
Sperling Nội tiết học Nhi khoa, Ấn bản thứ 5 – Biên dịch: Ths.Bs. Lê Đình Sáng
Sperling Pediatric Endocrinology, Fifth Edition
Tác giả: Sperling, Mark A., MD – Nhà xuất bản: Elsevier Inc.
PHẦN III. NỘI TIẾT HỌC TRẺ EM VÀ THANH THIẾU NIÊN
Chương 24: Béo phì, Hội chứng Chuyển hóa và Các Rối loạn Cân bằng Năng lượng
Joan C. Han; Ram Weiss
Obesity, Metabolic Syndrome and Disorders of Energy Balance
Sperling Pediatric Endocrinology, 24, 939-1003
Giới thiệu
Tỷ lệ béo phì ở trẻ em trên toàn cầu đã tăng gấp tám lần trong 4 thập kỷ qua, với ước tính hiện tại là 124 triệu trẻ em từ 5 đến 19 tuổi bị béo phì trên toàn thế giới. Quan niệm rằng tế bào mỡ (adipocyte) hoạt động như một cơ quan nội tiết thay vì chỉ đơn thuần là nơi dự trữ lipid đã xuất hiện từ một phần tư thế kỷ trước, khi leptin được xác định là adipokine đầu tiên trong số nhiều loại do mô mỡ sản xuất. Do đó, với cơ sở sinh lý bệnh của béo phì là một dạng rối loạn nội tiết, và vì nhiều biến chứng liên quan đến béo phì có bản chất là một yếu tố nội tiết, các bác sĩ nội tiết nhi khoa ngày càng nhận được nhiều lượt giới thiệu để điều trị béo phì và các bệnh đi kèm. Đái tháo đường tuýp 2, từng hiếm gặp ở trẻ em và thường liên quan đến béo phì, hiện chiếm khoảng một phần ba tổng số ca chẩn đoán đái tháo đường mới ở trẻ em và tiếp tục tăng gần 5% tỷ lệ mắc hàng năm ở thanh thiếu niên.
Chương này cung cấp một khuôn khổ để hiểu về sự điều hòa cân bằng năng lượng và tóm tắt các phương pháp tiếp cận để đánh giá và quản lý bệnh nhân nhi bị béo phì. Ở khía cạnh ngược lại của cán cân năng lượng, các rối loạn do thiếu hụt năng lượng cũng được thảo luận trong chương này, và chúng cung cấp cái nhìn sâu sắc về vấn đề ngược lại của tình trạng thiếu hụt dinh dưỡng. Mặc dù sự thiếu hụt tương đối kiến thức hiện tại về cách cải thiện tốt nhất các yếu tố sinh học, hành vi và môi trường phức tạp góp phần gây ra mất cân bằng năng lượng đặt ra một thách thức điều trị, những tiến bộ gần đây trong các phương pháp tiếp cận “y học chính xác” mới nhắm vào các khiếm khuyết cụ thể trong cân bằng nội môi năng lượng đã mang lại những hiểu biết về các phương pháp điều trị tiềm năng hiệu quả cho cả béo phì và các rối loạn nhẹ cân.
Cân bằng năng lượng
Các thành phần chính của cân bằng năng lượng là lượng năng lượng nạp vào, tiêu hao năng lượng và dự trữ năng lượng. Các nguồn năng lượng nạp vào bao gồm carbohydrate (4 kCal/g), protein (~4 kCal/g), chất béo (9 kCal/g) và cồn (7 kCal/g), thường không đáng kể ở trẻ em nhưng có thể có ý nghĩa ở một số thanh thiếu niên. Tiêu hao năng lượng bao gồm tỷ lệ chuyển hóa cơ bản (BMR – năng lượng cần thiết để duy trì các chức năng của cơ thể, có tương quan cao với khối lượng cơ nạc và các cơ quan, và bị ảnh hưởng bởi các tình trạng bệnh lý khác nhau), hiệu ứng nhiệt của thực phẩm (năng lượng được sử dụng để tiêu hóa thức ăn, sinh nhiệt do chế độ ăn [DIT]), và hoạt động thể chất (sinh nhiệt do tập thể dục và sinh nhiệt do các hoạt động không phải tập thể dục [NEAT], bao gồm các hoạt động sinh hoạt hàng ngày, bồn chồn và duy trì tư thế). Dự trữ năng lượng, chủ yếu ở dạng mỡ, xảy ra khi lượng nạp vào vượt quá lượng tiêu hao. Sử dụng hình ảnh một bồn tắm (Hình 24.1) như một phép ẩn dụ cho cân bằng năng lượng với mực nước đại diện cho tổng dự trữ năng lượng, thể tích nước (tổng lượng mỡ cơ thể) được quyết định bởi việc thêm nước không liên tục từ vòi (các bữa ăn), việc múc nước ra từng đợt (hoạt động thể chất gắng sức), nước liên tục thoát ra từ lỗ thoát đáy (BMR), và nước thoát ra từ lỗ thoát tràn phía trên (NEAT và DIT) được đặt ở điểm thiết lập cho mực nước tối đa (điểm thiết lập cân bằng nội môi của cá nhân). Những thay đổi về tốc độ dòng chảy của vòi, kích thước của các xô nước, đường kính của các lỗ thoát nước, và chiều cao của điểm thiết lập tràn cùng nhau quyết định mực nước trong bồn tắm.
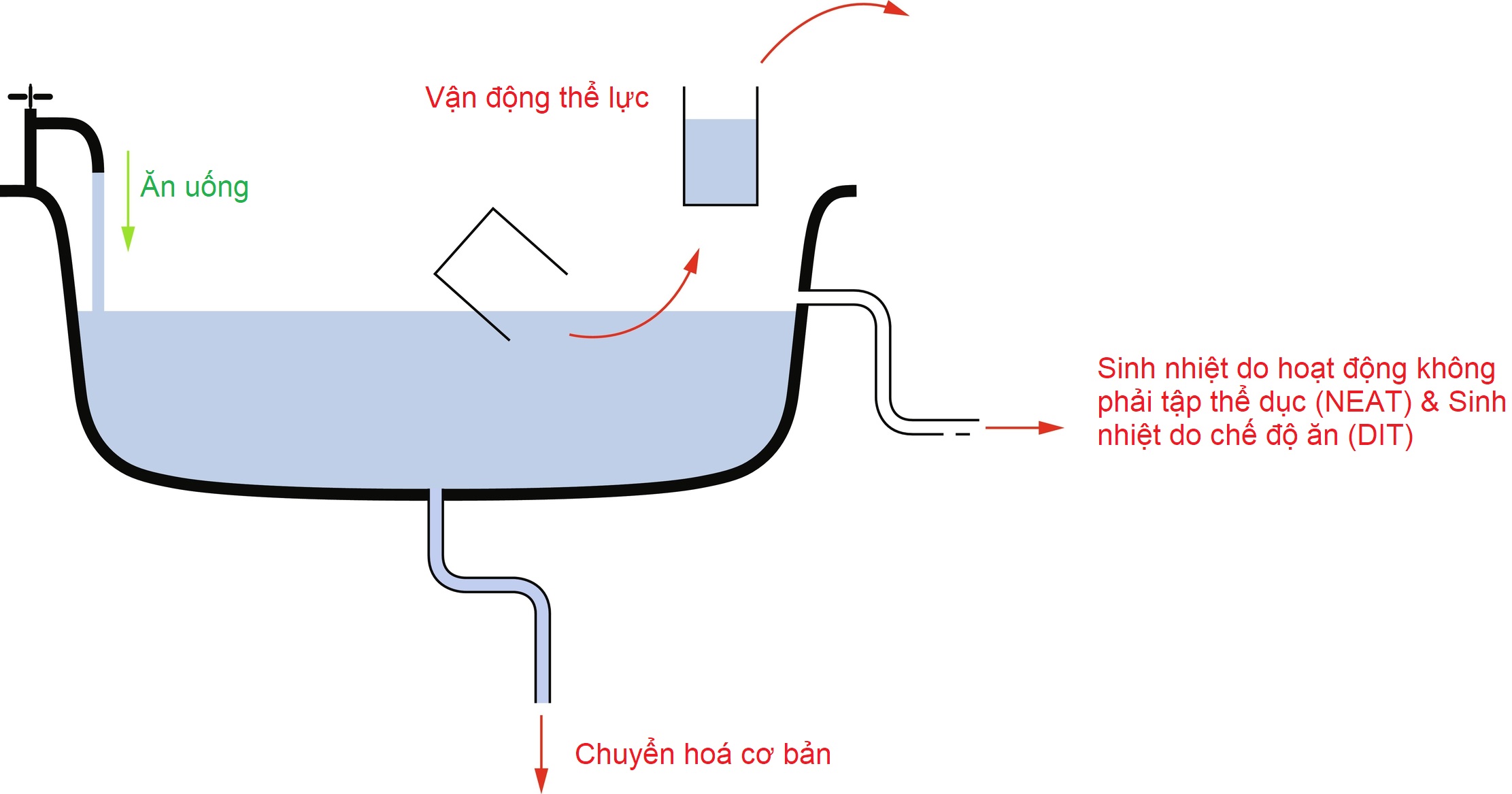
Hình 24.1 Các yếu tố quyết định tổng dự trữ năng lượng của cơ thể được biểu thị bằng nước trong bồn tắm.
Cân bằng năng lượng của con người được điều hòa bởi các cơ chế cân bằng nội môi và phi cân bằng nội môi kết nối phức tạp với nhau. Hệ thống cân bằng nội môi duy trì dự trữ mỡ cơ thể trong một phạm vi khá hẹp cho mỗi cá nhân, đáng chú ý là trong khoảng 10 kCal/ngày trung bình, sao cho ngay cả những xáo trộn nhỏ trong cân bằng cũng có thể dẫn đến tăng cân tích lũy theo thời gian. Hệ thống phi cân bằng nội môi trung gian cho các yếu tố môi trường và nhận thức thúc đẩy việc ăn uống, bao gồm các khía cạnh tưởng thưởng của việc ăn và các hành vi học được dẫn đến việc tiêu thụ thực phẩm vì những lý do khác ngoài việc đáp ứng nhu cầu dinh dưỡng. Mạng lưới thần kinh và các hormone thần kinh-nội tiết của các quá trình cân bằng nội môi và phi cân bằng nội môi tương tác và chồng chéo lên nhau để tạo thành một hệ thống phức tạp tích hợp các tín hiệu về thời gian, không gian và bối cảnh của tình trạng chuyển hóa và chức năng não bộ bậc cao. Bởi vì sự sống còn phụ thuộc vào nguồn cung cấp năng lượng đầy đủ, sự hiệu chỉnh tiến hóa của cân bằng năng lượng ưu tiên việc tiêu thụ thực phẩm và thúc đẩy dự trữ năng lượng dư thừa để bảo vệ chống lại nạn đói khi thực phẩm khan hiếm. Do đó, trong thời đại hiện đại của chúng ta, ở những nơi thực phẩm có sẵn dồi dào, đặc biệt là các loại thực phẩm ngon miệng, giàu năng lượng, các cơ chế ức chế điều hòa cảm giác thèm ăn có thể không đủ để ngăn chặn việc tiêu thụ quá mức.
Điều hòa việc ăn uống
Kiểu ăn uống của con người thường được đặc trưng bởi các đợt tiêu thụ thực phẩm rời rạc xen kẽ với các giai đoạn nhịn ăn. Diễn biến thời gian của mỗi bữa ăn tuân theo một chu kỳ, bao gồm các pha đầu, pha dạ dày, và pha ruột, theo sau là trạng thái sau hấp thu. Pha đầu bao gồm các phản ứng sinh lý đối với suy nghĩ, hình ảnh, mùi và vị của thức ăn để chuẩn bị cho việc ăn vào. Trong pha dạ dày, thức ăn được đưa vào dạ dày và quá trình tiêu hóa chính thức bắt đầu. Khi thức ăn rời khỏi dạ dày, pha ruột bắt đầu, nơi các enzyme tuyến tụy được tiết ra giúp tăng cường tiêu hóa và hấp thu chất dinh dưỡng. Cuối cùng, trong trạng thái sau hấp thu, việc hấp thu chất dinh dưỡng từ bữa ăn đã hoàn tất và nồng độ glucose trong tuần hoàn được duy trì bằng quá trình ly giải glycogen và tân tạo glucose cho đến khi bắt đầu bữa ăn tiếp theo.
Sự điều hòa cảm giác thèm ăn trong ngắn hạn quyết định cảm giác no (satiety – khoảng thời gian giữa các bữa ăn trước khi cảm giác đói thúc đẩy bắt đầu bữa ăn) và cảm giác thỏa mãn (satiation – cảm giác đã ăn đủ, dẫn đến kết thúc bữa ăn). Việc ăn uống được thúc đẩy bởi sự kết hợp của cảm giác đói, bối cảnh xã hội và các yếu tố đầu vào cảm quan. Cảm giác đói được trung gian bởi sự sụt giảm các tín hiệu gây chán ăn (ức chế cảm giác thèm ăn) trong trạng thái sau hấp thu sau bữa ăn trước, và bởi sự gia tăng của ghrelin, một peptide gây thèm ăn (kích thích cảm giác thèm ăn) được tiết ra bởi các tế bào nội tiết của dạ dày trong quá trình nhịn ăn. Việc ăn vào chất dinh dưỡng dẫn đến sự gia tăng các tín hiệu gây chán ăn, bao gồm sự căng giãn của thành dạ dày và sự tiếp xúc cơ học với thức ăn, sự tiết ra các peptide đường ruột (ví dụ: cholecystokinin [CCK], peptide YY, glucagon-like peptide 1 [GLP-1], v.v.), và sự đi vào của các chất dinh dưỡng đã tiêu hóa vào tuần hoàn. Dây thần kinh phế vị là kết nối thần kinh chính giữa đường tiêu hóa (GI) và hệ thần kinh trung ương (CNS). Đây là dây thần kinh sọ dài nhất và chứa cả các sợi cảm giác và vận động tham gia vào việc điều hòa cân bằng nội môi của hệ thần kinh đối giao cảm (“nghỉ ngơi và tiêu hóa”). Các sợi hướng tâm cảm giác của dây phế vị từ ruột kết thúc ở não sau, nơi có các kết nối đến các vùng vỏ não, não trước và não giữa tham gia vào việc điều hòa vận động ly tâm của dây phế vị đối với nhu động và các chức năng bài tiết của đường tiêu hóa để tiêu hóa. Điều quan trọng là, đường tiêu hóa cũng được chi phối bởi các dây thần kinh cột sống, truyền các tín hiệu cảm giác từ đường ruột đến các trung tâm cân bằng nội môi. Cảm giác thỏa mãn đạt được khi ghrelin giảm; sự căng dạ dày kích hoạt các sợi hướng tâm của dây phế vị gây chán ăn đến não sau; và sự gia tăng glucose, insulin và các peptide gây chán ăn dẫn đến làm chậm nhu động dạ dày và truyền tín hiệu cảm giác no đến CNS. Cảm giác no, quyết định khoảng thời gian cho đến bữa ăn tiếp theo, bị ảnh hưởng bởi số lượng và thành phần của bữa ăn trước đó và các yếu tố sinh lý bổ sung, chẳng hạn như hệ vi sinh vật đường ruột, các sản phẩm lên men và axit mật.
Các khía cạnh phi dinh dưỡng của việc điều hòa bữa ăn bao gồm độ ngon của thực phẩm và các yếu tố tâm thần kinh, chẳng hạn như sự tỉnh thức, căng thẳng, yêu cầu nhận thức và sự tỉnh táo. Cảm giác thèm ăn có liên quan đến các khía cạnh khoái lạc của việc ăn uống do sự kích hoạt của con đường tưởng thưởng vỏ não-vùng rìa (mesocorticolimbic reward pathway). Sự kích hoạt thụ thể opioid trung gian cho cảm giác tưởng thưởng của thức ăn và niềm vui khi ăn, trong khi sự kích hoạt hệ dopaminergic trung gian cho giá trị tưởng thưởng của thức ăn và động lực để có được thức ăn. Bối cảnh xã hội (các cuộc tụ họp và sự kiện nơi việc tiêu thụ thực phẩm được mong đợi) và các yếu tố đầu vào cảm quan (hình ảnh và mùi của thức ăn ngon miệng) có thể thúc đẩy việc ăn uống khi không có cảm giác đói, làm cho các yếu tố ngẫu nhiên của môi trường và việc kiểm soát kích thích trở nên quan trọng không kém các tín hiệu sinh lý trong việc quản lý béo phì.
Hệ thống cân bằng nội môi cho cân bằng năng lượng
Hệ thần kinh trung ương (CNS) là trung tâm kiểm soát cân bằng nội môi năng lượng. Các tín hiệu hướng tâm từ ngoại vi cung cấp thông tin về tình trạng năng lượng đến não, nơi xử lý các tín hiệu này và sau đó gửi các tín hiệu ly tâm để điều chỉnh lượng năng lượng nạp vào và tiêu hao. Hệ thống này được điều khiển bởi một số nhân vùng dưới đồi, nhận một loạt các tín hiệu đầu vào từ hormone, thần kinh, môi trường và vỏ não. Các tín hiệu này, dựa trên hướng của chúng, kích hoạt các con đường truyền tín hiệu gây chán ăn (anorexigenic) hoặc gây thèm ăn (orexigenic). Đầu ra của hệ thống được truyền qua hệ thần kinh tự chủ và được các cơ quan ngoại vi chuyển thành hành vi tìm kiếm thức ăn, bảo tồn/lãng phí năng lượng và các thích ứng sinh lý (một cái nhìn đơn giản hóa về các con đường này được trình bày trong Hình 24.2). Mục tiêu sinh lý của cân bằng nội môi năng lượng là duy trì mức độ ổn định của tổng lượng mỡ cơ thể hơn là trọng lượng tổng thể. Do đó, leptin, được tiết ra tương ứng với tổng khối lượng mỡ, hoạt động phù hợp như một tín hiệu tuần hoàn quan trọng để truyền đạt tình trạng mỡ đến CNS. Sự gắn kết của các hormone này với các thụ thể tương ứng của chúng trong nhân cung (ARC) của vùng dưới đồi làm tăng các tín hiệu gây chán ăn/dị hóa từ các tế bào thần kinh biểu hiện proopiomelanocortin (POMC)/cocaine-amphetamine–related transcript (CART) và làm giảm các tín hiệu gây thèm ăn/đồng hóa từ các tế bào thần kinh biểu hiện Agouti-related peptide (AgRP)/neuropeptide Y (NPY). Một sơ đồ về các con đường cân bằng nội môi năng lượng này được trình bày trong Hình 24.3, và các chi tiết hơn về từng hormone sẽ được mô tả sau.
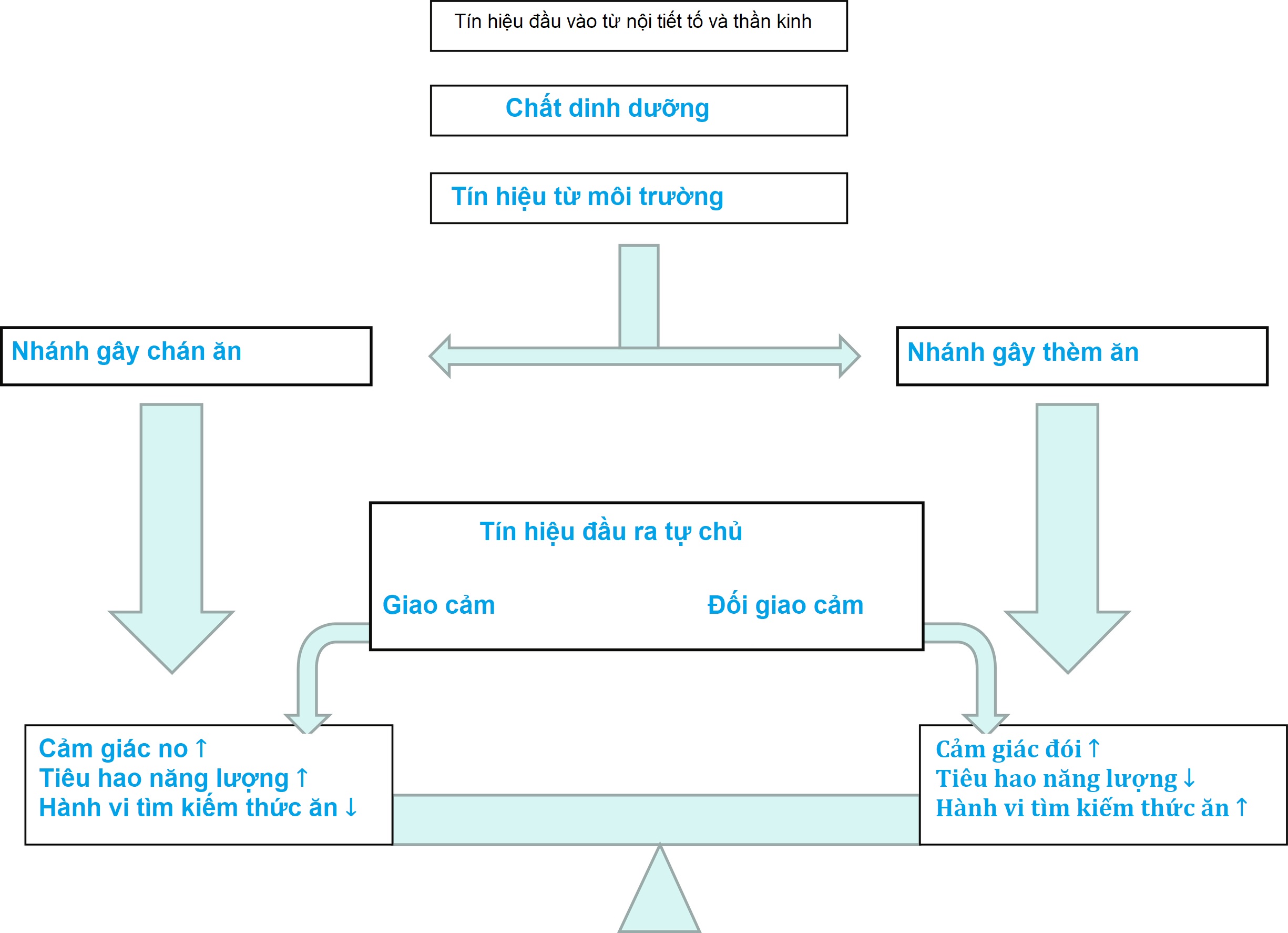
Hình 24.2 Cái nhìn đơn giản hóa về hệ thống năng lượng cân bằng nội môi và các đầu vào/đầu ra của nó. Các nhân vùng dưới đồi chi phối cân bằng nội môi. Các đầu vào của chúng bao gồm hormone (như leptin hoặc ghrelin), các tín hiệu thần kinh (trực tiếp từ đường tiêu hóa), chất dinh dưỡng và các tín hiệu môi trường. Sự tích hợp của các tín hiệu này dẫn đến một đầu ra tự chủ quyết định sự chiếm ưu thế của nhánh gây thèm ăn hoặc gây chán ăn.
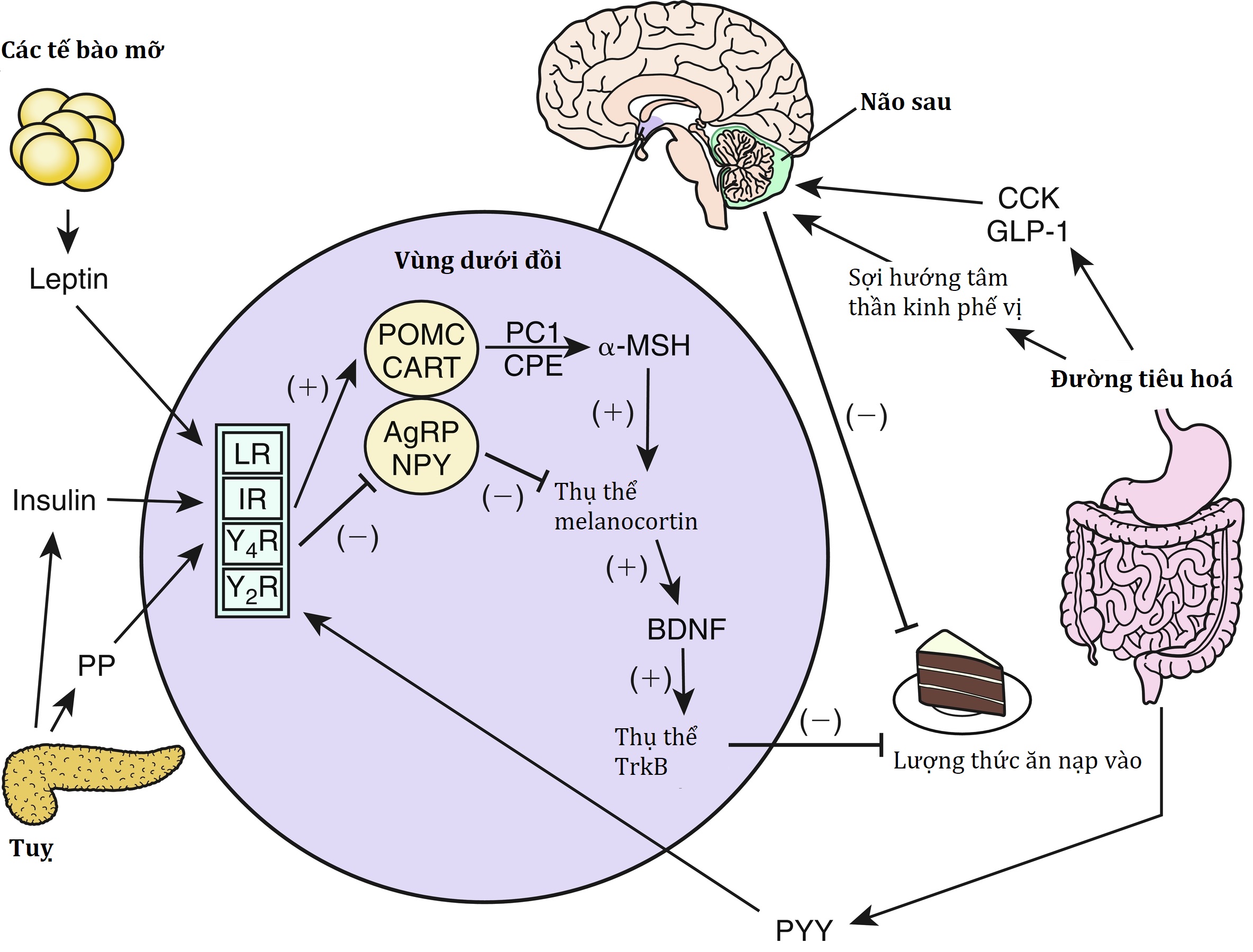
Hình 24.3 Sơ đồ các con đường cân bằng nội môi năng lượng. Các đường có mũi tên chỉ tác động kích thích. Các đường có vạch vuông góc ở cuối chỉ tác động ức chế. AgRP, peptide liên quan đến Agouti; BDNF, yếu tố dinh dưỡng thần kinh có nguồn gốc từ não; CART, transcript liên quan đến cocaine-amphetamine; CCK, cholecystokinin; CPE, carboxypeptidase E; GI, đường tiêu hóa; GLP-1, glucagon-like peptide 1; IR, thụ thể insulin; LR, thụ thể leptin; MSH, hormone kích thích tế bào hắc tố; NPY, neuropeptide Y; PC1, prohormone convertase 1; POMC, proopiomelanocortin; PP, polypeptide tuyến tụy; PYY, peptide YY; TrkB, tropomyosin-related kinase B; Y2R, thụ thể neuropeptide Y loại 2; Y4R, thụ thể neuropeptide Y loại 4.
Hệ thống hướng tâm
Việc theo dõi tình trạng chuyển hóa diễn ra ở nhiều vùng của cơ thể và đánh giá các dạng năng lượng tuần hoàn có sẵn để sử dụng ngay lập tức và dao động trong suốt chu kỳ bữa ăn, cũng như các dạng năng lượng dự trữ ổn định hơn, cụ thể là mỡ. Việc phát hiện trực tiếp các dạng năng lượng tuần hoàn sử dụng các thụ thể đặc hiệu cho vi chất dinh dưỡng hoặc các hệ thống phát hiện tế bào, chẳng hạn như dòng chảy đường phân tạo ra lượng adenosine triphosphate (ATP) khác nhau tùy thuộc vào sự sẵn có của glucose. Việc giám sát gián tiếp liên quan đến các hormone cận tiết và nội tiết phản ánh tình trạng năng lượng và cả các thụ thể căng giãn trong đường tiêu hóa phát hiện thể tích của vật chất trong ruột, dù có dinh dưỡng hay không, thay vì các vi chất dinh dưỡng cụ thể. Dòng máu và hệ thần kinh tự chủ, chủ yếu là dây thần kinh phế vị hướng tâm, là các tuyến đường chính để truyền thông tin chuyển hóa từ ngoại vi đến CNS. Vùng dưới đồi nền giữa (MBH, chứa nhân cung [ARC], nhân bụng giữa vùng dưới đồi [VMH], và lồi giữa [ME]) và phức hợp phế vị lưng (DVC, chứa nhân bó đơn độc [NTS], vùng postrema [AP], và nhân vận động lưng của dây thần kinh phế vị [DMV]) của hành não có một hàng rào máu não (BBB) dạng cửa sổ đặc biệt, cho phép các hormone và vi chất dinh dưỡng tuần hoàn khuếch tán vào các vùng não này để được phát hiện bởi các thụ thể và cơ chế cảm nhận đặc hiệu. DVC cũng nhận các sợi hướng tâm cảm giác từ dây thần kinh phế vị, chi phối đường tiêu hóa từ thực quản đến đại tràng và cung cấp thông tin từ các cảm biến hóa học và cơ học trong ruột đến CNS.
Điều hòa dương
Ngoài mạng lưới thần kinh nội tại mặc định ưu tiên cân bằng năng lượng dương, hiện tại, một tín hiệu điều hòa dương bổ sung từ ngoại vi được biết là có thể tăng cường xu hướng này.
Ghrelin. Ghrelin là một peptide 28 axit amin được octanoyl hóa, là một phối tử của thụ thể tiết hormone tăng trưởng (GHSR) và được dạ dày và tá tràng tiết ra một cách nội sinh trong trạng thái đói. Ghrelin lưu thông trong dòng máu đến CNS, nơi nó gắn vào các thụ thể GHSR ở vùng dưới đồi và thân não, tiếp cận qua các cửa sổ và vận chuyển chọn lọc qua BBB. Kích thích GHSR ở tuyến yên gây giải phóng hormone tăng trưởng (GH) trong khi kích thích GHSR ở VMH thúc đẩy cân bằng năng lượng dương thông qua việc tăng cảm giác đói, ăn vào, tích tụ mỡ và giảm tiêu hao năng lượng. Cho đến nay, ghrelin là hormone peptide gây thèm ăn duy nhất được biết đến sản xuất ở ngoại vi. Sự bài tiết ghrelin của dạ dày kết thúc khi chất dinh dưỡng đi vào dạ dày và ruột sau bữa ăn. Ghrelin tăng cùng với cảm giác đói tăng lên, và ghrelin đạt đỉnh khi cảm giác no kết thúc và việc ăn uống tự nguyện bắt đầu. Việc truyền ghrelin ngoại sinh gây ra việc ăn uống, hỗ trợ vai trò của ghrelin trong việc khởi xướng bữa ăn. Những người béo phì so với những người có cân nặng bình thường có nồng độ ghrelin huyết tương thấp hơn trên toàn cầu nhưng vẫn giữ được mô hình tăng ghrelin khi đói và giảm khi ăn theo nhịp sinh học tương tự, cho thấy ghrelin phản ứng với béo phì hơn là nguyên nhân gây ra nó. Giảm cân do hạn chế chế độ ăn dẫn đến tăng ghrelin trong khi phẫu thuật nối tắt dạ dày dẫn đến giảm ghrelin, điều này có thể góp phần vào sự khác biệt trong việc tăng cân trở lại sau khi giảm cân bằng các phương pháp khác nhau này. Điều quan trọng là, ghrelin được kích hoạt từ dạng tiền hormone của nó bằng cách gắn axit octanoic vào một gốc serine bởi ghrelin O-acetyltransferase (GOAT). Dạng không acyl hóa hoạt động như một tín hiệu cân bằng nội môi nhưng không gắn vào thụ thể GH.
Điều hòa âm
Cảm giác thỏa mãn, cảm giác no và việc tiêu hao năng lượng dư thừa được thực hiện bởi các yếu tố điều hòa âm phản ứng cấp tính với việc ăn vào và với các kho dự trữ năng lượng dài hạn.
Thụ thể căng giãn. Vật chất được ăn vào dạ dày tạo áp lực lên thành dạ dày mặc dù các thụ thể nhạy cảm với cơ học được hình thành bởi các sợi hướng tâm của dây phế vị có thân tế bào nằm ở hạch nốt và các đầu tận cùng nằm trong NTS của DVC ở thân não. Tín hiệu căng giãn làm chậm quá trình làm rỗng dạ dày, giúp giữ thức ăn trong dạ dày cho phép đạt được cảm giác thỏa mãn.
Glucose. Tiêu hóa carbohydrate dẫn đến giải phóng glucose và các loại đường khác, được hấp thu dễ dàng vào tĩnh mạch cửa gan và được tế bào gan tiếp nhận. Một phần đáng kể glucose đi vào cũng vào tuần hoàn hệ thống, và nồng độ glucose trong máu được phát hiện bởi các tế bào thần kinh cảm nhận glucose trong não có liên quan đến việc điều hòa cân bằng nội môi glucose, cũng như cân bằng năng lượng. Một tập hợp con các tế bào thần kinh được kích thích bởi glucose phản ứng với sự gia tăng glucose sau bữa ăn bằng cách kích thích tiêu hao năng lượng thông qua sinh nhiệt, trong khi một tập hợp con các tế bào thần kinh bị ức chế bởi glucose phản ứng với sự gia tăng glucose bằng cách giảm hoạt động của mạch thần kinh gây đói trong CNS.
Insulin và Amylin. Sự đi vào của glucose vào tuần hoàn từ sự hấp thu của bữa ăn kích hoạt sự giải phóng tương ứng của các hormone peptide insulin và amylin từ các tế bào β của tuyến tụy. Insulin tuần hoàn gây ra sự hấp thu và sử dụng glucose của tế bào và tổng hợp glycogen, và đi vào CNS thông qua vận chuyển tích cực qua BBB hoặc qua các cửa sổ trong BBB tại MBH và DVC để thúc đẩy cảm giác no và tăng tiêu hao năng lượng. Amylin tăng cường cảm giác thỏa mãn bằng cách làm chậm quá trình làm rỗng dạ dày và trung gian cảm giác no thông qua các tác động của nó tại AP của DVC.
Cholecystokinin. Khi thức ăn đi vào ruột non gần, các tế bào nội tiết I ở tá tràng và hỗng tràng gần phản ứng với các chất dinh dưỡng, mạnh nhất là với các axit béo tự do (FFA) chuỗi dài, bằng cách tiết ra CCK, một peptide 8 axit amin có tác dụng hormone cận tiết và nội tiết. Tại chỗ, CCK gắn vào các thụ thể ở môn vị để thúc đẩy làm chậm quá trình làm rỗng dạ dày, và kích thích các thụ thể hướng tâm của dây phế vị trong ruột gần truyền tín hiệu đến NTS và AP của DVC để gây ra cảm giác thỏa mãn. CCK cũng lưu thông trong dòng máu đến MBH và DVC có BBB dạng cửa sổ để ức chế cảm giác đói và thúc đẩy kết thúc bữa ăn.
Peptide YY, Glucagon-Like Peptide-1, và Oxyntomodulin. Khi thức ăn đi vào đường tiêu hóa dưới, các tế bào nội tiết L cư trú chủ yếu ở hồi tràng và đại tràng được kích hoạt và tiết ra các hormone peptide peptide YY (PYY), GLP-1, và oxyntomodulin vào tuần hoàn. Đoạn ruột tiết ra PYY, GLP-1, và oxyntomodulin vừa nằm ở hạ lưu vừa dài hơn đáng kể so với vùng tiết ra CCK, và do đó, việc thức ăn đi qua đoạn ruột sau này dẫn đến cảm giác no kéo dài giữa các bữa ăn.
PYY lưu thông trong dòng máu đến não dưới dạng các mảnh peptide, chủ yếu là PYY 3-36 34 axit amin, và gắn vào các thụ thể neuropeptide Y loại 2 (Y2Rs), chủ yếu ở ARC, nơi nó ức chế tín hiệu NPY gây thèm ăn và kích hoạt tín hiệu POMC gây chán ăn, cùng nhau gây ra cảm giác thỏa mãn và no. PYY cũng hoạt động theo cơ chế cận tiết bằng cách kích hoạt tại chỗ các tế bào thần kinh phế vị hướng tâm của ruột truyền tín hiệu gây chán ăn đến DVC.
GLP-1 được sản xuất bởi sự phân cắt protein qua trung gian prohormone convertase 1 (PC1) của preproglucagon trong các tế bào L, và tác động lên dạ dày để làm chậm quá trình làm rỗng dạ dày, góp phần vào cảm giác thỏa mãn thông qua việc giữ thức ăn. GLP-1 cũng lưu thông đến MBH và DVC, nơi nó kích hoạt thụ thể của mình để gây ra cảm giác thỏa mãn và no. Ngoài những tác động này lên cân bằng nội môi năng lượng, GLP-1 cũng hoạt động như một incretin (tăng cường bài tiết insulin phụ thuộc glucose) khi gắn vào các thụ thể GLP-1 biểu hiện ở các tế bào β của tuyến tụy, do đó tăng cường cân bằng năng lượng âm qua trung gian insulin.
Oxyntomodulin cũng được sản xuất thông qua sự phân cắt protein qua trung gian PC1 của preproglucagon như một sản phẩm riêng biệt với GLP-1. Oxyntomodulin kích hoạt thụ thể GLP-1, nhưng dường như hoạt động như một chất chủ vận thiên vị ưu tiên tham gia vào các con đường truyền tín hiệu nội bào khác với GLP-1, dẫn đến các tác động kéo dài đối với cảm giác thỏa mãn và no. Oxyntomodulin cũng kích hoạt thụ thể glucagon, kích thích tiêu hao năng lượng thông qua việc tăng sinh nhiệt.
Polypeptide tuyến tụy. Việc ăn vào thức ăn kích thích, thông qua CCK và các tín hiệu ly tâm của dây phế vị được gửi đến tuyến tụy, sự bài tiết polypeptide tuyến tụy (PP) từ các tế bào PP của tuyến tụy vào tuần hoàn. PP tuần hoàn làm chậm quá trình làm rỗng dạ dày và trung gian cảm giác no bằng cách kích hoạt các thụ thể NPY loại 4 (Y4Rs) ở vùng dưới đồi làm tăng hoạt động của tế bào thần kinh POMC gây chán ăn và giảm biểu hiện NPY gây thèm ăn. Nồng độ PP sau bữa ăn vẫn tăng cao trong vài giờ, có khả năng kéo dài đến pha sau hấp thu và trung gian cảm giác no.
Axit mật và Yếu tố tăng trưởng nguyên bào sợi 19. Axit mật được tổng hợp từ cholesterol trong gan và được tiết vào tá tràng sau bữa ăn, đóng vai trò là chất nhũ hóa lipid và các phân tử tín hiệu về tình trạng dinh dưỡng thông qua thụ thể kết hợp G-protein 19 (GPCR19) và thụ thể kích hoạt farnesoid X (FXR). GPCR19 và FXR được biểu hiện ở ruột non, gan và mô mỡ. Kích hoạt GPCR19 kích thích bài tiết GLP-1 và tăng nhu động đại tràng. Kích hoạt FXR gây ra bài tiết yếu tố tăng trưởng nguyên bào sợi 19 (FGF19) vào dòng máu. FGF19 điều hòa chuyển hóa lipid và glucose trong gan và cũng hoạt động trong CNS để giảm ăn và tăng tiêu hao năng lượng.
Leptin. Các tế bào mỡ tiết ra hormone peptide leptin 167 axit amin tương ứng với lượng mỡ cơ thể dự trữ. Thông thường, sự bài tiết này theo một mô hình nhịp sinh học với nồng độ cao hơn vào ban đêm khi ngủ. Tuy nhiên, khi pha sau hấp thu tiến triển thành nhịn ăn kéo dài và sau đó là đói, nồng độ leptin giảm nhanh chóng, dẫn đến hành vi tìm kiếm thức ăn mạnh mẽ. Hiệu ứng nổi bật này chỉ ra rằng sự sụt giảm cấp tính của leptin đóng vai trò như một tín hiệu đói và xác nhận vai trò chính của leptin là bảo vệ các kho dự trữ năng lượng hơn là ngăn ngừa béo phì. Điều quan trọng là, hiệu ứng cấp tính này đạt được bất kể nồng độ leptin cơ bản, do đó đói hoặc nhịn ăn kéo dài gây ra các thích ứng và hành vi chuyển hóa giống nhau ở những người có và không có béo phì. Sự bài tiết leptin được tăng cường bởi các dấu hiệu về sự sẵn có của chất dinh dưỡng, bao gồm glucose, insulin và cortisol, tất cả đều tăng khi ăn, và bị ức chế bởi các catecholamine được giải phóng khi kích hoạt hệ thần kinh giao cảm (SNS, phù hợp với phản ứng “chiến đấu hay bỏ chạy” đòi hỏi sự chuyển hướng chú ý khỏi các hành vi “nghỉ ngơi và tiêu hóa”). Leptin là một tín hiệu tiên quyết về dự trữ năng lượng đủ để cho phép bắt đầu các quá trình tiêu hao nhiều năng lượng, chẳng hạn như dậy thì và mang thai. Việc lập trình nồng độ leptin tương đối bằng lượng calo nạp vào sớm có thể là một cơ chế liên kết tình trạng dinh dưỡng thừa sớm với béo phì sau này.
Leptin tiếp cận CNS thông qua các cửa sổ BBB của MBH và DVC và cũng được vận chuyển tích cực qua BBB. Vị trí tác động chính của leptin là MBH nhưng nó cũng hoạt động ở các vùng khác trong CNS và ngoại vi. Các thụ thể leptin được biểu hiện bởi các tế bào mỡ trắng cho thấy sự tự điều hòa cận tiết, tế bào gan, tế bào tiểu đảo tụy bao gồm các tế bào β tiết insulin, các tế bào thần kinh trên khắp não bộ, và một phần tế bào nội mô mạch máu não tạo thành BBB. Thụ thể leptin (một thành viên của siêu họ thụ thể cytokine) có bốn đồng dạng, được hình thành bởi sự ghép nối khác nhau của RNA thông tin (mRNA): ObRa, một đồng dạng có vùng nội bào bị rút ngắn, có thể hoạt động như một chất vận chuyển; ObRb, thụ thể nguyên vẹn có chiều dài đầy đủ; ObRc, cũng có vùng nội bào ngắn; và ObRe, không có vùng nội bào, nhưng có thể hoạt động như một thụ thể hòa tan. Sự kích hoạt thụ thể leptin (LepR) dẫn đến ba tín hiệu thần kinh chính. Thứ nhất là việc mở một kênh kali nhạy cảm với ATP, làm tăng phân cực tế bào thần kinh và giảm tốc độ bắn của nó. Thứ hai là sự kích hoạt của Janus kinase 2 (JAK2) trong bào tương, phosphoryl hóa một gốc tyrosine trên các protein của một họ được gọi là bộ chuyển đổi tín hiệu và chất kích hoạt phiên mã (STAT-3). STAT-3 đã được phosphoryl hóa di chuyển đến nhân, nơi nó thúc đẩy phiên mã gen phụ thuộc leptin. Tuy nhiên, leptin cũng kích hoạt hệ thống truyền tin thứ hai của cơ chất thụ thể insulin 2/phosphatidyl inositol-3-kinase (IRS-2/PI3K), làm tăng sự dẫn truyền thần kinh của con đường truyền tín hiệu gây chán ăn trung ương.
Kích hoạt LepR ở các vùng dưới đồi và thân não ức chế hoạt động của các tế bào thần kinh gây thèm ăn và kích hoạt hoạt động gây chán ăn làm tăng tiêu hao năng lượng. Kích hoạt LepR làm tăng các đường ly tâm của SNS kết nối với các tế bào mỡ và có tác dụng làm giảm bài tiết leptin, cho thấy leptin, giống như các hormone cổ điển, có thể được tự điều hòa bởi một vòng phản hồi nội tiết. Leptin cũng dường như có vai trò trong việc điều hòa sinh nhiệt thông qua việc giảm khả năng chịu đựng nhiệt độ lạnh hơn của cơ chế điều nhiệt, về cơ bản là tăng bộ điều nhiệt của cơ thể lên một điểm đặt nhiệt độ cao hơn. Do đó, khi nồng độ leptin giảm trong quá trình nhịn ăn, nhiệt độ cơ thể không được bảo vệ mạnh mẽ và do đó tiêu hao năng lượng sinh nhiệt bị giảm để bảo tồn các kho dự trữ năng lượng.
Xử lý trung ương
Các tín hiệu hướng tâm ngoại vi được nêu ở trên đến CNS và tác động chủ yếu trong vùng dưới đồi và thân não, nơi chúng được tích hợp bởi một mạch thần kinh có cổng, được thiết kế để thúc đẩy các hiệu ứng dị hóa hoặc đồng hóa ròng (xem Hình 24.2).
Tín hiệu dị hóa trung ương
Tế bào thần kinh POMC/CART. Nhân cung chứa các tế bào thần kinh đồng biểu hiện POMC và CART. POMC là một peptide được phân cắt bởi PC1 và carboxypeptidase E (CPE) để tạo thành các peptide khác nhau tùy thuộc vào loại và vị trí của tế bào thần kinh. Các sản phẩm được phân cắt bao gồm β-endorphin, hormone vỏ thượng thận (ACTH), và, trong các tế bào thần kinh biểu hiện POMC ở nhân cung (ARC^POMC^), hormone kích thích tế bào hắc tố α (α-MSH). Cả việc ăn quá nhiều và truyền leptin ngoại vi đều gây ra sự tổng hợp POMC và α-MSH trong ARC. Tế bào thần kinh ARC^POMC^ cũng được kích hoạt trực tiếp bởi insulin, glucose và serotonin. Tế bào thần kinh ARC^POMC^ bị ức chế bởi ghrelin và các tế bào thần kinh AgRP. α-MSH gây chán ăn bằng cách gắn vào các thụ thể trong nhân cạnh não thất (PVN) và vùng dưới đồi bên (LHA). CART là một neuropeptide vùng dưới đồi được gây ra bởi leptin và giảm đi khi nhịn ăn. Truyền CART vào vùng dưới đồi ngăn chặn cảm giác thèm ăn, trong khi đối kháng CART nội sinh làm tăng lượng calo nạp vào.
Thụ thể Melanocortin. Các sản phẩm peptide được phân cắt của POMC gắn vào và kích hoạt các thụ thể melanocortin kết hợp G-protein 7-transmembrane (MCRs) khác nhau: MC1R ở da và tóc kích thích sản xuất sắc tố sẫm màu, melanin; MC2R ở tuyến thượng thận kích thích sản xuất glucocorticoid; và thụ thể melanocortin-4 (MC4R) và thụ thể melanocortin-3 (MC3R) ở CNS gây ra cân bằng năng lượng âm. Kích hoạt MC4R ở PVN và LHA dẫn đến trạng thái no, trong khi sử dụng các chất đối kháng MC4R vào não thất (ICV) ở loài gặm nhấm sẽ kích thích ăn. Chuột bị loại bỏ gen MC4R biểu hiện chứng ăn nhiều và béo phì nghiêm trọng. Chuột bị loại bỏ gen MC3R biểu hiện béo phì nhẹ hơn và không ăn nhiều nhưng dường như có hiệu quả ăn cao hơn và phân chia mỡ nhiều hơn.
Yếu tố dinh dưỡng thần kinh có nguồn gốc từ não. Yếu tố dinh dưỡng thần kinh có nguồn gốc từ não (BDNF) là một neurotrophin phụ thuộc vào hoạt động điều hòa tính dẻo của synap. BDNF còn được chứng minh là đóng một vai trò quan trọng trong cân bằng nội môi năng lượng như một chất trung gian hạ nguồn của con đường leptin-melanocortin. Biểu hiện BDNF ở VMH được điều hòa bởi tình trạng dinh dưỡng và tín hiệu MC4R. Xóa chọn lọc BDNF khỏi VMH và nhân dưới đồi lưng giữa (DMH) của chuột trưởng thành gây ra chứng ăn nhiều và béo phì, trong khi truyền BDNF vào VMH của chuột hoang dã (WT) làm giảm ăn và tăng tiêu hao năng lượng. Ở PVN trước, BDNF ức chế ăn và tăng hoạt động vận động, trong khi ở PVN giữa và sau, BDNF kích thích sinh nhiệt thông qua việc tăng dòng ra của SNS. Thiếu hụt một nửa gen BDNF ở cả người và loài gặm nhấm đều liên quan đến béo phì, mà ở chuột, có thể được ngăn ngừa bằng cách cho ăn theo cặp, cho thấy chứng ăn nhiều là nguyên nhân chính gây tăng cân. Rối loạn chức năng BDNF cũng có thể góp phần vào các hành vi ăn quá nhiều được tìm thấy trong hội chứng Prader-Willi (PWS) và hội chứng Smith-Magenis (SMS), cũng như trong các rối loạn ăn uống phổ biến. Đa hình BDNF Val66Met, làm suy giảm sự bài tiết BDNF phụ thuộc vào hoạt động, có liên quan đến các cơn ăn vô độ trong chứng cuồng ăn và rối loạn ăn uống vô độ, và sự tăng methyl hóa BDNF có liên quan đến chứng cuồng ăn. Biến thể intron BDNF rs12291063 (đồng hợp tử ở ~10% cá nhân gốc Phi) có liên quan đến việc giảm biểu hiện BDNF ở VMH và tăng tích tụ mỡ. Cùng nhau, những quan sát này chỉ ra rằng sự thiếu hụt BDNF có thể là nguyên nhân cơ bản của các nguyên nhân phổ biến cũng như hiếm gặp của hành vi ăn quá nhiều.
Thụ thể Tropomyosin-Related Kinase B. Tropomyosin-related kinase B (TrkB) được mã hóa bởi gen NTRK2 và là thụ thể tương ứng cho BDNF. Kích hoạt hóa học gen của các tế bào thần kinh biểu hiện TrkB ở DMH ức chế ăn trong chu kỳ tối khi chuột đói về mặt sinh lý, trong khi ức chế hóa học gen của các tế bào thần kinh này thúc đẩy ăn trong chu kỳ sáng khi chuột no về mặt sinh lý. Xóa chọn lọc Ntrk2 ở DMH của chuột trưởng thành gây ra chứng ăn nhiều, giảm tiêu hao năng lượng và béo phì.
Norepinephrine. Các tế bào thần kinh Norepinephrine (NE) trong locus coeruleus tạo synap với các tế bào thần kinh VMH để điều hòa việc ăn uống. Ở loài gặm nhấm, tác động của NE lên việc ăn uống dường như nghịch lý, vì truyền NE vào vùng dưới đồi kích thích ăn thông qua tác động lên các thụ thể α2- và β-adrenergic trung ương, trong khi truyền các chất chủ vận α1 trung ương làm giảm đáng kể việc ăn uống.
Trong các nghiên cứu trên người sử dụng chẩn đoán hình ảnh thần kinh phân tử, sự sẵn có của chất vận chuyển NE có liên quan tiêu cực với nhận thức về cảm giác đói và mối liên quan này mạnh hơn ở những người béo phì so với những người có cân nặng bình thường, cho thấy NE có vai trò trong việc điều chỉnh cảm giác đói.
Serotonin (5-hydroxytryptamine). 5-Hydroxytryptamine (5-HT) đã được liên quan đến nhận thức về cảm giác no dựa trên nhiều bằng chứng: (1) tiêm 5-HT vào vùng dưới đồi làm tăng cảm giác no, đặc biệt đối với carbohydrate; (2) sử dụng các chất chủ vận thụ thể 5-HT2c trung ương làm tăng cảm giác no, trong khi các chất đối kháng gây ra ăn uống; (3) sử dụng các chất ức chế tái hấp thu 5-HT chọn lọc gây ra cảm giác no sớm; (4) leptin làm tăng chu chuyển 5-HT; và (5) chuột bị loại bỏ gen 5-HT2c R biểu hiện tăng ăn và trọng lượng cơ thể. Vai trò của 5-HT trong việc truyền tín hiệu no có thể có cả thành phần trung ương và ngoại vi, vì 5-HT ở ruột được tiết vào dòng máu trong bữa ăn, nơi nó có thể có tác động đến chức năng thần kinh và trương lực cơ của đường tiêu hóa, và có thể gắn vào các thụ thể 5-HT trong NTS (đã thảo luận trước đó) để thúc đẩy cảm giác no. Chẩn đoán hình ảnh thần kinh phân tử ở người cho thấy béo phì được thúc đẩy bởi sự giảm phản hồi cân bằng nội môi qua trung gian serotonin để đáp ứng với việc ăn vào.
