
Sổ tay Phục hồi chức năng Lâm sàng Braddom, Ấn bản thứ hai (2026)
Nhà xuất bản: ELSEVIER, 2026
Tác giả: David Cifu, Henry L. Lew – Dịch và chú giải: Ths.Bs. Lê Đình Sáng
(C) Bản dịch tiếng Việt bởi THƯ VIỆN Y HỌC MEDIPHARM
Chương 40 – Bệnh lý Nơ-ron Vận động
Motor neuron diseases
Lydia Abdul Latif
Braddom’s Rehabilitation Care: A Clinical Handbook, 40, 393-400.e6
Hướng dẫn nhanh
Phân loại Bệnh lý Nơ-ron Vận động
Chẩn đoán và Tiêu chí cho ALS
Tác động cụ thể liên quan đến Bệnh
Các chiến lược Điều trị ALS
|
Chương này cung cấp một cái nhìn tổng quan về phổ của các bệnh lý nơ-ron vận động (MNDs), chủ yếu tập trung vào tác động của bệnh xơ cứng cột bên teo cơ (ALS) và phương pháp tiếp cận chung để đánh giá, chẩn đoán và quản lý bệnh này.
PHÂN LOẠI (BẢNG 40.1)
Bệnh lý nơ-ron vận động (MNDs) là các rối loạn di truyền hoặc mắc phải biểu hiện đặc trưng bằng sự thoái hóa của nơ-ron vận động. Các nguyên nhân mắc phải hoặc tản phát bao gồm bệnh bại liệt, ALS và các biến thể của nó, và các nguyên nhân di truyền như teo cơ tủy (SMA), ALS gia đình (fALS), và bệnh teo cơ tủy-hành não liên kết X (bệnh Kennedy [KD]). MNDs cũng có thể được chia thành mất nơ-ron vận động trên (UMNs, các bó vỏ-hành) hoặc nơ-ron vận động dưới (LMNs, các tế bào sừng trước tủy sống và các LMN của dây thần kinh sọ). ALS là bệnh lý NVĐ nguyên mẫu và là dạng phổ biến nhất ở người trưởng thành.
Bảng 40.1 Phân loại các Bệnh lý Nơ-ron Vận động
|
Xơ cứng cột bên teo cơ (Hình 40.2)
Tỷ lệ mắc ALS là 1.4/100,000 người với khởi phát ở thập kỷ thứ sáu đến thứ bảy. ALS gây ra sự thoái hóa của cả UMN và LMN mà không ảnh hưởng đến các nơ-ron khác trong hầu hết các trường hợp. Việc phát hiện các đột biến gen liên quan đến fALS đã cung cấp một số hiểu biết trong lĩnh vực này, và các đột biến gen tương tự đã được thấy trong ALS tản phát, cả hai đều có sự tương đồng về lâm sàng. Những quan sát này cho thấy ALS có thể là một con đường bệnh chung liên quan đến nhiều nguyên nhân ngược dòng. Khi ngày càng có nhiều đột biến gen được tìm thấy ở những bệnh nhân bị ALS tản phát, dường như ngày càng có nhiều khả năng nếu không phải là hầu hết các trường hợp đều có một đột biến cơ bản khiến một cá nhân dễ bị phát triển ALS. ALS theo định nghĩa liên quan đến cả UMN và LMN, nhưng một số dạng biến thể của ALS có thể bị giới hạn chỉ ở UMN hoặc LMN hoặc một số vùng cơ thể nhất định. Các biến thể như vậy bao gồm xơ cứng cột bên nguyên phát (PLS) và teo cơ tiến triển (PMA). Các biến thể khu vực bao gồm liệt hành não tiến triển (PBP) liên quan đến hệ cơ hành não, liệt teo cơ hai cánh tay (BAD) liên quan đến các chi trên gần, và liệt teo cơ hai chân (LAD) ảnh hưởng đến các chi dưới xa. Tất cả các biến thể bị giới hạn, ngoại trừ PBP, đều có tiên lượng tốt hơn ALS.
