
Sperling Nội tiết học Nhi khoa, Ấn bản thứ 5 – Biên dịch: Ths.Bs. Lê Đình Sáng
Sperling Pediatric Endocrinology, Fifth Edition
Tác giả: Sperling, Mark A., MD – Nhà xuất bản: Elsevier Inc.
PHẦN III. NỘI TIẾT HỌC TRẺ EM VÀ THANH THIẾU NIÊN
Chương 20. Rối loạn Chuyển hóa Khoáng chất II. Những bất thường về Nội môi Khoáng chất ở Trẻ sơ sinh, Trẻ nhỏ, Trẻ em và Vị thành niên
Allen W. Root; Michael A. Levine
Disorders of Mineral Metabolism II. Abnormalities of Mineral Homeostasis in the Newborn, Infant, Child, and Adolescent
Sperling Pediatric Endocrinology, 20, 705-813
Lời cảm ơn
Tưởng nhớ Bác sĩ Frank B. Diamond, Jr.
Các rối loạn chuyển hóa canxi, magiê, và phốt phát cũng như các rối loạn về sự hình thành, tích lũy và duy trì xương trong hai thập kỷ đầu đời là kết quả của việc ăn uống, hấp thu, hoặc giữ lại các chất dinh dưỡng thành phần dưới mức tối ưu, chuyển hóa hoặc hoạt tính sinh học của vitamin D bất thường, rối loạn tổng hợp, bài tiết, hoặc hoạt động của hormon cận giáp (PTH), và những sai lệch nội tại trong các tế bào sụn và xương. Nguồn gốc của những bệnh lý này có thể là nội tại do các biến thể bệnh lý trong các gen kiểm soát các quá trình này hoặc do các tác nhân mắc phải (Bảng 20.1). Nồng độ canxi huyết thanh thay đổi theo tuổi. Mặc dù nồng độ canxi toàn phần và canxi ion hóa (Ca2+) trong huyết thanh thường có mối liên hệ nội tại với nhau, sự phân ly giữa hai chất phân tích này có thể được quan sát thấy ở những bệnh nhân tăng hoặc giảm protein huyết và ở các mức pH huyết tương cực đoan—dù là nhiễm kiềm hay nhiễm toan. Hạ canxi máu hiện diện khi nồng độ Ca2+ huyết thanh dưới, và tăng canxi máu được xác định bởi các giá trị Ca2+ huyết thanh trên giới hạn bình thường theo tuổi, tương ứng. Để có một cái nhìn tổng quan tích hợp về cân bằng nội môi canxi, khoáng chất và xương, người đọc được giới thiệu đến Chương 9.
Bảng 20.1: Nguồn gốc Di truyền của các Rối loạn Chuyển hóa Khoáng chất, Sụn và Xương
| Gen | Nhiễm sắc thể | OMIM | Bệnh | OMIM |
|---|---|---|---|---|
| ACVR1 | 2q24.1 | 102576 | Loạn sản xương hóa đá tiến triển | 135100 |
| AIRE | 21q22.3 | 607358 | Hội chứng đa nội tiết tự miễn, type I | 240300 |
| ALPL | 1p36.12 | 171760 | Giảm phosphatase máu, thể nhũ nhi | 241500 |
| Giảm phosphatase máu, thể trẻ em | 241510 | |||
| Giảm phosphatase máu, thể người lớn | 146300 | |||
| AP2S1 | 19q13.31 | 602242 | Tăng canxi máu giảm canxi niệu di truyền 3 | 600740 |
| BAZ1B | 7q11.23 | 605681 | Hội chứng Williams-Beuren | 194050 |
| BMP1 | 8p21.3 | 112264 | Bệnh xương thủy tinh type XIII | 614856 |
| BSND | 1p32.2 | 606412 | Hội chứng Bartter type 4a | 602522 |
| CA2 | 8q22 | 611492 | Bệnh xương hóa đá – toan hóa ống thận | 259730 |
| CASR | 3q13.3-q21 | 601199 | Tăng canxi máu giảm canxi niệu di truyền 1 | 145980 |
| Cường cận giáp nặng ở trẻ sơ sinh | 239200 | |||
| Tăng canxi máu tăng canxi niệu | 601199 | |||
| Suy cận giáp, đơn độc có tính gia đình | 146200 | |||
| Tăng canxi máu giảm canxi niệu mắc phải | 145980 | |||
| CDC73 | 1q31.2 | 607393 | Suy cận giáp đơn độc có tính gia đình type 1 | 145000 |
| Hội chứng cường cận giáp – u xương hàm | 145001 | |||
| CDKN1B | 12p13.1 | 600778 | Đa u tuyến nội tiết, type 4 | 610755 |
| CHD7 | 8q12.2 | 608092 | Hội chứng CHARGE | 214800 |
| (Khuyết mống mắt, Dị tật tim, Tịt cửa mũi sau, Chậm phát triển, Bất thường sinh dục và tai) | ||||
| CLCN5 | Xp11.23 | 300008 | Còi xương giảm phosphat máu liên kết X lặn | 300554 |
| Bệnh Dent 1 | 300009 | |||
| Sỏi thận, liên kết X lặn | 310468 | |||
| CLCN7 | 16p13.3 | 602727 | Bệnh xương hóa đá, di truyền lặn trên NST thường Type IV | 611490 |
| Bệnh xương hóa đá, di truyền trội trên NST thường type II | 166600 | |||
| CLDN10 | 13q32.1 | 617579 | HELIX = Giảm tiết mồ hôi, Rối loạn điện giải, Rối loạn chức năng tuyến lệ, Bệnh vảy cá, Khô miệng | 617671 |
| CLDN16 | 3q27 | 603959 | Giảm magiê máu type 3 | 248250 |
| CLDN19 | 1p34.2 | 610036 | Giảm magiê máu type 5, tăng canxi niệu, suy giảm thị lực | 248190 |
| CLCNKB | 1p36.13 | 602023 | Hội chứng Bartter type 3 | 607364 |
| CNNM2 | 10q24.32 | 607803 | Giảm magiê máu type 6 với magiê niệu bình thường | 613882 |
| COL1A1 | 17q21.31-q22 | 120150 | Bệnh xương thủy tinh type I | 166200 |
| Bệnh xương thủy tinh type IIA | 166210 | |||
| Bệnh xương thủy tinh type III | 259420 | |||
| Bệnh xương thủy tinh type IV | 166220 | |||
| COL1A2 | 7q22.1 | 120160 | Bệnh xương thủy tinh type IIA | 166210 |
| Bệnh xương thủy tinh type III | 259420 | |||
| Bệnh xương thủy tinh type IV | 166220 | |||
| CREB3L1 | 11p11.2 | 616215 | Bệnh xương thủy tinh type XVI | 616229 |
| CRTAP | 3p22 | 605497 | Bệnh xương thủy tinh type IIB | 610854 |
| Bệnh xương thủy tinh type VII | 610682 | |||
| CTSK | 1q21.3 | 601105 | Bệnh Pycnodysostosis | 265800 |
| CYP2R1 | 11p15.2 | 608713 | Còi xương do thiếu hụt hydroxyl hóa vitamin D, type 1B (thiếu hụt 25-Hydroxylase) | 600081 |
| CYP3A4 | 7q22.1 | 124010 | Còi xương phụ thuộc vitamin D type 3 | |
| CYP24A1 | 12q13.2 | 126065 | Tăng canxi máu nhũ nhi type 1 | 143880 |
| CYP27B1 | 12q14.1 | 609506 | Còi xương do thiếu hụt hydroxyl hóa vitamin D, type IA (thiếu hụt 25α-Hydroxyvitamin D-1α-hydroxylase) | 264700 |
| DMP1 | 4q22.1 | 600980 | Còi xương giảm phosphat máu, di truyền lặn trên NST thường type 1 | 241520 |
| ELN | 7q13.23 | 120160 | Hội chứng Williams-Beuren | 194050 |
| ENPP1 | 6q23.2 | 173335 | Còi xương giảm phosphat máu, di truyền lặn trên NST thường type 2 | 613312 |
| FAM111A | 11q12.1 | 615292 | Hội chứng Kenny-Caffey 2 | 127000 |
| Loạn sản xương mảnh | 602361 | |||
| FAM20C | 7p22.3 | 611061 | Hội chứng Raine | 259775 |
| FERMT3 | 11q13.1 | 607901 | Bệnh xương hóa đá, di truyền lặn trên NST thường | |
| FGF23 | 12p13.3 | 605380 | Còi xương giảm phosphat máu, di truyền trội trên NST thường | 193100 |
| Bệnh vôi hóa dạng u có tính gia đình | 211900 | |||
| Hội chứng tăng xương-tăng phosphat máu | 610233 | |||
| FGFR3 | 4p16.3 | 134934 | Loạn sản sụn | 100800 |
| FKBP10 | 17q21.2 | 607063 | Bệnh xương thủy tinh type XI | 610968 |
| Hội chứng Bruck 1 | 259450 | |||
| FOXP3 | Xp11.23 | 300292 | Rối loạn điều hòa miễn dịch, bệnh đa nội tiết, bệnh ruột (IPEX) | 304790 |
| FXYD2 | 11q23.3 | 601814 | Giảm magiê máu di truyền trội trên NST thường type 2 với giảm canxi niệu | 154020 |
| GALNT3 | 2q24-q31 | 601756 | Bệnh vôi hóa dạng u có tính gia đình | 211900 |
| Hội chứng tăng xương-tăng phosphat máu | 610233 | |||
| GATA3 | 10p13-14 | 131320 | Suy cận giáp, điếc thần kinh giác quan, bệnh thận (suy cận giáp-điếc-loạn sản thận/hội chứng Barakat) | 146255 |
| GCM2 | 6p24.2 | 603716 | Suy cận giáp, đơn độc có tính gia đình | 146200 |
| GNA11 | 19p13.3 | 139313 | Tăng canxi máu giảm canxi niệu di truyền 2 | 145981 |
| GNAS | 20q13.32 | 139320 | Suy cận giáp giả, type 1A | 103580 |
| Suy cận giáp giả, type 1B | 603233 | |||
| Suy cận giáp giả, type 1C | 612462 | |||
| Suy cận giáp giả giả | 612463 | |||
| Loạn sản xương hóa đá tiến triển | 166350 | |||
| Loạn sản xơ/McCune-Albright | 174800 | |||
| GNPTAB | 12q23.2 | 607840 | Bệnh mucolipidosis type II | 252500 |
| GTF21 | 7q11.23 | 601679 | Hội chứng Williams-Beuren | 194050 |
| HADHB | 2p23.3 | 143450 | MELAS—Bệnh não-cơ ty thể, nhiễm toan lactic, đột quỵ, suy cận giáp | 540000 |
| HNF1B | 17q12 | 189907 | Giảm magiê máu với đái tháo đường khởi phát ở người trẻ type 5 và nang thận | 137920 |
| HNRNPC | 14q11.2 | 164020 | Còi xương phụ thuộc vitamin D type 2B | 600785 |
| HRPT2 | 1q24-q31 | 607393 | Cường cận giáp có tính gia đình 2 – hội chứng u xương hàm | 145001 |
| IKBKG | Xq28 | 300248 | Bệnh xương hóa đá, liên kết X | 300301 |
| IFITM5 | 11p15.5 | 6147577 | Bệnh xương thủy tinh type V | 610967 |
| KCNA1 | 12p13.32 | 176260 | Giảm magiê máu với rung giật cơ | 160120 |
| KCNJ1 | 11q24 | 600359 | Giảm magiê máu/Hội chứng Bartter trước sinh type 2 | 600839 |
| KCNJ10 | 1q23.2 | 612780 | Giảm magiê máu/hội chứng sesame | 612780 |
| KL | 13q13.1 | 604824 | Bệnh vôi hóa dạng u có tính gia đình | 211900 |
| Giảm phosphat máu & cường cận giáp | 612089 | |||
| LEPRE1 | 1p34 | 610339 | Bệnh xương thủy tinh type VIII | 610915 |
| LRP4 | 11p11.2 | 604270 | Xương đặc type 2 | 614305 |
| LRP5 | 11.13.4 | 603506 | Hội chứng loãng xương-giả u nguyên bào võng mạc | 259770 |
| Loãng xương vị thành niên tự phát | 259750 | |||
| Biến thể khối lượng xương cao | 601884 | |||
| Bệnh xương hóa đá di truyền trội trên NST thường type I | 607634 | |||
| Bệnh Van Buchem, type 2 | 607636 | |||
| MBTPS2 | Xp22.12 | 300294 | Bệnh xương thủy tinh type XIX | 301014 |
| MEN1 | 11q13 | 613733 | Đa u tuyến nội tiết type I | 131100 |
| NEBL | 10p12.31 | 605491 | Hội chứng DiGeorge type 2 | 605491 |
| Phức hợp hội chứng tim-mặt-vòm miệng 2 | ||||
| NPR2 | 9p21-p12 | 108961 | Loạn sản đầu chi-đoạn giữa (Maroteaux) | 602875 |
| OSTM1 | 6q21 | 607649 | Bệnh xương hóa đá di truyền lặn trên NST thường type V | 259700 |
| PCBD1 | 10q22.1 | 126090 | Giảm magiê máu ở thận, đái tháo đường khởi phát ở người trẻ, type 5 | |
| PDE4D | 5q11.2-q12.1 | 600129 | Loạn sản đầu xương type 2 | 614613 |
| PHEX | Xp22.2-p22.1 | 300550 | Còi xương giảm phosphat máu, di truyền trội liên kết X | 307800 |
| PLEKHM1 | 17q21.3 | 611466 | Bệnh xương hóa đá di truyền lặn trên NST thường type VI | 611497 |
| PPIB | 15q21-q22 | 123841 | Bệnh xương thủy tinh type IX | 259440 |
| PRKAR1A | 17q24.3 | 188830 | Loạn sản đầu xương type 1 | 101800 |
| PTH | 11p15.3 | 168450 | Suy cận giáp, đơn độc có tính gia đình | 146200 |
| PTH1R | 3p21.31 | 168468 | Loạn sản xương sụn Blomstrand | 215045 |
| Loạn sản sụn đầu xương Murk-Jansen | 156400 | |||
| U sụn (Bệnh Ollier) | 166000 | |||
| RET | 10q11.2 | 164761 | Đa u tuyến nội tiết type IIA | 171400 |
| Đa u tuyến nội tiết type IIB | 162300 | |||
| Ung thư biểu mô tuyến giáp thể tủy có tính gia đình | 155240 | |||
| SAMD9 | 7q21.2 | 610456 | Vôi hóa dạng u, phosphat máu bình thường | 610455 |
| SERPINF1 | 17p13.2 | 172860 | Gen bệnh xương thủy tinh type VI | 613982 |
| SERPINH1 | 11q13.5 | 600943 | Gen bệnh xương thủy tinh type X | 613848 |
| SLC2A2 | 3q26.2 | 138160 | Hội chứng Fanconi-Bickel | 227810 |
| SLC34A1 | 5q35.3 | 182309 | Giảm phosphat máu di truyền trội trên NST thường với sỏi niệu 1 | 612286 |
| Hội chứng Fanconi di truyền lặn trên NST thường với còi xương giảm phosphat máu | 613388 | |||
| Tăng canxi máu nhũ nhi, type 2 | 616963 | |||
| SLC34A3 | 9q34.3 | 609826 | Còi xương giảm phosphat máu với tăng canxi niệu | 241530 |
| SLC9A3R1 | 17q25.1 | 604990 | Giảm phosphat máu di truyền trội trên NST thường với sỏi niệu/loãng xương 2 | 612287 |
| SLC12A1 | 15q21.1 | 600839 | Hội chứng Bartter trước sinh type 1 | 601678 |
| SLC12A3 | 16q13 | 600968 | Giảm magiê máu/Hội chứng Gitelman | 263800 |
| SLC7A7 | 14q11.2 | 603593 | Không dung nạp protein lysinuric | 222700 |
| SNX10 | 7p15.2 | 614780 | Loãng xương di truyền lặn trên NST thường type VIII | 615085 |
| SOST | 17q12-q21 | 605740 | Xương đặc | 269500 |
| Tăng sinh xương vỏ toàn thể (Bệnh Van Buchem type 1) | 239100 | |||
| SOX3 | Xq26.3 | 313430 | Suy cận giáp, liên kết X | 307700 |
| SP7 | 12q13.13 | 606633 | Bệnh xương thủy tinh XII | 613849 |
| SPARC | 5q33.1 | 182120 | Bệnh xương thủy tinh XVII | 6616597 |
| STK3 | 11q23.3 | 614766 | Loạn sản đốt sống-đầu xương-đầu xương, Krakow | 618162 |
| STX16 | 20q13.32 | 603666 | Suy cận giáp giả, type 1B | 603233 |
| TBX1 | 22q11.12 | 602054 | Hội chứng DiGeorge | 188400 |
| TBCE | 1q42.3 | 604934 | Hội chứng Sanjad-Sakati (HRD) | 241410 |
| Hội chứng Kenney-Caffey, type 1 | 244460 | |||
| TCIRG1 | 11q13.2 | 604592 | Bệnh xương hóa đá di truyền lặn trên NST thường type I | 259700 |
| TENT5A | 6q14.1 | 611357 | Bệnh xương thủy tinh XVIII | 617592 |
| TGFB1 | 19q13.1 | 190180 | Loạn sản thân xương tiến triển | 131300 |
| TMEM38B | 9q31.1 | 611236 | Gen bệnh xương thủy tinh type XIV | 615066 |
| TNFRSF11A | 18q22.1 | 603499 | Bệnh xương hóa đá di truyền lặn trên NST thường type VII | 612301 |
| Loạn sản tiêu xương đa ổ, lan tỏa di truyền (có tính gia đình) | 174810 | |||
| TNFRSF11B | 8q24 | 602643 | Bệnh Paget, thể vị thành niên | 239000 |
| TNFSF11 | 13q14.11 | 602642 | Bệnh xương hóa đá di truyền lặn trên NST thường type III | 259730 |
| TRPM6 | 9q21.13 | 607009 | Giảm magiê máu type 1 với hạ canxi máu | 602014 |
| TRPV6 | 7q34 | 606680 | Cường cận giáp sơ sinh thoáng qua | 618188 |
| VDR | 12q13.11 | 601769 | Còi xương kháng vitamin D, type IIA | 277440 |
| WNT1 | 12q13.12 | 164820 | Bệnh xương thủy tinh type XV | 615220 |
Xem Bảng 20.13 và 20.14 về các gen liên quan đến loạn sản xương sụn.
Hạ canxi máu
Ở trẻ em trên 1 tuổi, tùy thuộc vào phòng xét nghiệm phân tích, hạ canxi máu được định nghĩa bởi sự giảm các giá trị Ca2+ dưới giới hạn dưới của mức bình thường theo tuổi là 4,64 đến 4,80 mg/dL = 1,16 đến 1,20 mmol/L; giới hạn dưới của mức bình thường theo tuổi của nồng độ canxi toàn phần là 8,5 đến 8,9 mg/dL = 2,20 đến 2,3 mmol/L. Nếu nồng độ albumin huyết thanh giảm 1 g/dL, giá trị canxi toàn phần sẽ giảm 0,8 mg/dL (0,2 mmol/L), trong khi nồng độ Ca2+ không thay đổi. Các triệu chứng của hạ canxi máu phản ánh sự tăng kích thích thần kinh cơ, chẳng hạn như dị cảm, tetany, co cứng bàn tay-bàn chân, co thắt thanh quản, chuột rút cơ và/hoặc co cứng cơ trương lực, và co giật khu trú hoặc toàn thể; các dấu hiệu thực thể của hạ canxi máu bao gồm sự hiện diện của dấu hiệu Chvostek (gõ vào dây thần kinh mặt gây ra co giật các cơ mặt cùng bên) và/hoặc dấu hiệu Trousseau (co cứng bàn tay-bàn chân sau khi duy trì băng đo huyết áp được bơm căng hơi cao hơn áp suất tâm thu trong 3 phút). Trong cơn hạ canxi máu, khoảng QT trên điện tâm đồ kéo dài.
Hạ canxi máu ở trẻ sơ sinh và trẻ nhỏ
Trong tử cung, nồng độ canxi toàn phần và canxi ion hóa của thai nhi cao hơn sau khi sinh; nồng độ canxi toàn phần vượt quá giá trị của mẹ khoảng 2,0 mg/dL = 0,5 mmol/L, vì sự vận chuyển canxi qua nhau thai được kích thích bởi protein liên quan đến hormon cận giáp (PTHrP); trong máu cuống rốn của trẻ đủ tháng, giá trị canxi toàn phần trung bình là 10 đến 11 mg/dL và nồng độ Ca2+ xấp xỉ 6,4 mg/dL = 1,6 mmol/L. Ở thai nhi, nồng độ phosphat, PTHrP và calcitonin trong huyết thanh cao hơn ở phụ nữ mang thai, trong khi nồng độ 1,25-dihydroxyvitamin D3 (calcitriol) và PTH của thai nhi thấp và nồng độ 25-hydroxyvitamin D3 (calcidiol) xấp xỉ giá trị của mẹ. Ở trẻ sơ sinh, nồng độ canxi toàn phần và Ca2+ giảm trong 24 giờ đầu sau sinh xuống các giá trị xấp xỉ 8 đến 9 mg/dL (2,0–2,75 mmol/L) và 4,4 đến 5,4 mg/dL (1,1–1,35 mmol/L), tương ứng; sau đó nồng độ canxi ổn định và sau đó tăng lên mức trung bình bình thường vào ngày thứ ba của cuộc đời. Nồng độ PTHrP và calcitonin ở trẻ sơ sinh giảm sau khi sinh, trong khi giá trị PTH và calcitriol tăng trong hai ngày đầu sau sinh. Sau đó, ở trẻ khỏe mạnh, nồng độ Ca2+ thay đổi theo tuổi sau sinh: 1 tháng—5,2 đến 6,1 mg/dL = 1,28 đến 1,52 mmol/L; 3 tháng—5,2 đến 6,0 mg/dL = 1,30 đến 1,49 mmol/L; 12 tháng—5,0 đến 5,6 mg/dL = 1,24 đến 1,39 mmol/L.
Các biểu hiện lâm sàng của hạ canxi máu xảy ra ở trẻ sơ sinh (được định nghĩa là giá trị canxi toàn phần < 7,5–8,0 mg/dL và/hoặc Ca2+ < 4,4 mg/dL [1,1 mmol/L] ở trẻ sơ sinh có cân nặng lúc sinh > 1500 g và < 7,0 mg/dL và/hoặc Ca2+ < 3,6 ng/dL [0,9 mmol/L] ở trẻ sơ sinh có cân nặng lúc sinh < 1500 g) chủ yếu là các biểu hiện của tăng kích thích thần kinh cơ: khó chịu, tăng thính lực, run rẩy, co giật mặt, tetany, co thắt thanh quản, và co giật khu trú hoặc toàn thể. Các triệu chứng không đặc hiệu, chẳng hạn như ngưng thở, nhịp tim nhanh, tím tái, nôn mửa, và các vấn đề về ăn uống cũng có thể xảy ra. Nguyên nhân của hạ canxi máu sơ sinh có thể được xem xét liên quan đến tuổi khởi phát (trước hoặc sau 72 giờ tuổi = sớm/muộn) (Bảng 20.2A, 20.2B).
Bảng 20.2A: Các nguyên nhân gây Hạ canxi máu
| I | Sơ sinh | ||||
|---|---|---|---|---|---|
| A | Rối loạn từ mẹ | 1 | Đái tháo đường | ||
| 2 | Tiền sản giật | ||||
| 3 | Thiếu vitamin D | ||||
| 4 | Nạp nhiều kiềm hoặc magiê sulfat | ||||
| 5 | Sử dụng thuốc chống co giật | ||||
| 6 | Cường cận giáp | ||||
| B | Rối loạn từ trẻ sơ sinh | 1 | Cân nặng lúc sinh thấp: sinh non, chậm tăng trưởng trong tử cung | ||
| 2 | Ngạt chu sinh, nhiễm trùng huyết, bệnh nặng | ||||
| 3 | Tăng bilirubin máu, chiếu đèn, thay máu | ||||
| 4 | Hạ magiê máu, tăng magiê máu | ||||
| 5 | Suy thận cấp/mạn | ||||
| 6 | Dinh dưỡng/thuốc—nạp nhiều phosphat, axit béo, phytate, truyền bicarbonate, máu có citrate, thuốc chống co giật, aminoglycosid | ||||
| 7 | Suy cận giáp | ||||
| 8 | Thiếu hoặc kháng vitamin D | ||||
| 9 | Bệnh xương hóa đá type II | ||||
| II | Suy cận giáp | ||||
| A | Bẩm sinh | 1 | Thoáng qua ở sơ sinh | ||
| 2 | Suy cận giáp bẩm sinh- xem thêm Bảng 20.2B | ||||
| a | |||||
| b | |||||
| c | |||||
| d | |||||
| e | |||||
| f | |||||
| 3 | Không nhạy cảm với hormon cận giáp | ||||
| a | |||||
| b | |||||
| c | |||||
| d | |||||
| B | Mắc phải | 1 | Hội chứng đa tuyến nội tiết tự miễn- type I (AIRE) | ||
| 2 | Kháng thể hoạt hóa thụ thể cảm nhận canxi | ||||
| 3 | Sau phẫu thuật, phá hủy do xạ trị | ||||
| 4 | Thâm nhiễm (lắng đọng sắt [bệnh huyết sắc tố, thalassemia] hoặc đồng [bệnh Wilson] quá mức; viêm u hạt, xâm lấn tân sinh; amyloidosis, sarcoidosis) | ||||
| 5 | Cường cận giáp từ mẹ | ||||
| 6 | Hạ magiê máu/tăng magiê máu | ||||
| III | Thiếu vitamin D (Xem Rối loạn Khoáng hóa) | ||||
| IV | Các nguyên nhân khác gây Hạ canxi máu | ||||
| A | Thiếu canxi | 1 | Thiếu hụt dinh dưỡng | ||
| 2 | Tăng canxi niệu | ||||
| B | Rối loạn nội môi magiê – xem Bảng 20.7A, 20.7B | 1 | Hạ magiê máu bẩm sinh | ||
| 2 | Mắc phải | ||||
| a | |||||
| b | |||||
| c | |||||
| C | Tăng phosphat máu | 1 | Suy thận | ||
| 2 | Dùng phosphat (tiêm tĩnh mạch, uống, trực tràng) | ||||
| 3 | Ly giải tế bào khối u | ||||
| 4 | Chấn thương cơ (dập nát, tiêu cơ vân) | ||||
| D | Các nguyên nhân khác | 1 | Giảm protein máu | ||
| 2 | Tăng thông khí | ||||
| 3 | Thuốc – furosemid, bisphosphonat, calcitonin, thuốc chống co giật, ketoconazol, thuốc chống ung thư (plicamycin, asparaginase, cisplatinum, cytosin arabinosid, doxorubicin), các sản phẩm máu chứa citrate | ||||
| 4 | Hội chứng xương đói | ||||
| 5 | Bệnh cấp tính và nặng – nhiễm trùng huyết, viêm tụy cấp, sốc nhiễm độc | ||||
| a |
Từ Carpenter T. Etiology of hypocalcemia in infants and children. UpToDate. 2018;1–17; Shaw N. A practical approach to hypocalcaemia in children. In: Allgrove J, Shaw N. (eds). Calcium and Bone Disorders in Children and Adolescents. Basel, Karger, Endocr Dev. 2018;16:73–92; Thakker R.V. Hypocalcemia: pathogenesis, differential diagnosis, and management. In: Favus, M.J. (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 6th ed. Washington, DC: American Society for Bone and Mineral Research; 2006:213–215.
Bảng 20.2B: Các biến thể di truyền liên quan đến Hạ canxi máu/Suy cận giáp
| Gen Protein | NST | OMIM | Rối loạn OMIM | Biểu hiện Lâm sàng/Hóa sinh | Chức năng Gen/Di truyền |
|---|---|---|---|---|---|
| Suy cận giáp di truyền trội trên NST thường | |||||
| CASR Thụ thể cảm nhận canxi | 3q13.3-q21.1 | 601199 | Hạ canxi máu di truyền trội trên NST thường type 1, (suy cận giáp đơn độc) 601198 | Tăng kích thích thần kinh cơ qua trung gian hạ canxi máu: dị cảm, tetany, co giật; cũng có thể dẫn đến hạ kali máu & tăng aldosteron thứ phát | Mã hóa thụ thể cảm nhận canxi biểu hiện trên màng plasma của tuyến cận giáp & ống thận; các biến thể tăng chức năng làm tăng độ nhạy và đáp ứng của CaSR với nồng độ canxi huyết thanh thấp, AD (có thể liên quan đến hội chứng Bartter type 5 do đồng suy giảm tái hấp thu natri clorua ở ống thận, AD 601198) |
| GNA11 Protein liên kết Guanin nucleotid, alpha-11 | 19p13.3 | 139313 | Hạ canxi máu di truyền trội trên NST thường type 2 (suy cận giáp đơn độc) 615361 | Tăng kích thích thần kinh cơ qua trung gian hạ canxi máu: dị cảm, tetany, co giật | Mã hóa tiểu đơn vị alpha của G-protein (Gα11) khởi phát truyền tín hiệu nội bào sau khi Ca2+ gắn vào CaSR, AD |
| Suy cận giáp đơn độc có tính gia đình | |||||
| PTH Hormon cận giáp | 11p15.3 | 168450 | Suy cận giáp đơn độc có tính gia đình 146200 | Tăng kích thích thần kinh cơ qua trung gian hạ canxi máu: dị cảm, tetany, co giật | Mã hóa hormon cận giáp, AD/AR |
| GCM2 Glial cells missing, drosophila, homolog of, 2 | 6p24.2 | 603716 | Suy cận giáp đơn độc có tính gia đình 146200 | Tăng kích thích thần kinh cơ qua trung gian hạ canxi máu: dị cảm, tetany, co giật | Mã hóa yếu tố phiên mã cần thiết cho sự biệt hóa của tuyến cận giáp, AD/AR |
| FHL1 Four-and-a-half Lim domains 1 | Xq26.3 | 300163 | Suy cận giáp đơn độc 146200 | Co giật do hạ canxi máu | Mã hóa gen cần thiết cho sự biệt hóa của tuyến cận giáp, liên kết X |
| Suy cận giáp phức hợp | |||||
| TBX1 T-box 1 | 22q11.21 | 602054 | Hội chứng DiGeorge (DGS) type 1 (hội chứng mất đoạn NST 22q11) 188400 | Suy cận giáp, thiểu sản tuyến ức, bất thường tim bẩm sinh, hở hàm ếch, suy giảm chức năng thận, đặc điểm khuôn mặt dị dạng | Yếu tố phiên mã T-box được biểu hiện trong phôi ở các mô bị ảnh hưởng xấu trong DGS; AD |
| NEBL Nebulette | 10p12.31 | 605491 | Hội chứng DiGeorge type (phức hợp) 2, 601362 | Bất sản tuyến ức, bất thường tim bẩm sinh, hở hàm ếch, suy giảm chức năng thận, đặc điểm khuôn mặt dị dạng | Protein biểu hiện trong cơ tim & cơ vân, liên quan đến actin, sợi cơ, & phức hợp bám dính tế bào, AD |
| CHD7 Protein liên kết DNA helicase chromodomain 7 | 8q12.2 | 608092 | Hội chứng CHARGE 214800 | CHARGE = Khuyết mống mắt, Dị tật tim, Tịt cửa mũi sau, Chậm phát triển, Bất thường Sinh dục, & Tai | Điều hòa biểu hiện gen mào thần kinh & hình thành RNA ribosome, AD |
| GATA3 Protein liên kết GATA 3 | 10p15 | 131320 | HDR (hội chứng Barakat) 146255 | Suy cận giáp, điếc thần kinh giác quan, loạn sản thận | Yếu tố phiên mã/yếu tố tăng cường cần thiết cho sự phát triển của tuyến cận giáp, hệ thống thính giác, thận & cho sự biểu hiện của các gen mã hóa các tiểu đơn vị của thụ thể tế bào T, AD |
| TBCE Chaperone đặc hiệu tubulin E | 1q42.3 | 604934 | HRD (hội chứng Sanjad-Sakati—SSS), 244460; Hội chứng Kenny-Caffey type 1 (KCS1) 241410 | HRD: Suy cận giáp, chậm phát triển, dị dạng; KCS1: các triệu chứng trên + xơ cứng xương & nhiễm trùng tái phát | Protein chaperone cần thiết cho sự cuộn gập đúng của các tiểu đơn vị tubulin & sự ổn định của khung xương tế bào, AR |
| FAM111A Family with sequence similarity 111, Member A | 11q12.1 | 615292 | Hội chứng Kenny-Caffey type 2 (KCS2), 127000; Loạn sản xương mảnh (GBD), 602381 | KCS2: Suy cận giáp, chậm phát triển, dị dạng; GBD: Hạ canxi máu, xương mỏng nhưng đặc & giòn, có thể gây tử vong chu sinh | Tác dụng chức năng ở người chưa rõ, (yếu tố giới hạn vật chủ trong virus), AD |
| Suy cận giáp do bệnh ty thể | |||||
| Mất đoạn NST ty thể | Hội chứng Kearns-Sayre (KSS) 530000 | Suy cận giáp, liệt vận nhãn, viêm võng mạc sắc tố, điếc thần kinh giác quan, thất điều tiểu não, dẫn truyền tim bất thường, bệnh cơ, chậm phát triển, rối loạn chức năng ống thận, suy vỏ thượng thận, suy sinh dục, đái tháo đường | Các gen ty thể mã hóa các protein vận chuyển điện tử tạo năng lượng, di truyền từ mẹ, AD | ||
| Mất đoạn NST ty thể | Bệnh não-cơ ty thể, nhiễm toan lactic, các cơn giống đột quỵ (MELAS) 540000 | Suy cận giáp, bệnh cơ, liệt vận nhãn, bệnh thần kinh, bệnh cơ tim, suy giảm nhận thức | Các gen ty thể mã hóa các protein vận chuyển điện tử tạo năng lượng, di truyền từ mẹ, AD | ||
| HADHB Hydroxylacyl-CoA dehydrogenase/3-Ketoacyl-CoA thiolase/Enoyl-CoA hydratase, tiểu đơn vị beta | 2p23.3 | 143450 | Hội chứng thiếu hụt protein ba chức năng ty thể, 609015 | Do không thể chuyển hóa nguồn năng lượng, biểu hiện lâm sàng có thể thay đổi từ cấp tính & gây tử vong trong giai đoạn chu sinh đến hội chứng giống Reye ở gan ở trẻ lớn hơn đến bệnh cơ xương ở thanh thiếu niên | Gen nhân mã hóa tiểu đơn vị beta của protein ba chức năng ty thể xúc tác quá trình oxy hóa beta của axit béo chuỗi dài, AR |
| Suy cận giáp tự miễn | |||||
| AIRE Bộ điều hòa tự miễn | 21q21.3 | 607358 | Hội chứng đa nội tiết tự miễn, type I 240300 | Suy cận giáp, suy vỏ thượng thận, nhiễm nấm candida niêm mạc da, rụng tóc, thiếu máu ác tính, suy sinh dục | Biểu hiện ở tuyến ức, cần thiết cho việc nhận dạng kháng nguyên bản thân, AD/trội âm tính/AR |
| Suy cận giáp giả | |||||
| GNAS Phức hợp locus GNAS | 20q13.32 | 139320 | Suy cận giáp giả type 1A, 103580 | Hạ canxi máu, tăng phosphat máu, đáp ứng cAMP ống thận dưới mức bình thường với PTH ngoại sinh, kiểu hình Loạn dưỡng xương di truyền Albright (AHO) | Các đột biến mất chức năng hoặc mất đoạn của gen GNAS mẹ hoặc biểu hiện hai alen GNAS từ cha (isochromosome) dẫn đến đề kháng PTH ở ống lượn gần và xương, AD |
| GNAS Phức hợp locus GNAS | 20q13.32 | 139320 | Suy cận giáp giả type 1B, 603233 | Hạ canxi máu, tăng phosphat máu, đáp ứng cAMP ống thận dưới mức bình thường với PTH ngoại sinh, hoạt động cAMP hồng cầu bình thường, kiểu hình bình thường | Khiếm khuyết methyl hóa biểu sinh di truyền từ mẹ của gen GNAS mẹ dẫn đến sự im lặng của nó và đề kháng PTH ở ống lượn gần, AD |
| GNASAS1 Phức hợp locus GNAS, bản sao đối nghĩa 1 | 20q13.32 | 610540 | Suy cận giáp giả type 1B 603233 | Hạ canxi máu, tăng phosphat máu, không có đáp ứng cAMP ống thận với PTH ngoại sinh, kiểu hình bình thường | Mất đoạn di truyền từ mẹ dẫn đến biểu hiện của GNASAS1 từ cha gây ra sự im lặng của GNAS mẹ do khiếm khuyết methyl hóa biểu sinh & đề kháng PTH ở ống lượn gần, AD |
| STX16 Syntaxin 16 | 20q13.32 | 603666 | Suy cận giáp giả type 1B 603233 | Hạ canxi máu, tăng phosphat máu, không có đáp ứng cAMP vòng ở thận với PTH ngoại sinh, kiểu hình bình thường | Khiếm khuyết methyl hóa biểu sinh di truyền từ mẹ của gen GNAS mẹ dẫn đến sự im lặng của nó và đề kháng PTH ở ống lượn gần, AD |
| GNAS Phức hợp locus GNAS | 20q13.32 | 139320 | Suy cận giáp giả type 1C | Hạ canxi máu, tăng phosphat máu, không có đáp ứng cAMP vòng ở thận với PTH ngoại sinh, kiểu hình AHO, hoạt động cAMP hồng cầu bình thường | Các đột biến mất chức năng hoặc mất đoạn ở exon 13 của GNAS mẹ dẫn đến đề kháng PTH ở ống lượn gần và xương, AD |
| ? | Suy cận giáp giả type 2, 203330 | Hạ canxi máu, tăng phosphat máu, đề kháng một phần với PTH, kiểu hình bình thường | Mặc dù tăng cAMP niệu bình thường sau khi dùng PTH, bệnh nhân vẫn đề kháng với tác dụng thải phosphat của PTH | ||
| GNAS Phức hợp locus GNAS | 20q13.32 | 139320 | Suy cận giáp giả giả, 612463 | Kiểu hình AHO nhưng canxi máu bình thường | Đề kháng PTH ở xương do các biến thể mất chức năng của GNAS cha |
| PRKAR1A Protein kinase, phụ thuộc cAMP, điều hòa, type 1 alpha | 17q24.2 | 188830 | Loạn sản đầu xương 1 101800 | Loạn sản xương: lùn, tật ngón ngắn, dị dạng mặt, thiểu sản mũi; có/không có đề kháng hormon | Mã hóa một tiểu đơn vị điều hòa của protein kinase A phụ thuộc cAMP cần thiết cho truyền tín hiệu nội bào |
| PDE4D Phosphodiesterase 4D, đặc hiệu cAMP | 5q11.2-q21.1 | 600129 | Loạn sản đầu xương 2 614613 | Vide supra, chậm phát triển | Mã hóa enzyme phân hủy cAMP, do đó ức chế truyền tín hiệu nội bào |
AD, Di truyền trội trên nhiễm sắc thể thường; AR, di truyền lặn trên nhiễm sắc thể thường; cAMP, cyclic adenosine monophosphate; PTH, hormon cận giáp; RNA, ribonucleic acid.
Từ Mannstadt M, Bilezikian JP, Thakker RV, et al. Hypoparathyroidism. Nat Rev Dis Primers. 2017:3.17055; Thiele S, Mantovani G, Barlier A, et al. From pseudohypoparathyroidism to inactivating PTH/PTHrP signalling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol. 2016;175:P1–P17; Vahe C, Benomar K, Espiard S, et al. Diseases associated with the calcium-sensing receptor. Orphanet J Rare Dis. 2017;12:19.
Hạ canxi máu sơ sinh sớm
Trong trường hợp không có giảm protein máu, hạ canxi máu xảy ra trong vòng 72 giờ đầu sau sinh được coi là “hạ canxi máu sơ sinh sớm”. Tình trạng này xảy ra phổ biến nhất ở trẻ sinh non hoặc nhỏ so với tuổi thai, trẻ nhẹ cân (LBW), hoặc trẻ bị ngạt, hoặc ở những trẻ được sinh ra từ các bà mẹ bị đái tháo đường thai kỳ hoặc đái tháo đường vĩnh viễn (hiếm khi là cường cận giáp không được nghi ngờ ở mẹ) và là hậu quả của việc tiết PTH dưới mức bình thường để đáp ứng với giá trị canxi huyết thanh giảm và đáp ứng thải phốt phát ở ống thận với PTH bị trì hoãn đặc trưng của trẻ sơ sinh, sự tiết calcitonin kéo dài bất thường, và/hoặc hạ magiê máu. Nồng độ canxi toàn phần và Ca2+ giảm nhanh hơn từ các giá trị cao trong tử cung xuống mức thấp nhất ở trẻ sinh non so với trẻ đủ tháng. Ở trẻ sơ sinh LBW, hạ canxi máu có thể được cho là do sự tích lũy nhanh chóng của canxi xương khi có sự đề kháng tương đối với tác dụng hấp thu và tái hấp thu canxi của calcitriol trên đường ruột và xương, tương ứng. Con của những bà mẹ thiếu vitamin D nghiêm trọng có thể bị hạ canxi máu ngay sau khi sinh. Hạ canxi máu phát triển ở khoảng một phần ba trẻ sơ sinh bị ngạt là sản phẩm của các cuộc sinh phức tạp và nguy cơ, bao gồm những trường hợp liên quan đến đái tháo đường ở mẹ, tiền sản giật, thiếu vitamin D, sử dụng thuốc chống co giật, và cường cận giáp (không được nghi ngờ) ở mẹ. Ở những trẻ này, tăng tải lượng phốt phát do tổn thương tế bào, giảm lượng canxi nạp vào, và tăng calcitonin máu là những yếu tố gây bệnh quan trọng trong sự phát triển của hạ canxi máu. Trẻ sinh non và những trẻ bị chậm tăng trưởng trong tử cung, ngạt, nhiễm trùng, hội chứng suy hô hấp, hoặc các bệnh nặng khác thường bị hạ canxi máu.
Khoảng 50% trẻ sơ sinh của các bà mẹ bị đái tháo đường phát triển hạ canxi máu sơ sinh sớm; tỷ lệ này có thể được giảm bớt bằng cách kiểm soát đường huyết nghiêm ngặt của mẹ. Nguyên nhân của nó là đa yếu tố và bao gồm giảm vận chuyển canxi qua nhau thai do mẹ bài tiết đáng kể canxi và magiê qua nước tiểu, giảm tiết PTH ở trẻ sơ sinh, tăng calcitonin máu, hạ magiê máu (xảy ra ở 40% con của phụ nữ đái tháo đường), và hạn chế nạp vào và suy giảm hấp thu canxi ăn vào. Tăng canxi máu ở mẹ do cường cận giáp không được nghi ngờ dẫn đến tăng vận chuyển canxi đến thai nhi và tăng thêm nồng độ canxi huyết thanh trong tử cung, điều này ức chế tổng hợp và giải phóng PTH của thai nhi và kích thích tiết calcitonin—những sai lệch trong cơ chế cân bằng nội môi này tồn tại sau sinh và có thể dẫn đến co giật/tetany do hạ canxi máu ở con của họ. Sự ức chế tiết PTH có thể kéo dài trong vài tháng và không được phát hiện cho đến khi hạ canxi máu có triệu chứng phát triển sau khi trẻ được cai sữa mẹ chuyển sang sữa công thức chứa nhiều phốt phát hơn. Việc mẹ uống một lượng lớn canxi cacbonat trong thuốc kháng axit cũng đã dẫn đến hạ canxi máu ở trẻ sơ sinh.
Hạ canxi máu có thể xảy ra ở trẻ sơ sinh bị tăng bilirubin máu đang được thay máu và ở những trẻ được chiếu đèn. Trẻ sơ sinh bị nhiễm rotavirus cấp và tiêu chảy nặng có thể biểu hiện co giật do hạ canxi máu. Thuốc kháng sinh aminoglycosid (ví dụ, gentamycin) làm tăng bài tiết canxi và magiê qua nước tiểu, do đó tạo điều kiện cho sự phát triển của hạ canxi máu sơ sinh. Các hợp chất tạo phức và cô lập canxi, chẳng hạn như citrate (có trong máu được truyền), phốt phát (làm thay đổi tích số canxi x phốt phát), và axit béo (được cung cấp dưới dạng bổ sung calo) làm giảm nồng độ Ca2+. Bicarbonate được dùng để điều chỉnh tình trạng nhiễm toan làm tăng liên kết canxi với albumin và do đó làm giảm giá trị Ca2+. Hạ magiê máu làm suy giảm sự giải phóng PTH từ các tuyến cận giáp. Hạ canxi máu cũng có thể xảy ra ở trẻ tăng thông khí bị nhiễm kiềm hô hấp nặng, cũng như ở những trẻ có các nguyên nhân khác gây nhiễm kiềm chuyển hóa. Phytate trong sữa đậu nành liên kết với canxi và phốt phát và cản trở sự hấp thu của chúng. Trẻ sơ sinh và trẻ nhỏ bị bệnh xương hóa đá ác tính type II và suy giảm tạo cốt bào có thể biểu hiện hạ canxi máu sơ sinh sớm hoặc muộn.
Hạ canxi máu sơ sinh muộn
Hạ canxi máu thoáng qua muộn (phát triển lần đầu khi trẻ sơ sinh > 72 giờ tuổi sau sinh) có thể do tăng lượng phốt phát nạp vào, hạ magiê máu, suy cận giáp, hoặc thiếu vitamin D (xem Bảng 20.2A, 20.2B). Hạ canxi máu sơ sinh có thể phát triển sau 3 ngày tuổi ở những đứa con được sinh ra vào cuối mùa đông-đầu mùa xuân của những bà mẹ đa sản không nạp đủ vitamin D hoặc không tiếp xúc với ánh nắng mặt trời. Hàm lượng phốt phát cao trong sữa đặc hoặc sữa công thức bò đã qua điều chỉnh có thể dẫn đến sự hình thành các muối canxi khó tan, hạn chế sự hấp thu canxi trong ruột trong khi làm tăng giá trị phốt phát huyết thanh. Việc cho trẻ ăn dặm quá sớm với các loại ngũ cốc chứa chất xơ cũng làm giảm sự hấp thu canxi. Trẻ bị ảnh hưởng có thể có khiếm khuyết liên quan đến bài tiết phốt phát ở thận hoặc thiếu vitamin D đồng thời. Tăng phốt phát máu và hạ canxi máu ban đầu có thể gợi ý suy cận giáp, nhưng nồng độ PTH huyết thanh thường bình thường hoặc tăng nhẹ ở trẻ bị quá tải phốt phát để đáp ứng với sự giảm tương hỗ của canxi huyết thanh; các giá trị PTH tăng rõ rệt hoặc tăng dai dẳng đặt ra câu hỏi liệu có thể có suy cận giáp giả (PHP, hiếm khi là loạn sản đầu xương—một loại loạn sản xương với đề kháng hormon liên quan đến các biến thể bất hoạt của PRKAR1A hoặc PDE4D, vide infra) hay không. Trẻ sơ sinh và trẻ nhỏ bị suy thận mạn do thiểu sản thận hoặc bệnh thận tắc nghẽn thường bị hạ canxi máu và tăng phốt phát máu với nồng độ PTH huyết thanh tăng cao, nhưng chúng cũng bị tăng nitơ máu. Hạ magiê máu dẫn đến suy giảm tiết PTH và giảm đáp ứng ngoại vi với PTH và có thể là thoáng qua hoặc liên quan đến các bất thường bẩm sinh về hấp thu ở ruột hoặc tái hấp thu ở ống thận của magiê. Tăng magiê máu đôi khi có thể liên quan đến hạ canxi máu sơ sinh.
Hạ canxi máu và hạ phốt phát máu do thiếu vitamin D ở thai nhi/sơ sinh xảy ra ở con của các bà mẹ thiếu hụt đáng kể vitamin D (vì lý do văn hóa hoặc kinh tế xã hội), suy giảm quá trình 25-hydroxyl hóa cholecalciferol ở gan hoặc hoạt động của 25-hydroxyvitamin D-1α hydroxylase ở thận hoặc các đột biến mất chức năng của thụ thể vitamin D (VDR). Thiếu vitamin D có thể phát triển ở một trẻ lớn hơn được bú mẹ của một bà mẹ ăn chay, người che chắn mình khỏi ánh nắng mặt trời và ăn một chế độ ăn ít vitamin D. Thiếu hụt vitamin D cận lâm sàng ở trẻ sơ sinh và trẻ nhỏ phổ biến hơn nhiều so với những gì đã được công nhận trước đây. Hạ canxi máu sơ sinh “rất muộn” xảy ra ở trẻ sinh non có khối lượng xương thấp ở 3 đến 4 tháng tuổi mà việc nạp canxi, phốt phát và vitamin D đã ở mức cận biên; có lẽ là do sự lắng đọng mạnh mẽ của canxi có sẵn vào xương. Hạ canxi máu do thiếu vitamin D có thể phát triển khá đột ngột và không có dấu hiệu lâm sàng hoặc X-quang của bệnh còi xương ở trẻ lớn hơn và trẻ nhỏ ăn chế độ ăn kiêng ít vitamin D do dị ứng nặng và/hoặc được giữ trong nhà với sự tiếp xúc hạn chế với ánh nắng mặt trời.
Hạ canxi máu biểu hiện lần đầu sau 72 giờ tuổi thường báo hiệu sự hiện diện của sự tổn thương đáng kể các cơ chế cân bằng nội môi canxi, chẳng hạn như những cơ chế liên quan đến suy cận giáp do dị tật của các tuyến cận giáp (ví dụ, hội chứng DiGeorge hoặc biến thể của một gen quan trọng đối với sự phát triển phôi thai của các cấu trúc này) hoặc lỗi chức năng trong việc tiết PTH (ví dụ, một bất thường trong hoạt động của thụ thể cảm nhận canxi [CaSR]).
Suy cận giáp
Suy cận giáp biểu hiện ở trẻ nhỏ thường là thoáng qua và liên quan đến sự trưởng thành phát triển chậm của chức năng tuyến cận giáp; nó có thể tự khỏi trong vài tuần đầu đời (xem Bảng 20.2A, 20.2B và Hình 20.1). Khi kéo dài, suy cận giáp có thể do một lỗi trong quá trình phát triển phôi thai của các tuyến cận giáp hoặc trong quá trình tổng hợp hoặc bài tiết PTH hoặc do không đáp ứng ngoại vi với PTH (suy cận giáp chức năng, tức là PHP, xảy ra ở những bệnh nhân kháng PTH [vide infra]). Suy cận giáp bẩm sinh đơn độc có tính gia đình có thể được di truyền theo kiểu trội trên nhiễm sắc thể thường, lặn trên nhiễm sắc thể thường, hoặc lặn liên kết X do các đột biến mất chức năng trong các gen cần thiết cho sự biệt hóa của các tuyến cận giáp dẫn đến bất sản hoặc thiểu sản bẩm sinh của các cấu trúc này xảy ra như một rối loạn đơn lẻ. Do đó, suy cận giáp đơn độc có tính gia đình (OMIM 146200) đã được liên kết với các đột biến bất hoạt trong PTH, glial cells missing (GCM2), và four-and-a-half Lim domains 1 (FHL1) và hạ canxi máu di truyền trội trên nhiễm sắc thể thường với sự ức chế bài tiết PTH do các biến thể tăng chức năng của CASR và guanine nucleotide binding protein alpha 11 (GNA11). Các đột biến bất hoạt trong các exon mã hóa peptide tín hiệu của PTH cản trở quá trình xử lý preproPTH thành phân tử PTH chức năng 84 aa có hoạt tính sinh học dẫn đến suy cận giáp có thể được di truyền như một đặc điểm trội hoặc lặn trên nhiễm sắc thể thường. Tùy thuộc vào độ đặc hiệu của xét nghiệm miễn dịch cho PTH, nồng độ PTH huyết thanh có thể thấp, bình thường, hoặc thậm chí cao ở những bệnh nhân này. GCM2 là một gen có năm exon mã hóa một yếu tố phiên mã liên kết DNA (deoxyribonucleic acid) 506 aa có biểu hiện bị giới hạn ở các tuyến cận giáp. Mất đoạn trong gen hoặc các đột biến sai nghĩa bất hoạt đồng hợp tử trong các exon 2, 3 và 5 của GCM2 dẫn đến suy cận giáp ở người. Các đột biến trong các exon 2 và 3 của GCM2 (mã hóa miền liên kết DNA và miền hoạt hóa phiên mã 1) dẫn đến suy giảm tổng hợp và ổn định protein và di truyền lặn trên nhiễm sắc thể thường của suy cận giáp bẩm sinh trong khi những đột biến trong exon 5 (mã hóa miền hoạt hóa phiên mã 2) dẫn đến các đột biến có tác dụng trội âm và di truyền trội trên nhiễm sắc thể thường của rối loạn này. (Biểu hiện của GCM2 diễn ra ngay sau khi các tế bào cận giáp được xác định và phụ thuộc vào chức năng phiên mã bình thường của GATA3, gen bị đột biến ở những bệnh nhân mắc hội chứng Barakat của suy cận giáp-điếc-loạn sản thận [HDR] [vide infra]. Các đột biến hoạt hóa của GCM2 có liên quan đến cường cận giáp [vide infra].) Suy cận giáp liên kết X có liên quan đến bất sản của các tuyến cận giáp; rối loạn này là do các biến thể mất chức năng của FHL1 (mã hóa Four-and-a-half Lim domains 1, OMIM 300163), một gen có sản phẩm cần thiết cho sự biệt hóa của các tuyến cận giáp. FHL1 là một protein 280 axit amin (aa) có một miền LIM (motif ngón tay kẽm kép) cũng được biểu hiện ở tinh hoàn, cơ tim và cơ xương.
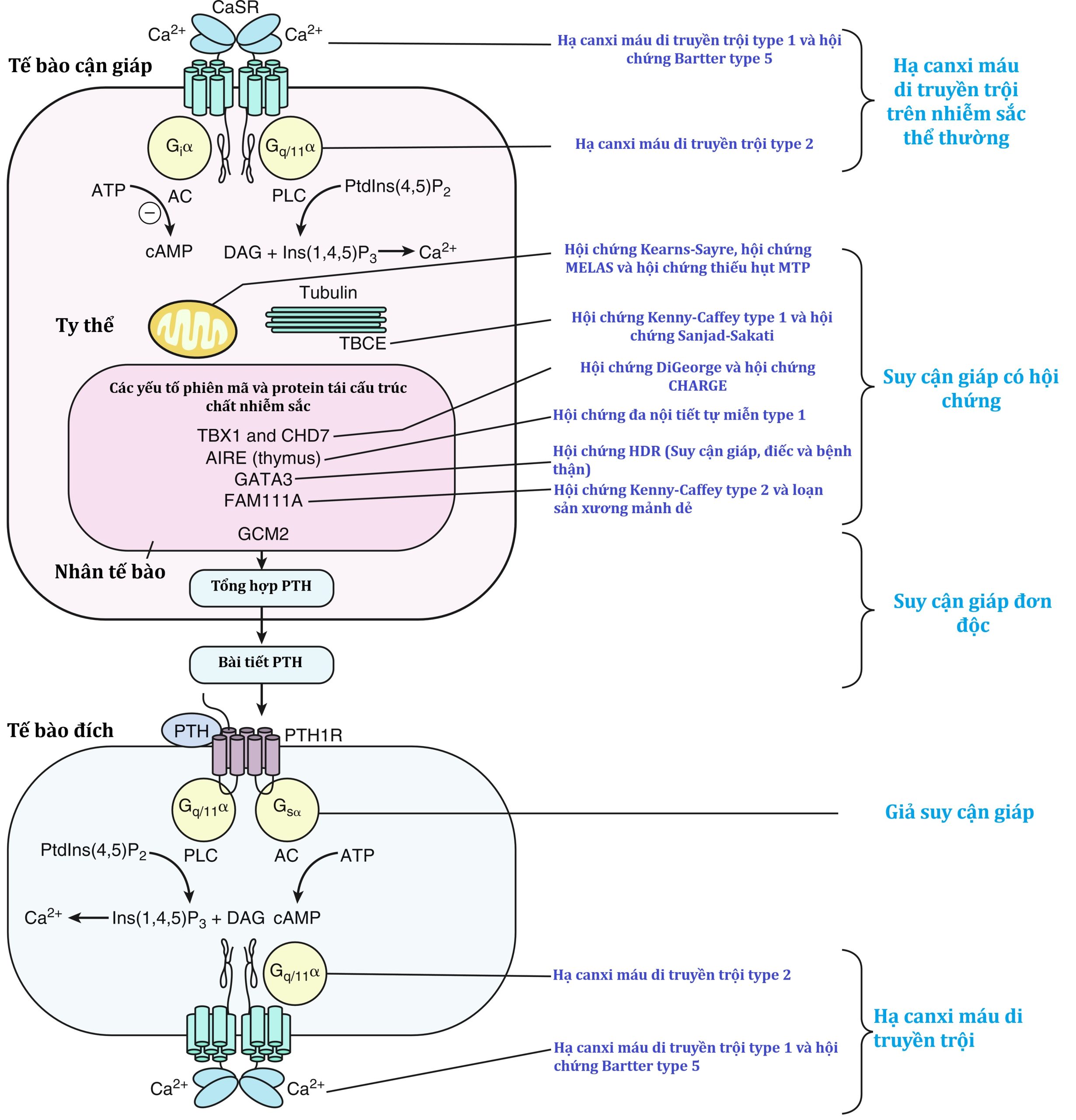
Hình 20.1: Sự điều hòa bài tiết hormon cận giáp (PTH) và các rối loạn di truyền dẫn đến suy cận giáp. Trong tuyến cận giáp, thụ thể cảm nhận canxi (CaSR) theo dõi nồng độ Ca2+ lưu hành và trung gian cho sự bài tiết PTH, hormon này tác động thông qua thụ thể PTH trên màng plasma của tế bào xương hoặc thận đích. Các lỗi di truyền trong con đường này dẫn đến suy cận giáp. AC, Adenylyl cyclase; AIRE, bộ điều hòa tự miễn; ATP, adenosine triphosphate; Ca2+, canxi ion hóa, CaSR, thụ thể cảm nhận canxi, cAMP, cyclic adenosine monophosphate; CHD, chromodomain; DAG, diacylglycerol; FAM111A, family with sequence similarity 111A; GATA3, GATA binding protein 3; GCM2, glial cell missing transcription factor 2; Ins(1,4,5)P3, inositol trisphosphate; PLC, phospholipase C; PtdIns, phosphatidylinositol; PTH, hormon cận giáp; PTH1R, thụ thể PTHrP; TBCE, tubulin-specific chaperone E; TBX1, T-box transcription factor 1.
(Từ Mannstadt M, Bilezikian JP, Thakker RV, et al. Hypoparathyroidism. Nat Rev Dis Primers. 2017;3.17055, với sự cho phép.)
Hạ canxi máu liên quan đến suy giảm bài tiết PTH do các đột biến hoạt hóa đơn alen trong CASR đã được xác định ở trẻ sơ sinh, trẻ nhỏ, trẻ em và người lớn. Hạ canxi máu tăng canxi niệu là một dạng suy cận giáp di truyền trội trên nhiễm sắc thể thường do các đột biến tăng chức năng trong CASR (hạ canxi máu di truyền trội trên nhiễm sắc thể thường type 1) hoặc GNA11 (hạ canxi máu di truyền trội trên nhiễm sắc thể thường type 2) dẫn đến tăng “độ nhạy” của CaSR đối với các tác dụng ức chế PTH của Ca2+ trong các tế bào chính của tuyến cận giáp và trong các tế bào thận ở nhánh lên dày của quai Henle (TALH). CaSR theo dõi nồng độ Ca2+ huyết tương, trong khi GNA11 là tiểu đơn vị alpha—protein liên kết guanine nucleotide của thụ thể liên kết protein guanosine ba tiểu đơn vị (GPCR) truyền tín hiệu của CaSR đến các con đường truyền tín hiệu nội bào. Một “ngưỡng” thấp hơn của CaSR hoặc sự tăng cường của chính hệ thống truyền tín hiệu sau thụ thể cho phép bài tiết PTH bị ức chế và tái hấp thu canxi ở ống thận bị giảm (do đó làm tăng bài tiết canxi qua nước tiểu) bởi nồng độ Ca2+ cực thấp dẫn đến tăng canxi niệu rõ rệt. Các đột biến hoạt hóa (ví dụ, p.Lys47Asn, p.Leu616Val, p.Phe788Leu) có thể nằm rải rác trong CASR nhưng xảy ra chủ yếu ở vòng peptide thứ hai của miền ngoại bào của nó. Một số biến thể đơn alen hoạt hóa (ví dụ, p.Cys141Trp, p.Leu125Pro, p.Ala843Glu) của CASR cũng có thể ức chế chức năng của kênh kali tủy thận ngoài (được mã hóa bởi KCNJ1, OMIM 600359), dẫn đến một hội chứng giống Bartter với nhiễm kiềm chuyển hóa hạ kali máu, tăng renin máu, tăng aldosteron máu, và hạ magiê máu, cũng như hạ canxi máu tăng canxi niệu (được gọi là hội chứng Bartter type 5, OMIM 601198). Những khiếm khuyết chuyển hóa kép này đáp ứng một phần với điều trị bằng hydrochlorothiazide và liều thấp calcitriol. Tuy nhiên, trẻ em bị hạ canxi máu tăng canxi niệu do các đột biến tăng chức năng trong CASR rất nhạy cảm với ngay cả liều thấp calcitriol, điều này có thể dẫn đến tăng canxi niệu rõ rệt hơn và nhiễm canxi thận. Do đó, việc quản lý những bệnh nhân này đã gặp nhiều khó khăn. Việc sử dụng PTH người tái tổ hợp 1-34 cho một bé trai 14 tháng tuổi bị hạ canxi máu với một đột biến vô nghĩa de novo trong CASR (p.Leu727Gln) trong 17 tháng đã phục hồi một phần cân bằng nội môi canxi với nồng độ canxi huyết thanh tăng nhưng vẫn dưới mức bình thường, trong khi bài tiết canxi qua nước tiểu giảm xuống trong giới hạn bình thường. Trong quá trình điều trị, trẻ không có triệu chứng lâm sàng, không phát triển nhiễm canxi thận, và dung nạp thuốc tốt. Tuy nhiên, việc sử dụng PTH cho một bệnh nhân như vậy có thể làm tăng bài tiết canxi qua nước tiểu và nguy cơ nhiễm canxi thận. Việc sử dụng các chất calcilytic (calcimimetics type II), chẳng hạn như cinacalcet đối kháng với tác dụng của canxi lên CaSR trong các tuyến cận giáp và ống thận có thể tỏ ra hữu ích về mặt điều trị ở những bệnh nhân này. Trẻ sơ sinh có các biến thể của GNA11 có thể không có mức độ tăng canxi niệu tương tự như những trẻ có CASR đột biến.
Các dạng suy cận giáp thường gặp nhất là những dạng liên quan đến các tập hợp bất thường bẩm sinh (xem Bảng 20.2A, 20.2B và xem Hình 20.1). Dạng suy cận giáp phức hợp thường gặp nhất là dạng liên quan đến hội chứng DiGeorge type 1 (OMIM 188400), một rối loạn xảy ra với tần suất 1:4000 ca sinh và hiện diện ở khoảng 70% trẻ em bị suy cận giáp đơn độc. Ở nhiều trẻ sơ sinh mắc hội chứng DiGeorge, suy cận giáp thuyên giảm một phần theo thời gian chỉ để tái xuất hiện trong các giai đoạn căng thẳng, chẳng hạn như nhiễm trùng hoặc chấn thương. Hội chứng DiGeorge là một bệnh lý mào thần kinh—kết quả của sự di chuyển bị xáo trộn của các tế bào mào thần kinh cổ và hậu quả là sự phát triển sai lệch của các mô có nguồn gốc mào thần kinh bắt nguồn từ các túi họng thứ ba và thứ tư và các cung mang thứ nhất đến thứ năm. Hội chứng DiGeorge type 1 có liên quan đến vi mất đoạn vùng nhiễm sắc thể 22q11.2 (del22q11.2—vùng nguy kịch DiGeorge), một đoạn trên đó có hơn 35 gen và do đó là một hội chứng gen liền kề (một rối loạn gây ra bởi sự mất đoạn của một số gen liền kề mà khi bị đột biến riêng lẻ có thể dẫn đến một đặc điểm lâm sàng riêng biệt nhưng khi bị mất chung sẽ dẫn đến một nhóm các phát hiện lâm sàng dường như không liên quan). Vi mất đoạn nhiễm sắc thể 22q11.2 nằm trong các vùng lặp lại số lượng bản sao thấp, và chính đặc điểm này dẫn đến sự trao đổi đoạn không đồng đều giữa các nhiễm sắc thể thứ 22 được ghép cặp trong quá trình giảm phân. Các đối tượng mắc hội chứng DiGeorge thường có bộ ba: (1) hạ canxi máu do thiểu sản các tuyến cận giáp thường biểu hiện trong giai đoạn sơ sinh nhưng có thể không được phát hiện cho đến tuổi lớn hơn, (2) chức năng tế bào lympho T bị khiếm khuyết và suy giảm miễn dịch qua trung gian tế bào do thiếu một phần hoặc hoàn toàn sự biệt hóa của tuyến ức dẫn đến tăng tần suất nhiễm virus và nấm và xu hướng mắc các rối loạn tự miễn, và (3) các khiếm khuyết nón thân của tim hoặc cung động mạch chủ (Tứ chứng Fallot, thông liên thất, gián đoạn hoặc cung động mạch chủ bên phải, thân chung động mạch, vòng mạch). Ở một mức độ đáng kể, hội chứng DiGeorge type 1 có liên quan đến sự mất hoặc các biến thể có hại của T-box 1 (TBX1, OMIM 602054) trong đoạn nhiễm sắc thể 22q11.2. Sự phá vỡ thực nghiệm của Tbx1 làm suy giảm sự phát triển của hệ mạch máu cung họng, trong khi việc đưa vào các đột biến null trong Tbx1 dẫn đến các bất thường của đường ra tim và thiểu sản của tuyến ức và tuyến cận giáp. Yếu tố phiên mã được mã hóa bởi TBX1 là một phần của một mạng lưới các sản phẩm gen (bao gồm cả những sản phẩm được mã hóa bởi ISL1, SHH, FOXA2, FOXC2) kiểm soát sự phát triển của các tuyến cận giáp và tuyến ức bằng cách điều chỉnh biểu hiện của GATA3, GCM2, và PAX9. Các đột biến của TBX1 đặc biệt giải thích cho năm biểu hiện chính của hội chứng DiGeorge: thiểu sản cận giáp và hạ canxi máu, bất sản tuyến ức, bất thường tim, các đặc điểm khuôn mặt bất thường (tai đóng thấp, cằm nhỏ, mắt xếch, khe mi ngắn và nhân trung, miệng nhỏ), và hở hàm ếch với hở vòm hầu-họng. Thiểu năng một nửa của TBX1 cũng có thể gây ra suy cận giáp đơn độc. Cũng trong vi mất đoạn hai megabase này tại nhiễm sắc thể 22q11.2 là HIRA (điều hòa chu kỳ tế bào histone, OMIM 600237), một yếu tố điều hòa phiên mã được biểu hiện trong tim đang phát triển và các yếu tố mào thần kinh phần trên cơ thể và cần thiết cho sự phát triển bình thường của tim. Một gen quan trọng khác nằm ở nhiễm sắc thể 22q11.2 là UFD1L (ubiquitin fusion degradation 1-like, OMIM 601754), sản phẩm của nó quan trọng cho quá trình xử lý sau dịch mã của protein và/hoặc sự phân hủy của chúng bằng cách tương tác với protein dung hợp ubiquitin. Về mặt thực nghiệm, hội chứng DiGeorge đã được liên kết với các gen mã hóa endothelin-1, yếu tố tăng trưởng nội mô mạch máu, và yếu tố tăng trưởng nguyên bào sợi-8 (Fgf8, một gen mục tiêu của TBX1). Ở chuột bị giảm hình thái Fgf8, có các khiếm khuyết tim mạch, sọ mặt, cận giáp và tuyến ức—một bản sao kiểu hình thực nghiệm của hội chứng del22q11.2 ở người. (Đái tháo đường ở mẹ, nghiện rượu, hoặc uống axit retinoic đôi khi có thể liên quan đến hội chứng DiGeorge ở con.)
Trước khi sinh, sự hiện diện của hội chứng DiGeorge có thể được xem xét khi siêu âm thai nhi cho thấy cung động mạch chủ bị gián đoạn hoặc thân chung động mạch và có thể được xác nhận bằng các nghiên cứu thích hợp (microarray, lai tại chỗ phát quang [FISH]) trên các mẫu gai rau hoặc nước ối. Các đặc điểm lâm sàng khác của hội chứng DiGeorge bao gồm: chậm tăng trưởng, loạn sản thận, dị tật đường tiêu hóa (teo thực quản, không có hậu môn), mất ổn định cột sống cổ, suy giảm thị lực và dị tật mắt, dị tật vỏ não (polymicrogyria quanh rãnh Sylvius), và chậm phát triển. Hội chứng DiGeorge type 1 (OMIM 188400) có thể xảy ra lẻ tẻ hoặc được di truyền như một đặc điểm trội trên nhiễm sắc thể thường. Phức hợp hội chứng DiGeorge type 2/hội chứng tim-mặt-vòm miệng 2 (OMIM 601362) đã được liên kết với mất đoạn kẽ của nhiễm sắc thể 10p13 nhưng được cho là, một phần, do mất NEBL (Nebulette, OMIM 605491), nằm ở nhiễm sắc thể 10p12.31. NEBL mã hóa một protein được biểu hiện trong cơ tim và cơ vân liên kết với actin, sợi cơ, và các phức hợp bám dính tế bào; hội chứng DiGeorge type 2 được di truyền như một đặc điểm trội trên nhiễm sắc thể thường. Hội chứng DiGeorge cũng đã được liên kết với vi mất đoạn của các nhiễm sắc thể 18q21.33 và 4q21.2-q25—cho thấy chuỗi các gen có khả năng liên quan đến việc tạo ra kiểu hình này. Ngoài hội chứng DiGeorge type 1, mất đoạn nhiễm sắc thể 22q11.2 đã được liên kết với hội chứng tim-mặt-vòm miệng và các hội chứng khác. Nói chung, các hội chứng này có các đặc điểm khuôn mặt tương tự (hai mắt xa nhau, dịch chuyển ra ngoài của góc mắt trong, khe mi ngắn, mí mắt sưng, mũi dị dạng “phân đoạn”, miệng nhỏ, tai đóng thấp với loa tai gấp bất thường, nhân trung ngắn, cằm nhỏ, thiểu sản xương gò má, hở vòm hầu-họng có/không có hở hàm ếch), rối loạn chức năng khứu giác, tầm vóc thấp, khuyết tật học tập không lời, và các bệnh tâm lý khác nhau. Hội chứng tim-mặt-vòm miệng Takao (bao gồm trong OMIM 188440) chủ yếu bao gồm các khuyết tật tim điển hình được mô tả trước đó cũng có thể liên quan đến hạ canxi máu; hội chứng tim-mặt-vòm miệng Shprintzen (OMIM 192430) được đặc trưng bởi các khuyết tật sọ mặt và vòm miệng và các bất thường tim; hội chứng tim-mặt Cayler (OMIM 125520) có liên quan đến liệt mặt một bên không hoàn toàn do thiểu sản cơ hạ góc miệng và các bất thường của tim và động mạch chủ. Các hội chứng này đã được nhóm lại thành các hội chứng CATCH-22 gồm các khuyết tật tim, khuôn mặt bất thường, thiểu sản tuyến ức, hở hàm ếch, hạ canxi máu.
Hạ canxi máu đã được quan sát thấy ở một số đối tượng có vi nhân đôi nhiễm sắc thể 22q11.2, một biến thể số lượng bản sao; các đặc điểm lâm sàng của các cá nhân có bất thường di truyền này thay đổi từ những người hoàn toàn bình thường đến những bệnh nhân có nhiều dị tật bẩm sinh, chậm phát triển nặng, tự kỷ và tâm thần phân liệt. Hội chứng nhân đôi nhiễm sắc thể 22q11.2 dường như được di truyền như một đặc điểm trội trên nhiễm sắc thể thường có biểu hiện được sửa đổi bởi các yếu tố khác. Sinh lý bệnh của hạ canxi máu ở các đối tượng bị ảnh hưởng là không chắc chắn. Trong một gia đình mà trong đó bệnh nhân chỉ điểm mắc hội chứng DiGeorge liên quan đến del22q11.2, người cha bình thường có cùng bất thường trên một trong các nhiễm sắc thể thứ 22 của mình và dup22q11.2 trên nhiễm sắc thể thứ 22 còn lại của mình; biểu hiện định lượng của người cha đối với các gen nằm trên nhiễm sắc thể 22q11.2 là bình thường cho thấy tác động bất lợi của mất đoạn 22q11.2 đã được bù đắp bởi nhân đôi 22q11.2.
Có một số hội chứng khác có sự tham gia của nhiều hệ cơ quan và suy cận giáp. Thiểu sản các tuyến cận giáp và hậu quả là suy cận giáp có thể được quan sát thấy ở những bệnh nhân mắc hội chứng CHARGE (khuyết mống mắt, dị tật tim, tịt cửa mũi sau, chậm phát triển, bất thường sinh dục và tai, OMIM 214800) là kết quả của việc mất đoạn nhiễm sắc thể 8q12 hoặc đặc biệt là do một biến thể bất hoạt dị hợp tử của CHD7 (protein liên kết DNA helicase chromodomain 7, OMIM 608092). CHD7 mã hóa một yếu tố điều hòa phiên mã cần thiết cho sự biệt hóa của mào thần kinh có thể liên kết với TBX1. CHD7 là một chất tái cấu trúc chromatin phụ thuộc adenosine triphosphate (ATP) điều chỉnh sự di chuyển của các nucleosome. Tỷ lệ mắc hội chứng CHARGE là khoảng 1/10.000 ca sinh. Các hội chứng CHARGE và DiGeorge có chung một số bất thường bao gồm suy cận giáp, bất thường tim và thận, hở hàm ếch, bất thường tai và chậm phát triển. Thật vậy, suy cận giáp phổ biến hơn (72%) ở trẻ sơ sinh mắc CHARGE so với trẻ mắc hội chứng DiGeorge (26%).
Hội chứng Barakat hoặc HDR của suy cận giáp, điếc thần kinh giác quan và bệnh thận (loạn sản, bệnh thận kháng steroid với suy thận tiến triển—OMIM 146255) đã được cho là do các biến thể có hại đơn alen của GATA3 mã hóa protein liên kết GATA-3 (OMIM 131320), một yếu tố phiên mã/yếu tố tăng cường ngón tay kẽm cần thiết cho sự phát triển của các tuyến cận giáp, hệ thống thính giác và thận và cho sự biểu hiện của các gen mã hóa bốn tiểu đơn vị của thụ thể tế bào T. Các đột biến bất hoạt dị hợp tử của GATA3 chủ yếu liên quan đến việc mất đoạn liên kết DNA ở đầu carboxyl của nó được di truyền như một rối loạn trội trên nhiễm sắc thể thường. Các đột biến chèn, sai nghĩa và vô nghĩa trong GATA3 cũng đã được xác định ở các bệnh nhân và gia đình mắc HDR. GATA3 là một yếu tố phiên mã ngón tay kẽm điều chỉnh biểu hiện của GCM2 và do đó rất quan trọng cho sự phát triển phôi thai của các tuyến cận giáp, cũng như cho thận, túi tai và tuyến ức. Các tuyến cận giáp của những đứa trẻ này bị thiểu sản hoặc không có. Hạ canxi máu có thể xuất hiện trong giai đoạn sơ sinh hoặc không được nhận ra cho đến sau này ở thời thơ ấu. Các dị tật của tử cung và âm đạo (tử cung đôi, vách ngăn âm đạo) có thể có ở nữ giới mắc rối loạn này.
Hội chứng Sanjad-Sakati của suy cận giáp bẩm sinh, chậm phát triển trí tuệ và dị dạng khuôn mặt (sống mũi lõm, nhân trung dài, môi trên mỏng) (HRD, OMIM 241410) là do các đột biến mất chức năng hai alen (thường là mất đoạn 12 cặp bazơ trong exon mã hóa thứ ba) trong TBCE (chaperone đặc hiệu tubulin E, OMIM 604934). TBCE là một protein chaperone cần thiết cho sự hình thành/cuộn gập và sự ổn định của các vi ống—các cấu trúc tế bào chất bao gồm các tiểu đơn vị heterodimer α- và β-tubulin tạo thành khung xương tế bào, bộ máy phân bào, lông mao và các thành phần tế bào khác; chaperonin này hỗ trợ sự cuộn gập chính xác của các tiểu đơn vị α- và β-tubulin và sự hình thành các heterodimer α-β-tubulin. Các tiểu đơn vị α- và β-tubulin và TBCE cần thiết cho sự phát triển phôi thai bình thường của các tuyến cận giáp. Các đột biến trong TBCE dẫn đến giảm sự hình thành vi ống và do đó làm giảm các thành phần dưới tế bào, chẳng hạn như khung xương tế bào, bộ máy Golgi và các khoang nội bào cần thiết cho sự di chuyển bình thường của protein trong tế bào, cũng như sự hình thành của bộ máy phân bào và lông mao. Đa số trẻ sơ sinh mắc HRD bị chậm tăng trưởng trong tử cung và biểu hiện co giật do hạ canxi máu với nồng độ PTH huyết thanh thấp và đáp ứng thải phốt phát bình thường với PTH ngoại sinh trong vài tuần và tháng đầu sau sinh. Trẻ em mắc HRD bị lùn, chậm phát triển và dễ bị co giật; chúng có hẹp tủy xương dài và các bất thường xương khác; chúng bị tật đầu nhỏ với khuôn mặt được đặc trưng bởi mắt lõm sâu hoặc tật mắt nhỏ, sống mũi lõm, mũi khoằm, nhân trung dài, viền môi trên mỏng, cằm nhỏ và dái tai to, mềm. Hệ tim mạch của những bệnh nhân này còn nguyên vẹn, nhưng khi còn nhỏ, chúng dễ bị nhiễm trùng phế cầu đe dọa tính mạng. Các đặc điểm bổ sung ở các đối tượng HRD được đánh giá bằng hình ảnh thần kinh bao gồm thiểu sản tuyến yên trước và teo cuống tuyến yên, phễu và thể chai.
Khi một bệnh nhân mắc bộ ba hội chứng Sanjad-Sakati—HRD cũng biểu hiện vỏ xương dày và hẹp tủy xương dài, xơ cứng xương sọ và dễ bị nhiễm trùng tái phát và có các biến thể hai alen của TBCE, phức hợp này được gọi là hội chứng Kenny-Caffey type 1 (KCS1, OMIM 244460). Trẻ sơ sinh mắc KCS1 thường bị hạ canxi máu nặng sớm trong giai đoạn sau sinh. Khi còn nhỏ, chúng bị lùn, với tật đầu nhỏ và các bất thường sọ mặt do không có khoang diploic trong hộp sọ, xơ cứng xương và dày vỏ xương dài với hẹp khoang tủy, phát triển bình thường hoặc chậm nhẹ, và tăng khả năng bị nhiễm trùng do vi khuẩn tái phát. Điều thú vị là, cùng một đột biến trong TBCE—một mất đoạn 12 bp đồng hợp tử trong exon 2—có thể dẫn đến kiểu hình HRD hoặc KCS1 trong một gia đình cụ thể. Các phát hiện lâm sàng, xét nghiệm và X-quang ở bệnh nhân mắc hội chứng Kenny-Caffey 2 (KCS2, OMIM 127000) tương tự như ở các đối tượng mắc KCS1, ngoại trừ bệnh nhân mắc KCS2 có trí thông minh bình thường. KCS2 là kết quả của các đột biến mất chức năng đơn alen trong FAM111A (family with sequence similarity 111, Member A, OMIM 615292). Cùng alen với KCS2 là loạn sản xương mảnh (OMIM 602361), trong đó xương mỏng, mảnh và giòn, thân xương đặc, các đường khớp sọ nền đóng sớm, có tật mắt nhỏ, và hạ canxi máu do suy cận giáp là phổ biến. Hạ magiê máu cũng thường xuất hiện. Các biến thể dị hợp tử trong FAM111A đã được xác định ở những bệnh nhân này. FAMIIIA mã hóa một yếu tố có biểu hiện thấp nhất trong giai đoạn G0 của chu kỳ tế bào nhưng chức năng chính của nó chưa được biết; người ta đã đề xuất rằng FAMIIIA có thể tương tác với TBCE để điều chỉnh biểu hiện hoặc chức năng gen.
Ty thể là các bào quan trong tế bào chất chủ yếu được tặng cho phôi trong tế bào chất của noãn được thụ tinh và do đó có nguồn gốc từ mẹ. Ty thể là nơi chính trong tế bào diễn ra quá trình hô hấp và sử dụng năng lượng. Bộ gen ty thể bao gồm một nhiễm sắc thể vòng đơn với 37 gen được nhúng. Mười ba gen ty thể mã hóa các protein cần thiết cho vận chuyển điện tử và tạo năng lượng; 22 gen ty thể mã hóa các axit ribonucleic chuyển (RNA) cần thiết cho quá trình tổng hợp protein. Ngoài ra, có các protein ty thể mà gen của chúng nằm trong nhân tế bào nhưng được biểu hiện trong ty thể. Do đó, rối loạn chức năng ty thể dẫn đến suy cận giáp có thể do mất đoạn và/hoặc nhân đôi DNA ty thể mã hóa các gen nội tại của chính ty thể hoặc do các biến thể của DNA nhân mã hóa một protein được biểu hiện trong ty thể. Các biểu hiện lâm sàng của các đột biến DNA ty thể hầu như luôn liên quan đến rối loạn chức năng cơ, cũng như các mô khác tùy thuộc vào tuổi sau thụ thai và (các) vị trí tế bào mà trong đó biến thể DNA ty thể xảy ra. Hội chứng Kearns-Sayre (OMIM 530000) có liên quan đến bất sản hoặc loạn sản các tuyến cận giáp dẫn đến suy cận giáp. Các biểu hiện chính của hội chứng này bao gồm liệt vận nhãn ngoài tiến triển, viêm võng mạc sắc tố, điếc thần kinh giác quan, thất điều tiểu não, dẫn truyền tim bất thường, bệnh cơ, chậm phát triển và rối loạn chức năng ống thận, cũng như suy vỏ thượng thận, suy sinh dục và đái tháo đường. Cả mất đoạn và nhân đôi DNA ty thể đều có thể được tìm thấy ở những bệnh nhân mắc hội chứng này. Hội chứng bệnh não-cơ ty thể, nhiễm toan lactic và các cơn giống đột quỵ (MELAS, OMIM 540000) cũng có liên quan đến suy cận giáp, cũng như bệnh cơ, liệt vận nhãn, bệnh thần kinh, bệnh cơ tim, suy giảm nhận thức và đái tháo đường. Các đột biến hai alen trong HADHB (Hydroxylacyl-CoA dehydrogenase/3-Ketoacyl-CoA thiolase/Enoyl-CoA hydratase, tiểu đơn vị beta, OMIM 143450) mã hóa protein ba chức năng ty thể làm suy giảm quá trình oxy hóa beta trong ty thể của các axit béo dẫn đến không thể sử dụng một nguồn năng lượng. Tùy thuộc vào mức độ thiếu hụt enzyme, các biểu hiện lâm sàng có thể thay đổi từ cấp tính và gây tử vong trong giai đoạn chu sinh đến hội chứng gan giống Reye ở trẻ lớn hơn đến bệnh cơ xương ở thanh thiếu niên. Đôi khi, suy cận giáp có thể xảy ra ở những bệnh nhân này.
PHP là một tập hợp các rối loạn lâm sàng thường được đặc trưng bởi sự mất nhạy cảm của cơ quan đích đối với các tác dụng sinh học của PTH (và PTHrP) dẫn đến hạ canxi máu mặc dù có sự bài tiết đáng kể PTH nội sinh. Thụ thể PTH (PTH1R, OMIM 168468) là một cấu trúc xuyên màng bảy vòng xoắn được ghép cặp với protein G, sau khi liên kết với PTH hoặc PTHrP ngoại bào sẽ khởi phát truyền tín hiệu nội bào bằng cách thay đổi cấu hình của nó, do đó cho phép protein liên kết guanine nucleotide kích thích (protein Gs) liên kết dưới màng trao đổi guanosine triphosphate (GTP) cho guanosine diphosphate (GDP) trên tiểu đơn vị alpha (GNAS, OMIM 139320) của dị tam hợp của các tiểu đơn vị alpha, beta và gamma cùng cấu thành nên protein Gs. Sau khi thay thế GTP cho GDP trên tiểu đơn vị α, Gsα phân ly khỏi các tiểu đơn vị βϐ được liên kết của nó. Gsα sau đó kích thích hoạt động enzyme adenylyl cyclase (ADCY3, OMIM 600291) nội bào, do đó chuyển đổi ATP thành cyclic adenosine monophosphate (AMP) và giải phóng nó khỏi bề mặt nội bào của màng plasma của tế bào đáp ứng với PTH. Cyclic AMP lần lượt liên kết với tiểu đơn vị alpha điều hòa của protein kinase A hoặc PKA (PRKAR1A, OMIM 188830), do đó khởi phát tín hiệu nội bào. Các tiểu đơn vị xúc tác của PKA sau đó phosphoryl hóa một số protein nội bào bao gồm protein liên kết đáp ứng cyclic AMP (CREB1, OMIM 123810) lần lượt khởi phát phiên mã của các gen mục tiêu cyclic AMP. CREB1 bị bất hoạt bởi một trong một số phosphodiesterase được mã hóa bởi PDE4D (OMIM 600129) và PDE3A. (OMIM 123805). Sau khi Gsα đã truyền tín hiệu thông qua việc kích hoạt adenylyl cyclase, hoạt động GTPase nội tại của Gsα thủy phân GTP được gắn vào thành GDP, do đó dừng việc truyền tín hiệu tiếp theo.
Trẻ sơ sinh có các đột biến mất chức năng hai alen trong PTH1R bị suy cận giáp chức năng mặc dù nồng độ PTH huyết thanh tăng cao và do đó đại diện cho một dạng “PHP”. Do không đáp ứng với PTHrP trong tử cung, sự hình thành xương của thai nhi là bất thường dẫn đến loạn sản sụn Blomstrand—một bệnh loạn dưỡng xương sụn được đặc trưng bởi các chi ngắn và sự trưởng thành xương và răng tiến triển—các bất thường có thể phát hiện được trong tử cung bằng siêu âm thai nhi. Về mặt mô học, vùng tăng sinh của sụn tăng trưởng bị thu hẹp với tương đối ít tế bào sụn nghỉ và tăng sinh, trong khi vùng phì đại bao gồm các cột tế bào sụn không đều. Được di truyền như một đặc điểm lặn trên nhiễm sắc thể thường, các đặc điểm lâm sàng của nó bao gồm đa ối, phù thai, lùn chi ngắn, các bất thường trên khuôn mặt, phát triển răng sai lệch, bất sản núm vú và vú, phổi thiểu sản, hẹp eo động mạch chủ trước ống động mạch, và hạ canxi máu và tăng phốt phát máu sơ sinh mặc dù nồng độ PTH huyết thanh tăng cao. Mặc dù loạn sản xương sụn Blomstrand (OMIM 215045) thường gây tử vong, các dị tật xương có thể nặng hơn (type I) hoặc nhẹ hơn (type II). Các đột biến trong PTH1R dẫn đến sự vắng mặt hoàn toàn của protein bình thường (ví dụ, Arg104Ter) được chỉ định là type I trong khi các đột biến cho phép tổng hợp một số PTH1R (Pro132Leu) dẫn đến loạn sản xương sụn Blomstrand type II. Loạn sản sụn Eiken (OMIM 600002) cũng do các đột biến mất chức năng hai alen trong PTH1R gây ra nhưng khác biệt về mặt lâm sàng và X-quang so với loạn sản xương sụn Blomstrand vì các đối tượng bị ảnh hưởng có chậm tăng trưởng nhẹ, cốt hóa đầu xương bị trì hoãn rõ rệt, loạn sản đa đầu xương, và các đảo sụn tồn tại trong xương chậu. Các biến thể của PTH1R xảy ra ở những bệnh nhân mắc hội chứng Eiken nằm ở phần đầu carboxyl của gen/protein (ví dụ, pArg485Ter). Các đột biến bất hoạt dị hợp tử của PTH1R có thể dẫn đến việc không mọc răng không kèm hội chứng di truyền trội trên nhiễm sắc thể thường (OMIM 125350).
Theo kinh điển, PHP là thuật ngữ được áp dụng cho tình trạng lâm sàng có liên quan đến các bất thường của con đường truyền tín hiệu truyền tải thông điệp được truyền đạt bởi sự tương tác của PTH/PTHrP với PTH1R. Trẻ sơ sinh và trẻ nhỏ mắc PHP thường bị hạ canxi máu và tăng phốt phát máu với nồng độ PTH huyết thanh tăng cao, trong khi những bệnh nhân lớn tuổi hơn mắc PHP biểu hiện kiểu hình đặc trưng và các dị tật xương của loạn dưỡng xương di truyền Albright ([AHO]; chậm tăng trưởng; tật ngón ngắn của xương bàn tay thứ ba, thứ tư và thứ năm; mặt tròn; suy giảm hình thành ngà răng; và hóa xương dưới da) có hoặc không có các bất thường sinh hóa. Kiểu hình AHO của chậm tăng trưởng và tật ngón ngắn là kết quả của việc đóng sớm các sụn tăng trưởng trong xương dài—hậu quả của sự thiếu hụt Gsα ở các tế bào sụn và các nguyên bào xương giai đoạn đầu làm tăng tốc độ trưởng thành của nguyên bào xương. Các khối hóa xương dị chỗ dưới da có thể sờ thấy được, đôi khi liên quan đến sự đổi màu xanh của da overlying, phát triển ở 70% bệnh nhân AHO liên quan đến PHP1a và suy cận giáp giả giả (PPHP); số lượng và kích thước của chúng có thể tăng theo thời gian và trở nên đau cấp tính hoặc mạn tính. Các hậu quả bất thường khác của PHP1a bao gồm hẹp ống sống, hội chứng ống cổ tay, mất thính lực thần kinh giác quan và dẫn truyền, suy giảm khứu giác, ngưng thở khi ngủ và hen suyễn. Phù hợp với sự đề kháng với PTH nội sinh, việc sử dụng PTH ngoại sinh không làm giảm số lượng các chất đồng vận chuyển natri-phốt phát (NaPi2a, NaPi2c) và do đó không làm tăng bài tiết phốt phát hoặc cyclic AMP qua thận.
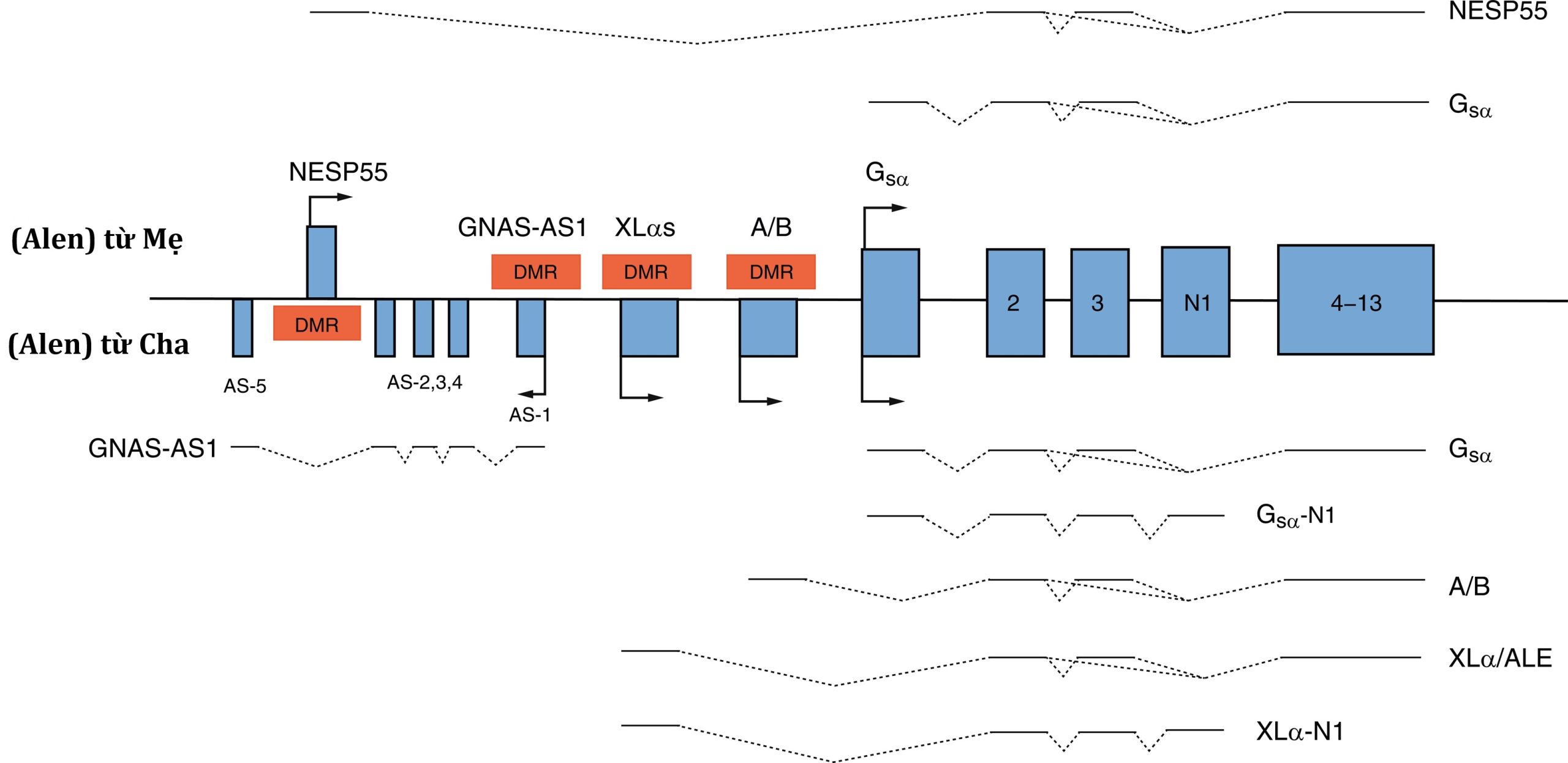
Hình 20.2: Sơ đồ của phức hợp gen GNAS. Ba bản sao chính mã hóa protein được bắt nguồn từ GNAS (protein tiết thần kinh nội tiết-55 [NESP55], XLαs, và Gsα) bằng cách liên kết exon đầu tiên của chúng với các exon chung 2–13. Bản sao GNAS-AS1 được phiên mã theo hướng ngược/đối nghĩa. Thông qua sự in dấu biểu sinh được điều hòa bởi các vùng methyl hóa khác biệt (DMR), các bản sao của XLαs và GNAS-AS1 chỉ được biểu hiện bởi alen của cha, trong khi NESP55 chỉ được biểu hiện bởi gen GNAS của mẹ. Gsα được biểu hiện từ cả hai alen GNAS của cha mẹ ngoại trừ ở ống lượn gần, tuyến giáp, tuyến sinh dục và tuyến yên trước nơi chỉ có alen Gsα của mẹ được biểu hiện. Các bản sao GNAS của mẹ được thể hiện phía trên đường ngang, trong khi các bản sao của cha được thể hiện phía dưới đường này. Các đường đậm thể hiện các exon; các đường đứt nét chỉ ra các intron.
(Từ Pignola RJ, Ramaswamy G, Fong JT, et al. Progressive osseous heteroplasia: diagnosis, treatment, and prognosis. Appl Clin Genet. 2015;8:37–48, với sự cho phép.)
GNAS là một gen được in dấu; mặc dù GNAS được biểu hiện từ cả gen GNAS của mẹ và cha trong nhiều mô, GNAS chỉ được biểu hiện từ gen của mẹ trong ống lượn gần, tuyến giáp, tuyến sinh dục và tuyến yên trước (vide infra). Do đó, trong ống lượn gần, biểu hiện của GNAS được “in dấu”; nghĩa là, có sự biểu hiện gen khác biệt tùy thuộc vào nguồn gốc của alen từ cha hay mẹ. GNAS bao gồm 13 exon; nó có nhiều bản sao phát sinh thông qua việc nối bốn exon đầu tiên duy nhất vào các exon chung từ 2 đến 13 (Hình 20.2). Các bản sao của GNAS bao gồm: (1) bản sao Gsα—một protein kích thích adenylyl cyclase và tạo ra cyclic AMP—Gsα được biểu hiện bởi cả alen của mẹ và cha trong hầu hết các mô (bao gồm da, mô mỡ trắng, tế bào sụn, xương); tuy nhiên, chỉ có alen của mẹ của GNAS được biểu hiện trong ống lượn gần, tuyến giáp, tuyến sinh dục và tuyến yên trước (do sự im lặng của GNAS của cha gây ra bởi một dấu ấn biểu sinh); (2) XLαs tạo ra một isoform Gsα được biểu hiện đặc biệt trong các mô thần kinh nội tiết và thần kinh và giống hệt Gsα ngoại trừ việc nó có một chuỗi axit amin ở đầu amino rất dài; nó chỉ được biểu hiện bởi alen của cha; (3) bản sao protein tiết thần kinh nội tiết-55 (NESP55) là một protein giống chromogranin được biểu hiện trong các mô thần kinh nội tiết nhưng chỉ bởi alen GNAS của mẹ; và (4) bản sao exon đầu tiên thay thế A/B (exon 1A) được biểu hiện ở khắp nơi nhưng chỉ ở mức độ thấp và bởi alen GNAS của cha và không được dịch mã. Các promoter của các bản sao XLαs, NESP55, và exon 1A nằm trong vùng methyl hóa khác biệt 5′ (DMR) của GNAS. Methyl hóa của promoter thường làm im lặng biểu hiện của bản sao GNAS đó.
Các đột biến mất chức năng trong GNAS hoặc các sai lệch biểu sinh (methyl hóa) dẫn đến thất bại trong việc biểu hiện một bản sao Gsα của cha mẹ dẫn đến PHP type IA, IB, và IC và PPHP. PHP type 1A (PHP1a, OMIM 103580) có liên quan đến sự đề kháng với các hormon protein truyền tín hiệu thông qua các GPCR và là hậu quả của các đột biến mất chức năng trong alen của mẹ mã hóa GNAS. Các biến thể bất hoạt của GNAS mẹ (về mặt chức năng dẫn đến sự bất hoạt hai alen của GNAS và mất gần như toàn bộ hoạt động Gsα trong ống lượn gần) dẫn đến sự đề kháng với các tác dụng sinh học của PTH trong ống lượn gần và giảm tái hấp thu canxi đã lọc dẫn đến hạ canxi máu, tăng tái hấp thu phốt phát đã lọc ở ống lượn gần dẫn đến tăng phốt phát máu, và nồng độ PTH huyết thanh tăng cao. Để đáp ứng với PTH ngoại sinh, cả bài tiết cyclic AMP qua nước tiểu và bài tiết phốt phát đều không tăng. Hoạt động Gsα của hồng cầu dưới mức bình thường ở bệnh nhân mắc PHP1a. (Sự đề kháng với hormon kích thích tuyến giáp [TSH] cũng xảy ra ở bệnh nhân mắc PHP1a. Do đó, PHP1a có thể được nghi ngờ ở một trẻ sơ sinh bị hạ canxi máu mà trong đó đã phát hiện tăng TSH máu trong nghiên cứu sàng lọc sơ sinh cho suy giáp bẩm sinh.) Hơn 200 đột biến mất chức năng dị hợp tử trong GNAS mẹ đã được mô tả. Một mất đoạn bốn cặp bazơ trong exon 7 (codon 188-189) trong GNAS dẫn đến dịch khung và codon dừng sớm đã được tìm thấy ở một số gia đình mắc PHP1a và dường như là một “điểm nóng đột biến” vì nó làm suy giảm quá trình trùng hợp và sao chép DNA. Các đột biến khác làm thay đổi sự di chuyển nội bào của protein GNAS (p.Leu99Pro, p.Ser250Arg), làm tăng tốc độ giải phóng GDP (p.Arg258Trp, p.Ala366Ser), hoặc làm suy giảm sự ghép cặp của protein G với PTH1R (p.Arg385His). Vì GNAS được biểu hiện bởi cả alen của mẹ và cha trong ống lượn xa nơi canxi đã lọc cũng được tái hấp thu, nhiễm canxi thận thường không phát triển ở bệnh nhân mắc PHP1a.
Mặc dù bệnh nhân mắc PHP type IB (PHP1b, OMIM 603233) thường có kiểu hình bình thường (ngoại trừ tật ngón ngắn nhẹ hoặc béo phì), họ bị hạ canxi máu, tăng phốt phát máu, và đề kháng với các tác dụng sinh học của PTH ngoại sinh và TSH. PHP type IB chỉ xảy ra ở con của những người mang gen nữ bắt buộc mà trong đó việc mất biểu hiện GNAS của mẹ trong thận dẫn đến sự đề kháng chọn lọc của ống lượn gần với PTH; vì biểu hiện xương của cả GNAS của mẹ và cha đều nguyên vẹn, sự hình thành xương là bình thường. PHP type IB là kết quả của các khiếm khuyết trong việc methyl hóa các nucleotide cytosine trong GNAS mẹ dẫn đến sự bất hoạt/chuyển đổi của nó thành một kiểu gen biểu sinh GNAS của cha và các hậu quả chức năng tương tự như những gì được quan sát thấy ở bệnh nhân mắc PHP type IA vì GNAS của mẹ bị “im lặng” trong các mô được in dấu, đặc biệt là thận. Trong một số trường hợp, lỗi biểu sinh này của mẹ đã được truyền từ một người mẹ mang gen cho con của bà. Ở một số bệnh nhân, PHP type IB là do mất đoạn trong một DMR của GNAS hoặc do mất một trung tâm kiểm soát in dấu tác động cis 5′ cần thiết cho việc methyl hóa DMR của GNAS điều chỉnh biểu hiện của GNAS mẹ trong ống lượn gần. PHP type IB cũng có thể do vi mất đoạn của các exon AS3-4 của GNAS mẹ hoặc mất methyl hóa trong các exon A/B của mẹ của vùng methyl hóa khác biệt dẫn đến sự im lặng của GNAS mẹ hoặc do các biến thể của GNAS-AS1 hoặc STX16. GNAS-AS1 (phức hợp locus GNAS, bản sao đối nghĩa 1, OMIM 610540) được nhúng trong vùng mã hóa GNAS, trong khi STX16 (Syntaxin16, OMIM 603666) được định vị ngay tâm (5′) so với GNAS trên nhánh dài (q) của nhiễm sắc thể 20; các đột biến (mất các exon 3-6) của STX16 mẹ có liên quan đến việc mất methyl hóa của các exon A/B của GNAS. Tuy nhiên, cơ sở di truyền cho hầu hết các bệnh nhân mắc PHP1b lẻ tẻ vẫn chưa được biết; một số bệnh nhân này dường như có các khiếm khuyết methyl hóa biểu sinh toàn cầu. Lưỡng bội đơn thân từ cha của nhánh dài nhiễm sắc thể 20 (vị trí của GNAS) có thể là nguyên nhân của PHP1b ở một số bệnh nhân. Những bệnh nhân này có hai nhiễm sắc thể thứ 20 bình thường từ cha nhưng không có GNAS chức năng trong các mô cụ thể (tức là ống lượn gần). Các khiếm khuyết in dấu liên quan đến GNAS cũng đã được xác định ở những bệnh nhân mắc AHO IB và vi mất đoạn nhiễm sắc thể 2q37. Vì xương đáp ứng với PTH ở bệnh nhân mắc AHO IA/IB, một đối tượng thỉnh thoảng có thể phát triển viêm xương xơ nang theo thời gian. Tuy nhiên, vì PTH cũng có tác dụng đồng hóa trên xương nội mạc, khoáng hóa xương ở một số bệnh nhân mắc PHP1a/1b có thể tăng lên.
Bệnh nhân mắc PHP type IC có kiểu hình AHO và bị hạ canxi máu và tăng phốt phát máu, nhưng hoạt động Gαs của hồng cầu là bình thường. PHP type I có liên quan đến các biến thể của alen GNAS mẹ trong exon 13 (xem Hình 20.2) gần đầu carboxyl của nó, thường để lại nguyên vẹn vùng hoạt động adenylate cyclase. Bệnh nhân mắc PHP type II (OMIM 203330) có hạ canxi máu, tăng phốt phát máu và nồng độ PTH huyết thanh tăng cao nhưng có kiểu hình bình thường. Để đáp ứng với PTH ngoại sinh, bệnh nhân mắc PHP type II tăng bài tiết cyclic AMP qua nước tiểu nhưng không tăng bài tiết phốt phát qua nước tiểu, cho thấy một khiếm khuyết trong tín hiệu nội bào ở hạ nguồn của việc tạo ra adenylyl cyclase. Hoạt động Gsα của hồng cầu là bình thường ở bệnh nhân mắc PHP type II. Sinh bệnh học của PHP type II vẫn chưa được xác định, nhưng ở một số đối tượng, người ta đã cho rằng nó có thể liên quan đến thiếu vitamin D và sự đề kháng PTH thứ phát sau đó; những người khác có thể đang dùng thuốc chống co giật làm tăng tốc độ phân hủy vitamin D và các chất chuyển hóa có hoạt tính sinh học của nó. Các đối tượng mắc PPHP (OMIM 612463) do các đột biến bất hoạt của GNAS cha có kiểu hình AHO do thiếu hụt biểu hiện GNAS trong xương nhưng không béo phì và trí tuệ bình thường; thận của họ, nơi GNAS mẹ được biểu hiện, đáp ứng bình thường với PTH nội sinh và ngoại sinh, và do đó những bệnh nhân này có canxi máu và phốt phát máu bình thường. Các đột biến dị hợp tử trong GNAS trên một trong hai alen của cha mẹ đã được liên kết với chậm tăng trưởng trong tử cung, với mức độ nghiêm trọng lớn nhất khi biến thể nằm trong alen của cha, cho thấy rằng một bản sao GNAS có nguồn gốc từ cha là cần thiết cho sự phát triển bình thường của thai nhi. Sự chậm tăng trưởng sau sinh của PHP và PPHP có thể là hậu quả kéo dài của sự đề kháng của sụn tăng trưởng với PTH trong tử cung và sau sinh. Bệnh nhân mắc PHP type IA đề kháng với nhiều hormon truyền tín hiệu thông qua GNAS bao gồm: TSH có thể biểu hiện như suy giáp bẩm sinh với tăng TSH máu, hormon giải phóng hormon tăng trưởng (GHRH) liên quan đến thiếu hormon tăng trưởng (GH), hormon luteinizing (LH) và hormon kích thích nang trứng (FSH) biểu hiện như dậy thì muộn, và melanocortin (MSH) liên quan đến béo phì khởi phát sớm.
Loạn sản đầu xương là một bệnh loạn sản sụn với nhiều đặc điểm của PHP1a (lùn, béo phì, tật ngón ngắn, khuôn mặt bất thường với thiểu sản mũi và hàm trên, hai mắt xa nhau, trưởng thành xương tiến triển rõ rệt) do các biến thể tăng chức năng của PRKAR1A (OMIM 188830) hoặc PDE4D (OMIM 600129) mã hóa các thành phần của hệ thống truyền tín hiệu được kích hoạt bởi Gsα (vide supra). PRKAR1A mã hóa tiểu đơn vị alpha điều hòa phụ thuộc cyclic AMP của PKA, protein kinase nằm ở hạ nguồn của Gsα và cyclic AMP và sự kích hoạt của nó dẫn đến chuỗi truyền tín hiệu nội bào điều chỉnh sự phân chia, biệt hóa, chuyển hóa, chức năng và apoptosis của tế bào. Nghịch lý thay, sự kích hoạt kéo dài của tiểu đơn vị alpha điều hòa của PKA dẫn đến sự suy giảm hoạt động chức năng của tiểu đơn vị xúc tác của PKA. Ở một bệnh nhân mắc loạn sản đầu xương type 1 (OMIM 101800), một đột biến tăng chức năng dòng mầm de novo (pArg368Stop) trong PRKAR1A hiện diện dẫn đến sự đề kháng chức năng với PTH và TSH. PRKAR1A đột biến là một protein bị rút ngắn có ái lực liên kết với tiểu đơn vị xúc tác của PKA tăng lên, vì nó thiếu một trong hai miền liên kết cyclic AMP; do đó, nó chỉ có thể được giải phóng chậm khỏi tiểu đơn vị xúc tác của PKA bởi cyclic AMP, do đó duy trì tiểu đơn vị xúc tác của PKA ở trạng thái không hoạt động. Ở một số bệnh nhân mắc loạn sản đầu xương type 1, sự đề kháng với PTH, GHRH, TSH và các gonadotropin hiện diện. PDE4D mã hóa phosphodiesterase 4D, một enzyme thủy phân và bất hoạt cyclic AMP; sự đề kháng hormon không hiện diện ở những bệnh nhân mắc loạn sản đầu xương type 2 (OMIM 614613) liên quan đến các biến thể của PDE4D, mặc dù những bệnh nhân này bị chậm phát triển. Ở những bệnh nhân có đột biến hoạt hóa trong PDE4D, tốc độ phân hủy cyclic AMP tăng lên.
(Một danh pháp thay thế cho PHP đã được mạng lưới EuroPHP trình bày và được gọi là phân loại rối loạn tín hiệu PTH/PTHrP bất hoạt [iPPSD]. Trong sơ đồ này, các rối loạn lâm sàng được liên kết với đột biến di truyền trong một thành phần của các con đường truyền tín hiệu truyền tải thông điệp của PTH và PTHrP khởi phát các phản ứng tế bào. Ví dụ, iPPSD1 có liên quan đến các đột biến bất hoạt hai alen của PTHR1 dẫn đến loạn sản sụn Blomstrand thường gây tử vong [OMIM 215045] của các chi ngắn, xương xơ cứng, trưởng thành xương tiến triển và dị dạng khuôn mặt liên quan đến đa ối và phù thai; hội chứng Eiken [OMIM 600002] là một bệnh loạn sản xương với sự trưởng thành đầu xương bị trì hoãn rõ rệt cũng liên quan đến các đột biến hai alen trong PTHR1; loạn sản sụn Murk-Jansen [OMIM 156400] là kết quả của các đột biến hoạt hóa đơn alen của PTHR1 và được đặc trưng bởi tầm vóc nhỏ, các chi ngắn và cong, ngón tay vẹo, khuôn mặt bất thường, tăng canxi máu và hạ phốt phát máu mặc dù nồng độ PTH huyết thanh bình thường đến thấp. Các đột biến bất hoạt của GNAS dẫn đến PHP1a được chỉ định là iPPSD2. Các khiếm khuyết methyl hóa liên quan đến GNAS làm phát sinh PHP1b được chỉ định là iPPSD3. Các đột biến dị hợp tử trong PRKAR1A [OMIM 188830] dẫn đến hoạt động PKA thiếu hụt dẫn đến loạn sản đầu xương type 1 [OMIM 101800] được đặc trưng bởi chậm tăng trưởng, dị dạng khuôn mặt biến đổi, tật ngón ngắn và trưởng thành xương tiến triển thường liên quan đến sự đề kháng của cơ quan đích với PTH và các hormon liên quan đến protein G khác được chỉ định là iPPSD4. Các biến thể đơn alen của PDE4D [OMIM 600129] dẫn đến các bất thường của hoạt động thủy phân phosphodiesterase đặc hiệu cyclic AMP dẫn đến loạn sản đầu xương type 2 [OMIM 614613] có kiểu hình giống với type 1 kết hợp với chậm phát triển được gọi là iPPSD5. Tật ngón ngắn với tăng huyết áp [OMIM 112410] đã được liên kết với các biến thể của PDE3A [OMIM 123805, chr. 12p12.2] và được gán cho iPPSD6. Ký hiệu iPPSD7 được sử dụng cho các rối loạn có sinh bệnh học di truyền chưa được biết.)
Loạn sản xương hóa đá tiến triển (POH, OMIM 166350) là một trong một nhóm các rối loạn liên quan đến hóa xương/vôi hóa trong hoặc dưới da, ngoài kiểu hình AHO của PHP, có liên quan đến các biến thể bất hoạt của GNAS cha (vide infra). POH là một rối loạn do các đột biến mất chức năng liên quan đến alen GNAS của cha được đặc trưng trên lâm sàng bởi các khối hóa xương ở da bắt đầu từ thời thơ ấu và tiến triển thành sự hình thành xương dị chỗ lan tỏa trong cơ xương và mạc sâu. POH cần được phân biệt với các hội chứng khác liên quan đến vôi hóa và hóa xương lạc chỗ, bao gồm loạn sản xương hóa đá tiến triển (OMIM 135100; ACVR1—OMIM 135100) và bệnh vôi hóa dạng u tăng phốt phát máu (OMIM 211900; GALNT3—OMIM 211900). Mặc dù chưa được mô tả trên lâm sàng, một đột biến bất hoạt trong LRP6 (mã hóa protein liên quan đến thụ thể lipoprotein 6—OMIM 603507) cũng có thể liên quan đến sự đề kháng với tác dụng sinh học của PTH. Ngoài vai trò chính trong quá trình nhập bào qua trung gian thụ thể của các lipoprotein, LRP6 cần thiết cho sự di chuyển của Gsα đến màng plasma và cho sự ghép cặp của nó với PTH1R.