Tín hiệu đồng hóa trung ương
Tế bào thần kinh AgRP/NPY. NPY và AgRP cùng khu trú ở một tập hợp tế bào thần kinh khác trong ARC, ngay cạnh các tế bào biểu hiện POMC/CART. ARC là nơi chứa quần thể tế bào thần kinh duy nhất biểu hiện AgRP, và phần lớn các tế bào thần kinh này đồng biểu hiện NPY. Hai peptide gây thèm ăn này được tiết ra từ các đầu dây thần kinh dưới dạng chất dẫn truyền thần kinh peptide, ngoài chất dẫn truyền thần kinh phân tử nhỏ ức chế là axit gamma-amino butyric (GABA). AgRP là tương đồng người của protein agouti, có mặt nhiều trong chuột vàng Agouti (Ay-a). AgRP là một chất đối kháng cạnh tranh nội sinh của tất cả các thụ thể MCR, giải thích cho màu vàng ở những con chuột này do tác động ức chế của nó tại các thụ thể MCR1 của lông. AgRP cũng đối kháng MC4R và MC3R, do đó làm giảm khả năng của α-MSH trong việc ức chế cảm giác thèm ăn và tích tụ mỡ. Các tế bào thần kinh AgRP cũng ức chế quá trình “nâu hóa” mô mỡ trắng qua trung gian SNS, quá trình này nếu không sẽ làm tăng tiêu hao năng lượng, do đó làm giảm sinh nhiệt và thúc đẩy cân bằng năng lượng dương. Các tế bào thần kinh AgRP được kích hoạt trực tiếp bởi hormone gây thèm ăn ghrelin; dopamine, có thể bắt nguồn từ các tế bào thần kinh dopamine cư trú trong ARC; và một số sợi chiếu glutamatergic từ các nhân dưới đồi khác bao gồm DMH (nơi thể hiện sự vận chuyển và hoạt động của leptin qua BBB) và PVH. Các tế bào thần kinh AgRP bị ức chế bởi leptin, insulin, PYY, và bởi các sợi chiếu GABAergic từ DMH và các tế bào thần kinh cung khác.
NPY có nhiều chức năng trong vùng dưới đồi, bao gồm khởi xướng ăn, điều hòa bài tiết gonadotropin, và điều chỉnh đáp ứng của tuyến thượng thận. NPY hoạt động như một chất gây thèm ăn và nó cũng kích thích sự hình thành mỡ. Truyền NPY vào não thất (ICV) ở chuột nhanh chóng dẫn đến chứng ăn nhiều, dự trữ năng lượng và béo phì, qua trung gian của các thụ thể Y1 và Y5. Nhịn ăn và giảm cân làm tăng biểu hiện NPY trong ARC, giải thích cho việc tăng cảm giác đói, trong khi PYY 3-36 (thông qua các thụ thể Y2) và leptin làm giảm mRNA của NPY.
Hormone cô đặc Melanin. Hormone cô đặc Melanin (MCH) là một peptide gây thèm ăn gồm 17 axit amin được biểu hiện trong vùng zona incerta và LHA. Chuột bị loại bỏ gen MCH ăn ít và gầy, trong khi chuột biến đổi gen biểu hiện quá mức MCH phát triển béo phì và kháng insulin. Sử dụng MCH vào não thất (ICV) kích thích ăn, tương tự như khi sử dụng NPY. Điều thú vị là, sự lưu thông MCH nội sinh trong dịch não tủy (CSF) thực sự có thể đại diện cho một cơ chế giao tiếp thần kinh thay thế trong việc điều hòa việc ăn uống với sự gia tăng nồng độ MCH trong CSF thúc đẩy việc bắt đầu ăn.
Orexin A và B. Orexin A và B là các peptide gây thèm ăn (lần lượt dài 33 và 28 axit amin) được sản xuất trong vùng dưới đồi và điều chỉnh cả cân bằng năng lượng và chức năng tự chủ ở chuột. Orexin kích thích giải phóng NPY, hormone giải phóng corticotropin (CRH), và dòng ra của SNS dẫn đến tăng ăn, tỉnh táo, huyết áp và tiêu hao năng lượng. Orexin cũng dường như là cầu nối giữa các cơ chế cân bằng nội môi và phi cân bằng nội môi điều hòa việc ăn uống và có thể đóng một vai trò trong việc học tập và trí nhớ dựa trên phần thưởng. Các tế bào thần kinh orexin ở LHA xử lý các khía cạnh khoái lạc của thực phẩm và các loại thuốc gây nghiện, trong khi các tế bào thần kinh orexin ở vùng quanh vòm não và DMH điều hòa sự tỉnh táo và phản ứng căng thẳng.
Endocannabinoids. Tetrahydrocannabinol, thành phần hướng thần chính của cần sa, từ lâu đã được biết đến là có tác dụng kích thích ăn. Thụ thể endocannabinoid (EC) nội sinh, CB1, được biểu hiện trong các tế bào thần kinh hormone giải phóng corticotropin (CRH) ở PVN, trong các tế bào thần kinh CART ở VMN, và trong các tế bào thần kinh dương tính với MCH và orexin ở LHA và vùng quanh vòm não. Nhịn ăn và ăn uống có liên quan đến nồng độ EC cao và thấp trong vùng dưới đồi, tương ứng. Ví dụ, chuột bị loại bỏ gen thụ thể CB1 có biểu hiện CRH tăng và CART giảm. Ở chuột ob/ob thiếu leptin, nồng độ EC ở vùng dưới đồi tăng lên, trong khi leptin được truyền tĩnh mạch làm giảm các nồng độ này, cho thấy leptin có tác dụng kiểm soát âm trực tiếp lên hệ thống EC. Glucocorticoid làm tăng việc ăn uống bằng cách kích thích tổng hợp và bài tiết EC, trong khi leptin ngăn chặn tác dụng này. Ngoài ra, sự hiện diện của các thụ thể CB1 trên các tế bào thần kinh phế vị hướng tâm cho thấy EC có thể liên quan đến việc trung gian các tín hiệu no bắt nguồn từ ruột.
Hệ thống ly tâm
Các thụ thể MCR trong PVN và LHA truyền thông tin gây chán ăn và gây thèm ăn đến từ VMH, để điều chỉnh hoạt động của SNS, và dây phế vị ly tâm, vốn thúc đẩy dự trữ năng lượng (Hình 24.4). Bằng cách này, cân bằng năng lượng ngoại vi có thể được điều chỉnh một cách cấp tính để cung cấp năng lượng cần thiết cho các nhu cầu chuyển hóa, và dự trữ phần còn lại.
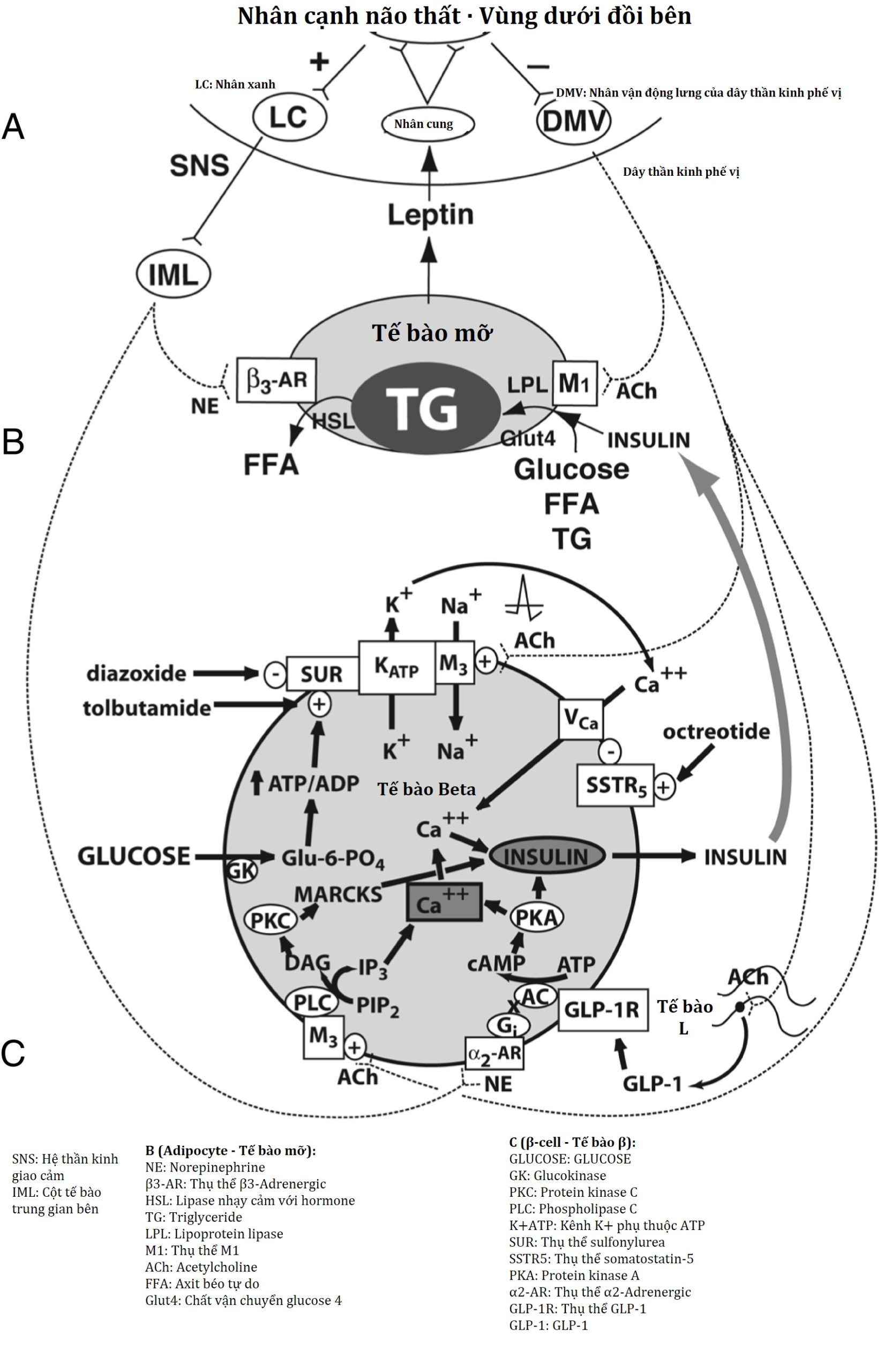
Hình 24.4 Sự điều hòa trung ương của tín hiệu leptin, sự chi phối tự chủ của tế bào mỡ và tế bào β, và phản ứng đói. (A) Nhân cung truyền tín hiệu leptin ngoại vi như là tín hiệu đủ hoặc thiếu. Trong tình trạng đủ leptin, các sợi ly tâm từ vùng dưới đồi tạo synap ở locus coeruleus, kích thích hệ thần kinh giao cảm. Trong tình trạng thiếu hoặc kháng leptin, các sợi ly tâm từ vùng dưới đồi kích thích nhân vận động lưng của dây phế vị. (B) Sự chi phối tự chủ và kích thích hormone của mô mỡ trắng. Trong tình trạng đủ leptin, norepinephrine gắn vào thụ thể β3-adrenergic, kích thích lipase nhạy cảm với hormone, thúc đẩy quá trình ly giải triglyceride dự trữ thành các axit béo tự do. Trong tình trạng thiếu hoặc kháng leptin, acetylcholine của dây phế vị làm tăng độ nhạy insulin của mô mỡ (chỉ được ghi nhận ở chuột cho đến nay), thúc đẩy sự hấp thu glucose và axit béo tự do để tạo mỡ, và thúc đẩy sự hấp thu triglyceride thông qua việc kích hoạt lipoprotein lipase. (C) Sự chi phối tự chủ và kích thích hormone của tế bào β. Glucose đi vào tế bào được chuyển đổi thành glucose-6-phosphate bởi enzyme glucokinase, tạo ra adenosine triphosphate (ATP), đóng một kênh kali phụ thuộc ATP, dẫn đến sự khử cực tế bào. Một kênh canxi phụ thuộc điện thế mở ra, cho phép dòng canxi vào nội bào, kích hoạt các cơ chế tiết thần kinh dẫn đến sự xuất bào của các túi insulin. Trong tình trạng đủ leptin, norepinephrine gắn vào các thụ thể α2-adrenergic trên màng tế bào β để kích thích các protein G ức chế, giảm adenyl cyclase và sản phẩm của nó là cyclic adenosine monophosphate (cAMP), và do đó làm giảm mức protein kinase A và giải phóng insulin. Trong tình trạng thiếu hoặc kháng leptin, dây phế vị kích thích bài tiết insulin thông qua ba cơ chế. Thứ nhất, acetylcholine gắn vào một thụ thể M3 muscarinic, mở một kênh natri, làm tăng sự khử cực tế bào phụ thuộc ATP, tăng dòng canxi vào và xuất bào insulin. Thứ hai, acetylcholine kích hoạt một con đường làm tăng protein kinase C, cũng thúc đẩy bài tiết insulin. Thứ ba, dây phế vị chi phối các tế bào L của ruột non, tiết ra glucagon-like peptide-1, kích hoạt protein kinase A, góp phần vào sự xuất bào insulin. Octreotide gắn vào một thụ thể somatostatin trên tế bào β, được kết hợp với kênh canxi phụ thuộc điện thế, hạn chế dòng canxi vào và lượng insulin được giải phóng để đáp ứng với glucose (in lại với sự cho phép của Springer Science and Business media). α2-AR, thụ thể α2-Adrenergic; β3-AR, thụ thể β3-adrenergic; AC, adenyl cyclase; ACh, acetylcholine; DAG, diacylglycerol; DMV, nhân vận động lưng của dây phế vị; FFA, axit béo tự do; Gi, protein G ức chế; GK, glucokinase; GLP-1, glucagon-like peptide-1; GLP-1R, thụ thể GLP-1; Glu-6-PO4, glucose-6-phosphate; GLUT4, chất vận chuyển glucose-4; HSL, lipase nhạy cảm với hormone; IML, cột tế bào trung gian bên; IP3, inositol triphosphate; LC, locus coeruleus; LHA, vùng dưới đồi bên; LPL, lipoprotein lipase; MARCKS, cơ chất protein kinase C giàu alanin được myristoyl hóa; NE, norepinephrine; PIP2, phosphatidylinositol pyrophosphate; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; PVN, nhân cạnh não thất; SUR, thụ thể sulfonylurea; SSTR5, thụ thể somatostatin-5; TG, triglyceride; VCa, kênh canxi phụ thuộc điện thế.
Hệ thần kinh giao cảm và tiêu hao năng lượng
Áp lực gây chán ăn làm tăng tiêu hao năng lượng thông qua việc kích hoạt SNS. Ví dụ, sử dụng leptin cho chuột ob/ob thiếu leptin thúc đẩy tăng ly giải mỡ ở mô mỡ nâu, sinh nhiệt, hoạt động mạch máu thận và tăng vận động, tất cả đều liên quan đến tăng tiêu hao năng lượng và tăng cường giảm cân. Tương tự, sử dụng insulin cấp tính làm tăng hoạt động SNS ở chuột bình thường và ở người. SNS làm tăng tiêu hao năng lượng theo bốn cách: (1) bằng cách chi phối vùng dưới đồi và các trung tâm thèm ăn ở hành não để giảm cảm giác thèm ăn, (2) bằng cách tăng bài tiết hormone kích thích tuyến giáp (TSH) để tăng giải phóng hormone tuyến giáp và tiêu hao năng lượng, (3) bằng cách chi phối cơ xương để tăng tiêu hao năng lượng, và (4) bằng cách chi phối các thụ thể β3-adrenergic trong mô mỡ trắng để thúc đẩy ly giải mỡ.
Kích hoạt SNS làm tăng tiêu hao năng lượng của cơ xương, bằng cách kích hoạt các thụ thể β2-adrenergic, từ đó làm tăng biểu hiện của nhiều gen trong cơ xương, đặc biệt là những gen liên quan đến chuyển hóa carbohydrate. Kích hoạt SNS kích thích ly giải glycogen, thúc đẩy tiêu hao năng lượng của cơ tim, tăng oxy hóa glucose và axit béo, và tăng tổng hợp protein.
Kích hoạt SNS ở loài gặm nhấm kích thích thụ thể β3-adrenergic của mô mỡ nâu để thúc đẩy ly giải mỡ. Ở người, việc kích hoạt thụ thể β3-adrenergic làm tăng cyclic adenosine monophosphate (cAMP), kích hoạt protein kinase A (PKA). PKA hoạt động trong hai con đường phân tử riêng biệt để tăng tiêu hao năng lượng. Đầu tiên, PKA phosphoryl hóa protein gắn yếu tố đáp ứng cAMP (CREB), gây ra biểu hiện của đồng kích hoạt-1α của thụ thể tăng sinh peroxisome (PPAR)γ (PGC-1α). PGC-1α sau đó gắn vào các yếu tố tăng cường trên gen protein không cặp đôi-1 (UCP1), làm tăng biểu hiện và hoạt động của các protein không cặp đôi (UCPs) 1 và 2. UCPs làm giảm gradient proton qua màng trong của ty thể, do đó chuyển hướng các proton từ việc lưu trữ dưới dạng ATP sang sản xuất nhiệt. Ban đầu, UCPs được phát hiện trong mô mỡ nâu và được cho là chịu trách nhiệm cho việc sinh nhiệt. UCP1 là một protein màng trong của ty thể làm mất cặp đôi sự đi vào của proton với quá trình tổng hợp ATP; do đó, biểu hiện UCP1 làm tiêu tán năng lượng dưới dạng nhiệt, do đó làm giảm hiệu suất năng lượng của mô mỡ. Tuy nhiên, UCP2 đã được tìm thấy trong hầu hết các mô và UCP3 trong cơ xương. Thứ hai, kích hoạt PKA kích hoạt enzyme lipase nhạy cảm với hormone (HSL), chịu trách nhiệm cho việc ly giải triglyceride nội bào thành các thành phần FFA của nó. Các FFA cũng gây ra UCP1, làm tăng thêm tiêu hao năng lượng. Các FFA được giải phóng từ tế bào mỡ cũng di chuyển đến gan, nơi chúng được sử dụng làm năng lượng bằng cách được chuyển hóa thành các mảnh hai carbon. Ly giải mỡ làm giảm biểu hiện leptin; do đó, một vòng phản hồi âm được hình thành giữa leptin và SNS (xem Hình 24.4).
Dây phế vị ly tâm và dự trữ năng lượng
Để đáp ứng với nồng độ leptin giảm hoặc áp lực gây thèm ăn kéo dài, LHA và PVN gửi các sợi chiếu ly tâm nằm trong bó dọc giữa đến DMV, kích hoạt dây phế vị ly tâm. Dây phế vị ly tâm đối kháng với SNS bằng cách thúc đẩy dự trữ năng lượng theo bốn cách: (1) bằng cách làm chậm nhịp tim, mức tiêu thụ oxy của cơ tim giảm; (2) dây thần kinh phế vị thúc đẩy nhu động đường tiêu hóa, mở môn vị và hấp thu chất nền năng lượng; (3) thông qua các tác động trực tiếp lên tế bào mỡ, dây thần kinh phế vị thúc đẩy độ nhạy insulin để tăng cường thanh thải chất nền năng lượng vào mô mỡ; và (4) thông qua các tác động lên tế bào β, dây phế vị làm tăng bài tiết insulin sau bữa ăn, thúc đẩy sự lắng đọng năng lượng vào mô mỡ.
Truy tìm ngược dòng của mô mỡ trắng cho thấy có nhiều sợi ly tâm bắt nguồn từ DMV. Các sợi ly tâm này tạo synap trên thụ thể muscarinic M1 trên tế bào mỡ, làm tăng độ nhạy insulin của tế bào mỡ. Việc cắt dây thần kinh của mô mỡ trắng dẫn đến giảm hấp thu glucose và FFA, và gây ra HSL, thúc đẩy ly giải mỡ—cả hai đều làm giảm hiệu quả của việc dự trữ năng lượng do insulin gây ra. Do đó, sự điều chỉnh của dây phế vị đối với tế bào mỡ làm tăng cường dự trữ cả glucose và FFA bằng cách cải thiện độ nhạy insulin của mỡ (xem Hình 24.4).
DMV cũng gửi các sợi chiếu ly tâm đến các tế bào β của tuyến tụy. Con đường này chịu trách nhiệm cho pha “đầu” hoặc pha trước hấp thu của bài tiết insulin, không phụ thuộc glucose và có thể bị chặn bởi atropine. Sự dẫn truyền thần kinh phế vị hoạt động quá mức làm tăng bài tiết insulin từ các tế bào β để đáp ứng với tải glucose đường uống thông qua ba cơ chế riêng biệt nhưng chồng chéo (xem Hình 24.4):
- Sự kích hoạt của dây phế vị làm tăng sự sẵn có của acetylcholine và sự gắn kết với thụ thể muscarinic M3 trên tế bào β, được kết hợp với một kênh natri trong màng tế bào β của tuyến tụy. Khi glucose đi vào tế bào β sau khi ăn, enzyme glucokinase phosphoryl hóa glucose để tạo thành glucose-6-phosphate, làm tăng ATP nội bào, gây ra sự đóng của kênh kali phụ thuộc ATP. Khi kênh đóng lại, tế bào β trải qua sự khử cực tế bào β phụ thuộc nồng độ ATP và sự mở của một kênh canxi phụ thuộc điện thế riêng biệt trong màng. Dòng canxi vào nội bào tăng lên một cách cấp tính, dẫn đến sự xuất bào nhanh chóng của các túi insulin. Sự mở đồng thời của kênh natri bởi acetylcholine qua trung gian dây phế vị làm tăng sự khử cực tế bào β, từ đó làm tăng dòng canxi vào nội bào và dẫn đến tăng tiết insulin.
- Acetylcholine qua trung gian dây phế vị làm tăng phospholipase A2, C và D trong tế bào β, thủy phân phosphatidylinositol nội bào thành diacylglycerol (DAG) và inositol triphosphate (IP3). DAG là một chất kích thích mạnh mẽ của protein kinase C (PKC), phosphoryl hóa cơ chất protein kinase C giàu alanin được myristoyl hóa (MARCKS), sau đó gắn vào actin và canxi-calmodulin và gây ra sự xuất bào của các túi insulin. IP3 làm tăng sự giải phóng canxi trong tế bào β từ các kho dự trữ nội bào, cũng thúc đẩy bài tiết insulin.
- Dây phế vị cũng kích thích giải phóng GLP-1 từ các tế bào L của ruột, lưu thông và gắn vào một thụ thể GLP-1 trong màng tế bào β. Sự kích hoạt của thụ thể này gây ra một adenyl cyclase nhạy cảm với canxi-calmodulin, với sự chuyển đổi ATP nội bào thành cAMP, sau đó kích hoạt PKA. PKA gây ra cả sự giải phóng canxi từ kho dự trữ nội bào và sự phosphoryl hóa các protein túi, mỗi yếu tố đều góp phần làm tăng sự xuất bào insulin.
Trong con đường ly tâm, insulin chịu trách nhiệm chuyển các chất dinh dưỡng trong máu vào mô mỡ để dự trữ. Thật vậy, tín hiệu hormone chính cho sự hình thành mỡ là insulin. Trong tế bào mỡ, insulin làm tăng: (1) biểu hiện chất vận chuyển glucose 4 (GLUT 4), (2) acetyl-CoA carboxylase, (3) fatty acid synthase, và (4) lipoprotein lipase. Do đó, tác dụng ròng của insulin lên tế bào mỡ là thanh thải và dự trữ nhanh chóng glucose và lipid tuần hoàn, do đó thúc đẩy dự trữ năng lượng.
Điều hòa phi cân bằng nội môi của cân bằng năng lượng
Các yếu tố quyết định phi cân bằng nội môi của cân bằng năng lượng là các chức năng bậc cao tích hợp hoạt động nhận thức và các tín hiệu môi trường thúc đẩy các lý do ăn uống phi dinh dưỡng. Việc ăn theo cân bằng nội môi là cần thiết cho sự sống còn, trong khi việc ăn theo khoái lạc được thúc đẩy bởi các khía cạnh tưởng thưởng của thực phẩm ngon miệng. Trái ngược với loài gặm nhấm hoặc các loài động vật có vú khác, các động lực khoái lạc ở người có thể lấn át các động lực cân bằng nội môi và chi phối hành vi ăn uống.
Nhân accumbens và con đường khoái lạc của phần thưởng thực phẩm
Con đường khoái lạc bao gồm vùng chéo bụng (VTA) và nhân accumbens (NA), với các đầu vào từ các thành phần khác nhau của hệ limbic, bao gồm thể vân, hạch hạnh nhân, vùng dưới đồi và hồi hải mã. Những con đường này cũng trung gian cho phản ứng khoái lạc với các loại thuốc gây nghiện, chẳng hạn như nicotine và morphine. Trên thực tế, việc sử dụng morphine cho NA làm tăng lượng thức ăn nạp vào theo cách phụ thuộc vào liều lượng. Khi hoạt động, con đường khoái lạc giúp hạn chế việc ăn uống trong các tình huống mà các kho dự trữ năng lượng đã đầy đủ; tuy nhiên, khi bị rối loạn chức năng, con đường này có thể làm tăng lượng thức ăn nạp vào dẫn đến béo phì.
VTA dường như trung gian cho việc ăn uống dựa trên độ ngon miệng hơn là nhu cầu năng lượng. Sợi chiếu dopaminergic từ VTA đến NA trung gian cho các đặc tính thúc đẩy, tưởng thưởng và củng cố của các kích thích khác nhau, chẳng hạn như thực phẩm và thuốc gây nghiện. Các thụ thể leptin và insulin được biểu hiện trong VTA, và cả hai hormone đều có liên quan đến việc điều chỉnh các phản ứng tưởng thưởng đối với thực phẩm và các kích thích thú vị khác. Ví dụ, nhịn ăn và hạn chế thực phẩm (khi nồng độ insulin và leptin thấp) làm tăng các đặc tính gây nghiện của các loại thuốc gây nghiện, trong khi leptin ICV có thể đảo ngược những tác động này. Trong các mô hình nghiện ở loài gặm nhấm, hành vi nghiện tăng lên (và phản ứng thú vị từ phần thưởng thực phẩm), được đo bằng sự giải phóng dopamine và tín hiệu thụ thể dopamine, lớn hơn sau khi thiếu ăn. Ở người bị thiếu hụt leptin, những thay đổi trong hoạt động ở NA có thể được nhìn thấy bằng cách sử dụng quét hình ảnh cộng hưởng từ chức năng (MRI), và những thay đổi này giảm đi khi sử dụng leptin ngoại sinh. Một cách cấp tính, insulin làm tăng biểu hiện và hoạt động của chất vận chuyển dopamine, giúp thanh thải và loại bỏ dopamine khỏi synap; do đó, việc tiếp xúc với insulin cấp tính làm giảm phần thưởng của thực phẩm. Hơn nữa, insulin dường như ức chế khả năng của các chất chủ vận VTA (ví dụ, opioid) trong việc tăng lượng sucrose nạp vào. Cuối cùng, insulin ngăn chặn khả năng của chuột trong việc hình thành một mối liên hệ ưu tiên vị trí có điều kiện với một loại thực phẩm ngon miệng. Tuy nhiên, sự kháng insulin của con đường này có thể dẫn đến tăng nhận thức về phần thưởng của thực phẩm thông qua việc giảm thanh thải dopamine khỏi synap và kéo dài phản ứng khoái lạc sau synap.
Một câu hỏi đã thu hút sự quan tâm ngày càng tăng là liệu bất kỳ chất dinh dưỡng đa lượng nào có đặc tính gây nghiện hay không. Trong các nghiên cứu trên động vật, đường đã được chứng minh là gây ra bốn tiêu chí cho nghiện: (1) ăn vô độ, (2) cai, (3) thèm, và (4) nhạy cảm chéo với các loại thuốc gây nghiện khác. Trong thức ăn nhanh, đường và caffeine đáp ứng các tiêu chí được trình bày trong ấn bản thứ năm của Sổ tay Chẩn đoán và Thống kê các Rối loạn Tâm thần (DSM-5) về sự phụ thuộc ở người. Tuy nhiên, câu hỏi liệu nghiện thực phẩm có tồn tại hay không, và liệu nó có thể giải thích cho những bệnh nhân béo phì hay không, vẫn còn gây tranh cãi, mặc dù một tổng quan hệ thống gần đây của y văn ủng hộ quan niệm về “nghiện thực phẩm có độ ngon cao” dựa trên các tiêu chí về suy giảm kiểm soát, suy giảm xã hội, sử dụng rủi ro, và dung nạp/cai, ít nhất là như một mô hình để xem xét trong các chiến lược điều trị.
Hạch hạnh nhân và phản ứng căng thẳng
VMH và VTA-NA trung gian cho cảm giác no khi các kho dự trữ năng lượng đã đầy đủ, nhưng chúng dường như dễ dàng bị lấn át bởi sự kích hoạt của hạch hạnh nhân và kết quả là căng thẳng, một trạng thái kháng insulin sinh lý (Hình 24.5). Nhiều bằng chứng cho thấy các glucocorticoid căng thẳng corticosterone (ở loài gặm nhấm) hoặc cortisol (ở người) là cần thiết cho sự biểu hiện đầy đủ của bệnh béo phì, điều này giúp giải thích vai trò gây rối của căng thẳng trong việc điều hòa cân nặng.
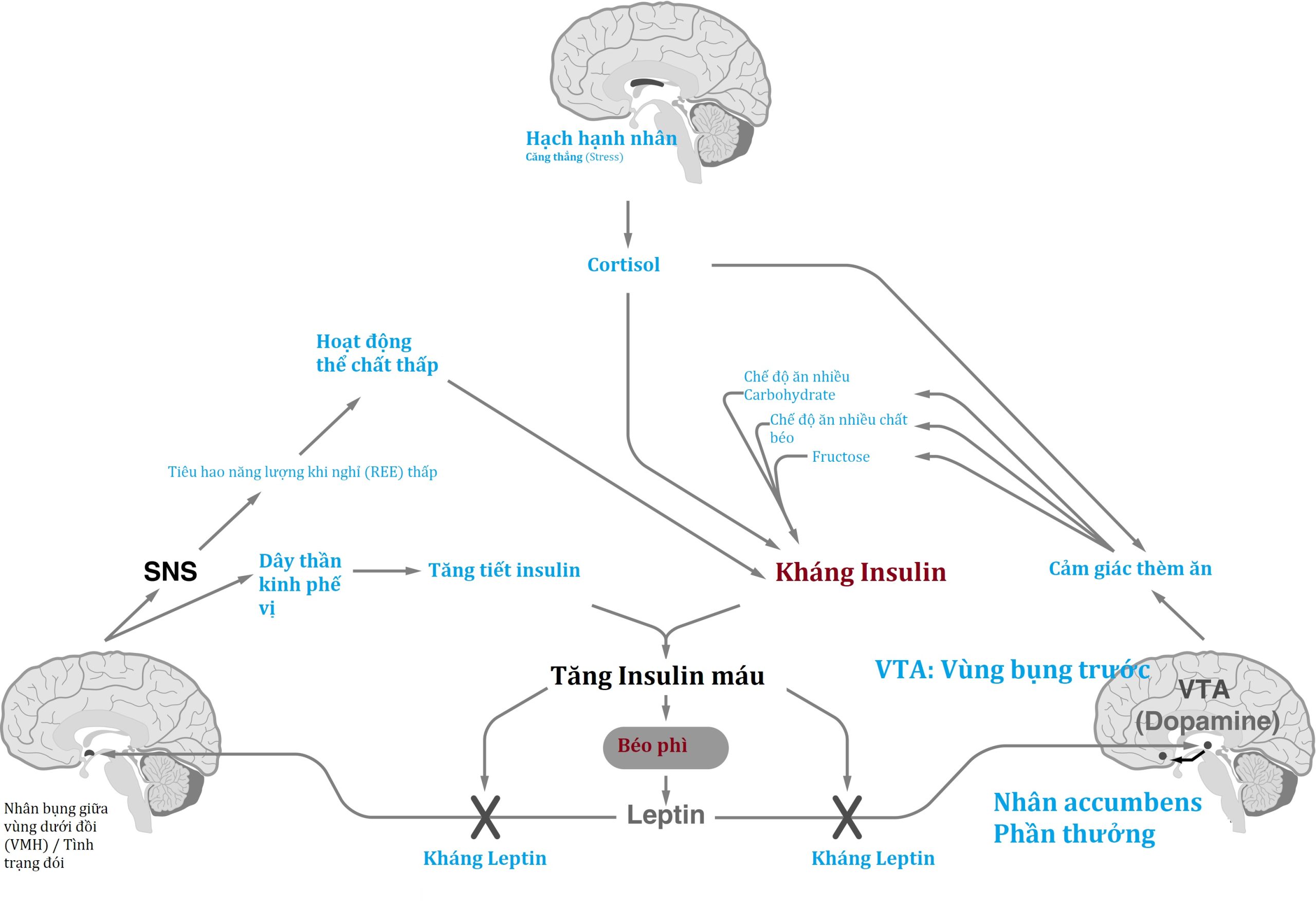
Hình 24.5 “Tam giác limbic (Hệ Viền).” Ba khu vực của CNS phối hợp để thúc đẩy việc ăn và giảm hoạt động thể chất, dẫn đến tăng cân kéo dài. Vùng dưới đồi bụng giữa (VMH) truyền tín hiệu leptin từ các tế bào mỡ để giảm lượng năng lượng nạp vào và tăng tiêu hao năng lượng; tuy nhiên, tăng insulin máu ngăn cản tín hiệu leptin, thúc đẩy “phản ứng đói.” Vùng chéo bụng (VTA) truyền tín hiệu leptin để giảm dẫn truyền thần kinh dopamine đến nhân accumbens, giảm ăn; tuy nhiên, tăng insulin máu cũng ngăn cản tín hiệu leptin ở đây, làm tăng dopamine và thúc đẩy “phần thưởng” của thực phẩm. Hạch hạnh nhân truyền nỗi sợ hãi và căng thẳng, dẫn đến tăng giải phóng cortisol từ vỏ thượng thận. Cortisol tăng cao cũng thúc đẩy việc ăn thực phẩm giàu năng lượng và thúc đẩy kháng insulin, can thiệp thêm vào tín hiệu leptin tại hai vị trí khác của hệ thần kinh trung ương. Do đó, sự kích hoạt của bất kỳ khía cạnh nào của tam giác limbic sẽ bật một vòng phản hồi dương, thúc đẩy tăng cân liên tục và béo phì.
Căng thẳng và glucocorticoid là một phần không thể thiếu trong việc thúc đẩy tích tụ mỡ và hội chứng chuyển hóa. Chuột bị cắt bỏ tuyến thượng thận được duy trì dược lý với nồng độ corticosterone cao cho thấy lượng mỡ ngoại sinh ăn vào tỷ lệ thuận với nồng độ corticosterone lưu thông, trong khi sự kích hoạt hạch hạnh nhân do căng thẳng bị giảm đi bởi việc ăn thực phẩm giàu năng lượng. Ở chuột nguyên vẹn, corticosterone kích thích ăn, đặc biệt là thực phẩm nhiều chất béo, và ở người, việc sử dụng cortisol làm tăng lượng thức ăn nạp vào. Nghiên cứu trên người cho thấy tăng lượng calo nạp vào từ “thực phẩm an ủi” (tức là những thực phẩm có mật độ năng lượng cao) sau căng thẳng cấp tính, và phản ứng căng thẳng góp phần vào tình trạng kháng leptin (sẽ được thảo luận sau). Một số nghiên cứu ở trẻ em đã quan sát thấy mối quan hệ giữa căng thẳng và các thói quen ăn uống không lành mạnh, bao gồm tăng ăn vặt, và nguy cơ cao gặp các vấn đề về cân nặng trong giai đoạn vị thành niên và trưởng thành. Trong một nghiên cứu có kiểm soát ở trẻ 9 tuổi có điểm số cao về hạn chế ăn kiêng và cảm thấy căng thẳng hơn bởi các thử thách trong phòng thí nghiệm có xu hướng ăn nhiều thực phẩm an ủi hơn. Những trải nghiệm bất lợi thời thơ ấu cũng liên quan đến sự phát triển sau này của bệnh béo phì và các yếu tố nguy cơ tim mạch chuyển hóa, cho thấy vai trò của căng thẳng trong việc tăng cân theo thời gian và sức khỏe chuyển hóa.
Rối loạn điều hòa cân bằng năng lượng
Kháng Leptin
Hầu hết trẻ em béo phì có nồng độ leptin cao nhưng không có đột biến thụ thể, biểu hiện tình trạng thường được gọi là kháng leptin chức năng. Tình trạng kháng leptin ngăn cản việc sử dụng leptin ngoại sinh để thúc đẩy giảm cân. Phản ứng với hầu hết các chế độ giảm cân nhanh chóng chững lại do sự sụt giảm nhanh chóng của nồng độ leptin ngoại vi, gây ra một “phản ứng đói” ngay lập tức, bất kể giá trị ban đầu, và có khả năng là do đạt đến một “ngưỡng leptin” cá nhân, mà có lẽ được xác định về mặt di truyền. Sự suy giảm leptin khiến nhân bụng giữa (VMH) cảm nhận được sự giảm sút dự trữ năng lượng ngoại vi, điều này điều chỉnh sự giảm tiêu hao năng lượng khi nghỉ (REE) để bảo tồn năng lượng, tương tự như một phản ứng đói, nhưng xảy ra ở nồng độ leptin tăng cao.
Nguyên nhân của kháng leptin vẫn chưa được biết rõ, nhưng nó có thể có nhiều nguyên nhân. Leptin đi qua hàng rào máu não (BBB) thông qua một chất vận chuyển có thể bão hòa, hạn chế lượng leptin đến được thụ thể của nó trong VMH; chất vận chuyển này hoạt động hiệu quả hơn ở nồng độ leptin thấp hơn, trong khi ngăn chặn sự gia tăng tín hiệu ở nồng độ cao hơn. Sự kích hoạt thụ thể leptin gây ra sự biểu hiện nội bào thần kinh của chất ức chế tín hiệu cytokine-3 (SOCS-3), hạn chế sự truyền tín hiệu leptin theo cơ chế tự điều hòa. Do sự hiện diện của tình trạng tăng leptin máu đã được chứng minh là một điều kiện tiên quyết cho sự phát triển của kháng leptin, người ta đã giả định rằng sự biểu hiện của các chất ức chế tín hiệu leptin do leptin gây ra có thể là một bước khởi đầu trong quá trình này. Các nghiên cứu khác cho thấy rằng chính bệnh béo phì gây ra viêm vùng dưới đồi, tăng sinh tế bào thần kinh đệm và stress lưới nội chất (ER), làm suy giảm khả năng đáp ứng với leptin.
Phương pháp tiêu chuẩn để gây kháng insulin và béo phì ở loài gặm nhấm là chế độ ăn nhiều chất béo. Chất béo trong chế độ ăn thúc đẩy kháng leptin thông qua tác động của nó lên tình trạng tăng triglyceride máu, làm hạn chế sự tiếp cận của leptin ngoại vi đến VMH, và cũng bằng cách can thiệp vào sự truyền tín hiệu leptin ở thượng nguồn của STAT-3, chất truyền tin thứ hai chính của nó. Một chất điều biến có khả năng của con đường này là enzyme PI3K, là chất tác động hạ nguồn của hoạt động insulin trong các tế bào thần kinh POMC và dường như giải thích cho các tác động của chất béo trong chế độ ăn lên kháng leptin và béo phì.
Hai mô hình lâm sàng đã được chứng minh là cải thiện độ nhạy leptin. Sau khi giảm cân thông qua hạn chế calo, việc sử dụng leptin ngoại sinh sau đó có thể làm tăng REE trở lại mức ban đầu và cho phép giảm cân thêm, cho thấy rằng bản thân việc giảm cân đã cải thiện độ nhạy leptin. Thứ hai, việc ức chế insulin tương quan với sự cải thiện độ nhạy leptin và thúc đẩy giảm cân, cho thấy rằng tình trạng tăng insulin máu thúc đẩy kháng leptin bằng cách can thiệp vào sự truyền tín hiệu leptin trong VMH và VTA. Thật vậy, các chiến lược giảm insulin có thể thúc đẩy giảm cân hiệu quả ở trẻ em bị tăng insulin máu bằng cách cải thiện độ nhạy leptin.
Các cơ chế điều hòa ngược chống lại việc giảm cân
Do hệ thống cân bằng nội môi cho cân bằng năng lượng được phát triển để bảo vệ chống lại nạn đói, các kho dự trữ mỡ cơ thể được bảo vệ một cách chặt chẽ. Do đó, việc hạn chế chế độ ăn, ngay cả trước khi bắt đầu giảm cân, cũng kích hoạt các cơ chế điều hòa ngược để chống lại mối đe dọa đói được nhận thấy, bất kể kho dự trữ mô mỡ ban đầu. Sự bài tiết ghrelin của dạ dày tăng lên một cách cấp tính, làm tăng giải phóng GH tuyến yên, để kích thích quá trình ly giải mỡ nhằm cung cấp chất nền năng lượng cho quá trình dị hóa. Ghrelin kích thích NPY/AgRP để đối kháng α-MSH/CART. Sự suy giảm leptin cũng làm giảm α-MSH/CART. Điều này dẫn đến giảm sự chiếm giữ của MC4R và MC3R, dẫn đến giảm tín hiệu gây chán ăn và dị hóa với hiệu quả tổng thể là tăng hành vi ăn uống và hiệu suất năng lượng cao hơn (với quá trình oxy hóa chất béo giảm). Cảm giác thèm ăn tăng lên tương ứng, dẫn đến việc tiêu thụ thực phẩm trên mức cơ bản khoảng 100 kCal/ngày cho mỗi kg trọng lượng bị mất. Trong khi đó, tổng tiêu hao năng lượng và tiêu hao năng lượng khi nghỉ ngơi giảm xuống trong nỗ lực bảo tồn năng lượng. Cụ thể, nồng độ UCP1 trong mô mỡ giảm do hoạt động của hệ thần kinh giao cảm (SNS) giảm. Mặc dù trương lực SNS tại tế bào mỡ giảm, rõ ràng có một quá trình ly giải mỡ bắt buộc (do ức chế insulin và tăng điều hòa HSL), cần thiết để duy trì việc cung cấp năng lượng cho hệ cơ và não dưới dạng các thể ketone có nguồn gốc từ gan. Ngoài ra, trong trạng thái giảm cân, trương lực phế vị tăng lên để làm chậm nhịp tim và mức tiêu thụ oxy của cơ tim, tăng bài tiết insulin của tế bào β để đáp ứng với glucose, và tăng độ nhạy insulin của mỡ—tất cả đều nhằm mục đích tăng cường dự trữ năng lượng. Các cơ chế điều hòa ngược này cùng nhau thúc đẩy việc tăng cân trở lại và thậm chí có thể tồn tại trong nhiều năm sau khi bắt đầu giảm cân, do đó khiến việc duy trì trọng lượng cơ thể đã giảm trở nên vô cùng khó khăn, đặc biệt là trong bối cảnh các cơ chế thúc đẩy tiến trình được mô tả sau này. Nói cách khác, các thích ứng chuyển hóa nhằm bảo tồn năng lượng và trở về trọng lượng cơ thể trước khi giảm cân được duy trì trong nhiều năm sau khi giảm cân và đạt được trạng thái cân nặng ổn định, khiến cá nhân dễ bị tăng cân. Khi so sánh hai cá nhân có cân nặng và thành phần cơ thể tương tự, một bệnh nhân có cân nặng ổn định và một bệnh nhân đã giảm cân để đạt được số đo này, để duy trì trọng lượng cơ thể hiện tại—bệnh nhân đã giảm cân sẽ phải tiêu thụ ít năng lượng hơn và chi tiêu nhiều năng lượng hơn so với người có cân nặng ổn định.
Cơ chế thúc đẩy tiến trình làm tăng cân
Tín hiệu thúc đẩy tiến trình (feed-forward) là cầu nối giữa các cơ chế cân bằng nội môi và khoái lạc trong việc điều hòa cảm giác thèm ăn, một khái niệm được phát triển dựa trên quan sát rằng một con chuột đói sẽ có hoạt động thần kinh AgRP tăng cao phù hợp trong trạng thái nhịn ăn nhưng sau đó các tế bào thần kinh AgRP này bị ức chế một cách cấp tính khi con chuột được cho ăn, ngay cả trước khi bắt đầu ăn. Ngoài ra, các tín hiệu từ ruột cũng được giải phóng để dự đoán sự xuất hiện sắp tới của thức ăn được ăn vào, khởi động các quá trình tiêu hóa trước khi thực sự ăn vào. Vì đói là một trải nghiệm khó chịu, sự giảm nhanh chóng hoạt động của AgRP và sự loại bỏ đột ngột cảm giác đói gây ra một trải nghiệm tưởng thưởng cấp tính, có lẽ đóng vai trò như một chất củng cố tích cực cho tín hiệu môi trường về sự sẵn có của thực phẩm. Ví dụ, bao bì hấp dẫn và dễ nhìn thấy của thực phẩm chế biến sẵn khuyến khích tiêu dùng theo khoái lạc và thiết lập một hành vi học được thúc đẩy sự trở lại sau đó đến môi trường nơi có sẵn các loại thực phẩm như vậy. Tương tự như vậy, các nhà hàng thức ăn nhanh và quảng cáo nhắm đến giới trẻ phụ thuộc nhiều vào việc kết nối hình ảnh thực phẩm với các kích thích thú vị khác (đồ chơi, trò chơi, niềm vui, v.v.), làm tăng thêm hiệu ứng tưởng thưởng của thực phẩm vốn đã rất ngon miệng. Cơ chế thúc đẩy tiến trình này có những ý nghĩa tiềm tàng khi chúng ta xem xét vai trò của môi trường xây dựng và vai trò của nó trong việc thúc đẩy tiêu dùng thực phẩm.
Năng lượng dư thừa—béo phì
Sự gia tăng tỷ lệ béo phì ở trẻ em và thanh thiếu niên là một trong những vấn đề sức khỏe cộng đồng đáng báo động nhất mà thế giới đang phải đối mặt hiện nay. Mặc dù sự gia tăng tỷ lệ béo phì ở trẻ em và thanh thiếu niên dường như đã chững lại ở một số nơi trên thế giới, nhiều nơi khác, đặc biệt là các nước đang phát triển và các quần thể di cư, vẫn đang trải qua sự gia tăng đều đặn. Béo phì liên quan đến các vấn đề sức khỏe đáng kể ở trẻ em và là một yếu tố nguy cơ sớm đối với phần lớn bệnh tật và tử vong do các bệnh không lây nhiễm ở người lớn, và là một yếu tố quan trọng làm tăng chi phí chăm sóc sức khỏe. Béo phì ở trẻ em có xu hướng kéo dài đến tuổi trưởng thành, và những người tiếp tục bị béo phì khi trưởng thành có nguy cơ đáng kể phát triển các bệnh lý do béo phì với nguy cơ tăng thêm liên quan đến thời gian tiếp xúc với béo phì. Ngược lại, những trẻ béo phì đã giảm cân và trở thành người lớn không béo phì không có nguy cơ gia tăng đối với các bệnh lý như vậy. Những quan sát này xác định béo phì ở thời thơ ấu là một cửa sổ cơ hội lớn cho các nỗ lực phòng chống béo phì với tác động tiềm năng suốt đời.
Định nghĩa
Định nghĩa lý thuyết của béo phì là một mức độ thừa cân về thể chất gây ra những hậu quả bất lợi cho sức khỏe. Dựa trên số liệu thống kê về bệnh tật và tử vong, và với mong muốn ngăn ngừa nguy cơ bệnh tật trong tương lai, chúng ta định nghĩa béo phì một cách thực tế là một mức độ thừa cân theo thống kê cho một quần thể, lưu ý rằng bệnh tật và tử vong thay đổi theo mức độ thừa cân ở các nhóm chủng tộc, dân tộc và kinh tế xã hội khác nhau.
Phần lớn các trường hợp béo phì ở tuổi trưởng thành có nguồn gốc từ thời thơ ấu, khiến béo phì trở thành một mối quan tâm của nhi khoa và việc phòng ngừa và điều trị béo phì trở thành một mục tiêu của nhi khoa. Chỉ số khối cơ thể (BMI) cũng là chỉ số được chấp nhận ở trẻ em. Ở trẻ em, việc so sánh BMI với các đường cong bình thường theo tuổi cho phép phân loại BMI trên bách phân vị thứ 85 là thừa cân, và trên bách phân vị thứ 95 là béo phì (Hình 24.6 A và B). Tổ chức Y tế Thế giới (WHO) phân loại thừa cân ở người lớn thành bốn phân nhóm dựa trên BMI (cân nặng [kg] ÷ chiều cao [m]²): BMI 25 đến 30 (thừa cân); BMI 30 đến 35, Độ 1 (béo phì vừa); BMI 35 đến 40, Độ 2 (béo phì nặng); và BMI trên 40, Độ 3 (béo phì bệnh lý). Một mức độ phân loại béo phì tương tự cũng có thể được sử dụng trong quần thể nhi khoa bằng cách sử dụng các bách phân vị BMI, trong đó bách phân vị thứ 95 theo tuổi và giới tính được sử dụng làm điểm tham chiếu và 100% đến 120% của bách phân vị thứ 95 theo tuổi và giới tính là độ 1, 120% đến 140% là độ 2, và trên 140% là độ 3. Sự phân loại như vậy cho thấy nguy cơ tim mạch chuyển hóa tăng lên cùng với mức độ béo phì tăng.
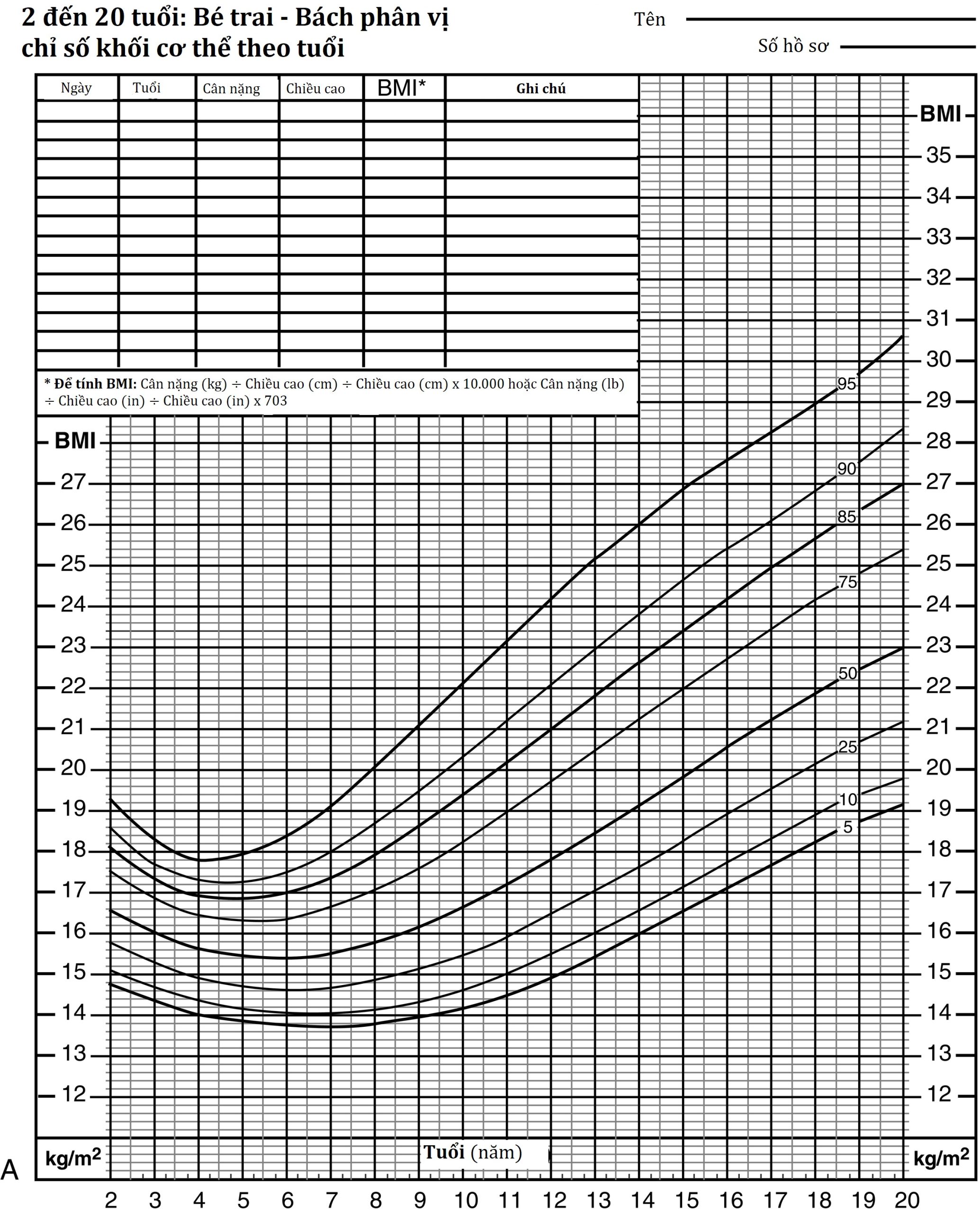
Hình 24.6 Bách phân vị chỉ số khối cơ thể (BMI) theo tuổi cho: (A) trẻ trai Hoa Kỳ, và (B) trẻ gái Hoa Kỳ, từ 2–20 tuổi. Lưu ý rằng bách phân vị thứ 95 biểu thị béo phì. Sự phục hồi mỡ (adiposity rebound) xảy ra vào khoảng 5 tuổi ở cả hai giới; sự phục hồi mỡ xảy ra càng sớm, khả năng suy ra một nguyên nhân hữu cơ cho việc tăng cân càng cao. Lấy từ http://www.cdc.gov/growthcharts.