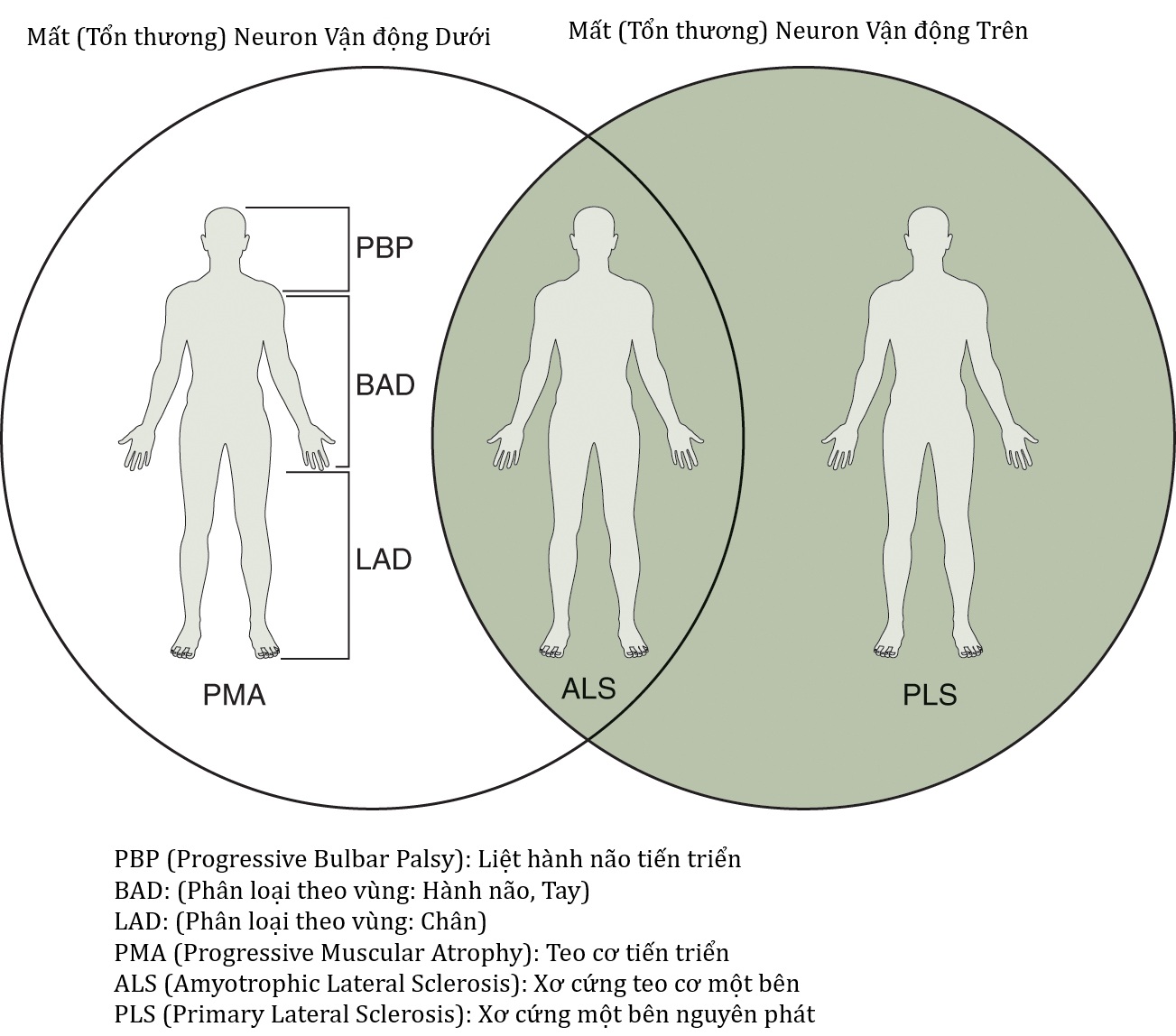
Hình 40.2 Phổ của Xơ cứng cột bên teo cơ và Biến thể Xơ cứng cột bên teo cơTeo cơ tiến triển (PMA), Xơ cứng cột bên nguyên phát (PLS), Liệt hành não tiến triển (PBP), Liệt teo cơ hai cánh tay (BAD), Liệt teo cơ hai chân (LAD).
Xơ cứng cột bên nguyên phát
PLS chủ yếu là sự thoái hóa của UMN và thường thấy ở thập kỷ thứ năm. Biểu hiện chính là co cứng tiến triển một bên, vụng về của bàn tay, và yếu chi và hành não. Liên quan đến bàng quang xuất hiện muộn trong PLS. Về mặt chẩn đoán, PLS được định nghĩa là có ít hoặc không có sự liên quan của LMN trên lâm sàng hoặc bằng điện cơ đồ (EMG). Sự tiến triển của bệnh chậm hơn ALS với tuổi thọ trung bình từ 8 đến 15 năm sau khi chẩn đoán. Không có điều trị thay đổi bệnh cho PLS, và không có nghiên cứu nào cho thấy hiệu quả của riluzole.
