
Sperling Nội tiết học Nhi khoa, Ấn bản thứ 5 – Biên dịch: Ths.Bs. Lê Đình Sáng
Sperling Pediatric Endocrinology, Fifth Edition
Tác giả: Sperling, Mark A., MD – Nhà xuất bản: Elsevier Inc.
PHẦN II: CÁC RỐI LOẠN NỘI TIẾT Ở TRẺ SƠ SINH
Chương 7. Hạ đường huyết ở trẻ sơ sinh và trẻ nhũ nhi
Diva D. De Leon; Paul Thornton; Charles A. Stanley; Mark A. Sperling
Sperling Nội tiết học Nhi khoa, 7, 175-201
Giới thiệu
Một trong những sự kiện chuyển hóa quan trọng nhất đánh dấu sự chuyển tiếp từ đời sống bào thai sang đời sống sơ sinh là sự thích nghi từ một môi trường có nguồn glucose liên tục và sẵn có—qua đường truyền nhau thai từ máu mẹ—sang một môi trường mà glucose được cung cấp một cách hạn chế và gián đoạn qua việc bú mẹ. Khi sinh ra, sau khi tách khỏi nhau thai, các cơ chế phức tạp liên quan đến việc duy trì nồng độ glucose huyết tương phải được kích hoạt và phối hợp để tránh hạ đường huyết và hậu quả là tổn thương hệ thần kinh trung ương. Một trẻ sơ sinh hoặc trẻ nhũ nhi bị hạ đường huyết đặt ra một thách thức chẩn đoán và điều trị khẩn cấp. Các đặc điểm lâm sàng phải được đánh giá nhanh chóng và một kế hoạch hành động phải được xây dựng dựa trên tuổi của trẻ, tiền sử của mẹ và quá trình sinh nở, mức độ nghiêm trọng và sự kéo dài của tình trạng hạ đường huyết, và tất cả các manh mối lâm sàng liên quan khác để chẩn đoán và điều trị xác định. Trong chương này, chúng tôi xem xét sinh lý bình thường của chuyển hóa glucose và sinh lý bệnh của các rối loạn phổ biến nhất gây hạ đường huyết ở trẻ sơ sinh (4 tuần đầu đời) và trẻ nhũ nhi (1 tháng đến 1 tuổi) với trọng tâm là cách tiếp cận chẩn đoán và quản lý các bệnh lý này.
Các nguyên lý của chuyển hóa glucose
Một cách tiếp cận hệ thống đối với tình trạng hạ đường huyết ở trẻ sơ sinh, trẻ nhũ nhi, hoặc trẻ em đòi hỏi sự hiểu biết về vai trò trung tâm của glucose trong nền kinh tế nhiên liệu của cơ thể. Chuyển hóa glucose chiếm khoảng một nửa nhu cầu năng lượng cơ bản hàng ngày và là nhiên liệu chuyển hóa chính của não người. Glucose có thể được lưu trữ để tạo năng lượng dưới dạng glycogen và chất béo, mặc dù chỉ có glucose được lưu trữ trong glycogen ở gan mới có thể được giải phóng vào tuần hoàn, và acid béo không thể được chuyển đổi trở lại thành glucose; carbon từ glucose có thể được sử dụng để tổng hợp protein và các thành phần cấu trúc (như màng tế bào). Quá trình oxy hóa hiếu khí của glucose tạo ra năng lượng cao bằng cách sản xuất 36 mol adenosine triphosphate (ATP) cho mỗi mol glucose. Mặc dù glucose là nhiên liệu chuyển hóa chủ yếu cho cả não và cơ thể trong giai đoạn ngay sau bữa ăn, nhưng trong thời gian nhịn ăn dài hơn, việc sử dụng glucose phải được giới hạn ở các mô ly giải đường, chẳng hạn như tế bào hồng cầu và não, và phần còn lại của cơ thể phải dựa vào quá trình oxy hóa acid béo được giải phóng từ các kho dự trữ triglyceride trong mô mỡ.
Tất cả glucose được não chiết xuất đều được oxy hóa, và do đó, việc sử dụng glucose của não song song với sự hấp thu oxy của não. Ở trẻ 5 tuần tuổi, việc sử dụng glucose của não đã chiếm từ 71% đến 93% mức độ của người lớn ở hầu hết các vùng não (dao động từ 13 đến 25 μmol/100 g/phút). Mức độ sử dụng glucose của não ở người lớn (19–33 μmol/100 g/phút) đạt được vào lúc 2 tuổi, và chúng tiếp tục tăng cho đến 3 đến 4 tuổi—khi chúng đạt đến các giá trị dao động từ 49 đến 65 μmol/100 g/phút, được duy trì cho đến khoảng 9 tuổi và sau đó giảm dần xuống mức của người lớn vào cuối thập kỷ thứ hai.
Sự hấp thu glucose của não xảy ra thông qua một quá trình khuếch tán thuận hóa qua trung gian chất mang, phụ thuộc vào nồng độ glucose, cũng như không phụ thuộc vào năng lượng, Na+, và insulin. Quá trình này được trung gian bởi các protein vận chuyển glucose thuận hóa (GLUT). Một số thành viên của họ GLUT đã được phát hiện trong não. GLUT1 nằm ở hàng rào máu não, và mặc dù một số tế bào thần kinh biểu hiện GLUT2 và GLUT4, phần lớn sử dụng GLUT3 làm chất vận chuyển chính. Dạng đồng phân chính có mặt trong các mô đáp ứng với insulin là GLUT4.
Điều quan trọng là sự xâm nhập của glucose vào các tế bào não và quá trình chuyển hóa sau đó của nó không phụ thuộc vào insulin mà phụ thuộc vào nồng độ glucose trong huyết tương động mạch tuần hoàn. Do đó, sự giảm nồng độ glucose động mạch hoặc một khiếm khuyết trong cơ chế vận chuyển glucose của não sẽ dẫn đến tình trạng thiếu glucose trong não (intracerebral glucopenia) và nồng độ glucose trong dịch não tủy thấp (hypoglycorrhachia)—với các triệu chứng và dấu hiệu kèm theo của tình trạng thiếu glucose não như được mô tả sau đây. Ví dụ, sự thiếu hụt di truyền của chất vận chuyển glucose GLUT1 gây ra tình trạng hạ glucose dịch não tủy, co giật và chậm phát triển nghiêm trọng.
Ở người lớn bình thường và ở trẻ sơ sinh đủ tháng bình thường sau vài ngày đầu đời, nồng độ glucose huyết tương dao động từ 3,9 đến 5,6 mmol/L (70–100 mg/dL). Trong các thí nghiệm trên chuột, mức tiêu thụ glucose của não vượt quá khả năng vận chuyển glucose, dẫn đến nồng độ glucose trong não gần bằng không ở nồng độ glucose huyết tương là 2 mmol/L (36 mg/dL). Để ngăn nồng độ glucose huyết tương động mạch tuần hoàn giảm đột ngột trong các điều kiện sinh lý bình thường, và do đó để ngăn ngừa sự suy giảm chức năng sống còn phụ thuộc vào chuyển hóa glucose của não, một cơ chế bảo vệ phức tạp đã được tiến hóa. Cơ chế bảo vệ chống lại hạ đường huyết này được tích hợp bởi hệ thần kinh tự chủ và bởi các hormone hoạt động để tăng cường sản xuất glucose thông qua sự điều chỉnh enzyme của quá trình ly giải glycogen và tân tạo glucose, đồng thời hạn chế việc sử dụng glucose ở ngoại vi. Các cơ chế bảo vệ tương tự chống lại hạ đường huyết cũng hoạt động ở trẻ sơ sinh đủ tháng bình thường sau khi tách khỏi nhau thai. Do đó, hạ đường huyết là kết quả của một khiếm khuyết trong một trong những hệ thống này duy trì một phạm vi bình thường của nồng độ glucose huyết tương, ngăn chặn sự sụt giảm của nó xuống dưới 3,9 mmol/L (70 mg/dL) trong khi nhịn ăn và sự gia tăng của nó lên hơn 7,8 mmol/L (140 mg/dL) trong khi ăn.
Các cơ chế này, quan trọng nhất là sự điều hòa bài tiết insulin của tế bào β, chưa trưởng thành hoàn toàn khi sinh ra, khi có một sự chuyển đổi đột ngột từ đời sống trong tử cung sang đời sống ngoài tử cung, và nồng độ glucose huyết tương trung bình tạm thời giảm 25% trước khi phục hồi sau 2 ngày tuổi về các giá trị bình thường của người lớn. Trẻ sơ sinh được sinh ra với các yếu tố nguy cơ khác nhau, bao gồm ngạt khi sinh, chậm tăng trưởng trong tử cung (IUGR), nhiễm độc thai nghén của mẹ, và sinh non có thể đặc biệt dễ bị hạ đường huyết, thường có hậu quả đối với sự phát triển hoặc chức năng não bộ sau này mà có thể được ngăn ngừa bằng cách phát hiện và điều trị sớm.
Sinh lý của cân bằng nội môi glucose chu sinh
Chuyển hóa glucose ở bào thai
Glucose cung cấp 80% năng lượng cho bào thai, 20% còn lại chủ yếu có nguồn gốc từ quá trình oxy hóa lactate và các acid amin. Các acid béo tự do được vận chuyển qua nhau thai với số lượng hạn chế đủ cho sự phát triển của thai nhi, nhưng không đóng góp đáng kể vào nhu cầu năng lượng của thai nhi. Glucose được thai nhi sử dụng hoàn toàn có nguồn gốc từ tuần hoàn của mẹ thông qua khuếch tán thuận hóa qua nhau thai, chủ yếu sử dụng chất vận chuyển glucose GLUT1. Việc vận chuyển glucose qua nhau thai không liên quan đến chất vận chuyển glucose GLUT4, và do đó không được điều hòa bởi nồng độ insulin của mẹ hoặc thai nhi. Bởi vì việc vận chuyển glucose qua nhau thai diễn ra rất nhanh, nồng độ glucose của mẹ và thai nhi hoạt động như một khoang duy nhất và không có sản xuất glucose nội sinh ở thai nhi. Điều này có những hậu quả lâm sàng quan trọng, ví dụ, hạ đường huyết cấp tính ở mẹ sẽ dẫn đến hạ đường huyết cấp tính ở thai nhi—với rất ít hoặc không có khả năng thai nhi bù đắp cấp tính cho sự giảm đột ngột nguồn cung cấp glucose.
Từ đầu thai kỳ đến khi sinh, nồng độ glucose huyết tương trong động mạch của mẹ và tĩnh mạch rốn của thai nhi rất giống nhau. Sự khác biệt trung bình về nồng độ glucose huyết tương giữa mẹ và thai nhi về cơ bản là bằng không ở tuần thứ 20 của thai kỳ và chỉ tăng nhẹ lên 0,75 mmol/L (13 mg/dL) vào cuối thai kỳ. Sự gia tăng nhỏ trong chênh lệch giữa mẹ và thai nhi có thể phản ánh sự gia tăng sử dụng của nhau thai đang phát triển, bởi vì khoảng 50% đến 60% glucose của mẹ được hấp thu bị tiêu thụ bởi quá trình oxy hóa trong nhau thai và chỉ 40% đến 50% được vận chuyển đến thai nhi. Khi thai kỳ tiến triển, nhu cầu ngày càng tăng của thai nhi về oxy hóa và lưu trữ glucose được thực hiện bằng cách tăng lưu lượng máu tử cung. Có sự dư thừa công suất trong lưu lượng máu tử cung, và nó có thể bị giảm tới 50% mà không gây hại cho thai nhi. Tuy nhiên, việc giảm lưu lượng máu từ nhau thai đến thai nhi gây ra tác động đáng kể đến việc cung cấp glucose và các chất dinh dưỡng khác cho thai nhi, dẫn đến IUGR.
Trong nửa sau của thai kỳ, đơn vị thai-nhau thai tiết ra một lượng lớn lactogen nhau thai, progesterone và estrogen, gây ra tình trạng kháng insulin của mẹ tăng lên và có thể, ở những bà mẹ nhạy cảm, dẫn đến đái tháo đường thai kỳ. Sự bài tiết insulin của mẹ chịu trách nhiệm kiểm soát nồng độ glucose huyết tương ở mẹ và, trong khi insulin không đi qua nhau thai (trừ khi liên kết với kháng thể), insulin của mẹ cũng kiểm soát nồng độ glucose của thai nhi, do sự vận chuyển thuận hóa nhanh chóng của glucose qua nhau thai. Mặc dù insulin của thai nhi không kiểm soát nồng độ glucose của thai nhi, sự bài tiết insulin của thai nhi đáp ứng với những thay đổi trong nồng độ glucose của thai nhi. Vai trò của insulin thai nhi là duy trì tốc độ tăng trưởng nhanh chóng của thai nhi. Ví dụ, tăng đường huyết của mẹ (ví dụ, mẹ bị đái tháo đường) dẫn đến tăng insulin máu của thai nhi và tăng sự phát triển của thai nhi; ngược lại, hạ đường huyết của mẹ (như trong tình trạng suy dinh dưỡng của mẹ hoặc tăng insulin máu của mẹ do đột biến kích hoạt glucokinase) có thể ức chế sự bài tiết insulin của thai nhi, dẫn đến suy giảm sự phát triển của thai nhi.
Những thay đổi khi sinh: Giai đoạn chuyển tiếp
Sự gián đoạn đột ngột của việc vận chuyển glucose từ mẹ sang thai nhi khi sinh đòi hỏi trẻ sơ sinh phải tự kiểm soát nồng độ glucose huyết tương bằng cách điều chỉnh sự bài tiết insulin và huy động các phản ứng điều hòa ngược. Có bằng chứng cho thấy hầu hết các cơ chế điều hòa ngược, bao gồm bài tiết glucagon và epinephrine và kích hoạt quá trình ly giải glycogen và tân tạo glucose ở gan, đều có chức năng khi sinh. Tuy nhiên, có một giai đoạn chuyển tiếp ngay sau khi sinh khi nồng độ glucose huyết tương trung bình giảm ở trẻ sơ sinh bình thường từ 70 đến 80 mg/dL (gần với giá trị glucose của mẹ) xuống 55 đến 60 mg/dL trước khi trở lại trong khoảng 70 đến 100 mg/dL vào ngày thứ 2 đến 3 của cuộc đời, là mức bình thường cho đời sống ngoài tử cung. Hiện tượng này, được gọi là hạ đường huyết sơ sinh chuyển tiếp, ở trẻ sơ sinh bình thường đã được công nhận trong nhiều năm, mặc dù ý nghĩa lâm sàng của nó vẫn chưa chắc chắn, thậm chí còn gây tranh cãi. Ngày càng có nhiều bằng chứng cho thấy cơ chế của hạ đường huyết chuyển tiếp liên quan đến sự điều hòa bài tiết insulin của tụy và phản ánh sự tồn tại của ngưỡng glucose thấp hơn cho sự bài tiết insulin của tế bào β ở thai nhi, điều này cần thiết để duy trì sự phát triển của thai nhi. Bằng chứng cho thấy hạ đường huyết sơ sinh chuyển tiếp nên được coi là “tăng insulin máu sơ sinh chuyển tiếp” bao gồm các quan sát cho thấy, trong thời gian glucose huyết tương thấp ở trẻ sơ sinh bình thường, quá trình ly giải mỡ và sinh ceton bị ức chế và dự trữ glycogen ở gan được duy trì, như được thể hiện bằng các phản ứng đường huyết lớn được tạo ra khi tiêm glucagon hoặc epinephrine. Như đã thảo luận sau trong chương này, những phát hiện này là dấu hiệu chẩn đoán đặc trưng của hạ đường huyết do insulin. Nồng độ glucose huyết tương thấp hơn ở trẻ sơ sinh bình thường khá ổn định và tương đối không bị ảnh hưởng bởi thời gian nhịn ăn sau sinh lên đến 24 giờ, cho thấy rằng hạ đường huyết sơ sinh chuyển tiếp phản ánh một ngưỡng glucose thấp hơn cho sự bài tiết insulin. Các nghiên cứu gần đây trên chuột xác nhận một ngưỡng glucose thấp hơn cho sự bài tiết insulin trong giai đoạn ngay sau sinh. Cơ chế chịu trách nhiệm cho ngưỡng glucose thấp ở tế bào beta của thai nhi vẫn chưa rõ ràng, nhưng các giải thích được đề xuất bao gồm sự biểu hiện của các gen thường bị “cấm” trong tế bào beta, chẳng hạn như hexokinase-1, chất vận chuyển màng huyết tương cho pyruvate và lactate, và lactate dehydrogenase hoặc tăng độ nhạy của việc giải phóng hạt insulin đối với glucose thấp. Hiện tượng tăng insulin máu sơ sinh chuyển tiếp ở trẻ sơ sinh bình thường dường như thường lành tính, nhưng thường làm phức tạp việc nhận biết những trẻ có rối loạn hạ đường huyết dai dẳng, bệnh lý (xem phần sau).
Những thách thức khác đối với việc điều hòa nồng độ glucose huyết tương sau khi sinh bao gồm nhu cầu chuyển từ nguồn cung cấp glucose và acid amin liên tục từ tuần hoàn của mẹ sang việc ăn các chất dinh dưỡng bằng đường miệng thay đổi và gián đoạn. Ở trẻ bú mẹ, thành phần của các cữ bú cũng thay đổi trong những ngày đầu đời, với sữa non có hàm lượng carbohydrate thấp hơn và chất béo cao hơn sữa trưởng thành. Trẻ bú mẹ thường có thể không được nuôi dưỡng đầy đủ ban đầu và, ngoài giai đoạn tăng insulin máu sơ sinh chuyển tiếp, có thể phát triển tình trạng tăng ceton máu nhẹ khi nhịn ăn. Mặc dù tình trạng tăng ceton máu này trước đây thường được cho là do hàm lượng chất béo cao của sữa non (“chứng nhiễm ceton do bú mẹ”), nó dường như phản ánh một phản ứng đối với stress nhịn ăn nhẹ. Những suy giảm trong việc thích ứng với những thay đổi này trong nguồn cung cấp nhiên liệu ở trẻ sơ sinh có thể biểu hiện dưới dạng hạ đường huyết có triệu chứng, như được mô tả trong các phần sau về các rối loạn hạ đường huyết cụ thể ở trẻ sơ sinh và trẻ nhũ nhi sớm. Sự bài tiết insulin đáp ứng với glucose trong giai đoạn cuối của sự phát triển thai nhi, nhưng công suất tối đa có thể bị hạn chế, đặc biệt là ở trẻ sinh cực non cần nuôi dưỡng qua đường tĩnh mạch (IV).
Tóm lại, nồng độ glucose huyết tương trong đời sống trong tử cung là không đổi và được duy trì ở mức lớn hơn 3,9 mmol/L (70 mg/dL). Sau khi sinh, glucose huyết tương giảm xuống mức glucose trung bình từ 3,1 đến 3,3 mmol/L (56–60 mg/dL), trong 12 giờ đầu tiên do hậu quả của ngưỡng glucose thấp của tế bào beta đối với sự bài tiết insulin. Khi sự điều hòa insulin thích nghi với đời sống ngoài tử cung, nồng độ glucose tăng lên mức trung bình 3,5 mmol/L (63 mg/dL) vào 12 đến 24 giờ tuổi; đến 48 đến 72 giờ, glucose huyết tương tăng lên mức trung bình 4,1 mmol/L (74 mg/dL). Sự phân bố nồng độ glucose huyết tương ở trẻ sơ sinh ngay sau khi sinh không “bình thường” mà bị lệch về các giá trị thấp hơn do những trẻ có các yếu tố nguy cơ khác nhau đối với hạ đường huyết, như được mô tả sau. Vì lý do này, khoảng 30% trẻ sơ sinh “bình thường” sẽ có nồng độ glucose huyết tương thấp hơn 2,8 mmol/L (50 mg/dL) trong 8 đến 12 giờ đầu đời, nhưng sau đó tần suất nồng độ glucose lớn hơn 50 mg/dL giảm xuống chỉ còn 0,5% ở trẻ sơ sinh trên 24 giờ tuổi. Sự sụt giảm nồng độ glucose huyết tương này trong 24 giờ đầu đời là hạ đường huyết chuyển tiếp (tăng insulin máu sơ sinh chuyển tiếp), theo định nghĩa xảy ra ở trẻ sơ sinh khỏe mạnh bình thường. Nó nên được phân biệt với cả các nguyên nhân bệnh lý thoáng qua và dai dẳng của hạ đường huyết.
Các bất thường của giai đoạn chuyển tiếp
Các nghiên cứu về nồng độ glucose huyết tương ngay sau khi sinh ở trẻ sơ sinh bình thường cho thấy không chỉ nồng độ glucose trung bình thấp hơn, mà còn có phương sai rộng hơn và, như đã lưu ý trước đó, sự lệch của phân phối về các giá trị glucose. Điều này có nghĩa là giá trị trung bình đơn giản ± 2 độ lệch chuẩn (SDs) không phải là một thước đo hợp lệ của phạm vi glucose bình thường ở trẻ sơ sinh, mặc dù điều này được sử dụng làm cơ sở cho nhiều hướng dẫn về hạ đường huyết sơ sinh. Sự dư thừa của các nồng độ glucose thấp phản ánh thực tế rằng một tỷ lệ lớn trẻ sơ sinh có nguy cơ gia tăng về hạ đường huyết có khả năng bệnh lý trong giai đoạn tăng insulin máu sơ sinh chuyển tiếp. Những trẻ có nguy cơ này có tình trạng hạ đường huyết nghiêm trọng hơn và kéo dài hơn sau khi sinh, do sự bài tiết insulin của tụy và đã được gọi là tăng insulin máu do stress chu sinh. Tăng insulin máu do stress chu sinh xảy ra với một số rối loạn của thai nhi và mẹ, chẳng hạn như IUGR, ngạt khi sinh, mẹ bị đái tháo đường và tăng huyết áp của mẹ (Hộp 7.1). Liệu tăng insulin máu do stress chu sinh có đơn giản là một dạng quá mức và kéo dài hơn của tăng insulin máu sơ sinh chuyển tiếp hay không vẫn chưa được biết. Trẻ sơ sinh bị ảnh hưởng có thể cần điều trị để kiểm soát hạ đường huyết trong vài tuần hoặc vài tháng sau khi sinh. Trẻ sinh non có thể chưa phát triển đủ kho dự trữ glycogen ở gan và cũng có thể có sự chưa trưởng thành của các hệ thống enzyme gan cho tân tạo glucose và tổng hợp ceton, điều này làm tăng nguy cơ hạ đường huyết nhưng ở mức độ thấp hơn so với những trẻ bị stress chu sinh. Điều quan trọng đối với bác sĩ lâm sàng là nhận biết những trẻ sơ sinh có nguy cơ này và những trẻ sơ sinh có khả năng kiểm soát glucose nằm ngoài phạm vi hạ đường huyết chuyển tiếp trong giai đoạn này và điều trị một cách thích hợp. Đặc biệt, điều quan trọng là phải nhận ra các tình trạng trong đó sự bài tiết insulin tăng lên, vì những trẻ này sẽ không huy động được glycogen cũng như không thể oxy hóa acid béo, do đó khiến não của chúng có nguy cơ bị tổn thương do sự kết hợp của hạ đường huyết và hạ ceton máu.
| Hộp 7.1 Trẻ sơ sinh có nguy cơ hạ đường huyết dai dẳng (sửa đổi từ hướng dẫn của PES)
Trẻ của các bà mẹ bị đái tháo đường (bao gồm cả đái tháo đường thai kỳ) Trẻ to so với tuổi thai (LGA) (ngay cả khi không có mẹ bị đái tháo đường) Tăng insulin máu do stress chu sinh Chậm tăng trưởng trong tử cung (IUGR), nhỏ so với tuổi thai (SGA) Ngạt khi sinh; mổ lấy thai vì suy thai Tiền sản giật hoặc tăng huyết áp của mẹ Hội chứng hít phân su Bệnh nguyên bào hồng cầu thai nhi (Erythroblastosis fetalis) Đa hồng cầu Sinh non hoặc già tháng Tiền sử gia đình có dạng hạ đường huyết di truyền Các hội chứng bẩm sinh liên quan đến hạ đường huyết Hội chứng Beckwith-Wiedemann (đặc biệt là 11pUPD) Hội chứng Turner Hội chứng Kabuki Dị tật mặt đường giữa, dương vật nhỏ |
Quản lý hạ đường huyết trong 24 đến 48 giờ đầu
Việc quản lý hạ đường huyết trong 24 giờ đầu đời là quan trọng từ hai quan điểm. Thứ nhất, kế hoạch quản lý nên tránh điều trị cho những trẻ không cần điều trị. Thứ hai, nó nên bao gồm một kế hoạch cho những trẻ hoặc có hoặc có nguy cơ mắc rối loạn hạ đường huyết dai dẳng, mà kế hoạch này vừa xác định được những trẻ sơ sinh đó vừa điều trị cho chúng một cách đầy đủ. Hiện tại không khuyến nghị rằng tất cả trẻ sơ sinh đều phải được đo nồng độ glucose huyết tương. Tuy nhiên, nếu một trẻ có các triệu chứng phù hợp với hạ đường huyết—chẳng hạn như lơ mơ, ngưng thở, hoặc co giật, hoặc trẻ không khỏe hoặc thuộc bất kỳ nhóm nguy cơ nào (ví dụ, trẻ có anh chị em mắc các rối loạn hạ đường huyết đã biết, trẻ sinh non, trẻ to so với tuổi thai, trẻ nhỏ so với tuổi thai, trẻ của mẹ bị đái tháo đường, trẻ bị ngạt khi sinh, và các trẻ khác có các rối loạn được nêu trong Hộp 7.1)—nồng độ glucose của trẻ nên được đo.
Các khuyến nghị gần đây của Hiệp hội Nội tiết Nhi khoa cho rằng ngoài giai đoạn hạ đường huyết sơ sinh chuyển tiếp bình thường trong 24 đến 48 giờ đầu sau sinh, các ngưỡng glucose và mục tiêu quản lý nồng độ glucose huyết tương thấp nên giống nhau ở trẻ sơ sinh như ở trẻ lớn hơn. Ngoài ra, các tiêu chuẩn tương tự này được khuyến nghị cho trẻ sơ sinh có hoặc có nguy cơ cao mắc rối loạn hạ đường huyết dai dẳng (ví dụ, tiền sử gia đình hoặc đặc điểm thể chất của rối loạn hạ đường huyết, chẳng hạn như cân nặng lúc sinh to so với tuổi thai, gợi ý tăng insulin máu hoặc lưỡi to, và phì đại nửa người, gợi ý hội chứng Beckwith-Wiedemann [BWS]). Đối với trẻ sơ sinh được nghi ngờ không có rối loạn dai dẳng và đang được cho ăn bình thường, mục tiêu glucose được khuyến nghị để quản lý là duy trì glucose huyết tương trên 50 mg/dL trong 48 giờ đầu đời và trên 60 mg/dL sau 48 giờ, nếu tình trạng hạ đường huyết có khả năng giải quyết trong vòng một hoặc hai ngày.
Điều trị cấp cứu hạ đường huyết ở trẻ sơ sinh nên được điều chỉnh tùy theo sự hiện diện hay không có triệu chứng và mức độ hạ đường huyết. Đối với hạ đường huyết nhẹ không triệu chứng trong 48 giờ đầu đời, có thể thử cho ăn sớm hoặc dùng glucose đường uống ban đầu. Tuy nhiên, hạ đường huyết có triệu chứng hoặc nghiêm trọng nên được điều trị bằng dextrose IV (ví dụ, bolus 0,2 g/kg, sau đó là tốc độ truyền glucose [GIR] ít nhất 4–6 mg/kg/phút ban đầu, và điều chỉnh khi cần thiết để duy trì glucose > 70 mg/dL). Nếu hạ đường huyết được biết là do tăng insulin máu, cũng có thể xem xét dùng glucagon (0,5–1 mg IV, tiêm dưới da [SQ], hoặc tiêm bắp [IM]). Nên theo dõi thường xuyên glucose huyết tương để xác định xem có cần đánh giá chẩn đoán chính thức hay không.