Mặc dù BMI là chỉ số tiêu chuẩn của béo phì cho các mục đích thống kê và trong các quần thể, cần lưu ý rằng BMI không tính đến các thông số thành phần cơ thể như tổng lượng mỡ cơ thể (cũng như cả mỡ dưới da và mỡ nội tạng), cơ và xương. Hơn nữa, BMI ở trẻ em phụ thuộc vào tuổi, giới tính và tuổi dậy thì, do đó điểm z-score của BMI là một đánh giá chính xác hơn về tình trạng tích mỡ ở trẻ em. Cuối cùng, chu vi vòng eo (một thước đo gián tiếp của mỡ nội tạng trong ổ bụng) đã nổi lên như một chỉ số chính xác hơn về rối loạn chuyển hóa ở trẻ em. Những hạn chế này của BMI chỉ ra rằng nó là một chỉ số hữu ích cho các nghiên cứu quần thể và dịch tễ học nhưng nên được sử dụng một cách thận trọng khi đánh giá một đứa trẻ riêng lẻ trong môi trường lâm sàng.
Tỷ lệ mắc và dịch tễ học
Tỷ lệ béo phì ở trẻ em ở Hoa Kỳ đã tăng lên đáng kể trong 30 năm qua, và vẫn tiếp tục như vậy, mặc dù việc so sánh dữ liệu dọc và ngang rất khó khăn do các định nghĩa và thông số đo lường khác nhau giữa các nghiên cứu dịch tễ học. Các ước tính gần đây nhất về tỷ lệ béo phì và xu hướng ở Hoa Kỳ dựa trên dữ liệu từ Khảo sát Kiểm tra Sức khỏe và Dinh dưỡng Quốc gia 2011 đến 2014 (NHANES V). NHANES cho thấy dịch bệnh béo phì ở trẻ em ở Hoa Kỳ dường như đã ổn định ở một số nhóm tuổi nhưng không ở những nhóm khác. Nhìn chung, trong năm 2013 đến 2014, 9,4% (khoảng tin cậy 95% [CI], 6,8–12,6) trẻ sơ sinh và trẻ mới biết đi và 17% (95% CI, 15,5–18,6) trẻ em và thanh thiếu niên từ 2 đến 19 tuổi bị béo phì. Đáng chú ý, tỷ lệ béo phì cực độ (>120% của bách phân vị thứ 95 theo tuổi và giới tính) là 5,8% (95% CI, 4,9–6,8). Phân tích xu hướng trong khoảng thời gian 25 năm cho thấy sự gia tăng đáng kể về tỷ lệ béo phì ở trẻ em từ 2 đến 5 tuổi trong khoảng thời gian từ 1988 đến 1994 và 2003 đến 2004, sau đó giảm nhẹ trong năm 2013 đến 2014 (lần lượt là 7,2%, 13,9% và 9,4%). Ở trẻ em từ 6 đến 11 tuổi, tỷ lệ béo phì tăng từ năm 1988 đến 1994 đến năm 2007 đến 2008 và duy trì ổn định trong năm 2013 đến 2014 (lần lượt là 11,3%, 19,6% và 17,4%). Ở thanh thiếu niên từ 12 đến 19 tuổi, tỷ lệ béo phì tăng đáng kể từ năm 1988 đến 1994 đến năm 2013 đến 2014 (lần lượt là 10,5% và 20,6%, P < 0,001). Đáng chú ý, tỷ lệ béo phì cực độ tăng ở trẻ em từ 6 đến 11 tuổi trong khoảng thời gian từ 1988 đến 1994 đến năm 2013 và 2014 (lần lượt là 3,6% và 4,3%, P = 0,02) và ở thanh thiếu niên từ 12 đến 19 tuổi (lần lượt là 2,6% và 9,1%, P < 0,001). Quan trọng là, không có xu hướng đáng kể nào được quan sát thấy trong khoảng thời gian từ 2005 đến 2006 và 2013 đến 2014. Ý nghĩa thực tế của các xu hướng này là, ví dụ, trong giai đoạn 1988 đến 1994, bách phân vị thứ 95 của BMI ở nam giới 17 tuổi là 31,5 kg/m² (tức là 5% nam giới có BMI > 31,5), và trong giai đoạn 2011 đến 2014, bách phân vị thứ 95 là 36,2 kg/m² (tức là 5% nam giới có BMI > 36,2). Do đó, trong khoảng thời gian từ 1988 đến 1994 và 2013 đến 2014, tỷ lệ béo phì tăng cho đến năm 2003 đến 2004 và sau đó giảm ở trẻ em từ 2 đến 5 tuổi, tăng cho đến năm 2007 đến 2008 và sau đó chững lại ở trẻ em từ 6 đến 11 tuổi, và tăng ở thanh thiếu niên từ 12 đến 19 tuổi. Trong năm 2013 đến 2014, 17,4% trẻ em đáp ứng tiêu chí béo phì loại I, bao gồm 6,3% cho loại II và 2,4% cho loại III. Một sự gia tăng rõ ràng, có ý nghĩa thống kê ở tất cả các loại béo phì tiếp tục từ năm 1999 đến năm 2014. Ở Hoa Kỳ, béo phì và béo phì nặng ở trẻ em tăng đáng kể với độ tuổi và trình độ học vấn của chủ hộ thấp hơn, và béo phì nặng tăng với mức độ đô thị hóa thấp hơn. So với thanh thiếu niên da trắng không phải gốc Tây Ban Nha, tỷ lệ béo phì và béo phì nặng cao hơn đáng kể ở thanh thiếu niên da đen không phải gốc Tây Ban Nha và thanh thiếu niên gốc Tây Ban Nha. Béo phì nặng, nhưng không phải béo phì, thấp hơn đáng kể ở thanh thiếu niên gốc Á không phải gốc Tây Ban Nha so với thanh thiếu niên da trắng không phải gốc Tây Ban Nha. Cuối cùng, các dự báo cho rằng đến năm 2030, 42% người trưởng thành ở Mỹ sẽ bị béo phì.
Tỷ lệ mắc toàn cầu
Béo phì đã vượt qua hội chứng suy giảm miễn dịch mắc phải và suy dinh dưỡng để trở thành vấn đề sức khỏe cộng đồng số một trên thế giới. Tỷ lệ béo phì ở trẻ em trên toàn cầu đã tăng lên với tốc độ đáng báo động trong 20 năm qua. Tỷ lệ này đã tăng từ 2,7 đến 3,8 lần trong 29 năm ở Hoa Kỳ, 2,0 đến 2,8 lần trong 10 năm ở Anh, 3,4 đến 4,6 lần trong 10 năm ở Úc, và 3,4 đến 3,6 lần trong 23 năm ở Brazil. Dữ liệu châu Âu, sử dụng các định nghĩa ngưỡng béo phì hơi khác một chút (BMI thời thơ ấu tương ứng với > 25 và 30 kg/m² ở người lớn biểu thị thừa cân và béo phì, tương ứng), cho thấy rằng trong số các nước châu Âu, tỷ lệ thừa cân/béo phì kết hợp dao động trong khoảng 16% đến 22% trong khi tỷ lệ béo phì dao động trong khoảng 4% đến 6% (tương ứng với 2,9–4,4 triệu trẻ em béo phì ở lục địa châu Âu). Sự gia tăng nhanh chóng về tỷ lệ trẻ em đi học thừa cân đang được thấy ở tất cả các nước châu Âu có dữ liệu. Các con số chỉ ra sự chậm trễ từ 10 đến 15 năm so với Hoa Kỳ. Sử dụng dữ liệu từ giữa những năm 70 đến 2016 ở 200 quốc gia, người ta đã chỉ ra rằng xu hướng BMI trung bình gần đây đã chững lại ở tây bắc châu Âu và các khu vực nói tiếng Anh và châu Á-Thái Bình Dương có thu nhập cao cho cả hai giới, tây nam châu Âu cho nam giới, và trung tâm và Andean Mỹ Latinh cho nữ giới. Ngược lại, sự gia tăng BMI đã tăng tốc ở đông và nam Á cho cả hai giới, và đông nam Á cho nam giới. Ở các nước phát triển, người nghèo ở thành thị dễ bị béo phì hơn, có lẽ do thói quen ăn uống kém và cơ hội hoạt động thể chất hạn chế. Ngược lại, béo phì thường xuyên hơn ở tầng lớp kinh tế xã hội cao hơn của các nước đang phát triển, có lẽ do sự chuyển đổi dinh dưỡng sang chế độ ăn phương Tây hơn với nhiều mặt hàng giàu năng lượng hơn bao gồm chất béo và đường cao hơn, có xu hướng ngon miệng hơn với chi phí thấp hơn. Điều này cũng có thể do các đặc tính cụ thể của thực phẩm chế biến sẵn, có thể thúc đẩy kháng leptin.
Các yếu tố về chủng tộc và dân tộc
Các cuộc khảo sát NHANES chỉ liệt kê tỷ lệ mắc ở người da trắng không phải gốc Tây Ban Nha, người da đen không phải gốc Tây Ban Nha và người châu Á, mặc dù thực tế là người Mỹ bản địa, người dân các đảo Thái Bình Dương và các nhóm chủng tộc/dân tộc khác cũng đang trải qua sự gia tăng nhanh chóng về tỷ lệ béo phì. Giữa các nhóm chủng tộc, có một sự phân đôi rõ rệt về tỷ lệ mắc và tốc độ gia tăng của béo phì ở trẻ em. Ví dụ, tỷ lệ mắc ở thanh thiếu niên người Mỹ gốc Phi (24,4%), gốc Tây Ban Nha (21,7%) và người Mỹ gốc Mexico (22,2%) cao hơn đáng kể so với thanh thiếu niên da trắng (15,6%). Quan trọng là, tỷ lệ béo phì nặng (BMI > bách phân vị thứ 97) ở thanh thiếu niên người Mỹ gốc Phi (18,5%), gốc Tây Ban Nha (15,2%) và người Mỹ gốc Mexico (15,2%) vượt xa tỷ lệ của thanh thiếu niên da trắng không phải gốc Tây Ban Nha (10,5%). Tốc độ gia tăng tỷ lệ béo phì ở thanh thiếu niên người Mỹ gốc Phi và gốc Tây Ban Nha gần như tăng gấp đôi trong khoảng thời gian từ 1988 đến 1994 và 1999 đến 2000, từ 13,4% lên 23,6% ở người Mỹ gốc Phi, và từ 13,8% lên 23,4% ở người gốc Tây Ban Nha. Hệ thống Giám sát Dinh dưỡng Nhi khoa (PedNSS) năm 1994 chỉ ra rằng 12% trẻ em người Mỹ bản địa từ 2 đến 4 tuổi bị thừa cân, tương tự như trẻ em gốc Tây Ban Nha ở cùng độ tuổi (12%) nhưng cao hơn nhiều so với trẻ em da trắng (6%). Tỷ lệ thừa cân ở độ tuổi 5 đến 6 ở người Mỹ bản địa cao gấp đôi so với thanh thiếu niên Hoa Kỳ nói chung, và tỷ lệ béo phì thậm chí còn cao hơn 3 lần. Nhìn chung, cả trẻ em và thanh thiếu niên người Mỹ da đỏ và người bản địa Alaska đều có tỷ lệ béo phì cao hơn so với trẻ em Hoa Kỳ nói chung. Ở trẻ sơ sinh và trẻ mới biết đi dưới 2 tuổi, tỷ lệ béo phì cao nhất ở người Mỹ gốc Phi (18,5%), so với 10,1% ở người da trắng không phải gốc Tây Ban Nha và 13,7% ở người gốc Tây Ban Nha. Có thể các thói quen ăn uống khác nhau có thể giải thích cho một số khác biệt này. Ví dụ, một nghiên cứu về trẻ em Latino 2 tuổi ở California đã cho thấy mối tương quan giữa béo phì và việc tiêu thụ sớm đồ uống có đường.
Trong các quần thể chủng tộc, sự biến đổi về dân tộc trong tỷ lệ béo phì ở trẻ em cũng đã được ghi nhận. Chỉ có 25% thanh thiếu niên gốc Tây Ban Nha thế hệ đầu tiên bị thừa cân dựa trên BMI ở bách phân vị thứ 85 hoặc cao hơn, so với 32% thanh thiếu niên gốc Tây Ban Nha thế hệ thứ hai và thứ ba. Tỷ lệ thừa cân ở thanh thiếu niên người Mỹ gốc Á trong nghiên cứu này là 20,6%, với tỷ lệ tương đương ở người Philippines (18,5%) và người Trung Quốc (15,3%). Một lần nữa, chỉ có 12% người Mỹ gốc Á thế hệ đầu tiên bị thừa cân, so với 27% và 28% ở thế hệ thứ hai và thứ ba, tương ứng. Ở người Mỹ bản địa, có sự thay đổi lớn về tỷ lệ béo phì từ 12% đến 77%, dựa trên các bộ lạc, nhóm tuổi, công cụ đo lường và giá trị ngưỡng, trong số các nghiên cứu được thực hiện từ năm 1990 đến năm 2000. Những nghiên cứu này chỉ ra rằng béo phì ở người Mỹ bản địa bắt đầu rất sớm từ thời thơ ấu.
Các yếu tố tiên lượng
BMI thời thơ ấu càng cao, khả năng béo phì ở tuổi trưởng thành càng lớn. Nhìn chung, trẻ em có BMI ở bách phân vị thứ 95 hoặc cao hơn có nguy cơ rất cao bị béo phì ở tuổi trưởng thành. Béo phì ở tuổi vị thành niên là một yếu tố nguy cơ chính gây béo phì ở tuổi trưởng thành, với tỷ số chênh tăng từ 1,3 đối với béo phì ở 1 đến 2 tuổi lên 17,5 đối với béo phì ở 15 đến 17 tuổi. Yếu tố dự báo mạnh nhất của béo phì ở tuổi vị thành niên là tăng cân nhanh trong khoảng từ 2 đến 6 tuổi. Sự thay đổi của BMI trong và sau tuổi vị thành niên là biến số dự báo quan trọng nhất đối với béo phì ở tuổi trưởng thành. Trẻ em và thanh thiếu niên có BMI ở bách phân vị thứ 95 hoặc cao hơn có 62% đến 98% khả năng bị béo phì ở tuổi 35, với 50% khả năng ở nam giới từ 13 tuổi trở lên và 66% khả năng ở nữ giới từ 13 tuổi trở lên. Quan trọng là, BMI tăng cao ở tuổi vị thành niên (ngay cả khi được coi là nằm trong phạm vi “bình thường”) cũng là một yếu tố nguy cơ đáng kể đối với các rối loạn liên quan đến béo phì ở tuổi trung niên. Mặc dù nguy cơ mắc bệnh đái tháo đường chủ yếu liên quan đến việc tăng BMI gần thời điểm chẩn đoán, nguy cơ mắc bệnh tim mạch vành lại liên quan đến việc tăng BMI cả ở tuổi vị thành niên và tuổi trưởng thành.
Tuổi phục hồi mỡ (adiposity rebound), tức là điểm thấp nhất của BMI trước khi lượng mỡ cơ thể bắt đầu tăng lên (trong khoảng từ 5 đến 6 tuổi), thường rõ rệt hơn ở nữ giới (xem Hình 24.6 A và B), cũng là một yếu tố dự báo quan trọng đối với béo phì ở tuổi trưởng thành. Trẻ em có sự phục hồi mỡ sớm có khả năng bị béo phì khi trưởng thành cao gấp năm lần so với những trẻ có sự phục hồi mỡ muộn. Ở độ tuổi phục hồi mỡ, trẻ em đã thừa cân có nguy cơ bị béo phì ở tuổi trưởng thành cao gấp sáu lần so với trẻ em gầy. Tích lũy cân nặng ở độ tuổi sớm hơn mang lại thời gian tiếp xúc lâu hơn với môi trường chuyển hóa liên quan đến béo phì và do đó làm tăng nguy cơ phát triển các bệnh lý liên quan đến béo phì. Do đó, béo phì ở trẻ em khởi phát càng sớm, nguy cơ béo phì ở tuổi trưởng thành càng lớn.
Dinh dưỡng thừa ở trẻ sơ sinh đóng một vai trò cực kỳ quan trọng trong sự phát triển của bệnh béo phì trong tương lai. Nhiều nghiên cứu đã chỉ ra việc bú bình là một yếu tố nguy cơ cụ thể. Tỷ lệ béo phì ở trẻ em chưa bao giờ được bú mẹ là 4,5%, so với 2,8% ở trẻ được bú mẹ, và một hiệu ứng thời gian-đáp ứng rõ ràng cũng được xác định đối với thời gian bú mẹ đối với sự suy giảm tỷ lệ béo phì. Dinh dưỡng thừa sớm có liên quan đến nồng độ leptin tăng cao trong cuộc sống sau này. Sự khác biệt về cả thể tích và thành phần của sữa công thức thương mại so với sữa mẹ đã được đề xuất là các yếu tố căn nguyên. Một mô hình mới nổi cho rằng hệ vi sinh vật đường ruột đóng một vai trò quan trọng trong sự phát triển của béo phì ở trẻ em cũng như ở người lớn. Sự tiếp xúc sớm với các yếu tố từ mẹ bao gồm sữa mẹ và các thành phần ăn kiêng khác trong giai đoạn sơ sinh (chẳng hạn như bắt đầu ăn dặm, tiếp xúc với chất làm ngọt nhân tạo, v.v.) có thể là những yếu tố quyết định chính đến hồ sơ của hệ vi sinh vật ảnh hưởng đến quá trình chuyển hóa và cân bằng cân nặng trong thời thơ ấu và tuổi trưởng thành.
Béo phì của cha mẹ cũng là một yếu tố dự báo quan trọng của béo phì ở trẻ em. Trẻ em có ít nhất một phụ huynh thừa cân ở độ tuổi phục hồi mỡ có khả năng trở thành người lớn béo phì cao gấp bốn đến năm lần. Trẻ gầy từ 5 tuổi trở xuống có nguy cơ béo phì ở tuổi trưởng thành cao gấp 13 lần nếu cả cha và mẹ đều béo phì. Việc tăng BMI quá mức của cha mẹ trong thời thơ ấu và tuổi trưởng thành cũng liên quan đến BMI cao hơn và nguy cơ béo phì ở con cái. Ngược lại, trẻ lớn hơn bị béo phì (10–14 tuổi) có nguy cơ trở thành người lớn béo phì tăng 22,3 lần bất kể cân nặng của cha mẹ, cho thấy rằng béo phì của cha mẹ quan trọng hơn trong việc tăng cân ở thời thơ ấu. Khi nghiên cứu mối liên quan giữa tình trạng béo phì của cha mẹ và con cái, các mối liên quan mạnh hơn đã được thể hiện ở trẻ lớn hơn so với trẻ nhỏ hơn, ở cả cha và mẹ so với chỉ cha hoặc mẹ, ở béo phì của cha mẹ và béo phì của con cái so với tình trạng thừa cân của cả hai. Béo phì của cha mẹ cũng liên quan đến sự phục hồi mỡ sớm, mặc dù vẫn chưa rõ liệu mối quan hệ giữa béo phì của cha mẹ và trẻ em là do di truyền, biểu sinh hay môi trường.
Tác động chuyển hóa của béo phì ở trẻ em
Nhiều biến chứng chuyển hóa và tim mạch (CV) của béo phì đã biểu hiện rõ ràng trong thời thơ ấu và liên quan chặt chẽ đến sự phát triển của kháng insulin-tăng insulin máu, bất thường sinh hóa phổ biến nhất thấy ở bệnh béo phì. Các bệnh đi kèm liên quan đến béo phì xuất hiện sớm ở trẻ em là những thay đổi trong chuyển hóa glucose, rối loạn lipid máu và tăng huyết áp. Mặc dù quá trình xơ vữa động mạch tăng tốc có mặt ở trẻ em béo phì, các biến cố tim mạch huyết khối thường không xuất hiện cho đến tuổi trưởng thành. Sự tập hợp của các biểu hiện này được gọi là hội chứng chuyển hóa, hoặc hội chứng kháng insulin, cho thấy rằng kháng insulin ngoại vi có thể là động lực thúc đẩy phần lớn các bệnh lý liên quan đến béo phì.
Kháng Insulin
Kháng insulin được định nghĩa là sự giảm đáp ứng của mô đối với các hoạt động tế bào qua trung gian insulin và là nghịch đảo của độ nhạy insulin. Thuật ngữ kháng insulin, như thường được áp dụng, đề cập đến việc giảm hấp thu glucose toàn thân để đáp ứng với nồng độ insulin sinh lý và các tác động do đó của nó lên chuyển hóa glucose và insulin. Tuy nhiên, hiện nay rõ ràng là không phải tất cả các mô đáp ứng với insulin đều nhạy cảm với insulin như nhau. Kháng insulin toàn thể sẽ dẫn đến rối loạn chuyển hóa toàn cầu, chẳng hạn như hội chứng leprechaunism hoặc hội chứng Rabson-Mendenhall. Do đó, kháng insulin của bệnh béo phì phải ảnh hưởng đến các mô khác nhau về mặt định lượng (xem Chương 3 và Chương 21 về đái tháo đường và đột biến thụ thể insulin).
Kháng Insulin tại gan. Gan đóng một vai trò chính trong chuyển hóa chất nền và là mục tiêu chính của hoạt động insulin. Sau khi insulin được giải phóng từ tế bào β sau một tải glucose, nó di chuyển trực tiếp đến gan qua tĩnh mạch cửa, nơi nó gắn vào thụ thể insulin và gây ra hai tác động chính ở cấp độ phiên mã gen. Đầu tiên, insulin kích thích sự phosphoryl hóa của FOXO1, ngăn không cho nó đi vào nhân, và do đó làm giảm sự biểu hiện của các gen cần thiết cho quá trình tân tạo glucose, chủ yếu là phosphoenolpyruvate carboxykinase (PEPCK) và glucose-6-phosphatase. Hiệu quả ròng là giảm sản xuất glucose của gan. Thứ hai, insulin kích hoạt yếu tố phiên mã sterol regulatory element-binding protein (SREBP)-1c, từ đó làm tăng phiên mã của các gen cần thiết cho quá trình sinh tổng hợp axit béo và triglyceride (TG), đáng chú ý nhất là ATP-citrate lyase, acetyl-coenzyme A carboxylase, và fatty acid synthase; cùng nhau tạo thành quá trình tân tạo mỡ (DNL). Các TG được tổng hợp bởi DNL sau đó được đóng gói với apolipoprotein B (apoB) vào các lipoprotein tỷ trọng rất thấp (VLDL) để xuất khẩu ra ngoại vi để lưu trữ hoặc sử dụng bằng cách kích hoạt đối ứng của lipoprotein lipase (LPL) trên bề mặt của các tế bào nội mô trong mô mỡ hoặc mô cơ.
Vì những lý do vẫn chưa rõ ràng, các đối tượng kháng insulin thường có kháng insulin gan “chọn lọc” hoặc “tách rời”; nghĩa là, họ bị suy giảm cân bằng nội môi glucose (qua trung gian của con đường FOXO1) nhưng DNL gan qua trung gian insulin bình thường (qua trung gian của con đường SREBP-1c và gen GCKR). Sự gia tăng dòng chảy FFA trong gan, do DNL hoặc do FFA được cung cấp qua tĩnh mạch cửa, làm suy giảm hoạt động insulin của gan thông qua các chất trung gian fatty acyl-CoA trong tế bào gan, dẫn đến tăng sản lượng glucose của gan, tổng hợp các cytokine gây viêm, và bài tiết TG dư thừa của gan, nồng độ cholesterol lipoprotein tỷ trọng cao (HDL) thấp, và sự gia tăng các hạt LDL tương đối bị suy giảm cholesterol. Hơn nữa, sự tích tụ FFA và lipid trong gan cũng có hại cho độ nhạy insulin của gan vì điều này dẫn đến việc tạo ra các chất chuyển hóa có nguồn gốc từ lipid độc hại, chẳng hạn như DAG, fatty acyl CoA và ceramide. Những chất này lần lượt kích hoạt protein kinase C-ɛ (PKCɛ, và sự phosphoryl hóa serine/threonine của cơ chất thụ thể insulin 1 (IRS-1), làm suy giảm sự truyền tín hiệu insulin của gan. Kháng insulin trong gan dẫn đến sự thanh thải insulin lần đầu ở gan lớn hơn, dẫn đến lượng insulin đến tuần hoàn hệ thống thấp hơn.
Kháng Insulin tại mô mỡ. Khối lượng mô mỡ mở rộng đi kèm với béo phì thường dẫn đến tăng ly giải mỡ và chu chuyển FFA. Thông thường, insulin ức chế ly giải mỡ ở mô mỡ; tuy nhiên, trong trạng thái kháng insulin, quá trình ly giải mỡ được tăng tốc, dẫn đến tăng giải phóng FFA vào tuần hoàn. Hơn nữa, các tế bào mỡ nội tạng nhạy cảm hơn với quá trình ly giải mỡ được kích thích bởi catecholamine so với các tế bào mỡ dưới da, làm tăng thêm dòng chảy FFA. Các đại thực bào cũng xâm nhập vào mô mỡ và góp phần vào cả sự phì đại của tế bào mỡ và giải phóng cytokine. Các cytokine tuần hoàn này cũng ảnh hưởng đến hoạt động của insulin ở các mô khác, chẳng hạn như gan và cơ. Trong phạm vi dung nạp glucose bình thường, sự gia tăng kháng insulin ở mô mỡ có liên quan đến sự gia tăng nồng độ glucose 2 giờ. Một mối quan hệ chặt chẽ tồn tại giữa mỡ nội tạng (r = 0,34; P < 0,001) và tỷ lệ mỡ nội tạng/dưới da và sự kháng insulin của mô mỡ. Nồng độ FFA lớn hơn sau một tải glucose đường uống cũng rõ ràng với tình trạng dung nạp glucose xấu đi, cho thấy sự ức chế ly giải mỡ giảm và thanh thải FFA thấp hơn.
Kháng Insulin tại cơ. Ở hạ nguồn của một gan kháng insulin, dòng chảy FFA huyết tương tăng vào cơ xương dẫn đến các dẫn xuất fatty acyl-CoA làm thay đổi con đường truyền tín hiệu insulin và dẫn đến giảm vận chuyển glucose qua trung gian insulin trong cơ xương, tạo điều kiện cho sự phát triển của tăng đường huyết. Sự lắng đọng lạc chỗ trong cơ xương của mỡ dưới dạng lipid nội bào cơ cũng có thể đóng một vai trò trực tiếp trong cơ chế bệnh sinh của kháng insulin toàn thân và hội chứng chuyển hóa thông qua việc kích hoạt PKCɛ do chất chuyển hóa lipid gây ra với sự suy giảm sau đó của tín hiệu insulin. Sự lắng đọng lipid nội bào cơ lớn hơn có liên quan chặt chẽ với kháng insulin và thường được phát hiện ở trẻ em béo phì có chuyển hóa glucose bị thay đổi. Biểu hiện của sự suy giảm truyền tín hiệu insulin trong cơ xương là giảm sự chuyển vị của GLUT4 đến màng tế bào dẫn đến giảm hấp thu glucose toàn thân.
Đánh giá kháng Insulin. Kỹ thuật kẹp glucose đẳng đường-tăng insulin máu (euglycemic hyperinsulinemic clamp) là tiêu chuẩn vàng để đo độ nhạy insulin; các phương pháp xét nghiệm dung nạp glucose tĩnh mạch lấy mẫu thường xuyên (FSIVGTT) và glucose huyết tương trạng thái ổn định (SSPG) cũng là các phép đo hợp lệ. Kỹ thuật kẹp được thực hiện bằng cách truyền liên tục insulin được điều chỉnh theo diện tích bề mặt cơ thể trong khi duy trì nồng độ glucose huyết tương lúc đói bằng cách điều chỉnh truyền glucose. Tốc độ truyền glucose lớn hơn cần thiết để duy trì đường huyết bình thường cho thấy độ nhạy insulin lớn hơn. Các nghiên cứu kẹp glucose đẳng đường-tăng insulin máu đã chỉ ra rằng kháng insulin được xác định chủ yếu bởi phản ứng của cơ xương, với hơn 75% glucose được truyền vào được cơ hấp thu và chỉ có 2% đến 3% được mô mỡ hấp thu. Cả ba phương pháp nói chung đều tốn thời gian, yêu cầu truyền tĩnh mạch và lấy mẫu máu thường xuyên, gây gánh nặng cho người tham gia, tốn kém và yêu cầu một môi trường nghiên cứu. Trong nỗ lực đơn giản hóa việc đo lường độ nhạy insulin, một số phương pháp sử dụng các mẫu insulin và glucose lúc đói được lấy đồng thời đã được phát triển, chẳng hạn như mô hình đánh giá cân bằng nội môi cho kháng insulin (HOMA-IR). Mỗi phương pháp này sử dụng một công thức toán học điều chỉnh cho sự thay đổi cá nhân trong bài tiết và thanh thải insulin và glucose. Mặc dù mục tiêu của các phương pháp này là cải thiện độ chính xác của chỉ riêng insulin lúc đói bằng cách thêm vào glucose lúc đói, hiện nay người ta đồng ý rằng chúng mang lại kết quả tương tự như insulin lúc đói. Khi được tương quan với các phương pháp tiêu chuẩn vàng ở trẻ em, insulin lúc đói là một thước đo kém về độ nhạy insulin toàn thân ở một đứa trẻ riêng lẻ. Mặc dù mối quan tâm chính là kháng insulin, các tác động bất lợi liên quan đến kháng insulin có nhiều khả năng được trung gian qua tình trạng tăng insulin máu bù trừ. Tỷ lệ triglyceride lúc đói trên HDL-cholesterol, một chỉ số thay thế cho kháng insulin không sử dụng các phép đo insulin, đã được đề xuất và tương quan khá tốt với kháng insulin có nguồn gốc từ kỹ thuật kẹp, tuy nhiên việc sử dụng nó cần được xác nhận trong các quần thể đa dạng về dân tộc.
Hai điều kiện sinh học quan trọng nhất liên quan đến kháng insulin ở trẻ em là dân tộc và tuổi dậy thì. Các nghiên cứu cho thấy trẻ em người Mỹ gốc Phi, gốc Tây Ban Nha, người da đỏ Pima và người châu Á ít nhạy cảm với insulin hơn so với trẻ em da trắng không phải gốc Tây Ban Nha có các số đo nhân trắc tương tự. Kháng insulin ở các nhóm dân tộc thiểu số được biểu hiện bằng việc hấp thu glucose được kích thích bởi insulin thấp hơn, đi kèm với tăng insulin máu, bằng chứng về việc tăng bài tiết insulin từ tế bào β, và giảm thanh thải insulin. Trong giai đoạn dậy thì, có sự suy giảm khoảng 25% đến 50% về độ nhạy insulin và sẽ phục hồi khi quá trình phát triển dậy thì hoàn tất. Sự gia tăng bù trừ trong bài tiết insulin trong giai đoạn dậy thì có thể bị suy giảm ở thanh thiếu niên người Mỹ gốc Phi và gốc Tây Ban Nha, do đó làm tăng nguy cơ mắc đái tháo đường tuýp 2 (T2DM) vào khoảng thời gian dậy thì. Sự phát triển của T2DM được đề cập sâu trong Chương 21, tuy nhiên, điều đáng chú ý là suy giảm dung nạp glucose (IGT), được gọi là tiền đái tháo đường, là một tình trạng tương đối phổ biến ở trẻ em và thanh thiếu niên béo phì. IGT ở thanh thiếu niên béo phì thường được đặc trưng bởi béo phì với một kiểu phân bố lipid không thuận lợi, với sự lắng đọng mỡ tăng lên ở các khoang nội tạng, gan và nội bào cơ.
Phân bố lipid
Thuật ngữ phân bố lipid đề cập đến sự phân bố mỡ cơ thể ở các cơ quan và khoang khác nhau. Phần lớn mỡ thừa được lưu trữ trong kho dưới da thông thường, tuy nhiên các vị trí lưu trữ tiềm năng khác cũng tồn tại, chẳng hạn như khoang mỡ trong ổ bụng (nội tạng) và các mô đáp ứng insulin, chẳng hạn như cơ và gan. Một giả thuyết để giải thích mối quan hệ giữa béo phì và kháng insulin là mô hình “cửa-nội tạng”. Giả thuyết này cho rằng tình trạng tích mỡ tăng lên gây ra sự tích tụ mỡ trong kho nội tạng, dẫn đến tăng dòng chảy FFA trong hệ cửa và toàn thân (Hình 24.7). Mối liên quan giữa béo phì nội tạng, kháng insulin và các bệnh đi kèm đã được chứng minh ở hầu hết các nhóm tuổi và dân tộc. Đáng chú ý, các nghiên cứu về dòng chảy FFA in vivo từ các kho nội tạng và thân mình và bụng dưới da đã không chứng minh được sự khác biệt đáng kể về dòng chảy ròng giữa các kho này.
Hình 24.7 Một giả thuyết về mối quan hệ giữa béo phì và hội chứng chuyển hóa. Tác động chuyển hóa của béo phì được quyết định bởi mô hình phân bố lipid. Việc lưu trữ lipid trong các mô nhạy cảm với insulin, chẳng hạn như gan hoặc cơ, và trong khoang nội tạng có liên quan đến một hồ sơ chuyển hóa đặc trưng bởi axit béo tự do và các cytokine gây viêm tăng cao cùng với nồng độ adiponectin giảm. Sự kết hợp này có thể độc lập dẫn đến kháng insulin ngoại vi và rối loạn chức năng nội mô. Sự kết hợp giữa kháng insulin và xơ vữa động mạch sớm (biểu hiện là rối loạn chức năng nội mô) thúc đẩy sự phát triển của rối loạn chuyển hóa glucose và bệnh tim mạch.
Mỡ dưới da, không đổ vào hệ cửa, có liên quan chặt chẽ đến kháng insulin ở nam giới khỏe mạnh bị béo phì và ở nam giới bị đái tháo đường. Tương tự, khối lượng mỡ dưới da ở thân mình đã được chứng minh là một yếu tố dự báo độc lập về kháng insulin ở phụ nữ béo phì. Mỡ nội tạng và dưới da khác nhau về phản ứng sinh học của chúng vì mỡ nội tạng kháng insulin hơn và có độ nhạy cảm tăng với catecholamine. Những quan sát này nhấn mạnh rằng cả mỡ nội tạng và dưới da ở bụng đều có thể góp phần vào kháng insulin, có thể bằng các cơ chế khác nhau. Các nghiên cứu được thực hiện ở thanh thiếu niên béo phì nhấn mạnh thực tế rằng tỷ lệ mỡ nội tạng trên mỡ dưới da có thể là yếu tố quyết định tác động chuyển hóa của chúng hơn là lượng mỡ tuyệt đối của chúng. Thật vậy, thanh thiếu niên béo phì có tỷ lệ mỡ nội tạng trên mỡ dưới da cao, mặc dù có mức độ béo phì tương đương, lại cho thấy một kiểu hình chuyển hóa bất lợi rõ rệt của kháng insulin nặng và những thay đổi trong chuyển hóa glucose và lipid. Hơn nữa, mỡ trong gan, mặc dù có liên quan chặt chẽ với nồng độ mỡ nội tạng cao, cũng có liên quan đến trạng thái kháng insulin ở thanh thiếu niên béo phì, độc lập với tất cả các kho mỡ khác.
Một mô hình thống nhất hơn là giả thuyết “khả năng mở rộng của mô mỡ” cho rằng tổng lượng mỡ cơ thể không phải là thủ phạm gây ra các tác động xấu đến sức khỏe trong bệnh béo phì mà là tỷ lệ tương đối của lipid trong các kho mỡ khác nhau mới là yếu tố quyết định kiểu hình chuyển hóa và nguy cơ chuyển hóa bắt nguồn từ đó. Lý thuyết này cho rằng khi mô mỡ mở rộng, “sự lắng đọng lipid lạc chỗ” trong các mô đáp ứng insulin là thủ phạm gây ra các tác động bất lợi đối với quá trình chuyển hóa. Lý thuyết này dựa trên những quan sát cho thấy hàm lượng lipid trong gan và/hoặc cơ tăng lên trong bệnh béo phì và T2DM và là một yếu tố dự báo mạnh mẽ về kháng insulin. Hơn nữa, trong các tình trạng như loạn dưỡng mỡ, tất cả mỡ đều được lưu trữ trong gan và cơ do thiếu mô mỡ dưới da, gây ra kháng insulin nặng và đái tháo đường. Ở người lớn béo phì (BMI > 30), sự suy giảm cơ trên chụp cắt lớp vi tính ([CT]; đại diện cho hàm lượng lipid) là một yếu tố dự báo kháng insulin mạnh hơn mỡ nội tạng. Các nghiên cứu được thực hiện in vivo bằng quang phổ cộng hưởng từ hạt nhân proton (¹H-NMR) đã chứng minh hàm lượng lipid nội bào cơ (IMCL) tăng là một yếu tố quyết định mạnh mẽ của kháng insulin ở người lớn và ở thanh thiếu niên béo phì. Tương tự, sự lắng đọng lipid trong tế bào gan để tạo ra lipid nội bào gan (IHCL) có khả năng dự báo cao về kháng insulin, thậm chí còn cao hơn mỡ nội tạng. Do đó, bệnh lý do béo phì có thể bắt đầu khi mô mỡ dưới da đạt đến khả năng lưu trữ mỡ thừa và bắt đầu chuyển lipid đến các mô lạc chỗ, chẳng hạn như gan và cơ, dẫn đến kháng insulin ngoại vi; hoặc có thể khi gan hoặc cơ tích lũy lipid được sản xuất mới để đáp ứng với sự điều chỉnh chế độ ăn (xem sau). Một nguyên nhân khác được giả định gây ra sự tích tụ IMCL và IHCL là sự giảm quá trình oxy hóa β của chất béo, liên quan đến khả năng hiếu khí thấp, số lượng ty thể giảm hoặc hoạt động kém, hoặc trương lực SNS giảm. Tác động của sự tích tụ IMCL hoặc IHCL lên độ nhạy ngoại vi được giả định là do sự thay đổi của con đường truyền tín hiệu insulin trong cơ, gây ra bởi các dẫn xuất của chất béo, chẳng hạn như axit béo acyl-CoA chuỗi dài và DAG trong tế bào gan hoặc tế bào cơ. Các dẫn xuất này kích hoạt chuỗi kinase serine/threonine và gây ra sự phosphoryl hóa serine của IRS-1, ức chế tín hiệu insulin. Một cơ chế tương đương đã được chứng minh trong gan, nơi sự tích tụ lipid, đặc biệt là DAG, kích hoạt chuỗi viêm bằng cách gây ra c-jun N-terminal kinase 1 (JNK-1), gây ra sự phosphoryl hóa serine thay vì tyrosine của IRS-1, dẫn đến ức chế tín hiệu insulin của gan.
Thay đổi mạch máu
Các giai đoạn đầu của quá trình xơ vữa động mạch có thể được phát hiện ở trẻ em béo phì. Trong những năm gần đây, đã trở nên rõ ràng rằng rối loạn chức năng nội mô là một bước sớm quan trọng trong sự phát triển của xơ vữa động mạch. Dấu hiệu và nguyên nhân của rối loạn chức năng nội mô là sự suy giảm trong giãn mạch qua trung gian nitric oxide (NO). Điều này là do sản xuất NO giảm bởi endothelial nitric oxide synthase (eNOS), được giả định là do nồng độ FFA và các cytokine gây viêm cao (interleukin [IL]-6, yếu tố hoại tử khối u [TNF]-α) ở những người béo phì kháng insulin, tăng các loại oxy phản ứng, hoặc tăng axit uric, ức chế hoạt động của eNOS. Sự khả dụng NO giảm dẫn đến sự mất cân bằng giữa các yếu tố giãn mạch và co mạch (chẳng hạn như endothelin), dẫn đến suy giảm sự giãn cơ trơn mạch máu, tăng sự bám dính của các tế bào viêm vào nội mô, tăng biểu hiện của chất ức chế hoạt hóa plasminogen–1 (PAI-1; một phân tử tiền huyết khối) và tăng sinh tế bào cơ trơn mạch máu. Do đó, sự khả dụng NO giảm được cho là tạo ra một môi trường tiền viêm, tiền huyết khối thúc đẩy xơ vữa động mạch. Chức năng nội mô đại diện cho một chỉ số tích hợp về gánh nặng nguy cơ tim mạch tổng thể ở bất kỳ cá nhân nào. Trong thập kỷ qua, các kỹ thuật không xâm lấn để đánh giá chức năng nội mô, bao gồm siêu âm mạch máu ngoài có độ phân giải cao để đo sự giãn nở phụ thuộc nội mô qua trung gian dòng chảy (FMD) của động mạch cánh tay trong quá trình sung huyết đã được phát triển. FMD suy giảm tương quan với độ cứng thành động mạch, sự giãn mạch vành và rối loạn chức năng nội mô ở trẻ em béo phì. Tương tự, những thay đổi về giải phẫu trong các mạch máu động mạch ngoại vi, chẳng hạn như tăng độ dày lớp nội trung mạc (IMT), cũng đã được chứng minh ở trẻ em và thanh thiếu niên béo phì, bắt chước bệnh lý mạch vành sớm và dự đoán các kết quả tim mạch bất lợi.
Không có nghiên cứu theo chiều dọc nào đo lường trực tiếp độ nhạy insulin in vivo và mối quan hệ của nó với sự phát triển của các bất thường xơ vữa động mạch ở trẻ em. Rất ít quan sát cho thấy mối quan hệ giữa HOMA-IR và độ cứng động mạch và nồng độ insulin lúc đói ở thanh thiếu niên. Tuy nhiên, một vai trò của kháng insulin trong các bất thường sớm của cơ trơn mạch máu được đề xuất dựa trên quan sát rằng các dấu ấn sinh học tuần hoàn của rối loạn chức năng nội mô (phân tử bám dính giữa các tế bào và E-selectin) là cao nhất, trong khi adiponectin, adipocytokine chống xơ vữa động mạch, là thấp nhất trong số những thanh thiếu niên kháng insulin nhất. Nghiên cứu mang tính bước ngoặt Bogalusa heart đã chứng minh rằng các yếu tố nguy cơ tim mạch có mặt ở thời thơ ấu có khả năng dự báo bệnh động mạch vành ở tuổi trưởng thành. Trong số các yếu tố nguy cơ này, LDL-cholesterol và BMI được đo ở thời thơ ấu được phát hiện là có khả năng dự báo IMT ở người trưởng thành trẻ tuổi. Hiện có bằng chứng đáng kể cho thấy kháng insulin của béo phì ở trẻ em tạo ra nền tảng chuyển hóa cho bệnh tim mạch ở người lớn. Ngưng thở tắc nghẽn khi ngủ (OSA), thường có ở trẻ em béo phì, cũng liên quan chặt chẽ đến sự hiện diện của rối loạn chức năng nội mô. Hơn nữa, sự kết hợp của kháng insulin ngoại vi, hồ sơ adipocytokine không thuận lợi, viêm bán cấp và rối loạn chức năng nội mô hoạt động song song để thúc đẩy các quá trình bệnh lý của lão hóa.
Adipocytokines
Leptin. Việc phát hiện ra leptin vào năm 1994 đã thay đổi đáng kể quan điểm về mô mỡ trong việc điều hòa cân bằng năng lượng. Các tế bào mỡ tiết ra một số protein hoạt động như các chất điều hòa cân bằng nội môi glucose và lipid. Những protein này đã được gọi chung là adipocytokines vì sự tương đồng về cấu trúc của chúng với các cytokine. Nồng độ leptin tuần hoàn tương quan với mức độ béo phì. Như đã nêu trước đó, vai trò chính của leptin là đóng vai trò như một cảm biến dự trữ năng lượng dài hạn để bảo vệ chống lại nạn đói. Leptin có lẽ có một vai trò cho phép trong các quá trình chuyển hóa năng lượng cao, chẳng hạn như dậy thì, rụng trứng và mang thai, nhưng vai trò của nó trong các trạng thái dư thừa năng lượng ít được biết đến hơn. Trong bệnh béo phì, sự phát triển của kháng leptin có thể dẫn đến sự phá vỡ sự phân bố bình thường của lipid dư thừa trong khoang tế bào mỡ.
Adiponectin. Cytokine adiponectin là một trường hợp đặc biệt trong bệnh béo phì vì, trái ngược với các adipocytokine khác, nồng độ của nó bị giảm ở những người béo phì. Gen adiponectin được biểu hiện độc quyền trong mô mỡ và mã hóa một miền đầu hình cầu ở đầu cuối carboxyl của protein và một miền collagen ở đầu cuối amino, có cấu trúc gợi nhớ đến yếu tố bổ thể 1q. Gen này nằm trên nhiễm sắc thể 3q27, một vị trí trước đây có liên quan đến sự phát triển của bệnh đái tháo đường tuýp 2 và hội chứng chuyển hóa. Một số đa hình đơn nucleotide (SNP) trong gen adiponectin đã được báo cáo là có liên quan đến sự phát triển của bệnh đái tháo đường tuýp 2 ở các quần thể trên khắp thế giới, cho thấy rằng adiponectin đóng một vai trò chính trong chuyển hóa glucose. Adiponectin lưu thông trong huyết tương ở ba dạng chính: một trimer có trọng lượng phân tử thấp, một hexamer có trọng lượng phân tử trung bình, và một 12- đến 18-mer có trọng lượng phân tử cao. Nồng độ adiponectin có trọng lượng phân tử cao trong huyết tương tuần hoàn cho thấy sự khác biệt về giới tính (nữ giới có nồng độ cao hơn), cho thấy vai trò của hormone sinh dục trong việc điều hòa sản xuất hoặc thanh thải adiponectin. Các yếu tố chế độ ăn uống, chẳng hạn như axit linoleic hoặc dầu cá so với chế độ ăn nhiều carbohydrate hoặc tăng stress oxy hóa, đã được chứng minh là làm tăng hoặc giảm nồng độ adiponectin, tương ứng. Những quan sát này cho thấy rằng nồng độ adiponectin trong tuần hoàn được điều hòa bởi các tương tác phức tạp giữa các yếu tố di truyền và môi trường.
Các thụ thể cho adiponectin đã được mô tả đặc điểm trong các mô hình gặm nhấm và được nhân bản. Hai thụ thể, được đặt tên là ADIPOR1 và ADIPOR2, đã được mô tả đặc điểm. ADIPOR1 được biểu hiện ở nhiều mô bao gồm cả cơ, trong khi ADIPOR2 chủ yếu giới hạn ở gan. Cả hai thụ thể đều gắn vào màng tế bào, nhưng độc đáo so với các thụ thể kết hợp G-protein khác ở chỗ đầu cuối C ở bên ngoài, trong khi đầu cuối N ở bên trong tế bào. Cả ADIPOR1 và ADIPOR2 đều là thụ thể cho đầu hình cầu của adiponectin và đóng vai trò là chất khởi đầu của các con đường truyền tín hiệu dẫn đến tăng PPARα và tăng hoạt động của kinase adenosine monophosphate (AMP), thúc đẩy sự hấp thu glucose và tăng quá trình oxy hóa axit béo. Adiponectin đã được chứng minh là có các chức năng chống xơ vữa động mạch mạnh mẽ, vì nó tích tụ trong không gian dưới nội mô của các thành mạch bị tổn thương để làm giảm sự biểu hiện của các phân tử bám dính và sự tuyển mộ các đại thực bào.
Các nghiên cứu ở trẻ em và thanh thiếu niên béo phì đã chỉ ra rằng adiponectin có mối quan hệ nghịch với mức độ béo phì, độ nhạy insulin, béo phì nội tạng, IHCL và IMCL, trong khi giảm cân làm tăng adiponectin. Ở thanh thiếu niên béo phì và đái tháo đường tuýp 2, adiponectin cơ bản thấp và sự gia tăng giảm đáp ứng với điều trị đã được chứng minh là dự báo thất bại điều trị. Tất cả những quan sát này cùng với dữ liệu lâm sàng trên người ủng hộ một vai trò then chốt của adiponectin trong việc ngăn ngừa các bệnh đi kèm của hội chứng chuyển hóa.
Các nghiên cứu gia đình sử dụng hồi quy cha mẹ-con cái cho thấy hầu hết các adipocytokine đều cho thấy bằng chứng về sự di truyền đáng kể. Có ba trục biến đổi chung chính trong tính di truyền của các adipocytokine. Trục chính, giải thích 21% sự biến đổi, chịu tải mạnh nhất ở các mức leptin, TNF-α, insulin và PAI-1, và nghịch với adiponectin. Trục này có liên quan đáng kể với BMI và mạnh hơn về mặt kiểu hình ở trẻ em, và cho thấy tính di truyền là 50%, sau khi điều chỉnh theo tuổi, giới tính và các hiệu ứng thế hệ. Do đó, các adipocytokine có tính di truyền cao và mô hình đồng biến đổi của chúng có tương quan đáng kể với BMI ngay từ những năm tiền thiếu niên.
Myokines và Peptide lợi niệu
Cơ xương và cơ tim cũng có thể đóng vai trò như một cơ quan nội tiết. Một số tác động của tập thể dục đối với cơ xương được trung gian bởi đồng kích hoạt phiên mã PPAR-γ coactivator 1α (PGC-1α). Ở chuột, biểu hiện PGC-1α trong cơ kích thích sự gia tăng biểu hiện của FNDC5, một protein màng được phân cắt và tiết ra dưới dạng một hormone mới được xác định, có tên là Irisin. Irisin tác động lên các tế bào mỡ trắng trong nuôi cấy và in vivo để kích thích biểu hiện UCP1 và gây ra sự “nâu hóa” của các tế bào mỡ trắng thành các tế bào hoạt động chuyển hóa mạnh hơn. Irisin được tạo ra khi tập thể dục ở chuột và người, và nồng độ irisin tăng nhẹ trong máu gây ra sự gia tăng tiêu hao năng lượng ở chuột mà không có thay đổi về vận động hoặc ăn uống, dẫn đến những cải thiện về béo phì và cân bằng nội môi glucose. Myokine mới này thực sự là mối liên kết nội tiết tố đầu tiên giữa tập thể dục và những thay đổi ở mô mỡ mà nó có thể gây ra. Quan trọng là, phân tử này có tương quan âm với chức năng điều hành của não và do đó có thể có nhiều tác động chưa được khám phá. Các peptide lợi niệu nhĩ (ANP) cũng có liên quan đến chuyển hóa chất béo. Các peptide lợi niệu này được sản xuất khi tập thể dục, căng thành tim, giảm cân và tiếp xúc với lạnh, và bị ức chế bởi béo phì và kháng insulin. ANP gắn vào thụ thể lợi niệu của nó, tạo điều kiện thuận lợi cho việc hình thành cyclic guanosine monophosphate (cGMP) từ guanosine triphosphate (GTP). cGMP phosphoryl hóa protein kinase phụ thuộc cGMP, kích hoạt quá trình ly giải mỡ, và phosphoryl hóa protein kinase được hoạt hóa bởi mitogen p38 để tăng cường sinh tổng hợp ty thể với việc tăng tiêu hao năng lượng và tăng sinh nhiệt, như một phần của chương trình sinh nhiệt của mỡ nâu. Do đó, cả cơ xương và cơ tim đều có thể đáp ứng với tập thể dục bằng cách gây ra những thay đổi trong chuyển hóa chất béo để tăng cường tiêu hao calo và hạn chế béo phì. Những phát hiện mới này có khả năng mở ra những con đường nghiên cứu lâm sàng mới để hạn chế hậu quả của “đại dịch béo phì”.
Các Cytokine gây viêm
Bằng chứng tích lũy cho thấy béo phì có liên quan đến viêm mãn tính cận lâm sàng. Mô mỡ không chỉ đóng vai trò là một kho chứa năng lượng đơn giản được lưu trữ dưới dạng TG, mà còn là một cơ quan bài tiết tích cực giải phóng nhiều peptide, bao gồm cả các cytokine gây viêm, vào tuần hoàn. Điều này có lẽ là do sự xâm nhập của mô mỡ bởi các tế bào của hệ miễn dịch, chủ yếu là các đại thực bào. Trong bệnh béo phì, sự cân bằng giữa nhiều peptide này bị thay đổi, sao cho các tế bào mỡ lớn hơn và các đại thực bào được nhúng trong chúng sản xuất nhiều cytokine gây viêm hơn (tức là TNF-α, IL-6) và ít peptide chống viêm hơn, chẳng hạn như adiponectin. Một giả thuyết cho rằng khi năng lượng tích tụ trong các tế bào mỡ, đường viền perilipin của không bào mỡ bị phá vỡ, gây ra cái chết của tế bào mỡ. Cái chết của tế bào tuyển mộ các đại thực bào vào mô mỡ, đặc biệt là trong khoang nội tạng, trong quá trình dọn dẹp các mảnh vụn, cũng tạo ra các cytokine gây viêm, khởi đầu một môi trường tiền viêm có trước và có thể thúc đẩy sự phát triển của kháng insulin toàn thân, đái tháo đường và rối loạn chức năng nội mô. Nồng độ toàn thân của protein phản ứng C (CRP) và IL-6, hai dấu hiệu và tác nhân chính của quá trình viêm, tăng lên ở trẻ em và thanh thiếu niên béo phì. Nồng độ CRP trong phạm vi “bình thường-cao” đã được chứng minh là dự báo bệnh tim mạch và sự phát triển của T2DM ở người lớn. Nồng độ CRP tăng cao tương quan với các thành phần khác của hội chứng chuyển hóa ở trẻ em béo phì. Do đó, viêm có thể là một trong những mối liên kết giữa béo phì và kháng insulin, có khả năng thúc đẩy sự kháng cự của tế bào cơ và gan trong con đường truyền tín hiệu insulin.