Đánh giá và Quản lý
Việc đánh giá trẻ sơ sinh bị hạ canxi máu bắt đầu bằng việc xem xét tiền sử trước khi thụ thai của mẹ, quá trình mang thai, và tiền sử chu sinh, sau sinh, và gia đình, sau đó là một cuộc kiểm tra thể chất toàn diện (Hình 20.3). Dữ liệu tiền sử bao gồm những dữ liệu liên quan đến số lần sinh của mẹ và các biến chứng của thai kỳ, chẳng hạn như đái tháo đường ở mẹ, tiền sản giật hoặc uống các chất có thể gây tăng canxi máu ở mẹ (quá nhiều kiềm), chậm tăng trưởng trong tử cung, các bất thường khi sinh, nhiễm trùng huyết sơ sinh, hoặc các bệnh sau sinh sớm khác. Tiền sử gia đình được kiểm tra để tìm các thành viên có bất thường về chuyển hóa khoáng chất, chẳng hang như sỏi thận, còi xương, hoặc hạ canxi máu (ví dụ, các rối loạn co giật). Tiền sử xã hội cung cấp thông tin về tình trạng kinh tế xã hội của người mẹ và môi trường văn hóa của bà có thể đã ảnh hưởng đến chế độ ăn uống và sự tiếp xúc với ánh nắng mặt trời của mẹ trong thời gian mang thai. Khám thực thể trẻ sơ sinh (khuôn mặt bất thường, tiếng thổi ở tim phù hợp với bệnh tim bẩm sinh) có thể gợi ý một dạng hạ canxi máu phức hợp. Việc xác định công thức máu toàn phần, nồng độ canxi toàn phần, Ca2+, magiê, phốt phát, creatinin, PTH nguyên vẹn, calcidiol, và calcitriol trong huyết thanh, và nồng độ canxi và creatinin trong nước tiểu trong một mẫu nước tiểu tại chỗ nên được thực hiện trước khi điều trị ban đầu cho trẻ sơ sinh bị hạ canxi máu bất cứ khi nào có thể. Nồng độ PTH huyết thanh giảm thường gặp ở trẻ sơ sinh bị hạ canxi máu khởi phát sớm, nhưng nồng độ PTH thấp dai dẳng gợi ý suy giảm bài tiết PTH, đôi khi do các biến thể bất hoạt của PTH hoặc do các đột biến tăng chức năng của CASR hoặc GNA11. Nồng độ PTH cao hiện diện ở những bệnh nhân thiếu hoặc không nhạy cảm với vitamin D, đề kháng PTH do các đột biến mất chức năng trong PTH1R, GNAS (PHP), hoặc PRKAR1A (loạn sản đầu xương type 1) hoặc suy giảm chức năng thận. Nồng độ calcidiol thấp biểu thị giảm dự trữ vitamin D của mẹ (và do đó của thai nhi) hoặc hiếm khi là một khiếm khuyết trong CYP27B1 mã hóa vitamin D-25 hydroxylase, trong khi nồng độ calcitriol thấp một cách không phù hợp ở những đối tượng có chức năng thận bị tổn thương nghiêm trọng, suy cận giáp, hoặc những người bị thiếu hụt 25OHD-1α-hydroxylase. Giá trị calcitriol tăng cao gợi ý đề kháng vitamin D có lẽ do một bất thường trong VDR, một rối loạn có thể liên quan đến rụng tóc. X-quang xương có thể cho thấy tình trạng loãng xương, trong khi X-quang ngực có thể không xác định được bóng tuyến ức (nhưng là một dấu hiệu không đáng tin cậy ở một trẻ sơ sinh bị bệnh nặng hoặc bị căng thẳng). Nồng độ canxi, Ca2+, phốt phát, và PTH nguyên vẹn trong huyết thanh nên được đo ở các bà mẹ của trẻ sơ sinh bị hạ canxi máu không giải thích được vì một số người có thể bị cường cận giáp không được nghi ngờ.
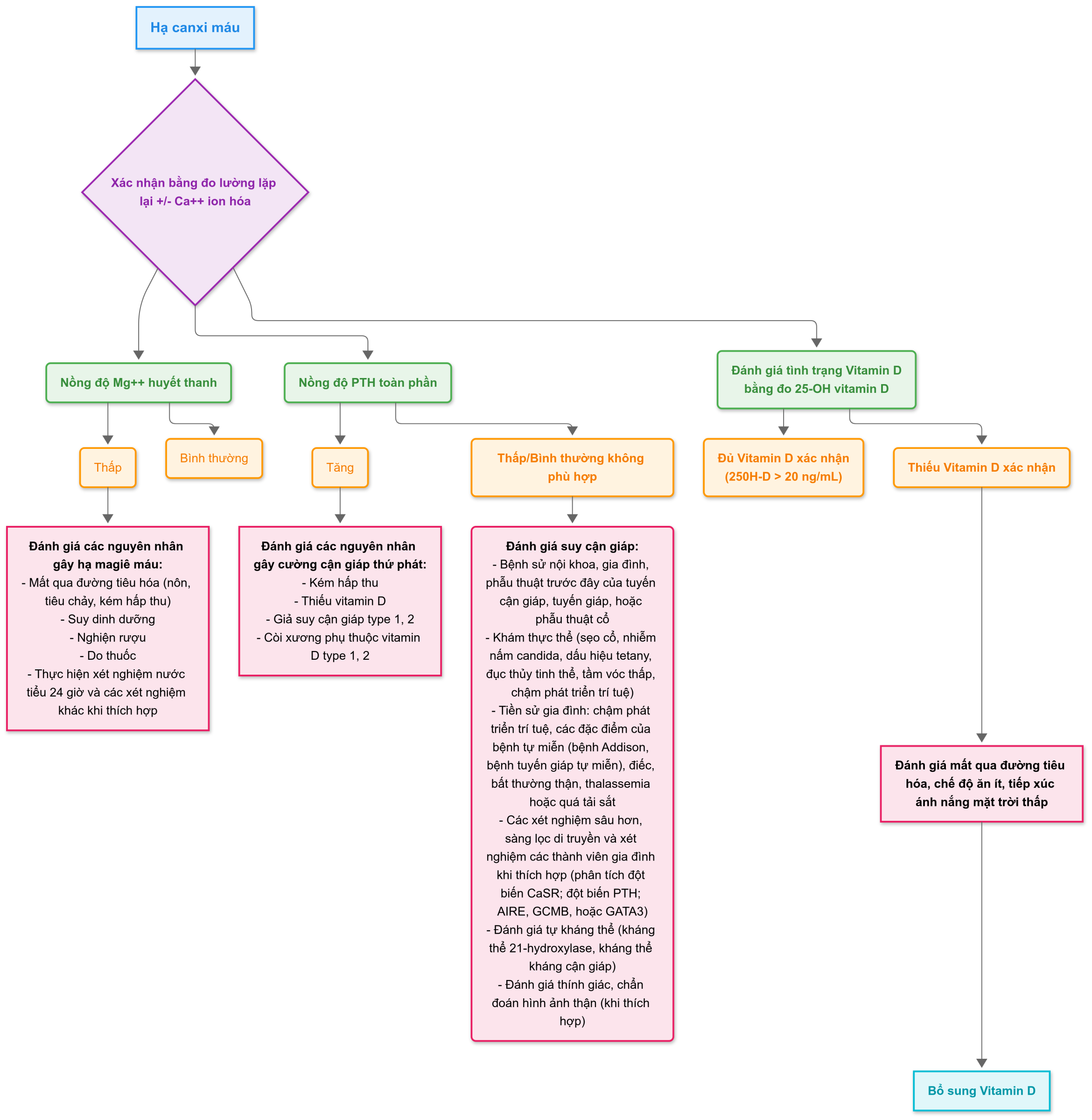
Hình 20.3: Đánh giá hạ canxi máu. (Từ Bilezikian JP, Khan A, Potts JT Jr, et al. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Mineral Res. 2011;26:2317–2337, với sự cho phép.)
Ở trẻ sơ sinh bị hạ canxi máu không giải thích được bằng cách khác, nên tiến hành đánh giá khả năng mắc hội chứng DiGeorge—đặc biệt khi khám thực thể cho thấy khuôn mặt bất thường, và có một bất thường bẩm sinh của đường ra của tim. Số lượng bạch cầu và tế bào lympho T (CD4) thấp và bóng tuyến ức thường không có ở những đối tượng này. Chẩn đoán hội chứng DiGeorge được xác nhận bởi sự hiện diện của một vi mất đoạn của nhiễm sắc thể 22q11.2 được chứng minh bằng microarray nhiễm sắc thể dựa trên đa hình đơn nucleotide hoặc FISH. Microarray đa hình đơn nucleotide cho phép phát hiện cả biến thể số lượng bản sao (mất đoạn hoặc nhân đôi) và các biến thể cấu trúc trung tính về số lượng bản sao, chẳng hạn như các vùng đồng hợp tử và lưỡng bội đơn thân. Đôi khi, có thể cần phân tích trình tự của TBX1 để xác lập chẩn đoán này, nếu các nghiên cứu khác là bình thường. Bởi vì hội chứng DiGeorge có thể di truyền, nên chỉ định đánh giá di truyền của cha mẹ và anh chị em của một trẻ sơ sinh bị ảnh hưởng. Cần lưu ý rằng phần lớn trẻ sơ sinh và trẻ nhỏ mắc hội chứng DiGeorge được nhận ra chủ yếu vì các bất thường về tim và những đối tượng không có các tổn thương này có thể không được xác định cho đến giữa hoặc cuối thời thơ ấu hoặc thanh thiếu niên. Trẻ sơ sinh mắc PHP1a có thể biểu hiện với nồng độ TSH huyết thanh tăng cao trong cuộc khảo sát sàng lọc chuyển hóa sơ sinh nhưng thường không có kiểu hình xương đặc trưng (xương bàn tay ngắn) của AHO; nếu không xác định được nguyên nhân gây suy giáp bẩm sinh, nên nghi ngờ chẩn đoán PHP1a và chỉ định đo nồng độ canxi huyết thanh và, nếu thích hợp, các nghiên cứu kiểu gen và methyl hóa của GNAS.
Hạ canxi máu sơ sinh sớm thường không có triệu chứng, nhưng dù sao cũng cần điều trị khi nồng độ canxi toàn phần trong huyết thanh dưới 8 mg/dL (2 mmol/L) (Ca2+ < 4,4 mg/dL [1,1 mmol/L]) ở trẻ đủ tháng hoặc trẻ sinh non có cân nặng lúc sinh trên 1500 g, hoặc ở trẻ sơ sinh rất nhẹ cân với giá trị canxi toàn phần dưới 7 mg/dL (1,75 mmol/L) (Ca2+ < 4,0 mg/dL [1,0 mmol/L]). Trẻ sơ sinh không có triệu chứng được quản lý dễ dàng nhất bằng cách tăng lượng canxi uống và thiết lập tỷ lệ tổng thể canxi:phốt phát nạp vào là 4:1 (bao gồm cả trong các cữ bú với sữa công thức ít phốt phát, chẳng hạn như Similac PM 60/40—tỷ lệ canxi:phốt phát 1,6:1) với canxi glubionate hoặc canxi cacbonat được dùng liều chia mỗi 4 đến 6 giờ (Bảng 20.3). Tình trạng canxi máu bình thường gần như luôn được phục hồi ở những đối tượng này trong vòng 3 tuần sau khi sinh và thường sớm hơn. Ở trẻ nhỏ bị hạ canxi máu có tetany hoặc co giật rõ ràng, có thể dùng 10% canxi gluconat (canxi nguyên tố 9,3 mg/mL) với liều 1 đến 3 mg/kg và tốc độ dưới 1 mL/phút và tổng liều không vượt quá 20 mg canxi nguyên tố/kg bằng cách truyền tĩnh mạch trong 15 phút; thường thì co giật sẽ ngừng sau khi đã dùng 1 đến 3 mL canxi gluconat 10%. Nhịp tim và nhịp điệu phải được theo dõi cẩn thận để ngăn ngừa nhịp tim chậm và vô tâm thu. Nên hạn chế sử dụng các liều canxi tiêm tĩnh mạch bolus tiếp theo (~ 10 mg/kg mỗi 6 giờ) vì chúng dẫn đến sự biến động lớn về giá trị canxi huyết thanh. Sau khi điều trị ban đầu hạ canxi máu sơ sinh, có thể dùng 500 mg canxi gluconat/kg/24 giờ bằng cách truyền tĩnh mạch liên tục, cẩn thận để ngăn ngừa thoát mạch hoặc thâm nhiễm vì canxi ngoại bào sẽ kết tủa trong các mô mềm. Những trẻ này có thể nhận canxi bổ sung qua đường uống nếu cần (vide supra). Tùy thuộc vào nguyên nhân của hạ canxi máu, cũng có thể cần bổ sung vitamin D hoặc calcitriol. Ở trẻ sơ sinh bị hạ canxi máu do suy cận giáp, đôi khi có thể cần dùng PTH tổng hợp 1-34 (teriparatide) hoặc PTH người tái tổ hợp 1-84 để phục hồi tình trạng canxi máu bình thường. Nồng độ canxi và creatinin trong huyết thanh và nước tiểu nên được xác định thường xuyên và điều trị được điều chỉnh để duy trì tình trạng canxi máu bình thường và tỷ lệ canxi/creatinin trong nước tiểu dưới 0,2 nhằm tránh tăng canxi máu, tăng canxi niệu, nhiễm canxi thận và suy thận do điều trị. Ở trẻ sơ sinh cần nuôi dưỡng qua đường tĩnh mạch, nên kết hợp 50 mg canxi nguyên tố/kg/24 giờ vào dung dịch truyền; phốt phát nguyên tố cũng phải được dùng nếu được phép và chỉ định nhưng riêng biệt với canxi.
Bảng 20.3: Các chế phẩm Calcitriol, Canxi, Magiê, và Phốt phát
| Hàm lượng | Khoáng chất Nguyên tố | |
|---|---|---|
| Vitamin D | ||
| Vitamin D | ||
| Calciferol | 8000 IU/mL | |
| 50.000 IU/viên nang | ||
| Calcidiol | 20 hoặc 50 mcg/viên nén | |
| Calcitriol | ||
| Rocaltrol | 1 mcg/mL (dung dịch uống) | |
| 0,25 mcg hoặc 0,5 mcg/viên nang | ||
| Dihydrotachysterol | ||
| Hytacherol | 0,2 mg/5 mL | |
| 0,125, 0,2, 0,4 mg/viên nén | ||
| Canxi | ||
| Canxi acetat | ||
| Phoslyra | 667 mg/5 mL, 667 mg/viên nang | |
| Canxi gluconat (iv) | 93 mg/10 mL | 93 mg/g |
| 500, 650 mg/viên nén | ||
| Canxi glubionat (dung dịch) | 64 mg/g | 115 mg/5 mL |
| Canxi carbonat | 400 mg/g | 500, 600, 750, 1250 mg/viên nén |
| Titralac | 420 mg viên | 168 mg/viên |
| 780 mg viên | 300 mg/viên | |
| Os-Cal | 650 mg viên | 260 mg/viên (Vitamin D) |
| 1250 mg viên | 500 mg/viên (Vitamin D) | |
| Tums | 500 mg viên | 200 mg/viên |
| 750 mg viên | 300 mg/viên | |
| 1000 mg viên | 400 mg/viên | |
| 1250 mg viên | 280 mg/viên | |
| Caltrate | 1500 mg viên | 600 mg/viên (Vitamin D) |
| Canxi carbonat (hỗn dịch) | 1250 mg/5 mL | 500 mg/5 mL |
| Canxi clorid | 100 mg/mL | |
| Canxi citrat | 210 mg/g | 200 mg/viên nén |
| Citracal | 950 mg viên | 200 mg/viên |
| Canxi lactat | 130 mg/g | 84 mg/viên |
| Magiê | ||
| Magiê sulfat | 49 mg/mL (dung dịch tiêm bắp 50%) | |
| 0,32, 0,64, 4 mEq/mL (dung dịch tiêm tĩnh mạch) | ||
| Magiê oxit | 603 mg/g | 241 mg/viên |
| Magiê gluconat | 54 mg/g | 27 mg/viên |
| Magiê clorid | 120 mg/g | 64 mg/viên |
| Phốt pho | ||
| Natri phốt phát (Phospha-Soda) | 127 mg/mL | |
| Natri/kali phốt phát (Phos-NaK) | 250 mg/gói (bột) | |
| Kali phốt phát (Neutraphos-K) | 250 mg/gói (bột) | |
| Kali phốt phát (K-Phos Original) | 114 mg/viên | |
| Natri/kali phốt phát (K-Phos #2) | 250 mg/viên bao | |
| Natri/kali phốt phát (K-Phos Neutral) | 250 mg/viên |
Từ Alon US. Hypophosphatemic vitamin D-resistant rickets. In: Favus MJ (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 6th ed. Washington, DC: American Society for Bone and Mineral Research; 2006:342–345; Shoback D. Hypoparathyroidism. N Engl J Med. 2008;359:391–403.
Sau khi phục hồi tình trạng canxi máu bình thường ở trẻ sơ sinh mắc hội chứng DiGeorge, các thành phần khác của rối loạn này phải được giải quyết. Các bất thường về tim thường cần phẫu thuật sửa chữa cũng như hở hàm ếch. Ở trẻ sơ sinh bị ảnh hưởng bị suy giảm miễn dịch và bị nhiễm trùng tái phát do bất sản tuyến ức, liệu pháp chống nhiễm trùng thích hợp là bắt buộc. Việc cấy ghép mô tuyến ức của thai nhi hoặc mô tuyến ức sau sinh được nuôi cấy, tủy xương, hoặc các tế bào đơn nhân máu ngoại vi đã phục hồi chức năng miễn dịch ở trẻ sơ sinh mắc rối loạn này. Bổ sung calcitriol (20–60 ng/kg/ngày) và canxi là cần thiết để phục hồi và duy trì tình trạng canxi máu bình thường ở trẻ sơ sinh bị suy cận giáp. Tăng trưởng kém do khó khăn trong việc ăn uống và khuyết tật học tập do chậm phát triển phải được quản lý trên cơ sở cá nhân và minh họa sự cần thiết của một phương pháp tiếp cận đa ngành đối với việc chăm sóc những bệnh nhân này. Hạ canxi máu do hạ magiê máu được quản lý cấp tính bằng cách truyền tĩnh mạch hoặc tiêm bắp 50% magiê sulfat với liều 0,1 đến 0,2 mL/kg trong khi theo dõi tình trạng tim mạch.
Hạ canxi máu ở Trẻ em và Vị thành niên
Nguyên nhân
Các nguyên nhân gây hạ canxi máu ở trẻ em và vị thành niên bao gồm nhiều nguyên nhân cũng dẫn đến hạ canxi máu sơ sinh và được liệt kê trong Bảng 20.2A, 20.2B và Hình 20.1. Hạ canxi máu được định nghĩa theo các chỉ số bình thường của phòng xét nghiệm phân tích; nồng độ canxi và phốt phát toàn phần trong huyết thanh thay đổi theo tuổi. (Hướng dẫn chung về nồng độ canxi toàn phần theo tuổi là: 1–5 tuổi: 9,4–10,8; 6–12 tuổi: 9,4–10,2; > 20 tuổi: 8,8–10,2 mg/dL.) Nồng độ canxi toàn phần thấp ở bệnh nhân giảm albumin máu—có thể tính toán hiệu chỉnh cho tình trạng giảm albumin máu bằng cách cộng 0,8 mg/dL vào nồng độ canxi toàn phần được ghi nhận cho mỗi 1 g/dL nồng độ albumin giảm. Do đó, việc đo cả giá trị canxi toàn phần và Ca2+ là phù hợp khi đánh giá trẻ bị hạ canxi máu. Điều đặc biệt quan trọng là theo dõi các giá trị Ca2+ ở trẻ bị bệnh nặng có hạ canxi máu. Ở trẻ/vị thành niên bị bệnh rất nặng, giá trị canxi toàn phần thấp đôi khi có thể liên quan đến giảm protein máu hoặc rối loạn cân bằng kiềm-toan và sự gắn kết thay đổi của canxi với protein huyết thanh, nhưng nó cũng có thể do suy giảm bài tiết PTH liên quan đến hạ magiê máu hoặc giảm tổng hợp vitamin D và hậu quả là hấp thu canxi ở ruột dưới mức bình thường. Về mặt cơ chế, hạ canxi máu phát triển là hậu quả của việc dòng canxi từ đường tiêu hóa, xương hoặc thận vào khoang ngoại bào và mạch máu quá ít hoặc mất canxi quá nhiều từ các khoang này vào nước tiểu, phân hoặc xương. Do đó, hạ canxi máu có thể do giảm lượng canxi ăn vào hoặc hấp thu hoặc mất quá nhiều, giảm sản xuất PTH có hoạt tính sinh học do các bất thường bẩm sinh trong sự phát triển của tuyến cận giáp hoặc tổng hợp PTH hoặc của CaSR, sự phá hủy các tuyến cận giáp bởi các tự kháng thể, quá tải kim loại (đồng, sắt), các tổn thương do phẫu thuật hoặc xạ trị, thâm nhiễm u hạt, hoặc do suy giảm đáp ứng tế bào với PTH. Hạn chế tiếp xúc với ánh nắng mặt trời hoặc giảm lượng ăn vào, hấp thu, chuyển hóa hoặc hoạt động của vitamin D dẫn đến hạ canxi máu. Hạ magiê máu làm suy giảm sự bài tiết (nhưng không phải là sự tổng hợp của PTH) và làm giảm đáp ứng của mô với PTH. Hạ canxi máu xảy ra sau khi tiếp xúc với một số loại thuốc. Do đó, co cứng do hạ canxi máu có thể phát triển sau khi dùng thuốc xổ chứa phốt phát qua đường trực tràng hoặc thuốc nhuận tràng qua đường uống. Đôi khi, trẻ hoặc vị thành niên bị hạ canxi máu có thể không có triệu chứng và được phát hiện qua sàng lọc hóa học cho một vấn đề không liên quan hoặc có thể biểu hiện bằng chuột rút cơ không liên tục khi nghỉ ngơi hoặc trong khi tập thể dục (khi sự gia tăng pH hệ thống do tăng thông khí làm giảm thêm nồng độ Ca2+); dị cảm ở ngón tay, ngón chân, hoặc vùng quanh miệng; co cứng (co cứng bàn tay-bàn chân, co thắt thanh quản, co thắt phế quản); hoặc co giật (động kinh cơn lớn, khu trú, cơn nhỏ, mất trương lực hoặc ngất). Hạ canxi máu kéo dài và nghiêm trọng có thể dẫn đến suy tim sung huyết. Khám thực thể thường cho thấy dấu hiệu Chvostek (co giật má khi gõ vào dây thần kinh sọ thứ bảy cùng bên) và/hoặc dấu hiệu Trousseau (co cứng bàn tay-bàn chân) dương tính và tăng phản xạ. Tuy nhiên, dấu hiệu Chvostek dương tính thường có ở thanh thiếu niên bình thường.
Suy cận giáp có thể xảy ra như một rối loạn đơn độc, như một phần của bệnh đa nội tiết tự miễn đa chiều, hoặc là một biểu hiện của một nhóm các dị tật bẩm sinh phức tạp (như được mô tả trong phần về hạ canxi máu sơ sinh). Suy cận giáp có thể là hậu quả của việc cắt bỏ toàn bộ tuyến giáp để cắt bỏ một tổn thương ác tính của tuyến giáp do vô tình cắt bỏ bốn tuyến cận giáp hoặc tổn thương nguồn cung cấp máu động mạch của chúng. Có các dạng suy cận giáp lẻ tẻ và có tính gia đình; khi có tính gia đình, suy cận giáp có thể được di truyền theo kiểu trội trên nhiễm sắc thể thường, lặn trên nhiễm sắc thể thường, hoặc lặn liên kết X (xem Bảng 20.2A, 20.2B và xem Hình 20.1). Các bất thường trong sự phát triển của tuyến cận giáp, phiên mã của PTH, và trong quá trình xử lý sản phẩm được dịch mã đã được liên kết với các dạng suy cận giáp di truyền. Suy cận giáp do rối loạn sinh tổng hợp hormon đơn độc/di truyền trội trên NST thường có tính gia đình có thể do một chuyển đoạn T ⇒ C đơn alen ở codon 18 (p.Cys18Arg) của peptide tín hiệu 25 aa của preproPTH làm suy giảm sự vận chuyển hiệu quả của protein từ ribosome và tương tác của preproPTH với hạt nhận dạng tín hiệu, sự di chuyển của peptide tiền chất vào và ra khỏi lưới nội chất hạt, sự phân cắt của nó bởi một peptidase tín hiệu, và sự kết hợp của nó vào một hạt bài tiết. Suy cận giáp do rối loạn sinh tổng hợp hormon đơn độc/di truyền lặn trên NST thường có tính gia đình cũng đã được liên kết với một chuyển đoạn G ⇒ C đồng hợp tử ở nucleotide 1 của intron 2 của PTH trong chuỗi tín hiệu đã ngăn cản sự phân cắt bình thường của preproPTH và làm giảm sự bài tiết PTH. Thỉnh thoảng, suy cận giáp đơn độc có thể được tìm thấy ở những bệnh nhân bị mất đoạn nhiễm sắc thể 22q11.2 mà không có các dấu hiệu hoặc triệu chứng khác của hội chứng DiGeorge hoặc các hội chứng liên quan.
Hạ canxi máu di truyền trội trên NST thường type 1 (OMIM 601198) là do các đột biến tăng chức năng dị hợp tử trong các miền ngoại bào, xuyên màng và nội bào của CASR phiên mã một CaSR không hoạt động cấu thành nội tại nhưng đặc biệt nhạy cảm và dễ dàng được kích hoạt bởi nồng độ Ca2+ huyết thanh rất thấp có thể được xác định ở trẻ sơ sinh hoặc ở các đối tượng lớn tuổi hơn (Hình 20.4). Trong rối loạn này, ngay cả ở mức hạ canxi máu, Ca2+ vẫn liên kết chặt chẽ với CaSR và kích hoạt phospholipase C-β1 làm tăng nồng độ inositol phosphate và Ca2+ trong tế bào chất và kích thích con đường truyền tín hiệu protein kinase được hoạt hóa bởi mitogen (MAPK) trong các tế bào chính của tuyến cận giáp, do đó ức chế tổng hợp và bài tiết PTH, và ở thận, làm giảm tái hấp thu canxi và magiê ở ống thận, dẫn đến mất các cation này qua nước tiểu (hạ canxi máu tăng canxi niệu) và giảm khả năng cô đặc nước tiểu. Ở những đối tượng này, nồng độ phốt phát huyết thanh tăng và giá trị magiê giảm; nồng độ PTH thấp hoặc bình thường một cách không phù hợp. Hạ canxi máu di truyền trội trên NST thường type 2 là do các biến thể dị hợp tử của GNA11 (OMIM 139313) mã hóa một tiểu đơn vị alpha của protein G (Gα11) khởi phát truyền tín hiệu nội bào sau khi Ca2+ liên kết với CaSR. Các biểu hiện lâm sàng và sinh hóa của nó tương tự như các biểu hiện do các biến thể của CASR gây ra với co giật, co thắt thanh quản, hạ canxi máu, tăng phốt phát máu và nồng độ PTH huyết thanh tương đối thấp. Vì bệnh nhân bị hạ canxi máu di truyền trội trên NST thường thường có các triệu chứng hạ canxi máu, chẳng hạn như co cứng và co giật, và rất nhạy cảm với vitamin D, liều calcitriol phải được giới hạn ở mức làm tăng giá trị canxi huyết thanh đến mức không có triệu chứng ngay cả khi không nằm trong giới hạn bình thường vì liều lớn hơn dẫn đến tăng canxi niệu (đôi khi ngay cả khi nồng độ canxi huyết thanh vẫn dưới mức bình thường), nhiễm canxi thận và suy thận chức năng. Việc sử dụng rhPTH 1-34 (teriparatide) tái tổ hợp ở người phục hồi cân bằng nội môi canxi trong rối loạn này nhưng không nhất thiết ngăn ngừa được nhiễm canxi thận. Tuy nhiên, rhPTH 1-34 không được dùng thường quy cho trẻ em vì về mặt thực nghiệm, có sự gia tăng tỷ lệ mắc sarcoma xương ở chuột non nhận lượng rất lớn chất này, mặc dù các loài linh trưởng dường như ít nhạy cảm hơn với sự hình thành khối u xương do PTH gây ra so với các loài gặm nhấm. Sự phát triển của các tự kháng thể kích thích đối với CaSR dẫn đến một biến thể mắc phải của suy cận giáp tự phát có thể là đơn độc hoặc là một phần của một bệnh nội tiết tự miễn phức tạp. Thật vậy, ở có lẽ tới một phần ba bệnh nhân bị suy cận giáp tự phát đơn độc mắc phải, các kháng thể chống lại các epitope trong miền ngoại bào của CaSR có thể hiện diện. Dạng suy cận giáp mắc phải này có thể hồi phục được, vì các kháng thể này không phá hủy các tuyến cận giáp. Ở những bệnh nhân mắc các dạng suy cận giáp tự miễn khác, các kháng thể gây độc tế bào và đi kèm với sự thâm nhiễm lympho bào, teo và thay thế bằng mô mỡ của mô cận giáp.
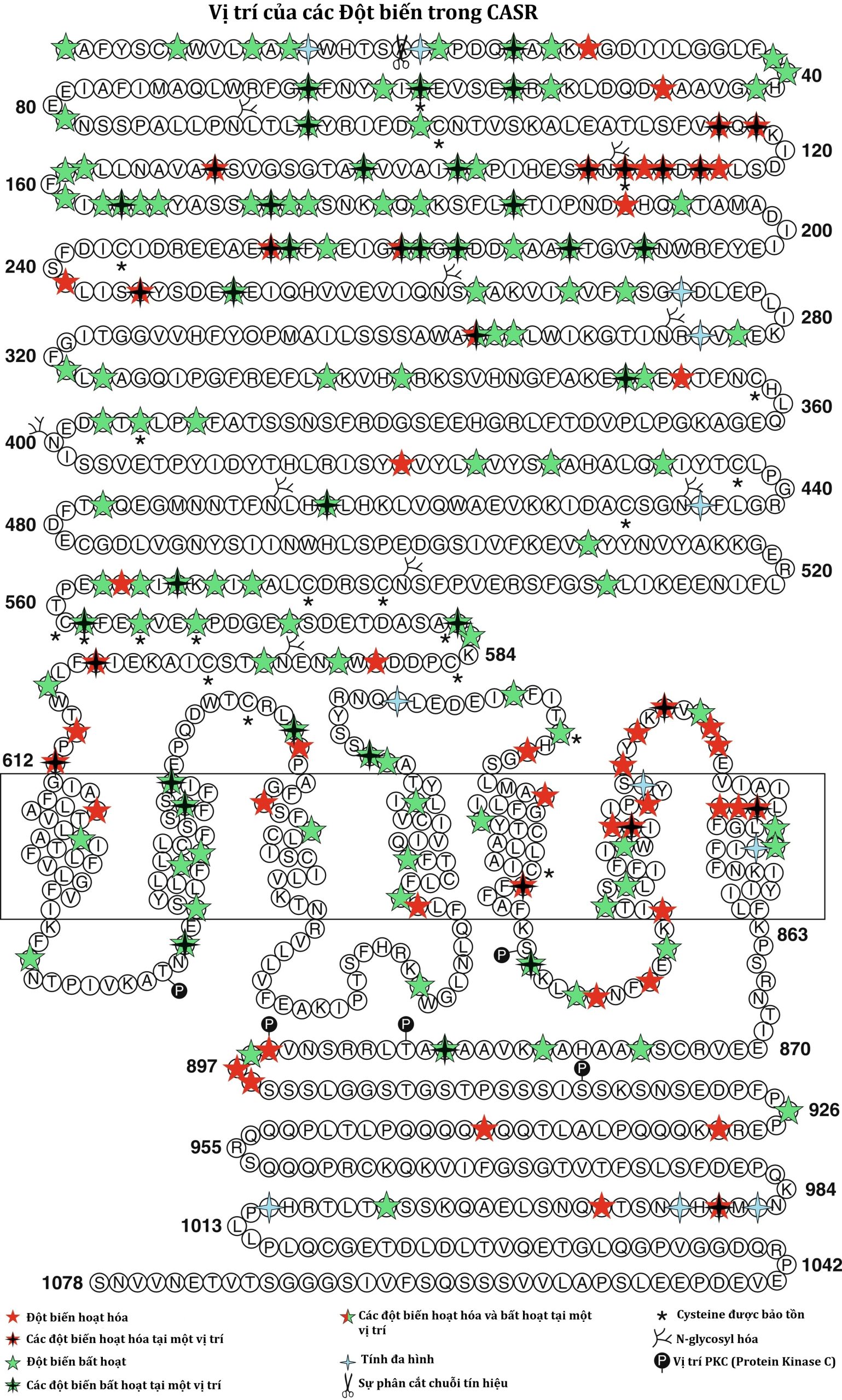
Hình 20.4: Sơ đồ của thụ thể cảm nhận canxi (CaSR) mô tả các đột biến bất hoạt (tăng canxi máu giảm canxi niệu di truyền/có tính gia đình) và hoạt hóa (suy cận giáp di truyền trội trên NST thường). Lưu ý rằng các đột biến ở các codon liền kề có thể dẫn đến tăng hoặc mất chức năng của sản phẩm CASR đột biến (ví dụ, codon 565, 567, 568). (Tải về từ Google ngày 27 tháng 4 năm 2013.)
Ở giữa thời thơ ấu và thanh thiếu niên, suy cận giáp mắc phải có thể là một biểu hiện muộn hoặc muộn của một biến thể di truyền (ví dụ, hội chứng DiGeorge), nhưng nó cũng có thể là kết quả của sự phá hủy các tuyến cận giáp do bệnh tự miễn hoặc phẫu thuật cắt bỏ hoặc chấn thương phẫu thuật đối với nguồn cung cấp mạch máu của các cấu trúc này. Các nguyên nhân bất thường của suy cận giáp mắc phải ở nhóm tuổi này bao gồm thâm nhiễm sắt (bệnh huyết sắc tố, truyền máu quá nhiều) hoặc đồng (bệnh thoái hóa gan-nhân đậu Wilson), các bệnh u hạt, hoặc xạ trị (xạ trị vùng trung thất cho u lympho Hodgkin/không Hodgkin hoặc điều trị bằng iốt phóng xạ cho bệnh cường giáp).
Suy cận giáp tự miễn có thể xảy ra như một rối loạn đơn độc hoặc như một phần của phức hợp hội chứng đa nội tiết tự miễn type I (APS1). Sự phát triển của một bệnh nội tiết tự miễn được khởi đầu bởi sự thất bại trong việc nhận dạng một peptide đặc hiệu cho một cơ quan đích bởi một phân nhóm tế bào T điều hòa thường nhận dạng peptide đó. Khi sự dung nạp miễn dịch đối với peptide đó bị mất, các dòng tế bào T điều hòa CD4 đối với peptide đó sẽ mở rộng; các tế bào T trợ giúp type 1 tiết ra các cytokine gây viêm, chẳng hạn như interferon-γ (IFN-γ), trong khi các tế bào T trợ giúp type 2 kích thích chức năng tế bào B và dẫn đến viêm qua trung gian tự kháng thể. Mất dung nạp miễn dịch có thể là hậu quả của tình trạng viêm sau nhiễm trùng do kích hoạt hệ thống miễn dịch bẩm sinh hoặc do một đột biến gen làm giảm dung nạp miễn dịch và cho phép mở rộng một dòng tế bào T điều hòa CD4+ sau khi tiếp xúc với một lượng nhỏ kháng nguyên. Các biến thể di truyền trong phức hợp tương hợp mô chính (kháng nguyên bạch cầu người [HLA]-DQ, HLA-DR) xác định sự trình diện peptide (kháng nguyên) cho các tế bào T điều hòa CD4+ kết hợp với các bất thường di truyền trong điều hòa miễn dịch để gây ra bệnh tự miễn. Ở 30% bệnh nhân bị suy cận giáp tự phát đơn độc, rối loạn này là do các kháng thể đối với miền ngoại bào của CaSR và do đó có chức năng ức chế nhưng có khả năng hồi phục; ở khoảng 33% bệnh nhân, các kháng thể huyết thanh đối với các thành phần khác của tế bào chính tuyến cận giáp có thể hiện diện. Có một số loại hội chứng đa nội tiết tự miễn nhưng chỉ ở type I, suy cận giáp là một thành phần chính.
APS1 (OMIM 240300) là một rối loạn di truyền lặn trên nhiễm sắc thể thường với bộ ba kinh điển của bệnh đa nội tiết tự miễn, nhiễm nấm candida niêm mạc da, và loạn dưỡng ngoại bì (APECED). Nó được đặc trưng bởi sự khởi phát ở thời thơ ấu của nhiều rối loạn hệ thống tự miễn và bệnh nội tiết bao gồm suy cận giáp, suy thượng thận nguyên phát, và nhiễm nấm candida niêm mạc da mạn tính, và ít gặp hơn là thiếu máu ác tính, viêm gan, suy buồng trứng và tinh hoàn nguyên phát, viêm giác mạc, viêm võng mạc, rụng tóc, và loạn sản hành xương có thể hồi phục. Hội chứng đa nội tiết tự miễn 1 là do các biến thể mất chức năng của bộ điều hòa tự miễn (AIRE, OMIM 607358). Trong một đoàn hệ Phần Lan gồm 91 bệnh nhân mắc APS1/APECED, các biểu hiện chính là nhiễm nấm candida niêm mạc da liên quan đến móng và miệng xảy ra ở 100% bệnh nhân—thường trong 2 năm đầu đời, suy cận giáp và suy vỏ thượng thận, cả hai bệnh này đều phát triển ở 80% đến 90% các đối tượng bị ảnh hưởng. Suy cận giáp xảy ra thường xuyên nhất trong độ tuổi từ 2 đến 10 và suy vỏ thượng thận phát triển trong độ tuổi từ 5 đến 15. Hầu hết tất cả phụ nữ mắc APECED đều phát triển suy cận giáp trong khi 80% nam giới bị ảnh hưởng cũng vậy. Hạ magiê máu, thường nghiêm trọng và khó điều trị, phổ biến ở những bệnh nhân bị suy cận giáp do APECED. Các biểu hiện khởi phát thường gặp nhất của APECED là nhiễm nấm candida niêm mạc da—60%, suy cận giáp—32%, và suy vỏ thượng thận—5%; bệnh lần đầu tiên trở nên rõ ràng trong độ tuổi từ 2 tháng đến 18 tuổi. Tuy nhiên, 10% bệnh nhân biểu hiện bằng một biểu hiện khác của APECED. Ngoài suy cận giáp và suy vỏ thượng thận, các bệnh nội tiết khác gặp trong APECED bao gồm viêm buồng trứng tự miễn dẫn đến suy buồng trứng (70%), viêm tinh hoàn dẫn đến suy tinh hoàn (30%), đái tháo đường (30%), viêm tuyến giáp (30%), và viêm tuyến yên (4%). Bên cạnh nhiễm nấm candida niêm mạc da, các biểu hiện và biến chứng da liễu của APECED trong đoàn hệ Phần Lan bao gồm rụng tóc (40%), bạch biến (30%), và phát ban kèm sốt (15%). Viêm kết giác mạc phát triển ở 20% các đối tượng bị ảnh hưởng, thiếu máu ác tính ở 30%, viêm gan ở 20%, và tiêu chảy mạn tính ở 20%. Trong một quần thể Na Uy gồm 36 bệnh nhân mắc APECED, 13 người có bằng chứng lâm sàng về bệnh ở hoặc trước 5 tuổi và thêm 15 đối tượng biểu hiện ở hoặc trước 15 tuổi. Trong một quần thể Nga gồm 46 bệnh nhân, nhiễm nấm candida niêm mạc da hiện diện ở 70%, suy cận giáp ở 83%, và suy thượng thận ở 54%. Các vấn đề phổ biến khác là rụng tóc (27%), rối loạn chức năng tuyến giáp (20%), và hội chứng kém hấp thu (18%). Các phát hiện bất thường trong đoàn hệ này là viêm võng mạc sắc tố và loạn sản hành xương—cả hai đều xảy ra ở 7% bệnh nhân. Các biểu hiện muộn hơn của APECED bao gồm ung thư biểu mô tế bào vảy ở thực quản và miệng, vô lách và viêm thận kẽ. Chẩn đoán APECED thường yêu cầu sự hiện diện của hai trong ba bệnh chính của nó (nhiễm nấm candida niêm mạc da, suy cận giáp, suy vỏ thượng thận), nhưng đôi khi nhiễm nấm candida mạn tính, suy cận giáp hoặc suy thượng thận nguyên phát đơn độc có thể là biểu hiện duy nhất của một đột biến bất hoạt trong AIRE.
APS1 là kết quả của các đột biến mất chức năng đồng hợp tử hoặc dị hợp tử phức hợp trong AIRE, một gen có 14 exon mã hóa một protein 545 aa với hai motif ngón tay kẽm được biểu hiện trong nhân của các tế bào biểu mô tủy tuyến ức và trong các hạch bạch huyết, lá lách và bạch cầu đơn nhân. AIRE cũng phục vụ như một E3 ubiquitin ligase, một thành phần thiết yếu của hệ thống ubiquitin-proteasome để sửa đổi và phá hủy protein liên quan đến sự phân chia và biệt hóa tế bào, vận chuyển protein và truyền tín hiệu nội bào. Về mặt cấu trúc, AIRE chứa hai homeodomain thực vật—một chuỗi các axit amin bao gồm một bộ tám cysteine và histidine phối hợp với hai ion kẽm; homeodomain thực vật đầu tiên là cần thiết cho hoạt động E3 ubiquitin ligase của protein và homeodomain thực vật thứ hai là cần thiết cho hành động điều hòa phiên mã của nó. AIRE cũng chứa một miền SAND, một cấu trúc cho phép nó liên kết với DNA. AIRE cho phép các tế bào tuyến ức (tế bào lympho T) phân biệt giữa các kháng nguyên bản thân bình thường và các protein lạ đối với vật chủ. AIRE làm như vậy bằng cách gây ra sự biểu hiện của các kháng nguyên bản thân bình thường trong tuyến ức và sau đó loại bỏ các tập hợp con dòng của các tế bào lympho T nhận ra chúng, do đó thiết lập sự dung nạp bản thân. Để phát triển một hệ thống miễn dịch có khả năng phân biệt giữa các kháng nguyên “bản thân” và “lạ”, các tế bào biểu mô tủy mô đệm tuyến ức phiên mã tại chỗ phần lớn các gen mã hóa protein được biểu hiện trong các mô ngoại vi khác nhau để cho phép các tế bào T đang phát triển nhận ra chúng trước khi các tế bào T này được giải phóng khỏi tuyến ức và do đó ngăn chặn phản ứng tự miễn khi các protein này được các tế bào T gặp ở ngoại vi. Gen điều hòa tự miễn AIRE cho phép phiên mã và biểu hiện của các gen này ở tuyến ức bằng cách tương tác với các chất vận chuyển nhân (ví dụ, exportin), các protein liên kết chromatin (ví dụ, histone), các yếu tố phiên mã và các quá trình phiên mã sau khởi đầu (ví dụ, CREB; phức hợp của protein kinase phụ thuộc DNA-polymerase–topoisomerase), và các yếu tố nối và xử lý tiền-sứ giả (m)RNA. AIRE không hoạt động như một yếu tố phiên mã cụ thể. Vì AIRE trong nhân liên kết với các đuôi không methyl hóa của histone-3 trong chromatin, người ta đã đề xuất rằng AIRE kích hoạt các gen im lặng bằng cách thu hút bộ máy phiên mã đến vị trí của chúng, cho phép bắt đầu phiên mã, kéo dài và xử lý tiền-mRNA. Trong các quần thể Phần Lan và Nga mắc APS1, đột biến mất chức năng phổ biến nhất trong AIRE là một đột biến cắt ngắn đồng hợp tử ở codon 257 (p.Arg257Ter). Các đột biến gây bệnh trong AIRE được phát hiện ở bệnh nhân mắc APECED bao gồm sai nghĩa, vô nghĩa (p.Arg139Ter), chèn, và mất đoạn (ví dụ, mất đoạn 13 cặp bazơ—964del13, NT 1094, exon 8) làm thay đổi sự phân bố dưới tế bào của AIRE và/hoặc làm giảm khả năng kích hoạt phiên mã và/hoặc hoạt động E3 ubiquitin ligase của nó. AIRE cũng kích thích sự phát triển của các tế bào T điều hòa FoxP3+ (Treg) của tuyến ức có khả năng ức chế các tế bào T tự phản ứng.
Nồng độ AIRE trong tuyến ức ở phụ nữ sau tuổi dậy thì đã được estrogen hóa bình thường thấp hơn so với nam giới sau tuổi dậy thì đã được nam hóa bình thường, có thể giải thích một phần cho sự nhạy cảm tăng lên của phụ nữ đối với sự phát triển muộn hơn của các rối loạn tự miễn. Mặc dù có sự biểu hiện của ba bản sao AIRE ở tuyến ức, các đối tượng mắc tam nhiễm sắc thể 21 (hội chứng Down) có nồng độ AIRE trong mô tuyến ức giảm, do đó có lẽ khiến quần thể bệnh nhân này có khuynh hướng phát triển các bệnh tự miễn. Khi AIRE bị bất hoạt bởi các đột biến mất chức năng hai alen (ví dụ, p.Arg257X, p.Cys322del13), sự tự nhận dạng của các tế bào T bị mất; các protein bản thân bình thường không thể phân biệt được với các protein lạ, và do đó một phản ứng (tự) miễn phá hủy đối với các kháng nguyên bản thân được khởi xướng. (Một số biến thể của AIRE được di truyền như các rối loạn trội trên NST thường hoặc trội âm tính.) Bệnh nhân có các đột biến của AIRE và hội chứng đa nội tiết tự miễn type 1 phát triển các kháng thể liên quan đến cơ quan đối với các protein tế bào chất (ví dụ, đối với NALRP5 đặc hiệu của tuyến cận giáp [protein chứa miền NACHT, lặp lại giàu leucine, miền pyrin 5, OMIM 609658] thường có thể chứng minh được ở những bệnh nhân bị suy cận giáp). Sự hiện diện của hội chứng đa nội tiết tự miễn type 1 được xác lập bằng các phát hiện lâm sàng, các bệnh nội tiết liên quan và các tự kháng thể có thể đo lường được đối với các mô nội tiết khác (ví dụ, glutamic acid decarboxylase 65 [đái tháo đường type 1], 21-hydroxylase [suy vỏ thượng thận]), và các mô không phải nội tiết (ví dụ, võng mạc) và được xác nhận bằng cách định kiểu gen của AIRE. Bệnh nhân mắc các bệnh nội tiết được cho là do hội chứng đa nội tiết tự miễn type 1 được điều trị thích hợp cho (các) thiếu hụt hormon hiện có, cũng như cho các biểu hiện khác của bệnh này. Bệnh nhân bị suy cận giáp cần bổ sung vitamin D (calcitriol), canxi và magiê; rhPTH 1-34 và PTH 1-84 đã được sử dụng ở người lớn để điều trị suy cận giáp (mặc dù không được Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ [FDA] chấp thuận để sử dụng ở trẻ em vì lo ngại về khả năng gây ung thư của chúng). Tất cả các họ hàng bậc một không có triệu chứng của bệnh nhân mắc hội chứng đa nội tiết tự miễn type 1 nên được khảo sát để tìm sự hiện diện của giai đoạn tiền triệu chứng của bệnh này.
Hội chứng đa nội tiết tự miễn type II là một rối loạn tự miễn đa gen biểu hiện bằng viêm thượng thận, viêm tuyến giáp và đái tháo đường type 1 (và ở một số bệnh nhân là thiếu máu ác tính, suy sinh dục tăng gonadotropin, viêm gan, bệnh celiac, nhược cơ, rụng tóc và bạch biến) và có liên quan đến các biến thể trong các phức hợp tương hợp mô chính, ví dụ, DR3-DQ2, DR4-DQ8, DRB104, DQB102, và các phức hợp khác. Suy cận giáp không xảy ra ở bệnh nhân mắc rối loạn này. Hội chứng Suy giảm miễn dịch, Đa nội tiết, và Bệnh ruột–Liên kết X (IPEX- OMIM 34790) có liên quan đến sự khởi phát sớm (sơ sinh và đầu thời thơ ấu) của bệnh ruột tự miễn, đái tháo đường phụ thuộc insulin, bệnh tuyến giáp tự miễn, chàm, viêm cầu thận, giảm tiểu cầu và thiếu máu tan máu chỉ xảy ra ở nam giới vì nó là kết quả của các đột biến mất chức năng trong FOXP3 (OMIM 300292). FOXP3 là một yếu tố phiên mã cần thiết cho sự phát triển bình thường của các tế bào T điều hòa tự nhiên duy trì sự dung nạp bản thân. PHP là một rối loạn với các biểu hiện lâm sàng đa dạng liên quan đến sự đề kháng với tác động của PTH mà các kiểu hình lâm sàng và các biến thể di truyền của nó đã được thảo luận trước đó.
Thiếu hụt lượng vitamin D ăn vào, chuyển hóa bất thường của cholecalciferol, hoặc giảm đáp ứng sinh học của VDR có thể dẫn đến hạ canxi máu. Ở đối tượng bị mất khoáng xương do thiếu vitamin D rõ rệt, nồng độ canxi huyết thanh có thể giảm đột ngột sau khi dùng ngay cả một lượng nhỏ vitamin D vì sự khoáng hóa trở lại của chất nền xương do hoạt động của nguyên bào xương tăng tốc tiêu thụ canxi và phốt phát (hội chứng “xương đói”). Sau khi cắt bỏ tuyến cận giáp vì cường cận giáp nguyên phát, nồng độ canxi thường giảm nhanh chóng theo cùng một cơ chế. Các loại thuốc ức chế bài tiết PTH (magiê quá mức), tái hấp thu xương của hủy cốt bào (bisphosphonat), hoặc tái hấp thu canxi ở thận (furosemide) có thể dẫn đến hạ canxi máu. Truyền tĩnh mạch hoặc dùng qua đường trực tràng phốt phát (trong thuốc xổ), sự phá hủy tế bào cấp tính do ly giải tế bào khối u hoặc tiêu cơ vân, và suy thận cấp và mạn tính làm tăng nồng độ phốt phát huyết thanh và dẫn đến sự giảm tương hỗ của các giá trị canxi. Nồng độ canxi huyết thanh giảm ở những bệnh nhân được truyền nhiều lần máu chứa citrate hoặc trong quá trình lọc huyết tương. Ở những đối tượng bị viêm tụy cấp, canxi tạo phức với các axit béo tự do được tạo ra bởi lipase tụy sẽ lắng đọng trong mô hoại tử. Bệnh nặng cấp tính do nhiều nguyên nhân gây bệnh khác nhau thường liên quan đến hạ canxi máu; điều này đã được cho là do giảm albumin máu, tăng phốt phát máu, nhiễm kiềm, suy cận giáp chức năng, tăng calcitonin máu, hạ magiê máu, thiếu vitamin D, giảm tổng hợp calcitriol, nhiễm kiềm và tăng nồng độ axit béo tự do trong huyết thanh (axit béo tự do làm tăng sự gắn kết của Ca2+ với albumin), tăng hoạt động của cytokine và các loại thuốc được sử dụng (ví dụ, aminoglycosid, máu chứa citrate, các chất chelat hóa canxi, florua).
Đánh giá
Hình 20.3 phác thảo việc đánh giá trẻ/vị thành niên bị hạ canxi máu. Hạ canxi máu có thể không có triệu chứng cho đến khi được phát hiện bằng một hồ sơ hóa học đa xét nghiệm được thực hiện cho một mục đích khác. Khoảng QT kéo dài được ghi nhận bằng điện tâm đồ được thực hiện để đánh giá tiếng thổi tim chức năng hoặc rối loạn nhịp tim gợi ý sự hiện diện của hạ canxi máu. X-quang được thực hiện để đánh giá đau bụng hoặc sau chấn thương đầu có thể cho thấy nhiễm canxi thận, sỏi thận hoặc vôi hóa hạch nền của não, tương ứng; những phát hiện như vậy cần đo nồng độ canxi huyết thanh. Các triệu chứng chính và dấu hiệu thực thể của hạ canxi máu phụ thuộc vào tốc độ khởi phát và mức độ của nó. Hạ canxi máu khởi phát càng nhanh và giá trị canxi đạt được càng thấp thì các triệu chứng càng đáng kể. Các triệu chứng có thể bao gồm dị cảm (tê và ngứa ran ở bàn tay, bàn chân, vùng quanh miệng), tăng kích thích cơ, bao gồm chuột rút, co cứng bàn tay-bàn chân (gấp khuỷu tay và cổ tay, khép ngón tay cái, gấp các khớp bàn-đốt ngón tay/bàn-đốt ngón chân, và duỗi các khớp liên đốt), co cứng—đặc biệt là trong khi tập thể dục gắng sức—đôi khi là co giật toàn thể, co thắt phế quản, và đôi khi là các triệu chứng tâm thần kinh. (Co thắt thanh quản là một biến chứng đe dọa tính mạng của hạ canxi máu.) Xem xét tiền sử bệnh và gia đình có thể tiết lộ các triệu chứng phù hợp với hoặc các bệnh liên quan đến hạ canxi máu (nhiễm trùng tái phát, dị tật tim bẩm sinh, các thủ thuật phẫu thuật ở cổ, xạ trị vùng cổ, các bệnh thâm nhiễm), hoặc có thể xác định các thành viên có liên quan bị suy cận giáp, các đặc điểm thể chất dị dạng, các bệnh nội tiết tự miễn hoặc hạ magiê máu.
Khám thực thể có thể cho thấy những bất thường đặc trưng ở trẻ em mắc PHP type IA có kiểu hình AHO (lùn, mặt tròn, các nốt cứng dưới da, tật ngắn xương bàn tay), hội chứng DiGeorge (khuôn mặt không điển hình, nhiễm trùng tái phát, tiếng thổi ở tim), hoặc APS1 có tính gia đình (nhiễm nấm candida niêm mạc da mạn tính và các bất thường ngoại bì khác, chẳng hạn như bạch biến, rụng tóc, viêm kết giác mạc), trong khi các biến dạng còi xương ở trẻ bị hạ canxi máu ngụ ý sự hiện diện của một dạng thiếu vitamin D. Tuy nhiên, phổ biến nhất, khám thực thể không cho thấy bất thường nổi bật nào ở trẻ bị hạ canxi máu ngoài những bất thường về tăng kích thích thần kinh cơ, chẳng hạn như tăng phản xạ, dấu hiệu Chvostek dương tính (co giật các cơ quanh miệng khi gõ nhẹ lên dây thần kinh sọ thứ bảy) hoặc dấu hiệu Trousseau (co cứng bàn tay-bàn chân khi duy trì băng đo huyết áp cao hơn huyết áp tâm thu 20 mm Hg trong 3 phút) và đôi khi là đục thủy tinh thể, phù gai thị hoặc răng bất thường. (Co cứng cũng xảy ra ở các đối tượng bị hạ và tăng natri máu, hạ và tăng kali máu, và hạ magiê máu, và dấu hiệu Chvostek dương tính có thể được tìm thấy ở nhiều thanh thiếu niên bình thường.)
Đánh giá xét nghiệm về hạ canxi máu bao gồm đo nồng độ canxi toàn phần, Ca2+, phốt phát, magiê, PTH, và 25-hydroxyvitamin D trong huyết thanh, và xác định bài tiết canxi qua nước tiểu. Ở hầu hết các bệnh nhân bị hạ canxi máu, bài tiết canxi qua nước tiểu thấp; tuy nhiên, nếu bài tiết canxi qua nước tiểu bình thường hoặc cao một cách không phù hợp, các rối loạn, chẳng hạn như hạ canxi máu di truyền trội trên NST thường type 1 (đột biến tăng chức năng trong CASR hoặc GNA11) có thể được xem xét; các gen này sau đó có thể được phân tích hoặc xác định các kháng thể đối với CaSR theo chỉ định lâm sàng. Các kháng thể đối với CaSR đã được phát hiện ở khoảng 50% đến 80% bệnh nhân mắc hội chứng đa nội tiết tự miễn type 1. Trẻ/vị thành niên bị hạ canxi máu, giảm canxi niệu, tăng phốt phát máu, và nồng độ PTH huyết thanh thấp hoặc không thể phát hiện được (và nồng độ magiê huyết thanh bình thường hoặc chỉ thấp nhẹ) có khả năng bị suy cận giáp do một khiếm khuyết nguyên phát trong tổng hợp hoặc bài tiết PTH liên quan đến dị tật bẩm sinh hoặc phá hủy mắc phải của các tuyến cận giáp. Bệnh nhân bị suy cận giáp thường có nồng độ calcitriol huyết thanh thấp, nồng độ calcidiol bình thường, giảm bài tiết cyclic AMP có nguồn gốc từ thận qua nước tiểu, và tăng tái hấp thu phốt phát ở ống thận. Hội chứng DiGeorge có thể được xác định bằng phân tích microarray hoặc FISH của nhiễm sắc thể 22q11.2 sử dụng các đầu dò hướng đến đoạn bị mất; đôi khi việc định kiểu gen TBX1 có thể hữu ích. Phân tích CASR, GNA11, PTH, GCM2, FHL1, và các gen khác liên quan đến suy cận giáp đơn độc hoặc hội chứng ( TCBE, GATA3 ) có thể được chỉ định trong bối cảnh lâm sàng thích hợp. Chẩn đoán APS1 dựa trên các phát hiện lâm sàng và xét nghiệm và định kiểu gen của AIRE. Sự hiện diện của hai trong ba biểu hiện chính của nó (nhiễm nấm candida và/hoặc loạn dưỡng ngoại bì, suy cận giáp, suy vỏ thượng thận) là các tiêu chí được chấp nhận cho chẩn đoán lâm sàng của nó, nhưng suy cận giáp đơn độc, suy thượng thận, hoặc nhiễm nấm candida niêm mạc mạn tính đôi khi có thể là biểu hiện duy nhất của nó. Các kháng thể đối với CaSR hoặc các thành phần tế bào khác của tuyến cận giáp, tuyến thượng thận (các enzyme phân cắt chuỗi bên, 21-hydroxylase, 17α-hydroxylase), các chất dẫn truyền thần kinh (aromatic L-amino acid decarboxylase, tryptophan hydroxylase), và IFN-α và -ω có thể được xác định; các kháng thể đối với IFN-ω thường hiện diện ở những bệnh nhân mắc APS1/APECED. Định kiểu gen của AIRE và xác định (các) đột biến xác nhận chẩn đoán APECED. Có sự thay đổi lớn trong biểu hiện lâm sàng của APECED cả giữa các gia đình và giữa các anh chị em vì kiểu hình không liên quan trực tiếp đến kiểu gen. Ở những bệnh nhân bị suy cận giáp tự phát, đơn độc, việc tìm kiếm các kháng thể đối với CaSR và/hoặc mô cận giáp là cần thiết. Ở các đối tượng hạ magiê máu, nồng độ magiê và PTH khá thấp, và bài tiết PTH tăng nhanh sau khi dùng magiê tiêm tĩnh mạch. Hạ magiê máu nguyên phát nên được xem xét khi hạ canxi máu và tăng canxi niệu trùng hợp và các giá trị PTH và magiê huyết thanh thấp; sau đó nên định lượng bài tiết magiê qua nước tiểu và định kiểu gen CLDN16 (OMIM 603959). Vì hạ magiê máu cũng có thể do một khiếm khuyết chọn lọc ở ruột non trong hấp thu magiê, các đột biến trong kênh cation, phân họ M, thành viên 6 của họ thụ thể tiềm năng thoáng qua (TRPM6, OMIM 607009) nên được kiểm tra khi cần thiết. Hạ magiê máu nguyên phát phải được phân biệt với hạ magiê máu do hội chứng Gittleman và Bartter (vide infra).
Nồng độ PTH huyết thanh tăng cao ở một đối tượng bị hạ canxi máu cho thấy bệnh nhân hoặc đang bài tiết một phân tử PTH bất thường hoặc kháng PTH hoặc có một phản ứng bài tiết PTH bù trừ (thứ phát) đối với hạ canxi máu (xem Hình 20.3). Việc mô tả hóa lý của phân tử PTH và phân tích PTH (OMIM 168450) cho phép xác định sự bất thường trong tổng hợp, xử lý sau dịch mã, bài tiết hoặc hoạt động của PTH dẫn đến tình trạng suy cận giáp chức năng. Phân tích GNAS (OMIM 139320) và kiểu hình in dấu của nó cho phép xác định khiếm khuyết di truyền cụ thể ở phần lớn bệnh nhân mắc PHP lâm sàng type IA và PPHP. Khả năng đáp ứng của xương và thận đối với PTH có thể được đánh giá nếu cần bằng cách đo những thay đổi về nồng độ canxi, Ca2+, phốt phát, cyclic AMP và calcitriol trong huyết thanh và bài tiết cyclic AMP có nguồn gốc từ thận và phốt phát qua nước tiểu sau khi dùng PTH 1-34 sinh tổng hợp (nghiệm pháp Elsworth-Howard). Ở đối tượng bình thường và ở bệnh nhân bị suy cận giáp nguyên phát, bài tiết cyclic AMP qua nước tiểu tăng 10 đến 20 lần và bài tiết phốt phát tăng vài lần; ở bệnh nhân mắc PHP type IA và IB, có sự gia tăng dưới ba lần trong bài tiết cyclic AMP sau khi dùng PTH 1-34. Chẩn đoán PHP type IB có thể được xác lập bằng cách kiểm tra các kiểu hình in dấu của GNAS (tức là, trạng thái methyl hóa của các vùng methyl hóa khác biệt của GNAS) và phân tích STX16 (OMIM 603666) và các chuỗi 5′ của GNAS theo chỉ định. Loạn sản đầu xương 1 và 2 (OMIM 101800; 614613) là các loạn sản xương có đề kháng hormon cũng nên được xem xét khi đánh giá một trẻ sơ sinh bị hạ canxi máu với nồng độ PTH huyết thanh tăng cao một cách không phù hợp. Bệnh loạn sản sụn này được cho là do các biến thể bất hoạt của PRKAR1A (OMIM 188830) hoặc PDE4D (OMIM 600129). PRKAR1A mã hóa một thành phần của PKA—chất trung gian chính của truyền tín hiệu nội bào sau thụ thể; PDE4D mã hóa một phosphodiesterase đặc hiệu cyclic AMP, sự mất mát của nó làm thay đổi chức năng cyclic AMP. Ở trẻ bị thiếu vitamin D, nồng độ calcidiol huyết thanh thấp. Ở những bệnh nhân bị giảm hoạt động 25OHD3-1α-hydroxylase ở thận, nồng độ calcidiol huyết thanh bình thường trong khi nồng độ calcitriol thấp một cách không phù hợp. Nồng độ calcitriol tăng cao cho thấy sự hiện diện của một khiếm khuyết trong VDR hạt nhân. Bệnh nhân bị suy thận được nhận biết bằng giá trị creatinin huyết thanh tăng. Các phát hiện khác ở các đối tượng bị hạ canxi máu bao gồm kéo dài khoảng QT bằng điện tâm đồ và vôi hóa hạch nền bằng chụp cắt lớp vi tính sọ não.
Quản lý
Mục tiêu chính trong việc chăm sóc trẻ em và vị thành niên bị hạ canxi máu là tăng nồng độ canxi huyết thanh lên mức mà bệnh nhân không có triệu chứng và càng gần giới hạn dưới của mức bình thường càng tốt mà không gây tăng canxi máu; mục tiêu thứ hai là xác định nguyên nhân của hạ canxi máu càng nhanh càng tốt để cung cấp quản lý đặc hiệu cho bệnh. Các mục tiêu chung của việc quản lý hạ canxi máu nói chung và suy cận giáp nói riêng là: (1) cải thiện các dấu hiệu và triệu chứng của hạ canxi máu, (2) ngăn ngừa tăng canxi máu bằng cách (3) duy trì nồng độ canxi huyết thanh ở hoặc hơi dưới giới hạn dưới của mức bình thường để (4) duy trì tích số canxi × phốt phát dưới 55 mg/dL và do đó (5) ngăn ngừa tăng canxi niệu và (6) lắng đọng canxi/phốt phát và vôi hóa ngoài xương.
Hạ canxi máu không triệu chứng (canxi toàn phần > 7,5 mg/dL = 1,88 mmol/L) có thể không cần can thiệp ngay lập tức. Với nồng độ canxi huyết thanh thấp hơn hoặc khi hạ canxi máu có triệu chứng (co cứng, co giật, co thắt thanh quản, co thắt phế quản), quản lý cấp tính có thể yêu cầu dùng canxi gluconat 10% tiêm tĩnh mạch (93 mg canxi nguyên tố/lọ 10 mL) với tốc độ chậm (không > 2 mL [1,86 mg canxi nguyên tố]/kg trong 10 phút) trong khi theo dõi chặt chẽ nhịp tim (và khoảng QT trên điện tâm đồ). Về mặt cấp tính, việc dùng canxi tiêm tĩnh mạch nhằm mục đích cải thiện các hậu quả nghiêm trọng hơn của hạ canxi máu, chẳng hạn như co giật, co thắt phế quản hoặc co thắt thanh quản, chứ không phải để phục hồi và duy trì tình trạng canxi máu bình thường. Tiêm tĩnh mạch, không nên dùng canxi cùng với phốt phát hoặc bicarbonate, vì các muối này có thể đồng kết tủa. Cần tránh thoát mạch canxi ra ngoài mạch máu vì nó có thể kết tủa và gây tổn thương mô tại chỗ. Sau khi các triệu chứng cấp tính đã được giải quyết, canxi gluconat (10 mL = 93 mg canxi nguyên tố trong 100 mL dextrose 5%/nước muối sinh lý 0,25) có thể được truyền tĩnh mạch với tốc độ đủ để duy trì nồng độ canxi trong khoảng thấp-bình thường không triệu chứng trong khi nguyên nhân của hạ canxi máu được xác định và liệu pháp cụ thể hơn cho hạ canxi máu dai dẳng được kê đơn. Ở trẻ bị tăng phốt phát máu rõ rệt là nguyên nhân gây hạ canxi máu, ngoài việc dùng canxi qua đường tiêm, cần truyền nước muối sinh lý đủ để duy trì lượng nước tiểu ở hoặc trên 2 mL/kg mỗi giờ. Việc đo thường xuyên nồng độ canxi và phốt phát huyết thanh cho phép điều chỉnh nhanh chóng liệu pháp dịch và điện giải. Việc đánh giá nên được tiến hành càng nhanh càng tốt và liệu pháp đường uống được bắt đầu một cách hợp lý và nhanh chóng.
Sau khi ổn định, bệnh nhân bị suy cận giáp hoặc PHP có thể được điều trị bằng calcitriol (20–60 ng/kg/ngày chia làm 2 liều bằng nhau) và bổ sung canxi (canxi glubionate, canxi citrate, canxi carbonat), 30 đến 75 mg canxi nguyên tố/kg/ngày chia làm 4 liều hàng ngày thường dùng trong bữa ăn—(xem Bảng 20.3) để phục hồi và duy trì tình trạng canxi máu bình thường. Nồng độ canxi huyết thanh nên được duy trì trong khoảng thấp-bình thường. Mỗi bệnh nhân phải được theo dõi cẩn thận để tránh tăng canxi máu, tăng canxi niệu (bài tiết canxi lớn hơn 4 mg/kg/24 giờ), nhiễm canxi thận và sỏi thận. Nên đo định kỳ nồng độ canxi, phốt phát, magiê và creatinin trong huyết thanh và bài tiết canxi và creatinin qua nước tiểu (mỗi 3 tháng). Siêu âm thận hai lần mỗi năm cũng được đề xuất để xác định sự phát triển của nhiễm canxi thận ở giai đoạn sớm nhất. Trẻ em bị hạ canxi máu di truyền trội trên NST thường do các đột biến tăng chức năng trong CASR (OMIM 601199) hoặc GNA11 (OMIM 139320) cực kỳ nhạy cảm với vitamin D và các chất chuyển hóa của nó. Ngay cả những liều nhỏ calcitriol cũng có thể dẫn đến tăng canxi niệu với sự gia tăng tối thiểu nồng độ canxi huyết thanh; trong trường hợp đó, việc bổ sung hydrochlorothiazide (0,5–2,0 mg/kg/ngày) có thể làm tăng tái hấp thu canxi ở ống thận và giảm yêu cầu calcitriol. Việc sử dụng rhPTH 1-34 đã có lợi ở các đối tượng cá nhân bị suy cận giáp. Ở người lớn và trẻ em bị suy cận giáp do nhiều nguyên nhân khác nhau, việc sử dụng rhPTH 1-34 (0,4–0,5 mcg/kg mỗi 12 giờ tiêm dưới da) cùng với canxi carbonat và vitamin D (1200 mg/ngày canxi nguyên tố và 800 IU/ngày cholecalciferol chia làm 4 liều bằng nhau) đã được chứng minh là hiệu quả và an toàn trong thời gian tới 3 năm. rhPTH 1-34 dẫn đến tăng tốc độ chu chuyển xương được phản ánh qua sự gia tăng phosphatase kiềm và osteocalcin huyết thanh và bài tiết pyridinoline và deoxypyridinoline qua nước tiểu, trong khi sự tích lũy khối lượng khoáng xương, tăng trưởng tuyến tính và tăng cân không bị ảnh hưởng xấu ở trẻ được điều trị. Ở sáu trẻ em bị suy cận giáp nguyên phát, việc tiêm dưới da rhPTH 1-34 (12,5 mcg hai lần mỗi ngày) được dung nạp tốt và cho phép ngừng hoặc giảm liều canxi và calcitriol bổ sung. Truyền liên tục rhPTH 1-34 cũng đã được chứng minh là hiệu quả trong việc duy trì nồng độ canxi huyết thanh hợp lý ở người lớn bị suy cận giáp sau phẫu thuật với ít biến động hơn về nồng độ canxi và giá trị canxi trong nước tiểu thấp hơn khi so sánh với tiêm dưới da rhPTH 1-34 hai lần mỗi ngày. Tuy nhiên, hiện tại rhPTH 1-34 không được chỉ định để điều trị thường quy cho trẻ em bị suy cận giáp vì những lo ngại về mối liên hệ của nó với sarcoma xương. Ở người lớn, rhPTH 1-84 cũng đã được sử dụng trong điều trị suy cận giáp với sự suy giảm đáng kể liều canxi và calcitriol bổ sung và sự ổn định của các giá trị canxi huyết thanh và nước tiểu trong hơn 6 năm. Ngoài ra, các dấu hiệu chu chuyển xương phản ánh sự gia tăng hình thành xương được ghi nhận bằng cách đo mật độ khoáng của xương (BMD) của cột sống thắt lưng. Mối quan hệ của rhPTH 1-34 với sự hình thành khối u xương, nếu có, hiện chưa được biết.
Khi tiến hành cắt bỏ toàn bộ tuyến giáp để điều trị ung thư tuyến giáp ác tính, hãy xem xét các tác động có thể có của thủ thuật này đối với chức năng của các tuyến cận giáp. Trước khi phẫu thuật, nên ghi lại nồng độ canxi, magiê, phốt phát và PTH trong huyết thanh. Trong quá trình phẫu thuật, nên nỗ lực xác định và bảo tồn các tuyến cận giáp và nguồn cung cấp máu của chúng. Khám thực thể tìm kiếm các dấu hiệu hạ canxi máu (Chvostek) và đo nối tiếp canxi, magiê và PTH nên bắt đầu ngay sau khi hoàn thành phẫu thuật và tiếp tục theo chỉ định lâm sàng.
Vì PHP type IA có liên quan đến sự đề kháng với một số hormon peptide hoạt động thông qua GPCR, việc đánh giá định kỳ chức năng tuyến yên-tuyến giáp và tuyến yên-buồng trứng và bài tiết GH là cần thiết và liệu pháp thay thế hormon được bắt đầu theo chỉ định. Nói chung, tầm vóc thấp của PHP type IA phản ánh kiểu hình AHO chứ không phải thiếu GH. Tuy nhiên, tình trạng giảm hormon tăng trưởng liên quan đến PHP có thể đáp ứng với GH người tái tổ hợp ngoại sinh. Suy cận giáp thoáng qua ở trẻ sơ sinh có thể là biểu hiện ban đầu của suy cận giáp khởi phát muộn hơn; do đó, hãy đánh giá cân bằng nội môi canxi ở những đối tượng như vậy trong suốt thời thơ ấu. Bệnh nhân bị suy cận giáp đơn độc rõ ràng không rõ nguyên nhân nên được đánh giá lại định kỳ để xác định sự phát triển của các rối loạn tự miễn ở bệnh nhân hoặc gia đình. Đánh giá chức năng tuyến ức rất quan trọng ở những đối tượng có những phát hiện gợi ý hội chứng DiGeorge. Việc quản lý bệnh nhân bị thiếu hoặc kháng vitamin D được thảo luận trong các phần tiếp theo.
Khi hạ magiê máu có triệu chứng, có thể cần dùng magiê sulfat qua đường tiêm (dung dịch 50%, 0,1–0,2 mL/kg tiêm bắp, lặp lại sau 12–24 giờ nếu cần). Bệnh nhân bị hạ magiê máu nguyên phát có thể cần các liều magiê sulfat hàng ngày qua đường tiêm (tiêm bắp, tiêm tĩnh mạch) để ngăn ngừa co cứng, co giật và các triệu chứng thần kinh khác (nói lắp, múa giật-múa vờn, yếu cơ), và để cho phép tăng trưởng và phát triển bình thường. Calcitriol đơn độc làm tăng nồng độ canxi huyết thanh ở các đối tượng hạ magiê máu nhưng không hiệu quả trong việc ngăn ngừa co cứng. Truyền magiê qua sonde dạ dày liên tục qua đêm có thể giúp giảm bớt các tác dụng phụ đường tiêu hóa của nhiều liều magiê uống lớn. Các dạng hạ magiê máu nhẹ hơn và thoáng qua có thể được điều trị bằng magiê gluconat hoặc magiê citrate tribasic uống (xem Bảng 20.3).
Tăng canxi máu
Tăng canxi máu hiện diện khi nồng độ canxi ion hóa trong huyết thanh cao hơn giới hạn bình thường theo tuổi. Nồng độ canxi toàn phần trong huyết thanh phản ánh cả nồng độ protein huyết thanh và pH huyết thanh; nhìn chung, cả nồng độ canxi toàn phần và canxi ion hóa đều nên được đo lường thường quy khi nghi ngờ có tăng canxi máu. Tăng canxi máu có thể được coi là nhẹ khi nồng độ canxi toàn phần trong huyết thanh cao hơn giới hạn trên của mức bình thường và dưới 12 mg/dL (3,0 mmol/L), trung bình với các giá trị canxi toàn phần từ 12 đến 14 mg/dL (3,00–3,50 mmol/L), hoặc nặng khi giá trị canxi toàn phần trong huyết thanh vượt quá 14 mg/dL (3,50 mmol/L). Về mặt sinh lý bệnh, tăng canxi máu có thể là kết quả của việc hấp thu quá mức canxi ăn vào từ ruột (ví dụ, thừa vitamin D), tăng tái hấp thu canxi xương do tăng cường hoạt động của PTH hoặc tổn thương tiêu xương, hoặc tăng tái hấp thu canxi ở ống thận do thuốc, chẳng hạn như thuốc lợi tiểu thiazide. Do đó, tăng canxi máu có thể do các sai lệch phụ thuộc hoặc không phụ thuộc vào PTH.
Tăng canxi máu ở Trẻ sơ sinh và Trẻ nhỏ
Tăng canxi máu ở trẻ sơ sinh và trẻ rất nhỏ được định nghĩa là nồng độ canxi toàn phần trong máu trên 10,8 đến 11,3 mg/dL (2,70–2,83 mmol/L) và Ca2+ lớn hơn 5,92 đến 6,40 mg/dL (1,48–1,60 mmol/L) (tùy thuộc vào phòng xét nghiệm phân tích). Tuy nhiên, các triệu chứng và dấu hiệu đáng kể của tăng canxi máu (ví dụ, lơ mơ, chán ăn, nôn ói, táo bón, đa niệu, mất nước, trào ngược dạ dày thực quản và nôn ói, táo bón, và lơ mơ và giảm trương lực cơ hoặc kích thích và co giật) có thể không xảy ra cho đến khi nồng độ canxi toàn phần trong huyết thanh vượt quá 12,5 đến 13 mg/dL (3,12–4,25 mmol/L). Trẻ sơ sinh bị tăng canxi máu thường bị đa niệu do thận đề kháng với hormon chống bài niệu và có thể bị mất nước nếu lượng dịch nạp vào bị hạn chế. Do tác dụng co mạch của canxi, trẻ sơ sinh bị tăng canxi máu có thể bị tăng huyết áp. Tăng canxi máu cũng làm ngắn đoạn S-T và có thể dẫn đến block tim và cuối cùng là vô tâm thu. Ở trẻ lớn hơn và trẻ nhỏ bị tăng canxi máu mạn tính, tăng trưởng kém và “chậm phát triển” thường là những biểu hiện khởi phát. Tăng canxi máu cũng dẫn đến tăng canxi niệu, nhiễm canxi thận và sỏi thận. Tăng canxi máu cũng có thể hiện diện ở những bệnh nhân có các bất thường về kiểu hình, ví dụ, hội chứng Williams-Beuren (WBS).
Nguyên nhân
Các nguyên nhân gây tăng canxi máu ở trẻ sơ sinh và trẻ nhỏ được liệt kê trong Bảng 20.4A, 20.4B. Tăng canxi máu ở trẻ sơ sinh/trẻ nhỏ có thể do nguyên nhân y tế—ví dụ, dùng quá nhiều canxi hoặc vitamin D—đôi khi cho người mẹ tiết cholecalciferol trong sữa mẹ; dùng thuốc lợi tiểu thiazide làm tăng tái hấp thu canxi ở ống thận; hoặc hạn chế phốt phát có chủ đích. Tăng canxi máu, hạ phốt phát máu, tăng phosphatase máu và bằng chứng X-quang về bệnh còi xương có thể phát triển ở trẻ sinh non đang được nuôi dưỡng qua đường tĩnh mạch thiếu phốt phát hoặc ở những trẻ chỉ bú sữa mẹ, có hàm lượng phốt phát thấp. Hạ phốt phát máu dẫn đến tăng tổng hợp calcitriol bằng cách: (1) kích thích trực tiếp hoạt động của 25OHD-1α hydroxylase ở ống thận, và (2) ức chế tổng hợp FGF23, do đó loại bỏ một tác nhân làm giảm hoạt động của 25OHD-1α hydroxylase; hai con đường này cộng gộp làm tăng hoạt động của 25OHD-1α hydroxylase, tổng hợp calcitriol và hấp thu canxi ở ruột. Vấn đề này có thể được khắc phục bằng cách tăng lượng phốt phát tiêm truyền ở mức có thể hoặc bằng cách sử dụng sữa mẹ được tăng cường phốt phát. (Để khoáng hóa xương ngoài tử cung đầy đủ cho trẻ sinh non, cần nạp cả canxi và phốt phát khoảng 200 mg/kg/ngày mỗi loại.) Oxy hóa qua màng ngoài cơ thể cũng có thể liên quan đến tăng canxi máu ở trẻ sơ sinh, có lẽ liên quan đến việc tăng tiết PTH ở trẻ.
Bảng 20.4A: Các nguyên nhân gây Tăng canxi máu
| I | Trẻ sơ sinh/Trẻ nhỏ | |||
|---|---|---|---|---|
| A | Rối loạn từ mẹ | 1 | ||
| B | Trẻ sơ sinh/trẻ nhỏ | 1 | ||
| 2 | ||||
| 3 | ||||
| 4 | ||||
| 5 | ||||
| 6 | ||||
| 7 | ||||
| 8 | ||||
| 9 | ||||
| 10 | ||||
| 11 | ||||
| 12 | ||||
| 13 | ||||
| 14 | ||||
| 15 | ||||
| 16 | ||||
| 17 | ||||
| 18 | ||||
| II | Cường cận giáp | |||
| A | Lẻ tẻ | 1 | ||
| B | Có tính gia đình | 1 | ||
| 2 | ||||
| 3 | ||||
| 4 | ||||
| 5 | ||||
| 6 | ||||
| 7 | ||||
| 8 | ||||
| 9 | ||||
| 10 | ||||
| 11 | ||||
| 12 | ||||
| C | Thứ phát/Tam phát | 1 | ||
| 2 | ||||
| D | Tăng canxi máu do ác tính | 1 | ||
| 2 | ||||
| III | Tăng canxi máu giảm canxi niệu có tính gia đình | |||
| A | Tăng canxi máu giảm canxi niệu có tính gia đình I (CASR) | 1 | ||
| 2 | ||||
| 3 | ||||
| B | Tăng canxi máu giảm canxi niệu có tính gia đình II—Liên kết với nhiễm sắc thể 19p13.3 | |||
| C | Tăng canxi máu giảm canxi niệu có tính gia đình III—Biến thể Oklahoma—Liên kết với nhiễm sắc thể 19q13 | |||
| D | Tự kháng thể khóa CaSR | |||
| IV | Thừa Canxi và/hoặc Vitamin D | |||
| A | Hội chứng sữa-kiềm | |||
| B | Uống canxi hoặc vitamin D ngoại sinh hoặc bôi vitamin D tại chỗ (calcitriol hoặc chất tương tự) | |||
| C | Sản xuất calcitriol lạc chỗ liên quan đến các bệnh u hạt: sarcoidosis, bệnh mèo cào; lao, histoplasmosis, coccidioidomycosis, bệnh phong; virus gây suy giảm miễn dịch ở người; cytomegalovirus; bệnh viêm ruột mạn tính | |||
| D | Tân sinh | 1 | ||
| 2 | ||||
| 3 | ||||
| 4 | ||||
| E | Hội chứng Williams-Beuren (mất đoạn 7q11.23) | |||
| V | Bất động | |||
| VI | Các nguyên nhân khác | |||
| A | Thuốc—thiazide, lithium, vitamin A & các chất tương tự, canxi, kiềm, chất kháng estrogen, aminophylline | |||
| B | Nuôi dưỡng qua đường tĩnh mạch toàn phần | |||
| C | Bệnh nội tiết: cường giáp, suy vỏ thượng thận, u tủy thượng thận | |||
| D | Khối u tiết polypeptide vận mạch đường ruột | |||
| E | Suy thận cấp hoặc mạn tính/dùng nhôm | |||
| F | Giảm phosphatase máu | |||
| G | Viêm khớp dạng thấp vị thành niên – qua trung gian cytokine |
CaSR, Thụ thể cảm nhận canxi; PTHrP, protein liên quan đến hormon cận giáp.
(Từ Lietman SA, Germain-Lee EL, Levine MA. Hypercalcemia in children and adolescents. Curr Opin Pediatr. 2010;22:508–515; Davies JH. A practical approach to the problems of hypercalcaemia. In: Allgrove J, Shaw N. (eds). Calcium and Bone Disorders in Children and Adolescents. Basel, Karger, Endocr Dev. 2009;16:93–114; Benjamin RW, Moats-Staats BM, Calikoglu A, et al. Hypercalcemia in children. Pediatr Endocrinol Rev. 2008;5:778–784.)
Bảng 20.4B: Các biến thể di truyền liên quan đến Hạ canxi máu/Cường cận giáp
| Gen Protein | NST | OMIM | Rối loạn OMIM | Biểu hiện Lâm sàng/Hóa sinh | Chức năng Gen/Di truyền |
|---|---|---|---|---|---|
| CASR Thụ thể cảm nhận canxi | 3q13.3-21.1 | 601199 | Tăng canxi máu giảm canxi niệu có tính gia đình (FHH) type I, 145980; Cường cận giáp nặng ở trẻ sơ sinh (NSHPT) 239200, Cường cận giáp nguyên phát khởi phát ở người lớn (AOPH) | Tăng canxi máu, NSHPT—đe dọa tính mạng AOPH—sỏi thận, khoáng hóa xương thấp | Thụ thể canxi điều hòa tổng hợp, bài tiết PTH, biến thể bất hoạt FHH1—AD NSHPT—AD/AR AOPH—AD/AR |
| GNA11 Protein liên kết Guanin nucleotid, alpha-11 | 19p13.3 | 139313 | Tăng canxi máu giảm canxi niệu có tính gia đình type II, 145981 | Tăng canxi máu | Mã hóa G-protein truyền tín hiệu của CaSR đến các con đường truyền tín hiệu nội bào, biến thể bất hoạt, AD |
| AP2S1 Phức hợp protein liên quan đến Adaptor 2, tiểu đơn vị sigma1 | 19q13.31 | 602242 | Tăng canxi máu giảm canxi niệu có tính gia đình type III, 600740 | Tăng canxi máu, khoáng hóa xương thấp, suy giảm nhận thức | Phức hợp protein liên quan đến adaptor 2 tương tác với β-arrestin (ARRB1, chr. 11q13.4, OMIM 107940) để tạo thành phức hợp tạo điều kiện cho sự di chuyển ngược (nhập bào) của CaSR từ bề mặt tế bào và sự trở lại của nó vào tế bào chất, biến thể bất hoạt AD |
| CYP24A1 Cytochrome P450, họ 24, phân họ A, polypeptide 1 | 20q13.2 | 143880 | Tăng canxi máu ở trẻ nhỏ 1 143880 | Tăng canxi máu, nôn ói, chán ăn, suy kiệt, nhiễm canxi thận với nồng độ calcitriol bình thường/cao không phù hợp | Mã hóa 1,25-dihydroxyvitamin D-24-hydroxylase bất hoạt calcitriol, AR |
| SLC34A1 Họ chất mang hòa tan 34 (Chất đồng vận chuyển natri phốt phát type II), thành viên 1 | 5q35 | 182309 | Tăng canxi máu ở trẻ nhỏ 2 616963 | Tăng canxi máu, nôn ói, chán ăn, suy kiệt, nhiễm canxi thận | Mã hóa một chất đồng vận chuyển natri phốt phát ở ống lượn gần, AR |
| TRPV6 Thụ thể tiềm năng thoáng qua, kênh cation, phân họ V, thành viên 6 | 7q34 | 606680 | Cường cận giáp sơ sinh thoáng qua 618188 | Mất khoáng xương trong tử cung & sau sinh, canxi máu bình thường hoặc hạ canxi máu | Mã hóa một kênh chọn lọc canxi hoạt động trong nguyên bào nuôi của nhau thai, ống thận và đường ruột, AR |
| Hội chứng mất đoạn 7q11.23 | 194050 | Hội chứng Williams-Beuren: Tăng canxi máu ở trẻ nhỏ | Tăng canxi máu, hẹp động mạch chủ trên van, hẹp động mạch phổi ngoại biên, khuôn mặt đặc trưng, chậm phát triển, tính cách thân thiện, kỹ năng ngôn ngữ & âm nhạc | Mất đoạn của 28 (±) gen liền kề bao gồm ELN | |
| MEN1 Gen Men1 | 11q13.1 | 613733 | Đa u tuyến nội tiết, type 1 131100 | Các khối u của tuyến cận giáp, tiểu đảo tụy, tế bào nội tiết ruột, tuyến yên trước | Gen ức chế khối u, protein giàn giáo nhân tương tác với một số yếu tố phiên mã, biến thể bất hoạt AD |
| RET Tiền-oncogene tái sắp xếp trong quá trình chuyển nhiễm | 10q11.21 | 164761 | Đa u tuyến nội tiết, type 2A 171400; type 2B 162300 | Ung thư biểu mô tuyến giáp thể tủy (2A, 2B), u tủy thượng thận (2A, 2B), u tuyến cận giáp (2A), thể tạng Marfanoid/u hạch thần kinh (2B) | Thụ thể tyrosine kinase xuyên màng, biến thể hoạt hóa, AD |
| CDKN1B Chất ức chế kinase phụ thuộc cyclin 1B | 12p13.1 | 600778 | Đa u tuyến nội tiết, type 4 610755 | U tuyến cận giáp, carcinoid dạ dày, u tuyến yên | Chặn chu kỳ tế bào ở pha G0/G1, điều hòa sự di chuyển của tế bào, apoptosis, biến thể bất hoạt AD |
| CDC73 Protein chu kỳ phân chia tế bào 73, S cervisiae, tương đồng của | 1q31.2 | 607391 | Cường cận giáp đơn độc có tính gia đình-type 1 (145000) & type 2 (145001—liên quan đến hội chứng cường cận giáp—u xương hàm) | U tuyến/ung thư cận giáp, u xơ hóa xương hàm trên & hàm dưới, u Wilms | Parafibromin—protein nội tại của chu kỳ phân chia tế bào; gen ức chế khối u, biến thể bất hoạt, AD |
| GCM2 Glial cells missing, drosophila, tương đồng của, 2 | 6p24.2 | 603716 | Cường cận giáp đơn độc có tính gia đình-type 4 617343 | Cường cận giáp đơn độc có tính gia đình với nhiều u tuyến cận giáp, ung thư | Mã hóa yếu tố phiên mã cần thiết cho sự biệt hóa của tuyến cận giáp, biến thể hoạt hóa AD |
| PTH1R Thụ thể hormon cận giáp 1 | 3p21.31 | 168468 | Loạn sản sụn đầu xương, type Murk-Jansen 156400 | Chậm tăng trưởng rõ rệt, chân vòng kiềng, hàm dưới nhỏ, tăng canxi máu, hạ phốt phát máu | Thụ thể ghép cặp protein G cho PTH & PTHrP, biến thể hoạt hóa, AD |
AD, Di truyền trội trên nhiễm sắc thể thường; AR, di truyền lặn trên nhiễm sắc thể thường; PTH, hormon cận giáp; PTHrP, protein liên quan đến hormon cận giáp.
(Từ Stokes VJ, Nielsen MF, Hannan FM, Thakker RV. Hypercalcemic disorders in children. J Bone Mineral Res. 2017;32:2157–2170; Hannan FM, Babinsky VN, Thakker RV. Disorders of the calcium-sensing receptor and partner proteins: insights into the molecular basis of calcium homeostasis. J Mol Endocrinol. 2016;57:R127–142; Bilezikian JP, Bandeira L, Khan A, Cusano NE. Hyperparathyroidism. Lancet. 2018;391:168–178; Marini F, Cianferotti L, Giusti F, Brandi ML. Molecular genetics in primary hyperparathyroidism: the role of genetic tests in differential diagnosis, disease prevention strategy, and therapeutic planning. A 2017 update. Clin Cases Miner Bone Metab. 2017;14:60–70; Suzuki Y, Chitayat D, Sawada H, et al. (2018). TRPV6 variants interfere with maternal-fetal calcium transport through the placental and cause transient neonatal hyperparathyroidism. Am J Human Genet. 2018;102: 1104–1114.)
Tăng vitamin D máu có thể do cho ăn kéo dài một loại sữa công thức được pha chế không đúng cách hoặc sữa bò thương mại chứa quá nhiều vitamin D, kê đơn vitamin D, calcidiol hoặc calcitriol do y tế, hoặc tăng sản xuất calcitriol nội sinh từ các vị trí viêm. Ở trẻ sơ sinh bị chấn thương khi sinh nặng, ngạt chu sinh hoặc hạ thân nhiệt, hoại tử mỡ dưới da có thể phát triển trong vòng 6 tuần đầu sau sinh, biểu hiện bằng các nốt cứng, chắc, đau, màu tím trên má, thân mình, vai, mông, tay và chân. Tăng canxi máu có thể hiện diện khi các tổn thương xuất hiện lần đầu hoặc phát triển khi các nốt này thoái lui vài tuần sau đó. Về mặt mô học, các tổn thương da bao gồm các tế bào mỡ, thâm nhiễm lympho-mô bào viêm, và các tế bào khổng lồ đa nhân trong một nền tinh thể canxi. Tăng canxi máu của hoại tử mỡ dưới da đã được cho là do tái hấp thu canxi kết tủa, tổng hợp calcitriol ngoài thận bởi các đại thực bào tại chỗ và hậu quả là tăng hấp thu canxi ăn vào, và tăng hoạt động của hủy cốt bào do tăng tiết prostaglandin. Hoạt động 25OHD-1α-hydroxylase của các đại thực bào viêm này không nằm dưới sự kiểm soát của PTH, canxi hoặc phốt phát, nhưng có thể bị ức chế bởi glucocorticoid. Việc sản xuất quá mức prostaglandin E và interleukin (IL)-1 và -6 góp phần thêm vào tình trạng tăng canxi máu trong rối loạn này bằng cách tăng tốc độ chu chuyển xương. Tăng canxi máu do hoại tử mỡ dưới da được quản lý bằng cách cho trẻ uống sữa công thức ít canxi, tránh vitamin D, và truyền dịch, calcitonin hoặc bisphosphonate (pamidronate), khi cần thiết. Nên tránh dùng thuốc lợi tiểu quai (ví dụ, furosemide) và glucocorticoid nếu có thể. Tăng canxi máu do hoại tử mỡ dưới da cũng đã được quan sát thấy ở trẻ lớn bị chấn thương nặng hoặc thủy đậu lan tỏa. Thiếu hụt lactase và các disaccharidase khác bẩm sinh cũng đã được liên kết với tăng canxi máu ở trẻ nhỏ, có khả năng là do tăng hấp thu canxi ở ruột được thúc đẩy bởi các disaccharide.
Cường cận giáp nặng ở trẻ sơ sinh (NSHPT) là một dạng có khả năng gây tử vong của tăng canxi máu giảm canxi niệu có tính gia đình (di truyền) (HHC). Nó thường do các đột biến bất hoạt đồng hợp tử hoặc dị hợp tử phức hợp của CASR làm tăng đáng kể nồng độ Ca2+ huyết thanh cần thiết để ức chế tổng hợp và bài tiết PTH. Tuy nhiên, ở một số trẻ sơ sinh mắc NSHPT, đã có các đột biến bất hoạt dị hợp tử của CASR (ví dụ, pArg185Gln, p.Arg227Leu) cho thấy các sản phẩm của các đột biến này có tác dụng trội âm tính đối với CaSR bình thường, có lẽ bằng cách cản trở sự di chuyển của thụ thể kiểu hoang dã đến bề mặt tế bào hoặc bất hoạt thụ thể kiểu hoang dã bằng cách liên kết với CaSR đột biến một khi đã được nhúng vào màng tế bào, hoặc bằng cách cô lập các protein G. Ngoài ra, thai nhi mang một đột biến dị hợp tử trong CASR được di truyền từ một người cha bị ảnh hưởng nhưng nằm trong bụng của một người mẹ bình thường có thể bị “hạ canxi máu” tương đối trong tử cung, dẫn đến tăng sản các tuyến cận giáp của thai nhi tồn tại sau khi sinh, gây ra NSHPT. Đôi khi, NSHPT có thể được di truyền như một rối loạn lặn trên NST thường bởi các bậc cha mẹ bình thường về mặt lâm sàng và sinh hóa. Ở các đối tượng có đột biến đồng hợp tử gần đầu amino của CASR (ví dụ, p.Leu13Pro), tăng canxi máu có thể không biểu hiện cho đến giữa thời thơ ấu hoặc thậm chí là tuổi trưởng thành. Do đó, phổ lâm sàng của NSHPT dao động từ nhẹ (táo bón, đa niệu) với nồng độ canxi từ 11 đến 13 mg/dL (2,75–3,25 mmol/L) đến nặng và đe dọa tính mạng (rối loạn nhịp tim, suy hô hấp do giảm trương lực cơ, mất khoáng và gãy xương sườn) khi nồng độ canxi vượt quá 15 mg/dL (3,75 mmol/L). Do đó, NSHPT có thể biểu hiện trong vòng vài ngày đầu đời đến vài tháng tuổi tùy thuộc vào mức độ tăng canxi máu. Việc tìm hiểu tiền sử gia đình có thể xác định các thành viên bị tăng canxi máu nhẹ do HHC di truyền trội trên NST thường. Khám thực thể thường cho thấy giảm trương lực cơ, khó khăn trong việc ăn uống và suy hô hấp. Nồng độ canxi huyết thanh thường tăng rõ rệt (trung bình 14 mg/dL, dao động từ 11 đến > 20 mg/dL; 3,5 mmol/L, 2,75 đến 5 mmol/L) cũng như giá trị PTH (trung bình 540 pg/mL, dao động từ 55 đến > 1000 pg/mL). Các phát hiện khác bao gồm: hạ phốt phát máu, tăng magiê máu, tăng phosphatase máu và giá trị calcitriol tăng cao, tái hấp thu phốt phát ở ống thận thấp và giảm canxi niệu tương đối. Về mặt X-quang, có bằng chứng về bệnh xương do cường cận giáp—loãng xương, đầu xương rộng và không đều, tái hấp thu dưới màng xương, biến dạng vẹo trong của hông và gãy xương.
Việc hạ thấp nồng độ canxi huyết thanh tăng cao hiện diện trong NSHPT chỉ bị ảnh hưởng một cách khiêm tốn bởi việc gây lợi tiểu bằng cách truyền natri clorid và dùng furosemide; có thể cần dùng calcitonin và một bisphosphonate (ví dụ, pamidronate/zolendronate) để ức chế tái hấp thu canxi xương và giảm nồng độ canxi huyết thanh. Tuy nhiên, các tác nhân này không làm giảm nồng độ PTH tăng cao và không giải quyết được tình trạng mất khoáng xương rõ rệt của cường cận giáp. Chất calcimimetic dị lập thể cinacalcet hydrochloride tác động trực tiếp lên CaSR cũng đã có hiệu quả trong việc hạ thấp nồng độ canxi huyết thanh ở trẻ sơ sinh mắc NSHPT. (Tuy nhiên, cinacalcet không được chấp thuận để sử dụng ở các đối tượng dưới 18 tuổi.) Cắt bỏ gần toàn bộ tuyến cận giáp đôi khi có thể là một biện pháp cứu sống cần thiết. Trẻ em mắc NSHPT vẫn bị tăng canxi máu sẽ chán ăn, chậm phát triển và có nguy cơ chậm phát triển.
Cường cận giáp thứ phát ở trẻ sơ sinh có thể là kết quả của tình trạng hạ canxi máu ở mẹ do suy cận giáp, PHP, toan hóa ống thận hoặc thiếu vitamin D. Hạ canxi máu ở mẹ làm giảm vận chuyển qua nhau thai và lượng canxi ròng được cung cấp cho thai nhi, dẫn đến hạ canxi máu tương đối ở thai nhi, gây tăng sản các tuyến cận giáp của thai nhi và cường cận giáp thứ phát tương xứng với sự thiếu hụt canxi của mẹ. Mặc dù chỉ có 25% trẻ sơ sinh của các bà mẹ bị hạ canxi máu bị tăng canxi máu, hầu hết đều có những thay đổi về xương phản ánh sự dư thừa PTH, từ mất khoáng nghiêm trọng có gãy xương đến loãng xương chỉ có thể phát hiện bằng phương pháp đo mật độ khoáng xương. Cường cận giáp thứ phát thường tự khỏi trong vòng vài tuần sau khi sinh khi trẻ ăn đủ canxi và phốt phát. Vận chuyển canxi qua nhau thai từ mẹ sang thai nhi bình thường phụ thuộc vào ba quá trình của nguyên bào nuôi nhau thai: (1) Ca2+ đi vào ở đỉnh qua các kênh Ca2+ thông qua một gradient điện hóa, (2) liên kết nội bào của Ca2+ với calbindin D9k, và (3) đẩy Ca2+ ra ngoài qua màng đáy của nguyên bào nuôi. Cường cận giáp sơ sinh thoáng qua (OMIM 618188) cũng có thể xảy ra ở trẻ sơ sinh có các biến thể bất hoạt hai alen của TRPV6 (OMIM 606680) mã hóa một kênh chọn lọc canxi thấm hoạt động trong nguyên bào nuôi của nhau thai, ống thận và đường ruột. Trong rối loạn này, việc giảm vận chuyển canxi qua nhau thai từ mẹ sang thai nhi dẫn đến hạ canxi máu ở thai nhi trong tử cung và cường cận giáp thứ phát biểu hiện bằng các bất thường về xương, bao gồm xương mỏng, ngắn, một số có gãy xương sẽ tự khỏi trong vòng 2 năm sau khi sinh. Bệnh mucolipidosis type II (OMIM 252500) cũng đã được liên kết với cường cận giáp thứ phát ở trẻ sơ sinh; rối loạn giống Hurler này được đặc trưng bởi các bất thường trên khuôn mặt (bất đối xứng, sống mũi phẳng), gan lách to, biến dạng xương (loạn dưỡng xương đa ổ) và chậm phát triển và là do các đột biến bất hoạt trong một gen (GNPTAB, OMIM 607040) mã hóa N-acetyglucosamine-1-phosphotransferase, các tiểu đơn vị α/β, một phosphotransferase lysosome cần thiết cho quá trình tổng hợp mannose 6-phosphate. Trong bệnh này, nồng độ canxi của mẹ bình thường, nhưng mô học nhau thai bất thường cho thấy sự suy giảm vận chuyển canxi qua nhau thai và hạ canxi máu ở thai nhi, dẫn đến sự gia tăng bù trừ trong việc tạo ra PTH của thai nhi. Đổi lại, các bằng chứng về xương của sự dư thừa PTH (loãng xương, gãy xương) phát triển; cường cận giáp thứ phát và các tác dụng bất lợi của nó sẽ thuyên giảm trong vài tuần đến vài tháng đầu sau khi sinh. Toan hóa ống thận xa sơ sinh thoáng qua cũng đã được liên kết với tăng canxi máu do cường cận giáp thứ phát. Thỉnh thoảng, trẻ sơ sinh bị suy giáp bẩm sinh nguyên phát cũng bị tăng canxi máu thoáng qua.
Loạn sản sụn đầu xương Murk-Jansen (OMIM 156400) là một bệnh loạn sản sụn di truyền trội trên NST thường liên quan đến tăng canxi máu rõ rệt là hậu quả của các đột biến dị hợp tử dẫn đến sự hoạt hóa cấu thành, không phụ thuộc vào phối tử của PTH1R được biểu hiện ở thận, xương và các tế bào sụn tăng trưởng. PTH1R chức năng tăng cường dẫn đến tăng sinh tế bào sụn, giảm tốc độ trưởng thành của tế bào sụn, chậm hình thành xương và tăng tái hấp thu xương. Về kiểu hình, bệnh nhân mắc loạn sản sụn Murk-Jansen biểu hiện lùn chi ngắn; biến dạng xương dài, các ngón, cột sống và xương chậu; tịt cửa mũi sau; vòm miệng cao; cằm nhỏ; các đường khớp sọ mở rộng (ở trẻ nhỏ); xơ cứng xương sọ nền; rối loạn tổ chức đầu xương (biệt hóa tế bào sụn chậm, sụn vôi hóa không đều nhô vào thân xương); và mất quá nhiều xương vỏ nhưng xương bè bình thường. Mặc dù chiều dài khi sinh và ngoại hình có thể bình thường ở trẻ sơ sinh bị ảnh hưởng, có bằng chứng X-quang về loạn sản sụn. Trẻ sơ sinh bị ảnh hưởng thường có canxi máu bình thường, nhưng sau đó (thời thơ ấu và trẻ em) phát triển tăng canxi máu, hạ phốt phát máu, tăng nồng độ calcitriol trong huyết thanh và tăng bài tiết cyclic AMP có nguồn gốc từ thận và canxi qua nước tiểu nhưng nồng độ PTH và PTHrP trong huyết thanh thấp hoặc không thể phát hiện được. (Người lớn mắc rối loạn này thường có canxi máu bình thường, cũng như rất lùn [chiều cao người lớn 100 cm]; sự trưởng thành của xương bị trì hoãn; đầu xương dài và có cấu trúc mô học bị rối loạn; và xương sọ bị xơ cứng.) Các đột biến hoạt hóa cấu thành của PTH1R ở những đối tượng này bao gồm phổ biến nhất là p.His223Arg ở điểm nối của vòng nội bào đầu tiên và miền xuyên màng thứ hai và p.Thr410Pro trong miền xuyên màng thứ sáu—những vị trí đặc biệt quan trọng trong việc mang lại hoạt động không phụ thuộc vào phối tử cho PTH1R. Kinase họ SLK 3 (SIK3, OMIM 614776) hoạt động trong con đường truyền tín hiệu nội bào PTH1R; một biến thể bất hoạt hai alen của SIK3 dẫn đến một bệnh loạn sản sụn đốt sống-đầu xương-đầu xương tăng canxi máu tương tự nhưng khác biệt với bệnh Murk-Jansen. Việc bài tiết quá mức PTHrP và hậu quả là tăng canxi máu cũng đã được ghi nhận ở trẻ sơ sinh mắc bệnh ứ sắt sơ sinh và các khối u thận phôi thai (Wilms, u trung bì thận).
Hội chứng Williams-Beuren (WBS) (OMIM 194050) là một hội chứng mất đoạn gen liền kề bán hợp tử (liên quan đến khoảng 26–28 gen trên nhiễm sắc thể 7q11.23) được di truyền như một rối loạn trội trên NST thường và có tỷ lệ hiện mắc là 1/7500 đến 1:15.000 ca sinh sống. WBS được đặc trưng bởi chậm tăng trưởng trong tử cung và sau sinh, tăng canxi máu ở trẻ nhỏ ở 15% bệnh nhân thường tự khỏi trước 2 tuổi nhưng đôi khi có thể tồn tại đến tuổi trưởng thành, tăng canxi niệu nhất quán, hẹp động mạch chủ trên van ở 30% đối tượng, hẹp và co thắt động mạch chủ ngực và các động mạch vành, phổi, thận, mạc treo và thân tạng, tăng huyết áp, tật đầu nhỏ, khuôn mặt “yêu tinh” (trán rộng, nếp quạt, mống mắt hình sao, lác trong, mũi ngắn với đầu mũi đầy, môi trên cong và môi dưới nhô ra, nhân trung dài, má phính với gò má phẳng, sai khớp cắn), giọng khàn, tăng thính lực ở thời thơ ấu liên quan đến điếc thần kinh, dính khớp quay-trụ, thiểu sản thận hoặc bất sản một bên, chậm phát triển (tích hợp thị giác-vận động kém, rối loạn thiếu tập trung, IQ trung bình 58, dao động 20–106), và tầm vóc thấp. Mặc dù hầu hết bệnh nhân mắc WBS đều bị chậm phát triển, họ có các kỹ năng ngôn ngữ độc đáo và thành thạo với vốn từ vựng phong phú và trí nhớ thính giác tăng cường đặc biệt đối với tên, các kỹ năng ngôn ngữ xã hội điêu luyện, và năng khiếu âm nhạc đặc biệt bao gồm khả năng ghi nhớ và hát nhiều tác phẩm âm nhạc và chơi nhiều loại nhạc cụ. Trẻ em mắc WBS thích ở cùng người lớn hơn là với bạn bè đồng trang lứa và không nhút nhát hay sợ hãi người lạ.
Tăng canxi máu ở các đối tượng mắc WBS có thể liên quan về mặt sinh bệnh học đến việc mất một phần chức năng của yếu tố phiên mã hội chứng Williams (WSTF) được mã hóa bởi BAZ1B (Bromodomain adjacent to zinc-finger domain, 1B, OMIM 605681). WSTF là một protein hạt nhân là một phần của một phức hợp tái cấu trúc chromatin đa phân, phụ thuộc vào ATP được gọi là WINAC (WSTF including nucleoside assembly complex). Độc lập với việc liên kết phối tử, VDR tương tác với WINAC. Trong các trường hợp thông thường, việc liên kết calcitriol với phức hợp VDR-WINAC (1) ức chế biểu hiện của CYP27B1 ở ống thận, gen mã hóa 25-hydroxyvitamin D-1α-hydroxylase, và (2) tăng cường phiên mã của CYP24A1, gen mã hóa 25-hydroxyvitamin D-24-hydroxylase. Do đó, sự thiếu hụt một nửa của WSTF có thể dẫn đến việc tăng và tổng hợp calcitriol tương đối không được điều hòa trong khi làm chậm tốc độ phân hủy của nó; để đáp ứng với sự gia tăng tạo ra calcitriol, sự hấp thu canxi ở ruột tăng lên và tăng canxi máu xảy ra sau đó. Các bệnh nội tiết khác liên quan đến WBS bao gồm không dung nạp glucose và suy giáp.
WBS là hậu quả của việc mất đoạn nhiễm sắc thể 7q11.23 do tái tổ hợp giảm phân không đồng đều (trao đổi chéo gen không đồng đều giữa các tương đồng của nhiễm sắc thể 7 trong quá trình giảm phân); lỗi di truyền này xảy ra với tần suất bằng nhau trong các giao tử của cả cha và mẹ và dẫn đến mất từ 26 đến 28 gen. Tình trạng bán hợp tử của ELN (OMIM 130160) mã hóa elastin được cho là nguyên nhân của các dị tật tim mạch và tăng huyết áp được quan sát thấy trong WBS, vì việc mất một phần ELN dẫn đến sự gia tăng bù trừ số lượng các vòng cơ trơn và các lá đàn hồi dẫn đến dày động mạch và tăng nguy cơ tắc nghẽn. Tình trạng bán hợp tử chức năng đối với yếu tố phiên mã chung II-I (GTF2I, OMIM 601679), GTF2IRD1 (OMIM 604238), HIP1 (OMIM 601767), và YWHAG (OMIM 605356) có thể là nguyên nhân cơ bản của các thiếu hụt và thế mạnh về thần kinh nhận thức đặc trưng cho WBS và tính cách đặc biệt của họ. Sự thiếu hụt một nửa của LIM domain kinase 1 (LIMK1, OMIM 601329) cũng có thể liên quan đến các khiếm khuyết thần kinh nhận thức ở bệnh nhân mắc WBS. Mất bán hợp tử của STX1A (OMIM 186590) có thể liên quan về mặt sinh bệnh học đến sự phát triển của tình trạng không dung nạp glucose ở các đối tượng WBS. Kiểu hình WBS cũng đã được liên kết với mất đoạn kẽ của nhiễm sắc thể 6q22.2-q23, cũng như các khiếm khuyết ở các nhiễm sắc thể 4, 11 và 22—ngụ ý rằng hội chứng này không đồng nhất về mặt di truyền. Chẩn đoán hội chứng WBS bị nghi ngờ trên cơ sở kiểu hình lâm sàng đặc trưng (có/không có tăng canxi máu) và được xác nhận bằng cách chứng minh sự vi mất đoạn tại nhiễm sắc thể 7q11.23 bằng microarray hoặc FISH, mặc dù một phân tích nhiễm sắc thể bình thường không loại trừ hoàn toàn chẩn đoán này. Việc định kiểu gen cụ thể đôi khi cũng có thể hữu ích trong chẩn đoán. Tăng canxi máu được quản lý bằng cách cho trẻ uống sữa công thức ít canxi, không chứa vitamin D; đôi khi có thể cần liệu pháp glucocorticoid ngắn hạn để phục hồi tình trạng canxi máu bình thường.
Một hội chứng mất đoạn gen liền kề được đặc trưng bởi chậm phát triển và co giật liên quan đến một đoạn của nhiễm sắc thể 7q11.23 ở phía xa so với đoạn mất liên quan đến WBS đã được mô tả (OMIM 613729). Sự nhân đôi của nhiễm sắc thể 7q11.23 dẫn đến một hội chứng liên quan đến các khuyết tật tim, thoát vị hoành, tinh hoàn ẩn, và các sai lệch về thần kinh nhận thức và hành vi. Các phát hiện bổ sung ở bệnh nhân có dup7q11.23 bao gồm tầm vóc thấp và các đặc điểm khuôn mặt dị dạng nhỏ (trán dô, đầu mũi dài, nhân trung ngắn, môi mỏng, cằm lẹm, vành tai gấp). Về mặt thần kinh nhận thức, có sự chậm nói nghiêm trọng và thường có hành vi tự kỷ nhưng các kỹ năng nhận thức thị giác còn nguyên vẹn—những đặc điểm trái ngược với những đặc điểm hiện diện ở bệnh nhân mắc WBS.
Mặc dù có kiểu hình bình thường, trẻ sơ sinh mắc tăng canxi máu ở trẻ nhỏ type 1 và 2 biểu hiện chán ăn, suy giảm tăng trưởng, chậm phát triển, các cơn sốt, đa niệu và nôn ói, dẫn đến mất nước, tăng canxi niệu và nhiễm canxi thận liên quan đến tăng canxi máu khi có nồng độ PTH huyết thanh giảm. Tăng canxi máu ở trẻ nhỏ type 1 (OMIM 143880) được đặc trưng bởi nồng độ calcitriol tăng cao hoặc bình thường một cách không phù hợp (so với nồng độ canxi huyết thanh) thường do các biến thể bất hoạt hai alen của CYP24A1 (OMIM 126065) mã hóa 24-hydroxylase, enzyme chuyển đổi 25-hydroxyvitamin D thành 24,25 dihydroxyvitamin D và 1,25-dihydroxyvitamin D thành 1,24,25-trihydroxyvitamin D, do đó làm tăng độ tan của chúng và bài tiết sau đó trong mật và nước tiểu. Việc trì hoãn sự phân hủy của 1,25-dihydroxyvitamin D kéo dài tuổi thọ sinh học hiệu quả của nó. Ở nhiều bệnh nhân mắc tăng canxi máu ở trẻ nhỏ type I, tăng canxi máu tự khỏi trong vài năm đầu đời nhưng nó có thể tồn tại đến tuổi lớn hơn. Các đột biến bất hoạt trong CYP24A1 cũng đã được tìm thấy ở người lớn bị tăng canxi máu, tăng canxi niệu và sỏi thận. Các đột biến CYP24A1 có thể làm giảm sự liên kết của enzyme với cơ chất của nó hoặc cản trở sự tương tác của heme và protein enzyme; cả hai loại đột biến đều làm giảm chức năng của nó. Việc tránh vitamin D, chế độ ăn ít canxi, tăng cường dịch, và đôi khi dùng glucocorticoid để giảm hấp thu canxi ở ruột là các phương thức điều trị cho rối loạn này. Tăng canxi máu ở trẻ nhỏ type 2 (OMIM 616963) là kết quả của các biến thể hai alen trong SLC34A1 mã hóa họ chất mang hòa tan 34 (Chất đồng vận chuyển natri phốt phát type II), thành viên 1 (OMIM 182309), một chất đồng vận chuyển natri phốt phát ở ống lượn gần, và được biểu hiện bằng nôn ói, chán ăn, suy kiệt, tăng canxi máu, hạ phốt phát máu, tăng canxi niệu và nhiễm canxi thận. Trong rối loạn này, việc giảm vận chuyển phốt phát ở ống thận dẫn đến tăng phốt phát niệu và hạ phốt phát máu, dẫn đến giảm tổng hợp FGF-23 và do đó làm tăng hoạt động chức năng của 25OHD-1α hydroxylase (CYP27B1) và sự tổng hợp calcitriol và tăng hấp thu canxi ở ruột sau đó. Việc hạn chế nạp canxi và vitamin D cũng đã được sử dụng trong việc quản lý bệnh nhân mắc tăng canxi máu ở trẻ nhỏ type 2, nhưng việc bổ sung phốt phát cũng có thể được xem xét. Khi trưởng thành, bệnh nhân có các biến thể của CYP24A1 hoặc SLC34A1 có chiều cao và cân nặng bình thường và sức khỏe tương đối tốt, mặc dù ở một số đối tượng, nhiễm canxi thận vẫn tồn tại. Tuy nhiên, một số bệnh nhân bị ảnh hưởng phát triển suy thận tiến triển khi trưởng thành.
Hội chứng Bartter trước sinh/sơ sinh type 1 (OMIM 601678) và type 2 (OMIM 241200) tương tự nhau về mặt lâm sàng và sinh hóa và là do các đột biến mất chức năng hai alen trong các gen kiểm soát một chất vận chuyển natri-kali-clorua xuyên biểu mô (SLC12A1, OMIM 600839) và một kênh kali chỉnh lưu vào trong ở đỉnh (KCNJ1, OMIM 600359), tương ứng, được biểu hiện ở nhánh lên dày của quai Henle ở ống thận (TALH). Các thai nhi bị ảnh hưởng phát triển đa ối, dẫn đến sinh non với mất muối sau sinh và tăng aldosteron thứ phát; sự gia tăng sản xuất prostaglandin E2 ở thận và toàn thân ức chế tái hấp thu natri và clorua trong TALH và tăng cường giải phóng renin ở bộ máy cận cầu thận, dẫn đến nhiễm toan chuyển hóa hạ kali máu, cường cận giáp, tăng canxi máu, tăng canxi niệu, nhiễm canxi thận, hạ magiê máu, loãng xương và thường tử vong ở hội chứng Bartter trước sinh type 1. Hội chứng Bartter sơ sinh type 2 do các biến thể của KCNJ1 gây ra được biểu hiện bằng hạ kali máu (đôi khi là tăng kali máu thoáng qua), giảm thể tích nội mạch và tăng nồng độ angiotensin dẫn đến sản xuất prostaglandin E2 ở thận và toàn thân cũng làm suy giảm tái hấp thu natri và clorua ở ống thận trong TALH. Nhiễm kiềm giảm clorua, hạ kali máu, tăng prostaglandin E, tăng canxi máu, tăng canxi niệu và nhiễm canxi thận gợi ý hội chứng Bartter sơ sinh. Bù dịch và điện giải và dùng thuốc lợi tiểu giữ kali (thiazide) và các chất ức chế cyclooxygenase (indomethacin) có thể có hiệu quả trong việc cải thiện các biểu hiện sinh hóa và lâm sàng của bệnh.