Đến 48 giờ tuổi, giai đoạn hạ đường huyết sơ sinh chuyển tiếp thường đã kết thúc và nồng độ glucose huyết tương nên tương tự như trẻ lớn và người lớn: một phạm vi bình thường từ 70 đến 100 mg/dL. Do đó, nên xem xét đánh giá chính thức để chẩn đoán nguyên nhân hạ đường huyết sau 48 giờ tuổi đối với trẻ sơ sinh mà khả năng có rối loạn hạ đường huyết dai dẳng không thể được loại trừ hoàn toàn. Một đánh giá như vậy để chẩn đoán nguyên nhân hạ đường huyết nên được thực hiện trước khi cho bé xuất viện về nhà. Để giải quyết vấn đề những em bé có thể chưa đạt được sự giải quyết hoàn toàn của hạ đường huyết chuyển tiếp trước thời điểm xuất viện, các khuyến nghị của Hiệp hội Nội tiết Nhi khoa đề nghị xem xét một thử thách nhịn ăn để đảm bảo rằng glucose huyết tương có thể được duy trì an toàn trên 60 mg/dL trong hơn 6 đến 8 giờ (thử nghiệm “bỏ một cữ bú”). Điều này chỉ nên được xem xét cho những trẻ sơ sinh không có đặc điểm nguy cơ nào đối với một rối loạn hạ đường huyết dai dẳng (tức là, không có đợt hạ đường huyết có triệu chứng, không cần dextrose IV, không có đặc điểm nào gợi ý dạng hạ đường huyết di truyền hoặc hội chứng, hoặc hạ đường huyết do stress chu sinh, v.v.). Đối với tất cả các trẻ sơ sinh khác, nên xem xét một nghiên cứu nhịn ăn chính thức hoàn chỉnh. Cần lưu ý rằng không có bằng chứng nào chứng minh rằng các phác đồ này được Hiệp hội Nội tiết Nhi khoa khuyến nghị sẽ xác định được 100% trẻ sơ sinh, vì vậy cần thận trọng nếu các tình huống lâm sàng đảm bảo cần phải suy nghĩ thêm. Cuối cùng, điều quan trọng là phải nhấn mạnh với các bậc cha mẹ lo lắng về hậu quả của việc bỏ sót chẩn đoán một rối loạn hạ đường huyết. Theo kinh nghiệm của chúng tôi, hầu hết các bậc cha mẹ sẵn sàng kéo dài thời gian nằm viện thêm 6 đến 9 giờ để đảm bảo sự an toàn lâu dài cho con của họ.
Các hệ thống hormone và chuyển hóa của sự thích nghi với nhịn ăn
Hạ đường huyết ở trẻ sơ sinh, trẻ nhũ nhi và trẻ em về cơ bản luôn là một vấn đề với sự thích nghi khi nhịn ăn. Hạ đường huyết sau ăn là cực kỳ hiếm và chỉ giới hạn trong một vài tình huống bất thường, chẳng hạn như hạ đường huyết sau ăn sau phẫu thuật Nissen, không dung nạp fructose di truyền, hoặc hạ đường huyết do protein gây ra ở một số dạng tăng insulin máu bẩm sinh. Do đó, việc xem xét bốn con đường hormone và chuyển hóa chính duy trì cân bằng nội môi nhiên liệu trong khi nhịn ăn cung cấp một khuôn khổ quan trọng để hiểu nguyên nhân, chẩn đoán và điều trị các dạng hạ đường huyết khác nhau.
Ba hệ thống chuyển hóa điều chỉnh phản ứng sinh lý đối với việc nhịn ăn: (1) ly giải glycogen ở gan, (2) tân tạo glucose ở gan, và (3) sinh ceton ở gan. Các bước enzyme chính trong các con đường này được thể hiện trong Hình 7.1. Các hệ thống chuyển hóa này được phối hợp bởi (4) hệ thống nội tiết, bao gồm sự ức chế insulin (phản ứng nội tiết quan trọng nhất đối với việc nhịn ăn, vì insulin ức chế cả ba hệ thống chuyển hóa), và sự bài tiết của các hormone điều hòa ngược: glucagon, epinephrine (một dấu hiệu của sự kích hoạt hệ thần kinh giao cảm), cortisol và hormone tăng trưởng (GH). Bảng 7.1 tóm tắt các tác động đối trọng của các hormone điều hòa ngược này lên ba hệ thống chuyển hóa chính. Có một sự dự phòng theo thứ bậc trong sự tương tác của các hormone điều hòa ngược, cung cấp một mức độ an toàn (“cơ chế dự phòng”) nếu chỉ có một hormone điều hòa ngược bị suy giảm. Epinephrine (kích hoạt giao cảm) và glucagon tác dụng nhanh, mỗi loại báo hiệu tác dụng của nó bằng cách kích thích cyclic adenosine monophosphate (AMP). Sự thiếu hụt glucagon, như xảy ra trong bệnh đái tháo đường type 1 lâu năm, phần lớn có thể được bù đắp bởi một hệ thần kinh tự chủ nguyên vẹn với các tác động α- và β-adrenergic và cholinergic thích hợp. Ngược lại, suy giảm hệ thần kinh tự chủ có thể được bù đắp phần lớn nếu sự bài tiết glucagon vẫn còn nguyên vẹn.
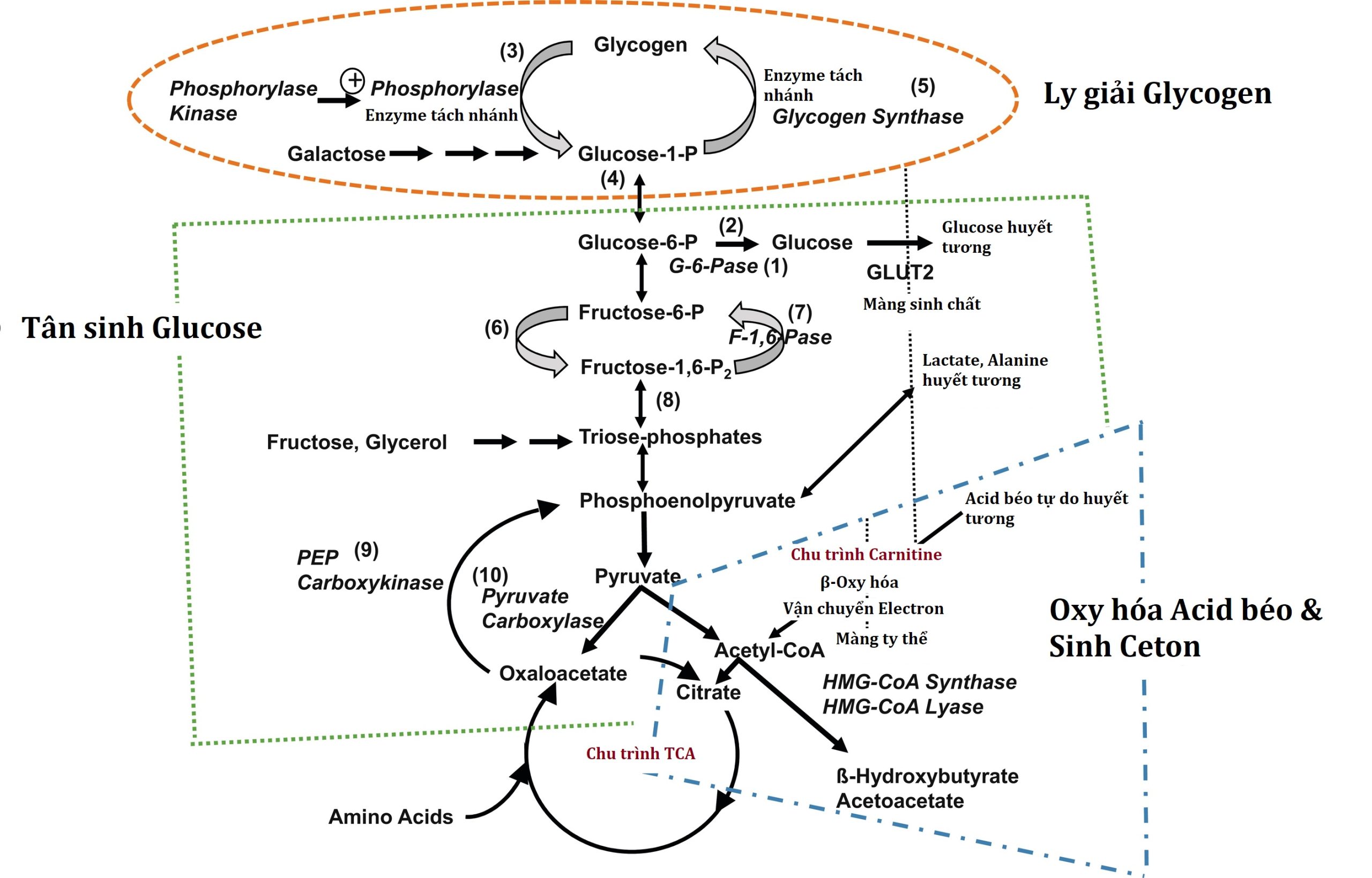
Hình 7.1 Các con đường chuyển hóa chính của chuyển hóa trung gian. Sự gián đoạn của các yếu tố trong các con đường này có thể là nguyên nhân gây bệnh trong sự phát triển của hạ đường huyết. Không hiển thị sự kiểm soát của hormone đối với các con đường này. Được chỉ ra là (1) glucose 6-phosphatase, (2) glucokinase, (3) phosphorylase, (4) phosphoglucomutase, (5) glycogen synthetase, (6) phosphofructokinase, (7) fructose 1,6-diphosphatase, (8) fructose 1,6-diphosphate aldolase, (9) phosphoenolpyruvate carboxykinase, và (10) pyruvate carboxylase.
Bảng 7.1 Sự điều hòa của Hormone đối với các Hệ thống Chuyển hóa khi Nhịn ăn
| Hormone điều hòa ngược | Ly giải Glycogen | Tân tạo Glucose | Ly giải Mỡ | Sinh Ceton |
|---|---|---|---|---|
| Insulin | Ức chế | Ức chế | Ức chế | Ức chế |
| Glucagon | Kích thích | Kích thích | Kích thích | |
| Cortisol | Kích thích | |||
| Hormone tăng trưởng | Kích thích | |||
| Epinephrine | Kích thích | Kích thích | Kích thích |
Quá trình ly giải glycogen ở gan chỉ đủ để đáp ứng nhu cầu năng lượng trong vài giờ. Sau thời gian đó, glucose phải được sản xuất bằng cách tân tạo glucose ở gan từ các tiền chất, chẳng hạn như acid amin, glycerol và lactate được tái chế từ quá trình ly giải đường. Nguồn chính của các tiền chất tân tạo glucose là các acid amin từ protein cơ. Mặc dù kho protein cơ lớn, nhưng nó cần thiết cho chức năng của cơ thể và do đó, trái ngược với các kho dự trữ glycogen trong gan và chất béo trong mô mỡ, không có “dự trữ” protein để sử dụng trong khi nhịn ăn. Để tiết kiệm việc sử dụng protein thiết yếu trong quá trình nhịn ăn kéo dài, việc tiêu thụ glucose phải bị ức chế bằng cách bật cơ chế huy động các acid béo từ các kho dự trữ triglyceride trong mô mỡ để oxy hóa trong cơ và các mô khác và glycerol làm chất nền cho quá trình tân tạo glucose ở gan. Các acid béo cũng được oxy hóa trong gan để tạo ra các thể ceton, β-hydroxybutyrate và acetoacetate, có thể được não sử dụng như một chất nền thay thế để tiết kiệm thêm việc tiêu thụ glucose.
Chức năng thiết yếu của sự thích nghi khi nhịn ăn là duy trì nguồn cung cấp nhiên liệu cho não. Cân bằng nội môi glucose rất hạn chế ở trẻ sơ sinh và trẻ nhũ nhi so với người lớn, một phần do dự trữ glycogen gan và protein cơ nhỏ hơn, nhưng chủ yếu là do tốc độ tiêu thụ glucose tương đối lớn hơn do tỷ lệ não trên khối lượng cơ thể lớn hơn. Ví dụ, kho dự trữ nhiên liệu của một trẻ 10 kg chỉ bằng 15% so với của người lớn. Tuy nhiên, nhu cầu calo bằng 60% của người lớn và tốc độ sử dụng glucose trên mỗi kg trọng lượng cơ thể lớn hơn 2 đến 3 lần. Như được thể hiện trong Hình 7.2, vào đầu giai đoạn nhịn ăn sau khi quá trình hấp thụ một bữa ăn hoàn tất, glucose là nhiên liệu chính của não và chiếm hơn 90% tổng lượng tiêu thụ oxy của cơ thể. Glucose ban đầu chủ yếu được cung cấp từ quá trình ly giải glycogen ở gan, được bổ sung bằng quá trình tân tạo glucose ở gan sử dụng các acid amin được giải phóng bởi sự luân chuyển protein cơ và lactate từ các mô ly giải đường, chẳng hạn như tế bào hồng cầu. Sau 8 đến 12 giờ ở trẻ nhũ nhi bình thường (24–36 giờ ở người lớn), sản xuất glucose giảm, vì nguồn cung cấp glycogen gan bị cạn kiệt và tốc độ tân tạo glucose từ các acid amin không đổi. Tại thời điểm này, một sự chuyển đổi sang chất béo làm nhiên liệu chính cho cơ thể bắt đầu, với sự ly giải mỡ trong mô mỡ tăng tốc và tăng quá trình oxy hóa acid béo trong cơ và sinh ceton trong gan. Quá trình ly giải mỡ cũng tạo ra glycerol, trở thành một chất nền tân tạo glucose quan trọng khi sự thích nghi với nhịn ăn hoạt động hoàn toàn. Não không thể sử dụng trực tiếp acid béo, vì chúng không đi qua hàng rào máu não. Tuy nhiên, não có thể thay thế việc tiêu thụ glucose bằng các ceton acetoacetate và β-hydroxybutyrate, được gan giải phóng như là sản phẩm cuối cùng của quá trình oxy hóa acid béo ở gan. Trong các giai đoạn cuối của sự thích nghi với nhịn ăn, quá trình oxy hóa acid béo và sử dụng ceton chiếm 90% tổng lượng tiêu thụ oxy của cơ thể.
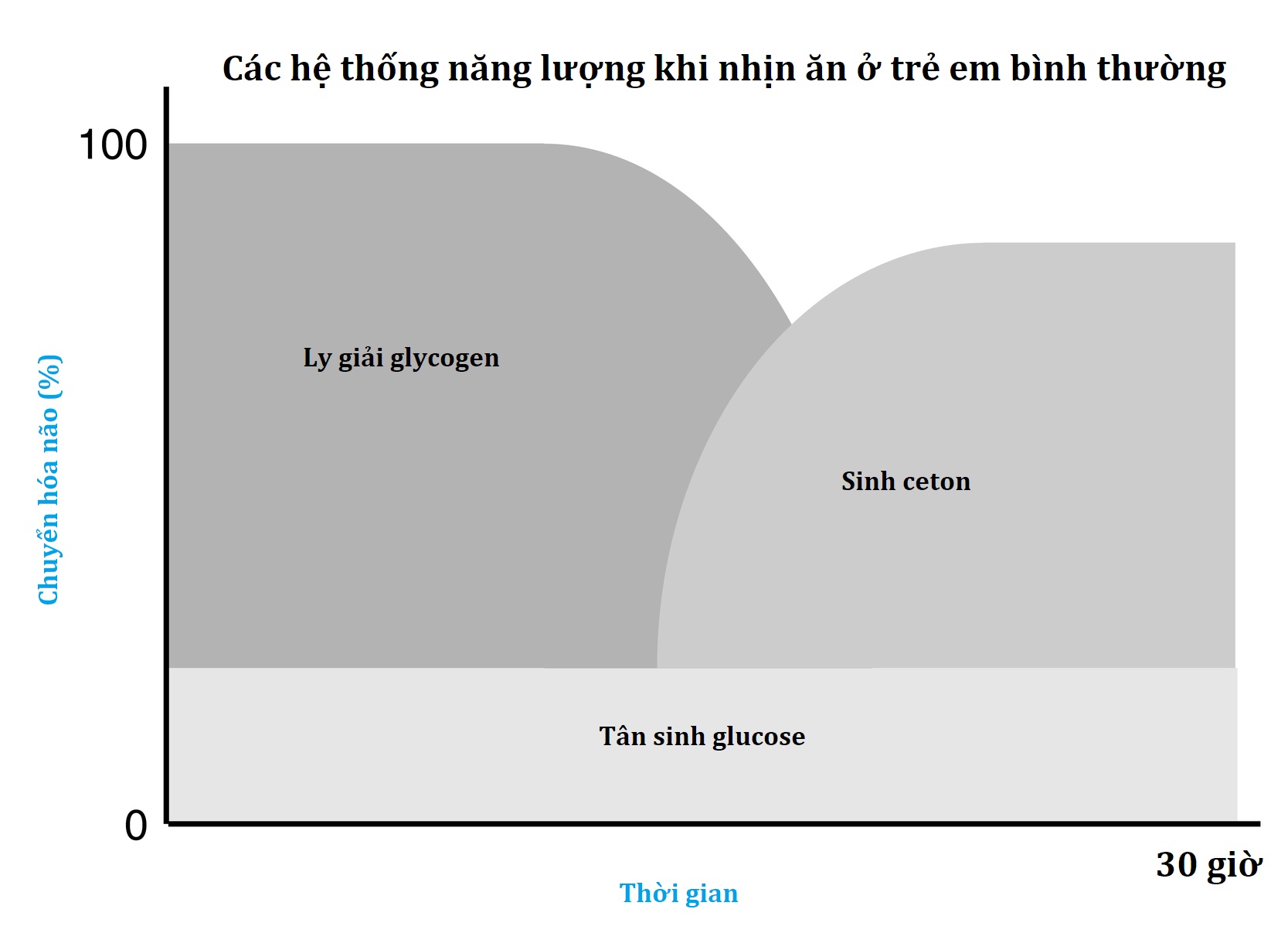
Hình 7.2 Sự đóng góp của các hệ thống nhịn ăn chính vào chuyển hóa não theo thời gian ở một trẻ nhũ nhi bình thường điển hình. Lưu ý rằng các kho dự trữ glycogen bị cạn kiệt sau 8 đến 12 giờ và quá trình sinh ceton trở thành nguồn chất nền chính cho não sau 24 giờ.
Hoạt động của các hệ thống chuyển hóa và nội tiết của sự thích nghi với nhịn ăn được chứng minh bằng những thay đổi trong nồng độ các nhiên liệu chuyển hóa và hormone trong tuần hoàn trong quá trình nhịn ăn. Như được thể hiện trong Hình 7.3, ở trẻ nhũ nhi, một cuộc nhịn ăn 24 giờ đi kèm với sự giảm dần nồng độ glucose huyết tương khi các kho dự trữ glycogen ở gan cạn kiệt, sự suy giảm dần nồng độ của chất nền tân tạo glucose (ví dụ, lactate, alanine) khi chúng được sử dụng cho quá trình tân tạo glucose ở gan, sự gia tăng nhanh chóng của các acid béo tự do khi quá trình ly giải mỡ được kích hoạt, và sự gia tăng đột ngột của β-hydroxybutyrate (ceton chính) khi quá trình sinh ceton ở gan được bật lên.
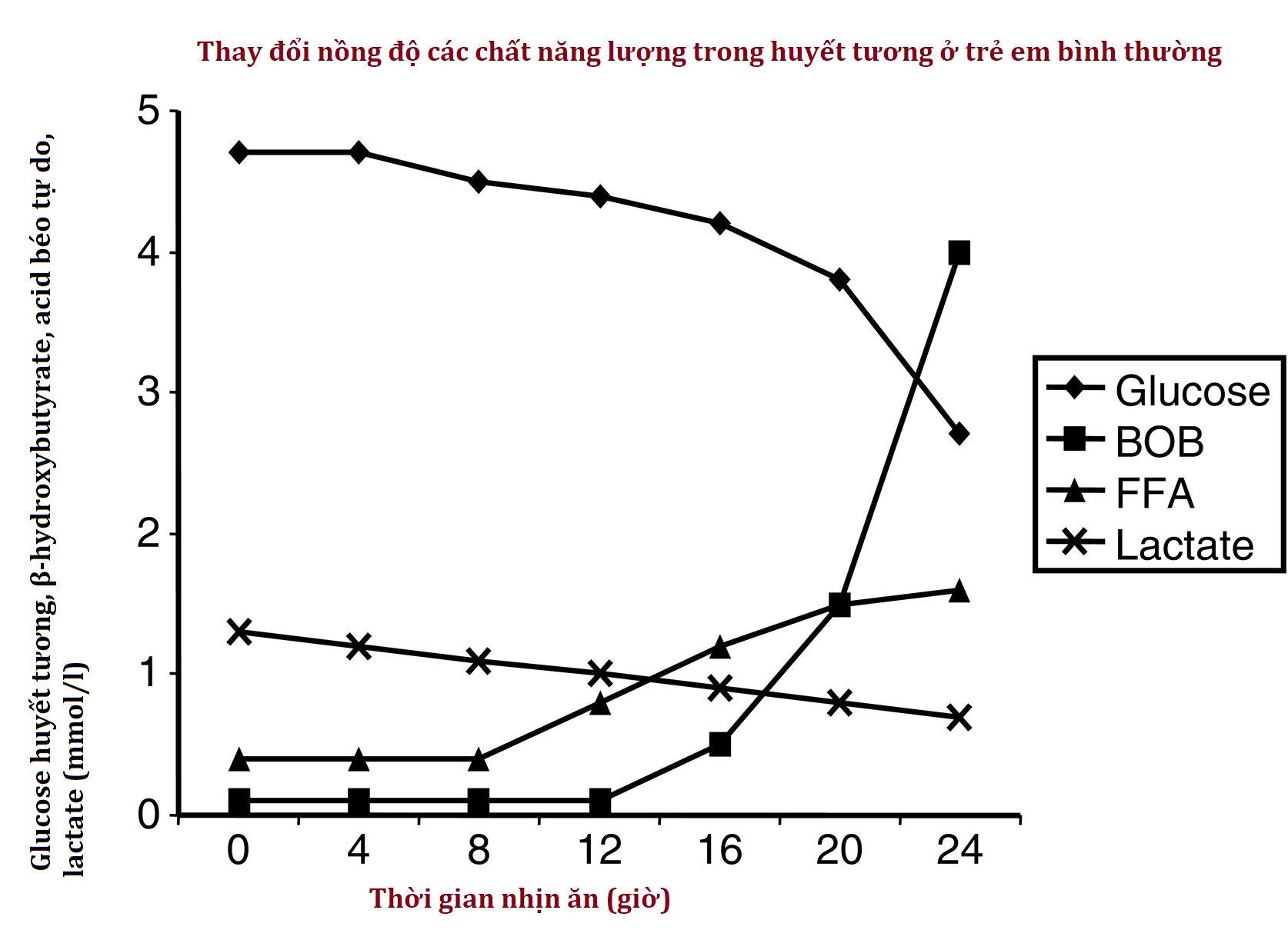
Hình 7.3 Ở trẻ nhũ nhi, một cuộc nhịn ăn 24 giờ đi kèm với sự giảm dần nồng độ glucose huyết tương khi các kho dự trữ glycogen ở gan cạn kiệt, sự suy giảm dần nồng độ của chất nền tân tạo glucose (ví dụ, lactate, alanine), khi chúng được sử dụng cho quá trình tân tạo glucose ở gan, sự gia tăng nhanh chóng của các acid béo tự do khi quá trình ly giải mỡ được kích hoạt, và sự gia tăng đột ngột của β-hydroxybutyrate (ceton chính) khi quá trình sinh ceton ở gan được bật lên.
Vào thời điểm nồng độ glucose huyết tương đã giảm xuống dưới 2,8 mmol/L (50 mg/dL), tất cả các hệ thống chuyển hóa và các phản ứng hormone được mô tả trước đó sẽ đã hoạt động hoàn toàn. Một “mẫu bệnh phẩm thời điểm quyết định” được lấy vào thời điểm này do đó sẽ cung cấp một cái nhìn tổng quan về sự toàn vẹn của bốn hệ thống chuyển hóa và nội tiết khi nhịn ăn (Bảng 7.2). Việc sử dụng các mẫu bệnh phẩm thời điểm quyết định này trong việc chẩn đoán nguyên nhân hạ đường huyết sẽ được thảo luận sau.
Bảng 7.2 Chẩn đoán phân biệt hạ đường huyết ở trẻ sơ sinh và trẻ nhũ nhi
| Rối loạn | Nhiên liệu huyết tương cuối giai đoạn nhịn ăn (mmol/L) | Hormone huyết tương cuối giai đoạn nhịn ăn | Khám thực thể |
|---|---|---|---|
| Glucose | Lactate | Acid béo tự do | |
| Trẻ nhũ nhi bình thường | <2.8 | 0.7–1.5 | 1.5–2.5 |
| Tăng insulin máu | <2.8 | B | < 1.7 |
| Thiếu hụt Cortisol | <2.8 | B | B |
| Thiếu hụt GH | <2.8 | B | B |
| Suy toàn bộ tuyến yên | <2.8 | B | B |
| Thiếu hụt Epinephrine (thuốc chẹn beta) | <2.8 | B | < 1.5 |
| Thiếu hụt enzyme debrancher (GSD III) | <2.8 | B | B |
| Thiếu hụt Phosphorylase (GSD VI) | <2.8 | B | B |
| Thiếu hụt Phosphorylase kinase (GSD IX) | <2.8 | B | B |
| Thiếu hụt Glycogen synthase (GSD 0) | <2.8 | B | B |
| Thiếu hụt Glucose 6-phosphatase (GSD Ia và Ib) | <2.8 | 4–8 + | B |
| “Thiếu hụt Fructose 1,6-diphosphatase” | <2.8 | 4–8 + | B |
| Thiếu hụt Pyruvate carboxylase | <2.8 | 4-8 + | B |
| Ly giải mỡ | |||
| “Loạn dưỡng mỡ bẩm sinh, rối loạn chức năng tự chủ gia đình, thuốc chẹn beta” | <2.8 | B | < 1.5 |
| Khiếm khuyết oxy hóa acid béo | <2.8 | B | > 2.5 |
GH, Hormone tăng trưởng; GSD, bệnh dự trữ glycogen; LGA, to so với tuổi thai; B, bình thường.
Định nghĩa hạ đường huyết ở trẻ sơ sinh và trẻ nhũ nhi
Định nghĩa về hạ đường huyết là một khái niệm quan trọng để hỗ trợ chẩn đoán và quản lý bệnh nhân nghi ngờ hoặc đã được chứng minh có rối loạn hạ đường huyết. Vì lý do này, chúng tôi khuyên bạn nên sử dụng các ngưỡng glucose huyết tương khác nhau cho các định nghĩa khác nhau. Ngưỡng chẩn đoán là mức glucose mà tại đó hoặc dưới mức đó có thể chẩn đoán được nguyên nhân của hạ đường huyết. Ngưỡng điều trị là mức glucose cho thấy việc điều trị một rối loạn điều hòa glucose là đầy đủ. Cuối cùng, có một vấn đề rất gây tranh cãi về mức glucose nào gây tổn thương não.
Một số yếu tố giả tạo có thể ảnh hưởng đến việc đo nồng độ glucose ở trẻ sơ sinh và trẻ nhũ nhi (Hộp 7.2). Nồng độ glucose trong máu toàn phần thấp hơn 10% đến 15% so với nồng độ glucose huyết tương vì hồng cầu có nồng độ protein (hemoglobin) cao hơn so với huyết tương có hàm lượng nước cao hơn, và do đó nồng độ glucose hòa tan cao hơn. Sự khác biệt có thể lớn hơn ở trẻ sơ sinh có hematocrit cao hơn. Các mẫu máu không được xử lý kịp thời có thể có mức glucose thấp sai lầm, do quá trình ly giải đường của tế bào hồng cầu và bạch cầu. Ở nhiệt độ phòng, sự suy giảm glucose trong máu toàn phần có thể là 5 đến 7 mg/dL/giờ. Việc sử dụng các chất ức chế, chẳng hạn như fluoride, trong các ống thu thập sẽ tránh được vấn đề này.
| Hộp 7.2. Các yếu tố ảnh hưởng đến việc đo nồng độ glucose huyết tương
• Nồng độ glucose máu toàn phần so với huyết tương (huyết tương cao hơn 10%–15%) • Thời gian giữa việc lấy mẫu và đo mẫu • Sự hiện diện hoặc không có các chất ức chế ly giải đường trong ống thu thập • Lấy mẫu từ các đường truyền nội mạch mà không rửa sạch đầy đủ (Sửa đổi từ Sacks, D.B. (1994). Carbohydrates. Trong: Burtis, C.A., Ashwood, E.R. (eds.), Sách giáo khoa Tietz về Hóa học lâm sàng, tái bản lần 2. WB Saunders, Philadelphia.) |
Các máy đo đường huyết tại giường bệnh viện và các máy đo đường huyết tại nhà tương tự kém chính xác hơn các phương pháp phòng thí nghiệm lâm sàng và có thể có sai số từ 10% đến 15%. Các phương pháp này cũng dễ bị lỗi, chẳng hạn như que thử hết hạn hoặc lấy mẫu không đủ—hầu hết các lỗi này dẫn đến giá trị glucose thấp giả. Vì lý do này, các máy theo dõi tại giường có thể được sử dụng cho mục đích sàng lọc—nhưng bất kỳ giá trị glucose nào dưới 3,3 mmol/L (60 mg/dL) đều phải được xác minh trong phòng thí nghiệm lâm sàng. Các giá trị glucose thấp (hoặc cao) giả có thể xảy ra với các mẫu được lấy từ các đường truyền nội mạch mà không rửa sạch đầy đủ dung dịch muối (hoặc glucose). Các mức thấp giả cũng có thể xảy ra ở trẻ sơ sinh bị bệnh nặng với tưới máu kém và việc sử dụng các máy đo tại giường trong môi trường chăm sóc đặc biệt chỉ được khuyến nghị cho các mẫu máu tĩnh mạch hoặc động mạch. Các máy theo dõi tại giường hiện đại sử dụng các mẫu máu toàn phần nhưng thường được hiệu chuẩn để đưa ra kết quả được biểu thị dưới dạng glucose huyết tương.