Teo cơ tiến triển
PMA là dạng tương tự LMN của MND. Nó hiếm gặp và ảnh hưởng chủ yếu đến nam giới lớn tuổi. Các đặc điểm lâm sàng bao gồm yếu và teo cơ tiến triển bắt đầu ở bàn tay và, ít phổ biến hơn, chi dưới, đai vai và hành não. Mặc dù nó chủ yếu là LMN, khoảng 70% cuối cùng sẽ biểu hiện các dấu hiệu thoái hóa UMN. Điều tra bằng kích thích từ xuyên sọ và quang phổ cộng hưởng từ cho thấy sự liên quan tinh tế của bó vỏ-tủy ở hơn 50% bệnh nhân bị PMA. Vẫn còn tranh cãi liệu PMA có đại diện cho một bệnh khác biệt với ALS hay không. Tuy nhiên, phương pháp tiếp cận chung đối với PMA tương tự như ALS. PMA cũng không đáp ứng với riluzole. Tiên lượng tốt hơn ALS với tỷ lệ sống sót sau 5 năm là hơn 50%.
Xơ cứng cột bên teo cơ khu vực
Các hội chứng biến thể bao gồm PBP, BAD và LAD liên quan đến các vùng nhất định. PBP là một biến thể của ALS với các triệu chứng hành não đơn thuần. Nó có tiên lượng xấu hơn ALS. Nó phổ biến hơn ở phụ nữ lớn tuổi. BAD thường liên quan đến yếu chi trên gần, dường như có liên quan chặt chẽ đến ALS. LAD là một dạng tương quan ở chi dưới của BAD, nhưng yếu cơ có xu hướng bắt đầu một cách không đối xứng, ưu thế ở ngọn chi với sự lan rộng sau đó đến chi dưới đối bên. Tương tự như BAD, LAD có thể có sự tiến triển chậm đến các vùng cột sống khác và do đó có tiên lượng tốt hơn.
Xơ cứng cột bên teo cơ gia đình
Có nhiều gen gây bệnh đang được xác định liên quan đến các kiểu hình của ALS bao gồm gen superoxide dismutase 1 (SOD1) và C9ORF72. Các đột biến SOD1 là những đột biến đầu tiên được xác định là một nguyên nhân gây bệnh ở 10% fALS và 1% ALS tản phát. Gen C9ORF72 là đột biến gây bệnh được xác định thường xuyên nhất ở 40% fALS và 5% ALS tản phát.
Hội chứng ALS-Plus
Hội chứng ALS-plus đáp ứng các tiêu chí lâm sàng và điện chẩn đoán cho ALS, nhưng cũng có các đặc điểm không phải của nơ-ron vận động, có thể bao gồm hội chứng Parkinson, sa sút trí tuệ trán-thái dương, các bất thường vận động mắt, các dấu hiệu ngoại tháp, rối loạn chức năng tự chủ và mất cảm giác.
Teo cơ tủy
SMA là một nhóm các rối loạn đa dạng về kiểu gen và kiểu hình liên quan đến các đặc điểm mất LMN. SMA gốc chi là một rối loạn LMN lặn trên nhiễm sắc thể thường với tần suất 1/11,000 ca sinh, và tần suất người mang gen là khoảng 1/50. Nó là tình trạng yếu ưu thế ở gốc chi. SMA gốc chi có thể được phân loại thành năm phân nhóm bệnh (Hình 40.3). Loại 0 là nghiêm trọng nhất và được đặc trưng bởi tình trạng yếu rất nặng, bắt đầu trước khi sinh; co rút khớp thường có mặt do giảm cử động trong tử cung. Loại 1 là dạng bệnh phổ biến nhất, chiếm 60% đến 70% bệnh nhân SMA. Nó được đặc trưng bởi khởi phát trước 6 tháng tuổi và không có khả năng tự ngồi. Khoảng 95% bệnh nhân loại 1 chết trước 2 tuổi trừ khi họ nhận được hỗ trợ thông khí và dinh dưỡng. Khởi phát ở bệnh nhân loại 2 xảy ra trong khoảng từ 6 đến 18 tháng tuổi, và khả năng tự ngồi thẳng được đạt được nhưng không thể đi lại. Bệnh nhân loại 3 có khởi phát sau 18 tháng tuổi và có thể tự đi lại. Loại 4 là dạng nhẹ nhất, với khởi phát ở tuổi trưởng thành và yếu chi gốc tương đối nhẹ.