Tăng canxi máu sơ sinh hoặc trẻ nhỏ có thể xảy ra ở những bệnh nhân có một trong số các lỗi chuyển hóa bẩm sinh. Cả thiếu hụt lactase bẩm sinh (OMIM 223000) do các đột biến trong LCT (OMIM 603202) và thiếu hụt sucrase-isomaltase (OMIM 222900) do các biến thể của SI (OMIM 609845) đã được liên kết với tăng canxi máu ở trẻ nhỏ, có khả năng do tăng hấp thu canxi ở ruột được thúc đẩy bởi các disaccharide. Tăng canxi máu cũng có thể gặp ở trẻ sơ sinh bị oxalosis nguyên phát (OMIM 259900) có thể là kết quả của các biến thể ở một trong ba gen (AGXT, OMIM 604285; GRHPR, OMIM 604296; DHDPSL, OMIM 613597). “Hội chứng tã xanh” (OMIM 211000) là kết quả của việc đường tiêu hóa không thể hấp thu tryptophan mà vi khuẩn đường ruột chuyển hóa thành một indole được hấp thu và bài tiết trong nước tiểu, dẫn đến indican niệu sau đó bị oxy hóa thành sắc tố “xanh indigo”, do đó làm nhuộm màu tã của trẻ sơ sinh và trẻ nhỏ bị ảnh hưởng. Tăng canxi máu, tăng canxi niệu và nhiễm canxi thận là phổ biến nhưng không phải là không đổi; sinh bệnh học của tăng canxi máu ở những bệnh nhân này chưa được biết. Rối loạn này đã được liên kết với một đột biến mất chức năng đồng hợp tử trong PCSK1 mã hóa một prohormone convertase/serine endoproteinase phụ thuộc canxi (proprotein convertase, subtilisin/kexin-type 1, OMIM 162150) chuyển đổi một protein đa thành phần thành các sản phẩm protein có hoạt tính sinh học cụ thể. Các biến thể của PCSK1 cũng đã được liên quan đến các khiếm khuyết trong tổng hợp insulin, cũng như một số bệnh nội tiết (suy sinh dục, suy vỏ thượng thận trung ương và đái tháo nhạt trung ương). Tăng canxi máu cũng có thể xảy ra ở những bệnh nhân mắc hội chứng IMAGe gồm chậm tăng trưởng trong tử cung, loạn sản hành xương, thiểu sản thượng thận bẩm sinh và các bất thường sinh dục (OMIM 614732) do các biến thể dị hợp tử của CDKN1C mã hóa chất ức chế kinase phụ thuộc cyclin 1C (OMIM 600586), một chất điều hòa sự phân chia tế bào sớm.
Giảm phosphatase máu (HPP) là một rối loạn bẩm sinh về khoáng hóa xương dẫn đến bệnh còi xương có mức độ nghiêm trọng khác nhau do giảm tổng hợp phosphatase kiềm không đặc hiệu của mô (TNSALP) ở nguyên bào xương—hậu quả của các đột biến bất hoạt đơn alen hoặc hai alen trong ALPL (OMIM 171760) (vide infra). Việc sản xuất phosphatase kiềm giảm dẫn đến thiếu hụt các ion phốt phát làm suy giảm quá trình tổng hợp hydroxyapatite dẫn đến bệnh còi xương, trong khi việc hấp thu canxi ở ruột tiếp tục có thể dẫn đến tăng canxi máu. Dạng giảm phosphatase máu ở trẻ nhỏ (OMIM 241500) là do các biến thể mất chức năng hai alen của ALPL. Nó trở nên rõ ràng về mặt lâm sàng sau tháng đầu tiên của cuộc đời nhưng thường trước 6 tháng tuổi và thường gây tử vong. Các triệu chứng ban đầu là chậm phát triển liên quan đến suy giảm ăn uống, yếu cơ và chậm đạt được các mốc vận động; các biến dạng còi xương của lồng ngực và các chi có thể nhìn thấy được. Về mặt X-quang, nó được đặc trưng bởi sự mất khoáng của vòm sọ và bộ xương ngoại vi, còi xương, các gai sụn và xương kéo dài từ hai bên khớp gối và khuỷu tay, và dính khớp sọ sớm, dẫn đến tăng áp lực nội sọ. Cũng có các cơn co giật phụ thuộc vitamin B6, tăng canxi máu và tăng canxi niệu. Sự tổng hợp TNSALP ở nguyên bào xương bị khiếm khuyết là kết quả của các đột biến sai nghĩa, vô nghĩa và vị trí nối donor của ALPL mất chức năng. Các đột biến đồng hợp tử hoặc dị hợp tử phức hợp gây chết người của ALPL nằm trong hoặc gần miền enzyme và/hoặc các giao diện homodimer và tetramer. Hoạt độ phosphatase kiềm xương trong huyết thanh thấp một cách không phù hợp phân biệt bệnh này với các tình trạng còi xương hoặc loãng xương khác (bệnh xương thủy tinh bẩm sinh [OIC] hoặc loạn sản sụn loại 1A) trong đó hoạt độ phosphatase kiềm thường bình thường hoặc tăng. Tăng phosphoethanolamine trong nước tiểu và pyrophosphat vô cơ trong huyết thanh (một chất ức chế sự kết tinh hydroxyapatite cũng làm suy giảm quá trình khoáng hóa xương) và các giá trị pyridoxal-5′-phosphate phù hợp với chẩn đoán giảm phosphatase máu, trong khi phân tích ALPL xác định (các) đột biến gen. Chẩn đoán giảm phosphatase máu có thể bị nghi ngờ trên siêu âm trước sinh và được xác nhận bằng định kiểu gen ALPL. Tăng canxi máu của giảm phosphatase máu ở trẻ nhỏ được quản lý bằng cách bù nước, dùng thuốc lợi tiểu tác động lên nhánh lên dày của quai Henle (ví dụ, furosemide), và dùng bisphosphonate (pamidronate), calcitonin hoặc glucocorticoid khi cần thiết. Lượng canxi ăn vào nên được hạn chế và nên tránh vitamin D và các chất chuyển hóa của nó. Ghép tủy xương và “tăng cường tế bào gốc” của các nguyên bào xương của người cho được truyền cũng đã được sử dụng để điều trị cho các bệnh nhân bị ảnh hưởng. Việc sử dụng một dạng TNSALP nhắm vào xương đã được sử dụng thành công để điều trị giảm phosphatase máu đe dọa tính mạng và hiện được FDA chấp thuận cho mục đích này.
Tăng canxi máu và tăng canxi niệu không rõ nguyên nhân đôi khi xảy ra ở các đối tượng mắc hội chứng IMAGe (OMIM 614732) do các biến thể tăng chức năng của CDKN1C (OMIM 600856, chr. 11p15.4) mã hóa một chất ức chế kinase phụ thuộc cyclin được in dấu từ cha (di truyền từ mẹ) rất quan trọng cho sự điều hòa bình thường của các quá trình phân bào của tế bào. Dính khớp sọ, hở hàm ếch và vẹo cột sống cũng có thể phát triển ở những bệnh nhân này.
Đánh giá và Quản lý
Sau khi xem xét tiền sử (tiền sử gia đình được khám phá để tìm các thành viên mắc các rối loạn khoáng chất và tình trạng của mẹ được xem xét sâu, và lượng canxi, phốt phát và vitamin D nạp vào của trẻ được ước tính) và khám thực thể (các biểu hiện không đặc hiệu của tăng canxi máu bao gồm—tăng huyết áp, tăng trưởng dưới mức bình thường, giảm trương lực cơ, yếu cơ, lơ mơ, sững sờ, đôi khi co giật; các phát hiện cụ thể liên quan đến tăng canxi máu bao gồm—các dấu hiệu trên khuôn mặt và tim mạch của WBS, các nốt dưới da phù hợp với hoại tử mỡ, các biến dạng xương gợi ý loạn sản sụn đầu xương hoặc giảm phosphatase máu ở trẻ nhỏ) đã được hoàn thành, việc đánh giá trẻ sơ sinh và trẻ nhỏ bị tăng canxi máu tiếp tục bằng cách đo canxi toàn phần và Ca2+, phốt phát, phosphatase kiềm, PTH, calcidiol và calcitriol trong huyết thanh và xác định nồng độ canxi và creatinin trong nước tiểu (Hình 20.5). Giảm canxi niệu khi có tăng canxi máu gợi ý sự hiện diện của NSHPT. Thỉnh thoảng, người ta có thể gặp một trẻ sơ sinh trong đó nồng độ canxi toàn phần tăng do tăng protein máu nhưng giá trị Ca2+ bình thường, một tình trạng được gọi là tăng canxi máu giả. Ở trẻ sơ sinh bị nghi ngờ mắc WBS, nên tiến hành phân tích microarray hoặc FISH để tìm kiếm sự mất đoạn của nhiễm sắc thể 7q11.23. Ở những trẻ mà chẩn đoán NSHPT đang được xem xét, việc xác định các giá trị canxi và creatinin trong huyết thanh và nước tiểu của cha mẹ thường hữu ích trừ khi rối loạn này được di truyền như một bệnh lặn trên NST thường. Nên tiến hành định kiểu gen của CASR khi nghi ngờ NSHPT.
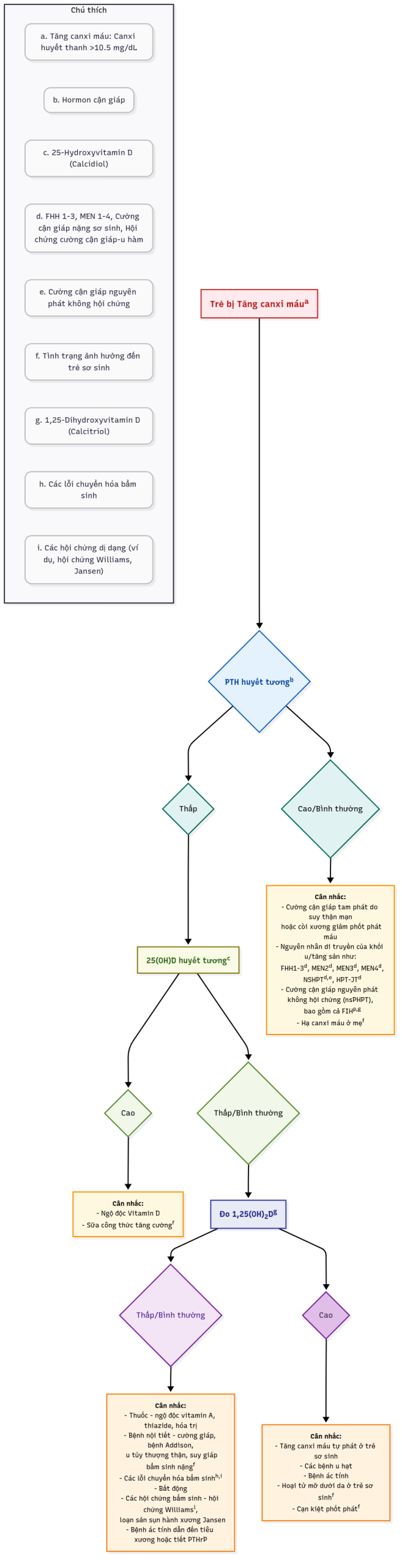
Hình 20.5: Đánh giá tăng canxi máu. a. Tăng canxi máu: Canxi huyết thanh >10,5 mg/dL (hoặc theo dữ liệu phòng xét nghiệm), b. Hormon cận giáp, c. 25-Hydroxyvitamin D (Calcidiol), d. Tăng canxi máu giảm canxi niệu có tính gia đình 1-3, Đa u tuyến nội tiết 1-4, Cường cận giáp nặng ở trẻ sơ sinh, Hội chứng cường cận giáp-u xương hàm, e. Cường cận giáp đơn độc có tính gia đình, f. Các tình trạng ảnh hưởng đến trẻ sơ sinh, g. 1,25-Dihydroxyvitamin D = Calcitriol, h. Các lỗi chuyển hóa bẩm sinh, I. Các hội chứng dị dạng (ví dụ, hội chứng William, Jansen).
(Từ Stokes VJ, Nielsen MF, Hannan FM, Thakker RV. Hypercalcemic disorders in children. J Bone Mineral Res. 2017;32:2157–2170, với sự cho phép.)
Điều trị tăng canxi máu ở trẻ sơ sinh và trẻ nhỏ phải hướng đến việc ngăn chặn sự tiến triển, xác định nguyên nhân và đánh giá mức độ nghiêm trọng. Việc sử dụng sữa công thức ít canxi và tránh vitamin D (nạp quá nhiều hoặc ánh nắng mặt trời) rất hữu ích ở phần lớn trẻ sơ sinh bị tăng canxi máu khiêm tốn. Trẻ sơ sinh có nồng độ canxi huyết thanh tăng đáng kể, đặc biệt nếu kéo dài, thường bị mất nước. Điều trị ngay lập tức bao gồm truyền natri clorid 0,9% với 30 mEq/L kali clorid (10–20 mL/kg trong 1 giờ); sau khi tình trạng mất nước đã được điều chỉnh và dòng nước tiểu đủ đã được thiết lập và nếu tình trạng tăng canxi máu đáng kể vẫn tồn tại, có thể tiêm tĩnh mạch một liều bolus furosemide (1–2 mg/kg). Hiện nay, bisphosphonate, các chất tương tự của pyrophosphat bám vào bề mặt của các tinh thể hydroxyapatite và được các hủy cốt bào kết hợp vào, do đó ức chế sự phân giải khoáng và chất nền xương qua trung gian hủy cốt bào, là các tác nhân được lựa chọn để điều trị tăng canxi máu đáng kể ở trẻ sơ sinh. Pamidronate (0,5–2,0 mg/kg trong 30 mL nước muối sinh lý tiêm tĩnh mạch trong 4 giờ) đã được sử dụng thành công ở trẻ sơ sinh bị tăng canxi máu do ngộ độc vitamin D, hoại tử mỡ dưới da, NSHPT và các nguyên nhân khác. Tác dụng của bisphosphonate kéo dài trong vài tuần đến vài tháng và khi dùng quá liều có thể dẫn đến hạ canxi máu, hạ phốt phát máu và hạ magiê máu. Các tác nhân khác có thể được xem xét trong điều trị trẻ sơ sinh bị tăng canxi máu bao gồm calcitonin cá hồi (2–4 đơn vị/kg tiêm dưới da mỗi 6–12 giờ) để ức chế sự huy động canxi từ xương. Mặc dù chất calcimimetic type II—cinacalcet—đã được chứng minh là cải thiện chức năng của nhiều dạng đột biến không hoạt động của CASR và có lợi ích lâm sàng ở người lớn bị tăng canxi máu giảm canxi niệu có tính gia đình, tác nhân này không được chấp thuận để sử dụng ở trẻ em tại Hoa Kỳ, một thử nghiệm điều trị của tác nhân này có thể được xem xét ở bệnh nhân mắc NSHPT với các biện pháp bảo vệ và sự cho phép thích hợp. Ở trẻ sơ sinh bị NSHPT đe dọa tính mạng, có thể cần phải cắt bỏ toàn bộ tuyến cận giáp khẩn cấp. Sau phẫu thuật, bệnh nhân có thể bị hạ canxi máu do canxi được lắng đọng mạnh mẽ vào xương (hội chứng xương đói) và cần một lượng lớn canxi bổ sung cho đến khi ổn định.
Trẻ sơ sinh bị tăng canxi máu do WBS (thường tự khỏi trước 2 tuổi) hoặc với một số dạng bệnh xương hóa đá có thể được quản lý bằng cách sử dụng sữa công thức ít canxi (ví dụ, Calcilo XD—2,9 mg canxi trên 100 calo; không chứa vitamin D) và ngừng bổ sung vitamin D. Trẻ sơ sinh bị tăng canxi máu “vô căn” ở trẻ nhỏ do các đột biến bất hoạt của CYP24A1 nên nhận sữa công thức ít canxi và nên ngừng bổ sung vitamin D. Một số trẻ bị tăng canxi máu do các đột biến bất hoạt của CYP24A1 đáp ứng với ketoconazole, chất ức chế sự chuyển đổi calcidiol thành calcitriol, hoặc rifampin, chất cũng bất hoạt các chất chuyển hóa của vitamin D. Bệnh nhân có đột biến trong SLC34A1 có thể đáp ứng với việc dùng bổ sung phốt phát. Cân bằng nội môi khoáng chất phải được theo dõi chặt chẽ ở trẻ sơ sinh đang nhận sữa công thức ít canxi và bị ngừng vitamin D để ngăn ngừa sự thiếu hụt cả hai chất dinh dưỡng và sự phát triển của cường cận giáp thứ phát.
Tăng canxi máu ở Trẻ em và Vị thành niên
Tăng canxi máu hiện diện ở trẻ em/vị thành niên khi nồng độ canxi huyết thanh vượt quá hai độ lệch chuẩn trên mức trung bình bình thường; đối với các đối tượng từ 4 đến 18 tuổi, các giá trị này là: canxi toàn phần 10,7 mg/dL (2,70 mmol/L); Ca2+ 5,52 mg/dL (1,38 mmol/L). Tăng canxi máu thường không có triệu chứng ở trẻ lớn và vị thành niên và được xác định bất ngờ qua phân tích hóa học huyết thanh thường quy cho một vấn đề không liên quan. Các biểu hiện lâm sàng của tăng canxi máu ở trẻ em/vị thành niên bao gồm: chán ăn, buồn nôn, nôn, táo bón, đau bụng hoặc đau hông, khát nhiều, tiểu nhiều, cơn đau quặn thận, thay đổi tỉnh táo, lơ mơ, giảm trương lực cơ, khó chịu hoặc co giật. Tăng canxi máu mạn tính không được nhận biết có thể làm suy giảm sự tăng trưởng, dẫn đến nhiễm canxi thận và sỏi thận trước khi suy thận, viêm tụy, suy giảm nhận thức hoặc các triệu chứng của bệnh tâm thần. Gãy xương dài qua một “u nâu” dạng nang xơ (viêm xương xơ nang) có thể là một phàn nàn khi nhập viện. Các dấu hiệu thực thể không đặc hiệu và có thể bao gồm tăng huyết áp, yếu cơ, giảm phản xạ, cảm xúc cùn mòn, rối loạn tâm thần hoặc đau hông.
Nguyên nhân và Sinh bệnh học
Các nguyên nhân gây tăng canxi máu ở trẻ em và vị thành niên được liệt kê trong Bảng 20.4A, 20.4B. Khi nồng độ protein huyết thanh bình thường, tăng canxi máu xảy ra khi “ngưỡng” của Ca2+ huyết thanh tăng lên do đột biến mất chức năng trong CASR hoặc ngưỡng này tăng lên một cách có thể đảo ngược do lithium, hoặc khi tốc độ đi vào của canxi vào các khoang ngoại bào và tuần hoàn từ xương, đường ruột hoặc thận vượt quá tốc độ mất đi của nó. Tốc độ tái hấp thu khoáng chất của xương có thể tăng lên do bài tiết quá mức PTH hoặc PTHrP, hoạt hóa cấu thành của PTH1R, nạp quá nhiều vitamin D hoặc các chất chuyển hóa, tăng sản xuất các cytokine viêm hoạt hóa hủy cốt bào hoặc các quá trình tiêu xương cục bộ, chẳng hạn như các khối u di căn, và sự phân ly giữa tốc độ hình thành và tái hấp thu xương—ví dụ, bất động. Tốc độ hấp thu canxi ở ruột có thể tăng lên do nạp quá nhiều canxi, do tăng vitamin D máu nguồn gốc ngoại sinh hoặc nội sinh, hoặc do tăng bài tiết PTH hoặc PTHrP. Tăng tái hấp thu canxi đã lọc ở ống thận có thể xảy ra khi dùng các thuốc lợi tiểu giữ canxi, chẳng hạn như thiazide, một tác dụng có thể “làm lộ ra” tình trạng tăng canxi máu ở một bệnh nhân trước đó có canxi máu bình thường bị cường cận giáp. Do đó, tăng canxi máu có thể được coi là qua trung gian PTH hoặc không phụ thuộc vào PTH. Khi có tăng albumin máu, nồng độ canxi toàn phần trong huyết thanh tăng nhưng nồng độ Ca2+ thì không (tăng canxi máu giả). Ứ trệ tĩnh mạch (ví dụ, do buộc garo trong khi lấy máu) dẫn đến các giá trị pH và Ca2+ tại chỗ bị thay đổi một cách giả tạo.
Cường cận giáp thường do một u tuyến cận giáp đơn độc gây ra nhưng cũng có thể do tăng sản cả bốn tuyến cận giáp như ở những bệnh nhân bị HHC do các biến thể mất chức năng của CASR, GNA11, hoặc AP2S1. Tương ứng, các gen này mã hóa CaSR bảy vòng xoắn xuyên màng plasma, yếu tố truyền tín hiệu được ghép cặp với guanosine triphosphate của nó (Gq/11α), và một phức hợp protein liên quan đến adaptor tạo điều kiện cho sự nhập bào của CaSR. Tăng canxi máu giảm canxi niệu có tính gia đình type 1 (HHC1, OMIM 145980) là một rối loạn di truyền trội trên NST thường có độ thấm hoàn toàn ở mọi lứa tuổi, được đặc trưng bởi tình trạng tăng canxi máu (cả toàn phần và ion hóa) phụ thuộc PTH, thường không có triệu chứng, với giảm canxi niệu (và do đó không có nhiễm canxi thận hoặc sỏi thận chứa canxi), tăng magiê máu, giảm magiê niệu và hạ phốt phát máu, là do các đột biến mất chức năng dị hợp tử trong CASR (OMIM 601199). Do đó, cần nồng độ Ca2+ huyết thanh cao hơn để kích hoạt CaSR và ức chế tổng hợp và bài tiết PTH. Nồng độ PTH huyết thanh có thể bình thường hoặc tăng nhẹ, nhưng cao một cách không phù hợp so với nồng độ Ca2+; các giá trị calcidiol và calcitriol bình thường. Ở trẻ em, HHC1 thường bị nghi ngờ ban đầu bởi sự hiện diện của tăng canxi máu không mong đợi (11–13 mg/dL) trong một hồ sơ hóa học hoặc thông qua sàng lọc gia đình của cha mẹ hoặc người thân khác bị tăng canxi máu. Mặc dù bệnh nhân mắc HHC1 bị tăng canxi máu, họ lại có tình trạng giảm canxi niệu một cách nghịch lý vì sự bất hoạt của CaSR dẫn đến tăng cường tái hấp thu canxi đã lọc ở ống lượn xa. Nồng độ PTH bình thường hoặc tăng nhẹ. Khi các đột biến trong CASR là hai alen hoặc khi một trẻ sơ sinh có biến thể đơn alen của CASR được sinh ra từ một phụ nữ có canxi máu bình thường, tình trạng tăng canxi máu có thể trở nên cực đoan và thậm chí đe dọa tính mạng (NSHPT). Các đối tượng lớn tuổi mắc HHC1 có thể phàn nàn về mệt mỏi, yếu cơ hoặc tiểu nhiều; có sự gia tăng nhẹ tỷ lệ viêm tụy tái phát, sỏi mật, vôi hóa sụn và vôi hóa mạch máu sớm ở các đối tượng mắc HHC1, nhưng khối lượng xương và tỷ lệ gãy xương là bình thường. Do giảm số lượng hoặc năng lực chức năng của các CaSR trên màng tế bào chính của tuyến cận giáp, ngưỡng ức chế bài tiết PTH của Ca2+ được thiết lập lại ở mức cao hơn. Các tuyến cận giáp tăng sản nhẹ. Trong các tế bào ống thận, sự giảm số lượng và hoạt động của các CaSR dẫn đến tăng tái hấp thu canxi đã lọc ở ống thận và giảm canxi niệu tương đối (tỷ lệ thanh thải canxi/thanh thải creatinin < 0,01 ở 80% bệnh nhân mắc HHC1); vì sự tái hấp thu magiê ở ống thận cũng tăng lên, nên có tình trạng tăng magiê máu và giảm magiê niệu; khả năng cô đặc nước tiểu và các chỉ số khác về chức năng thận là bình thường; trong tình trạng tăng canxi máu do các nguyên nhân khác, bài tiết canxi qua nước tiểu tăng và chức năng cô đặc của thận bị suy giảm. Thông thường, HHC1 không cần điều trị, nhưng phải được phân biệt với cường cận giáp nguyên phát nhẹ trong đó có hạ magiê máu và tăng canxi niệu. Cắt bỏ gần toàn bộ tuyến cận giáp trong HHC1 không làm giảm nồng độ canxi vì các tuyến cận giáp còn lại phì đại; cắt bỏ toàn bộ tuyến cận giáp là không cần thiết ngoại trừ ở một trẻ sơ sinh hiếm gặp mắc NSHPT. Hơn 100 đột biến vô nghĩa, sai nghĩa, chèn và mất đoạn trong CASR liên quan đến HHC1 hoặc NSHPT đã được xác định, chủ yếu ở miền liên kết Ca2+ ngoại bào của thụ thể; các đột biến này hoặc làm giảm ái lực của thụ thể đối với Ca2+ hoặc làm thay đổi quá trình xử lý nội bào của CaSR (glycosyl hóa, dimer hóa—ví dụ, p.Arg66His, p.Asn583Stop) trong lưới nội chất, ngăn cản sự chuyển vị của nó đến bề mặt màng tế bào (Hình 20.6). Nhiều đột biến nằm ở miền ngoại bào của CaSR giữa các codon 39 đến 300, một vùng giàu gốc aspartate và glutamate nơi Ca2+ có thể lồng vào. Glycosyl hóa là cần thiết cho cả quá trình dimer hóa và vận chuyển của CaSR. Các đột biến sai nghĩa liên quan đến arginine ở codon 66 (p.Arg66His/Cys) tạo ra một sản phẩm chỉ có thể được glycosyl hóa một phần; nó có thể tạo thành các homodimer trong lưới nội chất nhưng không thể đi vào bộ máy Golgi và được vận chuyển đến màng plasma của tế bào. Về mặt thực nghiệm, khoảng 50% sản phẩm của các đột biến bất hoạt của CASR có thể được “hộ tống” đến màng plasma và được ổn định về mặt chức năng bởi chất calcimimetic dị lập thể cinacalcet, đưa ra một tác nhân điều trị tiềm năng cho việc quản lý bệnh nhân bị tăng canxi máu có triệu chứng do các đột biến mất chức năng của CASR, đặc biệt là những người mắc NSHPT, nếu nó được chứng minh là an toàn ở trẻ em. Các đột biến trong CASR có xu hướng là duy nhất đối với gia đình bị ảnh hưởng. Ở khoảng một phần ba các gia đình mắc HHC1, không có đột biến nào trong vùng mã hóa của CASR được xác định; do đó, họ có thể có một đột biến ở vùng không mã hóa của CASR hoặc một biến thể của GNA11 hoặc AP2S1. Mặc dù các kiểu hình tương tự nhau, đây là hai nguyên nhân di truyền khác của tăng canxi máu giảm canxi niệu được chỉ định là type 2 và 3. Tăng canxi máu giảm canxi niệu di truyền type 2 (OMIM 145981) là do các biến thể mất chức năng của GNA11 (OMIM 139313), mã hóa một tín hiệu truyền G-protein của CaSR (Gα11) đến các con đường truyền tín hiệu nội bào. HHC di truyền type 3 (OMIM 600740) là hậu quả của các đột biến bất hoạt trong AP2S1 (OMIM 602242) mã hóa một phức hợp protein tương tác với β-arrestin (ARRB1, OMIM 107940, chr. 11q13.4) tạo thành một cấu trúc cho phép sự di chuyển của CaSR từ màng tế bào và sự trở lại của nó vào tế bào chất; khi trưởng thành, bệnh nhân mắc HHC3 có thể bị nhuyễn xương giảm phốt phát máu. Các tự kháng thể chống lại miền ngoại bào đầu amino của CaSR ức chế hoạt động của thụ thể một cách có thể đảo ngược đã được xác định ở những bệnh nhân mắc HHC mắc phải; rối loạn này có thể đáp ứng với liệu pháp glucocorticoid. Ở một số bệnh nhân bị cường cận giáp nguyên phát hoặc thứ phát do urê huyết, có sự giảm biểu hiện của CASR và tăng ngưỡng ức chế bài tiết PTH.
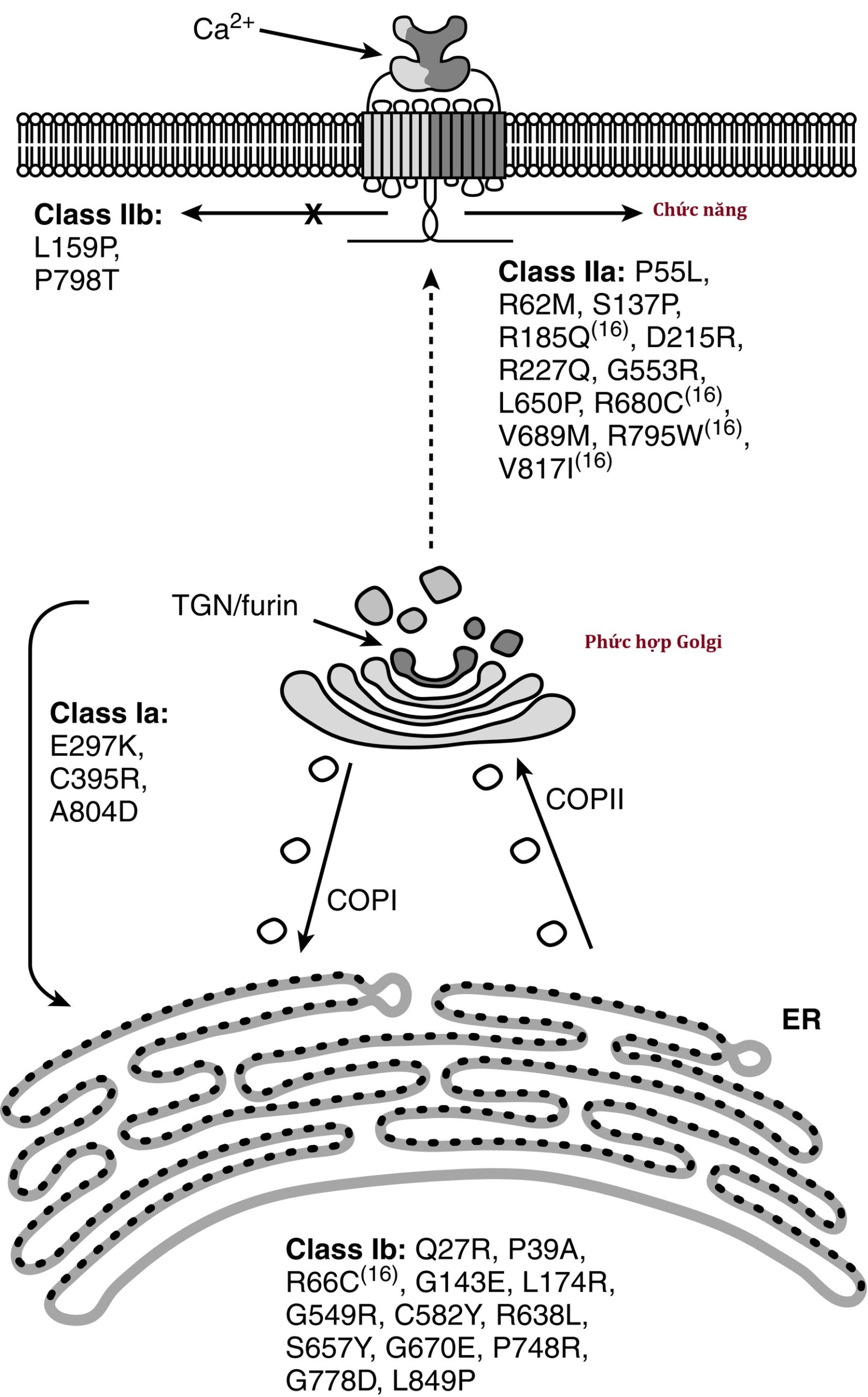
Hình 20.6: Sự di chuyển của thụ thể cảm nhận canxi (CASR) qua lưới nội chất (ER) và phức hợp Golgi đến màng tế bào trước khi chèn vào. Các đột biến bất hoạt của CASR có thể ngăn chặn sự di chuyển của CASR tại các vị trí khác nhau dọc theo con đường này, bao gồm cả ngay dưới màng tế bào. Các đột biến được chỉ định là các lớp Ia, Ib và IIb, tùy thuộc vào vị trí di chuyển bị ngăn chặn như được mô tả. Các đột biến lớp IIa có thể được chèn vào màng tế bào nhưng vẫn hoạt động dưới mức bình thường.
(Từ White E, McKenna J, Cavanaugh A, Breitwieser GE. Pharmacochaperone-mediated rescue of calcium sensing receptor loss-of-function mutants. Mol Endocrinol. 2009;23:1115–1123, với sự cho phép.)
Cường cận giáp nguyên phát là một rối loạn không phổ biến ở trẻ em với tỷ lệ mắc từ 2 đến 5/100.000 so với ở người lớn là khoảng 100/100.000. Ở người lớn bị cường cận giáp, nữ giới nhiều hơn nam giới với tỷ lệ 3:1; ở trẻ em, tỷ lệ nữ/nam gần bằng một. Ở trẻ lớn và vị thành niên, cường cận giáp nguyên phát thường là một bệnh lẻ tẻ và thường là kết quả của một u tuyến cận giáp đơn độc. Cường cận giáp có thể xảy ra như một rối loạn di truyền trội trên NST thường ở những bệnh nhân bị cường cận giáp nguyên phát đơn độc có tính gia đình liên quan đến các biến thể của CDC73 (OMIM 607391) và GCM2 (OMIM 603716) (vide infra). Cường cận giáp có tính gia đình cũng xảy ra ở những bệnh nhân có các biến thể của các gen chịu trách nhiệm cho các hội chứng đa u tuyến nội tiết (MEN) type 1 (MEN1, OMIM 613733), MEN type 2/3 (RET, OMIM 164761), và MEN type 4 (CDKN1B, OMIM 600778).
MEN type 1 và 4 tương tự nhau về mặt lâm sàng ở chỗ cả hai thực thể đều liên quan đến các tổn thương tân sinh của tuyến cận giáp, tuyến yên trước (u tiết somatotropin, u tiết prolactin), và các tế bào tiết nội tiết của đường ruột (u tiết gastrin, u tiết insulin) hoặc đường sinh dục-tiết niệu. MEN1 là hậu quả của các đột biến mất chức năng dị hợp tử trong MEN1 mã hóa một protein giàn giáo hạt nhân tương tác với nhiều yếu tố phiên mã và là một tác nhân ức chế khối u. MEN2A có liên quan đến các biến thể mất chức năng của RET mã hóa một thụ thể tyrosine kinase xuyên màng có liên quan đến u tuyến cận giáp, ung thư biểu mô tuyến giáp thể tủy và u tủy thượng thận; các biến thể RET liên quan đến MEN2A liên quan đến các gốc cysteine trong miền ngoại bào của nó. MEN2B có liên quan đến ung thư biểu mô tuyến giáp thể tủy (MCT) có tính xâm lấn cao, u tủy thượng thận, dày dây thần kinh giác mạc, u hạch thần kinh niêm mạc trên lưỡi, môi và mí mắt, và một thể tạng Marfanoid; về mặt di truyền, MEN2B liên quan đến biến thể RET p.Met918Thr trong exon 16, vị trí của miền tyrosine kinase của thụ thể. MEN4 có liên quan đến các u tuyến của tuyến cận giáp, tuyến yên trước và đường tiêu hóa là hậu quả của các biến thể bất hoạt đơn alen của CDKN1B, một chất ức chế kinase phụ thuộc cyclin chặn chu kỳ tế bào ở pha G0/G1. (Cyclin là các protein tế bào điều chỉnh sự tiến triển của chu kỳ tăng trưởng, phân chia và nhân đôi của tế bào bằng cách kích hoạt các kinase liên quan đến chu kỳ tế bào được chọn [phosphorylase].) Cường cận giáp đơn độc có tính gia đình type 1 (OMIM 145000) và cường cận giáp đơn độc có tính gia đình type 2 liên quan đến các khối u xơ của hàm trên và hàm dưới (OMIM 145001) là do các đột biến mất chức năng trong CDC73 (OMIM 607391) mã hóa một chất ức chế khối u. Cường cận giáp đơn độc có tính gia đình type 3 (610071) đã được xác định vị trí trên nhiễm sắc thể 2p14-p13.3. GCM2 (OMIM 603716) mã hóa một yếu tố phiên mã cần thiết cho sự biệt hóa của các tuyến cận giáp. Các biến thể tăng chức năng của GCM2 dẫn đến cường cận giáp đơn độc có tính gia đình type 4 (OMIM 239200) với sự phát triển của nhiều u tuyến cận giáp và đôi khi là ung thư. NSHPT (OMIM 239200) là hậu quả của các đột biến bất hoạt của CASR (OMIM 601199).
Trẻ em và vị thành niên bị tăng canxi máu do cường cận giáp có thể không có triệu chứng hoặc có thể biểu hiện những thay đổi về nhân cách và hành vi—đặc biệt là trầm cảm, đau đầu, khó chịu, yếu cơ gốc chi, chán ăn, đau quặn bụng, buồn nôn và nôn, táo bón, khát nhiều và tiểu nhiều hoặc có các triệu chứng phản ánh hậu quả của rối loạn này, chẳng hạn như đau hông và tiểu máu do sỏi thận (tăng canxi niệu), đau bụng (viêm tụy), hoặc gãy xương bệnh lý (qua các vùng loãng xương hoặc tổn thương của viêm xương xơ nang). Hiếm khi có thể sờ thấy một khối ở cổ ở những bệnh nhân này, mặc dù khám thực thể có thể cho thấy yếu nhẹ các cơ gốc chi. Tăng canxi máu, hạ phốt phát máu và nồng độ PTH nguyên vẹn trong huyết thanh tăng cao hiện diện ở phần lớn trẻ em bị cường cận giáp. Ở các đối tượng bị cường cận giáp nguyên phát, tăng canxi máu là kết quả của việc tăng tiết PTH do mất mối quan hệ bình thường giữa ngưỡng của Ca2+ huyết thanh và sự tổng hợp và giải phóng PTH và do sự bài tiết PTH độc lập với Ca2+ (cấu thành) liên quan đến khối lượng mô cận giáp. Siêu âm, chụp cộng hưởng từ, chụp cắt lớp vi tính (CT), và xạ hình (xạ hình CT phát xạ đơn photon sestamibi được đánh dấu bằng technicium-99m) đã được sử dụng để xác định vị trí của (các) tuyến cận giáp bất thường trước khi phẫu thuật cắt bỏ. (Sestamibi là methoxyisobutylisonitrile, một cation ưa mỡ.) Đôi khi, khối u cận giáp có thể nằm ở vị trí lạc chỗ trong tuyến ức, tuyến giáp hoặc trung thất. Các nghiên cứu sâu hơn có thể cho thấy sự tái hấp thu xương dưới màng xương, nhiễm canxi thận hoặc sỏi thận.
Về mặt bệnh học, cường cận giáp ở trẻ em thường do một u tuyến tế bào chính liên quan đến một tuyến cận giáp, nhưng các u tuyến ở nhiều tuyến cận giáp và tăng sản lan tỏa (đặc biệt ở bệnh nhân mắc MEN type 1), và hiếm khi là ung thư tế bào chính cũng có thể xảy ra. Phần lớn các u tuyến cận giáp có nguồn gốc đơn dòng; nghĩa là, một tế bào đột biến duy nhất phát triển thành một khối u. Ở thanh thiếu niên, các khối u cận giáp đôi khi có thể phát triển sau khi xạ trị ngoài vào cổ để điều trị u lympho. Trong một số khối u cận giáp, sự biểu hiện tăng lên của cyclin D1 (được mã hóa bởi CCND1, OMIM 168461, chr. 11q13.3) đã được chứng minh. Sự biểu hiện quá mức của CCND1 trong các tế bào chính của tuyến cận giáp đôi khi là kết quả của một đột biến nhiễm sắc thể soma—đảo đoạn (xoay) của các vùng 11p15 và 11q13 trong đó vùng promoter của PTH được định vị lại để phục vụ như một promoter cho CCND1, do đó làm tăng tốc độ phân chia tế bào chính bất cứ khi nào nhận được kích thích (hạ canxi máu) để tạo ra PTH và cuối cùng dẫn đến sự hình thành khối u (lành tính). Tuy nhiên, trong nhiều u tuyến cận giáp, có sự gia tăng hoạt động của cyclin D1 mà không có sự sắp xếp lại nhiễm sắc thể này. Ở những bệnh nhân bị u tuyến và ung thư cận giáp, sự biểu hiện quá mức của các gen ức chế khối u retinoblastoma và p53, mà sản phẩm của chúng thường ức chế chu kỳ tế bào ở bước G1/S, cũng đã được chứng minh cũng như các đột biến soma trong CDC73. Ở khoảng 35% các u tuyến cận giáp lẻ tẻ, một đột biến mất chức năng soma của yếu tố ức chế khối u menin—đột biến dòng mầm ở bệnh nhân mắc MEN type 1—có thể được xác định cùng với sự mất dị hợp tử trên nhiễm sắc thể 11. Các đột biến mất chức năng dòng mầm trong MEN1 và CASR và các biến thể tăng chức năng trong GCM2 cũng đã được phát hiện ở những bệnh nhân bị cường cận giáp nguyên phát đơn độc có liên quan đến sự tham gia của nhiều tuyến; trong trường hợp này, bệnh nhân có đột biến CASR có thể không có các phát hiện sinh hóa điển hình của HHC1 (xem Bảng 20.4B). Suy thận mạn tính dẫn đến cường cận giáp thứ phát do tăng sản các tuyến cận giáp và cũng dẫn đến các khối u cận giáp đơn dòng (cường cận giáp tam phát) liên quan đến các mất đoạn nhiễm sắc thể soma trong một số trường hợp.
Cường cận giáp đơn độc có tính gia đình type 1 (OMIM 145000) và type 2 (OMIM 145001—cường cận giáp liên quan đến các khối u cận giáp lành tính và ác tính có tính gia đình hoặc nhiều u xơ hóa xương của hàm trên và hàm dưới, tương ứng) đều do các đột biến dòng mầm trong CDC73 (OMIM 607393, chr. 1q31.2) mã hóa parafibromin, một protein nội tại của chu kỳ phân chia tế bào. Cường cận giáp xảy ra ở 80% đối tượng có đột biến trong CCD73 ở độ tuổi trung bình là 32 nhưng cũng có thể xuất hiện ở trẻ em trước 10 tuổi. Ở những bệnh nhân này, tổn thương cận giáp có thể là một u tuyến nang không điển hình, có khả năng tiền ác tính (65%), tăng sản (20%), hoặc thậm chí là ung thư (15%); khối u cận giáp có thể là một phát hiện đơn độc hoặc nó có thể liên quan đến các khối u xương hàm trên và/hoặc hàm dưới bao gồm các mô xơ bị hóa xương; các tổn thương thận (u Wilms, ung thư biểu mô tế bào thận nhú, u mô thừa, thận đa nang), tụy (ung thư), và tử cung (khối u) cũng có thể phát triển ở những bệnh nhân này. Các tổn thương trong các tuyến cận giáp có thể phát triển không đồng bộ. CDC73 là một gen có 17 exon mã hóa parafibromin, một protein hạt nhân 531 aa là một thành phần của một phức hợp các yếu tố phụ trợ điều chỉnh hoạt động của RNA polymerase II và của một histone methyltransferase và do đó điều chỉnh biểu hiện gen và sự tăng sinh tế bào—hoạt động như một chất hoạt hóa và ức chế phiên mã. Để một khối u cận giáp phát triển, một “cú đánh thứ hai” phải xảy ra dẫn đến mất dị hợp tử: nghĩa là, đột biến bất hoạt dòng mầm của CDC73 trên một alen phải được khớp với một đột biến trong hoặc mất đoạn của CDC73 trong alen bình thường còn lại. Khi một đột biến dòng mầm trong CDC73 đã được phát hiện, việc sàng lọc các thành viên trong gia đình để tìm đột biến này và đánh giá theo chiều dọc các đối tượng bị ảnh hưởng được khuyến nghị vì các cá nhân không có triệu chứng bị u tuyến không điển hình hoặc ung thư tuyến cận giáp có thể được xác định theo cách này. Các đột biến soma trong CDC73 cũng đã được xác định trong các u tuyến cận giáp không điển hình và nhiều ung thư cận giáp nhưng không phổ biến trong u tuyến cận giáp lẻ tẻ điển hình. Cường cận giáp đơn độc có tính gia đình type 3 (OMIM 610071) đã được liên kết với nhiễm sắc thể 2p14-13.3 nhưng một biến thể gen cụ thể tại vị trí này vẫn chưa được xác định. Cường cận giáp type 4 (OMIM 617343) là kết quả của các biến thể của GCM2 (OMIM 603716) mà sản phẩm của nó là cần thiết cho sự biệt hóa của các tuyến cận giáp.
Các hội chứng MEN là các bệnh di truyền trội trên NST thường có tính gia đình với độ thấm cao, liên quan đến sự phát triển của các khối u ở hai hoặc nhiều tuyến nội tiết trong một cá thể. Có bốn hội chứng MEN được xác định—1, 2A, 2B, và 4 (Bảng 20.5). MEN1 được đặc trưng bởi sự phát triển của các khối u của tuyến cận giáp (u tuyến), tuyến yên (u tiết prolactin, u tiết somatotropin), và đơn vị ruột-tụy (u tiết gastrin, u tiết insulin, u tiết polypeptide tụy, carcinoid). Ngoài ra, các khối u của vỏ thượng thận, các khối u thần kinh nội tiết của cây phế quản-phổi và tuyến ức, u mỡ, u xơ mạch, u collagen, và u màng não phát triển ở các đối tượng bị ảnh hưởng. Tỷ lệ mắc MEN1 sau tử vong là 0,25%; ở các đối tượng cường cận giáp là 1% đến 18%; và ở bệnh nhân bị u tiết gastrin là 16% đến 38%. Cường cận giáp (do một u tuyến cận giáp đơn độc hoặc các khối u trong nhiều tuyến cận giáp hoặc tăng sản cả bốn tuyến cận giáp) là biểu hiện phổ biến nhất của MEN1, xảy ra ở hơn 90% đến 95% bệnh nhân bị ảnh hưởng; nó là bệnh nội tiết thường gặp nhất ở trẻ em mắc MEN1, đôi khi phát triển trước 10 tuổi. Không giống như các dạng cường cận giáp khác, số lượng nam và nữ bị ảnh hưởng trong MEN1 là bằng nhau. Các khối u tuyến yên tiết prolactin và/hoặc GH thường phát triển (30%–40% đối tượng MEN1) cũng như các khối u tiết gastrin (hội chứng Zollinger-Ellison), insulin và glucagon của các tiểu đảo tụy và hệ tiêu hóa (30%–70% bệnh nhân); các khối u này cũng xảy ra ở trẻ em và vị thành niên mắc MEN1. Tăng cortisol máu ở bệnh nhân mắc MEN1 có thể do bài tiết quá mức adrenocorticotropin bởi một u tuyến yên hoặc lạc chỗ bởi một khối u hoặc do một khối u thượng thận nguyên phát. Các khối u tuyến giáp xảy ra ở 25% bệnh nhân mắc MEN1. Các khối u không phải nội tiết, chẳng hạn như u mỡ (34%), carcinoid ruột và phế quản, và các khối u ruột khác là khá phổ biến ở các đối tượng mắc MEN1. Thật vậy, các biểu hiện da liễu của MEN1, bao gồm u xơ mạch (85%), u collagen (70%) là những chỉ số cực kỳ nhạy cảm và đặc hiệu của bệnh này. Bệnh nhân mắc MEN1 cũng có thể phát triển u schwannoma hoặc u tủy thượng thận, loại u sau thường hiện diện ở bệnh nhân mắc MEN type 2A và 2B. Mặc dù hầu hết các khối u phát triển trong MEN1 là lành tính nhưng hoạt động quá mức, những khối u có nguồn gốc từ tụy, ruột và ruột trước có thể là ác tính.
Bảng 20.5: Các hội chứng Đa u tuyến nội tiết
| Phân loại | Gen | NST | Vị trí khối u thường gặp | Đột biến |
|---|---|---|---|---|
| MEN 1 | MEN1 | 11q13.1 | Cận giáp (90%) | Intron 4 N:5168 G ⇒ A (10%) |
| Ruột-tụy (30%–70%)—u gastrin, | Codon 83–84 (4%) | |||
| u insulin, u polypeptide tụy, | Codon 119 (3%) | |||
| không chức năng | Codon 209–211 (8%) | |||
| Tuyến yên trước (30%–40%)—u prolactin, | Codon 418 (4%) | |||
| GH, ACTH, không chức năng | Codon 516 (7%) | |||
| Vỏ thượng thận (40%)—tăng sản lan tỏa và nốt, u tuyến, ung thư | ||||
| Ruột—thần kinh nội tiết dạ dày (10%), | ||||
| Thần kinh nội tiết tuyến ức (2%), | ||||
| Khác: u xơ mạch mặt (85%), u collagen (70%), | ||||
| u mỡ (30%), u màng não (8%) | ||||
| MEN 2 | RET | 10q11.21 | ||
| MEN 2A | MCT (90%) | Codon 634 (Cys ⇒ Arg- 85%) | ||
| U tủy thượng thận (50%) | ||||
| Tăng sản cận giáp (20%–30%) | ||||
| Lichen amyloidosis da | ||||
| MEN 2B 3 | MCT (> 90%) | Codon 918 (Met ⇒ Thr- > 95%) | ||
| U tủy thượng thận (40%–50%) Kèm theo: | ||||
| Thể tạng Marfanoid | ||||
| U thần kinh niêm mạc (90%) | ||||
| Dây thần kinh giác mạc có myelin | ||||
| Phình đại tràng | ||||
| MEN 2C | MCT (100%) | Codon 618 (> 50%) | ||
| MEN 4 | CDKN1B | 12p13.1 | Cận giáp | |
| Tuyến yên | ||||
| Tinh hoàn/Cổ tử cung | ||||
| Thận/Thượng thận |
ACTH, Hormon vỏ thượng thận; GH, hormon tăng trưởng; MCT, ung thư biểu mô tuyến giáp thể tủy.
(Từ Thakker RV, Newey PJ, Walls GV, et al. Clinical Practice Guidelines for Multiple Endocrine Neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97:2990–3011; Marx SJ, Simonds WF. Hereditary hormone excess: genes, molecular pathways, and syndromes. Endocr Rev. 2005;26:615–661.)
Các đột biến dòng mầm trong MEN1, một gen có 10 exon mã hóa một protein tế bào chất và hạt nhân 610 aa được gọi là menin, đã được chứng minh ở phần lớn bệnh nhân mắc các dạng MEN1 có tính gia đình và lẻ tẻ. Menin điều hòa sự tăng trưởng, phân chia và chết của tế bào ở hai vị trí và thông qua nhiều con đường. Trong tế bào chất, menin liên kết với AKT1 (protein kinase B—OMIM 164730, chr. 14q32.33) ngăn cản sự chuyển vị của nó đến màng tế bào nơi nó thường được kích hoạt bằng cách phosphoryl hóa, cho phép AKT1 phát huy tác dụng tăng sinh và chống chết theo chương trình; bằng cách liên kết menin với AKT1, cả nồng độ AKT1 trong tế bào chất và hoạt động kinase của nó đều bị suy giảm, do đó ức chế tác dụng của nó đối với sự phân chia và tồn tại của tế bào. Menin đi vào nhân với sự hướng dẫn của hai tín hiệu định vị hạt nhân ở đầu carboxyl của nó (aa 479–498, 589–608) nơi nó tham gia trực tiếp vào việc điều hòa phiên mã, sao chép và tồn tại. Bằng cách liên kết trực tiếp với JunD, menin chặn sự ức chế qua trung gian JunD đối với phiên mã của protein hoạt hóa-1 (và do đó là sự phân chia tế bào); nhiều đột biến trong MEN1 ở bệnh nhân mắc MEN1 tập trung ở exon 4 và làm gián đoạn sự liên kết của hai protein này (giữa menin aa 139–142 và 323–428). Bằng cách tương tác với small mothers against decapentaplegic (SMAD)3 (OMIM 603109, chr. 15q22.3), menin ức chế tín hiệu của yếu tố tăng trưởng biến đổi (TGF) β và làm suy giảm sự kiểm soát ức chế qua trung gian TGFβ đối với sự sao chép tế bào. Sự tương tác của menin với phức hợp SMAD 1/5 ức chế tín hiệu của protein hình thái xương (BMP) 2; menin cũng ức chế protein điều hòa phiên mã—yếu tố hạt nhân κB (NF-κB). Hơn nữa, menin tương tác trực tiếp với một phức hợp histone methyltransferase và với các gen điều hòa sửa chữa DNA và sao chép và chết theo chương trình của tế bào, chẳng hạn như gen ức chế khối u TP53 (p53), CDKN1A, CDKN1B, GADD45A, POLR2A, BBC3, TP5313, và FAS. Các biến thể của CDKN1A (OMIM 116899), CDKN2B (600431), và CDKN2C (OMIM 603369), các protein điều chỉnh chu kỳ phân chia tế bào, đã được liên kết với cường cận giáp nguyên phát không kèm hội chứng.
Ở các đối tượng có đột biến mất chức năng dòng mầm dị hợp tử trong MEN1, sự tăng trưởng tế bào không được điều hòa và sự hình thành khối u xảy ra khi một tác động thứ hai dẫn đến mất MEN1 trên alen bình thường trong các mô nhạy cảm. Cũng như với CDC73 (vide supra), giả thuyết “hai cú đánh” về sự hình thành khối u trong MEN1 biểu thị rằng bệnh nhân thừa hưởng tính nhạy cảm dòng mầm với tân sinh, trên đó chồng chất một tác động thứ hai, dẫn đến mất dị hợp tử đối với đoạn nhiễm sắc thể 11q13 và mất hai alen của MEN1; cú đánh thứ hai có thể là mất đoạn của một đoạn nhiễm sắc thể 11 bao gồm 11q13 hoặc một đột biến (sai nghĩa, dịch khung) trong chính alen MEN1 kiểu hoang dã, một quan sát cũng mở rộng cho các khối u MEN type 1 có đột biến soma trong MEN1. Vài trăm đột biến dòng mầm trong MEN1 đã được xác định ở bệnh nhân mắc MEN1; 25% là đột biến vô nghĩa, 15% là đột biến sai nghĩa, 45% là chèn hoặc mất đoạn dịch khung, với hơn 80% dẫn đến việc tổng hợp một sản phẩm không hoạt động do mất tín hiệu định vị hạt nhân hoặc khả năng liên kết với JunD hoặc yếu tố hạ nguồn khác. Các “điểm nóng” đột biến dòng mầm đặc biệt nhạy cảm trong MEN1 là chuyển đoạn G ⇒ A ở nucleotide 5168 dẫn đến một vị trí nối mới trong intron 4, các codon 83 đến 84, 118 đến 119, 209 đến 211, 418 và 516, nơi các đột biến đã được xác định chung ở 37% bệnh nhân mắc MEN1. Ở hai bên của nhiều vị trí này là các đoạn trình tự DNA lặp lại của các axit nucleic đơn hoặc của các dinucleotide đến octanucleotide; cấu hình này có thể dẫn đến tăng tính nhạy cảm với “trượt sao chép” do sự sai lệch của các đoạn lặp nucleotide trong quá trình sao chép DNA, cho phép mất hoặc chèn các nucleotide tại các vị trí không phù hợp. Các đột biến trong vùng mã hóa vị trí nối exon/intron của MEN1 đã được phát hiện ở khoảng 70% bệnh nhân mắc MEN1 có tính gia đình; có khả năng ở 30% đối tượng còn lại, có hoặc là mất đoạn một alen MEN1 hoặc một đột biến ở một trong các vùng không mã hóa của nó. Các đột biến mới của MEN1 xảy ra lẻ tẻ ở 10% bệnh nhân mắc MEN1. Cường cận giáp nguyên phát đơn độc, di truyền trội trên NST thường có tính gia đình cũng đã được liên kết một cách khác nhau với các đột biến dòng mầm (ví dụ, Val184Glu, Glu255Lys, Gln260Pro) trong MEN1. Các đột biến soma trong MEN1 cũng đã được xác định ở những bệnh nhân bị các khối u đơn độc, lẻ tẻ của tuyến cận giáp, các tế bào tiểu đảo tụy, tuyến yên trước và vỏ thượng thận. Bệnh rõ ràng về mặt lâm sàng do các đột biến trong MEN1 tăng lên theo tuổi tác: ở 10 tuổi, 7% trẻ em có đột biến trong MEN1 có một bệnh nội tiết có thể phát hiện được (chủ yếu là cường cận giáp); 52% đối tượng 20 tuổi bị ảnh hưởng biểu hiện một hoặc nhiều khối u; độ thấm tăng lên 87% ở tuổi 30, 98% ở tuổi 40 và 100% ở tuổi 60.
Có một số dạng MEN type 2—các phân loại 2A, 2B, và ung thư biểu mô tuyến giáp thể tủy có tính gia đình (2C). MCT là khối u thường gặp nhất trong MEN 2A, xảy ra ở 90% bệnh nhân (OMIM 171400) (xem Bảng 20.5). Nó phát triển từ tình trạng tăng sản tế bào cận nang hoặc tế bào C có từ trước trong tuyến giáp. Những đối tượng này cũng phát triển u tủy thượng thận (50%), tăng sản hoặc u tuyến cận giáp (20%–30%), lichen amyloidosis da cục bộ (các lắng đọng keratin dưới biểu bì gây ngứa trên vai), và phình đại tràng một phần hoặc toàn bộ. Calcitonin, sản phẩm bài tiết của các tế bào C tuyến giáp, cũng được sản xuất với số lượng rất lớn bởi MCT; việc đo lường nó hỗ trợ chẩn đoán MCT và theo dõi đáp ứng của khối u này với điều trị. Ngoài MCT và u tủy thượng thận, bệnh nhân mắc MEN 2B (OMIM 162300) có thể tạng Marfanoid, u hạch thần kinh niêm mạc của môi và lưỡi, u hạch thần kinh đường tiêu hóa, các sợi thần kinh giác mạc có myelin, và phình đại tràng, nhưng họ không phát triển bệnh cận giáp. Ung thư biểu mô tuyến giáp thể tủy đơn độc có tính gia đình là một biến thể của các rối loạn này. Các đột biến tăng chức năng dị hợp tử dòng mầm trong tiền-oncogene RET là cơ sở sinh bệnh học của các MEN di truyền type 2. RET là một gen có 20 exon mã hóa một thụ thể tyrosine kinase màng tế bào được glycosyl hóa 860 aa với các miền ngoại bào, xuyên màng và nội bào được biểu hiện trong mô có nguồn gốc mào thần kinh (các hạch giao cảm, tủy thượng thận, các tế bào cận nang tuyến giáp) (Hình 20.7). Phối tử tự nhiên của thụ thể này là yếu tố dinh dưỡng thần kinh có nguồn gốc từ dòng tế bào thần kinh đệm (GDNF, OMIM 600837). Các đột biến hoạt hóa cấu thành của một trong sáu gốc cysteine trong miền ngoại bào của RET—các codon 609, 611, 618, 620, 630, và đặc biệt là codon 634 (Cys ⇒ Arg)—hiện diện ở bệnh nhân mắc MEN 2A. Mất đi chỉ một gốc cysteine tạo điều kiện cho sự dimer hóa đồng thể của thụ thể mà không cần liên kết phối tử, do đó kích hoạt miền tyrosine kinase nội bào của RET, dẫn đến tự phosphoryl hóa các gốc tyrosine quan trọng (đặc biệt là ở các codon 1015 và 1062), và truyền tín hiệu sau đó. Một đột biến hoạt hóa đã được xác định tại codon 918 (Met ⇒ Thr) ở hơn 95% bệnh nhân mắc MEN 2B; vị trí này nằm trong miền tyrosine kinase và đột biến này cho phép truyền tín hiệu và biến đổi và biệt hóa tế bào thần kinh khi không có cả liên kết phối tử và dimer hóa đồng thể của thụ thể. Đột biến RET liên quan đến MEN IIB p.Met918Thr thường phát sinh de novo từ các đột biến lẻ tẻ xảy ra trong dòng mầm của một người cha lớn tuổi, vì đột biến này mang lại một “lợi thế chọn lọc” cho tinh bào đột biến. Các đột biến sai nghĩa khác của RET liên quan đến MEN 2B đã được xác định tại các codon p.Val804, p.Ala883, và p.Ser904. Các đột biến sai nghĩa tại các codon RET 609, 611, 618, 620, và 634 trong miền ngoại bào và tại các codon 768, 790, 804, và 891 trong miền tyrosine kinase nội bào đã được tìm thấy ở những bệnh nhân bị MCT có tính gia đình. Ở trẻ em, cường cận giáp có thể là một biểu hiện bất thường của hội chứng McCune-Albright (OMIM 174800) do một đột biến hoạt hóa sau hợp tử trong GNAS. Cường cận giáp và đái tháo nhạt do thận đã được ghi nhận ở các đối tượng mắc hội chứng Bartter trước sinh (OMIM 601678) liên quan đến các biến thể hai alen của SLC12A1 (OMIM 600839), gen mã hóa một protein vận chuyển natri-kali-clorua.
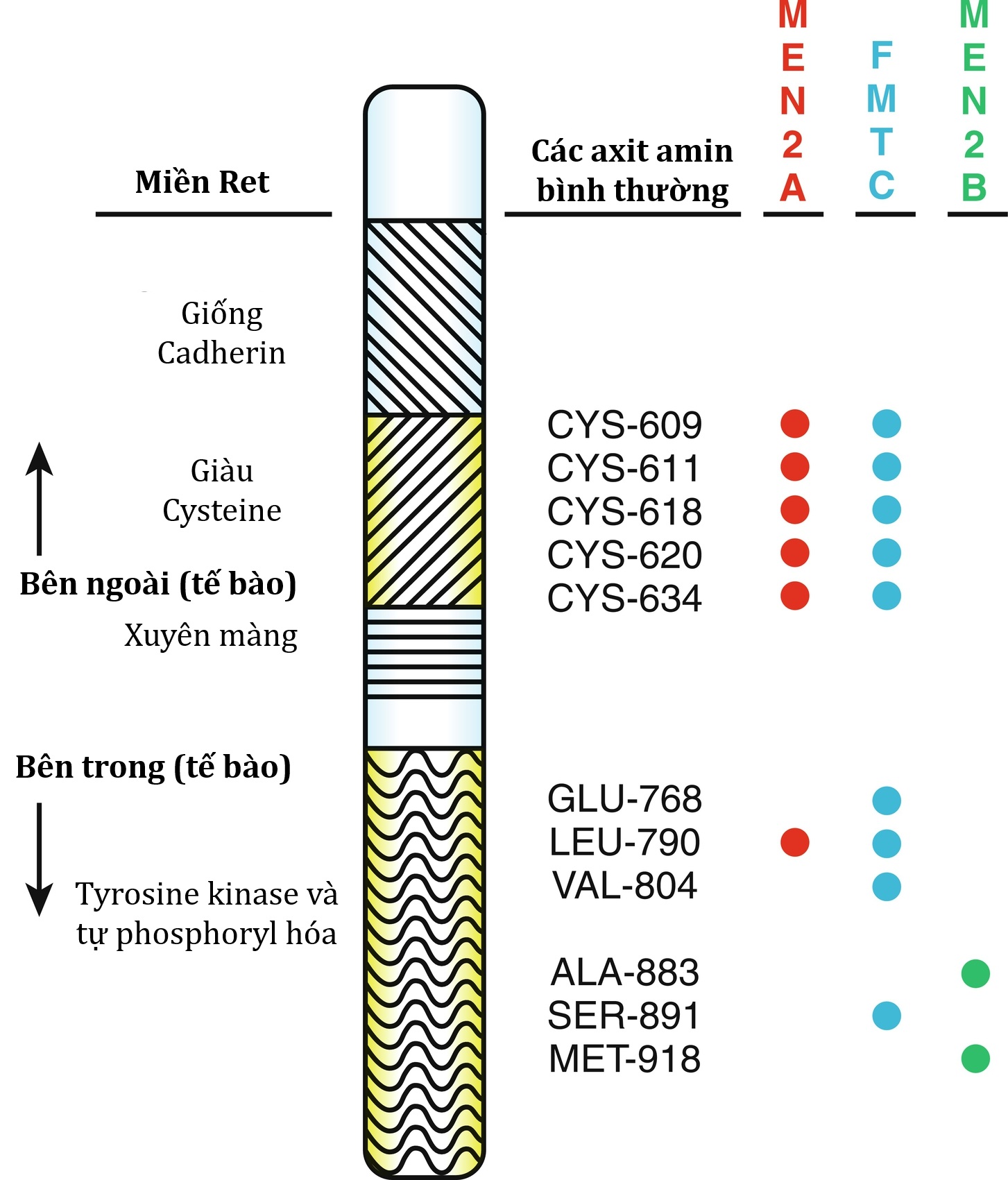
Hình 20.7: Sơ đồ mô tả các miền của tiền-oncogene RET với các vị trí của các đột biến hoạt hóa trong RET liên quan đến đa u tuyến nội tiết type 2A và 2B và ung thư biểu mô tuyến giáp thể tủy có tính gia đình.
(Từ Marx SJ, Simonds WF. Hereditary hormone excess: genes, molecular pathways, and syndromes. Endocr Rev. 2005;26:615–661, với sự cho phép.)
Việc ăn quá nhiều vitamin D hoặc calcitriol vì lý do điều trị (điều trị còi xương, suy cận giáp, hoặc các nguyên nhân khác gây hạ canxi máu), ăn nhiều vitamin, lỗi sản xuất trong việc pha chế các chất bổ sung vitamin, hoặc tăng cường sữa không phù hợp là những nguyên nhân đáng kể gây tăng canxi máu qua trung gian vitamin D. Việc bôi tại chỗ các loại kem chứa vitamin D hoặc một chất tương tự (ví dụ, 22-oxacalcitriol) để điều trị bệnh vẩy nến cũng có thể dẫn đến tăng canxi máu—đặc biệt nếu sự bài tiết canxi qua nước tiểu bị tổn hại. Bệnh nhân mắc các bệnh u hạt (không nhiễm trùng: sarcoidosis, berylliosis, u hạt ái toan, hoại tử mỡ dưới da, bệnh viêm ruột; nhiễm trùng: lao, histoplasmosis, coccidioidomycosis, candidiasis, bệnh mèo cào) và các rối loạn tân sinh (u lympho tế bào B, bệnh Hodgkin, u tế bào mầm) phát triển tăng canxi máu do sự biểu hiện liên quan của CYP27B1 ở bạch cầu đơn nhân (đại thực bào và các tế bào khác) và kết quả là sản xuất calcitriol. Không giống như các tế bào ống thận trong đó hoạt động của 25-hydroxyvitamin D-1α hydroxylase nằm trong ty thể và sự phiên mã của CYP27B1 được điều hòa chặt chẽ bởi PTH, calcitriol, canxi và phốt phát, trong các bạch cầu đơn nhân, enzyme này nằm ở vi thể và gen của nó được biểu hiện cấu thành và sự tổng hợp calcitriol được xác định về mặt định lượng bởi lượng cơ chất. Sự biểu hiện của CYP27B1 ở bạch cầu đơn nhân rất nhạy cảm với sự kích thích bởi IFN-γ và chất truyền tín hiệu sau thụ thể của nó là nitric oxide, cũng như bởi leukotriene C4; nó dễ bị ức chế bởi glucocorticoid, ketoconazole và chloroquine. Ở những bệnh nhân có đột biến bất hoạt của CYP24A1, calcitriol không thể được hydroxyl hóa thêm và bài tiết.
Bệnh nhân mắc bệnh suy giảm miễn dịch mắc phải có thể bị tăng canxi máu do nhiễm các sinh vật tạo u hạt hoặc do các cytokine hoạt hóa hủy cốt bào được tạo ra trong quá trình của rối loạn này. Nồng độ canxi huyết thanh tăng cao cũng đã được ghi nhận ở trẻ em bị suy giáp bẩm sinh, oxalosis nguyên phát, thiếu hụt lactase bẩm sinh, tam nhiễm sắc thể 21, và viêm khớp tự phát vị thành niên (dạng thấp). Ở một số trẻ bị tăng canxi máu, việc sản xuất quá mức prostaglandin có thể có ý nghĩa sinh bệnh học. Tăng canxi máu phát triển thường xuyên ở những đối tượng trẻ mắc dạng giảm phosphatase máu ở trẻ nhỏ, có khả năng là hậu quả của sự phân ly giữa tốc độ hình thành xương thấp và tái hấp thu xương bình thường. Bằng cùng một cơ chế sinh bệnh học, sự bất động cấp tính của trẻ đang phát triển nhanh bị gãy xương đùi hoặc chấn thương tủy sống dẫn đến giảm tích lũy khoáng chất xương và mất liên kết tương tác giữa nguyên bào xương và hủy cốt bào với tốc độ tái hấp thu xương tăng lên, dẫn đến tăng canxi niệu, tăng canxi máu và “loãng xương do không sử dụng cấp tính”. Loãng xương do không sử dụng cấp tính và tăng canxi máu thậm chí có thể xảy ra ở cá nhân bị suy cận giáp hoặc thiếu vitamin D bị bất động. Trẻ em và vị thành niên ăn chế độ ăn ketogenic trong nỗ lực quản lý bệnh động kinh khó chữa có thể phát triển tăng canxi niệu và tăng canxi máu và trải qua sự suy giảm khoáng hóa xương do sự phân ly giữa tốc độ tích lũy xương và tái hấp thu khoáng chất xương. Tăng canxi máu có thể xảy ra ở trẻ em nhận quá nhiều sữa và kiềm để điều trị viêm dạ dày. Nó có thể xảy ra sau khi ghép tủy xương thành công ở trẻ sơ sinh bị bệnh xương hóa đá khi các hủy cốt bào chức năng nhanh chóng tái hấp thu khoáng chất xương dư thừa.
Tăng canxi máu do ung thư hoặc liên quan đến ác tính có thể là hậu quả của việc tổng hợp và bài tiết các tác nhân hoạt hóa hủy cốt bào, chẳng hạn như PTHrP, (hiếm khi là PTH), calcitriol, hoặc các cytokine (IL, yếu tố hoại tử khối u [TNF], TGFβ), hoặc nó có thể do sự xâm lấn và phá hủy trực tiếp xương bởi khối u. Mặc dù tăng canxi máu xảy ra ở ít hơn 1% trẻ em bị ung thư, nó có thể phát triển ở bệnh nhân bị bệnh bạch cầu lympho và đơn nhân cấp, u lympho Hodgkin và không Hodgkin, sarcoma cơ vân, u nguyên bào gan, u nguyên bào thần kinh và sarcoma Ewing. Khi tốc độ tái hấp thu xương vượt quá khả năng bài tiết canxi của ống thận, tăng canxi máu xảy ra sau đó. Việc nạp nhiều canxi và kiềm có thể hấp thu (sữa hoặc các thuốc kháng axit chứa canxi, chẳng hạn như canxi carbonat) cho bệnh loét dạ dày tá tràng hoặc dưới dạng các chất bổ sung chế độ ăn dẫn đến tăng canxi máu do hấp thu, tăng canxi niệu và nhiễm canxi thận. Nuôi dưỡng qua đường tĩnh mạch với quá nhiều canxi hoặc nhôm hoặc quá ít phốt phát cũng có thể dẫn đến tăng canxi máu. Hạ phốt phát máu do nhiều nguyên nhân khác nhau dẫn đến tăng canxi máu khi cơ thể cố gắng duy trì tích số canxi × phốt phát trên 30. Các loại thuốc gây tăng canxi máu bao gồm: thuốc lợi tiểu thiazide làm tăng tái hấp thu canxi ở ống thận và giảm thể tích huyết tương; vitamin D và các chất tương tự làm tăng hấp thu canxi ở ruột; vitamin A và các chất tương tự axit retinoic của nó kích thích tái hấp thu xương; lithium làm tăng ngưỡng bài tiết PTH—do đó làm tăng nồng độ canxi huyết thanh trong khi làm giảm bài tiết canxi qua nước tiểu và do đó bắt chước HHC1. Ở đối tượng nhiễm độc giáp, tăng canxi máu là kết quả của sự kích thích qua trung gian hormon tuyến giáp đối với chức năng hủy cốt bào và sự gia tăng sau đó của tốc độ tái hấp thu xương. Các u tủy thượng thận và một số khối u tiểu đảo có thể liên quan đến tăng canxi máu, trong một số trường hợp là do đồng bài tiết PTHrP. Tăng canxi máu ở bệnh nhân bị suy vỏ thượng thận là hậu quả của việc huy động liên tục canxi xương, giảm lọc cầu thận của cation này và tăng tái hấp thu ở ống thận của nó và được đảo ngược bằng cách phục hồi trạng thái corticoid bình thường.
Trong quá trình hồi phục sau suy thận cấp, nồng độ canxi huyết thanh có thể tăng do sự huy động canxi từ các vị trí lạc chỗ, chẳng hạn như cơ nơi nó đã được lắng đọng trong giai đoạn tăng phốt phát máu của bệnh mà từ đó nó được giải phóng do tiêu cơ vân. Tăng canxi máu có thể phát triển ở bệnh nhân bị suy thận mạn tính do sự kết hợp của nhiều yếu tố bao gồm: bất động, ngộ độc nhôm, ăn quá nhiều thuốc kháng axit chứa canxi hoặc vitamin D hoặc các chất tương tự của nó, và cường cận giáp thứ phát. Sau khi ghép thận, tăng canxi máu thường là kết quả của cường cận giáp thứ phát do phì đại và tăng sản các tế bào chính của tuyến cận giáp xảy ra để đáp ứng với các tác dụng kích thích PTH của tăng phốt phát máu, hạ canxi máu và giảm tổng hợp và đáp ứng với calcitriol trong giai đoạn suy thận mạn tính. Ở những bệnh nhân có chức năng thận bị tổn hại, hạ canxi máu nhẹ và thiếu hụt calcitriol phát triển khi tốc độ lọc cầu thận giảm xuống dưới 80 đến 60 mL/phút/1,73 m2, trong khi sự giữ phốt phát xảy ra sau khi tốc độ lọc cầu thận đã giảm xuống 60 đến 30 mL/phút/1,73 m2. Sự bài tiết PTH tăng lên thứ phát ở những bệnh nhân này trong nỗ lực tăng tổng hợp calcitriol, tăng tái hấp thu canxi ở ống thận, tăng nồng độ canxi, giảm tái hấp thu phốt phát ở ống thận và giảm giá trị phốt phát. Cường cận giáp thứ phát kéo dài, không được kiểm soát có thể dẫn đến tăng chức năng cận giáp tương đối tự chủ (“cường cận giáp tam phát”) và tăng canxi máu, chủ yếu ở bệnh nhân bị suy thận mạn tính. Tăng sản các tế bào chính được theo sau bởi các khiếm khuyết trong chức năng của CaSR và mất sự điều hòa giảm hiệu quả của bài tiết PTH, khó điều trị đối với nồng độ Ca2+ huyết thanh tăng cao. Có một số lượng lớn các tế bào chính đơn dòng trong đó sự biểu hiện của CASR và số lượng các thụ thể hạt nhân của vitamin D đã giảm. Cường cận giáp thứ phát và tam phát đã xảy ra ở những bệnh nhân bị còi xương do thiếu vitamin D dinh dưỡng kéo dài và ở các đối tượng bị còi xương giảm phốt phát máu liên kết X đang nhận một lượng lớn phốt phát. Cường cận giáp thứ phát (trong đó theo định nghĩa, nồng độ canxi huyết thanh là bình thường) cũng phát triển ở những bệnh nhân có chế độ ăn không đủ canxi, suy giảm hấp thu canxi ở ruột (không dung nạp lactose, ăn phytate, hội chứng kém hấp thu do suy tụy hoặc bệnh celiac), hoặc mất canxi quá mức trong nước tiểu hoặc các mô mềm. Sự bài tiết PTH tăng cường nhưng thoáng qua có thể đi kèm với việc dùng GH cho thanh thiếu niên bị suy thận mạn tính, có khả năng là kết quả của việc chồng chất lên một tốc độ bài tiết PTH cơ bản cao sự gia tăng hơn nữa của tốc độ tái tạo xương liên quan đến somatotropin và các hormon sinh dục. Ở người lớn bị bệnh cấp tính, việc dùng GH cũng có liên quan đến tăng canxi máu.
Tăng canxi niệu đơn độc ở trẻ có canxi máu bình thường có thể là vô căn hoặc do rối loạn chức năng tủy hoặc ống thận hoặc tăng hấp thu canxi ở ruột, bao gồm các đột biến trong các gen mã hóa vitamin D và các thụ thể CaSR và adenylyl cyclase hòa tan, các rối loạn mất khoáng, chẳng hạn như viêm khớp tự phát vị thành niên, nuôi dưỡng qua đường tĩnh mạch, nhiễm toan chuyển hóa, ăn quá nhiều protein và đái tháo đường; tăng canxi niệu có/không có tăng canxi máu có thể được quan sát thấy ở những bệnh nhân mắc các dạng hạ magiê máu có tính gia đình, một số loại hội chứng Bartter và toan hóa ống thận xa.
Đánh giá
Đánh giá trẻ bị tăng canxi máu đòi hỏi phải kiểm tra có hệ thống từng nguyên nhân có thể có của nó (xem Hình 20.5). Việc xem xét cẩn thận tiền sử cá nhân và gia đình và khám thực thể kỹ lưỡng trước khi đo nồng độ canxi toàn phần và ion hóa, phốt phát, creatinin, PTH nguyên vẹn và 25-hydroxyvitamin D trong huyết thanh và bài tiết canxi và creatinin qua nước tiểu; siêu âm thận được sử dụng để phát hiện nhiễm canxi thận hoặc sỏi thận nếu có tăng canxi niệu. Nếu nồng độ PTH không bị ức chế ở bệnh nhân bị tăng canxi máu, sự hiện diện của cường cận giáp cần được xem xét thêm. Nếu các lần xác định lặp lại phù hợp với cường cận giáp, thì sinh bệnh học của nó cần được xác định. Các nghiên cứu hình ảnh của cổ cần được thực hiện có thể bao gồm siêu âm (một u tuyến cận giáp có độ phản âm kém), xạ hình technicium-99m sestamibi với chụp cắt lớp vi tính phát xạ đơn photon (SPECT), chụp CT động (4D) (hữu ích để phát hiện nhiều u tuyến cận giáp và u tuyến lạc chỗ), và chụp cộng hưởng từ (do đó tránh phơi nhiễm bức xạ).
Khi tăng canxi máu nhẹ (nồng độ canxi toàn phần < 12 mg/dL), có thể có ít, nếu có, triệu chứng; do đó, trẻ em/vị thành niên bị tăng canxi máu nhẹ thường được xác định bất ngờ bằng một bảng xét nghiệm hóa học máu sàng lọc được thực hiện cho một mục đích khác. Tăng canxi máu cũng có thể được phát hiện trong các nghiên cứu về sỏi thận, khối lượng xương bất thường, gãy xương bệnh lý, hoặc trong quá trình sàng lọc gia đình để tìm các vấn đề liên quan. Nồng độ canxi huyết thanh (toàn phần hoặc ion hóa) tăng cao trong một mẫu duy nhất cũng có thể phản ánh sự thay đổi của xét nghiệm và phải được xác minh bằng các lần xác định lặp lại trong một phòng xét nghiệm đáng tin cậy. Tăng canxi máu giả là sự hiện diện của nồng độ canxi toàn phần tăng cao dai dẳng trong khi Ca2+ bình thường và được tìm thấy ở các đối tượng bị tăng albumin máu và các trạng thái rối loạn protein máu khác. Các triệu chứng do tăng canxi máu là độc lập với nguyên nhân của nó và liên quan đến mức độ tăng canxi máu và bao gồm: đường ruột—chán ăn, buồn nôn, nôn, đau bụng (loét dạ dày tá tràng, viêm tụy cấp), và táo bón; đường tiểu—khát nhiều, tiểu đêm, và tiểu nhiều (canxi hoạt động như một thuốc lợi tiểu thẩm thấu trong khi tăng canxi máu làm suy giảm chức năng cô đặc của ống lượn xa); xương—đau xương; hệ thần kinh—đau đầu, yếu cơ, suy giảm khả năng tập trung, tăng nhu cầu ngủ, thay đổi ý thức (từ lơ mơ và lẫn lộn đến khó chịu, mê sảng, sững sờ và hôn mê); đôi khi, trầm cảm có thể là mối quan tâm chính khi nhập viện ở một vị thành niên bị tăng canxi máu. Ở trẻ mới biết đi và trẻ nhỏ, tăng canxi máu được biểu hiện bằng chán ăn, táo bón, tăng cân kém và suy giảm tăng trưởng tuyến tính (“chậm phát triển”). Trong một loạt 52 trẻ em và vị thành niên bị tăng canxi máu do cường cận giáp nguyên phát, 80% có triệu chứng; các triệu chứng phổ biến nhất là mệt mỏi/lơ mơ (35%), trầm cảm (14%), đau đầu (35%), buồn nôn (29%), nôn (23%), và khát nhiều (21%). Sự liên quan đến xương (khối lượng xương thấp, gãy xương) hiện diện ở 30%. Tất cả trẻ em (n = 17) bị sỏi thận trong loạt bài này đều có triệu chứng. Trong một loạt 44 trẻ em và vị thành niên khác (26 nữ) bị cường cận giáp nguyên phát, tuổi trung bình tại thời điểm chẩn đoán là 13 tuổi (dao động 6–18 tuổi), tổng cộng có 37 người có triệu chứng (chán ăn, sụt cân, khó chịu, trầm cảm) và có 18 bệnh nhân bị sỏi thận. Tại cuộc phẫu thuật, 29 bệnh nhân có một u tuyến cận giáp và 11 người có các tuyến cận giáp tăng sản—hai trong số đó mắc MEN. Một số đặc điểm của cường cận giáp ở trẻ em và vị thành niên khác với ở người lớn, bao gồm các u tuyến nặng hơn và lớn hơn và nồng độ canxi trong huyết thanh và nước tiểu cao hơn nhưng nồng độ iPTH, phốt phát và phosphatase kiềm trong huyết thanh tương đương.
Đánh giá trẻ bị tăng canxi máu bắt đầu bằng việc xem xét tiền sử, trong đó gia đình/bệnh nhân được hỏi không chỉ về các triệu chứng liên quan đến tăng canxi máu và các hậu quả của nó (sỏi thận) mà còn về việc có thể ăn quá nhiều vitamin D, vitamin A và các hợp chất liên quan (chẳng hạn như axit retinoic để điều trị mụn trứng cá), canxi (có lẽ để “ngăn ngừa” loãng xương), và kiềm, hoặc các loại thuốc ảnh hưởng đến chuyển hóa canxi (thuốc lợi tiểu thiazide có thể “làm lộ ra” cường cận giáp bằng cách tăng tái hấp thu canxi ở ống thận, do đó nâng nồng độ canxi giới hạn lên phạm vi tăng canxi máu). Tiền sử gia đình được khám phá để tìm các thành viên mắc các rối loạn chuyển hóa canxi đã biết (HHC1, cường cận giáp, sỏi thận) hoặc các khối u có tính gia đình (tiết sữa như một dấu hiệu của u tiết prolactin, bệnh loét dạ dày tá tràng nặng như một chỉ báo của u tiết gastrin). Ngoại trừ trong các trường hợp cực đoan khi có thể có tăng huyết áp (nếu được bù nước bình thường) hoặc nhịp tim chậm, mất nước, giảm sức mạnh cơ bắp hoặc thay đổi ý thức, hoặc ở đối tượng Marfanoid mắc MEN IIB, khám thực thể trẻ em và vị thành niên bị tăng canxi máu thường là bình thường. Hiếm khi có thể sờ thấy một khối cạnh khí quản (cận giáp) ở bệnh nhân cường cận giáp. (Các đối tượng bị tăng canxi máu do hoại tử mỡ dưới da có các khối cứng, không đều, di động rải rác trên thân và các chi. Những người mắc WBS có khuôn mặt điển hình trong khi những người mắc loạn sản sụn đầu xương Jansen có các biến dạng xương đặc trưng.)
Sau khi xác nhận sự hiện diện của tăng canxi máu toàn phần và ion hóa, bài tiết canxi qua nước tiểu được đo tiếp theo (Bảng 20.6). Nếu nồng độ PTH bình thường hoặc tăng và bài tiết canxi thấp, rất có thể bệnh nhân mắc HHC1; chẩn đoán này có thể được chứng minh bằng việc tìm thấy HHC không triệu chứng ở một trong hai cha mẹ và được xác định thêm bằng cách xác định đột biến bất hoạt trong CASR. Nếu bệnh nhân bị tăng canxi niệu, nên tìm kiếm các nguyên nhân khác gây tăng canxi máu. Với các xét nghiệm miễn dịch có độ nhạy và độ đặc hiệu cao cho PTH 1-84 nguyên vẹn so với các giá trị canxi huyết thanh, việc phân biệt bệnh nhân bị cường cận giáp với những người mắc các nguyên nhân khác gây tăng canxi máu trong đó giá trị PTH thấp hoặc dưới mức bình thường thường là có thể. Khi không có cường cận giáp thứ phát (suy thận mạn tính, hội chứng kém hấp thu, uống thuốc lợi tiểu thiazide hoặc lithium), nồng độ PTH tăng cao nhất quán ở trẻ em hoặc vị thành niên bị tăng canxi máu, hạ phốt phát máu, tăng canxi niệu phù hợp với cường cận giáp nguyên phát. Mặc dù chẩn đoán cường cận giáp nguyên phát thường khá rõ ràng ở trẻ em và vị thành niên, thỉnh thoảng có bệnh nhân mà trong đó các giá trị canxi huyết thanh và/hoặc PTH có thể không tăng trong một mẫu duy nhất và trong đó cần phải đo lặp lại các giá trị canxi và PTH trong huyết thanh và nước tiểu trước khi chẩn đoán này có thể được xác lập. Ở 52 trẻ em/vị thành niên bị cường cận giáp, các giá trị canxi huyết thanh bình thường ở 10% và nồng độ PTH ở 15%; tuy nhiên, ở tất cả các đối tượng, nồng độ PTH tăng một cách không phù hợp so với nồng độ canxi. Tăng canxi máu, hạ phốt phát máu và nồng độ PTH tăng cao đã được ghi nhận ở tất cả 44 trẻ em và vị thành niên (6–18 tuổi tại thời điểm chẩn đoán) bị cường cận giáp nguyên phát trong một loạt thứ hai. Viêm xương xơ nang, các khối u nâu (các vùng tái hấp thu xương cục bộ không phải tân sinh bao gồm các tế bào khổng lồ đa nhân giống hủy cốt bào, các tế bào hình thoi giống nguyên bào sợi và các thâm nhiễm xuất huyết), và sự tái hấp thu xương dưới màng xương và nội mạc xương có thể được phát hiện bằng X-quang ở hầu hết trẻ em bị cường cận giáp, trong khi BMD vỏ xương (đầu xa xương quay) có khả năng bị giảm ở những đối tượng này. Các phát hiện không đặc hiệu ở đối tượng bị tăng canxi máu do nhiều nguyên nhân khác nhau bao gồm rút ngắn khoảng QT bằng điện tâm đồ vì tốc độ tái cực của tim được tăng tốc bởi nồng độ canxi tăng, nhịp tim chậm và block nhĩ thất độ một, và nhiễm canxi thận và sỏi thận được phát hiện bằng siêu âm. Nồng độ PTHrP huyết thanh nên được đo khi các phát hiện lâm sàng và xét nghiệm phù hợp với cường cận giáp nguyên phát, nhưng giá trị PTH thấp và nghi ngờ tăng canxi máu thể dịch do ác tính. Khi nồng độ PTH thấp ở bệnh nhân bị tăng canxi máu, các chất chuyển hóa của vitamin D (calcidiol, calcitriol) nên được đo và tìm kiếm các nguyên nhân khác gây tăng canxi máu.
Bảng 20.6: Các phát hiện xét nghiệm ở bệnh nhân Tăng canxi máu
| Rối loạn | Canxi | Phốt phát | Canxi niệu | PTH | PTHrP | 25OHD | 1,25(OH)2 D |
|---|---|---|---|---|---|---|---|
| NSHPT | ⇑⇑⇑ | ⇓ | ⇓ | ⇑⇑ | ⇓ | N | ⇑ |
| FHH/HHC | ⇑, ⇑⇑ | ⇓ | ⇓ | ⇑ | ⇓ | N, ⇓ | ⇑ |
| Cường cận giáp nguyên phát | ⇑, ⇑⇑ | ⇓ | ⇑ | ⇑, ⇑⇑ | ⇓ | N | ⇑ |
| Ngộ độc vitamin D | ⇑ | ⇑ | ⇑⇑ | ⇓ | ⇓ | ⇑⇑ | ⇑ |
| WBS | ⇑ | ⇑ | ⇑ | ⇓ | ⇓ | N | ⇑ |
| Bất động | ⇑, ⇑⇑ | ⇑ | ⇑⇑ | ⇓ | ⇓ | N | ⇓ |
| Ác tính | ⇑⇑ | ⇓ | ⇑ | ⇓ | ⇑⇑ | N | N, ⇑, ⇓ |
| Bệnh u hạt | ⇑ | ⇑ | ⇑⇑ | ⇓ | ⇓ | N | ⇑⇑ |
| Hoại tử mỡ dưới da | ⇑ | ⇑ | ⇑ | ⇓ | ⇓ | N | ⇑⇑ |
⇑, Tăng; ⇑⇑, tăng nhiều; ⇓, giảm; FHH, tăng canxi máu giảm canxi niệu có tính gia đình; HHC, tăng canxi máu giảm canxi niệu; N, bình thường; NSHPT, cường cận giáp nặng ở trẻ sơ sinh; PTH, hormon cận giáp; PTHrP, peptide liên quan đến PTH; WBS, hội chứng Williams-Beuren.
(Từ Lietman SA, Germain-Lee EL, Levine MA. Hypercalcemia in children and adolescents. Curr Opin Pediatr. 2010;22:508–515; Benjamin RW, Moats-Staats BM, Calikoglu A, et al. Hypercalcemia in children. Pediatr Endocrinol Rev. 2008;5:778–784.)
Trước phẫu thuật, một u tuyến cận giáp có thể được xác định vị trí bằng siêu âm độ phân giải cao, chụp cắt lớp vi tính phát xạ đơn photon động (4D) (SPECT), xạ hình với 99mTc-sestamibi kết hợp với SPECT, hoặc chụp cộng hưởng từ. Phóng xạ 99mTc-sestamibi được cả tuyến giáp và tuyến cận giáp hấp thu nhưng nhanh chóng bị “rửa trôi” khỏi tuyến giáp; do đó, hai lần quét cách nhau 2 giờ cho phép phân biệt mô cận giáp với mô tuyến giáp. Xạ hình 99mTc-sestamibi có thể được thực hiện bằng các kỹ thuật hai chiều thông thường hoặc CT, kỹ thuật sau cung cấp hình ảnh ba chiều; các góc nhìn bên có thể xác định vị trí tuyến cận giáp bị phì đại tốt hơn. Việc kết hợp phương pháp SPECT 99mTc-sestamibi và CT đã thành công trong việc xác định vị trí các u tuyến cận giáp lạc chỗ. Việc kết hợp xạ hình 99mTc-sestamibi và siêu âm mang lại độ nhạy gần 90% ở trẻ em bị u tuyến cận giáp. Hiếm khi cần phải thực hiện thông tĩnh mạch chọn lọc với việc lấy mẫu nồng độ PTH tại chỗ và/hoặc chụp động mạch để xác định vị trí của một u tuyến cận giáp. Việc đánh giá các khối u nội tiết liên quan là cần thiết, nếu tiền sử gia đình gợi ý khả năng mắc MEN 1 hoặc MEN IIA hoặc nếu có các phát hiện lâm sàng (tiết sữa, tăng trưởng quá mức, tăng huyết áp) gợi ý sự hiện diện của u tiết prolactin, u tiết somatotropin, hoặc u tủy thượng thận. Bệnh nhân có nguy cơ mắc MEN có thể được sàng lọc bằng cách xác định nồng độ prolactin, GH/yếu tố tăng trưởng giống insulin 1 (IGF-1), gastrin, glucagon, polypeptide tụy, calcitonin, catecholamine, và các chất khác trong huyết thanh cơ bản và sau kích thích khi cần thiết. Sàng lọc di truyền các gen ứng cử viên MEN cũng có thể hữu ích. Thật vậy, việc xem xét sàng lọc trẻ em bị cường cận giáp trước phẫu thuật để tìm u tủy thượng thận liên quan hoặc các khối u của hàm trên và hàm dưới và để tìm các đột biến trong RET, MEN1, và CDC73 là hợp lý.
Ở bệnh nhân bị tăng canxi máu do ăn quá nhiều vitamin D, nồng độ calcidiol trong huyết thanh tăng rõ rệt; ở những người nhận calcitriol ngoại sinh hoặc ở những bệnh nhân bị tăng canxi máu mắc các bệnh u hạt, viêm mạn tính và u lympho, nồng độ calcitriol huyết thanh tăng. Các rối loạn khác liên quan đến tăng canxi máu (xem Bảng 20.4A) nên được loại trừ bằng các phát hiện tiền sử và các nghiên cứu xét nghiệm và di truyền thích hợp.
Quản lý
Việc quản lý thích hợp bệnh nhân bị tăng canxi máu phụ thuộc vào mức độ nghiêm trọng và nguyên nhân của nồng độ canxi cao. Điều quan trọng là phải phân biệt bệnh nhân bị tăng canxi máu giả (do tăng protein máu) với bệnh nhân có nguyên nhân bệnh lý gây tăng canxi máu để có thể bắt đầu liệu pháp y tế và phẫu thuật hiệu quả kịp thời. Điều quan trọng không kém là nhận ra trẻ em/vị thành niên mắc HHC1 để tránh điều trị tích cực. Khi nồng độ canxi dưới 12 mg/dL ở đối tượng không có triệu chứng, việc điều trị có thể bị trì hoãn cho đến khi nguyên nhân của tăng canxi máu được hiểu rõ; trong thời gian chờ đợi, việc khuyến nghị bệnh nhân tăng lượng dịch nạp vào, tránh các chất bổ sung canxi và vitamin D, và ngừng các loại thuốc liên quan đến tăng canxi máu nếu có liên quan là hợp lý. Trẻ mắc HHC1 thường có nồng độ canxi toàn phần trong huyết thanh từ 11 đến 13 mg/dL nhưng không có triệu chứng lâm sàng và không cần điều trị trong các trường hợp thông thường. Khi không có HHC1, nếu nồng độ canxi toàn phần trong huyết thanh vượt quá 12 mg/dL hoặc nếu trẻ có triệu chứng (chán ăn, buồn nôn, thay đổi tri giác, khát nhiều, tiểu nhiều, mất nước, yếu cơ, nhịp tim chậm với khoảng QT kéo dài), các nỗ lực để hạ nồng độ canxi huyết thanh thường là cần thiết vì các tác dụng bất lợi của tăng canxi máu đối với chức năng tim, thần kinh trung ương, thận và tiêu hóa. Ở những trẻ em và vị thành niên này, các nghiên cứu chẩn đoán như đã nêu ở trên và điều trị để hạ nồng độ canxi huyết thanh nên bắt đầu đồng thời. Các công cụ điều trị tăng canxi máu thường được sử dụng tuần tự.
Liệu pháp điều trị tăng canxi máu nặng bao gồm: (1) bù nước—bằng nước muối sinh lý 0,9% (gấp đôi thể tích duy trì trong 24–48 giờ)—do đó phục hồi thể tích nội mạch, pha loãng và giảm nồng độ Ca2+ huyết thanh, tăng lọc cầu thận của Ca2+, giảm tái hấp thu Ca2+ ở ống lượn gần và xa, và thúc đẩy bài niệu canxi; bù nước đơn độc thường làm giảm nồng độ canxi toàn phần trong huyết thanh từ 1 đến 3 mg/dL; (2) bài niệu canxi—đạt được bằng cách truyền tĩnh mạch thuốc lợi tiểu quai furosemide (1–2 mg/kg chậm) được bắt đầu sau khi phục hồi thể tích dịch ngoại bào bằng nước muối sinh lý làm giảm thêm nồng độ canxi bằng cách ức chế tái hấp thu canxi (và natri) bởi nhánh lên dày của quai Henle—thuốc lợi tiểu quai nên được giới hạn cho những bệnh nhân bị quá tải dịch trong khi nhận nước muối sinh lý vì các tác nhân này có thể dẫn đến mất nước và suy giảm tốc độ lọc cầu thận; bumetanide là một thuốc lợi tiểu quai tương tự nhưng mạnh hơn có thể được xem xét sử dụng ở bệnh nhân bị tăng canxi máu; (thuốc lợi tiểu thiazide cần được tránh vì chúng làm tăng tái hấp thu canxi ở ống thận và tăng nồng độ canxi huyết thanh và rõ ràng là chống chỉ định trong việc quản lý tăng canxi máu); (3) ức chế tái hấp thu xương—nếu tăng canxi máu không đáp ứng với các biện pháp trước đó, có thể sử dụng một bisphosphonate ức chế chức năng hủy cốt bào: pamidronate (0,5–2 mg/kg đến 90 mg truyền tĩnh mạch trong 4–6 giờ) và axit zoledronic (0,025–0,05 mg/kg đến 4 mg truyền tĩnh mạch trong 30–60 phút) đã được chứng minh là hữu ích trong việc quản lý cấp tính tăng canxi máu ở trẻ em và vị thành niên. Tác dụng hạ canxi máu đầy đủ của bisphosphonate được thiết lập trong vài ngày sau khi dùng và kéo dài trong nhiều ngày đến nhiều tuần; sau khi truyền bisphosphonate, các tác dụng toàn thân cấp tính (sốt, đau cơ) thường được quan sát thấy và có thể được quản lý theo triệu chứng. Calcitonin cá hồi làm giảm nồng độ canxi huyết thanh nhanh chóng nhưng thoáng qua bằng cách ức chế chức năng hủy cốt bào và gây bài niệu canxi; nó được tiêm dưới da (2–4 U/kg mỗi 6–12 giờ). Calcitonin và một bisphosphonate có thể được kết hợp để hạ nồng độ canxi huyết thanh nhanh hơn.
Ở người lớn bị tăng canxi máu do ác tính, denosumab, một kháng thể đơn dòng đối với phối tử của chất hoạt hóa thụ thể của yếu tố hạt nhân κB (RANK) cản trở sự hình thành, chức năng và tồn tại của hủy cốt bào, đã được sử dụng thành công và là một lựa chọn ở trẻ em bị tăng canxi máu, nhưng kinh nghiệm với tác nhân này ở trẻ em còn hạn chế. Tác dụng của denosumab đối với chu chuyển xương có thể đảo ngược nhanh chóng; do đó, tăng canxi máu có thể tái phát sau khi ngừng sử dụng tác nhân này. Mặc dù glucocorticoid không làm giảm nồng độ canxi ở bệnh nhân bị cường cận giáp hoặc các khối u đặc ác tính, chúng có hiệu quả trong việc quản lý tăng canxi máu do ăn quá nhiều vitamin D hoặc sản xuất calcitriol bởi các bạch cầu đơn nhân hoạt hóa hoặc các bệnh ác tính huyết học. Hiếm khi, có thể cần phải lọc máu (lọc màng bụng hoặc chạy thận nhân tạo với dịch lọc có nồng độ canxi thấp hoặc bằng không) cho bệnh nhân bị tăng canxi máu nặng kháng với liệu pháp thông thường. Trong quá trình điều trị cấp tính tăng canxi máu, nồng độ Ca2+ có thể được đánh giá gián tiếp bằng cách theo dõi khoảng QT (bị rút ngắn) bằng điện tâm đồ, nhưng việc đo nối tiếp nồng độ canxi toàn phần và Ca2+ là cần thiết để theo dõi hiệu quả của liệu pháp và ngăn ngừa hạ canxi máu.
Mặc dù ở người lớn tuổi (> 50 tuổi) bị cường cận giáp nguyên phát không triệu chứng (không có bệnh xương hoặc sỏi thận), đôi khi có thể không có lời khuyên can thiệp ngay lập tức, ở người trẻ tuổi, trẻ em và vị thành niên bị cường cận giáp, can thiệp phẫu thuật bởi một bác sĩ phẫu thuật có kinh nghiệm và chuyên môn được khuyến nghị khi chẩn đoán được xác lập. Trong khi cường cận giáp ở thanh thiếu niên thường (> 60%) là do một u tuyến, tăng sản các tuyến cận giáp cũng phổ biến (~ 30%). Trước khi can thiệp phẫu thuật ở trẻ em/vị thành niên bị cường cận giáp, một u tuyến cận giáp nên được xác định vị trí bằng một (hoặc nhiều) kỹ thuật hình ảnh, chẳng hạn như siêu âm có phân tích Doppler, chụp cộng hưởng từ, CT độ phân giải cao và bốn chiều hoặc 99mtechnetium-sestamibi kết hợp với CT (Hình 20.8). Việc xây dựng một mô hình ảo ba chiều của cổ bằng cách kết hợp thuốc cản quang iốt với chụp CT lát cắt mỏng với độ tương phản màu cho phép xác định rõ ràng hệ mạch máu cổ và các cấu trúc bình thường và bất thường của nó. Ở người lớn và vị thành niên lớn tuổi, các thủ thuật xâm lấn tối thiểu tập trung để loại bỏ một (các) u tuyến cận giáp được xác định về mặt giải phẫu có thể được sử dụng bằng cách dùng 99mtechnetium-sestamibi 2 giờ trước phẫu thuật, với việc mổ xẻ được hướng dẫn bằng cách đưa vào một đầu dò gamma cầm tay, cho đến khi u tuyến được định vị và loại bỏ. Hỗ trợ video là một phương pháp thay thế để xác định vị trí của một u tuyến cận giáp. Một kỹ thuật bổ sung để đánh giá tính đầy đủ của việc loại bỏ u tuyến trong mổ là đo nồng độ PTH ngoại vi bằng xét nghiệm miễn dịch nhanh trước và 10 phút sau khi loại bỏ u tuyến; sự suy giảm 50% nồng độ PTH cho thấy đã cắt bỏ hoàn toàn; nếu nồng độ PTH không giảm sau khi loại bỏ mô cận giáp bị nghi ngờ bất thường, việc thăm dò sâu hơn sẽ được thực hiện và mô bổ sung được loại bỏ theo hướng dẫn của nồng độ PTH huyết thanh. Nồng độ PTH huyết thanh đạt giá trị bình thường sau 15 đến 30 phút sau khi loại bỏ một u tuyến cận giáp hoạt động đơn độc. Các thủ thuật này hiệu quả về chi phí vì chúng làm giảm thời gian phẫu thuật và tỷ lệ mắc bệnh của bệnh nhân; nhiều bệnh nhân trở về nhà trong vòng vài giờ sau khi rời phòng mổ. Các kỹ thuật này đã được áp dụng vào việc quản lý phẫu thuật cho trẻ em và trẻ sơ sinh bị u tuyến cận giáp hoặc tăng sản nhiều tuyến cận giáp. Nếu có tăng sản cận giáp, sẽ thực hiện cắt bỏ toàn bộ tuyến cận giáp và tự cấy ghép các mảnh nhỏ của một tuyến vào một túi ở cẳng tay. Sau khi loại bỏ một u tuyến cận giáp, nhiều bệnh nhân phát triển hạ canxi máu thoáng qua có thể được quản lý bằng cách dùng bổ sung canxi đường uống. Khi có viêm xương xơ nang nặng và mất khoáng rõ rệt, hạ canxi máu đáng kể có thể xảy ra do hội chứng xương đói và cần quản lý tích cực hơn. Các biến chứng khác của phẫu thuật bao gồm rối loạn chức năng dây thanh (thoáng qua hoặc vĩnh viễn) và sự cần thiết phải phẫu thuật thêm do sự phát triển của một u tuyến thứ hai. Nếu suy cận giáp vĩnh viễn phát triển sau phẫu thuật, nó sẽ được điều trị bằng cách dùng calcitriol và bổ sung canxi khi cần thiết. Khi cường cận giáp do tăng sản tất cả các tuyến cận giáp, nên xem xét cắt bỏ gần toàn bộ tuyến cận giáp với việc cấy ghép mô cận giáp băm nhỏ vào một túi cơ ở cẳng tay.
Hình 20.8: Hình ảnh một u tuyến cận giáp bằng xạ hình technetium-99m sestamibi.
(Từ Steenkamp D, Lee SL. Hypercalcemic crisis in a young woman. Endocrine Today. 2011:Tháng 10, với sự cho phép của SLACK Incorporated.)
Bệnh nhân bị tăng canxi máu do HHC thường không có triệu chứng hoặc chỉ có triệu chứng nhẹ (mệt mỏi, yếu cơ) và thường không cần điều trị. Trẻ sơ sinh bị cường cận giáp sơ sinh nặng có thể cần cắt bỏ toàn bộ tuyến cận giáp khẩn cấp nếu không thể kiểm soát được tình trạng tăng canxi máu bằng các phương tiện khác (bù nước, bài niệu canxi bằng furosemide, dùng bisphosphonate hoặc có lẽ là denosumab). Các tác nhân calcimimetic (phenylalkylamine; ví dụ, cinacalcet) liên kết và kích hoạt CaSR trên màng của tế bào chính tuyến cận giáp, do đó làm tăng nồng độ Ca2+ trong tế bào chất và làm giảm bài tiết PTH ở người lớn bị cường cận giáp nguyên phát nhẹ. Khi được chỉ định ở một đối tượng lớn tuổi có triệu chứng mắc HHC, việc dùng một tác nhân calcimimetic (cinacalcet) có thể được xem xét, mặc dù các loại thuốc này chỉ được chấp thuận để dùng cho các đối tượng trên 18 tuổi. Việc sử dụng các tác nhân này ở trẻ em và vị thành niên mắc rối loạn này mang lại một con đường điều trị tiềm năng cho các đối tượng bị tăng sản cận giáp lan tỏa. Thật vậy, tác nhân này đã được sử dụng thành công trong việc quản lý trẻ sơ sinh mắc NSHPT. Tuy nhiên, vào năm 2013, FDA đã đình chỉ việc sử dụng cinacalcet ở trẻ em và vị thành niên để chờ đánh giá thêm về tính an toàn của nó ở quần thể này. Cường cận giáp thứ phát của bệnh thận mạn tính được quản lý tốt nhất bằng cách hạ nồng độ phốt phát huyết thanh đến mức có thể bằng cách hạn chế ăn vào và bằng cách dùng các chất liên kết phốt phát đường uống và bằng cách duy trì nồng độ Ca2+ huyết thanh trong khoảng thấp-bình thường bằng cách dùng calcitriol hoặc chất tương tự. Cắt bỏ tuyến cận giáp có thể cần thiết để quản lý hiệu quả cường cận giáp thứ phát và tam phát khó chữa được biểu hiện bằng loạn dưỡng xương do thận nặng, tăng canxi máu và các triệu chứng toàn thân, chẳng hạn như ngứa và đau xương.
Tăng canxi máu do tăng vitamin D máu hoặc sản xuất quá mức calcitriol bởi các mô u hạt và viêm mạn tính có thể được điều trị bằng glucocorticoid để ức chế hoạt động của 25OHD3-1α-hydroxylase. Ketoconazole (3–9 mg/kg/ngày chia làm 3 liều) là một tác nhân chống nấm cũng ức chế hoạt động 25OHD3-1α-hydroxylase ở thận và nhanh chóng làm giảm giá trị calcitriol và canxi ở trẻ em và người lớn mắc các rối loạn như vậy. Các tác dụng phụ của liệu pháp ketoconazole bao gồm buồn nôn, nôn, đau bụng, giảm bài tiết steroid sinh dục và suy giảm sản xuất cortisol của tuyến thượng thận. Do đó, việc theo dõi cẩn thận bệnh nhân đang dùng ketoconazole là cần thiết. Glucocorticoid cải thiện tình trạng tăng canxi máu liên quan đến sản xuất quá mức IL-1β ở thanh thiếu niên bị viêm khớp dạng thấp/tự phát vị thành niên. Cần cố gắng ngăn ngừa tăng canxi máu ở trẻ em hoặc vị thành niên bị bất động bằng cách cung cấp một chế độ ăn ít canxi, tránh vitamin D, uống nhiều nước và vận động sớm. Nồng độ canxi trong huyết thanh và nước tiểu nên được theo dõi thường xuyên, và tăng cường thêm dịch nếu xảy ra tăng canxi niệu. Một khi đã có, tăng canxi máu được điều trị tốt nhất bằng cách vận động; bài niệu bằng nước muối sinh lý và/hoặc dùng bisphosphonate có thể cần thiết cho đến khi tình trạng canxi máu bình thường được phục hồi. Hạn chế canxi trong chế độ ăn và hạn chế tiếp xúc với ánh nắng mặt trời có thể phù hợp trong việc quản lý lâu dài một số bệnh nhân bị tăng canxi máu không thể điều trị cụ thể hơn. Các tác nhân chống prostaglandin có thể hữu ích ở trẻ bị tăng canxi máu liên quan đến việc sản xuất quá mức các hợp chất này. Điều trị đặc hiệu các bệnh đi kèm với tăng canxi máu (nhiễm độc giáp, suy vỏ thượng thận) sẽ phục hồi trạng thái canxi máu bình thường.
Rối loạn chuyển hóa magiê
Magiê (Mg) là một cation thiết yếu để kiểm soát chức năng thần kinh-cơ và là một nguyên tố cần thiết cho các phản ứng enzyme liên quan đến ATP, cũng như duy trì cấu trúc của DNA và RNA. Magiê điều hòa sự bài tiết nhưng không điều hòa sự tổng hợp của PTH và sự tạo ra calcitriol. Trong huyết thanh, magiê vừa ở dạng phức hợp với protein, vừa ở dạng ion hóa hoặc tự do. Khoảng 50% lượng magiê dự trữ của cơ thể được lắng đọng trong xương, hấp phụ trên bề mặt của hydroxyapatite. Thụ thể cảm nhận canxi (CaSR) nhận biết và đáp ứng với Mg2+ cũng như với Ca2+. Nồng độ magiê trong huyết thanh được duy trì trong giới hạn xác định hẹp (1,6–2,4 mg/dL = 0,66–0,99 mmol/L) bằng sự phối hợp hấp thu ở ruột và tái hấp thu ở ống thận của cation này. Ở ống lượn gần và nhánh lên dày của quai Henle, sự tái hấp thu magiê chủ yếu là thụ động qua các kênh nối chặt gian bào được gọi là claudin. Trong đường ruột và ống lượn xa của thận, magiê được hấp thu/tái hấp thu chủ động qua tế bào thông qua một kênh bao gồm một dị-oligome của TRPM6 (OMIM 607009) liên kết với TRPM7 (OMIM 605692), một kênh cation thấm magiê. Dị-oligome TRPM6/TRPM7 bắt cặp được biểu hiện trên bề mặt các tế bào lót đường ruột và ống lượn xa của thận, cho phép hấp thu/tái hấp thu Mg2+ qua tế bào. Ở thận, 10% đến 20% magiê được lọc được tái hấp thu ở ống lượn gần và 65% đến 70% được tái hấp thu ở nhánh lên dày của quai Henle (TALH) bằng một quá trình thụ động gian bào; 10% đến 20% magiê được lọc được tái hấp thu ở ống lượn xa bằng một hệ thống chủ động qua tế bào.
Hạ magiê máu
Hạ magiê máu (nồng độ magiê toàn phần trong huyết thanh < 1,5 mg/dL = 0,62 mmol/L) có thể không có biểu hiện lâm sàng hoặc có thể biểu hiện bằng mệt mỏi, yếu cơ, dị cảm, tăng kích thích thần kinh-cơ (co cứng bàn tay-bàn chân, co cứng, run, co cứng tetany, co giật) và khi kéo dài và nặng có thể gây teo cơ, yếu cơ, thờ ơ, và thay đổi trạng thái ý thức; có thể gợi ra các dấu hiệu Chvostek và Trousseau; khoảng PR và QT kéo dài có thể hiện diện trên điện tâm đồ. Hạ magiê máu có thể dẫn đến hạ canxi máu bằng cách ức chế giải phóng PTH và cản trở hoạt động ngoại vi của nó. Hạ magiê máu có thể là hậu quả của việc giảm ăn vào hoặc dùng thuốc, suy giảm hấp thu ở ruột, hoặc tăng mất magiê trong phân hoặc nước tiểu, có thể là mắc phải hoặc bẩm sinh (Bảng 20.7A, 20.7B). Trẻ sơ sinh của các bà mẹ thiếu cation này, những người bị tiền sản giật hoặc đái tháo đường thai kỳ hoặc type 1 và ở trẻ sơ sinh có cân nặng khi sinh thấp—sinh non hoặc chậm tăng trưởng trong tử cung—có xu hướng phát triển hạ magiê máu. Hút dịch dạ dày kéo dài, các rối loạn kém hấp thu do cắt bỏ ruột rộng rãi (hội chứng “ruột ngắn”), rò ruột, hoặc các bệnh khác liên quan đến tiêu chảy mạn tính và tiêu phân mỡ cũng dẫn đến hạ magiê máu ở trẻ nhỏ. Thuốc lợi tiểu (furosemide, thiazide) làm giảm hấp thu magiê ở quai Henle của thận, thuốc ức chế bơm proton (omeprazole), thuốc ức chế calcineurin (cyclosporin A, tacrolimus), các dẫn xuất platin (cisplatin, carboplatin), kháng sinh (aminoglycosid, rapamycin), hoặc các tác nhân adrenergic chuyển magiê vào tế bào có thể dẫn đến hạ magiê máu cũng như các tác nhân độc với thận (ví dụ, thủy ngân, gentamycin); tăng magiê niệu dẫn đến hạ magiê máu. Ở trẻ sơ sinh, trẻ em và vị thành niên, hạ magiê máu có thể là nguyên phát và do một khiếm khuyết cụ thể trong việc hấp thu Mg2+ ở ruột hoặc trong việc tái hấp thu Mg2+ đã lọc ở ống thận (xem bên dưới) hoặc là thứ phát và do mất mát qua đường tiêu hóa (nôn mửa hoặc tiêu chảy mạn tính hoặc các trạng thái kém hấp thu do bệnh viêm ruột, cắt bỏ ruột hoặc rò, hoặc viêm tụy); do các bệnh ống thận liên quan (hội chứng Gitelman và Bartter); tiếp xúc với rượu, thuốc lợi tiểu và các tác nhân hóa trị liệu; và các bệnh nội tiết cụ thể (ví dụ, đái tháo đường, cường cận giáp nguyên phát, cường aldosteron).