Các loại oxy phản ứng (ROS)
“Thuyết gốc tự do” cho rằng sự mất cân bằng giữa việc tạo ra ROS và hệ thống phòng thủ chống oxy hóa là một yếu tố chính trong việc xác định quá trình peroxy hóa lipid và sai lệch cấu trúc protein, dẫn đến tổn thương deoxyribonucleic acid (DNA) và tế bào. Sự hình thành ROS nội bào quá mức xảy ra thông qua ba con đường: (1) các cytokine gây viêm có nguồn gốc từ sự tích tụ mỡ nội tạng, (2) năng lượng ty thể bị rối loạn chức năng, và (3) glycation. Việc xử lý chất dinh dưỡng quá mức của ty thể có thể dẫn đến sự mất cặp đôi của quá trình phosphoryl hóa oxy hóa và tăng tạo ra ROS; điều này, đến lượt nó, dẫn đến chức năng ty thể bị thay đổi và tạo ra ROS nhiều hơn. Sự tích tụ ROS cũng có thể làm suy giảm chức năng lưới nội chất (ER), gây ra stress ER và phản ứng protein không gấp nếp (UPR) bù trừ. Bản thân UPR có thể bị quá tải bởi việc xử lý chất dinh dưỡng quá mức và tạo ra ROS kéo dài, dẫn đến ngừng hoạt động của tế bào, bài tiết insulin bị lỗi và T2DM (Hình 24.8).
Hình 24.8 Các cơ chế của rối loạn chức năng chuyển hóa dưới tế bào, sử dụng fructose làm ví dụ. Sự hình thành acetyl-CoA dẫn đến lắng đọng lipid và kích hoạt các con đường viêm, dẫn đến sự phosphoryl hóa serine của IRS-1, dẫn đến kháng insulin. Hơn nữa, quá trình xử lý chuyển hóa trong ty thể, sự glycat hóa của các nhóm ɛ-amino của protein thông qua phản ứng Maillard, và các cytokine gây viêm tuần hoàn, do sự kích hoạt qua trung gian thụ thể của NADPH oxidase, tất cả đều làm tăng nồng độ các loại oxy phản ứng (ROS) nội bào. Trong trường hợp không có đủ sự dập tắt và phân hủy của peroxisome, các gốc ROS dẫn đến stress lưới nội chất (ER), thúc đẩy phản ứng protein không gấp nếp (UPR), và gây ra cái chết của tế bào (apoptosis) hoặc rối loạn chức năng tế bào/chuyển hóa. ATP, Adenosine triphosphate; CoA, coenzyme A; NADPH, nicotinamide adenine dinucleotide phosphate.
Vì ROS là sản phẩm phụ vốn có của quá trình chuyển hóa tế bào, các chất chống oxy hóa nội sinh của tế bào (ví dụ: catalase và glutathione) sẽ dập tắt ROS trước khi chúng có cơ hội thúc đẩy quá trình peroxy hóa. Các chất chống oxy hóa này được tìm thấy chủ yếu trong các peroxisome, hợp tác với ty thể trong việc xử lý ROS. Sự giảm hoạt động của peroxisome dẫn đến rối loạn chức năng ty thể và stress ER. Hơn nữa, các cytokine, chẳng hạn như TNF-α, có thể làm giảm số lượng và chức năng của peroxisome, khiến các tế bào càng dễ bị tổn thương hơn.
Các bệnh đi kèm liên quan đến kháng Insulin
Hội chứng chuyển hóa
Sự kết hợp và phân cụm của đái tháo đường tuýp 2 (T2DM), tăng huyết áp, rối loạn lipid máu và bệnh tim mạch (CV) ở người lớn đã dẫn đến giả thuyết rằng chúng có thể phát sinh từ một tiền đề chung. WHO cho rằng tiền đề này là kháng insulin, và định nghĩa sự kết hợp này là hội chứng chuyển hóa. Một định nghĩa đồng thuận về hội chứng chuyển hóa cho nhóm tuổi nhi khoa đã được công bố và tuyên bố rằng trẻ em dưới 10 tuổi không nên được xác định là mắc tình trạng này. Đối với trẻ em trên 10 tuổi, thành phần béo phì của định nghĩa là chu vi vòng eo chứ không phải BMI, cho thấy tầm quan trọng lâm sàng của mỡ trong ổ bụng. Hội chứng chuyển hóa ảnh hưởng đến khoảng 25% dân số trưởng thành ở Hoa Kỳ. Do tỷ lệ mắc rộng rãi, hội chứng chuyển hóa có tầm quan trọng to lớn về mặt lâm sàng và sức khỏe cộng đồng, ngay cả ở những giai đoạn sớm nhất. Mặc dù vẫn còn tranh cãi, một sơ đồ về sinh lý bệnh của hội chứng chuyển hóa được trình bày trong Hình 24.7. Theo mô hình này, tác động của béo phì được quyết định bởi mô hình phân bố lipid—nghĩa là, các kho cụ thể mà mỡ thừa được lưu trữ. Mô hình lưu trữ lipid này quyết định hồ sơ bài tiết adipocytokine, nồng độ cytokine gây viêm trong tuần hoàn và dòng chảy của FFA. Tác động kết hợp của các yếu tố này quyết định độ nhạy của các cơ quan đích của insulin (như cơ và gan) đối với insulin và tác động đến hệ mạch máu bằng cách ảnh hưởng đến chức năng nội mô. Kháng insulin ngoại vi và rối loạn chức năng nội mô là những yếu tố thúc đẩy sớm của bệnh lý rõ ràng, cuối cùng dẫn đến T2DM và bệnh tim mạch. Bất kể định nghĩa hội chứng chuyển hóa nào được sử dụng, kháng insulin và nồng độ insulin cao đều liên quan đến sự phân cụm của các nguy cơ tim mạch chuyển hóa liên quan đến hội chứng chuyển hóa ở nhiều nhóm dân tộc khác nhau. Cần lưu ý rằng khi nghiên cứu một quần thể (không nhất thiết là một cá nhân), mức độ béo phì ngày càng tăng ở trẻ em có liên quan đến nguy cơ lớn hơn về sự hiện diện của các yếu tố nguy cơ tim mạch, tuy nhiên nguy cơ này dường như chững lại trên một ngưỡng béo phì tương ứng với 40 kg/m² ở người lớn.
Bệnh gan nhiễm mỡ không do rượu
Bệnh gan nhiễm mỡ không do rượu (NAFLD) là tình trạng thâm nhiễm mỡ ở gan trong trường hợp không tiêu thụ rượu. Phổ của NAFLD bao gồm từ thâm nhiễm mỡ đơn thuần (gan nhiễm mỡ) đến viêm (viêm gan nhiễm mỡ không do rượu, hay NASH), đến xơ hóa và thậm chí xơ gan. NAFLD được phát hiện trong khảo sát NHANES III là phổ biến hơn ở nam giới người Mỹ gốc Phi và gốc Tây Ban Nha bị béo phì, T2DM, tăng huyết áp và tăng lipid máu. Những mối liên quan này đã dẫn đến giả thuyết rằng NAFLD là một dấu hiệu sớm của sự hiện diện kháng insulin và xuất hiện trước khi phát triển bệnh đái tháo đường rõ ràng. NAFLD hiện là bệnh gan phổ biến nhất ở trẻ em ở Bắc Mỹ. NAFLD ở trẻ em có liên quan đến sự lắng đọng mỡ nội tạng gia tăng, và có thể tiến triển thành xơ gan và các biến chứng liên quan. Mối liên quan giữa béo phì bụng và gan nhiễm mỡ có thể được giải thích một phần bởi sự tiếp xúc kéo dài của gan với dòng chảy FFA tăng lên từ kho nội tạng. NAFLD có thể là một biểu hiện sớm của sự lắng đọng lipid lạc chỗ trong gan và là một thách thức đối với bác sĩ lâm sàng do sự tương phản giữa các biểu hiện sớm tối thiểu và các kết quả nghiêm trọng tiềm tàng của nó. Các nghiên cứu sử dụng phương pháp kẹp insulin-đường huyết đẳng trương cho thấy NAFLD có liên quan đến kháng insulin ở gan và ngoại vi.
Insulin đóng một vai trò quan trọng trong việc điều hòa các yếu tố phiên mã, chẳng hạn như SREBP-1c, được biểu hiện nhiều ở gan. SREBP-1c là yếu tố then chốt trong việc kiểm soát quá trình tân tạo mỡ ở gan và tăng tỷ lệ thuận với nồng độ insulin trong tuần hoàn. Những dữ liệu này làm dấy lên khả năng rằng tình trạng tăng insulin máu lúc đói có thể góp phần gây ra gan nhiễm mỡ, thay vì ngược lại. Ngoài ra, các cytokine gây viêm do mỡ nội tạng hoặc các tế bào phản ứng miễn dịch của gan giải phóng có thể góp phần làm thay đổi chuyển hóa lipid ở gan. Người ta đã chỉ ra rằng các SNP cụ thể, chẳng hạn như SNP rs58542926 trong gen TM6SF2, có liên quan đến NAFLD ở trẻ em, giải thích tại sao một số trẻ béo phì chứ không phải tất cả đều phát triển kiểu hình này. Phần lớn bệnh nhân có lẽ bị NAFLD mà không tiến triển thành NASH. Có khả năng tình trạng viêm sau đó do sự hình thành ROS tăng lên mà không được dập tắt thích hợp là cần thiết để thúc đẩy sự tiến triển thành NASH (lý thuyết được gọi là cú đánh thứ hai). Hiện tại, NAFLD có thể được phỏng đoán bằng men gan alanine aminotransferase (ALT) tăng cao ở trẻ béo phì. Tuy nhiên, ALT không nhất thiết phải tăng rất cao; bách phân vị thứ 95 của ALT ở trẻ em là 25,8 U/mL đối với bé trai và 22,1 U/mL đối với bé gái. Quan trọng là, nồng độ ALT bình thường không loại trừ sự hiện diện của NAFLD.
Hội chứng buồng trứng đa nang
Sự kết hợp của cường androgen và thiểu kinh hoặc vô kinh ở nữ giới, được gọi là hội chứng buồng trứng đa nang (PCOS), là một bệnh đi kèm thường gặp của béo phì, có thể kéo dài đến thời thơ ấu. Rối loạn này được đề cập chi tiết trong Chương 16. Chẩn đoán PCOS phải dựa trên sự hiện diện của ít nhất hai trong ba tiêu chí sau: không rụng trứng mãn tính, cường androgen (lâm sàng hoặc sinh học), và buồng trứng đa nang. PCOS là nguyên nhân phổ biến nhất gây vô sinh do không rụng trứng và là một yếu tố nguy cơ chính cho sự phát triển của hội chứng chuyển hóa và rối loạn chuyển hóa glucose ở nữ giới. Các tiền đề của PCOS đã được xác định ở các bé gái tiền dậy thì, cho thấy một tổn thương phát triển.
Các bé gái vị thành niên mắc PCOS có thể bị kháng insulin từ trung bình đến nặng với nguy cơ gia tăng rối loạn chuyển hóa glucose và sự suy giảm độ nhạy insulin rõ rệt hơn ở những người béo phì so với những bé gái mắc PCOS nhưng gầy. Thông thường, các bé gái mắc PCOS và béo phì thể hiện sự phân bố lipid bị thay đổi bao gồm hàm lượng lipid nội tạng và gan cao, cũng như giảm huy động lipid, giảm oxy hóa chất béo và sự thiếu linh hoạt trong chuyển hóa. Ở một số nhóm dân tộc, các bé gái có dậy thì sớm, một tiền đề tiềm năng của PCOS, có nồng độ insulin tương đối tăng, do đó một mối liên hệ nhân quả giữa tăng insulin máu và tăng tiết androgen (có nguồn gốc từ tuyến thượng thận hoặc buồng trứng) đã được giả định. Các nghiên cứu quần thể về các bé gái bình thường đã chỉ ra rằng việc tăng cân nhanh có liên quan đến nồng độ androgen tuyến thượng thận và lượng mỡ cơ thể cao hơn, và tình trạng tăng insulin máu có liên quan đến kinh nguyệt sớm. Do đó, mối liên quan của nồng độ insulin cao hơn với dậy thì sớm và PCOS sau đó có thể được thúc đẩy, ít nhất một phần, bởi béo phì. Béo phì đặc trưng cho khoảng 50% phụ nữ mắc PCOS cổ điển, mặc dù nó thậm chí còn phổ biến hơn ở thanh thiếu niên. Kháng insulin ngoại vi tăng xảy ra ở khoảng 50% bệnh nhân mắc PCOS, và gần như chắc chắn đóng một vai trò trong cơ chế bệnh sinh của tình trạng này. Mặt khác, hầu hết các dạng kháng insulin nặng, chẳng hạn như T2DM hoặc các hội chứng loạn dưỡng mỡ hiếm gặp, cũng có liên quan đến PCOS. Đáng chú ý, kháng insulin chưa được đưa vào làm tiêu chí chẩn đoán cho PCOS chủ yếu do khó khăn trong việc đo lường nó. Tăng insulin máu lúc đói và đáp ứng bài tiết insulin tăng đối với tải glucose đường uống đã được chứng minh ở các bé gái mắc PCOS. Thật vậy, các bé gái vị thành niên mắc PCOS và béo phì đã được chứng minh là kháng insulin hơn 50% so với nhóm đối chứng có cùng cân nặng mà không mắc PCOS. Sự kết hợp của các bất thường chuyển hóa thường thấy ở những người kháng insulin thường gặp ở thanh thiếu niên mắc PCOS và béo phì, bao gồm NAFLD và T2DM. Tỷ lệ mắc hội chứng chuyển hóa gia tăng có thể liên quan đến tình trạng cường androgen độc lập với kháng insulin liên quan đến béo phì. Các dấu hiệu sớm của xơ vữa động mạch tăng tốc đã có mặt ở những phụ nữ trẻ mắc PCOS, cho thấy rằng can thiệp sớm nhằm giảm nguy cơ tim mạch có thể có lợi. Quan trọng là, giảm cân ở phụ nữ mắc PCOS có liên quan đến chức năng kinh nguyệt được cải thiện.
Kiểm tra chuyển hóa ở bệnh nhân PCOS cho thấy sự kháng cự ở gan và cơ, nhưng không có kháng insulin ở buồng trứng; có thể giải thích cho sự kích thích của insulin đối với việc sản xuất androgen của tế bào vỏ trong. Mối tương quan giữa kháng insulin và cường androgen đòi hỏi một giả thuyết thống nhất về cơ chế bệnh sinh của chúng, được đưa ra bởi “giả thuyết phosphoryl hóa serine”, cho rằng cả P450c17 và thụ thể insulin đều bị phosphoryl hóa serine một cách bất thường; trong trường hợp của P450c17, điều này dẫn đến hoạt động quá mức và tăng sản xuất androgen, và trong trường hợp của thụ thể insulin, điều này dẫn đến kháng insulin đặc hiệu cho mô. Tuy nhiên, giả thuyết này vẫn cần được chứng minh.
Các bệnh đi kèm nội tiết khác
Béo phì gây ra những thay đổi trong các hệ thống nội tiết tố khác, một số trong đó gây ra các bệnh lý cụ thể. Tuổi bắt đầu dậy thì đã ngày càng sớm hơn, đặc biệt là ở các bé gái người Mỹ gốc Phi. Sự tiến triển này được giải thích một phần bởi tình trạng dinh dưỡng thừa và chỉ số BMI ngày càng tăng trong quần thể này. Vô sinh ở thanh thiếu niên lớn tuổi và phụ nữ trưởng thành có thể xảy ra như một biểu hiện của PCOS do sản xuất androgen buồng trứng quá mức ở nữ giới, hoặc do sự thơm hóa quá mức của androgen thành estrogen bởi mô mỡ ngoại vi với sự ức chế trục hạ đồi-tuyến yên-tuyến sinh dục ở cả hai giới. Tăng estrogen máu cũng có thể thúc đẩy nữ hóa tuyến vú ở nam giới. Ngoài ra, tăng CO2 máu liên quan đến OSA có thể ức chế chức năng hormone gonadotropin (GnRH) của vùng dưới đồi, dẫn đến hội chứng dậy thì muộn.
Béo phì có liên quan đến giảm bài tiết GH, và thực tế hầu hết các đối tượng béo phì, mặc dù có sự tăng trưởng chiều cao bình thường hoặc quá mức, đều không đáp ứng với các xét nghiệm kích thích GH. Tuy nhiên, hạn chế calo trong 24 giờ có thể phục hồi khả năng đáp ứng GH bình thường. Mặc dù có sự thiếu hụt GH chức năng, sự tăng trưởng chiều cao được tăng tốc, tuổi xương tiến triển, và nồng độ yếu tố tăng trưởng giống insulin (IGF)-1 toàn phần và tự do ở ngoại vi là bình thường hoặc tăng cao trong béo phì, cho thấy độ nhạy GH bình thường hoặc tăng cường, hoặc có thể do sự ức chế protein gắn yếu tố tăng trưởng giống insulin (IGFBP)-1, và tác động của tăng insulin máu lên sự kích hoạt thụ thể IGF-1 của sụn tăng trưởng. Nồng độ thyroxine tự do có xu hướng thấp hơn và TSH cao hơn ở trẻ béo phì, mặc dù chủ yếu trong phạm vi bình thường cùng với một số mức TSH trong danh mục suy giáp cận lâm sàng; cơ chế vẫn chưa được biết. Nồng độ TSH tăng cao trong béo phì dường như là một hậu quả hơn là một nguyên nhân của béo phì. Do đó, điều trị tăng thyrotropin máu bằng thyroxine dường như không cần thiết ở trẻ béo phì. Cuối cùng, béo phì có thể liên quan đến việc tăng tiếp xúc với cortisol, có thể do sự chuyển đổi cortisone lưu thông thành cortisol bởi enzyme 11-β-hydroxysteroid dehydrogenase loại 1 (11β HSD1) nằm trong các tế bào mỡ.
Các bệnh đi kèm phi nội tiết khác
Béo phì ở trẻ em có liên quan đến nhiều bệnh đi kèm khác. U giả não là một tình trạng hiếm gặp và ít được hiểu rõ, dẫn đến tăng áp lực nội sọ, với các biểu hiện bao gồm phù gai thị và đau đầu. Điều trị bao gồm chọc dịch não tủy hàng loạt, acetazolamide để giảm sản xuất dịch não tủy, trong những trường hợp nặng cần có shunt tâm thất-ổ bụng, và đôi khi cần phải mở cửa sổ vỏ dây thần kinh thị giác để cứu thị lực. OSA xảy ra thường xuyên ở trẻ béo phì nặng; có lẽ do lượng lớn mỡ sau họng chèn ép đường thở trên trong khi ngủ. Bệnh nhân bị ảnh hưởng ngáy, thường ngừng thở hơn 20 giây trong khi ngủ và thức dậy vào ban đêm với cơn đau đầu. Điều trị bao gồm áp lực đường thở dương liên tục vào ban đêm và, khi thích hợp, cắt amidan-VA; tuy nhiên, các triệu chứng thường tái phát. Trẻ em béo phì biểu hiện nhiều khó khăn về chỉnh hình, bao gồm gãy xương, đau đầu gối, lệch trục chi dưới về mặt giải phẫu và suy giảm khả năng vận động. Sỏi mật xảy ra ở khoảng 2,5% thanh thiếu niên béo phì, đặc biệt là ở nữ giới, nhưng thường không thấy ở trẻ em béo phì tiền dậy thì. Cuối cùng, tình trạng đau khổ tâm lý, bao gồm trầm cảm lâm sàng, được biểu hiện rõ ràng ở trẻ béo phì và đặc biệt ở những trẻ béo phì nặng. Các bệnh đi kèm khác nhau này dường như đều có liên quan đến điểm z-score của BMI theo một kiểu đường cong; do đó, béo phì càng nặng, bệnh nhân càng có nhiều khả năng biểu hiện bệnh đi kèm.
Các yếu tố liên quan đến dịch béo phì hiện nay
Di truyền
Mối liên quan giữa béo phì và di truyền học bắt nguồn từ hai dòng điều tra riêng biệt: (1) những khám phá về các rối loạn đơn gen của con đường cân bằng năng lượng (xem sau) và (2) các nghiên cứu về các nhóm chủng tộc và dân tộc cụ thể, trong đó béo phì dường như phân ly, chẳng hạn như người da đỏ Pima và người gốc Tây Ban Nha ở Tây Nam Hoa Kỳ. Những quan sát này được kết hợp với một lý thuyết hấp dẫn về sự chọn lọc tự nhiên của các cá thể để đối phó với áp lực môi trường/sinh thái khắc nghiệt (tức là nạn đói), được gọi là giả thuyết gen tiết kiệm, để tạo ra một động lực rất mạnh mẽ cho việc làm sáng tỏ các vị trí di truyền cụ thể trong cơ chế bệnh sinh của béo phì. Tuy nhiên, quy mô thời gian nhanh chóng của sự gia tăng tỷ lệ béo phì ở trẻ em không thể phản ánh một sự thay đổi di truyền của quần thể. Do đó, mô hình hiện tại là béo phì là kết quả của sự tương tác giữa gen và môi trường; một sự chọn lọc di truyền cổ đại để tích trữ mỡ một cách hiệu quả có thể đã mang lại lợi thế sinh tồn trong thời kỳ trước đây nhưng lại không thích nghi với sự dư thừa thực phẩm hiện tại của chúng ta. Một cách tiếp cận tiến hóa đối với sự phát triển của béo phì liên quan đến một chiều kích bộ gen/nhân chủng học. Trong hàng triệu năm, lối sống của những người săn bắt-hái lượm bao gồm hoạt động thể chất cường độ cao và chế độ ăn nhiều protein/ít carbohydrate và bộ gen của tổ tiên chúng ta đã được thích nghi với độ nhạy insulin thấp. Khi nông nghiệp phát triển, một chế độ ăn nhiều carbohydrate xuất hiện và kể từ cuộc cách mạng công nghiệp, bộ gen của chúng ta đang nhanh chóng thích nghi với chế độ ăn như vậy. Bộ gen hiện tại của chúng ta đại diện cho sự pha trộn của cả hai. Khi tiếp xúc với sự dư thừa năng lượng do môi trường thực phẩm hiện tại gây ra, những bộ gen này có thể biểu hiện thành sự phát triển của béo phì ở các thời điểm khác nhau và có thể phản ứng khác nhau với các chế độ ăn cụ thể.
Trong các dạng béo phì phổ biến, việc liên kết các SNP với các nguy cơ liên quan đến béo phì là rất khó khăn vì các tác động không chắc chắn và kết quả không phải lúc nào cũng được xác nhận. Một số SNP trong các gen cụ thể đã được xác định, chẳng hạn như gen liên quan đến khối lượng mỡ và béo phì (FTO). Sự biến đổi trong gen FTO đã cung cấp các mối liên quan mạnh mẽ nhất với béo phì phổ biến cho đến nay, tuy nhiên biến thể FTO mang lại khuynh hướng béo phì dường như không liên quan đến việc điều hòa tiêu hao năng lượng mà có thể có vai trò trong việc kiểm soát lượng thức ăn nạp vào và lựa chọn thực phẩm, cho thấy một mối liên hệ với kiểu hình ăn nhiều hoặc sở thích đối với các loại thực phẩm giàu năng lượng. Các nghiên cứu liên kết toàn bộ bộ gen (GWAS) lớn cho đến nay đã mang lại hơn 300 locus có thể liên quan đến sự phát triển của béo phì ở người lớn, nhưng chỉ giải thích được chưa đến 5% kiểu hình.
Biểu sinh và Lập trình phát triển
Các nghiên cứu theo dõi trẻ sơ sinh sinh ra nhỏ so với tuổi thai (SGA), lớn so với tuổi thai (LGA), và sinh non, đã ghi nhận nguy cơ béo phì và hội chứng chuyển hóa tăng lên rõ rệt. “Giả thuyết nguồn gốc từ thai nhi” cho rằng một số khía cạnh của môi trường trong tử cung góp phần vào sự phát triển của béo phì và các bệnh mãn tính trong cuộc sống sau này. Quan trọng là, mỗi trong ba tình trạng trước sinh này đều có liên quan đến kháng insulin. “Mô hình phát triển” của các bệnh mãn tính cho rằng các sự kiện đầu đời ảnh hưởng đến sự khác biệt cá nhân về tính dễ bị tổn thương đối với lối sống và môi trường. Một cơ chế khả thi cho việc lập trình phát triển như vậy bao gồm các thay đổi biểu sinh, có thể góp phần vào những thay đổi trong biểu hiện gen. Ví dụ, mô hình methyl hóa DNA được ghi nhận trong máu cuống rốn dự đoán mức độ tích mỡ ở tuổi 9.
Việc ghi nhận mối quan hệ của SGA với béo phì và bệnh tim mạch ở người lớn bắt đầu bằng các nghiên cứu về nạn đói ở Hà Lan trong Thế chiến II và hậu quả của nó. Một số nghiên cứu về trẻ sơ sinh sinh ra SGA cho thấy chúng bị tăng insulin máu và kháng insulin khi sinh, thể hiện sự tăng trưởng bắt kịp nhanh chóng trong giai đoạn sau sinh sớm, và phát triển béo phì ở thời thơ ấu, tình trạng này vẫn tồn tại và thúc đẩy kháng insulin dai dẳng. Một phân tích về trẻ sơ sinh Ấn Độ sinh ra ở Ấn Độ so với Vương quốc Anh đã chứng minh rằng mặc dù những trẻ sinh ra ở Ấn Độ nhẹ hơn 700g khi sinh, nồng độ glucose và insulin của chúng tăng lên rõ rệt. Sau khi điều chỉnh theo cân nặng khi sinh, các em bé sinh ra ở Ấn Độ thể hiện tình trạng tích mỡ tăng, insulin cao hơn 4 lần, nồng độ leptin cao hơn 2 lần so với các em bé sinh ra ở Anh. Do đó, những em bé này đã kháng insulin ngay cả khi sinh, điều này dẫn đến tăng tích mỡ. Theo dõi những em bé như vậy đến thời thơ ấu, có rất nhiều nghiên cứu ghi nhận tình trạng kháng insulin trong thời thơ ấu.
Trẻ sinh ra LGA thường bị tăng insulin máu khi sinh. Mặc dù hầu hết trẻ LGA là do đái tháo đường thai kỳ (GDM) và tiếp xúc với béo phì và tăng đường huyết của mẹ trong suốt thai kỳ, nhưng đây không phải lúc nào cũng là nguyên nhân. Theo dõi trẻ LGA không có GDM cho thấy tỷ lệ kháng insulin và hội chứng chuyển hóa tăng gấp đôi, trong khi trẻ LGA do GDM biểu hiện sự gia tăng gấp ba lần. Người ta đã chỉ ra rằng thanh thiếu niên béo phì đã tiếp xúc trong tử cung với GDM có khả năng kháng insulin cao hơn và có chức năng tế bào β bị suy giảm so với những người cùng lứa tuổi có mức độ béo phì tương đương mà không có sự tiếp xúc như vậy. Thật vậy, sự lây truyền “theo chiều dọc” của bệnh đái tháo đường từ mẹ sang con dưới dạng béo phì và đái tháo đường sau này đã được ghi nhận trong các nghiên cứu về người da đỏ Pima. Cuối cùng, việc tăng cân trong thai kỳ làm tăng cân nặng khi sinh, nguy cơ LGA, và béo phì ở con cái (Hình 24.9).
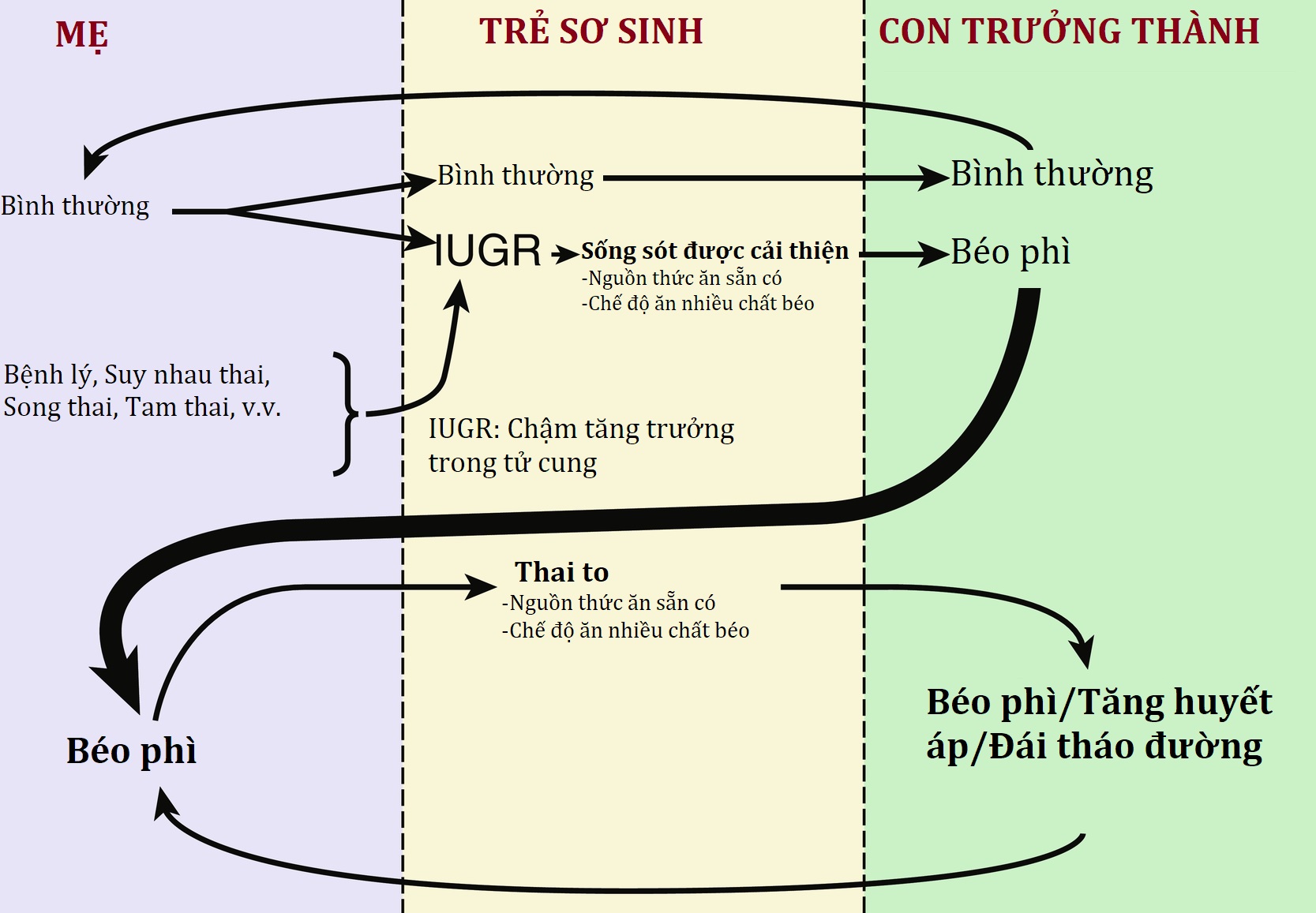
Hình 24.9 Các cơ chế được giả định mà qua đó việc lập trình phát triển (hoặc chậm phát triển trong tử cung hoặc thai to sơ sinh) có thể dẫn đến béo phì ở người lớn ở con cái.
Tác dụng bảo vệ của việc cho con bú chống lại sự phát triển của bệnh béo phì trong tương lai đã được biết đến từ lâu, và dường như có một mối quan hệ liều lượng-đáp ứng; cho con bú càng lâu, tác dụng bảo vệ càng lớn. Tuy nhiên, điều này có thể bị phức tạp hóa bởi các yếu tố gây nhiễu, chẳng hạn như tình trạng kinh tế xã hội, mẹ hút thuốc trong thai kỳ và chỉ số BMI của mẹ. Cơ chế tác dụng chống béo phì của việc cho con bú cũng chưa rõ ràng. Một số người cho rằng sự tự điều chỉnh việc ăn của trẻ sơ sinh là phù hợp nhất, trong khi một nghiên cứu gần đây cho thấy rằng leptin trong sữa mẹ có thể góp phần vào sự bảo vệ này, và cuối cùng, việc cho con bú của mẹ có thể định hình hệ vi sinh vật đường ruột của con cái thành một hồ sơ khác với hồ sơ do tiêu thụ sữa công thức gây ra.
Các yếu tố môi trường
Nhiều yếu tố môi trường cũng có liên quan đến dịch béo phì, đặc biệt là ở trẻ em. Tuy nhiên, hầu hết các mối liên quan này bắt nguồn từ các nghiên cứu cắt ngang hơn là các nghiên cứu theo chiều dọc, và trong nhiều trường hợp, cơ chế vẫn còn thiếu. Một số nghiên cứu theo chiều dọc ở người lớn đã chứng minh rõ ràng rằng các hành vi ăn kiêng và lối sống cụ thể khác có liên quan độc lập đến việc tăng cân dài hạn, với một tác động tổng hợp đáng kể. Ví dụ, dựa trên việc tăng khẩu phần hàng ngày của các thành phần ăn kiêng riêng lẻ, sự thay đổi cân nặng trong 4 năm có liên quan mạnh mẽ nhất với việc ăn khoai tây chiên (0,767 kg), khoai tây (0,58 kg), đồ uống có đường (0,45 kg), thịt đỏ chưa qua chế biến (0,43 kg), và thịt đã qua chế biến (0,42 kg) và có liên quan nghịch với việc ăn rau (-0,1 kg), ngũ cốc nguyên hạt (-0,168 kg), trái cây (-0,224 kg), các loại hạt (-0,25 kg), và sữa chua (-0,37 kg) (P ≤ 0,005 cho mỗi so sánh). Tương tự, môi trường kinh tế xã hội đã được chứng minh là ảnh hưởng đến khả năng bị béo phì ở tuổi trưởng thành. Thật vậy, cơ hội chuyển từ một khu phố có tỷ lệ nghèo đói cao sang một khu phố ít nghèo đói hơn có liên quan đến việc giảm khiêm tốn nhưng có thể quan trọng về tỷ lệ béo phì cực độ và đái tháo đường. Các mối quan hệ tương tự có thể xảy ra, nhưng chưa được chứng minh, đối với trẻ em và thanh thiếu niên.
Căng thẳng và Cortisol
Ở người, nồng độ cortisol tăng cao hoặc các dấu hiệu rối loạn điều hòa trục hạ đồi, tuyến yên và tuyến thượng thận (HPA) có tương quan với sự phân bố mỡ bụng và hội chứng chuyển hóa. Mặc dù cortisol trong tuần hoàn rõ ràng là quan trọng trong việc xác định mỡ nội tạng, việc xác định gần đây về sự giảm cortisone trong tuần hoàn thành cortisol trong mô mỡ nội tạng bởi enzyme 11βHSD1 cũng đã được liên kết với hội chứng chuyển hóa. Những dữ liệu này cho thấy rằng cortisol quan trọng cả trong việc tăng mỡ nội tạng và thúc đẩy hội chứng chuyển hóa. Mối liên hệ cơ chế giữa căng thẳng và béo phì chưa được làm rõ, một phần do sự phức tạp vốn có của việc đánh giá một tác động hai chiều tiềm tàng của căng thẳng đối với việc ăn uống và trọng lượng cơ thể. Các nghiên cứu tập trung vào chuyển hóa mô mỡ nâu ủng hộ một mối quan hệ lưỡng phân để giải thích tác động của căng thẳng đối với béo phì: căng thẳng thúc đẩy béo phì khi có tình trạng ăn nhiều và chức năng mô mỡ nâu ổn định, trong khi căng thẳng dẫn đến giảm cân hoặc bảo vệ khỏi sự phát triển của béo phì khi có tình trạng ăn ít hoặc khi lượng calo nạp vào tăng có liên quan đến việc huy động mô mỡ nâu và tăng cường sinh nhiệt (nâu hóa các tế bào mỡ trắng).
Bằng chứng về mối liên quan giữa cortisol tăng cao và tình trạng đau khổ tâm lý với sự phân bố mỡ bụng ở người lớn là rất thuyết phục. Ví dụ, sự bài tiết glucocorticoid trong nước tiểu có liên quan đến các khía cạnh của hội chứng chuyển hóa, bao gồm huyết áp, đường huyết lúc đói, insulin và chu vi vòng eo. Dường như một số cá nhân dường như là “người phản ứng cao” với một kích thích căng thẳng và thể hiện sự bài tiết cortisol cao hơn. Những cá nhân này dường như dễ bị thay đổi trong việc nhận biết cảm giác no và tiêu thụ lượng calo lớn hơn sau khi tiếp xúc với căng thẳng (xem Hình 24.5). Những dữ liệu này cho thấy rằng cortisol quan trọng cả trong việc tăng mỡ nội tạng và thúc đẩy kháng insulin, tương đương với “hội chứng Cushing của bụng”.
Thiếu ngủ
Người lớn ở Hoa Kỳ hiện trung bình ngủ ít hơn 7 giờ mỗi đêm—ít hơn gần 2 giờ so với năm 1980—và khoảng một phần ba trong số họ ngủ ít hơn 6 giờ mỗi đêm. Các phân tích dữ liệu từ NHANES I, cho thấy rằng những người trưởng thành (tuổi từ 32–49) ngủ ít hơn 7 giờ có nhiều khả năng bị béo phì hơn từ 5 đến 8 năm sau so với những người ngủ từ 7 giờ trở lên. Mối liên hệ giữa thời gian ngủ ngắn và béo phì cũng đã được quan sát thấy ở trẻ em. Các nghiên cứu theo chiều dọc ở trẻ em, với các đối tượng từ các hoàn cảnh đa dạng, cho thấy một mối liên quan nghịch giữa thời gian ngủ và chỉ số BMI. Trong 24.821 người tham gia, các đối tượng nhi khoa ngủ trong thời gian ngắn có nguy cơ bị thừa cân/béo phì cao gấp đôi so với các đối tượng ngủ trong thời gian dài (tỷ số chênh 2,15; 95% CI, 1,64-2,81). Giống như người lớn, ngày càng nhiều trẻ em bị thiếu ngủ mãn tính. Điều này đặc biệt đúng với trẻ béo phì, những trẻ được phát hiện là ngủ ít hơn những trẻ có cân nặng bình thường. Ngoài các tác động khác của nó, giấc ngủ là một trong những yếu tố dự báo mạnh mẽ nhất theo cả chiều ngang và chiều dọc về béo phì ở trẻ em tiền dậy thì.
Xem truyền hình và “Thời gian sử dụng thiết bị điện tử”
Xem truyền hình được coi là một trong những nguyên nhân có thể thay đổi nhất của béo phì ở trẻ em. Có bốn cơ chế khả thi liên kết việc xem truyền hình và béo phì. Thứ nhất, xem truyền hình có thể làm tăng mức độ căng thẳng và cortisol (xem ở trên), gây tăng lượng thức ăn nạp vào và thúc đẩy béo phì. Thứ hai, xem truyền hình thay thế hoạt động thể chất. Hầu hết, nhưng không phải tất cả các nghiên cứu đều tìm thấy mối tương quan nghịch giữa việc xem truyền hình và hoạt động thể chất cũng như thể lực. Thứ ba, xem truyền hình làm tăng lượng calo tiêu thụ từ việc ăn trong khi xem hoặc từ tác động của quảng cáo thực phẩm. Việc xem truyền hình cũng có liên quan đến việc tăng ăn thực phẩm nhiều chất béo, giảm tiêu thụ trái cây và rau quả, và tăng uống nước ngọt. “Thực phẩm rác” là loại sản phẩm được quảng cáo thường xuyên nhất trên truyền hình dành cho trẻ em. Cuối cùng, REE và NEAT dường như giảm trong khi xem truyền hình. Theo NHANES III, tỷ lệ béo phì ở trẻ em thấp nhất ở những trẻ xem truyền hình từ 1 giờ trở xuống mỗi ngày, và cao nhất ở những trẻ xem từ 4 giờ trở lên mỗi ngày. Một số nghiên cứu thực nghiệm về việc giảm xem truyền hình đã được tiến hành và kết quả của chúng ủng hộ gợi ý rằng việc giảm xem truyền hình có thể giúp giảm nguy cơ béo phì hoặc giúp thúc đẩy giảm cân ở trẻ béo phì. Những nghiên cứu này đại diện cho bằng chứng trực tiếp mạnh mẽ nhất cho thấy rằng chỉ riêng việc thay đổi việc xem truyền hình là một chiến lược đầy hứa hẹn để phòng ngừa béo phì ở trẻ em. Các hình thức sử dụng màn hình khác, chẳng hạn như điện thoại di động, cũng có liên quan đến cơ chế bệnh sinh của béo phì. Người ta đã chỉ ra rằng việc cha mẹ giám sát việc tiếp xúc với phương tiện truyền thông của con cái dự đoán điểm z-score BMI của trẻ thấp hơn ở tuổi 7 và điểm z-score BMI của trẻ tăng ít dốc hơn từ 5 đến 9 tuổi, do đó các hành vi của cha mẹ liên quan đến việc tiêu thụ phương tiện truyền thông của trẻ em có thể có tác động lâu dài đến chỉ số BMI của trẻ em ở tuổi giữa thời thơ ấu.
Các yếu tố ăn kiêng
Calo từ bất kỳ loại thực phẩm nào cũng có khả năng làm tăng nguy cơ béo phì và bệnh tim mạch chuyển hóa vì tất cả calo đều có thể trực tiếp góp phần vào cân bằng năng lượng dương và tăng mỡ. Tuy nhiên, các thành phần ăn kiêng khác nhau có thể thúc đẩy béo phì và bệnh tim mạch chuyển hóa bằng các cơ chế bổ sung không chỉ qua trung gian hàm lượng calo của chúng. Về tác động sức khỏe của các yếu tố ăn kiêng cụ thể, người ta đã chỉ ra rằng các axit béo bão hòa đặc trưng cho thực phẩm và đồ uống có đường thúc đẩy các bệnh tim mạch chuyển hóa bằng các cơ chế bổ sung cho sự đóng góp calo của chúng vào cân bằng năng lượng dương. Tác động và phản ứng chuyển hóa đối với một số thành phần ăn kiêng bị ảnh hưởng bởi tình trạng chuyển hóa, giai đoạn phát triển hoặc kiểu gen của cá nhân; bởi khả năng đáp ứng của các vùng não liên quan đến phần thưởng đối với các tín hiệu thực phẩm; và có thể bởi hệ vi sinh vật.
Chất béo và Carbohydrate trong chế độ ăn
Chất béo thường được coi là gây béo phì nhiều hơn các chất dinh dưỡng đa lượng khác, vì nó giàu năng lượng hơn, ngon miệng hơn và được chuyển hóa thành mỡ cơ thể hiệu quả hơn. Một bữa ăn nhiều chất béo gây ra ít sinh nhiệt hơn và cân bằng chất béo dương cao hơn so với một bữa ăn ít chất béo có cùng calo và protein. Lượng chất béo ăn vào quá mức được cho là gây tăng cân, nhưng mối quan hệ giữa lượng chất béo ăn vào và tình trạng tích mỡ ở trẻ em vẫn còn gây tranh cãi.
Tỷ lệ thừa cân ở Hoa Kỳ đã tăng lên mặc dù tỷ lệ năng lượng ăn kiêng có nguồn gốc từ chất béo đã giảm. Một phân tích tổng hợp của 12 nghiên cứu ở người lớn thừa cân hoặc béo phì được tư vấn về chế độ ăn ít chất béo và theo dõi trong 6 đến 18 tháng cho thấy rằng chế độ ăn ít chất béo không tốt hơn chế độ ăn hạn chế calo trong việc giảm cân dài hạn. Tương tự, ở trẻ em, tổng lượng chất béo tiêu thụ được biểu thị bằng phần trăm năng lượng nạp vào đã giảm. Sự giảm tiêu thụ chất béo này phần lớn là do sự gia tăng tổng năng lượng nạp vào dưới dạng carbohydrate. Phần lớn sự mất cân bằng này được cho là do sự thay đổi mô hình tiêu thụ đồ uống, được đặc trưng bởi việc giảm uống sữa và tăng đáng kể việc tiêu thụ nước ngọt, điều này có thể có cơ chế bệnh sinh riêng (xem sau). Hầu hết các can thiệp với chế độ ăn ít chất béo, tốt cho tim mạch, đều không thành công trong việc phòng ngừa thừa cân ở trẻ em.
Việc giảm lượng carbohydrate nạp vào được thực hiện đến mức cực đoan trong các chế độ ăn ketogenic, chẳng hạn như chế độ ăn Atkins, hạn chế người lớn ăn ít hơn 25 g carbohydrate mỗi ngày. Các đánh giá về chế độ ăn này ở người lớn đã gây thất vọng về lâu dài, và chế độ ăn phổ biến này gần đây đã bị từ bỏ. Hiện không có dữ liệu ở trẻ em hoặc thanh thiếu niên. Tuy nhiên, cần lưu ý rằng chế độ ăn ketogenic được sử dụng để kiểm soát co giật có thành phần tương tự như chế độ ăn Atkins. Một nghiên cứu kéo dài 2 năm về chế độ ăn ketogenic đã chứng minh sự giảm dai dẳng điểm z-score cân nặng ở trẻ em có cân nặng trên mức trung bình khi bắt đầu chế độ ăn, mà không ảnh hưởng đáng kể đến dinh dưỡng chung hoặc chiều cao.
Axit béo không bão hòa dạng Trans (Chất béo Trans)
Chất béo không bão hòa dạng trans trong thực phẩm chế biến sẵn đã là một mặt hàng chủ lực trong chế độ ăn phương Tây từ đầu thế kỷ 20. Điều này là do sự đồng phân hóa trans của liên kết đôi ngăn chặn sự phân hủy axit béo bởi vi khuẩn, kéo dài thời hạn sử dụng của các sản phẩm thực phẩm. Giống như các tiền thân vi khuẩn của nó, ty thể của con người không thể subjecting chất béo trans vào quá trình oxy hóa β trong gan, góp phần vào sự tích tụ lipid lạc chỗ trong gan. May mắn thay, do mối liên quan được công nhận giữa việc tiêu thụ chất béo trans và bệnh tim mạch vào giữa những năm 1980 và các yêu cầu ghi nhãn nghiêm ngặt hơn kể từ năm 2006, tỷ lệ calo từ chất béo trans được tiêu thụ trong chế độ ăn phương Tây đã giảm dần. Chất béo trans không có lợi cho sức khỏe và gây ra gan nhiễm mỡ và kháng insulin; tuy nhiên, xu hướng tiêu thụ hiện tại của chúng không tương xứng về mặt thời gian với sự gia tăng tỷ lệ hội chứng chuyển hóa hiện nay, cho thấy rằng các yếu tố khác có liên quan.
Chỉ số đường huyết và chất xơ
Không phải tất cả các loại đường đều gây ra phản ứng sinh insulin giống nhau. Carbohydrate phức tạp có thể có hai dạng: hoặc là sự kết hợp của các liên kết α1-4 và α1-6, tạo cho tinh bột một cấu trúc hình cầu được gọi là amylopectin, như thấy trong bánh mì, gạo, mì ống, khoai tây và glycogen; hoặc là một polyme tuyến tính của các liên kết α1-4 được gọi là amylose, như thấy trong đậu, đậu lăng và các loại đậu khác. Sự tiêu hóa và hấp thu của loại trước trong ruột diễn ra nhanh chóng do các hoạt động đồng thời của cả glucosidase α1-4 và α1-6, trong khi đó của loại sau chậm hơn nhiều vì glucosidase α1-4 chỉ có thể phân cắt các gốc glucose đơn lẻ ở hai bên của polyme. Hiện tượng này tạo thành cơ sở của chỉ số đường huyết (GIx), đề cập đến diện tích glucose tương đối dưới đường cong sau khi tiêu thụ (so với dextrose). Thực phẩm có GIx cao dẫn đến đáp ứng insulin tăng cao, có thể chuyển chất nền năng lượng đến mô mỡ. Ở trẻ em, các nghiên cứu có kiểm soát với chế độ ăn có GIx cao cho thấy lượng năng lượng nạp vào cao hơn 53% so với chế độ ăn có GIx thấp. Một nghiên cứu ở thanh thiếu niên đã chứng minh rằng một chế độ ăn có GIx thấp ad libitum hiệu quả hơn trong việc thúc đẩy giảm cân so với một chế độ ăn ít chất béo hạn chế năng lượng. Do đó, GIx có thể là một khái niệm đơn giản để thực hiện, mặc dù “môi trường độc hại” của các sản phẩm thực phẩm của Mỹ có thể khiến nó khó duy trì.
Chất xơ ăn kiêng bao gồm phần polysaccharide không phải tinh bột của thực phẩm thực vật, bao gồm cellulose, hemicellulose, pectin, β-glucan, fructan, gôm và polysaccharide tảo. Các nguồn chính của chất xơ ăn kiêng bao gồm ngũ cốc nguyên hạt, trái cây, rau, các loại đậu và các loại hạt. Hàm lượng chất xơ chiếm 50% sự thay đổi về tải lượng đường huyết (GL; GIx × thể tích) giữa các loại thực phẩm. Các nghiên cứu đoàn hệ ở người lớn cho thấy rằng lượng chất xơ nạp vào có liên quan nghịch với việc tăng cân, nồng độ insulin lúc đói và nguy cơ mắc T2DM. Chất xơ có thể ảnh hưởng đến việc điều hòa trọng lượng cơ thể bằng một số cơ chế liên quan đến các tác động nội tại, nội tiết tố và đại tràng, cuối cùng làm giảm lượng thức ăn nạp vào bằng cách thúc đẩy cảm giác no (hàm lượng năng lượng bữa ăn thấp hơn), cảm giác thỏa mãn (thời gian giữa các bữa ăn dài hơn), hoặc bằng cách tăng quá trình oxy hóa chất béo và giảm lưu trữ chất béo. Một bữa ăn giàu chất xơ được xử lý chậm hơn và có mật độ calo thấp hơn và ít chất béo và đường bổ sung hơn. Thực phẩm chứa chất xơ gây ra sự hấp thu glucose chậm hơn, làm giảm sự tăng vọt insulin sau bữa ăn và giảm quá trình tân tạo mỡ. Ngoài ra, các bữa ăn nhiều chất xơ cho phép cung cấp triglyceride chưa tiêu hóa đến đại tràng, nơi quá trình lên men thành các axit béo chuỗi ngắn và sự hấp thu của chúng cải thiện lipid và độ nhạy insulin. Các nhà khảo cổ học cho rằng tổ tiên của chúng ta đã tiêu thụ từ 100 đến 300 g chất xơ mỗi ngày. Tuy nhiên, lượng chất xơ ăn kiêng trong suốt thời thơ ấu và thanh thiếu niên hiện trung bình khoảng 12 g/ngày, và không thay đổi trong 30 năm qua. Do đó, cha mẹ và nhân viên phục vụ thực phẩm tại trường học nên cố gắng cung cấp các loại thực phẩm giàu chất xơ cho trẻ em để sự chấp nhận và tiêu thụ của chúng sẽ tăng lên.
Fructose
Chất làm ngọt được sử dụng phổ biến nhất trong chế độ ăn của Hoa Kỳ là disaccharide sucrose (ví dụ: đường ăn), chứa 50% fructose và 50% glucose. Tuy nhiên, ở Bắc Mỹ và nhiều quốc gia khác, nước ngọt không ăn kiêng được làm ngọt bằng xi-rô ngô có hàm lượng fructose cao (HFCS), chứa tới 55% monosaccharide fructose. Nhờ sự phong phú, độ ngọt và giá thành thấp, HFCS đã trở thành chất làm ngọt phổ biến nhất được sử dụng trong thực phẩm chế biến sẵn. Không phải HFCS có hại về mặt sinh học hơn sucrose; mà là, chi phí thấp của nó đã làm cho nó có sẵn cho tất cả mọi người, đặc biệt là các nhóm kinh tế xã hội thấp. HFCS được tìm thấy trong các loại thực phẩm chế biến sẵn từ nước ngọt và thanh kẹo đến bánh quy giòn đến bánh mì hot dog đến sốt cà chua. Mức tiêu thụ fructose trung bình hàng ngày đã tăng hơn 25% trong 30 năm qua. Sự phụ thuộc ngày càng tăng vào fructose trong chế độ ăn phương Tây có thể đang thúc đẩy dịch béo phì và T2DM. Lượng fructose cao nhất là soda (1,7 g/30 mL) và nước trái cây (1,8 g/30 mL). Mặc dù soda đã nhận được hầu hết sự chú ý, việc uống nhiều nước trái cây cũng có liên quan đến béo phì ở trẻ em, đặc biệt là ở các gia đình có thu nhập thấp. Các mô hình động vật cho thấy rằng chế độ ăn nhiều fructose dẫn đến tăng lượng năng lượng nạp vào, giảm tiêu hao năng lượng khi nghỉ, lắng đọng mỡ thừa và kháng insulin, cho thấy rằng việc tiêu thụ fructose đang đóng một vai trò trong dịch kháng insulin và béo phì và T2DM ở người.