Ngưỡng Glucose chẩn đoán
Định nghĩa kinh điển của hạ đường huyết có triệu chứng là tam chứng Whipple—các triệu chứng, dấu hiệu hoặc cả hai phù hợp với hạ đường huyết, nồng độ glucose huyết tương thấp, và sự biến mất của các triệu chứng/dấu hiệu sau khi nồng độ glucose huyết tương được nâng lên. Ba tiêu chí này ban đầu được sử dụng để chẩn đoán u insulin ở người lớn. Tuy nhiên, ở trẻ sơ sinh và trẻ nhũ nhi, những đối tượng không thể truyền đạt các triệu chứng của mình một cách đáng tin cậy và các dấu hiệu lâm sàng của hạ đường huyết không đặc hiệu, có thể không thể thỏa mãn tam chứng Whipple. Trong những trường hợp này, việc nhận biết hạ đường huyết có thể cần được xác nhận bằng các phép đo nồng độ glucose huyết tương lặp đi lặp lại, hoặc kết hợp với các triệu chứng hoặc dấu hiệu của hạ đường huyết hoặc trong quá trình sàng lọc những trẻ có nguy cơ. Nếu mức độ được phát hiện dưới mức glucose gây ra các triệu chứng thần kinh (< 3,3 mmol/L hoặc < 60 mg/dL), sau 48 giờ đầu đời, cần tiến hành các cuộc điều tra sâu hơn để xác định nguyên nhân. Trong những trường hợp này, cần thực hiện các xét nghiệm kích thích chính thức để xác định xem trẻ sơ sinh có an toàn để xuất viện hay không hoặc để xác định nguyên nhân của hạ đường huyết.
Sau 48 giờ đầu đời, nồng độ glucose huyết tương bình thường ở trẻ sơ sinh và trẻ nhũ nhi không khác biệt so với trẻ lớn hơn và người lớn. Do đó, nồng độ glucose huyết tương trong trạng thái sau hấp thu dao động trong khoảng từ 3,9 đến 5,6 mmol/L (70 và 100 mg/dL), với giá trị trung bình từ 4,4 đến 4,7 mmol/L (80–85 mg/dL). Mức glucose huyết tương 2,8 mmol/L (50 mg/dL) thường được sử dụng làm điểm kết thúc cho các xét nghiệm chẩn đoán kích thích đối với hạ đường huyết. Giá trị này đủ thấp để kích thích mạnh mẽ các cơ chế bảo vệ nội tiết và chuyển hóa chống lại hạ đường huyết nhằm xác định cơ chế gây ra hạ đường huyết. Mức glucose giảm dần gây ra một chuỗi các phản ứng điển hình: nồng độ insulin huyết tương bắt đầu giảm khi glucose huyết tương giảm xuống khoảng 4,4 đến 4,7 mmol/L (80–85 mg/dL) và sự bài tiết insulin thường được “tắt” ở nồng độ glucose dưới 2,5 đến 3 mmol/L (45–54 mg/dL); sự bài tiết glucagon tăng lên khi nồng độ glucose huyết tương ở khoảng 3,6 đến 3,9 mmol/L (65–70 mg/dL); các phản ứng epinephrine, cortisol và GH được kích hoạt trong khoảng 3,6 đến 3,9 mmol/L (65–70 mg/dL). Khi glucose huyết tương giảm xuống dưới 3,3 mmol/L (59 mg/dL), thời gian phản ứng thính giác và thị giác bị kéo dài và chức năng nhận thức bắt đầu suy giảm khi nồng độ glucose huyết tương giảm xuống dưới 2,5 đến 3,5 mmol/L (45–63 mg/dL), với một số sự thay đổi này phụ thuộc vào xét nghiệm được sử dụng.
Ngưỡng điều trị
Một khi chẩn đoán nguyên nhân của hạ đường huyết đã được thực hiện hoặc nghi ngờ, mục tiêu điều trị là duy trì nồng độ glucose huyết tương > 3,8 mmol/L (> 70 mg/dL). Mục tiêu này cho phép bệnh nhân mắc các rối loạn hạ đường huyết duy trì mức glucose trên ngưỡng cho các phản ứng điều hòa ngược và ngăn ngừa sự phát triển của tình trạng không nhận biết hạ đường huyết do hạ đường huyết lặp đi lặp lại. Đối với các rối loạn oxy hóa acid béo và tân tạo glucose, việc duy trì nồng độ glucose trong phạm vi này sẽ ngăn chặn sự tích tụ của các acid béo tự do và lactate, tương ứng.
Nồng độ glucose huyết tương trong khoảng từ 2,8 đến 3,9 mmol/L (50 và 70 mg/dL) nên được coi là dưới mức tối ưu và dưới mục tiêu điều trị hạ đường huyết và nên khiến người ta đánh giá xem có cần tăng cường điều trị hay không.
Tổn thương não do hạ đường huyết
“Mức độ glucose nào gây tổn thương não?” là một trong những câu hỏi thường được hỏi nhất. Thật không may, câu trả lời phức tạp hơn là glucose càng thấp, và thời gian glucose thấp càng lâu, nguy cơ càng tồi tệ. Tổn thương não có khả năng xảy ra do thiếu nhiên liệu trong các tế bào thần kinh. Tế bào thần kinh có nhiều nguồn năng lượng, phổ biến nhất là glucose, lactate và β-hydroxybutyrate. Các nghiên cứu về trẻ sơ sinh bình thường đã chỉ ra rằng sau 6 giờ nhịn ăn, ceton có thể chiếm 12% sản lượng năng lượng của não.
Để oxy hóa hoàn toàn các hợp chất này thành năng lượng, tế bào cần oxy và lưu lượng máu đầy đủ. Thật vậy, một trong những sự điều chỉnh sớm của não đối với hạ đường huyết (khoảng 30 mg/dL) là tăng lưu lượng máu não, cung cấp nhiều glucose và oxy hơn cho não. Các nghiên cứu đã chỉ ra rằng việc thêm tình trạng thiếu oxy, ngạt hoặc thiếu máu cục bộ vào hạ đường huyết làm tăng đáng kể mức độ tổn thương não so với chỉ hạ đường huyết đơn thuần, và sự hiện diện của hạ đường huyết làm tăng nguy cơ tổn thương não từ các mức độ hạ huyết áp thấp hơn. Có nhiều báo cáo cố gắng giải quyết các kết quả lâu dài liên quan đến nồng độ glucose 45 mg/dL so với 47 mg/dL so với 50 mg/dL và tất cả đều có sai sót vì không có báo cáo nào xem xét cả ba loại nhiên liệu. Do đó, người ta không thể đưa ra bất kỳ kết luận chắc chắn nào từ các bài báo hiện được công bố về mức độ glucose nào gây tổn thương não. Tuy nhiên, có bằng chứng cho thấy hạ đường huyết nặng có liên quan đến kết quả xấu với các nghiên cứu xem xét bệnh nhân có sự kết hợp của các bất thường hình ảnh não và kết quả thần kinh kém cho thấy 95% bệnh nhân có glucose dưới 30 mg/dL.
Điều tương tự cũng có thể đúng đối với sự phát triển của các triệu chứng thiếu glucose não, thường xảy ra trước khi tổn thương não xảy ra. Trong một số rối loạn, chẳng hạn như các khiếm khuyết trong quá trình sinh ceton, các dấu hiệu và triệu chứng có thể bắt đầu xuất hiện trong khi nhịn ăn ở nồng độ glucose huyết tương là 3,3 mmol/L (60 mg/dL). Mặt khác, một số bệnh nhân (những người bị thiếu hụt glucose 6-phosphatase gây ra GSD1) có thể có ít triệu chứng thiếu glucose não ở nồng độ glucose huyết tương thấp đến 1,1 đến 1,7 mmol/L (20–30 mg/dL) vì nồng độ lactate cao trong huyết tương của họ cung cấp một chất nền thay thế cho não.
Các tình trạng, chẳng hạn như tăng insulin máu có tỷ lệ tổn thương não rất cao (30%–50%), vì chúng bị thiếu glucose, β-hydroxybutyrate và lactate, trong khi trẻ sơ sinh bị GSD1 thường không được chẩn đoán cho đến khi chúng là trẻ mới biết đi và hiếm khi bị tổn thương não đáng kể.
Tóm lại, không có một ngưỡng cắt nào cho mức độ hạ đường huyết gây tổn thương não; thay vào đó, đó là một sự tương tác phức tạp của sự sẵn có của nhiên liệu, mức độ oxy và lưu lượng máu, tất cả kết hợp lại để duy trì sự toàn vẹn của não.
Các triệu chứng và dấu hiệu lâm sàng liên quan đến hạ đường huyết
Các đặc điểm lâm sàng của hạ đường huyết ở trẻ nhũ nhi có thể liên quan đến cả các thành phần thần kinh và thiếu glucose não (Hộp 7.3). Các triệu chứng thường tinh vi và không đặc hiệu, do đó phải duy trì một chỉ số nghi ngờ lâm sàng cao. Bất kỳ sự thay đổi nào trong tình trạng lâm sàng ở trẻ sơ sinh cho thấy sự thay đổi trong hành vi thần kinh, giảm nhiệt độ, thay đổi kiểu ăn, hoặc sự hiện diện của run rẩy phải được coi là một biểu hiện ban đầu có thể có của một đợt hạ đường huyết. Co giật hoặc ngưng thở phải luôn được coi là một biểu hiện có thể có của hạ đường huyết và glucose huyết tương nên được đo ngay lập tức.
| Hộp 7.3 Các dấu hiệu hạ đường huyết ở trẻ sơ sinh
Thần kinh (adrenergic hoặc cholinergic) Thiếu glucose não |
Tiếp cận chẩn đoán
Việc xác định nguyên nhân cơ bản cụ thể của hạ đường huyết là rất quan trọng để thiết lập phương pháp điều trị cụ thể và do đó ngăn ngừa các đợt hạ đường huyết tiếp theo. Chẩn đoán nên dựa trên sự kết hợp của dữ liệu thu được từ tiền sử, khám thực thể, các phát hiện xét nghiệm, và đặc biệt là các phản ứng hormone và nhiên liệu tại thời điểm hạ đường huyết.
Các yếu tố quan trọng từ tiền sử thai kỳ và chu sinh, ngoài tiền sử gia đình, là rất quan trọng để xác định sự tồn tại của các yếu tố nguy cơ có thể chỉ ra các chẩn đoán cụ thể, ví dụ như tăng insulin máu do stress chu sinh ở trẻ sơ sinh bị IUGR. Thông tin về thời gian nhịn ăn gây ra hạ đường huyết cũng hữu ích, ví dụ, khởi phát trong vòng vài giờ sau bữa ăn sẽ phù hợp với tăng insulin máu hoặc GSD I, trong khi khởi phát sau 10 đến 12 giờ sẽ phù hợp với một khiếm khuyết trong quá trình oxy hóa acid béo. Suy tuyến yên với thiếu hụt GH hoặc ACTH-cortisol có thể bị nghi ngờ bởi sự hiện diện của các dị tật mặt đường giữa, mắt nhỏ, hoặc dương vật nhỏ (thiếu hụt FSH/LH trong tử cung). Suy dinh dưỡng cũng là một đặc điểm nổi bật của GSD I hoặc bệnh dự trữ glycogen do thiếu hụt enzyme debrancher sau 3 tháng đầu đời. Cả hai rối loạn này đều liên quan đến gan to khổng lồ. Các kết quả bất thường của các xét nghiệm chức năng gan (transaminase) và tăng amoniac máu, có hoặc không có nồng độ creatine kinase tăng, sẽ gợi ý một rối loạn oxy hóa acid béo có thể xảy ra (xem Bảng 7.2).
Một cách tiếp cận chẩn đoán hợp lý là phân tích một biến cố hạ đường huyết như một sự thích nghi kém với việc nhịn ăn, và do đó, thông tin quan trọng nhất cần thiết để chẩn đoán, đến từ các xét nghiệm trên các mẫu máu và nước tiểu thu được tại thời điểm hạ đường huyết (còn được gọi là mẫu bệnh phẩm thời điểm quyết định). Hình 7.4 phác thảo một thuật toán để chẩn đoán các dạng hạ đường huyết khác nhau dựa trên các xét nghiệm phòng thí nghiệm có sẵn trên các mẫu bệnh phẩm thời điểm quyết định. Yếu tố phân biệt đầu tiên là đo lường tình trạng nhiễm toan tại thời điểm hạ đường huyết bằng cách sử dụng bicarbonate huyết thanh. Nếu tình trạng nhiễm toan là do sự gia tăng của các ketoacid (β-hydroxybutyrate và acetoacetate), các khả năng bao gồm một đứa trẻ bình thường bị nhịn ăn quá lâu, một khiếm khuyết trong quá trình ly giải glycogen (bệnh dự trữ glycogen loại 3), hoặc thiếu hụt hormone điều hòa ngược (suy tuyến yên). Nếu tình trạng nhiễm toan là do sự gia tăng của acid lactic, nên nghi ngờ một sự tắc nghẽn của quá trình tân tạo glucose (GSD I hoặc thiếu hụt fructose 1,6-diphosphatase hoặc uống rượu ethanol). Nếu không có tình trạng nhiễm toan (tức là, không có sự gia tăng bình thường của ceton) nhưng nồng độ acid béo tự do cao, nên nghi ngờ một khiếm khuyết trong quá trình oxy hóa acid béo và sinh ceton (ví dụ, thiếu hụt acyl-CoA dehydrogenase chuỗi trung bình [MCAD]). Nếu ceton không tăng một cách thích hợp nhưng nồng độ acid béo tự do cũng bị ức chế, nên nghi ngờ tăng insulin máu. Trong giai đoạn sơ sinh, các đặc điểm của tăng insulin máu có thể bị bắt chước bởi sự thiếu hụt hormone tuyến yên bẩm sinh.
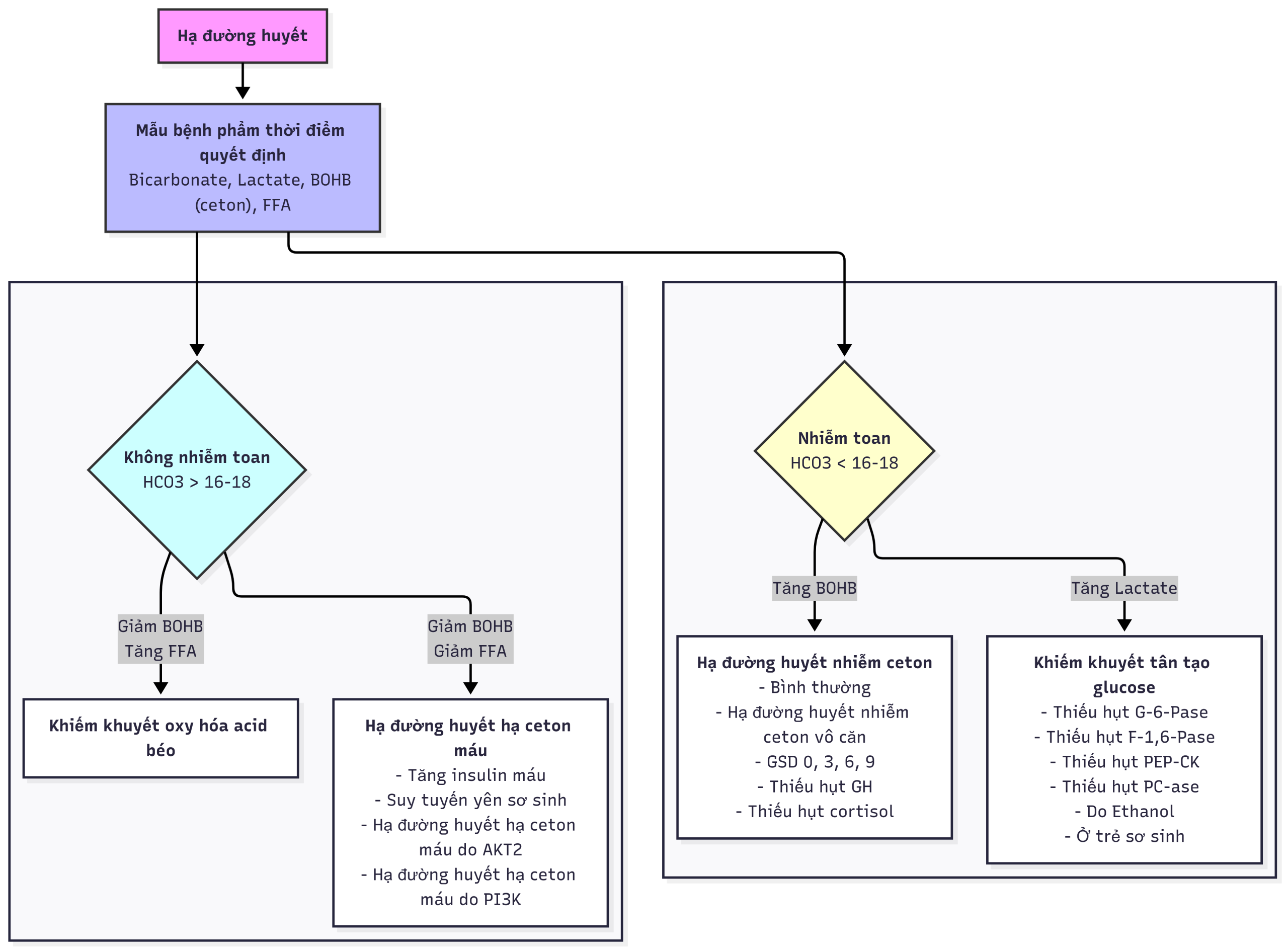
Hình 7.4 Thuật toán chẩn đoán hạ đường huyết dựa trên các xét nghiệm máu “thời điểm quyết định” thu được trong một đợt hạ đường huyết. BOHB, β-hydroxybutyrate; FFA, acid béo tự do; GSD, rối loạn dự trữ glycogen; HCO3, bicarbonate.
Các xét nghiệm sâu hơn sau đó có thể được lên kế hoạch bằng cách sử dụng các mẫu bệnh phẩm thời điểm quyết định ban đầu để xác nhận chẩn đoán nghi ngờ. Các xét nghiệm này có thể bao gồm các xét nghiệm sinh lý (chẳng hạn như xét nghiệm kích thích glucagon tại thời điểm hạ đường huyết để xác nhận tăng insulin máu) hoặc các xét nghiệm chuyên biệt (chẳng hạn như hồ sơ acylcarnitine huyết tương) để xác định một khiếm khuyết trong quá trình β-oxy hóa ở ty thể.
Nếu không có mẫu bệnh phẩm thời điểm quyết định thu được trong một đợt hạ đường huyết tự phát, có thể cần phải thực hiện một nghiệm pháp nhịn ăn chính thức để chẩn đoán nguyên nhân của hạ đường huyết. Mục tiêu của nghiệm pháp này là tái tạo bối cảnh mà hạ đường huyết xảy ra để xác định nguyên nhân cơ bản. Nghiệm pháp nhịn ăn nên được coi là một phương pháp kiểm tra một giả thuyết đã được phát triển, dựa trên dữ liệu lâm sàng và xét nghiệm có sẵn về nguyên nhân của hạ đường huyết. Do đó, nghiệm pháp có thể được sửa đổi với các bổ sung hoặc loại bỏ trong quy trình cơ bản. Điều này rất quan trọng, bởi vì việc thách thức một trẻ nhũ nhi bằng cách nhịn ăn không phải là không có rủi ro—đặc biệt nếu có một khiếm khuyết di truyền trong quá trình oxy hóa acid béo hoặc suy thượng thận. Do đó, việc nhịn ăn hoặc các thách thức chẩn đoán khác chỉ nên được thực hiện tại bệnh viện trong các điều kiện được kiểm soát cẩn thận với một bác sĩ và đội ngũ điều dưỡng có kinh nghiệm sẵn sàng. Trẻ nhũ nhi dưới 1 tuổi thường được nhịn ăn tối đa 24 giờ, trong khi ở trẻ lớn hơn, thời gian nhịn ăn tối đa có thể lên đến 36 giờ. Việc nhịn ăn được kết thúc khi glucose huyết tương giảm xuống dưới 2,8 mmol/L (50 mg/dL), nhưng nó có thể kết thúc sớm hơn nếu β-hydroxybutyrate huyết tương tăng lên hơn 2,5 mmol/L hoặc nếu có bất kỳ dấu hiệu hoặc triệu chứng bất lợi nào. Các mẫu máu định kỳ được lấy để phân tích các nhiên liệu và hormone chính và cho các xét nghiệm phụ trợ thích hợp (ví dụ, carnitine toàn phần trong huyết thanh, hồ sơ acylcarnitine, transaminase gan, creatine phosphokinase, hoặc các acid hữu cơ trong nước tiểu). Nếu nghi ngờ tăng insulin máu, nghiệm pháp nhịn ăn có thể được kết thúc bằng glucagon (1 mg tiêm IV) để đánh giá đáp ứng đường huyết.
Cần lưu ý đặc biệt rằng nguyên nhân thường gặp nhất của hạ đường huyết ở trẻ sơ sinh, trẻ nhũ nhi và trẻ em (tăng insulin máu) thường không thể được chẩn đoán chỉ dựa vào nồng độ insulin huyết tương. Với các xét nghiệm rất nhạy, nồng độ insulin huyết thanh sẽ nhỏ hơn 1 đến 2 μU/mL vào những thời điểm hạ đường huyết (tức là, dưới độ nhạy của hầu hết các xét nghiệm insulin). Tuy nhiên, nồng độ C-peptide huyết thanh có thể là một dấu hiệu hữu ích với các giá trị từ 0,5 ng/mL trở lên gợi ý mạnh mẽ đến tăng insulin máu, như được thể hiện trong Bảng 7.3. Do đó, chẩn đoán thường phải được thực hiện dựa trên bằng chứng về các tác động không phù hợp của insulin: hạ ceton máu, hạ acid béo tự do máu, và một đáp ứng đường huyết dương tính với glucagon (xem Bảng 7.3).
Bảng 7.3 Tiêu chí Chẩn đoán Tăng insulin máu Dựa trên các Mẫu bệnh phẩm “Thời điểm quyết định” (Lấy khi Glucose huyết tương nhỏ hơn 50 mg/dL)
|
Thông số |
Độ nhạy (%) | Độ đặc hiệu (%) |
|---|---|---|
| Insulin > 2 μU/mL | 82.2 | 100 |
| C-Peptide ≥ 0.5 ng/mL | 88.5 | 100 |
| β-hydroxybutyrate < 1.8 mmol/L | 100 | 100 |
| Acid béo tự do < 1.7 mmol/L | 87 | 100 |
| IGFBP-1 ≤ 110 ng/mL | 85 | 96.6 |
| Đáp ứng đường huyết với glucagon > 30 mg/dL | 89 | 100 |
IGFBP, Protein gắn yếu tố tăng trưởng giống insulin.
Phân loại các nguyên nhân gây hạ đường huyết dai dẳng ở trẻ sơ sinh và trẻ nhũ nhi (Hộp 7.4)
Tăng insulin máu và các rối loạn tương tự
Sự ức chế bài tiết insulin là rất quan trọng để duy trì đường huyết bình thường trong quá trình nhịn ăn thông qua việc kích hoạt quá trình ly giải glycogen, tân tạo glucose và oxy hóa acid béo ở gan. Thật vậy, việc không thể ức chế bài tiết insulin là nguyên nhân phổ biến nhất của hạ đường huyết dai dẳng ở trẻ sơ sinh và trẻ em. Các dấu hiệu lâm sàng và sinh hóa đặc trưng của sự gia tăng bài tiết/hành động của insulin ở trẻ sơ sinh là (1) cân nặng lúc sinh tăng do tác động thúc đẩy tăng trưởng của insulin trong tử cung; (2) tăng sử dụng glucose; và (3) bằng chứng cho thấy quá trình ly giải glycogen, ly giải mỡ và sinh ceton bị ức chế, được suy ra từ việc tìm thấy đáp ứng đường huyết với glucagon (tăng > 30 mg/dL), và sự ức chế nồng độ acid béo tự do và β-hydroxybutyrate trong huyết tương trong khi hạ đường huyết (xem Bảng 7.3).
| Hộp 7.4. Phân loại các nguyên nhân gây hạ đường huyết dai dẳng ở trẻ sơ sinh và trẻ nhũ nhi
A. Rối loạn do dư thừa Insulin hoặc hoạt động của Insulin
B. Khiếm khuyết trong phản ứng điều hòa ngược
C. Khiếm khuyết trong ly giải Glycogen và tân tạo Glucose 1. GSD Type 1 1. MCAD
ACTH, hormone vỏ thượng thận; FOXA2, forkhead box A2; GCK, glucokinase; GDH, glutamate dehydrogenase; GLUT, chất vận chuyển glucose; GSD, bệnh dự trữ glycogen; HNF, yếu tố nhân gan; MCAD, thiếu hụt acyl-CoA dehydrogenase chuỗi trung bình; SCHAD, 3-hydroxyacyl-CoA dehydrogenase chuỗi ngắn. |
Sự thiếu hụt các nhiên liệu thay thế (ceton) trong khi hạ đường huyết làm tăng nguy cơ tổn thương não ở trẻ nhũ nhi mắc các tình trạng này, do đó, điều cần thiết là phải nhanh chóng chẩn đoán và thiết lập điều trị. Chẩn đoán phân biệt và cơ chế bệnh sinh rất rộng, nhưng chúng có thể được nhóm thành hai loại: (1) tăng insulin máu do rối loạn điều hòa bài tiết insulin, và (2) các tình trạng bắt chước tăng insulin máu, trong đó sự bài tiết insulin của tế bào beta tụy được ức chế một cách thích hợp trong khi hạ đường huyết, nhưng tác động của insulin lên các mô đáp ứng với insulin lại bị bật lên một cách không phù hợp.
Tăng insulin máu
Tăng insulin máu là nguyên nhân phổ biến nhất của hạ đường huyết dai dẳng ở trẻ sơ sinh, trẻ nhũ nhi và trẻ em. Tăng insulin máu là do rối loạn điều hòa bài tiết insulin từ các tế bào β của tụy và có thể là kết quả của các yếu tố chu sinh (tăng insulin máu do stress chu sinh); đơn gen, do đột biến trong các gen quan trọng cho việc điều hòa bài tiết insulin; hoặc trong hội chứng (như trong BWS).
