bệnh tyrosin huyết |
| 125 | Herpes simplex virus | /ˈhɜːrpiːz ˈsɪmplɛks ˈvaɪrəs/ | Virus herpes simplex |
| 126 | Galactose-1-phosphate uridyltransferase (GALT) | /ɡəˈlæktəʊs wʌn ˈfɒsfeɪt jʊəˌrɪdɪlˈtrænsfəreɪs/ | Men GALT |
| 127 | E. coli sepsis | /iː ˈkoʊlaɪ ˈsɛpsɪs/ | Nhiễm trùng huyết do E. coli |
| 128 | Primary mitochondrial hepatopathies | /ˈpraɪməri ˌmaɪtəˈkɒndriəl ˌhɛpətɒˈpæθiz/ | Bệnh gan do ty thể nguyên phát |
| 129 | Neuromuscular symptoms | /ˌnjʊərəʊˈmʌskjʊlər ˈsɪmptəmz/ | Triệu chứng thần kinh-cơ |
| 130 | Fibrosing liver disease | /faɪˈbrəʊsɪŋ ˈlɪvər dɪˈziːz/ | Bệnh gan xơ hóa |
| 131 | Hyperammonemia | /ˌhaɪpərˌæməˈniːmiə/ | Tăng amoniac máu |
| 132 | Lactic acidosis | /ˈlæktɪk ˌæsɪˈdəʊsɪs/ | Nhiễm toan lactic |
| 133 | Fatty acid oxidation defects | /ˈfæti ˈæsɪd ˌɒksɪˈdeɪʃən ˈdiːfɛkts/ | Khiếm khuyết oxy hóa axit béo |
| 134 | Decompensated patients | /ˌdiːkɒmpənˈseɪtɪd ˈpeɪʃənts/ | Bệnh nhân mất bù |
| 135 | Hypoketosis | /ˌhaɪpəʊkiːˈtəʊsɪs/ | Giảm ceton máu |
| 136 | Respiratory chain defects | /rɪˈspɪrətəri tʃeɪn ˈdiːfɛkts/ | Khiếm khuyết chuỗi hô hấp |
| 137 | Alpers syndrome | /ˈælpərz ˈsɪndrəʊm/ | Hội chứng Alpers |
| 138 | Refractory seizures | /rɪˈfræktəri ˈsiːʒərz/ | Co giật kháng trị |
| 139 | Psychomotor regression | /ˌsaɪkəʊˈməʊtər rɪˈɡrɛʃən/ | Thoái triển tâm thần vận động |
| 140 | Mitochondrial DNA depletion syndrome | /ˌmaɪtəˈkɒndriəl diːɛnˈeɪ dɪˈpliːʃən ˈsɪndrəʊm/ | Hội chứng suy giảm DNA ty thể |
| 141 | Hepatocerebral form | /ˌhɛpətəʊsəˈriːbrəl fɔːrm/ | Dạng gan-não |
| 142 | Gestational alloimmune liver disease | /dʒɛˈsteɪʃənl ˌæləʊɪˈmjuːn ˈlɪvər dɪˈziːz/ | Bệnh gan đồng miễn dịch thai kỳ |
| 143 | Extrahepatic siderosis | /ˌɛkstrəhɪˈpætɪk ˌsɪdəˈrəʊsɪs/ | Nhiễm siderosis ngoài gan |
| 144 | Alpha fetoprotein (AFP) | /ˈælfə ˈfiːtəʊˈprəʊtiːn/ | Alpha fetoprotein (AFP) |
| 145 | Intravenous immunoglobulin | /ˌɪntrəˈviːnəs ˌɪmjʊnəʊˈɡlɒbjʊlɪn/ | Globulin miễn dịch tiêm tĩnh mạch |
| 146 | Citrin deficiency | /ˈsɪtrɪn dɪˈfɪʃənsi/ | Thiếu hụt citrin |
| 147 | Citrullinemia | /ˌsɪtrʊlɪˈniːmiə/ | Bệnh citrullin huyết |
| 148 | Reye syndrome | /raɪ ˈsɪndrəʊm/ | Hội chứng Reye |