Bảng 20.7A: Rối loạn Cân bằng nội môi Magiê
| I | Hạ magiê máu | ||
|---|---|---|---|
| A | Bẩm sinh | ||
| 1 | Trẻ sơ sinh của các bà mẹ bị đái tháo đường, sản giật, thiếu magiê | ||
| 2 | Chậm tăng trưởng trong tử cung, sinh non | ||
| 3 | Đột biến gen–xem Bảng 20.7B | ||
| B | Mắc phải | ||
| 1 | Thuốc—lợi tiểu quai và thiazide, kháng sinh (amphotericin, gentamycin), các tác nhân giống giao cảm, thuốc ức chế bơm proton (omeprazole), digitalis, các tác nhân chống tân sinh (cisplatin), cyclosporin | ||
| 2 | Hút dịch dạ dày | ||
| 3 | Kém hấp thu ở ruột (bệnh celiac, cắt bỏ ruột) | ||
| 4 | Tiêu chảy bài tiết (bệnh viêm ruột) | ||
| 5 | Viêm tụy cấp | ||
| 6 | Tăng magiê niệu do đái tháo đường hoặc nghiện rượu hoặc sau hoại tử ống thận hoặc ghép thận | ||
| 7 | Kết hợp với hạ kali máu | ||
| II | Tăng magiê máu | ||
| A | Mắc phải | ||
| 1 | Suy thận | ||
| 2 | Ăn quá nhiều muối magiê hoặc thuốc nhuận tràng/thuốc kháng axit có chứa magiê | ||
| 3 | Nhiễm toan ceton do đái tháo đường, suy vỏ thượng thận, cường cận giáp, ngộ độc lithium |
Bảng 20.7B: Các đột biến gen liên quan đến Hạ magiê máu
| Gen Protein | NST | OMIM | Rối loạn OMIM | Chức năng Gen/Di truyền |
|---|---|---|---|---|
| TRPM6 Thụ thể tiềm năng thoáng qua, kênh cation, phân họ M, thành viên 6 | 9q21.3 | 607009 | Hạ magiê máu 1, đường ruột 602014 | TRPM6 tạo thành dị-oligome với TRPM7 (chr. 15q21.2, OMIM 605692), một kênh cation thấm magiê, AR |
| FXYD2 Chất điều hòa vận chuyển ion chứa miền FXYD 2 | 11q23.3 | 601814 | Hạ magiê máu 2, thận 154020 | Tiểu đơn vị gamma của Na+-K+-ATPase thận, tăng magiê niệu & giảm canxi niệu do tăng tái hấp thu canxi ở ống thận, FXYD2 được hoạt hóa chuyển mã bởi HNF1B/PCBD1, AD |
| CLDN16 Claudin 16 | 3q28 | 603959 | Hạ magiê máu 3, thận 248250 | Kênh vận chuyển magiê gian bào, tăng magiê niệu, tăng canxi niệu, nhiễm canxi thận, thiểu sản men răng không hoàn toàn, AR |
| EGF Yếu tố tăng trưởng biểu bì | 4q25 | 131530 | Hạ magiê máu 4, thận 611718 | Chất hoạt hóa TRPM6, Tăng magiê niệu, Canxi niệu bình thường (các biến thể của EGFR cũng dẫn đến hạ magiê máu) |
| CLDN19 Claudin 19 | 1p34.2 | 610036 | Hạ magiê máu 5, thận 248190 | Kênh vận chuyển magiê gian bào, tăng magiê niệu, tăng canxi niệu, nhiễm canxi thận, khuyết màng mạch-võng mạc, AR |
| CNNM2 Cyclin M2 | 10q24.32 | 607803 | Hạ magiê máu 6, thận 613882 | Chất điều hòa TRPM7 & sự hình thành dị-oligome của nó với TRPM6, AD |
| HNF1B HNF1 homeobox B | 17q12 | 189907 | Hạ magiê máu, bệnh thận kẽ ống bẩm sinh, đái tháo đường khởi phát ở người trưởng thành trẻ, type 5 | Yếu tố phiên mã điều hòa biểu hiện của FXYD2 & tái hấp thu magiê ở ống lượn xa, AD |
| PCBD1 Pterin-4-alpha-carbinolamine dehydratase 1 | 10q22.1 | 126090 | Hạ magiê máu, đái tháo đường khởi phát ở người trưởng thành trẻ, type 5 | Yếu tố đồng dimer hóa hạt nhân cho HNF1B để tăng cường biểu hiện của tái hấp thu magiê ở ống lượn xa qua trung gian FXYD2, AR |
| KCNA1 Kênh kali, cổng điện thế, phân họ liên quan đến shaker, thành viên 1 | 12p13.32 | 178260 | Hạ magiê máu | Kênh kali cổng điện thế đồng định vị với TRPM6, biến thể bất hoạt dẫn đến tăng magiê niệu bằng cách gây ra hiệu ứng trội âm tính lên KCNA1 kiểu hoang dã, AD |
| CLCNKB Kênh clorua, thận, B | 1p36.13 | 602023 | Hội chứng Bartter (type 3) 241200 Hội chứng Bartter (type 4b), hai gen, 613090 | Kênh clorua ống thận, AR |
| SLC12A3 Họ chất mang hòa tan 12 (chất vận chuyển natri/clorua), thành viên 3 | 16q13 | 600968 | Hội chứng Gitelman—nhiễm toan chuyển hóa hạ kali máu, giảm canxi niệu, hạ magiê máu, 263800 | Chất đồng vận chuyển natri/clorua ở ống lượn xa của thận, AR |
AD, Di truyền trội trên nhiễm sắc thể thường; AR, di truyền lặn trên nhiễm sắc thể thường; ATPase; adenosine triphosphatase.
Hạ magiê máu có thể liên quan đến hạ canxi máu và tăng canxi niệu như ở một số bệnh nhân có các biến thể hoạt hóa của CASR dẫn đến suy cận giáp di truyền trội trên NST thường (OMIM 601198). Hạ magiê máu xảy ra ở những bệnh nhân bị rối loạn ống thận, chẳng hạn như hội chứng Gitelman và Bartter (xem bên dưới) và là kết quả của các lỗi di truyền trong các con đường ảnh hưởng xấu đến sự hấp thu ở ruột hoặc tái hấp thu ở ống thận của magiê (xem Bảng 20.7B). Hạ magiê máu type 1 với hạ canxi máu thứ phát là hậu quả của các đột biến bất hoạt hai alen của TRPM6 mã hóa kênh ion tạo thành một dị-oligome với TRPM7, một kênh vận chuyển ion thấm Mg2+ và Ca2+ hiện diện trong đường ruột và ống thận. Trẻ sơ sinh và trẻ nhỏ có các biến thể bệnh lý của TRPM6 bị tăng kích thích thần kinh-cơ (run, co thắt và co giật) trong vài tháng đầu sau khi sinh. Về mặt sinh lý bệnh, rối loạn này là do một khiếm khuyết chọn lọc ở ruột non trong việc hấp thu Mg2+ qua tế bào (ví dụ, biến thể p.Ser141Leu làm suy giảm sự liên kết của TRPM6 và TRPM7 do đó ngăn cản sự hấp thu Mg2+ ở ruột). Trẻ sơ sinh bị ảnh hưởng biểu hiện co cứng và/hoặc co giật do hạ magiê máu/hạ canxi máu dẫn đến vôi hóa cơ tim, thận và động mạch. Bài tiết magiê qua thận là bình thường ở các đối tượng mắc bệnh này. Hạ canxi máu được cho là do giảm bài tiết và giảm nhạy cảm ngoại vi với PTH. Uống một lượng lớn magiê có hiệu quả điều trị cho bệnh này. Các đột biến bất hoạt đơn alen trong FXYD2 (OMIM 601814) mã hóa một tiểu đơn vị gamma của Na+/K+-ATPase thận dẫn đến hạ magiê máu di truyền trội trên NST thường type 2 với giảm canxi niệu. FXYD2 mã hóa một tiểu đơn vị gamma của một Na+/K+-ATPase được biểu hiện ở ống lượn xa của thận. Các đột biến mất chức năng đơn alen của FXYD2 dẫn đến việc định tuyến sai sản phẩm protein của nó đến màng đáy của các tế bào biểu mô của nephron, dẫn đến tăng mất magiê qua nước tiểu nhưng nghịch lý là lại tăng tái hấp thu Ca2+ ở ống thận trong quai Henle dẫn đến giảm canxi niệu. Sự bất hoạt của các yếu tố phiên mã điều hòa của FXYD2 được mã hóa bởi HNF1B (OMIM 189907) và pterin-4-alpha-carbinolamine dehydratase 1 (PCBD1, OMIM 126090) cũng dẫn đến hạ magiê máu. HNF1B mã hóa yếu tố hạt nhân tế bào gan 1 homeobox B, một yếu tố phiên mã cần thiết cho sự phát triển phôi bình thường của thận và tụy. Các đột biến bất hoạt trong HNF1B dẫn đến các dị tật của thận (nang, dị tật thận), hạ magiê máu và đái tháo đường khởi phát ở người trưởng thành trẻ. PCBD1 mã hóa một protein liên kết với HNF1B và làm tăng khả năng phiên mã của nó.
Các đột biến mất chức năng trong các gen mã hóa các kênh vận chuyển nối chặt gian bào claudin-16 và -19 (CLDN16, OMIM 603959; CLDN19, OMIM 610036) dẫn đến hạ magiê máu ở thận type 3 và type 5, tương ứng. Các bệnh này thường biểu hiện ở trẻ nhỏ và có liên quan đến co cứng, tăng magiê niệu, tăng canxi niệu, hạ canxi máu nhẹ, nhiễm canxi thận, suy giảm chức năng thận và cường cận giáp thứ phát. Claudin-16 (còn được gọi là paracellin 1) là một protein 305 aa với bốn miền xuyên màng và các đầu amino và carboxyl nội bào được biểu hiện trong các mối nối chặt gian bào của các tế bào biểu mô thận ở TALH và ống lượn xa, nơi nó tạo điều kiện cho sự vận chuyển và tái hấp thu gian bào của Mg2+ và Ca2+ từ ống thận. Vòng ngoại bào đầu tiên của claudin-16 bắc qua không gian gian bào và là vị trí dẫn truyền ion gian bào; một số đột biến sai nghĩa trong CLDN16 đã được xác định—đặc biệt là ở vị trí leucine 151 (p.Leu151Phe, Trp, Pro). Hầu hết các biến thể của CLDN16 làm suy giảm sự di chuyển bình thường của nó đến bề mặt bên của tế bào biểu mô thận; trong các đột biến khác (p.Ala62Val, p.His71Asp), các sản phẩm định vị tại các mối nối chặt nhưng bị khiếm khuyết về chức năng. Việc dùng đường uống 20 lần nhu cầu magiê hàng ngày bình thường đã là liệu pháp thành công ở những đối tượng này. CLDN19 mã hóa claudin-19, một protein 224 aa được biểu hiện ở các kẽ giữa các tế bào của TALH thận và các ống lượn xa, các biến thể bất hoạt hai alen của nó gây ra hạ magiê máu type 5. CLDN19 cần thiết cho sự vận chuyển gian bào của cả canxi và magiê. CLDN19 cũng được biểu hiện ở mắt; ngoài các bất thường về mắt (khuyết màng mạch-võng mạc, rung giật nhãn cầu, cận thị nặng), các đột biến trong CLDN19 dẫn đến hạ magiê máu, tăng canxi niệu, nhiễm canxi thận và suy thận. Các đột biến bất hoạt hai alen trong EGF (OMIM 131530), gen mã hóa yếu tố tăng trưởng biểu bì, dẫn đến hạ magiê máu ở thận type 4, một rối loạn liên quan đến co giật và chậm phát triển nhưng trong đó nồng độ canxi trong huyết thanh và nước tiểu là bình thường. Yếu tố tăng trưởng biểu bì (EGF) là một đồng yếu tố cần thiết cho sự tái hấp thu Mg2+ bình thường ở ống lượn xa của thận bởi TRPM6. Một biến thể bất hoạt của thụ thể EGF (EGFR, OMIM 131550) cũng đã được liên kết với hạ magiê máu, cũng như bệnh viêm da và ruột sơ sinh (OMIM 616069). Các đột biến bất hoạt đơn alen của CNNM2 mã hóa cyclin M2 (OMIM 607803), một protein rất quan trọng cho sự hình thành dị-oligome TRPM6-TRPM7, dẫn đến hạ magiê máu type 6. Mặc dù bị hạ magiê máu, bài tiết magiê qua nước tiểu là bình thường một cách không phù hợp; các giá trị canxi trong huyết thanh và nước tiểu là bình thường ở những đối tượng này. CNNM2 mã hóa một protein định vị ở màng đáy của các tế bào cấu thành nhánh lên dày của quai Henle và ống lượn xa của thận, nơi sự biểu hiện của nó được tăng lên do thiếu magiê. Một chức năng của CNMM2 có thể là để “cảm nhận” nồng độ Mg2+ xung quanh. Hạ magiê máu liên quan đến giật rung cơ (myokymia) đã được liên kết với một đột biến mất chức năng dị hợp tử (p.Asn255Asp) trong KCNA1 (OMIM 178260) mã hóa một kênh kali thận đồng định vị với TRPM6 ở ống lượn xa của thận và cũng rất quan trọng cho sự vận chuyển Mg2+.
Hạ magiê máu gặp ở những bệnh nhân bị nhiễm toan chuyển hóa hạ kali máu kết hợp với hội chứng Sesame (co giật, điếc thần kinh giác quan, mất điều hòa, chậm phát triển trí tuệ và mất cân bằng điện giải) do các đột biến bất hoạt trong KCNJ10 (OMIM 602208—mã hóa một kênh kali), và hội chứng Bartter trước sinh type 2 (đa ối, sinh non, tăng trưởng sau sinh kém, mất muối và tăng canxi niệu do các đột biến bất hoạt trong một gen mã hóa một kênh kali thứ hai—KCNJ1 (OMIM 600359). Hội chứng Bartter type 1 (OMIM 601678) là một rối loạn di truyền lặn trên NST thường biểu hiện bằng mất muối do suy giảm tái hấp thu natri và kali ở nhánh lên của quai Henle, nhiễm kiềm chuyển hóa hạ kali máu, tăng canxi niệu và đôi khi tăng magiê niệu do các biến thể của SLC12A1 (OMIM 600839). (Các biến thể bất hoạt hai alen trong CLCNKB [OMIM 602023] mã hóa một kênh clorua [OMIM 602023] hiện diện ở những bệnh nhân mắc dạng kinh điển của bệnh này—hội chứng Bartter type 3 [OMIM 607364].) Hội chứng Bartter type 4a có liên quan đến các biến thể của BSND (OMIM 606412) mã hóa barttin, một tiểu đơn vị beta cho các kênh vận chuyển clorua được mã hóa bởi CLCNKA (OMIM 602024) và CLCNKB (OMIM 602023). Ở trẻ em và vị thành niên, hội chứng Gitelman di truyền lặn trên NST thường (OMIM 263800) gồm nhiễm kiềm toàn thân, hạ kali máu, hạ magiê máu và giảm canxi niệu là kết quả của các biến thể của các gen cấu thành nên chất đồng vận chuyển natri-clorua nhạy cảm với thiazide ở ống lượn xa của thận—thường là SLC12A3 (OMIM 600968). Hạ magiê máu cũng đã được liên kết với các rối loạn ty thể bao gồm bệnh cơ tim mô bào ở trẻ nhỏ (OMIM 50000), hội chứng Kearns-Sayre (OMIM 530000), và hội chứng tăng axit uric máu—tăng áp phổi—suy thận—nhiễm kiềm (OMIM 613845).
Sự hiện diện của hạ magiê máu được xác định bằng cách đo nồng độ magiê huyết thanh, trong khi nguyên nhân sinh lý bệnh của nó được xác định bằng xét nghiệm đồng thời nồng độ canxi, phốt phát, natri, kali, clorua, bicarbonate, creatinin, PTH và vitamin D và đánh giá sự mất mát qua nước tiểu và hấp thu ở ruột của nó. Khi có đồng thời hạ magiê máu và tăng canxi niệu, các biến thể của CLDN16, CLDN19, CLCNKB, và CASR có thể được xem xét. Ở những bệnh nhân hạ magiê máu không có tăng canxi niệu, các biến thể của TRPM6, CNNM2, EGF/EGFR, KCNA1, và FAM111A có thể được tìm kiếm.
Các cơn co giật do hạ magiê máu, hạ canxi máu chỉ đáp ứng thoáng qua và đôi khi kháng với việc dùng canxi nguyên tố đơn độc qua đường tiêm. Việc dùng dung dịch magiê sulfat 50% (MgSO4.7H2O—0,05–0,1 mL/kg hoặc 2,5–5,0 mg/kg magiê nguyên tố có theo dõi tim) tiêm tĩnh mạch hoặc tiêm bắp có thể cần thiết để kiểm soát co giật ở trẻ sơ sinh bị hạ magiê máu. Ở các đối tượng lớn tuổi, hạ magiê máu có triệu chứng được quản lý bằng cách dùng thận trọng magiê sulfat 20% (800 mmol/L) tiêm tĩnh mạch (30 mL trong 500 mL nước muối sinh lý hoặc dextrose 5%) với theo dõi lâm sàng và điện tâm đồ chặt chẽ và đo thường xuyên nồng độ Ca2+ và magiê trong huyết thanh. Các chất bổ sung magiê đường uống cũng có thể hữu ích (50% MgSO4.7H2O—0,2 mL/kg/ngày). Ở bệnh nhân người lớn bị hạ magiê máu mạn tính, các chế phẩm uống của magiê clorua hoặc glycerophosphate (120 mg ba lần mỗi ngày) có thể được xem xét nếu dung nạp được; liều magiê quá lớn có thể dẫn đến tiêu chảy và nên tránh.
Tăng magiê máu
Tăng magiê máu (magiê > 2,6 mg/dL = > 1,05 mmol/L) được ghi nhận trong giai đoạn sơ sinh sau khi magiê sulfat đã được dùng cho phụ nữ mang thai bị tăng huyết áp, tiền sản giật hoặc nhiễm độc thai nghén. Hầu hết trẻ sơ sinh bị tăng magiê máu đều không có triệu chứng; tuy nhiên, khi nồng độ magiê huyết thanh đặc biệt cao, có thể có giảm trương lực cơ và ức chế hệ thần kinh trung ương và khi lan rộng, bệnh xương chuyển hóa có thể phát triển. Do đó, việc dùng magiê sulfat tiêm tĩnh mạch kéo dài (9–10 tuần) cho những phụ nữ chuyển dạ sớm không chỉ liên quan đến tăng magiê máu mà còn với hạ canxi máu rõ rệt và loãng xương đáng kể ở con. Tăng magiê máu cũng có thể là kết quả của việc dùng qua đường tiêm hoặc uống các thuốc kháng axit hoặc thuốc xổ chứa magiê. Tăng magiê máu cũng có thể phát triển ở những bệnh nhân bị suy thận đang dùng các thuốc kháng axit chứa magiê. Nồng độ magiê huyết thanh tăng nhẹ ở những bệnh nhân bị HHC có tính gia đình do các đột biến mất chức năng trong CASR. Với liều lượng lớn, magiê sulfat ức chế bài tiết PTH và làm giảm tái hấp thu canxi ở ống thận, các yếu tố góp phần gây hạ canxi máu. Các biểu hiện lâm sàng của tăng magiê máu nặng (magiê > 4,8 mg/dL = > 2 mmol/L) tương tự như các biểu hiện của tăng canxi máu và bao gồm thay đổi tri giác, buồn nôn, nôn, mệt mỏi và yếu cơ; nhịp tim và huyết áp có thể thấp; có thể có giảm phản xạ. Cả hạ và tăng magiê máu đều làm suy giảm sự bài tiết PTH dẫn đến hạ canxi máu. Các biến thể của claudin-10 (CLDN10, OMIM 617579) dẫn đến tăng vận chuyển canxi và magiê gian bào và tăng magiê máu được xác định ở các đối tượng mắc hội chứng HELIX (giảm tiết mồ hôi, mất cân bằng điện giải, rối loạn chức năng tuyến lệ, bệnh vảy cá, khô miệng—OMIM 617671). Trẻ sơ sinh và trẻ em bị tăng magiê máu được quản lý thích hợp nhất bằng cách ngừng magiê ngoại sinh, lợi tiểu bằng nước muối sinh lý, hoặc dùng furosemide nếu cần thiết; nếu bệnh nhân cũng bị hạ canxi máu và loãng xương, việc dùng canxi và calcitriol được chỉ định.
Các rối loạn về xương
Các bất thường bẩm sinh và mắc phải trong quá trình hình thành chất dạng xương (osteoid), phần hữu cơ của chất nền xương bao gồm các sợi collagen type 1, và/hoặc sự lắng đọng có trật tự của các tinh thể hydroxyapatite vào chất nền xương dẫn đến suy giảm sức mạnh của xương, gây ra các dị tật và biến dạng xương và tăng nguy cơ gãy xương.
Rối loạn Hình thành và Khoáng hóa Xương ở Trẻ sơ sinh và Trẻ nhỏ
Khối lượng xương thấp
Ở thai nhi, quá trình hình thành phần hữu cơ của xương và quá trình khoáng hóa sau đó diễn ra tuần tự, các quá trình này được kiểm soát bởi GH; PTH; PTHrP; vitamin A, D, và C; và các cytokine khác nhau. Khoảng 80% tổng lượng canxi trong xương của trẻ sơ sinh đủ tháng được tích lũy trong ba tháng cuối của thai kỳ khi tốc độ lắng đọng canxi trong tử cung tăng hơn hai lần từ tuần 28 đến 36 của thai kỳ (lượng canxi tích lũy trong ba tháng cuối xấp xỉ 100–130 mg/kg/ngày). Ở trẻ sinh non, tuổi thai, cân nặng khi sinh và tốc độ tăng cân sau sinh là những yếu tố quyết định quan trọng đến tốc độ tích lũy khối lượng xương sau sinh. Do đó, trẻ sinh non, cân nặng khi sinh rất thấp (VLBW < 1500 g), và cực kỳ thấp (ELBW < 1000 g) đặc biệt dễ bị bệnh xương chuyển hóa liên quan đến giảm khối lượng xương (được định nghĩa là hàm lượng khoáng chất trong xương [BMC] sau sinh thấp hơn BMC của thai nhi trong tử cung ở cùng tuổi sau thụ thai, có hoặc không có bằng chứng X-quang về giảm khoáng hóa xương). Sau khi sinh, trẻ sinh cực non không thể duy trì tốc độ tăng trưởng xương nhanh như trong tử cung, vốn đòi hỏi sự tổng hợp chất nền xương hữu cơ và sự lắng đọng canxi và phốt phát vào chất dạng xương từ lượng chất dinh dưỡng có thể được cung cấp qua đường ăn uống và hấp thu qua đường tiêu hóa hoặc qua dinh dưỡng đường tĩnh mạch. Hạ canxi máu sau sinh và giảm vận động tự phát chống lại lực tác động của thành cơ tử cung cũng làm giảm tốc độ khoáng hóa xương, trong khi tốc độ tái hấp thu xương tăng lên càng làm suy giảm sự tích lũy khối lượng xương ở trẻ sinh non. Xoay ruột bất toàn hoặc viêm ruột hoại tử thảm khốc, dẫn đến nhồi máu ruột đòi hỏi phải cắt bỏ ruột non trên diện rộng, làm tăng đáng kể nguy cơ kém hấp thu và hậu quả là khối lượng xương thấp. Nuôi dưỡng qua đường tĩnh mạch ở trẻ VLBW hoặc ELBW hạn chế việc cung cấp dịch, protein, canxi và phốt phát; nhôm dư thừa trong dịch truyền tĩnh mạch cũng ảnh hưởng xấu đến sự hình thành xương. Tỷ lệ sinh đẻ của người mẹ tăng, giới tính nam, bệnh hệ thống nặng (loạn sản phế quản phổi), bất động, và các tác nhân dược lý (glucocorticoid, methylxanthine như theophylline, thuốc lợi tiểu như furosemide) cũng ảnh hưởng xấu đến sự hình thành xương ở những trẻ sơ sinh này. Theophylline và furosemide làm tăng bài tiết canxi qua nước tiểu. Các yếu tố trước sinh góp phần làm giảm khối lượng xương ở trẻ VLBW và LBW là: chậm tăng trưởng trong tử cung—có thể do giảm vận chuyển canxi qua nhau thai và giảm tốc độ hình thành xương; phơi nhiễm trước sinh với lượng lớn magiê sulfat được dùng lặp đi lặp lại cho người mẹ trong chuyển dạ sớm dẫn đến hạ canxi máu và loãng xương bằng cách ức chế bài tiết PTH thông qua tương tác với CaSR và bằng cách cạnh tranh với canxi để lắng đọng trên bề mặt xương, tương ứng. Trong quần thể trẻ sơ sinh dễ bị tổn thương này, việc giảm vôi hóa chất nền xương dẫn đến BMC thấp và dễ bị gãy xương, trong khi việc giảm vôi hóa sụn tăng trưởng có thể dẫn đến bệnh còi xương và các biến dạng đặc trưng của nó.
Khoảng 30% (16%–40%) trẻ sinh non có cân nặng khi sinh dưới 1500 g phát triển bệnh xương chuyển hóa trong vòng 6 đến 16 tuần sau sinh. Bệnh xương chuyển hóa do sinh non có liên quan đến giảm BMC; mỏng vỏ xương; giảm khối lượng xương bè; loe, tua và lõm hành xương; và tăng tần suất gãy xương. Về mặt sinh hóa, hạ phốt phát máu do thiếu phốt phát và tăng phosphatase kiềm, bằng chứng về nỗ lực của nguyên bào xương để tăng lắng đọng khoáng chất trong xương, thường được ghi nhận. Một số trẻ sơ sinh bị bệnh xương chuyển hóa bị thiếu canxi, bằng chứng là sự gia tăng nồng độ PTH trong huyết thanh (cường cận giáp thứ phát); ở những trẻ sơ sinh được nuôi dưỡng qua đường ruột như vậy, việc dùng calcitriol có thể làm tăng hấp thu canxi ở ruột, do đó làm tăng nồng độ canxi trong huyết thanh và giảm nồng độ PTH.
Ở trẻ sinh non, nồng độ phosphatase kiềm toàn phần và đặc hiệu cho xương, propeptide đầu carboxyl của collagen type I (PICP), và osteocalcin (các dấu hiệu của hoạt động nguyên bào xương và hình thành xương) tăng cao so với trẻ sơ sinh đủ tháng và trẻ lớn hơn và tiếp tục tăng trong 10 tuần đầu đời. Người ta đã khuyến cáo rằng ở trẻ VLBW, nếu giá trị phosphatase kiềm huyết thanh vượt quá 500 IU/L và nồng độ phốt phát huyết thanh giảm xuống dưới 5,6 mg/dL (1,8 mmol/L), nên xác định sự tái hấp thu phốt phát ở ống thận; các giá trị lớn hơn 95% cho thấy lượng phốt phát dự trữ của trẻ thấp và cần chỉ định bổ sung phốt phát. Các giá trị hydroxyproline và pyridinoline/deoxypyridinoline trong nước tiểu (các dấu hiệu của hoạt động hủy cốt bào và tái hấp thu xương) cũng tăng ở trẻ sinh non, mặc dù nồng độ telopeptide đầu carboxyl của collagen type I (ICTP) trong huyết thanh, một dấu hiệu khác của tái hấp thu xương, giảm trong 10 tuần đầu sau khi sinh non. Do đó, tốc độ chu chuyển xương trong tử cung và sau sinh ở trẻ sơ sinh non tháng là nhanh và tăng cao liên tục cho đến 40 tuần tuổi sau thụ thai, một kết luận được xác nhận bằng phương pháp đo hình thái xương. Bằng phương pháp đo hấp thụ photon và siêu âm định lượng (QUS), khối lượng xương của trẻ sinh non dường như giảm trong vài tuần đầu sau khi sinh. Nồng độ phosphatase kiềm và osteocalcin trong huyết thanh và bài tiết pyridinoline và canxi qua nước tiểu cao và có thể vẫn tăng ở trẻ LBW có khối lượng xương thấp do sinh non so với các giá trị ở trẻ LBW không bị bệnh xương trong năm đầu đời, mặc dù sự cải thiện về khoáng hóa xương trên X-quang thường rõ ràng sau 6 tháng tuổi.
Bởi vì phim X-quang tiêu chuẩn có thể không phát hiện được tình trạng khoáng hóa xương thấp trước khi thiếu hụt từ 20% đến 30% trở lên xảy ra trong tháng thứ hai của cuộc đời, việc ước tính BMC bằng phương pháp đo hấp thụ tia X năng lượng kép (DXA) đã trở thành phương pháp ưa thích để đánh giá khoáng hóa xương ở trẻ nhỏ vì độ chính xác, khả năng lặp lại, thực hiện nhanh chóng và phơi nhiễm bức xạ thấp (2–3 mrem). BMC toàn thân trung bình trong 2 ngày đầu đời dao động từ 21,7 g ở trẻ sơ sinh có cân nặng khi sinh từ 1001 đến 1500 g đến 78,8 g ở trẻ sơ sinh có cân nặng khi sinh từ 3501 đến 4000 g, trong khi BMD toàn thân thay đổi từ 0,146 mg/cm2 (1001–1500 g) đến 0,234 gm/cm2 (3501–4000 g). BMC và BMD tăng trong suốt năm đầu đời và sau đó được đo bằng DXA. Ở độ tuổi này, khối lượng xương tương quan tốt nhất với trọng lượng cơ thể. QUS cũng có thể được sử dụng để đánh giá tính toàn vẹn và sức mạnh của xương ở trẻ sinh non và các trẻ LBW khác; QUS có thể được thực hiện tại giường, không phơi nhiễm bức xạ, và có thể được lặp lại thường xuyên khi cần thiết. QUS đo tốc độ âm thanh (SOS) truyền qua xương (xương cánh tay, xương chày, xương quay, xương bánh chè, xương gót, xương bàn tay, xương ngón tay), một phép đo tương quan với sức mạnh của xương; hàm lượng khoáng chất chỉ là một trong số các thành phần xương (độ đàn hồi, độ dày vỏ xương, vi cấu trúc) cùng góp phần tạo nên sức mạnh của xương. QUS cũng cho phép tính toán thời gian truyền xương, một phép đo xác định sự khác biệt về vận tốc âm thanh khi nó di chuyển qua xương và các mô mềm xung quanh. Có sự chồng chéo đáng kể của các giá trị SOS ở các độ tuổi và kích thước cơ thể khác nhau; tuy nhiên, thời gian truyền xương có thể phân biệt ở mức độ lớn hơn giữa các thông số này. Các phép đo QUS ở xương cánh tay và xương chày thấp hơn ở trẻ sinh non so với trẻ đủ tháng và tương quan dương với tuổi thai, cân nặng khi sinh, chiều dài và cân nặng sau sinh. Ở trẻ sơ sinh bị chậm tăng trưởng trong tử cung, mức SOS ở xương chày có thể thay đổi. Mối quan hệ giữa BMD được xác định bằng DXA và sức mạnh xương được ước tính bằng QUS ở trẻ sơ sinh non tháng và đủ tháng là yếu nhưng có ý nghĩa.
Với tình trạng thiếu hụt canxi, phốt phát và vitamin D kéo dài, trẻ LBW không chỉ chậm tích lũy khối lượng xương mà còn phát triển các bằng chứng lâm sàng và X-quang của bệnh còi xương (một rối loạn khoáng hóa xương, xem bên dưới)—thường từ tuần thứ 6 đến 12 sau sinh—và gãy xương có thể xảy ra ở 24% trẻ VLBW. Trẻ sơ sinh LBW có nguy cơ khối lượng xương thấp và/hoặc còi xương được quản lý phòng ngừa tốt nhất bằng cách duy trì nồng độ canxi huyết thanh từ 8 đến 11 mg/dL và giá trị phốt phát huyết thanh từ 5,8 đến 9 mg/dL và bằng cách cung cấp qua đường ruột hàng ngày càng nhiều càng tốt lượng canxi cần thiết (140–160 mg/100 kcal), phốt phát (95–108 mg/100 kcal sữa công thức), và vitamin D (400–1000 U), cũng như protein (để tổng hợp collagen) và năng lượng (carbohydrate, lipid). Khi cần nuôi dưỡng qua đường tĩnh mạch ở trẻ LBW hoặc VLBW, cần cố gắng cung cấp lượng canxi và phốt phát tối đa có thể đạt được một cách an toàn. Độ hòa tan của canxi và phốt phát không chỉ phụ thuộc vào số lượng của chúng, mà còn vào các dạng được chọn để truyền, tức là, canxi clorua, -gluconate, -glycerophosphate, phốt phát monobasic, hoặc phốt phát dibasic. Sử dụng phốt phát monobasic và glycerophosphate, có thể truyền tới 86 mg canxi/kg/dL và 46 mg/kg/dL phốt phát. Tuy nhiên, các chế phẩm này làm tăng nguy cơ nhiễm toan chuyển hóa và tăng canxi niệu. Việc truyền các dung dịch dinh dưỡng qua đường tĩnh mạch chứa 60 đến 80 mg/kg/ngày canxi và 40 đến 50 mg/kg/ngày phốt phát chỉ cho phép đạt được 60% đến 70% tốc độ tích lũy khoáng chất xương ước tính trong tử cung. Do đó, nên bắt đầu cho ăn qua đường ruột càng sớm càng tốt ở trẻ VLBW và LBW bằng cách sử dụng sữa mẹ được tăng cường canxi glycerophosphate (canxi 80–120 mg/kg/ngày, phốt phát 45–60 mg/kg/ngày). Các loại sữa công thức được chuẩn bị để cho trẻ LBW ăn cung cấp 200 mg/kg/ngày canxi và 100 mg/kg/ngày phốt phát có thể dẫn đến giữ lại tới 90 mg/kg/ngày canxi và 40 mg/kg/ngày phốt phát. Loại sữa công thức được chuẩn bị (nguồn protein, chất béo và carbohydrate) và các chất phụ gia lipid và khoáng chất của nó quyết định tốc độ hấp thu và giữ lại canxi ở ruột; do đó, việc lựa chọn sữa công thức phải được xem xét cẩn thận trước khi chọn. Tuy nhiên, dinh dưỡng qua đường tĩnh mạch, sữa mẹ được tăng cường và các loại sữa công thức cho trẻ sinh non hiện có không thể cung cấp đủ lượng canxi và đặc biệt là phốt phát mà thai nhi thường tích lũy trong tử cung. Vitamin D 400 đến 1000 IU/ngày cũng nên được cung cấp cho trẻ sinh non qua đường ruột hoặc đường tĩnh mạch. Việc theo dõi nồng độ canxi, phốt phát, creatinin và phosphatase kiềm trong huyết thanh và bài tiết canxi, phốt phát và creatinin qua nước tiểu là cần thiết để ngăn ngừa tăng canxi máu và tăng canxi niệu và nhiễm canxi thận. Điều quan trọng là phải tránh hạ canxi máu do ái lực của chất nền xương đối với canxi một khi quá trình tái khoáng hóa đã bắt đầu (hội chứng xương đói). Hoạt động thể chất thụ động (tập vận động thụ động hàng ngày với duỗi/gấp tất cả các khớp của mỗi chi ở trẻ nằm ngửa trong 4 tuần, bắt đầu sau khi trẻ sơ sinh đã ổn định trong khoảng từ 2 đến 6 tuần tuổi sau sinh) có hoặc không có xoa bóp nhẹ nhàng cho trẻ nằm sấp từ đầu đến chân làm tăng nồng độ các dấu hiệu hình thành xương trong huyết thanh, cũng như khoáng hóa xương theo DXA và những thay đổi QUS phù hợp với việc tăng cường sức mạnh của xương. Mặc dù ở trẻ sinh non, khối lượng xương có thể vẫn thấp trong suốt thời thơ ấu và đầu thời thơ ấu, khoáng hóa xương được điều chỉnh theo kích thước (chiều cao, khối lượng nạc, khối lượng mỡ) cuối cùng sẽ bình thường hóa ở hầu hết các đối tượng trưởng thành ngoại trừ có lẽ ở cổ xương đùi.
Bệnh xương thủy tinh gây chết bẩm sinh (type lâm sàng 2—xem bên dưới) (OMIM 166210) là một rối loạn trước/chu sinh được đặc trưng bởi sự hiện diện của các xương sườn “hình chuỗi hạt” và các biến dạng và gãy xương dài có thể chứng minh được trong tử cung và rõ ràng khi sinh, tật đốt sống dẹt, và giảm khoáng hóa vòm sọ. Dạng bệnh xương thủy tinh nặng nhất này thường là kết quả của các đột biến mất chức năng dị hợp tử xảy ra tự phát trong các gen mã hóa collagen-α1(I) (COL1A1, OMIM 120150) hoặc collagen-α2(I) (COL1A2, 120160). Các đột biến có thể là mất đoạn gen một phần dẫn đến giảm tổng hợp chuỗi xoắn ba của collagen type I hoặc các đột biến sai nghĩa dẫn đến thay thế axit amin (ví dụ, arginine, axit aspartic, cysteine) cho các gốc glycine cần thiết cho cấu trúc không gian ba chiều bình thường của collagen-α1(I)/collagen-α2(I) và sự tổng hợp chuỗi xoắn ba và tính toàn vẹn cấu trúc của collagen type I trong chất nền ngoại bào của xương, nơi hydroxyapatite được lắng đọng. Bệnh xương thủy tinh gây chết type lâm sàng 2 hiếm khi có thể được di truyền như một tính trạng trội trên NST thường bởi một phụ huynh là thể khảm cho một đột biến dị hợp tử trong collagen-α1(I) hoặc -α2(I).
Các dạng bệnh xương thủy tinh gây chết được di truyền như các rối loạn lặn trên NST thường cũng được cho là do các đột biến bất hoạt trong CRTAP (OMIM 605497), PPIB (OMIM259440), hoặc LEPRE1 (OMIM 610339), ba yếu tố cần thiết cho quá trình hydroxyl hóa proline-986 trong COL1A1 và proline-707 trong COL1A2, những sửa đổi thiết yếu của các protein này. Ngoài ra, các biến thể của CREB3L1—mã hóa protein liên kết yếu tố đáp ứng cyclic AMP 3-like 1 (OMIM 616215) có thể dẫn đến một dạng bệnh xương thủy tinh gây chết, trước/chu sinh. Khi sinh, thường là sinh non, các biểu hiện lâm sàng của bệnh xương thủy tinh gây chết bao gồm chậm tăng trưởng trong tử cung, biến dạng và gãy xương dài, và vỏ sọ mềm với các thóp lớn; tử vong thường xảy ra trong giai đoạn sơ sinh do suy hô hấp. Về mặt X-quang, bệnh xương thủy tinh bẩm sinh được biểu hiện bằng các xương dài ngắn, rộng và “nhàu nát”, biến dạng gập góc của xương chày, và các xương sườn hình chuỗi hạt. Chẩn đoán bệnh này thường được thực hiện trong tử cung; nó phải được phân biệt với loạn sản sụn loại I, loạn sản tử vong, và giảm phosphatase máu chu sinh, và được xác nhận bằng cách định lượng lượng collagen dưới mức bình thường được tổng hợp bởi các nguyên bào sợi trong ống nghiệm và xác định đột biến gen. Các đột biến gây chết của COL1A1 tập trung tại các vị trí mà monome collagen liên kết với integrin, metalloproteinase chất nền, fibronectin, và protein chất nền sụn oligomeric; các đột biến khác làm suy giảm quá trình xử lý sau dịch mã của collagen α1(I) và do đó cản trở sự kết hợp của các chuỗi collagen hoặc sự lan truyền của cấu trúc chuỗi xoắn ba. Các đột biến trong COL1A2 mã hóa collagen α2(I) trùng với các vị trí liên kết của proteoglycan. Thật vậy, các đột biến gây chết trong COL1A1 dẫn đến các dạng bệnh xương thủy tinh nặng nhất về mặt lâm sàng làm mất chức năng của protein nhiều hơn so với sự vắng mặt hoàn toàn của một alen COL1A1; sự thiếu hụt một nửa của COL1A1 dẫn đến bệnh xương thủy tinh type I ít nghiêm trọng hơn về mặt lâm sàng. Mặc dù việc dùng bisphosphonate pamidronate đã hữu ích ở trẻ sơ sinh có các biểu hiện nặng của bệnh xương thủy tinh type III và IV, nó đã không hiệu quả trong dạng gây chết của bệnh xương thủy tinh (xem bên dưới).
Khối lượng xương cực kỳ thấp hiện diện ở trẻ sơ sinh và trẻ em không dung nạp protein lysinuric (OMIM 222700), một rối loạn di truyền lặn trên NST thường về vận chuyển các axit amin dibasic—lysine, arginine, ornithine—ở ống thận và ruột, là do các biến thể mất chức năng trong SLC7A7 (OMIM 603593) mã hóa một tiểu đơn vị vận chuyển axit amin cationic. Các biểu hiện lâm sàng bao gồm nôn, tiêu chảy, suy hô hấp, chậm phát triển, chậm phát triển trí tuệ, gan to và xơ gan. Các bệnh nhân bị ảnh hưởng bị suy giảm tổng hợp urê do giảm hấp thu ornithine ở gan nhưng có các đợt tăng amoniac máu với tăng bài tiết các axit amin dibasic qua nước tiểu; có khối lượng xương cực kỳ thấp do thiếu hụt protein rõ rệt và có lẽ do tăng tái hấp thu xương do cytokine gây ra. Việc dùng citrulline đã được báo cáo là làm tăng sự tăng trưởng và khối lượng xương ở một số bệnh nhân này. Các biến thể của SLC7A7 bao gồm mất đoạn nhiều exon, nhân đôi, sai nghĩa, vô nghĩa và các đột biến vị trí nối phân tán khắp gen; những bệnh nhân có các mất đoạn lớn nhất có kiểu hình nặng nhất. Khoáng hóa xương dưới mức bình thường cũng có thể được quan sát thấy ở trẻ sơ sinh bị giảm phosphatase máu ở trẻ nhỏ (OMIM 241500) và ở những trẻ bị rối loạn cơ bắp hạn chế vận động, chẳng hạn như hội chứng Prader-Willi (OMIM 176270), bệnh dự trữ glycogen type II (bệnh Pompe—OMIM 232300), và các dạng cứng khớp.
Khối lượng xương tăng
Khối lượng xương tăng có thể là toàn thể hoặc cục bộ; xơ cứng xương (osteosclerosis) đề cập đến sự dày lên của xương bè và tăng sinh xương (hyperostosis) đề cập đến sự gia tăng khối lượng xương vỏ. Dạng bệnh xương hóa đá “ác tính” ở trẻ nhỏ (OMIM 259700) là một trong số các rối loạn di truyền lặn trên NST thường do sự biệt hóa hoặc chức năng của hủy cốt bào bất thường dẫn đến sự tái hấp thu khiếm khuyết của pha khoáng của xương (xem bên dưới). Bệnh xương hóa đá ở trẻ nhỏ được biểu hiện lâm sàng ở trẻ sơ sinh bị ảnh hưởng bằng chậm phát triển, chậm phát triển trí tuệ, tắc nghẽn mũi, mất thị lực, thính giác và các chức năng thần kinh sọ khác, và sự phát triển quá mức của xương, dẫn đến giảm ba dòng tế bào máu, gan lách to tại các vị trí tạo máu ngoài tủy, và tăng nhạy cảm với nhiễm trùng (ví dụ, viêm tủy xương hàm dưới) và gãy xương do giảm sức mạnh của xương mặc dù khối lượng xương cao. Tử vong thường xảy ra trong vài năm đầu đời do nhiễm trùng huyết, thiếu máu hoặc xuất huyết. Các phát hiện thực thể bổ sung ở trẻ sơ sinh và trẻ nhỏ mắc dạng bệnh xương hóa đá nặng nhất bao gồm suy giảm tăng trưởng tuyến tính, đầu to, trán dô, rung giật nhãn cầu, mọc răng sữa chậm và các vết bầm tím. Dấu hiệu X-quang đặc trưng của bệnh xương hóa đá ở trẻ nhỏ là sự gia tăng tương đối đồng đều mật độ xương của hộp sọ, các đốt sống và bộ xương trục với xương vỏ và xương bè dày lên, các biến dạng “bình Ehrlenmeyer” ở đầu xa của các xương dài ở trẻ lớn, và các dải xơ cứng và thấu quang xen kẽ ở cánh chậu và hành xương. Các dấu ấn sinh học cho bệnh xương hóa đá là nồng độ phosphatase axit và đồng dạng não của creatine kinase trong huyết thanh tăng. Bệnh xương hóa đá gây chết ở trẻ nhỏ có thể do các đột biến mất chức năng đồng hợp tử hoặc dị hợp tử phức hợp trong các gen mà bản phiên mã của chúng là cần thiết cho chức năng bình thường của nguyên bào xương: chất điều hòa miễn dịch tế bào T 1 (TCIRG1, OMIM 604592) mã hóa một tiểu đơn vị của bơm proton không bào trong rìa bàn chải của hủy cốt bào, qua đó các ion hydro được vận chuyển từ tế bào chất vào hốc tái hấp thu dưới hủy cốt bào; kênh clorua 7 (CLCN7, OMIM 602727) mã hóa một kênh clorua trong rìa bàn chải của hủy cốt bào cần thiết cho việc vận chuyển cation này vào và do đó axit hóa hốc tái hấp thu; protein xuyên màng liên quan đến bệnh xương hóa đá 1 (OSTM1, OMIM 607649) mã hóa tiểu đơn vị beta của CLCN7 cần thiết cho quá trình xử lý sau dịch mã bình thường của nó (xem bên dưới). Ở những đối tượng được chọn, ghép tủy xương cung cấp các tế bào tiền thân của hủy cốt bào đã có hiệu quả trong việc ngăn chặn sự tiến triển của rối loạn này, mặc dù thường có những thiếu sót còn lại đáng kể. Tăng canxi máu thoáng qua nhưng đáng kể có thể xảy ra sau thủ thuật này. Bệnh xương hóa đá do thiếu hụt carbonic anhydrase II (CA2, OMIM 611492) mã hóa carbonic anhydrase II cũng có thể biểu hiện ở trẻ nhỏ bằng chậm phát triển hoặc gãy xương với chấn thương không đáng kể.
Tầm vóc thấp không cân đối là biểu hiện chính của bệnh pycnodysostosis (OMIM 265800) và được biểu hiện trong thời thơ ấu hoặc đầu thời thơ ấu bằng một chu vi vòng đầu tương đối lớn với các thóp và đường khớp sọ mở, các đặc điểm khuôn mặt dị dạng (trán-chẩm dô, lồi mắt, củng mạc xanh, hàm trên thiểu sản, cằm nhỏ, vòm miệng cao, sai khớp cắn, mũi khoằm), các ngón tay ngắn và dùi trống với móng tay thiểu sản, lồng ngực hẹp, ngực ức gà, ưỡn cột sống thắt lưng, gù vẹo cột sống, và tăng nguy cơ gãy xương. Về mặt X-quang, có xơ cứng xương rõ rệt tăng theo tuổi, các thóp và đường khớp sọ mở, xương đòn mỏng với các đầu bên thiểu sản, ăn mòn và thiểu sản các đốt ngón tay xa và xương sườn, và các đốt sống đặc nhưng các mỏm ngang bình thường. Về mặt mô học, có sự giảm hoạt động của nguyên bào xương và hủy cốt bào. Pycnodysostosis là do các đột biến mất chức năng đồng hợp tử hoặc dị hợp tử phức hợp (dừng, sai nghĩa, vô nghĩa) trong CTSK (OMIM 601105)—gen mã hóa cathepsin K, một protease cysteine lysosome được biểu hiện trong các hủy cốt bào mà sự mất đi của nó làm suy giảm sự phân hủy collagen và sự tái hấp thu của chất dạng xương, thành phần hữu cơ của chất nền xương, nhưng không phải thành phần khoáng chất của nó. Hội chứng Raine (OMIM 259775) gồm xơ cứng xương toàn thể trước sinh với sự hình thành xương màng xương, dị dạng khuôn mặt (mũi thiểu sản, tai đóng thấp, lồi mắt, hở hàm ếch), và lồng ngực thiểu sản thường dẫn đến tử vong chu sinh là hậu quả của các biến thể bất hoạt hai alen của FAM20C (OMIM 611061) mã hóa một phosphorylase tác động lên các glycoprotein tiết liên kết N-ligand liên kết integrin nhỏ (SIBLING) liên kết canxi và điều hòa sự khoáng hóa của chất dạng xương, sự mất đi của nó làm tăng cường quá trình này.
Rối loạn Khoáng hóa và Hình thành Xương ở Trẻ em và Vị thành niên
Sự hình thành xương có thể bị suy giảm do thiếu khoáng chất (canxi và/hoặc phốt phát) hoặc do sản xuất thiếu chất nền xương. Khoáng hóa xương có thể quá mức do tăng tốc độ lắng đọng khoáng chất hoặc giảm tốc độ tái hấp thu pha khoáng của xương. Vôi hóa lạc chỗ của các mô ngoài xương có thể xảy ra khi nồng độ canxi và phốt phát tại chỗ cao, trong khi hóa xương ngoài xương có thể xảy ra khi sự điều hòa hình thành xương bị rối loạn.
Còi xương
Còi xương ở trẻ em là một rối loạn của sụn tăng trưởng và sự thất bại trong việc lắng đọng đủ khoáng chất xương (hydroxyapatite) để đạt được sức mạnh xương cần thiết cho phép chịu trọng lượng thẳng đứng và đi lại do thiếu canxi, phốt phát, vitamin D, hoặc một trong nhiều đồng yếu tố đảm bảo sự đồng hóa và sử dụng thích hợp các vật liệu này. Do đó, còi xương là một rối loạn ở trẻ em về giảm khoáng hóa sụn và xương xốp nguyên phát của các đĩa tăng trưởng ở đầu các xương dài và của chất nền xương collagen type I bên dưới đĩa tăng trưởng, dẫn đến suy giảm sức mạnh của xương và tăng trưởng tuyến tính và các biến dạng xương (mềm sọ, loe hành xương, chân vòng kiềng chữ X, chân vòng kiềng chữ O), và tăng nguy cơ gãy xương. Còi xương có thể do thiếu canxi và/hoặc phốt phát do hạn chế ăn vào hoặc giảm hấp thu ở ruột của các chất dinh dưỡng này; loại sau có thể liên quan đến các bất thường nội tại của chức năng ruột (hội chứng kém hấp thu, chiều dài ruột ngắn) hoặc do không đủ vitamin D ăn vào hoặc giảm hấp thu hoặc hiệu quả chức năng dưới mức bình thường của nó; suy giảm tổng hợp vitamin D nội sinh ở da có thể do điều kiện khí hậu dẫn đến giảm tiếp xúc với ánh sáng mặt trời, việc bôi các tác nhân dược phẩm chặn sóng tia cực tím mạnh lên da, hoặc việc mặc quần áo rộng rãi hạn chế đáng kể diện tích bề mặt da tiếp xúc với ánh sáng mặt trời; thiếu vitamin D cũng có thể là hậu quả của việc không xử lý cholecalciferol thành chất chuyển hóa có hoạt tính sinh học của nó, 1,25-dihydroxyvitamin D (calcitriol), tăng tốc chuyển đổi 1,25-dihydroxyvitamin D thành 1,24,25-trihydroxyvitamin D tan trong nước và mất nó qua nước tiểu, hoặc do suy giảm hoạt động chức năng của calcitriol do bất thường của thụ thể hạt nhân VDR của nó; giảm hoạt động của phosphatase kiềm cũng dẫn đến suy giảm hình thành xương (Bảng 20.8A, 20.8B). Ở người lớn, sự thiếu hụt vitamin D, canxi, hoặc phốt phát dẫn đến nhuyễn xương, sự thất bại trong việc khoáng hóa vỏ và các bè xương.
Bảng 20.8A: Các rối loạn khoáng hóa xương: Còi xương
| I | Thiếu Vitamin D | |||
|---|---|---|---|---|
| A | Giảm ăn vào, tổng hợp nội sinh, giữ lại, hoặc cô lập | |||
| 1 | Thiếu vitamin D ở mẹ—cho con bú | |||
| 2 | Giảm tổng hợp ở da—thiếu ánh nắng mặt trời, sử dụng kem chống nắng, tăng sắc tố da | |||
| 3 | Kém hấp thu—bệnh celiac, rối loạn chức năng gan mật, hội chứng ruột ngắn, xơ nang, bệnh viêm ruột, phẫu thuật nối tắt dạ dày | |||
| 4 | Thuốc—thuốc chống co giật, glucocorticoid, cholestyramine, thuốc kháng retrovirus | |||
| 5 | Hội chứng thận hư | |||
| 6 | Béo phì | |||
| B | Lỗi chuyển hóa | |||
| 1 | Thiếu 25-hydroxylase | |||
| a | ||||
| b | ||||
| 2 | Thiếu 25OH-vitamin D3-1-hydroxylase | |||
| a | ||||
| b | ||||
| 3 | Suy giảm đáp ứng với vitamin D | |||
| a | ||||
| b | ||||
| II | Thiếu Canxi—Nguyên phát | |||
| A | Thiếu hụt dinh dưỡng | |||
| B | Tăng canxi niệu—hội chứng tăng prostaglandin E2 | |||
| III | Thiếu Phốt phát/Hạ phốt phát máu | |||
| A | Dịch chuyển qua tế bào | |||
| 1 | Truyền glucose hoặc insulin | |||
| 2 | Nuôi ăn lại | |||
| 3 | Nhiễm kiềm hô hấp; tăng thông khí | |||
| 4 | Ngộ độc salicylate | |||
| 5 | Catecholamine | |||
| B | Giảm hấp thu ở ruột | |||
| 1 | Thiếu hụt dinh dưỡng | |||
| a | ||||
| 2 | Giảm hấp thu ở ruột | |||
| a | ||||
| b | ||||
| c | ||||
| C | Tăng phốt phát niệu | |||
| 1 | Còi xương giảm phốt phát máu gia đình di truyền trội liên kết X (PHEX) a | |||
| 2 | Còi xương giảm phốt phát máu di truyền lặn liên kết X (CLCN5) | |||
| 3 | Còi xương giảm phốt phát máu di truyền trội trên NST thường (FGF23) a | |||
| 4 | Giảm phốt phát máu di truyền trội trên NST thường có sỏi niệu 1 (SLC34A1) b | |||
| 5 | Giảm phốt phát máu di truyền trội trên NST thường có sỏi niệu 2 (SLC9A3R1) | |||
| 6 | Còi xương giảm phốt phát máu di truyền lặn trên NST thường 1 (DMP1) a | |||
| 7 | Còi xương giảm phốt phát máu di truyền lặn trên NST thường 2 (ENPP1) a | |||
| 8 | Còi xương giảm phốt phát máu di truyền lặn trên NST thường có tăng canxi niệu (SLC34A3) | |||
| 9 | Còi xương giảm phốt phát máu có cường cận giáp (KL) | |||
| 10 | Còi xương giảm phốt phát máu có hội chứng ống thận Fanconi (SLC34A1) c | |||
| 11 | Còi xương/nhuyễn xương giảm phốt phát máu do ung thư (FGF23, sFRP4, MEPE, FGF7) | |||
| 12 | Thừa FGF23 không rõ nguyên nhân (hội chứng McCune-Albright [GNAS] a, loạn sản sụn đầu xương type Jansen [PTH1R] a, loạn sản xương-sụn-răng [FGFR1] a, hội chứng nevus bã nhờn dạng dải a, sắt polymaltose a) | |||
| 13 | Toan hóa ống thận/Hội chứng Fanconi | |||
| a | ||||
| b | ||||
| D | Các nguyên nhân khác | |||
| 1 | Cường cận giáp | |||
| 2 | Thuốc—lợi tiểu (thiazide, acetazolamide, lợi tiểu quai); thuốc kháng retrovirus (tenovir, adefovir); thuốc chống tân sinh (ifosfamide, cisplatin); aminoglycosid, imatinib | |||
| IV | Giảm phosphatase máu (ALPL) | |||
| A | Các dạng chu sinh, trẻ nhỏ, trẻ em, người lớn | |||
| B | Giảm phosphatase máu ở răng | |||
| C | Giảm phosphatase máu giả | |||
| V | Các chất ức chế Khoáng hóa | |||
| A | Nhôm—đường tiêm | |||
| B | Bisphosphonate | |||
| C | Florua |
(Từ Gattineni J, Baum M. Genetic disorders of phosphate regulation. Pediatr Nephrol. 2012;27:1477–1487; Hori M, Shimizu Y, Fukumoto S. Minireview: fibroblast growth factor 23 in phosphate homeostasis and bone metabolism. Endocrinology. 2011;152:4–10; Imel EA, Econs MJ. Approach to the hypophosphatemic patient. J Clin Endocrinol Metab. 2012;97:696–706.)
Bảng 20.8B: Rối loạn di truyền về khoáng hóa xương: Còi xương
| Gen | NST | OMIM | Sinh lý bệnh | Đột biến—Biểu hiện lâm sàng |
|---|---|---|---|---|
| Rối loạn chuyển hóa vitamin D | ||||
| CYP2R1—Cytochrome P450, phân họ IIR, polypeptide 1 | 11p15.2 | 608713 | 25-hydroxylase gan—enzyme chuyển đổi vitamin D3 thành 25OHD3 (calcidiol) | Đột biến mất chức năng hai alen dẫn đến còi xương thiếu hydroxyl hóa vitamin D type 1B (còn gọi là còi xương phụ thuộc vitamin D type 1B), AR |
| CYP27B1—Cytochrome P450, phân họ XXVII, polypeptide 1 | 12q13.1-q13.3 | 609506 | 25OHD3-1α hydroxylase—enzyme chuyển đổi 25OHD3 thành 1,25(OH)2D3 (calcitriol) | Các đột biến bất hoạt hai alen dẫn đến còi xương phụ thuộc vitamin D type 1A, AR |
| VDR—Thụ thể Vitamin D | 12q12-q14 | 601769 | Thụ thể Vitamin D—yếu tố phiên mã truyền tải tác dụng của calcitriol lên sự hoạt hóa hoặc ức chế gen | Các đột biến mất chức năng hai alen dẫn đến không nhạy cảm với calcitriol & còi xương phụ thuộc vitamin D type 2A, AR |
| HNRNPC—Ribonucleoprotein hạt nhân không đồng nhất C | 14q11.2 | 164020 | Mã hóa một ribonucleoprotein điều hòa phiên mã gen bằng cách chiếm giữ VDRE một cách tương hỗ & thoáng qua trong vùng promoter thượng nguồn của các gen đích đáp ứng vitamin D | Biểu hiện quá mức trong các mô đáp ứng vitamin D dẫn đến chiếm giữ VDRE kéo dài cản trở tương tác của VDR/RXR với VDRE dẫn đến còi xương phụ thuộc vitamin D type 2B |
| CYP3A4—Cytochrome P450, phân họ IIIA, polypeptide 4 | 7q22.1 | 124010 | Mã hóa một enzyme CYP450 gan chuyển hóa oxy hóa & bất hoạt calcidiol & calcitriol | Biến thể đơn alen tăng chức năng dẫn đến dị hóa nhanh calcitriol đòi hỏi liều cao calcitriol để điều trị còi xương (còi xương phụ thuộc vitamin D type 3) (AD) |
| Rối loạn chuyển hóa phốt phát | ||||
| SLC34A1—Họ chất mang hòa tan 34 (chất đồng vận chuyển natri phốt phát), thành viên 1 | 5q35 | 182309 | Mã hóa NPT2a—chất đồng vận chuyển natri/phốt phát biểu hiện trên màng đỉnh của ống lượn gần; dưới sự kiểm soát ức chế của PTH | Đột biến mất chức năng dẫn đến còi xương giảm phốt phát máu có sỏi thận, type 1, AD; hội chứng Fanconi type 2, AR |
| SLC34A2—Họ chất mang hòa tan 34 (chất đồng vận chuyển natri phốt phát), thành viên 2 | 2p15.31-p15.2 | 604217 | Mã hóa NPT2b—chất đồng vận chuyển natri/phốt phát biểu hiện ở ruột non, phổi và tinh hoàn | Các đột biến mất chức năng liên quan đến lắng đọng vi sỏi phế nang & lắng đọng vi sỏi tinh hoàn, AR |
| SLC34A3—Họ chất mang hòa tan 34 (chất đồng vận chuyển natri phốt phát), thành viên 3 | 9q34 | 609826 | Mã hóa NPT2c—chất đồng vận chuyển natri/phốt phát biểu hiện trên màng đỉnh của ống lượn gần | Đột biến mất chức năng dẫn đến còi xương giảm phốt phát máu di truyền có tăng canxi niệu, AR |
| SLC9A3R1—Họ chất mang hòa tan 9, thành viên 3, chất điều hòa 1 | 17q25.1 | 604990 | Mã hóa NHERF1—một yếu tố điều hòa trao đổi natri/hydro ở ống thận liên kết với NPT2a neo nó vào màng lòng ống của ống lượn gần; phosphoryl hóa bởi PTH dẫn đến sự phân ly của nó khỏi & nhập bào của NPT2a | Các đột biến mất chức năng dẫn đến còi xương giảm phốt phát máu có sỏi thận, type 2, AD |
| CLCN5—Kênh clorua 5 | Xp11.23-p11.22 | 300008 | Mã hóa một chất trao đổi ion clorua & hydro ở ống lượn gần | Các đột biến mất chức năng dẫn đến còi xương giảm phốt phát máu di truyền lặn liên kết X, tăng canxi niệu, nhiễm canxi thận, XLR |
| PHEX—Tương đồng endopeptidase điều hòa phốt phát, liên kết X | Xp22.2-p22.1 | 300550 | Ectoenzyme biểu hiện trên màng tế bào của nguyên bào xương; cơ chất sinh lý của nó có thể là MEPE & pASARM, sản phẩm phân hủy được phosphoryl hóa của nó bao phủ hydroxyapatite & cản trở sự lắng đọng khoáng chất | Đột biến mất chức năng dẫn đến còi xương giảm phốt phát máu liên kết X; liên quan đến tăng biểu hiện FGF23; trội liên kết X |
| DMP1—Phosphoprotein axit chất nền ngà răng 1 | 4q22.1 | 600980 | Protein chất nền xương không phải collagen, giàu serine; một glycoprotein tiết liên kết N-ligand liên kết integrin nhỏ (SIBLING) biểu hiện ở tế bào xương | Các đột biến mất chức năng dẫn đến tăng tổng hợp FGF23 ở tế bào xương, tăng phốt phát niệu, & còi xương giảm phốt phát máu di truyền lặn trên NST thường, AR |
| ENPP1—Ectonucleotide pyrophosphatase/phosphodiesterase 1 | 6q23.2 | 173335 | Ectoenzyme được biểu hiện bởi các tế bào sụn, xương, & tương bào thủy phân ATP thành pyrophosphat, một chất ức chế khoáng hóa xương | Các đột biến mất chức năng dẫn đến còi xương giảm phốt phát máu di truyền lặn trên NST thường với tăng biểu hiện FGF23, AR |
| ANKH– ANK, chuột, tương đồng của | 5p15.2-141 | 605145 | Protein bề mặt tế bào xuyên màng điều hòa bài tiết pyrophosphat | Các đột biến bất hoạt dẫn đến giảm phốt phát máu nhẹ & cứng khớp, chậm phát triển, điếc, & loạn sản ngà răng không hoàn toàn; AR |
| FGF23—Yếu tố tăng trưởng nguyên bào sợi 23 | 12p13.2 | 605380 | Sản phẩm của nguyên bào xương & tế bào xương làm giảm tái hấp thu phốt phát ở ống thận & ức chế tổng hợp calcitriol | Đột biến tăng chức năng làm giảm tốc độ phân hủy FGF23 dẫn đến còi xương giảm phốt phát máu di truyền trội trên NST thường, AD; tổng hợp lạc chỗ quá mức bởi các khối u dẫn đến còi xương giảm phốt phát máu; đột biến mất chức năng dẫn đến giảm tổng hợp FGF23 dẫn đến vôi hóa u gia đình tăng phốt phát máu di truyền lặn trên NST thường, AR |
| GALNT3—UDP-N-Acetyl-alpha-D-Galactosamine: polypeptide N-acetylglactosaminyl transferase 3 | 2q24.3 | 601756 | Mã hóa một enzyme cần thiết cho O-glycosyl hóa Thr178 của FGF23 trong quá trình xử lý sau dịch mã; thất bại ở bước này dẫn đến phân hủy FGF23 trước khi bài tiết | Đột biến mất chức năng dẫn đến vôi hóa u gia đình tăng phốt phát máu di truyền lặn trên NST thường, AR |
| KL—α-Klotho | 13q13.1 | 604824 | Đồng thụ thể với FGFR1(IIIc) cho FGF23 chuyển đổi FGFR1(IIIc) thành thụ thể FGF23 đặc hiệu cho phép truyền tín hiệu | Đột biến mất chức năng dẫn đến vôi hóa u gia đình tăng phốt phát máu di truyền lặn trên NST thường, AR; Chuyển vị sang (9;13(q21.13;q13.1) dẫn đến hình thành Klotho quá mức dẫn đến còi xương giảm phốt phát máu & cường cận giáp |
| Giảm phosphatase máu | ||||
| ALPL—Phosphatase kiềm, gan | 1p36.12 | 171760 | Phosphatase kiềm không đặc hiệu của mô—ectoenzyme biểu hiện trên màng tế bào của nguyên bào xương, loại bỏ phốt phát liên kết hữu cơ; các cơ chất chính là pyrophosphat, phosphoethanolamine, pyridoxal-5′-phosphate | Các đột biến mất chức năng dẫn đến giảm phosphatase máu & còi xương có mức độ nghiêm trọng & khởi phát khác nhau: chu sinh, trẻ nhỏ, chuyển tiếp, trẻ em, người lớn, giảm phosphatase máu ở răng, giảm phosphatase máu giả; AR, AD |
AD, Di truyền trội trên nhiễm sắc thể thường; AR, di truyền lặn trên nhiễm sắc thể thường; ATP, adenosine triphosphate; MEPE, phosphoglycoprotein ngoại bào chất nền; pASARM, mô-típ liên quan đến MEPE giàu aspartate serine axit được phosphoryl hóa.
(Từ Farrow EG, White KE. Recent advances in renal phosphate handling. Nature Rev/Nephrol. 2010;6:207–217; Roizen JD, Li D, O’Lear L, et al. CYP3A4 mutation causes vitamin D-dependent rickets type 3. J Clin Invest. 2018;128:1013–1918.)
Trong quá trình hình thành xương nội sụn ở trẻ em đang lớn, chất nền sụn được tạo ra và sau đó được khoáng hóa. Khi chất nền nội sụn không được khoáng hóa hoàn toàn, sụn tích tụ, có sự dày lên của đĩa tăng trưởng và sự mất tổ chức của các tế bào sụn; các đầu của xương dài (đặc biệt là những xương chịu trọng lượng) bị biến dạng dẫn đến các biến dạng còi xương. Trong các quá trình tạo hình và tái tạo xương bè và các bề mặt màng xương và nội mạc xương của xương vỏ, chất dạng xương được hình thành bởi các nguyên bào xương; sự thất bại trong việc khoáng hóa chất nền xương ở những vùng này dẫn đến nhuyễn xương. Trong các khoảng thời gian thiếu hụt canxi và/hoặc phốt phát, trẻ đang lớn tích cực, chịu trọng lượng với các đĩa tăng trưởng sụn mở sẽ phát triển bệnh còi xương và nhuyễn xương, trong khi trong các trường hợp tương tự, người lớn chỉ phát triển nhuyễn xương trong quá trình tái tạo khi chất nền xương không được khoáng hóa tích tụ. Do đó, còi xương là biểu hiện của sự khoáng hóa nội sụn khiếm khuyết tại đĩa tăng trưởng và nhuyễn xương là sự thất bại trong việc khoáng hóa vỏ và các bè xương.
Về mặt lâm sàng, còi xương được biểu hiện bằng các bất thường về xương, chẳng hạn như chậm đóng thóp, mềm sọ (lún có thể đảo ngược của bản ngoài hộp sọ), trán dô (giãn nở xương sọ), và đôi khi dính khớp sọ ở trẻ nhỏ; cong cẳng tay ở trẻ chưa biết đi và chân vòng kiềng chữ O và/hoặc chữ X ở trẻ biết đi và trẻ em; mọc răng chậm với sự hình thành men răng kém và dễ bị sâu răng; ngực ức gà, nổi rõ các khớp sụn sườn, lồng ngực dưới loe, vẹo cột sống và gù; loe hành xương của các xương dài, và xoắn xương chày hoặc xương đùi (Hình 20.9). Về mặt X-quang, còi xương ban đầu được đặc trưng bởi sự mở rộng sụn tiếp hợp và mất vùng vôi hóa tạm thời được xác định, sau đó là hình ảnh lõm chén, bè ra và tua ở hành xương của các xương dài (đặc biệt là đầu xa xương trụ, đầu xa xương đùi và đầu gần xương chày), mất khoáng, biến dạng các xương dài và gãy xương. “Vùng Looser” là các đường thấu quang có bờ đặc, là các gãy xương giả nằm ở cổ và thân xương đùi. Hệ thống tính điểm X-quang Thacher để đánh giá mức độ nghiêm trọng của bệnh còi xương cung cấp một phương pháp khách quan và hữu ích hợp lý để xác định mức độ còi xương và đáp ứng của nó với điều trị. Về mặt mô học, do hậu quả của sự suy giảm vôi hóa trong đĩa tăng trưởng sụn, mô hình biệt hóa và trưởng thành của tế bào sụn bị phá vỡ và mất tổ chức, trong khi các đường viền chất dạng xương bị mở rộng. Ở các đối tượng thiếu phốt phát, chủ yếu là các bè xương bị giảm khoáng.
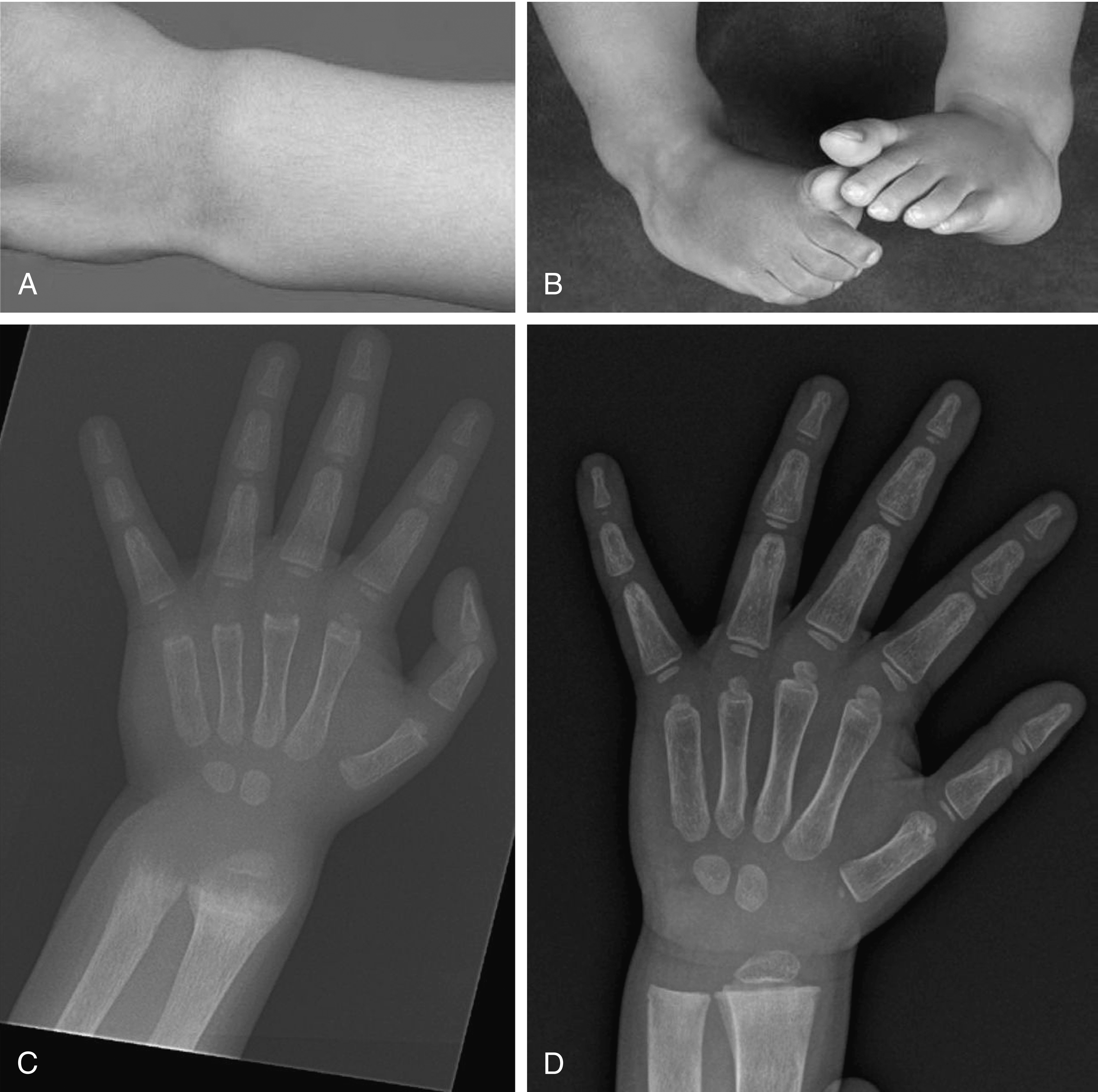
Hình 20.9: Các biểu hiện lâm sàng và X-quang của bệnh còi xương do thiếu vitamin D ở một đứa trẻ hai tuổi. Lưu ý sự loe ra của các hành xương xa của cổ tay trái (A) và cả hai mắt cá chân (B), hình ảnh X-quang cho thấy sự mở rộng của các hành xương của xương quay và xương trụ (C) với sự khoáng hóa tăng lên sau khi điều trị bằng cholecalciferol trong ba tháng (D).
(Từ Girgis CM, Clifton-Bligh RJ, Hamrick MW, et al. The roles of vitamin D in skeletal muscle: form, function, and metabolism. Endocr Rev. 2013;34:33–83. Hình ảnh được cung cấp bởi Phó Giáo sư Craig Munns, Bệnh viện Nhi đồng Westmead, Sydney, Úc.)
Sự suy giảm khoáng hóa chất dạng xương có thể do thiếu hụt dinh dưỡng hoặc giảm hấp thu canxi, phốt phát hoặc vitamin D ở ruột, lượng không đủ các chất dinh dưỡng này trong các dịch được sử dụng trong dinh dưỡng qua đường tĩnh mạch toàn phần, các lỗi chuyển hóa trong quá trình chuyển hóa hoặc hoạt động của vitamin D, các khiếm khuyết trong việc bảo tồn phốt phát hoặc canxi ở ống thận, hoặc các bất thường trong việc tạo ra và chức năng của phosphatase kiềm (xem Bảng 20.8A, 20.8B). Nhìn chung, còi xương có thể được coi là do thiếu canxi (thường liên quan đến thiếu hụt dinh dưỡng vitamin D hoặc hiếm khi do một khiếm khuyết trong quá trình chuyển hóa của nó thành chất chuyển hóa hoạt động—calcitriol—hoặc trong hoạt động tế bào của nó hoặc do giảm ăn vào canxi hoặc mất quá nhiều qua nước tiểu) hoặc do thiếu phốt phát (liên quan đến mất phốt phát qua thận do các khiếm khuyết ống thận nguyên phát trong việc tái hấp thu phốt phát hoặc do tạo ra quá nhiều phosphatonin—các hợp chất ức chế tái hấp thu phốt phát ở ống thận). Do đó, còi xương dinh dưỡng có thể do giảm ăn vào vitamin D (hoặc không tiếp xúc đủ với ánh nắng mặt trời) hoặc canxi hoặc do ăn vào ở mức giới hạn cả hai chất dinh dưỡng. Thiếu hụt phốt phát trong chế độ ăn là không phổ biến do sự sẵn có rộng rãi của nó, nhưng chất dinh dưỡng này có thể bị thiếu hụt trong các dịch truyền tĩnh mạch. Khoáng hóa xương cũng có thể bị suy giảm trực tiếp bởi các bất thường trong việc tạo ra phosphatase kiềm hoặc bởi các tác nhân, chẳng hạn như nhôm hoặc florua.
Còi xương do thiếu canxi
Mặc dù thiếu canxi trong chế độ ăn là một nguyên nhân hiếm gặp của bệnh còi xương, nhưng tỷ lệ thiếu vitamin D và còi xương ở trẻ em là đáng kể—dao động từ 2,9 trên 100.000 trẻ em Canada đến 7,5, 24 và 95 trên 100.000 trẻ em ở Vương quốc Anh, Hoa Kỳ và Châu Phi, tương ứng. Tình trạng không đủ/thiếu vitamin D có thể phát sinh do giảm lượng ăn vào trong điều kiện tiếp xúc với ánh sáng mặt trời không tối ưu, các hội chứng kém hấp thu ở ruột (bệnh celiac, “ruột ngắn”, xơ nang, bệnh viêm ruột, dị ứng thực phẩm), suy giảm tổng hợp calcitriol ở thận do bệnh thận mạn tính hoặc do các biến thể của CYP27B1 (OMIM 609506), hoặc do tiếp xúc với thuốc (phenytoin, carbamazepine, phenobarbital, glucocorticoid—loại sau làm tăng hoạt động của vitamin D-24-hydroxylase ở thận). Ở trẻ sơ sinh, tình trạng vitamin D liên quan đến tuổi thai và sự tăng trưởng trong tử cung và lượng chất dinh dưỡng này của mẹ và liệu trang phục của mẹ có cho phép tiếp xúc đủ với ánh sáng mặt trời và tổng hợp nội sinh cholecalciferol hay không. Mặc dù béo phì có liên quan đến nồng độ calcidiol trong huyết thanh thấp hơn do khả năng hòa tan của nó trong mô mỡ và do đó bị cô lập trong lipid, béo phì không làm suy giảm các quá trình hình thành hoặc khoáng hóa xương. Da của trẻ càng sẫm màu và trẻ sống ở độ cao càng cao thì nguy cơ thiếu vitamin D càng lớn nếu không được bổ sung. Ngoài ra, lượng tia cực tím chiếu tới bề mặt trái đất bị ảnh hưởng bởi vĩ độ nơi trẻ ở, mùa trong năm, thời gian trong ngày, lượng thời gian ở ngoài trời và quần áo mặc khi ra ngoài, mây che phủ và cường độ ô nhiễm không khí. Còi xương dinh dưỡng do thiếu vitamin D hoặc giảm lượng canxi ăn vào dẫn đến các khiếm khuyết trong sự biệt hóa và chức năng của tế bào sụn và suy giảm sự lắng đọng canxi vào chất nền sụn và chất dạng xương. Trong huyết thanh, hơn 90% cholecalciferol và các chất chuyển hóa của nó (calcidiol, calcitriol) được liên kết bởi các protein huyết thanh, chủ yếu bởi protein liên kết vitamin D (DBP) được mã hóa bởi GC (thành phần đặc hiệu nhóm, OMIM 139200). Sự vắng mặt bẩm sinh của DBP do mất đoạn hai alen của nhánh dài nhiễm sắc thể 4 có liên quan đến nồng độ calcidiol và calcitriol toàn phần trong huyết tương cực kỳ thấp nhưng có thể đo được mà không có bằng chứng về bệnh còi xương hoặc nhuyễn xương—có lẽ là do nồng độ calcitriol tự do đủ để hình thành xương qua trung gian nguyên bào xương. Do hậu quả của việc thiếu vitamin D, cường cận giáp thứ phát phát triển dẫn đến suy giảm tái hấp thu phốt phát đã lọc ở ống thận và hạ phốt phát máu. Mặc dù khiếm khuyết gần nhất ở đối tượng thiếu vitamin D là thiếu canxi, sự tăng trưởng và phát triển suy giảm của sụn tăng trưởng là do hạ phốt phát máu ức chế sự biểu hiện của CASP9 (OMIM 602234) mã hóa caspase 9, đến lượt nó, làm suy giảm quá trình chết theo chương trình được điều hòa bởi phốt phát của các tế bào sụn phì đại, cho phép chúng tiếp tục tăng sinh.
Do đó, còi xương do thiếu canxi thường do thiếu vitamin D, làm giảm hấp thu canxi trong chế độ ăn ở ruột và tái hấp thu canxi đã lọc ở thận. Được xác định bằng cách đo nồng độ calcidiol (25OHD) trong huyết thanh, tình trạng thiếu vitamin D hiện diện khi các giá trị dưới 12 đến 15 ng/mL. Nồng độ calcidiol từ 15 đến 19,5 ng/mL cho thấy tình trạng “không đủ” vitamin D, trong khi những nồng độ từ 20 ng/mL trở lên là đủ. (Tuy nhiên, điều quan trọng là phải đánh giá tính hợp lệ và sự thay đổi của một xét nghiệm 25OHD/calcidiol cụ thể khi giải thích dữ liệu do xét nghiệm đó cung cấp vì có thể có sự khác biệt định lượng đáng kể giữa các bộ thuốc thử riêng lẻ. Ngoài ra, vì nồng độ đáng kể của các đồng phân epime C-3 của calcidiol thường hiện diện ở trẻ sơ sinh làm tăng nồng độ biểu kiến của chất phân tích này, điều quan trọng là các phép đo giá trị calcidiol huyết thanh ở trẻ em dưới 1 tuổi phải được thực hiện bằng một xét nghiệm định lượng chính xác calcidiol khi có mặt các đồng phân epime C-3.)
Trẻ sơ sinh của các bà mẹ thiếu vitamin D nặng có thể biểu hiện các dấu hiệu còi xương khi sinh, bao gồm gãy xương và co giật do hạ canxi máu; sự mềm đi của bản ngoài hộp sọ ở trẻ sơ sinh và trẻ rất nhỏ cho phép nó bị lõm có thể đảo ngược (mềm sọ). Các biểu hiện lâm sàng của bệnh còi xương ở trẻ sơ sinh chưa biết đi bao gồm cong cẳng tay, mềm sọ, trán dô và chậm đóng thóp sọ. Ở trẻ em bị còi xương, cong xương đùi ra trước, lệch trong hoặc ngoài của đầu gối dẫn đến chân vòng kiềng chữ O hoặc chữ X (tương ứng) hoặc biến dạng “gió thổi” liên quan đến các biến dạng chân đối diện, loe (mở rộng) hành xương của các xương dài với cổ tay to rõ rệt, nổi rõ các khớp sụn sườn (chuỗi hạt còi xương), và lõm thành ngực trước dưới (rãnh Harrison) được ghi nhận. Tầm vóc thấp và cân nặng dưới mức tối ưu cũng thường xuyên hiện diện. Mọc răng có thể bị trì hoãn và men răng thiểu sản—dẫn đến sâu răng. Yếu cơ biểu hiện bằng giảm trương lực cơ và chậm đi có thể được quan sát thấy. Co cứng và co giật do hạ canxi máu có thể xảy ra ở trẻ sơ sinh thiếu vitamin D nặng mà không có các dấu hiệu lâm sàng hoặc X-quang rõ rệt của bệnh còi xương. Khi thiếu vitamin D cực kỳ nghiêm trọng và kéo dài ở một thanh thiếu niên, co giật và gãy xương do hạ canxi máu có thể xảy ra.
Tình trạng thiếu vitamin D thường được quan sát thấy ở trẻ em và thanh thiếu niên da sẫm màu có ít tiếp xúc với ánh sáng mặt trời, đặc biệt là trong và cuối những tháng mùa đông, và/hoặc ở những người ăn chay, hoặc những người đang được điều trị bằng thuốc chống co giật hoặc thuốc kháng retrovirus. Bởi vì vitamin D có nhiều vị trí hoạt động ngoài xương do sự biểu hiện đa tế bào của VDR, người ta cho rằng thiếu vitamin D có thể đóng một vai trò trong sự phát triển của các rối loạn tự miễn, nhiễm trùng và hệ thống (ví dụ, tăng nhạy cảm với nhiễm trùng—đặc biệt là viêm phổi—do cả sự yếu của thành ngực và thiếu tác dụng kích thích của vitamin D đối với hệ miễn dịch; hen suyễn và đáp ứng của nó với điều trị; viêm da cơ địa, đái tháo đường type 1, bệnh viêm ruột và các rối loạn tim mạch). Về mặt X-quang, giảm khoáng hóa xương với mỏng vỏ xương và các đường gãy xương do căng thẳng mỏng, cũng như hình ảnh lõm chén, mở rộng và không đều (tua) của các hành xương xa của các xương dài được quan sát thấy ở đối tượng bị còi xương. Các vùng viêm xương xơ nang liên quan đến cường cận giáp thứ phát đôi khi có thể phát triển.
Sau khi dầu gan cá tuyết được giới thiệu như một chất bổ sung chế độ ăn vào năm 1918 (và sau đó là việc tăng cường các loại sữa công thức cho trẻ sơ sinh và sữa và các thực phẩm khác bằng ergosterol được chiếu xạ) và khám phá vào năm 1919 rằng việc tiếp xúc với ánh sáng mặt trời ngăn ngừa sự phát triển của bệnh còi xương, tình trạng thiếu vitamin D do dinh dưỡng trở nên tương đối hiếm ở Bắc Mỹ chỉ để tái xuất hiện vài thập kỷ sau đó. Bởi vì thai nhi được cung cấp vitamin D bằng cách chuyển calcidiol của mẹ qua nhau thai, tình trạng thiếu vitamin D xảy ra chủ yếu ở trẻ sơ sinh được sinh ra từ những bà mẹ có dự trữ vitamin D thấp (phụ nữ da màu hoặc những người ăn chay hoặc dinh dưỡng kém, hoặc những người ít tiếp xúc với ánh sáng mặt trời). Bệnh còi xương phát triển ở trẻ sơ sinh và trẻ nhỏ ăn chế độ ăn ít vitamin D (ít hoặc không có sữa, thịt, trứng hoặc cá) mà không bổ sung vitamin D, những người ít tiếp xúc với ánh sáng mặt trời vì chúng bị nhốt trong nhà do bệnh tật, khí hậu, hoặc lựa chọn của cha mẹ hoặc vì chúng mặc quần áo che toàn bộ cơ thể khỏi ánh sáng mặt trời. Tình trạng thiếu vitamin D phổ biến hơn ở trẻ bú mẹ vì sữa mẹ chứa ít vitamin D; trẻ sơ sinh da đen có nguy cơ cao hơn trẻ sơ sinh da trắng một phần do sắc tố da tăng lên dẫn đến giảm tổng hợp nội sinh cholecalciferol ở da (vì melanin hấp thụ tia cực tím). Sữa mẹ bình thường chỉ chứa 25 IU/L vitamin D, do đó, khuyến cáo rằng tất cả trẻ bú mẹ đều nhận được bổ sung vitamin D 400 IU/ngày. Vì hầu hết các loại sữa công thức thương mại cho trẻ sơ sinh đều chứa 400 IU/L vitamin D, cũng khuyến cáo rằng trẻ sơ sinh được nuôi bằng sữa công thức tiêu thụ ít hơn 1 L sữa công thức hàng ngày cũng nhận được một chất bổ sung 400 IU/ngày vitamin D. Trẻ em trên 1 tuổi, trẻ em và thanh thiếu niên nên ăn hoặc nhận bổ sung vitamin D 600 IU hàng ngày.
Mặc dù các định nghĩa về giảm vitamin D máu thay đổi giữa các nghiên cứu riêng lẻ, các dạng thiếu hụt vitamin D tinh vi hoặc “không đủ” có khả năng phổ biến trong dân số Bắc Mỹ, đặc biệt là trong những tháng mùa đông khi có ít tiếp xúc với ánh sáng mặt trời và tia cực tím cần thiết cho việc tổng hợp vitamin D nội sinh bị hạn chế. Mặc dù tình trạng thiếu vitamin D rõ ràng trên lâm sàng là không phổ biến ở thanh thiếu niên Hoa Kỳ, tình trạng không đủ vitamin D có thể tương đối thường xuyên. Ngoài sắc tố da và vĩ độ phía bắc, các giá trị calcidiol huyết thanh thấp đã được cho là do tiêu thụ ít sữa và vitamin tổng hợp, uống nhiều nước giải khát chứa phốt phát và tăng khối lượng mỡ mà vitamin D được lắng đọng vào và có lẽ không dễ dàng có sẵn về mặt sinh học. Sử dụng một xét nghiệm đáng tin cậy cho 25-hydroxyvitamin D, chẳng hạn như những xét nghiệm sử dụng sắc ký lỏng-khối phổ song song, nồng độ calcidiol dưới 12 đến 15 ng/mL ở trẻ em cho thấy rõ tình trạng thiếu vitamin D trong khi các giá trị từ 12 đến 20 ng/mL phù hợp với tình trạng không đủ vitamin D. Nồng độ calcidiol huyết thanh vượt quá 100 ng/mL là dấu hiệu của thừa vitamin D. Thiếu hụt canxi trong chế độ ăn có thể làm trầm trọng thêm các tác động bất lợi của việc dự trữ vitamin D ở mức giới hạn.
Thiếu vitamin D cũng có thể là hậu quả của các rối loạn kém hấp thu ở ruột, chẳng hang như các bệnh celiac hoặc viêm ruột, tắc mật, cắt bỏ dạ dày và ruột, hoặc suy tụy ngoại tiết (ví dụ, xơ nang), hoặc uống các chất liên kết canxi, chẳng hạn như cholestyramine, hoặc do tăng hydroxyl hóa vitamin D thành các dạng tan trong nước làm tăng sự mất mát qua nước tiểu do các thuốc chống co giật, chẳng hạn như phenytoin. Liều điều trị của glucocorticoid cũng liên quan đến nồng độ calcidiol huyết thanh thấp hơn. Ở phần lớn trẻ sơ sinh và trẻ em bị còi xương do thiếu vitamin D, nồng độ canxi toàn phần trong huyết thanh ở mức giới hạn-bình thường hoặc thấp, nồng độ phốt phát thấp, và hoạt độ phosphatase kiềm và nồng độ PTH tăng (Bảng 20.9). Cường cận giáp thứ phát phát triển khi sự hấp thu canxi ở ruột bị giảm và nồng độ trong huyết thanh của nó bị giảm; PTH làm tăng mất phốt phát qua nước tiểu và làm giảm nồng độ phốt phát huyết thanh, trong khi tăng cường tốc độ tái hấp thu và chu chuyển xương. PTH cũng làm tăng tổng hợp calcitriol, có thể làm tăng tốc độ dị hóa calcidiol và làm cạn kiệt thêm dự trữ vitamin D của cơ thể. Thông thường, ở những bệnh nhân thiếu vitamin D, nồng độ calcidiol huyết thanh thấp, trong khi các giá trị calcitriol có thể bình thường, cao hoặc thấp tùy thuộc vào việc thiếu vitamin D là nhẹ, trung bình hay nặng. Nồng độ osteocalcin huyết thanh thấp; nồng độ PICP (một dấu hiệu của sự hình thành xương) và ICTP, và sự bài tiết telopeptide đầu amino của collagen type I (NTx) qua nước tiểu—cả hai đều là dấu hiệu của sự tái hấp thu xương—tăng đáng kể ở các đối tượng bị còi xương do thiếu vitamin D, cho thấy sự gia tăng chu chuyển collagen trong rối loạn này. Với điều trị, các giá trị này tăng lên thoáng qua và sau đó giảm xuống mức bình thường theo tuổi trước khi quá trình lành các tổn thương còi xương trên X-quang hoàn tất.
Bảng 20.9: Dữ liệu xét nghiệm trong bệnh Còi xương do các nguyên nhân khác nhau
| Loại | Canxi | Phốt phát | Phosphatase Kiềm | Calcidiol | Calcitriol | PTH | FGF23 |
|---|---|---|---|---|---|---|---|
| Thiếu canxi | N, ⇓ | ⇓ | ⇑⇑ | N | ⇑ | ⇑ | N, ⇓, ⇑ |
| Thiếu phốt phát | N, ⇑ | ⇓⇓ | ⇑⇑ | N | ⇑ | N, ⇓, ⇑ | ⇑ |
| Thiếu vitamin D | N, ⇓ | N, ⇓ | ⇑⇑ | ⇓⇓ | N, ⇓ | ⇑ | N, ⇓ |
| Mất chức năng CYP2R1 (25-hydroxylase) | ⇓ | ⇓ | ⇑ | ⇓ | ⇓ | ⇑ | N, ⇓ |
| Mất chức năng CYP27B1 (25OHD-1α-hydroxylase) | ⇓⇓ | ⇓⇓ | ⇑⇑⇑ | N | ⇓⇓⇓ | ⇑⇑⇑ | N, ⇓ |
| Mất chức năng VDR (kháng calcitriol) | ⇓⇓ | ⇓⇓ | ⇑⇑⇑ | N | ⇑⇑⇑ | ⇑⇑⇑ | N, ⇓ |
| Mất chức năng PHEX (còi xương giảm phốt phát máu liên kết X) | N, ⇓ | ⇓⇓ | ⇑ | N | N, ⇓ | N, ⇑ | ⇑ |
| Giảm phosphatase máu | N, ⇑ | N, ⇑ | ⇓ | N | N | N, ⇓ |
⇓, loại thấp; ⇑, loại cao; FGF2, yếu tố tăng trưởng nguyên bào sợi 2; N, bình thường; PTH, hormon cận giáp; VDR, thụ thể vitamin D.
(Từ Mannstadt M, Bilezikian JP, Thakker RV, et al. Hypoparathyroidism. Nat Rev Dis Primers. 2017:3.17055; Minisola S, Peacock M, Fukumoto S, et al. Tumour-induced osteomalacia. Nat Rev. 2017;3:17044.)
Phòng ngừa thiếu vitamin D là cách quản lý hiệu quả nhất. Bởi vì lượng vitamin D trong sữa mẹ khoảng 5 đến 80 IU/L và việc bổ sung vitamin D cho người mẹ cho con bú có thể không đủ để đảm bảo nồng độ calcidiol bình thường ở con (trừ khi bà mẹ nhận được khoảng 4000 IU vitamin D3 mỗi ngày), điều quan trọng là tất cả trẻ bú mẹ đều nhận được một chất bổ sung vitamin D3 hàng ngày (400 IU/ngày); mở rộng ra, khuyến nghị này cũng phù hợp cho những trẻ sơ sinh không nhận đủ lượng vitamin D3 trong các loại sữa công thức được chuẩn bị hoặc chế độ ăn hoặc có ít tiếp xúc với ánh sáng mặt trời. Ở trẻ da trắng năng động, ăn mặc thoáng mát có làn da thường rám nắng khi tiếp xúc với ánh sáng mặt trời và chơi ngoài trời 30 phút ba lần mỗi tuần hoặc 10 đến 15 phút hàng ngày từ 1000 đến 1500 giờ trong mùa xuân, mùa hè và mùa thu, việc tổng hợp vitamin D nội sinh thường đủ để đạt được nồng độ calcidiol huyết thanh thỏa đáng. Một đứa trẻ da vàng hoặc da nâu cần 30 đến 45 phút chơi ngoài trời trong khi một đứa trẻ da đen cần 60 đến 150 phút tiếp xúc với ánh sáng mặt trời để đạt được nồng độ calcidiol huyết thanh tương đương với trẻ da trắng. Vĩ độ nơi trẻ sống, mùa trong năm, thời gian trong ngày, các chất ô nhiễm môi trường tại địa phương, lượng quần áo và việc sử dụng kem chống nắng ảnh hưởng đến thời gian cần thiết để tiếp xúc với ánh sáng mặt trời để tạo ra đủ vitamin D tổng hợp ở từng trẻ. Từ tháng 11 đến tháng 2, có rất ít hoặc không có vitamin D có thể được tổng hợp ở vĩ độ trên 35 độ (Atlanta, GA, Hoa Kỳ), nhưng tình trạng không đủ vitamin D cũng phổ biến ở các vĩ độ thấp hơn.
Ở thanh thiếu niên béo phì, nồng độ calcidiol huyết thanh có mối tương quan nghịch và các giá trị PTH có mối tương quan thuận với khối lượng mỡ. Trong một nghiên cứu trên 236 ứng cử viên thanh thiếu niên phẫu thuật giảm cân, hơn 50% bị thiếu vitamin D theo định nghĩa là nồng độ calcidiol huyết thanh thấp. Mặc dù 43% trong số các đối tượng này là người da trắng và 15% là người Mỹ gốc Phi, 82% bệnh nhân thiếu vitamin D là người Mỹ gốc Phi so với 37% thanh thiếu niên da trắng. Do đó, béo phì là một yếu tố nguy cơ tiềm ẩn đối với nồng độ 25-hydroxyvitamin D trong huyết thanh dưới mức bình thường; tuy nhiên, khoáng hóa xương bằng DXA là bình thường ở trẻ em và người lớn béo phì. Tuy nhiên, khoáng hóa xương bình thường của trẻ em và thanh thiếu niên béo phì có thể không đủ để đáp ứng các yêu cầu về sức mạnh của xương mà khối lượng cơ thể cao hơn của chúng có thể cần. Do đó, béo phì không chỉ không bảo vệ chống lại gãy xương ở quần thể này mà còn có thể làm tăng nguy cơ gãy xương do tải trọng cơ học cao mà trẻ em/thanh thiếu niên béo phì phải mang. Sau khi giảm cân do chế độ ăn và tập thể dục, các bất thường về nồng độ calcidiol và PTH trong huyết thanh được đảo ngược, cho thấy các sai lệch thấy ở trẻ em béo phì là hậu quả chứ không phải là nguyên nhân của bệnh béo phì. Điều thú vị là tế bào mỡ và nguyên bào xương có nguồn gốc từ một tế bào gốc trung mô chung dưới sự chỉ đạo của các kích thích khác nhau. Sự hình thành nguyên bào xương xảy ra khi tế bào gốc tiến triển dọc theo con đường truyền tín hiệu WNT-Frizzled-β-catenin, trong khi sự hình thành tế bào mỡ xảy ra sau khi kích thích tế bào gốc bởi thụ thể kích hoạt tăng sinh peroxisome gamma (PPAR-γ). Liệu sự phân đôi này trong sự biệt hóa tế bào có đóng một vai trò trong việc điều chỉnh tác động của béo phì đối với khoáng hóa xương ở trẻ em hay không vẫn chưa được biết đến. Vị trí lắng đọng mỡ cũng có thể ảnh hưởng đến khoáng hóa xương; ở người lớn, mỡ bụng và mỡ tủy xương có thể gây tác động bất lợi lên quá trình này.
Đánh giá trẻ bị còi xương dinh dưỡng bao gồm xem xét tiền sử về chế độ ăn và tiếp xúc với ánh sáng mặt trời và các bệnh đã gặp, khám thực thể (ghi nhận các biến dạng của ngực và các chi), chụp X-quang bộ xương (mở rộng sụn tiếp hợp, các vùng thấu quang của chất dạng xương không được khoáng hóa kéo dài vào hành xương), và đánh giá xét nghiệm: công thức máu toàn phần (hemoglobin, hematocrit), các phép đo hóa học toàn diện (bao gồm canxi toàn phần, magiê, phốt pho, phosphatase kiềm xương, creatinin, calcidiol, PTH); xác định canxi, creatinin trong nước tiểu tại chỗ. Còi xương dinh dưỡng được điều trị hiệu quả ở hầu hết các đối tượng bằng liều vitamin D đường uống (cholecalciferol hoặc ergocalciferol): trẻ sơ sinh—2000 IU/ngày trong 6 tuần; trẻ nhỏ dưới 1 tuổi—2000/ngày trong 6 tuần; trẻ em trên 1 tuổi—2000 IU/ngày trong 8 tuần với liều duy trì sau đó là 600 đến 1000 IU/ngày. Khi bắt đầu điều trị, canxi nguyên tố (30–75 mg/kg/ngày chia làm 3 liều mỗi ngày) cũng phải được dùng cho trẻ thiếu vitamin D đang nhận vitamin D để tránh hạ canxi máu đi kèm với quá trình tái khoáng hóa nhanh chóng của chất nền xương (hội chứng xương đói). Một phác đồ điều trị thay thế cho bệnh còi xương do thiếu vitamin D là dùng một liều duy nhất đường uống (hoặc tiêm bắp) từ 150.000 đến 600.000 đơn vị vitamin D3 tùy thuộc vào tuổi của bệnh nhân và các hoàn cảnh cá nhân khác. Trong quá trình điều trị, việc xác định định kỳ các giá trị phosphatase kiềm toàn phần và/hoặc đặc hiệu cho xương là một công cụ hiệu quả để theo dõi hiệu quả điều trị vì nồng độ giảm dần khi các tổn thương còi xương lành lại. Để tránh hạ canxi máu hoặc tăng canxi máu, tăng canxi niệu và nhiễm canxi thận, cần xác định nối tiếp các giá trị canxi, phốt phát và creatinin trong huyết thanh và bài tiết canxi và creatinin qua nước tiểu. Các phim X-quang nối tiếp ghi nhận sự đảo ngược của các thay đổi còi xương (xem Hình 20.9). Sau khi hoàn thành liệu pháp và có bằng chứng X-quang về sự lành bệnh của bệnh còi xương, liều vitamin D có thể được giảm xuống 400 đến 600 IU/ngày; các giá trị calcidiol sau đó nên được đo lường trước để chắc chắn rằng trẻ tiếp tục nhận đủ lượng vitamin D.
Còi xương chủ yếu do lượng canxi trong chế độ ăn thấp đã xảy ra ở những trẻ sơ sinh uống các loại sữa công thức chứa ít canxi và ở trẻ em từ các nước đang phát triển nhận chế độ ăn với 200 mg (hoặc ít hơn) canxi nguyên tố mỗi ngày mặc dù ăn đủ phốt phát và có đủ dự trữ vitamin D nội sinh được xác định bằng nồng độ calcidiol huyết thanh. Còi xương do thiếu canxi cũng xảy ra ở Hoa Kỳ nếu trẻ sơ sinh được cai sữa sang các loại thực phẩm chứa ít canxi sau khi kết thúc bú mẹ. Còi xương do thiếu canxi cũng có thể phát triển do hậu quả của việc suy giảm hấp thu canxi trong chế độ ăn ở ruột do các yếu tố nội tại hoặc do nó đã bị liên kết bởi việc ăn các loại ngũ cốc chứa nhiều chất xơ và phytate. Thiếu canxi được giải quyết tốt nhất bằng cách phòng ngừa, đảm bảo ăn đủ nguyên tố này theo các hướng dẫn đã được thiết lập cho trẻ sơ sinh, trẻ em và thanh thiếu niên đang lớn (lượng canxi khuyến nghị: 400–600 mg/ngày ở trẻ sơ sinh; 700 mg/ngày ở trẻ em dưới 4 tuổi; 1000–1300 mg/ngày ở trẻ lớn và thanh thiếu niên). Khi có, còi xương do thiếu canxi có thể được điều trị hiệu quả bằng cách đảm bảo ăn 1000 đến 1500 mg canxi nguyên tố hàng ngày trong 6 tháng với việc cung cấp lượng vitamin D bình thường bằng cách tiếp xúc với ánh sáng mặt trời hoặc bổ sung. Thiếu hụt đồng thời canxi và vitamin D có thể xảy ra ở một số trẻ em.
Thiếu hụt phốt phát trong chế độ ăn là không phổ biến vì anion này có mặt với số lượng lớn trong hầu hết các loại thực phẩm. Hạ phốt phát máu có thể do giảm hấp thu phốt phát ở ruột (ví dụ, đói, trẻ sinh non uống sữa mẹ không bổ sung phốt phát, trẻ lớn uống sữa công thức có hàm lượng phốt phát rất thấp, các đối tượng uống một lượng lớn thuốc kháng axit chứa nhôm vì nhôm và phốt phát đồng kết tủa trong đường ruột, bệnh nhân bị hội chứng ruột ngắn, hoặc thiếu vitamin D), dùng dinh dưỡng qua đường tĩnh mạch với các dịch có hàm lượng phốt phát thấp, truyền tĩnh mạch sắt, tăng bài tiết phốt phát qua thận ở những bệnh nhân bị suy giảm tái hấp thu anion này ở ống thận, và sự di chuyển của phốt phát từ khoang dịch ngoại bào vào khoang nội bào (xem bên dưới). Ở trẻ sinh cực non nhận dinh dưỡng qua đường tĩnh mạch dài hạn, sự phát triển của bệnh xương chuyển hóa là thường xuyên và không chỉ liên quan đến sự thiếu hụt canxi, phốt phát và vitamin D mà còn do dư thừa nhôm trong dịch truyền. Trẻ sơ sinh nhận một lượng lớn thuốc kháng axit chứa nhôm trong thời gian dài để điều trị trào ngược dạ dày thực quản cũng có thể có khối lượng xương thấp. Nhôm làm giảm tốc độ hình thành xương bằng nhiều cơ chế: dùng đường uống nhôm liên kết với phốt phát ở ruột, do đó cản trở sự hấp thu của nó dẫn đến cạn kiệt phốt phát; dùng đường tĩnh mạch trong dinh dưỡng qua đường tĩnh mạch toàn phần hoặc trong quá trình chạy thận nhân tạo, nhôm ức chế chức năng của nguyên bào xương và ngăn cản sự khoáng hóa của chất dạng xương; nó cũng làm suy giảm sự bài tiết PTH và làm giảm hoạt động 25OHD-1α hydroxylase. Bệnh nhân cần dinh dưỡng qua đường tĩnh mạch toàn phần nên nhận càng nhiều canxi và phốt phát càng tốt một cách an toàn, cũng như bổ sung vitamin D. Việc đo lường định kỳ nồng độ canxi, phốt phát, phosphatase kiềm, PTH và calcidiol trong huyết thanh, và chụp X-quang xương nối tiếp và ước tính khoáng hóa xương được khuyến nghị ở những bệnh nhân nhận dinh dưỡng qua đường tĩnh mạch toàn phần; nếu bệnh xương chuyển hóa phát triển mặc dù đã có những nỗ lực này, nên đo nồng độ nhôm huyết thanh và nếu tăng (> 100 mcg/L), nên bắt đầu tìm kiếm nguồn nhôm và loại bỏ sản phẩm đó khỏi dịch truyền nếu có thể. Cadmium, florua và oxit sắt saccharated cũng có thể cản trở quá trình khoáng hóa xương bình thường.
Các khiếm khuyết chuyển hóa và chức năng của vitamin D dẫn đến các dạng còi xương hiếm gặp hơn (xem Bảng 20.8A, 20.8B). Các đột biến mất chức năng hai alen trong CYP2R1 (OMIM 608713), enzyme 25-hydroxylase ở gan chuyển đổi cholecalciferol (vitamin D3) thành 25-hydroxycholecalciferol (calcidiol), dẫn đến còi xương thiếu hydroxyl hóa vitamin D type 1B (còn gọi là còi xương phụ thuộc vitamin D type 1B—OMIM 600081). Còi xương do khiếm khuyết trong 25-hydroxyl hóa đã được mô tả ở hai anh em gốc Nigeria, trong đó một đột biến mất chức năng đồng hợp tử (p.Leu99Pro) trong CYP2R1 đã loại bỏ hoạt động hydroxylase của protein 501 aa này. Đột biến đồng hợp tử có hại (p.Gly42_LeudelinsArg) trong CYP2R1 cũng đã được xác định. Còi xương thiếu 25-hydroxyl hóa vitamin D được di truyền như một rối loạn trội không phải do một đột biến được xác định trong các exon mã hóa hoặc các vùng intron của CYP2R1 cũng đã được báo cáo. Còi xương thiếu hydroxyl hóa vitamin D 25 type 1B được biểu hiện bằng các dấu hiệu lâm sàng và X-quang của bệnh còi xương kết hợp với hạ canxi máu, hạ phốt phát máu, tăng phosphatase kiềm, PTH cao, nồng độ calcitriol thấp-bình thường, bình thường hoặc tăng, và các giá trị calcidiol rất thấp; những người mang dị hợp tử của một đột biến có hại trong CYP2R1 có nồng độ calcidiol giảm nhưng không phát triển bệnh còi xương. Các đối tượng bị ảnh hưởng đáp ứng với việc dùng calcidiol (cũng như calcitriol) với sự cải thiện các triệu chứng lâm sàng, bình thường hóa các bất thường sinh hóa và tăng khoáng hóa xương cột sống.
Còi xương thiếu hydroxyl hóa vitamin D, type 1A ([OMIM 264700]—còi xương phụ thuộc vitamin D type 1A [VDDR1A] hoặc còi xương giả thiếu vitamin D type 1A [PDDR1A]) là do các đột biến bất hoạt đồng hợp tử hoặc dị hợp tử phức hợp của CYP27B1 (OMIM 609506) mã hóa enzyme trong ống lượn gần của thận xúc tác quá trình 1α-hydroxyl hóa carbon-1 của calcidiol, chuyển nó thành 1,25-dihydroxyvitamin D3 (calcitriol), chất chuyển hóa có hoạt tính sinh học của vitamin D. Mặc dù có vẻ bình thường khi sinh, các biến dạng xương (cong cẳng tay), chậm tăng trưởng, yếu cơ hoặc co thắt, chậm các mốc vận động (đi bộ), và/hoặc co giật do hạ canxi máu phát triển từ 6 đến 30 tháng tuổi ở bệnh nhân mắc VDDR1A; các dấu hiệu thực thể của bệnh còi xương (nổi rõ hành xương, chuỗi hạt còi xương, mềm sọ), và thiểu sản men răng thường hiện diện; về mặt sinh hóa, hạ canxi máu, hạ phốt phát máu, tăng phosphatase máu, nồng độ PTH huyết thanh tăng rõ rệt, nồng độ calcidiol huyết thanh bình thường và nồng độ calcitriol thấp một cách không phù hợp được ghi nhận; X-quang cho thấy các biến dạng còi xương của các xương dài. Chẩn đoán VDDR1A được xác lập bằng sự hiện diện của nồng độ calcidiol huyết thanh bình thường trong khi các giá trị calcitriol cực kỳ thấp và không tăng sau khi dùng vitamin D hoặc calcidiol và được xác nhận bằng cách xác định đột biến trong CYP27B1. Mục tiêu điều trị là phục hồi tình trạng canxi máu bình thường mà không gây tăng canxi niệu, dẫn đến tăng sức mạnh cơ bắp và lành các tổn thương còi xương, cho phép tăng trưởng bình thường. Các biểu hiện lâm sàng, sinh hóa và X-quang của VDDR1A giải quyết hoàn toàn và hợp lý nhanh chóng sau khi điều trị bằng một lượng calcitriol sinh lý (10–20 ng/kg/ngày). Cần điều trị suốt đời, nhưng việc điều trị hiệu quả và tuân thủ dẫn đến bình thường hóa tất cả các thông số sinh hóa, tăng trưởng tuyến tính và chiều cao người lớn bình thường, và khoáng hóa xương bình thường. Liều calcitriol thường cần được tăng lên trong thời kỳ mang thai; tuy nhiên, thai kỳ không phức tạp và con của các bà mẹ bị ảnh hưởng là bình thường. VDDR1A là một bệnh di truyền lặn trên NST thường được tìm thấy với tần suất cao ở một quần thể người Canada gốc Pháp ở Quebec nhưng xảy ra ở tất cả các chủng tộc và ở các vùng địa lý đa dạng. CYP27B1 mã hóa một cytochrome P450 hydroxylase ty thể 508 aa với các vị trí được bảo tồn liên kết với ferrodoxin và heme và sử dụng các electron từ nicotinamide adenine dinucleotide phosphate khử và oxy để chuyển đổi calcidiol thành calcitriol. Nhiều đột biến mất chức năng sai nghĩa, vô nghĩa, nối, và nhân đôi hoặc mất đoạn/dịch khung trong CYP27B1 dẫn đến các sản phẩm protein không hoạt động hoặc bị cắt ngắn không thể liên kết với cơ chất (calcidiol) hoặc heme, khiếm khuyết sau ngăn cản sự chuyển điện tử và ức chế quá trình xúc tác. Đột biến phổ biến nhất trong CYP27B1 ở quần thể người Canada gốc Pháp ở Quebec có nguy cơ mắc VDDR1A là mất đoạn guanine tại nucleotide 958 (exon 2, codon 88) (c.958delG) làm thay đổi khung đọc dẫn đến kết thúc dịch mã sớm và một sản phẩm không hoạt động (đột biến Charlevoix). Một đột biến phổ biến thứ hai trong quần thể này là nhân đôi một chuỗi 7 cặp bazơ trong exon 8; tuy nhiên, đột biến này cũng được tìm thấy ở những bệnh nhân có nguồn gốc châu Á và Tây Ban Nha.
Các đột biến mất chức năng hai alen trong VDR (OMIM 601769) mã hóa VDR dẫn đến kháng lại các tác dụng sinh lý của calcitriol và còi xương kháng vitamin D type 2A (OMIM 277440). Các biểu hiện lâm sàng và sinh hóa của tình trạng kháng calcitriol bao gồm các biến dạng xương khởi phát ở trẻ nhỏ nặng đặc trưng của bệnh còi xương (cong các chi, mở rộng các đầu xa của xương dài, trán dô, chuỗi hạt ở các khớp sụn sườn của lồng ngực), chậm tăng trưởng, rụng tóc ở mức độ nghiêm trọng khác nhau, hạ canxi máu, hạ phốt phát máu, tăng phosphatase kiềm, và nồng độ calcitriol (300–1000 pg/mL) và PTH trong huyết thanh cực kỳ cao; nồng độ 25-hydroxyvitamin D trong huyết thanh bình thường trong khi nồng độ 24,25-dihydroxyvitamin D thường thấp (xem Bảng 20.9). Các giá trị calcitriol cao phản ánh các tác dụng kích thích kết hợp của hạ canxi máu, hạ phốt phát máu và cường cận giáp thứ phát đối với hoạt động của 25-hydroxyvitamin D3-1α-hydroxylase cùng với sự suy giảm tốc độ dị hóa của nó do hoạt động suy giảm của 1,25α-dihydroxyvitamin D3 24-hydroxylase phụ thuộc calcitriol. X-quang cho thấy các dấu hiệu của bệnh còi xương, bao gồm hình ảnh lõm chén và tua ở hành xương và cong xương chày và xương đùi. Phân tích gen cho thấy các đột biến đồng hợp tử hoặc dị hợp tử phức hợp trong VDR (Hình 20.10). Truyền tĩnh mạch canxi (0,4–1,4 g/m2/ngày) đã hữu ích trong việc phục hồi sự tăng trưởng bình thường và lành bệnh còi xương nhưng đòi hỏi phải theo dõi cẩn thận, thường là trong bệnh viện, để tránh rối loạn nhịp tim, tăng canxi niệu, nhiễm canxi thận và các biến chứng liên quan. Dùng đường uống liều cao calcitriol (1–6 mcg/ngày chia làm 2 liều) cùng với canxi nguyên tố (1–3 g/ngày) đã được sử dụng trong điều trị những bệnh nhân này, những người cũng cần theo dõi chặt chẽ và đo lường định kỳ các giá trị canxi trong huyết thanh và nước tiểu. Trong quá trình điều trị, các giá trị canxi, phốt phát, phosphatase kiềm, creatinin và PTH trong huyết thanh, bài tiết canxi và creatinin qua nước tiểu, X-quang xương và siêu âm thận để phát hiện sự phát triển của nhiễm canxi thận được theo dõi nối tiếp. Ở những bệnh nhân kháng với liệu pháp đường uống, việc dùng một lượng lớn canxi (0,4–1,4 g canxi nguyên tố/m2/ngày) liên tục qua đường tĩnh mạch hoặc trong tĩnh mạch chủ sẽ bình thường hóa các giá trị canxi, phốt phát, phosphatase kiềm và PTH, chữa lành bệnh còi xương và tăng tốc độ tăng trưởng ở một số trẻ được chọn. Nếu sử dụng phương thức trị liệu này, việc theo dõi cẩn thận nhiễm trùng huyết do catheter và rối loạn nhịp tim, cũng như tăng canxi máu, tăng canxi niệu và nhiễm canxi thận là bắt buộc. Sau khi chữa lành bệnh còi xương bằng canxi đường tiêm, liệu pháp duy trì với liều lớn canxi đường uống (3,5–9 g canxi nguyên tố/m2/ngày) là phù hợp. Ở trẻ sơ sinh nhỏ tuổi bị còi xương kháng vitamin D trước khi phát triển bệnh còi xương toàn phát, liều cao canxi đường uống có thể cải thiện quá trình còi xương. Những quan sát lâm sàng này cho thấy khiếm khuyết chính ở những bệnh nhân bị còi xương kháng vitamin D là thiếu canxi. Với điều trị, sự tăng trưởng có thể bình thường hóa ở những bệnh nhân kháng vitamin D, nhưng rụng tóc nếu có thì không có khả năng giải quyết. Một phương pháp điều trị thay thế cho tình trạng kháng vitamin D là dùng cinacalcet, một chất calcimimetic kích hoạt CaSR trong tuyến cận giáp do đó làm giảm sự bài tiết PTH; việc dùng cinacalcet ngắn hạn (2 tháng) cho một đứa trẻ 3 tuổi bị kháng vitamin D đã cho phép giải quyết tương đối lâu dài các biểu hiện lâm sàng, sinh hóa và X-quang của bệnh này với liệu pháp duy trì là truyền canxi ba lần mỗi tuần. Điều thú vị là, mặc dù việc liên kết calcitriol với VDR là cần thiết cho sự biểu hiện chức năng của nó ở hầu hết các mô và tóc phụ thuộc vào VDR cho chức năng bình thường của các tế bào sừng da và các tế bào bao rễ ngoài và chu kỳ sinh lý của nang tóc, chu kỳ nang tóc không cần calcitriol; do đó, việc duy trì mô hình bình thường của chu kỳ nang tóc giữa các giai đoạn nghỉ và tăng trưởng có khả năng là một chức năng độc lập với phối tử của VDR đạt được bằng cách tương tác với β-catenin.