Tăng insulin máu thoáng qua do các yếu tố từ mẹ
Tăng insulin máu thoáng qua là một biến chứng được công nhận rõ ràng ở trẻ sơ sinh sau một thai kỳ có biến chứng đái tháo đường thai kỳ. Khi sinh, trẻ sinh ra từ những bà mẹ này có thể to và hồng hào—và các kho dự trữ glycogen, protein và chất béo trong cơ thể của chúng rất đầy đủ.
| Mô tả lâm sàng kinh điển về tác động của tăng insulin máu liên quan đến trẻ của mẹ bị đái tháo đường:
Những đứa trẻ này đáng chú ý không chỉ vì giống như những phiên bản bào thai của Shadrack, Meshack và Abednego, chúng ít nhất cũng sống sót ra khỏi lò lửa chuyển hóa của bệnh đái tháo đường, mà còn vì chúng giống nhau đến mức có thể là họ hàng. Chúng mũm mĩm, bóng bẩy, được phủ một lớp vernix caseosa dày, mặt tròn trịa và hồng hào. Dây rốn và nhau thai cũng to ra. Trong 24 giờ đầu đời hoặc hơn, chúng nằm ngửa, phù nề và đỏ bừng, chân gập và dạng ra, bàn tay nắm hờ hai bên đầu, bụng nổi rõ và hơi thở dài. Chúng tạo ấn tượng rõ rệt về việc đã được một bà chủ nhà nhiệt tình ép ăn uống quá nhiều đến mức chúng chỉ muốn yên tĩnh để có thể phục hồi sau những dư thừa của mình. Vào ngày thứ hai, sự khó chịu của chúng đối với tiếng ồn nhỏ nhất càng làm tăng thêm sự tương tự khi sự lo lắng run rẩy của chúng dường như nói lên những điều không thận trọng trong tử cung mà chúng ta không biết gì. |
Hạ đường huyết ở trẻ của mẹ bị đái tháo đường là do tăng insulin máu được kích hoạt bởi tăng đường huyết của mẹ. Trẻ sinh ra từ các bà mẹ bị đái tháo đường cũng có một sự tăng vọt glucagon huyết tương dưới mức bình thường ngay sau khi sinh, bài tiết glucagon dưới mức bình thường để đáp ứng với các kích thích, và (ban đầu) hoạt động giao cảm quá mức có thể dẫn đến suy kiệt tủy thượng thận vì sự bài tiết epinephrine qua nước tiểu bị giảm. Do đó, mặc dù có nhiều kho dự trữ chất nền trong mô, mô hình hormone huyết tương bình thường là insulin thấp, glucagon và catecholamine cao lại bị đảo ngược. Sản xuất glucose nội sinh của chúng bị ức chế và việc sử dụng glucose tăng lên so với trẻ bình thường, do đó khiến chúng dễ bị hạ đường huyết.
Các bà mẹ có bệnh đái tháo đường được kiểm soát tốt trong thai kỳ nói chung, có con gần như bình thường về kích thước, ít có khả năng bị hạ đường huyết sơ sinh và các biến chứng khác trước đây được coi là điển hình của bệnh đái tháo đường ở mẹ. Tuy nhiên, việc điều trị cho trẻ sinh ra từ các bà mẹ bị đái tháo đường thường đòi hỏi cung cấp glucose IV trong vài ngày, cho đến khi tình trạng tăng insulin máu giảm bớt.
Trong quá trình chuyển dạ và sinh, nên tránh tăng đường huyết của mẹ vì nó có thể dẫn đến tăng đường huyết của thai nhi—điều này dẫn đến hạ đường huyết phản ứng khi nguồn cung cấp glucose bị gián đoạn khi sinh. Các yếu tố khác của mẹ có thể dẫn đến tăng insulin máu sơ sinh thoáng qua bao gồm thuốc hạ đường huyết đường uống (chẳng hạn như sulfonylurea) hoặc các loại thuốc khác (terbutaline hoặc propranolol).
Theo định nghĩa, tăng insulin máu thoáng qua, là nguyên nhân gây hạ đường huyết sơ sinh ở trẻ của mẹ bị đái tháo đường, sẽ giảm bớt trong 1 hoặc 2 ngày. Nếu tình trạng này kéo dài, phải xem xét tăng insulin máu kéo dài hoặc bẩm sinh.
Tăng insulin máu do stress chu sinh
Quá trình trưởng thành của tế bào beta sau khi sinh có thể bị ảnh hưởng bởi các yếu tố chu sinh dẫn đến tăng insulin máu do stress chu sinh, một dạng tăng insulin máu riêng biệt tự khỏi trong vài tuần đầu đời. Các yếu tố chu sinh liên quan đến tăng insulin máu do stress chu sinh bao gồm ngạt khi sinh, tiền sản giật của mẹ, sinh non, chậm tăng trưởng trong tử cung và các căng thẳng khác trong giai đoạn chu sinh. Có tới 50% trẻ sơ sinh trong các nhóm nguy cơ này có thể bị ảnh hưởng. Tỷ lệ ước tính của tăng insulin máu do stress chu sinh là 1:6.000 đến 12.000 ca sinh sống.
Biểu hiện lâm sàng của tăng insulin máu do stress chu sinh được đặc trưng bởi việc sử dụng glucose cao, và phản ứng với hạ đường huyết khi nhịn ăn cho thấy insulin huyết tương có thể phát hiện được một cách không phù hợp, nồng độ β-hydroxybutyrate và acid béo tự do trong huyết tương thấp, và đáp ứng đường huyết không phù hợp với glucagon tại thời điểm hạ đường huyết. Không giống như tăng insulin máu thoáng qua thấy ở trẻ của mẹ bị đái tháo đường, tăng insulin máu do stress chu sinh có thể kéo dài trong vài tuần. Trong một loạt các trẻ sơ sinh bị tăng insulin máu do stress chu sinh, tuổi trung vị khỏi bệnh là 6 tháng. Cơ chế gây ra rối loạn điều hòa bài tiết insulin vẫn chưa được biết. Các phản ứng insulin cấp tính cho thấy, nói chung, các mô hình phản ứng insulin với các chất kích thích tiết (canxi, tolbutamide, glucose và leucine) ở trẻ bị tăng insulin máu do stress chu sinh giống với những người đối chứng bình thường.
Trẻ bị tăng insulin máu do stress chu sinh thường đáp ứng rất tốt với liệu pháp y tế bằng diazoxide. Trước đây, việc sử dụng liều dược lý của glucocorticoid để điều trị cho những trẻ sơ sinh bị hạ đường huyết dai dẳng là phổ biến. Tuy nhiên, việc sử dụng glucocorticoid như một liệu pháp không đặc hiệu cho hạ đường huyết sơ sinh không được khuyến khích vì chúng không chỉ không hiệu quả mà còn có thể ức chế trục hạ đồi-tuyến yên-thượng thận.
Tăng insulin máu đơn gen
Tăng insulin máu bẩm sinh, còn được gọi là hạ đường huyết tăng insulin máu dai dẳng ở trẻ nhũ nhi, đại diện cho một nhóm các rối loạn không đồng nhất về mặt lâm sàng và di truyền, đặc trưng bởi rối loạn điều hòa bài tiết insulin và dẫn đến hạ đường huyết nghiêm trọng và dai dẳng. Lần đầu tiên được mô tả vào năm 1954 bởi MacQuarrie là “hạ đường huyết vô căn ở trẻ nhũ nhi,” tăng insulin máu bẩm sinh là nguyên nhân phổ biến nhất của hạ đường huyết dai dẳng ở trẻ sơ sinh, trẻ nhũ nhi và trẻ em. Trên toàn thế giới, tỷ lệ mắc tăng insulin máu bẩm sinh được ước tính là 1 trên 50.000 ca sinh sống, với tỷ lệ cao hơn lên đến 1 trên 2500 ở những khu vực có tỷ lệ hôn nhân cận huyết cao.
Để hiểu sinh lý bệnh của tăng insulin máu bẩm sinh, kiến thức về các con đường chính điều chỉnh sự bài tiết insulin của tế bào β tụy là rất quan trọng (được phác thảo trong Hình 7.5). Sự bài tiết insulin được kích thích bởi glucose bao gồm sự xâm nhập của glucose vào tế bào beta bằng cách khuếch tán thuận hóa thông qua các chất vận chuyển glucose (chủ yếu là GLUT1 trong tế bào beta của người) và quá trình phosphoryl hóa bởi glucokinase (GK), dẫn đến quá trình oxy hóa glucose và sự gia tăng tỷ lệ ATP/adenosine diphosphate (ADP), dẫn đến việc đóng kênh kali phụ thuộc ATP (KATP) trên màng huyết tương. Kênh KATP của tế bào β là một phức hợp dị tám phân tử bao gồm hai tiểu đơn vị: một tiểu đơn vị tạo lỗ chọn lọc K+ (Kir6.2), và một tiểu đơn vị điều hòa (thụ thể sulfonylurea 1 [SUR-1]). Bốn tiểu đơn vị Kir6.2 tạo thành lỗ trung tâm, kết hợp với bốn tiểu đơn vị SUR-1. Kênh KATP bị ức chế (đóng) bởi các loại thuốc sulfonylurea (được sử dụng điều trị để kích thích bài tiết insulin trong bệnh đái tháo đường type 2), và được kích hoạt (mở) bởi diazoxide (phương pháp điều trị y tế hàng đầu cho tăng insulin máu bẩm sinh). Ở trạng thái không bị kích thích, các kênh kali nhạy cảm với ATP của tế bào β mở—giữ một điện thế màng nghỉ khoảng –65 mV. Sau khi hấp thu và chuyển hóa glucose, sự gia tăng tỷ lệ ATP/ADP nội bào dẫn đến việc đóng các kênh kali nhạy cảm với ATP, khử cực màng tế bào, mở các kênh Ca2+ phụ thuộc điện thế, dòng Ca2+ đi vào, tăng nồng độ Ca2+ tự do trong bào tương, và kích hoạt bộ máy xuất bào. Sự kích thích bài tiết insulin bởi các acid amin xảy ra thông qua sự hoạt hóa dị lập thể của glutamate dehydrogenase (GDH) bởi leucine, dẫn đến tăng quá trình oxy hóa glutamate—dẫn đến tăng tỷ lệ ATP/ADP, ức chế hoạt động của kênh KATP, và khử cực màng.
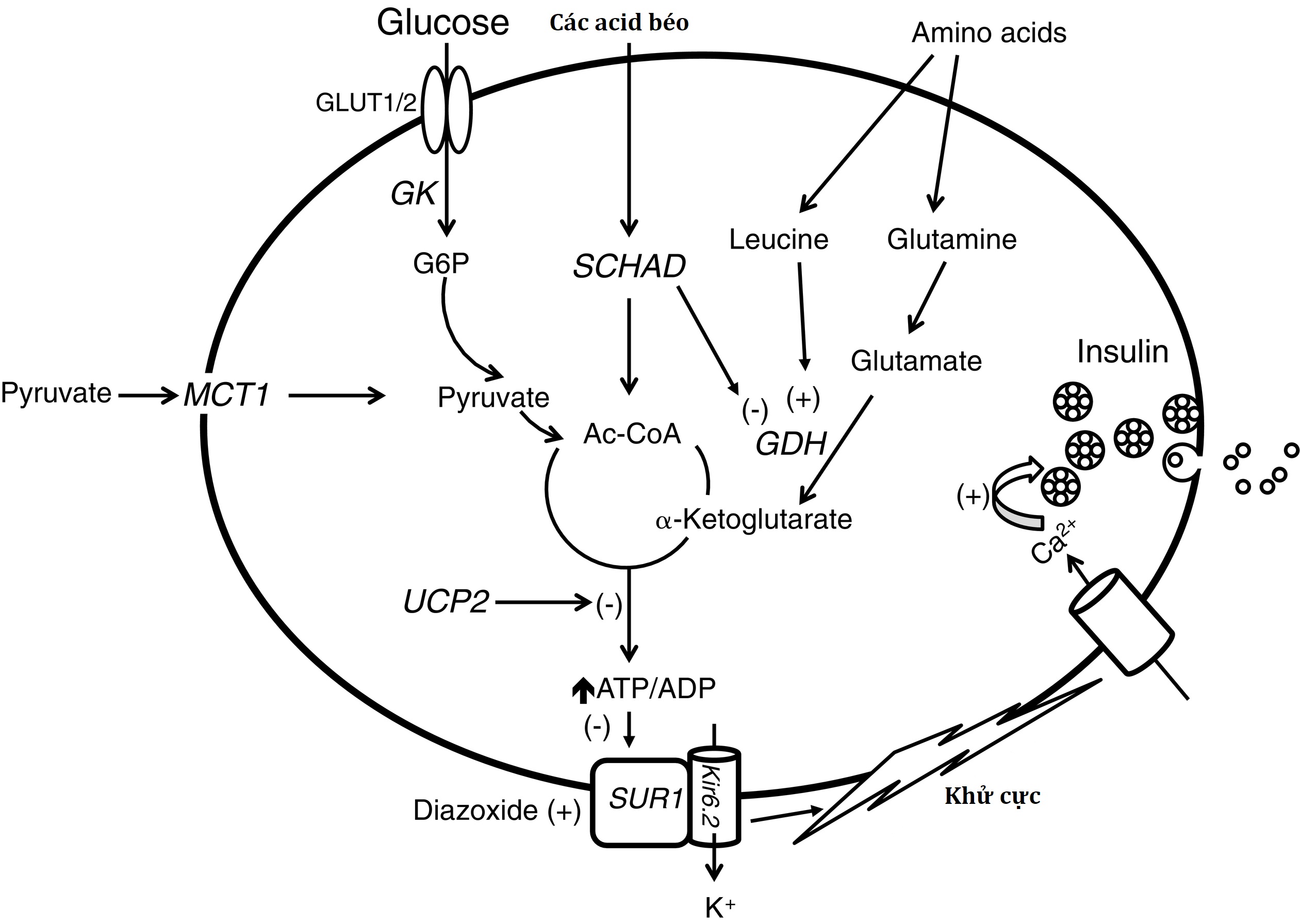
Hình 7.5 Mô hình hiện tại về các cơ chế bài tiết insulin của tế bào beta của tụy. Glucose được vận chuyển vào tế bào beta bởi chất vận chuyển glucose không phụ thuộc insulin GLUT2/GLUT1 trải qua quá trình phosphoryl hóa bởi glucokinase và sau đó được chuyển hóa, dẫn đến sự gia tăng tỷ lệ adenosine triphosphate/adenosine diphosphate (ATP/ADP). Sự gia tăng tỷ lệ ATP/ADP làm đóng kênh KATP và khởi đầu chuỗi các sự kiện được đặc trưng bởi sự gia tăng nồng độ kali nội bào, khử cực màng, dòng canxi vào, và giải phóng insulin từ các hạt lưu trữ. Leucine kích thích bài tiết insulin bằng cách hoạt hóa dị lập thể glutamate dehydrogenase (GDH) và bằng cách tăng quá trình oxy hóa glutamate, do đó làm tăng tỷ lệ ATP/ADP và đóng kênh KATP. Diazoxide ức chế bài tiết insulin bằng cách kích thích kênh KATP. Các khiếm khuyết gen được biết là gây ra tăng insulin máu được hiển thị bằng chữ nghiêng. Sáu là các đột biến bất hoạt: SUR1 (thụ thể sulfonylurea), Kir6.2 (kênh kali), SCHAD (3-OH acyl-CoA dehydrogenase chuỗi ngắn), UCP2 (protein tách cặp 2), HNF4A (yếu tố phiên mã nhân gan 4 alpha), và HNF1A (yếu tố phiên mã nhân gan 1 alpha). Ba là các đột biến kích hoạt: GK (glucokinase), GDH (glutamate dehydrogenase), và MCT1 (chất vận chuyển monocarboxylate 1). (–), Ức chế; (+), kích thích.
Các đột biến trong các gen mã hóa 10 yếu tố liên quan đến các bước khác nhau của con đường này đã được liên kết với tăng insulin máu bẩm sinh: SUR-1, một thành viên của siêu họ protein cassette gắn ATP, được mã hóa bởi ABCC8; Kir6.2, được mã hóa bởi KCNJ11; GK, được mã hóa bởi GCK; GDH, được mã hóa bởi GLUD1; enzyme ty thể 3-hydroxyacyl-CoA dehydrogenase chuỗi ngắn (SCHAD), được mã hóa bởi HADH; chất vận chuyển monocarboxylate-1 (MCT-1), được mã hóa bởi SLC16A1; protein tách cặp-2 (UCP2), được mã hóa bởi Ucp2; yếu tố nhân gan 4 alpha (HNF4A), được mã hóa bởi HNF4A; yếu tố nhân gan 1 alpha (HNF1A), được mã hóa bởi HNF1A; và forkhead box A2 (Foxa2), được mã hóa bởi FOXA2. Trong chương này, chúng tôi xem xét các dạng có nhiều khả năng xuất hiện trong giai đoạn sơ sinh. Để thảo luận về tăng insulin máu MCT-1, xem Chương 23.
Tăng insulin máu do kênh KATP
Các đột biến bất hoạt của các kênh KATP tế bào beta là nguyên nhân của dạng tăng insulin máu bẩm sinh phổ biến và nghiêm trọng nhất. Theo mức độ nghiêm trọng của khiếm khuyết phân tử và kiểu hình, KATP HI có thể được phân thành ba loại phụ: (1) lặn, không đáp ứng với diazoxide; (2) trội, không đáp ứng với diazoxide; và (3) trội, đáp ứng với diazoxide. Các đột biến lặn cản trở sự biểu hiện protein hoặc vận chuyển kênh, về cơ bản dẫn đến sự vắng mặt hoàn toàn của các kênh trên màng huyết tương, và do đó diazoxide không hiệu quả. Các đột biến lặn là các khiếm khuyết phổ biến nhất được xác định ở trẻ em bị tăng insulin máu bẩm sinh. Các đột biến trội cho phép vận chuyển bình thường các kênh đến màng huyết tương, nhưng làm suy giảm hoạt động của kênh hoàn toàn hoặc một phần, dẫn đến một phổ kiểu hình từ nặng, không đáp ứng với diazoxide đến nhẹ, đáp ứng với diazoxide.
Biểu hiện lâm sàng của KATP HI sau đó phụ thuộc vào mức độ nghiêm trọng của tác động đột biến. Thông thường hơn, những đứa trẻ này to so với tuổi thai và bị hạ đường huyết sơ sinh nghiêm trọng không đáp ứng với diazoxide, ngoại trừ các trường hợp có kiểu hình phụ trội đáp ứng với diazoxide trong đó cân nặng lúc sinh là bình thường và biểu hiện lâm sàng có xu hướng xảy ra muộn hơn trong cuộc đời.
Sinh lý bệnh của KATP HI đã được minh họa bằng đánh giá chức năng của tiểu đảo tụy trong cơ thể sống và trong ống nghiệm. Sự vắng mặt của các kênh KATP chức năng dẫn đến sự tách rời giữa glucose huyết tương và sự bài tiết insulin hoặc “sự mù glucose” của tế bào β. Do đó, sự bài tiết insulin không bị tắt, khi glucose huyết tương giảm và không tăng để đáp ứng với sự tăng nhanh glucose huyết tương (Hình 7.6). Khiếm khuyết đầu tiên chi phối các biểu hiện lâm sàng ở trẻ nhũ nhi sớm, trong khi khiếm khuyết sau có thể đóng một vai trò trong sự phát triển sau này của tình trạng không dung nạp glucose và, có thể, bệnh đái tháo đường. Trái ngược hoàn toàn với việc không đáp ứng với sự thay đổi nồng độ glucose huyết tương, các tế bào β này lại phản ứng quá mức với sự kích thích bằng các acid amin, dẫn đến hạ đường huyết do protein gây ra.
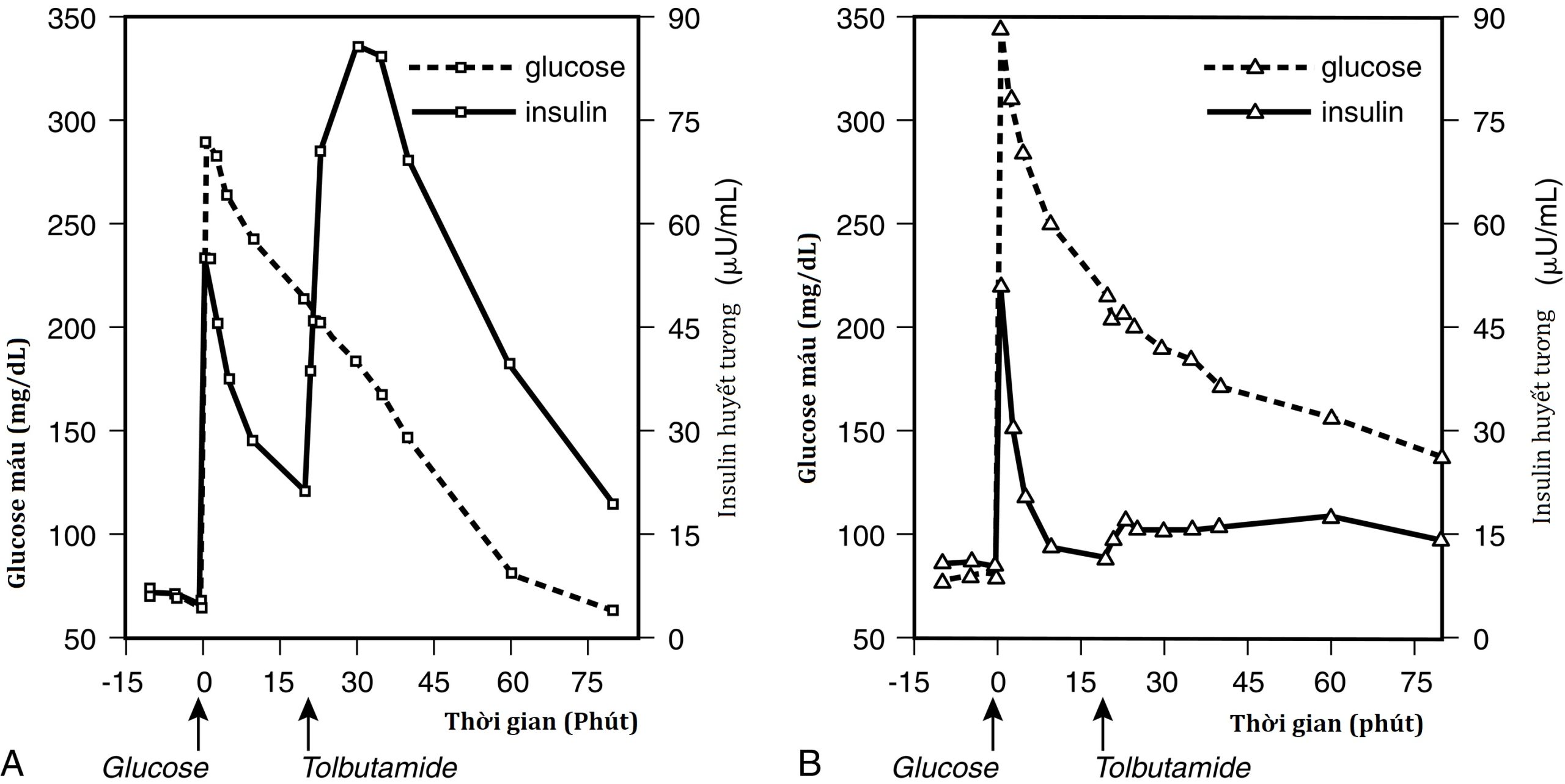
Hình 7.6 Đáp ứng insulin cấp tính với glucose và tolbutamide ở trẻ em bị tăng insulin máu KATP lan tỏa (gia số trung bình 11 và 13 phút). A, Người lớn đối chứng bình thường. B, Bệnh nhân bị tăng insulin máu KATP lan tỏa.
Các đặc điểm lâm sàng khác của trẻ nhũ nhi bị KATP HI bao gồm nhu cầu glucose cực kỳ cao, thường cao gấp 4 đến 5 lần so với bình thường để kiểm soát glucose huyết tương (mặc dù trong một số trường hợp nhu cầu có thể bình thường), biếng ăn và trào ngược dạ dày thực quản, có thể là hậu quả của việc ép ăn. Các lựa chọn điều trị cho các trường hợp không đáp ứng với diazoxide bị hạn chế. Những trẻ nhũ nhi này có thể cần phải cắt bỏ tụy, trong vòng vài tuần đầu sau khi sinh, để quản lý tình trạng hạ đường huyết. Mặt khác, diazoxide rất hiệu quả trong việc kiểm soát hạ đường huyết trong các trường hợp KATP HI trội đáp ứng với diazoxide. Octreotide, một chất tương tự somatostatin tác dụng kéo dài, là một liệu pháp y tế hàng thứ hai cho trẻ nhũ nhi không đáp ứng với diazoxide, mặc dù Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA) chưa chấp thuận nó cho chỉ định này, và do những lo ngại về mối liên quan có thể có với viêm ruột hoại tử, việc sử dụng nó cần được xem xét cẩn thận. Một phương pháp tiếp cận không phẫu thuật trong tăng insulin máu không đáp ứng với diazoxide bao gồm octreotide kết hợp với việc cho ăn qua đường ruột liên tục hoặc thường xuyên hoặc dextrose liên tục qua ống thông dạ dày. Phương pháp tiếp cận này là một thách thức đối với việc quản lý tại nhà của những đứa trẻ này, và nó đòi hỏi sự giám sát chặt chẽ và kiểm tra glucose thường xuyên.
Có hai dạng mô học đặc trưng của tăng insulin máu do kênh KATP, tăng insulin máu khu trú và lan tỏa.
Tăng insulin máu KATP khu trú (U tuyến khu trú)
Khoảng 40% đến 60% các trường hợp KATP HI (cần phẫu thuật) có bệnh khu trú. Các tổn thương khu trú phát sinh bởi một cơ chế “hai đòn”: (1) một đột biến được thừa hưởng từ cha của gen ABCC8 hoặc KCNJ11 (nằm trên nhiễm sắc thể 11p15), và (2) một sự mất đoạn của vùng nhiễm sắc thể 11p15 được thừa hưởng từ mẹ, được bù đắp bởi một disomy đơn cha, như quan sát thấy trong một số trường hợp BWS. Vùng 11p15, mang các gen ABCC8 và KCNJ11, chứa một số gen được in dấu: yếu tố tăng trưởng giống insulin 2 (IGF2), một protein thúc đẩy tăng trưởng được biểu hiện từ nhiễm sắc thể của cha, trong khi đó, H19, một ribonucleic acid (RNA) không mã hóa dài ức chế tăng trưởng và CDKN1C, một chất điều hòa chu kỳ tế bào và ức chế tăng trưởng, được biểu hiện từ nhiễm sắc thể của mẹ. Mất các gen ức chế tăng trưởng này, trong khi gen thúc đẩy tăng trưởng của cha hoạt động, đóng một vai trò cho phép quan trọng trong sự mở rộng dòng của các tế bào biểu hiện kênh bị đột biến.
Tăng insulin máu lan tỏa
Trong tăng insulin máu lan tỏa, tất cả các tế bào β trong tụy đều bị ảnh hưởng. Nó là kết quả của việc thừa hưởng hai đột biến lặn trong ABCC8 hoặc KCNJ11 hoặc một đột biến trội trong các gen này.
Trẻ sơ sinh bị tăng insulin máu lan tỏa có nhiều khả năng biểu hiện ngay khi sinh, trong khi những trẻ bị dạng khu trú có thể không được phát hiện trong giai đoạn sơ sinh và biểu hiện sau vài tuần đến vài tháng tuổi với co giật do hạ đường huyết. Tuy nhiên, do sự chồng chéo đáng kể trong biểu hiện lâm sàng, chỉ riêng các đặc điểm lâm sàng không thể được sử dụng để phân biệt giữa hai dạng. Xét nghiệm di truyền cung cấp phương tiện tốt nhất để xác định trẻ nhũ nhi bị KATP-HI khu trú: một đột biến lặn duy nhất được thừa hưởng từ cha trong ABCC8 hoặc KCNJ11 có giá trị tiên đoán dương tính là 94% đối với tăng insulin máu khu trú.
Về mặt mô học, tăng insulin máu lan tỏa và khu trú là khác biệt. Tăng insulin máu lan tỏa được đặc trưng bởi sự hiện diện của các nhân tế bào tiểu đảo lớn bất thường phân bố khắp tụy. Ngược lại, mô học tụy trong tăng insulin máu khu trú được đặc trưng bởi một tổn thương được hình thành bởi sự hợp lưu của các tiểu đảo tăng sản chiếm hơn 40% diện tích mặt cắt ngang của các tiểu thùy tụy. Trái ngược với các u tuyến thực sự, tăng sản dạng u tuyến khu trú bao gồm các tế bào nang ngoại tiết xen kẽ trong tổn thương. Hình thái của các tiểu đảo cách xa tổn thương khu trú là bình thường. Khả năng giải thích các đặc điểm mô học này đòi hỏi đào tạo chuyên biệt và chỉ có ở các trung tâm có các đội ngũ đa ngành chuyên biệt để đánh giá và điều trị tăng insulin máu.