Fructose trong ruột được vận chuyển vào tế bào ruột thông qua chất vận chuyển fructose GLUT5, độc lập với sự thủy phân ATP và hấp thu natri. Khi vào trong tế bào ruột, một phần nhỏ lượng fructose được chuyển thành axit lactic và giải phóng vào tuần hoàn cửa, một phần nhỏ khác cũng có thể được chuyển thành glucose. Tuy nhiên, phần lớn fructose ăn vào được tiết vào tuần hoàn cửa và được đưa đến gan. Ở đó, fructose được chuyển hóa nhanh chóng thành fructose-1-phosphate (F1P) thông qua fructokinase, một quá trình độc lập với insulin, cũng bỏ qua sự điều hòa phản hồi âm của phosphofructokinase trong con đường đường phân. Do đó, quá trình chuyển hóa fructose tạo ra các chất nền tạo mỡ (ví dụ: glyceraldehyde-3-phosphate và acetyl-CoA) một cách không điều hòa, được đưa trực tiếp vào ty thể. Lượng chất nền ty thể quá mức này sau đó thúc đẩy DNL ở gan, dẫn đến lắng đọng lipid trong gan và gan nhiễm mỡ. DNL ở gan cũng hạn chế quá trình oxy hóa axit béo tiếp theo trong gan thông qua việc sản xuất quá mức malonyl-CoA, làm giảm sự xâm nhập của axit béo vào ty thể bằng cách ức chế carnitine palmitoyl transferase 1 (CPT-1). F1P cũng kích thích SREBP-1c thông qua peroxisome proliferator-activated receptor gamma coactivator (PGC)-1β, độc lập với insulin, kích hoạt các gen liên quan đến DNL; hơn nữa, fructose đã được chứng minh là gây ra sự kích hoạt của carbohydrate-response element binding protein (ChREBP), cũng làm tăng sự biểu hiện của tất cả các enzyme của DNL. Hơn nữa, F1P kích hoạt dual-specificity mitogen-activated protein kinase 7 (MKK7), sau đó kích thích Janus kinase 1 (JAK1), một enzyme gan được coi là đóng vai trò cầu nối giữa chuyển hóa gan và viêm. Ngoài ra, chất trung gian tạo mỡ DAG (được hình thành trong quá trình chuyển hóa fructose trong gan) kích hoạt PKCɛ, phosphoryl hóa các gốc serine trên IRS-1, làm bất hoạt nó, và dẫn đến kháng insulin ở gan. Điều này làm suy giảm sự phosphoryl hóa qua trung gian insulin của FOXO1, dẫn đến tăng biểu hiện của các gen cần thiết cho quá trình tân tạo glucose và thúc đẩy tăng sản lượng glucose của gan, cũng góp phần vào tăng đường huyết và sự phát triển của T2DM. Các TG dư thừa được gan tiết ra vào tuần hoàn dưới dạng các hạt VLDL chứa đầy chất béo sau khi ăn fructose, cùng với sự giảm hoạt động LPL do fructose gây ra, gây ra rối loạn lipid máu sau bữa ăn kéo dài, do đó làm tăng nguy cơ mắc bệnh tim mạch (Hình 24.10 A,B).
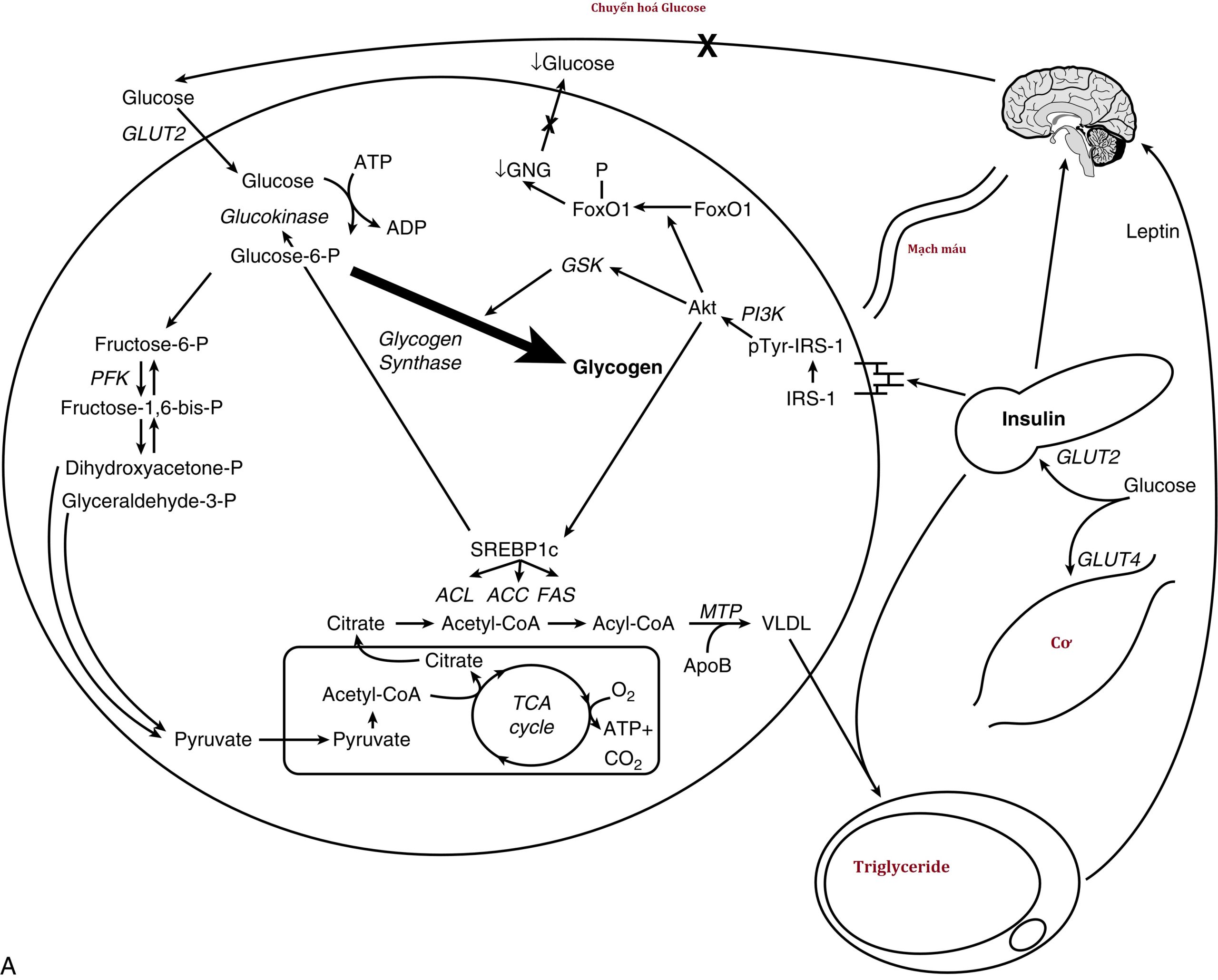
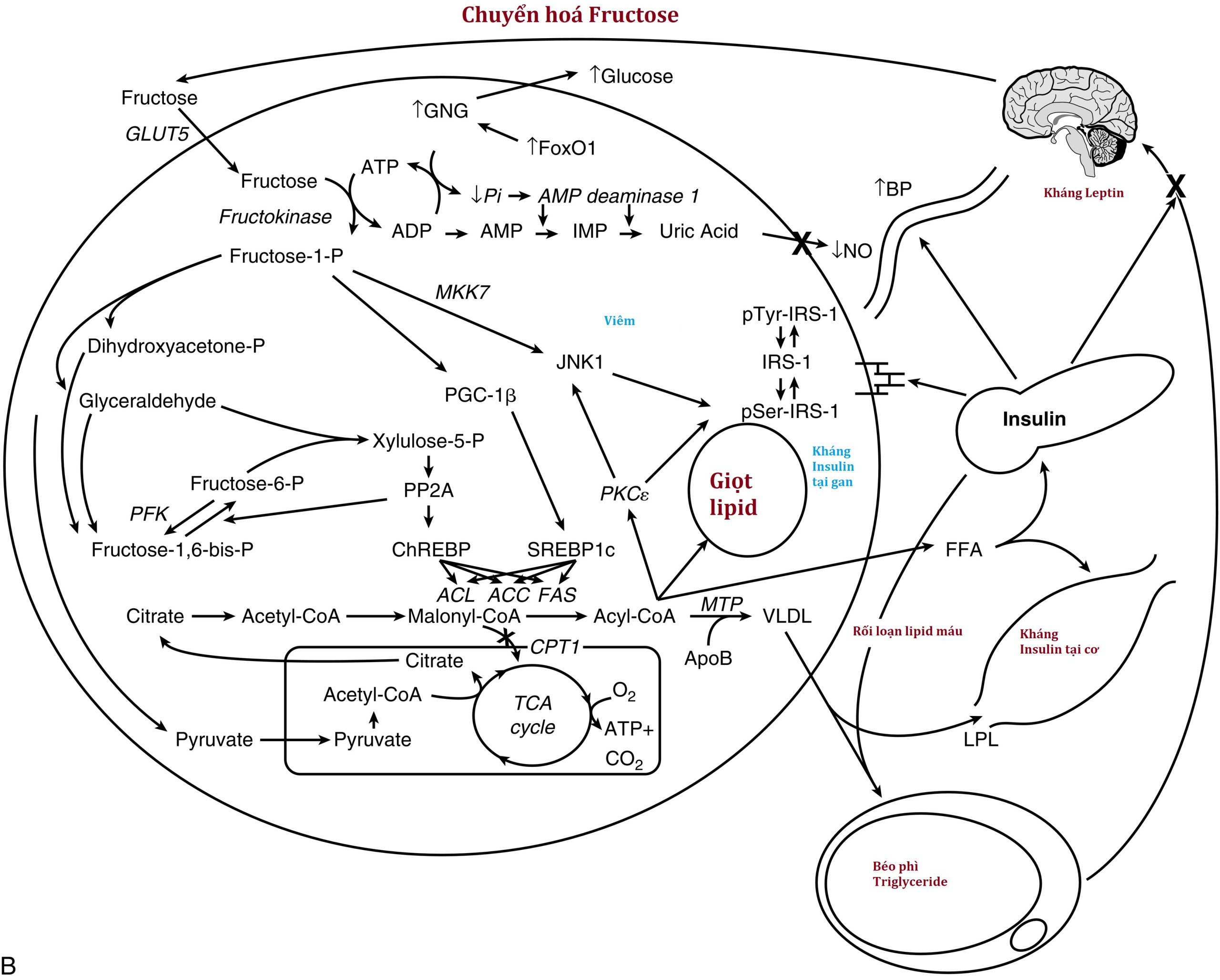
Hình 24.10 Chuyển hóa (A) glucose và (B) fructose ở gan. Trong một lượng glucose ăn vào, 20% được gan chuyển hóa; do đó, trong một lượng glucose 120 kCal (hai lát bánh mì trắng), 24 calo được gan chuyển hóa. Dưới tác động của insulin, glycogen synthase được tăng lên, và phần lớn lượng glucose được lưu trữ dưới dạng glycogen. Mặc dù sự kích hoạt của insulin đối với sterol regulatory element-binding protein 1c (SREBP-1c) kích hoạt con đường tạo mỡ, có rất ít citrate được hình thành để hoạt động như chất nền cho quá trình tân tạo mỡ. Ngoài ra, tác động của insulin lên gan phosphoryl hóa forkhead protein-1 (FOXO1), loại trừ nó khỏi nhân, và ức chế các enzyme liên quan đến quá trình tân tạo glucose (GNG). So sánh, gần như 100% lượng fructose được gan chuyển hóa; do đó, trong một lượng sucrose 120 kCal (một ly nước cam 8 oz.), một lượng lớn 72 calo đến gan. Trái ngược với glucose, fructose gây ra: (1) sự suy giảm phosphate tế bào gan phụ thuộc chất nền, làm tăng axit uric và góp phần gây tăng huyết áp thông qua việc ức chế endothelial nitric oxide synthase và giảm nitric oxide (NO); (2) kích thích tân tạo mỡ và sản xuất dư thừa VLDL và triglyceride huyết thanh, thúc đẩy rối loạn lipid máu; (3) tích tụ các giọt lipid trong gan, thúc đẩy gan nhiễm mỡ; (4) sản xuất FFA, thúc đẩy kháng insulin ở cơ; (5) kích hoạt c-jun N-terminal kinase (JNK-1), phosphoryl hóa serine và thụ thể insulin của gan, làm cho nó không hoạt động, và góp phần vào kháng insulin ở gan, thúc đẩy tăng insulin máu và ảnh hưởng đến sự lắng đọng chất nền vào mỡ; và (6) tăng insulin máu ở hệ thần kinh trung ương, đối kháng với tín hiệu leptin (xem Hình 24.5) và thúc đẩy việc tiếp tục nạp năng lượng. ACC, Acetyl-CoA carboxylase; ACL, ATP citrate lyase; ACSS2, acyl-CoA synthetase short-chain family member 2; ApoB, apolipoprotein B; ChREBP, carbohydrate-response element binding protein; CPT-1, carnitine palmitoyl transferase-1; FAS, fatty acid synthase; FFA, axit béo tự do; GLUT2, chất vận chuyển glucose 2; GLUT4, chất vận chuyển glucose 4; GLUT5, chất vận chuyển glucose 5; GSK, glycogen synthase kinase; IR, kháng insulin; IRS-1, cơ chất thụ thể insulin-1; LPL, lipoprotein lipase; MKK7, MAP kinase 7; MTP, microsomal transfer protein; PFK, phosphofructokinase; PGC-1β, peroxisome proliferator-activated receptor-γ coactivator-1β; PI3K, phosphatidyl inositol-3-kinase; PKCɛ, protein kinase C-ɛ; PP2A, protein phosphatase 2a; SREBP-1c, sterol regulatory element binding protein-1c; VLDL, very low-density lipoprotein.
Do hóa học lập thể độc đáo của nó, dạng vòng của fructose (một furan năm cạnh với các nhóm hydroxymethyl ở vị trí trục) chịu rất nhiều sức căng ion, ưu tiên dạng tuyến tính của phân tử, để lộ nhóm 2-keto phản ứng, có thể dễ dàng tham gia vào quá trình fructosyl hóa không enzyme của các gốc amino tiếp xúc của protein thông qua phản ứng Maillard, theo cách tương tự như vị trí 1-aldehyde của glucose có tính phản ứng. Mỗi phản ứng Maillard tạo ra một ROS, phải được dập tắt bởi một chất chống oxy hóa hoặc có nguy cơ gây tổn thương tế bào. Trong một nghiên cứu in vitro, việc ủ tế bào gan với fructose không gây tổn thương trực tiếp; tuy nhiên, khi những tế bào gan này được ủ trước với liều hydrogen peroxide dưới ngưỡng gây chết để giảm khả năng dập tắt ROS của peroxisome, fructose sau đó trở nên độc hại cho gan như các aldehyde hữu cơ khác. Việc giảm nhẹ fructose trong một can thiệp chế độ ăn đẳng calo trong vài ngày đã được chứng minh là làm giảm mỡ trong gan, nồng độ glucose và insulin, cho thấy tác động chuyển hóa độc đáo của nó, độc lập với giá trị calo của nó.
Axit amin chuỗi nhánh
Các axit amin chuỗi nhánh (BCAA: valine, leucine, và isoleucine) là các axit amin thiết yếu chiếm hơn 20% các axit amin trong “chế độ ăn phương Tây” điển hình. Mặc dù thường được sử dụng để sinh tổng hợp protein và tăng trưởng tế bào, khi được cung cấp dư thừa, chúng bị chuyển hướng khỏi quá trình tổng hợp protein và hướng tới việc sử dụng năng lượng.
Trong gan, BCAA làm tăng phiên mã của ChREBP và SREBP-1c, tạo điều kiện thuận lợi cho DNL. Hơn nữa, BCAA hạn chế tín hiệu PI3K do insulin gây ra và kích thích sự kích hoạt của mục tiêu phân tử của rapamycin ở động vật có vú (mTOR), thúc đẩy sự phosphoryl hóa serine của IRS-1 và làm suy giảm tín hiệu insulin. Ngoài ra, cũng như có những thay đổi liên quan đến béo phì ở các adipokine và các dấu hiệu nguy cơ tim mạch, dường như cũng có những thay đổi liên quan đến béo phì trong chuyển hóa BCAA và nồng độ huyết thanh sau đó. Cụ thể, nồng độ valine và leucine/isoleucine đã được báo cáo là cao hơn lần lượt 20% và 14% ở các đối tượng béo phì so với những người có cân nặng bình thường. Về mặt cơ chế, điều này dường như được giải thích bởi một tốc độ dòng chảy cao thông qua con đường dị hóa BCAA, dẫn đến việc sản xuất alanine tăng lên. Vì alanine là một axit amin tân tạo glucose cao, sự dị hóa BCAA tăng lên do đó có thể góp phần làm tăng sản lượng glucose của gan. Hơn nữa, các axit α-keto tăng lên được tạo ra bởi dòng chảy BCAA tăng lên thông qua các con đường dị hóa của chúng cũng có khả năng ức chế quá trình oxy hóa β của ty thể.
Hơn nữa, sự tăng BCAA mãn tính làm suy giảm sự vận chuyển của các axit amin thơm vào não; việc sản xuất serotonin (có nguồn gốc từ tryptophan) và catecholamine (có nguồn gốc từ phenylalanine và tyrosine) giảm có thể thúc đẩy cảm giác đói. Giả thuyết “quá tải BCAA” cho rằng trong bối cảnh một mô hình ăn kiêng bao gồm tiêu thụ nhiều chất béo, BCAA có thể có một đóng góp độc lập vào sự phát triển của kháng insulin, một giả thuyết được hỗ trợ bởi các nghiên cứu chuyển hóa học chứng minh nồng độ BCAA cao ở những người có đường huyết bình thường sau đó phát triển kháng insulin và đái tháo đường.
Ethanol
Mặc dù các nghiên cứu dịch tễ học ở người lớn liên kết việc tiêu thụ ethanol từ nhẹ đến vừa phải với việc cải thiện độ nhạy insulin và việc tiêu thụ rượu vang đỏ với việc giảm nguy cơ tim mạch, các nghiên cứu cắt ngang và tiền cứu khác lại cho thấy một tác động phụ thuộc vào liều lượng của rượu trong hội chứng chuyển hóa, và cho thấy rằng việc tiêu thụ mãn tính một lượng lớn ethanol làm xấu đi độ nhạy insulin. Ethanol bỏ qua quá trình đường phân bằng cách được chuyển đổi bởi alcohol dehydrogenase-1B để tạo thành acetaldehyde, thúc đẩy sự hình thành ROS và cũng phải được dập tắt bởi các chất chống oxy hóa của gan, chẳng hạn như glutathione hoặc axit ascorbic. Acetaldehyde sau đó được chuyển hóa bởi enzyme aldehyde dehydrogenase-2 thành axit axetic, đến lượt nó được chuyển hóa bởi enzyme acyl-CoA synthase short-chain family member 2 để tạo thành acetyl-CoA. Acetyl-CoA sau đó có thể đi vào ty thể, hoặc, khi có các chất nền calo khác, nó được ưu tiên sử dụng để tổng hợp axit béo thông qua DNL. Malonyl-CoA dư thừa được sản xuất từ quá trình chuyển hóa ethanol ức chế CPT-1 và do đó hạn chế quá trình oxy hóa β của axit béo trong ty thể. Ethanol cũng ngăn chặn quá trình oxy hóa β của axit béo bằng cách ức chế PPAR-γ, ức chế protein chuyển triglyceride vi thể, do đó làm thay đổi bộ máy xuất khẩu lipid của gan. Sự tích tụ các chất chuyển hóa lipid trong gan dẫn đến sự kích hoạt sau đó của enzyme JNK-1 và sự phosphoryl hóa serine của IRS-1, thúc đẩy thêm kháng insulin ở gan. Do đó, quá trình chuyển hóa ethanol dẫn đến sự tích tụ lipid trong gan và tổn thương gan, thúc đẩy kháng insulin ở gan và thúc đẩy hội chứng chuyển hóa. Tuy nhiên, mặc dù rõ ràng là một mối quan tâm ở người lớn, không có khả năng ethanol đóng góp đáng kể vào hội chứng chuyển hóa ở trẻ em.
Canxi và sữa
Đã có một số báo cáo về mối quan hệ nghịch giữa canxi trong chế độ ăn và các chỉ số béo phì. Canxi trong chế độ ăn đóng một vai trò quan trọng trong việc điều hòa chuyển hóa năng lượng. Tăng calcitriol (1,25-dihydroxyvitamin D) để đáp ứng với chế độ ăn ít canxi kích thích dòng Ca²⁺ vào các tế bào mỡ của người, có thể dẫn đến kích thích biểu hiện gen tạo mỡ và tân tạo mỡ, cũng như ức chế ly giải mỡ. Điều này có thể dẫn đến sự mở rộng các kho dự trữ triglyceride của tế bào mỡ, có thể thúc đẩy tình trạng tích mỡ. Tăng canxi trong chế độ ăn làm giảm nồng độ calcitriol và dẫn đến giảm khối lượng mỡ mà không cần hạn chế calo ở chuột, và tác dụng chống béo phì này của canxi trong chế độ ăn được hỗ trợ bởi các nghiên cứu lâm sàng và dịch tễ học ở người. Thiếu vitamin D tương quan với việc tăng BMI, đặc biệt là ở người Mỹ gốc Phi; tuy nhiên, không biết liệu điều này có phải do việc thay thế nước ngọt bằng sữa, không dung nạp lactose, hay các yếu tố khác. Một nghiên cứu ở người lớn đã tiết lộ một tác động nhất quán của việc hấp thu canxi cao hơn đối với trọng lượng cơ thể và mỡ cơ thể thấp hơn; tuy nhiên, các nghiên cứu ở trẻ em còn thiếu.
Hệ vi sinh vật đường ruột
Mặc dù đã thiết lập một mối liên quan mạnh mẽ giữa hệ vi sinh vật đường ruột và béo phì ở người, một mối quan hệ nhân quả và việc khám phá các cơ chế cơ bản vẫn cần được xác định. Các nghiên cứu đã chỉ ra rằng việc cấy ghép phân từ người béo phì và người có cân nặng bình thường cho chuột gnotobiotic dẫn đến việc các loài gặm nhấm trước đây không có vi khuẩn áp dụng kiểu hình béo phì của người cho. Điều này cho thấy rằng hệ vi sinh vật thực sự có liên quan đến sự phát triển của béo phì, và có lẽ trong sự phát triển của các bệnh đi kèm liên quan đến tích mỡ. Ruột có được hồ sơ vi sinh vật của nó bằng cách vi khuẩn xâm chiếm từ khi sinh ra và trong năm đầu đời. Sự tương tác chéo của ruột với các vi khuẩn xâm chiếm trong ruột đang phát triển ảnh hưởng đến sự thích nghi của trẻ sơ sinh với cuộc sống ngoài tử cung (cân bằng nội môi miễn dịch) và có thể cung cấp sự bảo vệ chống lại sự phát triển của bệnh (chẳng hạn như béo phì) sau này trong cuộc sống. Sự xâm chiếm bị gián đoạn (loạn khuẩn) do loạn khuẩn của mẹ, sinh mổ, và việc sử dụng kháng sinh chu sinh và sơ sinh có thể ảnh hưởng xấu đến sự phát triển hệ thống phòng thủ của ruột của vật chủ và dẫn đến khuynh hướng viêm thay vì cân bằng nội môi, dẫn đến tăng tính nhạy cảm với sự phát triển của các bệnh sau này trong cuộc sống. Người ta đã chỉ ra rằng hệ vi sinh vật đường ruột phản ứng với chế độ ăn, kháng sinh và các kích thích bên ngoài khác theo những cách ảnh hưởng đến một loạt các tình trạng chuyển hóa, bao gồm béo phì và NAFLD. Sự chiếm ưu thế của một số loài hệ thực vật đường ruột của người (Firmicutes so với Bacteroides) có thể dẫn đến khuynh hướng béo phì ở cả động vật và con người, có thể bằng cách tăng hiệu quả hấp thu năng lượng; tuy nhiên, các yếu tố quyết định sự chiếm ưu thế của chúng vẫn chưa được biết.
Thuốc
Nhiều loại thuốc thúc đẩy tăng cân quá mức ở trẻ em. Các loại thuốc được kê đơn phổ biến nhất là các liều dược lý của glucocorticoid (ví dụ: prednisone, methylprednisolone, dexamethasone) được sử dụng cho các hoạt động chống viêm và chống tân sinh của chúng. Bệnh nhân được điều trị như vậy thường phát triển béo phì, và phát triển nhiều đặc điểm của hội chứng Cushing (ví dụ: béo phì nội tạng, tăng lipid máu, tăng huyết áp, không dung nạp glucose), đặc trưng cho hội chứng chuyển hóa. Việc sử dụng hormone sinh dục cũng thúc đẩy tăng cân quá mức, có lẽ bằng cách gây ra kháng insulin. Ở bệnh nhân đái tháo đường tuýp 1, việc kiểm soát đường huyết nghiêm ngặt thường đi kèm với việc sử dụng insulin hơi quá mức cho bệnh nhân, dẫn đến tỷ lệ các cơn hạ đường huyết nhẹ cao hơn đòi hỏi tiêu thụ calo không do đói thúc đẩy, và có khả năng dẫn đến tăng cân quá mức. Cuối cùng, ngày càng có nhiều trẻ em được cho dùng các thuốc chống loạn thần không điển hình risperidone, olanzapine, quetiapine, clozapine, aripiprazole, và ziprasidone để ảnh hưởng đến tâm trạng và hành vi. Các thuốc chống loạn thần này thường liên quan đến việc tăng cân. Các nghiên cứu đánh giá tác dụng bảo vệ cân nặng của liệu pháp tăng cường với metformin hoặc topiramate cho thấy sự tăng cân thấp hơn nhưng vẫn đáng kể khi bổ sung các thuốc này.
Các rối loạn béo phì
Quan niệm rằng béo phì là một kiểu hình của nhiều bệnh lý được thể hiện rõ qua việc xem xét các rối loạn cụ thể dẫn đến béo phì ở thời thơ ấu (Hộp 24.1, Hình 24.11). Một số liên quan đến các cơ chế thần kinh, một số khác liên quan đến các cơ chế nội tiết tố cổ điển, trong khi những rối loạn khác liên quan đến sự rối loạn điều hòa của việc tăng lượng năng lượng nạp vào, giảm tiêu hao năng lượng, hoặc tăng dự trữ năng lượng tại tế bào mỡ. Cần nhớ rằng ngay cả trong các trung tâm chuyên khoa về béo phì nhi khoa, trẻ em có nguyên nhân “hữu cơ” gây béo phì chỉ chiếm một thiểu số nhỏ trong quần thể. Ít hơn 1% sẽ có một bệnh lý nội tiết “cổ điển” và ít hơn 3% sẽ có một nguyên nhân di truyền có thể xác định được trong các đoàn hệ béo phì nói chung, và khoảng 7% sẽ có một bệnh lý nội tiết hoặc nguyên nhân di truyền có thể xác định được trong các đoàn hệ nhi khoa bị béo phì nặng.
| Hộp 24.1. Phân loại các Rối loạn Béo phì ở Trẻ em
Các Rối loạn Nội tiết Cổ điển (lùn/chậm tăng trưởng là nổi bật) a. Suy giáp 1. Nguyên phát 2. Trung ương b. Hội chứng Cushing (thừa glucocorticoid) 1. U tuyến/ung thư biểu mô tuyến thượng thận 2. Tăng sản nốt nhỏ tuyến thượng thận 3. U tuyến yên tiết ACTH 4. U tiết ACTH lạc chỗ 5. Sử dụng glucocorticoid ngoại sinh c. Thiếu hụt hormone tăng trưởng d. Giả suy cận giáp type 1a 1. Di truyền từ mẹ (AHO + kháng đa nội tiết tố) 2. Di truyền từ cha (Giả-giả suy cận giáp, chỉ có AHO) Khiếm khuyết con đường Leptin-Melanocortin (chứng ăn nhiều là nổi bật) a. Thiếu hụt leptin b. Thiếu hụt thụ thể leptin c. Đột biến POMC (suy thượng thận và giảm sắc tố/tóc đỏ) d. Thiếu hụt Prohormone convertase-1 (thừa proinsulin) e. Thiếu hụt Carboxypeptidase E f. Đột biến MC3R g. Đột biến MC4R (tầm vóc cao) h. Đột biến SIM1 (rối loạn chức năng tự chủ, khuyết tật trí tuệ) i. Thiếu hụt protein phụ trợ thụ thể Melanocortin 2 (MRAP2) j. Thiếu hụt một nửa gen yếu tố dinh dưỡng thần kinh có nguồn gốc từ não (BDNF)/mất đoạn 11p14.1 k. Đột biến NTRK2 l. Thiếu hụt một nửa gen SH2B1/mất đoạn 16p11.2 Các Rối loạn Béo phì dạng Hội chứng (liên quan đến nhiều hệ cơ quan và có các đặc điểm thể chất riêng biệt) a. Hội chứng Prader-Willi b. Hội chứng Bardet-Biedl c. Hội chứng Alström d. Hội chứng Smith-Magenis e. Hội chứng WAGR f. Hội chứng Cohen g. Hội chứng Carpenter Béo phì do vùng dưới đồi Các rối loạn động lực insulin • khối u • phẫu thuật • xạ trị • chấn thương • bệnh thâm nhiễm • viêm • Hội chứng ROHHAD (béo phì khởi phát nhanh với rối loạn chức năng vùng dưới đồi, giảm thông khí, rối loạn điều hòa tự chủ, u mào thần kinh) ACTH, Hormone vỏ thượng thận; AHO, Loạn dưỡng xương di truyền Albright; POMC, proopiomelanocortin. |
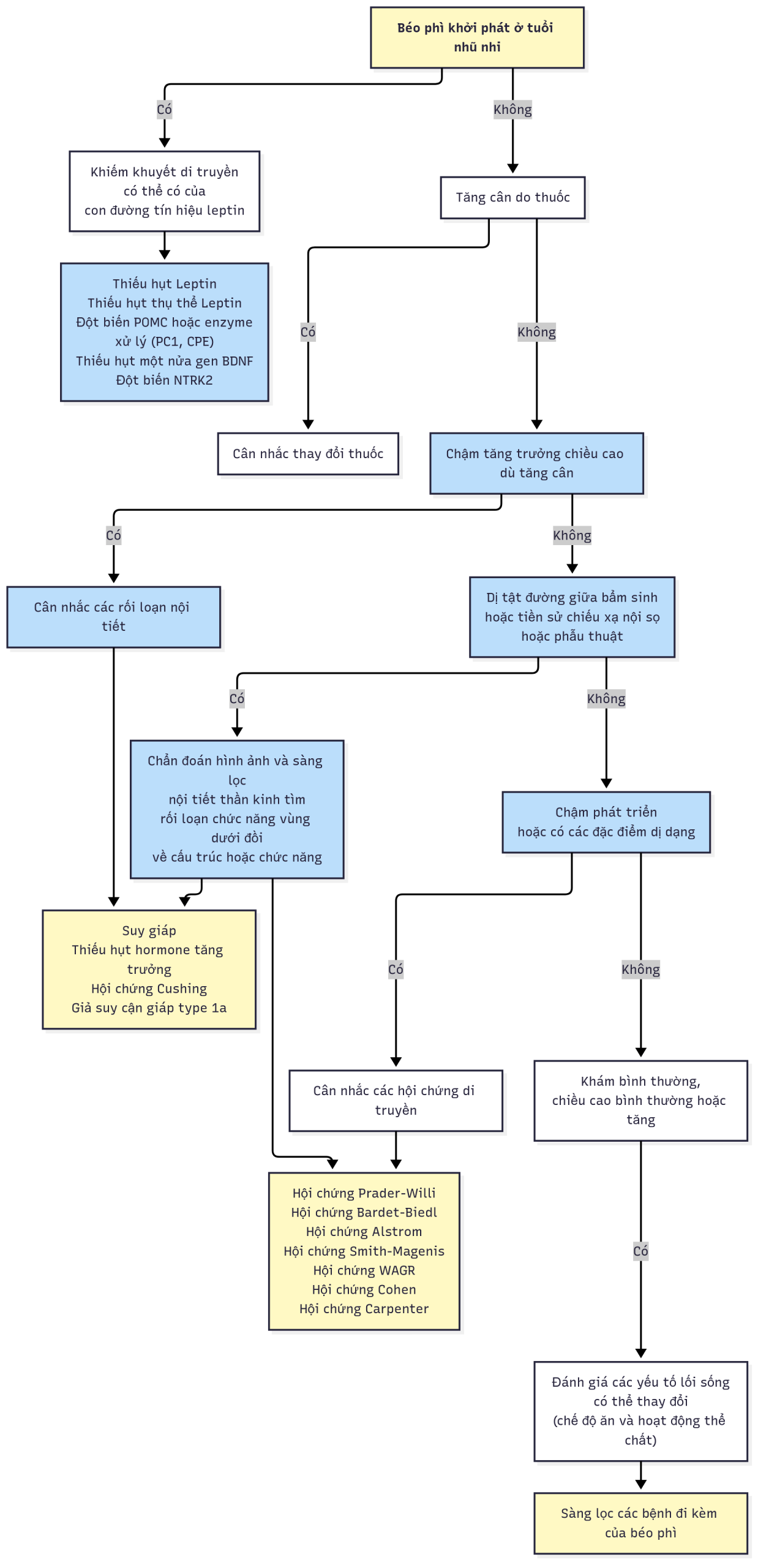
Hình 24.11 Đánh giá béo phì khởi phát ở trẻ em.
Các Rối loạn Nội tiết “Cổ điển” với Kiểu hình Béo phì
Ở trẻ em, sự tăng trưởng theo chiều dài hoặc chiều cao chiếm tới 20% lượng calo nạp vào. Các tình trạng nội tiết cho phép lượng năng lượng nạp vào bình thường theo tuổi, nhưng ức chế sự tăng trưởng theo chiều dài, tất yếu sẽ dẫn đến dự trữ năng lượng quá mức. Đây là trường hợp của bốn rối loạn nội tiết “cổ điển” liên quan đến béo phì. Chúng có thể được phân biệt với các nguyên nhân béo phì nhi khoa khác dựa trên tốc độ tăng trưởng dưới mức tối ưu, trái ngược với tình trạng dinh dưỡng thừa, có xu hướng làm tăng tốc độ cả tăng trưởng và trưởng thành xương, có lẽ một phần là do insulin dư thừa phản ứng chéo với thụ thể IGF-1.
Suy giáp. Thiếu hụt hormone triiodothyronine (T3) gây ra giảm REE và giảm hoạt động thể chất do mệt mỏi. Hơn nữa, T3 là yếu tố cho phép các tác động đồng hóa của GH. Sự giảm tổng tiêu hao năng lượng, mặc dù lượng calo nạp vào tương đối thấp, thúc đẩy dự trữ năng lượng dai dẳng và tăng tích mỡ. Các dấu hiệu, triệu chứng và đánh giá chẩn đoán suy giáp được thảo luận trong Chương 13. Liệu pháp thay thế hormone tuyến giáp đủ để tăng trưởng, REE và hoạt động thể chất để giải quyết tình trạng béo phì theo thời gian. Đáng chú ý, suy giáp là một nguyên nhân gây tăng cân không nên bị nhầm lẫn với sự tăng nhẹ TSH xảy ra như một hậu quả của béo phì, được cho là qua trung gian của sự tăng tiết hormone giải phóng thyrotropin (TRH) do leptin gây ra. Bổ sung hormone tuyến giáp thường không được chỉ định trong những trường hợp như vậy, và TSH thường trở lại bình thường khi giảm cân và giảm tích mỡ, từ đó làm giảm nồng độ leptin trong tuần hoàn. Liệu việc bổ sung hormone tuyến giáp có thể hỗ trợ giảm cân hoặc cải thiện các bệnh đi kèm tim mạch và gan nhiễm mỡ hay không vẫn chưa chắc chắn.
Thừa Glucocorticoid. Hội chứng Cushing là tình trạng thừa glucocorticoid làm ngừng tăng trưởng và gây ra chứng ăn nhiều, cùng với sự giảm REE và hoạt động thể chất do teo cơ. Liệu pháp glucocorticoid ngoại sinh có thể dẫn đến một kiểu hình béo phì tương tự. Hội chứng Cushing được thảo luận thêm trong Chương 14. Việc giảm glucocorticoid trong tuần hoàn thông qua điều trị nội khoa hoặc phẫu thuật sẽ đảo ngược tình trạng béo phì, nhưng béo phì trung tâm thường vẫn tồn tại. Mặc dù hội chứng Cushing là một nguyên nhân gây béo phì rất hiếm gặp, tăng cortisol chức năng (hội chứng giả Cushing) lại phổ biến trong béo phì và được cho là do tăng hoạt động của trục HPA và sự chuyển đổi cortisone không hoạt động thành cortisol bởi 11βHSD1 trong mô mỡ nhiều hơn. Tông màu glucocorticoid cao hơn mãn tính này trong béo phì và các tình trạng giả Cushing khác, chẳng hạn như trầm cảm, đái tháo đường, thêm rối loạn giấc ngủ, có thể góp phần vào nguy cơ sức khỏe chuyển hóa và làm trầm trọng thêm tình trạng béo phì nội tạng, dẫn đến một vòng luẩn quẩn làm béo phì trung tâm ngày càng nặng hơn. Chuột biến đổi gen biểu hiện quá mức 11βHSD1 chọn lọc trong mô mỡ phát triển đái tháo đường kháng insulin, tăng lipid máu và chứng ăn nhiều. Hoạt động của enzyme 11βHSD1 cao hơn ở mô mỡ nội tạng so với mô mỡ dưới da và có tương quan với BMI ở trẻ em tiền dậy thì có cân nặng bình thường. Tuy nhiên, ở người lớn, mối liên quan giữa các đa hình 11βHSD1 và BMI hoặc tỷ lệ eo:hông là yếu nhất, và hoạt động của enzyme không tăng trong béo phì. Tuy nhiên, các chất ức chế đặc hiệu của 11βHSD1 đang được nghiên cứu như là các mục tiêu thuốc tiềm năng để điều trị béo phì và đái tháo đường tuýp 2.
Thiếu hụt Hormone Tăng trưởng. Thiếu hụt GH ngăn cản quá trình ly giải mỡ và thúc đẩy béo phì nội tạng, mặc dù mức độ béo phì thường nhẹ, do đó chẩn đoán thiếu hụt GH thường có trước khi béo phì đáng kể khởi phát. Thiếu hụt GH cũng liên quan đến mệt mỏi và giảm hoạt động thể chất. Thiếu hụt GH thường đi kèm với các thiếu hụt hormone tuyến yên khác (ví dụ: suy giáp trung ương), cũng có thể làm giảm REE. Liệu pháp GH có thể đảo ngược những thiếu hụt tiêu hao năng lượng này, tăng khối lượng cơ và thúc đẩy giảm cân. Các xét nghiệm chẩn đoán thiếu hụt GH được thảo luận trong Chương 11, nhưng cần đề cập ở đây rằng bản thân béo phì ảnh hưởng đến sự bài tiết GH, do đó các phản ứng đỉnh trong các xét nghiệm mô phỏng thấp hơn ở trẻ béo phì so với những trẻ có cân nặng bình thường. Cơ chế giảm bài tiết GH trong béo phì chưa được biết rõ nhưng các yếu tố góp phần được giả định bao gồm tăng insulin máu và tăng nồng độ FFA trong tuần hoàn. Do đó, việc giải thích kết quả xét nghiệm kích thích GH nên tính đến các ngưỡng bị thay đổi trong béo phì. Hơn nữa, lợi ích tiềm năng của việc bổ sung GH trong tình trạng thiếu GH tương đối vẫn cần được xác định.
Giả suy cận giáp Type 1a (PHP1a) và Loạn dưỡng xương di truyền Albright (AHO). PHP1a và AHO là do đột biến trội trên nhiễm sắc thể thường của gen GNAS1, mã hóa cho tiểu đơn vị Gsα cần thiết cho sự truyền tín hiệu hormone peptide của các thụ thể kết hợp G-protein (GCPRs) (các phối tử bị ảnh hưởng bao gồm hormone cận giáp, TSH, hormone giải phóng hormone tăng trưởng, và α-MSH). Sự di truyền từ mẹ của đột biến GNAS1 dẫn đến PHP1a (kháng đa nội tiết tố, được mô tả thêm trong Chương 3 và Chương 20) cùng với AHO, trong đó một đặc điểm chính là béo phì, gây ra bởi sự giảm tín hiệu gây chán ăn/dị hóa của MC3R và MC4R trong vùng dưới đồi và giảm khả năng kích thích cAMP để đáp ứng với kích thích β-adrenergic trong các tế bào mỡ. Ngoài béo phì, các biểu hiện điển hình khác bao gồm hạ canxi máu, lùn, khuyết tật trí tuệ nhẹ, mặt tròn, sống mũi thấp, mũi và cổ ngắn, mọc răng chậm với thiểu sản men răng, xương bàn tay và bàn chân thứ tư và thứ năm ngắn, và đốt ngón tay xa của ngón cái ngắn. Sự di truyền từ cha dẫn đến AHO mà không có kháng đa nội tiết tố, còn được gọi là giả-giả suy cận giáp. Ngoài việc thay thế hormone và phòng ngừa hạ canxi máu, hiện không có phương pháp điều trị cụ thể nào cho khía cạnh béo phì của tình trạng này.
Các Rối loạn Béo phì di truyền
Khoảng 5% đến 10% trẻ em bị chứng ăn nhiều và béo phì khởi phát sớm (3 năm đầu đời) được ước tính là có một tình trạng di truyền có thể xác định được, và tỷ lệ này tăng lên tới 22% khi sàng lọc trẻ em bị béo phì dạng hội chứng. Các rối loạn béo phì di truyền có thể được phân loại thành các tình trạng đơn gen với béo phì là kiểu hình chính và các hội chứng đa hiệu bao gồm các đặc điểm khác, chẳng hạn như khuyết tật trí tuệ và các đặc điểm thể chất dị dạng. Có khoảng 80 hội chứng di truyền bao gồm béo phì là một đặc điểm, nhưng chưa đến một phần tư trong số các hội chứng này đã được làm sáng tỏ hoàn toàn về nguyên nhân di truyền. Trong chương này, chúng ta sẽ tập trung chủ yếu vào các rối loạn đơn gen của con đường leptin-melanocortin và một vài trong số các hội chứng đa hiệu được làm sáng tỏ rõ hơn. Các mô tả thêm về nhiều hội chứng khác được đánh giá chi tiết ở những nơi khác.
Khiếm khuyết con đường Leptin-Melanocortin
Kể từ khám phá năm 1994 về sự thiếu hụt leptin là nguyên nhân gây béo phì ở chuột ob/ob, con đường leptin-melanocortin đã được làm sáng tỏ rộng rãi trong các mô hình động vật, trong khi song song đó, các khiếm khuyết đơn gen dọc theo cùng con đường này đã được xác định trong các rối loạn béo phì ở người. Mỗi rối loạn sẽ được mô tả theo thứ tự tuần tự dựa trên trình tự của các bước của con đường như được trình bày trong Hình 24.3. Một số hội chứng béo phì đa hiệu dường như cũng hội tụ trên con đường leptin-melanocortin, và những tình trạng này sẽ được liệt kê cùng với đối tác rối loạn đơn gen của chúng.
Thiếu hụt Leptin. Các đột biến lặn trên nhiễm sắc thể thường của gen leptin ở người tái tạo lại kiểu hình của chuột ob/ob thiếu leptin. Chỉ có một vài bệnh nhân như vậy đã được mô tả, chủ yếu có nguồn gốc từ Pakistan và Thổ Nhĩ Kỳ, và phần lớn họ được sinh ra từ cha mẹ cùng huyết thống. Cân nặng khi sinh bình thường sau đó là tăng cân nhanh trong giai đoạn nhũ nhi và béo phì ngay từ những tháng đầu đời. Chứng ăn nhiều biểu hiện khi sinh như cảm giác đói không thể thỏa mãn với yêu cầu được cho ăn liên tục ở trẻ sơ sinh và hành vi tìm kiếm thức ăn hung hăng ở trẻ em. Sự thiếu hụt leptin gây ra phản ứng đói dưới dạng suy giáp trung ương, nhiệt độ cơ thể thấp hơn, suy sinh dục do thiếu gonadotropin, và suy giảm miễn dịch qua trung gian tế bào T. Mặc dù bị suy giáp, tầm vóc và sự trưởng thành xương bình thường thường được bảo tồn cho đến tuổi dậy thì thông thường vì tình trạng tăng insulin máu do béo phì cho phép insulin dư thừa phản ứng chéo với thụ thể IGF-1 để duy trì sự tăng trưởng. Tuy nhiên, do vai trò quan trọng của leptin trong việc khởi đầu và duy trì tuổi dậy thì, những bệnh nhân trưởng thành không được điều trị bị thiếu hụt leptin sẽ bị lùn do thiếu đợt tăng trưởng vượt bậc ở tuổi dậy thì. Chẩn đoán được thực hiện bằng cách chứng minh nồng độ leptin trong huyết thanh cực thấp hoặc không thể đo được. Cần lưu ý rằng một số đột biến của gen leptin có thể tạo ra protein không chức năng nhưng vẫn được phát hiện trong các xét nghiệm dựa trên kháng thể tiêu chuẩn, do đó có thể cần phải đo hoạt tính sinh học. Trong cả hai trường hợp leptin giảm hoặc rối loạn chức năng, điều trị bằng leptin tái tổ hợp có hiệu quả phục hồi tín hiệu leptin, do đó làm giảm chứng ăn nhiều, giải quyết béo phì, và phục hồi sự tiến triển dậy thì và chức năng miễn dịch bình thường. Những người mang gen đột biến leptin dị hợp tử biểu hiện một kiểu hình trung gian với nồng độ leptin huyết thanh thấp hơn và khuynh hướng béo phì.
Thiếu hụt Thụ thể Leptin (LEPR). Các đột biến lặn trên nhiễm sắc thể thường của gen thụ thể leptin tạo ra các triệu chứng và dấu hiệu tương tự như thiếu hụt leptin. Trong một đoàn hệ lớn các bệnh nhân bị béo phì nặng, khởi phát sớm, tỷ lệ các biến thể gây bệnh đồng hợp tử hoặc dị hợp tử phức hợp của LEPR là 3%. Các cá nhân bị ảnh hưởng bị chứng ăn nhiều, béo phì nặng, dậy thì muộn do suy sinh dục do thiếu gonadotropin và chức năng miễn dịch bị thay đổi. Tuy nhiên, nhìn chung, các đặc điểm lâm sàng của họ ít nghiêm trọng hơn so với những bệnh nhân bị thiếu hụt leptin bẩm sinh. Nồng độ leptin huyết thanh tăng cao và tương quan với khối lượng mỡ nhưng dịch chuyển cao hơn đối với mức độ tích mỡ khi so sánh với các đối tượng đối chứng bị béo phì có LEPR bình thường. Nồng độ leptin trong tuần hoàn thường cao hơn trong tình trạng thiếu hụt LEPR được cho là do sự gián đoạn trong vòng phản hồi leptin-SNS kết nối CNS với các tế bào mỡ ngoại vi. Tuy nhiên, do mức độ chồng chéo khá cao về giá trị, nồng độ leptin không thể được sử dụng để chẩn đoán đáng tin cậy tình trạng thiếu hụt LEPR, do đó cần phải giải trình tự gen. Những người mang gen dị hợp tử có tỷ lệ tích mỡ cao hơn nhưng ngoài ra không có triệu chứng. Các biểu hiện khác nhau tùy thuộc vào loại đột biến LEPR đã được báo cáo. Ba thành viên của một gia đình ở Pháp gốc Algeria với một đột biến cắt ngắn đồng hợp tử của LEPR (thiếu cả miền xuyên màng và miền nội bào) có các triệu chứng bổ sung là IGF-1, IGFBP-3 thấp và chậm tăng trưởng; nguyên nhân của những đặc điểm bổ sung này chưa được biết. Điều trị thiếu hụt LEPR đòi hỏi nhắm vào các chất trung gian hạ nguồn của con đường leptin-melanocortin. Bởi vì sự kích hoạt LEPR thường sẽ làm tăng POMC và do đó là sản phẩm phân cắt của POMC, α-MSH, gắn vào MC4R, một mục tiêu điều trị hợp lý sẽ là các chất chủ vận thụ thể melanocortin. Một loại thuốc ứng cử viên đầy hứa hẹn là setmelanotide, hoạt động như một chất chủ vận thiên vị cho MC4R, ưu tiên tín hiệu Gαq được điều hòa bởi protein kinase được hoạt hóa bởi mitogen (MAPK), do đó tránh được tác dụng phụ tăng huyết áp của các chất chủ vận MC4R được phát triển trước đây chủ yếu làm tăng tín hiệu Gαs. Một báo cáo trường hợp ban đầu về ba bệnh nhân có đột biến LEPR đồng hợp tử đã nhận setmelanotide mô tả sự giảm cân đáng kể với sự tăng sắc tố da và tóc (do kích hoạt MC1R) là tác dụng phụ chính duy nhất. Liệu loại thuốc này có thể có lợi ở những người có đột biến LEPR dị hợp tử hay không vẫn cần được xác định.
Thiếu hụt POMC và Đột biến Vị trí Nối. Các đột biến lặn trên nhiễm sắc thể thường trong gen POMC cản trở việc dịch mã protein hoặc phân cắt protein thành các sản phẩm ACTH và α-MSH dẫn đến da trắng và tóc đỏ (do thiếu sự kích hoạt MC1R bởi α-MSH), thiểu sản tuyến thượng thận và thiếu hụt glucocorticoid (do thiếu sự kích hoạt MC2R bởi ACTH) với sự sản xuất aldosterone và catecholamine được bảo tồn, và béo phì (do thiếu sự kích hoạt MC4R và MC3R bởi α-MSH). Cân nặng khi sinh bình thường nhưng chứng ăn nhiều thúc đẩy tăng cân nhanh bắt đầu từ giai đoạn nhũ nhi. Mặc dù tóc đỏ là một đặc điểm phổ biến, một bệnh nhân người Thổ Nhĩ Kỳ với một đột biến vô nghĩa sớm của POMC được báo cáo là có tóc sẫm màu, cho thấy rằng sắc tố có thể thay đổi. Chẩn đoán có thể được thiết lập dựa trên biểu hiện lâm sàng, thiếu hụt ACTH và giảm cortisol máu. Những người mang gen dị hợp tử có sắc tố và cortisol bình thường, nhưng có khuynh hướng béo phì cao hơn. Các biến thể methyl hóa của POMC trong dân số nói chung có tương quan với trọng lượng cơ thể cá nhân, cho thấy một vai trò rộng hơn của POMC trong việc xác định nguy cơ béo phì. Một báo cáo trường hợp ở hai bệnh nhân bị thiếu hụt POMC cho thấy sự giảm cân đáng kể với điều trị setmelanotide, cho thấy rằng chủ vận MC4R cũng có thể có lợi cho tình trạng này.
Thiếu hụt Prohormone Convertase-1. Các đột biến lặn trên nhiễm sắc thể thường trong gen PSCK1 mã hóa enzyme PC1 dẫn đến không có khả năng xử lý các tiền hormone khác nhau thành các phối tử hoạt động của chúng, chẳng hạn như POMC thành ACTH và α-MSH, proinsulin thành insulin, và các tiền peptide đường ruột khác nhau thành các hormone hoạt động. Chỉ có một số ít bệnh nhân đã được mô tả trong y văn, và họ được báo cáo là biểu hiện béo phì khởi phát sớm nghiêm trọng, thiếu hụt ACTH, suy sinh dục do thiếu gonadotropin, tăng proinsulin máu và rối loạn chức năng ruột non do không có khả năng phân cắt các tiền peptide đường ruột thành dạng trưởng thành của chúng. Suy giáp và đái tháo nhạt cũng đã được báo cáo. Do sự phân cắt proinsulin bị suy giảm, tăng đường huyết sau bữa ăn sau đó là hạ đường huyết vài giờ sau bữa ăn được quan sát thấy. Điều này là do ban đầu không đủ bài tiết insulin sau đó là giảm thanh thải proinsulin, có hiệu lực nhẹ tại thụ thể insulin, gây hạ đường huyết trong trạng thái sau hấp thu. Chẩn đoán có thể được thực hiện bằng cách tìm thấy nồng độ proinsulin cực cao, và có thể cần chẩn đoán phân tử để xác nhận khiếm khuyết gen. Lợi ích tiềm năng của việc sử dụng chất chủ vận MC4R setmelanotide để điều trị béo phì ở bệnh nhân có đột biến PCSK1 hiện đang được điều tra.