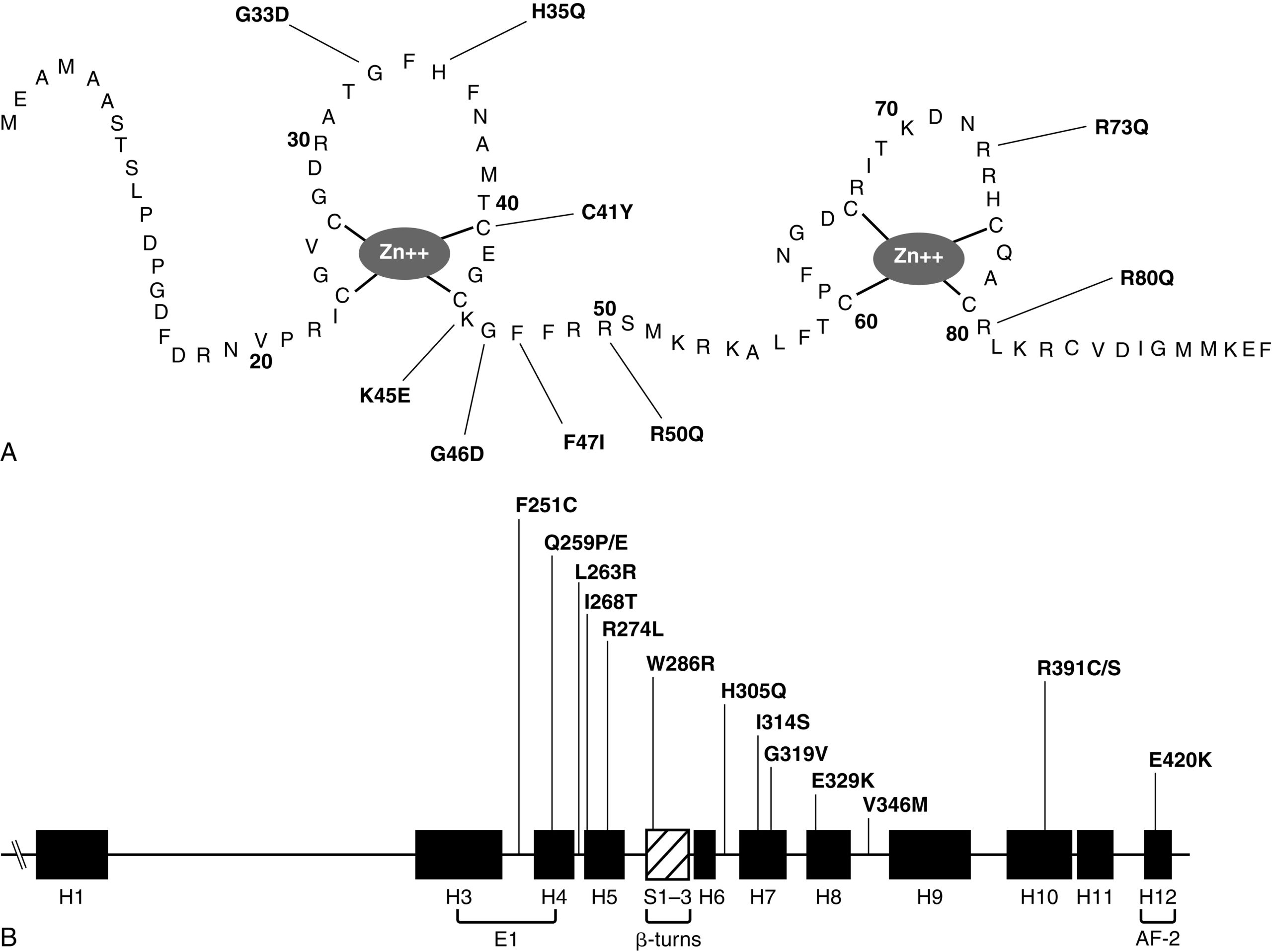
Hình 20.10: Các đột biến trong thụ thể vitamin D (VDR) ở bệnh nhân không nhạy cảm với vitamin D ở cơ quan đích. (Từ Malloy PJ, Feldman D. Genetic disorders and defects in vitamin D actions. Endocrinol Metab Clin North Am. 2010;39:333–346.)
Mặc dù nổi bật ở trẻ sơ sinh và đầu thời thơ ấu, các biểu hiện lâm sàng của tình trạng kháng vitamin D có thể khác nhau, và những bệnh nhân có các khiếm khuyết nhẹ hơn trong VDR có thể không được xác định cho đến tuổi vị thành niên hoặc trưởng thành. Ngoài ra, sự thuyên giảm tự phát của quá trình còi xương có thể xảy ra, thường xuyên nhất là từ 7 đến 15 tuổi hoặc khi bệnh nhân bước vào tuổi dậy thì. Thật vậy, sau tuổi dậy thì ở nhiều bệnh nhân bị còi xương kháng vitamin D, nồng độ canxi, phốt phát và phosphatase kiềm trong huyết thanh bình thường hóa và việc bổ sung canxi không còn cần thiết nữa. Rõ ràng, nhu cầu canxi của xương giảm sau khi hoàn thành tăng trưởng xương, và các cơ chế hấp thu canxi ở ruột không phụ thuộc vitamin D phát triển. Trong một nghiên cứu trên 17 bệnh nhân bị còi xương kháng vitamin D trong độ tuổi từ 1,5 đến 37, các thông số sinh hóa được cải thiện, nhu cầu bổ sung canxi giảm, và BMD theo DXA bình thường hóa trong nửa sau của thập kỷ thứ hai của cuộc đời, trong khi các giá trị calcitriol huyết thanh vẫn tăng cao. Việc kiểm tra sự hấp thu canxi ở ruột cho thấy sự hấp thu phân đoạn của canxi, trong khi ăn chế độ ăn ít canxi, thấp hơn đáng kể so với các đối tượng đối chứng ở những bệnh nhân trẻ tuổi; ở những bệnh nhân từ 18 đến 26 tuổi, sự hấp thu phân đoạn của canxi cao hơn đáng kể so với các giá trị đối chứng; ở những bệnh nhân trưởng thành, sự hấp thu phân đoạn của canxi tương tự như dữ liệu đối chứng. Tuy nhiên, các tác động suy giảm tăng trưởng của bệnh còi xương nặng ở trẻ em vẫn tồn tại đến tuổi trưởng thành ở phần lớn bệnh nhân. VDR bao gồm các miền liên kết DNA-, phối tử-, và thụ thể X retinoic (RXR) và một miền hoạt hóa chuyển mã mà nhiều chất đồng điều hòa chức năng VDR được tuyển dụng đến. VDR và RXR liên kết như một phức hợp hoạt hóa phiên mã dị nhị phân. Các đột biến mất chức năng đã được tìm thấy trong mỗi miền VDR: do đó, VDR bị đột biến có thể không thể liên kết với calcitriol do số lượng thụ thể giảm hoặc ái lực với phối tử, không có khả năng hình thành các dị nhị phân với RXR hoặc chuyển vị đến gen mục tiêu trong nhân, không thể liên kết với yếu tố đáp ứng vitamin D (VDRE) hoặc bắt đầu phiên mã gen một khi đã liên kết với VDRE. Hơn 30 đột biến dị hợp tử riêng biệt trong VDR đã được xác định rải rác trong toàn bộ protein VDR nhưng với một số lượng lớn các biến thể trong miền liên kết phối tử. Nói chung, những bệnh nhân bị còi xương kháng vitamin D không bị rụng tóc có thể đáp ứng tốt hơn với điều trị.
Còi xương phụ thuộc vitamin D, type 2B (VDRR2B, OMIM 600785) là một dạng hiếm, thứ hai của còi xương kháng vitamin D với một kiểu hình tương tự như của bệnh nhân có VDR bị đột biến nhưng thụ thể hạt nhân vitamin D của họ còn nguyên vẹn. Sự biểu hiện quá mức của HNRNPC (OMIM 164020), mã hóa một ribonucleoprotein hạt nhân C liên kết với yếu tố đáp ứng vitamin D VDR/RXR (5′) ở thượng nguồn của gen mục tiêu vitamin D và làm suy giảm phiên mã gen mục tiêu, đã được quan sát thấy ở một bệnh nhân mắc VDRR2B. Cơ chế mà sự biểu hiện quá mức của một ribonucleoprotein bình thường khác xảy ra và mối quan hệ của quan sát này với sinh bệnh học của VDRR2B vẫn chưa được biết. Một biến thể của CYP3A4 (OMIM 124010) mã hóa một enzyme CYP450 ở gan chuyển hóa oxy hóa nhanh chóng và do đó bất hoạt calcidiol và calcitriol cũng dẫn đến giảm hoạt động chức năng của các chất chuyển hóa này của vitamin D, dẫn đến còi xương ở những bệnh nhân bị ảnh hưởng, những người cần liều cao calcitriol để quản lý điều trị. (Rối loạn này đã được chỉ định là còi xương phụ thuộc vitamin D type 3.)
Còi xương do thiếu phốt phát
Nồng độ phốt phát trong huyết thanh giảm theo tuổi trong suốt thời thơ ấu và vị thành niên; do đó, từ 0 đến 5 ngày, nồng độ phốt phát huyết thanh dao động trong khoảng 4,8 đến 8,2 mg/dL; 1 và 3 tuổi—3,8 đến 6,5 mg/dL; 4 và 11 tuổi—3,7 đến 5,6 mg/dL; 12 và 15 tuổi—2,9 đến 5,4 mg/dL; 16 và 19 tuổi—2,7 đến 4,7 mg/dL. Hạ phốt phát máu cấp tính có liên quan đến khó chịu, lú lẫn, dị cảm, yếu cơ và liệt ruột; hạ phốt phát máu mạn tính dẫn đến còi xương ở trẻ đang lớn. Hạ phốt phát máu ở trẻ em có thể do các rối loạn di truyền hoặc mắc phải (xem Bảng 20.8A, 20.8B và xem Hình 20.12). Hạ phốt phát máu có thể là hậu quả của sự di chuyển của phốt phát ngoại bào vào tế bào (trong quá trình điều trị nhiễm toan ceton do đái tháo đường bằng insulin khi quá trình phosphoryl hóa carbohydrate nội bào tăng lên, nhiễm kiềm hô hấp cấp do tăng thông khí, nhiễm trùng huyết), suy giảm hấp thu phốt phát ở ruột (thiếu vitamin D, ăn mạn tính các thuốc kháng axit chứa magiê hoặc nhôm, tiêu chảy mạn tính và/hoặc tiêu phân mỡ), hoặc tăng mất phốt phát qua thận (thứ phát sau tăng tiết PTH hoặc nguyên phát do các bất thường nội tại trong các cơ chế kiểm soát tái hấp thu anion này ở ống thận). Hạ phốt phát máu được quan sát thấy ở các đối tượng thiếu dinh dưỡng, chẳng hạn như trẻ sơ sinh LBW hoặc bệnh nhân lớn tuổi phụ thuộc vào dinh dưỡng qua đường tĩnh mạch, những người bị chán ăn nặng, hoặc những người nghiện rượu. Hạ phốt phát máu và bệnh xương chuyển hóa có thể xảy ra ở trẻ sơ sinh và trẻ em nhận một loại sữa công thức nguyên tố dựa trên axit amin với phốt phát có khả dụng sinh học giảm. Hạ phốt phát máu xảy ra ở nhiều bệnh nhân nặng, đặc biệt là những người bị nhiễm trùng huyết. Tái khoáng hóa nhanh chóng của xương bị loãng xương đi kèm với hạ phốt phát máu và hạ canxi máu (hội chứng xương đói). Thường xuyên nhất, hạ phốt phát máu mạn tính là kết quả của tăng phốt phát niệu—hoặc do một khiếm khuyết nguyên phát hoặc thứ phát trong việc điều hòa tái hấp thu phốt phát ở ống thận ở ống lượn gần (nơi 60%–70% lượng phốt phát được lọc được tái hấp thu) hoặc xa (nơi 10%–15% phốt phát được lọc được tái hấp thu). Do đó, tăng tiết PTH do cường cận giáp nguyên phát hoặc thứ phát sau thiếu vitamin D hoặc nguyên nhân khác có thể dẫn đến tăng phốt phát niệu và hạ phốt phát máu. Các hội chứng Fanconi được đặc trưng bởi nhiều khiếm khuyết chức năng ống thận bao gồm các bất thường về tái hấp thu phốt phát. Các khiếm khuyết ống thận đặc biệt cản trở các quá trình mà qua đó phốt phát được tái hấp thu bởi các ống thận bao gồm các bất thường trong vận chuyển phốt phát qua tế bào và sản xuất quá mức các chất gây bài tiết phốt phát khác ngoài PTH, chẳng hạn như FGF23 và các phosphatonin khác và các đồng yếu tố của chúng.
Hình 20.12: Đánh giá trẻ bị hạ phốt phát máu. (Từ Minisola, S., Peacock, M., Fukumoto, S., et al. (2017). Tumour-induced osteomalacia. Nat Rev, 3, 17044. Với sự cho phép.)
Ở các nước phát triển nơi bệnh còi xương do thiếu vitamin D nặng không phải là một vấn đề phổ biến, còi xương giảm phốt phát máu di truyền trội liên kết X (XLHR, OMIM 307800) do các biến thể bất hoạt của tương đồng Endopeptidase điều hòa PHosphate (PHEX, liên kết X—OMIM 300550) là dạng còi xương thường gặp nhất (1:20.000 ca sinh). Còi xương giảm phốt phát máu di truyền trội liên kết X ảnh hưởng đến nam giới bán hợp tử và nữ giới dị hợp tử tương tự nhau do sự bất hoạt ngẫu nhiên của nhiễm sắc thể X, mặc dù có sự thay đổi đáng kể trong biểu hiện lâm sàng giữa các gia đình và trong cùng một gia đình. Còi xương giảm phốt phát máu di truyền trội liên kết X thường được biểu hiện trong năm đầu đời, ban đầu với sự cong của xương trụ và xương quay. Các phát hiện thực thể ở trẻ em mắc XLHR bao gồm tầm vóc thấp, chân vòng kiềng chữ O hoặc chữ X phát triển khi trẻ bắt đầu đi, loe hành xương, chuỗi hạt còi xương, trán dô; tăng tần suất sâu răng và/hoặc áp xe quanh chân răng ở những chiếc răng không bị sâu; cứng, đau nhức và đôi khi đau xương, cơ và khớp. Mềm sọ, co cứng và yếu cơ không được tìm thấy ở bệnh nhân mắc XLHR như ở những người bị thiếu vitamin D. Mặc dù chúng không còn phát triển, người lớn mắc XLHR bị nhuyễn xương, đau xương, tăng tỷ lệ gãy xương và thường xuyên bị áp xe răng, nhiễm canxi thận và suy giảm thính lực; họ cũng có thể phát triển hẹp ống sống và cứng khớp cột sống tiến triển; phẫu thuật chỉnh hình, phẫu thuật giải nén bản sống và thay khớp háng và khớp gối thường cần thiết ở quần thể này. Bệnh điểm bám gân—vôi hóa gân, dây chằng và bao khớp—phổ biến ở người lớn mắc XLHR nhưng cũng có thể xảy ra ở trẻ em mắc rối loạn này. Biểu hiện lâm sàng của XLHR có thể thay đổi rất nhiều giữa các gia đình bị ảnh hưởng và giữa các trẻ em bị ảnh hưởng trong cùng một gia đình.
Hạ phốt phát máu, tăng phốt phát niệu, canxi máu bình thường, tăng phosphatase kiềm, nồng độ PTH bình thường hoặc chỉ tăng nhẹ, giá trị calcidiol bình thường và nồng độ calcitriol thấp một cách không phù hợp so với mức độ hạ phốt phát máu hiện diện ở những bệnh nhân này trong vài tháng đầu sau khi sinh. Bởi vì phốt phát gây ra sự biệt hóa, trưởng thành và chết theo chương trình của các tế bào sụn phì đại trong sụn tăng trưởng của các xương dài, hạ phốt phát máu có liên quan đến sự phát triển quá mức không có tổ chức của các tế bào sụn. Do đó, về mặt X-quang, loe hành xương đùi xa và hành xương chày gần là một phát hiện nổi bật cũng như sự cong của xương đùi và xương chày kết hợp với đau đầu gối. Trẻ em mắc XLHR bị đau đầu gối nhưng X-quang bình thường có thể có các bất thường về xương có thể xác định được bằng chụp cộng hưởng từ. Bộ xương trục có thể có vẻ ngoài đặc.
PHEX mã hóa một metallopeptidase kẽm điều hòa nồng độ huyết thanh và bài tiết phốt phát qua nước tiểu. PHEX là một gen có 22 exon mã hóa một protein màng tích hợp 749 aa với một miền ngoại bào rất dài (702 aa) và các miền xuyên màng (27 aa) và nội bào (20 aa) ngắn. Miền ngoại bào có 10 gốc cysteine được bảo tồn và một mô-típ pentapeptide (His-Glu-Phe-Thr-His) đặc trưng của các metallopeptidase kẽm có thể chuyển đổi các propeptide thành các dạng hoạt động hoặc phân hủy và bất hoạt các cơ chất của chúng. PHEX được biểu hiện trên bề mặt của các nguyên bào xương và tế bào xương, cũng như bởi răng, cơ, phổi, gan, tinh hoàn và buồng trứng; phiên mã PHEX bởi các nguyên bào xương bị điều hòa giảm bởi calcitriol. Ở hầu hết bệnh nhân mắc XLHR, các đột biến bất hoạt của PHEX đã được tìm thấy chủ yếu ở miền ngoại bào và bao gồm các mất đoạn dịch khung (16%), nhân đôi, chèn (8%), mất-chèn, vị trí nối (15%), vô nghĩa (27%), và sai nghĩa (34%), nhiều trong số đó ở exon 15 và 17; một đột biến cũng đã được xác định ở vùng không dịch mã 5′ (chuyển đoạn A ⇒ G 429 bp ở thượng nguồn của vị trí khởi đầu ATG) (Hình 20.11). Bởi vì PHEX là một protein được glycosyl hóa, sự thất bại trong quá trình glycosyl hóa dẫn đến sự cô lập của nó trong lưới nội chất do đó cản trở sự di chuyển (vận chuyển) của nó đến màng tế bào; các đột biến khác cản trở chức năng xúc tác của protein hoặc cấu trúc không gian ba chiều của nó. Các đột biến trong PHEX phát sinh tự phát ở hơn 20% bệnh nhân mắc XLHR. Không có mối tương quan giữa vị trí hoặc loại đột biến trong PHEX và các biểu hiện lâm sàng hoặc mức độ nghiêm trọng của bệnh.
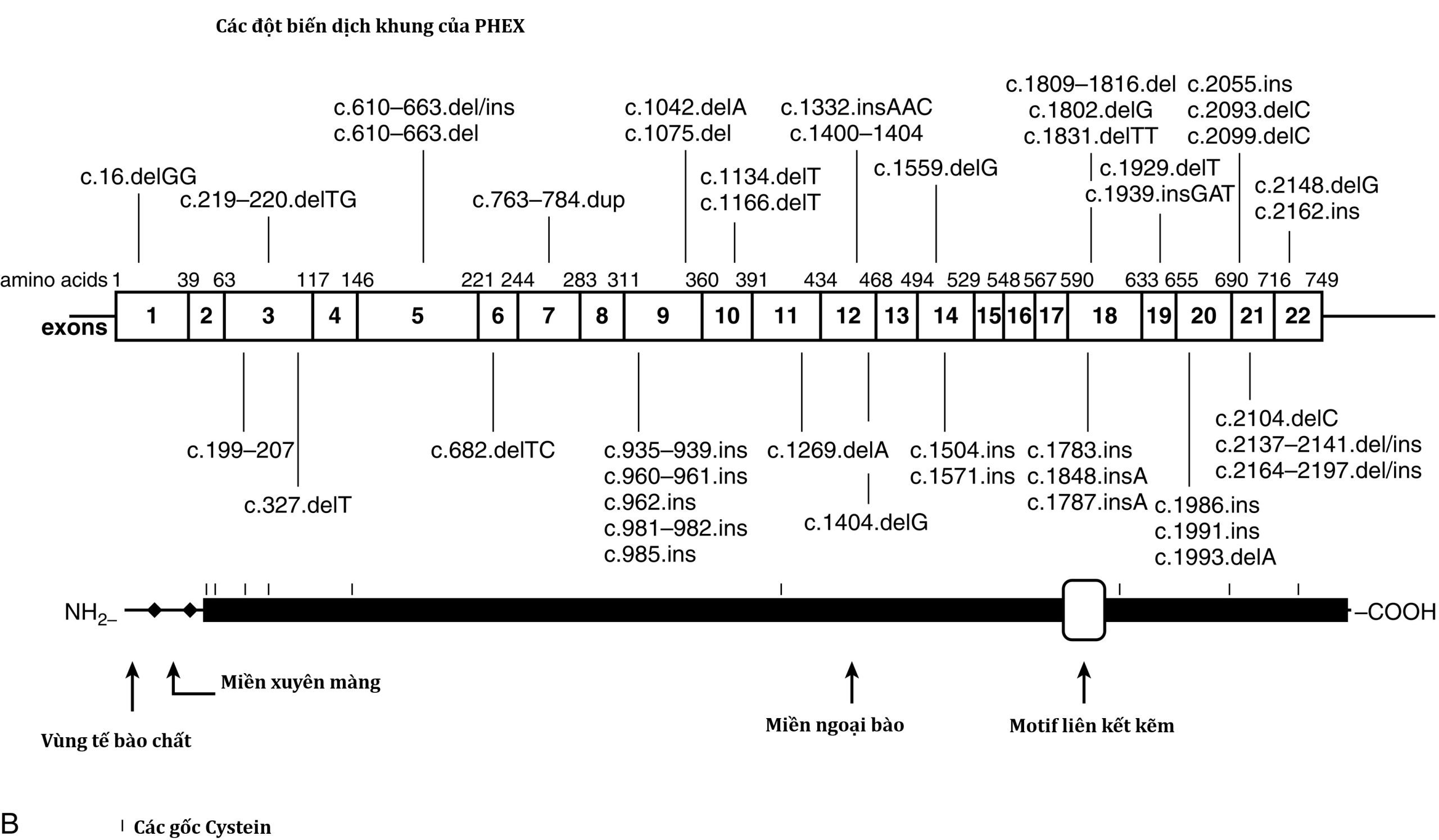
Hình 20.11: Các đột biến sai nghĩa (A) và dịch khung (B) trong PHEX dẫn đến còi xương giảm phốt phát máu di truyền lặn liên kết X.
(Từ PHEXdb, Đại học McGill, Montreal, Quebec, www.phexdb.mcgill.ca/Missense.html và www.phexdb.mcgill.ca/Frameshift.html.)
PHEX là một endopeptidase trung tính mà trong số các cơ chất chính của nó có họ protein SIBLING có nguồn gốc từ xương bao gồm phosphoglycoprotein ngoại bào chất nền (MEPE, OMIM 605912), phosphoprotein axit chất nền ngà răng 1 (DMP1, OMIM 600980), osteopontin/phosphoprotein tiết 1 (OMIM 166490), sialoprotein liên kết integrin (OMIM 147563), và sialophosphoprotein ngà răng (OMIM 125485), tất cả các gen của chúng đều tập trung trên nhánh dài của nhiễm sắc thể 4. Các protein SIBLING được tiết vào chất nền ngoại bào trong quá trình khoáng hóa, liên kết với canxi và điều hòa quá trình khoáng hóa sinh học. MEPE là một protein không phải collagen 525 aa được tiết bởi các nguyên bào xương, có mặt trong chất nền xương ngoại bào, răng và não. Cũng như ảnh hưởng đến quá trình khoáng hóa sinh học, MEPE ức chế sự hấp thu phốt phát ở ống thận và làm giảm tổng hợp calcitriol và do đó là một phosphatonin, các tác nhân có các đặc tính kép này. MEPE và các protein SIBLING khác chứa một mô-típ liên quan đến MEPE giàu aspartate serine axit (ASARM = minhibin) trong các vùng đầu carboxyl của chúng được giải phóng bởi cathepsin B và gốc serine của nó sau đó có thể được phosphoryl hóa; ASARM được phosphoryl hóa (p) liên kết chặt chẽ với hydroxyapatite. PHEX liên kết với MEPE và ngăn cản sự tiêu hóa proteolytic của nó bởi cathepsin B, do đó ức chế sự giải phóng và phosphoryl hóa của ASARM. Khi không có sự liên kết PHEX-MEPE bình thường, ASARM được giải phóng khỏi MEPE bởi cathepsin B; ở những bệnh nhân mắc XLHR, nồng độ ASARM huyết thanh tăng. pASARM được phosphoryl hóa serine bao phủ bề mặt của hydroxyapatite bằng cách tương tác với các nguyên tử canxi của nó và do đó làm suy giảm sự lắng đọng thêm của canxi và phốt phát và do đó ức chế sự khoáng hóa. pASARM cũng là một cơ chất cho hoạt động enzyme của PHEX, phân cắt proteolytic peptide giữa các gốc serine-glutamate và serine-aspartate; bằng cách đó, PHEX phá hủy pASARM và ngăn cản sự ức chế khoáng hóa xương qua trung gian pASARM. Mặc dù ASARM không được phosphoryl hóa cũng liên kết (yếu) với hydroxyapatite và ức chế sự khoáng hóa và có thể bị phân cắt bằng enzyme bởi PHEX, vai trò của ASARM không được phosphoryl hóa trong quá trình khoáng hóa vẫn chưa được hiểu đầy đủ. Điều thú vị là, các peptide ASARM cũng làm suy giảm sự tái hấp thu phốt phát ở ống thận vì pASARM làm tăng biểu hiện của FGF23 (OMIM 605380) trong các tế bào xương. Vì các mô-típ ASARM có mặt trong tất cả các protein SIBLING, pASARM cũng có nguồn gốc từ osteopontin, cung cấp một nguồn peptide khác có khả năng liên kết và ức chế sự khoáng hóa của hydroxyapatite; pASARM có nguồn gốc từ osteopontin cũng là một cơ chất của PHEX. Theo đó, trong XLHR, các bất thường sinh lý bệnh nguyên phát dường như là sự thất bại của PHEX bị đột biến, không có hoạt tính sinh học trong việc: (1) liên kết với MEPE, do đó ức chế sự phân cắt proteolytic qua trung gian cathepsin B của MEPE và giải phóng ASARM; (2) phá hủy pASARM có nguồn gốc từ MEPE, do đó cho phép pASARM bao phủ hydroxyapatite và ức chế sự khoáng hóa; (3) phân hủy các peptide ASARM do đó cho phép chúng làm giảm tái hấp thu phốt phát ở ống thận; và (4) ức chế biểu hiện của FGF23 (xem bên dưới).
Mặc dù nồng độ canxi toàn phần và ion hóa trong huyết thanh là bình thường ở bệnh nhân mắc XLHR, hạ phốt phát máu rõ rệt do lãng phí qua nước tiểu vì sự tái hấp thu phốt phát đã lọc ở ống thận giảm đáng kể và sự hấp thu anion này ở ruột bị hạn chế. Nồng độ PTH và calcidiol trong huyết thanh bình thường, nhưng các giá trị calcitriol thấp một cách không phù hợp so với mức độ hạ phốt phát máu; hoạt độ phosphatase kiềm huyết thanh tăng cũng như nồng độ FGF23 trong huyết thanh (Hình 20.12). Nhiều trong số những bất thường sinh hóa này hiện diện ở bệnh nhân bị ảnh hưởng trong vài tháng đầu sau khi sinh. Bởi vì nồng độ canxi ion hóa trong huyết thanh là bình thường, cường cận giáp thứ phát không xảy ra trừ khi bệnh nhân mắc XLHR nhận một lượng lớn phốt phát bổ sung. Thông thường, phốt phát gây ra sự biệt hóa và chết của các tế bào sụn phì đại trong sụn tăng trưởng bằng cách kích hoạt con đường chết theo chương trình của ty thể phụ thuộc vào caspase 9. Hạ phốt phát máu dẫn đến sự mất chậm và số lượng tế bào sụn phì đại tăng và sự mở rộng của đĩa tăng trưởng đặc trưng của bệnh còi xương. Phép đo hình thái xương trong XLHR cho thấy chất dạng xương không được khoáng hóa tích tụ dọc theo các bè trong xương xốp (tức là, nhuyễn xương) và các tổn thương quanh tế bào xương bị giảm khoáng đặc trưng của rối loạn này.
FGF23 (OMIM 605380) là một tác nhân gây bài tiết phốt phát mạnh được sản xuất bởi các nguyên bào xương và tế bào xương có nồng độ huyết thanh tăng ở bệnh nhân mắc XLHR và có liên quan nghịch với nồng độ phốt phát. Về mặt sinh lý bệnh, FGF23 chịu trách nhiệm cho sự bài tiết phốt phát qua nước tiểu tăng lên đặc trưng của bệnh này. Bệnh nhân mắc các rối loạn bẩm sinh hoặc mắc phải có liên quan đến tăng tổng hợp FGF23 thường bị hạ phốt phát máu cùng với nồng độ calcitriol bình thường hoặc thấp, loại sau dẫn đến cường cận giáp thứ phát. Vì sự biểu hiện của FGF23 bị điều hòa giảm bởi PHEX, các biến thể bất hoạt của PHEX dẫn đến tăng sản xuất FGF23. Ngoài ra, FGF23 ức chế 1α-hydroxyl hóa calcidiol và do đó làm giảm tổng hợp nội sinh calcitriol, cản trở sự hấp thu canxi trong chế độ ăn ở ruột. FGF23 ức chế tái hấp thu phốt phát bởi ống thận và khi có mặt với lượng dư thừa sẽ dẫn đến tăng phốt phát niệu và hậu quả là hạ phốt phát máu. FGF23 được tổng hợp dưới dạng một protein 251 aa; sau khi loại bỏ chuỗi tín hiệu 24 aa, FGF23 trưởng thành lưu hành dưới dạng cả peptide glycosyl hóa 227 và 154 aa. Hoạt động thông qua thụ thể yếu tố tăng trưởng nguyên bào sợi 1 (FGFR1) và đồng thụ thể của nó (α-klotho), FGF23 ức chế sự hấp thu phốt phát phụ thuộc natri bởi các tế bào ống lượn gần của thận và làm giảm hoạt động 25OHD-1α hydroxylase và do đó làm giảm tổng hợp calcitriol; do đó FGF23 cũng là một phosphatonin, một họ các tác nhân gây bài tiết phốt phát, cũng như MEPE và protein-4 liên quan đến frizzled trong huyết thanh (sFRP4). FGF23 ức chế tái hấp thu phốt phát bằng cách điều hòa giảm biểu hiện của SLC34A1 và SLC34A3, tương ứng mã hóa các chất đồng vận chuyển natri-phốt phát type II (a và c) (NPT2a, NPT2c) trong các màng đỉnh của ống lượn gần của thận bằng cách tạo điều kiện cho sự nhập bào và loại bỏ chúng khỏi bờ bàn chải của ống thận, do đó làm giảm tái hấp thu phốt phát đã lọc. FGF23 cũng điều hòa giảm biểu hiện của CYP27B1 mã hóa 25-hydroxyvitamin D-1α hydroxylase và do đó làm giảm tổng hợp calcitriol và cảm ứng CYP24A1 làm tăng tốc độ bất hoạt các chất chuyển hóa của vitamin D. FGF23 được biểu hiện và FGF23 được tiết ra chủ yếu bởi các nguyên bào xương và tế bào xương; nó được đồng biểu hiện với PHEX trong các tế bào này, và PHEX ức chế biểu hiện của FGF23. Mặc dù FGF23 bị bất hoạt bằng cách phân cắt giữa các axit amin Arg179 và Ser180, nó không phải là một cơ chất sinh học cho PHEX. Thay vào đó, FGF23 bị phân cắt bởi các proprotein giống subtilisin và các convertase giống furin. Vì PHEX làm giảm biểu hiện của FGF23, khi không có PHEX, sự biểu hiện của FGF23 sẽ tăng lên.
Chẩn đoán XLHR được xác lập khi có tiền sử gia đình điển hình (nếu bệnh nhân không phải là người đột biến ban đầu), các phát hiện lâm sàng (biến dạng các chi dưới, loe hành xương), dữ liệu X-quang (các thay đổi còi xương) và xét nghiệm (hạ phốt phát máu, tăng phốt phát niệu, nồng độ calcitriol huyết thanh thấp một cách không phù hợp, các giá trị FGF23 tăng, nồng độ PTH, canxi, creatinin và 25OHD huyết thanh bình thường) hiện diện và khi các nguyên nhân khác gây hạ phốt phát máu và tăng phốt phát niệu đã được loại trừ (xem Bảng 20.8A, 20.8B, 20.9 và xem Hình 20.12). Sự bài tiết phốt phát qua thận có thể được đánh giá bằng cách đo sự tái hấp thu phốt phát ở ống thận (TRP) (1-[phốt phát nước tiểu x creatinin huyết thanh]/[phốt phát huyết thanh × creatinin nước tiểu]; giới hạn dưới của mức bình thường dao động trong khoảng 75% đến 85%). Khi có hạ phốt phát máu, TRP thường lớn hơn 95%. Thông thường, các giá trị FGF23 huyết thanh cao nhất ở trẻ sơ sinh và giảm trong thời thơ ấu và vị thành niên xuống các giá trị người lớn (khoảng 30 pg/mL sử dụng một xét nghiệm miễn dịch phát hiện FGF23 nguyên vẹn). Nồng độ FGF23 tăng khi nạp phốt phát và giảm để đáp ứng với tình trạng thiếu hụt phốt phát. Tuy nhiên, nồng độ FGF23 huyết thanh tăng hiện diện ở những bệnh nhân mắc nhiều dạng còi xương giảm phốt phát máu bao gồm XLHR, còi xương giảm phốt phát máu di truyền trội và lặn trên NST thường, các rối loạn McCune-Albright, hội chứng Raine, opismodysplasia, loạn sản xương-sụn-răng, và loạn sản sụn đầu xương type Jansen, hội chứng nevus bã nhờn dạng dải, và còi xương/nhuyễn xương do khối u (xem bên dưới). Vì các biến thể của các gen khác cũng dẫn đến còi xương giảm phốt phát máu (xem bên dưới), việc phân tích PHEX đôi khi cần thiết để xác nhận chẩn đoán XLHR. Hiếm khi, thể khảm soma và dòng mầm cho một đột biến trong PHEX có thể bắt chước sự di truyền trội trên NST thường của còi xương giảm phốt phát máu.
Mục tiêu điều trị trong XLHR là điều chỉnh hoặc giảm thiểu còi xương/nhuyễn xương được đánh giá bằng sự giải quyết các biến dạng xương và các bất thường X-quang và bình thường hóa nồng độ phosphatase kiềm. Việc phục hồi nồng độ phốt phát huyết thanh không phải là mục tiêu điều trị vì làm như vậy sẽ làm tăng nguy cơ cường cận giáp thứ phát và nhiễm canxi thận. Theo truyền thống, bệnh nhân mắc XLHR đã được điều trị bằng calcitriol (20–40 ng/kg/ngày) dùng làm 2 liều mỗi ngày với liều lớn hơn được dùng vào ban đêm khi sự bài tiết PTH có xu hướng tăng và một chế phẩm phốt phát (với liều dao động từ 40–60 mg/kg/ngày bắt đầu với liều thấp hơn để tránh tiêu chảy, dùng làm 4 đến 6 liều chia đều mỗi ngày tùy thuộc vào tuổi, kích thước, sự tuân thủ và đáp ứng với điều trị). Trẻ sơ sinh và trẻ nhỏ có thể dung nạp dung dịch phospho-soda dễ dàng hơn so với các chế phẩm khác; khi có thể, hầu hết trẻ lớn thích một viên nén phốt phát nhai được; các sản phẩm phốt phát kali axit được ưa thích vì chúng làm axit hóa nước tiểu và tăng độ hòa tan của canxi phốt phát. Calcitriol có sẵn dưới dạng dung dịch uống (1 mcg/mL) và dưới dạng viên nang 025 và 0,5 mcg. Nếu tăng canxi máu hoặc tăng canxi niệu phát triển (bài tiết canxi qua nước tiểu hơn 4 mg/kg/ngày), liều calcitriol nên được giảm xuống; nếu quá trình còi xương trầm trọng hơn, việc bổ sung một tác nhân làm tăng tái hấp thu canxi ở ống thận (ví dụ, amiloride) có thể được thêm vào một cách thận trọng vào chương trình điều trị. Tuy nhiên, việc tuân thủ quy trình điều trị chuyên sâu là có vấn đề và các biến chứng (nhiễm canxi thận) thường xuyên. Cần đánh giá lâm sàng và xét nghiệm lặp lại để tránh tăng canxi máu, tăng canxi niệu và nhiễm canxi thận, và cường cận giáp thứ phát vì liều cao phốt phát có thể dẫn đến cường cận giáp thứ phát (và đôi khi tam phát) phản tác dụng. Ít nhất hàng quý, cần tiến hành đánh giá lâm sàng và đo lường nồng độ canxi huyết thanh và nước tiểu, phốt phát, phosphatase kiềm, creatinin và PTH nguyên vẹn. Siêu âm thận trước khi điều trị và cách quãng 12 tháng trong quá trình điều trị để xác định giai đoạn sớm của nhiễm canxi thận và chụp X-quang xương hàng năm để đánh giá mức độ lành bệnh của bệnh còi xương được khuyến nghị. Sự phát triển của nhiễm canxi thận (và sự tổn hại chức năng thận ở một số bệnh nhân) liên quan trực tiếp đến lượng phốt phát mà bệnh nhân nhận được vì tác nhân này làm tăng tổng hợp FGF23 một cách phản tác dụng; tăng oxalat niệu cũng có liên quan đến sinh bệnh học của nhiễm canxi thận. Ở một vài trẻ em mắc XLHR, việc dùng cinacalcet ngắn hạn, một tác nhân calcimimetic ức chế sự bài tiết PTH, đã hữu ích trong việc cải thiện cường cận giáp thứ phát có thể xảy ra trong quá trình điều trị những đứa trẻ này. Việc đồng quản lý một bệnh nhân mắc XLHR với một bác sĩ chỉnh hình có kinh nghiệm là quan trọng, vì bác sĩ chỉnh hình có thể kê đơn nẹp hoặc ở những bệnh nhân có các biến dạng cực đoan và tiến triển, thực hiện phẫu thuật chỉnh hình, chẳng hạn như cắt nửa sụn tiếp hợp xương đùi và xương chày. Các khiếm khuyết trong sự khoáng hóa của ngà răng dẫn đến áp xe răng và mất răng sớm; do đó, những bệnh nhân này phải thực hành vệ sinh răng miệng và được theo dõi bởi một nha sĩ có kinh nghiệm.
Vào năm 2018, việc quản lý XLHR đã thay đổi đáng kể khi có báo cáo rằng việc trung hòa FGF23 bằng một kháng thể đơn dòng immunoglobin (Ig)G1 tái tổ hợp của người đối với FGF23 (burosumab—dùng tiêm dưới da mỗi 2 đến 4 tuần) có hiệu quả điều trị ở trẻ em mắc rối loạn này mà liệu pháp thông thường đã bị rút lại. Cải thiện về lâm sàng (giảm đau, tăng hoạt động thể chất, tăng trưởng), X-quang (giảm điểm mức độ nghiêm trọng của bệnh còi xương Thacher), và sinh hóa (tăng nồng độ phốt phát huyết thanh [do tăng tái hấp thu ở ống thận] và calcitriol và giảm các giá trị phosphatase kiềm) đã được ghi nhận trong quá trình dùng burosumab. Liệu burosumab có trở thành liệu pháp hàng đầu cho trẻ em mắc XLHR hay sẽ được sử dụng sau một thử nghiệm calcitriol/phốt phát vẫn chưa chắc chắn. Một tác nhân khác có thể hữu ích trong điều trị bệnh nhân mắc XLHR là peptide polyarginine hexa-D-arginine; trong một mô hình chuột thực nghiệm của XLHR, hexa-D-arginine tăng cường sự phân hủy của FGF23 thành các mảnh không hoạt động và ức chế sản xuất FGF23 do đó cải thiện rối loạn xương nguyên phát. Cũng về mặt thực nghiệm, sự ức chế CYP24A1, enzyme chịu trách nhiệm cho sự bất hoạt của calcidiol và calcitriol, đã cải thiện đáng kể các khiếm khuyết xương trong một mô hình chuột của XLHR mà không làm thay đổi nồng độ canxi hoặc phốt phát trong huyết thanh.
Trước khi giới thiệu burosumab, lịch sử tự nhiên của bệnh nhân mắc XLHR là mặc dù chiều dài khi sinh của họ bình thường, tốc độ tăng trưởng chậm lại trong vài năm đầu đời dẫn đến sự rút ngắn chiều cao tiến triển. Nhiều trẻ em mắc XLHR bị thấp đáng kể khi 5 tuổi, mặc dù một số bệnh nhân (đặc biệt là các bé gái) bị ảnh hưởng tối thiểu vẫn phát triển bình thường. Điều trị trẻ lớn bị XLHR bằng calcitriol và phốt phát đã cải thiện tốc độ tăng trưởng, một phần là do sự điều chỉnh các biến dạng của các chi dưới. Trong tuổi dậy thì, sự tăng chiều cao là bình thường ở các bé trai (+ 28,2 cm) và các bé gái (+ 24,2 cm) mắc XLHR; do đó, tầm vóc người lớn bị tổn hại trong XLHR liên quan đến sự tăng trưởng suy giảm ở đầu thời thơ ấu và tiền vị thành niên. Trong một loạt 19 trẻ em được theo dõi chặt chẽ mắc XLHR, việc bắt đầu điều trị bằng calcitriol và phốt phát ở độ tuổi trung bình là 4,2 tháng (dao động từ 7 tuần đến 6 tháng, n = 8) đã dẫn đến tầm vóc người lớn cao hơn (–0,2 so với –1,2 SD) so với khi bắt đầu điều trị ở độ tuổi trung bình là 2,1 tuổi (dao động 1,3–8,0 tuổi, n = 11) mà không có sự khác biệt về tỷ lệ biến chứng (cường cận giáp thứ phát, nhiễm canxi thận, dính khớp sọ) giữa hai nhóm. Mặc dù tuân thủ điều trị chặt chẽ, nhiều trẻ em mắc XLHR cho đến nay vẫn bị còi cọc về chiều cao chủ yếu do các chi dưới ngắn và giữ lại các dấu hiệu X-quang từ nhẹ đến trung bình của bệnh còi xương và hoạt độ phosphatase kiềm huyết thanh tăng nhẹ, mặc dù mức độ biến dạng chi dưới được cải thiện một phần nhờ sự tuân thủ tốt. GH của người làm tăng tốc độ lọc cầu thận, tái hấp thu ở ống thận và nồng độ phốt phát trong huyết thanh, và tốc độ tích lũy khoáng chất xương; nó làm tăng tốc độ chiều cao và tốc độ tăng chiều dài chi của trẻ em mắc XLHR và có thể làm tăng nhẹ tầm vóc người lớn. Nhiều người lớn không được điều trị mắc XLHR bị hạ phốt phát máu; hoạt độ phosphatase kiềm huyết thanh tăng và nhuyễn xương hiện diện trên sinh thiết xương; những bệnh nhân này bị áp xe răng thường xuyên, bệnh thoái hóa khớp háng do các biến dạng của các chi dưới, nhiễm canxi thận và suy giảm thính lực. Đôi khi, một phụ nữ mắc XLHR có thể khỏe mạnh về mặt lâm sàng mặc dù bị hạ phốt phát máu đơn độc. Các biểu hiện X-quang của XLHR ở người lớn bao gồm các thay đổi thoái hóa ở các khớp lớn, dày các mỏm gai và hợp nhất các đốt sống, và hẹp ống sống. Khoáng hóa xương được xác định bằng phương pháp đo hấp thụ photon đơn và kép có xu hướng bình thường ở người lớn mắc XLHR (mặc dù có các bất thường về hình thái mô học), cho thấy hầu hết các đối tượng này không có nguy cơ gãy xương do loãng xương tăng lên. Tuy nhiên, ở khoảng 25% người lớn mắc XLHR, có bằng chứng lâm sàng về nhuyễn xương, chẳng hạn như các biến dạng chi dưới tiến triển, đau xương, gãy xương, bệnh điểm bám gân và gãy xương giả. Điều trị một số bệnh nhân XLHR trưởng thành được chọn bằng calcitriol và phốt phát có thể có lợi. Nhiều người lớn cần phẫu thuật chỉnh hình, thay khớp háng và khớp gối, và phẫu thuật giải nén bản sống cho hẹp ống sống. Burosumab, kháng thể đơn dòng đối với FGF23, cũng có thể hữu ích trong việc quản lý lâu dài người lớn mắc XLHR, mặc dù vai trò của nó trong điều trị người lớn mắc rối loạn này vẫn chưa chắc chắn.
Còi xương giảm phốt phát máu di truyền lặn liên kết X (OMIM 300554) có liên quan đến tăng canxi niệu, nhiễm canxi thận và suy thận; nó là hậu quả của các đột biến mất chức năng trong CLCN5 (OMIM 300008) mã hóa một chất vận chuyển trao đổi hydro/clorua xuyên màng có cổng điện thế được biểu hiện ở ống thận và độc lập với FGF23; p.Ser244Leu là một biến thể phổ biến của CLCN5 liên quan đến rối loạn này. Các biến thể bất hoạt của CLCN5 cũng đã được xác định ở những bệnh nhân mắc bệnh Dent 1 (OMIM 30000—với aminoacid niệu, protein niệu, glycosuria, tăng canxi niệu và nhiễm canxi thận) và các bệnh ống thận khác.
Các rối loạn bẩm sinh liên quan đến còi xương giảm phốt phát máu và tăng sản xuất FGF23 có thể liên quan đến các biến thể của một trong số các gen trên NST thường hoặc liên kết X. Trẻ em bị còi xương giảm phốt phát máu di truyền trội trên NST thường (ADHR, OMIM 193100) do các biến thể hoạt hóa của chính FGF23 có bằng chứng lâm sàng, sinh hóa và X-quang của bệnh còi xương tương tự như ở bệnh nhân mắc XLHR, mặc dù ở một số đối tượng, mức độ biểu hiện bệnh có thể là tối thiểu hoặc thậm chí suy giảm theo tuổi; bệnh nhân mắc ADHR cũng biểu hiện yếu cơ là hậu quả của hạ phốt phát máu. Thông thường, FGF23 bị bất hoạt bằng cách phân cắt giữa aa Arg179 và Ser180 bởi các protease giống subtilisin, nhưng các biến thể tăng chức năng của FGF23 (p.Arg176Gln, p.Arg179Trp) ngăn cản sự phân cắt proteolytic của protein nguyên vẹn và do đó kéo dài hoạt động sinh học của nó, dẫn đến tăng phốt phát niệu dai dẳng và hạ phốt phát máu với nồng độ canxi huyết thanh bình thường hoặc hơi thấp và nồng độ calcitriol huyết thanh thấp hoặc bình thường. Bởi vì thiếu sắt làm tăng cường biểu hiện của FGF23, nó là một kích thích cho sự phát triển của các biểu hiện lâm sàng của ADHR. Điều trị bao gồm dùng calcitriol và phốt phát với việc theo dõi nối tiếp chặt chẽ cả về sự an toàn và vì tăng phốt phát niệu đôi khi có thể tự khỏi. Tổng hợp lạc chỗ quá mức FGF23 bởi các khối u cũng dẫn đến còi xương giảm phốt phát máu (xem bên dưới).
Còi xương giảm phốt phát máu có cường cận giáp (OMIM 612089) là một rối loạn được đặc trưng bởi sự khởi phát của hạ phốt phát máu và tăng sản tuyến cận giáp lan tỏa ở trẻ nhỏ. Về mặt sinh bệnh học, nó có thể là kết quả của việc sản xuất quá mức α-klotho (KL, OMIM 604824), đồng thụ thể với FGFR1(IIIc) cho FGF23 chuyển đổi FGFR1(IIIc) thành thụ thể FGF23 đặc hiệu kích thích con đường truyền tín hiệu nội bào FGF23 và hoạt tính sinh học của nó. Lượng α-Klotho dư thừa dường như làm tăng cường sản xuất và hoạt tính sinh học của FGF23 dẫn đến tăng phốt phát niệu và hạ phốt phát máu và cũng làm trung gian cho sự tăng sản tuyến cận giáp và tăng cường tổng hợp PTH. Ở một bệnh nhân mắc rối loạn này, việc sản xuất quá mức α-klotho đã được cho là do một chuyển vị cân bằng xảy ra tự phát giữa các nhánh dài của nhiễm sắc thể 9 và 13 (–t[9;13][q21.13;q13.1]) có thể liên quan đến một vùng promoter cho phép biểu hiện quá mức của KL và tổng hợp α-klotho.
Các đột biến trong hai gen dẫn đến còi xương giảm phốt phát máu di truyền lặn trên NST thường (ARHR) có các đặc điểm lâm sàng, sinh hóa, sinh bệnh học và hình thái mô học tương tự như XLHR nhưng được phân biệt bởi phương thức di truyền và sự phát triển của xơ cứng xương ở nền sọ và trong các xương vòm sọ. ARHR 1 (OMIM 241520) là do các đột biến hai alen mất chức năng trong phosphoprotein-1 axit chất nền ngà răng (DMP1, OMIM 600980), một protein SIBLING liên kết canxi chất nền xương giàu serine, không phải collagen được tổng hợp bởi các tế bào xương, cũng làm suy giảm biểu hiện của FGF23. Không có DMP1, các tế bào xương không thể trưởng thành và sự tạo mầm hydroxyapatite bị cản trở; sự gia tăng thứ phát trong tổng hợp FGF23 bởi các tế bào xương dẫn đến tăng phốt phát niệu, hạ phốt phát máu và khoáng hóa xương khiếm khuyết. Ở các đối tượng bị ARHR1 hoặc XLHR, sinh thiết xương cho thấy một vùng chất dạng xương không khoáng hóa riêng biệt xung quanh mỗi tế bào xương. Nghịch lý thay, một số bệnh nhân lớn tuổi mắc ARHR1 phát triển khoáng hóa xương quá mức ở các xương sọ nền và vòm sọ không khác gì so với những gì được quan sát thấy ở bệnh nhân bị bệnh xương hóa đá. Type 2 ARHR (OMIM 613312) là kết quả của các đột biến bất hoạt hai alen trong ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1, OMIM 173335), một enzyme thủy phân ATP ngoại bào thành cyclic AMP, giải phóng phốt phát để tạo thành pyrophosphat, một chất ức chế khoáng hóa chất nền xương. (ENPP1 và TNSALP cùng nhau điều hòa nồng độ pyrophosphat lưu hành. Do đó, bệnh xương do thiếu ENPP1 giống với bệnh giảm phosphatase máu [xem bên dưới].) Các biến thể bất hoạt của ENPP1 dẫn đến tăng tiết thứ phát FGF23 bởi một cơ chế không rõ ràng dẫn đến tăng phốt phát niệu và hạ phốt phát máu, còi xương và vôi hóa lạc chỗ. (Các biến thể mất chức năng của ENPP1 cũng dẫn đến vôi hóa động mạch toàn thể di truyền lặn trên NST thường ở trẻ nhỏ [OMIM 208000].) ENPP1 làm tăng biểu hiện KL ở thận và cần thiết cho sự tổng hợp α-klotho trong bối cảnh tải lượng phốt phát cao, một quan sát có thể có liên quan về mặt sinh lý bệnh ở bệnh nhân mắc ARHR2. Tăng tiết FGF23 và còi xương giảm phốt phát máu cũng đã được ghi nhận ở các đối tượng bị loạn sản xương-sụn-răng (OMIM 166250—dính khớp sọ, rút ngắn các chi ở gốc, các tổn thương xương không vôi hóa) liên quan đến các đột biến hoạt hóa đơn alen trong FGFR1 (OMIM 136350); loạn sản sụn đầu xương type Jansen (OMIM 156400) do các đột biến hoạt hóa cấu thành trong PTH1R (OMIM 168468); loạn sản xơ/hội chứng McCune Albright (OMIM 174800) kết hợp với các đột biến của GNAS (OMIM 139320); hội chứng Schimmelpenning-Feuerstein-Mims/hội chứng nevus bã nhờn dạng dải (OMIM 163200) liên quan đến các biến thể tăng chức năng soma sau hợp tử của NRAS (OMIM 164790), HRAS (OMIM 190020), hoặc KRAS (OMIM 190070); và opismodysplasia (một loạn sản cột sống[sụn tiếp hợp]đầu xương với sự hóa xương chậm, các chi nhỏ, đốt sống dẹt, thiểu sản đốt sống, tầm vóc rất thấp—OMIM 258480) do các biến thể bất hoạt hai alen của INPPL1 (OMIM 600829) mã hóa một inositol-1,4,5-trisphosphatase thủy phân inositol-1,4,5-trisphosphate thành inositol-4,5-bisphosphate, một chất truyền tín hiệu nội bào quan trọng. Họ có trình tự tương tự 20, thành viên C (FAM20C, OMIM 611061) mã hóa một phosphorylase có cơ chất là họ protein SIBLING bao gồm DMP1. Các đột biến mất chức năng trong FAM20C dẫn đến nồng độ FGF23 tăng, dẫn đến còi xương giảm phốt phát máu hoặc rối loạn thường gây tử vong được gọi là hội chứng Raine có liên quan đến nhuyễn xương xơ cứng bẩm sinh với vôi hóa não (OMIM 259775). Hội chứng Raine được đặc trưng bởi sự gia tăng vôi hóa bẩm sinh của tất cả các xương (đặc biệt là các xương sọ dẫn đến lồi mắt và thiểu sản giữa mặt) kết hợp với sự hình thành xương màng xương—một đặc điểm không thấy ở bệnh nhân bị bệnh xương hóa đá hoặc các rối loạn xương xơ cứng khác và vôi hóa não. Nhiều bệnh nhân mắc hội chứng Raine chết trong giai đoạn sơ sinh. FAM20C mã hóa một kinase (protein chất nền ngà răng 4) thường trực tiếp phosphoryl hóa DMP1 sau đó ức chế phiên mã của FGF23; FAM20C cũng phosphoryl hóa gốc Ser180 của FGF23, do đó ức chế sự glycosyl hóa của nó và thúc đẩy sự phân cắt và bất hoạt của FGF23; do đó, sự mất FAM20C dẫn đến giảm ức chế và bất hoạt FGF23 dẫn đến hoạt động chức năng kéo dài của nó.
Nhuyễn xương/còi xương do khối u là một rối loạn mắc phải do sự tổng hợp quá mức của một trong số các phosphatonin bởi một khối u có nguồn gốc trung mô. Nhiều khối u trung mô tổng hợp và tiết ra FGF23, dẫn đến hạ phốt phát máu rõ rệt và suy giảm sự hình thành xương (nhuyễn xương do khối u). Các khối u khác biểu hiện một dạng của FGFR1, thụ thể cho FGF23. Những khối u này cũng đã tổng hợp protein-4 liên quan đến frizzled, MEPE và FGF 7, tất cả đều có thể làm tăng bài tiết phốt phát qua nước tiểu và ức chế tổng hợp calcitriol ở thận, mặc dù có lẽ không đến mức độ tương tự như FGF23. Việc xác định một khối u trung mô tiết FGF23 phụ thuộc vào độ nhạy của xét nghiệm FGF23 được sử dụng. Mặc dù không phổ biến ở trẻ em, một bé gái 11 tuổi bị đau xương đáng kể và hạn chế chức năng liên quan đến nhuyễn xương/còi xương giảm phốt phát máu đã được chứng minh bằng sinh thiết và nồng độ FGF23 trong huyết thanh tăng rõ rệt. Sau khi loại bỏ một khối u xơ xương lành tính khỏi một khối u xương nhỏ trên hành xương trụ xa, nồng độ FGF23 huyết thanh bình thường hóa trong vòng 7 giờ sau phẫu thuật và nồng độ phốt phát bình thường 2 tuần sau đó. Các triệu chứng lâm sàng giảm bớt và các bất thường về X-quang và hình thái mô học được giải quyết trong vòng 1 năm sau phẫu thuật. Ở một cậu bé 11 tuổi bị đau xương dữ dội, yếu cơ tiến triển đến mức phải ngồi xe lăn, tăng phốt phát niệu và hạ phốt phát máu, nồng độ FGF23 tăng cao (1874 RU/mL) đã giảm xuống giá trị bình thường (43 RU/mL) trong vòng 48 giờ sau khi loại bỏ một khối u mạch máu ngoại bào chứa FGF23 khỏi cánh chậu trái của mình. Tổn thương này đã không được xác định bằng X-quang thông thường, CT hoặc chụp cộng hưởng từ, hoặc xạ hình xương technetium; khối u chỉ được chứng minh bằng chụp cộng hưởng từ dội gradient ( Hình 20.13). Trong vòng 2 tuần sau phẫu thuật, cậu bé đã đi lại được mà không cần trợ giúp, và vài tuần sau đó, cậu đã hoạt động bình thường trở lại.
Hình 20.13: Hình ảnh chụp cộng hưởng từ dội gradient của một khối u mạch máu ngoại bào sản xuất FGF23 ở cánh chậu trái của một cậu bé 11 tuổi bị còi xương do khối u.
(Từ Shulman DI, Hahn G, Benator R, et al. Tumor-induced rickets: usefulness of MR gradient echo recall imaging for tumor localization. J Pediatr. 2004;144:381–385.)
Mặc dù tăng sản xuất FGF23 là cơ chế sinh bệnh phổ biến nhất của hạ phốt phát máu, nó cũng có thể là kết quả của các rối loạn không liên quan đến tăng tổng hợp FGF23 (xem Bảng 20.8B). ADHR có nhiễm canxi thận/sỏi thận/loãng xương type 1 (OMIM 612286) là do các đột biến bất hoạt đơn alen trong SLC34A1 (OMIM 182309) mã hóa chất đồng vận chuyển natri-phốt phát NPT2a (còn được chỉ định là NaPi-IIa). Sự thiếu hụt một nửa của NPT2a trong màng đỉnh bờ bàn chải của ống lượn gần của thận mà sự biểu hiện của nó nằm dưới sự kiểm soát ức chế của PTH và FGF23 cản trở sự tái hấp thu phốt phát. Rối loạn này được đặc trưng bởi sự giảm tái hấp thu phốt phát, tăng phốt phát niệu, hạ phốt phát máu, tăng tổng hợp calcitriol với tăng hấp thu canxi ở ruột, tăng canxi máu với ức chế bài tiết PTH, tăng canxi niệu, hình thành sỏi thận và loãng xương. Hội chứng Fanconi di truyền lặn trên NST thường có còi xương giảm phốt phát máu (OMIM 613388) (còn được chỉ định là hội chứng ống thận Fanconi 2) là do các đột biến bất hoạt hai alen trong SLC34A1 cũng như tăng canxi máu ở trẻ nhỏ type 2 (OMIM 616963). Giảm phốt phát máu di truyền trội trên NST thường có sỏi niệu type 2 (OMIM 612287) được cho là do các đột biến mất chức năng dị hợp tử trong SLC9A3R1 (OMIM 604990) mã hóa NHERF1—một yếu tố điều hòa trao đổi natri/hydro ở ống thận; một trong những chức năng của nó là liên kết với NPT2a và neo nó vào màng lòng ống của ống lượn gần; một chức năng khác là điều chỉnh tín hiệu PTH1R để đáp ứng với PTH. Mất chức năng NHERF1 dẫn đến sự phân ly của nó khỏi NPT2a và sự nhập bào sau đó của NPT2a dẫn đến giảm tái hấp thu phốt phát ở ống thận, tăng phốt phát niệu và hạ phốt phát máu. Bệnh nhân có đột biến trong SCL9A3R1 cũng bị loãng xương; nồng độ PTH và FGF23 huyết thanh bình thường ở những đối tượng này. Sinh bệnh học và các đặc điểm lâm sàng và xét nghiệm của bệnh nhân bị còi xương giảm phốt phát máu có nhiễm canxi thận/sỏi thận/loãng xương type 2 tương tự như dạng type 1 của rối loạn này. Hạ phốt phát máu dẫn đến tăng tổng hợp calcitriol, hấp thu canxi ở ruột, tăng canxi máu nhẹ, tăng canxi niệu và sỏi thận. Bệnh nhân có đột biến trong SCL9A3R1 cũng bị loãng xương. Các đột biến mất chức năng hai alen trong SLC34A3 (OMIM 609826) mã hóa NPT2c, chất vận chuyển phốt phát ở ống thận, dẫn đến còi xương giảm phốt phát máu có tăng canxi niệu (OMIM 241530) độc lập với FGF23. Protein 599 aa này với tám miền xuyên màng nằm ở bờ bàn chải của các tế bào ống lượn gần cạnh tủy thận. Mất hoạt động của NPT2c dẫn đến tăng phốt phát niệu, hạ phốt phát máu, tăng tổng hợp calcitriol với tăng hấp thu canxi ở ruột, tăng canxi niệu, hình thành sỏi thận và còi xương do thiếu canxi. Hạ phốt phát máu dẫn đến tăng tổng hợp calcitriol và do đó tăng cường hấp thu canxi ăn vào, tăng canxi niệu và nhiễm canxi thận. Những người mang dị hợp tử của các đột biến bất hoạt trong SLC34A3 cũng có nồng độ calcitriol huyết thanh tăng vừa phải, tăng phốt phát niệu và tăng canxi niệu nhưng không có bệnh xương chuyển hóa được xác định. Bệnh nhân có các biến thể của SLC34A1, SLC9A3R1, hoặc SLC34A3 có thể được điều trị thành công chỉ bằng phốt phát đường uống kết hợp với bù nước và tránh chế độ ăn nhiều natri, trong khi việc dùng calcitriol bị chống chỉ định.
Các biến thể có hại của kênh clorua 5 (CLCN5, OMIM 30008) dẫn đến bệnh Dent 1 (OMIM 300009) do rối loạn chức năng ống lượn gần của thận tiến triển dẫn đến suy thận liên quan đến aminoacid niệu, protein niệu, tăng phốt phát niệu, hạ phốt phát máu, tăng canxi niệu, glycosuria, và uricosuria và các mối liên quan thay đổi với bệnh xương chuyển hóa (còi xương giảm phốt phát máu—OMIM 300554) và nhiễm canxi thận (OMIM 310468). Các đột biến ở các miền khác nhau của kênh clorua xuyên màng 12 exon, 746 aa này dẫn đến các biểu hiện lâm sàng và sinh hóa khác nhau; đột biến p.Ser244Leu trong CLCN5 đã được liên kết với còi xương giảm phốt phát máu di truyền lặn liên kết X. Các đặc điểm lâm sàng của bệnh Dent 2 (OMIM 300555) tương tự như của bệnh Dent 1, nhưng rối loạn này là kết quả của các biến thể bất hoạt của OCRL1 (OMIM 300535) mã hóa một phosphatidylinositol 4,5-bisphosphate-5-phosphatase ảnh hưởng đến sự nhập bào và có thể phá vỡ sự di chuyển của tế bào của sản phẩm của CLCN5. Hạ phốt phát máu của bệnh nhân bệnh Dent thường có thể được điều chỉnh bằng các chất bổ sung phốt phát đường uống. Tăng bài tiết phốt phát qua nước tiểu do các rối loạn mắc phải và di truyền của ống lượn gần của thận là đặc trưng của bệnh xương chuyển hóa đi kèm với các dạng khác nhau của hội chứng Fanconi gồm toan hóa ống thận, glycosuria và aminoacid niệu (di truyền: bệnh cystinosis, tyrosinemia, galactosemia, hội chứng mắt-não-thận, không dung nạp fructose, bệnh Wilson; mắc phải: ghép thận, hội chứng thận hư, huyết khối tĩnh mạch thận, ngộ độc thủy ngân, chì và đồng, tetracycline hết hạn). Ngoài hạ phốt phát máu, nhiễm toan góp phần vào sinh bệnh học của bệnh xương trong hội chứng Fanconi bằng cách làm tăng độ hòa tan của pha khoáng của xương và tăng mất canxi qua nước tiểu và bằng cách làm suy giảm sự chuyển đổi calcidiol thành calcitriol. Ở những bệnh nhân mắc hội chứng Fanconi-Bickel di truyền lặn trên NST thường (OMIM 227810), các đột biến trong SLC2A2 (OMIM 138160) dẫn đến giảm tổng hợp một chất vận chuyển glucose (GLUT2) dẫn đến suy giảm sử dụng glucose và galactose và hậu quả là tích tụ glycogen trong gan và thận cùng với một bệnh thận ống lượn gần. Ở chuột chuyển gen, việc xóa Slc2a2 dẫn đến giảm biểu hiện của Npt2c trong các ống lượn gần của thận. Bệnh nhân có một đột biến sai nghĩa dị hợp tử cụ thể trong HNF4A (OMIM 600281) (p.Arg76Trp, c.226C ⇒ T) cũng phát triển kiểu hình Fanconi-Bickel cũng như ở những con chuột mà HNF4A đã bị loại bỏ.
Tăng phốt phát máu có thể do các biến thể gen nội tại hoặc do các nguyên nhân ngoại sinh, chẳng hạn như dùng quá nhiều phốt phát qua đường tiêm hoặc dùng thuốc xổ chứa phốt phát, tăng tốc độ phá hủy tế bào cấp tính (chấn thương do đè ép, tiêu cơ vân, trong quá trình hóa trị liệu cho bệnh ác tính), giảm tốc độ bài tiết phốt phát qua thận (suy thận), suy giảm bài tiết hoặc hoạt động của PTH, và trong quá trình dùng một số bisphosphonate. Suy giảm tổng hợp FGF23 và/hoặc α-klotho làm giảm bài tiết phốt phát ở ống thận và làm suy giảm sản xuất calcitriol ở thận, dẫn đến tăng phốt phát máu, hạ canxi máu biên và cường cận giáp thứ phát. Kết quả là, có sự vôi hóa lạc chỗ của các mô mềm—đặc biệt là ở vùng quanh khớp, một rối loạn được gọi là vôi hóa u tăng phốt phát máu có tính gia đình (FHTC). Vôi hóa u tăng phốt phát máu có tính gia đình (OMIM 211900) là một rối loạn di truyền lặn trên NST thường được đặc trưng bởi sự lắng đọng tiến triển của canxi phốt phát trong các mô mềm và ở các vùng quanh khớp của các xương dài, biểu hiện lâm sàng bằng các khối quanh khớp do canxi lắng đọng lạc chỗ và đôi khi bằng các phàn nàn về đau xương. Các vị trí khác của vôi hóa lạc chỗ bao gồm não và các mạch máu; về mặt X-quang, có tăng sinh xương vỏ và phản ứng màng xương liên quan đến các xương dài. Do đó, FHTC là hình ảnh phản chiếu của ADHR. Rối loạn này là do giảm tổng hợp FGF23, sự phân hủy nhanh chóng của nó, hoặc sự kháng ngoại vi đối với chức năng của nó liên quan đến các biến thể bất hoạt hai alen của FGF23, GALNT3, và KL, tương ứng (OMIM 605380, 601756, 604824). Do đó, có thể có sự giảm tổng hợp FGF23 do các đột biến mất chức năng trong chính FGF23, sự phân hủy nhanh chóng của FGF23 vì nó chưa được glycosyl hóa do một đột biến bất hoạt trong GALNT3 mã hóa UDP-N-acetyl-alpha-D-galactosamine: polypeptide N-acetylgalactosaminyl transferase 3, hoặc sự không đáp ứng của mô đối với FGF23 do một đột biến bất hoạt trong KL, mã hóa đồng thụ thể α-klotho của nó. GALNT3 là một enzyme glycosyl hóa FGF23 một cách bảo vệ; sự mất mát của nó cho phép mất nhanh FGF23. Trong các tế bào biểu mô của ống lượn gần của thận, FGF23 tương tác với α-klotho-FGFR1(IIIc)-heparan sulfate để tăng bài tiết phốt phát qua thận bằng cách giảm biểu hiện của các chất vận chuyển phốt phát. Quản lý FHTC được giới hạn trong việc ăn chế độ ăn ít phốt phát, dùng các tác nhân liên kết phốt phát ở ruột (nhôm hydroxit), và tăng bài tiết phốt phát qua nước tiểu bằng cách sử dụng một loại thuốc lợi tiểu, chẳng hạn như acetazolamide; việc bôi tại chỗ natri thiosulfat đã được báo cáo là làm giảm các vôi hóa lạc chỗ trên bề mặt da có lẽ bằng cách tạo phức và do đó hòa tan canxi trong các lắng đọng này. Cắt bỏ phẫu thuật các khối u làm suy giảm chức năng, dùng các chất liên kết phốt phát ở ruột, các tác nhân làm tăng bài tiết phốt phát qua thận (acetazolamide), và chế độ ăn ít canxi và phốt phát đã là các chiến lược điều trị chính được sử dụng trong điều trị những bệnh nhân này.
Các rối loạn về Hoạt động của Phosphatase Kiềm
Ở các dạng nặng nhất, giảm phosphatase máu (OMIM 241500) là một rối loạn di truyền lặn trên nhiễm sắc thể thường do các đột biến mất chức năng trong ALPL (OMIM 171760), gen mã hóa phosphatase kiềm không đặc hiệu của mô (xương/gan/thận) (TNSALP); các dạng ít gây tổn hại hơn của bệnh này được di truyền như các tính trạng trội trên nhiễm sắc thể thường (OMIM 241510, 146300). TNSALP là một phosphohydrolase được biểu hiện trên bề mặt của các tế bào xương, có chức năng thủy phân các hợp chất được phosphoryl hóa, chẳng hạn như pyrophosphate, pyridoxyl-5-phosphate, và phosphoethanolamine. Nếu không có TNSALP, nhóm phosphate của pyrophosphate vô cơ không thể bị loại bỏ; vì pyrophosphate liên kết và cản trở sự chuyển đổi của canxi/phosphate vô định hình thành hydroxyapatite, sự hiện diện dai dẳng của nó ức chế sự lớn lên của tinh thể hydroxyapatite và do đó ức chế sự khoáng hóa của chất nền xương collagen. Phosphatase kiềm là các phosphohydrolase monoester orthophosphoric tối ưu ở môi trường kiềm, phụ thuộc vào kẽm và magiê. Các đồng dạng phosphatase kiềm của xương, gan và thận không khác nhau về cấu trúc axit amin mà khác nhau về kiểu glycosyl hóa sau dịch mã. TNSALP xương dạng homotetramer được neo qua đầu carboxyl của nó vào màng tế bào bên ngoài của các tế bào sụn phì đại và nguyên bào xương (tức là, nó là một ectophosphatase) bởi một phân tử phosphatidylinositol-glycan. Về mặt sinh lý bệnh, hoạt động TNSALP giảm dẫn đến sự tích tụ các cơ chất nội sinh của nó—pyrophosphate vô cơ, pyridoxal 5′-phosphate, và phosphoethanolamine. Bởi vì pyrophosphate là một chất ức chế khoáng hóa chất dạng xương, sự thất bại trong việc phân hủy dẫn đến sự tích tụ của nó và sau đó là sự hình thành không đầy đủ của hydroxyapatite dẫn đến còi xương hoặc nhuyễn xương, cũng như tăng canxi máu, tăng canxi niệu, ức chế tổng hợp và bài tiết PTH, tăng tái hấp thu phosphate ở thận và tăng phốt phát máu. (Sinh lý bệnh của bệnh xương do thiếu TNSALP tương tự như bệnh do thiếu ENPP1, trong đó pyrophosphate cũng tích tụ nhưng theo một cơ chế hoàn toàn khác [xem ở trên].) Sự thất bại trong việc chuyển hóa pyridoxal 5′-phosphate thành pyridoxine dẫn đến co giật phụ thuộc pyridoxine. Mức độ nghiêm trọng lâm sàng của giảm phosphatase máu thay đổi từ tử vong sớm đến rụng răng sữa sớm đơn độc và phụ thuộc vào mức độ thiếu hụt TNSALP chức năng được xác định bởi (các) vị trí của các biến thể trong ALPL. Tuổi khởi phát càng trẻ, bệnh càng có khả năng nghiêm trọng hơn. Tỷ lệ mắc các dạng giảm phosphatase máu nặng dao động từ 1/2500 trẻ sơ sinh trong các gia đình Mennonite Canada đến 1/300.000 trong các quần thể châu Âu. Hiện tại, có bảy dạng hoặc loại giảm phosphatase máu được công nhận—sáu dạng cổ điển (được đánh số 1–6) và một loại chuyển tiếp (ở đây được chỉ định là loại 1A):
- Dạng giảm phosphatase máu chu sinh có thể được xác định ở thai nhi trong tử cung và biểu hiện lâm sàng khi sinh với các dị tật và gãy xương sườn, đốt sống và các chi; suy giảm khoáng hóa các xương dài (tua hành xương) và hộp sọ (mặc dù vậy, sự hợp nhất rõ ràng của các đường khớp của xương sọ màng có thể xảy ra dẫn đến dính khớp sọ chức năng mà không có bằng chứng X-quang về nó); và co giật phụ thuộc pyridoxine. Các dị tật của hộp sọ có liên quan đến xuất huyết nội sọ, trong khi gãy xương sườn và các dị tật của thành ngực dẫn đến suy hô hấp sau sinh và ngưng thở. Các phát hiện X-quang bao gồm không có hoặc khoáng hóa xương cực kỳ kém với các dị tật còi xương, các phần mở rộng thấu quang không đều vào hành xương, các đốt sống dường như “mất tích”, và các gai xương nhô ra ở giữa thân xương mác và xương trụ. Dạng giảm phosphatase máu chu sinh gần như luôn gây tử vong, mặc dù hiệu quả của liệu pháp thay thế enzyme (xem bên dưới) ở những bệnh nhân này vẫn chưa được xác định;
1A. Ở dạng giảm phosphatase máu lành tính trước sinh hoặc chuyển tiếp, thai nhi và trẻ sơ sinh biểu hiện các dị tật xương nhưng không có dấu hiệu X-quang của bệnh còi xương; ngay sau khi sinh, có sự cải thiện tự phát và đáng kể; các phát hiện phân biệt loại giảm phosphatase máu ít nghiêm trọng này là sự chen chúc của thai nhi, khoáng hóa xương thai nhi bình thường và thể tích lồng ngực phù hợp; dạng giảm phosphatase máu này có thể liên quan đến các đột biến đơn alen hoặc hai alen trong ALPL;
- Dạng trẻ nhỏ (OMIM 241500) trở nên rõ ràng về mặt lâm sàng trong 6 tháng đầu đời và được đặc trưng bởi chán ăn, suy giảm tăng trưởng tuyến tính và tăng cân, các dị tật của xương dài và lồng ngực, các thóp và đường khớp “mở” rộng (giảm khoáng hóa vòm sọ) với dính khớp sọ chức năng và tăng áp lực nội sọ (biểu hiện bằng sự nhô ra của thóp trước, lồi mắt và phù gai thị), yếu cơ, chậm phát triển, co giật phụ thuộc pyridoxine, giảm trương lực cơ, táo bón, tăng canxi máu, tăng canxi niệu, nhiễm canxi thận, rụng răng sữa sớm và bằng chứng X-quang về bệnh còi xương với giảm khoáng hóa xương rõ rệt; các dạng giảm phosphatase máu ở trẻ sơ sinh và trẻ nhỏ được di truyền như các rối loạn lặn trên nhiễm sắc thể thường; cho đến khi phát triển liệu pháp thay thế enzyme, phần lớn bệnh nhân bị giảm phosphatase máu chu sinh và 50% những người bị giảm phosphatase máu ở trẻ nhỏ đã chết vì suy hô hấp trước sinh nhật đầu tiên của họ (xem bên dưới);
- Bệnh nhân mắc dạng trẻ em của giảm phosphatase máu (OMIM 241510) không đồng nhất về mặt lâm sàng; họ khỏe mạnh khi còn là trẻ sơ sinh, nhưng ở và sau 6 tháng tuổi, các bất thường ở các mức độ nghiêm trọng khác nhau có thể trở nên rõ ràng bao gồm: rụng răng sữa sớm đơn độc, mềm sọ, nổi rõ các khớp sụn sườn, loe hành xương, và chân vòng kiềng chữ X và/hoặc chữ O. Về mặt X-quang, có thể có các dấu hiệu còi xương với loe hành xương, sự xâm nhập của các vùng thấu quang vào hành xương, biến dạng các chi, loãng xương và dính khớp sọ. Những bệnh nhân này có thể cải thiện tự phát trong tuổi dậy thì và hoặc có thể tiến triển thành dạng người lớn của bệnh này. Những bệnh nhân này đã đáp ứng tốt với liệu pháp thay thế enzyme (xem bên dưới);
- Dạng người lớn của giảm phosphatase máu (OMIM 146300) có thể không có triệu chứng lâm sàng và chỉ được xác định trong quá trình nghiên cứu một gia đình có con mắc dạng giảm phosphatase máu nặng hơn; cha mẹ có thể có tiền sử rụng răng sữa sớm, nhuyễn xương với tăng nhạy cảm với gãy xương tái phát chủ yếu ở xương bàn chân, gãy xương giả ở đầu gần xương đùi, vôi hóa sụn khớp, hoặc bệnh gút giả do lắng đọng các tinh thể canxi pyrophosphate dihydrate ở khớp; gãy xương giả dưới mấu chuyển xương đùi có thể được chứng minh bằng X-quang;
- Giảm phosphatase máu ở răng (bao gồm trong OMIM 146300) chỉ biểu hiện bằng rụng răng sữa sớm trước 5 tuổi do giảm khoáng hóa xi măng răng mà không có bất thường xương trên X-quang; viêm nha chu có thể phát triển; các dạng giảm phosphatase máu ở trẻ em và người lớn và giảm phosphatase máu ở răng thường (nhưng không phải luôn luôn) được di truyền như các rối loạn trội đơn alen, trên nhiễm sắc thể thường;
- Giảm phosphatase máu giả có kiểu hình và sinh hóa tương tự như giảm phosphatase máu cổ điển type 2 ở trẻ nhỏ nhưng hoạt độ phosphatase kiềm huyết thanh trong ống nghiệm là bình thường, cho thấy rằng trong rối loạn này, hoạt động enzyme đối với các cơ chất nhân tạo trong dung dịch pH cao được bảo tồn nhưng hoạt động phosphatase kiềm đối với các cơ chất nội sinh ở pH 7,4 là bất thường.
Ngoài (các) vị trí đột biến của ALPL, mức độ nghiêm trọng lâm sàng của giảm phosphatase máu có liên quan nghịch với độ tuổi mà bệnh xương biểu hiện rõ và với các yếu tố sinh học cá nhân (có thể là biểu sinh) ảnh hưởng đến sự biểu hiện của bệnh với sự thay đổi cá nhân rộng rãi ngay cả ở các thành viên trong gia đình có cùng (các) đột biến ALPL. Ở dạng giảm phosphatase máu chu sinh gây tử vong, các đột biến sai nghĩa, vô nghĩa, và vị trí nối donor và mất đoạn dịch khung trong ALPL tập trung ở các đoạn quan trọng của protein trong (p.Ala94Thr, p.Arg167Trp) hoặc gần (p.Gly103Arg, p.Gly317Asp) vị trí hoạt động xúc tác, chức năng enzyme của nó hoặc tại giao diện dimer (p.Ala23Val, p.Arg374Cys) hoặc chúng có thể làm suy giảm sự di chuyển của TNSALP đến vị trí xuyên màng hoạt động của nó. Các đột biến có thể làm gián đoạn sự liên kết của TNSALP với phối tử được phosphoryl hóa hoặc làm mất ổn định sự gắn kết của các đồng yếu tố cần thiết, chẳng hạn như kẽm hoặc magiê. Các đột biến ALPL liên quan đến dạng trẻ em của giảm phosphatase máu (p.Arg119His, p.Asp361Val) có xu hướng tập trung trên bề mặt ba chiều của phân tử enzyme tại các vị trí liên quan đến việc neo nó vào bề mặt tế bào hoặc sự hình thành các tetramer của nó; các biến thể như vậy có thể giữ lại hoạt tính sinh học còn lại đáng kể. Ở một số bệnh nhân bị giảm phosphatase máu mức độ trung bình, các đột biến dị hợp tử trong ALPL (p.Gly46Val, p.Arg167Trp, p.Asn461Ile) có tác dụng trội-âm đối với dimer TNSALP nguyên vẹn; các đột biến này đã được tập trung tại vị trí hoạt động của enzyme hoặc tại (các) miền liên quan đến sự dimer hóa, tetramer hóa, hoặc neo màng. Giảm phosphatase máu ở răng đã được liên kết với các đột biến dị hợp tử trong ALPL (p.Pro91Leu, p.Ala99Thr).
Sau khi xác định các phát hiện lâm sàng và X-quang của bệnh còi xương, chẩn đoán giảm phosphatase máu được gợi ý bởi sự hiện diện ở bệnh nhân của các giá trị phosphatase kiềm lưu hành thấp một cách nghịch lý (liên quan đến các tiêu chuẩn tuổi) và được xác nhận bằng cách xác định biến thể di truyền cá nhân của ALPL gây ra bệnh. Ở các dạng còi xương khác, các giá trị phosphatase kiềm tăng đáng kể (xem Bảng 20.9). Ngoài ra, có nồng độ pyrophosphate huyết thanh tăng (đối chứng < 200 μmol/g creatinin) và pyridoxal-5′-phosphate (~ 5000 nM) và bài tiết phosphoethanolamine qua nước tiểu tăng (đối chứng < 15 tuổi 83–222 μmol/g creatinin). Nồng độ canxi và phốt phát trong huyết thanh thường tăng ở những bệnh nhân mắc các dạng chu sinh và trẻ nhỏ của bệnh, vì sự hấp thu canxi ở ruột và tái hấp thu phốt phát ở ống thận là bình thường trong khi tốc độ hình thành xương bị giảm; các giá trị canxi thường bình thường ở các dạng trẻ em và người lớn của giảm phosphatase máu. Nồng độ PTH huyết thanh bình thường hoặc thấp; không có bằng chứng về cường cận giáp thứ phát. Mặc dù hoạt độ phosphatase kiềm huyết thanh trong ống nghiệm là bình thường ở bệnh nhân bị giảm phosphatase máu giả, nó không đủ chức năng trong cơ thể—có lẽ vì protein TNSALP bị cô lập trong tế bào do một khiếm khuyết trong việc vận chuyển nó đến bề mặt tế bào hoặc vì protein hoạt động với cơ chất nhân tạo nhưng không hoạt động với các cơ chất tự nhiên ở pH sinh lý. Các rối loạn khác liên quan đến giảm phosphatase máu bao gồm hội chứng sữa-kiềm, suy giáp, tăng cortisol máu, ngộ độc vitamin D, thiếu vitamin C, đói, thiếu magiê, kẽm hoặc đồng, bệnh celiac, thiếu máu nặng và truyền máu ồ ạt. Chẩn đoán trước sinh của giảm phosphatase máu có thể được xem xét ở một thai nhi mà siêu âm đã chứng minh xương sườn và xương dài ngắn, lõm hình chén ở hành xương, gai xương, hóa xương loang lổ, và mất khoáng của hộp sọ và cột sống ngực và được chứng thực bằng phân tích DNA của thai nhi có trong tuần hoàn của mẹ. Cần được đưa vào chẩn đoán phân biệt của một siêu âm thai nhi với những phát hiện này là loạn sản sụn và bệnh xương thủy tinh type II.
Điều trị giảm phosphatase máu phụ thuộc vào mức độ nghiêm trọng của bệnh. Các biện pháp hỗ trợ là phù hợp, nhưng việc dùng các tác nhân bisphosphonate thường bị chống chỉ định. Nên tránh dùng ngay cả liều nhỏ vitamin D hoặc các chất chuyển hóa của nó vì những bệnh nhân này dễ bị tăng canxi máu. Tăng canxi máu ở trẻ sơ sinh bị giảm phosphatase máu đáp ứng với bisphosphonate và tạm thời với calcitonin. Co giật có thể đáp ứng với việc dùng pyridoxine. Các dạng giảm phosphatase máu ở người lớn nhẹ hơn có thể đáp ứng với teriparatide (rhPTH 1-34). TNSALP tái tổ hợp của người, nhắm mục tiêu vào khoáng chất (alfotase alfa) đã được sử dụng thành công để điều trị các dạng giảm phosphatase máu chu sinh, trẻ nhỏ và trẻ em. Alfatase alpha là một protein tái tổ hợp sinh tổng hợp của người bao gồm hai chuỗi axit amin giống hệt nhau được liên kết bởi hai cầu nối disulfide chứa ectodomain xúc tác của TNSALP được hợp nhất với miền Fc IgG1 của người và một mô-típ deca-aspartate cuối cùng nhắm mục tiêu vào xương. Nó đã được chứng minh là cực kỳ hiệu quả trong điều trị bệnh nhân mắc các dạng giảm phosphatase máu chu sinh và trẻ nhỏ trong tối đa 7 năm. Việc dùng hợp chất này cho 11 bệnh nhân như vậy đã được theo sau bởi sự lành bệnh còi xương trên X-quang sau 24 tuần điều trị; ngoài ra, sự phát triển vận động và chức năng phổi được cải thiện. Không có tác dụng phụ nghiêm trọng nào được ghi nhận trong nghiên cứu này. Tác nhân này đã được chứng minh là có hiệu quả và đã được phê duyệt để điều trị trẻ em, thanh thiếu niên và người lớn bị giảm phosphatase máu, cũng như trẻ sơ sinh. Một hệ thống tính điểm đã được phát triển để đánh giá những thay đổi theo thời gian trong các bất thường xương của bệnh nhân bị giảm phosphatase máu được điều trị bằng alfatase alpha.
Tăng phosphatase máu có thể là một biến thể sinh lý bình thường hoặc một dấu hiệu sinh hóa của bệnh. Hoạt độ phosphatase kiềm đặc hiệu cho xương trong huyết thanh thường tăng trong tuổi dậy thì và để đáp ứng với các chấn thương xương và phản ánh hoạt động của nguyên bào xương tăng lên. Sự gia tăng thoáng qua hoạt độ phosphatase kiềm trong huyết thanh cũng có thể được quan sát thấy ở trẻ em bị nhiễm virus (bao gồm cả virus gây suy giảm miễn dịch ở người), sau khi ghép gan hoặc thận, hoặc trong quá trình điều trị bệnh bạch cầu/lymphoma. Tăng phosphatase máu cũng hiện diện ở bệnh nhân mắc bệnh Paget vị thành niên/bệnh biến dạng vỏ xương tăng sinh ở tuổi vị thành niên (OMIM 239000), bệnh tiêu xương lan rộng có tính gia đình (OMIM 174810), và bệnh tăng sinh vỏ xương xơ cứng toàn thể/bệnh van Buchem (OMIM 239100). Tăng phosphatase máu thoáng qua lành tính ở trẻ sơ sinh và đầu thời thơ ấu được gặp ở trẻ sơ sinh và trẻ em khỏe mạnh về mặt lâm sàng dưới 5 tuổi (tuổi trung bình 16 tháng); hoạt độ huyết thanh của cả hai đồng dạng phosphatase kiềm của xương và gan đều tăng (mặc dù không có bệnh gan hoặc xương); đó là một quá trình tự giới hạn với các giá trị phosphatase kiềm trở lại bình thường trong vài tháng. Tăng phosphatase máu thoáng qua (giá trị phosphatase kiềm > 1000 U/L) đã được báo cáo xảy ra ở khoảng 2% đến 3% trẻ em dưới 2 tuổi. Người ta đã gợi ý rằng tăng phosphatase máu lành tính và thoáng qua là hậu quả của việc sialyl hóa quá mức của phosphatase kiềm làm chậm quá trình phân hủy và kéo dài thời gian bán hủy của nó, nhưng lý do cho hàm lượng axit sialic quá mức của phân tử là không chắc chắn. Hiếm khi, tăng phosphatase máu có thể có tính gia đình và được di truyền như một tính trạng trội trên nhiễm sắc thể thường lành tính. Đôi khi, một thanh thiếu niên bị tăng phosphatase máu dai dẳng, không có tính gia đình, lành tính có thể được gặp. Tăng phosphatase máu kết hợp với chậm phát triển trí tuệ là một rối loạn được đặc trưng bởi chậm phát triển, co giật, giảm trương lực cơ và tăng phosphatase máu có nguồn gốc di truyền đa dạng (OMIM 239300). Dạng type 1 (trong số 6) của rối loạn này là do các đột biến mất chức năng hai alen trong protein V thuộc lớp sinh tổng hợp neo glycan phosphatidylinositol (PIGV, OMIM 610274), một glycolipid gắn các protein bao gồm phosphatase kiềm vào bề mặt của một tế bào.
Bệnh thận mạn tính – Rối loạn Khoáng chất và Xương
Bệnh thận mạn tính – rối loạn khoáng chất và xương (CKD-MBD) là thuật ngữ được sử dụng để mô tả các sai lệch trong chuyển hóa canxi và phốt phát, tổng hợp và bài tiết PTH và vitamin D, cân bằng nội môi xương, và vôi hóa lạc chỗ ở mạch máu, ngoài mạch máu và tim gặp ở bệnh nhân bị suy giảm chức năng thận. CKD-MBD thay thế cho tên gọi loạn dưỡng xương do thận chỉ liên quan đến những thay đổi bệnh lý có trong xương của bệnh nhân suy thận mạn. Các hậu quả chính về xương của bệnh thận mạn tính liên quan đến sự đề kháng của xương đối với các tác động của PTH, tổng hợp calcidiol và calcitriol dưới mức bình thường, và các tác động bất lợi của nồng độ FGF23, sclerostin và protein 1 liên quan đến Dickkopf (DKK1) tăng cao đối với sự hình thành xương. Bệnh xương do suy thận mạn dẫn đến giảm tăng trưởng, khoáng hóa và sức mạnh của xương, và tăng nguy cơ gãy xương. Nó có thể được đặc trưng là tăng động—liên quan đến chu chuyển xương cao và viêm xương xơ do cường cận giáp thứ phát, giảm động hoặc chu chuyển thấp, hoặc bất động—được đặc trưng về mặt mô học bởi ít xơ hóa, số lượng tế bào dưới mức bình thường, tốc độ chu chuyển xương và thể tích xương, nhưng nghịch lý là khoáng hóa xương và giá trị PTH bình thường liên quan đến liệu pháp tích cực với các tác nhân chống tái hấp thu canxi (ví dụ, bisphosphonate hoặc kháng thể đối với RANKL, hoặc các tác nhân tăng đồng hóa [ví dụ, rhPTH 1-34, kháng thể đối với sclerostin]), hoặc phổ biến nhất là một hỗn hợp của các dạng chu chuyển cao và thấp, được gọi là loạn dưỡng xương do thận hỗn hợp.
Khi chức năng thận suy giảm, việc sản xuất cả FGF23 và PTH đều tăng lên. Sự bài tiết FGF23 tăng tương đối sớm ở bệnh nhân CKD khi tình trạng giữ phốt phát lần đầu tiên xảy ra—ngay cả khi mức lọc cầu thận vượt quá 80 mL/phút/1,73 m2. Do đó, ở bệnh nhân CKD-MBD, nồng độ FGF23 trong huyết thanh tăng trước khi chức năng thận suy giảm và trước khi nồng độ phốt phát huyết thanh tăng, bài tiết phốt phát qua nước tiểu tăng—ban đầu duy trì các giá trị phốt phát huyết thanh bình thường. Việc tăng sản xuất FGF23 khi suy thận tiến triển góp phần làm giảm tổng hợp calcitriol ở ống thận và giảm hấp thu canxi ở ruột. Khi mức lọc cầu thận suy giảm dần, bài tiết phốt phát qua nước tiểu bị suy giảm, dẫn đến sự tích tụ nội bào và ngoại bào của nó, tăng phốt phát máu (khi mức lọc cầu thận giảm < 20 mL/phút/1,73 m2), và hạ canxi máu nhẹ. Sự bài tiết PTH tăng lên khi mức lọc cầu thận giảm xuống các giá trị nhỏ hơn 70 mL/phút/1,73 m2. Nồng độ phốt phát trong huyết thanh tăng dẫn đến tăng phốt phát máu (khi mức lọc cầu thận giảm < 20 mL/phút/1,73 m2), nồng độ canxi giảm (do cả giảm hấp thu cation này ở ruột do thiếu cả calcidiol và calcitriol, và nhu cầu duy trì tích số canxi x phốt phát không đổi), và nồng độ FGF23 tăng có tác dụng kích thích trực tiếp lên sự biểu hiện của PTH dẫn đến tăng sản xuất PTH thứ phát và khi không được kiểm soát sẽ dẫn đến cường cận giáp tam phát. Các yếu tố khác góp phần vào sự phát triển của tăng sản tế bào chính tuyến cận giáp và cường cận giáp thứ phát trong CKD-MBD bao gồm sự điều hòa giảm biểu hiện của CASR trong các tuyến cận giáp urê huyết (dẫn đến tăng nồng độ ngưỡng mà tại đó canxi làm giảm biểu hiện PTH) và sự không nhạy cảm của xương đối với PTH. Phốt phát cũng có thể làm chậm tốc độ phân hủy mRNA của PTH trong tuyến cận giáp và có tác dụng tăng cường trực tiếp lên sự phát triển của các tuyến cận giáp. Bởi vì calcitriol ức chế sự phát triển và chức năng của tuyến cận giáp, sự giảm tổng hợp của nó cũng dẫn đến tăng sinh của các tế bào chính tuyến cận giáp và tổng hợp PTH. Khi có sự bài tiết PTH tăng cao và các cytokine khác nhau (IL-1,-6,-11, TNF, yếu tố kích thích dòng đại thực bào [M-CSF]), sự hình thành hủy cốt bào và tốc độ tái hấp thu và hình thành xương tăng lên. Nhiễm toan góp phần vào sự hòa tan pha khoáng của xương một cách trực tiếp và bằng cách làm suy giảm sự ức chế tạo hủy cốt bào qua trung gian osteoprotegerin. Vôi hóa mạch máu và mô mềm cũng xảy ra ở các đối tượng bị suy thận mạn.
Bệnh thận mạn tính và bệnh xương chuyển hóa liên quan ở trẻ em có thể không có triệu chứng lâm sàng ngoại trừ sự thất bại trong tăng trưởng tuyến tính; khi bệnh tiến triển, các dị tật của các chi, trượt sụn tiếp hợp, gãy xương, đau xương và khớp, yếu cơ và mệt mỏi phát triển. Ở những bệnh nhân có tốc độ hình thành xương giảm và thể tích xương không được khoáng hóa (tức là nhuyễn xương) tăng, quá trình này là do sự tích tụ nhôm ở mặt trận khoáng hóa; với việc ngừng sử dụng các chất kết dính phốt phát chứa nhôm, sự phát triển của nhuyễn xương trong suy thận hiện nay là không phổ biến. Khi không có nhuyễn xương, bệnh xương bất động trong suy thận mạn là kết quả của việc giảm sản xuất PTH do kiểm soát tốt hơn nồng độ phốt phát huyết thanh, tăng dự trữ canxi, nồng độ PTH 7-84 và các mảnh đầu carboxyl khác của PTH ức chế tái hấp thu xương cao hơn, và các yếu tố khác ảnh hưởng đến đáp ứng của mô đối với PTH. Các biến chứng tim mạch (tăng huyết áp, vôi hóa mạch máu lạc chỗ, bệnh mạch máu do urê huyết, bệnh cơ tim) phát triển ở trẻ em bị CKD-MBD bắt đầu trước khi cần và tiến triển trong quá trình lọc máu và là nguyên nhân tử vong chính ở trẻ em và thanh thiếu niên bị CKD. Tăng phốt phát máu, cường cận giáp thứ phát và nồng độ FGF23 tăng cao ảnh hưởng xấu đến hệ tim mạch. Trẻ em bị bệnh thận đa nang di truyền trội trên nhiễm sắc thể thường có thể bị hạ phốt phát máu với giảm tái hấp thu phốt phát đã lọc ở ống thận có lẽ liên quan đến sự đề kháng với hoạt động sinh học của FGF23.
Về mặt sinh hóa, CKD-MBD được đánh dấu chủ yếu bởi tăng phốt phát máu, nồng độ canxi huyết thanh thấp-bình thường, và nồng độ PTH và FGF23 huyết thanh và hoạt độ TNSALP tăng. Nồng độ các dấu hiệu chu chuyển xương—phosphatase kiềm xương, phosphatase axit kháng tartrate (TRAP) 5b, và FGF23 đầu carboxyl—tăng, trong khi nồng độ sclerostin giảm ở trẻ em bị CKD. Các dấu hiệu X-quang của bệnh còi xương, khối lượng xương thấp và gãy xương giả thường hiện diện ở trẻ em bị loạn dưỡng xương do thận. Sau khi đánh giá lâm sàng, việc đánh giá bệnh xương ở trẻ em bị bệnh thận mạn tính bao gồm kiểm tra X-quang xương và định lượng BMC và BMD bằng DXA và/hoặc chụp cắt lớp vi tính định lượng ngoại vi (pQCT) và có thể yêu cầu sinh thiết mào chậu sau đó là phân tích hình thái mô học của mô. Về mặt điều trị, mục tiêu điều trị cho một đứa trẻ bị suy thận mạn là giảm thiểu các tác động toàn thân bất lợi của CKD-MBD; duy trì các giá trị canxi, phốt phát và phosphatase kiềm trong huyết thanh (gần) bình thường; và ngăn chặn sự phát triển hoặc tiến triển của cường cận giáp thứ phát. Điều trị bệnh xương do thận phải được cá nhân hóa; nó bao gồm lượng canxi trong chế độ ăn phù hợp, bổ sung vitamin D hoặc calcidiol và calcitriol, giảm lượng phốt phát trong chế độ ăn và dùng các tác nhân liên kết phốt phát ở ruột và ngăn chặn sự hấp thu của nó ở ruột, thuốc tăng bài tiết phốt phát, và đôi khi cắt bỏ tuyến cận giáp bán phần. Calcitriol làm giảm tốc độ hình thành xương ở bệnh nhân suy thận mạn bằng cách ức chế sự biệt hóa/chức năng của nguyên bào xương, giảm tổng hợp PTH, thay đổi sự phân hủy của PTH trong các tuyến cận giáp, và giảm biểu hiện của PTH1R. Các chất kết dính phốt phát đường uống chứa canxi cũng có thể hữu ích. Dịch lọc máu phải được chuẩn bị bằng nước không chứa nhôm. Khi được chỉ định, sự ức chế bài tiết PTH có thể được thực hiện thêm bằng cách sử dụng các chất tương tự calcitriol, chẳng hạn như paricalcitol hoặc một phối tử tổng hợp của CaSR—cinacalcet hydrochloride. Về mặt thực nghiệm, mặc dù việc trung hòa miễn dịch FGF23 trong một mô hình chuột của CKD-MBD đã dẫn đến giảm bài tiết PTH, tăng nồng độ calcitriol và canxi, và cải thiện sự hình thành xương, đã có sự gia tăng vôi hóa động mạch chủ và tỷ lệ tử vong ở những con vật được điều trị bằng kháng-FGF23. Do đó, việc ức chế bài tiết FGF23 ở bệnh nhân CKD có thể có những hậu quả không mong muốn một cách nghịch lý. Ngay cả sau khi ghép thận thành công, cường cận giáp thứ phát có thể tồn tại trong nhiều tháng hoặc nhiều năm—mức độ và cường độ của nó phản ánh mức độ nghiêm trọng và thời gian suy thận mạn trước khi ghép thận, sự phát triển của tăng sản nốt hoặc đơn dòng của các tuyến cận giáp, và tuổi thọ 20 năm của tế bào chính tuyến cận giáp. Năm năm sau khi ghép thận trong thời thơ ấu, nồng độ PICP, osteocalcin và ICTP trong huyết thanh vẫn tăng đáng kể cho thấy tốc độ chu chuyển xương nhanh, trong khi BMD theo diện tích và thể tích ở một phần ba xa của xương quay không thuận tay là bình thường theo chiều cao nhưng dưới mức bình thường theo tuổi. Tăng canxi máu và cường cận giáp thứ phát hoặc tam phát dai dẳng cần cắt bỏ tuyến cận giáp có thể trở nên rõ ràng ở trẻ em sau khi ghép thận. Các tác động gây loãng xương của glucocorticoid và các tác nhân ức chế miễn dịch, chẳng hạn như cyclosporin, cũng được quan sát thấy ở bệnh nhân sau ghép thận.
Các rối loạn Khoáng hóa Xương
Sự khoáng hóa xương có thể bị suy giảm hoặc tăng cường do rối loạn chức năng của nguyên bào xương hoặc hủy cốt bào. Các bất thường của chức năng nguyên bào xương dẫn đến suy giảm (ví dụ, loãng xương, bệnh xương thủy tinh) hoặc tăng cường khoáng hóa xương (ví dụ, xơ cứng xương, bệnh xương hóa đá). Do đó, ở những bệnh nhân mắc rối loạn xơ cứng xương, bệnh tăng sinh xương vỏ toàn thể/bệnh van Buchem type 1 (OMIM 239100), sự biểu hiện giảm của SOST mã hóa sclerostin (OMIM 605740, chr. 17q21.31), một chất ức chế tín hiệu WNT bằng cách tương tác với các đồng thụ thể WNT là các protein liên quan đến thụ thể lipoprotein mật độ thấp (LRP) 5 và 6, có liên quan đến chức năng nguyên bào xương tăng cường và khoáng hóa xương tăng lên. Mặc dù có nguồn gốc từ một tế bào gốc tạo máu chung, có một số phân nhóm hủy cốt bào với các hồ sơ sinh lý độc đáo phản ánh: vị trí chúng hoạt động (xương nội màng hoặc nội sụn, sụn); quá trình tái tạo mà chúng tham gia (có mục tiêu—đến các vị trí thay thế xương, ngẫu nhiên—qua trung gian hormone [PTH, calcitonin, calcitriol] để duy trì tình trạng canxi máu bình thường); loại xương (vỏ, bè) mà chúng tác động; và thời gian trong ngày (biến thiên ngày đêm) mà hủy cốt bào hoạt động mạnh nhất. Sự hình thành hoặc chức năng của hủy cốt bào giảm dẫn đến khối lượng xương tăng và các dạng bệnh xương hóa đá khác nhau, trong khi hoạt động của hủy cốt bào tăng dẫn đến khối lượng xương giảm do tiêu xương. Trong một số rối loạn xương, các vùng xương có cả khoáng hóa tăng và giảm có thể cùng tồn tại. Bệnh Paget vị thành niên/bệnh biến dạng vỏ xương tăng sinh ở tuổi vị thành niên (OMIM 239000) là một rối loạn tăng phosphatase máu thường bắt đầu ở đầu thời thơ ấu và được đặc trưng về mặt lâm sàng bởi các chi mở rộng và cong, gãy xương dài không do chấn thương, gù lưng, đầu to và yếu cơ; rối loạn này có thể tiến triển đến mức phải ngồi xe lăn. Nồng độ phosphatase kiềm trong huyết thanh tăng rõ rệt, một phản ứng đi kèm với hoạt động của hủy cốt bào tăng lên. Các bất thường xương trên X-quang bao gồm dày vỏ xương, cả xơ cứng xương và loãng xương, các bè xương thô và các dị tật xương tiến triển. Rối loạn này là do các đột biến mất chức năng hai alen của TNFRSF11B (OMIM 602643) mã hóa osteoprotegerin, một thành viên của siêu họ thụ thể TNF có chức năng như một thụ thể mồi nhử và do đó là một chất ức chế RANKL (TNFSF11, OMIM 602642), yếu tố tạo hủy cốt bào có nguồn gốc từ tế bào đệm-nguyên bào xương. Thông thường, osteoprotegerin ức chế sự hình thành hủy cốt bào; do đó, các đột biến bất hoạt của TNFRSF11B có liên quan đến sự hình thành hủy cốt bào và hoạt động của hủy cốt bào tăng cường. Mức độ nghiêm trọng của bệnh Paget vị thành niên phụ thuộc vào vị trí đột biến trong TNFRSF11B—những đột biến dẫn đến mất toàn bộ gen hoặc những đột biến trong miền liên kết phối tử liên quan đến mất các gốc cysteine dẫn đến bệnh lâm sàng rõ rệt. Điều trị bằng osteoprotegerin tái tổ hợp đã dẫn đến cải thiện về lâm sàng và X-quang cũng như việc dùng bisphosphonate, nhưng các tác nhân sau có liên quan đến nguy cơ hạ canxi máu đáng kể. Bệnh tiêu xương lan rộng có tính gia đình (OMIM 174810) và bệnh Paget xương khởi phát sớm có tính gia đình (OMIM 602080) là các rối loạn đơn alen, trội trên nhiễm sắc thể thường có sinh lý bệnh tương tự như bệnh Paget vị thành niên mặc dù khác biệt về lâm sàng và nguyên nhân. Hai rối loạn này là do các đột biến tăng chức năng đơn alen của gen (TNFRSF11A, OMIM 603499) mã hóa RANK dẫn đến sự gia tăng tín hiệu NF-κB, hậu quả là sự hình thành hủy cốt bào tăng cường. Ở thanh thiếu niên bị bệnh tiêu xương lan rộng có tính gia đình, các vùng tiêu điểm của chu chuyển xương tăng ở bộ xương phụ xuất hiện trong thập kỷ thứ hai của cuộc đời, sau đó là sự mở rộng tủy, gãy xương bệnh lý và các dị tật xương; điếc và mất răng sớm có thể xảy ra ở những đối tượng này. Bệnh nhân bị bệnh tiêu xương lan rộng có tính gia đình hoặc bệnh Paget xương khởi phát sớm cho thấy các vùng tiêu điểm của xương tiêu và tăng sinh xương, mất răng sớm và điếc. Hoạt động tăng của RANK bị đột biến được cho là do sự nhân đôi chèn dị hợp tử của 18 hoặc 27 cặp bazơ trong vùng peptide tín hiệu của exon 1 của TNFRSF11A. Tuy nhiên, vì các dạng đột biến của RANK bị kẹt trong lưới nội chất và không thể tương tác trực tiếp với RANKL, (các) cơ chế mà qua đó các đột biến dị hợp tử này làm tăng sự hình thành hủy cốt bào qua trung gian NF-κB là không chắc chắn. Người ta đã giả thuyết rằng RANK bị đột biến có thể tương tác với RANK nguyên vẹn còn lại và hoặc làm chậm tốc độ phân hủy của nó hoặc kéo dài sự tương tác của nó với RANKL. Ngược lại, các đột biến mất chức năng của TNFRSF11A có liên quan đến bệnh xương hóa đá loại 7 nghèo hủy cốt bào, di truyền lặn trên nhiễm sắc thể thường và giảm gammaglobulin máu (xem bên dưới). Các đột biến bất hoạt trong TNFSF11 mã hóa RANKL có liên quan đến bệnh xương hóa đá loại 2 nghèo hủy cốt bào (xem bên dưới).
Khối lượng xương thấp
Khối lượng xương thấp hoặc loãng xương thường liên quan đến tăng nguy cơ gãy xương, mặc dù trong một số rối loạn có khối lượng xương cao, tỷ lệ gãy xương tăng lên do vi kiến trúc xương bất thường dẫn đến giảm sức mạnh của xương (ví dụ, bệnh xương hóa đá, bệnh pycnodysostosis—xem bên dưới). Khoảng 50% bé trai và 40% bé gái sẽ bị gãy xương do chấn thương trong thời thơ ấu hoặc thanh thiếu niên (tỷ lệ mắc cao nhất từ 11 đến 12 tuổi ở bé gái và 13 đến 14 tuổi ở bé trai), thường xuyên nhất là ở đầu dưới xương quay do sự suy giảm tạm thời sức mạnh vỏ xương ở vị trí này. Điều quan trọng là phải phân biệt gãy xương do chấn thương mạnh với gãy xương là kết quả của sự mong manh của xương tăng lên, chẳng hạn như gãy lún đốt sống, gãy xương do tác động thấp và gãy xương đùi “tự phát” do một rối loạn nguyên phát hoặc thứ phát của khoáng hóa xương (Bảng 20.10A, 20.10B, 20.10C). Ở người lớn, loãng xương và giảm xương (“xương xốp”) là các thuật ngữ chỉ các trạng thái giảm khối lượng và chất nền xương, vi kiến trúc xương bất thường và giảm sức mạnh xương làm tăng nguy cơ gãy xương; nhuyễn xương đề cập đến sự giảm pha khoáng của xương. Loãng xương ở người lớn (chủ yếu là phụ nữ sau mãn kinh) được Tổ chức Y tế Thế giới (WHO) định nghĩa là BMD theo diện tích tại một vị trí xương cụ thể thấp hơn hoặc bằng –2,5 độ lệch chuẩn so với giá trị đỉnh trung bình của người trưởng thành trẻ cùng giới (điểm T ≤ –2,5). Loãng xương sau mãn kinh thường là hậu quả của sự gia tăng tốc độ tái hấp thu xương so với tốc độ hình thành xương do thiếu hụt estrogen. Ở người lớn, giảm xương hiện diện khi BMD theo diện tích nằm trong khoảng từ –1,1 đến –2,4 độ lệch chuẩn dưới giá trị đỉnh trung bình của người trưởng thành trẻ cùng giới (điểm T –1,1 đến –2,4), và BMD theo diện tích bình thường là BMD không thấp hơn một độ lệch chuẩn so với hoặc cao hơn giá trị đỉnh trung bình của người trưởng thành trẻ (điểm T –1,0 đến +1,0). Khi BMD theo diện tích cao hơn +2 độ lệch chuẩn so với trung bình theo tuổi và giới (điểm T > +2), khối lượng xương được coi là cao. Bởi vì ở phụ nữ tiền mãn kinh, chỉ riêng BMD theo diện tích không phải là một công cụ đánh giá tốt nguy cơ gãy xương, Hiệp hội Đo mật độ xương lâm sàng Quốc tế (ISCD) khuyến nghị rằng thuật ngữ loãng xương nên được giới hạn cho những bệnh nhân có cả BMD theo diện tích thấp (được định nghĩa bằng điểm Z của BMD < –2,0, tức là, thấp hơn –2 độ lệch chuẩn so với các đối tượng đối chứng phù hợp theo tuổi và giới) và tiền sử gãy xương do tác động thấp (mong manh). Phụ nữ có BMD theo diện tích dưới –2 độ lệch chuẩn nhưng không có tiền sử gãy xương được chỉ định là có BMD theo diện tích “dưới phạm vi dự kiến cho tuổi;” việc sử dụng thuật ngữ giảm xương bị tránh ở quần thể này. Tổ chức Loãng xương Quốc tế (IOF) đã định nghĩa loãng xương ở người trưởng thành trẻ là điểm T của BMD theo diện tích ở đốt sống hoặc hông dưới –2,5 và có liên quan đến một rối loạn ảnh hưởng xấu đến khoáng hóa xương. Cả hai nhóm đều đồng ý rằng ở người trưởng thành trẻ, sự hiện diện của loãng xương không chỉ được định nghĩa bằng BMD thấp. Nhiều bệnh dẫn đến loãng xương ở người trưởng thành trẻ bắt đầu trong thời thơ ấu và thanh thiếu niên. Các tiêu chí do WHO và IOF chỉ định cho khối lượng xương thấp hoặc cao không áp dụng cho trẻ em và thanh thiếu niên, ở những đối tượng này, sự thay đổi về tuổi theo thời gian và tuổi xương, chiều cao, cân nặng và giai đoạn trưởng thành sinh dục, cũng như giới tính và dân tộc ảnh hưởng đến khoáng hóa xương. (Thuật ngữ giảm xương không được sử dụng khi thảo luận về khoáng hóa xương ở trẻ sơ sinh, trẻ em và thanh thiếu niên.)
Bảng 20.10A: Các rối loạn Khoáng hóa Xương: Khối lượng xương thấp
| I | Nguyên phát/Di truyền | |
| A | Bệnh xương thủy tinh (Types I– XIII—xem Bảng 20.10B) | |
| B | Hội chứng loãng xương-giả u thần kinh thị giác (LRP5) | |
| C | Loãng xương vị thành niên tự phát (LRP5) | |
| D | Hội chứng Marfan (FBN1) | |
| E | Hội chứng Ehlers-Danlos (COL1A1) | |
| F | Bệnh homocystin niệu (CBS) | |
| G | Loạn sản xơ (GNAS) | |
| H | Bệnh dự trữ glycogen type I (G6PC) | |
| I | Bệnh galactosemia (GALT) | |
| J | Hội chứng tóc xoắn Menkes (ATP7A) | |
| K | Giảm phosphatase máu (ALPL) | |
| L | Còi xương (các dạng di truyền—xem Bảng 20.8B) | |
| II | Thứ phát | |
| A | Dinh dưỡng dưới mức tối ưu | |
| 1 | ||
| 2 | ||
| 3 | ||
| 4 | ||
| 5 | ||
| 6 | ||
| 7 | ||
| B | Các bệnh nội tiết/chuyển hóa | |
| 1 | ||
| 2 | ||
| 3 | ||
| 4 | ||
| 5 | ||
| 6 | ||
| 7 | ||
| 8 | ||
| C | Không sử dụng/bất động | |
| 1 | ||
| 2 | ||
| 3 | ||
| 4 | ||
| 5 | ||
| 6 | ||
| D | Các bệnh viêm | |
| 1 | ||
| 2 | ||
| 3 | ||
| 4 | ||
| E | Thuốc | |
| 1 | ||
| 2 | ||
| 3 | ||
| 4 | ||
| F | Bệnh mạn tính | |
| 1 | ||
| 2 | ||
| 3 | ||
| 4 | ||
| 5 | ||
| 6 | ||
| 7 |
Biên soạn từ: Boyce AM, Gafni RI. Approach to the child with fractures. J Clin Endocrinol Metab. 2011;96:1943–1952; Ferrari S, Bianchi ML, Eisman JA, et al. Osteoporosis in young adults: pathophysiology, diagnosis, and management. Osteoporos Int. 2012;23:2735–2748; Rauch F, Bishop N. Juvenile osteoporosis. In: Rosen CJ (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 7th ed. Washington, DC: The American Society of Bone and Mineral Metabolism; 2008:264–267; Bachrach LK, Ward LM. Clinical review: bisphosphonate use in childhood oteoporosis. J Clin Endocrinol Metab. 2009;94:400–409.