KATP HI khu trú có thể chữa khỏi bằng phẫu thuật, trong khi phẫu thuật chỉ có tính chất giảm nhẹ đối với KATP HI lan tỏa. Do đó, những nỗ lực để chẩn đoán và định vị các tổn thương khu trú ở trẻ nhũ nhi bị tăng insulin máu không đáp ứng với diazoxide trước khi phẫu thuật là rất quan trọng. Các kỹ thuật hình ảnh thông thường, chẳng hạn như chụp cắt lớp vi tính (CT) hoặc chụp cộng hưởng từ, không thể phát hiện các tổn thương khu trú. Các nghiên cứu X-quang can thiệp, chẳng hạn như lấy mẫu insulin tĩnh mạch cửa qua gan và kích thích canxi động mạch tụy chọn lọc, chỉ có thành công khiêm tốn, khó khăn về mặt kỹ thuật và có tính xâm lấn cao. Kỹ thuật tiêu chuẩn vàng để định vị các tổn thương khu trú là chụp cắt lớp phát xạ positron (PET) với flurorine-18 L-3, 4-dihydroxyphenylalanine (18F-fluoro-L-DOPA). Tế bào β của tụy hấp thụ L-DOPA, và DOPA decarboxylase hoạt động trong các tế bào tiểu đảo tụy. Ở trẻ em bị tăng insulin máu khu trú, có sự tích tụ cục bộ của 18F-fluoro-L-DOPA (Hình 7.7). Việc đồng đăng ký hình ảnh PET và CT cho phép định vị giải phẫu của tổn thương (Hình 7.8).
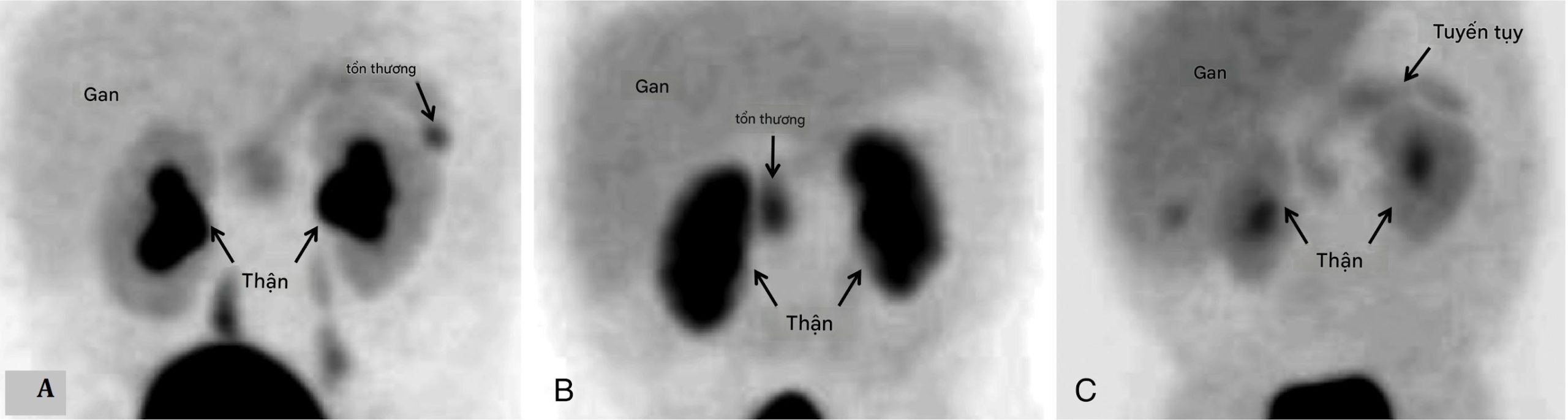
Hình 7.7 Hình ảnh chụp PET 18Fluoro-L-DOPA cho thấy sự hấp thu L-DOPA ở gan, thận và tụy. Lưu ý sự hấp thu tăng lên ở đuôi (A) và đầu (B) của tụy cho thấy các tổn thương khu trú. Ngược lại, trong tăng insulin máu lan tỏa, sự hấp thu trên toàn bộ tụy là đồng nhất (C).
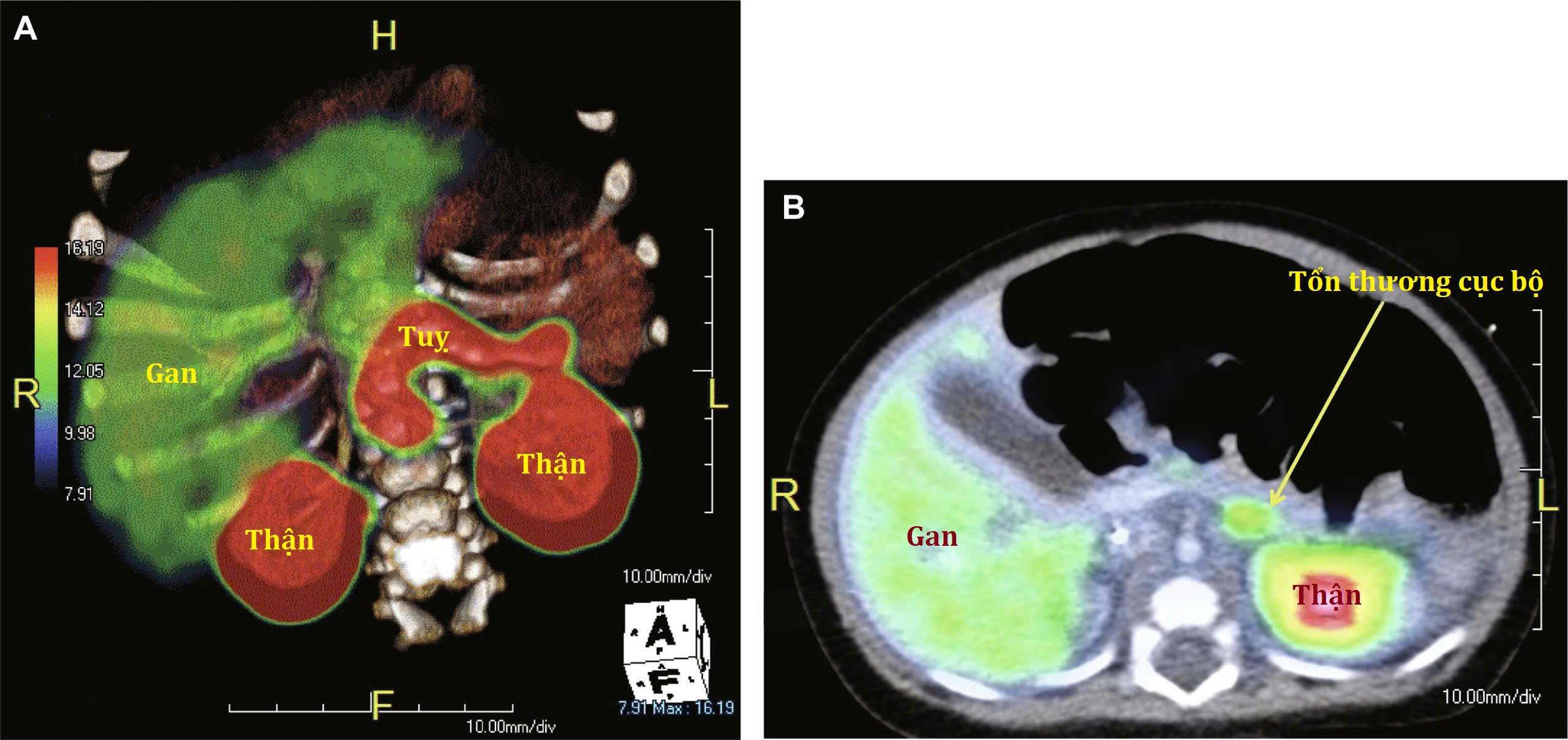
Hình 7.8 A, Chế độ xem phía trước của hình ảnh chụp cắt lớp phát xạ positron (PET)/cắt lớp vi tính (CT) hợp nhất của một trường hợp tăng insulin máu lan tỏa cho thấy sự hấp thu L-3, 4-dihydroxyphenylalanine (L-DOPA) ở gan, thận và trên toàn bộ tụy với cường độ bằng nhau. B, Hình ảnh trục của một chụp CT cản quang hợp nhất với một chụp PET 18Fluoro-L-DOPA cho thấy một ổ tăng hấp thu ở thân sau của tụy. Lưu ý sự hấp thu chất đánh dấu bình thường ở gan và thận trái. F, chân; H, đầu; L, trái; R, phải.
Tăng insulin máu do Glutamate Dehydrogenase: Hội chứng tăng insulin máu tăng amoniac máu
Tăng insulin máu bẩm sinh do đột biến tăng chức năng của GLUD1 (mã hóa GDH) là dạng tăng insulin máu di truyền phổ biến thứ hai và là dạng phổ biến nhất đáp ứng với điều trị bằng diazoxide. Trong khoảng 70% các trường hợp, các đột biến là de novo. Trong số 30% các trường hợp gia đình, một mô hình di truyền trội trên nhiễm sắc thể thường rõ ràng là rõ ràng.
GDH là một enzyme nền ty thể, là một chất điều hòa chính của chuyển hóa acid amin và amoniac trong các tế bào β của tụy, gan, thận và não. Như được thể hiện trong Hình 7.5, GDH được hoạt hóa dị lập thể bởi leucine để oxy hóa glutamate thành α-ketoglutarate, đi vào chu trình Krebs, dẫn đến tăng tỷ lệ ATP/ADP—kích hoạt giải phóng insulin. Các đột biến HI/HA ảnh hưởng đến vị trí gắn guanosine-5′-triphosphate (GTP) ức chế hoặc vòng ăng-ten của enzyme, giao tiếp giữa các tiểu đơn vị liền kề, làm suy yếu tác động dị lập thể ức chế của GTP lên hoạt động của enzyme GDH, do đó dẫn đến giải phóng insulin quá mức.
Biểu hiện lâm sàng của HI/HA được đặc trưng bởi hạ đường huyết khi nhịn ăn và do protein gây ra và sự tăng amoniac huyết tương dai dẳng nhưng không có triệu chứng. Trẻ nhũ nhi bị HI/HA có cân nặng lúc sinh bình thường và thường không được chẩn đoán cho đến giai đoạn sau của thời thơ ấu. Nồng độ amoniac huyết tương trong HI/HA tăng gấp 3 đến 5 lần so với phạm vi bình thường, lên khoảng 60 đến 150 μmol/L. Nồng độ amoniac khá ổn định, và trái ngược với các khiếm khuyết enzyme chu trình urê, không tăng khi cho ăn protein. Tình trạng tăng amoniac máu dường như không gây ra các triệu chứng và không cần điều trị.
Kiểu hình lâm sàng của HI/HA bị chi phối bởi tác động của các đột biến kích hoạt trong các tế bào β của tụy. Các tiểu đảo tụy cô lập từ chuột chuyển gen biểu hiện GDH người đột biến cho thấy sự bài tiết insulin được kích thích bởi glucose bình thường nhưng sự bài tiết insulin được kích thích bởi leucine và acid amin tăng cường. Ở trẻ em bị HI/HA, có sự gia tăng đáng kể insulin sau một liều bolus IV của leucine, nhưng trái ngược với trẻ em bị KATP HI, chúng không đáp ứng với kích thích canxi. Hạ đường huyết ở trẻ em bị HI/HA bị kích động bởi việc nhịn ăn và bởi các bữa ăn giàu protein. Hạ đường huyết khi nhịn ăn có thể tương đối nhẹ. Trẻ em có thể nhịn ăn trong 8 đến 12 giờ trước khi bị hạ đường huyết. Tuy nhiên, những bệnh nhân này bị hạ đường huyết do protein gây ra một cách đáng kể—với nồng độ glucose huyết tương giảm mạnh từ 30 đến 90 phút sau một bữa ăn protein (Hình 7.9). Liệu pháp diazoxide, 5 đến 15 mg/kg/ngày, thường có hiệu quả trong việc kiểm soát cả hạ đường huyết khi nhịn ăn và do protein gây ra trong HI/HA. Việc nạp carbohydrate trước có thể hữu ích trong việc tránh tình trạng sau.
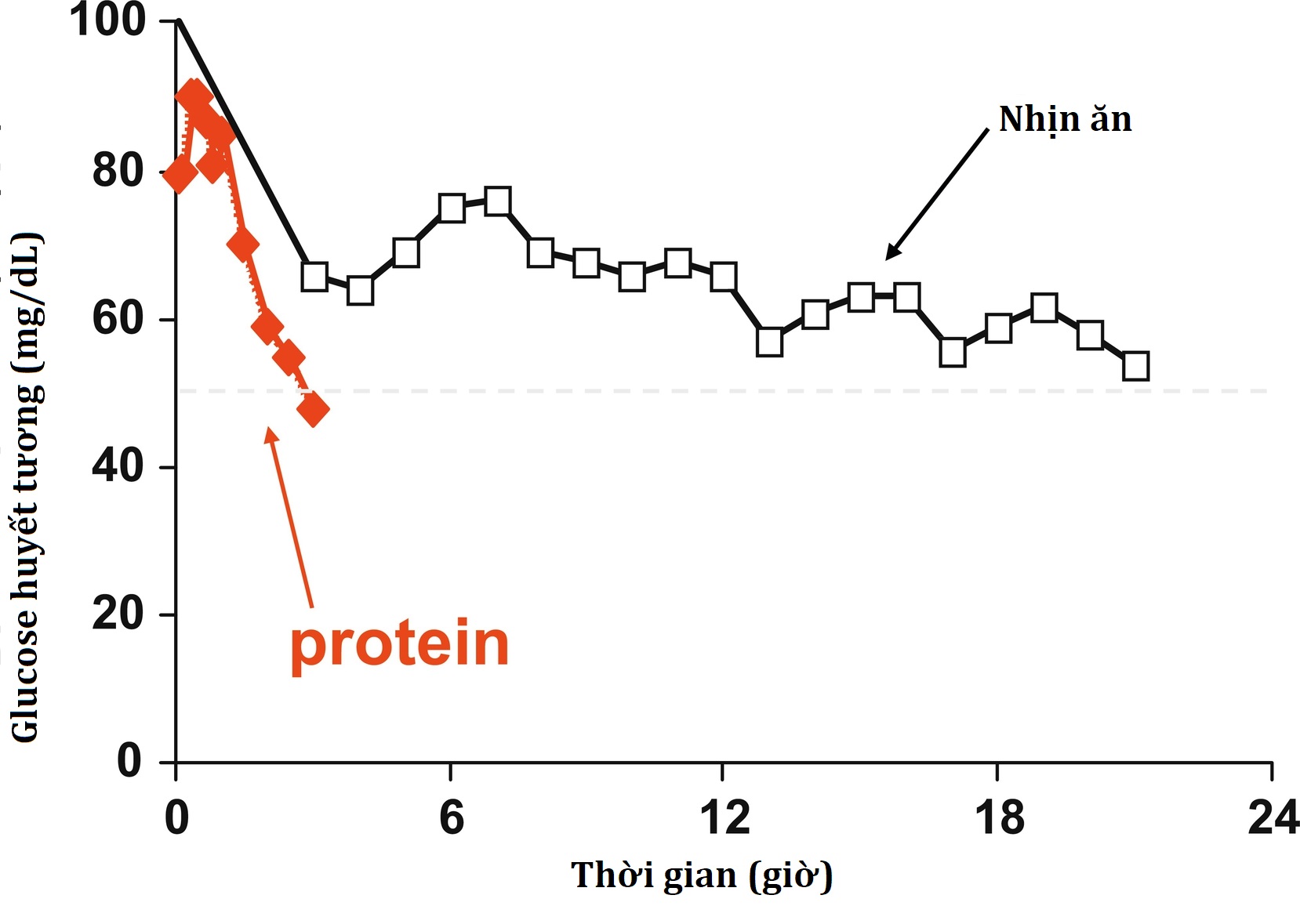
Hình 7.9 Phản ứng glucose huyết tương với việc nhịn ăn (hình vuông mở) và cho ăn protein (hình thoi đặc) ở một cô gái 16 tuổi mắc hội chứng tăng insulin máu/tăng amoniac máu do một đột biến điều hòa R269H biểu hiện trội của glutamate dehydrogenase.
Ban đầu, tình trạng tăng amoniac máu được cho là do tác động của enzyme đột biến trong gan; tuy nhiên, một cơ chế thay thế liên quan đến việc kích hoạt GDH ở thận dường như có nhiều khả năng hơn. Tình trạng tăng amoniac máu dai dẳng và không liên quan đến các dấu hiệu kinh điển của ngộ độc amoniac. Nó xảy ra ở cả trạng thái ăn no và nhịn đói và không bị ảnh hưởng bởi nồng độ glucose huyết tương hoặc lượng protein ăn vào.
Ngoài hạ đường huyết và tăng amoniac máu, bệnh nhân mắc hội chứng HI/HA có tần suất co giật, khuyết tật học tập và rối loạn hành vi tăng lên mà không liên quan trực tiếp đến hạ đường huyết. Các nghiên cứu mô tả các biểu hiện thần kinh của HI/HA còn hạn chế; tuy nhiên, bằng chứng có sẵn cho thấy 42% đến 64% bệnh nhân bị động kinh toàn thể được đặc trưng bởi các đợt phóng điện gai và sóng toàn thể biên độ cao trên điện não đồ (EEG). Sinh lý bệnh của các biểu hiện thần kinh vẫn chưa được làm sáng tỏ. Một cơ chế được đề xuất liên quan đến sự hoạt động quá mức của GDH trong các tế bào não, dẫn đến sự gián đoạn của các kho glutamate của hệ thần kinh trung ương, dẫn đến sự mất cân bằng giữa các chất dẫn truyền thần kinh kích thích và ức chế chính, glutamine và acid γ-aminobutyric (GABA), tương ứng.
Tăng insulin máu do SCHAD
Một dạng tăng insulin máu bẩm sinh ít phổ biến hơn, cũng liên quan đến việc mất sự điều hòa hoạt động của GDH, là do các đột biến bất hoạt trong HADH (gen mã hóa SCHAD ty thể). SCHAD-HI là một rối loạn lặn trên nhiễm sắc thể thường được đặc trưng bởi hạ đường huyết khi nhịn ăn do điều hòa insulin không phù hợp. Nguyên nhân của rối loạn điều hòa bài tiết insulin đã được làm sáng tỏ với khám phá rằng trong tế bào β, SCHAD đóng một vai trò ức chế hoạt động của GDH. Do đó, rối loạn điều hòa insulin quan sát thấy trong thiếu hụt SCHAD là do mất sự ức chế này, giải thích sự tương đồng lâm sàng giữa GDH-HI và SCHAD-HI.
Trái ngược với tất cả các khiếm khuyết khác trong quá trình oxy hóa acid béo, trẻ em bị SCHAD-HI không có dấu hiệu rối loạn chức năng gan hoặc bệnh cơ tim, hoặc các ảnh hưởng đến cơ xương. Biểu hiện lâm sàng của SCHAD-HI không đồng nhất, từ khởi phát muộn của hạ đường huyết nhẹ đến khởi phát sớm nghiêm trọng của hạ đường huyết trong giai đoạn sơ sinh. Ngoài hạ đường huyết khi nhịn ăn, trẻ em bị SCHAD-HI còn có biểu hiện hạ đường huyết do protein gây ra, tương tự như trẻ em bị GDH-HI. Các dấu hiệu sinh hóa đặc trưng, ngoài các dấu hiệu của tăng hoạt động insulin, là tăng nồng độ 3-hydroxybutyryl-carnitine huyết tương và tăng nồng độ 3-hydroxyglutarate trong nước tiểu, những phát hiện phù hợp với hoạt động enzyme SCHAD giảm. Trái ngược với GDH-HI, trẻ em bị SCHAD-HI không bị tăng amoniac, có lẽ do sự biểu hiện thấp hơn của SCHAD trong các mô khác nơi GDH được biểu hiện. Trẻ em bị ảnh hưởng đáp ứng với liệu pháp y tế bằng diazoxide.
Tăng insulin máu do Glucokinase
Một dạng tăng insulin máu bẩm sinh ít gặp hơn là do các đột biến kích hoạt trong GCK (mã hóa glucokinase-GK). Glucokinase là một hexokinase đóng vai trò là cảm biến glucose trong các tế bào β của tụy (xem Hình 7.5). Trái ngược với các hexokinase khác, GK có ái lực thấp hơn nhiều đối với cơ chất của nó với hoạt động bán tối đa (S0.5) ở nồng độ glucose 7,6 mM. Các đặc tính của GK và tác động hợp tác dương của glucose lên hoạt động của enzyme, với một đường cong dốc trong phạm vi glucose 5 mM, làm cho GK rất phù hợp để kiểm soát chặt chẽ glucose huyết tương trong phạm vi sinh lý bình thường từ 70 đến 100 mg/dL. Các đột biến trong GCK làm thay đổi động học của glucokinase bằng cách tăng ái lực của enzyme với glucose và gây hạ đường huyết bằng cách hạ thấp ngưỡng bài tiết insulin được kích thích bởi glucose trong tế bào beta. Ngược lại, các đột biến GCK bất hoạt dị hợp tử dẫn đến bệnh đái tháo đường khởi phát ở người trưởng thành trẻ type 2 (MODY 2) và các đột biến GCK bất hoạt đồng hợp tử là một nguyên nhân của bệnh đái tháo đường sơ sinh vĩnh viễn. Một số trường hợp GK-HI là lẻ tẻ, và một số là di truyền trội. Tất cả các đột biến đều có S0.5 glucose giảm, dao động từ 1,1 đến 4,5 mM.
Biểu hiện lâm sàng của GK-HI được đặc trưng bởi cân nặng lúc sinh to so với tuổi thai, phản ánh tác động thúc đẩy tăng trưởng của sự tăng bài tiết insulin của thai nhi. Hạ đường huyết có thể xuất hiện trong giai đoạn sơ sinh, nhưng thường không được nhận ra cho đến giai đoạn sau của thời thơ ấu hoặc thời niên thiếu. Mức độ nghiêm trọng của kiểu hình là khác nhau, với một số đột biến có kiểu hình nhẹ, với hạ đường huyết khi nhịn ăn đáp ứng với diazoxide, trong khi những đột biến khác dường như làm giảm ngưỡng glucose hơn nữa và có thể khó điều trị hơn. Dựa trên kinh nghiệm của chúng tôi, diazoxide đồng loạt không thể ngăn ngừa hạ đường huyết ở trẻ em bị GK-HI. Điều trị bằng các chất tương tự somatostatin đã được thử ở một số trẻ em bị GK-HI, với nhiều thành công khác nhau. Mặc dù một số trường hợp đã được cắt bỏ tụy, hầu hết trẻ em vẫn tiếp tục bị hạ đường huyết và cần quản lý y tế bổ sung sau phẫu thuật. Trong một số trường hợp, cần phải cung cấp hỗ trợ liên tục bằng cách cho ăn qua đêm hoặc glucose để ngăn ngừa hạ đường huyết.
Từ một vài trường hợp đã được cắt bỏ tụy, mô học tụy đã được báo cáo là có ít hoặc không có đặc điểm bất thường. Những người khác đã báo cáo sự phì đại nhân và tăng kích thước tiểu đảo và tăng sinh tế bào β.
Tăng insulin máu do Protein tách cặp-2
Các đột biến mất chức năng trong protein tách cặp ty thể UCP2 có liên quan đến một dạng tăng insulin máu bẩm sinh trội, đáp ứng với diazoxide. UCP2 vận chuyển proton và phosphate từ bào tương qua màng ty thể trong, để đổi lấy oxaloacetate và malate. Bằng cách làm cạn kiệt kho các chất trung gian của chu trình acid tricarboxylic trong ty thể, hoạt động của UCP2 hạn chế quá trình oxy hóa glucose để ủng hộ quá trình oxy hóa glutamine và các acid amin. Do đó, các đột biến bất hoạt của UCP2 làm tăng cường quá trình oxy hóa glucose trong các tế bào beta của tụy, dẫn đến sự khuếch đại phản ứng insulin với glucose. Thật vậy, kiểu hình của UCP2-HI được đặc trưng bởi hạ đường huyết do glucose gây ra ngoài hạ đường huyết khi nhịn ăn.
Các yếu tố nhân gan và Tăng insulin máu: Tăng insulin máu do HNF1A và HNF4A
HNF1A và 4A là các yếu tố phiên mã được biểu hiện trong các tế bào gan, tế bào β của tụy, tế bào biểu mô ruột và tế bào ống thận. Trong các tế bào β và tế bào gan, chúng tạo thành một vòng lặp tiến, sao cho sự thiếu hụt đơn bội của một trong hai gây ra sự giảm biểu hiện của cái còn lại. Cả HNF1A và HNF4A đều đóng một vai trò quan trọng đối với chức năng tế bào β của tụy và các đột biến dị hợp tử trong các gen mã hóa chúng: HNF1A và HNF4A, tương ứng, được biết là gây ra bệnh đái tháo đường đơn gen gia đình (MODY 3 và MODY 1). Các dạng đái tháo đường trội trên nhiễm sắc thể thường này được đặc trưng bởi sự suy giảm tiến triển trong bài tiết insulin được kích thích bởi glucose dẫn đến tăng đường huyết rõ ràng, thường biểu hiện trước 25 tuổi.
Gần đây hơn, người ta đã chứng minh rằng kiểu hình ở những người có đột biến bất hoạt trong HNF1A và HNF4A là hai pha, đặc trưng bởi hạ đường huyết dai dẳng do tăng insulin máu sớm trong cuộc đời, sau đó là giảm bài tiết insulin dẫn đến bệnh đái tháo đường ở tuổi vị thành niên và thanh niên sớm. Cùng với nhau, các đột biến trong HNF1A và HNF4A chiếm khoảng 5,9% tất cả các trường hợp tăng insulin máu trong đó một đột biến được xác định.
Kiểu hình lâm sàng của HNF1A- và HNF4A-HI được đặc trưng bởi cân nặng lúc sinh lớn, mặc dù một số trường hợp có cân nặng lúc sinh bình thường, và hạ đường huyết dai dẳng phần lớn đáp ứng với diazoxide. Tăng insulin máu là thoáng qua, với một số trường hợp khỏi trong vài tháng đầu đời, trong khi ở những trường hợp khác, giai đoạn tăng insulin máu có thể kéo dài đến thời thơ ấu.
Kiểu hình ở trẻ em có một đột biến đặc biệt trong HNF4A (p.Arg76Trp) liên quan đến các đặc điểm ngoài tụy, bao gồm gan to với transaminase tăng và bệnh ống thận Fanconi, tương tự như hội chứng Fanconi-Bickel.
Cơ chế chính xác mà các đột biến trong HNF1A và HNF4A dẫn đến tăng insulin máu hiện chưa được biết, nhưng có khả năng là các khiếm khuyết trong các yếu tố phiên mã này làm thay đổi các mô hình biểu hiện gen khác nhau trong giai đoạn đầu và sau này của cuộc đời.
Tăng insulin máu do FOXA2
Một dạng tăng insulin máu mới liên quan đến suy tuyến yên gần đây đã được mô tả ở những người có đột biến bất hoạt trong FOXA2. Foxa2 là một yếu tố phiên mã tham gia vào sự phát triển của nhiều mô, bao gồm cả tuyến yên, và cũng đóng một vai trò quan trọng trong việc bài tiết insulin của các tế bào beta trưởng thành. Các trường hợp được báo cáo có biểu hiện hạ đường huyết hạ ceton máu trong giai đoạn sơ sinh, đáp ứng với việc thay thế các hormone tuyến yên và điều trị bằng diazoxide.
Các dạng tăng insulin máu trong hội chứng
Một số hội chứng di truyền hiện được công nhận là có liên quan đến tăng insulin máu, bao gồm BWS, hội chứng Kabuki và hội chứng Turner. Các đặc điểm lâm sàng điển hình để phân biệt các hội chứng này có thể tinh vi hơn trong giai đoạn sơ sinh, do đó, chỉ số nghi ngờ cao và đánh giá lâm sàng cẩn thận là rất cần thiết để đưa ra chẩn đoán.