Đột biến Carboxypeptidase E. Một bệnh nhân trưởng thành với đột biến cắt ngắn đồng hợp tử của gen CPE đã được báo cáo. CPE xử lý các neuropeptide và tiền hormone khác nhau, bao gồm POMC và gonadotropin. Bệnh nhân này biểu hiện chứng ăn nhiều, béo phì nặng từ thời thơ ấu, đái tháo đường tuýp 2, và suy sinh dục do thiếu gonadotropin. Cô cũng bị khuyết tật trí tuệ, đây không phải là một đặc điểm điển hình của các khiếm khuyết con đường leptin-melanocortin gần khác đã được thảo luận cho đến nay. Chuột bị thiếu hụt CPE biểu hiện thoái hóa hồi hải mã và suy giảm trí nhớ, cho thấy vai trò của CPE trong chức năng nhận thức.
Đột biến Thụ thể Melanocortin-3. α-MSH là phối tử nội sinh cho cả MC3R và MC4R, với MC4R là thụ thể quan trọng hơn để điều hòa cảm giác thèm ăn và cân bằng năng lượng trong khi MC3R chỉ có một vai trò khiêm tốn. Một đột biến sai nghĩa dị hợp tử của MC3R đã được xác định ở hai thành viên gia đình của một gia đình người gốc Ấn Độ ở Singapore, biểu hiện là béo phì khởi phát sớm nghiêm trọng ở đứa trẻ nhưng chỉ béo phì nhẹ ở người cha. Biến thể MC3R phổ biến T6K + V81I có liên quan đến tình trạng tích mỡ nhiều hơn ở trẻ em và người lớn. In vitro, biến thể này dường như làm giảm biểu hiện MC3R. Chuột knock-in được nhân hóa biểu hiện hiệu quả ăn tăng và lưu trữ triglyceride cao hơn trong các tế bào mỡ. Chẩn đoán các biến thể MC3R chỉ có thể được thực hiện bằng cách giải trình tự gen. Hiện không có phương pháp điều trị cụ thể nào.
Đột biến Thụ thể Melanocortin-4. Các đột biến trong gen MC4R dường như chiếm khoảng 5% các trường hợp béo phì khởi phát sớm nghiêm trọng. Các cá nhân có đột biến dị hợp tử, béo phì nặng, tăng khối lượng nạc, tăng trưởng theo chiều dài, chứng ăn nhiều, và tăng insulin máu nghiêm trọng; các cá nhân có đột biến đồng hợp tử hoặc dị hợp tử phức hợp thậm chí còn bị ảnh hưởng nghiêm trọng hơn. Mức độ nghiêm trọng về chức năng thay đổi tùy theo đột biến cụ thể, với những bệnh nhân có đột biến vẫn giữ được khả năng truyền tín hiệu còn lại có kiểu hình béo phì ít nghiêm trọng hơn. Do đó, các đột biến trong MC4R dẫn đến một hội chứng béo phì riêng biệt được di truyền theo kiểu đồng trội. Huyết áp thường thấp hơn so với các đối tượng đối chứng bị béo phì và có MC4R bình thường, phù hợp với vai trò của hệ thống melanocortin trong việc trung gian dòng ra của SNS đến các hệ thống thận và tim mạch. Chức năng trí tuệ thường bình thường, mặc dù một mối liên quan có thể có với rối loạn tăng động giảm chú ý đã được báo cáo ở trẻ em có đột biến đồng hợp tử. Chẩn đoán được thực hiện bằng cách giải trình tự gen. Hiện không có phương pháp điều trị cụ thể nào cho rối loạn này, nhưng các công nghệ kích hoạt cụm lặp lại palindromic ngắn cách đều nhau (CRISPR) ở chuột đã thành công trong việc điều hòa tăng biểu hiện của alen nguyên vẹn còn lại trong knockout Mc4r dị hợp tử để phục hồi trọng lượng cơ thể bình thường. Liệu loại phương pháp chỉnh sửa gen này có thể được chuyển sang người hay không vẫn cần được xác định.
Đột biến SIM1. Single-minded 1 (SIM1) là một tương đồng người của gen single-minded của ruồi giấm có liên quan đến sự hình thành thần kinh ở cả hai loài. Ở người, SIM1 có liên quan đến sự phát triển và chức năng của PVN, vùng dưới đồi biểu hiện các tế bào thần kinh MC4R và prodynorphin. SIM1 được cho là hoạt động như một chất trung gian hạ nguồn của các chức năng gây chán ăn của cả hai phân nhóm tế bào thần kinh này. Trong một đoàn hệ gồm 2100 cá nhân bị béo phì nặng, tỷ lệ đột biến SIM1 dị hợp tử là 1,3%, và phân tích vi mảng của 279 cá nhân bị béo phì dạng hội chứng đã xác định được một đối tượng có vi mất đoạn liên quan đến SIM1. Bệnh nhân bị thiếu hụt một nửa gen SIM1 biểu hiện chứng ăn nhiều, béo phì khởi phát sớm nghiêm trọng, tỷ lệ chuyển hóa cơ bản bình thường, rối loạn chức năng tự chủ (huyết áp tâm thu thấp hơn, giảm trương lực SNS, giảm biến thiên nhịp tim giữa trạng thái ngủ và thức), và các mức độ khác nhau của các bất thường thần kinh hành vi và khuyết tật trí tuệ. Sự tăng methyl hóa tại locus SIM1 có liên quan đến chỉ số BMI cao hơn ở thanh thiếu niên, cho thấy rằng các kiểu hình trung gian tồn tại trong dân số nói chung. Chẩn đoán đột biến SIM1 được thực hiện bằng xét nghiệm gen. Hiện không có phương pháp điều trị cụ thể nào cho rối loạn này, nhưng các công nghệ kích hoạt CRISPR ở chuột đã thành công trong việc điều hòa tăng biểu hiện của alen nguyên vẹn còn lại trong knockout Sim1 dị hợp tử để phục hồi trọng lượng cơ thể bình thường. Liệu loại phương pháp chỉnh sửa gen này có thể được chuyển sang người hay không vẫn cần được xác định.
Đột biến Protein Phụ trợ Thụ thể Melanocortin 2 (MRAP2). Protein MRAP2 tương tác trực tiếp với MC4R và tăng cường tạo ra cAMP khi phối tử gắn vào MC4R. Knockout toàn thân và đặc hiệu cho não của Mrap2 ở chuột gây ra béo phì nặng. Các đột biến gây bệnh dị hợp tử của MRAP2 đã được xác định ở bốn bệnh nhân bị béo phì khởi phát sớm nghiêm trọng không có hội chứng mà không có các đặc điểm hội chứng bổ sung.
Thiếu hụt một nửa gen Yếu tố Dinh dưỡng Thần kinh có nguồn gốc từ não. Gen BDNF nằm ở vùng 11p14. Mất chức năng đồng hợp tử không tương thích với sự sống nhưng mất BDNF dị hợp tử được quan sát thấy trong hội chứng WAGR, vi mất đoạn 11p14, và đảo đoạn trung tâm nhiễm sắc thể 11, và các biến thể gen BDNF.
Hội chứng WAGR/Mất đoạn 11p: Hội chứng u nguyên bào thận Wilms, vô mống mắt, bất thường sinh dục-tiết niệu, và một loạt các chậm phát triển là một rối loạn di truyền hiếm gặp (tỷ lệ mắc ~ 1 trên 1.000.000) gây ra bởi sự mất đoạn gen liền kề trên nhánh ngắn của nhiễm sắc thể 11. Thiếu hụt một nửa gen của WT1 và PAX6, có liên quan đến sự phát triển sinh dục-tiết niệu và mắt, tương ứng, gây ra các đặc điểm cốt lõi của hội chứng. BDNF nằm ở 11p14.1, cách vùng WAGR quan trọng 11p13 chứa WT1 và PAX6 4 Mb, và được bao gồm trong đoạn bị xóa của khoảng một nửa số bệnh nhân mắc hội chứng WAGR. Trong một đoàn hệ gồm 33 bệnh nhân mắc hội chứng WAGR, thiếu hụt một nửa gen BDNF có liên quan đến điểm z-score BMI cao hơn, tần suất phát triển béo phì ở trẻ em cao hơn năm lần (100% so với 20% đối với bệnh nhân có và không có mất đoạn BDNF, tương ứng), nồng độ BDNF huyết thanh giảm, và điểm số cao hơn trên bảng câu hỏi về chứng ăn nhiều do cha mẹ báo cáo. Trong đoàn hệ bệnh nhân mắc hội chứng WAGR này, việc có thiếu hụt một nửa gen BDNF so với BDNF nguyên vẹn cũng có liên quan đến chỉ số IQ thấp hơn 20 điểm, suy giảm xã hội lớn hơn và tỷ lệ đáp ứng tiêu chí tự kỷ trên Phỏng vấn Chẩn đoán Tự kỷ-Sửa đổi cao hơn, phản ứng hành vi do cha mẹ báo cáo thấp hơn đối với các kích thích thường gây đau, và nhận thức tự báo cáo về cơn đau thấp hơn để đáp ứng với các kích thích nhiệt độ nóng và lạnh.
Vi mất đoạn 11p14: Vi mất đoạn gây thiếu hụt một nửa gen BDNF trong khi không ảnh hưởng đến vùng WAGR đã được báo cáo ở 11 cá nhân bị béo phì và các bất thường về phát triển thần kinh. Kết hợp các báo cáo, vùng chung 1 Mb được chia sẻ bởi các mất đoạn của tất cả 11 cá nhân chỉ bao gồm một gen, BDNF.
Đảo đoạn trung tâm 11p: Một dạng khác của thiếu hụt một nửa gen BDNF có thể được gây ra bởi sự mất biểu hiện từ một alen BDNF ngay cả khi alen đó vẫn còn hiện diện. Gray và cộng sự đã mô tả một bé gái 8 tuổi với một đảo đoạn trung tâm dị hợp tử 11p13p15.3 bao gồm BDNF mà không làm gián đoạn trình tự của chính gen đó nhưng lại cản trở sự biểu hiện của alen BDNF bị đảo ngược. Nồng độ BDNF huyết thanh của cô bé giảm đáng kể so với các đối tượng đối chứng bị béo phì và có cân nặng bình thường, và cô bé biểu hiện chứng ăn nhiều, béo phì, suy giảm cảm giác đau và khuyết tật trí tuệ.
Đột biến/biến thể gen BDNF: Giải trình tự 765 trẻ em bị béo phì khởi phát sớm không có hội chứng đã cho thấy năm biến thể hiếm BDNF dị hợp tử được dự đoán là có hại in silico không có mặt ở 480 đối tượng đối chứng không bị béo phì. Đa hình BDNF Val66Met phổ biến, làm suy giảm sự bài tiết BDNF phụ thuộc vào hoạt động, có liên quan đến các cơn ăn vô độ trong chứng cuồng ăn và rối loạn ăn uống vô độ, và sự tăng methyl hóa BDNF có liên quan đến chứng cuồng ăn. Các mối liên quan của BDNF Val66Met với BMI và béo phì chưa có kết luận, nhưng phần lớn cho thấy rằng alen Met có tác dụng bảo vệ. Biến thể intron BDNF rs12291063 (đồng hợp tử ở ~10% cá nhân gốc Phi hoặc Latino; hiếm ở người da trắng không phải gốc Tây Ban Nha ở châu Âu) có liên quan đến việc giảm biểu hiện BDNF ở VMH và tăng tích mỡ. Cùng nhau, những quan sát này chỉ ra rằng sự thiếu hụt BDNF có thể là nguyên nhân cơ bản của các nguyên nhân phổ biến, cũng như hiếm gặp, của chứng ăn nhiều và béo phì.
Đột biến NTRK2. Gen NTRK2 mã hóa tiểu đơn vị đặc hiệu cho phối tử của TrkB, là thụ thể cho BDNF. Một báo cáo trường hợp mô tả một cậu bé 8 tuổi với một đột biến dị hợp tử của NTRK2 ngăn cản sự tự phosphoryl hóa của thụ thể khi phối tử gắn vào. Đứa trẻ biểu hiện cân nặng bình thường khi sinh, chứng ăn nhiều và tăng cân nghiêm trọng bắt đầu từ 6 tháng tuổi, và các bất thường thần kinh bao gồm co giật, giảm trương lực cơ, chậm phát triển và giảm cảm giác đau.
Thiếu hụt một nửa gen SH2B1. Nhiễm sắc thể 16p11.2 đã được chứng minh là chiếm 0,7% trong một đoàn hệ lớn các bệnh nhân bị béo phì nặng và 4% trong một đoàn hệ nhi khoa bao gồm trẻ em bị chậm phát triển ngoài béo phì. Một gen ứng cử viên cho locus này là SH2B1, mã hóa một protein thích ứng tín hiệu cần thiết cho sự phát triển của sợi trục được kích thích bởi BDNF. Các đột biến dị hợp tử trong SH2B1 đã được mô tả ở 1,7% trong một đoàn hệ các bệnh nhân bị béo phì nặng. Các bệnh nhân bị ảnh hưởng cũng biểu hiện các bất thường về thần kinh-tâm thần.
Các Rối loạn Béo phì dạng Hội chứng
Một số hội chứng béo phì đã được bao gồm trong phần trước về các rối loạn con đường leptin-melanocortin vì các gen gây bệnh được cho là có liên quan đến con đường đó. Sau đó, các hội chứng bổ sung có nguyên nhân chưa được phân định rõ ràng.
Hội chứng Prader-Willi. PWS là một rối loạn béo phì do ăn nhiều (tỷ lệ mắc ~ 1 trên 20.000) gây ra bởi sự thiếu biểu hiện của các gen có nguồn gốc từ cha trên nhiễm sắc thể 15q11-13, do mất đoạn dị hợp tử của các alen của cha (70%), dị nhiễm sắc thể đơn bội trong đó hai bản sao của các alen của mẹ được thừa hưởng (20%–30%), hoặc các khiếm khuyết in dấu trong đó các alen của cha bị im lặng do methyl hóa không phù hợp (2%–5%). Trong PWS cổ điển, các đặc điểm chẩn đoán chính bao gồm giảm trương lực cơ sơ sinh, các vấn đề về ăn uống ở trẻ sơ sinh, tăng cân nhanh sau giai đoạn nhũ nhi, chứng ăn nhiều, chậm phát triển, suy sinh dục do thiếu gonadotropin, và các đặc điểm khuôn mặt đặc trưng (mặt hẹp, mắt hình quả hạnh, miệng nhỏ, môi trên mỏng, và khóe miệng trễ xuống). Các đặc điểm chẩn đoán phụ bao gồm giảm cử động của thai nhi, khóc yếu và hôn mê ở trẻ sơ sinh, các vấn đề về hành vi, rối loạn giấc ngủ, lùn (thiếu hụt GH), giảm sắc tố (trong các trường hợp mất đoạn liên quan đến OCA2, một gen liên quan đến bệnh bạch tạng trong khu vực), bàn tay và bàn chân nhỏ, và cạy da. Các đặc điểm phổ biến khác bao gồm ngưỡng đau cao, giảm nôn, bất ổn nhiệt độ, vẹo cột sống, dậy thì sớm, và kỹ năng khác thường với các câu đố ghép hình.
Các nỗ lực để phân lập các gen gây bệnh cho PWS cho thấy rằng sự liên quan của nhiều hơn một gen có thể là cần thiết để hội chứng đầy đủ được biểu hiện. Nhiều đặc điểm của PWS có thể được quan sát thấy ở những bệnh nhân có đột biến bất hoạt của MAGEL2, và việc sàng lọc các đột biến MAGEL2 được khuyến nghị cho những bệnh nhân có biểu hiện PWS không điển hình, còn được gọi là hội chứng Schaaf-Yang. Hội chứng này được đặc trưng bởi giảm trương lực cơ ở trẻ sơ sinh, khó khăn trong việc ăn uống, chậm phát triển, và rối loạn phổ tự kỷ, nhưng chỉ một phần ba số bệnh nhân phát triển chứng ăn nhiều. Chuột bị loại bỏ gen Magel2 bị chậm phát triển sơ sinh, tăng cân quá mức sau khi cai sữa, và phát triển tăng tích mỡ, do đó những con chuột này có thể được sử dụng như một mô hình để nghiên cứu các thông số béo phì trong PWS. Nguyên nhân của chứng ăn nhiều trong PWS chưa được biết và có khả năng là đa yếu tố. Tăng ghrelin máu được quan sát thấy ở những bệnh nhân mắc PWS, nhưng việc ức chế ghrelin bằng dược lý đã không thành công trong việc giảm chứng ăn nhiều hoặc trọng lượng cơ thể, đặt ra câu hỏi về vai trò sinh lý bệnh của ghrelin trong PWS. Nồng độ EC trong tuần hoàn cao hơn cũng đã được quan sát thấy ở những bệnh nhân mắc PWS. Chuột bị loại bỏ gen Magel2 có biểu hiện tăng của các thụ thể EC và việc sử dụng một chất đối kháng EC gây ra giảm cân.
Nồng độ leptin huyết thanh ở bệnh nhân PWS tăng tương ứng với khối lượng mỡ cao hơn của họ và ở mức tương tự như bệnh nhân bị các dạng béo phì không có hội chứng. Do đó, việc sản xuất leptin được bảo tồn trong PWS. Biểu hiện của NPY và AgRP dường như bị ức chế một cách thích hợp trong mô dưới đồi sau khi chết từ bệnh nhân PWS, cho thấy rằng sự biểu hiện quá mức của các peptide gây thèm ăn này không có khả năng là nguyên nhân gây ra chứng ăn nhiều trong PWS. Tuy nhiên, một số bằng chứng chỉ ra sự truyền tín hiệu bị lỗi dọc theo nhánh POMC/CART của con đường leptin-melanocortin. Chuột bị loại bỏ gen Magel2 kháng lại tác dụng ức chế cảm giác thèm ăn của việc sử dụng leptin, với sự phát triển của sự không nhạy cảm với leptin xảy ra trong khoảng từ 4 đến 6 tuần tuổi, đồng thời với sự suy giảm số lượng tế bào thần kinh POMC ở nhân cung, cho thấy rằng sự chuyển đổi từ chậm phát triển ở trẻ sơ sinh sang chứng ăn nhiều ở thời thơ ấu đối với bệnh nhân PWS có thể là do một quá trình thoái hóa thần kinh. Biểu hiện PC1 giảm đã được báo cáo trong các tế bào gốc vạn năng cảm ứng có nguồn gốc từ bệnh nhân người mắc PWS, nếu điều này cũng đúng in vivo, sẽ làm trầm trọng thêm bất kỳ sự thiếu hụt nào trong POMC bằng cách giảm thêm α-MSH được xử lý từ sự phân cắt POMC của PC1. Phù hợp với giả thuyết rằng sự gián đoạn của tín hiệu POMC có thể là nguyên nhân gây ra chứng ăn nhiều trong PWS là quan sát rằng bệnh nhân PWS có nồng độ BDNF huyết thanh và huyết tương giảm so với các đối tượng đối chứng bị béo phì và có cân nặng bình thường. Xem xét vai trò của BDNF như một chất trung gian hạ nguồn của tín hiệu MC4R và chức năng của nó trong nhận thức thần kinh và nhận thức đau, sự thiếu hụt BDNF có thể giải thích cho chứng ăn nhiều, khuyết tật trí tuệ, các bất thường về hành vi, và khả năng chịu đau cao liên quan đến PWS. Mặc dù chứng ăn nhiều là nguyên nhân chính gây tăng cân, khối lượng cơ nạc thấp hơn trong PWS làm giảm REE 40%, góp phần thêm vào sự mất cân bằng năng lượng. Thiếu hụt GH cũng dẫn đến ly giải mỡ bị lỗi, thúc đẩy thêm tình trạng tích mỡ, nhưng điều này có thể được đảo ngược bằng cách bổ sung GH.
Hội chứng Bardet-Biedl (BBS). BBS là một hội chứng lặn trên nhiễm sắc thể thường (tỷ lệ mắc ~ 1 trên 100.000) được đặc trưng bởi chứng ăn nhiều, béo phì, loạn dưỡng võng mạc, thừa ngón, suy giảm nhận thức, bệnh thận, và suy sinh dục ở nam giới. Hơn 20 gen mã hóa các protein liên quan đến sự hình thành, ổn định, và chức năng của lông mao đã được liên quan đến BBS. Các mô hình chuột của BBS có lông mao bị lỗi và suy giảm vận chuyển và truyền tín hiệu của thụ thể leptin. Những con chuột này không đáp ứng với việc sử dụng leptin với sự thiếu phosphoryl hóa STAT-3 và thiếu sự giảm lượng thức ăn nạp vào. Tăng leptin máu có trước khi béo phì khởi phát trong một số nhưng không phải tất cả các nghiên cứu, đặt ra câu hỏi liệu kháng leptin có phát triển trước hay sau khi béo phì khởi phát hay không, và vai trò của lông mao trong sự biệt hóa của tế bào mỡ cho thấy rằng sự lắng đọng mỡ nguyên phát cũng có thể là một nguyên nhân góp phần gây ra béo phì trong BBS. Ở bệnh nhân BBS, nồng độ leptin huyết thanh cao hơn so với các đối tượng đối chứng có cùng chỉ số BMI, cho thấy rằng ở người, kháng leptin không tương xứng với mức độ tích mỡ có thể đóng một vai trò trong sinh lý bệnh của béo phì liên quan đến bệnh lông mao. Hỗ trợ giả thuyết rằng tín hiệu melanocortin không đủ góp phần vào chứng ăn nhiều trong BBS, một nghiên cứu thí điểm về chất chủ vận MC4R setmelanotide cho thấy sự ức chế cảm giác đói và gây ra giảm cân ở bệnh nhân BBS.
Hội chứng Alström. Hội chứng Alström (AS) là một dạng béo phì đơn gen hiếm gặp (< 500 trường hợp được báo cáo) gây ra bởi các đột biến lặn trong gen liên quan đến trung thể và thể gốc ALMS1. AS được đặc trưng bởi loạn dưỡng võng mạc, mất thính lực thần kinh giác quan, bệnh cơ tim, xơ phổi, bệnh thận, béo phì ở trẻ em, kháng insulin nặng, khả năng mắc đái tháo đường tuýp 2 cao hơn, triglyceride tăng cao, và viêm gan nhiễm mỡ. Các bệnh nội tiết thường gặp bao gồm suy giáp (một phần ba trung ương, hai phần ba nguyên phát), suy thượng thận trung ương, suy sinh dục ở nam giới (một phần ba trung ương, hai phần ba nguyên phát), cường androgen ở nữ giới, và lùn/nồng độ IGF-1 thấp. Trái ngược với BBS, bệnh nhân AS không bị thừa ngón và thường duy trì chức năng trí tuệ bình thường. Chức năng của protein ALMS1 chưa được biết rõ nhưng được cho là quan trọng đối với sự hình thành, ổn định, và chức năng của lông mao. Chuột bị loại bỏ gen Alms1 có số lượng lông mao thần kinh vùng dưới đồi giảm, và bị chứng ăn nhiều và béo phì. Bởi vì chức năng của tế bào thần kinh POMC phụ thuộc vào lông mao và sự vận chuyển trong lông mao, chức năng POMC giảm được giả định là góp phần vào chứng ăn nhiều của AS. Phù hợp với giả thuyết này, một nghiên cứu thí điểm về chất chủ vận MC4R setmelanotide cho thấy sự ức chế cảm giác đói, gây ra giảm cân, và cải thiện cân bằng nội môi glucose ở bệnh nhân AS.
Hội chứng Smith-Magenis. SMS (tỷ lệ mắc ~ 1 trên 25.000) là do đột biến hoặc mất đoạn dị hợp tử của gen cảm ứng bởi axit retinoic 1 (RAI1). RAI1 là một chất điều hòa phiên mã của BDNF liên quan đến sự phát triển của sọ mặt và hệ thần kinh. Ở ếch, việc hạ gục Rai1 bằng cách sử dụng các morpholino antisense dẫn đến biểu hiện mRNA của Bdnf thấp hơn và sự phát triển não và mặt bất thường. Chuột bị loại bỏ gen Rai1 dị hợp tử có biểu hiện Bdnf ở vùng dưới đồi giảm và biểu hiện chứng ăn nhiều và béo phì sau 20 tuần tuổi với chế độ ăn bình thường, và trước 16 tuần với chế độ ăn nhiều chất béo hoặc nhiều carbohydrate. SMS ở bệnh nhân người được đặc trưng bởi khuyết tật trí tuệ, các hành vi kém thích nghi và tự gây thương tích, rối loạn giấc ngủ, và các đặc điểm khuôn mặt dị dạng (đầu ngắn, mặt rộng, trán dô, dính chân mày, hai mắt xa nhau, mắt xếch lên, thiểu sản giữa mặt với sống mũi lõm, môi trên hình lều, hàm nhô, và tai thấp hoặc có hình dạng bất thường). Chứng ăn nhiều và béo phì thường không được quan sát thấy cho đến cuối thời thơ ấu hoặc thanh thiếu niên, bắt chước sự khởi phát muộn hơn của các triệu chứng này ở chuột, và rõ rệt hơn ở những bệnh nhân có đột biến Rai1 so với những người bị mất đoạn. Việc kiểm tra các tác động chức năng của các đột biến được tìm thấy ở bệnh nhân đã cho thấy rằng protein RA1 bị đột biến không thể định vị đến nhân và không kích hoạt biểu hiện của một gen báo cáo được điều khiển bởi một promoter BDNF nội sinh. Sự hiện diện của protein bị đột biến có thể có một tác động trội âm lên protein bình thường còn lại, do đó dẫn đến một kiểu hình béo phì nghiêm trọng hơn ở những bệnh nhân có đột biến.
Hội chứng Cohen. Hội chứng Cohen (< 1000 trường hợp được báo cáo) là do các đột biến lặn trên nhiễm sắc thể thường trong gen VPS13B và được đặc trưng bởi đầu nhỏ, giảm trương lực cơ, khuyết tật trí tuệ, cận thị tiến triển, loạn dưỡng võng mạc, tăng động khớp, giảm bạch cầu trung tính, béo phì thân mình phát triển ở cuối thời thơ ấu, kháng insulin/đái tháo đường tuýp 2, và các đặc điểm dị dạng (tóc và lông mày dày, lông mi dài, khe mi mắt xếch xuống và có hình sóng, đầu mũi hình củ hành, nhân trung trơn hoặc ngắn, răng cửa trên trung tâm nhô, miệng mở, bàn tay và bàn chân hẹp, và ngón tay thon). VPS13B mã hóa một protein của bộ máy Golgi có liên quan đến quá trình glycosyl hóa protein và vận chuyển nội bào trong các tế bào thần kinh và tế bào mỡ.
Hội chứng Carpenter. Hội chứng Carpenter (< 100 trường hợp được báo cáo) là do các đột biến lặn trên nhiễm sắc thể thường trong gen RAB23 (liên quan đến vận chuyển túi) hoặc MEGF8 (liên quan đến sự kết dính tế bào) và được đặc trưng bởi dính khớp sọ hình chóp hoặc hình lá cỏ ba lá, khuyết tật trí tuệ, béo phì khởi phát ở trẻ em, sống mũi phẳng, khe mi mắt xếch xuống, tai thấp và có hình dạng bất thường, hàm nhỏ, răng sữa nhỏ, dính da ngón tay thứ ba và thứ tư, ngón ngắn, thừa ngón, thoát vị rốn, mất thính lực, dị tật tim, hông biến dạng, gù vẹo cột sống, chân vòng kiềng, tinh hoàn ẩn, phủ tạng đảo ngược, tim lệch phải, và chuyển vị các động mạch lớn.
Béo phì do vùng dưới đồi
Tổn thương vùng dưới đồi có thể xảy ra do khối u hệ thần kinh trung ương, phẫu thuật, xạ trị, chấn thương, viêm, hoặc các bệnh thâm nhiễm. Tổn thương VMH và ARC của MBH là những vùng quan trọng dẫn đến hiện tượng béo phì do vùng dưới đồi, được đặc trưng bởi sự tăng cân cực kỳ nhanh chóng. Ví dụ, điều điển hình trong 6 đến 12 tháng đầu tiên sau phẫu thuật u sọ hầu đối với các khối u liên quan đến sàn não thất ba (các trung tâm cân bằng nội môi vùng dưới đồi được mô tả ở trên) là bệnh nhân tăng cân hơn 50%. Về mặt cơ chế, người ta đã biết rõ rằng các tổn thương điện giải hai bên hoặc sự mất kết nối của VMH ở chuột dẫn đến tăng cân khó chữa, ngay cả khi hạn chế thức ăn. Ban đầu, việc tăng cân được cho là chỉ là một vấn đề về chứng ăn nhiều và tăng dự trữ năng lượng. Tuy nhiên, bây giờ chúng ta hiểu rằng một rối loạn chức năng của tín hiệu con đường leptin-melanocortin có thể làm thay đổi cả con đường hướng tâm và ly tâm của cân bằng năng lượng và dẫn đến tăng cân nghiêm trọng và khó chữa do nhiều hơn chỉ là ăn quá nhiều. Sự tổn thương vùng dưới đồi ngăn cản sự tích hợp của các tín hiệu hướng tâm ngoại vi, và do đó hệ thần kinh trung ương không thể phát hiện sự đủ chất dinh dưỡng ở ngoại vi và do đó phản ứng như thể đang ở trong trạng thái đói. Chỉ riêng việc hạn chế calo là không đủ vì bệnh nhân cũng bị giảm hoạt động giao cảm, làm giảm tiêu hao năng lượng và ly giải mỡ, và tăng trương lực đối giao cảm (phế vị), thúc đẩy tăng tiết insulin và dự trữ năng lượng. Ở cả động vật và con người, sự tăng phản ứng của phế vị có thể được ngăn ngừa bằng cách cắt dây phế vị tuyến tụy. Điều trị cho rối loạn này vẫn còn cực kỳ khó khăn, vì não dường như bị “khóa” trong một sự cân bằng gây thèm ăn ủng hộ việc tiêu thụ và dự trữ năng lượng. Vì giai đoạn tăng cân quan trọng là trong năm đầu tiên sau tổn thương não, điều quan trọng là phải tập trung vào các biện pháp phòng ngừa trong cửa sổ cơ hội hẹp này. Điều trị toàn diện nên bao gồm chăm sóc tâm lý để giải quyết các rối loạn nhận thức, hành vi và giấc ngủ thường xảy ra; thay thế hormone nội tiết cho các thiếu hụt; và quản lý lối sống chuyên sâu (dinh dưỡng và tập thể dục). Các liệu pháp y tế, chẳng hạn như thuốc làm tăng nhạy cảm insulin, thuốc chẹn bài tiết insulin, các chất tương tự GLP-1, chất kích thích và oxytocin đều đã được điều tra và mỗi loại đều cho thấy những lợi ích khiêm tốn, nhưng bản chất khó chữa của bệnh khiến việc quản lý trở nên vô cùng khó khăn. Ngày nay, người ta chấp nhận rằng mọi nỗ lực nên được thực hiện để tránh chấn thương phẫu thuật đối với các trung tâm cân bằng nội môi vùng dưới đồi, ngay cả với cái giá phải trả là để lại mô khối u còn sót lại (trong các trường hợp u sọ hầu thường liên quan đến khu vực này).
Hội chứng ROHHAD (béo phì khởi phát nhanh với rối loạn chức năng vùng dưới đồi, giảm thông khí và rối loạn chức năng tự chủ) là một dạng béo phì do vùng dưới đồi chưa được hiểu rõ và có tiên lượng rất xấu. Tuổi chẩn đoán trung bình là 4 tuổi và biểu hiện là tăng cân cực nhanh ở một đứa trẻ trước đây khỏe mạnh. ROHHAD dường như là một tình trạng thoái hóa thần kinh tiến triển từ béo phì do ăn nhiều đến rối loạn chức năng vùng dưới đồi toàn cầu (bao gồm thiếu hụt GH, suy sinh dục do thiếu gonadotropin, suy giáp trung ương, và các bất thường ACTH), giảm thông khí trung ương hoặc kiểm soát hô hấp bị thay đổi, rối loạn chức năng tự chủ (nhịp tim chậm cần đặt máy tạo nhịp), rối loạn điều hòa nhiệt, và nguy cơ phát triển các khối u có nguồn gốc từ mào thần kinh (ví dụ: u hạch thần kinh và u nguyên bào hạch thần kinh). Mặc dù BDNF và NTRK2 đã được sàng lọc như là các gen ứng cử viên cho ROHHAD, cho đến nay vẫn chưa xác định được đột biến nào trong các gen này. Tuy nhiên, một bệnh nhân, một cậu bé 11 tuổi có chỉ số BMI là 62 kg/m² với chẩn đoán lâm sàng là ROHHAD, được phát hiện có một đột biến cắt ngắn dị hợp tử của RAI1, là một chất điều hòa phiên mã của BDNF và có liên quan là gen gây bệnh cho SMS. Ngoài béo phì nặng, bệnh nhân này còn biểu hiện khuyết tật trí tuệ, rối loạn phổ tự kỷ, các đặc điểm khuôn mặt dị dạng (đầu to, hai mắt xa nhau, sống mũi phẳng, trán dô, và lỗ mũi hếch), khả năng chịu đau cao, đổ mồ hôi quá nhiều, không sốt khi bị nhiễm trùng, ngưng thở khi ngủ do tắc nghẽn và trung ương, và giảm thông khí dẫn đến phải mở khí quản. Các bất thường về hormone tuyến yên và các khối u mào thần kinh không có ở bệnh nhân này. Do đó, anh ta có thể được phân loại là ROHHAD không điển hình hoặc có thể là SMS không điển hình, và việc sàng lọc thiếu hụt một nửa gen RAI1 nên được xem xét trong chẩn đoán phân biệt cho những bệnh nhân có các đặc điểm của ROHHAD. Nếu không được điều trị, ROHHAD có tỷ lệ tử vong cao do suy hô hấp. Các liệu pháp điều hòa miễn dịch đã được thử nghiệm và cho thấy một số lợi ích trong việc làm chậm quá trình tiến triển của bệnh nhưng không có phương pháp điều trị chữa khỏi nào được biết đến.
Đánh giá và điều trị béo phì ở trẻ em
Tiếp cận chẩn đoán
Chìa khóa để điều trị béo phì thành công là chẩn đoán chính xác. Các công cụ chẩn đoán của chúng ta chưa được phát triển đầy đủ, vì vậy việc kết hợp điều trị với chẩn đoán vẫn còn chưa chắc chắn. Các điểm cụ thể trong quá trình đánh giá và lý do của chúng được liệt kê trong Bảng 24.1. Khi khai thác bệnh sử, cân nặng lúc sinh, chỉ số BMI của cha mẹ, tiếp xúc với đái tháo đường thai kỳ, sinh non, tiền sử bú mẹ và các biến chứng sơ sinh (đặc biệt là tổn thương hệ thần kinh trung ương) đều có liên quan. Béo phì của bệnh nhân được ghi nhận càng sớm, khả năng xác định được nguyên nhân hữu cơ càng cao. Các bất thường về phát triển thần kinh và các dấu hiệu dị dạng có thể báo hiệu sự cần thiết của việc chuyển khám di truyền. Danh sách thuốc phải được xem xét, đặc biệt là đối với glucocorticoid và thuốc chống loạn thần không điển hình. Đau xương khớp, đau đầu và ngáy phải được đánh giá. Tiền sử ăn uống phải bao gồm việc bỏ bữa sáng, uống soda và nước trái cây hàng ngày, tần suất và loại đồ ăn vặt. Một hệ quả là số lượng người chăm sóc trẻ vì điều này làm tăng căng thẳng, sự hỗn loạn trong gia đình và thiếu sự giám sát trẻ.
Bảng 24.1 Đánh giá Chẩn đoán Béo phì ở Trẻ em và các Bệnh đi kèm
| Bệnh sử | |
| Nguyên nhân di truyền | “BMI của cha mẹ, chủng tộc, khuyết tật trí tuệ” |
| Nguyên nhân biểu sinh | “Cân nặng lúc sinh, các khó khăn trong thai kỳ, sinh non” |
| Nguyên nhân TKTW | “Tổn thương TKTW, khuyết tật trí tuệ hoặc chậm phát triển” |
| Nguyên nhân nội tiết | “Chậm tăng trưởng theo chiều dài, tóc đỏ” |
| Nguyên nhân do thuốc | “Tiền sử dùng thuốc, đặc biệt là thuốc chống loạn thần không điển hình” |
| Nguyên nhân ăn uống | “Hồi cứu lượng calo, đặc biệt là chất lỏng chứa đường, tiền sử bú mẹ” |
| Nguyên nhân hoạt động thể chất | “Tiền sử tập thể dục, hồi cứu thời gian xem tivi, máy tính và điện thoại di động” |
| Nguyên nhân căng thẳng | “Tình trạng kinh tế xã hội, số lượng người chăm sóc, hồi cứu thời gian xem tivi, tình trạng giấc ngủ, trầm cảm không điển hình” |
| Ngưng thở khi ngủ | “Tiền sử ngáy, đau đầu, thức dậy với cơn đau đầu” |
| PCOS | “Rậm lông, thiểu kinh, vô kinh” |
| Đái tháo đường tuýp 2 | “Tiểu nhiều, uống nhiều, tiểu đêm, sụt cân gần đây” |
| Bệnh lý xương khớp | “Đau đầu gối hoặc hông, hạn chế vận động” |
| Trầm cảm | “Cảm xúc, mức độ hoạt động, kết quả học tập” |
| Khám thực thể | |
| Kháng insulin | “Gai đen, u mềm treo, chu vi vòng eo” |
| Tăng huyết áp | HA tâm thu hoặc HA tâm trương > bách phân vị thứ 90 theo tuổi |
| U giả não | Phù gai thị |
| Gan nhiễm mỡ | Gan to |
| PCOS | Rậm lông |
| Dậy thì sớm/muộn | Tình trạng tuyến sinh dục và lông mu |
| Ngưng thở khi ngủ | Phì đại amidan |
| Bệnh cơ | “Giảm trương lực cơ, giảm phản xạ” |
| Béo phì dạng hội chứng | “Các dấu hiệu thần kinh-da đặc hiệu (xem Hộp 24.1), khuyết tật trí tuệ” |
| Xét nghiệm | |
| Gan nhiễm mỡ | “ALT, siêu âm gan” |
| Rối loạn dung nạp glucose | Glucose lúc đói > 100 hoặc glucose 2 giờ > 140 |
| Đái tháo đường tuýp 2 | Glucose lúc đói > 125 hoặc glucose 2 giờ > 200; HbA1c > 6.5% |
| Rối loạn lipid máu | “Hồ sơ lipid với VLDL tăng, TG:HDL > 2.5” |
| Kháng insulin | “Insulin lúc đói, glucose” |
| Tăng tiết insulin | NGDPĐU 3 giờ với nồng độ insulin |
| Tổn thương TKTW | “MRI, đặc biệt với các hình ảnh tập trung vào vùng dưới đồi” |
Cũng cần xét nghiệm axit uric: > 5.5 như một chỉ số đại diện cho việc tiêu thụ đường Thyroxine tự do, hormone kích thích tuyến giáp để loại trừ suy giáp ALT, Alanine aminotransferase; BMI, chỉ số khối cơ thể; TKTW, hệ thần kinh trung ương; HA tâm trương, huyết áp tâm trương; HbA1c, hemoglobin A1c; HDL, lipoprotein tỷ trọng cao; MRI, chụp cộng hưởng từ; NGDPĐU, nghiệm pháp dung nạp glucose đường uống; PCOS, hội chứng buồng trứng đa nang; HA tâm thu, huyết áp tâm thu; TG, triglyceride; VLDL, lipoprotein tỷ trọng rất thấp.
Khi khám thực thể, sự tăng trưởng theo chiều dài là yếu tố then chốt, vì đánh giá nội tiết cổ điển (ví dụ: suy giáp, Cushing, thiếu GH, PHP) là không cần thiết nếu sự tăng trưởng theo chiều dài không bị suy giảm. Tuy nhiên, tăng insulin máu và kháng insulin có thể gây ra tăng trưởng nhanh, do sự phản ứng chéo của insulin với thụ thể IGF-1. Các đặc điểm thể chất quan trọng cần đánh giá bao gồm gai đen và chu vi vòng eo (cả hai đều liên quan đến kháng insulin và hội chứng chuyển hóa); soi đáy mắt để loại trừ u giả não nếu có các triệu chứng gợi ý; gan to để gợi ý gan nhiễm mỡ; rậm lông để gợi ý PCOS; và trương lực cơ để đánh giá giảm trương lực và bệnh cơ, làm giảm tiêu hao năng lượng. Béo phì khởi phát sớm (trong giai đoạn nhũ nhi và mới biết đi) cùng với các đặc điểm dị dạng nên làm dấy lên nghi ngờ về một rối loạn di truyền tiềm ẩn.
Đánh giá xét nghiệm bao gồm các xét nghiệm về các bệnh đi kèm liên quan đến béo phì (ví dụ: aspartate aminotransferase, ALT, lipid, glucose lúc đói và hemoglobin A1c) và chụp X-quang đầu gối và hông (trong các trường hợp nghi ngờ biến dạng Blount). Các nghiên cứu chẩn đoán cụ thể phải được điều chỉnh cho phù hợp với từng bệnh nhân. Ví dụ, sự suy giảm tăng trưởng (thường là tăng cân nhanh cùng với giảm tốc độ tăng trưởng theo chiều dài) đòi hỏi phải đánh giá nội tiết, bao gồm các xét nghiệm chức năng tuyến giáp, IGF-1 và IGFBP-3, cortisol nước tiểu 24 giờ hoặc cortisol huyết thanh lúc nửa đêm, và có thể là MRI vùng dưới đồi và tuyến yên. Nồng độ hormone luteinizing (LH), hormone kích thích nang trứng (FSH) và testosterone có thể phù hợp khi đánh giá dậy thì muộn ở nam giới hoặc PCOS ở nữ giới. Bệnh nhân chậm phát triển sẽ cần xét nghiệm nhiễm sắc thể và MRI. Béo phì nặng ở trẻ mới biết đi có thể cần xét nghiệm nồng độ leptin và xét nghiệm di truyền cho đột biến thụ thể MC4R hoặc leptin. Mối tương quan giữa insulin lúc đói và độ nhạy insulin toàn thân có nguồn gốc từ kỹ thuật kẹp glucose đẳng đường-tăng insulin máu ở trẻ em là kém, cho thấy rằng nó là một chỉ số thay thế không phù hợp cho độ nhạy insulin toàn thân. Điều này có thể liên quan đến thực tế là nồng độ insulin lúc đói có khả năng phản ánh độ nhạy insulin của gan hơn là của toàn bộ cơ thể. Nghiệm pháp dung nạp glucose đường uống (NGDPĐU) có thể trở nên cần thiết ở một đứa trẻ béo phì, để đánh giá bệnh đái tháo đường tuýp 2 trong các trường hợp có tiền sử gợi ý và sự hiện diện của các yếu tố nguy cơ gia đình.
Thay đổi lối sống
Thay đổi lối sống vẫn là nền tảng của liệu pháp điều trị béo phì, đặc biệt là ở trẻ em. Cách tiếp cận này dựa trên lẽ thường, và một số ít các nghiên cứu ban đầu chứng minh hiệu quả của lối sống trong một nhóm trẻ em được lựa chọn cẩn thận với sự theo dõi chuyên sâu. Dữ liệu hỗ trợ hiệu quả lâu dài của việc thay đổi lối sống trong “thế giới thực” không đặc biệt thuyết phục. Một phân tích gần đây về nhiều phương pháp đã kết luận rằng mặc dù có dữ liệu chất lượng hạn chế để khuyến nghị một chương trình điều trị được ưa chuộng hơn một chương trình khác, các can thiệp lối sống hành vi kết hợp so với chăm sóc tiêu chuẩn hoặc tự giúp đỡ có thể tạo ra sự giảm thừa cân đáng kể và có ý nghĩa lâm sàng ở trẻ em và thanh thiếu niên. Quan trọng là, các nghiên cứu theo dõi của nhiều can thiệp quy mô lớn thường tìm thấy một hiệu quả giảm nhanh chóng sau khi nghiên cứu hoàn thành. Cuối cùng, một phân tích tổng hợp của 39 nghiên cứu can thiệp được công bố được thiết kế để ngăn ngừa béo phì ở trẻ em cho thấy rằng 40% trong số 33.852 trẻ em tham gia đã giảm BMI, trong khi 60% còn lại không có hiệu quả.
Người ta đã chỉ ra rằng 1 năm sau khi giảm cân ban đầu, nồng độ của các chất trung gian tuần hoàn của cảm giác thèm ăn khuyến khích tăng cân trở lại sau khi giảm cân do chế độ ăn không trở lại mức được ghi nhận trước khi giảm cân. Hơn nữa, sau khi giảm cân đáng kể, dù được gây ra bởi các biện pháp bảo tồn hay xâm lấn, hồ sơ chuyển hóa và nội tiết tố vẫn duy trì một “phản ứng đói” (một sự thích ứng chuyển hóa nhằm trở lại cân nặng trước đó) ngay cả nhiều năm sau can thiệp. Điều này nhấn mạnh vấn đề duy trì giảm cân lâu dài và những khó khăn mà những người muốn giảm cân và duy trì nó phải đối mặt. Một cách lạc quan, hiệu quả của các can thiệp để ngăn ngừa, thay vì đảo ngược, béo phì ở trẻ em (đặc biệt là đối với các chương trình nhắm đến trẻ em từ 6–12 tuổi) dường như hiệu quả hơn nhiều.
Mặc dù giảm cân là mục tiêu thông thường cho can thiệp ở người lớn, duy trì cân nặng là mục tiêu được khuyến nghị cho phần lớn trẻ em. Việc ngăn ngừa tăng cân dễ dàng hơn, ít tốn kém hơn và hiệu quả hơn so với việc điều trị chính bệnh béo phì, và việc ngăn ngừa thừa cân ở trẻ em trước khi có các hành vi liên quan đến nguy cơ là rất quan trọng để ngăn chặn dịch béo phì. Mục tiêu chính của phòng chống béo phì nên là thúc đẩy hoạt động thể chất và chế độ ăn uống lành mạnh với sự nhấn mạnh vào việc cải thiện sức khỏe tổng thể, thay vì giảm cân. Có ít nhất bốn lý do để thúc đẩy các can thiệp nhằm cải thiện dinh dưỡng và hoạt động thể chất ở trẻ em. Thứ nhất, đứa trẻ có thể nhận được lợi ích ngay lập tức, chẳng hạn như thể lực tốt hơn hoặc năng lượng hoặc lượng vi chất dinh dưỡng nạp vào. Thứ hai, can thiệp vào các giai đoạn quan trọng có thể cải thiện sức khỏe người lớn. Thứ ba, sửa đổi các nguy cơ bệnh mãn tính ở trẻ em có thể dẫn đến tỷ lệ và các yếu tố nguy cơ thấp hơn ở người lớn. Cuối cùng, việc sửa đổi hành vi của trẻ em có thể dẫn đến các hành vi được cải thiện ở tuổi trưởng thành sẽ bảo vệ chống lại các bệnh mãn tính.
Liệu pháp hành vi-nhận thức được thiết kế để đối phó với cả phụ huynh và bệnh nhân, với việc tái cấu trúc và củng cố hành vi. Các thay đổi hành vi bao gồm các buổi tư vấn, dạy kỹ năng làm cha mẹ, khen ngợi và hợp đồng, các công cụ tự theo dõi, kiểm soát kích thích trong nhà, làm gương về hành vi của cha mẹ, và các chương trình tập thể dục mạnh mẽ và lâu dài. Các chương trình này đã thành công trong các nghiên cứu nhỏ với các đối tượng được lựa chọn cẩn thận bởi các nhà điều tra cụ thể, nhưng vẫn chưa thành công khi được thử nghiệm trong các quần thể phòng khám. Một phương pháp lâm sàng mới bao gồm phỏng vấn tạo động lực, một phương pháp giúp bệnh nhân giải quyết sự mâu thuẫn của họ về việc thay đổi hành vi. Phương pháp này đã được chứng minh là có hiệu quả đối với lạm dụng chất và trong bệnh đái tháo đường ở người lớn; liệu nó có thành công trong bệnh béo phì nhi khoa hay không vẫn còn phải xem xét.
Can thiệp chế độ ăn
Can thiệp chế độ ăn là cần thiết để không chỉ giảm lượng calo nạp vào, mà còn để giảm phản ứng insulin thúc đẩy sự lắng đọng năng lượng quá mức vào mô mỡ. Vô số nghiên cứu chứng minh mối liên quan giữa việc tiêu thụ thực phẩm nhiều calo, nhiều chất béo, nhiều carbohydrate, ít chất xơ và sự phát triển của béo phì ở trẻ em. Các biện pháp cụ thể đã thành công trong việc giảm phản ứng insulin và thúc đẩy giảm cân hoặc ổn định cân nặng ở trẻ em bao gồm (1) loại bỏ đồ uống chứa đường (bao gồm cả soda và nước trái cây) và (2) chuyển sang chế độ ăn có tải lượng đường huyết thấp. Không chỉ việc sửa đổi chế độ ăn để giảm phản ứng insulin hỗ trợ thúc đẩy giảm cân, mà nó còn thúc đẩy tăng tiêu hao năng lượng trong giai đoạn duy trì cân nặng, có lẽ bằng cách cải thiện độ nhạy leptin, điều này sẽ giúp ngăn ngừa tăng cân trở lại. Tuy nhiên, chỉ riêng can thiệp chế độ ăn không phải là một chiến lược thành công để giảm thừa cân ở trẻ em, trừ khi việc điều trị rất chuyên sâu, trong trường hợp đó, phần lớn sẽ bỏ cuộc. Hơn nữa, các nghiên cứu về chế độ ăn kiêng không có giám sát đã chứng minh tác dụng ngược lại, tức là, nỗ lực gia tăng ở nữ thanh thiếu niên dự đoán sự gia tăng cân nặng lớn hơn và nguy cơ béo phì.
Quan trọng là, chế độ ăn là một phần quan trọng của việc duy trì cân nặng và không chỉ nhằm mục đích giảm cân. Cụ thể, sau khi giảm cân thành công—một chế độ ăn duy trì cân nặng là rất quan trọng để ngăn ngừa tăng cân trở lại do “phản ứng đói” chuyển hóa đã đề cập trước đó. Do đó, những thay đổi chế độ ăn được đề xuất cho trẻ nên được điều chỉnh cho cả đời như một chiến lược “ăn uống lành mạnh” thay vì một kế hoạch giảm cân ngắn hạn. So sánh các phương pháp tiếp cận chế độ ăn để duy trì cân nặng sau khi giảm cân ở trẻ em đã chỉ ra rằng trong số những người trưởng thành trẻ tuổi bị thừa cân/béo phì so với tiêu hao năng lượng trước khi giảm cân, việc cho ăn đẳng calo sau khi giảm 10% đến 15% cân nặng đã dẫn đến sự giảm REE và tổng tiêu hao năng lượng (TEE) lớn nhất với chế độ ăn ít chất béo, trung gian với chế độ ăn có chỉ số đường huyết thấp, và ít nhất với chế độ ăn rất ít carbohydrate. Điều này cho thấy rằng một chế độ ăn ít carbohydrate có thể làm giảm “phản ứng đói” do giảm cân gây ra và giúp duy trì cân nặng mới đạt được.
Can thiệp hoạt động thể chất
Ở người lớn, tập thể dục không phải là một phương tiện hiệu quả để gây giảm cân trừ khi kết hợp với việc giảm lượng calo nạp vào. Vai trò của tập thể dục có thể có tác động lớn hơn đến việc duy trì cân nặng, hơn là giảm cân. Tương tự, có câu hỏi về việc liệu các chế độ hoạt động thể chất cho trẻ em có thể ổn định hoặc giảm BMI hay không. Các nghiên cứu ngắn hạn cho thấy rằng tập thể dục mạnh có thể dẫn đến giảm tích mỡ ngắn hạn và cải thiện độ nhạy insulin. Điều này tương đương với tối thiểu 30 phút tập thể dục mạnh, 5 ngày mỗi tuần, như các hướng dẫn hoạt động được đề xuất gần đây từ Viện Y học. Tuy nhiên, các can thiệp như vậy cuối cùng cũng chững lại về hiệu quả, và ngay cả việc ngừng tập trong thời gian ngắn cũng nhanh chóng đảo ngược bất kỳ lợi ích nào đã tích lũy được. Để thành công, các can thiệp hoạt động thể chất phải lâu dài, bền vững và được tích hợp vào các sửa đổi hành vi nhắm vào cá nhân, gia đình, trường học và trong cộng đồng nói chung. Nói cách khác, để hoạt động thể chất có hiệu quả, nó phải trở thành một ưu tiên.
Các chương trình lối sống bao gồm tập thể dục có giám sát có thể cải thiện nồng độ insulin lúc đói nhanh nhất là 2 tuần trước khi xảy ra sụt cân có thể đo được. Hơn nữa, can thiệp lối sống đã cải thiện thành phần cơ thể mà không thay đổi trọng lượng cơ thể. Các nghiên cứu hiện có cho thấy rằng thể lực có thể đóng một vai trò quan trọng hơn việc giảm BMI đối với việc cải thiện độ nhạy insulin ở thanh thiếu niên béo phì. Các can thiệp phù hợp bao gồm (1) bắt buộc giáo dục thể chất tại trường học cho mọi trẻ em và trường học; (2) tăng khả năng tiếp cận các hoạt động giải trí sau giờ học; (3) tăng các hoạt động phù hợp với văn hóa; (4) giảm tính cạnh tranh của thể thao, để nhiều trẻ em tham gia hơn; (5) tăng cường kết hợp hoạt động thể chất vào cuộc sống hàng ngày (ví dụ: cầu thang bộ, đi bộ đến trường nếu phù hợp); và (6) tăng sự tham gia của cha mẹ vào các hoạt động giải trí thể chất, để quản lý cân nặng của chính họ và làm gương cho con cái của họ. Cuối cùng, tác động của việc giảm hành vi ít vận động bằng cách hạn chế xem truyền hình đã có hiệu quả trong cả các nghiên cứu nhỏ và lớn, và ở các nhóm dân tộc đa dạng, mặc dù thể lực không được cải thiện. Việc hạn chế xem truyền hình cũng có một tác động thứ cấp là giảm lượng calo nạp vào trong khi xem truyền hình.