| 149 | Fulminant hepatic failure | /ˈfʊlmɪnənt hɪˈpætɪk ˈfeɪljər/ | Suy gan tối cấp |
| 150 | Varicella infection | /ˌværɪˈsɛlə ɪnˈfɛkʃən/ | Nhiễm thủy đậu |
| 151 | Intracranial pressure | /ˌɪntrəˈkreɪniəl ˈprɛʃər/ | Áp lực nội sọ |
| 152 | Cell hyperplasia | /sɛl ˌhaɪpərˈpleɪʒə/ | Tăng sản tế bào |
| 153 | Hypertrophy | /haɪˈpɜːrtrəfi/ | Phì đại |
| 154 | Fibrosis | /faɪˈbrəʊsɪs/ | Xơ hóa |
| 155 | Venous congestion | /ˈviːnəs kənˈdʒɛstʃən/ | Sung huyết tĩnh mạch |
| 156 | Fibrocystic liver disease | /ˌfaɪbrəʊˈsɪstɪk ˈlɪvər dɪˈziːz/ | Bệnh gan xơ nang |
| 157 | Congenital hepatic fibrosis | /kənˈdʒɛnɪtl hɪˈpætɪk faɪˈbrəʊsɪs/ | Xơ gan bẩm sinh |
| 158 | Autosomal recessive polycystic kidney disease (ARPKD) | /ˌɔːtəˈsəʊməl rɪˈsɛsɪv ˌpɒliˈsɪstɪk ˈkɪdni dɪˈziːz/ | Bệnh thận đa nang di truyền lặn trên NST thường (ARPKD) |
| 159 | Caroli disease | /kəˈroʊli dɪˈziːz/ | Bệnh Caroli |
| 160 | Cholangitis | /ˌkoʊlænˈdʒaɪtɪs/ | Viêm đường mật |
| 161 | Budd-Chiari syndrome | /bʌd kiˈɑːri ˈsɪndrəʊm/ | Hội chứng Budd-Chiari |
| 162 | Ascites | /əˈsaɪtiːz/ | Cổ trướng |
| 163 | Steatosis | /ˌstiːəˈtəʊsɪs/ | Nhiễm mỡ |
| 164 | Nonalcoholic fatty liver disease (NAFLD) | /ˌnɒnˌælkəˈhɒlɪk ˈfæti ˈlɪvər dɪˈziːz/ | Bệnh gan nhiễm mỡ không do rượu (NAFLD) |
| 165 | Steatohepatitis | /ˌstiːətəʊˌhɛpəˈtaɪtɪs/ | Viêm gan nhiễm mỡ |
| 166 | Metabolic syndrome | /ˌmɛtəˈbɒlɪk ˈsɪndrəʊm/ | Hội chứng chuyển hóa |
| 167 | Lysosomal acid lipase deficiency (LAL-D) | /ˌlaɪsəˈsəʊməl ˈæsɪd ˈlaɪpeɪs dɪˈfɪʃənsi/ | Thiếu hụt lipase axit lysosome (LAL-D) |
| 168 | Cholesteryl ester storage disease (CESD) | /kəˌlɛstəˈrɪl ˈɛstər ˈstɔːrɪdʒ dɪˈziːz/ | Bệnh dự trữ este cholesteryl (CESD) |
| 169 | Wolman disease | /ˈwʊlmən dɪˈziːz/ | Bệnh Wolman |
| 170 | Adrenal calcification | /əˈdriːnl ˌkælsɪfɪˈkeɪʃən/ | Vôi hóa tuyến thượng thận |
| 171 | Glycogen storage disease (GSD) | /ˈɡlaɪkədʒən ˈstɔːrɪdʒ dɪˈziːz/ | Bệnh dự trữ glycogen (GSD) |
| 172 | Hyperuricemia | /ˌhaɪpərˌjʊərɪˈsiːmiə/ | Tăng axit uric máu |
| 173 | Hypophosphatemia | /ˌhaɪpəʊˌfɒsfəˈtiːmiə/ | Giảm phosphat máu |
| 174 | Hepatic adenomas | /hɪˈpætɪk ˌædɪˈnəʊməz/ | U tuyến gan |