Tăng insulin máu trong hội chứng Beckwith-Wiedemann
BWS là một hội chứng tăng trưởng quá mức và có khuynh hướng ung thư, là kết quả của các thay đổi di truyền và biểu sinh khác nhau liên quan đến hai trung tâm kiểm soát được in dấu trên nhiễm sắc thể 11p15.5. Vùng này có thể được chia thành hai miền được in dấu riêng biệt được ngăn cách bởi một vùng không được in dấu. Miền xa 1 chứa IGF2, một protein thúc đẩy tăng trưởng được biểu hiện từ cha, và H19, một RNA không mã hóa dài ức chế tăng trưởng được biểu hiện từ alen của mẹ. Trong miền gần 2, vùng được in dấu chứa ba gen có liên quan đến BWS: KCNQ1OT1, một bản phiên mã không dịch được biểu hiện từ cha; CDKN1C, được biểu hiện từ mẹ và mã hóa một chất điều hòa âm của sự tăng sinh tế bào; và KCNQ1, một kênh kali tái cực được biểu hiện từ mẹ (Hình 7.10). Năm mươi phần trăm tất cả các trường hợp BWS là do mất methyl hóa tại vùng kiểm soát được in dấu 2, 20% trường hợp là do disomy đơn cha liên quan đến cả hai cụm gen được in dấu, 5% là do tăng methyl hóa tại vùng kiểm soát được in dấu 1, và 5% là do các đột biến được thừa hưởng từ mẹ trong CDKN1C.
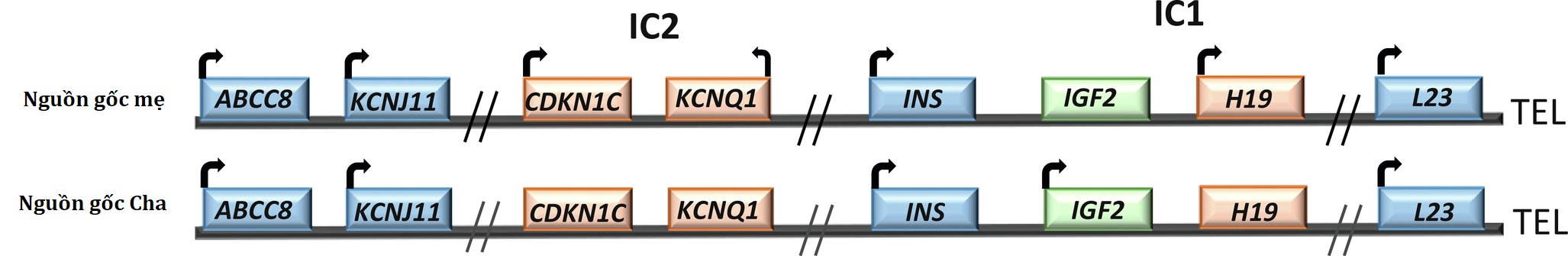
Hình 7.10 Locus hội chứng Beckwith-Wiedemann trên 11p15.5 phác thảo sự biểu hiện alen đặc hiệu theo nguồn gốc cha mẹ bình thường. Các gen được biểu hiện được ghi chú bằng một mũi tên. Các gen mã hóa các kênh KATP, ABCC8 và KCNJ11, cũng như insulin (INS) và L23 được biểu hiện từ cả hai alen, trong khi CDKN1C, KCNQ1, IGF2 và H19 được in dấu. IC1 được methyl hóa trên alen của cha, dẫn đến sự biểu hiện của IGF2 từ cha và không được methyl hóa trên alen của mẹ dẫn đến sự biểu hiện của H19 từ mẹ. IC2 được methyl hóa trên alen của mẹ dẫn đến sự biểu hiện của KCNQ1 và CDKN1C và không được methyl hóa trên alen của cha. ABCC8, ATP-binding cassette, subfamily C, member 8; CDKN1C, cyclin-dependent kinase inhibitor 1C; IC, imprinting center; IGF2, insulin-like growth factor 2; INS, insulin; KCNJ11, potassium channel, inwardly rectifying, subfamily J, member 11; KCNQ1, potassium channel, voltage-gated, KQT-like subfamily, member 1; L23, ribosomal protein L23- like; TEL, telemore.
Các đặc điểm kinh điển của BWS bao gồm thai to, lưỡi to, tạng to, các khiếm khuyết thành bụng, nếp/lỗ tai, bất đối xứng cơ thể, và tăng nguy cơ phát triển khối u phôi. Hạ đường huyết do tăng insulin máu có mặt ở tới 50% các trường hợp. Cơ chế gây ra tăng insulin máu trong BWS vẫn chưa được làm sáng tỏ, nhưng có thể liên quan đến rối loạn điều hòa bài tiết insulin và tăng sản tế bào beta.
Tăng insulin máu trong BWS có thể nhẹ và thoáng qua, mặc dù trong một số trường hợp, nó có thể nghiêm trọng và dai dẳng. Một số trẻ BWS đáp ứng với diazoxide, trong khi những trẻ khác cần cắt bỏ tụy để kiểm soát hạ đường huyết. Vì tăng insulin máu sẽ khỏi trong năm đầu đời ở hầu hết các trường hợp, việc cắt bỏ tụy nên được dành cho những trường hợp mà hạ đường huyết không thể kiểm soát được bằng y tế.
Tăng insulin máu trong hội chứng Kabuki
Hội chứng Kabuki là nguyên nhân phổ biến thứ hai của tăng insulin máu trong hội chứng. Khoảng 70% đến 75% các trường hợp hội chứng Kabuki là do các đột biến lặn trên nhiễm sắc thể thường trong KMT2D (lysine-specific methyltransferase 2D), và các đột biến liên kết X trong KDM6A (lysine-specific demethylase 6A) chiếm 1% đến 9% các trường hợp. Các đặc điểm chính của hội chứng Kabuki bao gồm các đặc điểm khuôn mặt “Mặt nạ Kabuki” điển hình (khe mi dài, lông mày cong, lật ra ngoài của một phần ba ngoài của mí dưới); các bất thường xương; các bất thường vân da; chậm phát triển thần kinh và khuyết tật trí tuệ; và chậm tăng trưởng sau sinh. Các biểu hiện nội tiết bao gồm thiếu hụt GH (22% các trường hợp), suy giáp (≤ 10% các trường hợp), dậy thì sớm ở vú (20%), và hạ đường huyết sơ sinh (11%). Cơ chế gây ra hạ đường huyết sơ sinh có thể là đa yếu tố: bao gồm tăng insulin máu và thiếu hụt GH. Tăng insulin máu là một đặc điểm ở tới 6% các trường hợp Kabuki, nó thường xuất hiện từ khi sinh, đáp ứng với diazoxide, và thường dường như khỏi trong thập kỷ đầu tiên của cuộc đời.
Tăng insulin máu trong hội chứng Turner
Người ta biết rõ rằng các bé gái mắc hội chứng Turner có khuynh hướng cao hơn đối với cả bệnh đái tháo đường type 1 và type 2, nhưng ít được công nhận rằng họ cũng có khuynh hướng bị hạ đường huyết. Ngày càng có nhiều bằng chứng cho thấy hạ đường huyết ở các bé gái mắc hội chứng Turner là do tăng insulin máu. Thật vậy, một tần suất cao hơn dự kiến của hội chứng Turner trong số trẻ em bị tăng insulin máu đã được báo cáo.
Tương tự như các dạng tăng insulin máu khác, tăng insulin máu ở các bé gái mắc hội chứng Turner thường xuất hiện ngay sau khi sinh và thường đáp ứng với điều trị bằng diazoxide, mặc dù, một số trường hợp được báo cáo đã không đáp ứng và nghiêm trọng cần phải cắt bỏ tụy. Mặc dù cơ chế gây ra tăng insulin máu trong hội chứng Turner chưa được thiết lập rõ ràng, một gen ứng cử viên tiềm năng trên nhiễm sắc thể X là KDM6A, một histone demethylase liên quan đến hội chứng Kabuki. Kiểm tra mô học của tụy của một bé gái mắc hội chứng Turner và tăng insulin máu cho thấy sự gia tăng kích thước nhân tế bào tiểu đảo, tương tự như tăng insulin máu lan tỏa do các đột biến bất hoạt kênh KATP. Các tiểu đảo được phân lập từ tụy này cho thấy một mô hình chức năng bất thường nhưng khác biệt, với sự gia tăng canxi và insulin trong bào tương cơ bản, ngưỡng thấp hơn cho sự bài tiết insulin được kích thích bởi glucose, tăng độ nhạy với sự kích thích bởi một hỗn hợp các acid amin, và phản ứng được bảo tồn với sự kích thích bằng glyburide.
Quản lý và Điều trị Tăng insulin máu
Việc điều trị cho trẻ sơ sinh nghi ngờ bị tăng insulin máu bẩm sinh nên được bắt đầu ngay lập tức, vì nguy cơ tổn thương não của chúng cao do não không chỉ bị thiếu hụt glucose mà còn cả các nhiên liệu thay thế (ceton). Các báo cáo từ nhiều trung tâm đã chứng minh tỷ lệ chậm phát triển cao ở bệnh nhân bị tăng insulin máu bẩm sinh, dao động từ 30% đến 50%. Bệnh nhân bị tăng insulin máu, cần điều trị phẫu thuật, có tỷ lệ mắc các vấn đề về phát triển thần kinh cao hơn so với bệnh nhân đáp ứng với diazoxide; tuy nhiên, sự phát triển thần kinh bất thường cũng được thấy ở tới 30% trẻ nhũ nhi bị tăng insulin máu thoáng qua. Nhìn chung, nguy cơ tổn thương não cao dường như là do sự chậm trễ trong chẩn đoán và điều trị hơn là hậu quả của chính khiếm khuyết phân tử, và do đó có khả năng phòng ngừa được.
Mục tiêu của liệu pháp là khôi phục glucose huyết tương về phạm vi bình thường (> 70 mg/dL) và duy trì nó ở đó. Có thể cần truyền glucose IV với tốc độ cao, lên đến 5 lần nhu cầu glucose bình thường, và có thể cần đặt đường truyền trung tâm để truyền dextrose nồng độ cao. Glucagon, được truyền IV liên tục (1 mg/ngày), có thể được sử dụng ở trẻ nhũ nhi cần tốc độ truyền glucose cao như một biện pháp tạm thời để giảm nguy cơ quá tải dịch.
Liệu pháp hàng đầu cho tăng insulin máu là diazoxide, một chất mở kênh KATP, đòi hỏi các kênh chức năng phải có mặt trên bề mặt tế bào để có tác dụng; do đó, hầu hết bệnh nhân bị KATP HI không đáp ứng với diazoxide. Ngược lại, bệnh nhân bị GDH-HI, HNF1A-HI, HNF4A-HI, SCHAD-HI, và những người bị tăng insulin máu do stress chu sinh đều đáp ứng với diazoxide. Bệnh nhân bị GK-HI đáp ứng một phần với diazoxide và hầu hết cần điều trị bổ sung. Phạm vi liều dược lý của diazoxide là 5 đến 15 mg/kg/ngày, uống hai lần một ngày. Biến cố bất lợi chính với diazoxide ở trẻ sơ sinh là giữ nước và do đó, nên sử dụng đồng thời một loại thuốc lợi tiểu (chlorothiazide hoặc furosemide), đặc biệt là ở trẻ nhũ nhi đang truyền dịch IV. Tăng áp động mạch phổi là biến chứng tiềm ẩn nghiêm trọng nhất; tuy nhiên, sự xuất hiện của nó rất hiếm nếu các bước thích hợp được thực hiện để ngăn chặn nó, đặc biệt là liệu pháp với thuốc lợi tiểu. Các tác dụng phụ khác liên quan đến diazoxide bao gồm giảm bạch cầu trung tính, giảm tiểu cầu và tăng acid uric máu. Liệu pháp diazoxide cũng liên quan đến chứng rậm lông đáng kể và làm thô các đặc điểm trên khuôn mặt.
Việc đánh giá xem tình trạng tăng insulin máu có đáp ứng với diazoxide hay không là rất quan trọng vì điều này quyết định con đường điều trị và cũng cung cấp manh mối về nguyên nhân di truyền tiềm tàng của tăng insulin máu. Thời gian bán thải của diazoxide là 24 đến 36 giờ ở người lớn; dữ liệu hạn chế cho thấy ở trẻ em, thời gian bán thải là 9,5 đến 24 giờ, do đó, đáp ứng với diazoxide nên được đánh giá sau ít nhất 5 ngày điều trị. Đáp ứng thành công nên được chứng minh bằng cách cho thấy việc duy trì glucose huyết tương trên 3,9 mmol/L (70 mg/dL) sau khi nhịn ăn. Thời gian nhịn ăn nên được xác định theo từng trường hợp, một phần dựa trên tuổi của trẻ.
Việc không đáp ứng với diazoxide gợi ý khả năng bị KATP-HI và do đó, xét nghiệm di truyền là rất quan trọng để xác định khả năng bị KATP-HI khu trú. Do đó, một khi chẩn đoán được thiết lập và liệu pháp ban đầu được bắt đầu, nên lấy mẫu của trẻ và cha mẹ của trẻ để phân tích phân tử deoxyribonucleic acid. Hình 7.11 mô tả cách tiếp cận chẩn đoán và điều trị cho trẻ em bị tăng insulin máu. Cũng nên xem xét đánh giá tim ở trẻ em bị tăng insulin máu, vì một tỷ lệ lớn các trường hợp có các bất thường cấu trúc tim, phổ biến hơn là phì đại thất.
Hình 7.11 Tiếp cận chẩn đoán và điều trị trong các trường hợp tăng insulin máu.
Đối với trẻ em bị tăng insulin máu không đáp ứng với diazoxide mà đã loại trừ được tăng insulin máu khu trú, nên xem xét liệu pháp y tế với các tác nhân hàng thứ hai, bao gồm các chất tương tự somatostatin. Các chất tương tự somatostatin ức chế bài tiết insulin bằng cách gây tăng phân cực tế bào β, ức chế trực tiếp các kênh canxi phụ thuộc điện thế, và các sự kiện xa hơn trong con đường bài tiết insulin. Chất tương tự somatostatin được sử dụng thường xuyên nhất là octreotide, được tiêm SQ mỗi 6 đến 8 giờ, với liều 5 đến 20 μg/kg/ngày. Đáp ứng ban đầu với octreotide là tốt ở hầu hết trẻ nhũ nhi bị tăng insulin máu, nhưng hiện tượng quen thuốc nhanh (tachyphylaxis) phát triển sau một vài liều, làm cho liệu pháp không đủ hiệu quả để sử dụng lâu dài trong hầu hết các trường hợp. Một cách tiếp cận để tránh nhu cầu tiêm nhiều lần là sử dụng bơm SQ để truyền octreotide liên tục (ví dụ, sử dụng ngoài nhãn các bơm insulin chứa đầy octreotide). Một cách tiếp cận khác giới hạn liều octreotide xuống hai lần một ngày, được sử dụng kết hợp với dextrose liên tục qua đêm qua ống thông dạ dày, trong nỗ lực tránh hiện tượng quen thuốc nhanh. Các tác dụng phụ thường gặp nhất của octreotide là tăng men gan thoáng qua, bệnh lý túi mật, và ức chế GH. Các tác dụng phụ nghiêm trọng hơn cũng đã được báo cáo, bao gồm viêm ruột hoại tử. Do mối liên quan này, nên tránh sử dụng octreotide ở trẻ nhũ nhi dưới 4 tuần tuổi hoặc trẻ nhũ nhi có các yếu tố nguy cơ khác. Các chất tương tự somatostatin tác dụng kéo dài, chẳng hạn như octreotide tác dụng kéo dài và lanreotide autogel được ưa chuộng để điều trị cho trẻ lớn hơn. Việc sử dụng chúng ở trẻ nhỏ hơn bị hạn chế vì liều lượng không thể dễ dàng điều chỉnh với các công thức hiện tại.
Hạ đường huyết hạ ceton máu do các đột biến kích hoạt trong con đường truyền tín hiệu Insulin
Hạ đường huyết hạ ceton máu do AKT2
Các đột biến tăng chức năng lặn trong AKT2, một thành phần quan trọng của chuỗi truyền tín hiệu insulin, gây ra một kiểu hình tương tự như hạ đường huyết do tăng insulin máu, nhưng không có nồng độ insulin huyết tương có thể phát hiện được, mặc dù có các dấu hiệu kinh điển của tăng hoạt động insulin (ức chế acid béo tự do và ceton). Kiểu hình ở những đứa trẻ này được đặc trưng bởi hạ đường huyết nghiêm trọng, dai dẳng, không đáp ứng với liệu pháp y tế và cần cho ăn liên tục qua ống thông dạ dày. Những đứa trẻ này cũng bị phát triển quá mức với sự bất đối xứng cơ thể.
Hạ đường huyết hạ ceton máu do PI3K
Các đột biến kích hoạt dạng khảm trong phosphatidylinositol-3-kinase (PI3K) loại 1A, nằm ở thượng nguồn của AKT2 trong truyền tín hiệu insulin, cũng đã được mô tả là một nguyên nhân của hạ đường huyết hạ ceton máu. Kiểu hình của các trường hợp được báo cáo được đặc trưng bởi khởi phát sớm và hạ đường huyết hạ ceton máu nghiêm trọng với insulin không thể phát hiện được.
Hạ đường huyết tăng insulin máu sau ăn mắc phải sau phẫu thuật Fundoplication
Phẫu thuật Nissen fundoplication và các thủ thuật fundoplication sửa đổi khác, thường được thực hiện để điều trị trào ngược dạ dày thực quản ở trẻ sơ sinh và trẻ em, đã được liên kết với hội chứng dumping muộn, đặc biệt là hạ đường huyết tăng insulin máu sau ăn. Trong một nghiên cứu ở trẻ sơ sinh, người ta ước tính rằng khoảng 24% những trẻ trải qua phẫu thuật fundoplication đã phát triển hạ đường huyết sau ăn sau phẫu thuật. Kinh điển, hội chứng dumping được đặc trưng bởi các triệu chứng sớm hoặc “dumping sớm”, được cho là do sự dịch chuyển chất lỏng gây ra bởi tải lượng thẩm thấu trong ruột non, và “dumping muộn” hoặc hạ đường huyết sau ăn. Theo kinh nghiệm của chúng tôi, hội chứng dumping ở trẻ em được đặc trưng bởi hạ đường huyết sau ăn nghiêm trọng mà không có các triệu chứng tiêu hóa đáng kể quan sát thấy ở người lớn. Rất thường xuyên, trẻ em bị hạ đường huyết tăng insulin máu sau ăn, sau phẫu thuật fundoplication, bị co giật, lơ mơ, và các triệu chứng khác của hạ đường huyết trong vài tháng trước khi chẩn đoán được đưa ra. Đặc biệt ở trẻ sơ sinh, hạ đường huyết có thể không được nhận ra trừ khi những trẻ này được sàng lọc bằng cách theo dõi glucose huyết tương sau ăn sau phẫu thuật.
Sinh lý bệnh của hạ đường huyết tăng insulin máu sau phẫu thuật fundoplication có thể là đa yếu tố. Các nghiên cứu về chức năng vận động và cảm giác của dạ dày sau phẫu thuật fundoplication đã cho thấy sự giãn nở dạ dày sau ăn giảm đáng kể và làm rỗng dạ dày nhanh hơn đáng kể. Việc làm rỗng nhanh một bữa ăn vào ruột non dẫn đến sự hấp thu nhanh chóng glucose vào máu, với tăng đường huyết sớm sau đó là một đợt tăng insulin quá mức và hạ đường huyết sau đó. Sự tăng tiết của hormone kích thích insulin mạnh, glucagon-like peptide-1 (GLP-1), sau một bữa ăn có thể một phần chịu trách nhiệm cho tình trạng tăng insulin máu sau ăn. Điều này được hỗ trợ bởi bằng chứng chứng minh rằng việc đối kháng thụ thể GLP-1 với exendin-(9-39) làm giảm phản ứng insulin, cho thấy rằng phản ứng insulin quá mức với một bữa ăn ít nhất một phần là do tác động của GLP-1 lên tế bào beta của tụy.
Chẩn đoán hạ đường huyết tăng insulin máu ở trẻ sơ sinh và trẻ em sau phẫu thuật fundoplication được thiết lập dựa trên hồ sơ glucose và insulin sau một nghiệm pháp dung nạp bữa ăn hỗn hợp hoặc một nghiệm pháp dung nạp glucose đường uống. Phản ứng điển hình được đặc trưng bởi tăng đường huyết có thể đạt tới hơn 11,1 mmol/L (200 mg/dL) trong giờ đầu tiên sau bữa ăn, sau đó là hạ đường huyết sau 90 đến 120 phút. Insulin huyết tương đạt đến đỉnh lớn hơn 200 μU/mL khoảng 30 phút sau bữa ăn. Một loạt các liệu pháp đã được sử dụng với nhiều thành công khác nhau để điều trị hội chứng dumping, bao gồm tinh bột ngô sống, pectin, octreotide, acarbose, và các thay đổi chế độ ăn uống. Nhiều bệnh nhân bị hạ đường huyết tăng insulin máu sau ăn tiếp tục có các triệu chứng nghiêm trọng, mặc dù đã có những can thiệp này, và cần một chế độ ăn liên tục qua đường ruột để tránh hạ đường huyết, nhưng vẫn có nguy cơ cao bị các biến cố hạ đường huyết nếu việc cho ăn bị dừng đột ngột. Trong hầu hết các trường hợp, hạ đường huyết tăng insulin máu sau ăn cải thiện theo thời gian, đặc biệt là sau khi thức ăn đặc đã được đưa vào chế độ ăn.
Khiếm khuyết trong phản ứng điều hòa ngược
Hạ đường huyết liên quan đến thiếu hụt nội tiết thường do thiếu hụt glucocorticoid hoặc GH, phổ biến hơn là sự kết hợp của cả hai. Ở trẻ em bị suy toàn bộ tuyến yên, thiếu hụt GH đơn độc, hoặc sự kết hợp của thiếu hụt ACTH và thiếu hụt GH, tỷ lệ hạ đường huyết cao tới 20%. Trong giai đoạn sơ sinh, hạ đường huyết có thể là biểu hiện đầu tiên của suy tuyến yên. Ở nam giới, dương vật nhỏ và tinh hoàn nhỏ có thể cung cấp một manh mối về sự thiếu hụt gonadotropin đồng thời (Hình 7.12). Trẻ sơ sinh bị suy tuyến yên cũng có thể bị rối loạn chức năng gan giống như bệnh gan ứ mật, và một số có các dị tật đường giữa, chẳng hạn như hội chứng loạn sản vách-thị. Về mặt lâm sàng, những đứa trẻ như vậy có thể biểu hiện rung giật nhãn cầu, hoặc có các hội chứng thiểu sản giữa mặt, bao gồm cả hở hàm ếch.
Hình 7.12 Dương vật nhỏ và tinh hoàn ẩn ở một trẻ nhũ nhi bị suy tuyến yên bẩm sinh. Trẻ bị hạ đường huyết lúc 12 giờ tuổi (glucose, 24 mg/dL). Lúc 72 giờ tuổi, trẻ bị vàng da—và sinh thiết gan cho thấy viêm gan sơ sinh. Đánh giá nội tiết của trẻ dương tính với suy giáp, nồng độ cortisol thấp, nồng độ hormone tăng trưởng không thể phát hiện được, và nồng độ prolactin tăng cao (xác nhận suy tuyến yên do vùng dưới đồi).
Mặc dù trẻ nhũ nhi và trẻ em lớn hơn bị thiếu hụt tuyến yên có biểu hiện hạ đường huyết nhiễm ceton trong giai đoạn sơ sinh, hạ đường huyết có thể bắt chước tăng insulin máu, vì các cơ chế sinh ceton chưa phát triển đầy đủ trong giai đoạn ngay sau sinh. Tuy nhiên, hạ đường huyết của chúng không đáp ứng với diazoxide và chỉ thuyên giảm khi thay thế các hormone bị thiếu hụt (bao gồm cả thyroxine, cũng như GH và cortisol). Đáng chú ý, các đột biến trong yếu tố phiên mã Foxa2 gây ra cả suy tuyến yên và tăng insulin máu và biểu hiện trong giai đoạn sơ sinh với hạ đường huyết dai dẳng.
Khi bệnh thượng thận nghiêm trọng, như trong tăng sản thượng thận bẩm sinh, xuất huyết thượng thận hai bên, không có hoặc thiểu sản thượng thận bẩm sinh, hoặc các hội chứng kháng ACTH khác nhau, tăng sắc tố da, rối loạn điện giải huyết thanh với hạ natri máu và tăng kali máu, hoặc bộ phận sinh dục không rõ ràng có thể cung cấp các manh mối chẩn đoán. Các bất thường của thụ thể ACTH hoặc thiểu sản thượng thận có thể khó phân biệt về mặt kiểu hình với thiếu hụt cortisol do các nguyên nhân khác, ngoại trừ sự tăng đáng kể nồng độ ACTH huyết thanh, được ghi nhận nếu thụ thể ACTH hoặc tuyến thượng thận bị trục trặc. Tuy nhiên, tất cả các trạng thái có tăng ACTH đều có thể bị nghi ngờ trên lâm sàng nhờ vào tình trạng tăng sắc tố da liên quan (xem Chương 14). Những trường hợp suy thượng thận đơn độc này rất hiếm khi gây hạ đường huyết ở trẻ sơ sinh, trong khi ở trẻ nhũ nhi và trẻ em lớn hơn, tăng sản thượng thận bẩm sinh được điều trị và thiếu hụt ACTH đơn độc có thể gây hạ đường huyết nghiêm trọng do stress. Thiếu hụt ACTH bẩm sinh do đột biến trong TPIT, mã hóa TBX19, một yếu tố phiên mã cần thiết cho sự biểu hiện của gen proopiomelanocortin (POMC) và cho sự biệt hóa cuối cùng của dòng corticotroph tuyến yên, là một ngoại lệ, biểu hiện trong giai đoạn sơ sinh với hạ đường huyết nghiêm trọng và dai dẳng, cho đến khi việc thay thế hormone được thiết lập. Điều quan trọng cần lưu ý là nồng độ GH và cortisol thấp tại thời điểm hạ đường huyết không phải là chẩn đoán xác định cho một sự thiếu hụt thực sự và có thể cần các nghiệm pháp kích thích để thiết lập chẩn đoán.
Cơ chế hạ đường huyết trong thiếu hụt GH có thể là kết quả của việc giảm ly giải mỡ. Cơ chế hạ đường huyết với thiếu hụt cortisol có thể là do giảm dự trữ glycogen ở gan cộng với giảm tân tạo glucose, do không cung cấp được chất nền tân tạo glucose nội sinh dưới dạng các acid amin từ quá trình phân giải protein cơ. Do đó, việc điều tra một đứa trẻ bị hạ đường huyết đòi hỏi phải loại trừ thiếu hụt ACTH, cortisol, hoặc GH—và nếu được chẩn đoán, phải thay thế thích hợp bằng cortisol hoặc GH. Việc thay thế cortisol ở liều stress nên là phương pháp điều trị hàng đầu và việc thay thế GH chỉ được thực hiện nếu chỉ riêng liệu pháp glucocorticoid không kiểm soát được hạ đường huyết. Có bằng chứng cho thấy sự tổng hợp và bài tiết GH ở trẻ sơ sinh cần có cortisol, do đó tình trạng thiếu hụt GH rõ ràng sẽ được giải quyết chỉ bằng điều trị cortisol.
Mặc dù thiếu hụt glucagon đã được mô tả là một nguyên nhân của hạ đường huyết, các tác giả đã rút lại kết luận của họ và hiện tin rằng những bệnh nhân này bị tăng insulin máu do SCHAD; do đó, rối loạn này cực kỳ hiếm và có thể không tồn tại. Thiếu hụt epinephrine hoặc thiếu hụt tác dụng của nó cũng hiếm gặp nhưng phải được xem xét trong bệnh rối loạn chức năng tự chủ gia đình hoặc ở trẻ em được điều trị bằng thuốc chẹn thụ thể adrenergic, chẳng hạn như thuốc chẹn beta.
Khiếm khuyết trong ly giải Glycogen và tân tạo Glucose
Hầu hết các rối loạn về ly giải glycogen và tân tạo glucose dẫn đến hạ đường huyết thường được chẩn đoán ở thời thơ ấu và sẽ được thảo luận trong Chương 23. Ngoại lệ là GSD I, làm suy giảm sự giải phóng glucose từ quá trình ly giải glycogen và tân tạo glucose và biểu hiện trong giai đoạn sơ sinh, mặc dù trong một số trường hợp, nó được chẩn đoán muộn hơn.
Thiếu hụt Glucose 6-Phosphatase (GSD Type 1)
Bệnh dự trữ glycogen type 1 là một rối loạn lặn trên nhiễm sắc thể thường do các khiếm khuyết trong phức hợp glucose 6-phosphatase (G6Pase), xúc tác cho các bước cuối cùng của cả quá trình tân tạo glucose và ly giải glycogen ở gan, quá trình thủy phân glucose 6-phosphate thành glucose và phosphate vô cơ. Sự thiếu hụt hoạt động của G6Pase trong gan, thận và ruột dẫn đến sự tích tụ glycogen trong các cơ quan này, hạ đường huyết khi nhịn ăn do sản xuất glucose không đủ, và các bất thường sinh hóa thứ phát khác, bao gồm tăng acid lactic máu, tăng acid uric máu và tăng lipid máu.