Kết quả của nghiên cứu TODAY đã chỉ ra rằng việc bổ sung một can thiệp tập thể dục chuyên sâu vào các chế độ dược lý và ăn kiêng ở thanh thiếu niên mắc bệnh đái tháo đường tuýp 2 không có lợi ích bổ sung nào trong bất kỳ kết quả lâm sàng nào. Quan sát này có lẽ phản ánh sự tuân thủ “thực tế” đối với các can thiệp như vậy trong các quần thể mục tiêu có liên quan. Kết quả sử dụng chỉ riêng can thiệp chế độ ăn hoặc tập thể dục không mấy khả quan. Những nghiên cứu sử dụng các thành phần hành vi, chế độ ăn và tập thể dục cùng nhau dường như thành công hơn, mặc dù hiệu quả lâu dài còn thiếu.
Can thiệp gia đình
Không thể tránh khỏi, bệnh nhân không phải là thành viên duy nhất trong gia đình bị béo phì. Thường có một môi trường gia đình thúc đẩy béo phì ở các thành viên khác trong gia đình. Nhiều người chăm sóc khác nhau (ví dụ: bà, người giữ trẻ, v.v.) sẽ cho trẻ ăn và cho phép xem truyền hình không hạn chế như một phương pháp để hạn chế các hoạt động của chúng trong nhà, đặc biệt là ở những khu vực nguy hiểm. Một số cha mẹ sẽ không thay đổi việc mua sắm của họ để mua thực phẩm kém lành mạnh hơn vì họ tin rằng các anh chị em khác không nên bị tước đoạt nó. Cha mẹ ly hôn thường sẽ sử dụng thức ăn như một phần thưởng để mua chuộc tình yêu và lòng trung thành của đứa trẻ. Do đó, chính gia đình phải là mục tiêu của can thiệp lối sống. Sự tham gia của cha mẹ là rất quan trọng, và khái niệm “thức ăn không phải là tình yêu” phải được nhấn mạnh. Các gia đình cần được đào tạo để sửa đổi hành vi để đưa ra các lựa chọn ăn uống lành mạnh, tăng cường hoạt động, và giảm căng thẳng được nhận thấy. Một phân tích tổng hợp gần đây về các thử nghiệm ngẫu nhiên của các can thiệp lối sống kết hợp để điều trị béo phì ở trẻ em đã mang lại một sự giảm BMI đáng kể mặc dù không quá ấn tượng là 1,5 với can thiệp nhắm vào gia đình, và một sự giảm BMI không đáng kể là 0,4 ở những người chỉ nhắm vào bệnh nhân.
Điều trị bằng thuốc
Chỉ định điều trị bằng thuốc
Các liệu pháp dược lý ở trẻ em hiện phải được coi là các biện pháp bổ trợ cho việc thay đổi lối sống tiêu chuẩn. Vấn đề chính là hiện tại hầu hết các loại thuốc không được chấp thuận để sử dụng cho trẻ em. Ngoài ra, một số hạn chế ngăn cản các bác sĩ thực hiện sớm các liệu pháp thuốc để điều trị béo phì ở trẻ em, bao gồm (1) đứa trẻ nhỏ nhất mà bất kỳ liệu pháp dược lý béo phì nào hiện được Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA) chấp thuận là 10 tuổi; (2) việc sử dụng can thiệp dược lý lâu dài không phải lúc nào cũng được chứng minh là hiệu quả hơn việc sửa đổi hành vi; (3) có một số lượng hạn chế các nghiên cứu được kiểm soát tốt về can thiệp dược lý an toàn và hiệu quả ở trẻ béo phì; (4) nguy cơ tương đối đối với sự phát triển của các biến cố bất lợi ở trẻ em phải được cân nhắc với tiềm năng lâu dài để cải thiện bệnh tật và tử vong, điều này khó ước tính ở trẻ em; và (5) việc nhắm mục tiêu vào bệnh lý vẫn còn ở giai đoạn sơ khai. Ngoài ra, chúng ta không được quên rằng nhiều loại thuốc được sử dụng để điều trị béo phì ở người lớn đã dẫn đến các biến chứng không lường trước được, dẫn đến việc chúng bị hạn chế (hormone tuyến giáp, amphetamine) hoặc thu hồi (ví dụ: sibutramine, rimonabant, dinitrophenol, fenfluramine, dexfenfluramine, phenylpropanolamine, ephedra). Mặc dù có những lo ngại này, tác động tiêu cực đến sức khỏe của béo phì ở trẻ em có thể biện minh cho việc dùng thuốc lâu dài để kiểm soát sự tiến triển của nó.
Trong dược điển hiện tại về béo phì ở trẻ em (Bảng 24.2), chỉ có một phương pháp tiếp cận giảm hấp thu chất béo không đặc hiệu dành cho chính bệnh béo phì. Việc nhắm vào tình trạng kháng insulin liên quan đến béo phì bổ sung thêm các tác nhân khác nhưng hiệu quả của chúng về việc giảm cân là đáng thất vọng.
Bảng 24.2 Các loại thuốc điều trị béo phì ở trẻ em
| Thuốc | Liều lượng | Hiệu quả | Tác dụng phụ | Theo dõi và Chống chỉ định |
|---|---|---|---|---|
| Orlistat Không được FDA chấp thuận cho < 12 tuổi | 120 mg uống 3 lần/ngày | “Nhãn mở: Cân nặng–5.4 kg, BMI–2.0 kg/m² sau 6 tháng; TNNCT: Cân nặng–2.6 kg, BMI–0.85, Vòng eo–2.7 cm so với giả dược sau 12 tháng” | “Sôi bụng, đầy hơi, đau quặn bụng, đại tiện không tự chủ, đốm dầu, kém hấp thu vitamin” | Theo dõi nồng độ 25OHD3. Bổ sung đa vitamin được khuyến nghị mạnh mẽ. Một chế phẩm liều thấp hơn đã được chấp thuận để bán không kê đơn |
| Metformin Không được FDA chấp thuận để điều trị béo phì. Được chấp thuận cho ≥ 10 tuổi đối với đái tháo đường tuýp 2 | 250–1000 mg uống 2 lần/ngày | “TNNCT: Điểm z-score BMI–0.35 SD so với giả dược sau 6 tháng. TNNCT: Cân nặng–2.7% so với giả dược sau 6 tháng; Phân tích post-hoc: hiệu quả phụ thuộc vào mức độ kháng insulin; điểm z-score BMI–0.23 SD trong 4 tháng đầu, –0.12 SD trong năm tiếp theo” | “Buồn nôn, đầy hơi, chướng bụng, tiêu chảy; thường tự khỏi. Nhiễm toan lactic chưa được báo cáo ở trẻ em” | Không sử dụng trong suy thận hoặc với thuốc cản quang tiêm tĩnh mạch. Bổ sung đa vitamin được khuyến nghị mạnh mẽ |
| “Octreotide Không được FDA chấp thuận để điều trị béo phì, ngoài ra ≥ 18 tuổi” | 5–15 mcg/kg/ngày tiêm dưới da chia 3 lần | “Nhãn mở: Cân nặng–4.8 kg, BMI–2.0 kg/m² trong 6 tháng; TNNCT: –7.6 kg, BMI–2.5 kg/m² so với giả dược sau 6 tháng. Phân tích posthoc: điểm z-score BMI–0.70 SD trong 6 tháng, phụ thuộc vào bài tiết và độ nhạy insulin” | “Sỏi mật, tiêu chảy, phù, đau quặn bụng, buồn nôn, chướng bụng, giảm nồng độ thyroxine” | “Theo dõi glucose lúc đói, FT4, HbA1c. Chỉ hữu ích cho béo phì do vùng dưới đồi. Dùng đồng thời Ursodiol được khuyến nghị mạnh mẽ” |
| Leptin Không được FDA chấp thuận | “Chỉnh liều theo nồng độ huyết thanh, tiêm dưới da” | Giai thoại: BMI–19.0 kg/m² trong 4 năm | Phản ứng tại chỗ | Chỉ hữu ích trong thiếu hụt leptin |
| Topiramate Không được FDA chấp thuận để điều trị béo phì | 96–256 mg uống mỗi ngày | TNNCT: Cân nặng–8.0% so với giả dược sau 6 tháng | “Dị cảm, khó tập trung/chú ý, trầm cảm, khó ghi nhớ, các vấn đề về ngôn ngữ, căng thẳng, chậm vận động tâm thần” | Không có dữ liệu ở trẻ em |
| Hormone tăng trưởng Không được FDA chấp thuận để điều trị béo phì | 1–3 mg/m² tiêm dưới da mỗi ngày | “Giảm tỷ lệ phần trăm mỡ cơ thể, với sự gia tăng khối lượng nạc tuyệt đối” | “Phù, hội chứng ống cổ tay, tử vong ở bệnh nhân có ngưng thở khi ngủ do tắc nghẽn từ trước” | “Chỉ được khuyến nghị trong hội chứng Prader-Willi chủ yếu để tăng tốc độ chiều cao. Nó cũng làm giảm khối lượng mỡ nhưng chỉ nên được sử dụng ở những người đã được sàng lọc để loại trừ ngưng thở khi ngủ do tắc nghẽn. Phải theo dõi chặt chẽ chức năng phổi, glucose, HbA1c” |
Nên được xem xét chỉ sau một thử nghiệm 6 tháng không thành công của can thiệp lối sống. Tất cả các loại thuốc chỉ có hiệu quả khi được kết hợp với can thiệp lối sống phù hợp. 2 lần/ngày, hai lần một ngày; BMI, chỉ số khối cơ thể; FDA, Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ; FT4, thyroxine tự do; HbA1c, hemoglobin A1c; MVI, đa vitamin; uống, bằng đường uống; mỗi ngày, mỗi ngày; TNNCT, thử nghiệm ngẫu nhiên có đối chứng; SD, độ lệch chuẩn; dưới da, tiêm dưới da; 3 lần/ngày, ba lần một ngày; cân nặng, trọng lượng.
Giảm hấp thu năng lượng: Orlistat
Thuốc này là một sản phẩm vi khuẩn đã được sửa đổi, đặc biệt ức chế lipase đường ruột và có thể làm giảm hấp thu chất béo và cholesterol khoảng 30% ở các đối tượng ăn chế độ ăn 30% chất béo. Orlistat gắn không thuận nghịch vào vị trí hoạt động của lipase, ngăn chặn quá trình khử acyl của triglyceride trong lòng ruột, dẫn đến sự gia tăng bài tiết chất béo qua phân khoảng 16 g/ngày. Orlistat không ức chế các enzyme đường ruột khác. Nó có sự hấp thu tối thiểu và không có tác dụng đối với các lipase toàn thân.
Mặc dù đã có một số thử nghiệm nhãn mở về orlistat ở thanh thiếu niên, chỉ có hai thử nghiệm ngẫu nhiên có đối chứng (TNNCT) đã được công bố. Các tác dụng phụ với orlistat có thể dự đoán được từ cơ chế hoạt động của nó đối với lipase đường ruột. Orlistat dường như được dung nạp tốt ở người lớn, với các phàn nàn chính là sôi bụng, đầy hơi và đau quặn bụng. Các tác dụng phụ đáng lo ngại nhất là đại tiện không tự chủ, đốm dầu và đầy hơi kèm theo tiết dịch, điều này rất khó chịu trong quần thể nhi khoa. Orlistat không ảnh hưởng đến các đặc tính dược động học của hầu hết các tác nhân dược phẩm khác. Sự hấp thu của vitamin A và E và ß-carotene có thể bị giảm nhẹ, và điều này có thể đòi hỏi liệu pháp vitamin ở một số ít bệnh nhân. Orlistat phải được dùng trong mỗi bữa ăn, điều này làm giảm sức hấp dẫn của nó ở trẻ em, những người đang đi học vào giờ ăn trưa. Orlistat hiện được chấp thuận để điều trị cho trẻ em từ 12 tuổi. Một chế phẩm liều thấp hơn không kê đơn gần đây đã được FDA chấp thuận, và sẽ sớm có mặt trên thị trường.
Cải thiện kháng Insulin: Metformin
Metformin là một dẫn xuất guanidine hydrophilic chuỗi ngắn, được thay thế hai lần, được sử dụng để điều trị cho trẻ em và người lớn mắc T2DM. Metformin cũng làm giảm tăng insulin máu lúc đói, có thể ngăn ngừa T2DM và thúc đẩy giảm cân ở một số nhưng không phải tất cả các cá nhân béo phì, bằng cách cải thiện độ nhạy insulin của gan và cơ. Metformin có ít tác dụng đối với tiêu hao năng lượng. Mặc dù một số người tin rằng metformin thúc đẩy giảm cân thông qua một tác dụng gây chán ăn chính (vì các tác dụng phụ ban đầu là buồn nôn và khó chịu đường tiêu hóa hạn chế lượng calo nạp vào một cách cấp tính), hầu hết tin rằng sự suy giảm lượng calo nạp vào được quan sát thấy với metformin có liên quan đến việc tăng cường thanh thải glucose, thông qua việc giảm sản lượng glucose của gan, và giảm tăng insulin máu lúc đói. Metformin cải thiện kháng insulin ở gan bằng cách gây ra AMP kinase ở gan, làm giảm tân tạo glucose ở gan; do đó bài tiết insulin của tuyến tụy và nồng độ insulin ngoại vi giảm. Metformin cũng phục hồi hoạt động của PI3K và MAPK trong các tế bào cơ, cải thiện độ nhạy insulin của cơ. Một cơ chế hoạt động khác có thể có của metformin là thông qua việc kích thích GLP-1, có thể ức chế lượng thức ăn nạp vào thông qua các tác động trung ương lên VMH.
Cho đến nay, kết quả của hai TNNCT ở trẻ em và thanh thiếu niên đã được báo cáo. Việc kiểm tra các phản ứng nhãn mở đối với metformin trong một phân tích đa biến đã chứng minh hai yếu tố dự báo hiệu quả: chủng tộc (da trắng > người Mỹ da đen) và mức độ kháng insulin trước khi điều trị (trong số thanh thiếu niên béo phì, những người kháng insulin đáp ứng tốt hơn những người nhạy cảm với insulin). Metformin cũng đã được sử dụng “ngoài nhãn” để điều trị PCOS và NASH, với các mức độ thành công khác nhau. Một công dụng đặc biệt của metformin có thể là để chống lại sự tăng cân liên quan đến thuốc chống loạn thần không điển hình. Tuy nhiên, việc ngừng điều trị bằng metformin dẫn đến tăng insulin máu trở lại và tăng cân nhanh chóng, có thể phủ nhận bất kỳ tác dụng có lợi nào đã thấy trong khoảng thời gian dùng thuốc.
Các tác dụng phụ với metformin bao gồm buồn nôn, đầy hơi, chướng bụng và tiêu chảy khi bắt đầu điều trị, dường như tự giới hạn và hết trong vòng 3 đến 4 tuần sau khi bắt đầu dùng thuốc. Khoảng 5% bệnh nhân nhi ngừng điều trị bằng metformin do mức độ nghiêm trọng của các tác dụng phụ. Biến chứng đáng sợ nhất của metformin ở người lớn là nhiễm toan lactic, được ước tính xảy ra với tỷ lệ 3 trên 100.000 bệnh nhân-năm phơi nhiễm, chủ yếu ở những bệnh nhân có chống chỉ định sử dụng metformin; tuy nhiên, chưa có trường hợp nào được ghi nhận ở trẻ em. Metformin làm tăng bài tiết qua nước tiểu của vitamin B1 và B6, rất quan trọng trong chu trình axit tricarboxylic, và có thể đẩy nhanh quá trình nhiễm toan lactic. Thiếu hụt vitamin B12 cũng đã được báo cáo ở 9% đối tượng người lớn sử dụng metformin. Do đó, việc bổ sung đa vitamin dự phòng được khuyến nghị khi sử dụng metformin. Chống chỉ định sử dụng metformin bao gồm suy thận, suy tim sung huyết hoặc suy phổi, bệnh gan cấp tính và sử dụng rượu đủ để gây độc tính gan cấp tính. Metformin cũng nên được giữ lại khi bệnh nhân nhập viện với bất kỳ tình trạng nào có thể gây giảm tưới máu toàn thân, hoặc khi dự kiến sử dụng các chất cản quang. Cần lưu ý rằng metformin được FDA chấp thuận để điều trị T2DM ở trẻ em nhưng không có khả năng được chấp thuận cho béo phì hoặc kháng insulin ở trẻ em.
Ức chế tăng tiết Insulin: Octreotide
Người ta đã biết rõ rằng các tổn thương điện giải hai bên hoặc sự mất kết nối của VMH ở chuột dẫn đến tăng cân khó chữa, ngay cả khi hạn chế thức ăn. Ở người, tổn thương vùng dưới đồi, do khối u TKTW, phẫu thuật, xạ trị hoặc chấn thương, có thể làm thay đổi cả con đường hướng tâm và ly tâm của cân bằng năng lượng, và dẫn đến tăng cân nghiêm trọng và khó chữa. Trong hội chứng “béo phì do vùng dưới đồi” này, tổn thương vùng dưới đồi gây ra một “kháng leptin hữu cơ” khi VMH cảm nhận được nạn đói; do đó lượng năng lượng nạp vào cao, và tiêu hao thấp. Trẻ em bị béo phì do vùng dưới đồi biểu hiện tăng cân, ngay cả khi đáp ứng với việc hạn chế calo bắt buộc, thứ phát sau (1) sự hoạt động quá mức của dây thần kinh phế vị, thúc đẩy tăng tiết insulin bắt buộc và dự trữ năng lượng và (2) sự kích hoạt khiếm khuyết của SNS, làm chậm quá trình ly giải mỡ và tiêu hao năng lượng. Tăng tiết insulin với độ nhạy insulin bình thường được ghi nhận trên xét nghiệm dung nạp glucose đường uống ở những trẻ này. Hiện tượng tăng tiết insulin tương tự này cũng đã được ghi nhận ở một phân nhóm người lớn bị béo phì mà không có tổn thương TKTW.
Kênh canxi cổng điện thế của tế bào β được kết hợp với một thụ thể somatostatin-5 (SSTR5). Octreotide gắn vào thụ thể này, hạn chế sự mở của kênh canxi này, giảm dòng canxi vào các tế bào β, và đến lượt nó làm giảm sự kích hoạt calmodulin và sự xuất bào của các túi, do đó làm giảm cấp tính mức độ đáp ứng insulin với glucose (xem Hình 24.3), dẫn đến giảm cân hoặc ổn định cân nặng. Hai TNNCT và một nghiên cứu dự đoán quan sát sử dụng octreotide cho bệnh béo phì đã được thực hiện. Một cuộc kiểm tra các phản ứng BMI đối với octreotide trong béo phì do vùng dưới đồi ở trẻ em trong một phân tích đa biến đã chứng minh rằng sự tăng tiết insulin với sự duy trì đồng thời độ nhạy insulin trước khi điều trị đã báo hiệu sự thành công. Một nghiên cứu quy mô lớn hơn về các bệnh nhân sau một tổn thương não cho thấy hiệu quả hạn chế, có lẽ do thời điểm can thiệp—phần lớn sự tăng cân trong các trường hợp béo phì do vùng dưới đồi xảy ra sớm sau tổn thương. Điều trị bằng thuốc bắt đầu quá muộn có thể không đảo ngược được tình trạng này trong khi việc bắt đầu điều trị sớm có thể ngăn ngừa tăng cân quá mức.
Octreotide thường được dung nạp tốt. Các tác dụng phụ phổ biến nhất bao gồm tiêu chảy, đau quặn bụng, buồn nôn và chướng bụng, tự giới hạn và thường hết sau 3 đến 4 tuần. Các biến cố bất lợi khác bao gồm sỏi mật (có thể phòng ngừa bằng cách dùng đồng thời ursodiol), phù, phát triển áp xe vô trùng tại các vị trí tiêm, thiếu hụt B12, ức chế bài tiết GH và TSH, và tăng đường huyết nhẹ, đặc biệt ở những người bị kháng insulin nặng.
Các liệu pháp nhắm mục tiêu khác
Leptin. Các đột biến của gen leptin ở người tái tạo lại kiểu hình của chuột ob/ob thiếu leptin. Khoảng 11 bệnh nhân như vậy đã được mô tả; họ biểu hiện chứng ăn nhiều từ khi sinh ra, với béo phì có thể ghi nhận được ngay từ 6 tháng tuổi. Thiếu hụt leptin gây ra phản ứng đói, với sự tăng lượng năng lượng nạp vào và giảm REE. Chẩn đoán được thực hiện bằng nồng độ leptin huyết thanh cực thấp hoặc không thể đo được. Ở trẻ em bị thiếu hụt leptin, liệu pháp leptin dẫn đến sự sụt giảm phi thường về trọng lượng và khối lượng mỡ, cùng với việc giảm chứng ăn nhiều, giải quyết béo phì, gây ra dậy thì và cải thiện khả năng miễn dịch. Mặc dù việc sử dụng leptin ở người lớn không chứng tỏ hiệu quả tự nó do kháng leptin, leptin có thể đóng vai trò như một chất bổ trợ kết hợp với các loại thuốc khác sau khi độ nhạy leptin được cải thiện thông qua việc giảm cân.
Các chất tương tự GLP-1. Một số chất tương tự GLP-1 tác dụng kéo dài gần đây đã được giới thiệu, một số có chỉ định chống béo phì. GLP-1 là một incretin có nguồn gốc từ ruột cũng đóng vai trò là một tín hiệu no như đã mô tả trước đó. Vì thời gian bán hủy của nó trong tuần hoàn ngắn (~ 2 phút) do sự phân hủy bởi enzyme dipeptidyl peptidase-4 (DPP-4), các chất tương tự ổn định đã được thiết kế, ban đầu nhằm vào việc tăng cường tế bào β nhưng cũng cho thấy một số tác dụng giảm cân đáng kể. Tính an toàn của các chất tương tự GLP-1 đã được chứng minh ở trẻ béo phì, các thử nghiệm hiệu quả hiện đang được thực hiện và sẽ sớm được công bố.
Tương lai của Dược lý điều trị béo phì ở trẻ em
Để đối phó với sự thiếu hiệu quả tương đối của các can thiệp lối sống, kiến thức ngày càng mở rộng về sinh lý học cân bằng năng lượng, và đặc biệt là một quyết định kinh doanh về phần thưởng tài chính tiềm năng, nhiều công ty dược phẩm đã khởi động các chương trình nghiên cứu về béo phì. Các tác nhân sau đây hiện đang được nghiên cứu trên người; tuy nhiên, việc sử dụng bất kỳ tác nhân mới nào trong số này ở trẻ em sẽ phụ thuộc vào bằng chứng về tính an toàn và hiệu quả dựa trên kinh nghiệm ở người lớn. Topiramate là một thuốc chống co giật ngăn chặn các kênh natri phụ thuộc điện thế, tăng cường hoạt động của thụ thể GABAA, và đối kháng một thụ thể glutamate khác với thụ thể N-methyl-D-aspartate (NMDA). Topiramate thúc đẩy giảm cân theo kiểu phụ thuộc vào liều. Một TNNCT ở người lớn đã chứng minh giảm 9,1% cân nặng ở các đối tượng dùng topiramate 192 mg/ngày cùng với những cải thiện đáng kể về huyết áp, chu vi vòng eo, và glucose và insulin lúc đói. Tuy nhiên, gần 33% đối tượng đã bỏ cuộc do các biến cố bất lợi, bao gồm dị cảm, buồn ngủ, chán ăn, mệt mỏi, căng thẳng, giảm tập trung, khó ghi nhớ và hung hăng. Hiện chưa có nghiên cứu nào về topiramate trong bệnh béo phì ở trẻ em. Oxyntomodulin là một chất tương tự của PYY(3-36), đã được chứng minh trong một TNNCT kéo dài 4 tuần là làm giảm lượng năng lượng nạp vào và trọng lượng ở người lớn. Chưa có nghiên cứu nào ở trẻ em. Các công thức giảm cân hiện tại đã được chấp thuận ở người lớn bao gồm kết hợp phentermine-topiramate (QSymia®); kết hợp naltrexone-bupropion (Contrave®); lorcaserin, một chất chủ vận serotonin 2c (Belviq®); và liraglutide, một chất chủ vận GLP-1 (Saxenda®). Chưa có thử nghiệm hiệu quả lâm sàng nào được công bố ở trẻ em cho đến nay.
Phẫu thuật giảm cân
So với người lớn, các tiêu chí nghiêm ngặt và bảo thủ hơn phải được áp dụng cho thanh thiếu niên vì chỉ có 85% thanh thiếu niên béo phì sẽ trở thành người lớn béo phì bao gồm cả tỷ lệ hiệu quả của lối sống và liệu pháp dược lý được cải thiện một chút so với người lớn; một khoảng thời gian dài hơn trước khi các bệnh đi kèm trở nên đe dọa tính mạng; và họ không có khả năng đưa ra sự đồng ý hợp pháp. Vì tất cả những lý do này, một hội đồng chuyên gia với đại diện từ Hiệp hội Phẫu thuật Nhi khoa Hoa Kỳ và Viện Hàn lâm Nhi khoa Hoa Kỳ đã đề nghị rằng phẫu thuật giảm cân cho thanh thiếu niên chỉ nên được thực hiện tại các cơ sở cam kết quản lý lâu dài cho những bệnh nhân này và được biện minh trong các tình huống khi các tình trạng bệnh đi kèm liên quan đến béo phì (như OSA) đe dọa sức khỏe của trẻ. Họ đã đưa ra các khuyến nghị nghiêm ngặt rằng phẫu thuật giảm cân nên được giới hạn ở những thanh thiếu niên có chỉ số BMI trên 35 hoặc 120% của bách phân vị thứ 95 với sự hiện diện của bệnh đi kèm nghiêm trọng (OSA nặng, u giả não, đái tháo đường tuýp 2 hoặc viêm gan nhiễm mỡ) hoặc chỉ số BMI trên 40 hoặc 140% của bách phân vị thứ 95 với một bệnh đi kèm ít nghiêm trọng hơn. Dữ liệu được công bố ủng hộ quan điểm rằng phẫu thuật giảm cân có hiệu quả ở thanh thiếu niên béo phì về mặt giảm cân và cải thiện các bệnh đi kèm tương tự như người lớn. Phần lớn cũng cải thiện các thông số tâm lý xã hội, tuy nhiên, điều quan trọng là một số có thể phát triển trầm cảm, ý định tự tử và sức khỏe tâm lý xã hội tổng thể thấp hơn. Vẫn chưa rõ quy trình phẫu thuật giảm cân được lựa chọn nên là gì trong quần thể này và cách thức chuẩn bị và theo dõi nào sẽ cho phép tối ưu hóa kết quả lâu dài.
Nối tắt dạ dày đã được thực hiện rộng rãi hơn ở trẻ em và thanh thiếu niên từ những năm 1990, trong khi cắt dạ dày hình ống đã được áp dụng từ thực hành người lớn từ năm 2010 và hiện được thực hiện thường xuyên gấp đôi so với nối tắt dạ dày. Các quy trình này dường như an toàn và hiệu quả khi các ứng cử viên được lựa chọn cẩn thận, và bác sĩ phẫu thuật giảm cân có các kỹ năng nội soi tiên tiến. Cho đến nay, chúng ta có rất ít dữ liệu về các biến chứng lâu dài, bao gồm cả những biến chứng có thể liên quan đến mang thai trong tương lai, sức khỏe của xương và kết quả của các quy trình phẫu thuật lặp lại trong trường hợp tăng cân trở lại. Tuy nhiên, dữ liệu cho đến nay cho thấy lợi ích tiềm năng đối với các bệnh đi kèm do béo phì và cải thiện chất lượng cuộc sống ở thanh thiếu niên bị béo phì nặng và các biến chứng liên quan đến béo phì. Tuy nhiên, phẫu thuật giảm cân nên được sử dụng rộng rãi như một giải pháp như thế nào vẫn cần được xác định cho đến khi chúng ta có thêm dữ liệu kết quả lâu dài hơn.
Chỉ định Phẫu thuật Giảm cân
Một hội đồng chuyên gia với đại diện từ Hiệp hội Phẫu thuật Nhi khoa Hoa Kỳ và Viện Hàn lâm Nhi khoa Hoa Kỳ đã đề nghị rằng phẫu thuật giảm cân ở tuổi vị thành niên có thể được biện minh trong các tình huống khi các tình trạng bệnh đi kèm liên quan đến béo phì đe dọa sức khỏe của trẻ. Họ đã đưa ra các khuyến nghị nghiêm ngặt rằng phẫu thuật giảm cân nên được giới hạn ở những thanh thiếu niên có chỉ số BMI trên 35 với sự hiện diện của bệnh đi kèm nghiêm trọng, hoặc chỉ số BMI trên 40 với một bệnh đi kèm ít nghiêm trọng hơn. Các quy trình như vậy chỉ nên được thực hiện tại các trung tâm xuất sắc bao gồm một đội ngũ đa ngành có thể cung cấp sự hỗ trợ nội tiết, tâm thần, ăn kiêng và tâm lý cho bệnh nhân và gia đình trước và sau quy trình phẫu thuật giảm cân.
Cần đặc biệt cân nhắc để tránh phẫu thuật giảm cân ở các giai đoạn béo phì rất muộn, khi sự hiện diện của các bệnh đi kèm liên quan đến béo phì, và sự không thể tiếp cận của hình ảnh (hầu hết các máy quét MRI có giới hạn trọng lượng là 450 lbs hoặc 200 kg) có thể ảnh hưởng đến kết quả phẫu thuật. Thật vậy, một tổng quan của tám nghiên cứu hồi cứu ở thanh thiếu niên đã phát hiện ra rằng phẫu thuật giảm cân ở thanh thiếu niên có thể thúc đẩy giảm cân bền vững ở hầu hết bệnh nhân, nhưng dường như có một tỷ lệ biến chứng và tử vong đáng kể. Do đó, cần có hướng dẫn để xác định các trường hợp lý tưởng mà sự cân bằng giữa rủi ro và lợi ích ủng hộ việc bảo tồn sức khỏe và đảo ngược các biến chứng với nguy cơ bệnh tật và tử vong thấp nhất từ quy trình.
Các quy trình phẫu thuật giảm cân có thể được chia thành các quy trình hoàn toàn hạn chế, những quy trình bỏ qua một đoạn của ruột trước, và các quy trình kết hợp. Các quy trình hoàn toàn kém hấp thu nhằm mục đích giảm chiều dài chức năng hoặc hiệu quả của niêm mạc ruột thông qua sự sắp xếp lại giải phẫu của ruột. Các quy trình này bao gồm nối tắt hỗng-hồi tràng, và chuyển lưu mật-tụy với chuyển vị tá tràng. Do tỷ lệ bệnh tật và tử vong cao của các quy trình này, chúng không thể được khuyến nghị ở trẻ em, và sẽ không được thảo luận thêm. Các quy trình hạn chế làm giảm thể tích dạ dày để giảm thể tích thức ăn ăn vào. Chúng bao gồm bóng dạ dày nội soi (không có dữ liệu ở trẻ em) và thắt đai dạ dày có thể điều chỉnh qua nội soi (LAGB). Nối tắt dạ dày Roux-en-Y (RYGB) là một quy trình kết hợp. Cắt dạ dày hình ống dường như là một quy trình hạn chế nhưng nó có lẽ chia sẻ một số tác động nội tiết tố của RYGB và đang ngày càng phổ biến.
Hạn chế: Thắt đai dạ dày có thể điều chỉnh qua nội soi
LAGB sử dụng một đai nhân tạo để bao quanh và phân chia phần gần của dạ dày thành một túi nhỏ và một phần còn lại lớn. Lợi thế lý thuyết của kỹ thuật này là giảm nguy cơ bung đường khâu. Việc giới thiệu gần đây hơn của một phương pháp nội soi mới và việc sử dụng một đai có thể điều chỉnh (cho phép kích thước dạ dày thay đổi) làm cho quy trình này hấp dẫn hơn. Cuối cùng, quy trình này có thể đảo ngược (ít nhất là về mặt lý thuyết; có một số bác sĩ phẫu thuật chế giễu quan niệm này), hoặc nó có thể được sửa đổi thành RYGB vào một ngày sau đó. Kết quả thay đổi rất nhiều ở người lớn. Một TNNCT duy nhất so sánh LAGB với quản lý béo phì bảo tồn ở trẻ em béo phì đã cho thấy sự giảm cân vượt trội được duy trì trong 2 năm. Trong số những người tham gia là thanh thiếu niên béo phì, việc sử dụng thắt đai dạ dày so với can thiệp lối sống đã dẫn đến một tỷ lệ lớn hơn đạt được mức giảm 50% trọng lượng thừa, được điều chỉnh theo tuổi. Có những lợi ích liên quan đến sức khỏe và chất lượng cuộc sống. Các tác dụng phụ đáng kể và việc duy trì cân nặng lâu dài hạn chế sau LAGB đã làm giảm sự phổ biến của nó trong những năm gần đây.
Kết hợp: Nối tắt dạ dày Roux-en-Y
RYGB bao gồm việc chia dạ dày để tạo ra một túi dạ dày nhỏ (15–30 mL) vào đó một đoạn hỗng tràng khoảng 15 đến 60 cm dưới dây chằng Treitz được đưa vào, trong khi phần gần của hỗng tràng dẫn lưu phần dạ dày và tá tràng dưới bị bỏ qua được nối lại 75 đến 150 cm dưới chỗ nối dạ dày-hỗng tràng. Quy trình này kết hợp bản chất hạn chế của phẫu thuật cắt dạ dày với hậu quả của sinh lý học dumping như một phản ứng điều kiện tiêu cực khi các bữa ăn lỏng có hàm lượng calo cao được ăn vào. Ngoài ra, RYGB có liên quan đến sự suy giảm nồng độ ghrelin trong tuần hoàn, có thể là một phần nguyên nhân gây ra sự giảm cảm giác đói liên quan đến quy trình này. Quy trình không chỉ có thể dẫn đến giảm cân phi thường, mà còn có thể đảo ngược bệnh đái tháo đường tuýp 2.
RYGB dường như dẫn đến sự giảm cân sớm đáng kể ở người lớn; tuy nhiên, các nghiên cứu dài hạn cho thấy sự tăng cân trở lại ở nhiều bệnh nhân. RYGB dường như cung cấp sự thuyên giảm hoàn toàn T2DM ở thanh thiếu niên béo phì. Quan trọng là, khi được thực hiện ở thanh thiếu niên bị béo phì nặng, chỉ số BMI thấp nhất đạt được thường là khoảng 37% so với ban đầu. Nếu người ta thực hiện quy trình trên một bệnh nhân có chỉ số BMI ở mức thấp 40—kết quả có thể là một chỉ số BMI trong phạm vi lành mạnh chấp nhận được. Ngược lại, việc thực hiện quy trình ở những bệnh nhân có chỉ số BMI trên 50 khiến họ, ngay cả sau một kết quả tối ưu, vẫn ở trong phạm vi BMI béo phì mà không có sự giảm thêm.
Các biến chứng được báo cáo phổ biến nhất của RYGB bao gồm thiếu máu do thiếu sắt (50%), thiếu folate thoáng qua (30%), và các biến cố cần can thiệp phẫu thuật (40%: cắt túi mật ở 20%, tắc ruột non ở 10%, và thoát vị vết mổ ở 10%). Vì hầu hết dạ dày và tá tràng bị bỏ qua trong quy trình này, có nguy cơ gia tăng thiếu hụt vitamin B12, sắt, canxi và thiamine. Mặc dù bệnh beriberi đã được báo cáo ở thanh thiếu niên sau RYGB, việc tuân thủ bổ sung hàng ngày suốt đời và theo dõi thường xuyên bệnh nhân có thể ngăn ngừa các thiếu hụt dinh dưỡng như vậy.
Cắt dạ dày hình ống qua nội soi
Quy trình cắt dạ dày hình ống qua nội soi (LSG) đang được sử dụng rộng rãi hơn. Nó làm giảm dung tích dạ dày, bằng cách cắt bỏ bờ cong lớn của dạ dày, nói cách khác, biến dạ dày thành một cái ống. Cắt dạ dày hình ống đang ngày càng phổ biến ở trẻ em cũng như người lớn béo phì vì nó dường như là một quy trình đơn giản hơn về mặt kỹ thuật với kết quả lâm sàng tương đương với kết quả của RYGB. Dữ liệu hạn chế ở thanh thiếu niên thực sự cho thấy sự giảm cân tương tự so với RYGB. Tỷ lệ thất bại 5 năm về việc tăng cân trở lại và sự trở lại của các bệnh đi kèm của quy trình này ở người lớn béo phì là khoảng 50%, nhưng dữ liệu như vậy ở trẻ em vẫn chưa có.
Ai nên thực hiện Phẫu thuật Giảm cân ở Trẻ em?
Kết quả phẫu thuật ở người lớn thay đổi rất nhiều giữa các bác sĩ phẫu thuật và các cơ sở. Hơn nữa, có một đường cong học tập rất rõ ràng vì tỷ lệ bệnh tật của phẫu thuật giảm cân thay đổi nghịch với số lượng các quy trình được thực hiện. Nguy cơ tái nhập viện gia tăng sau phẫu thuật giảm cân ở người lớn cho thấy sự cần thiết của việc theo dõi và giám sát chặt chẽ và cẩn thận ở thanh thiếu niên. Do đó, điều cần thiết là phẫu thuật giảm cân ở thanh thiếu niên phải được thực hiện tại các trung tâm học thuật nhi khoa khu vực với các chương trình được trang bị để xử lý việc thu thập dữ liệu, theo dõi lâu dài, bản chất đa ngành của những bệnh nhân khó khăn này.
Một đội ngũ đa ngành với chuyên môn y tế, phẫu thuật, dinh dưỡng và tâm lý nên lựa chọn cẩn thận những thanh thiếu niên được thông báo đầy đủ và có động lực để trở thành ứng cử viên tiềm năng cho phẫu thuật giảm cân. Chú ý đến các nguyên tắc tăng trưởng, phát triển và tuân thủ là cần thiết để tránh các kết quả bất lợi về thể chất, nhận thức và tâm lý xã hội sau phẫu thuật giảm cân. Phải rõ ràng với đối tượng và phụ huynh rằng phẫu thuật giảm cân thực chất là một biện pháp bổ trợ cho một cam kết chân thành với lối sống, chứ không phải là một “viên đạn thần kỳ”. Thật vậy, bằng chứng về sự tái phát ở người lớn sau RYGB hiện nay là phổ biến.
Các đối tượng và gia đình phải được thông báo đầy đủ về các rủi ro và biến chứng của phẫu thuật như vậy. Đội ngũ y tế sẽ cần sự hỗ trợ của các chuyên khoa nội tiết, tiêu hóa, tim mạch, phổi và tai mũi họng. Mở khí quản dự phòng hiếm khi được yêu cầu để duy trì sự thông thoáng của đường thở và cho phép giải quyết tình trạng tăng CO2 máu trước phẫu thuật. Thanh thiếu niên trải qua phẫu thuật giảm cân cần được giám sát y tế và dinh dưỡng suốt đời sau phẫu thuật. Cần có sự tư vấn, giáo dục và hỗ trợ sâu rộng cả trước và sau phẫu thuật giảm cân; những bệnh nhân tự lo liệu có xu hướng tăng cân trở lại theo thời gian. Thật vậy, các nghiên cứu ở người lớn ghi nhận nguy cơ nhập viện gia tăng sau RYGB, do những khó khăn từ quy trình. Việc theo dõi duy trì cân nặng lâu dài, những cải thiện về bệnh tật tim mạch và tuổi thọ đều cần thiết để xác định hiệu quả chi phí của phẫu thuật giảm cân trong quần thể nhi khoa.
Thiếu hụt năng lượng
Đói so với Suy mòn
Mặc dù cả hai đều là hội chứng sụt cân, việc hiểu rõ các cơ chế nội tiết thần kinh phân biệt giữa đói và suy mòn là rất cần thiết để hiểu và điều trị đúng cách các rối loạn này. Trong tình trạng đói, con đường cân bằng năng lượng phản hồi âm vẫn còn nguyên vẹn. Tín hiệu về sự thiếu hụt leptin do sụt cân được tế bào thần kinh VMH chuyển thành hoạt động giao cảm giảm (để bảo tồn năng lượng) và hoạt động phế vị tăng (để dự trữ năng lượng). Tuy nhiên, trong tình trạng suy mòn, con đường này bị đoản mạch bởi tác động của cytokine lên vùng dưới đồi. Tế bào thần kinh POMC của VMH biểu hiện các thụ thể cho các cytokine khác nhau, bao gồm IL-1 và TNF-α. Để đáp ứng với việc tiếp xúc với cytokine, các tế bào thần kinh POMC được kích hoạt, dẫn đến chán ăn, tăng hoạt động giao cảm, giảm hoạt động phế vị và lãng phí năng lượng. Các cytokine tiền viêm làm tăng epinephrine, GH và cortisol, và làm giảm insulin. Những thay đổi nội tiết tố dài hạn này làm tăng tốc độ phân giải protein cơ (cortisol), tăng tiêu hao năng lượng khi nghỉ (SNS), góp phần vào tình trạng kháng insulin (cả epinephrine và cortisol), tăng dị hóa (cortisol), và ức chế cảm giác thèm ăn và sự vận chuyển của ruột (phế vị). Điều này rõ ràng là thích nghi trong ngắn hạn trong thời gian nhiễm trùng (để tạo ra nhiệt độ cơ thể để tiêu diệt sinh vật), nhưng lại không thích nghi trong dài hạn, khi tín hiệu cytokine mãn tính có thể dẫn đến suy mòn. Do đó, ngay cả trong các tình huống suy giảm hoặc thiếu hụt leptin, sự kích hoạt của cytokine đối với các tế bào thần kinh POMC sẽ thúc đẩy tình trạng suy mòn và sụt cân liên tục thông qua sự kích hoạt SNS dai dẳng.
Chậm phát triển
Chậm phát triển (FTT) không phải là một bệnh tự thân mà là một dấu hiệu của nhiều tình trạng hữu cơ và phi hữu cơ và sự tương tác giữa chúng dẫn đến sự tăng trưởng bị tổn hại ở độ tuổi trẻ. FTT vẫn là một vấn-đề y tế nhi khoa phổ biến chủ yếu được quản lý trong môi trường ngoại trú. Mặc dù FTT hiếm khi có thể là một biểu hiện của bệnh hiểm nghèo, phần lớn các trường hợp là kết quả của tình trạng suy dinh dưỡng do sự kết hợp của các yếu tố sinh học, môi trường và tâm lý. Chẩn đoán FTT đòi hỏi người chăm sóc phải khai thác bệnh sử kỹ lưỡng, thận trọng và có định hướng và không phải lúc nào cũng đơn giản để thiết lập. Hơn nữa, chẩn đoán này có thể mang một số ý nghĩa pháp lý không nằm trong phạm vi của văn bản này.
Định nghĩa
Không có sự đồng thuận về một định nghĩa duy nhất cho FTT. Tình trạng này phản ánh sự tăng trưởng thể chất không đầy đủ được ghi nhận theo thời gian bằng cách sử dụng biểu đồ tăng trưởng tiêu chuẩn. Các định nghĩa thường được sử dụng trong thực hành lâm sàng bao gồm chiều dài và/hoặc cân nặng dưới bách phân vị thứ 5 theo tuổi và giới tính, sự đi xuống cắt qua hai đường bách phân vị chính của biểu đồ tăng trưởng theo thời gian, hoặc cân nặng theo chiều cao dưới bách phân vị thứ 10 so với dự kiến. Tính thực tiễn của việc thường sử dụng đường cong cân nặng chứ không phải chiều dài bắt nguồn từ thực tế là suy dinh dưỡng và bệnh mãn tính có xu hướng chủ yếu ảnh hưởng đến việc tăng cân trong khi vẫn bảo tồn sự tăng trưởng theo chiều dài. Cuối cùng, sự tăng trưởng theo chiều dài cũng bị ảnh hưởng nếu những tình trạng này kéo dài. Điều quan trọng cần nhấn mạnh là các phép đo đơn lẻ mà không có các điểm tăng trưởng theo dõi theo chiều dọc là không đủ để chẩn đoán FTT, và một chẩn đoán sai lầm có thể được thiết lập ở những trẻ sinh ra nhỏ so với tuổi thai hoặc sinh non, và ở một số trẻ sơ sinh khỏe mạnh đang phát triển dọc theo các bách phân vị thấp hơn.
Phân loại và Nguyên nhân
Phân loại truyền thống của FTT là phân biệt giữa các nguyên nhân hữu cơ và phi hữu cơ. Các nguyên nhân phi hữu cơ đề cập đến các yếu tố môi trường và tâm lý, chẳng hạn như thiếu hụt cảm giác, thiếu thốn tình cảm và cha mẹ, và những khó khăn trong việc cho ăn không có nguồn gốc hữu cơ xảy ra ở trẻ sơ sinh. Phân loại truyền thống này dường như thiếu cái nhìn sâu sắc rằng phần lớn các trường hợp mắc phải sự kết hợp của cả hai, phản ánh một nguyên nhân hỗn hợp. Một cách tiếp cận khác là phân loại rối loạn dựa trên sinh lý bệnh của rối loạn, tức là, lượng calo nạp vào không đủ, hấp thu không đủ, yêu cầu chuyển hóa dư thừa, sử dụng lượng nạp vào bị lỗi, và tiềm năng tăng trưởng giảm. Các nguyên nhân phổ biến của FTT dựa trên sơ đồ phân loại này được trình bày trong Hộp 24.2.
Hộp 24.2 TKTW, Hệ thần kinh trung ương; ALNS, Áp lực nội sọ. Chẩn đoán Phân biệt Chậm phát triển
Lượng calo nạp vào không đủ • Nghèo đói và nguồn thực phẩm thấp • Khó khăn trong việc cho ăn cơ học (khả năng nuốt bị thay đổi, dị tật bẩm sinh, tổn thương hệ thần kinh trung ương, trào ngược dạ dày thực quản nặng) • Pha sữa công thức sai cách (quá loãng, quá đặc) • Thói quen cho ăn không phù hợp của cha mẹ • Các vấn đề hành vi ảnh hưởng đến việc ăn uống • Bỏ mặc trẻ em • Tương tác cha mẹ-con cái kém 2. Hấp thu không đủ lượng calo nạp vào • Diện tích bề mặt hấp thu giảm (hội chứng ruột ngắn, sau viêm ruột hoại tử) • Bệnh gan mãn tính, teo đường mật • Bệnh celiac • Xơ nang • Dị ứng sữa bò • Tiêu chảy mãn tính • Thiếu hụt vitamin hoặc khoáng chất (viêm da đầu chi do ruột) • Nôn do các bất thường của TKTW (khối u, tăng ALNS) 3. Tăng chuyển hóa • Cường giáp • Nhiễm trùng mãn tính (do suy giảm miễn dịch) • Ung thư tiềm ẩn • Dị tật tim bẩm sinh hoặc bệnh tim mắc phải (chủ yếu là shunt phải-trái và suy tim) • Bệnh phổi mãn tính với tình trạng thiếu oxy máu (loạn sản phế quản-phổi) • Bỏng 4. Sử dụng calo bị lỗi • Suy thận, nhiễm toan ống thận • Các lỗi chuyển hóa bẩm sinh (bệnh dự trữ, rối loạn axit amin) 5. Tiềm năng tăng trưởng giảm • Các rối loạn di truyền (trisomy, loạn sản xương, hội chứng Russell-Silver) • Các hội chứng di truyền đặc hiệu • Còi cọc nguyên thủy
Đáng chú ý, sự biến đổi tăng trưởng bình thường có thể làm nhiễu chẩn đoán FTT vì một số trẻ sơ sinh có thể sinh ra lớn so với tuổi thai do các nguyên nhân trong tử cung (chẳng hạn như đái tháo đường thai kỳ), và sau đó trải qua một mô hình tăng trưởng “bắt kịp xuống” trong giai đoạn sơ sinh hướng tới các đường cong tiềm năng tăng trưởng thực tế. Một nguyên nhân khác của việc cắt ngang các bách phân vị tiêu cực như vậy có thể là chậm tăng trưởng thể chất. Người ta ước tính rằng có tới 25% trẻ em có thể cắt ngang các đường cong hơn 25 vạch bách phân vị (đại diện cho một sự cắt ngang của hai đường bách phân vị tăng trưởng chính) do các lý do đã đề cập ở trên. Những trẻ sơ sinh này đạt đến một điểm mới từ đó chúng thể hiện một tốc độ tăng trưởng và mô hình tăng cân bình thường, nhưng chúng không bị FTT.
Các nguyên nhân nội tiết của FTT không phổ biến, vì các thiếu hụt nội tiết tố điển hình, chẳng hạn như thiếu hụt GH hoặc suy giáp, biểu hiện là chậm tăng trưởng nhưng với việc tăng cân được bảo tồn hoặc tăng lên. Cường giáp (đại diện cho một trạng thái tăng nhu cầu chuyển hóa và đặc trưng bởi sự tăng trưởng theo chiều dài tăng lên) và các rối loạn chuyển hóa muối, chẳng hạn như giảm aldosteron và giả-giảm aldosteron, có thể có FTT như một phần của các biểu hiện lâm sàng của chúng. Còi xương do giảm phốt phát máu cũng có thể biểu hiện là FTT.
Chẩn đoán và Đánh giá
Chìa khóa để chẩn đoán FTT là vẽ biểu đồ dữ liệu nhân trắc học (cân nặng và chiều dài) trong một khoảng thời gian theo dõi hợp lý. Mặc dù một bệnh sử và khám thực thể thận trọng và tập trung là chìa khóa để chẩn đoán, nhưng thường thì chẩn đoán đúng được thực hiện sau khi nhìn lại. Việc thiếu một nguyên nhân hữu cơ để giải thích các phát hiện không đủ để thiết lập chẩn đoán FTT phi hữu cơ. Một phản ứng với một can thiệp tích cực, được biểu hiện ít nhất là một giai đoạn tăng trưởng đầy đủ hạn chế trong khi thay đổi một yếu tố hành vi của người chăm sóc hoặc trẻ em, có thể giúp thiết lập chẩn đoán FTT phi hữu cơ.
Bệnh sử nên tập trung vào tiền sử ăn uống và cho ăn, tiền sử y tế quá khứ và hiện tại, môi trường xã hội và tiền sử gia đình. Tiền sử ăn uống nhằm mục đích đánh giá càng chính xác càng tốt lượng calo thực tế mà bệnh nhân nạp vào. Một công cụ quan trọng cho việc đánh giá này có thể là việc sử dụng nhật ký thực phẩm trong vài ngày. Các chi tiết quan trọng liên quan đến lượng thực phẩm thực tế, cách chế biến thực phẩm (đặc biệt liên quan đến kỹ thuật pha loãng sữa công thức và ngũ cốc thêm vào sữa công thức), và đồ uống được tiêu thụ với sự nhấn mạnh cụ thể vào nước trái cây có đường và sữa công thức. Những chi tiết này sẽ cho phép người hành nghề ước tính lượng calo nạp vào.
Các chi tiết quan trọng liên quan đến việc cho ăn bắt đầu bằng vị trí của các bữa ăn và thời gian của chúng trong ngày. Ai cho bệnh nhân ăn hoặc giám sát quá trình cho ăn là rất quan trọng. Kỹ thuật cho ăn phải phù hợp với giai đoạn phát triển của trẻ. Thời gian có liên quan đến việc ăn vặt thường xuyên giữa các bữa ăn có thể gây no sớm trong giờ ăn.
Một bệnh sử nhi khoa tiêu chuẩn nên được lấy từ tất cả các bệnh nhân, nhưng nó nên được tập trung vào các chi tiết có thể liên quan đến chẩn đoán FTT. Tiền sử mang thai và sinh nở rất quan trọng để phân biệt trẻ sơ sinh sinh ra nhỏ so với tuổi thai so với những trẻ bị FTT. Thời điểm bắt đầu tăng cân kém, đặc biệt là liên quan đến những thay đổi trong việc cho ăn, là cực kỳ quan trọng cho chẩn đoán. Các tình trạng y tế mãn tính, chẳng hạn như bệnh tim bẩm sinh, hen suyễn, nhiễm trùng tái phát nhiều lần và thiếu máu đều có thể là nguyên nhân của FTT hữu cơ. Nhiều lần nhập viện và tiền sử chấn thương có thể làm dấy lên nghi ngờ về sự bỏ mặc của cha mẹ. Các biểu hiện tiêu hóa của các tình trạng y tế liên quan, chẳng hạn như nôn thường xuyên (trong các trường hợp dị ứng sữa hoặc trào ngược dạ dày thực quản) và tần suất và tính chất của phân (để loại trừ kém hấp thu, bệnh celiac, bệnh viêm ruột hoặc xơ nang), nên được khai thác chi tiết.