Bảng 20.10B: Phân loại lâm sàng/kiểu hình của Bệnh xương thủy tinh (OI)
| Loại | Biểu hiện lâm sàng & xương | Di truyền | Các biến thể gen 1 |
|---|---|---|---|
| 1 | Nhẹ—tầm vóc gần như bình thường; xương sọ có các xương wormian, đốt sống lõm hai mặt & dẹt, đôi khi cong xương đùi, tăng khả năng vận động khớp, củng mạc xanh, răng giòn (bệnh tạo ngà không hoàn hảo), điếc | Trội trên NST thường | COL1A1, COL1A2 |
| 2 | Tử vong chu sinh, đa gãy xương, xương sọ có các xương wormian, thân đốt sống dẹt khắp cột sống, xương dài cong, cánh chậu và ổ cối dẹt | Trội hoặc lặn trên NST thường | COL1A1, COL1A2, CRTAP, LEPRE1, PPIB, CREB3L1 |
| 3 | Các dị tật và gãy xương dài tiến triển, tầm vóc thấp, xương sọ mềm, gãy lún đốt sống, vẹo cột sống, củng mạc xanh, điếc | Trội hoặc lặn trên NST thường | COL1A1, COL1A2, CRTAP, LEPRE1, PPIB, FKBP10, SERPINH1, SERPINF1, BMP, BMP1, WNT1, SP7, TENT5A, MBTPS2 |
| 4 | Xu hướng gãy và biến dạng xương dài thay đổi, tầm vóc thấp, củng mạc xanh nhạt, có thể có bệnh tạo ngà không hoàn hảo, điếc | Trội hoặc lặn trên NST thường | COL1A1, COL1A2, CRTAP, PPIB, WNT1, TMEM38B, SP7 |
| 5 | Tương tự OI type IV với sự hình thành callus quá mức tại các vị trí gãy xương đang lành & vôi hóa màng gian cốt | Trội trên NST thường | IFITM5 |
Vui lòng xem Bảng 20.10C để biết thêm chi tiết về các biến thể gen cụ thể liên quan đến bệnh xương thủy tinh.
Biên soạn từ: Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet. 2015;167A:2869–2892; Vierucci F, Saggese G, Cimaz R. Osteoporosis in childhood. Curr Opin Rheumatol. 2017;29(5):539–546.
Bảng 20.10C: Các biến thể/đột biến gen liên quan đến Bệnh xương thủy tinh
| Số OMIM | Bệnh—Gen | Nhiễm sắc thể/OMIM | Mức độ nghiêm trọng/ Loại | Đặc điểm lâm sàng | Suy giảm tăng trưởng | Củng mạc xanh | Di truyền | Khiếm khuyết chức năng |
|---|---|---|---|---|---|---|---|---|
| I- 166200 | COL1A1 – Collagen type I, alpha-1 | 17q21.33 1120150 hoặc COL1A2 – Collagen type I, alpha-2 | 7q22.3 120160 | Nhẹ – 1 | Ít gãy xương (thường là đốt sống), ít biến dạng, mất thính lực ở 50%; hiếm khi có bệnh tạo ngà không hoàn hảo | Tối thiểu | Hiện diện-mạnh | AD |
| II- 166210 | COL1A1 hoặc COL1A2 | Tử vong chu sinh- 2 bẩm sinh (xem thêm Types VII, VIII, IX, X) | Nhiều gãy xương sườn & xương dài trong tử cung & khi sinh, dị tật xương dài nghiêm trọng, xương sườn “hình chuỗi hạt”, vòm sọ không khoáng hóa | Nghiêm trọng | Hiện diện | AD, thể khảm của cha mẹ | Thay thế Glycine trong COL1A1 hoặc COL1A2 dẫn đến collagen type I có cấu trúc bất thường | |
| III- 259420 | COL1A1 hoặc COL1A2 | Nghiêm trọng, tiến triển & biến dạng– 3 | Cong vừa đến nặng, đa gãy xương dài & đốt sống, bệnh tạo ngà không hoàn hảo, mất thính lực | Nghiêm trọng | Hiện diện nhưng nhạt dần theo tuổi | AD | Thay thế Glycine trong COL1A1 hoặc COL1A2 dẫn đến collagen type I có cấu trúc bất thường | |
| IV-166220 | COL1A2 hoặc COL1A1 | Biến dạng vừa– 4 | Cong nhẹ đến vừa, gãy xương, bệnh tạo ngà không hoàn hảo | Vừa, thay đổi | Xám hoặc không có | AD | Thay thế Glycine trong COL1A1 hoặc COL1A2 dẫn đến collagen type I có cấu trúc bất thường | |
| V- 610967 | IFITM5 – Protein xuyên màng-5 do interferon gây ra | 11p15.5 614757 | Biến dạng vừa- 5 | Mong manh xương nhẹ đến vừa, hóa xương màng gian cốt của cẳng tay, hình thành callus tăng sản tại các vị trí gãy xương; kiểu phiến xương “giống lưới” | Nhẹ đến vừa | Không có | AD | Protein màng đặc hiệu của nguyên bào xương cần thiết cho sự biệt hóa của nguyên bào xương; hậu quả chức năng của đột biến hằng định (c.-14C > T) trong IFITM5 là không chắc chắn |
| VI- 613982 | SERPINF1 – Chất ức chế serpin peptidase, nhánh F, thành viên 1 | 17p13.3 172860 | Biến dạng vừa đến nặng- 3 | Khởi phát gãy xương dài & đốt sống ở trẻ sơ sinh, tăng hoạt độ phosphatase kiềm & chất dạng xương, kiểu phiến “vảy cá” | Vừa đến nặng | Xám | AR | Đột biến mất chức năng trong yếu tố có nguồn gốc từ biểu mô sắc tố (PEDF)—chất điều hòa khoáng hóa xương & biệt hóa hủy cốt bào |
| VII- 610682 a | CRTAP – Protein liên quan đến sụn | 3p22.3 605497 | Biến dạng vừa đến nặng hoặc tử vong- 2, 3, 4 | Gãy xương có thể có khi sinh, ngắn chi ở gốc, dị tật chi | Vừa | Không có hoặc mờ nhạt | AR | Đột biến bất hoạt (nhân đôi) của CRTAP làm suy giảm quá trình 3-hydroxyl hóa prolyl-986 của procollagen α1(I) |
| VIII- 610915 a | P3HI (LEPRE1) – Leucine- và proline-enriched proteoglycan 1 (Prolyl hydroxylase 1) | 1p34 610339 | Biến dạng nặng đến tử vong- 2, 3 | Khoáng hóa xương giảm rõ rệt, vẹo cột sống, đốt sống dẹt, hành xương nổi rõ, các đốt ngón tay dài | Nghiêm trọng | Không có | AR | Đột biến bất hoạt của LEPRE mã hóa prolyl-3-hydroxylase 1 (leprecan) làm suy giảm quá trình 3-hydroxyl hóa prolyl-986 của procollagen α1(I) |
| IX- 259440 a | PPIB – Cyclophilin B | 15q22.31 123841 | Tử vong đến biến dạng nặng, xem- 2, 3, 4 | Xương dài ngắn, cong, & gãy ở giữa thai kỳ | Nghiêm trọng | Xám | AR | Đột biến bất hoạt của PPIB mã hóa peptidyl-prolyl isomerase B làm suy giảm quá trình 3-hydroxyl hóa prolyl-986 của procollagen α1(I) |
| X- 613848 | SERPINH1 – Chất ức chế serpin peptidase, nhánh H, thành viên 1 | 11q13.5 600943 | Nghiêm trọng- 3 | Xương đùi ngắn cong trong tử cung, đa gãy xương trong tháng đầu đời, bệnh tạo ngà không hoàn hảo | Nghiêm trọng | Hiện diện | AR | Đột biến mất chức năng trong CBP2 (HSP47)—chaperone, cần thiết để duy trì tính toàn vẹn của cấu trúc xoắn ba của collagen type 1 & khả năng chống lại sự phân hủy proteolytic của nó |
| XI- 610968 | FKBP10 – Protein liên kết FK506 10 | 17q21.2 607063 | Dị tật nặng & tiến triển- 3 | Các chi ngắn & cong, co cứng khớp; bệnh tạo ngà không hoàn hảo; kiểu phiến xương vảy cá (xem Type VI) | Vừa | Xám | AR | Đột biến mất chức năng trong một protein chaperone—FKBP65—cần thiết cho quá trình xử lý sau dịch mã của procollagen type I; cũng bị đột biến trong hội chứng Bruck type 1 |
| XII- 613849 | SP7 – Yếu tố đặc hiệu phiên mã (Sp)7 | 12q13.13 606633 | Vừa đến nặng- 4 | Đa gãy xương ở đầu thời thơ ấu; xương dài ngắn, cong, vòm miệng cao | Nghiêm trọng | Không có | AR | Đột biến bất hoạt trong Osterix, một yếu tố phiên mã cần thiết cho sự biệt hóa của nguyên bào xương & khoáng hóa xương |
| XIII- 614856 | BMP1 – Protein hình thái xương 1 | 8p21.3 112264 | Vừa đến nặng- 3 | Dị tật toàn thể của tất cả các xương, đa gãy xương mặc dù mật độ khoáng xương tăng | Nghiêm trọng | Xanh nhạt | AR | Đột biến bất hoạt của endoproteinase propeptide đầu C của procollagen type I làm suy giảm sự hình thành collagen |
| XIV- 615066 | TMEM38B – Protein xuyên màng 38B | 9q31.1 611236 | Vừa- 3 | Mức độ nghiêm trọng thay đổi; gãy xương xảy ra trong tử cung hoặc trong thời thơ ấu; cong vừa các chi dưới | Vừa | Không có | AR | Đột biến bất hoạt của một thành phần của một kênh cation đơn hóa trị nội bào cần thiết để giải phóng Ca2+ từ các vị trí lưu trữ |
| XV- 615220 | WNT1 – Họ vị trí tích hợp không cánh, thành viên 1 | 12q13.12 164820 | Biến dạng tiến triển- 3 | Đa gãy xương & dị tật xương ở trẻ em; thiểu sản tiểu não, chậm phát triển sâu sắc | Hiện diện | Hiện diện | AR | WNT1 bị đột biến không kích hoạt được tín hiệu LRP5-beta catenin dẫn đến suy giảm sự hình thành & chức năng của nguyên bào xương |
| XVI- 616229 | CREB3L1 – Protein 3-like liên kết yếu tố đáp ứng AMP vòng | 11p11.2 616215 | Tử vong trước sinh- 2 | Gãy xương có trong tử cung | Hiện diện | AR | Mất kích hoạt phiên mã của COL1A1 | |
| XVII- 616597 | SPARC – Protein tiết ra, giàu cysteine axit | 5q33.1 182120 | Mức độ nghiêm trọng vừa- 4 | Gãy xương chi & đốt sống ở đầu thời thơ ấu, lỏng lẻo khớp, yếu cơ | Hiện diện | Không nêu rõ | AR | Mã hóa osteonectin—phosphoprotein được tiết bởi nguyên bào xương cho phép vôi hóa collagen xương |
| XVIII- 617592 | TENT5A – Terminal nucleotidyltransferase 5A | 611357 6q14.1 | Mức độ nghiêm trọng vừa- 3 | Cong bẩm sinh, đa gãy xương trước 1 tuổi, xẹp đốt sống, vẹo cột sống tiến triển, bệnh tạo ngà không hoàn hảo | Hiện diện | Hiện diện | AR | Nucleotidyltransferase xúc tác quá trình chuyển monophosphate nucleoside đến nhóm hydroxyl của phân tử nhận; được biểu hiện trong nguyên bào xương |
| XIX 301014 | MBTPS2 – Vị trí protease yếu tố phiên mã liên kết màng 2 | Xp22.12 300294 | Nghiêm trọng- 3 | Gãy xương xảy ra trước và sau sinh, cong xương dài, ngực ức gà | Vừa | Hiện diện | XLR | Metalloproteinase kẽm; điều hòa sự phân giải protein của phiên mã & các yếu tố khác cần thiết cho sự bài tiết & liên kết chéo của collagen type 1 |
| Không được chỉ định | ||||||||
| PLOD2 – Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 | 3q24 601865 | Nghiêm trọng- 3; mong manh xương với co cứng khớp bẩm sinh | Gãy xương ở trẻ sơ sinh, màng bơi, vẹo cột sống | Nghiêm trọng | Không có | AR | Mã hóa một procollagen lysyl hydroxylase cần thiết cho sự hình thành telopeptide collagen type 1; hội chứng Bruck 2 | |
| 300131 | PLS3 Plastin 3 | Xq23 300131 | Nhẹ- 1 | Giảm xương- phát hiện tình cờ | Tối thiểu | Không có | XLR | Protein liên kết actin & canxi cần thiết cho sự toàn vẹn của bộ xương tế bào xương |
| 176790 | P4HB Procollagen-proline, 2-oxoglutarate-4-dioxygenase, tiểu đơn vị beta | 17q25.3 | Nghiêm trọng- 3 | Hội chứng Cole-Carpenter 1- Mong manh xương, dính khớp sọ, não úng thủy, lồi mắt | Khiêm tốn | Hiện diện | AD | Tiểu đơn vị beta của prolyl 4-hydroxylase- xúc tác quá trình tổng hợp 4-hydroxyproline trong preprocollagen |
| 607186 | SEC24D SEC24-related gene family, member D | 4q26 | Nghiêm trọng- 3 | Hội chứng Cole-Carpenter 2 | Hiện diện | AR | Thành phần của các túi tạo điều kiện cho việc vận chuyển protein từ lưới nội chất | |
| 603506 | LRP5 Protein 5 liên quan đến thụ thể lipoprotein mật độ thấp | 11q13.2 | Nghiêm trọng- 3 | Hội chứng loãng xương-giả u thần kinh thị giác (259770)- mong manh xương, giả u thần kinh thị giác trong mắt, đục giác mạc, tăng nhãn áp | Nghiêm trọng | Đục giác mạc | AR | Đồng thụ thể với LRP6 & Frizzled cho WNT1; các biến thể bất hoạt làm suy giảm sự hình thành nguyên bào xương |
AD, Trội trên NST thường; AR, Lặn trên NST thường.
(Từ Marini JC. Osteogenesis imperfecta. In: Rosen CJ (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 7th ed. Washington, DC: The American Society of Bone and Mineral Metabolism; 2008:446–450; Barnes AM, Chang W, Morello R, et al. Deficiency of cartilage-associated protein in lethal osteogenesis imperfecta. N Engl J Med. 2006;355, 2757–2764; Bishop N. Characterising and treating osteogenesis imperfecta. Early Hum Dev. 2010;86:743–746; Rauch F, Lalic L, Roughley P, Glorieux FH. Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J Bone Miner Res. 2010;25:1367–1374.)
Ở trẻ em và thanh thiếu niên, loãng xương hoặc khối lượng xương thấp được coi là hiện diện khi trẻ/thanh thiếu niên đã bị: (1) một hoặc nhiều gãy lún đốt sống do chấn thương thấp trong trường hợp không có bệnh tại chỗ hoặc chấn thương mạnh hoặc (2) hai hoặc nhiều gãy xương dài do tác động thấp (ngã từ độ cao đứng hoặc thấp hơn) nếu dưới 10 tuổi hoặc ba hoặc nhiều gãy xương do tác động thấp trước 19 tuổi cùng với BMC/BMD toàn thân trừ (–) đầu và cột sống thắt lưng sau-trước được đánh giá bằng DXA thấp hơn (–) 2 độ lệch chuẩn (ví dụ, điểm Z–2) so với trung bình theo dân tộc, giới tính, tuổi theo thời gian và chiều cao/tuổi theo chiều cao; khi giải thích một nghiên cứu DXA, người ta cũng nên xem xét giai đoạn trưởng thành sinh dục so với tuổi theo thời gian của đối tượng. (Dữ liệu DXA toàn thân bao gồm cả đầu không được coi là một thước đo tối ưu của BMC/BMD vì hộp sọ không đáp ứng trực tiếp với các yếu tố môi trường, chẳng hạn như tập thể dục chịu trọng lượng.) Do đó, loãng xương ở trẻ em và thanh thiếu niên được gợi ý bởi biểu hiện lâm sàng (đau xương, ngừng tăng trưởng, bất động) do gãy xương dài không/do chấn thương thấp và/hoặc gãy lún đốt sống được xác định bằng X-quang xương dài và/hoặc các phát hiện trên phim X-quang nghiêng của cột sống ngực-thắt lưng. Điều cần thiết là phải phân biệt gãy lún đốt sống với các biến thể của hình thái đốt sống bình thường bằng cách sử dụng các phương pháp bán định lượng phù hợp. Ở trẻ em và thanh thiếu niên, khối lượng xương có thể được định lượng bằng cách xác định BMD theo diện tích của “toàn thân trừ đầu” và của các vùng cơ thể (cột sống thắt lưng, hông, một phần ba xa xương quay) bằng DXA. (Phân tích DXA khối lượng xương hông thay đổi ở trẻ em và thanh thiếu niên và thường không được sử dụng để giải thích dữ liệu BMC hoặc BMD.) Một đơn vị DXA di động đo BMD theo diện tích ở các vùng cực xa, giữa xa và một phần ba xa của cẳng tay không thuận cũng có thể được sử dụng để xác định BMD theo diện tích; nó cung cấp dữ liệu tương đương với một máy DXA cố định. Mặc dù có những hạn chế (cung cấp dữ liệu BMD theo diện tích thay vì thể tích và không phân biệt được giữa xương bè và xương vỏ), DXA là phương pháp đo mật độ xương được sử dụng rộng rãi nhất ở trẻ em tại thời điểm này. Các phương pháp khác được sử dụng để xác định khoáng hóa xương bao gồm QCT, pQCT của xương chày và xương quay, pQCT độ phân giải cao (HRpQCT), và QUS. Ước tính khối lượng xương bằng QCT cung cấp thông tin ba chiều định lượng về thể tích và khối lượng xương vỏ và xương bè. So với sức mạnh của xương được xác định bằng cách tính chỉ số ứng suất-biến dạng của một xương dài với dữ liệu thu được bằng pQCT, DXA BMC theo diện tích toàn thân (trừ đầu) theo chiều cao cũng cung cấp một phép đo đáng tin cậy để xác định sức mạnh của xương vỏ và do đó nguy cơ gãy xương. Báo cáo về BMD/hàm lượng nên bao gồm không chỉ phương pháp được sử dụng mà còn cả thiết bị cụ thể được sử dụng, phiên bản phần mềm được sử dụng để phân tích dữ liệu và sự pha trộn dân tộc của quần thể tham chiếu. Điều thú vị là, ở những bé trai trước tuổi dậy thì và tuổi dậy thì khỏe mạnh khác bị gãy xương do chấn thương mạnh, BMD vùng có xu hướng nằm trong nửa dưới của phạm vi bình thường; ngoài ra, vi kiến trúc xương có xu hướng bất thường và sức mạnh của xương giảm—có lẽ phản ánh một khiếm khuyết xương nội tại.
Nói chung, sức mạnh xương suy giảm có thể phát triển như là hậu quả của các rối loạn bẩm sinh hoặc mắc phải ảnh hưởng xấu đến sự hình thành hoặc khoáng hóa xương. Các rối loạn này có thể liên quan đến lượng dinh dưỡng hoặc hấp thu chất dinh dưỡng dưới mức tối ưu (chán ăn tâm thần, xơ nang, bệnh celiac), bệnh viêm mạn tính (ruột, khớp), bất động (liệt, loạn dưỡng cơ, bại não), các bất thường huyết học (bệnh bạch cầu và hóa trị liệu của nó, bệnh hồng cầu hình liềm, thalassemia), thuốc (glucocorticoid, thuốc ức chế miễn dịch, thuốc chống co giật), hoặc các bệnh nội tiết (thiếu hụt hormone tăng trưởng, tuyến giáp hoặc sinh dục hoặc bài tiết quá mức glucocorticoid). Ở những bệnh nhân bị dư thừa cortisol và các glucocorticoid liên quan nội sinh hoặc do điều trị, khối lượng và khoáng hóa xương giảm trong khi nguy cơ gãy xương tăng lên do tác dụng ức chế của các tác nhân này đối với sự hình thành xương và tăng tốc tái hấp thu xương với hậu quả là tăng sự mong manh của xương. Thiếu hụt GH có liên quan đến giảm tốc độ chu chuyển xương và khoáng hóa dưới mức bình thường của bộ xương, cũng như tăng nguy cơ gãy xương với sự cải thiện trong quá trình điều trị bằng GH tái tổ hợp của người.
Mặc dù các yếu tố di truyền chiếm 60% đến 80% khoáng hóa xương tối ưu, các yếu tố có thể thay đổi góp phần vào sự phát triển của giảm xương và loãng xương ở tuổi trưởng thành (tập thể dục chịu trọng lượng, dinh dưỡng, khối lượng cơ thể, môi trường nội tiết tố) có nguồn gốc từ trong tử cung, trẻ sơ sinh, thời thơ ấu và thanh thiếu niên. Ở trẻ em cũng như người lớn, khối lượng, thành phần, vi kiến trúc và kích thước của xương quyết định sức mạnh của xương. Bởi vì nhiều thanh thiếu niên tiêu thụ quá nhiều đồ uống có ga và nước trái cây pha loãng do đó hạn chế lượng sữa của họ, trẻ em và thanh thiếu niên chỉ ăn 55% đến 70% lượng canxi khuyến nghị hàng ngày (1300 mg/ngày), mặc dù nam giới cuối tuổi dậy thì có xu hướng tiêu thụ nhiều hơn nữ giới tuổi dậy thì. Ở thanh thiếu niên và phụ nữ trưởng thành, việc uống quá nhiều đồ uống cola có hàm lượng axit photphoric cao làm giảm hàm lượng canxi trong cơ thể bằng cách cô lập canxi trong chế độ ăn ở đường ruột và tăng sự hòa tan khoáng chất xương để trung hòa axit với hậu quả có thể là sự phát triển của cường cận giáp thứ phát nhẹ. Các hoạt động tĩnh tại, không chịu trọng lượng được khuyến khích bởi truyền hình, video và trò chơi máy tính cũng làm suy giảm khoáng hóa xương. Trong một nghiên cứu trên 300 thanh thiếu niên nam và nữ và người trưởng thành trẻ (13–21 tuổi) sử dụng cả đánh giá DXA về thành phần cơ thể và đo QCT của bộ xương trục và phụ, một mối tương quan thuận giữa khối lượng nạc và tất cả các phép đo xương đã được báo cáo ở cả hai giới. Ở trẻ em và thanh thiếu niên, khối lượng mỡ, chu vi vòng eo và kháng insulin có liên quan tiêu cực đến DXA-BMC của cột sống thắt lưng và BMD toàn thân. Những quan sát này cho thấy khối lượng và sức mạnh của xương được quyết định bởi các lực cơ động—không chỉ bởi tải trọng tĩnh. Tỷ lệ chênh cho gãy xương bàn chân, mắt cá chân, chân và đầu gối tăng lên khi chỉ số khối cơ thể tăng đặc biệt ở trẻ em từ 6 đến 11 tuổi.
Thiếu hụt dinh dưỡng làm giảm tốc độ tích lũy xương; giảm cân cực độ, chẳng hạn như ở bệnh nhân chán ăn tâm thần có nhiều tác động bất lợi lên khối lượng xương, bao gồm giảm BMD theo diện tích được đánh giá bằng DXA, và BMD thể tích toàn phần và bè ở đầu dưới xương quay thấp, giảm độ dày và diện tích vỏ xương, và độ xốp vỏ cao hơn được xác định bằng HRpQCT dẫn đến giảm sức mạnh của xương và tăng nguy cơ gãy xương. Chán ăn tâm thần là một rối loạn tâm thần trong đó suy dinh dưỡng mạn tính là do tự gây ra vì một rối loạn cảm xúc tiềm ẩn. Nó xảy ra thường xuyên hơn ở nữ giới so với nam giới và thường bắt đầu ở tuổi vị thành niên (tuổi khởi phát cao nhất của chán ăn tâm thần là từ 13 đến 18 tuổi) và có thể tồn tại đến tuổi trưởng thành. Nguy cơ gãy xương ở những bệnh nhân này tăng đáng kể do không tích lũy đủ khối lượng xương vì độ dày và diện tích vỏ xương, số lượng và độ dày bè xương, tốc độ chu chuyển và sức mạnh của xương giảm so với các đối tượng bình thường. Về mặt sinh lý bệnh, khối lượng xương thấp và sức mạnh xương giảm là hậu quả không chỉ của sự khan hiếm các thành phần protein và khoáng chất của xương và nhiễm toan mạn tính, mà còn của sự thích ứng nội tiết tố với tình trạng đói. Do đó, sự đề kháng với các tác dụng sinh học của GH biểu hiện bằng giảm tổng hợp IGF-1 và đề kháng với các tác dụng kích thích sự thèm ăn của ghrelin, một chất kích thích tiết GH, tăng sản xuất glucocorticoid liên quan đến căng thẳng dẫn đến tăng cortisol máu, và giảm tổng hợp estrogen và androgen (cả testosterone và dehydroepiandrosterone) do giảm gonadotropin hiệp đồng để cản trở quá trình tạo xương và sức mạnh của xương, do đó làm tăng nguy cơ gãy xương. Bệnh nhân chán ăn tâm thần cũng bị giảm leptin máu do khan hiếm mô mỡ. Leptin là một adipokine làm giảm sự thèm ăn, tăng cường bài tiết gonadotropin, và có thể ảnh hưởng đến khối lượng xương vì nó trực tiếp làm tăng sự hình thành sụn và nguyên bào xương và ức chế sự hình thành hủy cốt bào. Việc dùng estrogen/progestin cho nữ giới bị chán ăn tâm thần không làm tăng khối lượng xương hoặc ngăn chặn sự mất mát của nó; bisphosphonate, IGF-1 và dehydroepiandrosterone có thể duy trì hoặc tăng BMD ở những bệnh nhân như vậy nhưng việc sử dụng chúng nên được hạn chế. Ngay cả ở thanh thiếu niên bị chán ăn tâm thần mà rối loạn của họ được quản lý thành công, nguy cơ gãy xương tăng vẫn tồn tại đến tuổi trưởng thành. “Bộ ba thể thao” gồm khối lượng mỡ cơ thể dưới mức tối ưu, vô kinh ở phụ nữ và khối lượng xương giảm do sản xuất hormone sinh dục thấp được gặp ở các vận động viên nữ được đào tạo chuyên sâu và các vận động viên chạy đường dài nam ưu tú. Có một quần thể trẻ em, thanh thiếu niên và người trưởng thành trẻ khỏe mạnh có “thể trạng gầy” được đặc trưng bởi cân nặng dưới mức tối ưu kéo dài so với chiều cao nhưng tăng trưởng, trưởng thành sinh dục và khả năng sinh sản bình thường, ở những người này nguy cơ gãy xương không tăng mặc dù BMD theo diện tích thấp.
Mặc dù hoạt động thể thao hàng tuần càng lâu và càng cường độ cao ở trẻ em và thanh thiếu niên, BMD theo diện tích của đốt sống càng lớn độc lập với lượng canxi ăn vào, việc tham gia quá lâu vào các hoạt động có tác động mạnh, chẳng hạn như bóng rổ, thể dục dụng cụ và cổ vũ, khả năng bị gãy xương ở nữ giới càng lớn. Ở trẻ em trước và trong tuổi dậy thì, các chương trình giáo dục thể chất đơn giản ở trường học sử dụng các bài tập nhảy, bật và nhảy lò cò 2 đến 3 lần mỗi tuần làm tăng đáng kể BMD theo diện tích ở mấu chuyển xương đùi chỉ trong 8 tháng so với trẻ em tham gia vào một chương trình giáo dục thể chất tiêu chuẩn. Dinh dưỡng dưới mức tối ưu và các hoạt động tĩnh tại trong thời thơ ấu và thanh thiếu niên, cũng như tiêu thụ cola và rượu và hút thuốc lá, ngăn chặn khoáng hóa xương tối ưu và làm tăng khả năng phát triển loãng xương sau này và các biến chứng của nó. Bởi vì nguy cơ phát triển gãy xương do loãng xương khi trưởng thành giảm 40% cho mỗi 5% tăng khối lượng khoáng xương đỉnh, nền tảng để phòng ngừa loãng xương ở người lớn phải được xây dựng ở trẻ em và thanh thiếu niên bằng cách duy trì lượng canxi đầy đủ, dự trữ vitamin D (nồng độ calcidiol huyết thanh > 20 ng/mL), và hoạt động chịu trọng lượng bổ sung trong những năm hình thành này. Các chất dinh dưỡng khác, ví dụ, magiê, protein, vitamin C và K, và đồng cũng phải được tiêu thụ để tổng hợp chất nền xương tối ưu.
Bất động cấp tính của trẻ em và thanh thiếu niên khỏe mạnh, năng động và hậu quả là giảm chịu trọng lượng và giảm tải trọng cơ học lên xương dẫn đến tốc độ hình thành xương giảm; khi có sự tái hấp thu xương liên tục, tăng canxi niệu và sau đó là tăng canxi máu và khối lượng xương giảm phát triển. Ở trẻ em hoặc thanh thiếu niên bị bất động mạn tính (bại não, liệt tứ chi co cứng, loạn dưỡng cơ), tỷ lệ gãy xương (chủ yếu là xương đùi) cao trong các thao tác đơn giản, chẳng hạn như xoay người, mặc quần áo hoặc cho ăn. Trong nhóm đối tượng này, không chỉ thiếu chịu trọng lượng mà còn mức độ nghiêm trọng của bệnh chính, kích thước cơ thể và tình trạng dậy thì, tình trạng dinh dưỡng chung, lượng vitamin D và canxi ăn vào, các trạng thái viêm cùng tồn tại và thuốc (ví dụ, thuốc chống co giật, glucocorticoid), cũng như việc bị giam trong nhà ảnh hưởng xấu đến khối lượng xương và nguy cơ gãy xương. BMD theo diện tích của đầu dưới xương đùi và đốt sống thắt lưng tăng với tốc độ chậm hơn bình thường ở trẻ bị bại não co cứng, dẫn đến giảm điểm Z của BMD theo diện tích khi là đối tượng lớn tuổi hơn. Việc dùng pamidronate và axit zoledronic qua đường tĩnh mạch và tiêm dưới da GH tái tổ hợp của người đã làm tăng BMD theo diện tích ở các nhóm nhỏ được chọn của trẻ em bị bại não liệt tứ chi và co cứng. BMD theo diện tích tăng đáng kể ở 26 bệnh nhân bất động bị bại não liệt tứ chi (3–17 tuổi) trong quá trình dùng bisphosphonate alendronate (1 mg/kg/tuần đường uống), canxi (600 mg/ngày), và vitamin D (400 IU/ngày) trong 1 năm điều trị. Đứng có trợ giúp với việc chịu trọng lượng tự nó cũng có thể làm tăng BMD ở trẻ em bị bại não nặng.
Tầm quan trọng của sự bài tiết steroid sinh dục tuyến sinh dục bình thường trong quá trình trưởng thành sinh dục phù hợp với lứa tuổi được nhấn mạnh bởi quan sát rằng ở nam giới trưởng thành bị chậm phát triển sinh dục, BMD theo diện tích của xương quay, đốt sống và xương đùi thấp hơn so với nam giới có thời gian trưởng thành dậy thì bình thường. Về mặt hình thái mô học, ở xương bị thiếu steroid sinh dục ở người lớn, có sự giảm độ rộng vỏ xương, số lượng bè xương, chất dạng xương và hoạt động khoáng hóa; khối lượng xương giảm và vi kiến trúc bất thường dẫn đến giảm sức mạnh của xương và tăng nguy cơ gãy xương. Do hậu quả của sự thiếu hụt estrogen (và androgen), có sự gia tăng sản xuất nhưng giảm tuổi thọ của nguyên bào xương và tế bào xương, trong khi sự hình thành hủy cốt bào được kích thích và tuổi thọ của hủy cốt bào kéo dài; những sự kiện này phản ánh tổng hợp các hoạt động của nhiều cytokine tiền tạo hủy cốt bào mà sự tổng hợp của chúng được điều hòa bởi estrogen, bao gồm M-CSF, IL-1 và-6, TNFα, RANKL và osteoprotegerin. Mặc dù giảm xương đáp ứng với estrogen đã được ghi nhận ở nam giới trưởng thành bị thiếu hụt aromatase, phụ nữ trưởng thành bị mất cảm giác hoàn toàn với androgen cũng bị giảm xương mặc dù sản xuất estrogen bình thường đến tăng, rõ ràng cho thấy rằng androgen cũng quan trọng đối với khoáng hóa xương bình thường.
Nguy cơ gãy xương tăng ở các bé gái trước tuổi dậy thì và vị thành niên và phụ nữ trưởng thành mắc hội chứng Turner (loạn sản tuyến sinh dục) đã được cho là do cả các bất thường nội tại của chính xương biểu hiện bằng các dị tật đặc trưng có trong rối loạn này, bao gồm dị tật Madelung của cổ tay, ngắn chi ở giữa, vẹo cột sống, khuỷu tay vẹo ngoài, và xương bàn tay và bàn chân ngắn—hậu quả của sự thiếu hụt một nửa gen chứa hộp đồng dạng tầm vóc thấp (SHOX, OMIM 312865) và của sự giảm khoáng hóa xương do thiếu hụt estrogen mạn tính. Ở bệnh nhân mắc hội chứng Turner, có sự thiếu hụt chọn lọc của xương vỏ, trong khi mật độ xương bè có thể bình thường cũng như BMD theo diện tích và thể tích khi được điều chỉnh theo chiều cao. Vì FSH kích thích sự hình thành hủy cốt bào cả trực tiếp và bằng cách tăng cường biểu hiện của RANKL qua trung gian TNFα và sự biệt hóa hủy cốt bào sau đó, nồng độ cao của gonadotropin này có ở phụ nữ bị suy buồng trứng có thể góp phần vào sự khoáng hóa xương dưới mức tối ưu của bệnh nhân mắc hội chứng Turner. Ngoài ra, các rối loạn có thể có tác động bất lợi lên khoáng hóa xương (ví dụ, bệnh viêm ruột, bệnh celiac, suy giáp) phổ biến hơn ở bệnh nhân mắc hội chứng Turner. Tần suất gãy xương cổ tay tăng trong thời thơ ấu cũng như nguy cơ gãy xương nói chung ở người lớn mắc hội chứng Turner. Ở các đối tượng bị loạn sản tuyến sinh dục, liệu pháp thay thế estrogen trong đầu tuổi vị thành niên làm tăng lắng đọng canxi xương và BMD theo diện tích và khối lượng xương vỏ và bè, trong khi thay thế estrogen muộn có liên quan đến sự giảm dai dẳng khối lượng xương vỏ mặc dù khối lượng xương bè bình thường hóa. GH tái tổ hợp của người được dùng trong thời thơ ấu làm tăng tốc độ tăng trưởng, tầm vóc người lớn và kích thước xương ở nhiều bệnh nhân mắc hội chứng Turner, và có lẽ làm tăng sức mạnh của xương và giảm nguy cơ gãy xương. Ở các bé gái mắc hội chứng Turner, lượng canxi và vitamin D đầy đủ và các bài tập chịu trọng lượng cũng cần thiết. Bisphosphonate không được khuyến nghị để điều trị thanh thiếu niên mắc hội chứng Turner vì các tác nhân này chủ yếu tác động lên xương bè, mục tiêu chính của estrogen. Ở trẻ em bị dậy thì sớm trung ương, việc dùng một tác nhân ức chế gonadotropin có thể làm giảm tốc độ tích lũy khoáng chất xương, một quá trình dường như có thể đảo ngược sau khi hoàn thành liệu pháp. Lượng canxi và vitamin D đầy đủ và các bài tập chịu trọng lượng nên được kê đơn cho các đối tượng đang nhận một tác nhân ức chế gonadotropin.
Các đối tượng tuổi dậy thì bị suy sinh dục nguyên phát (ví dụ, hội chứng Klinefelter, galactosemia, sau xạ trị hoặc hóa trị) hoặc thứ phát (chán ăn tâm thần, tập luyện thể chất quá mức, giảm gonadotropin) cũng bị giảm khoáng hóa xương. Bằng cách giảm sản xuất estrogen, ngay cả việc sử dụng ngắn hạn (6 tháng) của thuốc tránh thai tiêm bắp depot medroxyprogesterone acetate (MPA) cũng dẫn đến mất khối lượng xương đáng kể ở nữ thanh thiếu niên và phụ nữ trẻ (18–21 tuổi) khi BMD thường sẽ tăng lên; tuy nhiên, khối lượng xương tăng theo thời gian sau khi ngừng sử dụng tác nhân này. Tác động bất lợi của depot MPA lên BMD có thể được ngăn chặn bằng cách dùng đồng thời estrogen.
Khối lượng xương thấp do dư thừa glucocorticoid là kết quả của sự ức chế hình thành nguyên bào xương và tăng tốc độ chết theo chương trình của nguyên bào xương và tế bào xương—dẫn đến giảm tốc độ hình thành chất nền xương và sửa chữa vi gãy xương; chúng tăng cường sự hình thành hủy cốt bào và tăng tiêu xương—kết quả của sự gia tăng biểu hiện của RANKL kết hợp với sự giảm tốc độ chết theo chương trình của hủy cốt bào—cho phép tái hấp thu xương kéo dài và quá mức. Bởi vì glucocorticoid làm giảm sự hình thành xương qua trung gian tế bào xương, trong mỗi chu kỳ tái tạo, lượng xương được thay thế ít hơn nhiều so với lượng bị loại bỏ và vi kiến trúc xương bị suy thoái dẫn đến giảm khối lượng và sức mạnh của xương theo thời gian. Một cách gián tiếp, glucocorticoid làm suy giảm sự tích lũy xương bằng cách ức chế bài tiết IGF-1 và bằng cách giảm hấp thu canxi ở ruột. Bằng cách giảm sức mạnh cơ bắp, glucocorticoid làm giảm các lực tác động lên xương, làm suy giảm thêm sự hình thành của nó. Trẻ em đang nhận glucocorticoid để điều trị hen suyễn hoặc các rối loạn thấp khớp cũng nên nhận 2000 IU vitamin D hàng ngày.
Ở cấp độ phân tử, glucocorticoid ức chế sự biểu hiện và tổng hợp của RUNX2 (OMIM 600211, chr. 6p21.1) và BMP2 (OMIM 112261, chr. 20p12.3), các yếu tố cần thiết cho sự biệt hóa của nguyên bào xương trước và sau sinh, tương ứng, và làm tăng biểu hiện của nguyên bào xương của TNFSF11 mã hóa RANKL và làm giảm biểu hiện của TNFRSF11B mã hóa osteoprotegerin—thụ thể mồi nhử cho RANKL, những thay đổi có lợi cho sự hình thành hủy cốt bào. Glucocorticoid làm suy giảm tín hiệu WNT/β-catenin và do đó ức chế tổng hợp collagen type I, cũng như làm tăng tốc độ phân hủy của nó; chúng làm suy giảm sự hình thành và chức năng của IGF-1. Chúng cũng hướng tế bào tiền thân nguyên bào xương trung mô vào con đường biệt hóa của tế bào mỡ. Ở một mức độ hạn chế, glucocorticoid ức chế chuyển hóa vitamin D bình thường và do đó hấp thu canxi ở ruột phụ thuộc vitamin D. Chúng làm tăng mất canxi qua thận bằng một tác động trực tiếp lên ống thận, dẫn đến cường cận giáp thứ phát. Glucocorticoid cũng làm giảm sản xuất hormone sinh dục ở thanh thiếu niên và người lớn. Sự yếu cơ của việc tiếp xúc với glucocorticoid mạn tính làm giảm tác động của các lực cơ học lên sự hình thành xương. Sự ức chế tăng trưởng tuyến tính và mở rộng xương qua trung gian glucocorticoid góp phần làm suy giảm khoáng hóa xương ở trẻ em. Glucocorticoid ảnh hưởng xấu đến sự hình thành xương bè ban đầu và sau đó làm suy giảm sự tích lũy xương vỏ. Bệnh (ví dụ, hen suyễn, hội chứng thận hư, bệnh bạch cầu nguyên bào lympho cấp, các rối loạn viêm mạn tính, chẳng hạn như viêm khớp vị thành niên) mà liều dược lý của glucocorticoid đã được kê đơn góp phần làm giảm khối lượng xương bằng cách làm suy giảm khả năng vận động và bằng cách sản xuất các cytokine tạo hủy cốt bào. Mặc dù sự tích lũy khối lượng xương do glucocorticoid giảm nhiều hơn với các đợt bùng phát ngắt quãng thường xuyên của glucocorticoid đường uống so với glucocorticoid dạng hít ở các bé trai bị hen suyễn, ở liều cao trong thời gian dài, glucocorticoid dạng hít cũng có thể dẫn đến giảm khối lượng xương. Ở người trưởng thành trẻ bị hen suyễn, có một mối quan hệ nghịch giữa BMD theo diện tích của đốt sống và xương đùi và liều tích lũy của glucocorticoid dạng hít với nguy cơ gãy xương tăng lên khi liều và thời gian dùng glucocorticoid tăng lên; liều tích lũy 5000 mg hydrocortisone dẫn đến giảm 1 độ lệch chuẩn BMD của đốt sống. Ở phụ nữ trưởng thành bị tăng sản tuyến thượng thận bẩm sinh do thiếu 21-hydroxylase được điều trị bằng glucocorticoid, khoáng hóa xương giảm nhẹ, một phần liên quan đến mức độ ức chế sản xuất androgen của tuyến thượng thận. Người ta đề nghị rằng khi trẻ em bắt đầu nhận glucocorticoid đường uống hoặc dạng hít, chúng cũng nên được hướng dẫn ăn đủ lượng canxi và vitamin D phù hợp với lứa tuổi và tham gia các bài tập chịu trọng lượng. Ở trẻ em gặp phải các tác dụng bất lợi của glucocorticoid đối với sự tăng trưởng và khoáng hóa xương, điều quan trọng là phải giảm liều steroid của chúng đến mức tối đa có thể và ngừng chúng nếu có thể. Mặc dù thường được khuyến nghị cho người lớn được điều trị bằng glucocorticoid, việc dùng bisphosphonate thường quy cho trẻ em đang nhận các tác nhân này không được khuyến nghị trừ khi có sự suy giảm ổn định về khối lượng xương hoặc bệnh nhân đã bị gãy xương do tác động thấp hoặc mong manh do khối lượng xương thấp. Tuy nhiên, việc sử dụng sớm bisphosphonate (ví dụ, alendronate) có thể ngăn ngừa mất xương ở bệnh nhân được điều trị bằng glucocorticoid mắc bệnh thấp khớp khởi phát ở trẻ em.
GH có tác động trực tiếp và gián tiếp lên sự hình thành xương. Trong sụn đĩa tăng trưởng, GH kích thích tổng hợp IGF-1 tại chỗ, đến lượt nó, tăng cường sự tăng sinh của các tế bào sụn nghỉ và hóa xương nội sụn; trong xương, GH tăng cường sự biệt hóa, tăng sinh và chức năng của nguyên bào xương dẫn đến tăng tổng hợp IGF-1, IGFBP-3, osteocalcin, phosphatase kiềm đặc hiệu cho xương và procollagen type I của nguyên bào xương; GH kích thích tổng hợp osteoprotegerin của nguyên bào xương, do đó làm suy giảm sự hình thành hủy cốt bào nhưng nó cũng kích thích sự hình thành hủy cốt bào và tái hấp thu xương một cách gián tiếp thông qua sự tổng hợp RANKL qua trung gian IGF-1. GH chủ yếu kích thích sự hình thành xương vỏ. IGF-1 điều hòa sự biệt hóa của các tế bào sụn và duy trì tốc độ tăng sinh của chúng. IGF-1 do nguyên bào xương và tế bào xương tổng hợp ảnh hưởng đến sự trưởng thành của nguyên bào xương và tốc độ khoáng hóa chất nền và chu chuyển xương và làm trung gian cho phản ứng của xương đối với tải trọng cơ học và PTH. PTH, hormone tuyến giáp và estrogen kích thích và glucocorticoid ức chế bài tiết IGF-1 của nguyên bào xương. Việc dùng GH cho trẻ em và người lớn thiếu GH làm tăng tốc độ cả hình thành và phá hủy xương, loại sau chiếm ưu thế ban đầu; trong thời gian điều trị dài (12–18 tháng), GH làm tăng BMD theo diện tích ở những bệnh nhân này; tuy nhiên, để đạt được khối lượng xương đỉnh, liệu pháp GH phải được tiếp tục đến tuổi trưởng thành. Tuy nhiên, ở nhiều người lớn không được điều trị bị thiếu GH, BMD thể tích thường bình thường mặc dù các giá trị BMD theo diện tích thấp phản ánh kích thước xương nhỏ hơn của họ. Một phần, khối lượng xương thấp ở trẻ thiếu GH cũng có thể được cho là do kích thước xương nhỏ của trẻ thấp so với các bạn cùng lứa tuổi khi được kiểm tra bằng BMD theo diện tích bằng DXA. Đo khối lượng xương bằng BMD theo diện tích được điều chỉnh theo kích thước hoặc các phương pháp thể tích (ví dụ, pQCT) cho thấy BMD thể tích là bình thường hoặc hơi thấp ở phần lớn các đối tượng thiếu GH; tuy nhiên, nguy cơ gãy xương tăng ở người lớn bị thiếu GH khởi phát ở trẻ em. Ở những bệnh nhân bị suy tuyến yên khởi phát ở tuổi trưởng thành liên quan đến thiếu hụt nhiều hormone tuyến yên, nguy cơ gãy xương tăng lên, nhưng điều này có thể là do mất bài tiết hormone sinh dục được kích thích bởi gonadotropin và do các tác động cơ học giảm của khối lượng cơ giảm lên sự tích lũy xương ở những đối tượng đã phát triển đầy đủ này. GH chủ yếu tăng cường sự phát triển của xương vỏ; do đó, trong tình trạng bài tiết GH quá mức (ví dụ, bệnh to đầu chi), sự phát triển xương màng tăng lên dẫn đến các đặc điểm khuôn mặt đặc trưng và sự phát triển quá mức của xương liên quan của những người bị ảnh hưởng; tuy nhiên, dư thừa GH cũng có thể làm giảm BMD theo diện tích của đốt sống và làm tăng nguy cơ gãy đốt sống ở những bệnh nhân này.
Triiodothyronine, hormone tuyến giáp có hoạt tính sinh học, điều hòa giảm sự tăng sinh của các tế bào sụn và làm tăng tốc độ biệt hóa của chúng thành các tế bào sụn tiền phì đại và phì đại. Triiodothyronine kích thích sự biệt hóa của nguyên bào xương và tổng hợp osteocalcin, osteopontin, collagen type I, phosphatase kiềm và IGF-1; nó cũng tăng cường sự hình thành hủy cốt bào và, do đó, tốc độ tái hấp thu xương, nhưng có khả năng làm như vậy một cách gián tiếp bằng cách tăng tổng hợp RANKL của nguyên bào xương. Ở trẻ em bị cường giáp, tốc độ trưởng thành của sụn được tăng tốc, sự hợp nhất của các đường khớp sọ được tăng cường, và các tác động tạo hủy cốt bào là nổi bật. Do đó, ở bệnh nhân nhiễm độc giáp, có sự gia tăng tốc độ chu chuyển xương và giảm độ dài của chu kỳ tái tạo xương (chủ yếu là do rút ngắn giai đoạn hình thành xương) dẫn đến mất ròng xương được khoáng hóa. Giống như ở người lớn bị nhiễm độc giáp, BMD toàn thân, đốt sống và xương đùi thấp ở trẻ em và thanh thiếu niên bị cường giáp, nhưng chúng tăng đáng kể trong 12 đến 24 tháng đầu sau khi phục hồi trạng thái bình giáp. Ở trẻ em bị suy giáp, sự trưởng thành nội sụn bị trì hoãn và vôi hóa bất thường; việc dùng liều thay thế sinh lý của thyroxine cho trẻ em bị suy giáp mắc phải hoặc bẩm sinh thúc đẩy sự trưởng thành của sụn và hình thành xương nội sụn; điều trị không ảnh hưởng xấu đến khoáng hóa xương trong thời thơ ấu, mặc dù người lớn bị suy giáp bẩm sinh có thể bị giảm 10% khối lượng xương quay.
Tăng đường huyết có liên quan đến sự tích tụ các protein bị glycated hóa cao (protein liên kết glucose/fructose) làm suy giảm sự biệt hóa của các tế bào gốc trung mô thành các tế bào sụn và nguyên bào xương với hậu quả là giảm tổng hợp osteocalcin và các sản phẩm khác của nguyên bào xương; sự hình thành hủy cốt bào cũng bị suy giảm. Ở bệnh nhân đái tháo đường type 1, BMD theo diện tích giảm, kích thước xương giảm và nguy cơ gãy xương tăng, phản ánh các tác động bất lợi của tăng đường huyết mạn tính và thiếu hụt insulin đối với sự hình thành xương. Ở thanh thiếu niên bị đái tháo đường type 1, khối lượng xương toàn thân, trục và phụ được đánh giá bằng DXA giảm so với các đối tượng đối chứng và có liên quan nghịch với các giá trị Hb A1c. Sử dụng pQCT ở các đối tượng trẻ, trước tuổi dậy thì bị đái tháo đường type 1, diện tích mặt cắt ngang và BMD của xương vỏ được phát hiện là giảm, ngụ ý nguy cơ gãy xương tăng lên. Mặc dù bệnh nhân đái tháo đường type 2 có xu hướng có BMD theo diện tích tăng (liên quan đến sự suy giảm tốc độ chu chuyển xương có lẽ liên quan đến tăng insulin máu), nguy cơ gãy xương của họ vẫn tăng lên.
Khối lượng xương giảm ở phần lớn trẻ em bị bệnh bạch cầu nguyên bào lympho cấp và 26% trong số những bệnh nhân này bị gãy xương thường là đốt sống trước hoặc trong bốn năm đầu điều trị. Các yếu tố sinh bệnh liên quan đến sự phát triển của khối lượng xương thấp ở những đối tượng này bao gồm: các tác động bất lợi của chính bệnh trực tiếp lên xương—qua trung gian sự gia tăng hình thành và hoạt động của hủy cốt bào do cytokine và ức chế tái tạo xương, tổn thương xương do bức xạ; tác dụng ức chế của glucocorticoid, hóa trị liệu, kháng sinh và các tác nhân ức chế miễn dịch đối với sự hình thành xương; giảm hoạt động thể chất; giảm lượng calo, protein và vitamin D ăn vào; thiếu hụt hormone sinh dục do chậm hoặc ngừng phát triển ở tuổi vị thành niên; và thiếu hụt GH ở trẻ em đã được xạ trị sọ não. Sau khi điều trị ban đầu khối u và đạt được sự thuyên giảm, việc dùng bisphosphonate, chẳng hạn như axit zoledronic, có thể làm tăng đáng kể khoáng hóa xương ở những trẻ em và thanh thiếu niên này. Ở những người nhận ghép tạng đặc, cyclosporine A gây mất xương bằng cách tăng biểu hiện RANKL của nguyên bào xương và giảm sản xuất osteoprotegerin, do đó làm tăng sự hình thành hủy cốt bào. Methotrexate và hóa trị liệu nội tủy có tác dụng ức chế đáng kể đối với khoáng hóa xương ở trẻ em bị bệnh ác tính. Những bệnh nhân này nên nhận đủ canxi và vitamin D bổ sung và các bài tập chịu trọng lượng đến mức có thể. Những người bị thiếu hụt GH sau xạ trị sọ não có thể được điều trị bằng hGH tái tổ hợp nếu họ vẫn thiếu GH sau khi bệnh chính đã thuyên giảm kéo dài. Giảm khoáng hóa xương là phổ biến ở đối tượng sau ghép tủy xương hoặc tạng đặc; sinh bệnh học đa dạng của nó bao gồm chính bệnh chính và bệnh mạn tính có thể đi kèm, việc sử dụng glucocorticoid liều cao và thuốc chống thải ghép, cũng như chức năng ruột, thận, gan và tuyến sinh dục bị thay đổi. Ngoài việc cung cấp dinh dưỡng, canxi và vitamin D đầy đủ, ở người lớn, các tác động của việc cấy ghép lên sự tích lũy xương có thể được cải thiện một phần bằng cách dùng bisphosphonate.
Ở những bệnh nhân bị bỏng nặng và trẻ em bị bệnh ưa chảy máu, bệnh hồng cầu hình liềm, đái tháo nhạt trung ương, hội chứng Marfan, bệnh homocystin niệu, không dung nạp protein lysinuric, và bệnh acid propionic và methylmalonic niệu, BMD theo diện tích cũng giảm. Trẻ em bị xơ nang có thể có BMD theo diện tích cổ xương đùi và đốt sống thắt lưng thấp do hậu quả của dinh dưỡng dưới mức tối ưu, thiếu vitamin D, viêm mạn tính, đái tháo đường đồng thời, chậm dậy thì và điều trị bằng thuốc. Tuy nhiên, khoảng một phần ba bệnh nhân xơ nang được quản lý tối ưu với sự kiểm soát lâm sàng tốt vẫn có thể có BMD dưới mức bình thường, mặc dù điều này không nhất thiết chuyển thành nguy cơ gãy xương tăng lên. Khối lượng xương thấp và xẹp đốt sống có thể là những biểu hiện sớm của bệnh viêm ruột mạn tính. Thiếu vitamin D và cường cận giáp thứ phát, cũng như tình trạng viêm mạn tính và các tác nhân điều trị, chẳng hạn như glucocorticoid, có khả năng góp phần làm giảm sự hình thành xương và tăng tái hấp thu xương trong bệnh này. Việc BMD theo diện tích toàn thân ở trẻ em, thanh thiếu niên và người trưởng thành trẻ bị bệnh viêm ruột mạn tính có thể bình thường so với khối lượng nạc (mặc dù giảm so với các tiêu chuẩn dân tộc, tuổi và chiều cao) không nhất thiết ngụ ý rằng sức mạnh của xương ở những bệnh nhân này là bình thường như bằng chứng là nguy cơ gãy xương tăng lên của người lớn mắc rối loạn này. Khối lượng xương thấp là phổ biến ở trẻ em và người lớn bị bệnh celiac. Các yếu tố sinh bệnh liên quan đến khối lượng xương thấp ở trẻ em bị bệnh celiac bao gồm kém hấp thu vitamin D, canxi, protein và các chất dinh dưỡng khác, tổng hợp các cytokine kích hoạt hủy cốt bào và tiền viêm, chẳng hạn như IFN-γ và IL-15,-18, và-21, và cường cận giáp thứ phát. Ở trẻ em và thanh thiếu niên bị nhiễm virus gây suy giảm miễn dịch ở người, BMD theo diện tích toàn thân có thể giảm do hậu quả của chính tác nhân lây nhiễm, tình trạng viêm mạn tính mà nó gây ra, dinh dưỡng dưới mức tối ưu bao gồm thiếu vitamin D tinh vi, và việc dùng liệu pháp kháng retrovirus hoạt tính cao có thể có tác động trực tiếp lên sự tạo ra và chức năng của nguyên bào xương và hủy cốt bào. Mặc dù khỏe mạnh về mặt lâm sàng và tăng trưởng tuyến tính bình thường, tốc độ tích lũy khối lượng xương giảm ở những đối tượng này trong khi tốc độ tái hấp thu xương tăng lên. Khối lượng xương giảm ở trẻ em mắc nhiều loại bệnh mô liên kết (viêm khớp vị thành niên tự phát, lupus ban đỏ hệ thống, viêm da cơ vị thành niên) do tình trạng viêm mạn tính và sản xuất các cytokine tiền hủy cốt bào và do điều trị bằng glucocorticoid.
Loãng xương vị thành niên tự phát (OMIM 259750) là một bệnh không rõ nguyên nhân sinh bệnh biểu hiện bằng đau xương và gãy xương với chấn thương tối thiểu xảy ra ở bé gái hoặc bé trai cuối tuổi trước dậy thì-đầu tuổi dậy thì (8–12 tuổi) và tự khỏi theo thời gian với các biểu hiện còn lại thay đổi. Ở các đối tượng bị ảnh hưởng, X-quang được thực hiện để đánh giá đau khớp, cơ và/hoặc lưng, khó đi lại, rút ngắn thân mình và/hoặc sự hiện diện của gù lưng cho thấy đốt sống lõm hai mặt và/hoặc gãy lún đốt sống, các vùng thấu quang trong xương dài và gãy hành xương. Các phân tích bằng DXA và QCT cho thấy khoáng hóa xương giảm. Các nghiên cứu hóa học là bình thường. Các nghiên cứu hình thái mô học động cho thấy tốc độ hình thành và chu chuyển xương thấp với sự giảm thể tích xương xốp và độ dày bè xương chủ yếu do hoạt động của nguyên bào xương giảm trên bề mặt nội mạc nhưng không phải bề mặt màng xương; không có bằng chứng về sự tái hấp thu xương tăng lên. Loãng xương vị thành niên tự phát rất có khả năng có nguồn gốc di truyền không đồng nhất. Các phân tích đột biến của COL1A1 và COL1A2 là bình thường ở những đối tượng này. Ở 15% bệnh nhân bị loãng xương vị thành niên, một đột biến mất chức năng dị hợp tử có tính gia đình trong gen mã hóa protein 5 liên quan đến thụ thể LDL (LRP5, OMIM 603506) đã được phát hiện. Các đột biến mất chức năng hai alen của LRP5 dẫn đến hội chứng loãng xương-giả u thần kinh thị giác, trong khi các biến thể tăng chức năng dị hợp tử dẫn đến các dạng bệnh xương hóa đá và xơ cứng xương (xem bên dưới). Một trong những thách thức chẩn đoán khó khăn nhất là sự phân biệt lâm sàng giữa loãng xương vị thành niên tự phát và bệnh xương thủy tinh type I (xem bên dưới). Bệnh sau được đặc trưng về mặt lâm sàng bởi tiền sử gia đình dương tính, khởi phát ở đầu thời thơ ấu, tồn tại suốt đời, gãy thân xương, xương wormian, dây chằng lỏng lẻo và sức mạnh cơ giảm, củng mạc xanh, răng bất thường, mất thính lực và tốc độ chu chuyển xương cao. Ngoài ra, có một số trẻ em có khối lượng xương thấp và gãy xương tái phát mà bức tranh lâm sàng không nghiêm trọng như những trẻ bị loãng xương vị thành niên tự phát, cho thấy có một phổ rộng các phát hiện lâm sàng ở trẻ em và thanh thiếu niên có khoáng hóa xương ở mức giới hạn. Ở trẻ em bị loãng xương vị thành niên tự phát cổ điển, điều trị triệu chứng được đưa ra; ở một số bệnh nhân, calcitriol hoặc bổ sung natri florua đã có lợi. Mặc dù rối loạn này cải thiện và thậm chí biến mất ở tuổi dậy thì, việc điều trị bệnh nhân trước tuổi dậy thì bằng steroid sinh dục dường như không làm tăng tốc quá trình chữa lành. Việc dùng bisphosphonate pamidronate đã hữu ích trong việc giảm đau xương và tăng BMD của đốt sống ở một số ít trẻ em bị loãng xương vị thành niên tự phát.
LPR5 (xem ở trên) là một protein màng tế bào được biểu hiện bởi các nguyên bào xương có vai trò như một đồng thụ thể cho sự truyền tín hiệu thông qua con đường WNT-Frizzled-β-Catenin dẫn đến sự biệt hóa và chức năng của các nguyên bào xương. Tín hiệu WNT tăng cường sự biệt hóa của các tế bào tiền thân trung mô đa năng vào con đường hình thành sụn và xương và cản trở sự biệt hóa của nó thành các tế bào mỡ. Bằng cách kích thích RUNX2, β-catenin tiếp tục hướng tế bào tiền thân xương sụn vào con đường nguyên bào xương, và ở nguyên bào xương trưởng thành, β-catenin tăng cường biểu hiện của osteoprotegerin và do đó làm giảm sự hình thành hủy cốt bào. Sclerostin (được mã hóa bởi SOST, OMIM 605740) liên kết với miền ngoại bào của LRP5 và do đó ức chế tín hiệu WNT. Các bất thường nội tại của biểu hiện LRP5 đã được liên kết về mặt lâm sàng với các rối loạn cả khoáng hóa xương suy giảm và quá mức. Hội chứng loãng xương-giả u thần kinh thị giác (OMIM 259770) được đặc trưng về mặt lâm sàng bởi sự khởi phát bẩm sinh hoặc đầu thời thơ ấu của suy giảm thị lực nghiêm trọng do mắt nhỏ và tăng sản dịch kính (giả u thần kinh thị giác có thể bị xác định nhầm là u nguyên bào võng mạc), dẫn đến bong võng mạc, tăng nhãn áp và mù lòa, sự mong manh của xương rõ rệt với mềm sọ và gãy xương trong cuối thời thơ ấu, thời thơ ấu hoặc thanh thiếu niên, và suy giảm nhận thức, lỏng lẻo dây chằng và giảm trương lực cơ thay đổi. Rối loạn này được di truyền như một tính trạng lặn trên nhiễm sắc thể thường và là do các đột biến bất hoạt hai alen (đồng hợp tử hoặc dị hợp tử phức hợp) (sai nghĩa [p.Val336Met, p.Arg494Gln], vô nghĩa [p.Arg428Ter, p.Arg1002Ter], dịch khung, vị trí nối) trong LRP5—chủ yếu nằm ở miền ngoại bào của LRP5. Các đột biến sai nghĩa có khả năng ngăn chặn sự liên kết bình thường của LRP5 với sản phẩm của gen phát triển trung bì (MESDC2, OMIM 607783, chr. 15q25.1), một protein chaperone hướng LRP5 đến màng tế bào. Mặc dù thường không có triệu chứng, những người mang dị hợp tử có thể bị giảm xương; tuy nhiên, thị lực không bị suy giảm. Việc dùng pamidronate qua đường tĩnh mạch trong vài năm có thể làm tăng khối lượng xương ở trẻ em mắc rối loạn này. Các đột biến mất chức năng trong LRP5 cũng đã được liên kết với bệnh võng mạc dịch kính xuất tiết có tính gia đình (OMIM 133780), một rối loạn phát triển của mạch máu võng mạc có thể được di truyền như một tính trạng trội trên nhiễm sắc thể thường (p.Leu145Phe) hoặc lặn trên nhiễm sắc thể thường (p.Arg570Gly, p.Arg752Gly); những bệnh nhân này cũng bị giảm khối lượng xương. LRP5 truyền tín hiệu không chỉ của WNT mà còn của NDP (OMIM 300658) mã hóa Norrin mà tín hiệu của nó điều chỉnh sự hình thành võng mạc dịch kính mắt. Các đột biến tăng chức năng trong LRP5 có liên quan đến khối lượng xương tăng (xem bên dưới).
Bệnh xương thủy tinh
Bệnh xương thủy tinh hay “bệnh xương giòn” là một rối loạn tăng sự mong manh của xương thường liên quan đến khối lượng xương thấp, có mức độ nghiêm trọng lâm sàng thay đổi từ tử vong trong tử cung hoặc chu sinh đến tăng nhẹ nhạy cảm với gãy xương ở cuộc sống sau này và có thể được di truyền như các tính trạng trội hoặc lặn trên nhiễm sắc thể thường. Phân loại Sillence ban đầu gồm bốn loại bệnh xương thủy tinh dựa trên sinh bệnh học cơ bản, các đặc điểm lâm sàng và diễn biến bệnh đã được sửa đổi bằng việc thiết lập năm dạng lâm sàng của bệnh này (được chỉ định là các loại lâm sàng 1–5) (xem Bảng 20.10B) mà các biểu hiện lâm sàng và X-quang của chúng có thể được cho là do các biến thể của một số trong số 19 gen bị đột biến hiện đang liên quan đến bệnh xương thủy tinh (được chỉ định là các loại gen I-XIX) (xem Bảng 20.10C). Do đó, bệnh xương thủy tinh là một dạng loãng xương nguyên phát là do các lỗi di truyền của sự hình thành và khoáng hóa xương có mức độ nghiêm trọng thay đổi từ nhẹ đến tử vong là do các biến thể di truyền quan trọng đối với sự biệt hóa của nguyên bào xương (hình thành nguyên bào xương) và chức năng, tổng hợp và xử lý collagen, và sự hình thành hydroxyapatite. Các loại (dạng) của bệnh xương thủy tinh có thể được phân loại theo kiểu hình, tức là, mức độ mong manh của xương và mức độ nghiêm trọng lâm sàng kết quả (xem Bảng 20.10B) hoặc theo vị trí của (các) lỗi di truyền trong sự hình thành nguyên bào xương hoặc sự hình thành collagen và các sửa đổi của nó (xem Bảng 20.10C). (19 loại di truyền hiện được chỉ định [tức là, các gen biến thể] của bệnh xương thủy tinh được đánh số theo thứ tự mô tả lâm sàng ban đầu của chúng. Ngoài ra, có một số gen mà các biến thể của chúng có liên quan đến sự mong manh của xương tăng lên mà sau này có thể được chỉ định là các dạng di truyền khác của bệnh xương thủy tinh [xem Bảng 20.10C].) Dấu hiệu của mỗi trong năm loại lâm sàng của bệnh xương thủy tinh là sự mong manh của xương tăng lên, nhưng mức độ nghiêm trọng thay đổi với loại lâm sàng 1 là ít nghiêm trọng nhất vì những người được chỉ định như vậy có các chi thẳng và chiều cao trong phạm vi bình thường thấp và loại lâm sàng 2 gây tử vong trong giai đoạn chu sinh do suy hô hấp là hậu quả của nhiều gãy xương sườn làm giảm thể tích lồng ngực. Bệnh xương thủy tinh loại lâm sàng 3 có liên quan đến các dị tật đáng kể của các chi, nhưng bệnh nhân bị ảnh hưởng sống qua tuổi thơ ấu và thời thơ ấu mặc dù bị hạn chế tăng trưởng rõ rệt và khả năng vận động hạn chế. Mức độ nghiêm trọng của bệnh xương thủy tinh loại lâm sàng 4 là trung gian giữa các loại lâm sàng 1 và 3; tầm vóc người lớn bị hạn chế đáng kể với chiều cao từ –3,6 đến –4,6 độ lệch chuẩn dưới trung bình theo giới. Kiểu hình của bệnh nhân bị bệnh xương thủy tinh loại lâm sàng 5 (OMIM 610967) tương tự như của loại lâm sàng 4 nhưng độc đáo ở chỗ dạng này có liên quan đến sự hình thành callus quá mức tại các vị trí gãy xương đang lành và vôi hóa màng gian cốt chỉ xảy ra ở bệnh nhân có đột biến trong protein xuyên màng-5 do IFN gây ra hoặc protein giống IFITM5 giới hạn ở xương (IFITM5, OMIM 614757) (xem bên dưới). Phân loại sau đây của bệnh xương thủy tinh dựa trên vị trí chức năng của lỗi di truyền trong sự hình thành nguyên bào xương, hoạt động của nguyên bào xương, hoặc tổng hợp, xử lý và/hoặc bài tiết collagen.
Các lỗi trong sự biệt hóa, trưởng thành, chức năng của Nguyên bào xương
Các biến thể mất chức năng của WNT1 (họ vị trí tích hợp virus u vú chuột type không cánh [MMTV], thành viên 1, OMIM 164820) và LRP5 (OMIM 603506) có tác động bất lợi đến sự biệt hóa của nguyên bào xương và do đó các đột biến bất hoạt trong các gen này làm suy giảm sự hình thành xương dẫn đến bệnh xương thủy tinh (WNT1—gen type XV, các type lâm sàng 3 và 4) và hội chứng loãng xương-giả u thần kinh thị giác (OMIM 259770) do các biến thể của LRP5. WNT1 là một phối tử quan trọng cho con đường tín hiệu WNT chính tắc của sự biệt hóa nguyên bào xương từ các tế bào gốc trung mô. WNT1 kích thích sự hình thành nguyên bào xương bằng cách liên kết với phức hợp thụ thể LRP5-Frizzled sau đó kích hoạt một con đường truyền tín hiệu nội bào ức chế sự phân hủy của β-catenin (CTNNB1, OMIM 116806) trong tế bào chất qua trung gian proteasome. β-catenin tích lũy sẽ chuyển vị vào nhân của tế bào gốc trung mô, nơi nó ức chế sự biệt hóa thành tế bào sụn hoặc tế bào mỡ và kích thích sự hình thành nguyên bào xương, dẫn đến tăng hình thành xương và giảm tái hấp thu xương. WNT1 cũng gián tiếp kích thích sự hình thành hủy cốt bào. Các đột biến bất hoạt trong WNT1 có thể là đơn alen hoặc hai alen; mức độ nghiêm trọng lâm sàng của các đột biến WNT1 có hại có thể khác nhau và có thể bao gồm các dị tật ngoài xương, chẳng hạn như thiểu sản một bán cầu tiểu não.
Bệnh xương thủy tinh gen type XII (OMIM 613849) là một dạng bệnh nặng vừa (type lâm sàng 4) được đặc trưng bởi đa gãy xương ở đầu thời thơ ấu với xương dài ngắn, cong và vòm miệng cao nhưng răng, thính giác và củng mạc bình thường. Nó được di truyền như một rối loạn lặn trên nhiễm sắc thể thường và là do các đột biến bất hoạt trong yếu tố đặc hiệu phiên mã 7 hoặc osterix (SP7, OMIM 606633). Osterix là một yếu tố phiên mã ngón tay kẽm 431 aa phụ thuộc WNT1 cần thiết cho sự trưởng thành của tiền nguyên bào xương thành nguyên bào xương và cho sự biểu hiện chức năng của COL1A1; nó kích thích cả sự phát triển xương nội sụn và nội màng; sự biểu hiện của nó được điều hòa bởi RUNX2 (OMIM 600211). Mặc dù chức năng liên quan của nó trong nguyên bào xương chưa được biết, các đột biến bất hoạt hai alen trong TENTA5 (OMIM 611357) mã hóa một nucleotidyltransferase được gọi là FAM46A xúc tác quá trình chuyển nucleoside triphosphate đến một nhóm hydroxyl của một phân tử nhận có liên quan đến bệnh xương thủy tinh gen type XVIII. Sau khi phiên mã hạt nhân và bắt đầu dịch mã COL1A1 và COL1A2, các sản phẩm peptide non của chúng rời khỏi nhân và đi vào lưới nội chất, nơi chúng trải qua các sửa đổi tiếp theo đòi hỏi các chaperone để di chuyển qua hệ thống này. Do đó, các đột biến mất chức năng hai alen trong chất ức chế serpin peptidase, nhánh F, thành viên 1 (SERPINF1, OMIM 172860) mã hóa chaperone collagen protein sốc nhiệt 47 dẫn đến bệnh xương thủy tinh nặng (gen type VI, type lâm sàng 3). Các biến thể của IFITM5 mã hóa protein xuyên màng-5 do IFN gây ra (OMIM 614757), một protein được tổng hợp bởi các nguyên bào xương có chức năng chính có thể là ổn định SERPINF1, có liên quan đến một dạng riêng biệt của bệnh xương thủy tinh được đặc trưng bởi sự hóa xương của màng gian cốt của cẳng tay, sự hình thành callus tăng sản tại các vị trí gãy xương, các dải hành xương đặc và một kiểu phiến xương “giống lưới” (gen type V, type lâm sàng 5). Sự mất đoạn hoặc các đột biến trong PLS3 (OMIM 300131) nằm trên nhiễm sắc thể Xq23, mã hóa plastin 3—một protein liên kết actin và canxi cần thiết cho sự toàn vẹn và tái tạo của các sợi actin tạo thành bộ xương tế bào của tế bào xương, dẫn đến một dạng bệnh xương thủy tinh liên kết X có mức độ nghiêm trọng khiêm tốn (type lâm sàng 1). Nam giới có các biến thể có hại đơn alen của PLS3 bị giảm khoáng hóa xương và bị gãy lún đốt sống và gãy xương dài ở đầu thời thơ ấu, nhưng không biểu hiện củng mạc xanh, bệnh tạo ngà không hoàn hảo hoặc tăng độ mềm dẻo của khớp. BMD thấp có thể hiện diện ở những người mang nữ dị hợp tử của các biến thể PL3, ở những người này các biểu hiện lâm sàng có thể thay đổi từ gãy xương do mong manh khởi phát ở trẻ em như ở nam giới đến loãng xương sau mãn kinh. Plastin 3 cũng có thể điều hòa sự hình thành hủy cốt bào và quá trình cảm nhận cơ học.
Các lỗi trong tổng hợp và xử lý Collagen Type 1
Collagen type 1 là một trimer bao gồm hai phân tử liên kết chéo của collagen type 1A1 (COL1A1, Collagen type I, alpha-1, OMIM 120150) và một phân tử collagen type 1A2 (COL1A2, Collagen type I, alpha-2, OMIM 120160) quấn vào nhau để tạo thành collagen type I trong xương, da, dây chằng, gân, củng mạc và răng. Các sợi của cả collagen type 1A1 và 1A2 đều bao gồm các đoạn lặp ba của glycine và hai axit amin bổ sung—thường là proline, hydroxyproline hoặc lysine. Các đột biến trong COL1A1 (gen type I) hoặc COL1A2 (gen type II) dẫn đến một codon dừng sẽ tạo ra một sản phẩm procollagen bị cắt ngắn và nhanh chóng bị phân hủy; do đó, chỉ có collagen type I bình thường được sản xuất nhưng với số lượng giảm. Bệnh nhân có các đột biến trong COL1A1 hoặc COL1A2 làm thay đổi (ví dụ, thay thế cysteine cho glycine ở các vị trí quan trọng) cấu hình ba chiều của nó dẫn đến sự hình thành các sợi collagen 1α1 bất thường và do đó bệnh xương nặng hơn. Thông thường, các biến thể của COL1A1 làm tổn hại đến quá trình tổng hợp collagen nghiêm trọng hơn so với các biến thể của COL1A2. Do đó, bệnh xương thủy tinh gen type I (OMIM 166200) là một rối loạn trội trên nhiễm sắc thể thường (đột biến mới ở 33% bệnh nhân) chủ yếu do “các alen rỗng chức năng”—kết quả của các khiếm khuyết nối hoặc các đột biến điểm dẫn đến lỗi chèn hoặc cắt ngắn collagen type 1A1 (COL1A1 – p.Gly178Cys, p.Arg963Ter), các đột biến dẫn đến giảm vận chuyển procollagen-α1(I) vào tế bào chất hoặc giải phóng nó vào chất nền—do đó làm giảm nhẹ sản xuất procollagen type I nguyên vẹn. Các biểu hiện lâm sàng của nó tương đối lành tính: củng mạc xanh đậm có khi sinh và tồn tại suốt tuổi trưởng thành, khối lượng xương giảm nhẹ, gãy xương không thường xuyên với ít biến dạng, tầm vóc người lớn bình thường thấp, mất thính lực ở 50%, sa van hai lá ở 18%, và hiếm khi có bệnh tạo ngà không hoàn hảo (bệnh xương thủy tinh gen type IB). (Nghịch lý thay, những bệnh nhân bị bệnh xương thủy tinh gen type I nhẹ do các đột biến tại các vị trí phân cắt propeptide đầu carboxyl của procollagen type I được chọn lọc [COL1A1—p.Asp1219Asn; COL1A2—p.Ala1119Thr] có thể có BMD cột sống thắt lưng bình thường hoặc tăng theo cả kiểm tra DXA và QCT mặc dù độ mong manh của xương tăng). Các đột biến gây tử vong trong COL1A1 là những đột biến làm thay đổi một axit amin có chuỗi bên phân nhánh hoặc mang điện tích và những đột biến trong các vị trí liên kết của monomer collagen đối với integrin, metalloproteinase chất nền, fibronectin và protein chất nền sụn oligomeric và những đột biến dẫn đến liên kết và phân hủy các tiểu đơn vị procollagen nguyên vẹn. Các đột biến gây tử vong trong COL1A2 là những đột biến cản trở sự liên kết của nó với proteoglycan. Các đột biến (p.Arg134Cys) trong COL1A1 cũng có thể được tìm thấy ở những bệnh nhân mắc hội chứng Ehlers-Danlos cổ điển (OMIM 130000) với da quá mức co giãn và lỏng lẻo các dây chằng của cột sống và các khớp lớn và nhỏ. Trẻ em có các đặc điểm lâm sàng của cả bệnh xương thủy tinh (mong manh xương) và hội chứng Ehlers-Danlos đã được mô tả. Ở những bệnh nhân này, các đột biến đã tập trung trong 90 aa đầu tiên của vùng xoắn của collagen 1α1 và ngăn chặn sự loại bỏ sau dịch mã bình thường của propeptide amino-procollagen; mặc dù protein bị đột biến có thể được kết hợp vào collagen, tính toàn vẹn cấu trúc của sản phẩm bị suy giảm vì các sợi của nó mỏng và yếu. Bệnh xương thủy tinh gen types I-IV mô tả các dạng có mức độ nghiêm trọng thay đổi của bệnh này liên quan đến các đột biến mất chức năng trong COL1A1 hoặc COL1A2 (xem Bảng 20.10B).
Các đột biến của COL1A1 và COL1A2 thường là dị hợp tử (đơn alen) và được di truyền như các bệnh trội trên nhiễm sắc thể thường; tiểu đơn vị collagen type 1 bị đột biến có thể có tác dụng trội-âm khi nó tương tác với tiểu đơn vị nguyên vẹn (gen types I–IV; các type lâm sàng 1–4). Sau khi tổng hợp, COL1α1 và Col1α2 trải qua các sửa đổi sau dịch mã, bao gồm hydroxyl hóa các gốc lysine và proline được chọn lọc, liên kết chéo các chuỗi collagen, giải phóng khỏi nguyên bào xương vào chất nền xung quanh, loại bỏ các propeptide đầu amino và carboxyl, và lắp ráp thành các sợi collagen. Trong số các sửa đổi sau dịch mã quan trọng nhất của procollagen type I là hydroxyl hóa các gốc proline và lysine bởi các prolyl và lysyl hydroxylase. Hydroxyl hóa các gốc proline ở carbon 4 mang lại sự ổn định nhiệt, trong khi hydroxyl hóa các gốc lysyl cho phép liên kết carbohydrate—galactose hoặc glucosyl-galactose—và sự hình thành các liên kết chéo trong hoặc giữa các chuỗi procollagen. Hydroxyl hóa proline ở carbon 3 ở vị trí 986 của COL1A1 và ở vị trí 707 của COL1A2 đặc biệt quan trọng đối với sự ổn định của cấu hình ba chiều của procollagen type I và sự trưởng thành sau đó của nó thành collagen type I. Sau khi tổng hợp, COL1α1 và COL1α2 được sửa đổi thêm bằng cách hydroxyl hóa trên các gốc proline và lysine được chọn lọc. Thất bại trong việc hình thành 3-hydroxyproline ở aa 986 của collagen 1α1 và aa 707 của collagen 1α2 có thể là do các biến thể mất chức năng của một trong ba gen liên quan đến quá trình này—prolyl 3-hydroxylase 1 (P3HI, OMIM 610339)—còn được gọi là leprecan, protein liên quan đến sụn (CRTAP, OMIM 605497), và cyclophilin B (CypB, Peptidyl-prolyl isomerase B, PPIB, OMIM 123841). Khi không có bất kỳ yếu tố nào trong số này, miền xoắn của collagen 1α1 bị sửa đổi bởi các enzyme khác dẫn đến những thay đổi về độ bền của nó. Các biến thể mất chức năng hai alen trong bất kỳ một trong ba gen này dẫn đến các dạng bệnh xương thủy tinh từ nặng đến tử vong (các type lâm sàng 2, 3, 4; bệnh xương thủy tinh gen types VII, VIII, IX; xem Bảng 20.10B, 20.10C). Bệnh xương thủy tinh gen type VIII (OMIM 610915) do P3H1 bị đột biến xảy ra chủ yếu ở các đối tượng người Mỹ gốc Phi và Trung Đông. Prolyl 3-hydroxylase 1 đặc biệt hydroxyl hóa carbon 3 của gốc proline ở codon 986 trong collagen 1A1 và ở codon 707 trong collagen 1A2. Kiểu hình của bệnh xương thủy tinh gen type VIII trùng lặp với các type lâm sàng 2 và 3; ngoài sự mong manh của xương, nó còn liên quan đến sự co rút tăng trưởng đáng kể, củng mạc trắng và hành xương hình củ. CRTAP và CypB ổn định phức hợp hydroxyl hóa collagen prolyl 3 và cũng là chaperone cho sự di chuyển của collagen từ lưới nội chất. Các đột biến mất chức năng trong PPIB đã được chỉ định là bệnh xương thủy tinh type gen type IX, một rối loạn thường gây tử vong trong đó sự mong manh của xương là rõ ràng trong tử cung (xem Bảng 20.10B). CRTAP được biểu hiện trong vùng tăng sinh của sụn đang phát triển và tại khớp sụn-xương. Chuột “knock-out” Crtap phát triển một bệnh loạn sản sụn-xương (gù vẹo cột sống, rút ngắn phần gần của các chi) và giảm xương nặng, loại sau là do giảm sản xuất và thay đổi chất lượng của chất dạng xương và hậu quả là giảm tốc độ lắng đọng khoáng chất. Chuột thiếu Crtap không thể 3-hydroxyl hóa gốc proline gần đầu carboxyl của collagen xương 1A1, dẫn đến tăng hydroxyl hóa các gốc lysine và cấu trúc bất thường của sợi collagen—những thay đổi dẫn đến khoáng hóa khiếm khuyết của collagen xương type I. Proline không được hydroxyl hóa ở codon 986 trong collagen xương 1A1 đã được chứng minh ở 3/11 bệnh nhân mắc bệnh xương thủy tinh di truyền lặn gây tử vong hoặc nặng được đặc trưng bởi đa gãy xương dài dẫn đến rút ngắn các chi ở gốc với chân xoay ngoài và dạng, vòm sọ và xương sườn khoáng hóa kém, mắt lồi và củng mạc trắng hoặc xanh nhạt. Điều này được chứng minh là do các đột biến mất chức năng đồng hợp tử hoặc dị hợp tử phức hợp trong CRTAP (dịch khung ([c.879delT], nhân đôi 16-bp trong exon 1, vô nghĩa [p.Gly276Ter], sai nghĩa [p.Met1Ile], vị trí nối donor của exon 1 tại nucleotide intron đầu tiên [IVS1 + 1G ⇒ C]) cản trở quá trình hydroxyl hóa hiệu quả của gốc proline ở codon 986 của procollagen xương 1A1. Rối loạn này đã được chỉ định là bệnh xương thủy tinh gen type VII (OMIM 610682). Ở những bệnh nhân khác bị bệnh xương thủy tinh gen type VII, một đột biến đồng hợp tử trong CRTAP đã được xác định, trong đó sự thay đổi của một nucleotide (c.472–1021C ⇒ G) tạo ra một vị trí nối donor ẩn dẫn đến việc bao gồm 73 bp của intron 1 vào bộ gen của CRTAP do đó kéo dài exon 2. Đột biến này dẫn đến sự phân hủy nhanh hơn của CRTAP và do đó dẫn đến giảm 3-hydroxyl hóa proline 986 trong collagen xương 1A1. Bệnh xương thủy tinh gen type VII đã được xác định ở một quần thể người Mỹ bản địa ở phía bắc Quebec. Về mặt lâm sàng, đây là một rối loạn lặn trên nhiễm sắc thể thường trong đó gãy xương có khi sinh; tần suất gãy xương thường giảm dần theo tuổi—đặc biệt là sau tuổi vị thành niên; củng mạc hơi xanh; có sự biến dạng xương tiến triển dẫn đến rút ngắn các chi ở gốc và hạn chế đi lại.
Các biến thể mất chức năng trong PLOD1 (OMIM 153454) làm suy giảm quá trình hydroxyl hóa lysine trong các vùng xoắn của collagen type I và đã được liên kết với hội chứng Ehlers-Danlos, type 1 gù vẹo cột sống (OMIM 225400). Sự bất hoạt của PLOD2 (OMIM 601865) dẫn đến sự liên kết của bệnh xương thủy tinh, co cứng khớp bẩm sinh và màng bơi (hội chứng Bruck 2, OMIM 609222). (Hội chứng Bruck 1 [OMIM 259450] đã được cho là do các biến thể không hoạt động của FKBP10 [OMIM 607063], một protein chaperone cần thiết cho sự di chuyển của các propeptide collagen type I qua nguyên bào xương.) Các đột biến hai alen có hại (p.Phe249Leu; c.34G > C) trong BMP1 (OMIM 112264) ngăn chặn sự loại bỏ các propeptide đầu carboxyl của collagen 1α1 và collagen 1α2 và do đó sự hình thành sợi collagen type I, dẫn đến một dạng bệnh xương thủy tinh nặng vừa (gen type XIII; type lâm sàng 3). Bệnh xương thủy tinh gen type XIII có liên quan đến đa gãy xương hàng năm, khối lượng xương ở mức giới hạn thấp hoặc tăng theo kiểm tra DXA, chậm tăng trưởng đáng kể, củng mạc xanh nhạt và răng bình thường. BMP1 là một protein đa chức năng; một trong những hoạt động quan trọng nhất của nó là một endoproteinase propeptide đầu C của procollagen type I. Sự bất hoạt của BMP1 làm suy giảm sự loại bỏ proteolytic của propeptide đầu carboxyl khỏi procollagen type I và sự lắp ráp bình thường của các sợi collagen type I trưởng thành. Điều thú vị là, dạng bệnh xương thủy tinh này có thể liên quan đến BMD theo diện tích cột sống thắt lưng ở mức giới hạn thấp hoặc thậm chí tăng theo DXA mặc dù việc điều trị bằng bisphosphonate đã hữu ích về mặt lâm sàng và X-quang. Về vấn đề này, cần lưu ý rằng những bệnh nhân bị bệnh xương thủy tinh gen type I/type lâm sàng 1 nhẹ do các đột biến ở các vị trí phân cắt propeptide đầu carboxyl của procollagen type I, cũng có BMD cột sống thắt lưng bình thường hoặc tăng theo cả kiểm tra DXA và pQCT. Các đột biến dị hợp tử trong IFITM5 (OMIM 614757) dẫn đến bệnh xương thủy tinh có mức độ nghiêm trọng vừa (gen type V; type lâm sàng 5) được đặc trưng bởi sự hình thành callus tăng sản đau tại các vị trí gãy xương, vôi hóa màng giữa các xương quay và trụ liền kề, và một dải hành xương đặc bên dưới các đĩa tăng trưởng của các xương dài.
Bệnh xương thủy tinh gen type II/type lâm sàng 2 là một rối loạn gây tử vong trong giai đoạn chu sinh hoặc đầu thời thơ ấu. Nó thường là kết quả của các đột biến dị hợp tử mới trong COL1A1 hoặc COL1A2 với sự thay thế một axit amin khác cho glycine trong các miền xoắn ba của các chuỗi procollagen α1(I)/α2(I) (COL1A1—p.Gly94Cys, p.Gly391Arg, p.Gly1003Ser; COL1A2—p.Gly547Asp, p.Gly865Ser, p.Gly976Asp); các đột biến này dẫn đến sự tổng hợp các chuỗi procollagen bất thường liên kết và do đó bất hoạt các peptide procollagen nguyên vẹn theo một cách trội-âm, làm giảm nghiêm trọng sự tổng hợp collagen type I nguyên vẹn. Bệnh xương thủy tinh type lâm sàng 2 được biểu hiện bằng gãy xương trong tử cung, dị tật xương dài, khoáng hóa vòm sọ rất ít, và tử vong do suy hô hấp. Các kiểu hình gây tử vong của bệnh xương thủy tinh cũng liên quan đến các đột biến mất chức năng đồng hợp tử trong LEPRE, CRTAP, PPIB, và CREB3L1, gen sau mã hóa protein 3-like liên kết yếu tố đáp ứng cAMP (OMIM 616215) (xem Bảng 20.10C). Bệnh xương thủy tinh gen type III (OMIM 259420) là một tính trạng trội trên nhiễm sắc thể thường do các đột biến điểm hoặc dịch khung trong COL1A1 (p.Gly154Arg, p.Gly844Ser) hoặc COL1A2 (p.Gly526Cys). Bệnh xương thủy tinh type lâm sàng 3 được đặc trưng bởi gãy xương tái phát dẫn đến các dị tật xương tiến triển thường rõ ràng khi sinh, gù vẹo cột sống, tầm vóc cực kỳ thấp, củng mạc xanh nhạt dần theo tuổi, răng bất thường (ở 80% trẻ em < 10 tuổi), và mất thính lực; nó có thể do các biến thể của nhiều gen gây ra (xem Bảng 20.10B). Bệnh xương thủy tinh type lâm sàng 4 (gen type IV, OMIM 166220) là một bệnh trội trên nhiễm sắc thể thường thường liên quan nhất đến các đột biến điểm hoặc mất đoạn nhỏ trong COL1A2 (Gly586Val, Gly646Cys, Gly1012Arg) và đôi khi trong COL1A1 (p.Gly175Cys, p.Gly832Ser). Nó có mức độ nghiêm trọng thay đổi với thời gian sống kéo dài, dị tật xương nhẹ đến vừa, tầm vóc thấp, củng mạc bình thường, bệnh tạo ngà không hoàn hảo và mất thính lực. Bệnh xương thủy tinh type lâm sàng 4 cũng có thể do các biến thể trong CRTAP, PPIB, WNT1, TMEM38B, hoặc SP7 gây ra. Bệnh xương thủy tinh type lâm sàng 5 (gen type V, OMIM 610967) không chỉ biểu hiện bằng sự mong manh của xương mà còn bởi sự trật khớp của đầu xương quay, sự hình thành callus quá mức tại các vị trí gãy xương đang lành và vôi hóa màng gian cốt giữa các xương quay/trụ và chày/mác liền kề và hiện chỉ liên quan đến các biến thể của IFITM5 mã hóa protein xuyên màng-5 do IFN gây ra (OMIM 614757, chr. 11p15.5). Các đặc điểm lâm sàng khác của bệnh xương thủy tinh gen type V là di truyền trội trên nhiễm sắc thể thường, độ mong manh của xương từ vừa đến nặng (điểm Z của BMD theo diện tích cột sống thắt lưng dao động từ –7,7 đến –0,7), chậm tăng trưởng từ nặng đến nhẹ (điểm Z của chiều cao người lớn thay đổi từ –8,7 đến –0,1), trật khớp đầu xương quay, củng mạc trắng và phát triển răng bình thường. Về mặt mô học, có sự sắp xếp không đều của các phiến. Đột biến thường gặp nhất trong IFITM5 bằng giải trình tự toàn bộ exome là một chuyển đoạn dị hợp tử c.–14C > T trong vùng không dịch mã 5′ của nó. Đột biến này nằm ở 14 bp thượng nguồn của codon khởi đầu tham chiếu và tạo ra một tín hiệu khởi đầu mới bổ sung năm aa (Met-Ala-Leu-Glu-Pro) vào đầu amino của IFITM5, làm tăng chiều dài của nó từ 132 lên 137 aa. Biến thể IFITM5 p.Ser40Leu cũng đã được xác định ở những bệnh nhân bị bệnh xương thủy tinh gen type V, mặc dù không có các đặc điểm cổ điển của nó là vôi hóa gian cốt và hình thành callus quá mức. IFITM5 là một protein được biểu hiện nhiều trong màng nguyên bào xương trong quá trình phát triển phôi và sau sinh của bộ xương; nó có các đầu amino và carboxyl ngoại bào, hai miền xuyên màng và một miền xoắn nội bào. IFITM5 mã hóa một yếu tố biệt hóa nguyên bào xương có liên quan đến việc vận chuyển và gấp nếp protein; về mặt thực nghiệm, sự mất Ifitm5 dẫn đến giảm sự hình thành xương, đặc biệt là trong tử cung và giảm biểu hiện của SERPINF1 (OMIM 172860). Sự biểu hiện giảm của SERPINF1 dẫn đến bệnh xương thủy tinh gen type VI di truyền lặn trên nhiễm sắc thể thường (OMIM 610968) (type lâm sàng 3) và giảm tổng hợp sản phẩm của nó là yếu tố có nguồn gốc từ biểu mô sắc tố (PEDF). PEDF là một protein chaperone, có nồng độ có thể được đo trong huyết thanh, điều hòa sự biệt hóa của nguyên bào xương và khoáng hóa xương; nó được tổng hợp bởi các tế bào sụn, nguyên bào xương và hủy cốt bào; PEDF cản trở chức năng của yếu tố tăng trưởng nội mô mạch máu (VEGF), một protein tăng cường sự di chuyển của các nguyên bào xương và hủy cốt bào vào sụn; PEDF có thể điều hòa khoáng hóa xương bằng cách tăng tổng hợp osteoprotegerin, do đó cản trở sự hình thành hủy cốt bào. Bệnh xương thủy tinh gen type VI (OMIM 610968) có kiểu hình tương tự như gen type IV và được đặc trưng về mặt lâm sàng bởi sự mong manh của xương nghiêm trọng lần đầu tiên biểu hiện sau 6 tháng tuổi và về mặt vi thể bởi dư thừa chất dạng xương không được khoáng hóa, và một kiểu phiến của chất nền xương “vảy cá” phù hợp với một khiếm khuyết trong khoáng hóa xương. Các đột biến mất chức năng trong SERPINF1 có thể cản trở sự hình thành xương trong khi tăng tạo hủy cốt bào và hòa tan xương. Nồng độ PEDF trong huyết thanh không thể phát hiện được ở những bệnh nhân bị bệnh xương thủy tinh gen type VI, cung cấp một công cụ chẩn đoán cho bệnh này. Điều thú vị là, những bệnh nhân bị bệnh xương thủy tinh gen type VI không đáp ứng với điều trị bằng bisphosphonate cũng như những bệnh nhân mắc các dạng khác của bệnh không đồng nhất này.
Bệnh xương thủy tinh gen type XI (type lâm sàng 3) là kết quả của các đột biến bất hoạt trong FKBP10 (protein liên kết FK506 10, OMIM 607063) mã hóa một protein lưới nội chất (FKBP65) cũng là một chaperone cho procollagen type I—một protein cần thiết cho quá trình hydroxyl hóa các gốc lysine trong telopeptide của nó, một sửa đổi cần thiết cho sự liên kết chéo, cũng như sự di chuyển và bài tiết qua tế bào của nó. Khi không có FKBP65 chức năng, procollagen type I tích tụ trong lưới nội chất của nguyên bào xương. Rối loạn này được di truyền như một tính trạng lặn trên nhiễm sắc thể thường bắt đầu ở trẻ sơ sinh; bệnh nhân bị ảnh hưởng bị gãy xương dài lặp đi lặp lại và tiến triển đến mức phải ngồi xe lăn ở đầu thời thơ ấu; họ có thể hoặc không thể phát triển co cứng khớp nhưng không biểu hiện bệnh tạo ngà không hoàn hảo. Các đột biến trong FKBP10 cũng đã được ghi nhận ở những bệnh nhân mắc hội chứng Bruck 1 (OMIM 259450—màng bơi, co cứng bẩm sinh, gãy xương ở trẻ sơ sinh dẫn đến dị tật chi, chậm tăng trưởng, vẹo cột sống) cho thấy rằng bệnh xương thủy tinh gen type XI và hội chứng Bruck 1 có khả năng là các rối loạn alen mà các biểu hiện lâm sàng phụ thuộc vào vị trí đột biến và các yếu tố sửa đổi khác.
Sự tiết Collagen và Căng thẳng của Lưới nội chất
Bệnh xương thủy tinh gen type X là một rối loạn di truyền lặn trên nhiễm sắc thể thường do các đột biến bất hoạt hai alen trong SERPINH1 (OMIM 600943) mã hóa một peptide đa chức năng 418 aa được gọi là protein liên kết collagen type 1 (CBP) 2 còn được gọi là protein sốc nhiệt (HSP) 47. Bệnh xương thủy tinh gen type X (type lâm sàng 3) là một rối loạn nghiêm trọng với các gãy xương xảy ra khắp bộ xương bắt đầu từ trong tử cung. CBP2/HSP47 cần thiết cho tổ chức sụn, sự hình thành xương nội sụn, và sự chế tạo và duy trì tính toàn vẹn của cấu trúc xoắn ba của procollagen type I, sự di chuyển và bài tiết qua tế bào của nó, và khả năng chống lại sự phân hủy proteolytic của nó. Các biến thể hai alen của CREB3L1 (OMIM 616215), một thành phần của phản ứng căng thẳng lưới nội chất trong các nguyên bào xương, đã được xác định ở các anh chị em thai nhi bị bệnh xương thủy tinh nặng (gen type XVI, type lâm sàng 2). Các đột biến trong vị trí protease yếu tố phiên mã liên kết màng 2 (MBTPS2, OMIM 300294) (mã hóa một metalloproteinase kẽm điều hòa sự phân giải protein trong màng của một số yếu tố phiên mã điều hòa sự bài tiết collagen type 1 và cũng tham gia vào phản ứng căng thẳng của lưới nội chất) được di truyền như một tính trạng liên kết X và dẫn đến bệnh xương thủy tinh có mức độ nghiêm trọng vừa (gen type XIX, type lâm sàng 3). Các biến thể hai alen của protein xuyên màng 38B (TMEM38B, OMIM 611236) mã hóa một kênh kali nội bào làm thay đổi nồng độ Ca2+ nội bào ảnh hưởng xấu đến việc giải phóng procollagen type I từ các nguyên bào xương (và do đó làm tăng sự tích tụ procollagen type I trong lưới nội chất của nguyên bào xương, do đó làm suy giảm chức năng tiếp theo của nó và dẫn đến căng thẳng lưới nội chất) đã được chỉ định là bệnh xương thủy tinh gen type XIV. TMEM38B mã hóa kênh cation nội bào trimeric type B (TRICB) 291 aa, một thành phần của TRIC, một kênh cation đơn hóa trị quan trọng đối với việc giải phóng Ca2+ từ các vị trí lưu trữ nội bào, chẳng hạn như mạng lưới sarcoplasmic và lưới nội chất và do đó duy trì nồng độ Ca2+ trong tế bào chất, một quá trình cần thiết cho sự biệt hóa, phân chia và chức năng của tế bào bình thường (bao gồm cả của nguyên bào xương). Bệnh xương thủy tinh gen type XIV là một rối loạn lặn trên nhiễm sắc thể thường có mức độ nghiêm trọng thay đổi (các type lâm sàng 3, 4) với các gãy xương xảy ra trong tử cung hoặc trong thời thơ ấu nhưng với răng, thính giác và củng mạc bình thường đã được xác định ở các gia đình Trung Đông.
Mặc dù không được chỉ định là nguyên nhân của bệnh xương thủy tinh, một số biến thể gen dẫn đến sự mong manh của xương khác với những biến thể được định nghĩa như vậy (xem Bảng 20.10C), bao gồm những biến thể dẫn đến hội chứng Cole-Carpenter 1 (OMIM 112240) và 2 (OMIM 616294) được đặc trưng về mặt lâm sàng bởi gãy xương dài trước và sau sinh, dính khớp sọ trán và vành, lồi mắt, não úng thủy và đầu to. Hội chứng Cole-Carpenter 1 là kết quả của các biến thể bất hoạt của procollagen-proline, 2-oxoglutarate-4-dioxygenase, tiểu đơn vị beta (P4HB, OMIM 176790) dẫn đến giảm hydroxyl hóa các gốc proline trong preprocollagen type 1. Hội chứng Cole-Carpenter 2 là hậu quả của các biến thể bất hoạt của SEC24D (họ gen liên quan đến SEC24, thành viên D, OMIM 607186) mã hóa một thành phần của các túi nguyên bào xương vận chuyển/xuất khẩu protein từ lưới nội chất trong nguyên bào xương, tạo điều kiện cho sự di chuyển của các phân tử procollagen type 1.
Đánh giá Trẻ em và Thanh thiếu niên có Khối lượng xương thấp
Việc xác định sinh bệnh học của sự giảm khoáng hóa xương ở trẻ em được bắt đầu bằng việc xem xét cẩn thận tiền sử y khoa chung của bệnh nhân, cũng như tiền sử liên quan đến hệ xương bắt đầu từ trước khi sinh cùng với tiền sử của các thành viên gia đình trực tiếp qua nhiều thế hệ, sau đó là ghi nhận các phát hiện thực thể (tầm vóc, dị tật xương, màu củng mạc và tình trạng răng), tình trạng sinh hóa (đo canxi, magiê, phốt phát, creatinin, phosphatase kiềm, calcidiol, calcitriol huyết thanh và các nghiên cứu lâm sàng liên quan khác), các phim X-quang thích hợp, đánh giá khoáng hóa xương, và cuối cùng là kiểm tra tính toàn vẹn của các gen liên quan đến sự hình thành và duy trì bộ xương. Các dạng còi xương khác nhau nên được loại trừ cũng như các rối loạn được biết là có liên quan đến khoáng hóa xương dưới mức tối ưu, chẳng hạn như chán ăn tâm thần, hen suyễn được điều trị bằng glucocorticoid, các bệnh khớp và xương thấp khớp, các rối loạn viêm mạn tính (ví dụ, bệnh viêm ruột), và các rối loạn tân sinh và điều trị của chúng. Giảm khoáng hóa xương cũng có thể xảy ra ở những bệnh nhân bị rối loạn vận động (ví dụ, bại não, loạn dưỡng cơ, teo cơ tủy sống) và những người bị các bệnh nội tiết (thiếu GH, tiếp xúc quá mức hoặc sản xuất quá mức glucocorticoid, thiếu hụt hormone sinh dục, đái tháo đường type 1), các bệnh huyết học (thalassemia, bệnh hồng cầu hình liềm, bệnh bạch cầu), các bất thường toàn thân (xơ nang), và suy thận mạn trước và/hoặc sau khi ghép thận. Loãng xương do bệnh xương thủy tinh thường được biểu hiện lâm sàng rõ ràng nhất bởi sự xuất hiện của các gãy xương không do chấn thương xảy ra trong tử cung, sau sinh, trong thời thơ ấu, hoặc ở tuổi vị thành niên và được xác nhận bằng đánh giá X-quang của bộ xương và xác định BMC và nồng độ.
Do đó, việc đánh giá trẻ/thanh thiếu niên có tiền sử gãy xương do tác động thấp (được định nghĩa là hai hoặc nhiều gãy xương dài do ngã từ khoảng cách nhỏ hơn chiều cao đứng của bệnh nhân trước 10 tuổi, hoặc ba hoặc nhiều hơn trước 19 tuổi, hoặc gãy đốt sống không có bệnh tại chỗ hoặc chấn thương đáng kể) bắt đầu bằng việc xem xét tiền sử và khám thực thể nhằm xác định các yếu tố có thể ảnh hưởng xấu đến sự hình thành, khoáng hóa và sức mạnh của xương. Gãy xương chày và thân xương đùi thường xảy ra ở trẻ em bị bệnh xương thủy tinh. Ngoài các ảnh hưởng di truyền và nội tiết tố, các yếu tố quan trọng nhất cần thiết cho sự tích lũy và duy trì khối lượng xương bình thường cần được đánh giá là những yếu tố liên quan đến chế độ ăn (ăn đủ canxi và protein; không bị chán ăn tâm thần), duy trì dự trữ vitamin D bình thường bằng cách tiếp xúc với ánh sáng mặt trời hoặc uống thuốc bổ, vận động bình thường với các bài tập chịu trọng lượng nhất quán, và sự hiện diện của các bệnh mạn tính, đặc biệt là những bệnh có thể được điều trị bằng glucocorticoid (ví dụ, hen suyễn, các trạng thái viêm mạn tính) có thể làm suy giảm sự phát triển của xương. Việc kiểm tra mô hình tăng trưởng của bệnh nhân để xác định xem sự tăng trưởng tầm vóc có bình thường hay không và cân nặng bình thường theo tầm vóc và giới tính đã đạt được và duy trì hay chưa và để đánh giá bằng các giai đoạn khám thực thể của sự trưởng thành sinh dục nguyên phát và thứ phát là rất cần thiết. Việc xác định sự trưởng thành của xương (tuổi xương) hữu ích để xác định xem trẻ có đang phát triển phù hợp với tiềm năng di truyền của mình hay không. Nếu có liên quan, các bệnh toàn thân, chẳng hạn như bệnh thận mạn tính, bệnh celiac, bệnh viêm ruột, và các bệnh nội tiết nên được loại trừ. Đánh giá khoáng hóa xương theo diện tích và mật độ khoáng xương biểu kiến thường được thực hiện bằng DXA ở trẻ em với việc tham chiếu đến dữ liệu tham chiếu cụ thể cho tuổi theo thời gian, giới tính và dân tộc/tổ tiên, đồng thời cũng xem xét các yếu tố như chiều cao, giai đoạn dậy thì và sự trưởng thành của xương. Trong một loạt 304 trẻ em và thanh thiếu niên trải qua các nghiên cứu khoáng hóa xương bằng DXA, 36% đang thực hiện vì tiền sử gãy xương, 27% vì suy sinh dục, và 22% vì bệnh đường tiêu hóa (celiac, viêm ruột). Chỉ số khối cơ thể thấp và dự trữ vitamin D thấp là những yếu tố dự báo quan trọng nhất của BMD dưới mức bình thường (<–2 độ lệch chuẩn theo tuổi và giới). Tùy thuộc vào từng bệnh nhân, việc đo canxi, phốt phát, phosphatase kiềm, creatinin, PTH, calcidiol (25OHD) và các hormone sinh sản trong huyết thanh, cũng như các chất phân tích đặc hiệu cho bệnh (ví dụ, đối với bệnh viêm ruột hoặc bệnh celiac) có thể được chỉ định. Các dấu hiệu chu chuyển xương có giá trị chẩn đoán ở mức giới hạn ở trẻ em, mặc dù hữu ích như các chỉ số đáp ứng điều trị.
Các biểu hiện lâm sàng của bệnh xương thủy tinh thay đổi từ nhẹ đến vừa, nặng hoặc gây tử vong. Các phát hiện X-quang ở các đối tượng bị bệnh xương thủy tinh bao gồm, ngoài khối lượng xương thấp lan tỏa, vỏ xương mỏng, loe hành xương, gãy xương và các dị tật xương do đó, xương sọ wormian (thường xuyên nhưng không phải là dấu hiệu bệnh lý của bệnh xương thủy tinh), nền sọ dẹt có thể chèn ép tiểu não trên, lún đốt sống và khung chậu ba tia. Ở trẻ em bị bệnh xương thủy tinh, đo mật độ xương, thường nhưng không phải luôn luôn, cho thấy khoáng hóa giảm, mức độ của nó tương quan ở một mức độ nào đó với các biểu hiện lâm sàng. Chẩn đoán bệnh xương thủy tinh được thiết lập bằng các tiêu chí lâm sàng và được xác nhận bằng việc xác định kiểu gen của COL1A1 và/hoặc COL1A2 hoặc (các) gen thích hợp khác, mặc dù việc không phát hiện ra một đột biến di truyền không nhất thiết loại trừ rối loạn này nhưng gợi ý rằng cần có sự điều tra di truyền sâu rộng hơn (giải trình tự toàn bộ exome hoặc toàn bộ bộ gen). Nồng độ PEDF trong huyết thanh không thể phát hiện được là dấu hiệu của bệnh xương thủy tinh do sự bất hoạt của SERPINF1. Đôi khi, sinh thiết mào chậu và kiểm tra mô học của xương có thể cần thiết để phân loại phụ của rối loạn. Bệnh xương thủy tinh loại lâm sàng 2 có thể được xác định trước sinh bằng siêu âm thai nhi; các loại khác có thể được xác định trước sinh bằng cách phân tích collagen được tổng hợp bởi các tế bào được nuôi cấy từ các sinh thiết nhung mao màng đệm và bằng các phân tích di truyền. Được bao gồm trong chẩn đoán phân biệt của gãy xương tái phát ở trẻ em là lạm dụng trẻ em, các dạng còi xương khác nhau bao gồm giảm phosphatase máu, hội chứng McCune-Albright của loạn sản xơ (OMIM 174800), bệnh Paget vị thành niên và loãng xương vị thành niên.
Các phát hiện lâm sàng đáng chú ý khác ở bệnh nhân bị bệnh xương thủy tinh bao gồm: suy giảm thính lực tiến triển—có ở 40% đến 60% đối tượng, tăng độ mềm dẻo của khớp ở có lẽ 60% đến 70% bệnh nhân có thể dẫn đến trật khớp và/hoặc đứt gân, và các tổn thương khớp sọ-cổ. Các dị tật sọ-cổ xảy ra ở khoảng 30% bệnh nhân bị bệnh xương thủy tinh và có thể được phân loại thành: lún nền sọ, ấn nền sọ và nền sọ dẹt—biến chứng phổ biến nhất trong số này. Sự hiện diện của bệnh tạo ngà không hoàn hảo, tầm vóc thấp đáng kể và BMD trục thắt lưng rất thấp có liên quan chặt chẽ đến các bất thường nền sọ. Các triệu chứng/dấu hiệu của sự liên quan đến sọ là đau đầu khi cử động, ho hoặc hắt hơi; đau dây thần kinh sinh ba; yếu cánh tay/chân; và khó giữ thăng bằng. Việc sàng lọc các đối tượng bị bệnh xương thủy tinh cho các biến chứng này là rất cần thiết. Cần nhấn mạnh lại rằng khi không có gãy đốt sống, chẩn đoán loãng xương không được thiết lập chỉ bằng dữ liệu DXA mà cần có tiền sử lâm sàng tương ứng và phù hợp của bệnh liên quan và nguy cơ gãy xương được ghi nhận tăng lên. Các phép đo BMD theo diện tích nối tiếp trong khoảng thời gian từ 6 đến 12 tháng hữu ích trong việc xác định xu hướng khoáng hóa xương ở trẻ em mắc các bệnh có nguy cơ phát triển loãng xương.
Quản lý Trẻ em và Thanh thiếu niên có Khối lượng xương thấp/Bệnh xương thủy tinh
Chìa khóa để ngăn ngừa trẻ em và thanh thiếu niên phát triển khối lượng xương thấp là cung cấp một chế độ ăn đủ canxi và vitamin D, khuyến khích các hoạt động và tập thể dục chịu trọng lượng, và tránh tiếp xúc với các tác nhân có thể cản trở sự tích lũy khoáng chất xương bình thường. Sau khi đánh giá và khi cần thiết và phù hợp, các can thiệp có thể bao gồm điều trị một bệnh hệ thống hoặc bệnh nội tiết đi kèm có thể có ý nghĩa sinh bệnh học trong sự phát triển của khối lượng xương thấp, chẳng hạn như loại bỏ, chẳng hạn như loại bỏ hoặc giảm liều glucocorticoid ở trẻ bị hen suyễn. Các tác nhân điều trị được sử dụng trong điều trị bệnh nhân có khối lượng xương thấp làm tăng khối lượng xương bằng cách ức chế tái hấp thu xương (thuốc chống tái hấp thu hoặc chống tái tạo) hoặc bằng cách kích thích hình thành xương (các tác nhân đồng hóa). Ở người lớn, các loại thuốc chống tái hấp thu được sử dụng rộng rãi nhất là hormone sinh dục, các chất điều biến thụ thể estrogen chọn lọc, denosumab (một kháng thể đơn dòng liên kết và ức chế sự hình thành hủy cốt bào qua trung gian RANKL—do đó bắt chước tác dụng của osteoprotegerin), và bisphosphonate. Các chất điều biến thụ thể estrogen chọn lọc (SERMS), chẳng hạn như triphenylethylene, benzothiophene, hoặc các hợp chất liên quan đến naphthalene (ví dụ, raloxifene) liên kết với ái lực cao với thụ thể estrogen α ở các mô cụ thể, nơi chúng làm thay đổi cấu hình ba chiều của thụ thể và tuyển dụng các nhóm đồng yếu tố chọn lọc theo mô, do đó hoặc làm giảm (vú, não) hoặc gây ra (xương) chức năng của thụ thể ở các vị trí được nhắm mục tiêu. SERM làm giảm sự hình thành hủy cốt bào chủ yếu ở các vị trí xương bè, nhưng hiệu quả của chúng trong việc tăng BMD thấp hơn so với chính estrogen. Mặc dù calcitonin cá hồi dạng xịt mũi ức chế trực tiếp chức năng của hủy cốt bào và có tác dụng phục hồi xương khiêm tốn, nó hiếm khi được sử dụng để điều trị loãng xương ở người lớn hoặc trẻ em. Các loại thuốc đồng hóa xương bao gồm teriparatide (PTH 1-34), abaloparatide (PTH 1-34), và romosozumab (một kháng thể đơn dòng được nhân hóa chống lại sclerostin, một chất ức chế sự biệt hóa của nguyên bào xương qua trung gian LRP5/6- WNT-β-catenin). Mặc dù được phê duyệt để điều trị loãng xương ở người lớn, denosumab và romosozumab đang chờ phát triển và đánh giá thêm ở trẻ em bị loãng xương do các nguyên nhân sinh bệnh khác nhau. Mặc dù đã được chứng minh là hữu ích trong điều trị người lớn bị loãng xương, cả teriparatide (PTH 1-34) và abaloparatide (PTH 1-34) đều có nguy cơ tiềm ẩn gây ra u xương ác tính và không được sử dụng ở trẻ em.
Trẻ sơ sinh/trẻ em/thanh thiếu niên bị bệnh xương thủy tinh cần được chăm sóc bởi một đội ngũ các nhà nội tiết học, bác sĩ phẫu thuật chỉnh hình và bác sĩ phục hồi chức năng có kinh nghiệm và các cộng sự chăm sóc sức khỏe tương ứng của họ. Các dịch vụ phục hồi chức năng và vật lý trị liệu để cải thiện sức mạnh cơ bắp và khả năng vận động trong giới hạn của sự mong manh của xương được khuyến khích cũng như các bài tập được bảo vệ—chẳng hạn như đi bộ và bơi lội. Bệnh nhân bị bệnh xương thủy tinh nguyên phát (và các dạng loãng xương thứ phát) có gãy đốt sống và/hoặc xương dài được ghi nhận thường được điều trị bằng bisphosphonate. Bisphosphonate là các hợp chất liên quan đến pyrophosphate, trong đó cầu nối phân tử oxy liên kết giữa hai gốc phosphate được thay thế bằng một liên kết carbon mà hai chuỗi bên được gắn vào; một chuỗi bên thường là một nhóm hydroxyl hoặc nguyên tử clo cùng với các gốc phosphate liên kết chặt chẽ với hydroxyapatite và bao phủ bề mặt xương; chuỗi bên thứ hai có thể “đơn giản” và chứa các nguyên tử clo hoặc lưu huỳnh hoặc phức tạp và nặng hơn với các nguyên tử nitơ và các cấu trúc vòng bao gồm carbon và oxy (Hình 20.14). Khi một chuỗi bên gắn vào nguyên tử carbon trung tâm là một nhóm hydroxyl (OH–), sự liên kết của bisphosphonate với hydroxyapatite tăng lên và độ hòa tan của pha khoáng của xương giảm, do đó làm giảm sự tái hấp thu hydroxyapatite. Chuỗi bên thứ hai quyết định hiệu lực và tuổi thọ của bisphosphonate. Các bisphosphonate chứa nitơ như một thành phần trong một trong các chuỗi bên có hiệu lực cao hơn đáng kể so với các bisphosphonate “đơn giản”. Trong axit zoledronic, một bisphosphonate thường được sử dụng ở trẻ em do hồ sơ an toàn và thời gian tác dụng kéo dài, chuỗi bên thứ hai là CH2-imidazole.