G6Pase là một phức hợp đa thành phần bao gồm một đơn vị xúc tác, G6Pase, nằm ở phía lòng của mạng lưới nội chất, và một chất vận chuyển hai chiều đặc hiệu cho G6P (G6PT) cho phép glucose 6-phosphate đi vào đơn vị xúc tác. Gen của đơn vị xúc tác nằm trên nhiễm sắc thể 17, trong khi gen của chất vận chuyển glucose 6-phosphate nằm trên nhiễm sắc thể 11. Các đột biến trong đơn vị xúc tác gây ra GSD type 1a, trong khi các đột biến trong G6PT gây ra GSD type 1b. Kiểu hình của hai loại phụ này giống hệt nhau, ngoại trừ ở type 1b, ngoài kiểu hình gan, còn có các bệnh nhiễm khuẩn tái phát và bệnh viêm ruột liên quan đến giảm bạch cầu trung tính và rối loạn chức năng bạch cầu trung tính.
Tỷ lệ mắc GSD type 1 ước tính là 1 trên 100.000 ca sinh sống, trong đó GSD type 1a chiếm khoảng 80% các trường hợp. Về mặt lâm sàng, GSD type 1 có thể biểu hiện trong giai đoạn sơ sinh với hạ đường huyết nghiêm trọng, xảy ra từ 2 đến 2,5 giờ sau bữa ăn, và thở nhanh thứ phát sau bù trừ hô hấp cho tình trạng nhiễm toan chuyển hóa. Tuy nhiên, vì lactate và ceton có thể cung cấp đủ chất nền cho não để bảo vệ chức năng hệ thần kinh trung ương (và vì trong giai đoạn đầu của thời thơ ấu, việc cho ăn đều đặn được cung cấp một cách nhất quán), chẩn đoán có thể bị trì hoãn trong nhiều tháng cho đến khi gan to khổng lồ đưa trẻ đến khám, mặc dù gan to có thể bị bỏ sót vì gan mềm. Sau giai đoạn nhũ nhi, bệnh nhân bị ảnh hưởng có thể được thấy đi với dáng đi lạch bạch, thứ phát sau bụng nổi bật và yếu cơ. Các đặc điểm nhất quán khác là tăng acid uric máu, hạ phosphat máu, một xu hướng chảy máu, thứ phát sau suy giảm độ dính của tiểu cầu, và chậm phát triển.
Hạ đường huyết có thể xảy ra bất cứ lúc nào những đứa trẻ này tiếp xúc với ngay cả những khoảng thời gian nhịn ăn ngắn. Chúng hoàn toàn phụ thuộc vào việc cung cấp glucose từ các nguồn ngoại sinh, ngoại trừ một lượng nhỏ glucose tự do—được giải phóng như một phần của quá trình tách nhánh glycogen. Vì ít hơn 10% glycogen bao gồm các điểm nhánh, cơ chế này cung cấp rất ít sự bảo vệ chống lại hạ đường huyết trong khi nhịn ăn. Hạ đường huyết trong bối cảnh insulin bị ức chế và glucagon tăng lên thúc đẩy quá trình ly giải glycogen, nhưng sự vắng mặt của G6Pase buộc glucose-1-phosphate được sản xuất bởi phosphorylase phải dị hóa qua đường phân, dẫn đến tăng sản xuất lactate (xem Hình 7.1). Một hậu quả khác của hoạt động suy giảm của G6Pase là sự chuyển hướng của G6P qua con đường pentose phosphate để tạo ra ribose-6-phosphate, cuối cùng tạo ra acid uric, dẫn đến tăng acid uric máu. Tăng triglycerid máu là kết quả của việc tăng hình thành triglyceride như một con đường chính để xử lý pyruvate từ lactate và các acid amin, khi việc tạo ra glucose bị chặn trong thiếu hụt G6Pase. Sự tích tụ lớn chất béo trong gan là nguyên nhân gây ra gan to khổng lồ đặc trưng của GSD type 1.
Bệnh thận là một biến chứng thường gặp của GSD type 1 (với tỷ lệ ước tính là 30%). Các biểu hiện bao gồm rối loạn chức năng ống thận gần (hội chứng giống Fanconi), khiếm khuyết toan hóa ống lượn xa, và tăng canxi niệu. Có một mối quan hệ nghịch đảo giữa tuổi và sự bài tiết citrate, và sự kết hợp của bài tiết citrate thấp và tăng canxi niệu khiến những đứa trẻ này dễ bị nhiễm canxi thận và sỏi thận. Bổ sung citrate có thể ngăn ngừa hoặc cải thiện các biến chứng này. Tỷ lệ mắc bệnh rộng rãi và tiên lượng nghiêm trọng của tổn thương thận được biểu hiện bằng sự tăng lọc cầu thận nghiêm trọng và albumin niệu vi thể theo thời gian, tăng huyết áp động mạch hệ thống, và hậu quả là suy thận. Việc thực hiện sớm điều trị bằng thuốc ức chế men chuyển angiotensin đã được chứng minh là làm chậm sự tiến triển của tổn thương thận. Các phát hiện bệnh lý bao gồm xơ cứng cầu thận khu trú từng phần với xơ hóa kẽ. Nguyên nhân của tổn thương thận chưa rõ ràng, nhưng nó tương quan nghịch với sự kiểm soát chuyển hóa. Người ta đã đề xuất rằng rối loạn lipid máu góp phần gây ra tổn thương thận.
Ngoài gan to đặc trưng, gan còn trải qua những thay đổi dạng u tuyến. Các u tuyến thường được quan sát lần đầu tiên trong thập kỷ thứ hai và thứ ba của cuộc đời nhưng có thể xuất hiện trước tuổi dậy thì. Các u tuyến có thể bị thoái hóa ác tính hoặc xuất huyết và thường liên quan đến thiếu máu kháng sắt mãn tính. Các biến chứng khác của GSD1 bao gồm loãng xương và chậm phát triển.
Chẩn đoán GSD type 1 dựa trên các đặc điểm lâm sàng và sinh hóa: hạ đường huyết sau một thời gian ngắn nhịn ăn, gan to, nhiễm toan lactic, và tăng acid uric và triglyceride. Kích thích bằng glucagon, từ 2 đến 4 giờ sau một bữa ăn chứa carbohydrate, dẫn đến tăng nồng độ lactate, nhưng glucose huyết tương không tăng. Chẩn đoán có thể được xác nhận bằng phân tích đột biến.
Mục tiêu điều trị của trẻ em bị GSD I là loại bỏ hoàn toàn hạ đường huyết và ức chế sự mất bù chuyển hóa thứ phát. Việc cho ăn liên tục qua sonde mũi-dạ dày hoặc trong dạ dày vào ban đêm đã được chứng minh là làm giảm hoặc loại bỏ các phát hiện chuyển hóa và lâm sàng thông qua việc tránh hoàn toàn hạ đường huyết. Tuy nhiên, cách tiếp cận này đặt trẻ em vào nguy cơ bị hạ đường huyết nghiêm trọng, nếu việc cho ăn bị dừng đột ngột. Một cách tiếp cận an toàn hơn là giới thiệu tinh bột ngô sống đường uống, giúp kéo dài khả năng chịu đựng nhịn ăn, và có thể được sử dụng ở trẻ nhũ nhi lớn hơn, thường lớn hơn 6 tháng tuổi. Liều tinh bột ngô được sử dụng là khoảng 1,6 g/kg mỗi liều mỗi 4 giờ ở trẻ nhũ nhi và 1,7 đến 2,5 g/kg mỗi liều mỗi 6 giờ ở bệnh nhân lớn tuổi hơn. Một chế độ điển hình bao gồm các bữa ăn ban ngày mỗi 3 đến 4 giờ được tính toán để cung cấp đủ calo carbohydrate để tránh nhu cầu sản xuất glucose ở gan. Hầu hết các calo này bao gồm carbohydrate, chủ yếu cung cấp glucose tinh khiết như một nguồn năng lượng và tránh các disaccharide chứa fructose hoặc galactose. Vào ban đêm, chế độ bao gồm một đợt truyền glucose trong dạ dày có hoặc không có protein được thiết kế để truyền với tốc độ khoảng 125% sản lượng glucose gan được tính toán cho trẻ nhũ nhi bình thường. Đối với trẻ lớn hơn, có thể thực hiện một chế độ ăn tinh bột ngô sống vào ban đêm.
Kiểm soát chế độ ăn uống tỉ mỉ có thể dẫn đến sự cải thiện lâm sàng và chuyển hóa đáng kể và phòng ngừa các biến chứng. Các liệu pháp bổ trợ nên bao gồm theo dõi cẩn thận nồng độ acid uric và điều trị bằng allopurinol nếu nồng độ acid uric tăng. Với nhận thức ngày càng tăng về rối loạn chức năng ống thận, điều trị tình trạng tăng lọc bằng thuốc ức chế men chuyển angiotensin nên được bắt đầu ngay lập tức. Điều trị bằng yếu tố kích thích dòng tế bào hạt-đại thực bào để tăng cường sản xuất bạch cầu trung tính đã được chứng minh là cải thiện loét miệng và viêm ruột ở type 1b.
Rối loạn oxy hóa Acid béo: Thiếu hụt Acyl-Coenzyme A Dehydrogenase chuỗi trung bình
Các lỗi chuyển hóa bẩm sinh liên quan đến sự thiếu hụt quá trình oxy hóa acid béo và sinh ceton (Hình 7.13) được di truyền theo kiểu lặn trên nhiễm sắc thể thường. Tất cả các rối loạn này có thể bị kích động bởi việc nhịn ăn và biểu hiện các biến cố cấp tính đe dọa tính mạng được đặc trưng bởi hôn mê, hạ đường huyết hạ ceton máu, trụy tim mạch-hô hấp, và gan to nhiễm mỡ; những rối loạn làm suy giảm các bước đầu trong quá trình oxy hóa các acid béo chuỗi dài hơn cũng có thể bao gồm bệnh cơ tim phì đại hoặc giãn mạn tính và yếu cơ hoặc tiêu cơ vân do tập thể dục. Các khiếm khuyết chuyển hóa của quá trình oxy hóa acid béo có thể được chia theo vị trí của khiếm khuyết enzyme (xem Hình 7.13): (1) khiếm khuyết của chu trình carnitine ty thể để vận chuyển acid béo vào chất nền ty thể, (2) khiếm khuyết của chu trình β-oxy hóa, (3) khiếm khuyết của việc chuyển điện tử đến chuỗi vận chuyển điện tử, và (4) khiếm khuyết trong con đường tổng hợp ceton. Tuổi biểu hiện thường ngoài giai đoạn sơ sinh, nhưng biểu hiện sơ sinh có thể xảy ra ở những trẻ bú mẹ chưa bắt đầu nhận đủ sữa. Do tính chất đe dọa tính mạng và phần lớn có thể điều trị được của các rối loạn này, sàng lọc sơ sinh được áp dụng rộng rãi ở tất cả các tiểu bang của Hoa Kỳ và nhiều quốc gia khác bằng cách sử dụng các đốm máu trên giấy thấm và phân tích khối phổ song song của các hồ sơ acylcarnitine. Điều này phát hiện hầu hết các rối loạn oxy hóa acid béo, ngoại trừ sự thiếu hụt β-hydroxy-methylglutaryl-CoA synthase. Phần này xem xét chẩn đoán, các đặc điểm lâm sàng và điều trị của thiếu hụt MCAD; các khiếm khuyết oxy hóa acid béo khác được xem xét trong Chương 23.
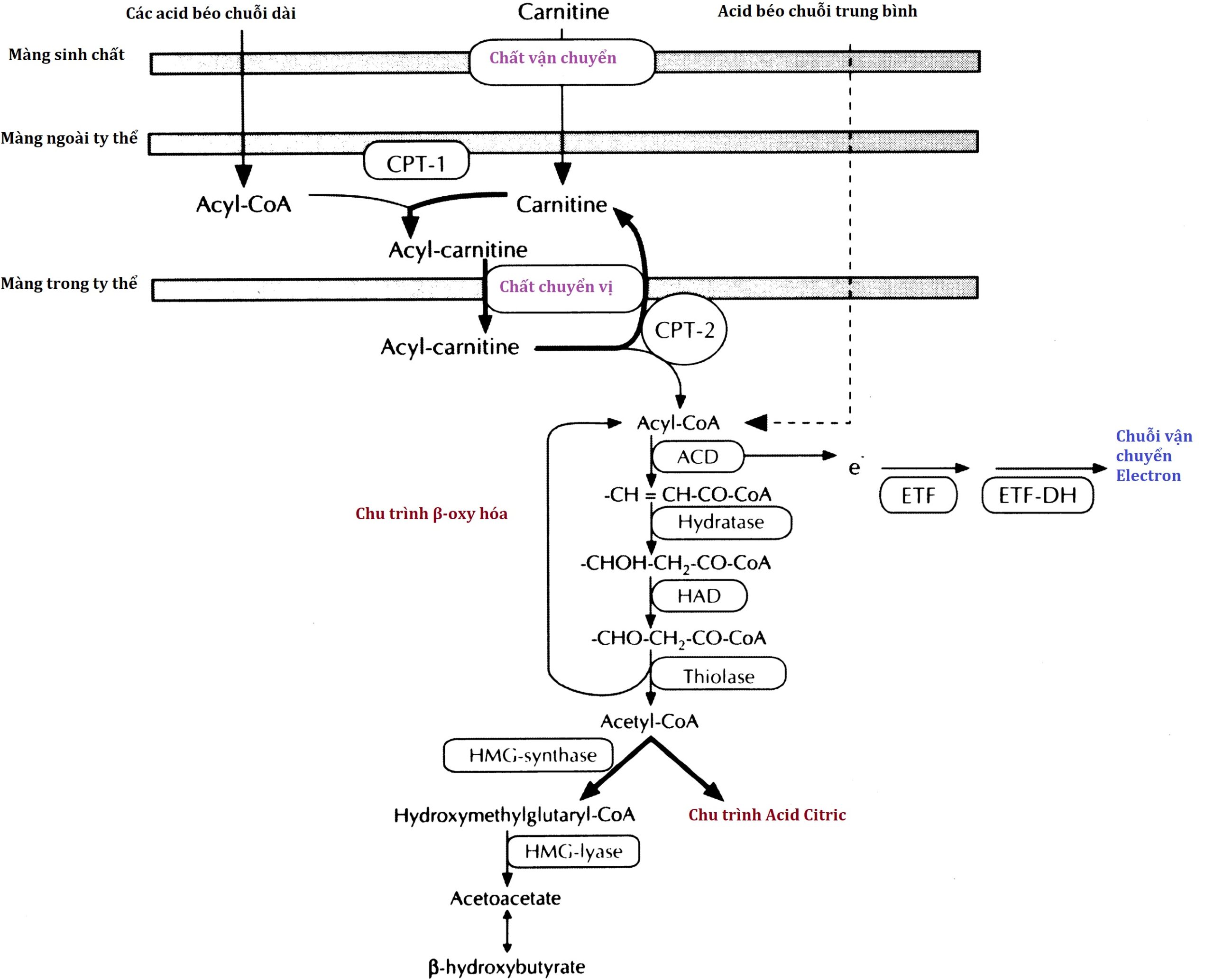
Hình 7.13 Các con đường oxy hóa acid béo ở ty thể và tổng hợp thể ceton. ACD, Acyl-CoA dehydrogenase; CPT-1 và CPT-2, carnitine palmitoyltransferase I và II; ETF 5, flavoprotein chuyển điện tử; ETF-DH, dehydrogenase flavoprotein chuyển điện tử; HAD, hydroxyacyl-CoA dehydrogenase.
Thiếu hụt MCAD là khiếm khuyết thường gặp nhất của quá trình oxy hóa acid béo và là một trong những lỗi chuyển hóa bẩm sinh phổ biến hơn. Sàng lọc sơ sinh ở Pennsylvania đã cho thấy tỷ lệ mắc là 1 trên 15.700 trẻ sơ sinh và rối loạn này đặc biệt phổ biến ở các nước Bắc Âu.
MCAD (được mã hóa bởi ACADM trên 1p31) chịu trách nhiệm cho bước đầu tiên trong chu trình β-oxy hóa đối với các acyl-CoA chuỗi trung bình, với chiều dài chuỗi từ 4 đến 12 nguyên tử carbon. Sự thiếu hụt hoạt động của enzyme MCAD dẫn đến sự tích tụ các chất chuyển hóa acid béo chuỗi trung bình (từ 6 đến 10 carbon) trong huyết tương (octanoylcarnitine–C8, hexanoylcarnitine–C6, decanoylcarnitine–C10, decenoylcarnitine–C10:1; và các acylglycine bất thường—hexanoylglycine và suberylglycine). Các bất thường chuyển hóa trong nước tiểu bao gồm tăng bài tiết các acid dicarboxylic. Hồ sơ chuyển hóa thường là chẩn đoán, nhưng có thể được xác nhận bằng xét nghiệm trình tự gen. Đột biến phổ biến nhất ở người Bắc Âu, 985A > G (p.Lys304Glu), là đồng hợp tử ở 80% các trường hợp biểu hiện lâm sàng, và ở 60% các trường hợp được xác định bằng sàng lọc sơ sinh.
Trẻ em bị thiếu hụt MCAD có thể trông bình thường khi sinh, mặc dù các triệu chứng có thể biểu hiện trước khi có kết quả từ sàng lọc sơ sinh, đặc biệt là ở trẻ sơ sinh bú mẹ. Biểu hiện lâm sàng điển hình (nếu không được phát hiện bằng sàng lọc sơ sinh) xảy ra trong khoảng từ 3 đến 24 tháng tuổi và được thúc đẩy để đáp ứng với việc nhịn ăn kéo dài (ví dụ, tại thời điểm cai sữa các cữ bú ban đêm hoặc trong một bệnh xen kẽ). Các biểu hiện có thể bao gồm nôn mửa, lơ mơ, hôn mê và bệnh não gan. Các đợt mất bù cấp tính có thể nhanh chóng tiến triển thành hôn mê và tử vong và có thể giống với hội chứng đột tử ở trẻ sơ sinh (SIDS) hoặc hội chứng Reye. Ở những trẻ chưa được chẩn đoán trước đó, tỷ lệ tử vong từ đợt đầu tiên cao tới 18%.
Mặc dù có sự không đồng nhất đáng kể trong biểu hiện của MCAD, biểu hiện lâm sàng thường gặp nhất là hạ đường huyết hạ ceton máu không liên tục với ít hoặc không có nhiễm toan, tăng urê, amoniac, và acid uric huyết thanh, các bất thường chức năng gan, và gan nhiễm mỡ. Nguy cơ biến chứng nặng, tổn thương não và tử vong là rất cao trừ khi việc điều trị thích hợp để đảo ngược trạng thái dị hóa được thực hiện. Chẩn đoán các trường hợp này có thể bị nhầm lẫn với hội chứng Reye. Mặc dù hạ đường huyết có thể là một đặc điểm nổi bật muộn với MCAD, kiểu hình của một khiếm khuyết của một rối loạn oxy hóa acid béo có thể không biểu hiện nếu tránh được tình trạng nhịn ăn. Một chỉ số nghi ngờ cao đối với các khiếm khuyết oxy hóa acid béo là quan trọng vì liệu pháp thích hợp có thể dẫn đến sự gián đoạn và phòng ngừa các đợt đe dọa tính mạng này. Bệnh nhân bị ảnh hưởng cũng đã bị chẩn đoán nhầm là SIDS vô căn. Nồng độ carnitine huyết tương giảm và sự gia tăng tỷ lệ carnitine este hóa trên carnitine tự do là một phát hiện xét nghiệm thường liên quan.
Đánh giá nghi ngờ thiếu hụt MCAD hoặc các lỗi khác trong quá trình oxy hóa acid béo trước tiên nên bao gồm việc xác định hồ sơ acylcarnitine huyết tương bằng khối phổ và đo lường carnitine toàn phần, este hóa và tự do trong huyết tương. Bảng 7.4 cho thấy các bất thường của acylcarnitine huyết tương liên quan đến các rối loạn oxy hóa acid béo cụ thể có liên quan đến các bất thường cụ thể của acylcarnitine huyết tương—octanoylcarnitine trong các trường hợp thiếu hụt MCAD. Việc xác định các acid hữu cơ trong nước tiểu với việc đánh giá sự hiện diện hoặc không có acid dicarboxylic niệu cũng có thể hữu ích. Các xét nghiệm khác có thể hữu ích ở những bệnh nhân cần đánh giá thêm bao gồm các xét nghiệm oxy hóa acid béo và các xét nghiệm enzyme cụ thể trong các nguyên bào sợi da hoặc lympho bào được nuôi cấy. Kể từ đầu những năm 1990, việc sử dụng khối phổ song song đã giúp sàng lọc sơ sinh cho hầu hết các rối loạn oxy hóa acid béo, dựa trên hồ sơ acylcarnitine trong các đốm máu. Việc xác định trước triệu chứng của những cá nhân này có thể ngăn ngừa các sự kiện thảm khốc, chẳng hạn như đột tử. Phân tích đột biến gen có thể được thực hiện cho tất cả các khiếm khuyết này, điều này đặc biệt hữu ích trong MCAD, trong đó hầu hết các trường hợp là do một đột biến phổ biến duy nhất (Lys304Gly).
Bảng 7.4 Các rối loạn oxy hóa Acid béo với các Dấu hiệu Chuyển hóa Phân biệt
| Rối loạn | Acylcarnitine huyết tương | Acylglycine nước tiểu | Acid hữu cơ nước tiểu |
|---|---|---|---|
| VLCAD | Tetradecenoyl- | ||
| MCAD | Octanoyl | Hexanoyl-Suberyl-Phenylpropionyl- | |
| SCAD | Butyryl- | Butyryl- | Ethylmalonic |
| LCHAD | 3-Hydroxy-palmitoyl-3-Hydroxy-oleoyl-3-Hydroxy-linoleoyl- | 3-Hydroxydicarboxylic | |
| DER | Dodecadienoyl- | ||
| ETF và ETF-DH | Butyryl-Isovaleryl-Glutaryl- | Isovaleryl- | Ethylmalonic |
| Hexanoyl- | Glutaric | ||
| Isovaleric | |||
| HMG-CoA lyase | Methylglutaryl- | 3-Hydroxy-3-methyl-glutaric |
DER, 2,4-dienoyl-coenzyme A reductase; ETF, flavoprotein chuyển điện tử; ETF-DH, dehydrogenase ETF; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; LCHAD, 3-hydroxyacyl-coenzyme A dehydrogenase chuỗi dài; MCAD, acyl-coenzyme A dehydrogenase chuỗi trung bình; SCAD, acyl-coenzyme A dehydrogenase chuỗi ngắn; VLCAD, acyl-coenzyme A dehydrogenase chuỗi rất dài.
Phương pháp điều trị chính của các rối loạn oxy hóa acid béo là tránh nhịn ăn một cách tận tâm. Đối với trẻ nhũ nhi dưới 1 tuổi, 6 đến 8 giờ nhịn ăn có thể đủ để gây ra một đợt. Mặt khác, khi trẻ lớn hơn, chúng có thể chịu được các khoảng thời gian nhịn ăn lên đến 10 đến 12 giờ mà không bị mất bù. Một chỉ số nghi ngờ cao và việc bắt đầu truyền glucose IV nhanh chóng thường sẽ đảo ngược một đợt đang tiến triển. Theo dõi glucose không hữu ích, vì sự hiện diện của hạ đường huyết thường là một sự kiện xảy ra muộn trong sự tiến triển của một đợt mất bù chuyển hóa. Nên tránh chế độ ăn nhiều chất béo (ví dụ, ketogenic), mặc dù lượng chất béo trong chế độ ăn bình thường là được phép. Việc sử dụng tinh bột ngô sống (như được sử dụng để điều trị bệnh dự trữ glycogen type 1) hiếm khi được chỉ định.
Khiếm khuyết của các chất vận chuyển Glucose
Thiếu hụt GLUT1
Hội chứng thiếu hụt GLUT1 là một lỗi bẩm sinh di truyền trội trên nhiễm sắc thể thường của vận chuyển glucose, đặc trưng bởi co giật, chậm phát triển, co cứng, tật đầu nhỏ mắc phải và mất điều hòa. Dấu hiệu sinh hóa đặc trưng là phát hiện hạ glucose dịch não tủy (nồng độ glucose trong dịch não tủy thấp), mặc dù nồng độ glucose huyết tương bình thường.
GLUT1, được mã hóa bởi SLC2A1, là phương tiện cơ bản tạo điều kiện cho glucose đi vào não. Chẩn đoán thiếu hụt GLUT1 dựa trên việc tìm thấy glucose trong dịch não tủy thấp, trong trường hợp không có hạ đường huyết, và xác định đột biến SLC2A1 (nằm trên nhiễm sắc thể 1). Kiểu hình kinh điển là một dạng nghiêm trọng của bệnh não động kinh khởi phát sớm ở khoảng 90% các trường hợp (dạng kinh điển). Một dạng không động kinh chiếm 10% các trường hợp, bao gồm một phổ kiểu hình rộng có thể biểu hiện với các biểu hiện kịch phát không động kinh đã được báo cáo bao gồm mất điều hòa không liên tục, múa giật-múa vờn, loạn trương lực cơ và liệt nửa người xen kẽ.
Các nỗ lực điều trị đã dựa trên việc cung cấp các nguồn nhiên liệu não thay thế bằng chế độ ăn ketogenic. Chế độ ăn ketogenic kiểm soát hiệu quả các cơn co giật và các hoạt động kịch phát khác, nhưng nó có ít ảnh hưởng hơn đến chức năng nhận thức.
Thiếu hụt GLUT2
Ban đầu được mô tả bởi Fanconi và Bickel như một hội chứng được đặc trưng bởi hạ đường huyết và ceton niệu trong trạng thái nhịn ăn và tăng đường huyết trong trạng thái sau hấp thu, hội chứng Fanconi-Bickel là do các đột biến lặn của chất vận chuyển glucose màng huyết tương GLUT2 (được mã hóa bởi SLC2A2). Khiếm khuyết trên GLUT2 dẫn đến sự tích tụ glycogen ở gan-thận, rối loạn chức năng ống thận gần, và suy giảm việc sử dụng glucose và galactose. GLUT2 được biểu hiện trong tế bào gan, tế bào β của tụy, và màng đáy của các tế bào biểu mô ruột và ống thận. Các biểu hiện lâm sàng phản ánh sự suy giảm giải phóng glucose từ gan và tái hấp thu glucose từ các tế bào ống thận. Sự thanh thải galactose và chuyển đổi thành glucose bị trì hoãn.
Bệnh nhân thiếu hụt GLUT2 thường biểu hiện ở độ tuổi từ 3 đến 10 tháng. Các dấu hiệu lâm sàng điển hình là gan to do tích tụ glycogen, một bệnh ống thận loại Fanconi nghiêm trọng với glucose niệu nặng không tương xứng, không dung nạp glucose và galactose, còi xương hạ phosphat máu, và chậm phát triển nghiêm trọng. Những bệnh nhân này có thể biểu hiện với sự kết hợp của hạ đường huyết khi nhịn ăn và tăng đường huyết sau ăn. Hạ đường huyết trong khi nhịn ăn được giải thích bằng sự thay đổi vận chuyển glucose ra khỏi gan, dẫn đến tăng nồng độ glucose nội bào có thể ức chế sự phân hủy glycogen, dẫn đến lưu trữ glycogen và gan to. Hạ đường huyết trầm trọng hơn do mất glucose qua thận do khiếm khuyết vận chuyển glucose và galactose qua màng đáy của các tế bào ống thận. Tăng đường huyết (và tăng galactose máu) trong trạng thái no được giải thích bằng việc giảm hấp thu monosaccharide của gan và được tăng cường bởi sự bài tiết insulin thấp không phù hợp do suy giảm cảm nhận glucose của các tế bào β của tụy.
Mục tiêu điều trị cho bệnh nhân thiếu hụt GLUT2 là cải thiện hậu quả của bệnh ống thận bằng cách thay thế nước, điện giải và kiềm—và bằng cách cung cấp bổ sung phosphate và vitamin D. Về chế độ ăn uống, nên có một lượng calo đầy đủ để bù đắp cho sự mất glucose qua thận và ruột—được cho ăn thường xuyên chứa carbohydrate hấp thu chậm (chẳng hạn như tinh bột ngô) để tránh hạ đường huyết khi nhịn ăn.