Tiền sử xã hội nên tập trung vào việc xác định những người chăm sóc thực tế của bệnh nhân trong phần lớn thời gian và liệu có các vấn đề kinh tế có thể ảnh hưởng đến khả năng nuôi dưỡng bệnh nhân một cách đầy đủ hay không. Các yếu tố gây căng thẳng bên ngoài và trong gia đình tiềm ẩn có thể ảnh hưởng đến việc cung cấp thực phẩm cho trẻ nên được tìm kiếm (bất kỳ yếu tố gây căng thẳng hoặc sự kiện cuộc sống nào có thể ảnh hưởng đến chức năng của người chăm sóc theo cách có thể làm tổn hại đến sức khỏe của trẻ). Tiền sử gia đình nên tập trung vào thể trạng của cha mẹ và anh chị em để có được manh mối về tiềm năng di truyền về chiều cao và cân nặng. Các tình trạng y tế ở anh chị em và người thân có thể gợi ý một khuynh hướng mắc các rối loạn di truyền. Người chăm sóc nên được hỏi về các bệnh tâm thần, chẳng hạn như trầm cảm, có thể cản trở khả năng cung cấp sự chăm sóc đầy đủ cho trẻ. Tiền sử gia đình về những đứa trẻ trước đây bị FTT cũng nên được điều tra. Một cuộc gọi đến Sở Dịch vụ Gia đình địa phương có thể được bảo đảm.
Việc khám thực thể bắt đầu bằng việc vẽ biểu đồ chiều dài, cân nặng và chu vi vòng đầu của trẻ trên các biểu đồ tăng trưởng tiêu chuẩn, cùng với các phép đo trước đó (nếu có). Mức độ nghiêm trọng của FTT có thể được ước tính bằng cách đánh giá cân nặng hiện tại so với cân nặng dự kiến theo tuổi. Nếu cân nặng nhỏ hơn 60% so với dự kiến theo bách phân vị thứ 50 theo tuổi và chiều dài, tình trạng là nghiêm trọng, trong khi cân nặng từ 60 đến 75 bách phân vị so với dự kiến được coi là FTT trung bình. Đầu nhỏ kèm theo các dấu hiệu thần kinh có thể gợi ý một tổn thương TKTW. Cần nhớ rằng chu vi vòng đầu là thông số cuối cùng thay đổi trong FTT, và chỉ trong những trường hợp nặng nhất. Việc phát hiện các dị dạng có thể gợi ý một nguyên nhân di truyền cho sự tăng trưởng và phát triển bị suy giảm. Các biện pháp về tình trạng dinh dưỡng (chẳng hạn như độ dày của nếp gấp da và sự phân bố mỡ cơ thể) có thể được kiểm tra. Điều quan trọng là phải quan sát cẩn thận sự tương tác của người chăm sóc và đứa trẻ trong khi cho ăn. Sự tương tác cha mẹ-con cái bị suy giảm có thể có tác động lớn đến thói quen ăn uống, và việc xác định chúng là rất quan trọng để thiết kế các can thiệp hành vi hiệu quả phù hợp với bệnh nhân và gia đình.
Phần lớn trẻ em bị FTT không có bất thường về xét nghiệm, và không có thay đổi nội tiết tố. Có rất ít tài liệu về các xét nghiệm toàn diện ở trẻ em bị FTT, mặc dù một bản thảo kinh điển về việc xét nghiệm hơn 180 trẻ sơ sinh trong một môi trường nội trú đã phát hiện ra các bất thường trong phòng thí nghiệm ở ít hơn 1,4% các xét nghiệm được thực hiện. Việc lựa chọn các xét nghiệm có thể có lợi nên dựa trên bệnh sử và khám thực thể và thường tập trung vào việc đánh giá suy dinh dưỡng trong các trường hợp nặng. Một xét nghiệm tối thiểu, mặc dù không hiệu quả về chi phí, có thể bao gồm công thức máu, bảng hóa học (bao gồm các xét nghiệm chức năng gan và thận, điện giải, nồng độ protein và albumin huyết thanh, cũng như tình trạng axit-bazơ trong máu) và xét nghiệm nước tiểu với pH. Các xét nghiệm bổ sung nên được định hướng vào các phát hiện cụ thể từ bệnh sử và khám thực thể. Không có xét nghiệm nội tiết tố nào được bảo đảm ban đầu trừ khi có nghi ngờ lâm sàng về một rối loạn cụ thể. Ở trẻ em trên 6 tháng tuổi, việc sàng lọc thiếu sắt và ngộ độc chì là cần thiết. Việc nhập viện và xét nghiệm nội trú không mang lại thêm lợi ích nào cho việc xét nghiệm, trừ khi mức độ FTT nghiêm trọng, hoặc nếu có lo ngại về sự an toàn và bỏ mặc của trẻ.
Quản lý
Việc quản lý FTT dựa trên việc xác định nguyên nhân cơ bản và khắc phục nó. Phần lớn các trường hợp được xử lý bằng sự kết hợp của can thiệp dinh dưỡng và hành vi. Quan trọng là, can thiệp nên bắt đầu trước khi quá trình xét nghiệm hoàn tất, tức là, từ lần đánh giá đầu tiên. Tất cả các vấn đề y tế được điều trị độc lập với các can thiệp dinh dưỡng và hành vi và không nên trì hoãn hoặc cản trở chúng. Nền tảng của việc điều trị tất cả trẻ sơ sinh bị FTT là một chế độ ăn giàu calo kèm theo việc theo dõi chặt chẽ và thường xuyên đáp ứng cân nặng. Một can thiệp hiệu quả sẽ ghi nhận được sự tăng cân và chiều cao bắt kịp được duy trì theo thời gian.
Các hành vi cho ăn và ăn uống nên được giải quyết bằng cách đi trên ranh giới mong manh giữa sự khuyến khích và áp lực để thúc đẩy việc ăn uống. Việc định giờ các bữa ăn và đồ ăn nhẹ và ăn uống như một gia đình trong một môi trường dễ chịu, ít căng thẳng có thể quan trọng để có được các thói quen ăn uống và cho ăn được cải thiện. Can thiệp cho ăn phụ thuộc vào độ tuổi của trẻ sơ sinh khi đến khám. Đối với trẻ sơ sinh bú mẹ, việc cố gắng tăng nguồn cung cấp sữa mẹ bằng cách vắt sữa, điều trị bằng metoclopramide để gây tiết oxytocin, cải thiện dinh dưỡng và lượng chất lỏng của mẹ, và thực hiện các điều chỉnh tại nhà và nơi làm việc có thể thúc đẩy và đơn giản hóa quá trình cho con bú là có lợi. Các vấn đề về bú ở trẻ sơ sinh bị suy giảm thần kinh có thể được giải quyết bằng cách cung cấp sữa mẹ đã vắt qua bình sữa. Trẻ bú bình và trẻ lớn hơn cho phép nhiều can thiệp hơn để thúc đẩy tăng hàm lượng calo của chế độ ăn. Trẻ sơ sinh bị FTT nên nhận được khoảng 150% lượng calo khuyến nghị hàng ngày dựa trên cân nặng dự kiến của chúng (thay vì dựa trên cân nặng thực tế của chúng). Việc làm giàu sữa công thức có thể đạt được bằng cách thêm ngũ cốc, và trẻ mới biết đi có thể được hưởng lợi từ việc bổ sung các loại thực phẩm ngon miệng có mật độ năng lượng cao (chẳng hạn như phô mai và bơ đậu phộng) vào chế độ ăn của chúng. Các loại đồ uống làm từ sữa có hàm lượng calo cao (chẳng hạn như PediaSure cung cấp 30 calo mỗi ounce, so với sữa nguyên chất cung cấp 19 calo mỗi ounce) có thể được bổ sung cùng với việc bổ sung vitamin. Việc bổ sung kẽm đã được chứng minh là làm tăng nồng độ IGF-1 mà không ảnh hưởng đến IGFBP-3 ở trẻ sơ sinh bị FTT phi hữu cơ, nhưng tác dụng này không thực sự thúc đẩy tăng trưởng.
Tiên lượng
Phần lớn trẻ sơ sinh và trẻ em bị FTT cho thấy sự cải thiện với can thiệp. Những trẻ khác thậm chí có thể cho thấy sự tiến bộ khi chúng đạt đến một giai đoạn phát triển độc lập hơn, nơi chúng có thể tự kiếm thức ăn. Những trẻ cần cho ăn qua ống thông dạ dày do rối loạn chức năng thần kinh có thể cần dinh dưỡng qua đường ruột được hỗ trợ suốt đời. Kết quả chức năng nhận thức và trí tuệ của những người bị FTT dường như tồi tệ hơn so với các bạn cùng lứa, mặc dù mối liên quan này chỉ được thiết lập rõ ràng trong các trường hợp thiếu máu do thiếu sắt. Có vẻ hợp lý rằng sự thiếu hụt các yếu tố khác quan trọng cho sự phát triển của não trong giai đoạn sơ sinh có thể có tác động bất lợi tương tự đối với các đặc tính trí tuệ ở các lứa tuổi sau này, mặc dù điều này chưa được nghiên cứu một cách có hệ thống. Tác động của các yếu tố phi hữu cơ (chẳng hạn như thiếu thốn tình cảm) đối với sự phát triển trí tuệ, thường cùng tồn tại với các yếu tố hữu cơ, cũng có thể góp phần làm giảm khả năng nhận thức ở các lứa tuổi sau này.
Suy mòn do ung thư
Suy mòn do ung thư được đặc trưng bởi một hội chứng suy kiệt mãn tính, liên quan đến sự mất mát của cơ xương và mô mỡ, và thường kháng lại sự hỗ trợ dinh dưỡng thông thường. Suy mòn chịu trách nhiệm cho sự giảm đáng kể về chất lượng và thời gian sống của bệnh nhân ung thư, cũng như ở nhóm tuổi nhi khoa. Tương tự như suy mòn do nhiễm trùng mãn tính hoặc các bệnh viêm, bệnh nhân ung thư có thể biểu hiện cả suy dinh dưỡng và căng thẳng chuyển hóa. Cả hai con đường sử dụng carbohydrate, cũng như sự kết hợp axit amin, đều bị giảm trong cơ của bệnh nhân suy mòn do ung thư. Các tế bào khối u có thể ảnh hưởng đến quá trình chuyển hóa của bệnh nhân theo hai cách: quá trình chuyển hóa chất dinh dưỡng của chính chúng thành các chất chuyển hóa khác và bởi các yếu tố tuần hoàn mà chúng tiết ra hoặc gây ra cho vật chủ tiết ra. Tăng tốc độ đường phân và sản xuất lactate, và sự gia tăng hoạt động của chu trình Cori resultant, là những tác động chuyển hóa thường được quan sát thấy. Một số lượng lớn các chất chuyển hóa và yếu tố tuần hoàn gây suy mòn đã được tìm thấy, chẳng hạn như TNF-α, IL-6, myostatin và protein liên quan đến PTH, nhưng chưa có một phân tử nào được chứng minh là một yếu tố quyết định có thể được nhắm mục tiêu riêng lẻ để điều trị suy mòn do ung thư ở người. Ung thư kích hoạt một tập hợp phức tạp các con đường chuyển hóa của TKTW, dẫn đến suy mòn (Hộp 24.3). Các cytokine ngoại vi xâm nhập vào TKTW thông qua các cơ quan quanh não thất trung ương, bỏ qua hàng rào máu-não, hoặc bằng cách kích thích và khuếch đại cytokine vi mô hoặc sản xuất eicosanoid của TKTW. Ví dụ, TNF-α kích thích các tế bào thần kinh POMC của VMH, kích thích SNS, đến lượt nó làm tăng tiêu hao năng lượng khi nghỉ, tăng nồng độ cortisol và glucagon, và góp phần vào tình trạng kháng insulin. IL-1 làm giảm neuropeptide Y trong VMH và do đó làm giảm cảm giác thèm ăn. IL-1 cũng làm tăng CRH, gián tiếp ức chế cảm giác thèm ăn. IL-6 có sự tương đồng đáng kinh ngạc với yếu tố dinh dưỡng thần kinh lông mi (CNTF), đã được chứng minh là làm giảm cân bằng cách kích hoạt các tế bào thần kinh POMC của VMH thông qua một cơ chế độc lập với leptin. Ngược lại, do sự giảm hoạt động của phế vị, khả năng vận động của đường tiêu hóa bị suy giảm trong tình trạng suy mòn do ung thư và được biểu hiện lâm sàng bằng cảm giác no sớm và không phù hợp, xảy ra ở 40% đến 60% bệnh nhân ung thư.
Hộp 24.3 Những thay đổi chuyển hóa trong Suy mòn
Biểu hiện của cytokine • Tăng sản xuất protein pha cấp (APP) • Điều hòa tăng yếu tố phiên mã NF Kappa B và AP-1 • Tăng interleukin (IL)-1, IL-6, và yếu tố hoại tử khối u-α và interferon-γ • Tăng biểu hiện của các cachexin đặc hiệu cho khối u (yếu tố gây phân giải protein, yếu tố huy động lipid, và chất gây thiếu máu) • Tăng biểu hiện của ubiquitin, E1, E2, E3, và các thành phần của proteasome (chết và loại bỏ tế bào) Tăng trương lực hệ thần kinh giao cảm • Tăng biểu hiện của lipase nhạy cảm với hormone trong mô mỡ • Điều hòa tăng các protein tách cặp (UCP2 và UCP3) trong cơ và mô mỡ • Tăng tân tạo glucose ở gan Giảm trương lực phế vị • Giảm biểu hiện của lipoprotein lipase trong mô mỡ • Giảm vận chuyển qua ruột • Giảm cảm giác đói
Ngoài ra, cytokine có tác động ngoại vi bất lợi. Các protein tách cặp được điều hòa tăng bởi cytokine và góp phần làm tăng tiêu hao năng lượng. Suy mòn do ung thư dẫn đến sự biểu hiện quá mức của UCP1 trong mô mỡ nâu; UCP2 trong não, cơ xương và gan; và UCP3 trong cơ xương. Mức độ của UCP2 và UCP3 trong gan và cơ được điều hòa bởi prostaglandin, và UCP3 cũng được điều hòa bởi TG, tất cả đều tăng trong ung thư.
Cytokine gây kháng insulin ở cơ xương, gan và mô mỡ. TNF-α làm giảm thụ thể insulin và hoạt động của PPAR. TNF-α có tác dụng dị hóa trực tiếp lên cơ xương và gây teo cơ bằng cách cảm ứng hệ thống ubiquitin-proteasome (UPS). TNF-α thúc đẩy sự gia tăng tân tạo glucose ở gan, mất mô mỡ và phân giải protein, đồng thời gây ra sự giảm tổng hợp protein, lipid và glycogen. Nó đã được liên kết với sự hình thành của IL-1 và làm tăng biểu hiện của UCP2 và UCP3 trong cơ xương ở trạng thái suy mòn. Ngoài ra, có một mối tương quan nghịch giữa nồng độ IL-6 và độ nhạy insulin. Kháng insulin của mô mỡ làm tăng quá trình oxy hóa chất béo, và làm giảm hoạt động của lipoprotein lipase, dẫn đến quá trình ly giải mỡ liên tục. Kháng insulin của ung thư không liên quan đến sự thanh thải insulin bị lỗi, và do đó khác với các dạng kháng insulin nguyên phát khác.
Các bệnh ung thư đồng nhất là kỵ khí và phụ thuộc vào glucose để tồn tại; do đó, glucose được sản xuất từ quá trình tân tạo glucose thứ phát sau kháng insulin ở gan là cần thiết cho sự phát triển của khối u. Các bệnh ung thư giải phóng một lượng lớn lactate, được chuyển đổi ở gan trở lại thành glucose. Quá trình tân tạo glucose như vậy tiêu thụ ATP, cũng làm tăng tiêu hao năng lượng. Các nguyên liệu thô bổ sung cho quá trình tân tạo glucose là alanin có nguồn gốc từ sự phân giải protein cơ xương và glycerol từ quá trình ly giải mỡ.
Với bản chất đa yếu tố phức tạp của hồ sơ nội tiết tố/chuyển hóa liên quan đến suy mòn và sự khác biệt cá nhân giữa các bệnh nhân ung thư, việc nhắm mục tiêu vào bất kỳ yếu tố tuần hoàn đơn lẻ nào sẽ luôn không đủ để điều trị suy mòn cho tất cả các bệnh nhân ung thư. Hiện không có tác nhân dược lý cụ thể nào có thể giải quyết hiệu quả tình trạng suy mòn do ung thư, và việc điều trị dựa vào việc bổ sung chế độ ăn và hỗ trợ tâm lý.
Hội chứng Diencephalic
Hội chứng diencephalic bao gồm các đặc điểm lâm sàng là suy kiệt nặng, tăng trưởng theo chiều dài bình thường, và phát triển trí tuệ bình thường hoặc sớm phát triển liên quan đến các khối u TKTW. Được Russell mô tả lần đầu tiên vào năm 1951, rối loạn hiếm gặp này xuất hiện ở trẻ sơ sinh dưới 1 tuổi, và là một dấu hiệu của một tổn thương vùng dưới đồi, thường là một u thần kinh đệm vùng dưới đồi trước, hoặc một khối u khác ảnh hưởng đến chức năng vùng dưới đồi. Mặc dù phổ lâm sàng có thể thay đổi, suy kiệt với sự khan hiếm của mỡ dưới da, nhưng với sự tăng trưởng theo chiều dài và chu vi vòng đầu bình thường là bất biến. Các đặc điểm thường gặp khác bao gồm sự tỉnh táo quá mức, tăng động, rung giật nhãn cầu, và nôn.
Nhiều bệnh nhân đã được báo cáo và mô tả theo giai thoại; tuy nhiên, nguyên nhân của sự suy kiệt vẫn chưa rõ ràng. Các đối tượng mắc hội chứng diencephalic có nồng độ GH cơ bản cực cao, nhưng với nồng độ IGF-1 bình thường, cho thấy một biến thể của kháng GH. Người ta đã gợi ý rằng GH cao dẫn đến ly giải mỡ và giải thích cho sự suy kiệt, nhưng phát hiện này không nhất quán ở tất cả các bệnh nhân. Chỉ có một đánh giá về cân bằng năng lượng đã được thực hiện, cho thấy REE cao hơn 30% đến 50% so với trẻ sơ sinh bình thường, và tiêu hao năng lượng cao hơn 13% so với lượng nạp vào.
Điều trị được khuyến nghị là cắt bỏ tổn thương bằng phẫu thuật bất cứ khi nào có thể. Xạ trị thường được dành riêng cho bệnh nhân rất trẻ. Thông thường, những bệnh nhân này sau phẫu thuật biểu hiện suy tuyến yên, và cuối cùng phát triển béo phì do vùng dưới đồi.
Chán ăn tâm thần
Định nghĩa
Chán ăn tâm thần (AN) là một rối loạn ăn uống thường bắt đầu trong giai đoạn thanh thiếu niên và bao gồm việc ăn kiêng dai dẳng và hoạt động thể chất cường độ cao, thường đi kèm với các đặc điểm hành vi cưỡng chế và đôi khi với các hành vi ăn uống vô độ và thanh lọc. Các cá nhân mắc AN cũng biểu hiện một hình ảnh cơ thể bị xáo trộn và một nỗi sợ hãi dai dẳng về việc tăng cân, cả hai đều thúc đẩy việc giảm cân thêm. Kết quả của hành vi này là một sự sụt cân bệnh lý, với các hậu quả sinh lý bệnh. Nguy cơ phát triển AN ở nữ giới trong các xã hội phương Tây được ước tính là từ 1,7% đến 3,6%. Có hai loại phụ của chứng chán ăn: loại hạn chế, đặc trưng bởi lượng calo nạp vào rất thấp và/hoặc tập thể dục quá mức, và loại ăn uống vô độ/thanh lọc, đặc trưng bởi các mức độ thanh lọc thức ăn khác nhau, thường bằng cách tự gây nôn và/hoặc lạm dụng thuốc nhuận tràng. Cùng với các yếu tố cảm xúc và hành vi, định nghĩa trong DSM-5 bao gồm việc có một cân nặng thấp đáng kể (được định nghĩa là BMI < bách phân vị thứ 5 theo tuổi và giới tính ở trẻ em hoặc BMI ≤ 17 ở người lớn).
Các biến chứng y tế do việc giảm lượng calo nạp vào mãn tính, hành vi thanh lọc và tập thể dục quá mức có thể ảnh hưởng đến một số hệ cơ quan. Thông thường, bệnh nhân phát triển một sự mất mát rõ rệt của mô mỡ dưới da, suy giảm chức năng kinh nguyệt, nhịp tim chậm và hạ huyết áp tư thế đứng, hạ thân nhiệt, và tăng rụng tóc. Quan trọng là, AN phát triển trong giai đoạn thanh thiếu niên có thể tạo ra các tác động lâm sàng bất lợi kéo dài đến tuổi trưởng thành, bao gồm loãng xương và xốp xương, tỷ lệ sẩy thai cao hơn, và giảm cân nặng khi sinh của con cái. Chán ăn cũng có thể làm thay đổi khả năng nhận thức vì trong quá trình sụt cân cực độ trong các trường hợp xảy ra sự giảm đáng kể cả chất xám và chất trắng của não; ngược lại, trong quá trình phục hồi cân nặng, chất trắng của não trở lại mức trước khi mắc bệnh, nhưng chất xám thì không. Chán ăn mang một nguy cơ tử vong gia tăng, đặc biệt là do tự tử, nhưng cũng từ các nguyên nhân y tế, chẳng hạn như chính tình trạng đói, cũng như rối loạn nhịp tim qua trung gian điện giải do thanh lọc gây ra. Sự phục hồi hoàn toàn về thể trạng và sự tăng trưởng và phát triển xảy ra ở 50% đến 70% thanh thiếu niên được điều trị; nhưng để đạt được sự phục hồi sinh lý và tâm lý hoàn toàn có thể mất một can thiệp điều trị toàn diện có thể kéo dài từ 5 đến 7 năm. Sụt cân nhiều hơn, duy trì cân nặng thấp hơn, và sự cùng tồn tại của các rối loạn tâm thần khác ảnh hưởng xấu đến khả năng phục hồi. Kết quả cho người lớn mắc AN kém hơn so với những người được chẩn đoán và điều trị ở tuổi vị thành niên.
Các biểu hiện nội tiết của Chán ăn Tâm thần
Béo phì và suy dinh dưỡng thường dẫn đến các tác động trái ngược nhau đối với sinh lý bình thường và cả hai đều liên quan đến những thay đổi trong hồ sơ nội tiết tố. AN có liên quan đến nhiều thay đổi nội tiết sâu sắc có thể là thích nghi, phản ứng, hoặc có lẽ là nguyên nhân. Phần lớn những thay đổi này đại diện cho một phản ứng thích nghi nhưng nên được xem xét như một phần của chẩn đoán phân biệt của các rối loạn thừa hoặc thiếu nội tiết tố cụ thể. Rối loạn chức năng của trục hạ đồi-tuyến yên bao gồm suy sinh dục do thiếu gonadotropin với sự thiếu hụt tương đối estrogen và androgen, kháng GH, cortisol tăng, hội chứng “euthyroid bệnh”, và oxytocin giảm. Nồng độ leptin bị ức chế ở bệnh nhân AN phản ánh sự dự trữ năng lượng giảm và nồng độ ghrelin, một peptide đường ruột gây thèm ăn, tăng cao. Hầu hết, nhưng không phải tất cả, những rối loạn nội tiết này là thích nghi với trạng thái năng lượng thấp của tình trạng đói mãn tính và đảo ngược với việc điều trị rối loạn ăn uống. Mặc dù vậy, những thay đổi này góp phần làm suy giảm tính toàn vẹn của xương, cũng như các bệnh đi kèm về thần kinh-tâm thần, ở các cá nhân mắc AN. Quan trọng là, chỉ có 5% đến 15% bệnh nhân AN là nam giới, và chỉ có dữ liệu hạn chế tồn tại về tác động nội tiết của bệnh ở các bé trai vị thành niên. Hình 24.12 minh họa hồ sơ nội tiết tố điển hình của bệnh nhân AN, phản ánh những nỗ lực của cơ thể nhằm bảo tồn năng lượng và ngừng các quá trình tốn kém năng lượng và không quan trọng.
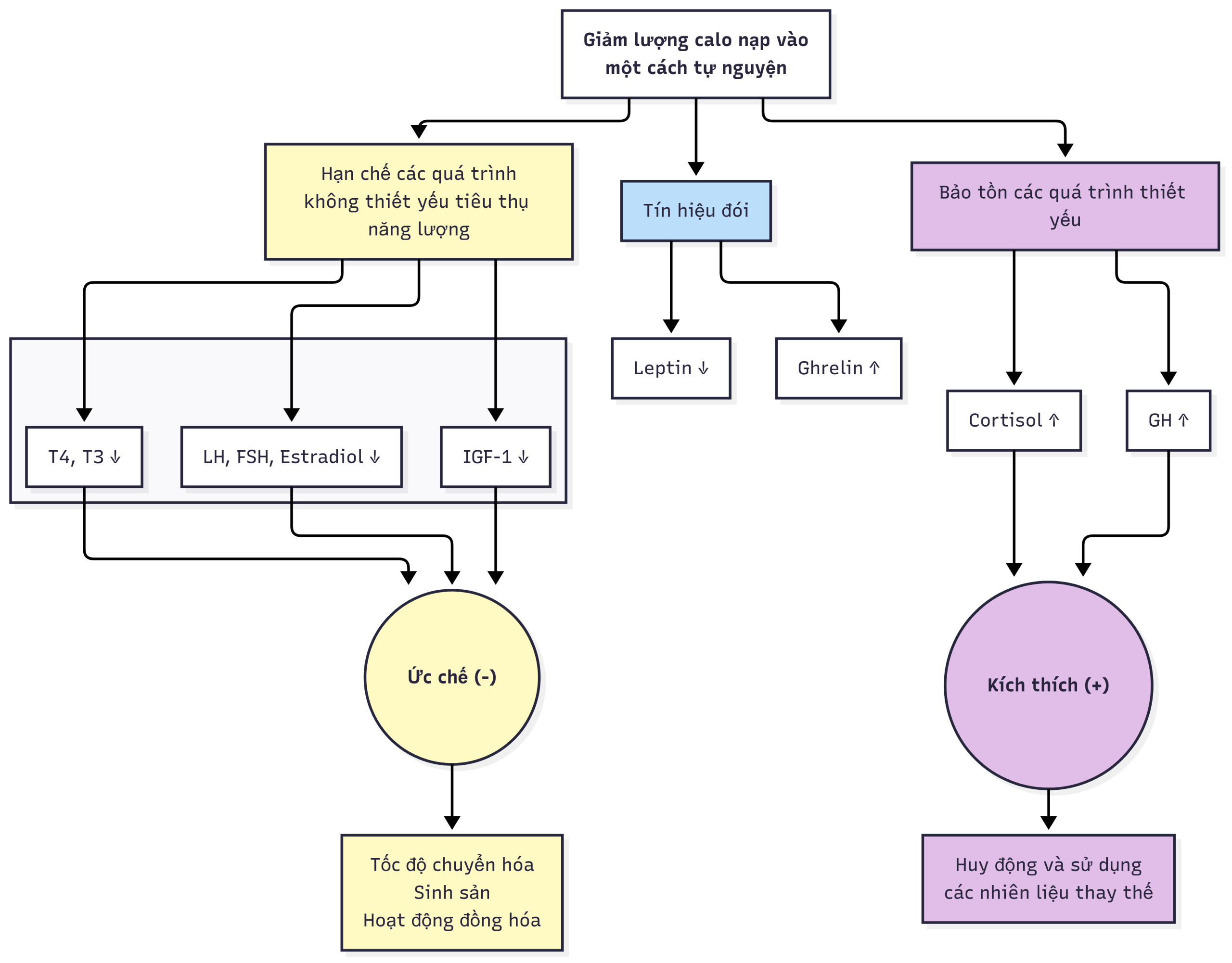
Hình 24.12 Những thay đổi nội tiết tố trong chán ăn tâm thần. Hồ sơ nội tiết tố của bệnh nhân chán ăn tâm thần đại diện cho một phản ứng thích nghi nhằm bảo tồn năng lượng. Các quá trình đòi hỏi năng lượng đáng kể, chẳng hạn như sinh sản và tăng trưởng, bị hạn chế bởi sự ức chế hoàn toàn của trục gonadotropic và bởi nồng độ yếu tố tăng trưởng giống insulin (IGF)-1 giảm, tương ứng. Một phản ứng căng thẳng dường như mãn tính được đặc trưng bởi sự gia tăng hormone tăng trưởng (GH) và cortisol nhằm mục đích sử dụng hiệu quả các nguồn năng lượng hạn chế hiện có. Leptin giảm đóng vai trò là một tín hiệu để điều hòa giảm trục hạ đồi-tuyến yên-tuyến sinh dục. Leptin giảm cũng đóng vai trò là một tín hiệu đói, mặc dù tín hiệu này dường như bị bỏ qua trong rối loạn này.
Trục Hạ đồi-Tuyến yên-Tuyến giáp. Tình trạng đói của AN có thể giống với hội chứng euthyroid bệnh. Ở bệnh nhân AN, nồng độ T4 toàn phần và tự do, T3 toàn phần và tự do, TSH và globulin gắn thyroxine trong huyết thanh thấp hơn đáng kể so với bình thường, trong khi nồng độ triiodothyronine đảo ngược (rT3) lớn hơn đáng kể so với nhóm đối chứng khỏe mạnh. Hầu hết bệnh nhân AN có đáp ứng TSH giảm hoặc chậm đối với kích thích TRH. Đáng chú ý, việc tăng cân trở lại sẽ đảo ngược tác động của AN lên trục hạ đồi-tuyến yên-tuyến giáp trở lại bình thường; do đó, việc giảm thyroxinemia thực sự có thể là một phản ứng sinh lý thích nghi bình thường đối với tình trạng đói. Chẩn đoán phân biệt suy giáp nên được xem xét ở bệnh nhân AN vì cả hai rối loạn đều được đặc trưng bởi nồng độ T4 và T3 thấp. Trong suy giáp nguyên phát, nồng độ TSH sẽ lớn hơn so với những gì thấy ở bệnh nhân AN, mặc dù trong suy giáp thứ phát nhẹ, sự phân biệt này có thể khó khăn. Việc lấy nồng độ rT3 trong huyết thanh có thể hữu ích trong việc phân biệt một rối loạn tuyến giáp thực sự với hội chứng euthyroid bệnh liên quan đến bệnh toàn thân.
Trục Hormone Tăng trưởng-IGF-1. Bệnh nhân AN thường có sự tăng tiết GH kèm theo nồng độ IGF-1 thấp. Liệu hồ sơ này có phải do rối loạn chức năng vùng dưới đồi nguyên phát, kháng cơ quan đích ngoại vi đối với GH, hay một cơ chế phản hồi trung ương IGF-1 âm bị suy giảm vẫn chưa rõ ràng. Một phản ứng GH tăng đối với GHRH đã được chứng minh trong AN, có thể phản ánh sự suy giảm của sự ức chế beta-adrenergic đối với sự bài tiết GH. Biểu hiện lâm sàng của sụt cân, ngừng kinh nguyệt và không dung nạp lạnh cùng với nồng độ hormone có nguồn gốc từ tuyến yên thấp có thể giống với suy toàn bộ tuyến yên, nhưng bệnh nhân AN lại biểu hiện tăng tiết GH.
Trục Hạ đồi-Tuyến yên-Tuyến thượng thận. Bệnh nhân AN thường biểu hiện với nồng độ cortisol tăng cao trong sự hiện diện của nồng độ ACTH bình thường. Nồng độ cortisol tăng cao rõ ràng là do sự bài tiết cortisol tăng cùng với sự giảm thanh thải cortisol. Nồng độ CRH tăng cao được tìm thấy trong dịch não tủy của bệnh nhân AN cho thấy rằng AN đại diện cho một trạng thái kích hoạt tổng thể của phản ứng căng thẳng vùng dưới đồi, được biểu hiện ở ngoại vi bởi tăng cortisol máu. Một phản ứng bất thường trong xét nghiệm ức chế dexamethasone, chủ yếu là sự ức chế giảm, cho thấy rằng một yếu tố của sự giảm nhạy cảm phản hồi xảy ra trong rối loạn này.
Chuyển hóa xương. Thanh thiếu niên đại diện cho một giai đoạn quan trọng cho sự tích lũy khoáng chất xương, do đó xây dựng sức mạnh và mật độ xương cho những năm sau này. Việc đạt được khối lượng xương đỉnh phụ thuộc vào một số tác động nội tiết tố đặc trưng của tuổi dậy thì, chẳng hạn như tăng nồng độ estradiol và IGF-1, cũng như phụ thuộc vào dinh dưỡng đầy đủ; tất cả đều bị tổn hại ở bệnh nhân AN. Thanh thiếu niên và người lớn mắc AN có mật độ khoáng xương thấp. Ở người lớn, điều này là do sự tăng tái hấp thu xương và giảm hình thành xương; thanh thiếu niên được đặc trưng bởi sự giảm tổng thể chu chuyển xương. Mật độ xương giảm là do hồ sơ nội tiết tố điển hình của AN, tức là, giảm estrogen và androgen, IGF-1 thấp, và tăng cortisol máu tương đối. Hơn nữa, khối lượng nạc cơ thể giảm và các lực cơ học thấp hơn tác động lên các xương dài cũng có thể góp phần vào việc giảm tổng thể MĐX. Các ấn phẩm gần đây cũng cho thấy rằng nồng độ hormone có nguồn gốc từ ruột PYY(3-36) và ghrelin tăng cao cũng có thể góp phần làm suy giảm chuyển hóa xương trong AN. Quan trọng là, việc giải quyết AN với việc tăng cân thường không mang lại sự phục hồi hoàn toàn tình trạng mật độ khoáng xương. Loãng xương do suy dinh dưỡng và động lực nội tiết tố điển hình của AN là một trong những biến chứng lâu dài nghiêm trọng của AN trong giai đoạn thanh thiếu niên, và do đó là một mục tiêu điều trị chính.
Trục Hạ đồi-Tuyến yên-Tuyến sinh dục. Vô kinh là một trong những dấu hiệu đặc trưng của AN, nhưng nó không phải lúc nào cũng được giải thích bởi sự sụt cân nghiêm trọng; do đó, vô kinh do vùng dưới đồi trong AN thường có trước sụt cân và có thể tồn tại sau khi ăn lại và đạt được cân nặng bình thường. Nồng độ gonadotropin bị giảm ở bệnh nhân AN, và xét nghiệm kích thích GnRH ở bệnh nhân AN cho thấy một phản ứng LH bị cùn với một phản ứng FSH được bảo tồn và nồng độ estradiol rất thấp. Mô hình dao động của nồng độ LH ở tuổi dậy thì bình thường có thể trở lại các mô hình tiền dậy thì ở thanh thiếu niên đã đạt được trạng thái dậy thì trước đó. Vô kinh của AN cũng được đánh dấu bằng nồng độ leptin giảm đáng kể. Leptin đóng vai trò là một tín hiệu chuyển hóa về tình trạng năng lượng và dự trữ dinh dưỡng và do đó có thể có vai trò cho phép đối với việc khởi đầu các động lực nội tiết tố phức tạp cần thiết cho chức năng sinh sản bình thường. Về mặt mục đích học, việc bảo tồn năng lượng cho các nhu cầu chuyển hóa tức thời và cần thiết trong khi ức chế các quá trình tốn kém năng lượng, chẳng hạn như sinh sản, đóng vai trò là một biện pháp bảo vệ ở những người bị suy dinh dưỡng nặng, như thấy trong AN. Sự gia tăng leptin khi tăng cân có liên quan đến sự gia tăng bài tiết gonadotropin, cho thấy rằng leptin đóng một vai trò cho phép đối với việc kích hoạt trục hạ đồi-tuyến yên-tuyến sinh dục.
Các Hormone có nguồn gốc từ mỡ. Các tế bào mỡ tiết ra một loạt các adipocytokine, hồ sơ bình thường của chúng bị thay đổi trong AN. Bệnh nhân AN thường có nồng độ leptin huyết tương rất thấp cùng với sự xáo trộn rõ rệt của hồ sơ bài tiết leptin hàng ngày. Nồng độ của protein gắn leptin (dạng hòa tan của thụ thể leptin) đã được báo cáo là tăng ở bệnh nhân AN, góp phần làm giảm thêm nồng độ leptin tự do. Leptin có một vai trò chính trong các thích ứng nội tiết thần kinh đối với tình trạng đói mãn tính và suy dinh dưỡng điển hình của AN, chẳng hạn như giảm gonadotropin, giảm hormone tuyến giáp để bảo tồn năng lượng, một phản ứng căng thẳng đã được sửa đổi được biểu hiện bằng tăng cortisol máu, và GH tăng nhằm huy động và sử dụng các nguồn năng lượng thay thế. Việc tăng cân ở bệnh nhân AN có thể gây ra tăng leptin máu tương đối so với nhóm đối chứng phù hợp với BMI; do đó, nồng độ leptin trong tuần hoàn ở bệnh nhân AN đi từ mức dưới bình thường đến mức trên bình thường trong vòng vài tuần.
Adiponectin là adipocytokine duy nhất có nồng độ huyết tương có tương quan nghịch với khối lượng mỡ. Các kết quả mâu thuẫn về nồng độ adiponectin trong AN, từ tăng adiponectin máu đến giảm adiponectin máu, đã được báo cáo. Điều thú vị là, việc tăng cân trong AN không nhất thiết liên quan đến những thay đổi đối ứng trong nồng độ adiponectin.
Điều trị
Các chỉ định nhập viện và điều trị nội trú AN bao gồm mất nước, rối loạn điện giải (chủ yếu là hạ kali máu), rối loạn nhịp tim, bất ổn tim mạch (nhịp tim chậm đáng kể, hạ huyết áp, và thay đổi tư thế đứng), hạ thân nhiệt, từ chối thức ăn cấp tính, các biến chứng cấp tính của suy dinh dưỡng (co giật, viêm tụy, suy tim), và các trường hợp cấp cứu tâm thần.
Cùng với các can thiệp tâm lý có thể bao gồm liệu pháp tâm lý và/hoặc liệu pháp dược lý (dựa trên phổ bệnh lý tâm thần), việc tăng lượng calo nạp vào phải là một phần của phác đồ điều trị. Bắt đầu với 1200 đến 1500 kcal mỗi ngày và tăng thêm 500 kcal nhằm mục đích tăng từ 0,5 đến 1 kg mỗi tuần. Không có một chế độ ăn uống ưu việt duy nhất nào, miễn là lượng calo đầy đủ được tiêu thụ. Các trường hợp cực đoan có thể được xử lý thông qua việc nhập viện tại các đơn vị nội trú chuyên dụng; lượng calo nạp vào trong môi trường này ban đầu có thể được cung cấp bằng ống thông mũi-dạ dày và trong các trường hợp cực đoan thông qua nuôi dưỡng qua đường tĩnh mạch.
Một quyết định điều trị chính mà người chăm sóc bệnh nhân AN phải đối mặt là liệu có nên sử dụng liệu pháp thay thế estrogen để điều trị vô kinh và bảo vệ bộ xương khỏi loãng xương hay không. Thiếu một tác dụng có lợi hiệu quả đã được chứng minh của liệu pháp thay thế estrogen về mặt cải thiện khối lượng xương trong AN so với giả dược, mặc dù phương thức điều trị này vẫn thường được sử dụng. Cách tiếp cận điều trị này với estrogen có thể có một số tác động bất lợi đối với việc điều trị thanh thiếu niên vì nó che giấu tác dụng có lợi của việc tăng cân đối với sự trở lại của kinh nguyệt và có thể cung cấp một cảm giác an toàn sai lầm ở những bệnh nhân vẫn còn ở trạng thái cân nặng thấp nghiêm trọng. Việc tăng lượng canxi nạp vào cùng với 400 IU vitamin D để tăng tốc độ hấp thu nên được khuyến khích ở tất cả các bệnh nhân AN như một biện pháp bảo vệ tiềm năng khác cho tình trạng xương.
Kết luận
Cân bằng năng lượng rất phức tạp và được điều hòa bởi các cơ chế phức tạp đã tiến hóa để tinh chỉnh và duy trì trọng lượng cơ thể trong giới hạn bình thường được chấp nhận. Một số cơ chế này đã tiến hóa để duy trì và bảo tồn sự sống ngay cả khi đối mặt với nạn đói. Tác động của ngay cả những sự mất cân bằng tương đối nhỏ trong lượng nạp vào so với chi tiêu có thể có những tác động tích lũy đáng kể đối với trọng lượng cơ thể theo thời gian, dẫn đến béo phì hoặc thiếu cân. Mặc dù quan trọng cần xem xét trong chẩn đoán phân biệt béo phì ở trẻ em, một bệnh nội tiết cổ điển (thừa glucocorticoid hoặc thiếu hụt hormone tuyến giáp hoặc GH), hội chứng béo phì di truyền, hoặc tổn thương vùng dưới đồi hiếm khi là nguyên nhân. Tuy nhiên, trong một kỷ nguyên thừa dinh dưỡng dồi dào, các bất thường nội tiết tinh vi khác, chẳng hạn như nồng độ hoặc hoạt động của leptin và kháng insulin, là những đặc điểm nổi bật của béo phì, và các biến thể phổ biến của các gen liên quan đến con đường truyền tín hiệu leptin có thể góp phần vào nguy cơ béo phì ở một bộ phận dân số đáng kể. Hơn nữa, béo phì, do sự hiện diện của nó, cho thấy một mức độ rối loạn chức năng nào đó trong các cơ chế cân bằng nội môi thần kinh nội tiết của TKTW điều hòa cân bằng năng lượng. Do đó, béo phì và các hậu quả của nó nên được công nhận là một phần của một phổ rối loạn nội tiết mãn tính. Quan niệm rằng béo phì là do lượng calo nạp vào quá mức với sự tiêu hao calo không đủ đồng thời thông qua công việc và tập thể dục là đơn giản và không giải quyết được vấn đề cơ bản của sự không phù hợp trong các cơ chế điều hòa lượng năng lượng nạp vào và tiêu tán. Chính sự không phù hợp này, được bộc lộ trong kỷ nguyên của chúng ta về lượng năng lượng nạp vào dồi dào, đã duy trì tình trạng mất cân bằng năng lượng, và kỳ thị béo phì chỉ đơn thuần là do thiếu kỷ luật hoặc ý chí. Trên thực tế, béo phì là một tình trạng bệnh lý, ở hầu hết các bệnh nhân, đòi hỏi phải quản lý suốt đời. Tương tự, trong suy mòn, việc điều tra phải vượt ra ngoài sự thiếu thèm ăn để hiểu được lý do của sự suy kiệt và bệnh tật. Việc hiểu rõ con đường cân bằng năng lượng, và nơi các rối loạn khác nhau này làm suy giảm sự điều hòa của nó, cùng với việc giáo dục sức khỏe cộng đồng về hậu quả của béo phì, là chìa khóa để nghiên cứu thêm, và phòng ngừa và điều trị thành công.
Các chữ viết tắt
| Viết tắt | Định nghĩa |
|---|---|
| α-MSH | hormone kích thích tế bào hắc tố alpha |
| 11βHSD1 | 11-beta-hydroxysteroid dehydrogenase loại 1 |
| 5-HT | 5-hydroxytryptamine (serotonin) |
| Ay-a | Chuột vàng Agouti |
| ACTH | hormone vỏ thượng thận |
| AgRP | peptide liên quan đến Agouti |
| AHO | loạn dưỡng xương di truyền Albright |
| ALT | alanine aminotransferase |
| AMP | adenosine monophosphate |
| AN | chán ăn tâm thần |
| ANP | peptide natri lợi niệu nhĩ |
| AP | vùng postrema |
| apoB | apolipoprotein B |
| APP | protein pha cấp |
| ARC | nhân cung |
| ARCPOMC | tế bào thần kinh biểu hiện proopiomelanocortin của nhân cung |
| AS | hội chứng Alström |
| ATP | adenosine triphosphate |
| BBB | hàng rào máu-não |
| BBS | hội chứng Bardet-Biedl |
| BCAA | axit amin chuỗi nhánh |
| BDNF | yếu tố dinh dưỡng thần kinh có nguồn gốc từ não |
| BMI | chỉ số khối cơ thể |
| BMR | tỷ lệ chuyển hóa cơ bản |
| cAMP | cyclic adenosine monophosphate |
| CART | bản ghi liên quan đến cocaine-amphetamine |
| CCK | cholecystokinin |
| cGMP | cyclic guanosine monophosphate |
| ChREBP | protein gắn yếu tố đáp ứng carbohydrate |
| CI | khoảng tin cậy |
| TKTW | hệ thần kinh trung ương |
| CNTF | yếu tố dinh dưỡng thần kinh lông mi |
| CoA | coenzyme A |
| CPE | carboxypeptidase E |
| CPT-1 | carnitine palmitoyl transferase 1 |
| CREB | protein gắn yếu tố đáp ứng cyclic adenosine monophosphate (AMP) |
| CRH | hormone giải phóng corticotropin |
| CRISPR | cụm lặp lại palindromic ngắn cách đều nhau |
| CRP | protein phản ứng C |
| DNT | dịch não tủy |
| CT | chụp cắt lớp vi tính |
| TM | tim mạch |
| DAG | diacylglycerol |
| DIT | sinh nhiệt do chế độ ăn |
| DMH | nhân dưới đồi lưng giữa |
| DMV | nhân vận động lưng của dây thần kinh phế vị |
| DNA | axit deoxyribonucleic |
| DNL | tân tạo mỡ |
| DSM-5 | “Sổ tay Chẩn đoán và Thống kê các Rối loạn Tâm thần, ấn bản thứ năm” |
| DVC | phức hợp phế vị lưng |
| EC | endocannabinoid |
| eNOS | endothelial nitric oxide synthase |
| LNM | lưới nội chất |
| F1P | fructose-1-phosphate |
| FDA | Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ |
| FFA | axit béo tự do |
| FGF19 | yếu tố tăng trưởng nguyên bào sợi 19 |
| FMD | giãn mạch qua trung gian dòng chảy phụ thuộc vào nội mô |
| FOXO1 | protein forkhead-1 |
| FSH | hormone kích thích nang trứng |
| FSIVGTT | nghiệm pháp dung nạp glucose tĩnh mạch lấy mẫu thường xuyên |
| FTO | gen liên quan đến khối lượng mỡ và béo phì |
| FTT | chậm phát triển |
| FXR | thụ thể X farnesoid |
| GABA | axit gamma-amino butyric |
| GDM | đái tháo đường thai kỳ |
| GH | hormone tăng trưởng |
| GHSR | thụ thể tiết hormone tăng trưởng |
| TT | tiêu hóa |
| GIx | chỉ số đường huyết |
| GL | tải lượng đường huyết |
| GLP-1 | peptide giống glucagon 1 |
| GLUT4 | chất vận chuyển glucose 4 |
| GNG | tân tạo glucose |
| GnRH | hormone gonadotropin |
| GOAT | ghrelin O-acetyltransferase |
| GPCR19 | thụ thể kết hợp G-protein 19 |
| GTP | guanosine triphosphate |
| GWAS | nghiên cứu liên kết toàn bộ bộ gen |
| HDL | lipoprotein tỷ trọng cao |
| HFCS | xi-rô ngô có hàm lượng fructose cao |
| HGP | Sản xuất glucose ở gan |
| HOMA-IR | mô hình cân bằng nội môi để đánh giá kháng insulin |
| HPA | “hạ đồi, tuyến yên, và tuyến thượng thận” |
| HSL | lipase nhạy cảm với hormone |
| NNT | trong não thất |
| IGF | yếu tố tăng trưởng giống insulin |
| IGFBP | protein gắn yếu tố tăng trưởng giống insulin |
| IGT | rối loạn dung nạp glucose |
| IHCL | Lipid trong gan |
| IHCL | lipid trong tế bào gan |
| IL-6 | interleukin-6 |
| IMCL | lipid trong cơ |
| IMT | độ dày lớp nội trung mạc |
| IP3 | inositol triphosphate |
| IRS-1 | cơ chất thụ thể insulin 1 |
| IRS-2 | cơ chất thụ thể insulin 2 |
| JAK1 | Janus kinase 1 |
| JAK2 | Janus kinase 2 |
| JNK-1 | c-jun N-terminal kinase 1 |
| LAGB | thắt đai dạ dày có thể điều chỉnh qua nội soi |
| LEPR | thụ thể leptin |
| LGA | lớn so với tuổi thai |
| LH | hormone luteinizing |
| LHA | vùng dưới đồi bên |
| LPL | lipoprotein lipase |
| LSG | cắt dạ dày hình ống qua nội soi |
| MAPK | protein kinase được hoạt hóa bởi mitogen |
| MARCKS | cơ chất protein kinase C giàu alanin được myristoyl hóa |
| MBH | vùng dưới đồi giữa nền |
| MC3R | thụ thể melanocortin-3 |
| MC4R | thụ thể melanocortin-4 |
| MCH | hormone cô đặc melanin |
| MCR | thụ thể melanocortin |
| ME | lồi giữa |
| MKK7 | mitogen-activated protein kinase 7 |
| MRAP2 | protein phụ trợ thụ thể melanocortin 2 |
| MRI | chụp cộng hưởng từ |
| mRNA | axit ribonucleic thông tin |
| mTOR | mục tiêu của rapamycin ở động vật có vú |
| NA | nhân accumbens |
| NAFLD | bệnh gan nhiễm mỡ không do rượu |
| NASH | viêm gan nhiễm mỡ không do rượu |
| NE | norepinephrine |
| NEAT | sinh nhiệt do hoạt động không phải tập thể dục |
| NHANES | Khảo sát Sức khỏe và Dinh dưỡng Quốc gia |
| NMDA | N-methyl-d-aspartate |
| NO | nitric oxide |
| NPY | neuropeptide Y |
| NTS | nhân của bó đơn độc |
| NGDPĐU | nghiệm pháp dung nạp glucose đường uống |
| OSA | ngưng thở khi ngủ do tắc nghẽn |
| PAI-1 | chất ức chế hoạt hóa plasminogen–1 |
| PC1 | prohormone convertase-1 |
| PCOS | hội chứng buồng trứng đa nang |
| PedNSS | Hệ thống Giám sát Dinh dưỡng Nhi khoa |
| PEPCK | phosphoenolpyruvate carboxykinase |
| PGC | đồng hoạt hóa gamma thụ thể được hoạt hóa bởi peroxisome proliferator |
| PGC-1α | đồng hoạt hóa alpha 1 thụ thể gamma được hoạt hóa bởi peroxisome proliferator |
| PHP1a | giả suy cận giáp type 1a |
| PI3K | phosphatidyl inositol-3-kinase |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PKCε | protein kinase C epsilon |
| POMC | proopiomelanocortin |
| PP | polypeptide tuyến tụy |
| PPAR | thụ thể được hoạt hóa bởi peroxisome proliferator |
| PVN | nhân cạnh não thất của vùng dưới đồi |
| PWS | hội chứng Prader-Willi |
| PYY | peptide YY |
| RAI1 | cảm ứng bởi axit retinoic 1 |
| TNNCT | thử nghiệm ngẫu nhiên có đối chứng |
| REE | tiêu hao năng lượng khi nghỉ |
| ROHHAD | “Béo phì khởi phát nhanh với rối loạn chức năng vùng dưới đồi, giảm thông khí, và rối loạn chức năng tự chủ” |
| ROS | các loại oxy phản ứng |
| rT3 | triiodothyronine đảo ngược |
| RYGB | nối tắt dạ dày Roux-en-Y |
| SGA | nhỏ so với tuổi thai |
| SIM1 | single-minded 1 |
| SMS | hội chứng Smith-Magenis |
| SNP | đa hình đơn nucleotide |
| SNS | hệ thần kinh giao cảm |
| SOCS-3 | chất ức chế tín hiệu cytokine 3 |
| SREBP-1c | protein gắn yếu tố điều hòa sterol 1c |
| SSPG | glucose huyết tương trạng thái ổn định |
| SSTR5 | thụ thể somatostatin-5 |
| STAT-3 | chất truyền tín hiệu và chất hoạt hóa phiên mã |
| ĐTĐ2 | đái tháo đường tuýp 2 |
| T3 | triiodothyronine |
| TEE | tổng tiêu hao năng lượng |
| TG | triglyceride |
| TNF-α | yếu tố hoại tử khối u alpha |
| chất béo trans | axit béo không bão hòa dạng trans |
| TRH | hormone giải phóng thyrotropin |
| TrkB | kinase B liên quan đến tropomyosin |
| TSH | hormone kích thích tuyến giáp |
| UCP | protein tách cặp |
| UPR | phản ứng protein không gấp khúc |
| UPS | hệ thống ubiquitin-proteasome |
| VLDL | lipoprotein tỷ trọng rất thấp |
| VMH | nhân dưới đồi bụng giữa |
| VTA | vùng bụng trước |
| WHO | Tổ chức Y tế Thế giới |
| WT | kiểu hoang dã |
| Y2R | thụ thể neuropeptide Y loại 2 |
| Y4R | thụ thể neuropeptide Y loại 4 |