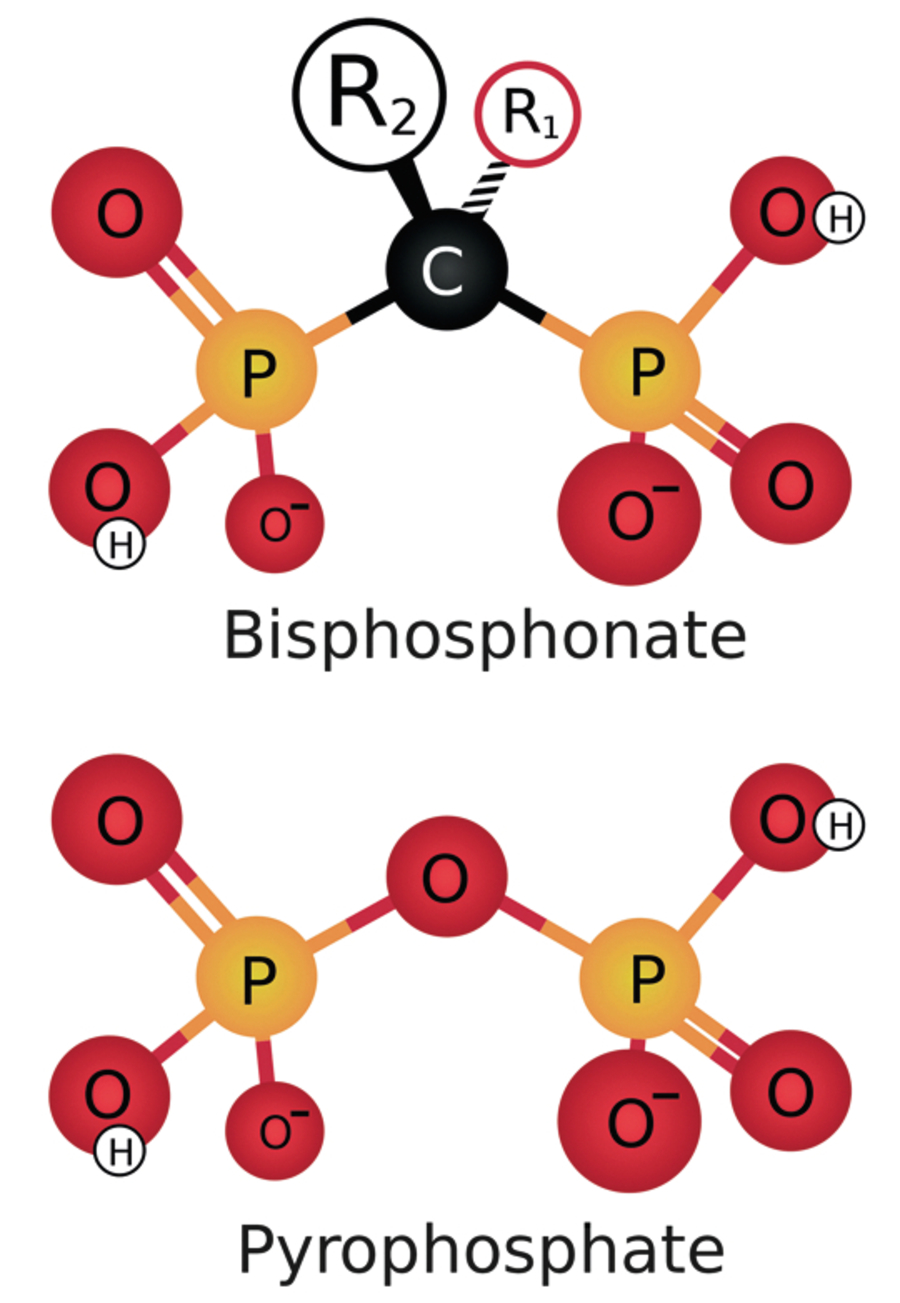
Hình 20.14: Bisphosphonate là các chất tương tự của pyrophosphate trong đó carbon đã được thay thế cho cầu nối oxy giữa hai nhóm phosphate; hai chuỗi bên (R1, R2) được gắn vào nguyên tử carbon: R1 có thể là một nhóm hydroxyl hoặc nguyên tử clo cùng với các gốc phosphate liên kết chặt chẽ với hydroxyapatite và bao phủ bề mặt xương; R2 có thể chứa các nguyên tử clo hoặc lưu huỳnh hoặc nó có thể bao gồm các cấu trúc vòng chứa carbon, oxy và nitơ.
Bisphosphonate tạo phức (liên kết) với các ion canxi của hydroxyapatite và do đó được nhắm mục tiêu vào xương; trong hốc tái hấp thu bên dưới một hủy cốt bào, bisphosphonate phân ly khỏi hydroxyapatite khi pH bị hạ thấp do sự bài tiết H+ của hủy cốt bào và sau đó được nhập bào vào bên trong hủy cốt bào. Sau khi bisphosphonate được hủy cốt bào hấp thụ, chúng ức chế khả năng sản xuất năng lượng của tế bào, dẫn đến cái chết của chúng và do đó làm giảm sự tái hấp thu và tái tạo xương mà không làm suy giảm sự tổng hợp xương mới qua trung gian nguyên bào xương với hậu quả là tăng khối lượng và sức mạnh của xương. Bên trong hủy cốt bào, bisphosphonate cản trở chức năng của hủy cốt bào bằng cách đẩy nhanh cái chết của chúng bằng hai cơ chế: (1) các bisphosphonate “đơn giản”, chẳng hạn như medronate, etidronate và clodronate được chuyển hóa thành các chất tương tự của ATP cản trở việc giải phóng phosphate và do đó cản trở việc tạo ra năng lượng; ngoài ra, các bisphosphonate đơn giản ngăn cản sự di chuyển của ATP vào ty thể của hủy cốt bào, hậu quả là ty thể tan rã và quá trình chết theo chương trình của hủy cốt bào được bắt đầu—do đó cản trở thêm sự tái hấp thu xương. (2) Các bisphosphonate lớn, chứa nitơ (pamidronate, axit zoledronic) hoạt động bằng cách làm suy giảm quá trình tổng hợp cholesterol bằng cách ức chế hoạt động của farnesyl pyrophosphate synthase trong các hủy cốt bào. Enzyme này cần thiết cho quá trình tổng hợp cholesterol thông qua con đường mevalonate; sự ức chế của nó ngăn cản quá trình prenyl hóa protein, một sửa đổi sau dịch mã cho phép các protein được prenyl hóa tương tác với các protein và liên kết với màng tế bào. Sự ức chế con đường này trong các hủy cốt bào làm suy giảm sự hình thành của bờ diềm và các vòng actin, sự di chuyển của các sản phẩm của hủy cốt bào vào hốc tái hấp thu, và sự tái hấp thu các sản phẩm xương bị phân hủy—tất cả các chức năng chuyển hóa của hủy cốt bào cần thiết cho sự tái hấp thu xương. Hoạt động sinh học của một bisphosphonate đối với chức năng của hủy cốt bào được quan sát thấy ngay sau khi dùng vì nồng độ canxi huyết thanh giảm nhanh chóng; thực tế, tác dụng nhanh chóng này đã được sử dụng trong điều trị bệnh nhân tăng canxi máu. Các tác dụng của bisphosphonate đối với xương kéo dài rất lâu sau khi ngừng sử dụng tác nhân (thời gian “lưu trú”), cho phép một số hợp chất được dùng không thường xuyên như mỗi năm một lần hoặc nửa năm một lần (ví dụ, axit zolendronic). Bởi vì các bisphosphonate tồn tại trong xương trong những khoảng thời gian cực kỳ dài, các tác dụng lâu dài của chúng là tích lũy. Phân tích hình thái mô học đã cho thấy rằng sự ức chế tái hấp thu xương được kích thích bởi hủy cốt bào qua trung gian bisphosphonate làm tăng khoáng hóa xương bằng cách giảm số lượng các hốc tái hấp thu và do đó không gian tái tạo, bảo tồn kiến trúc xương xốp (bè), và giảm độ xốp của xương vỏ.
Bisphosphonate đã hữu ích trong việc cải thiện khoáng hóa ở trẻ em bị bệnh xương thủy tinh và các hội chứng liên quan, cũng như ở những trẻ bị loãng xương do glucocorticoid và những trẻ bị hạn chế vận động, chẳng hạn như loạn dưỡng cơ và bại não. Ở nhiều trẻ sơ sinh và trẻ em, pamidronate tiêm tĩnh mạch (0,5–1 mg/kg/liều trong ba ngày liên tiếp lên đến 2–15 mg/kg/năm được dùng mỗi 3 đến 6 tháng một lần cho một liều hàng năm là 9 mg/kg/năm) đã được sử dụng. Truyền tĩnh mạch axit zoledronic (liều đầu tiên 0,025 mg/kg trong 30 phút; liều 0,05 mg/kg ở + 6, 12, 18, 24 tháng; tiếp tục liều nếu cần 0,0125 mg/kg mỗi 6 tháng) đã được chứng minh là có hiệu quả trong việc tăng BMD, giảm tỷ lệ gãy xương và cải thiện khả năng vận động và sức khỏe ở bệnh nhân bị bệnh xương thủy tinh. Phân tích hình thái mô học của sinh thiết xương chậu sau khi dùng bisphosphonate cho trẻ em bị bệnh xương thủy tinh cho thấy sự gia tăng độ dày của xương vỏ và số lượng bè xương nhưng không tăng độ dày bè xương. Việc ngừng dùng bisphosphonate có thể dẫn đến tăng nguy cơ gãy xương giữa xương mới yếu hơn và xương được điều trị mạnh hơn. Do đó, nếu không có phản ứng bất lợi và có lợi ích lâm sàng tích cực (không có gãy xương mới, giảm đau xương, tăng khả năng vận động) và tăng BMD/BMC bằng các phép đo DXA nối tiếp, việc điều trị bằng bisphosphonate ở nửa liều có thể được tiếp tục cho đến khi đạt được chiều cao người lớn, tại thời điểm đó việc dùng bisphosphonate được ngừng lại và đánh giá lại toàn diện được thực hiện; kế hoạch này có thể được sửa đổi tùy thuộc vào phản ứng của cá nhân với thuốc điều trị. Các phác đồ điều trị cho trẻ em/thanh thiếu niên bị các dạng loãng xương thứ phát cũng đã được trình bày. Bisphosphonate cũng đã được sử dụng trong điều trị trẻ em bị tăng canxi máu kháng trị với các phương pháp quản lý khác (ví dụ, bù nước, thuốc lợi tiểu). Một tác dụng phụ dự kiến của việc dùng bisphosphonate là hạ canxi máu—được dự đoán bằng việc cung cấp canxi và vitamin D bổ sung trước và trong vài ngày sau khi dùng bisphosphonate điều trị. Để tránh các tác dụng toàn thân liên quan đến thuốc, chẳng hạn như sốt, khó chịu và đau cơ, việc dùng một tác nhân giảm đau và hạ sốt trong 48 đến 72 giờ sau khi dùng bisphosphonate thường được sử dụng. Các tác dụng phụ nghiêm trọng của bisphosphonate là không phổ biến nhưng bao gồm viêm mống mắt, gãy xương đùi không điển hình do giảm tái tạo xương, và hoại tử xương hàm, biến chứng sau không phổ biến ở trẻ em. Dữ liệu hạn chế cho thấy các bisphosphonate đường uống (alendronate, olpadronate) được dùng hàng ngày cũng làm tăng BMD theo diện tích ở trẻ em bị bệnh xương thủy tinh và bệnh mô liên kết nhưng với hiệu quả thấp hơn so với dùng đường tĩnh mạch. Tuy nhiên, hiện tại mặc dù bisphosphonate không được FDA chấp thuận để sử dụng ở trẻ em, chúng được sử dụng rộng rãi.
Các tác dụng phụ của bisphosphonate đã được ghi nhận cả cấp tính (sốt, đau cơ, đau bụng, nôn, hạ canxi máu) và mạn tính (các rối loạn viêm của mắt, hoại tử xương hàm ở người già, và bệnh “xương hóa đá” do thuốc gây ra) nhưng nhìn chung không xảy ra ở quần thể nhi khoa. Về mặt thực nghiệm và ở người lớn nhận liệu pháp dài hạn, bisphosphonate có thể ức chế chu chuyển xương và góp phần vào sự tăng khoáng hóa, loại sau dẫn đến giảm sức mạnh cơ học một cách nghịch lý và tăng nguy cơ gãy xương. Do đó, khi xem xét một đứa trẻ để điều trị bằng bisphosphonate, người ta phải đánh giá cẩn thận chẩn đoán chính và liệu khối lượng xương thấp và tần suất gãy xương của bệnh nhân có đáng để điều trị hay không khi xem xét các tác dụng phụ tiềm ẩn, mặc dù hiếm gặp, của bisphosphonate. Việc điều trị bằng bisphosphonate vài năm trước khi thụ thai dường như không có tác động bất lợi đến kết quả của thai nhi, nhưng việc điều trị trong khi mang thai bị chống chỉ định do độc tính có thể xảy ra.
Mặc dù không phù hợp để sử dụng ở trẻ em và thanh thiếu niên do lo ngại về khả năng gây ra u xương ác tính, PTH 1-34 (teriparatide) là một tác nhân đồng hóa xương được sử dụng rộng rãi ở người lớn. Khi được dùng liên tục, PTH 1-34 làm tăng sự hòa tan xương qua trung gian hủy cốt bào, nhưng khi được dùng ngắt quãng, nó có tác dụng đồng hóa đối với chức năng của nguyên bào xương và sự hình thành xương. Đặc tính này được sử dụng trên lâm sàng thông qua việc sử dụng teriparatide (PTH 1-34) và abaloparatide (PTHrP 1-34) trong điều trị phụ nữ sau mãn kinh bị loãng xương. Khi được dùng ngắt quãng với lượng nhỏ, cả hai tác nhân đều ưu tiên tăng tốc độ tái tạo xương và hình thành xương so với tốc độ tái hấp thu xương bằng các tác động trực tiếp lên sự biệt hóa, trưởng thành và tuổi thọ của nguyên bào xương. PTH 1-34 cũng tác động lên tế bào xương để làm giảm sản xuất sclerostin, một chất ức chế tổng hợp xương hoạt động bằng cách kìm hãm sự hình thành xương qua trung gian LRP5/6-WNT- và BMP. Lượng xương được hình thành trong mỗi đơn vị tái tạo tăng lên với sự gia tăng độ dày và kết nối của bè xương và sự hình thành xương màng xương mới và độ dày vỏ xương dẫn đến tăng kích thước, khối lượng và sức mạnh của xương. Các tác dụng phụ của việc dùng PTH 1-34 bao gồm tăng canxi máu thoáng qua, tăng canxi niệu và sự phát triển của các kháng thể đối với peptide. Mặc dù u xương ác tính đã được quan sát thấy ở chuột nhận liều rất cao của các tác nhân này, không có rối loạn ác tính nào được ghi nhận ở người lớn nhận một trong hai tác nhân. Việc sử dụng PTH 1-34 ở trẻ em chủ yếu được giới hạn ở trẻ em bị hạ canxi máu do một đột biến tăng chức năng trong CASR và hậu quả là hạ canxi máu có tăng canxi niệu—một dạng suy cận giáp đơn độc có tính gia đình.
Việc quản lý cơ bản bệnh nhân bị bệnh xương thủy tinh nhằm mục đích ngăn ngừa gãy xương đến mức có thể và điều trị các gãy xương xảy ra bằng các thủ thuật chỉnh hình hợp lý và bởi các bác sĩ chỉnh hình quen thuộc với rối loạn này. Việc quản lý chỉnh hình chuyên nghiệp là rất cần thiết cho trẻ bị bệnh xương thủy tinh, vì việc sử dụng các thanh nội tủy để điều chỉnh các dị tật xương dài và cho sự phát triển tuyến tính của chúng và sự điều chỉnh đúng đắn của vẹo cột sống đòi hỏi kinh nghiệm đáng kể. Các dịch vụ nha khoa và phục hồi chức năng và vật lý trị liệu để cải thiện tình trạng răng, sức mạnh cơ bắp và khả năng vận động trong giới hạn của sự mong manh của xương được khuyến khích cũng như việc đi lại và các bài tập được bảo vệ, chẳng hạn như bơi lội. Việc dùng ngắt quãng bisphosphonate cho trẻ sơ sinh, trẻ em và thanh thiếu niên mắc các dạng bệnh xương thủy tinh nặng vừa (type lâm sàng I, III, IV) đã mang lại lợi ích đáng kể. Bệnh nhân mắc các loại bệnh xương thủy tinh này đã đáp ứng về mặt triệu chứng (giảm đau cơ xương, tăng khả năng vận động) với việc dùng bisphosphonate tiêm tĩnh mạch ngắt quãng—ví dụ, pamidronate, axit zoledronic; khối lượng xương đã tăng, nguy cơ gãy xương đã giảm, và kích thước đốt sống có thể được bình thường hóa mặc dù tỷ lệ vẹo cột sống không giảm.
Người ta đã khuyến nghị rằng bisphosphonate được sử dụng ở trẻ sơ sinh bị bệnh xương thủy tinh có gãy xương bẩm sinh và tái phát, các dị tật của xương dài và giảm khối lượng xương. Trẻ sơ sinh nhỏ đến 2 tháng tuổi đã dung nạp an toàn các đợt truyền tĩnh mạch pamidronate trong 4 giờ (0,5 mg/kg/ngày trong 3 ngày liên tiếp mỗi 6–8 tuần) nhận thấy sự cải thiện lâm sàng, chẳng hạn như giảm đau xương rõ rệt, tăng BMD của đốt sống thắt lưng từ 86% đến 227%, và giảm tỷ lệ gãy xương sau 1 năm điều trị. Việc dùng bisphosphonate cũng được khuyến nghị cho trẻ em bị bệnh xương thủy tinh và gãy xương tái phát ở các chi hoặc xẹp đốt sống có triệu chứng kết hợp với việc chứng minh giảm khoáng hóa xương. Ở trẻ em (3–16 tuổi), pamidronate được dùng dưới dạng truyền trong 4 giờ (1,5–3,0 mg/kg/ngày) trong 3 ngày liên tiếp mỗi 4 tháng, dẫn đến tăng BMD cột sống thắt lưng 42% mỗi năm, chiều rộng vỏ xương bàn tay 27% mỗi năm, và kích thước đốt sống, cũng như giảm tỷ lệ gãy xương và cải thiện triệu chứng. Trong 2 đến 4 năm dùng pamidronate tiêm tĩnh mạch, sự gia tăng khối lượng và kích thước xương đốt sống (bè) đi kèm với sự suy giảm mức độ lún đốt sống và ít đốt sống bị lún hơn so với bệnh nhân không được điều trị. Ở mào chậu, bisphosphonate làm tăng độ dày xương vỏ và số lượng bè xương nhưng không tăng độ dày bè xương; ở xương bàn tay, bisphosphonate tăng cường độ dày vỏ. Việc dùng bisphosphonate cũng đi kèm với hạ canxi máu thoáng qua (được quản lý theo triệu chứng), tăng nồng độ PTH và calcitriol, và giảm giá trị các dấu hiệu chu chuyển xương. Lợi ích gần như tối đa của bisphosphonate đối với BMD theo diện tích cột sống thắt lưng bằng DXA và đối với chiều rộng vỏ trung bình, thể tích xương xốp và tốc độ hình thành xương bè bằng phân tích hình thái mô học của sinh thiết xương mào chậu đạt được trong 2 đến 4 năm đầu điều trị với ít thay đổi hơn nữa với liệu pháp kéo dài hơn. Việc sử dụng các kháng thể đơn dòng đối với sclerostin (romososumab) hoặc RANKL (denosumab) cũng có thể hữu ích trong điều trị trẻ em bị bệnh xương thủy tinh. Việc cấy ghép các tế bào gốc trung mô, các tế bào gốc trung mô của thai nhi người hoặc các tế bào gốc màng đệm, hoặc các tế bào đệm tủy xương đã được thực hiện ở những bệnh nhân bị bệnh xương thủy tinh nhưng vẫn là liệu pháp thử nghiệm cũng như các chiến lược thay thế gen và làm im lặng đột biến cụ thể.
Loạn sản xơ
Loạn sản xơ là một rối loạn xơ-xương thường không ác tính liên quan đến các xương dài, xương sườn và hộp sọ, trong đó xương và tủy xương bình thường được thay thế bằng mô xơ; nó có thể là đơn ổ, đa ổ hoặc toàn ổ. Rối loạn này có thể là đơn ổ hoặc đa ổ và biểu hiện như nguyên nhân của một gãy xương bệnh lý với chấn thương nhẹ. Mặc dù loạn sản xơ có thể là một bất thường đơn độc, nó xảy ra ở những bệnh nhân mắc hội chứng McCune-Albright (OMIM 174800) kết hợp với các đốm sắc tố café-au-lait rất lớn, có bờ không đều và các bệnh nội tiết khác nhau, bao gồm dậy thì sớm đồng giới, cường somatotropin, nhiễm độc giáp và cường vỏ thượng thận, cũng như rối loạn chức năng ở nhiều mô khác (tim, gan, tụy). Các tổn thương xương và da thường ở cùng một bên của cơ thể. Loạn sản xơ là do thể khảm của các đột biến sai nghĩa tăng chức năng soma, phôi sớm, sau hợp tử (p.Arg201Cys/His/Ser/Gly; p.Gln227Leu) trong GNAS (OMIM 139320), gen mã hóa tiểu đơn vị α của protein Gs làm cho Gsα hoạt động cấu thành bằng cách kéo dài tuổi thọ sinh học của nó. Mức độ và mức độ nghiêm trọng của bệnh được quyết định bởi thời điểm phát triển của thai nhi mà đột biến xảy ra và sự phân bố mô của nó. Các đột biến dẫn đến mất hoạt động guanosine triphosphatase nội tại trong tiểu đơn vị Gsα; do đó, tác dụng kích thích của Gsα đối với adenylyl cyclase được kéo dài do đó làm tăng tạo ra AMP vòng. Trong số các con đường tín hiệu được nhắm mục tiêu của AMP vòng là con đường liên quan đến WNT/β-catenin trong các tế bào tiền thân nguyên bào xương. Để đáp ứng với AMP vòng dư thừa, các dòng tiền nguyên bào xương trung mô bị đột biến tăng sinh nhưng sự biệt hóa của chúng thành các nguyên bào xương trưởng thành là không hoàn chỉnh và chất nền tế bào xơ do chúng tiết ra là bất thường; sự mở rộng liên tục của các tế bào tiền thân tạo xương trong tủy xương dẫn đến xơ hóa tại chỗ. Khi các tế bào tạo xương tăng về số lượng, chúng dần dần ăn mòn xương liền kề. Những tổn thương này cũng có thể tổng hợp FGF23 và do đó dẫn đến tăng phốt phát niệu, hạ phốt phát máu và dư thừa chất dạng xương không khoáng hóa và một tình trạng lâm sàng giống còi xương. Các tổn thương loạn sản xơ ban đầu im lặng trong khi các hủy cốt bào ở ngoại vi của các tổn thương tích cực chèn ép và làm mỏng vỏ xương, cuối cùng dẫn đến đau xương và gãy xương bệnh lý của các xương dài, đặc biệt là hành xương đùi gần. Trẻ em từ 6 đến 10 tuổi có tỷ lệ gãy xương cao nhất (0,4 gãy xương mỗi năm). Trong nền sọ và các xương mặt, sự mở rộng của các tổn thương loạn sản xơ dẫn đến biến dạng và chèn ép các dây thần kinh sọ. Về mặt X-quang, tổn thương loạn sản xơ được xem như một cấu trúc tủy “giống nang” với tính nhất quán “kính mờ” không có mô hình bè xương. Về mặt mô học, loạn sản xơ được đặc trưng bởi nhiều tế bào đệm tủy xương chưa trưởng thành, các nguyên bào xương biệt hóa không hoàn toàn, các bè xương được hình thành không đều có thể giống với các ký tự Trung Quốc, nhiều đường nối chất dạng xương không được khoáng hóa đặc trưng của nhuyễn xương và các đảo sụn. Các biểu hiện lâm sàng của loạn sản xơ phụ thuộc vào các vị trí và mức độ liên quan của xương và các bệnh nội tiết liên quan. Chẩn đoán loạn sản xơ dựa trên các đặc điểm lâm sàng, hình ảnh của các tổn thương xương (ลักษณะ “kính mờ”), và xác nhận đột biến di truyền trong GNAS. Ngoài việc quản lý nhiều bệnh nội tiết và khiếm khuyết cơ quan, cần chú ý đến các tổn thương xương. Gãy xương được sửa chữa bằng các kỹ thuật tiêu chuẩn, bao gồm cả đóng đinh nội tủy khi được chỉ định; đôi khi có thể khả thi để sơ tán một tổn thương loạn sản xơ bằng phẫu thuật và lấp đầy khoang bằng các mảnh ghép xương. Ở những bệnh nhân bị loạn sản xơ đa ổ không có hoặc liên quan đến hội chứng McCune-Albright, việc dùng bisphosphonate alendronate đường uống không làm thay đổi diện mạo X-quang của các tổn thương xương, giảm đau xương hoặc cải thiện chức năng. Tuy nhiên, ở những bệnh nhân bị loạn sản xơ đa ổ đơn độc hoặc liên quan đến hội chứng McCune-Albright, điều trị bằng pamidronate hoặc axit zoledronic tiêm tĩnh mạch đã làm giảm đau xương và giảm nồng độ phosphatase kiềm trong huyết thanh phản ánh sự suy giảm tốc độ chu chuyển xương nhưng không sửa chữa được các tổn thương xương. Ở một cậu bé 9 tuổi có một khối u đùi phát triển nhanh do loạn sản xơ (và một đột biến soma trong GNAS1), điều trị bằng denosumab đã dẫn đến sự suy giảm nhanh chóng tốc độ phát triển của khối u và làm giảm nồng độ các dấu hiệu chu chuyển xương. Tuy nhiên, đứa trẻ bị hạ canxi máu và cường cận giáp thứ phát cần bổ sung canxi, phốt phát và calcitriol. Việc ngừng denosumab được đánh dấu bằng sự gia tăng trở lại các giá trị của các dấu hiệu chu chuyển xương và tăng canxi máu.
Khối lượng xương cao
Khối lượng xương tăng bất thường là hậu quả của sự gián đoạn cân bằng bình thường giữa các quá trình kết hợp của sự hình thành và tái hấp thu xương. Xơ cứng xương là tên gọi được áp dụng cho một rối loạn di truyền hoặc mắc phải được đặc trưng bởi hoặc liên quan đến sự gia tăng khối lượng xương và BMD. Cụ thể, sự gia tăng chiều rộng xương vỏ được gọi là tăng sinh xương; sự dày lên của xương bè được gọi là xơ cứng xương. Bệnh xương hóa đá hay “bệnh xương cẩm thạch” là một nhóm các bệnh liên quan đến xương bị vôi hóa tăng lên nhưng nghịch lý là phức tạp bởi sự mong manh của xương, dẫn đến gãy xương với chấn thương nhẹ hoặc không có chấn thương, chủ yếu là do giảm sự hình thành hủy cốt bào hoặc suy giảm chức năng hủy cốt bào hoặc hiếm khi do sự gia tăng tốc độ hình thành xương do hoạt động quá mức của nguyên bào xương. Thất bại trong việc tái hấp thu xương qua trung gian hủy cốt bào dẫn đến bệnh xương hóa đá có thể liên quan đến một số lượng lớn các hủy cốt bào hoạt động kém (“bệnh xương hóa đá giàu hủy cốt bào”) hoặc với một số lượng bình thường hoặc ít ỏi các hủy cốt bào (“bệnh xương hóa đá nghèo hủy cốt bào”).
Các nguyên nhân di truyền được chỉ định năm 2019 của bệnh xương hóa đá và các dạng xơ cứng xương liên quan được liệt kê trong Bảng 20.11A, 20.11B và được minh họa một cách sơ đồ trong Hình 20.15. Các rối loạn lâm sàng này được di truyền như các rối loạn lặn hoặc trội trên nhiễm sắc thể thường hoặc liên kết X; bệnh xương hóa đá do các đột biến của CLCN7 và PLEKHM1 có thể được di truyền như các bất thường hai alen hoặc đơn alen. Các bệnh xương hóa đá là do các lỗi trong sự biệt hóa của hủy cốt bào và liên quan đến bệnh xương hóa đá “nghèo hủy cốt bào” có liên quan đến các đột biến trong TNFRSF11A, TNFSF11, và IKBKG. Những bệnh liên quan đến bệnh xương hóa đá “giàu hủy cốt bào” là do các biến thể của TCIRG1, CLCN7, OSTM1, SNX10, PLEKHM1, và CAII. Về mặt vi thể, xương của bệnh nhân bị bệnh xương hóa đá di truyền lặn trên nhiễm sắc thể thường type 1, 3, 4, 5, 6 và 8 có nhiều hủy cốt bào nhưng chức năng bị suy giảm (giàu hủy cốt bào); ở bệnh xương hóa đá di truyền lặn trên nhiễm sắc thể thường type 2 và 7 và dạng liên kết X, có ít hủy cốt bào do thiếu RANKL hoặc RANK (“nghèo hủy cốt bào”). Bệnh xương hóa đá di truyền trội trên nhiễm sắc thể thường type 1 không liên quan đến một lỗi trong sự biệt hóa hoặc chức năng của hủy cốt bào và tái hấp thu xương khiếm khuyết mà thay vào đó liên quan đến sự hình thành nguyên bào xương tăng lên dẫn đến sự hình thành và khoáng hóa xương tăng cường. Dạng bệnh xương hóa đá này là do một biến thể tăng chức năng (p.Gly171Val) của LRP5 (OMIM 603506) dẫn đến khối lượng xương tăng lên bằng cách làm suy giảm sự liên kết của LRP5 với sclerostin (SOST, OMIM 605740) và với DKK1 (Dickkopf, xenopus, tương đồng của, OMIM 605189), các đồng yếu tố mà về mặt sinh lý liên kết và ức chế sự liên kết của LRP5 với phối tử của nó. Do đó, khi có LRP5 bị đột biến, con đường tín hiệu WNT1/Frizzled receptor/LRP5 coreceptor/β-catenin trong tế bào gốc trung mô được lập trình theo hướng hình thành nguyên bào xương. Biến thể hoạt hóa này của LRP5 được biểu hiện bởi nguyên bào xương có liên quan đến sự hình thành xương tăng lên (bệnh xương hóa đá di truyền trội trên nhiễm sắc thể thường type I)—chủ yếu là của hộp sọ; rối loạn này có thể không có triệu chứng hoặc liên quan đến các tổn thương dây thần kinh sọ dẫn đến đau dây thần kinh sinh ba, liệt dây thần kinh mặt hoặc rối loạn chức năng dây thần kinh thính giác, mặc dù không có các hậu quả có hại nghiêm trọng hơn của các dạng bệnh xương hóa đá do hủy cốt bào.
Bảng 20.11A: Các biến thể/Đột biến gen liên quan đến Sinh bệnh học của Bệnh xương hóa đá
| Loại | OMIM | Di truyền: Lặn trên NST thường (AR) Trội trên NST thường (AD) | Gen | Protein | OMIM | Nhiễm sắc thể | Khiếm khuyết chức năng | Bất thường lâm sàng |
|---|---|---|---|---|---|---|---|---|
| AR1 | 259700 | TCIRG1 | T cell immune regulator 1 | 604592 | 11q13.2 | Mã hóa tiểu đơn vị a3 của V-H+ ATPase, bơm chịu trách nhiệm cho sự di chuyển của H+ từ tế bào chất của hủy cốt bào đến hốc dưới tế bào xương nơi H+ hòa tan pha khoáng của xương; cũng được biểu hiện ở niêm mạc dạ dày nơi nó cần thiết cho sự hấp thu Ca2+ ở ruột | Giàu hủy cốt bào Bệnh xương hóa đá “ác tính” nặng với tử vong sớm; kiểu hình xương nghịch lý giống còi xương | |
| AR2 | 259710 | TNFSF11 | Tumor necrosis factor ligand superfamily, Member 11 | 602642 | 13q14.11 | Yếu tố nguyên bào xương (còn được gọi là phối tử yếu tố hạt nhân-κB hoạt hóa thụ thể [RANKL]) kích thích sự hình thành hủy cốt bào | Nghèo hủy cốt bào Bệnh xương hóa đá nặng | |
| AR3 | 259730 | CAII | Carbonic anhydrase II | 611492 | 8q22 | Enzyme xúc tác quá trình sản xuất H+ bằng cách tổng hợp và ion hóa axit carbonic (H2CO3) của hủy cốt bào; HCO3– dư thừa được loại bỏ qua bộ trao đổi ion Cl–/HCO3– | Giàu hủy cốt bào Bệnh xương hóa đá mức độ nghiêm trọng trung bình; liên quan đến toan hóa ống thận, vôi hóa não, khiếm khuyết nhận thức | |
| AR4 | 611490 | AD2 166600 | CLCN7 | Chloride channel 7 | 602727 | 16p13 | Kênh clorua được biểu hiện trong các hủy cốt bào tạo điều kiện cho sự axit hóa của hốc dưới tế bào xương và sự hòa tan pha khoáng của xương | Giàu hủy cốt bào AR4—Các biến thể hai alen dẫn đến bệnh xương hóa đá nặng, giảm nhận thức liên quan đến thoái hóa thần kinh do bệnh dự trữ lysosome; AD2—Các biến thể đơn alen dẫn đến xơ cứng nền sọ, xương chậu và các đĩa cuối đốt sống dẫn đến hình ảnh X-quang cột sống “sandwich”; không liên quan đến sự gia tăng tỷ lệ gãy xương nhưng chèn ép và làm suy giảm chức năng dây thần kinh sọ (thính giác, thị giác) |
| AR5 | 259720 | OSTM1 | Osteopetrosis-associated transmembrane protein 1 | 607649 | 6q21 | Yếu tố giống chaperone liên kết với CLCN7 trong quá trình trưởng thành và chuyển vị của nó | Giàu hủy cốt bào Liên quan đến bệnh xương hóa đá nặng, thoái hóa thần kinh do bệnh dự trữ lysosome | |
| AR6 | 611497 | AD3 618017 | PLEKHM1 | Pleckstrin homology domain containing protein, Family M, Member 1 | 611466 | 17q21.31 | Yếu tố cần thiết cho việc vận chuyển các protein trong túi ở hủy cốt bào | Giàu hủy cốt bào Bệnh xương hóa đá mức độ nghiêm trọng vừa phải; các hủy cốt bào không hình thành bờ diềm |
| AR7 | 612301 | TNFRSF11A | Tumor necrosis factor receptor superfamily, Member 11A | 603499 | 18q21.33 | Thụ thể xuyên màng cho RANKL—được biểu hiện bởi tế bào tiền thân của hủy cốt bào | Nghèo hủy cốt bào Còn được gọi là yếu tố hạt nhân-κB hoạt hóa thụ thể (RANK); nặng, liên quan đến giảm gammaglobulin máu | |
| AR8 | 615085 | SNX10 | Sorting nexin 10 | 614780 | 7p15.2 | Chất điều hòa sự hình thành không bào và sự di chuyển của protein liên quan đến túi ở các hủy cốt bào và dạ dày | Nghèo hủy cốt bào (±) Bệnh xương hóa đá nặng, thiếu máu và giảm bạch cầu do xóa khoang tủy của xương; kiểu hình xương nghịch lý giống còi xương | |
| AR osteopetrosis | FERMT3 | Fermitin family, member 3 | 607901 | 11q13.1 | Tín hiệu integrin, kết tập tiểu cầu | Giàu hủy cốt bào. Mã hóa protein kindlin-3; các đột biến liên quan đến bệnh xương hóa đá, nhiễm trùng, xuất huyết | ||
| AD1 | LRP5 | Low-density lipoprotein receptor-related protein 5 | 603506 | 11q13.2 | Đồng thụ thể cho WNT1 với Frizzled cần thiết cho sự hình thành nguyên bào xương | Các biến thể tăng chức năng của protein liên quan đến thụ thể WNT1-Frizzled này dẫn đến xơ cứng vòm sọ nhưng không liên quan đến tăng tần suất gãy xương | ||
| AD2 | CLCN7 | |||||||
| AD3 | PLEKHM1 | |||||||
| Liên kết X | IKBKG | Inhibitor of kappa light polypeptide gene enhancer in B cells, Kinase of, Gamma | 300248 | Xq28 | Serine/threonine kinase tăng cường chức năng của NF-κB do đó kích thích sự hình thành hủy cốt bào | Nghèo hủy cốt bào (±) Còn được gọi là NF-κB essential modulator (NEMO); liên quan đến bệnh xương hóa đá vừa, loạn sản ngoại bì, suy giảm miễn dịch |
(Từ Teti, A., Econs, M.J. (2017). Osteopetroses, emphasizing potential approach to treatment. Bone, 102:50–59; Penna, S., Capo, V., Palagano, E., et al. (2019). One disease, many genes: Implications for the treatment of osteopetroses. Front Endocrinol, 10:85.)
Bảng 20.11B: Các đột biến gen liên quan đến việc Tăng khối lượng xương/Xơ cứng xương
| Rối loạn | OMIM | Gen | Protein gen | OMIM | Nhiễm sắc thể | Chức năng | Khiếm khuyết chức năng |
|---|---|---|---|---|---|---|---|
| Pycnodysostosis | 265800 | CTSK | Cathepsin K | 601105 | 1q21.2 | Cysteine proteinase | Thất bại trong việc hòa tan enzyme của chất nền hữu cơ của xương trong quá trình tái tạo xương, AR |
| Bệnh Van Buchem 1 | 239100 | SOST | Sclerostin | 605740 | 17q21.31 | Yếu tố liên kết với miền ngoại bào của LRP5 và LRP6 ức chế sự liên kết của chúng với WNT1 & sự truyền tín hiệu của nó | Tăng sinh xương vỏ toàn thể—tăng khoáng hóa xương do tăng hình thành nguyên bào xương liên quan đến vỏ sọ và xương dài dẫn đến chèn ép dây thần kinh sọ và mất chức năng, AR |
| Xơ cứng xương 1 | 269500 | SOST | Sclerostin | 605740 | 17q21.31 | Xơ cứng xương và tăng sinh xương toàn thể—tăng khoáng hóa xương với tăng hình thành nguyên bào xương liên quan đến chứng khổng lồ, dính ngón, tăng áp lực nội sọ, tử vong sớm, AR | |
| Bệnh Van Buchem 2 | 607636 | LRP5 | Protein 5 liên quan đến thụ thể lipoprotein mật độ thấp | 603506 | 11q13.2 | Yếu tố liên kết với phức hợp WNT1-β-catenin và tăng cường sự hình thành nguyên bào xương | Tăng sinh xương vỏ toàn thể, AD |
| Xơ cứng xương 2 | 614305 | LRP4 | 604270 | 11p11.2 | Yếu tố liên kết với sclerostin và tạo điều kiện cho tác dụng ức chế của nó đối với sự hình thành nguyên bào xương | Xơ cứng xương và tăng sinh xương toàn thể, AR/AD | |
| Hội chứng Raine | 259775 | FAM20C | Họ có trình tự tương tự 20, thành viên C | 611061 | 7p22.3 | Phosphorylase của các protein SIBLING cản trở quá trình khoáng hóa sinh học | Xơ cứng xương toàn thể trước sinh với sự hình thành xương màng xương thường dẫn đến tử vong chu sinh, AR |
AD, Trội trên NST thường; AR, Lặn trên NST thường, SIBLING, các glycoprotein tiết ra liên kết N, phối tử liên kết integrin nhỏ.
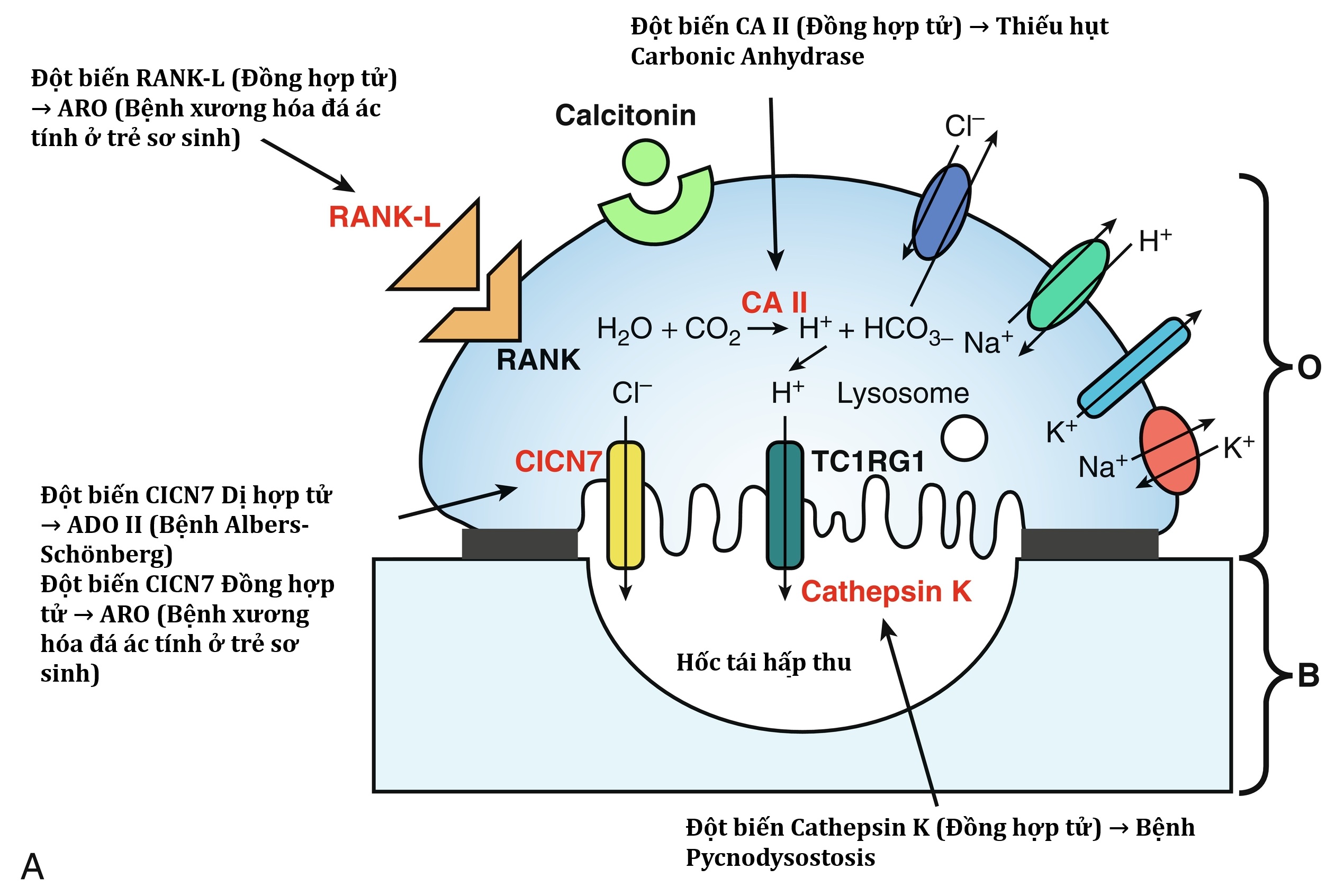
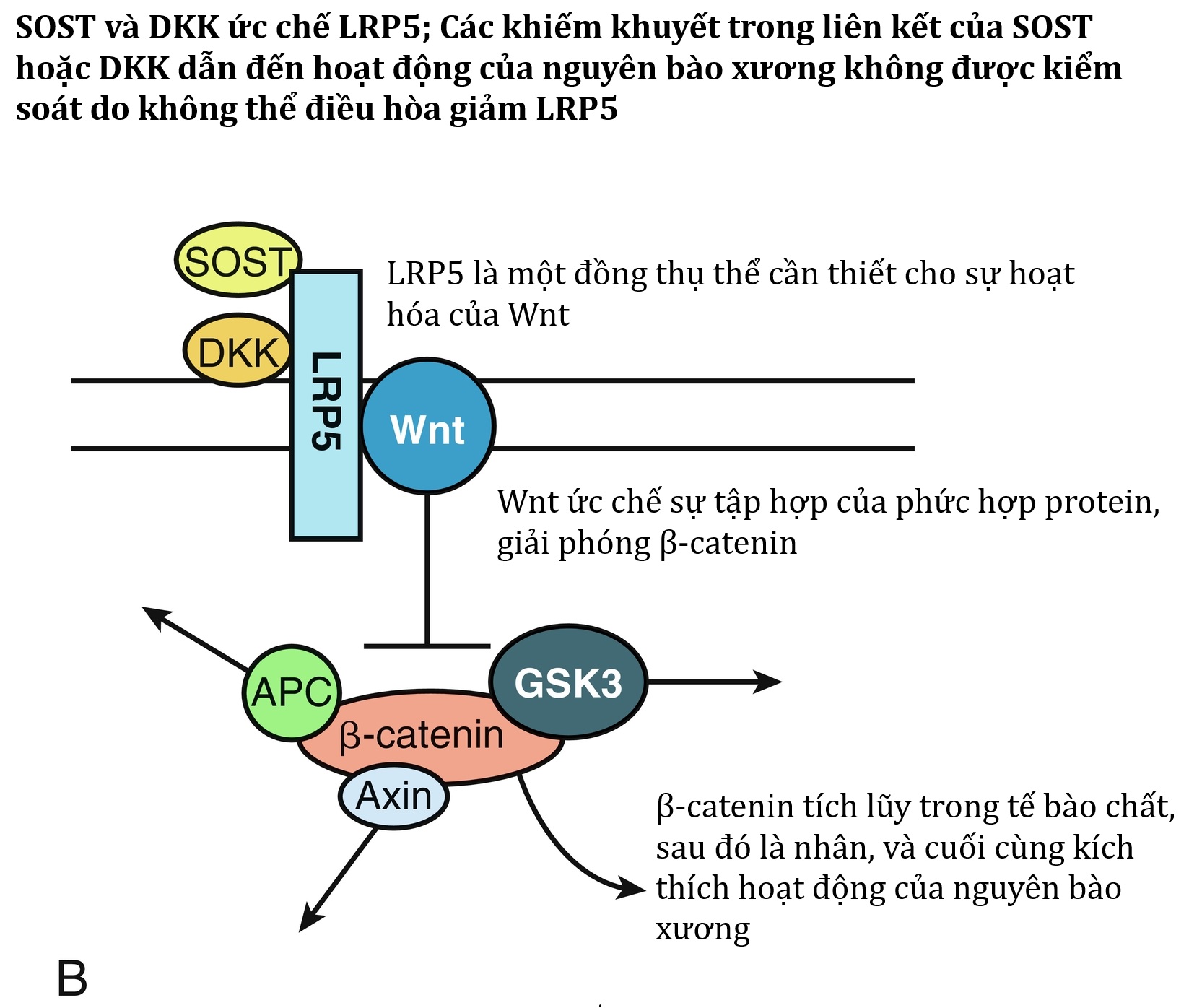
Hình 20.15: Các khiếm khuyết sinh hóa trong hủy cốt bào (A) và nguyên bào xương (B) liên quan đến bệnh xương hóa đá (xem chi tiết trong văn bản).
(Phỏng theo sự cho phép, từ de Vernejoul MC. Sclerosing bone disorders. Best Pract Res Clin Rheumatol 2008;22(1):71–83.)
Các dạng bệnh xương hóa đá di truyền lặn (xem Bảng 20.11A) thường nặng hơn về mặt lâm sàng so với các dạng được di truyền theo kiểu trội. Các biểu hiện của sự tăng khoáng hóa xương có thể có trong tử cung (gãy xương) hoặc phát triển trong vài tháng đầu đời (gãy xương, chèn ép các dây thần kinh sọ trong các lỗ hẹp với hậu quả là teo do chèn ép của các dây thần kinh này và mất chức năng—đặc biệt là thị giác và thính giác; sự chèn ép của não đang phát triển có thể dẫn đến chậm phát triển). Bệnh xương hóa đá di truyền lặn trên nhiễm sắc thể thường type 1 là do các biến thể của T cell immune regulator 1 (TCIRG1, OMIM 604592) và có liên quan đến sự gia tăng hình thành nguyên bào xương, dẫn đến sự hình thành và khoáng hóa xương tăng cường. Các biến thể trong CLCN7 dẫn đến một loạt các biểu hiện lâm sàng từ tình trạng người mang lành tính đến các biểu hiện khá nặng của bệnh xương hóa đá. Các đột biến hai alen bất hoạt của OSTM1 hoặc CLCN7 thường liên quan đến bệnh dự trữ lysosome, thoái hóa thần kinh và suy giảm nhận thức. Do mất tủy xương, quá trình tạo máu bị tổn hại ở hầu hết các đối tượng bị bệnh xương hóa đá. Sự phát triển suy giảm của các hủy cốt bào dẫn đến bệnh xương hóa đá nghèo hủy cốt bào và là do các đột biến trong các gen mã hóa RANKL (TNFRSF11) và RANK (TNFSF11A). IKBKG là một gen liên kết X mã hóa một tiểu đơn vị của phức hợp kinase inhibitor of kappa B (IκB) cũng cần thiết cho sự kích hoạt NF-κB và sự hình thành hủy cốt bào. Mặc dù có mặt với số lượng bình thường hoặc dồi dào ở những bệnh nhân bị bệnh xương hóa đá giàu hủy cốt bào, hoạt động của hủy cốt bào có thể bị giảm do giảm tổng hợp axit (do các đột biến mất chức năng trong CAII, gen mã hóa carbonic anhydrase), giảm số lượng hoặc hoạt động của các kênh ion vận chuyển Cl– và H+ vào các hốc dưới tế bào xương (các protein kênh được mã hóa bởi CLCN7 và TCIRG1 hoặc đồng yếu tố của nó OSTM1, tương ứng), hoặc sự tạo ra dưới mức bình thường của các proteinase phân hủy chất nền xương, chẳng hạn như cathepsin K (do các đột biến bất hoạt trong CTSK). Hiếm khi, sự lắng đọng quá mức của khoáng chất xương do chức năng nguyên bào xương tăng lên có thể là hậu quả của sự tăng cường con đường truyền tín hiệu WNT/β-catenin do các đột biến tăng chức năng trong LRP5 như đã thảo luận hoặc do sự ức chế giảm của quá trình này do các biến thể bất hoạt của SOST (mã hóa sclerostin). Điều thú vị là, ở những bệnh nhân có đột biến trong LRP5 và bệnh xương hóa đá di truyền trội trên nhiễm sắc thể thường type I (xem Bảng 20.11A), cũng có sự khan hiếm các hủy cốt bào. Sự bất hoạt của SOST (OMIM 605740) dẫn đến xơ cứng xương type 1 (OMIM 269500), được đặc trưng bởi sự phát triển quá mức của xương và chèn ép các dây thần kinh sọ kết hợp với dính ngón do hoạt động không được điều hòa của sclerostin của con đường kích thích nguyên bào xương LRP5-Frizzled receptor-β-catenin. Các biến thể bất hoạt của LRP4 (OMIM 604270) mã hóa một protein liên kết với sclerostin cũng liên quan đến khối lượng xương cao (xơ cứng xương type 2, OMIM 614305)—hậu quả có thể là do hoạt động của sclerostin được hỗ trợ bởi LRP4 bị suy giảm trong xương hoặc do sự cô lập ngoài xương của sclerostin bởi LRP4 bị đột biến.
Về mặt bệnh học mô, xương ở phần lớn các dạng bệnh xương hóa đá được đặc trưng bởi sự hiện diện của các hủy cốt bào không hoạt động—có số lượng tăng, bình thường hoặc ít—và các “đảo” sụn vôi hóa được giữ lại hình thành trong quá trình hóa xương nội sụn (xốp nguyên phát) do thất bại trong việc tái hấp thu xương chưa trưởng thành. Mặc dù được đóng gói dày đặc với các khoáng chất, xương bị bệnh xương hóa đá khá mong manh vì sự bất thường trong việc tái tạo xương do giảm tái hấp thu xương của hủy cốt bào dẫn đến sự kết hợp của sụn đĩa tăng trưởng vôi hóa yếu vào xương và sự chậm trễ trong việc sửa chữa các vi gãy xương. Về mặt X-quang, bệnh xương hóa đá được đặc trưng bởi sự gia tăng khối lượng xương lan tỏa liên quan đến cả xương vỏ và xương bè, sự mở rộng thân xương/hành xương với hình dạng “bình Ehrlenmeyer”, các dải xương xơ cứng và thấu quang xen kẽ ở các đầu của xương dài (“xương-trong-xương”), mào chậu và đốt sống (“đốt sống áo len” hoặc “sandwich”), các thay đổi xơ cứng ở nền sọ, các khoang tủy hẹp và các gãy xương bệnh lý. CT sọ não thường cho thấy sự thu hẹp của các ống xương mà qua đó các dây thần kinh sọ II, III, IV, VII và VIII đi qua. Mặc dù theo kinh điển, ba dạng lâm sàng của bệnh xương hóa đá có mức độ nghiêm trọng thay đổi đã được xác định—trẻ nhỏ ác tính (lặn trên nhiễm sắc thể thường), trung gian (lặn trên nhiễm sắc thể thường) và người lớn (trội trên nhiễm sắc thể thường)—khi thông tin về các đột biến di truyền gây ra bệnh đã được xác định, phân loại này đã được thay thế bằng một phân loại xác định phương thức di truyền của nó (trội hoặc lặn trên nhiễm sắc thể thường hoặc lặn liên kết X) và phù hợp với gen bị đột biến gây ra (xem Bảng 20.11A, 20.11B). Do đó, dạng bệnh xương hóa đá ở trẻ nhỏ/ác tính (OMIM 259700) có thể do các đột biến hai alen trong TCIRG1, CLCN7, hoặc OSTM1—các gen mã hóa các protein kênh vận chuyển hydro và clorua của màng hủy cốt bào hoặc một đồng điều biến mà qua đó hủy cốt bào tiết ra axit vào hốc dưới hủy cốt bào để hòa tan hydroxyapatite, pha khoáng của xương. Một dạng bệnh xương hóa đá nặng liên quan đến giảm gammaglobulin máu là do các biến thể có hại trong TNFRSF11A mã hóa RANKL. Các dạng bệnh xương hóa đá có mức độ nghiêm trọng trung bình là kết quả của các đột biến trong TNFSF11, CAII, PLEKHM1—các gen mã hóa RANK, carbonic anhydrase và một protein tham gia vào việc vận chuyển lysosome trong hủy cốt bào, tương ứng. Bệnh xương hóa đá liên kết X là một rối loạn có mức độ nghiêm trọng vừa phải liên quan đến các biến thể của IKBKG, một serine/threonine kinase tăng cường chức năng của NF-κB, do đó kích thích sự hình thành nguyên bào xương (còn được gọi là NF-κB essential modulator [NEMO]; đây là một dạng bệnh xương hóa đá nghèo (±) hủy cốt bào có mức độ nghiêm trọng vừa phải kết hợp với loạn sản ngoại bì và suy giảm miễn dịch. Các đột biến hoạt hóa trong LRP5 và các biến thể bất hoạt dị hợp tử trong CLCN7 có liên quan đến bệnh xương hóa đá có mức độ nghiêm trọng từ nhẹ đến trung bình được di truyền như các rối loạn trội trên nhiễm sắc thể thường.
Dạng bệnh xương hóa đá ở trẻ nhỏ/ác tính là một rối loạn di truyền lặn trên nhiễm sắc thể thường (AR type 4) với sự tăng trưởng bị suy giảm đặc biệt là ở các chi, chậm phát triển, tăng tỷ lệ gãy xương và không mọc được răng do các biến thể của CLCN7 (xem Bảng 20.11A). Sự phát triển quá mức của xương dẫn đến đầu to, đôi khi não úng thủy, sự phát triển kém của các xoang cạnh mũi và nghẹt mũi có triệu chứng. Sự thu hẹp của các lỗ sọ làm tổn hại đến chức năng của các dây thần kinh sọ (II, III, VII, VIII) với hậu quả là mù, điếc hoặc liệt dây thần kinh sọ. Sự giảm thể tích tủy xương dẫn đến suy giảm tạo máu trong tủy, thiếu máu và giảm bạch cầu được bù trừ một phần bằng cách tạo máu ngoài tủy và hậu quả là gan lách to với hậu quả là tăng nhạy cảm với nhiễm trùng và xuất huyết. Sự lưu giữ của răng trong hàm bị xơ cứng dẫn đến viêm tủy xương hàm trên và hàm dưới tái phát và dai dẳng. Khám thực thể cho thấy tầm vóc thấp, đầu to, trán dô, các đặc điểm khuôn mặt nhỏ và hai mắt xa nhau; thị lực có thể bị tổn hại do lồi mắt và teo võng mạc. Trẻ sơ sinh và trẻ nhỏ bị bệnh xương hóa đá thường bị hạ canxi máu (do không thể tái hấp thu canxi lắng đọng trong xương) với cường cận giáp thứ phát và các giá trị calcitriol tăng; nồng độ phosphatase axit huyết thanh và isoenzyme não của creatine kinase cũng tăng. Tử vong thường xảy ra trong thập kỷ đầu của cuộc đời do nhiễm trùng huyết, thiếu máu hoặc xuất huyết. Bệnh xương hóa đá di truyền lặn trên nhiễm sắc thể thường có thể do không chỉ các đột biến hai alen trong kênh clorua của hủy cốt bào—CLCN7, mà còn trong tiểu đơn vị α3 của bơm proton không bào của hủy cốt bào—TCIRG1, protein xuyên màng-1 liên quan đến bệnh xương hóa đá (OSTM1), hoặc một protein hướng TCIRG1 đến màng diềm của hủy cốt bào (SNX10) (xem bên dưới). Các đột biến hai alen trong TNFRSF11A mã hóa RANK dẫn đến một dạng bệnh xương hóa đá nặng thường kết hợp với giảm gammaglobulin máu.
Các dạng lâm sàng trung gian của bệnh xương hóa đá cũng được di truyền như các tính trạng lặn trên nhiễm sắc thể thường liên quan đến tầm vóc thấp, đầu to, tổn hại chức năng dây thần kinh sọ thay đổi, gãy xương tái phát, phát triển răng bất thường dễ dẫn đến viêm tủy xương hàm trên hoặc hàm dưới và thiếu máu. Về mặt sinh bệnh học, chúng đại diện cho sự biểu hiện thay đổi của một trong những đột biến di truyền có thể liên quan đến dạng bệnh xương hóa đá ở trẻ nhỏ, chủ yếu là của CLCN7, nhưng cũng có của CAII, TNFSF11, và PLEKHM1. Có ba dạng lâm sàng và X-quang được chỉ định của bệnh xương hóa đá di truyền trội đơn alen trên nhiễm sắc thể thường: type 1 (OMIM 607634) được đặc trưng bởi vòm sọ to và đặc và xơ cứng đốt sống lan tỏa và có liên quan đến các đột biến hoạt hóa của LRP5; nó không liên quan đến sự gia tăng tỷ lệ gãy xương vì sức mạnh của xương thực sự tăng lên—do đó nó còn được gọi là bệnh khối lượng xương cao. Bệnh xương hóa đá di truyền trội trên nhiễm sắc thể thường type 2 (OMIM 166600) được đặc trưng bởi sự dày lên của các đĩa cuối đốt sống giống như “xương-trong-xương” và dẫn đến “cột sống áo len” và các dải xương xơ cứng ở xương chậu và nền sọ. Đây là một biến thể của bệnh Albers-Schonberg là kết quả của các đột biến mất chức năng dị hợp tử trong CLCN7. Các đối tượng bị ảnh hưởng biểu hiện tổn hại dây thần kinh sọ (16%), viêm tủy xương hàm và không hàm (19%), viêm xương khớp háng (27%) và gãy xương (78%). Bằng chứng lâm sàng của bệnh có xu hướng xấu đi theo thời gian. Tuy nhiên, sự biểu hiện của tính trạng là thay đổi. Do đó, một phần ba số người mang một đột biến bất hoạt trong CLCN7 không có biểu hiện X-quang hoặc lâm sàng, mặc dù họ có BMD cao hơn đáng kể so với các đối tượng có gen kiểu hoang dã. Ở một phần tư số bệnh nhân có biểu hiện lâm sàng với một đột biến mất chức năng dị hợp tử trong CLCN7, sự biểu hiện của bệnh (gãy xương, viêm tủy xương, thị lực bị tổn hại) có thể xác định được khi sinh hoặc đầu thời thơ ấu hoặc thời thơ ấu. Bệnh nhân có các biểu hiện X-quang/lâm sàng của rối loạn này có nồng độ TRAP và đồng dạng BB của creatine kinase trong huyết thanh tăng do các hủy cốt bào sản xuất; các giá trị này bình thường ở những người mang không bị ảnh hưởng. Bệnh xương hóa đá di truyền trội trên nhiễm sắc thể thường type 3 là kết quả của các biến thể đơn alen trong PLEKHM1 (OMIM 611466) mã hóa một yếu tố cần thiết cho việc vận chuyển các bào quan trong hủy cốt bào. Bệnh xương hóa đá liên kết X có liên quan đến các biến thể bất hoạt của IKBKG (OMIM 300248) mã hóa một serine/threonine kinase tăng cường chức năng của NF-κB, do đó kích thích sự hình thành hủy cốt bào; sự mất mát của nó có liên quan đến bệnh xương hóa đá vừa, loạn sản ngoại bì và suy giảm miễn dịch.
Thường xuyên nhất, các biến thể di truyền cản trở sự tái hấp thu xương ở các đối tượng bị bệnh xương hóa đá có liên quan đến các bất thường trong sự hình thành hủy cốt bào hoặc chức năng của hủy cốt bào—đặc biệt là hiệu quả của việc axit hóa hốc tái hấp thu bên dưới bờ diềm của hủy cốt bào cho phép hòa tan khoáng chất hoặc phân hủy enzyme của chất nền xương hữu cơ. TNFSF11 mã hóa RANKL, một protein xuyên màng 317 aa được biểu hiện trên bề mặt của các tế bào đệm và nguyên bào xương tương tác như một trimer với thụ thể của nó—RANK (được mã hóa bởi TNFRSF11A), một protein màng plasma được biểu hiện bởi các tế bào tiền thân của hủy cốt bào để tạo thành một phức hợp dị lục phân kích thích NF-κB và do đó gây ra sự hình thành hủy cốt bào. Các đột biến bất hoạt trong TNFSF11 và TNFRSF11A dẫn đến sự hình thành hủy cốt bào bị suy giảm và do đó gây ra bệnh xương hóa đá nghèo hủy cốt bào. Các biến thể mất chức năng trong cấu trúc của RANKL liên quan đến bệnh xương hóa đá bao gồm một đột biến sai nghĩa (p.Met199Lys) và các mất đoạn 2-bp (828delCG) và 5-bp (intron 7 − 532 + 4_532 + 8) của TNFSF11. Bệnh nhân bị bệnh xương hóa đá do các đột biến bất hoạt trong TNFSF11 không đáp ứng với cấy ghép tế bào gốc tạo máu vì sự bất thường trong sự hình thành hủy cốt bào nằm ngoài dòng tế bào hủy cốt bào; vì protein RANKL tái tổ hợp có thể biến đổi các monocyte của bệnh nhân bị ảnh hưởng thành các hủy cốt bào chức năng trong ống nghiệm, một con đường điều trị thay thế tiềm năng có thể khả thi. Các đột biến mất chức năng sai nghĩa (p.Gly53Arg, p.Arg 170Cys), vô nghĩa (p.Trp244Stop, p.Gly280Stop) và chèn hai alen trong miền ngoại bào của RANK cũng làm suy giảm sự liên kết với RANKL và hậu quả là kích thích NF-κB và sự hình thành hủy cốt bào. Nhiều bệnh nhân có đột biến bất hoạt trong TNFRSF11A cũng phát triển chức năng tế bào lympho B bị suy giảm và giảm gammaglobulin máu. Các monocyte từ những đối tượng này không đáp ứng về mặt chức năng với RANKL và M-CSF tái tổ hợp trong ống nghiệm; cấy ghép tế bào gốc tạo máu có thể chữa khỏi ở các đối tượng bị bệnh xương hóa đá có các đột biến có hại trong TNFRSF11A. Các đột biến bất hoạt trong IKBKG, mã hóa chất ức chế kinase của gen tăng cường chuỗi nhẹ kappa trong các tế bào B (tiểu đơn vị gamma), còn được gọi là NEMO, cũng làm suy giảm sự tạo ra NF-κB và sự hình thành hủy cốt bào dẫn đến hội chứng bệnh xương hóa đá, phù bạch huyết, loạn sản ngoại bì không có mồ hôi và suy giảm miễn dịch (OL-EDA-ID—OMIM 300301). IKBKG là một thành phần 419 aa của phức hợp kinase IκB kích hoạt NF-κB. IKBKG nằm trên nhánh dài của nhiễm sắc thể X, và do đó sự mất mát của nó được di truyền như một tính trạng liên kết X.
CAII (một trong số các metalloisoenzyme kẽm) là một protein được biểu hiện trong các hủy cốt bào, hồng cầu, não và thận; nó điều hòa sự hình thành axit carbonic từ nước và carbon dioxide (CO2 + H2O ⇒ H2CO3) sau đó phân ly để tạo thành các ion proton/hydro (H+) và bicarbonate (HCO3–). Các đột biến mất chức năng đồng hợp tử hoặc dị hợp tử phức hợp (p.Lys17Glu, p.Tyr40Ter, p.His107Tyr, p.Asn252Asp) trong CAII dẫn đến bệnh xương hóa đá di truyền lặn trên nhiễm sắc thể thường (type 3) biểu hiện ở trẻ em với sự chậm phát triển, tầm vóc thấp, suy giảm thị lực và chậm phát triển kết hợp với toan hóa ống thận gần nhẹ và xa nặng, vôi hóa não trong vỏ não và các hạch nền, và bệnh xương hóa đá với nguy cơ gãy xương tăng. Bệnh xương hóa đá có mức độ nghiêm trọng khiêm tốn và thường không tiến triển; nó thậm chí có thể cải thiện ở tuổi dậy thì. Bicarbonate có thể được sử dụng để bình thường hóa cân bằng axit-bazơ. Sau khi được tạo ra bởi CAII, H+ được đẩy ra khỏi hủy cốt bào vào hốc tái hấp thu dưới hủy cốt bào thông qua các chất vận chuyển và bơm proton. TCIRG1 (OMIM 604592) mã hóa một protein 822 aa, 116 kDa là một tiểu đơn vị của bơm proton không bào của hủy cốt bào (H+-ATPase). (Bằng cách nối thay thế, gen này cũng mã hóa một protein 614 aa—TIRC7—cần thiết cho sự kích hoạt của các tế bào lympho T.) Các đột biến bất hoạt hai alen (sai nghĩa, vô nghĩa, mất đoạn, chèn, vị trí nối) trong TCIRG1 mà sự mất mát của nó làm suy giảm sự vận chuyển H+ và do đó làm giảm sự tái hấp thu khoáng chất xương đã được tìm thấy ở 50% đối tượng mắc dạng bệnh xương hóa đá gây tử vong ở trẻ sơ sinh/trẻ nhỏ (AR type 1) (OMIM 259700). TCIRG1 có thể được hướng đến màng diềm của hủy cốt bào phía trên hốc dưới hủy cốt bào bởi sản phẩm của SNX10; các đột biến hai alen (p.Arg51Gln; c.212 + 1G > T) trong SNX10 cũng dẫn đến bệnh xương hóa đá ác tính ở trẻ nhỏ (di truyền lặn trên nhiễm sắc thể thường type 8) cũng liên quan đến hẹp lỗ chẩm, thoát vị hạnh nhân tiểu não và viêm tủy xương hàm dưới. Các hủy cốt bào trong rối loạn này được phân biệt bởi số lượng túi tế bào chất tăng lên mà chúng chứa, kích thước lớn của chúng và không có khả năng hình thành bờ diềm do các khiếm khuyết trong các mô hình vận chuyển của các endosome và lysosome cần thiết để hình thành các cấu trúc này. Các đột biến mất chức năng trong CLCN7, một kênh clorua được biểu hiện trong màng diềm của hủy cốt bào được kích hoạt và các lysosome của nó, cũng làm suy giảm sự axit hóa của không gian tái hấp thu dưới hủy cốt bào và do đó hòa tan khoáng chất. Các đột biến bất hoạt dị hợp tử (p.Arg767Trp; mất đoạn 2-bp—1423AG) của CLCN7 dẫn đến một dạng bệnh xương hóa đá di truyền trội trên nhiễm sắc thể thường (type 2), trong khi các đột biến mất chức năng hai alen (p.Ile261Phe, p.Arg762Gln, p.Leu766Pro) được tìm thấy ở trẻ sơ sinh mắc các dạng gây tử vong, di truyền lặn trên nhiễm sắc thể thường và trung gian của bệnh này (type 4). Các đột biến trong CLCN7 chiếm 15% đối tượng bị bệnh xương hóa đá di truyền lặn trên nhiễm sắc thể thường nặng và 40% bệnh nhân bị bệnh xương hóa đá di truyền lặn trên nhiễm sắc thể thường trung gian và 75% người lớn bị bệnh xương hóa đá di truyền trội trên nhiễm sắc thể thường type 2. CLCN7 được đồng biểu hiện và tạo phức với Protein xuyên màng 1 liên quan đến bệnh xương hóa đá (OSTM1, OMIM 607649) trong các endosome và lysosome và trong màng diềm của các hủy cốt bào được kích hoạt. Bằng cách giảm sự ổn định sau dịch mã của CLCN7, các đột biến mất chức năng đồng hợp tử (vô nghĩa, mất đoạn) trong OSTM1 đã có liên quan về mặt sinh bệnh học đến bệnh xương hóa đá gây tử vong di truyền lặn trên nhiễm sắc thể thường (type 5); các biến thể của OSTM1 có liên quan đến một quá trình thoái hóa thần kinh nghiêm trọng được đặc trưng bởi teo não và mất myelin. Ngoài ra, một chuỗi tiết ra của OSTM1 liên kết với màng tế bào của các tiền thân của các hủy cốt bào, do đó ức chế sự hợp nhất và do đó sự hình thành của các hủy cốt bào đa nhân. Sản phẩm của PLEKHM1 là một protein cần thiết cho sự di chuyển và phân phối lysosome trong hủy cốt bào, liên kết các lysosome với các vi ống, hợp nhất với màng plasma của hủy cốt bào và bài tiết từ hủy cốt bào và cho sự hình thành các bờ diềm trên hốc của chúng; các đột biến bất hoạt hai alen của PLEKHM1 dẫn đến bệnh xương hóa đá có mức độ nghiêm trọng trung gian hoặc đáng kể (di truyền lặn trên nhiễm sắc thể thường type 6).
Nếu không được xác định trước khi sinh, các phát hiện lâm sàng ở trẻ sơ sinh bị bệnh xương hóa đá có thể bao gồm chậm phát triển, lồi mắt và các cử động ngoài nhãn cầu bất thường, mất chức năng thị giác và thính giác, liệt/liệt dây thần kinh mặt và các dấu hiệu tăng áp lực nội sọ. Ở trẻ lớn hơn mắc dạng trung gian của bệnh xương hóa đá, gãy xương không do chấn thương có thể là một khiếu nại khởi phát của bệnh xương hóa đá; có thể có chân vòng kiềng chữ X, cũng như các bất thường về chức năng dây thần kinh sọ, răng sữa bị kẹt và gan lách to. Các biểu hiện khác của bệnh này bao gồm thiếu máu, hạ canxi máu, chậm mọc răng sữa, sâu răng, nhiễm trùng hàm dưới, gan lách to và chậm tăng trưởng. Các phát hiện X-quang ở bệnh nhân bị bệnh xương hóa đá có thể có trong tử cung hoặc đầu thời thơ ấu và bao gồm: vòm sọ rộng, mật độ xương dài tăng đồng đều, vỏ và tủy xương rộng dẫn đến hẹp khoang tủy, các dải hành xương của xương đặc xen kẽ với các vùng xương thấu quang dẫn đến hình ảnh “xương-trong-xương”, mô hình hành xương “hình chùy” (dị tật bình Ehrlenmeyer), chiều rộng xương sườn tăng, khuyết ở phần trước của các đốt sống đặc đồng đều hoặc các đốt sống có bờ trên và dưới đặc (đốt sống áo len hoặc sandwich), các gãy xương mới hoặc đã lành. Các triệu chứng và dấu hiệu ở bệnh nhân bị bệnh xương hóa đá di truyền trội trên nhiễm sắc thể thường bao gồm: gãy xương liên quan đến chấn thương tối thiểu, áp xe răng và viêm tủy xương hàm dưới. Do đó, ở hầu hết bệnh nhân, chẩn đoán bệnh xương thủy tinh bị nghi ngờ và được xác lập trên cơ sở các phát hiện lâm sàng và X-quang. Đứng đầu trong số các xét nghiệm chẩn đoán ở bệnh nhân bị bệnh xương hóa đá là phân tích các gen được biết là có liên quan đến bệnh này (xem Bảng 20.11A) hoặc như đang nhanh chóng trở nên phổ biến hơn—giải trình tự toàn bộ bộ gen. Nhóm công tác về Bệnh xương hóa đá cũng khuyến nghị trong việc đánh giá và theo dõi trẻ em bị nghi ngờ/chẩn đoán mắc bệnh xương hóa đá: xác định công thức máu toàn phần và đo nồng độ canxi, phốt phát, creatinin, calcidiol, PTH, isozyme BB của creatine kinase, lactate dehydrogenase trong huyết thanh; canxi/creatinin trong nước tiểu; siêu âm thận; và chụp cộng hưởng từ các lỗ/ống dây thần kinh sọ.
Một đội ngũ đa ngành có kỹ năng trong việc quản lý bệnh nhân bị bệnh xương hóa đá là rất cần thiết cho việc chăm sóc tối ưu trẻ sơ sinh và trẻ em mắc rối loạn này. Điều trị triệu chứng của những bệnh nhân này bao gồm tránh các hoạt động gây gãy xương, truyền các thành phần máu cần thiết, và dùng canxi và vitamin D/calcitriol bổ sung để quản lý hạ canxi máu và còi xương (“còi xương hóa đá”). Còi xương hóa đá thường phát triển ở những bệnh nhân có các biến thể của TCIRG1 do giảm axit dạ dày và hậu quả là giảm hấp thu canxi ở ruột. Việc dùng IFN-γ (một chất kích thích gián tiếp sự hình thành hủy cốt bào) có thể có lợi ích hạn chế ở những bệnh nhân mắc các dạng bệnh xương hóa đá di truyền lặn trên nhiễm sắc thể thường đang chờ liệu pháp tế bào gốc. Việc dùng liều cao calcitriol cho bệnh nhân bị bệnh xương hóa đá không được khuyến nghị. Khi được thực hiện ở đầu thời thơ ấu, việc truyền tế bào gốc tạo máu từ các nhà tài trợ HLA-giống hệt nhau với việc thay thế các hủy cốt bào khiếm khuyết bằng các tế bào tiền thân hủy cốt bào bình thường, mặc dù nguy hiểm, đã có giá trị ở những bệnh nhân mắc các dạng bệnh xương hóa đá giàu hủy cốt bào, mặc dù không có khả năng đảo ngược các hậu quả đã phải chịu của bệnh. Cấy ghép tế bào gốc tạo máu cho phép sự cấy ghép của các tiền thân hủy cốt bào có nguồn gốc từ người cho biệt hóa thành các hủy cốt bào chức năng trưởng thành cho phép tái tạo xương và tạo máu. Việc truyền tế bào gốc tạo máu thường không được khuyến nghị ở những bệnh nhân bị bệnh xương hóa đá cũng bị thoái hóa thần kinh nguyên phát (CLCN7, OSTM1) hoặc ở các dạng bệnh xương hóa đá nghèo hủy cốt bào liên quan đến các đột biến bất hoạt trong TNFSF11, TNFRSF11A, và IKBKG. Khả năng cấy ghép tế bào gốc tạo máu trong tử cung cũng có thể được xem xét. Tăng canxi máu thường làm phức tạp giai đoạn sau cấy ghép khi chức năng của hủy cốt bào hoạt động trở lại, đặc biệt là ở những bệnh nhân có các đột biến bất hoạt trong TNFRSF11A. Tăng canxi máu đã được quản lý bằng cách hạn chế chế độ ăn, calcitonin và dùng bisphosphonate và/hoặc bằng cách sử dụng một chất ức chế RANKL–denosumab, một kháng thể đơn dòng được tạo ra chống lại protein này. Việc thay thế gen khiếm khuyết bằng cách chỉnh sửa bộ gen hoặc các kỹ thuật di truyền tiên tiến khác với việc phục hồi chức năng gen bình thường có thể có lợi cho bệnh nhân bị bệnh xương hóa đá trong tương lai. Ngoài việc chăm sóc lâm sàng chung bởi bác sĩ chăm sóc chính của trẻ, bác sĩ nội tiết, bác sĩ huyết học, bác sĩ nhãn khoa, bác sĩ chỉnh hình, bác sĩ thần kinh và nhà di truyền học có vai trò cơ bản trong việc quản lý và phối hợp chăm sóc bệnh nhân bị bệnh xương hóa đá. Với việc điều trị được cải thiện, tuổi thọ đã tăng lên và sự tiến bộ về phát triển đã được cải thiện. Bệnh xương hóa đá do thiếu CAII không được điều chỉnh bằng cách phục hồi cân bằng axit-bazơ toàn thân bình thường, nhưng cấy ghép tủy xương có thể hữu ích.
Ngoài các dạng bệnh xương hóa đá giàu hủy cốt bào được chỉ định OMIM đã được thảo luận trước đó, các đột biến bất hoạt trong FERMT3 (OMIM 607901) cũng dẫn đến một dạng bệnh xương hóa đá nặng ở trẻ nhỏ liên quan đến tăng nhạy cảm với xuất huyết và nhiễm trùng. Các biến thể của MITF mã hóa yếu tố tăng trưởng liên quan đến mắt nhỏ (OMIM 15684), LRKK1 mã hóa kinase 1 lặp lại giàu leucine (OMIM 610986), và USB1 mã hóa phosphodiesterase 1 sinh tổng hợp RNA hạt nhân nhỏ U6 (OMIM 613276) cũng đã được liên kết với các hội chứng lâm sàng giống bệnh xương hóa đá/xơ cứng xương. Ở một bệnh nhân hiếm gặp, các biến thể di truyền của SLC29A3 mã hóa thành viên 3 của họ chất vận chuyển chất tan 29 (OMIM 612373) và CSF mã hóa yếu tố kích thích dòng đại thực bào (OMIM 120420) đã được liên kết với các dạng bệnh xương hóa đá nghèo hủy cốt bào. Liên quan đến các tác dụng ức chế kéo dài của bisphosphonate đối với việc tạo hình và tái tạo xương, việc dùng liều cao pamidronate tiêm tĩnh mạch (2800 mg) trong khoảng thời gian 3 năm đã dẫn đến một rối loạn “giống bệnh xương hóa đá” mắc phải ở một cậu bé 12 tuổi bị tăng phosphatase kiềm không giải thích được—một bất thường tồn tại trong vài năm sau khi ngừng sử dụng tác nhân này. Hành xương của cậu bé này cực kỳ đặc và hình chùy, nền sọ bị xơ cứng và các đĩa cuối đốt sống dày lên. Về mặt mô học, sinh thiết mào chậu cho thấy các thanh sụn vôi hóa và các hủy cốt bào không hoạt động. Mặc dù mức độ nghiêm trọng của các phát hiện X-quang và vi thể, bệnh nhân khỏe mạnh về mặt lâm sàng với sự tăng trưởng bình thường và không có bằng chứng về sự ức chế tủy xương hoặc tạo máu ngoài tủy, mặc dù nguy cơ gãy xương trong tương lai có thể đã tăng lên.
Ngoài bệnh xương hóa đá, còn có các dạng lâm sàng và các biến thể di truyền khác gây ra sự tăng khoáng hóa xương ở trẻ em (xem Bảng 20.11B). Pycnodysostosis (OMIM 265800) được biểu hiện lâm sàng bằng tầm vóc thấp không cân đối trong thời thơ ấu và thời thơ ấu với đầu to và các đường khớp sọ mở, trán cao, các đặc điểm khuôn mặt nhỏ, lồi mắt, củng mạc xanh, mũi khoằm và nhọn, hàm dưới nhỏ, vòm miệng cao, răng sữa tồn tại, ngón tay ngắn với móng tay thiểu sản, lồng ngực hẹp, ngực ức gà và gù vẹo cột sống với ưỡn cột sống thắt lưng. Về mặt X-quang, có sự gia tăng mật độ xương ngày càng xấu đi theo tuổi mặc dù nhạy cảm với gãy xương tăng lên, các thóp mở, sự kẹt của các răng vĩnh viễn và răng thừa, xương đòn mỏng và thiểu sản hai bên, sự vắng mặt một phần hoặc toàn bộ của các xương sườn và xương móng, và tiêu xương đầu ngón của các đốt ngón tay xa—một đặc điểm bệnh lý của bệnh pycnodysostosis. Dữ liệu xét nghiệm và mô học xương thường bình thường, mặc dù có thể có bằng chứng sinh hóa về giảm hoạt động của nguyên bào xương và hủy cốt bào. Quan sát vi thể rằng sự phong phú của các hủy cốt bào có bờ diềm được bao quanh bởi các vùng trong suốt mở rộng cho thấy sự hòa tan khoáng chất xương là bình thường nhưng sự phân hủy chất nền là bất thường ở những bệnh nhân này. Thực tế, có một lượng lớn chất nền không vôi hóa cả trong các hốc dưới hủy cốt bào và các hủy cốt bào. Pycnodysostosis là do các đột biến mất chức năng hai alen trong CTSK mã hóa cathepsin K, protease cysteine lysosome của hủy cốt bào phân hủy chất nền hữu cơ sau khi pha khoáng của xương đã được tái hấp thu. Trong số các biến thể trong CTSK được xác định ở bệnh nhân bị pycnodysostosis có: đồng hợp tử đơn cha cho nhiễm sắc thể 1 với CTSK của người cha mang một đột biến bất hoạt p.Ala277Val, thay thế p.Leu9Pro trong peptide tín hiệu của dạng tiền thân của protein enzyme ngăn cản sự hoàn thành quá trình xử lý sau dịch mã của nó, và thay thế p.Ter330Trp cho phép bổ sung 19 aa vào đầu carboxyl của protein enzyme này.
Trái ngược với các bệnh làm tăng khối lượng xương bằng cách giảm tái hấp thu xương là những rối loạn chủ yếu làm tăng sự hình thành xương. Một ví dụ về loại sau là dạng bệnh khối lượng xương cao di truyền trội trên nhiễm sắc thể thường, tương đối lành tính (OMIM 601884) có liên quan đến một đột biến tăng chức năng dị hợp tử (p.Gly171Val) trong LRP5 đã được thảo luận. Dickkopf và sclerostin là các protein liên kết với miền ngoại bào của LRP5 và nhập bào phức hợp thụ thể do đó ngăn chặn tín hiệu WNT. Mặc dù nhìn chung là lành tính, các đột biến hoạt hóa của LRP5 cũng có thể liên quan đến các biến chứng thần kinh, chẳng hạn như mất thính lực, đau đầu và đau ở các chi. Ở một số gia đình, các đột biến hoạt hóa dị hợp tử (p.Ala242Thr) của LRP5 và sự hình thành xương quá mức đã được liên kết với bệnh xơ cứng xương nội mạc toàn thể di truyền trội trên nhiễm sắc thể thường (bệnh van Buchem, type 2, OMIM 607636). Các biến thể của SOST (mã hóa sclerostin) có liên quan đến bệnh van Buchem type 1 (OMIM 239100) đã được cho là do một mất đoạn 52-kb ba phẩy (3′) của SOST với hậu quả là mất một yếu tố điều hòa của gen; rối loạn này được đặc trưng bởi hộp sọ dày lên rõ rệt—dẫn đến mất chức năng dây thần kinh sọ, hàm dưới và các xương dài, và xơ cứng xương di truyền lặn trên nhiễm sắc thể thường type 1 (OMIM 269500), một rối loạn phát triển quá mức của xương toàn thể, tiến triển kết hợp với dính ngón; sự liên quan của hộp sọ dẫn đến chèn ép và suy giảm chức năng của các dây thần kinh sọ, cũng như tăng áp lực nội sọ. Xơ cứng xương type 2 (OMIM 614305) có liên quan đến các biến thể bất hoạt hai alen của LRP4 (OMIM 604270) mã hóa đồng thụ thể 4 liên quan đến lipoprotein mật độ thấp ngăn cản sự tương tác của nó với sclerostin do đó làm mất ức chế sclerostin. Xơ cứng xương type 1 lần đầu tiên biểu hiện ở thời thơ ấu và được đặc trưng bởi các xương ngoại vi và sọ rất dày với sự phát triển quá mức của vòm sọ, dẫn đến biến dạng khuôn mặt, chèn ép các dây thần kinh sọ II, VII và VIII, tăng áp lực nội sọ và chèn ép thân não. Bệnh nhân bị ảnh hưởng cũng có dính ngón da hoặc xương không đối xứng thay đổi của ngón trỏ và ngón giữa và tăng trưởng cơ thể quá mức; họ cực kỳ kháng lại gãy xương. Nồng độ phosphatase kiềm và propeptide đầu N của procollagen type 1 (P1NP) trong huyết thanh tăng ở những đối tượng này trong khi nồng độ sclerostin thấp hoặc không thể đo được. Xơ cứng xương type 1 là do các đột biến mất chức năng hai alen trong SOST mã hóa sclerostin, một peptide 213 aa được tiết ra chủ yếu bởi các tế bào xương nằm trong xương. Về mặt sinh lý, sclerostin ức chế sự hình thành xương qua trung gian WNT bằng cách liên kết và nhập bào LRP5, đồng thụ thể cho WNT. Khi hoạt động của sclerostin giảm, sự hình thành xương tăng lên. Sclerostin cũng làm tăng sự hình thành hủy cốt bào bằng cách tăng tổng hợp RANKL của tế bào xương. Ở những người mang dị hợp tử của các đột biến bất hoạt trong SOST, khối lượng xương tăng nhưng không đến mức bệnh lý. Bệnh tăng sinh xương vỏ toàn thể/bệnh van Buchem type 1 là một rối loạn di truyền lặn trên nhiễm sắc thể thường được đặc trưng bởi sự dày lên và to ra của các xương sọ, hàm dưới, xương sườn và thân các xương dài dẫn đến tăng mật độ xương và lấn chiếm các lối đi của dây thần kinh sọ dẫn đến các bất thường về thị giác, thính giác và cử động của mặt. Tuy nhiên, bệnh nhân mắc bệnh van Buchem type 1 không bị dính ngón hoặc tầm vóc cao do đó phân biệt nó với xơ cứng xương type 1. Rối loạn này cũng là do mất biểu hiện hai alen của SOST nhưng là kết quả của việc mất đoạn 52-kb của một yếu tố điều hòa nằm ở 35 kb hạ nguồn (3′) của SOST thay vì do một đột biến trong cấu trúc exon/intron của chính SOST. Do đó, bệnh này là alen với xơ cứng xương; nó khác biệt về mặt lâm sàng ở chỗ chứng khổng lồ và các bất thường về tay có ở bệnh nhân bị xơ cứng xương type 1 nhưng không có ở những người bị bệnh van Buchem type 1. Các biểu hiện lâm sàng của xơ cứng xương type 2 tương tự như của type 1 nhưng có liên quan về mặt sinh bệnh học đến các đột biến mất chức năng hai alen trong LRP4. Tác dụng ức chế đối với sự hình thành xương của sự tương tác giữa sclerostin và LRP5/LRP6 được tạo điều kiện thuận lợi bằng cách liên kết của sclerostin với LRP4. Do đó, các đột biến bất hoạt (p.Arg1170Trp, p.Trp1186Ser) trong LRP4 cản trở sự tương tác của sclerostin với LRP5/6 và do đó tác dụng ức chế của sclerostin đối với sự hình thành nguyên bào xương. Điều trị các rối loạn này chủ yếu là triệu chứng.
Loạn sản thân xương tiến triển (bệnh Camurati-Englemann; OMIM 131300) là một rối loạn tăng sinh xương sọ-ngoại vi, trội trên nhiễm sắc thể thường với sự biểu hiện thay đổi, biểu hiện ở trẻ em với các vấn đề như đi khập khiễng, dáng đi lạch bạch và/hoặc đau chân, mệt mỏi và yếu cơ không tiến triển. Về mặt X-quang, có sự dày vỏ xương đối xứng (tăng sinh xương) do sự hình thành xương màng xương và nội mạc tăng lên ở thân các xương dài, bộ xương trục và hộp sọ, chân vòng kiềng chữ X và vẹo cột sống. Về mặt sinh bệnh học, rối loạn này là do các đột biến sai nghĩa hoạt hóa trong miền “peptide liên quan đến độ trễ” của propeptide tiền thân của TGFB1 (OMIM 190180). Thông thường, sau khi xử lý sau dịch mã, hai peptide liên quan đến độ trễ được liên kết không cộng hóa trị với hai peptide TGFβ1 trưởng thành để tạo thành một “phức hợp độ trễ”. Các đột biến tăng chức năng trong miền peptide liên quan đến độ trễ của TGFB1 (đặc biệt là ở codon 218—p.Arg218His, p.Arg218Cys—một điểm nóng đột biến) làm suy giảm sự liên kết này dẫn đến sự kích hoạt sớm của TGF-β1 và hậu quả là kích thích sự hình thành xương nội màng và kìm hãm sự tái hấp thu xương. TGF-β1 cũng ức chế sự hình thành cơ và mỡ. Do tác dụng ức chế của glucocorticoid đối với sự hình thành xương và tác dụng kích thích của chúng đối với sự tái hấp thu xương, các tác nhân này đã hữu ích trong việc làm giảm nhiều triệu chứng lâm sàng và các bất thường X-quang ở bệnh nhân bị loạn sản thân xương tiến triển cũng như các chất ức chế thụ thể angiotensin II (ví dụ, losartan). Các loạn sản xương xơ cứng di truyền khác bao gồm bệnh đốm xương (OMIM 166700)—các nốt phát triển quá mức của xương trong các xương chậu và xương dài, hộp sọ và/hoặc xương sườn do các đột biến bất hoạt dị hợp tử trong LEMD3 (OMIM 607844) mã hóa một protein màng nhân điều chỉnh sự truyền tín hiệu bởi các BMP và TGFβ; bệnh đốm xương có liên quan đến các biến dạng mật độ cao của xương vỏ—bệnh vằn xương; và bệnh xương sọc (OMIM 300373) là một rối loạn liên kết X gây ra bởi các đột biến mất chức năng trong AMER1 (OMIM 300647) mã hóa một protein thúc đẩy sự phân hủy của β-catenin qua trung gian proteasome—sự mất mát của nó do đó thúc đẩy sự chuyển β-catenin đến nhân và kéo dài hoạt động sinh học của hệ thống truyền tín hiệu WNT/LRP5,6/β-catenin thúc đẩy sự trưởng thành và chức năng của nguyên bào xương dẫn đến tăng sinh xương nội mạc. Các loạn sản xương xơ cứng di truyền và không di truyền (ví dụ, xơ cứng xương nội tủy) phải được phân biệt với các bệnh xơ cứng mắc phải, chẳng hạn như bệnh hồng cầu hình liềm và xơ tủy.
Hình thành xương lạc chỗ/Vôi hóa lạc chỗ
Các rối loạn hình thành xương lạc chỗ là những rối loạn trong đó xương phát triển bên ngoài bộ xương và trong các mô mềm (Bảng 20.12). Sự rối loạn điều hòa của các quá trình biệt hóa và trưởng thành cho phép các tế bào tiền thân phát triển thành các nguyên bào xương sau đó sản xuất xương nội sụn hoặc màng bình thường nhưng ở các vị trí ngoài xương bất thường. Sự hình thành xương lạc chỗ lẻ tẻ xảy ra ở các vị trí vết thương và bỏng nặng, sau chấn thương tủy sống và ở các vùng loét do tỳ đè. Loạn sản xơ hóa cốt hóa tiến triển (OMIM 135100) là một rối loạn gây tàn tật của sự hình thành xương lạc chỗ có thể phát triển tự phát hoặc tại các vị trí chấn thương và dẫn đến cứng khớp của tất cả các khớp chính làm hạn chế nghiêm trọng khả năng vận động. Nó được đặc trưng bởi sự hóa xương lạc chỗ tiến triển của cơ xương và mô liên kết (cân, gân, dây chằng) dẫn đến bất động và hợp nhất của hàm dưới, cổ, cột sống, hông và các khớp khác và sự phát triển của một “bộ xương thứ hai” bao bọc và giam cầm cơ thể. Rối loạn này có thể có khi sinh và thường biểu hiện trước 5 tuổi. Loạn sản xơ hóa cốt hóa tiến triển cũng liên quan đến các bất thường bẩm sinh của ngón chân cái (ngón chân cái vẹo ngoài, các xương bàn chân thứ nhất bị dị dạng, một đốt ngón), các đặc điểm khuôn mặt đặc trưng (mặt dài hẹp, hàm dưới nhỏ, tai thấp), điếc, hói da đầu và chậm phát triển nhẹ. Về mặt vi thể, có sự hình thành xương nội sụn bình thường nhưng lạc chỗ xảy ra sau một giai đoạn viêm trước đó phát triển hoặc khi không có chấn thương hoặc sau một chấn thương nhỏ, chẳng hạn như tiêm chủng; quá trình bệnh lý tiến triển qua các giai đoạn thâm nhiễm bạch cầu đơn nhân, thoái hóa sợi cơ, tăng sinh xơ, tạo mạch, hình thành sụn và hình thành xương. Mặc dù thường là lẻ tẻ vì các đối tượng bị ảnh hưởng hiếm khi sinh sản, loạn sản xơ hóa cốt hóa tiến triển có thể được di truyền như một tính trạng trội trên nhiễm sắc thể thường. Bệnh này chủ yếu là do một biến thể rất đặc hiệu (chuyển đoạn c.617G > A dẫn đến p.Arg206His) của ACVR1 (mã hóa thụ thể activin A, type-1, OMIM 102576); một đột biến ACVR1 khác được mô tả ở bệnh nhân bị loạn sản xơ hóa cốt hóa tiến triển cổ điển là chuyển đoạn c.744G > C dẫn đến p.Arg258Ser. Activin là thành viên của siêu họ TGFβ bao gồm các BMP cùng với các inhibin và yếu tố ức chế ống Mullerian. ACVR1 mã hóa một thụ thể BMP type I (thụ thể activin type Ia) được biểu hiện trong các tế bào sụn và nguyên bào xương. Đột biến p.Arg206His trong ACVR1 nằm ở điểm nối của các miền kích hoạt glycine-serine và tyrosine kinase trong tế bào chất của thụ thể và dẫn đến một thụ thể BMP type I hoạt động cấu thành. Sự kích hoạt quá mức của các con đường truyền tín hiệu nội bào xảy ra khi các BMP liên kết với thụ thể ACVR1 bị đột biến. Ngoài ra, biến thể p.Arg206His dường như thay đổi tính đặc hiệu tín hiệu của thụ thể ACVR1 đối với activin A, dẫn đến sự phosphoryl hóa của SMAD 1/5/8 thay vì SMAD 2/3, gây ra sự hình thành xương nội sụn lạc chỗ. Chỉ riêng các BMP có thể kích thích sự hình thành xương nội sụn hoàn chỉnh ở các vị trí lạc chỗ.
Bảng 20.12: Các nguyên nhân di truyền của sự hình thành xương lạc chỗ và vôi hóa lạc chỗ
| Loại bệnh OMIM | Gen Nhiễm sắc thể OMIM | Biểu hiện lâm sàng | Sinh lý bệnh |
|---|---|---|---|
| Hình thành xương lạc chỗ | |||
| Loạn sản xơ hóa cốt hóa tiến triển 135100 | ACVR1: Thụ thể Activin A, type I 2q24.1 102576 | Hóa xương tiến triển của các cơ xương, cân, gân và dây chằng | Đột biến hoạt hóa đơn alen đặc hiệu (Arg206His) trong miền tế bào chất của một thụ thể BMP type 1 làm tăng sự hình thành sụn và xương ngoài xương |
| Tăng sinh xương lạc chỗ tiến triển 166350 | GNAS: Phức hợp gen GNAS 20q13.32 139320 | Hóa xương của lớp bì bắt đầu ở trẻ sơ sinh, sau đó là sự hình thành xương màng trong cơ sâu và cân ở trẻ em | Các đột biến bất hoạt đơn alen trong tiểu đơn vị Gsα được mã hóa bởi người cha của protein G cho phép sự hình thành xương dưới da; liên quan đến giả giả suy cận giáp |
| Vôi hóa lạc chỗ | |||
| Vôi hóa u, tăng phốt phát máu, có tính gia đình 211900 | GALNT3: UDP-N-acetyl-alpha-D-galactosamine: polypeptide N-acetylgalactosaminyl-transferase 3 2q24.3 601756 | Lắng đọng tiến triển của các tinh thể canxi phốt phát ở vùng quanh khớp, mô mềm và xương | Các đột biến hai alen trong một đồng yếu tố cần thiết cho sự tổng hợp FGF23 có hoạt tính sinh học và bài tiết phốt phát ở ống thận |
| Vôi hóa u, tăng phốt phát máu, có tính gia đình 211900 | FGF23: Yếu tố tăng trưởng nguyên bào sợi 23 12p13.32 605380 | Lắng đọng tiến triển của các tinh thể canxi phốt phát ở vùng quanh khớp, mô mềm và xương | Các đột biến bất hoạt hai alen trong một yếu tố ức chế sự tái hấp thu phốt phát ở ống thận |
| Vôi hóa u, tăng phốt phát máu, có tính gia đình 211900 | KL: Klotho 13q13.1 604824 | Lắng đọng tiến triển của các tinh thể canxi phốt phát ở vùng quanh khớp, mô mềm và xương | Đột biến bất hoạt hai alen trong một đồng yếu tố cho phép FGF23 ức chế sự tái hấp thu phốt phát ở ống thận |
| Vôi hóa u, phốt phát máu bình thường, có tính gia đình 610455 | SAMD9: Protein 9 chứa miền alpha motif vô trùng 7q21.2 610456 | Các khối vôi hóa quanh khớp và ngoại vi sau viêm | Các đột biến bất hoạt hai alen trong một protein điều hòa sự phân chia, vận động, tuổi thọ và viêm của tế bào |
BMP, Protein hình thái xương; GNAS, guanosine diphosphate trên tiểu đơn vị alpha.
Bệnh nhân có các biến thể lâm sàng của loạn sản xơ hóa cốt hóa tiến triển và các đột biến ACVR1 khác ngoài p.Arg206His và p.Arg258Ser đã được mô tả; họ đã được phân loại thành những người bị loạn sản xơ hóa cốt hóa tiến triển cộng với các bất thường của não, mắt và/hoặc tủy xương, hoặc những người có các biến thể loạn sản xơ hóa cốt hóa tiến triển—hoặc không có bất thường ở ngón chân cái hoặc hình thành xương lạc chỗ ít nghiêm trọng hơn; bệnh nhân có đột biến ở codon 328 có mức độ nghiêm trọng thay đổi từ cổ điển đến bệnh khởi phát muộn; bệnh nhân có đột biến p.Arg201Ile trong ACVR1 có sự hóa xương ngoài xương khởi phát ở tuổi trưởng thành và ngón chân bình thường. Việc quản lý những bệnh nhân này chủ yếu là triệu chứng và giảm nhẹ đến mức có thể, mặc dù ức chế miễn dịch có thể làm giảm cường độ của sự hóa xương ngoài xương.
Glucocorticoid đã được sử dụng trong điều trị bệnh nhân bị loạn sản xơ hóa cốt hóa tiến triển, nhưng hiệu quả kém và tác dụng phụ đáng kể. Các chất ức chế tế bào mast và leukotriene cũng đã được sử dụng trong nỗ lực ức chế viêm cũng như bisphosphonate; tuy nhiên, không có thử nghiệm lâm sàng kết luận nào để hỗ trợ việc sử dụng các tác nhân này. Vì chấn thương có thể gây ra sự hình thành xương mới ở những bệnh nhân này, việc phòng ngừa chấn thương là nguyên tắc quản lý hàng đầu. Ngay cả một mũi tiêm bắp cũng có thể gây ra sự hóa xương đau tại chỗ. Việc tránh phẫu thuật là tối quan trọng vì sự xúc phạm này có thể kích thích không chỉ sự phát triển xương mới tại chỗ mà còn ở xa. Mặc dù hiện tại, không có liệu pháp nào cho bệnh loạn sản xơ hóa cốt hóa tiến triển, các phương pháp điều trị nhằm mục đích ngắt quãng (các) con đường sinh lý bệnh chính của bệnh này, chẳng hạn như trung hòa qua trung gian kháng thể đơn dòng của thụ thể activin A hoặc ngắt quãng con đường truyền tín hiệu SMAD 1/5/8 (mục tiêu của rapamycin ở động vật có vú [mTOR]) có thể có lợi.
POH (OMIM 166350) được đặc trưng bởi nhiều ổ (thân, các chi hoặc các ngón tay) hình thành xương nội màng ở da liên quan đến mô mỡ (u xương ở da) bắt đầu ở trẻ sơ sinh khi không có bất kỳ chấn thương tại chỗ hoặc sự xúc phạm viêm nào. Các tổn thương có thể không có triệu chứng hoặc đau. Theo thời gian, sự hóa xương lạc chỗ chui vào cơ xương và mô liên kết sâu và có thể được kết hợp vào xương của bộ xương. POH được di truyền như một tính trạng trội trên nhiễm sắc thể thường và xảy ra ở cả bé trai và bé gái; nó là do các đột biến bất hoạt của alen GNAS thường được di truyền từ người cha. Sự di truyền từ người cha của GNAS bị đột biến có liên quan đến sự chậm phát triển trong tử cung đáng kể và các biểu hiện lâm sàng của POH nghiêm trọng hơn so với khi đột biến GNAS được di truyền bởi người mẹ. Các đột biến giống hệt nhau trong GNAS có thể được biểu hiện lâm sàng như POH, PHP, PPHP hoặc u xương ở da nguyên phát ở các thành viên khác nhau trong cùng một gia đình (ví dụ, mất 1-bp, 725C), tất cả các rối loạn này đều liên quan đến sự hóa xương dưới da (lớp bì). Tuy nhiên, bệnh nhân bị POH không có các đặc điểm thực thể của AHO cũng như không kháng hormone. Do đó, các tiêu chí chẩn đoán cho POH là: các tổn thương xương bề mặt, không có các đặc điểm của AHO (PPHP) và đáp ứng bình thường với PTH. Hiện tại chỉ có điều trị triệu chứng cho bệnh nhân bị POH.
Vôi hóa u có tính gia đình được đặc trưng bởi sự lắng đọng của canxi phốt phát cơ bản vào các không gian quanh khớp xung quanh các khớp vai, khuỷu tay, cổ tay, hông và đầu gối và đôi khi trong chính xương xảy ra tự phát hoặc sau chấn thương tối thiểu nhưng lặp đi lặp lại đối với các mô mềm. Các biến thể của một số gen (xem bên dưới) dẫn đến vôi hóa u có tính gia đình với phốt phát máu bình thường hoặc tăng phốt phát máu—tất cả đều có các biểu hiện lâm sàng tương tự. Vôi hóa u có tính gia đình biểu hiện ở trẻ em với đau xương tái phát, các khối lớn; cứng, không đau, dưới da; các khối quanh khớp rộng có thể làm tổn hại đến khả năng vận động của khớp; và các lắng đọng mạch máu của canxi phốt phát. Ở một số bệnh nhân, các vôi hóa lạc chỗ có thể chỉ giới hạn ở mí mắt; nó được đặc trưng về mặt X-quang bởi sự tăng sinh xương vỏ, phản ứng màng xương và các lắng đọng khoáng chất xung quanh các khớp lớn, đặc biệt là hông và vai. Về mặt vi thể, có một phản ứng mô bào với sự hình thành các cấu trúc giống bao hoạt dịch bị vôi hóa. Các nghiên cứu xét nghiệm cho thấy hoặc phốt phát máu bình thường hoặc tăng phốt phát máu rõ rệt và giảm phốt phát niệu tương đối, loại sau là do tăng tái hấp thu phốt phát ở ống thận và nồng độ calcitriol huyết thanh bình thường hoặc tăng một cách không phù hợp, vì mặc dù có tăng phốt phát máu, sự bài tiết PTH không tăng và sự tổng hợp calcitriol và hấp thu canxi ở ruột vẫn tồn tại. Có một loại phốt phát máu bình thường (OMIM 610455) và ba loại tăng phốt phát máu được chỉ định (OMIM 211900, 617993, 617994) của vôi hóa u có tính gia đình và cũng có các dạng thứ phát của bệnh này. Rối loạn này là do mất chức năng của hoạt động FGF23 và hậu quả là sự tái hấp thu phốt phát ở ống thận không bị cản trở. Do đó, sinh lý bệnh của rối loạn này là hình ảnh phản chiếu của sinh lý bệnh liên quan đến các dạng còi xương giảm phốt phát máu liên kết X và trội trên nhiễm sắc thể thường và nhuyễn xương do khối u trong đó có sản xuất FGF23 dư thừa và hoạt động quá mức, dẫn đến tăng phốt phát niệu và hậu quả là hạ phốt phát máu, còi xương và nhuyễn xương. Vôi hóa u tăng phốt phát máu có tính gia đình không đồng nhất về mặt di truyền: type 1 (OMIM 211900) có liên quan đến các biến thể bất hoạt của GALNT3 (OMIM 601756), type 2 (OMIM 617993) do các lỗi trong FGF23 (OMIM 605380), và type 3 (OMIM 617994) do các đột biến trong KL (OMIM 604824). Thường gặp nhất ở bệnh nhân bị vôi hóa u tăng phốt phát máu có tính gia đình là các mất đoạn nhỏ, vị trí nối, và các đột biến sai nghĩa hoặc vô nghĩa hai alen mất chức năng trong GALNT3 (p.Arg162Stop, p.Thr272Lys, p.Cys574Gly, p.Gln592Stop). Sản phẩm của GALNT3 là một glycosyl transferase khởi đầu quá trình glycosyl hóa O trong đó N-acetylgalactosamine là đường đầu tiên trong chuỗi bên, một bước cần thiết cho sự bài tiết FGF23 nguyên vẹn, ổn định và chức năng. Thất bại trong việc glycosyl hóa O FGF23 tại Thr178 trong bộ máy Golgi cho phép sự phân cắt nội bào nhanh chóng của nó giữa Arg179 và Ser180 thành các mảnh đầu amino và carboxyl không hoạt động sinh học. Các đột biến bất hoạt đồng hợp tử trong FGF23 (p.Ser71Gly; p.Met96Thr, p.Ser129Phe) đã được xác định ở những bệnh nhân mắc bệnh vôi hóa u có tính gia đình type; khi không có FGF23, sự tái hấp thu phốt phát đã lọc ở ống thận không bị cản trở. Nồng độ FGF23 nguyên vẹn trong huyết thanh thấp hoặc không thể phát hiện được trong khi nồng độ FGF23 đầu carboxyl tăng ở những bệnh nhân bị vôi hóa u có tính gia đình do các đột biến trong GALNT3 hoặc FGF23. Liệu pháp với một chất kết dính phốt phát đường uống và chất ức chế carbonic anhydrase acetazolamide đã dẫn đến tăng phốt phát niệu và tái hấp thu các vôi hóa lạc chỗ mà không có sự thay đổi nồng độ phốt phát hoặc canxi trong huyết thanh. Hội chứng tăng sinh xương tăng phốt phát máu (OMIM 610233) là một biến thể lâm sàng của vôi hóa u có tính gia đình và cũng là do các đột biến trong FGF23 hoặc GALNT3; các triệu chứng và dấu hiệu có thể có trước sự phát triển của kiểu hình điển hình hơn của vôi hóa u có tính gia đình. Vôi hóa u tăng phốt phát máu có tính gia đình type 3 đã được cho là do một đột biến mất chức năng đồng hợp tử trong KL (p.His193Arg) mã hóa α-klotho, một đồng yếu tố cần thiết cho sự tương tác của FGF23 với thụ thể của nó trong ống thận. Vôi hóa u có tính gia đình có phốt phát máu bình thường là một dạng vôi hóa loạn dưỡng vì các tổn thương viêm có trước vôi hóa lạc chỗ; nó có liên quan đến các đột biến bất hoạt (p.Arg344Stop, p.Lys1495Glu) trong SAMD9 (mã hóa protein 9 chứa miền alpha motif vô trùng, OMIM 610456), một protein 1589 aa điều hòa sự phân chia, vận động và tuổi thọ của tế bào. SAMD9 được tạo ra để đáp ứng với TNFα và IFN-γ và điều hòa sự biểu hiện của EGR1 (OMIM 128990) mã hóa một yếu tố phiên mã kiểm soát sự biểu hiện của TGFB1 và tham gia vào sự di chuyển, tăng sinh, vận động và sống sót của tế bào. Xơ cứng bì, bệnh thận mạn tính có cường cận giáp thứ phát, cường cận giáp nguyên phát, thừa vitamin D, sau khi ly giải tế bào do hóa trị liệu ung thư, hoại tử mỡ dưới da, viêm da cơ và xơ vữa động mạch có thể là những nguyên nhân thứ phát của các vôi hóa dưới da lớn với các phát hiện lâm sàng tương đương. Điều trị phải được cá nhân hóa và có thể bao gồm việc làm cạn kiệt phốt phát bằng cách hạn chế chế độ ăn, liên kết phốt phát ở ruột và gây ra tăng phốt phát niệu (acetazolamide), cũng như cắt bỏ phẫu thuật các khối đặc biệt gây tổn hại.
Các bệnh loạn sản sụn xương
Các bệnh loạn sản sụn xương là các dị tật không đồng nhất về mặt sinh bệnh học của sụn và xương được phân nhóm theo các biến thể di truyền (ví dụ, nhóm loạn sản sụn FGFR3; nhóm collagen Type 2—COL2A1) hoặc các đặc điểm lâm sàng, chẳng hạn như những đặc điểm liên quan đến sự phát triển của xương dài (loạn sản đầu xương, hành xương và/hoặc thân xương), xương dài và đốt sống (loạn sản đầu xương-đốt sống và/hoặc loạn sản đầu xương-hành xương-đốt sống), hộp sọ (loạn sản đòn-sọ), bàn tay và bàn chân (các tật thiếu ngón), và các thành phần xương khác (Hình 20.16). Do đó, nhiều biến thể di truyền (ví dụ, FGF23, COLA1A) có thể ảnh hưởng xấu đến sự phát triển/hình thành hoặc khoáng hóa xương; các đột biến trong một gen có thể gây ra một số rối loạn được xác định lâm sàng khác nhau cùng với các rối loạn được đặc trưng bởi các phát hiện lâm sàng và X-quang có thể là do các biến thể ở nhiều gen khác nhau (ví dụ, các bệnh loạn sản đầu chi-giữa chi). Các kiểu hình xương bất thường đã được phân loại thành 42 nhóm dựa trên các biến thể sinh bệnh học của 436 gen và/hoặc các biểu hiện lâm sàng và X-quang của các rối loạn. Các nhóm từ 1 đến 8 là các rối loạn do các biến thể trong một gen duy nhất (ví dụ, nhóm 1 FGFR3; nhóm 2 COL2A1; nhóm 3 COL11A1; nhóm 4 DTDST; nhóm 5 PLC; nhóm 6 AGC1; nhóm 7 FLNA/FLNB; nhóm 8 TRPV4). Các nhóm từ 9 đến 42 dựa vào các phát hiện lâm sàng, X-quang và di truyền để phân loại; trong mỗi nhóm, các biến thể ở các gen khác nhau có thể gây ra (các) bất thường xương giống nhau hoặc tương tự (ví dụ, nhóm 10—loạn sản nhiều đầu xương COMP, COL9A2, MATN3, COL9A1, SMARCAL1; nhóm 13—Loạn sản đốt sống-đầu-(hành)-xương MATN3, RAB33B, SEDL, DYM; nhóm 17—loạn sản giữa chi/gốc-giữa chi SHOX, GPC6, RDR2, FZD2, WNT5A). Nhóm 23 bao gồm các biến thể của bệnh xương hóa đá, nhóm 25 là các biến thể của bệnh xương thủy tinh, nhóm 27 là các rối loạn lysosome, và nhóm 30 là các hội chứng phát triển quá mức. Các rối loạn về phát triển và chức năng của xương được quan tâm không chỉ vì những thách thức lâm sàng về chẩn đoán và điều trị mà chúng đặt ra, mà còn vì chúng đã xác định được nhiều yếu tố sinh lý cơ bản thường điều hòa sự phát triển và chức năng của sụn và xương. Việc áp dụng các nghiên cứu liên kết toàn bộ bộ gen vào việc phân loại các rối loạn xương đã bổ sung thêm những hiểu biết sâu sắc đáng kể về vô số gen và các con đường truyền tín hiệu nội bào ảnh hưởng đến sự biệt hóa, hình thái và chức năng của xương.
Hình 20.16: Đặc điểm giải phẫu của các bệnh loạn sản sụn xương.
(Từ Alanay Y, Rimoin DL. Chondrodysplasias. Trong: Rosen CJ (Ed.). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism (tái bản lần thứ 7). Washington, DC: American Society of Bone and Mineral Metabolism; 2008:428–429.)