Điều trị
Một cách tiếp cận điều trị hợp lý đối với việc điều trị hạ đường huyết trước hết dựa vào một đánh giá chẩn đoán hệ thống dẫn đến một chẩn đoán cụ thể. Trước khi chẩn đoán cụ thể đó được đưa ra, và trước khi thực hiện các kế hoạch điều trị cá nhân, mục tiêu của liệu pháp là bảo tồn chức năng hệ thần kinh trung ương và ngăn ngừa các trạng thái dị hóa trong đó các chất chuyển hóa trung gian, chẳng hạn như acid béo tự do và các acylcarnitine của chúng, ceton, hoặc lactate có thể gây hại thứ phát. Truyền glucose IV vẫn là phương pháp chính của liệu pháp cấp cứu, đặc biệt là đối với trẻ bị hạ đường huyết nặng. Điều quan trọng là phải theo sau một liều bolus glucose ban đầu bằng một đợt truyền glucose liên tục để ngăn ngừa các đợt hạ đường huyết tiếp theo, cho đến khi các liệu pháp cụ thể được thiết lập. Đối với trẻ sơ sinh trong 24 đến 48 giờ đầu đời, xem phần trước (Sinh lý Cân bằng nội môi Glucose Chu sinh).
Liều bolus glucose ban đầu có thể dao động từ 0,2 đến 0,5 g/kg glucose (2–5 mL/kg dextrose 10%) dưới dạng bolus IV, sau đó là truyền 8 mg/kg/phút (khoảng 5 mL/kg/giờ dextrose 10%). Liều này có thể được giảm nếu glucose huyết tương lớn hơn 5 mmol/L (> 90 mg/dL), nhưng nên được tăng nhanh nếu các kiểm tra glucose thường xuyên cho thấy không thể nâng glucose huyết tương lên lớn hơn 3,8 mmol/L (> 70 mg/dL). Điều này ban đầu có thể được thực hiện theo từng bước tăng 1,6 mg/kg/phút (1 mL/kg/giờ dextrose 10%), nhưng nếu glucose không tăng lên lớn hơn 3,8 mmol/L (> 70 mg/dL) sau 2 lần tăng mỗi 15 phút, thì hãy tăng thêm 3,2 mg/kg/giờ (2 mL/kg/giờ dextrose 10%).
Lý tưởng nhất, một khi chẩn đoán cụ thể được đưa ra, việc điều trị nên duy trì đường huyết bình thường theo lịch ăn bình thường theo tuổi của bệnh nhân. Ngoài ra, cần tính đến ảnh hưởng của bệnh xen kẽ đối với việc điều hòa glucose và khoảng thời gian nhịn ăn dài nhất mà bệnh nhân nên trải qua thường xuyên, nên được xác định cho cả trạng thái khỏe mạnh và trong khi bị bệnh xen kẽ. Nên định kỳ đánh giá lại hiệu quả của việc điều trị cho bất kỳ dạng hạ đường huyết nào bằng một nghiên cứu nhịn ăn chính thức trong quá trình điều trị. Tất cả bệnh nhân nên có một lá thư cấp cứu nêu rõ chẩn đoán cụ thể của họ, lời khuyên về phòng ngừa hạ đường huyết và thông tin về điều trị cấp tính một đợt hạ đường huyết, ngoài thông tin liên lạc của bác sĩ điều trị để được hỗ trợ liên tục.
Kết luận
Kể từ những báo cáo đầu tiên về hạ đường huyết sơ sinh vào những năm 1950 của McQuarrie và các đồng nghiệp của Cornblath, sự hiểu biết của chúng ta về di truyền phân tử và sinh lý bệnh của các rối loạn khác nhau gây hạ đường huyết ở trẻ sơ sinh và trẻ nhũ nhi đã có những tiến bộ đáng kể. Các khám phá kể từ ấn bản trước của cuốn sách này bao gồm việc mô tả các dạng tăng insulin máu trong hội chứng mới, đặc biệt là hội chứng Kabuki và hội chứng Turner; một kiểu hình kết hợp của tăng insulin máu và suy tuyến yên do các đột biến bất hoạt trong FOXA2; và sự kích hoạt truyền tín hiệu thụ thể insulin thông qua các đột biến trong PI3K, bổ sung cho những đột biến trong AKT2 biểu hiện với một kiểu hình hạ đường huyết hạ ceton máu không có tăng insulin máu. Với sự tiến bộ nhanh chóng trong việc hiểu biết về di truyền phân tử và các mối tương quan kiểu gen-kiểu hình cho các loại tăng insulin máu bẩm sinh khác nhau, nguyên nhân phổ biến nhất của hạ đường huyết dai dẳng ở trẻ sơ sinh, một mô hình y học cá nhân hóa đang bắt đầu xuất hiện. Ví dụ điển hình nhất là cách tiếp cận hiện tại đối với trẻ em nghi ngờ bị tăng insulin máu khu trú—từ phân tích di truyền để xác định khả năng bị tăng insulin máu khu trú, đến việc định vị tổn thương bằng hình ảnh không xâm lấn, sử dụng PET 18F-fluoro-L-DOPA, và phẫu thuật cắt bỏ tổn thương bảo tồn tụy bình thường và chữa khỏi bệnh. Những khám phá này và trong tương lai sẽ tiếp tục cải thiện kết quả điều trị.
Bảng chú giải thuật ngưc Anh – Việt – Chương 7
| STT | Tên thuật ngữ tiếng Anh | Phiên âm (IPA) | Nghĩa tiếng Việt |
|---|---|---|---|
| 1 | Hypoglycemia | /ˌhaɪpoʊɡlaɪˈsiːmiə/ | Hạ đường huyết (Tình trạng nồng độ glucose trong máu thấp bất thường) |
| 2 | Newborn | /ˈnuːbɔːrn/ | Trẻ sơ sinh (Trẻ từ lúc sinh ra đến 28 ngày tuổi) |
| 3 | Infant | /ˈɪnfənt/ | Trẻ nhũ nhi (Trẻ từ 1 tháng đến 1 tuổi) |
| 4 | Metabolic | /ˌmɛtəˈbɒlɪk/ | (Thuộc về) Chuyển hóa |
| 5 | Fetal | /ˈfiːtl/ | (Thuộc về) Bào thai |
| 6 | Neonatal | /ˌniːoʊˈneɪtl/ | (Thuộc về) Sơ sinh |
| 7 | Transplacental passage | /ˌtrænspləˈsɛntəl ˈpæsɪdʒ/ | Sự vận chuyển qua nhau thai |
| 8 | Plasma glucose concentration | /ˈplæzmə ˈɡluːkoʊs ˌkɒnsənˈtreɪʃən/ | Nồng độ glucose huyết tương |
| 9 | Central nervous system | /ˈsɛntrəl ˈnɜːrvəs ˈsɪstəm/ | Hệ thần kinh trung ương |
| 10 | Diagnostic | /ˌdaɪəɡˈnɒstɪk/ | (Thuộc về) Chẩn đoán |
| 11 | Therapeutic | /ˌθɛrəˈpjuːtɪk/ | (Thuộc về) Điều trị |
| 12 | Parturition history | /ˌpɑːrtjʊˈrɪʃən ˈhɪstəri/ | Bệnh sử sinh đẻ |
| 13 | Physiology | /ˌfɪziˈɒlədʒi/ | Sinh lý học |
| 14 | Pathophysiology | /ˌpæθoʊˌfɪziˈɒlədʒi/ | Sinh lý bệnh học |
| 15 | Glucose metabolism | /ˈɡluːkoʊs məˈtæbəlɪzəm/ | Chuyển hóa glucose |
| 16 | Fuel economy | /ˈfjuːəl ɪˈkɒnəmi/ | Cân bằng năng lượng |
| 17 | Basal daily energy | /ˈbeɪsəl ˈdeɪli ˈɛnərdʒi/ | Năng lượng cơ bản hàng ngày |
| 18 | Glycogen | /ˈɡlaɪkədʒən/ | Glycogen (Dạng dự trữ glucose trong cơ thể) |
| 19 | Fatty acids | /ˈfæti ˈæsɪdz/ | Acid béo |
| 20 | Aerobic oxidation | /ɛəˈroʊbɪk ˌɒksɪˈdeɪʃən/ | Oxy hóa hiếu khí |
| 21 | Adenosine triphosphate (ATP) | /əˈdɛnəˌsiːn traɪˈfɒsfeɪt/ | Adenosine triphosphate (Đơn vị năng lượng của tế bào) |
| 22 | Glycolytic tissues | /ˌɡlaɪkəˈlɪtɪk ˈtɪʃuːz/ | Các mô đường phân |
| 23 | Adipose tissue | /ˈædɪpoʊs ˈtɪʃuː/ | Mô mỡ |
| 24 | Triglyceride | /traɪˈɡlɪsəraɪd/ | Triglyceride (Một dạng chất béo) |
| 25 | Cerebral glucose utilization | /ˈsɛrəbrəl ˈɡluːkoʊs ˌjuːtɪlaɪˈzeɪʃən/ | Sự sử dụng glucose ở não |
| 26 | Carrier-mediated facilitated diffusion | /ˈkæriər ˈmiːdieɪtɪd fəˈsɪlɪteɪtɪd dɪˈfjuːʒən/ | Khuếch tán được thuận hóa qua chất mang |
| 27 | Glucose transporter (GLUT) | /ˈɡluːkoʊs trænsˈpɔːrtər/ | Chất vận chuyển glucose |
| 28 | Blood-brain barrier | /blʌd breɪn ˈbæriər/ | Hàng rào máu-não |
| 29 | Neurons | /ˈnʊərɒnz/ | Tế bào thần kinh (Neuron) |
| 30 | Insulin-responsive tissues | /ˈɪnsəlɪn rɪˈspɒnsɪv ˈtɪʃuːz/ | Các mô nhạy cảm với insulin |
| 31 | Intracerebral glucopenia | /ˌɪntrəˈsɛrəbrəl ˌɡluːkoʊˈpiːniə/ | Tình trạng thiếu glucose trong não |
| 32 | Cerebrospinal fluid | /səˌriːbroʊˈspaɪnəl ˈfluːɪd/ | Dịch não tủy |
| 33 | Hypoglycorrhachia | /ˌhaɪpoʊˌɡlaɪkəˈreɪkiə/ | Hạ đường huyết dịch não tủy |
| 34 | Seizures | /ˈsiːʒərz/ | Cơn co giật |
| 35 | Developmental delay | /dɪˌvɛləpˈmɛntəl dɪˈleɪ/ | Chậm phát triển |
| 36 | Autonomic nervous system | /ˌɔːtəˈnɒmɪk ˈnɜːrvəs ˈsɪstəm/ | Hệ thần kinh tự chủ (thực vật) |
| 37 | Glycogenolysis | /ˌɡlaɪkəʊdʒəˈnɒlɪsɪs/ | Ly giải glycogen (Quá trình phân giải glycogen thành glucose) |
| 38 | Gluconeogenesis | /ˌɡluːkoʊˌniːoʊˈdʒɛnɪsɪs/ | Tân tạo glucose (Quá trình tạo glucose từ các nguồn không phải carbohydrate) |
| 39 | Perinatal glucose homeostasis | /ˌpɛrɪˈneɪtəl ˈɡluːkoʊs ˌhoʊmiəˈsteɪsɪs/ | Cân bằng nội môi glucose chu sinh |
| 40 | Lactate | /ˈlækteɪt/ | Lactate |
| 41 | Uterine blood flow | /ˈjuːtəraɪn blʌd floʊ/ | Lưu lượng máu tử cung |
| 42 | Intrauterine growth restriction (IUGR) | /ˌɪntrəˈjuːtərɪn ɡroʊθ rɪˈstrɪkʃən/ | Chậm tăng trưởng trong tử cung |
| 43 | Placental lactogen | /pləˈsɛntəl ˈlæktədʒən/ | Lactogen nhau thai |
| 44 | Insulin resistance | /ˈɪnsəlɪn rɪˈzɪstəns/ | Đề kháng insulin |
| 45 | Gestational diabetes | /dʒɛˈsteɪʃənəl ˌdaɪəˈbiːtiːz/ | Đái tháo đường thai kỳ |
| 46 | Fetal hyperinsulinemia | /ˈfiːtl ˌhaɪpərˌɪnsəlɪˈniːmiə/ | Tăng insulin máu ở bào thai |
| 47 | Counterregulatory responses | /ˌkaʊntərˈrɛɡjələtɔːri rɪˈspɒnsɪz/ | Các phản ứng điều hòa ngược |
| 48 | Transitional neonatal hypoglycemia | /trænˈzɪʃənəl ˌniːoʊˈneɪtəl ˌhaɪpoʊɡlaɪˈsiːmiə/ | Hạ đường huyết sơ sinh chuyển tiếp |
| 49 | Lipolysis | /laɪˈpɒlɪsɪs/ | Ly giải mỡ (Quá trình phân giải chất béo) |
| 50 | Ketogenesis | /ˌkiːtoʊˈdʒɛnɪsɪs/ | Sinh ceton (Quá trình tạo ra thể ceton) |
| 51 | Hyperketonemia | /ˌhaɪpərˌkiːtoʊˈniːmiə/ | Tăng ceton máu |
| 52 | Suckling ketosis | /ˈsʌklɪŋ kiːˈtoʊsɪs/ | Nhiễm ceton do bú mẹ |
| 53 | Intravenous (IV) feedings | /ˌɪntrəˈviːnəs ˈfiːdɪŋz/ | Nuôi ăn đường tĩnh mạch |
| 54 | Beta-cell | /ˈbeɪtə sɛl/ | Tế bào beta (Tế bào tụy sản xuất insulin) |
| 55 | Perinatal stress-induced hyperinsulinism | /ˌpɛrɪˈneɪtəl strɛs ɪnˈdjuːst ˌhaɪpərˈɪnsəlɪnɪzəm/ | Tăng insulin máu do stress chu sinh |
| 56 | Birth asphyxia | /bɜːrθ æsˈfɪksiə/ | Ngạt khi sinh |
| 57 | Maternal toxemia | /məˈtɜːrnəl tɒkˈsiːmiə/ | Nhiễm độc thai nghén |
| 58 | Maternal hypertension | /məˈtɜːrnəl ˌhaɪpərˈtɛnʃən/ | Tăng huyết áp ở mẹ |
| 59 | Meconium aspiration syndrome | /məˈkoʊniəm ˌæspəˈreɪʃən ˈsɪndroʊm/ | Hội chứng hít phân su |
| 60 | Erythroblastosis fetalis | /ɪˌrɪθroʊblæˈstoʊsɪs fiːˈtælɪs/ | Bệnh nguyên hồng cầu bào thai |
| 61 | Polycythemia | /ˌpɒlisaɪˈθiːmiə/ | Đa hồng cầu |
| 62 | Postmature delivery | /ˌpoʊstməˈtʃʊər dɪˈlɪvəri/ | Sinh già tháng |
| 63 | Congenital Syndromes | /kənˈdʒɛnɪtl ˈsɪndroʊmz/ | Các hội chứng bẩm sinh |
| 64 | Midline facial malformations | /ˈmɪdlaɪn ˈfeɪʃəl ˌmælfɔːrˈmeɪʃənz/ | Dị tật đường giữa của mặt |
| 65 | Microphallus | /ˌmaɪkroʊˈfæləs/ | Dương vật nhỏ |
| 66 | Glucose infusion rate (GIR) | /ˈɡluːkoʊs ɪnˈfjuːʒən reɪt/ | Tốc độ truyền glucose |
| 67 | Subcutaneous (SQ) | /ˌsʌbkjuːˈteɪniəs/ | (Đường) Dưới da |
| 68 | Intramuscular (IM) | /ˌɪntrəˈmʌskjələr/ | (Đường) Tiêm bắp |
| 69 | Fasting challenge | /ˈfæstɪŋ ˈtʃælɪndʒ/ | Nghiệm pháp nhịn ăn |
| 70 | Fasting adaptation | /ˈfæstɪŋ ˌædæpˈteɪʃən/ | Thích ứng với nhịn ăn |
| 71 | Postprandial hypoglycemia | /ˌpoʊstˈprændiəl ˌhaɪpoʊɡlaɪˈsiːmiə/ | Hạ đường huyết sau ăn |
| 72 | Nissen fundoplication | /ˈnɪsən ˌfʌndoʊplɪˈkeɪʃən/ | Phẫu thuật Nissen (thắt đáy vị) |
| 73 | Hereditary fructose intolerance | /həˈrɛdɪtəri ˈfrʊktoʊs ɪnˈtɒlərəns/ | Bất dung nạp fructose di truyền |
| 74 | Congenital hyperinsulinism | /kənˈdʒɛnɪtl ˌhaɪpərˈɪnsəlɪnɪzəm/ | Tăng insulin máu bẩm sinh |
| 75 | Counterregulatory hormones | /ˌkaʊntərˈrɛɡjələtɔːri ˈhɔːrmoʊnz/ | Các hormone điều hòa ngược |
| 76 | Type 1 diabetes mellitus | /taɪp wʌn ˌdaɪəˈbiːtiːz ˈmɛlɪtəs/ | Đái tháo đường type 1 |
| 77 | Autonomic failure | /ˌɔːtəˈnɒmɪk ˈfeɪljər/ | Suy hệ thần kinh tự chủ |
| 78 | Ketone bodies | /ˈkiːtoʊn ˈbɒdiz/ | Thể ceton |
| 79 | Whipple’s triad | /ˈwɪpəlz ˈtraɪæd/ | Tam chứng Whipple |
| 80 | Neurogenic symptoms | /ˌnʊəroʊˈdʒɛnɪk ˈsɪmptəmz/ | Các triệu chứng thần kinh |
| 81 | Erythrocytes | /ɪˈrɪθrəsaɪts/ | Hồng cầu |
| 82 | Hematocrit | /hɪˈmætəkrɪt/ | Hematocrit (Dung tích hồng cầu) |
| 83 | Indwelling lines | /ɪnˈdwɛlɪŋ laɪnz/ | Các đường truyền lưu |
| 84 | Venous | /ˈviːnəs/ | (Thuộc về) Tĩnh mạch |
| 85 | Arterial | /ɑːrˈtɪəriəl/ | (Thuộc về) Động mạch |
| 86 | Neuroglycopenic | /ˌnʊəroʊˌɡlaɪkoʊˈpiːnɪk/ | (Thuộc về) Thiếu glucose thần kinh |
| 87 | Jitteriness | /ˈdʒɪtərɪnəs/ | Tình trạng run rẩy |
| 88 | Tachycardia | /ˌtækɪˈkɑːrdiə/ | Nhịp tim nhanh |
| 89 | Pallor | /ˈpælər/ | Da xanh xao |
| 90 | Hypothermia | /ˌhaɪpoʊˈθɜːrmiə/ | Hạ thân nhiệt |
| 91 | Lethargy | /ˈlɛθərdʒi/ | Lơ mơ |
| 92 | Irritability | /ˌɪrɪtəˈbɪləti/ | Dễ bị kích thích |
| 93 | Cyanosis | /ˌsaɪəˈnoʊsɪs/ | Tím tái |
| 94 | Tachypnea | /ˌtækɪpˈniːə/ | Thở nhanh |
| 95 | Apneic episodes | /ˈæpniɪk ˈɛpɪsoʊdz/ | Cơn ngưng thở |
| 96 | Floppiness | /ˈflɒpɪnəs/ | Trương lực cơ giảm (mềm nhũn) |
| 97 | Microphthalmia | /ˌmaɪkrɒfˈθælmiə/ | Mắt nhỏ |
| 98 | Transaminases | /trænsˈæmɪneɪsɪz/ | Transaminase (Men gan) |
| 99 | Hyperammonemia | /ˌhaɪpərˌæmoʊˈniːmiə/ | Tăng amoniac máu |
| 100 | Creatine kinase | /ˈkriːətiːn ˈkaɪneɪs/ | Creatine kinase (Men cơ) |
| 101 | Acidemia | /ˌæsɪˈdiːmiə/ | Nhiễm toan máu |
| 102 | Serum bicarbonate | /ˈsɪərəm baɪˈkɑːrbənət/ | Bicarbonate huyết thanh |
| 103 | Glucagon stimulation test | /ˈɡluːkəɡɒn ˌstɪmjəˈleɪʃən tɛst/ | Nghiệm pháp kích thích bằng glucagon |
| 104 | Plasma acylcarnitine profile | /ˈplæzmə ˌeɪsɪlˈkɑːrnɪtiːn ˈproʊfaɪl/ | Hồ sơ acylcarnitine huyết tương |
| 105 | Mitochondrial β-oxidation | /ˌmaɪtəˈkɒndriəl ˈbeɪtə ˌɒksɪˈdeɪʃən/ | β-oxy hóa tại ty thể |
| 106 | Provocative diagnostic tests | /prəˈvɒkətɪv ˌdaɪəɡˈnɒstɪk tɛsts/ | Các nghiệm pháp chẩn đoán kích thích |
| 107 | Hypoketonemia | /ˌhaɪpoʊˌkiːtoʊˈniːmiə/ | Hạ ceton máu |
| 108 | Hypo-free fatty acidemia | /ˈhaɪpoʊ friː ˈfæti ˌæsɪˈdiːmiə/ | Hạ acid béo tự do trong máu |
| 109 | Dysregulated insulin secretion | /dɪsˈrɛɡjəleɪtɪd ˈɪnsəlɪn sɪˈkriːʃən/ | Rối loạn điều hòa bài tiết insulin |
| 110 | Monogenic hyperinsulinism | /ˌmɒnoʊˈdʒɛnɪk ˌhaɪpərˈɪnsəlɪnɪzəm/ | Tăng insulin máu đơn gen |
| 111 | Syndromic hyperinsulinism | /sɪnˈdroʊmɪk ˌhaɪpərˈɪnsəlɪnɪzəm/ | Tăng insulin máu trong hội chứng |
| 112 | Plethoric | /pləˈθɔːrɪk/ | (Da) Ứ huyết, đỏ bừng |
| 113 | Vernix caseosa | /ˈvɜːrnɪks keɪˈsoʊsə/ | Chất gây (Lớp sáp trắng bao phủ da trẻ sơ sinh) |
| 114 | Adrenomedullary exhaustion | /əˌdriːnoʊˈmɛdʒʊləri ɪɡˈzɔːstʃən/ | Suy kiệt tủy thượng thận |
| 115 | Glucocorticoids | /ˌɡluːkoʊˈkɔːrtɪkɔɪdz/ | Glucocorticoid (Một nhóm hormone steroid) |
| 116 | Hypothalamic-pituitary-adrenal axis | /ˌhaɪpoʊθəˈlæmɪk pɪˈtjuːɪtəri əˈdriːnəl ˈæksɪs/ | Trục dưới đồi-tuyến yên-thượng thận |
| 117 | Consanguinity | /ˌkɒnsæŋˈɡwɪnəti/ | Hôn nhân cận huyết |
| 118 | Phosphorylation | /ˌfɒsfərɪˈleɪʃən/ | Phosphoryl hóa |
| 119 | ATP-dependent potassium (KATP) channel | /eɪ tiː piː dɪˈpɛndənt pəˈtæsiəm ˈtʃænəl/ | Kênh Kali phụ thuộc ATP (KATP) |
| 120 | Heterooctameric complex | /ˌhɛtəroʊˌɒktəˈmɛrɪk ˈkɒmplɛks/ | Phức hợp tám tiểu đơn vị khác nhau |
| 121 | Pore-forming subunit | /pɔːr ˈfɔːrmɪŋ ˈsʌbˌjuːnɪt/ | Tiểu đơn vị tạo lỗ |
| 122 | Regulatory subunit | /ˈrɛɡjələtɔːri ˈsʌbˌjuːnɪt/ | Tiểu đơn vị điều hòa |
| 123 | Resting membrane potential | /ˈrɛstɪŋ ˈmɛmbreɪn pəˈtɛnʃəl/ | Điện thế màng lúc nghỉ |
| 124 | Voltage-dependent Ca2+ channels | /ˈvoʊltɪdʒ dɪˈpɛndənt ˈkælsiəm ˈtʃænəlz/ | Kênh Canxi phụ thuộc điện thế |
| 125 | Exocytotic machinery | /ˌɛksoʊsaɪˈtɒtɪk məˈʃiːnəri/ | Bộ máy xuất bào |
| 126 | Allosteric activation | /ˌæloʊˈstɛrɪk ˌæktɪˈveɪʃən/ | Hoạt hóa dị lập thể |
| 127 | Diazoxide-unresponsive | /daɪəˈzɒksaɪd ʌnrɪˈspɒnsɪv/ | Không đáp ứng với Diazoxide |
| 128 | Diazoxide-responsive | /daɪəˈzɒksaɪd rɪˈspɒnsɪv/ | Đáp ứng với Diazoxide |
| 129 | Paternal uniparental disomy | /pəˈtɜːrnəl ˌjuːnɪpəˈrɛntəl ˈdaɪsoʊmi/ | Lưỡng bội thể đơn cha |
| 130 | Clonal expansion | /ˈkloʊnəl ɪkˈspænʃən/ | Sự tăng sinh dòng |
| 131 | Focal adenomatous hyperplasia | /ˈfoʊkəl ˌædɪˈnɒmətəs ˌhaɪpərˈpleɪʒə/ | Tăng sản dạng tuyến khu trú |
| 132 | Pancreatectomy | /ˌpæŋkriəˈtɛktəmi/ | Phẫu thuật cắt bỏ tụy |
| 133 | Histologically | /ˌhɪstəˈlɒdʒɪkəli/ | Về mặt mô học |
| 134 | Islet cell nuclei | /ˈaɪlət sɛl ˈnuːkliaɪ/ | Nhân tế bào đảo tụy |
| 135 | Transhepatic portal venous insulin sampling | /ˌtræns hɛˈpætɪk ˈpɔːrtl ˈviːnəs ˈɪnsəlɪn ˈsæmplɪŋ/ | Lấy mẫu insulin tĩnh mạch cửa qua gan |
| 136 | Positron emission tomography (PET) | /ˈpɒzɪtrɒn ɪˈmɪʃən təˈmɒɡrəfi/ | Chụp cắt lớp phát xạ Positron (PET) |
| 137 | Guanosine-5’-triphosphate (GTP) | /ˈɡwɑːnəˌsiːn traɪˈfɒsfeɪt/ | Guanosine-5’-triphosphate |
| 138 | Haploinsufficiency | /ˌhæploʊˌɪnsəˈfɪʃənsi/ | Thiểu năng đơn bội |
| 139 | Neurodevelopmental | /ˌnʊəroʊdɪˌvɛləpˈmɛntəl/ | (Thuộc về) Phát triển thần kinh |
| 140 | Electroencephalogram (EEG) | /ɪˌlɛktroʊɛnˈsɛfələˌɡræm/ | Điện não đồ |
| 141 | Histone demethylase | /ˈhɪstoʊn diːˈmɛθɪleɪs/ | Histone demethylase (Enzyme khử methyl histone) |
| 142 | Nucleomegaly | /ˌnuːklioʊˈmɛɡəli/ | Nhân to |
| 143 | Tachyphylaxis | /ˌtækɪfɪˈlæksɪs/ | Hiện tượng quen thuốc nhanh |
| 144 | Necrotizing enterocolitis | /ˈnɛkrəˌtaɪzɪŋ ˌɛntəroʊkəˈlaɪtɪs/ | Viêm ruột hoại tử |
| 145 | Gain-of-function mutations | /ɡeɪn ʌv ˈfʌŋkʃən mjuːˈteɪʃənz/ | Đột biến tăng chức năng |
| 146 | Dumping syndrome | /ˈdʌmpɪŋ ˈsɪndroʊm/ | Hội chứng Dumping (hội chứng dạ dày rỗng nhanh) |
| 147 | Osmotic load | /ɒzˈmɒtɪk loʊd/ | Tải lượng thẩm thấu |
| 148 | Insulinotropic hormone | /ˌɪnsəlɪnoʊˈtroʊpɪk ˈhɔːrmoʊn/ | Hormone hướng insulin |
| 149 | Septooptic dysplasia | /ˌsɛptoʊˈɒptɪk dɪsˈpleɪʒə/ | Loạn sản vách-thị |
| 150 | Panhypopituitarism | /ˌpænˌhaɪpoʊpɪˈtjuːɪtərɪzəm/ | Suy toàn bộ tuyến yên |