| 175 | Mauriac syndrome | /ˈmɔːriæk ˈsɪndrəʊm/ | Hội chứng Mauriac |
| 176 | Gaucher’s disease | /ˈɡaʊʃərz dɪˈziːz/ | Bệnh Gaucher |
| 177 | Glucocerebrosidase | /ˌɡluːkəʊˌsɛrəˈbrɒsɪdeɪs/ | Glucocerebrosidase |
| 178 | Niemann-Pick disease | /ˈniːmən pɪk dɪˈziːz/ | Bệnh Niemann-Pick |
| 179 | Sphingomyelinase | /ˌsfɪŋɡəʊˈmaɪəlɪneɪs/ | Sphingomyelinase |
| 180 | Reticuloendothelial system | /rɪˌtɪkjʊləʊˌɛndəʊˈθiːliəl ˈsɪstəm/ | Hệ thống võng nội mô |
| 181 | Foam cells | /fəʊm sɛlz/ | Tế bào bọt |
| 182 | Alpha-1 antitrypsin deficiency | /ˈælfə wʌn ˌæntiˈtrɪpsɪn dɪˈfɪʃənsi/ | Thiếu hụt alpha-1 antitrypsin |
| 183 | Endoplasmic reticulum | /ˌɛndəʊˈplæzmɪk rɪˈtɪkjʊləm/ | Lưới nội chất |
| 184 | Wilson disease | /ˈwɪlsən dɪˈziːz/ | Bệnh Wilson |
| 185 | Ceruloplasmin | /səˌruːləʊˈplæzmɪn/ | Ceruloplasmin |
| 186 | D-penicillamine | /diː ˌpɛnɪˈsɪləmiːn/ | D-penicillamine |
| 187 | Trientine | /ˈtraɪɛntiːn/ | Trientine |
| 188 | Embryonal rhabdomyosarcoma | /ˌɛmbriˈɒnəl ˌræbdəʊˌmaɪəʊsɑːrˈkəʊmə/ | Rhabdomyosarcoma phôi thai |
| 189 | Hepatoblastoma | /ˌhɛpətəʊblæˈstəʊmə/ | Hepatoblastoma |
| 190 | Hemangioendothelioma | /hɪˌmændʒiəʊˌɛndəʊˌθiːliˈəʊmə/ | Hemangioendothelioma |
| 191 | Neuroblastoma | /ˌnjʊərəʊblæˈstəʊmə/ | Neuroblastoma |
| 192 | Wilms tumor | /vɪlmz ˈtjuːmər/ | U Wilms |
| 193 | Cystic fibrosis (CF) | /ˈsɪstɪk faɪˈbrəʊsɪs/ | Xơ nang (CF) |
| 194 | Sclerosing cholangitis | /sklɪəˈrəʊsɪŋ ˌkoʊlænˈdʒaɪtɪs/ | Viêm đường mật xơ hóa |
| 195 | Langerhans cell histiocytosis (LCH) | /ˈlæŋərhænz sɛl ˌhɪstiəʊsaɪˈtəʊsɪs/ | Bệnh mô bào Langerhans (LCH) |
| 196 | Hemophagocytic lymphohistiocytosis (HLH) | /ˌhiːməʊˌfæɡəˈsɪtɪk ˌlɪmfəʊˌhɪstiəʊsaɪˈtəʊsɪs/ | Bệnh thực bào máu lympho mô bào (HLH) |
| 197 | Muscular dystrophies | /ˈmʌskjʊlər ˈdɪstrəfiz/ | Loạn dưỡng cơ |
| 198 | Inborn errors of glycosylation | /ˈɪnbɔːrn ˈɛrərz əv ˌɡlaɪkɒsɪˈleɪʃən/ | Lỗi bẩm sinh của quá trình glycosyl hóa |
| 199 | Carbohydrate-deficient glycoprotein syndromes | /ˌkɑːrbəʊˈhaɪdreɪt dɪˈfɪʃənt ˌɡlaɪkəʊˈprəʊtiːn ˈsɪndrəʊmz/ | Các hội chứng glycoprotein thiếu carbohydrate |
| 200 | Isoelectric focusing | /ˌaɪsəʊɪˈlɛktrɪk ˈfəʊkəsɪŋ/ | Điện di đẳng điện |