Bảng 20.13 mô tả một số biến thể di truyền thường gặp hơn liên quan đến các bệnh loạn sản sụn xương. Bệnh loạn sản sụn (OMIM 100800), bệnh loạn sản sụn phổ biến nhất ở người (1/15.000–1/40.000 ca sinh sống) và các bệnh loạn sản sụn xương liên quan (loạn sản sụn nhẹ, loạn sản tử vong—type I và II, hội chứng loạn sản sụn nặng-chậm phát triển-bệnh gai đen [SADDAN]), là do các đột biến tăng chức năng trong FGFR3 (OMIM 134934), gen mã hóa thụ thể yếu tố tăng trưởng nguyên bào sợi 3. FGFR3 là một protein xuyên màng có ba miền immunoglobulin ở vùng ngoại bào của thụ thể và hai miền tyrosine kinase ở phần nội bào của nó; biến thể p.Gly380Arg trong miền xuyên màng của FGFR3 có mặt ở 98% bệnh nhân loạn sản sụn. Biến thể p.Gly380Arg của FGFR3 làm thay đổi cấu dạng của sản phẩm gen cho phép tự phosphoryl hóa không phụ thuộc phối tử của các gốc tyrosine 647 và 648 trong miền tế bào chất của nó, sau đó có thể truyền tín hiệu qua các con đường MAPK và STAT dẫn đến ức chế sự phân bào của tế bào sụn, tổng hợp chất nền và biệt hóa cuối cùng (phì đại). Về mặt thực nghiệm, ở chuột biểu hiện đột biến tăng chức năng trong Fgfr3 liên quan đến bệnh loạn sản sụn, có sự phát triển xương tăng cường nhưng khối lượng xương giảm và hoạt động của hủy cốt bào tăng lên. Do đó, FGFR3 thường hoạt động như một chất điều hòa âm tính của sự hình thành sụn và xương. Là một rối loạn đơn gen, có thể xác định một đột biến trong FGFR3 ở thai nhi bị ảnh hưởng bằng cách phân tích DNA tự do của thai nhi trong huyết tương của mẹ, nếu người mẹ không bị ảnh hưởng. Bệnh loạn sản sụn cũng liên quan đến biến thể p.Gly375Cys trong FGFR3. Các rối loạn này được di truyền như các tính trạng trội trên nhiễm sắc thể thường nhưng với tỷ lệ đột biến tự phát cao (chủ yếu là của alen FGFR3 của người cha). Về mặt lâm sàng, bệnh loạn sản sụn được biểu hiện bằng sự rút ngắn của các chi gần và thân, đầu to, trán dô và sống mũi lõm, và phức tạp bởi tăng nguy cơ chèn ép tủy cổ và hẹp ống sống. Việc sinh ra đứa con thứ hai bị loạn sản sụn từ cha mẹ bình thường có thể phản ánh thể khảm dòng mầm của người cha đối với đột biến p/Gly380Arg trong FGFR3. Loạn sản sụn nhẹ có liên quan đến biến thể p.Asn540Lys (60% bệnh nhân) có trong đoạn tyrosine kinase gần của miền nội bào của FGFR3; đây là một rối loạn ít nghiêm trọng hơn về mặt lâm sàng với tầm vóc thấp (ngắn chi ở gốc) thường khiêm tốn biểu hiện ở giữa thời thơ ấu. Loạn sản sụn nhẹ cũng có liên quan đến các đột biến trong miền tyrosine kinase xa (p.Lys650Gln), cũng như trong các miền immunoglobulin của vùng ngoại bào (p.Ser84Leu, p.Arg200Cys) và miền xuyên màng (p.Val381Glu) của FGFR3. Loạn sản tử vong type I (OMIM 187600) có liên quan đến các đột biến (p.Arg248Cys, p.Gly370Cys) ở vùng liên kết phối tử ngoại bào bên cạnh miền xuyên màng của FGFR3 và với các thay thế ở codon cuối cùng bình thường 807—p.Ter807Gly/Cys/Arg—tất cả đều dẫn đến việc bổ sung 141 aa vào đầu carboxyl của protein FGFR3. Loạn sản tử vong type I được biểu hiện bằng các dị tật xương nghiêm trọng bao gồm xương đùi cong ngắn, dị dạng hộp sọ và xương sườn ngắn, loại sau dẫn đến suy hô hấp và tử vong sớm; đó là hậu quả của biến thể p.Arg248Cys trong miền ngoại bào của FGFR3. Loạn sản tử vong type II (OMIM 187601) có liên quan đến một đột biến (p.Lys650Glu) trong miền tyrosine kinase xa của đoạn nội bào của FGFR3 và được đặc trưng bởi hộp sọ hình lá cỏ ba lá và xương đùi thẳng ngắn. Kiểu hình lâm sàng của hội chứng SADDAN (OMIM 616482) cũng liên quan đến biến thể p.Lys650Met của FGFR3 có các biểu hiện lâm sàng ở mức độ nghiêm trọng trung gian giữa loạn sản tử vong và loạn sản sụn. Nó liên quan đến tầm vóc thấp ngắn chi ở gốc đáng kể, bệnh gai đen và những thách thức phát triển nhẹ; bệnh gai đen cũng có thể phát triển ở những bệnh nhân bị loạn sản sụn cổ điển và ở những người có đột biến trong FGFR2. Đáng chú ý là các đột biến khác nhau tại p.Lys650 dẫn đến ba kiểu hình riêng biệt—loạn sản sụn nhẹ, loạn sản tử vong type II và hội chứng SADDAN. Các đột biến hoạt hóa FGFR3 cũng đã được gặp ở những bệnh nhân bị dính khớp vành Muenke (OMIM 602849; p.Pro250Arg) và hội chứng Crouzon (OMIM 123500)—bằng chứng về sự không đồng nhất di truyền của các hội chứng lâm sàng này. Một đột biến mất chức năng (p.Arg621His trong miền tyrosine kinase xa) trong FGFR3 đã được xác định ở một gia đình có các thành viên biểu hiện kiểu hình tầm vóc cao, ngón tay co quắp và mất thính lực. Các đột biến hoạt hóa trong FGFR1 và FGFR2 đã được liên kết với các bệnh loạn sản sụn phức tạp bởi sự dính khớp sọ sớm (hội chứng Pfeiffer, Apert, Crouzon, Jackson-White, Antley-Bixler và Beare-Stevenson cutis gyrata). Điều thú vị là, các đột biến trong FGFR1 cũng đã được liên kết với suy sinh dục do giảm gonadotropin (OMIM 147950) và mất khứu giác, vì FGFR1 là một yếu tố cần thiết cho sự di chuyển của tế bào thần kinh và sự tạo mạch. Cho đến nay, các đột biến trong FGFR4 chưa được liên kết với các bệnh loạn sản sụn xương.
Bảng 20.13: Các biến thể di truyền liên quan đến các bệnh Loạn sản sụn xương (Chọn lọc)
| Gen Nhiễm sắc thể OMIM | Sinh lý bệnh | Rối loạn lâm sàng (OMIM) |
|---|---|---|
| TRIP11: Thyroid hormone interactor 11 14q31-q32 604505 | Đồng kích hoạt của thụ thể triiodothyronine hạt nhân; tương tác với các vi ống và bộ máy Golgi | Loạn sản sụn, type IA (200600), AR |
| Các bệnh Collagen | ||
| COL2A1: Collagen, type II, alpha 1 12q13.11 120140 | Tiểu đơn vị của collagen type II, collagen chính của sụn bao gồm ba chuỗi 3-alpha 1(II) | Loạn sản sụn, type II/ Loạn sản sụn nhẹ (200610), AD Loạn sản đầu xương-đốt sống bẩm sinh (183900), AD Loạn sản đầu xương-hành xương-đốt sống (184250), AD |
| COL9A1: Collagen, type IX, alpha 1 6q13 120210 | Mã hóa một collagen là một thành phần của sụn hyaline | Loạn sản nhiều đầu xương, type 6 (614135), AD Hội chứng Stickler type 4 (614134), AR |
| COL10A1: Collagen, type X, alpha 1 6p22.1 120110 | Thành phần của sụn được biểu hiện ở các giai đoạn muộn của sự hình thành xương nội sụn | Loạn sản sụn hành xương, type Schmid (156500), AD |
| COL11A1: Collagen, type XI, alpha 1 1p21.1 120280 | Tiểu đơn vị của collagen type XI bao gồm hai tiểu đơn vị alpha 1 và một tiểu đơn vị COL2A1 đã được sửa đổi; quan trọng cho sự hình thành sợi | Hội chứng Stickler type 2 (604841), AD Hội chứng Marshall (154780), AD |
| FLNB: Filamin B 3p14.3 603381 | Protein liên kết actin tế bào chất cho phép hình thành và giao tiếp với bộ xương tế bào; ảnh hưởng đến sự phân đoạn đốt sống, hóa xương nội sụn, hình thành khớp | Atelosteogenesis I (108720), AD Atelosteogenesis III (108721), AD |
| Các thụ thể yếu tố tăng trưởng nguyên bào sợi | ||
| FGFR1: Thụ thể yếu tố tăng trưởng nguyên bào sợi 1 8p11.22 136350 | Thụ thể tyrosine kinase xuyên màng cho các FGF | Hội chứng Pfeiffer (101600), AD |
| FGFR2: Thụ thể yếu tố tăng trưởng nguyên bào sợi 2 10q26.13 176943 | Thụ thể tyrosine kinase xuyên màng cho các FGF | Hội chứng Apert (101200), AD Hội chứng Crouzon (123500), AD Hội chứng Jackson-Weiss (123150), AD Hội chứng Antley-Bixler với steroidogenesis bình thường (207410), AD |
| FGFR3: Thụ thể yếu tố tăng trưởng nguyên bào sợi 3 4p16.3 134934 | Thụ thể tyrosine kinase xuyên màng cho các FGF | Loạn sản sụn (100800), AD Loạn sản sụn nhẹ (146100), AD Loạn sản tử vong types I (187600) và II (187601), AD |
| Các rối loạn Sulfat hóa | ||
| SLC26A2 – Họ chất vận chuyển chất tan 26 (chất vận chuyển sulfat) 5q32 606718 | Còn được gọi là DTDST; mã hóa chất vận chuyển sulfat cần thiết cho sự tổng hợp collagen bình thường | Loạn sản sụn, type IB (600972), AR Atelosteogenesis II (256050), AR Loạn sản gập (222600), AR Loạn sản nhiều đầu xương, type 4 (226900), AR |
| HSPG2: Heparan sulfate proteoglycan của màng đáy 1p36.12 142461 | Còn được gọi là perlecan, một heparan sulfate proteoglycan; đồng thụ thể cho FGFR2; ổn định màng đáy và điều hòa tính thấm của chúng | Loạn sản sụn nhược cơ type 1 Schwartz-Jampel (255800), AR |
| PAPSS2: 3-Prime-phosphoadenosine 5-prime-phosphosulfate synthase 2 10q23.2-23.3 603005 | Enzyme tổng hợp chất cho sulfat (3′-phosphoadenosine 5′-phosphosulfate) từ ATP và sulfat; đồng yếu tố cho sulfotransferase vỏ thượng thận | Loạn sản đầu xương-hành xương-đốt sống (Brachyolmia type 4) (612847), AR |
| ARSE: Aryl sulfatase E Xp22.33 300180 | Enzyme loại bỏ một nhóm sulfat khỏi protein của nó | Loạn sản sụn đốm type 1 (302950), lặn liên kết X |
| PTPN11: Protein tyrosine phosphatase, không phải thụ thể, type 11 12q24.1 176876 | Mã hóa SHP2: một protein tyrosine phosphatase không phải thụ thể hoạt động ở thượng nguồn của RAS | Các đột biến mất chức năng đơn alen dẫn đến metachondromatosis (156250), AD Hội chứng Noonan với nhiều nốt ruồi: các đột biến tăng chức năng đơn alen (163950), AD |
| Khác | ||
| COMP: Protein chất nền sụn oligomeric 19p13.1 600310 | Protein tế bào sụn liên kết canxi và các loại collagen I, II và IX | Giả loạn sản sụn (177170), AD Loạn sản nhiều đầu xương (132400), AD |
| TRPV4: Kênh cation tiềm năng thụ thể thoáng qua, phân họ V, thành viên 4 12q24.11 605427 | Kênh cation làm trung gian cho dòng canxi vào | Loạn sản biến hình (156530), AD Loạn sản đầu xương-hành xương-đốt sống: Maroteaux (184095), AD |
| SHOX: Hộp đồng dạng tầm vóc thấp Xp22.33 312865 | Yếu tố phiên mã gen hộp đồng dạng nằm trên vùng giả nhiễm sắc thể thường của Yp | Loạn sản sụn xương Leri-Weill (127300), trội liên kết X Loạn sản Langer (249700), hai alen/X và Y |
| SOX9: SRY-box 9 17q24.3 608160 | Yếu tố phiên mã cần thiết cho sự hình thành sụn và biệt hóa tinh hoàn | Loạn sản campomelic (114290), Đơn alen do thiếu hụt một nửa |
| RUNX2: Yếu tố phiên mã liên quan đến Runt 2 6p21 600211 | Yếu tố phiên mã đặc hiệu của nguyên bào xương | Loạn sản đòn-sọ (119600), AD |
| PTH1R: Thụ thể PTH 1 Xp22.33 168468 | GPCR nhận biết PTH và PTHrP với ái lực bằng nhau | Các đột biến bất hoạt dẫn đến hạ canxi máu và loạn sản sụn xương Blomstrand (215045), AR Hội chứng Eiken (600002), AR; các đột biến hoạt hóa dẫn đến tăng canxi máu và loạn sản sụn hành xương Murk-Jansen (156400), AD |
| PRKAR1A: Protein kinase, phụ thuộc cAMP, điều hòa, type 1, alpha 17q24.2 188830 | Thành phần của đáp ứng PKA với AMP vòng dẫn đến chuỗi các tín hiệu truyền tín hiệu nội bào để đáp ứng với Gαs điều hòa sự phân chia, biệt hóa, chuyển hóa, chết theo chương trình của tế bào | Đột biến tăng chức năng dẫn đến acrodysostosis (101800) và đề kháng ngoại vi với các tác dụng sinh học của PTHrP, PTH và TSH (de novo) |
| POR: Cytochrome P450 oxidoreductase 7q11.2 124015 | Đồng yếu tố Flavoprotein cho electron cho tất cả các enzyme P450 bao gồm P450c17, P450c21 và P450arom | Các đột biến bất hoạt dẫn đến tăng sản tuyến thượng thận bẩm sinh thường có sự không rõ ràng về giới tính ở cả các đối tượng 46XX và 46XY (613571), AR; đôi khi kết hợp với các bất thường về xương (Hội chứng Antley-Bixler: dính khớp sọ, thiểu sản giữa mặt, hẹp lỗ mũi sau, cong xương đùi, dính xương quay-trụ, 201750, AR) |
| RMRP: Endoribonuclease xử lý RNA ty thể, thành phần RNA của 9p13.3 157660 | Tiểu đơn vị RNA không được dịch mã của endoribonuclease xử lý RNA ty thể: RNase MRP | Tùy thuộc vào mức độ nghiêm trọng của (các) đột biến mất chức năng hai alen, phổ của loạn sản không tăng trưởng (607095), thiểu sản tóc sụn (250250) xuất hiện |
| INPPL1: Inositol polyphosphate phosphatase like-1 11q13.4 600829 | Enzyme thủy phân inositol-1,4,5-trisphosphate thành inositol-4,5-bisphosphate: chất truyền tín hiệu nội bào | Các đột biến bất hoạt dẫn đến opsismodysplasia (258480); liên quan đến tăng tổng hợp FGF23 của xương, AR |
AD, Trội trên NST thường; AR, Lặn trên NST thường, ATP, adenosine triphosphate; AMP vòng, cyclic adenosine monophosphate; GPCR, thụ thể kết hợp với protein G; PKA, protein kinase A; PTH, hormon cận giáp; PTHrP, protein liên quan đến PTH; RNA, ribonucleic acid; TSH, hormon kích thích tuyến giáp.
(Sửa đổi từ Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet. 2015;167A:2869–2892.)
Giả loạn sản sụn (OMIM 177170) là một bệnh loạn sản sụn xương không biểu hiện cho đến sau năm đầu đời vì thai nhi và trẻ sơ sinh mắc rối loạn này có vẻ bình thường. Chậm tăng trưởng trở nên rõ ràng trong năm thứ hai của cuộc đời; tầm vóc ngày càng bị tổn hại trong thời thơ ấu kết hợp với chân vòng kiềng chữ O hoặc chữ X, ngón tay ngắn và lỏng lẻo khớp và đau xương và khớp nghiêm trọng làm suy giảm vận động. X-quang cho thấy sự ức chế sự phát triển của xương dài; các bất thường của hành xương; và các đầu xương nhỏ, kém hóa xương. Giả loạn sản sụn là hậu quả của các biến thể bất hoạt đơn alen trong protein chất nền sụn oligomeric (COMP, OMIM 600310). COMP là một glycoprotein tiết ra được sản xuất bởi các tế bào sụn có bốn miền—một trong số đó là một miền liên kết canxi; các biến thể trong các chuỗi bazơ của miền này dẫn đến giả loạn sản sụn do sự gấp nếp protein bị gián đoạn và hậu quả là rối loạn chức năng tế bào sụn đồng thời cũng có tác dụng trội âm tính đối với sản phẩm nguyên vẹn của alen COMP bình thường và sự lưu giữ của protein COMP trong lưới nội chất của tế bào sụn. COMP được tiết ra bình thường góp phần vào sức mạnh của collagen bằng cách liên kết chéo một số collagen chất nền khác nhau (types I, II, IX, XII, XIV) và sự mất mát của nó làm suy giảm đặc tính này. COMP bị đột biến được giữ lại trong lưới nội chất của tế bào sụn dẫn đến viêm nội bào và sau đó là cái chết của tế bào sụn. Về mặt thực nghiệm ở chuột, phản ứng viêm đối với COMP bị đột biến, được giữ lại có thể được cải thiện bằng cách dùng các tác nhân chống viêm, chẳng hạn như axit acetylsalicylic với sự phục hồi sự tăng trưởng của động vật. Liệu những quan sát thực nghiệm này có thể được nhân đôi ở trẻ em bị giả loạn sản sụn hay không vẫn chưa được biết.
Ngoài bốn loại di truyền (I–IV) của bệnh xương thủy tinh do các biến thể của COL1A1 và COL1A2, các khiếm khuyết trong sự hình thành của một số loại collagen khác do các đột biến trong COL2A1, COL9A1, COL9A2, COL10A1, COL11A1, và COL11A2 dẫn đến một số lượng lớn các dị tật xương tùy thuộc vào vị trí và thời điểm phát triển của lỗi tổng hợp (xem Bảng 20.13). Các đột biến trong một số gen mã hóa collagen khác nhau (COL2A1, COL9A1, COL9A2, và COL11A1) dẫn đến các biến thể của hội chứng Stickler (OMIM 609508, 604841, 614134, 614284) được đặc trưng bởi các bất thường của bộ xương (loạn sản đầu xương), mặt (hàm dưới nhỏ, hở hàm ếch), thị lực (cận thị, bong võng mạc), và suy giảm thính lực có thể được di truyền như các tính trạng trội hoặc lặn trên nhiễm sắc thể thường. Các tật thiếu ngón hoặc dị tật bàn tay/bàn chân chẻ được đặc trưng bởi sự vắng mặt của các xương bàn tay/bàn chân thứ hai và thứ ba và các ngón tay/ngón chân tương ứng với ngón thứ nhất còn lại và các ngón thứ tư và thứ năm hợp nhất (dẫn đến hình dạng “giống móng vuốt” của chi xa) có thể là do các bất thường cấu trúc (sắp xếp lại, mất đoạn, nhân đôi) của nhiễm sắc thể 7q21.3 trong đó đoạn này có một số gen ảnh hưởng đến sự hình thành phôi của các ngón tay, cũng như các bất thường của các nhiễm sắc thể khác (Xq26, 2q31, 10q24).
Atelosteogenesis type I (OMIM 108720) là một rối loạn xương gây tử vong được đặc trưng bởi sự vắng mặt, ngắn hoặc thon nhọn ở đầu xa của xương đùi và xương cánh tay, xương chày và xương trụ cong ngắn, không có xương mác, thiểu sản đốt sống và hóa xương dưới mức bình thường của xương bàn tay; rối loạn này có một số biến thể; nó là kết quả của các đột biến mất chức năng đơn alen trong FLNB (OMIM 603381) mã hóa một protein tế bào chất liên kết với actin, do đó cho phép actin hình thành bộ xương tế bào, và cũng tạo điều kiện cho sự giao tiếp nội bào giữa màng tế bào và bộ xương tế bào của nó. Các bất thường về vận chuyển sulfat, sulfat hóa protein chất nền collagen và hoạt động của sulfatase dẫn đến một số bệnh loạn sản sụn. SLC26A2 (OMIM 606718) mã hóa một chất vận chuyển sulfat bị đột biến ở những bệnh nhân bị loạn sản gập (DTD, OMIM 222600) và loạn sản sụn type IB (OMIM 600972). Loạn sản đầu xương-hành xương-đốt sống/brachyolmia type 4 là do các đột biến mất chức năng hai alen (p.Thr48Arg, p.Ser438Ter) trong PAPSS2 (OMIM 603005) mã hóa 3′-phosphoadenosine-5′-phosphosulfate synthase 2, một enzyme có hai hoạt động; nó xúc tác cả quá trình tổng hợp adenosine 5′-phosphosulfate và quá trình phosphoryl hóa của nó thành 3′-phospho-adenosine 5′-phosphosulfate, chất cho sulfat phổ quát cần thiết cho quá trình sulfat hóa của các protein chất nền sụn và xương. Các biểu hiện lâm sàng của rối loạn này bao gồm các chi ngắn, gù vẹo cột sống, ngón tay ngắn và các khớp gối to. Các đột biến bất hoạt trong ARSE (mã hóa arylsufatase E, OMIM 300180) dẫn đến loạn sản sụn đốm loại 1 brachytelephalangic di truyền lặn liên kết X. Rối loạn này được đánh dấu về mặt lâm sàng bởi tầm vóc bị tổn hại do sự rút ngắn của các chi ở gốc kết hợp với sự lốm đốm của các đầu xương, các khiếm khuyết sọ mặt, các tổn thương da dạng vảy cá teo và có sắc tố, rụng tóc, đục thủy tinh thể và chậm phát triển.
Các đột biến mất chức năng (chèn, sai nghĩa, vô nghĩa) trong SOX9 (OMIM 608160), một yếu tố phiên mã được biểu hiện ở cả các tế bào sụn đang phát triển, nơi nó được đồng biểu hiện với COL2A1 và ở các gờ sinh dục trong quá trình biệt hóa tuyến sinh dục, gây ra cả loạn sản campomelic (OMIM 114290) và đảo ngược giới tính từ nam sang nữ ở 75% các đối tượng 46XY bị ảnh hưởng. Vì SOX9 cần thiết cho sự biểu hiện và chức năng bình thường của COL2A1 trong quá trình hình thành sụn, sự vắng mặt của nó dẫn đến sự hình thành chất nền sụn bị suy giảm và dị dạng của các xương có nguồn gốc phôi học từ xương nội sụn. Ở tuyến sinh dục lưỡng tính đang phát triển của thai nhi 46XY, sự biểu hiện của SOX9 được điều chỉnh bởi SRY (OMIM 480000); sự biểu hiện của SOX9 cũng cần thiết cho sự biệt hóa và phát triển bình thường của các tế bào Sertoli và các thành phần khác của tinh hoàn. Về mặt lâm sàng, loạn sản campomelic được đặc trưng bởi sự khởi phát trước sinh của sự cong của các xương ống, xương bả vai thiểu sản, 11 xương sườn, hở hàm ếch và hàm dưới nhỏ thường dẫn đến tử vong sơ sinh, cũng như thất bại trong việc nam hóa của thai nhi 46XY.
Gen giả nhiễm sắc thể thường liên kết X và Y SHOX (OMIM 312865) mã hóa hai protein hạt nhân—SHOXa với 292 aa và SHOXb với 225 aa. SHOX là một yếu tố phiên mã được biểu hiện ở các tế bào sụn tăng sinh muộn, tiền phì đại và phì đại, một trong những gen mục tiêu của nó là NPPB mã hóa tiền chất peptide lợi niệu natri B (OMIM 600295) cũng được biểu hiện ở các tế bào sụn tăng sinh muộn, tiền phì đại và phì đại. Sự vắng mặt của một gen chức năng SHOX là nguyên nhân gần của tầm vóc thấp của các bé gái mắc hội chứng Turner và một số trẻ em có tầm vóc thấp “tự phát”. Các mất đoạn nhỏ dị hợp tử và các đột biến bất hoạt trong gen (p.Leu132Val, p.Ala170Pro, p.Arg195Stop) của SHOX dẫn đến thiếu hụt một nửa có ở những bệnh nhân bị loạn sản sụn xương Leri-Weill (OMIM 127300) được đặc trưng bởi sự rút ngắn chi ở giữa và chậm tăng trưởng, dị tật Madelung của cổ tay, cong xương quay và trật khớp trụ. Kiểu hình Leri-Weill đã được ghi nhận ở những bệnh nhân có SHOX nguyên vẹn nhưng có các mất đoạn nhỏ của các đoạn hạ nguồn của vùng giả nhiễm sắc thể thường của nhiễm sắc thể X, ngụ ý sự hiện diện của các gen sửa đổi trong vùng này. Mất hai alen (nhiễm sắc thể X và Y) của SHOX dẫn đến loạn sản giữa chi Langer (OMIM 249700) được đặc trưng bởi sự thiểu sản nặng của xương trụ và xương mác và một xương quay và xương chày dày, cong.
Cả các biến thể tăng và mất chức năng của PTH1R (OMIM 168468) đều dẫn đến các bất thường về sự hình thành và tăng trưởng của xương. Loạn sản sụn Blomstrand (OMIM 215045) là một rối loạn di truyền lặn trên nhiễm sắc thể thường gây tử vong trong tử cung có thể được xác định ở thai nhi với các xương dài ngắn, cực kỳ đặc và sự trưởng thành xương tiến triển rõ rệt, cũng như các bất thường về cơ thể, chẳng hạn như hẹp eo động mạch chủ và các bất thường về mặt. Về mặt bệnh học, sụn đầu xương bị giảm, và có các cột không đều và sự phân bố thất thường của các tế bào sụn trong chất nền. Bất thường này là kết quả của các đột biến bất hoạt (mất đoạn, đột biến sai nghĩa—p.Pro132Leu, p.Arg383Gln) của gen mã hóa thụ thể PTH/PTHrP. Hội chứng Eiken (OMIM 600002) của loạn sản đầu xương với sự hóa xương bị trì hoãn rõ rệt của các trung tâm hóa xương chính của các xương ống, các đầu xương và khớp mu cũng là do các đột biến mất chức năng hai alen (p.Glu35Lys, p.Arg485Stop) trong PTH1R. Bệnh nhân bị loạn sản sụn hành xương Murk-Jansen (OMIM 156400) có các chi cong ngắn, ngón tay vẹo, hàm dưới nhỏ và các dị tật của cột sống và xương chậu, nhưng sống sót đến tuổi trưởng thành, nơi chiều cao trung bình của người lớn là 125 cm và có thể sinh con. Đặc trưng, những bệnh nhân này bị tăng canxi máu, hạ phốt phát máu và nồng độ PTH hoặc PTHrP trong huyết thanh thấp hoặc không thể phát hiện được, hậu quả của hoạt động không được điều hòa của thụ thể PTH do các đột biến hoạt hóa đơn alen (p.His223Arg, p.Thr410Pro) của PTH1R. Hai rối loạn này phản ánh các tác động chức năng thay đổi của PTHrP/PTH hoạt động thông qua PTH1R trong sụn đang phát triển, nơi các phối tử này thường làm chậm sự biệt hóa của tế bào sụn và làm giảm tốc độ chết theo chương trình của tế bào sụn, do đó kéo dài sự tăng sinh của tế bào sụn và tăng cường sự phát triển của xương dài.
Loạn sản không tăng trưởng (OMIM 607095) là một loạn sản đầu xương-hành xương-đốt sống được di truyền như một rối loạn lặn trên nhiễm sắc thể thường được đặc trưng bởi sự chậm phát triển trong tử cung (chiều dài khi sinh < 40 cm), chiều cao người lớn bị tổn hại nghiêm trọng (< 85 cm), thiếu răng và chậm phát triển trí tuệ nhẹ. Tất cả các xương đều bị dị dạng; có ít tế bào sụn trong các đĩa tăng trưởng sụn. Nó là do các đột biến mất chức năng (chèn) trong RMRP (OMIM 157660), một gen mã hóa tiểu đơn vị RNA không được dịch mã của một endoribonuclease xử lý RNA ty thể. Enzyme này tham gia vào: (1) sự lắp ráp của các ribosome—các đơn vị cấu trúc trong đó quá trình tổng hợp protein diễn ra, (2) sự tạo ra các mồi RNA để sao chép DNA ty thể, và (3) sự điều hòa của chu kỳ tế bào phụ thuộc cyclin. Các đột biến dẫn đến loạn sản không tăng trưởng làm suy giảm sự lắp ráp ribosome và tổng hợp protein. Các đột biến bất hoạt (nhân đôi, chèn) trong RMRP làm suy giảm khiêm tốn cả sự lắp ráp ribosome và sự điều hòa của chu kỳ tế bào có ở những bệnh nhân bị thiểu sản tóc sụn (OMIM 250250) và loạn sản hành xương không có rụng tóc (OMIM 250460). Do đó, thiểu sản tóc sụn và loạn sản không tăng trưởng đại diện cho các biểu hiện và mức độ nghiêm trọng khác nhau của sự bất hoạt của RMRP.
Các lỗi trong quá trình sinh tổng hợp cholesterol đã được liên kết với một số rối loạn ảnh hưởng xấu đến sự phát triển của xương, cũng như nhiều hệ thống khác (Bảng 20.14; Hình 20.17). Cholesterol là một thành phần của màng plasma của tế bào, cũng như các màng của các bào quan nội bào; nó liên kết cộng hóa trị với đầu amino và cần thiết cho chức năng của Indian hedgehog, Sonic hedgehog và Desert hedgehog, các yếu tố cảm ứng cần thiết cho sự phát triển bình thường của sụn và xương, não và tinh hoàn, tương ứng. Nó là tiền chất của các steroid và axit mật. Có khả năng là sự thiếu hụt cholesterol có tác dụng gây quái thai bằng cách làm suy giảm chức năng màng và/hoặc (các) phản ứng truyền tín hiệu nội bào đối với các kích thích bình thường. Ngoài ra, sự tích tụ các tiền chất của cholesterol có thể gây độc cho thai nhi đang phát triển. Ở những bệnh nhân mắc hội chứng Smith-Lemli-Opitz (OMIM 270400), các đột biến bất hoạt của enzyme microsome Δ7-dehydrocholesterol reductase (DHCR7; OMIM 602858) làm suy giảm bước cuối cùng trong con đường tổng hợp cholesterol từ 7-dehydrocholesterol và dẫn đến chậm phát triển trong tử cung và sau sinh, các chi ngắn, dính ngón và thừa ngón, các đặc điểm khuôn mặt đặc trưng (sụp mí, mũi hếch, gờ ổ răng rộng, hở hàm ếch), các dị tật bẩm sinh của tim và hệ thần kinh trung ương, đầu nhỏ, nam hóa không hoàn toàn của cơ quan sinh dục ngoài của nam giới, ngón tay cái thiểu sản, chậm phát triển, tự kỷ và chức năng vỏ thượng thận bị tổn hại, một hội chứng dị tật với tỷ lệ mắc từ 1/15000 đến 1/40000 ca sinh. Nồng độ cholesterol trong huyết thanh bị suy giảm trong khi nồng độ 7- và 8-dehydrocholesterol tăng. Nồng độ 7-dehydrocholesterol và 8-dehydrocholesterol trong huyết thanh có liên quan nghịch với mức độ nghiêm trọng lâm sàng và thành tích trí tuệ của các đối tượng có kiểu hình Smith-Lemli-Opitz.
Bảng 20.14: Các bệnh loạn sản sụn xương do các biến thể di truyền của các Protein cần thiết cho quá trình tổng hợp Cholesterol
| Gen Nhiễm sắc thể OMIM | Sinh lý bệnh | Rối loạn lâm sàng (OMIM) | ||
|---|---|---|---|---|
| DHCR7: 7-Dehydrocholesterol reductase 11q12-q13 602858 | 3β-Hydroxysteroid △7 reductase chuyển đổi 7-dehydrocholesterol thành cholesterol | Hội chứng Smith-Lemli-Opitz (270400), AR | ||
| DHCR24: 24-Dehydrocholesterol reductase 1p32.3 606418 | 3β-Hydroxysterol △24 reductase chuyển đổi desmosterol thành cholesterol | Desmosterolosis (602398), AR | ||
| SC5DL: Sterol C5-desaturase-like 11q23.3 602286 | Enzyme microsome (3β-hydroxysteroid- △5-desaturase) chuyển đổi lathosterol thành 7-dehydrocholesterol, tiền chất của cholesterol và cholecalciferol (vitamin D) | Lathosterolosis (607330), AR | ||
| EBP: Protein liên kết Emopamil Xp11.23 300205 | Protein chức năng kép: protein liên kết; 3β-Hydroxysteroid △7, △8 isomerase chuyển đổi cholest-8(9)-en-3β-ol thành lathosterol | Loạn sản sụn đốm type 2 (302960), trội liên kết X | ||
| NSDHL: Protein giống NAD(P)H steroid dehydrogenase Xq28 300275 | Phức hợp demethylase 3β-Hydroxysteroid C4 chuyển đổi 4,4-dimethylcholest-8(9)-en-3β-ol thành cholest 8(9)-en-3β-ol; các thành viên khác của phức hợp là SC4MOL (607545) và HSD17B7 (606756) | Hội chứng CHILD (loạn sản nửa người bẩm sinh, đỏ da dạng vảy cá, khiếm khuyết chi) (308050), trội liên kết X | ||
| LBR: Thụ thể Lamin B 1q42.12 600024 | Protein chức năng kép: thúc đẩy sự liên kết của heterochromatin với màng nhân trong; 3β-hydroxysteroid △14 reductase quan trọng cho quá trình tổng hợp cholesterol | Loạn sản HEM (phù thai-vôi hóa lạc chỗ-”mọt ăn” hoặc Greenberg) (215140), AR | ||
| POR: Cytochrome P450 oxidoreductase 1q11.2 124015 | Chất cho electron Flavoprotein cho tất cả các enzyme P450 bao gồm P450c17, P450c21, P450arom | Hội chứng Antley-Bixler (ABS) với các bất thường sinh dục và rối loạn steroidogenesis, (201750), AR (ABS chỉ có các bất thường về xương ( |
AD, Trội trên NST thường; AR, Lặn trên NST thường.
(Sửa đổi từ Forbes FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52:6–34.)
Hình 20.17: Sinh tổng hợp cholesterol mô tả các vị trí hoạt động của enzyme bị mất do các đột biến bất hoạt dẫn đến các dị tật xương, sinh dục và toàn thân. Việc chuyển đổi lanosterol thành cholesterol có thể tiến hành qua hai con đường như được mô tả (xem chi tiết trong văn bản).
(Từ Forbes FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52:6–34, với sự cho phép.)
Desmosterolosis (OMIM 602398) là một rối loạn di truyền lặn trên nhiễm sắc thể thường với các biểu hiện lâm sàng đa dạng bao gồm suy giảm tăng trưởng, xơ cứng xương, các chi ngắn, đầu to hoặc nhỏ, hở hàm ếch, hàm dưới nhỏ, gờ ổ răng dày, chậm phát triển và co cứng. Chụp cộng hưởng từ sọ não cho thấy ít chất trắng và mỏng đến gần như không có thể chai. Desmosterolosis là do các đột biến bất hoạt hai alen của gen (DHCR24, OMIM 606418) mã hóa 3β-hydroxysterol-Δ24 reductase, một enzyme chuyển đổi desmosterol thành cholesterol. Lathosterolosis (OMIM 607330) được đặc trưng bởi đầu nhỏ, hẹp thái dương của hộp sọ, đục thủy tinh thể, lỗ mũi hếch và các bất thường thực thể khác được ghi nhận ở bệnh nhân mắc hội chứng Smith-Lemli-Opitz. Nó là kết quả của các đột biến mất chức năng trong gen (SC5DL, OMIM 602286) mã hóa 3β-hydroxysteroid-Δ5-desaturase, một enzyme chuyển đổi lathosterol thành 7-dehydrocholesterol. Loạn sản sụn đốm 2 (OMIM 302960) là một rối loạn trội liên kết X (hội chứng Conradi-Hunermann-Happle) thường gây tử vong ở nam giới bị ảnh hưởng. Nó được đặc trưng về mặt lâm sàng ở nữ giới bị ảnh hưởng bởi tầm vóc thấp với các chi gần ngắn không đối xứng (lùn gốc chi), trán dô, các tổn thương da dạng vảy và đỏ giống như bệnh vảy cá ở trẻ em và các tổn thương sắc tố teo ở người lớn, tóc thô với rụng tóc và đục thủy tinh thể; phản ánh sự khảm nhiễm sắc thể X chức năng, ở nữ giới, kiểu hình có thể thay đổi từ thai chết lưu đến bị ảnh hưởng nhẹ. Khám X-quang cho thấy xơ cứng xương toàn thể, vôi hóa đốm không đều (lốm đốm) của các đầu xương của các xương dài và đốt sống ở trẻ em. Rối loạn này là do các đột biến mất chức năng trong protein liên kết emopamil (EBP, OMIM 300205), gen mã hóa 3β-hydroxysteroid-Δ8,Δ7 isomerase, một enzyme chuyển đổi cholesta-8(9)-en-3β-ol thành lathosterol; trong huyết thanh của các đối tượng bị loạn sản sụn đốm 2, nồng độ 8-dehydrocholesterol cholest-8(9)-en-3β-ol trong huyết tương tăng. Protein này cũng liên kết với nhiều phân tử không liên quan; tên gọi di truyền của nó bắt nguồn từ khả năng liên kết với emopamil (tức là, EBP)—một chất đối kháng ion canxi. Các dạng lặn liên kết X và lặn trên nhiễm sắc thể thường của hội chứng Conradi-Hunermann-Happle cũng đã được báo cáo, cho thấy sự không đồng nhất di truyền đối với kiểu hình này. Hội chứng CHILD của loạn sản nửa người bẩm sinh với đỏ da dạng vảy cá hoặc nevus và các khiếm khuyết chi là đáng chú ý vì sự phân bố một bên của các bất thường chỉ giới hạn ở một nửa cơ thể. Đây là một rối loạn liên kết X gây ra bởi các đột biến mất chức năng trong NADPH steroid dehydrogenase-like (NSDHL, OMIM 300275) mã hóa một sterol dehydrogenase hoặc decarboxylase là một phần của phức hợp demethylase 3β-hydroxysteroid C-4 sterol cũng bao gồm các sản phẩm của SC4MOL và HSD17B7. Phức hợp này chuyển đổi 4,4-dimethylcholesta-8-en-3β.ol thành cholesta-8(9)-en-3β-ol.
Loạn sản Greenberg (OMIM 215140) thường gây tử vong của phù thai-vôi hóa lạc chỗ-loạn sản xương “mọt ăn” (HEM) được di truyền như một tính trạng lặn trên nhiễm sắc thể thường liên quan đến lùn chi ngắn, thừa ngón và vôi hóa giảm không đều của các xương dài cùng với vôi hóa thanh quản và khí quản. Nó là do một đột biến mất chức năng trong gen (LBR, OMIM 600024) mã hóa thụ thể lamin B, một protein màng trong của vỏ nhân không chỉ liên kết với lamin B mà còn có hoạt động 3β-hydroxysteroid-Δ14 reductase, một enzyme chuyển đổi 4,4-dimethylcholesta-8(9)-dien-3β-ol thành 4,4-dimethylcholesta-8-en-3β-ol, một bước cần thiết cho quá trình sinh tổng hợp cholesterol bình thường. Hoạt động 3β-Hydroxysteroid-Δ14 reductase cũng được mã hóa bởi DHCR14, một enzyme lưới nội chất cũng xúc tác quá trình khử các sterol trung gian C14-C15 không bão hòa. Loạn sản xương HEM được coi là một bệnh laminopathy hơn là một lỗi bẩm sinh trong quá trình sinh tổng hợp cholesterol. Hội chứng Antley-Bixler (OMIM 207410) của dính khớp sọ, dính xương cánh tay-quay, cong xương đùi, thiểu sản giữa mặt, hẹp/tịt lỗ mũi sau và co cứng khớp đã được liên kết với các đột biến dị hợp tử trong FGFR2 (OMIM 176943). Khi các bất thường về xương này cùng tồn tại với các dị tật sinh dục và các khiếm khuyết trong steroidogenesis (OMIM 201750), các đột biến mất chức năng hai alen đã được xác định trong POR (OMIM 124015), mã hóa P450 oxidoreductase, một chất cho electron cho các enzyme P450 cần thiết cho quá trình tổng hợp các steroid vỏ thượng thận và tuyến sinh dục. Do đó, khi hoạt động của POR giảm, chức năng của enzyme được mã hóa bởi CYP51A1 (OMIM 601637) chuyển đổi dihydrolathosterol thành 4,4-dimethylcholesta-8(9)14-dien-3β-ol bị suy giảm.
Lời kết
Việc giải mã các cơ chế phức tạp làm nền tảng cho sinh lý bệnh của các bệnh ảnh hưởng xấu đến sự điều hòa chuyển hóa canxi, phốt phát và magiê, sự biệt hóa và tăng trưởng của tế bào sụn, và sự hình thành, khoáng hóa và sức mạnh của xương đã được cung cấp thêm thông tin bởi những hiểu biết sâu sắc mà những tiến bộ trong di truyền học, biểu sinh và proteomics đã mang lại cho những vấn đề lâm sàng này. Có thể dự đoán rằng việc làm rõ thêm các cơ chế cơ bản nhất làm nền tảng cho các rối loạn này sẽ cho phép chuyển những phát hiện này vào việc chăm sóc nhiều rối loạn ảnh hưởng đến các bệnh nhân trẻ của chúng ta.
Các chữ viết tắt
| ADHR | Còi xương giảm phốt phát máu di truyền trội trên NST thường |
| AHO | Loạn dưỡng xương di truyền Albright |
| AMP | Adenosine monophosphate |
| APECED | Bệnh đa nội tiết tự miễn-nhiễm nấm candida-loạn sản ngoại bì |
| APS | Hội chứng đa nội tiết tự miễn |
| ARHR | Còi xương giảm phốt phát máu di truyền lặn trên NST thường |
| ASARM | Motif liên quan đến MEPE giàu aspartate serine axit |
| ATP | Adenosine triphosphate |
| BMC | Hàm lượng khoáng chất trong xương |
| BMD | Mật độ khoáng chất trong xương |
| BMP | Protein hình thái xương |
| CA | Carbonic anhydrase |
| CaSR | Thụ thể cảm nhận canxi |
| CBP | Protein liên kết collagen (HSP) |
| CKD-MBD | Bệnh thận mạn tính-rối loạn khoáng chất và xương |
| COL1A1 | Tiểu đơn vị collagen xương α1(I) |
| CRTAP | Protein liên quan đến sụn |
| DXA | Đo hấp thụ tia X năng lượng kép |
| DGS | Hội chứng DiGeorge |
| DMR | Vùng methyl hóa khác biệt |
| FGF | Yếu tố tăng trưởng nguyên bào sợi |
| FGFR | Thụ thể FGR |
| FISH | Lai tại chỗ phát huỳnh quang |
| GDNF | Yếu tố dinh dưỡng thần kinh có nguồn gốc từ tế bào thần kinh đệm |
| GH | Hormone tăng trưởng |
| GPCR | Thụ thể kết hợp với protein G |
| GDP | Guanosine diphosphate |
| Gsα | Tiểu đơn vị α của protein G kích thích (Gsα) |
| GTP | Guanosine triphosphate |
| H+ | Ion hydro |
| HDR | Hội chứng suy cận giáp, điếc, bệnh thận |
| HHC | Tăng canxi máu giảm canxi niệu có tính gia đình |
| HRD | Hội chứng suy cận giáp, chậm phát triển, dị dạng |
| HSP | Protein sốc nhiệt (CBP) |
| ICTP | Telopeptide đầu carboxyl của collagen type I |
| IFITM5 | Protein xuyên màng-5 do interferon gây ra hoặc protein giống Itifm5 giới hạn ở xương |
| IGF | Yếu tố tăng trưởng giống insulin |
| IL | Interleukin |
| IOF | Hiệp hội Loãng xương Quốc tế |
| ISCD | Hiệp hội Quốc tế về Đo mật độ xương lâm sàng |
| KCS | Hội chứng Kenny-Caffey |
| LBW | Cân nặng khi sinh thấp |
| MAPK | Kinase protein hoạt hóa bởi mitogen |
| M-CSF | Yếu tố kích thích dòng đại thực bào |
| MCT | Ung thư biểu mô tủy của tuyến giáp |
| MEN | Đa u tuyến nội tiết |
| MEPE | Phosphoglycoprotein ngoại bào chất nền |
| Mg2+ | Magiê |
| MPA | Medroxyprogesterone acetate |
| NF-κB | Yếu tố hạt nhân κB |
| NHERF1 | Yếu tố điều hòa trao đổi natri hydro 1 |
| NSHPT | Cường cận giáp nặng ở trẻ sơ sinh |
| NTx | Telopeptide đầu amino của collagen type I |
| OIC | Bệnh xương thủy tinh bẩm sinh |
| OMIM | Di truyền Mendel ở người trực tuyến |
| OSTM1 | Protein xuyên màng-1 liên quan đến bệnh xương hóa đá |
| P3H1 | Prolyl 3-hydroxylase 1 |
| PDDR | Còi xương giả thiếu vitamin D |
| PEDF | Yếu tố có nguồn gốc từ biểu mô sắc tố |
| PHEX | Tương đồng Endopeptidase điều hòa phốt phát, liên kết X |
| PHP | Giả suy cận giáp |
| PICP | Propeptide đầu carboxyl của collagen type I |
| POH | Tăng sinh xương lạc chỗ tiến triển |
| PPHP | Giả giả suy cận giáp |
| PTH | Hormone cận giáp |
| PTHrP | Protein liên quan đến PTH |
| PTHR1 | Thụ thể PTH/PTHrP 1 |
| QCT | Chụp cắt lớp vi tính định lượng |
| QUS | Siêu âm định lượng |
| RANK | Thụ thể hoạt hóa của yếu tố hạt nhân κB |
| RANKL | Phối tử RANK |
| SDS | Điểm độ lệch chuẩn |
| SERM | Chất điều biến thụ thể estrogen chọn lọc |
| sFRP4 | Protein-4 liên quan đến frizzled trong huyết thanh |
| SIBLING | Glycoprotein tương tác phối tử liên kết integrin ngắn |
| SOS | Tốc độ âm thanh |
| TALH | Nhánh lên dày của quai Henle |
| TGFβ | Yếu tố tăng trưởng biến đổi β |
| TNF | Yếu tố hoại tử khối u |
| TNSALP | Phosphatase kiềm không đặc hiệu mô |
| TRAP | Phosphatase axit kháng tartrate |
| VDDR1A | Còi xương phụ thuộc vitamin D type 1A |
| VDR | Thụ thể vitamin D |
| VDRE | Yếu tố đáp ứng vitamin D |
| VEGF | Yếu tố tăng trưởng nội mô mạch máu |
| VLBW | Cân nặng khi sinh rất thấp |
| WBS | Hội chứng Williams-Beuren |
| WINAC | Phức hợp lắp ráp nucleotide bao gồm WSTF |
| WHO | Tổ chức Y tế Thế giới |
| WSTF | Yếu tố phiên mã hội chứng Williams |
| XLHR | Còi xương giảm phốt phát máu di truyền trội liên kết X |
| 1,25(OH)2D3 | 1,25-Dihydroxyvitamin D3 (calcitriol) |
| 25OHD3 | 25-Hydroxyvitamin D3 (calcidiol) |
A, Adenine; C, cytidine; G, guanine; T, thymine.
BẢNG CHÚ GIẢI THUẬT NGỮ ANH VIỆT – CHƯƠNG 20
| STT | Tên thuật ngữ tiếng Anh | Phiên âm IPA | Nghĩa tiếng Việt |
|---|---|---|---|
| 1 | Mineral Metabolism | /ˈmɪnərəl məˈtæbəˌlɪzəm/ | Chuyển hóa Khoáng chất |
| 2 | Mineral Homeostasis | /ˈmɪnərəl ˌhoʊmiəˈsteɪsɪs/ | Nội môi Khoáng chất |
| 3 | Newborn | /ˈnuːbɔːrn/ | Sơ sinh |
| 4 | Infant | /ˈɪnfənt/ | Nhũ nhi |
| 5 | Adolescent | /ˌædəˈlɛsənt/ | Vị thành niên |
| 6 | Pediatric Endocrinology | /ˌpiːdiˈætrɪk ˌɛndoʊkrɪˈnɒlədʒi/ | Nội tiết học Nhi khoa |
| 7 | Calcium | /ˈkælsiəm/ | Canxi |
| 8 | Magnesium | /mæɡˈniːziəm/ | Magiê |
| 9 | Phosphate | /ˈfɒsfeɪt/ | Phốt phát |
| 10 | Bone formation | /boʊn fɔːrˈmeɪʃən/ | Sự hình thành xương |
| 11 | Accrual | /əˈkruːəl/ | Sự tích lũy |
| 12 | Maintenance | /ˈmeɪntənəns/ | Sự duy trì |
| 13 | Ingestion | /ɪnˈdʒɛstʃən/ | Sự ăn vào |
| 14 | Absorption | /əbˈsɔːrpʃən/ | Sự hấp thu |
| 15 | Retention | /rɪˈtɛnʃən/ | Sự giữ lại |
| 16 | Constituent nutrients | /kənˈstɪtʃuənt ˈnuːtriənts/ | Các chất dinh dưỡng cấu thành |
| 17 | Vitamin D metabolism | /ˈvaɪtəmɪn diː məˈtæbəˌlɪzəm/ | Chuyển hóa vitamin D |
| 18 | Bioactivity | /ˌbaɪoʊækˈtɪvɪti/ | Hoạt tính sinh học |
| 19 | Parathyroid hormone (PTH) | /ˌpærəˈθaɪrɔɪd ˈhɔːrmoʊn/ | Hormon cận giáp (PTH) |
| 20 | Synthesis | /ˈsɪnθəsɪs/ | Sự tổng hợp |
| 21 | Secretion | /sɪˈkriːʃən/ | Sự bài tiết |
| 22 | Intrinsic aberrations | /ɪnˈtrɪnsɪk ˌæbəˈreɪʃənz/ | Những sai lệch nội tại |
| 23 | Cartilage | /ˈkɑːrtəlɪdʒ/ | Sụn |
| 24 | Bone cells | /boʊn sɛlz/ | Tế bào xương |
| 25 | Pathological variations | /ˌpæθəˈlɒdʒɪkəl ˌvɛəriˈeɪʃənz/ | Các biến thể bệnh lý |
| 26 | Genes | /dʒiːnz/ | Gen |
| 27 | Acquired insults | /əˈkwaɪərd ˈɪnsʌlts/ | Các tổn thương mắc phải |
| 28 | Serum concentrations | /ˈsɪərəm ˌkɒnsənˈtreɪʃənz/ | Nồng độ huyết thanh |
| 29 | Total calcium | /ˈtoʊtəl ˈkælsiəm/ | Canxi toàn phần |
| 30 | Ionized calcium | /ˈaɪənaɪzd ˈkælsiəm/ | Canxi ion hóa |
| 31 | Hyperproteinemia | /ˌhaɪpərˌproʊtiːˈniːmiə/ | Tăng protein máu |
| 32 | Hypoproteinemia | /ˌhaɪpoʊˌproʊtiːˈniːmiə/ | Giảm protein máu |
| 33 | Plasma pH | /ˈplæzmə piː eɪtʃ/ | pH huyết tương |
| 34 | Alkalosis | /ˌælkəˈloʊsɪs/ | Nhiễm kiềm |
| 35 | Acidosis | /ˌæsɪˈdoʊsɪs/ | Nhiễm toan |
| 36 | Hypocalcemia | /ˌhaɪpoʊkælˈsiːmiə/ | Hạ canxi máu |
| 37 | Hypercalcemia | /ˌhaɪpərkælˈsiːmiə/ | Tăng canxi máu |
| 38 | Skeletal homeostasis | /ˈskɛlɪtəl ˌhoʊmiəˈsteɪsɪs/ | Nội môi xương |
| 39 | Hypophosphatasia | /ˌhaɪpoʊˌfɒsfəˈteɪʒə/ | Bệnh giảm phosphatase máu |
| 40 | Neonatal severe hyperparathyroidism | /ˌniːəˈneɪtəl sɪˈvɪər ˌhaɪpərˌpærəˈθaɪrɔɪdɪzəm/ | Cường cận giáp nặng ở trẻ sơ sinh |
| 41 | Fibrous dysplasia | /ˈfaɪbrəs dɪsˈpleɪʒə/ | Loạn sản xơ |
| 42 | Williams-Beuren syndrome | /ˈwɪliəmz ˈbɔɪrən ˈsɪndroʊm/ | Hội chứng Williams-Beuren |
| 43 | Osteogenesis imperfecta | /ˌɒstioʊˈdʒɛnɪsɪs ˌɪmpərˈfɛktə/ | Bệnh xương thủy tinh |
| 44 | Pycnodysostosis | /ˌpɪknoʊdɪsɒsˈtoʊsɪs/ | Bệnh pycnodysostosis (bệnh lùn Toulouse-Lautrec) |
| 45 | Vitamin D hydroxylation-deficient rickets | /ˈvaɪtəmɪn diː haɪˌdrɒksɪˈleɪʃən dɪˈfɪʃənt ˈrɪkɪts/ | Còi xương do thiếu hydroxyl hóa vitamin D |
| 46 | Autosomal dominant | /ˌɔːtəˈsoʊməl ˈdɒmɪnənt/ | Trội trên nhiễm sắc thể thường |
| 47 | Autosomal recessive | /ˌɔːtəˈsoʊməl rɪˈsɛsɪv/ | Lặn trên nhiễm sắc thể thường |
| 48 | X-linked | /ɛks-lɪŋkt/ | Liên kết nhiễm sắc thể X |
| 49 | Paresthesias | /ˌpærəsˈθiːʒəz/ | Dị cảm |
| 50 | Tetany | /ˈtɛtəni/ | Cơn tetany (co cứng) |
| 51 | Carpopedal spasm | /ˌkɑːrpoʊˈpiːdəl ˈspæzəm/ | Co thắt bàn tay-bàn chân |
| 52 | Laryngospasm | /ləˈrɪŋɡoʊˌspæzəm/ | Co thắt thanh quản |
| 53 | Muscular cramps | /ˈmʌskjələr kræmps/ | Chuột rút cơ |
| 54 | Myotonic spasms | /ˌmaɪəˈtɒnɪk ˈspæzəmz/ | Co thắt trương lực cơ |
| 55 | Convulsions | /kənˈvʌlʃənz/ | Co giật |
| 56 | Chvostek sign | /ˈ(t)ʃvɒstɛk saɪn/ | Dấu hiệu Chvostek |
| 57 | Trousseau sign | /truːˈsoʊ saɪn/ | Dấu hiệu Trousseau |
| 58 | Electrocardiographic QT interval | /ɪˌlɛktroʊˌkɑːrdioʊˈɡræfɪk kjuː tiː ˈɪntərvəl/ | Khoảng QT trên điện tâm đồ |
| 59 | Fetus | /ˈfiːtəs/ | Thai nhi |
| 60 | Postnatally | /poʊstˈneɪtəli/ | Sau sinh |
| 61 | Placental calcium transport | /pləˈsɛntəl ˈkælsiəm ˈtrænspɔːrt/ | Vận chuyển canxi qua nhau thai |
| 62 | Parathyroid hormone–related protein (PTHrP) | /ˌpærəˈθaɪrɔɪd ˈhɔːrmoʊn rɪˈleɪtɪd ˈproʊtiːn/ | Protein liên quan đến hormon cận giáp (PTHrP) |
| 63 | Umbilical cord blood | /ʌmˈbɪlɪkəl kɔːrd blʌd/ | Máu cuống rốn |
| 64 | Calcitonin | /ˌkælsɪˈtoʊnɪn/ | Calcitonin |
| 65 | 1,25-dihydroxyvitamin D3 (calcitriol) | /… ˌdaɪhaɪˈdrɒksiˌvaɪtəmɪn diː θriː (ˌkælsɪˈtraɪɒl)/ | 1,25-dihydroxyvitamin D3 (calcitriol) |
| 66 | 25-hydroxyvitamin D3 (calcidiol) | /… ˌhaɪˈdrɒksiˌvaɪtəmɪn diː θriː (ˌkælsɪˈdaɪɒl)/ | 25-hydroxyvitamin D3 (calcidiol) |
| 67 | Neuromuscular hyperexcitability | /ˌnʊəroʊˈmʌskjələr ˌhaɪpərɛkˌsaɪtəˈbɪlɪti/ | Tăng kích thích thần kinh-cơ |
| 68 | Hyperacusis | /ˌhaɪpərˈækjusɪs/ | Tăng thính lực |
| 69 | Jitteriness | /ˈdʒɪtərɪnəs/ | Run rẩy, bồn chồn |
| 70 | Tremulousness | /ˈtrɛmjələsnəs/ | Run |
| 71 | Seizures | /ˈsiːʒərz/ | Cơn co giật |
| 72 | Apnea | /ˈæpniə/ | Ngưng thở |
| 73 | Tachycardia | /ˌtækɪˈkɑːrdiə/ | Nhịp tim nhanh |
| 74 | Cyanosis | /ˌsaɪəˈnoʊsɪs/ | Tím tái |
| 75 | Early neonatal hypocalcemia | /ˈɜːrli ˌniːəˈneɪtəl ˌhaɪpoʊkælˈsiːmiə/ | Hạ canxi máu sơ sinh sớm |
| 76 | Low birth weight (LBW) | /loʊ bɜːrθ weɪt/ | Cân nặng khi sinh thấp |
| 77 | Small-for-gestational-age | /smɔːl fər dʒɛsˈteɪʃənəl eɪdʒ/ | Nhỏ so với tuổi thai |
| 78 | Asphyxiated neonates | /əsˈfɪksieɪtɪd ˈniːəneɪts/ | Trẻ sơ sinh bị ngạt |
| 79 | Gestational diabetes mellitus | /dʒɛsˈteɪʃənəl ˌdaɪəˈbiːtiːz ˈmɛlɪtəs/ | Đái tháo đường thai kỳ |
| 80 | Maternal hyperparathyroidism | /məˈtɜːrnəl ˌhaɪpərˌpærəˈθaɪrɔɪdɪzəm/ | Cường cận giáp ở mẹ |
| 81 | Renal tubular phosphaturic response | /ˈriːnəl ˈtjuːbjələr ˌfɒsfəˈtjʊərɪk rɪˈspɒns/ | Đáp ứng bài tiết phốt phát ở ống thận |
| 82 | Skeletal calcium | /ˈskɛlɪtəl ˈkælsiəm/ | Canxi xương |
| 83 | Calcium absorptive effects | /ˈkælsiəm əbˈsɔːrptɪv ɪˈfɛkts/ | Tác dụng hấp thu canxi |
| 84 | Reabsorptive effects | /ˌriːəbˈsɔːrptɪv ɪˈfɛkts/ | Tác dụng tái hấp thu |
| 85 | Intestinal tract | /ɪnˈtɛstɪnəl trækt/ | Đường ruột |
| 86 | Asphyxiated newborns | /əsˈfɪksieɪtɪd ˈnuːbɔːrnz/ | Trẻ sơ sinh bị ngạt |
| 87 | Toxemia of pregnancy | /tɒkˈsiːmiə əv ˈprɛɡnənsi/ | Nhiễm độc thai nghén |
| 88 | Anticonvulsant drugs | /ˌæntikənˈvʌlsənt drʌɡz/ | Thuốc chống co giật |
| 89 | Hypercalcitonemia | /ˌhaɪpərˌkælsɪtoʊˈniːmiə/ | Tăng calcitonin máu |
| 90 | Intrauterine growth restriction | /ˌɪntrəˈjuːtərɪn ɡroʊθ rɪˈstrɪkʃən/ | Chậm tăng trưởng trong tử cung |
| 91 | Respiratory distress syndrome | /ˈrɛspərətɔːri dɪˈstrɛs ˈsɪndroʊm/ | Hội chứng suy hô hấp |
| 92 | Placental transfer | /pləˈsɛntəl ˈtrænsfər/ | Sự vận chuyển qua nhau thai |
| 93 | Urinary excretion | /ˈjʊərɪnəri ɛkˈskriːʃən/ | Sự bài tiết qua nước tiểu |
| 94 | Hypomagnesemia | /ˌhaɪpoʊˌmæɡnɪˈsiːmiə/ | Hạ magiê máu |
| 95 | Hyperbilirubinemia | /ˌhaɪpərˌbɪlɪˌruːbɪˈniːmiə/ | Tăng bilirubin máu |
| 96 | Exchange transfusion | /ɪksˈtʃeɪndʒ trænsˈfjuːʒən/ | Truyền máu thay thế |
| 97 | Phototherapy | /ˌfoʊtoʊˈθɛrəpi/ | Quang trị liệu (chiếu đèn) |
| 98 | Rotavirus infection | /ˈroʊtəˌvaɪrəs ɪnˈfɛkʃən/ | Nhiễm Rotavirus |
| 99 | Diarrhea | /ˌdaɪəˈriːə/ | Tiêu chảy |
| 100 | Aminoglycoside antibiotics | /əˌmiːnoʊˈɡlaɪkəˌsaɪd ˌæntibaɪˈɒtɪks/ | Kháng sinh nhóm Aminoglycoside |
| 101 | Citrate | /ˈsɪtreɪt/ | Citrat |
| 102 | Transfused blood | /trænsˈfjuːzd blʌd/ | Máu được truyền |
| 103 | Fatty acids | /ˈfæti ˈæsɪdz/ | Axit béo |
| 104 | Bicarbonate | /ˌbaɪˈkɑːrbənət/ | Bicarbonat |
| 105 | Respiratory alkalosis | /ˈrɛspərətɔːri ˌælkəˈloʊsɪs/ | Nhiễm kiềm hô hấp |
| 106 | Metabolic alkalosis | /ˌmɛtəˈbɒlɪk ˌælkəˈloʊsɪs/ | Nhiễm kiềm chuyển hóa |
| 107 | Phytates | /ˈfaɪteɪts/ | Phytat |
| 108 | Malignant osteopetrosis | /məˈlɪɡnənt ˌɒstioʊpɛˈtroʊsɪs/ | Bệnh xương hóa đá ác tính |
| 109 | Osteoclastogenesis | /ˌɒstioʊklæstoʊˈdʒɛnɪsɪs/ | Sự tạo hủy cốt bào |
| 110 | Late neonatal hypocalcemia | /leɪt ˌniːəˈneɪtəl ˌhaɪpoʊkælˈsiːmiə/ | Hạ canxi máu sơ sinh muộn |
| 111 | Modified cow milk formulas | /ˈmɒdɪfaɪd kaʊ mɪlk ˈfɔːrmjələz/ | Sữa công thức bò đã điều chỉnh |
| 112 | Renal phosphate excretion | /ˈriːnəl ˈfɒsfeɪt ɛkˈskriːʃən/ | Sự bài tiết phốt phát qua thận |
| 113 | Chronic renal insufficiency | /ˈkrɒnɪk ˈriːnəl ˌɪnsəˈfɪʃənsi/ | Suy thận mạn |
| 114 | Renal hypoplasia | /ˈriːnəl ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản thận |
| 115 | Obstructive nephropathies | /əbˈstrʌktɪv nɛˈfrɒpəθiz/ | Bệnh thận tắc nghẽn |
| 116 | Azotemic | /ˌæzoʊˈtiːmɪk/ | Tăng nitơ máu |
| 117 | Peripheral responsiveness | /pəˈrɪfərəl rɪˈspɒnsɪvnəs/ | Đáp ứng ngoại vi |
| 118 | Hypermagnesemia | /ˌhaɪpərˌmæɡnɪˈsiːmiə/ | Tăng magiê máu |
| 119 | Hepatic 25-hydroxylation | /hɪˈpætɪk … haɪˌdrɒksɪˈleɪʃən/ | 25-hydroxyl hóa tại gan |
| 120 | 25-hydroxyvitamin D-1α hydroxylase activity | /… haɪˈdrɒksiˌvaɪtəmɪn diː … haɪˈdrɒksɪleɪz ækˈtɪvɪti/ | Hoạt độ 25-hydroxyvitamin D-1α hydroxylase |
| 121 | Vitamin D receptor (VDR) | /ˈvaɪtəmɪn diː rɪˈsɛptər/ | Thụ thể vitamin D (VDR) |
| 122 | Hypovitaminosis D | /ˌhaɪpoʊˌvaɪtəmɪˈnoʊsɪs diː/ | Tình trạng thiếu vitamin D |
| 123 | Low bone mass | /loʊ boʊn mæs/ | Khối lượng xương thấp |
| 124 | Hypoparathyroidism | /ˌhaɪpoʊˌpærəˈθaɪrɔɪdɪzəm/ | Suy cận giáp |
| 125 | Embryogenesis | /ˌɛmbrioʊˈdʒɛnɪsɪs/ | Sự hình thành phôi |
| 126 | Calcium sensing receptor (CaSR) | /ˈkælsiəm ˈsɛnsɪŋ rɪˈsɛptər/ | Thụ thể cảm nhận canxi (CaSR) |
| 127 | Developmental maturation | /dɪˌvɛləpˈmɛntəl ˌmætʃəˈreɪʃən/ | Sự trưởng thành về phát triển |
| 128 | Functional hypoparathyroidism | /ˈfʌŋkʃənəl ˌhaɪpoʊˌpærəˈθaɪrɔɪdɪzəm/ | Suy cận giáp chức năng |
| 129 | Pseudohypoparathyroidism (PHP) | /ˌsuːdoʊˌhaɪpoʊˌpærəˈθaɪrɔɪdɪzəm/ | Giả suy cận giáp (PHP) |
| 130 | Congenital aplasia | /kənˈdʒɛnɪtəl eɪˈpleɪʒə/ | Bất sản bẩm sinh |
| 131 | Congenital hypoplasia | /kənˈdʒɛnɪtəl ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản bẩm sinh |
| 132 | DNA-binding transcription factor | /diː ɛn eɪ ˈbaɪndɪŋ trænsˈkrɪpʃən ˈfæktər/ | Yếu tố phiên mã liên kết DNA |
| 133 | Autosomal dominant hypocalcemia | /ˌɔːtəˈsoʊməl ˈdɒmɪnənt ˌhaɪpoʊkælˈsiːmiə/ | Hạ canxi máu di truyền trội trên NST thường |
| 134 | Guanine nucleotide binding protein alpha 11 (GNA11) | /ˈɡwɑːniːn ˈnuːkliəˌtaɪd ˈbaɪndɪŋ ˈproʊtiːn ælfə ɪˈlɛvən/ | Protein liên kết Guanine nucleotide alpha 11 (GNA11) |
| 135 | Thick ascending limb of the loop of Henle (TALH) | /θɪk əˈsɛndɪŋ lɪm əv ðə luːp əv ˈhɛnli/ | Nhánh lên dày của quai Henle (TALH) |
| 136 | Guanosine protein binding receptor (GPCR) | /ˈɡwɑːnəsiːn ˈproʊtiːn ˈbaɪndɪŋ rɪˈsɛptər/ | Thụ thể kết hợp với protein G (GPCR) |
| 137 | Intracellular signal transduction pathways | /ˌɪntrəˈsɛljələr ˈsɪɡnəl trænsˈdʌkʃən ˈpæθweɪz/ | Các con đường truyền tín hiệu nội bào |
| 138 | Hypercalciuria | /ˌhaɪpərˌkælsɪˈjʊəriə/ | Tăng canxi niệu |
| 139 | Renal outer medullary potassium channel | /ˈriːnəl ˈaʊtər məˈdʌləri pəˈtæsiəm ˈtʃænəl/ | Kênh kali tủy thận ngoài |
| 140 | Bartter-like syndrome | /ˈbɑːrtər laɪk ˈsɪndroʊm/ | Hội chứng giống Bartter |
| 141 | Hypokalemic metabolic alkalosis | /ˌhaɪpoʊkəˈliːmɪk ˌmɛtəˈbɒlɪk ˌælkəˈloʊsɪs/ | Nhiễm kiềm chuyển hóa hạ kali máu |
| 142 | Hyperreninemia | /ˌhaɪpərˌriːnɪˈniːmiə/ | Tăng renin máu |
| 143 | Hyperaldosteronism | /ˌhaɪpərælˈdɒstəroʊˌnɪzəm/ | Cường aldosteron |
| 144 | Nephrocalcinosis | /ˌnɛfroʊˌkælsɪˈnoʊsɪs/ | Nhiễm canxi thận |
| 145 | Recombinant human PTH | /riːˈkɒmbɪnənt ˈhjuːmən piː tiː eɪtʃ/ | PTH tái tổ hợp của người |
| 146 | Calcilytic agents | /ˌkælsɪˈlɪtɪk ˈeɪdʒənts/ | Các tác nhân tiêu canxi |
| 147 | DiGeorge syndrome | /diːˈdʒɔːrdʒ ˈsɪndroʊm/ | Hội chứng DiGeorge |
| 148 | Neurocristopathy | /ˌnʊəroʊkrɪsˈtɒpəθi/ | Bệnh lý mào thần kinh |
| 149 | Cervical neural crest cells | /ˈsɜːrvɪkəl ˈnʊərəl krɛst sɛlz/ | Tế bào mào thần kinh cổ |
| 150 | Pharyngeal pouches | /fəˈrɪndʒiəl ˈpaʊtʃɪz/ | Các túi hầu |
| 151 | Branchial arches | /ˈbræŋkiəl ˈɑːrtʃɪz/ | Các cung mang |
| 152 | Contiguous gene syndrome | /kənˈtɪɡjuəs dʒiːn ˈsɪndroʊm/ | Hội chứng gen liền kề |
| 153 | Meiosis | /maɪˈoʊsɪs/ | Giảm phân |
| 154 | Thymic differentiation | /ˈθaɪmɪk ˌdɪfəˌrɛnʃiˈeɪʃən/ | Sự biệt hóa của tuyến ức |
| 155 | T lymphocyte function | /tiː ˈlɪmfəˌsaɪt ˈfʌŋkʃən/ | Chức năng tế bào lympho T |
| 156 | Cell-mediated immunity | /sɛl ˈmiːdieɪtɪd ɪˈmjuːnəti/ | Miễn dịch qua trung gian tế bào |
| 157 | Conotruncal defects | /ˌkoʊnoʊˈtrʌŋkəl ˈdiːfɛkts/ | Dị tật nón-thân động mạch |
| 158 | Aortic arch | /eɪˈɔːrtɪk ɑːrtʃ/ | Cung động mạch chủ |
| 159 | Tetralogy of Fallot | /tɛˈtrælədʒi əv fəˈloʊ/ | Tứ chứng Fallot |
| 160 | Ventricular septal defect | /vɛnˈtrɪkjələr ˈsɛptəl ˈdiːfɛkt/ | Thông liên thất |
| 161 | Interrupted aortic arch | /ˌɪntəˈrʌptɪd eɪˈɔːrtɪk ɑːrtʃ/ | Cung động mạch chủ gián đoạn |
| 162 | Right aortic arch | /raɪt eɪˈɔːrtɪk ɑːrtʃ/ | Cung động mạch chủ bên phải |
| 163 | Truncus arteriosus | /ˈtrʌŋkəs ɑːrˌtɪərioʊsəs/ | Thân chung động mạch |
| 164 | Vascular ring | /ˈvæskjələr rɪŋ/ | Vòng mạch máu |
| 165 | T-box 1 (TBX1) | /tiː bɒks wʌn/ | T-box 1 (TBX1) |
| 166 | Haploinsufficiency | /ˌhæploʊɪnsəˈfɪʃənsi/ | Thiểu năng một nửa |
| 167 | Histone | /ˈhɪstoʊn/ | Histone |
| 168 | Ubiquitin | /juːˈbɪkwɪtɪn/ | Ubiquitin |
| 169 | Posttranslational processing | /poʊsttrænsˈleɪʃənəl ˈprɒsɛsɪŋ/ | Xử lý sau dịch mã |
| 170 | Velocardiofacial syndrome | /ˌviːloʊˌkɑːrdioʊˈfeɪʃəl ˈsɪndroʊm/ | Hội chứng vòm-tim-mặt |
| 171 | Ocular hypertelorism | /ˈɒkjələr ˌhaɪpərˈtɛlərɪzəm/ | Hai mắt xa nhau |
| 172 | Cleft palate | /klɛft ˈpælət/ | Hở hàm ếch |
| 173 | Olfactory dysfunction | /ɒlˈfæktəri dɪsˈfʌŋkʃən/ | Rối loạn chức năng khứu giác |
| 174 | Learning disabilities | /ˈlɜːrnɪŋ ˌdɪsəˈbɪlɪtiz/ | Khuyết tật học tập |
| 175 | Unilateral facial paresis | /ˌjuːnɪˈlætərəl ˈfeɪʃəl pəˈriːsɪs/ | Liệt mặt một bên |
| 176 | Depressor anguli oris muscle | /dɪˈprɛsər ˈæŋɡjəlaɪ ˈɔːrɪs ˈmʌsəl/ | Cơ hạ góc miệng |
| 177 | Microduplication | /ˌmaɪkroʊˌduːplɪˈkeɪʃən/ | Vi nhân đôi |
| 178 | Copy number variant | /ˈkɒpi ˈnʌmbər ˈvɛəriənt/ | Biến thể số lượng bản sao |
| 179 | Autism | /ˈɔːtɪzəm/ | Tự kỷ |
| 180 | Schizophrenia | /ˌskɪtsəˈfriːniə/ | Tâm thần phân liệt |
| 181 | CHARGE syndrome | /tʃɑːrdʒ ˈsɪndroʊm/ | Hội chứng CHARGE |
| 182 | Coloboma | /ˌkɒləˈboʊmə/ | Khuyết mống mắt |
| 183 | Choanal atresia | /ˈkoʊənəl əˈtriːʒə/ | Tịt lỗ mũi sau |
| 184 | Barakat syndrome (HDR) | /bəˈrɑːkæt ˈsɪndroʊm/ | Hội chứng Barakat (HDR) |
| 185 | Sensorineural deafness | /ˌsɛnsəriˈnʊərəl ˈdɛfnəs/ | Điếc thần kinh giác quan |
| 186 | Renal disease | /ˈriːnəl dɪˈziːz/ | Bệnh thận |
| 187 | Zinc-finger transcription factor | /zɪŋk ˈfɪŋɡər trænsˈkrɪpʃən ˈfæktər/ | Yếu tố phiên mã ngón tay kẽm |
| 188 | Sanjad-Sakati syndrome | /sænˈdʒɑːd səˈkɑːti ˈsɪndroʊm/ | Hội chứng Sanjad-Sakati |
| 189 | Facial dysmorphism | /ˈfeɪʃəl dɪsˈmɔːrfɪzəm/ | Dị dạng khuôn mặt |
| 190 | Medullary stenosis | /məˈdʌləri stəˈnoʊsɪs/ | Hẹp tủy xương |
| 191 | Microphthalmia | /ˌmaɪkrɒfˈθælmiə/ | Mắt nhỏ |
| 192 | Adenohypophyseal hypoplasia | /ˌædɪnoʊˌhaɪpoʊfɪˈsiːəl ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản thùy trước tuyến yên |
| 193 | Pituitary stalk | /pɪˈtjuːɪtəri stɔːk/ | Cuống tuyến yên |
| 194 | Infundibulum | /ˌɪnfʌnˈdɪbjələm/ | Phễu (não) |
| 195 | Corpus callosum | /ˈkɔːrpəs kəˈloʊsəm/ | Thể chai |
| 196 | Kenny-Caffey syndrome | /ˈkɛni ˈkæfi ˈsɪndroʊm/ | Hội chứng Kenny-Caffey |
| 197 | Osteosclerosis | /ˌɒstioʊskləˈroʊsɪs/ | Xơ cứng xương |
| 198 | Diploic space | /dɪˈploʊɪk speɪs/ | Khoang lưỡng bản (xương sọ) |
| 199 | Gracile bone dysplasia | /ˈɡræsɪl boʊn dɪsˈpleɪʒə/ | Loạn sản xương mảnh |
| 200 | Basal cranial sutures | /ˈbeɪsəl ˈkreɪniəl ˈsuːtʃərz/ | Các đường khớp sọ nền |
| 201 | Mitochondria | /ˌmaɪtəˈkɒndriə/ | Ty thể |
| 202 | Mitochondrial genome | /ˌmaɪtəˈkɒndriəl ˈdʒiːnoʊm/ | Bộ gen ty thể |
| 203 | Electron transport | /ɪˈlɛktrɒn ˈtrænspɔːrt/ | Vận chuyển điện tử |
| 204 | Transfer ribonucleic acids (RNAs) | /ˈtrænsfər ˌraɪboʊnjuːˈkliːɪk ˈæsɪdz/ | Axit ribonucleic vận chuyển (tRNA) |
| 205 | Kearns-Sayre syndrome | /kɜːrnz seər ˈsɪndroʊm/ | Hội chứng Kearns-Sayre |
| 206 | Progressive external ophthalmoplegia | /prəˈɡrɛsɪv ɛkˈstɜːrnəl ˌɒfθælmoʊˈpliːdʒə/ | Liệt mắt ngoài tiến triển |
| 207 | Pigmentary retinopathy | /ˈpɪɡməntəri ˌrɛtɪˈnɒpəθi/ | Bệnh võng mạc sắc tố |
| 208 | Cerebellar ataxia | /ˌsɛrəˈbɛlər əˈtæksiə/ | Thất điều tiểu não |
| 209 | Cardiac conductivity | /ˈkɑːrdiæk ˌkɒndʌkˈtɪvɪti/ | Dẫn truyền tim |
| 210 | Myopathy | /maɪˈɒpəθi/ | Bệnh cơ |
| 211 | Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) | /… ɛnˌsɛfəloʊmaɪˈɒpəθi, ˈlæktɪk ˌæsɪˈdoʊsɪs, ənd stroʊk laɪk ˈɛpɪsoʊdz/ | Bệnh não cơ ty thể, nhiễm toan lactic và các cơn giống đột quỵ (MELAS) |
| 212 | Neuropathy | /nʊəˈrɒpəθi/ | Bệnh thần kinh |
| 213 | Cardiomyopathy | /ˌkɑːrdioʊmaɪˈɒpəθi/ | Bệnh cơ tim |
| 214 | End-organ insensitivity | /ɛnd ˈɔːrɡən ɪnˌsɛnsɪˈtɪvɪti/ | Mất nhạy cảm của cơ quan đích |
| 215 | PTH receptor (PTH1R) | /piː tiː eɪtʃ rɪˈsɛptər/ | Thụ thể PTH (PTH1R) |
| 216 | G-protein–coupled structure | /dʒiː ˈproʊtiːn ˈkʌpəld ˈstrʌktʃər/ | Cấu trúc kết hợp với protein G |
| 217 | Stimulatory guanine nucleotide-binding protein (Gs-protein) | /ˈstɪmjələˌtɔːri ˈɡwɑːniːn ˈnuːkliəˌtaɪd ˈbaɪndɪŋ ˈproʊtiːn/ | Protein G kích thích liên kết guanine nucleotide (protein Gs) |
| 218 | Guanosine triphosphate (GTP) | /ˌɡwɑːnəsiːn traɪˈfɒsfeɪt/ | Guanosine triphosphate (GTP) |
| 219 | Guanosine diphosphate (GDP) | /ˌɡwɑːnəsiːn daɪˈfɒsfeɪt/ | Guanosine diphosphate (GDP) |
| 220 | Adenylyl cyclase | /əˈdɛnəlɪl ˈsaɪkleɪz/ | Adenylyl cyclase |
| 221 | Cyclic adenosine monophosphate (AMP) | /ˈsaɪklɪk əˈdɛnəsiːn ˌmɒnoʊˈfɒsfeɪt/ | Cyclic adenosine monophosphate (AMP vòng) |
| 222 | Protein kinase A (PKA) | /ˈproʊtiːn ˈkaɪneɪs eɪ/ | Protein kinase A (PKA) |
| 223 | Cyclic AMP- responsive binding protein (CREB1) | /ˈsaɪklɪk æmp rɪˈspɒnsɪv ˈbaɪndɪŋ ˈproʊtiːn/ | Protein liên kết yếu tố đáp ứng AMP vòng (CREB1) |
| 224 | Phosphodiesterases | /ˌfɒsfoʊdaɪˈɛstəreɪsɪz/ | Phosphodiesterase |
| 225 | Blomstrand chondrodysplasia | /ˈblɒmstrænd ˌkɒndroʊdɪsˈpleɪʒə/ | Loạn sản sụn Blomstrand |
| 226 | Osteochondrodystrophy | /ˌɒstioʊˌkɒndroʊˈdɪstrəfi/ | Bệnh loạn sản sụn xương |
| 227 | Polyhydramnios | /ˌpɒlihaɪˈdræmniɒs/ | Đa ối |
| 228 | Hydrops fetalis | /ˈhaɪdrɒps fiːˈtælɪs/ | Phù thai |
| 229 | Short-limbed dwarfism | /ʃɔːrt lɪmd dwɔːrfɪzəm/ | Lùn chi ngắn |
| 230 | Aortic coarctation | /eɪˈɔːrtɪk ˌkoʊɑːrkˈteɪʃən/ | Hẹp eo động mạch chủ |
| 231 | Eiken chondrodysplasia | /ˈaɪkən ˌkɒndroʊdɪsˈpleɪʒə/ | Loạn sản sụn Eiken |
| 232 | Epiphyseal ossification | /ˌɛpɪˈfɪziəl ˌɒsɪfɪˈkeɪʃən/ | Hóa xương đầu xương |
| 233 | Multiple epiphyseal dysplasia | /ˈmʌltəpəl ˌɛpɪˈfɪziəl dɪsˈpleɪʒə/ | Loạn sản nhiều đầu xương |
| 234 | Albright hereditary osteodystrophy (AHO) | /ˈɔːlbraɪt həˈrɛdɪtəri ˌɒstioʊˈdɪstrəfi/ | Loạn dưỡng xương di truyền Albright (AHO) |
| 235 | Brachydactyly | /ˌbrækɪˈdæktəli/ | Ngón ngắn |
| 236 | Dentinogenesis | /ˌdɛntɪnoʊˈdʒɛnɪsɪs/ | Sự tạo ngà |
| 237 | Subcutaneous ossifications | /ˌsʌbkjuːˈteɪniəs ˌɒsɪfɪˈkeɪʃənz/ | Sự hóa xương dưới da |
| 238 | Chondrocytes | /ˈkɒndrəˌsaɪts/ | Tế bào sụn |
| 239 | Osteoblasts | /ˈɒstioʊˌblæsts/ | Nguyên bào xương (tế bào tạo xương) |
| 240 | Heterotopic subcutaneous ossifications | /ˌhɛtəroʊˈtɒpɪk ˌsʌbkjuːˈteɪniəs ˌɒsɪfɪˈkeɪʃənz/ | Sự hóa xương lạc chỗ dưới da |
| 241 | Spinal stenosis | /ˈspaɪnəl stəˈnoʊsɪs/ | Hẹp ống sống |
| 242 | Carpal tunnel syndrome | /ˈkɑːrpəl ˈtʌnəl ˈsɪndroʊm/ | Hội chứng ống cổ tay |
| 243 | Hearing loss | /ˈhɪərɪŋ lɒs/ | Mất thính lực |
| 244 | Olfactory impairment | /ɒlˈfæktəri ɪmˈpɛərmənt/ | Suy giảm khứu giác |
| 245 | Sleep apnea | /sliːp ˈæpniə/ | Ngưng thở khi ngủ |
| 246 | Asthma | /ˈæzmə/ | Hen suyễn |
| 247 | Sodium-phosphate cotransporters | /ˈsoʊdiəm ˈfɒsfeɪt ˌkoʊtrænsˈpɔːrtərz/ | Các chất đồng vận chuyển natri-phốt phát |
| 248 | Imprinted gene | /ɪmˈprɪntɪd dʒiːn/ | Gen được in dấu |
| 249 | Allele | /əˈliːl/ | Alen (gen tương ứng) |
| 250 | Exons | /ˈɛksɒnz/ | Exon |
| 251 | Transcripts | /ˈtrænskrɪpts/ | Bản phiên mã |
| 252 | Neuroendocrine secretory protein-55 (NESP55) | /ˌnʊəroʊˈɛndoʊkrɪn ˈsɛkrətəri ˈproʊtiːn …/ | Protein tiết ra thần kinh nội tiết-55 (NESP55) |
| 253 | Chromogranin | /ˌkroʊmoʊˈɡrænɪn/ | Chromogranin |
| 254 | Differentially methylated region (DMR) | /ˌdɪfəˈrɛnʃəli ˈmɛθəleɪtɪd ˈriːdʒən/ | Vùng methyl hóa khác biệt (DMR) |
| 255 | Promoter | /prəˈmoʊtər/ | Vùng khởi động (của gen) |
| 256 | Epigenetic | /ˌɛpɪdʒəˈnɛtɪk/ | Biểu sinh |
| 257 | Uniparental isodisomy | /ˌjuːnɪpəˈrɛntəl ˌaɪsoʊˈdaɪsəmi/ | Lưỡng bội đồng nguồn đơn cha mẹ |
| 258 | Osteitis fibrosa cystica | /ˌɒstiˈaɪtɪs faɪˈbroʊsə ˈsɪstɪkə/ | Viêm xương xơ nang |
| 259 | Anabolic effects | /ˌænəˈbɒlɪk ɪˈfɛkts/ | Tác dụng đồng hóa |
| 260 | Endocortical bone | /ˌɛndoʊˈkɔːrtɪkəl boʊn/ | Xương nội mạc |
| 261 | Pseudopseudohypoparathyroidism (PPHP) | /ˌsuːdoʊˌsuːdoʊˌhaɪpoʊˌpærəˈθaɪrɔɪdɪzəm/ | Giả giả suy cận giáp (PPHP) |
| 262 | Intrauterine growth retardation | /ˌɪntrəˈjuːtərɪn ɡroʊθ ˌriːtɑːrˈdeɪʃən/ | Chậm tăng trưởng trong tử cung |
| 263 | Congenital hypothyroidism | /kənˈdʒɛnɪtəl ˌhaɪpoʊˈθaɪrɔɪdɪzəm/ | Suy giáp bẩm sinh |
| 264 | Hyperthyrotropinemia | /ˌhaɪpərˌθaɪroʊtroʊpɪˈniːmiə/ | Tăng thyrotropin máu |
| 265 | Growth hormone releasing hormone (GHRH) | /ɡroʊθ ˈhɔːrmoʊn rɪˈliːsɪŋ ˈhɔːrmoʊn/ | Hormon giải phóng hormon tăng trưởng (GHRH) |
| 266 | Growth hormone (GH) deficiency | /ɡroʊθ ˈhɔːrmoʊn dɪˈfɪʃənsi/ | Thiếu hụt hormon tăng trưởng (GH) |
| 267 | Luteinizing (LH) hormone | /ˈluːtiənaɪzɪŋ ˈhɔːrmoʊn/ | Hormon hoàng thể hóa (LH) |
| 268 | Follicle-stimulating (FSH) hormone | /ˈfɒlɪkəl ˈstɪmjəleɪtɪŋ ˈhɔːrmoʊn/ | Hormon kích thích nang trứng (FSH) |
| 269 | Melanocortin (MSH) | /məˈlænoʊˌkɔːrtɪn/ | Melanocortin (MSH) |
| 270 | Obesity | /oʊˈbiːsəti/ | Béo phì |
| 271 | Acrodysostosis | /ˌækroʊdɪsɒsˈtoʊsɪs/ | Bệnh acrodysostosis |
| 272 | Maxillary hypoplasia | /mækˈsɪləri ˌhaɪpoʊˈpleɪʒə/ | Thiểu sản xương hàm trên |
| 273 | Hypertelorism | /ˌhaɪpərˈtɛlərɪzəm/ | Hai mắt xa nhau |
| 274 | Inactivating PTH/PTHrP signaling disorder (iPPSD) | /ɪnˈæktɪveɪtɪŋ piː tiː eɪtʃ piː tiː eɪtʃ ɑːr piː ˈsɪɡnəlɪŋ dɪsˈɔːrdər/ | Rối loạn tín hiệu PTH/PTHrP bất hoạt (iPPSD) |
| 275 | Murk-Jansen chondrodysplasia | /mɜːrk ˈjænsən ˌkɒndroʊdɪsˈpleɪʒə/ | Loạn sản sụn Murk-Jansen |
| 276 | Genu varum | /ˈdʒiːnuː ˈvɛərəm/ | Chân vòng kiềng |
| 277 | Clinodactyly | /ˌklaɪnoʊˈdæktəli/ | Ngón tay vẹo |
| 278 | Brachydactyly with hypertension | /ˌbrækɪˈdæktəli wɪθ ˌhaɪpərˈtɛnʃən/ | Ngón ngắn kèm tăng huyết áp |
| 279 | Progressive osseous heteroplasia (POH) | /prəˈɡrɛsɪv ˈɒsiəs ˌhɛtəroʊˈpleɪʒə/ | Tăng sinh xương lạc chỗ tiến triển (POH) |
| 280 | Intra- or subdermal ossification/calcification | /ˈɪntrə ɔːr ˌsʌbˈdɜːrməl ˌɒsɪfɪˈkeɪʃən/ˌkælsɪfɪˈkeɪʃən/ | Hóa xương/vôi hóa trong hoặc dưới da |
| 281 | Dermal ossifications | /ˈdɜːrməl ˌɒsɪfɪˈkeɪʃənz/ | Hóa xương ở da |
| 282 | Deep fascia | /diːp ˈfæʃiə/ | Mạc sâu |
| 283 | Ectopic calcification | /ɛkˈtɒpɪk ˌkælsɪfɪˈkeɪʃən/ | Vôi hóa lạc chỗ |
| 284 | Hyperphosphatemic tumoral calcinosis | /ˌhaɪpərˌfɒsfəˈtiːmɪk ˈtjuːmərəl ˌkælsɪˈnoʊsɪs/ | Vôi hóa dạng u tăng phốt phát máu |
| 285 | Lipoprotein receptor-related protein 6 (LRP6) | /ˈlaɪpoʊˌproʊtiːn rɪˈsɛptər rɪˈleɪtɪd ˈproʊtiːn sɪks/ | Protein 6 liên quan đến thụ thể lipoprotein (LRP6) |
| 286 | Endocytosis | /ˌɛndoʊsaɪˈtoʊsɪs/ | Nhập bào |
| 287 | Preconceptual history | /ˌpriːkənˈsɛptʃuəl ˈhɪstəri/ | Tiền sử trước khi thụ thai |
| 288 | Gestation | /dʒɛsˈteɪʃən/ | Thai kỳ |
| 289 | Peripartum | /ˌpɛrɪˈpɑːrtəm/ | Chu sinh |
| 290 | Postnatal | /poʊstˈneɪtəl/ | Sau sinh |
| 291 | Parity | /ˈpærəti/ | Số lần sinh |
| 292 | Renal calculi | /ˈriːnəl ˈkælkjəlaɪ/ | Sỏi thận |
| 293 | Rickets | /ˈrɪkɪts/ | Bệnh còi xương |
| 294 | Socioeconomic status | /ˌsoʊsioʊˌɛkəˈnɒmɪk ˈsteɪtəs/ | Tình trạng kinh tế-xã hội |
| 295 | Thymic shadow | /ˈθaɪmɪk ˈʃædoʊ/ | Bóng tuyến ức |
| 296 | Single nucleotide polymorphism | /ˈsɪŋɡəl ˈnuːkliəˌtaɪd ˌpɒliˈmɔːrfɪzəm/ | Đa hình đơn nucleotide (SNP) |
| 297 | Microarray | /ˈmaɪkroʊəˌreɪ/ | Vi mảng (microarray) |
| 298 | Fluorescent in situ hybridization (FISH) | /flʊəˈrɛsənt ɪn ˈsaɪtuː ˌhaɪbrɪdaɪˈzeɪʃən/ | Lai tại chỗ phát huỳnh quang (FISH) |
| 299 | Regions of homozygosity | /ˈriːdʒənz əv ˌhoʊmoʊzaɪˈɡɒsɪti/ | Các vùng đồng hợp tử |
| 300 | Sequence analysis | /ˈsiːkwəns əˈnæləsɪs/ | Phân tích trình tự |
| 301 | Brachymetacarpals | /ˌbrækɪˌmɛtəˈkɑːrpəlz/ | Xương bàn tay ngắn |
| 302 | Genotyping | /ˈdʒiːnoʊˌtaɪpɪŋ/ | Xác định kiểu gen |
| 303 | Methylation studies | /ˌmɛθəˈleɪʃən ˈstʌdiz/ | Các nghiên cứu methyl hóa |
| 304 | Elemental calcium | /ˌɛlɪˈmɛntəl ˈkælsiəm/ | Canxi nguyên tố |
| 305 | Bradycardia | /ˌbrædɪˈkɑːrdiə/ | Nhịp tim chậm |
| 306 | Asystole | /eɪˈsɪstəli/ | Vô tâm thu |
| 307 | Bolus doses | /ˈboʊləs doʊsɪz/ | Liều tiêm nhanh |
| 308 | Extravasation | /ɪkˌstrævəˈseɪʃən/ | Sự thoát mạch |
| 309 | Infiltration | /ˌɪnfɪlˈtreɪʃən/ | Sự thâm nhiễm |
| 310 | Parenteral alimentation | /pəˈrɛntərəl ˌælɪmɛnˈteɪʃən/ | Nuôi dưỡng qua đường tĩnh mạch |
| 311 | Antiinfectious therapy | /ˌæntiɪnˈfɛkʃəs ˈθɛrəpi/ | Liệu pháp chống nhiễm trùng |
| 312 | Transplantation | /ˌtrænsplænˈteɪʃən/ | Cấy ghép |
| 313 | Mononuclear cells | /ˌmɒnoʊˈnuːkliər sɛlz/ | Tế bào đơn nhân |
| 314 | Immunocompromised | /ɪˌmjuːnoʊˈkɒmprəmaɪzd/ | Suy giảm miễn dịch |
| 315 | Hypoalbuminemia | /ˌhaɪpoʊælbjuːmɪˈniːmiə/ | Giảm albumin máu |
| 316 | Critically ill | /ˈkrɪtɪkəli ɪl/ | Bệnh nặng, nguy kịch |
| 317 | Acid-base balance | /ˈæsɪd beɪs ˈbæləns/ | Cân bằng kiềm toan |
| 318 | Granulomatous infiltration | /ˌɡrænjəˈloʊmətəs ˌɪnfɪlˈtreɪʃən/ | Thâm nhiễm dạng u hạt |
| 319 | Grand mal seizure | /ɡrænd mæl ˈsiːʒər/ | Cơn co giật toàn thể (cơn động kinh lớn) |
| 320 | Petit mal seizure | /ˈpɛti mæl ˈsiːʒər/ | Cơn vắng ý thức (cơn động kinh nhỏ) |
| 321 | Syncopal seizure | /ˈsɪŋkəpəl ˈsiːʒər/ | Cơn co giật do ngất |
| 322 | Congestive heart failure | /kənˈdʒɛstɪv hɑːrt ˈfeɪljər/ | Suy tim sung huyết |
| 323 | Hyperreflexia | /ˌhaɪpərriːˈflɛksiə/ | Tăng phản xạ |
| 324 | Autoimmune polyendocrinopathy | /ˌɔːtoʊɪˈmjuːn ˌpɒliˌɛndoʊkrɪˈnɒpəθi/ | Bệnh đa nội tiết tự miễn |
| 325 | Thyroidectomy | /ˌθaɪrɔɪˈdɛktəmi/ | Phẫu thuật cắt bỏ tuyến giáp |
| 326 | Arterial blood supply | /ɑːrˈtɪəriəl blʌd səˈplaɪ/ | Nguồn cung cấp máu động mạch |
| 327 | Wilson hepatolenticular degeneration | /ˈwɪlsən həˌpætoʊlɛnˈtɪkjələr diːˌdʒɛnəˈreɪʃən/ | Bệnh thoái hóa gan-nhân bèo Wilson |
| 328 | Hodgkin lymphoma | /ˈhɒdʒkɪn lɪmˈfoʊmə/ | U lympho Hodgkin |
| 329 | Non-Hodgkin lymphoma | /nɒn ˈhɒdʒkɪn lɪmˈfoʊmə/ | U lympho không Hodgkin |
| 330 | Radioiodine therapy | /ˌreɪdioʊˈaɪədaɪn ˈθɛrəpi/ | Liệu pháp iod phóng xạ |
| 331 | Hyperthyroidism | /ˌhaɪpərˈθaɪrɔɪdɪzəm/ | Cường giáp |
| 332 | Autoimmune polyendocrinopathy syndrome type I (APS1) | /… ˌpɒliˌɛndoʊkrɪˈnɒpəθi ˈsɪndroʊm taɪp wʌn/ | Hội chứng đa nội tiết tự miễn type I (APS1) |
| 333 | Regulatory T cells | /ˈrɛɡjələˌtɔːri tiː sɛlz/ | Tế bào T điều hòa |
| 334 | Immunological tolerance | /ˌɪmjənoʊˈlɒdʒɪkəl ˈtɒlərəns/ | Dung nạp miễn dịch |
| 335 | Inflammatory cytokines | /ɪnˈflæmətəri ˈsaɪtəˌkaɪnz/ | Các cytokine gây viêm |
| 336 | Interferon-γ (IFN-γ) | /ˌɪntərˈfɪərɒn ˈɡæmə/ | Interferon-γ (IFN-γ) |
| 337 | Helper T cells | /ˈhɛlpər tiː sɛlz/ | Tế bào T hỗ trợ |
| 338 | B cell function | /biː sɛl ˈfʌŋkʃən/ | Chức năng tế bào B |
| 339 | Autoantibody-mediated inflammation | /ˌɔːtoʊˈæntɪˌbɒdi ˈmiːdieɪtɪd ˌɪnfləˈmeɪʃən/ | Viêm qua trung gian tự kháng thể |
| 340 | Innate immune system | /ɪˈneɪt ɪˈmjuːn ˈsɪstəm/ | Hệ miễn dịch bẩm sinh |
| 341 | Major histocompatibility complex (MHC) | /ˈmeɪdʒər ˌhɪstoʊkəmˌpætəˈbɪlɪti ˈkɒmplɛks/ | Phức hợp tương hợp mô chính (MHC) |
| 342 | Human leukocyte antigen (HLA) | /ˈhjuːmən ˈluːkəˌsaɪt ˈæntɪdʒən/ | Kháng nguyên bạch cầu người (HLA) |
| 343 | Parathyroid chief cell | /ˌpærəˈθaɪrɔɪd tʃiːf sɛl/ | Tế bào chính tuyến cận giáp |
| 344 | Cytotoxic | /ˌsaɪtoʊˈtɒksɪk/ | Gây độc tế bào |
| 345 | Lymphocytic infiltration | /ˌlɪmfoʊˈsɪtɪk ˌɪnfɪlˈtreɪʃən/ | Thâm nhiễm lympho bào |
| 346 | Atrophy | /ˈætrəfi/ | Teo |
| 347 | Fatty replacement | /ˈfæti rɪˈpleɪsmənt/ | Thay thế bằng mô mỡ |
| 348 | Mucocutaneous candidiasis | /ˌmjuːkoʊkjuːˈteɪniəs ˌkændɪˈdaɪəsɪs/ | Nhiễm nấm Candida da niêm mạc |
| 349 | Ectodermal dystrophy | /ˌɛktoʊˈdɜːrməl ˈdɪstrəfi/ | Loạn sản ngoại bì |
| 350 | Primary adrenal insufficiency | /ˈpraɪməri əˈdriːnəl ˌɪnsəˈfɪʃənsi/ | Suy thượng thận nguyên phát |
| 351 | Pernicious anemia | /pərˈnɪʃəs əˈniːmiə/ | Thiếu máu ác tính |
| 352 | Hepatitis | /ˌhɛpəˈtaɪtɪs/ | Viêm gan |
| 353 | Primary ovarian failure | /ˈpraɪməri oʊˈvɛəriən ˈfeɪljər/ | Suy buồng trứng nguyên phát |
| 354 | Primary testicular failure | /ˈpraɪməri tɛsˈtɪkjələr ˈfeɪljər/ | Suy tinh hoàn nguyên phát |
| 355 | Keratitis | /ˌkɛrəˈtaɪtɪs/ | Viêm giác mạc |
| 356 | Retinitis | /ˌrɛtɪˈnaɪtɪs/ | Viêm võng mạc |
| 357 | Alopecia | /ˌæləˈpiːʃə/ | Rụng tóc |
| 358 | Metaphyseal dysplasia | /ˌmɛtəˈfɪziəl dɪsˈpleɪʒə/ | Loạn sản hành xương |
| 359 | Hypoadrenocorticism | /ˌhaɪpoʊəˌdriːnoʊˈkɔːrtɪˌsɪzəm/ | Suy vỏ thượng thận |
| 360 | Autoimmune oophoritis | /ˌɔːtoʊɪˈmjuːn ˌoʊəfəˈraɪtɪs/ | Viêm buồng trứng tự miễn |
| 361 | Orchitis | /ɔːrˈkaɪtɪs/ | Viêm tinh hoàn |
| 362 | Thyroiditis | /ˌθaɪrɔɪˈdaɪtɪs/ | Viêm tuyến giáp |
| 363 | Hypophysitis | /ˌhaɪpoʊfɪˈsaɪtɪs/ | Viêm tuyến yên |
| 364 | Vitiligo | /ˌvɪtɪˈlaɪɡoʊ/ | Bệnh bạch biến |
| 365 | Keratoconjunctivitis | /ˌkɛrətoʊkənˌdʒʌŋktɪˈvaɪtɪs/ | Viêm kết giác mạc |
| 366 | Esophageal squamous cell carcinoma | /ɪˌsɒfəˈdʒiːəl ˈskweɪməs sɛl ˌkɑːrsɪˈnoʊmə/ | Ung thư biểu mô tế bào vảy thực quản |
| 367 | Oral squamous cell carcinoma | /ˈɔːrəl ˈskweɪməs sɛl ˌkɑːrsɪˈnoʊmə/ | Ung thư biểu mô tế bào vảy miệng |
| 368 | Asplenia | /eɪˈspliːniə/ | Vô lách |
| 369 | Interstitial nephritis | /ˌɪntərˈstɪʃəl nɛˈfraɪtɪs/ | Viêm thận kẽ |
| 370 | Autoimmune regulator (AIRE) | /ˌɔːtoʊɪˈmjuːn ˈrɛɡjəleɪtər/ | Chất điều hòa tự miễn (AIRE) |
| 371 | Zinc-finger motifs | /zɪŋk ˈfɪŋɡər moʊˈtiːfs/ | Các motif ngón tay kẽm |
| 372 | Thymic medullary epithelial cells | /ˈθaɪmɪk məˈdʌləri ˌɛpɪˈθiːliəl sɛlz/ | Tế bào biểu mô tủy tuyến ức |
| 373 | Lymph nodes | /lɪmf noʊdz/ | Hạch bạch huyết |
| 374 | Spleen | /spliːn/ | Lách |
| 375 | Monocytes | /ˈmɒnəˌsaɪts/ | Bạch cầu đơn nhân |
| 376 | E3 ubiquitin ligase | /iː θriː juːˈbɪkwɪtɪn ˈlaɪɡeɪz/ | E3 ubiquitin ligase |
| 377 | Ubiquitin-proteasomal system | /juːˈbɪkwɪtɪn proʊtiəˈsoʊməl ˈsɪstəm/ | Hệ thống ubiquitin-proteasome |
| 378 | Plant homeodomains | /plænt ˈhoʊmioʊdoʊˌmeɪnz/ | Các miền homeodomain thực vật |
| 379 | SAND domain | /sænd doʊˈmeɪn/ | Miền SAND |
| 380 | Self-antigens | /sɛlf ˈæntɪdʒənz/ | Tự kháng nguyên |
| 381 | Thymic stromal medullary epithelial cells | /ˈθaɪmɪk ˈstroʊməl məˈdʌləri ˌɛpɪˈθiːliəl sɛlz/ | Tế bào biểu mô tủy đệm tuyến ức |
| 382 | Nuclear transporters | /ˈnuːkliər trænsˈpɔːrtərz/ | Các chất vận chuyển hạt nhân |
| 383 | Chromatin | /ˈkroʊmətɪn/ | Chất nhiễm sắc |
| 384 | Transcription factors | /trænsˈkrɪpʃən ˈfæktərz/ | Các yếu tố phiên mã |
| 385 | Pre-messenger (m)RNA | /priː ˈmɛsɪndʒər …/ | tiền RNA thông tin (pre-mRNA) |
| 386 | Splicing | /ˈsplaɪsɪŋ/ | Cắt nối (splicing) |
| 387 | FoxP3+ regulatory T cells (Treg) | /fɒks piː θriː ˈrɛɡjələˌtɔːri tiː sɛlz/ | Tế bào T điều hòa FoxP3+ (Treg) |
| 388 | Autoreactive T cells | /ˌɔːtoʊriˈæktɪv tiː sɛlz/ | Tế bào T tự phản ứng |
| 389 | Trisomy 21 (Down syndrome) | /ˈtraɪsoʊmi … daʊn ˈsɪndroʊm/ | Tam nhiễm sắc thể 21 (Hội chứng Down) |
| 390 | NALRP5 | /…/ | NALRP5 |
| 391 | Glutamic acid decarboxylase 65 | /ɡluːˈtæmɪk ˈæsɪd diːkɑːrˈbɒksɪleɪz …/ | Glutamic acid decarboxylase 65 |
| 392 | 21-hydroxylase | /… haɪˈdrɒksɪleɪz/ | 21-hydroxylase |
| 393 | Adrenocortical failure | /əˌdriːnoʊˈkɔːrtɪkəl ˈfeɪljər/ | Suy vỏ thượng thận |
| 394 | Aromatic L-amino acid decarboxylase | /ˌærəˈmætɪk … əˈmiːnoʊ ˈæsɪd diːkɑːrˈbɒksɪleɪz/ | Aromatic L-amino acid decarboxylase |
| 395 | Tryptophan hydroxylase | /ˈtrɪptəfæn haɪˈdrɒksɪleɪz/ | Tryptophan hydroxylase |
| 396 | Autoimmune polyendocrine syndrome type II | /ˌɔːtoʊɪˈmjuːn ˌpɒliˌɛndoʊkrɪn ˈsɪndroʊm taɪp tuː/ | Hội chứng đa nội tiết tự miễn type II |
| 397 | Adrenalitis | /əˌdriːnəˈlaɪtɪs/ | Viêm tuyến thượng thận |
| 398 | Celiac disease | /ˈsiːliæk dɪˈziːz/ | Bệnh celiac |
| 399 | Myasthenia gravis | /ˌmaɪəsˈθiːniə ˈɡrɑːvɪs/ | Bệnh nhược cơ |
| 400 | Immunodeficiency, Polyendocrinopathy, and Enteropathy–X-linked (IPEX) | /ˌɪmjənoʊdɪˈfɪʃənsi, ˌpɒliˌɛndoʊkrɪˈnɒpəθi, ənd ˌɛntəˈrɒpəθi ɛks lɪŋkt/ | Suy giảm miễn dịch, Bệnh đa nội tiết, và Bệnh ruột–Liên kết X (IPEX) |
| 401 | Autoimmune enteropathy | /ˌɔːtoʊɪˈmjuːn ˌɛntəˈrɒpəθi/ | Bệnh ruột tự miễn |
| 402 | Insulin-dependent diabetes mellitus | /ˈɪnsəlɪn dɪˈpɛndənt ˌdaɪəˈbiːtiːz ˈmɛlɪtəs/ | Đái tháo đường phụ thuộc insulin |
| 403 | Eczema | /ˈɛksɪmə/ | Chàm (Eczema) |
| 404 | Glomerulonephritis | /ɡloʊˌmɛrələʊnɛˈfraɪtɪs/ | Viêm cầu thận |
| 405 | Thrombocytopenia | /ˌθrɒmboʊˌsaɪtoʊˈpiːniə/ | Giảm tiểu cầu |
| 406 | Hemolytic anemia | /ˌhiːməˈlɪtɪk əˈniːmiə/ | Thiếu máu tan máu |
| 407 | Hungry bone syndrome | /ˈhʌŋɡri boʊn ˈsɪndroʊm/ | Hội chứng xương đói |
| 408 | Parathyroidectomy | /ˌpærəˌθaɪrɔɪˈdɛktəmi/ | Phẫu thuật cắt bỏ tuyến cận giáp |
| 409 | Bisphosphonates | /ˌbɪsˈfɒsfəneɪts/ | Bisphosphonat |
| 410 | Osteoclast | /ˈɒstioʊˌklæst/ | Hủy cốt bào (tế bào hủy xương) |
| 411 | Phosphate containing enemas | /ˈfɒsfeɪt kənˈteɪnɪŋ ˈɛnəməz/ | Thuốc xổ chứa phốt phát |
| 412 | Laxatives | /ˈlæksətɪvz/ | Thuốc nhuận tràng |
| 413 | Tumor cell lysis | /ˈtjuːmər sɛl ˈlaɪsɪs/ | Ly giải tế bào khối u |
| 414 | Rhabdomyolysis | /ˌræbdoʊmaɪˈɒlɪsɪs/ | Tiêu cơ vân |
| 415 | Plasmapheresis | /ˌplæzməfəˈriːsɪs/ | Lọc huyết tương |
| 416 | Pancreatitis | /ˌpæŋkriəˈtaɪtɪs/ | Viêm tụy |
| 417 | Pancreatic lipase | /ˌpæŋkriˈætɪk ˈlaɪpeɪz/ | Lipase tụy |
| 418 | Cytokine | /ˈsaɪtəˌkaɪn/ | Cytokine |
| 419 | Fluoride | /ˈflʊəraɪd/ | Florua |
| 420 | Multiassay chemical profile | /ˈmʌltiəˌseɪ ˈkɛmɪkəl ˈproʊfaɪl/ | Hồ sơ hóa học đa xét nghiệm |
| 421 | Heart murmur | /hɑːrt ˈmɜːrmər/ | Tiếng thổi ở tim |
| 422 | Arrhythmia | /əˈrɪðmiə/ | Loạn nhịp tim |
| 423 | Basal ganglia | /ˈbeɪsəl ˈɡæŋɡliə/ | Các hạch nền |
| 424 | Bronchospasm | /ˈbrɒŋkoʊˌspæzəm/ | Co thắt phế quản |
| 425 | Neuropsychiatric symptoms | /ˌnʊəroʊˌsaɪkiˈætrɪk ˈsɪmptəmz/ | Triệu chứng tâm thần kinh |
| 426 | Cataracts | /ˈkætərækts/ | Đục thủy tinh thể |
| 427 | Papilledema | /ˌpæpɪlɪˈdiːmə/ | Phù gai thị |
| 428 | Dentition | /dɛnˈtɪʃən/ | Bộ răng |
| 429 | Hyponatremia | /ˌhaɪpoʊnəˈtriːmiə/ | Hạ natri máu |
| 430 | Hypernatremia | /ˌhaɪpərnəˈtriːmiə/ | Tăng natri máu |
| 431 | Hypokalemia | /ˌhaɪpoʊkəˈliːmiə/ | Hạ kali máu |
| 432 | Hyperkalemia | /ˌhaɪpərkəˈliːmiə/ | Tăng kali máu |
| 433 | Immunoassays | /ˌɪmjənoʊˈæseɪz/ | Xét nghiệm miễn dịch |
| 434 | Elsworth-Howard test | /ˈɛlzwɜːrθ ˈhaʊərd tɛst/ | Nghiệm pháp Elsworth-Howard |
| 435 | Nephrogenous cyclic AMP | /nɛˈfrɒdʒɪnəs ˈsaɪklɪk æmp/ | AMP vòng nguồn gốc từ thận |
| 436 | Chemotherapeutic agents | /ˌkiːmoʊˌθɛrəˈpjuːtɪk ˈeɪdʒənts/ | Các tác nhân hóa trị liệu |
| 437 | Gitelman syndrome | /ˈɡɪtəlmən ˈsɪndroʊm/ | Hội chứng Gitelman |
| 438 | Bartter syndrome | /ˈbɑːrtər ˈsɪndroʊm/ | Hội chứng Bartter |
| 439 | Magnesium sulfate | /mæɡˈniːziəm ˈsʌlfeɪt/ | Magiê sulfat |
| 440 | Choreoathetoid movements | /ˌkɔːrioʊˈæθɪtɔɪd ˈmuːvmənts/ | Các cử động múa giật-múa vờn |
| 441 | Iatrogenic | /ˌaɪətroʊˈdʒɛnɪk/ | Do thầy thuốc gây ra |
| 442 | Thiazide diuretics | /ˈθaɪəzaɪd ˌdaɪjʊˈrɛtɪks/ | Thuốc lợi tiểu thiazide |
| 443 | Phosphate depletion | /ˈfɒsfeɪt dɪˈpliːʃən/ | Sự cạn kiệt phốt phát |
| 444 | Hyperphosphatasemia | /ˌhaɪpərˌfɒsfəˈteɪʒə/ | Tăng phosphatase máu |
| 445 | Intravenous alimentation | /ˌɪntrəˈviːnəs ˌælɪmɛnˈteɪʃən/ | Nuôi dưỡng qua đường tĩnh mạch |
| 446 | Fibroblast growth factor 23 (FGF23) | /ˈfaɪbroʊˌblæst ɡroʊθ ˈfæktər …/ | Yếu tố tăng trưởng nguyên bào sợi 23 (FGF23) |
| 447 | Breast milk fortified with phosphate | /brɛst mɪlk ˈfɔːrtɪfaɪd wɪθ ˈfɒsfeɪt/ | Sữa mẹ được tăng cường phốt phát |
| 448 | Extracorporeal membrane oxygenation | /ˌɛkstrəkɔːrˈpɔːriəl ˈmɛmbreɪn ˌɒksɪdʒəˈneɪʃən/ | Oxy hóa qua màng ngoài cơ thể (ECMO) |
| 449 | Subcutaneous fat necrosis | /ˌsʌbkjuːˈteɪniəs fæt nɛˈkroʊsɪs/ | Hoại tử mỡ dưới da |
| 450 | Violaceous nodules | /ˌvaɪəˈleɪʃəs ˈnɒdjuːlz/ | Các nốt sần màu tím |
| 451 | Adipocytes | /ˈædɪpoʊˌsaɪts/ | Tế bào mỡ |
| 452 | Lymphohistiocytic infiltrate | /ˌlɪmfoʊˌhɪstioʊˈsɪtɪk ˈɪnfɪlˌtreɪt/ | Thâm nhiễm lympho-mô bào |
| 453 | Multinucleated giant cells | /ˌmʌltiˈnuːkliˌeɪtɪd ˈdʒaɪənt sɛlz/ | Tế bào khổng lồ đa nhân |
| 454 | Prostaglandin E | /ˌprɒstəˈɡlændɪn iː/ | Prostaglandin E |
| 455 | Interleukins (IL) | /ˌɪntərˈluːkɪnz/ | Interleukin (IL) |
| 456 | Disaccharidases | /ˌdaɪˈsækəraɪdeɪsɪz/ | Disaccharidase |
| 457 | Familial (hereditary) hypocalciuric hypercalcemia (HHC) | /fəˈmɪliəl (həˈrɛdɪtəri) ˌhaɪpoʊˌkælsɪˈjʊərɪk ˌhaɪpərkælˈsiːmiə/ | Tăng canxi máu giảm canxi niệu có tính gia đình (HHC) |
| 458 | Dominant-negative effect | /ˈdɒmɪnənt ˈnɛɡətɪv ɪˈfɛkt/ | Tác dụng trội âm tính |
| 459 | Wild-type receptor | /waɪld taɪp rɪˈsɛptər/ | Thụ thể kiểu hoang dã |
| 460 | Dysrhythmia | /dɪsˈrɪðmiə/ | Loạn nhịp |
| 461 | Respiratory distress | /ˈrɛspərətɔːri dɪˈstrɛs/ | Suy hô hấp |