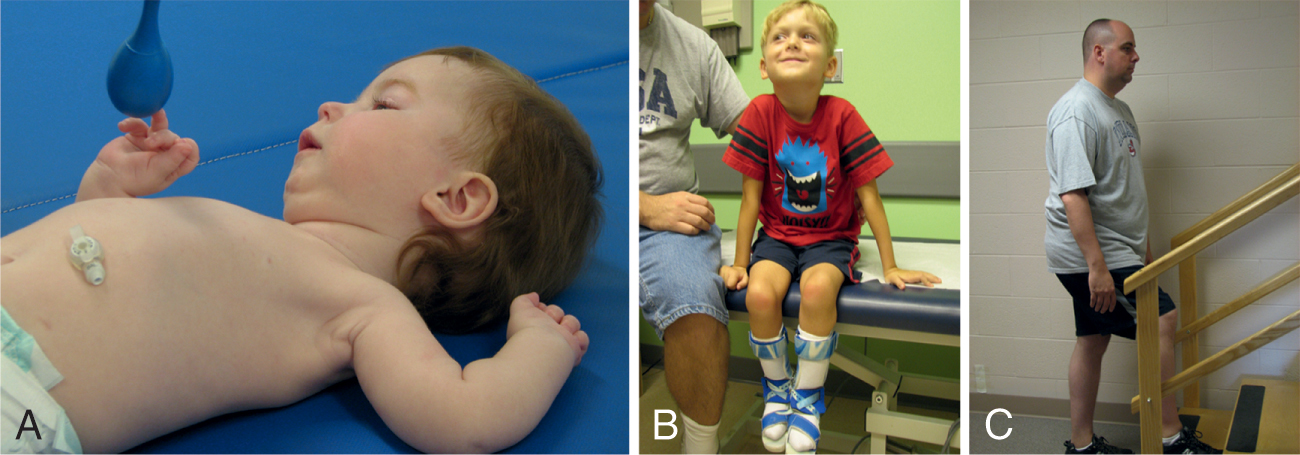
Hình 40.3 Các Phân nhóm của Teo cơ tủy (SMA) Gốc chi(A) SMA loại 1, dạng phổ biến nhất, khởi phát <6 tháng tuổi, và không bao giờ đạt được khả năng tự ngồi. (B) SMA loại 2 khởi phát trong khoảng 6 đến 18 tháng; đạt được khả năng tự ngồi, nhưng không thể đứng. SMA loại 3 có liên quan đến khởi phát >18 tháng, và bệnh nhân có thể đứng hoặc đi lại ít nhất là tạm thời. (C) SMA loại 4 là phân nhóm nhẹ nhất với khởi phát >30 tuổi. Loại 0 với khởi phát trước khi sinh không được hiển thị.
Biểu hiện lâm sàng của SMA là yếu và giảm trương lực cơ ưu thế ở gốc chi với các phản xạ vắng mặt hoặc giảm. Chẩn đoán dựa trên nghiên cứu di truyền về việc xóa đồng hợp tử của gen SMN1, được thấy ở 95% bệnh nhân SMA. Một số dạng SMA có liên quan đến một kiểu yếu ưu thế ở ngọn chi và do đó đã được mô tả là SMA ngọn chi (Hình 40.4). Một dạng hiếm hơn của SMA, SMA vai-mác hoặc hội chứng Davidenkow, có liên quan đến các đặc điểm của nơ-ron vận động và mất sợi trục trong một sự phân bố quanh xương vai và cẳng chân xa, bắt chước kiểu của các khiếm khuyết trong loạn dưỡng cơ mặt-vai-cánh tay (Hình 40.5). Không có liệu pháp hiệu quả nào cho bất kỳ dạng SMA nào, nhưng chăm sóc hỗ trợ có thể làm giảm hiệu quả tác động và gánh nặng của bệnh. Một nghiên cứu gần đây hiện cho thấy nusinersen có thể hiệu quả ở trẻ em và đã được Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ phê duyệt. Bệnh phổi là nguồn tử vong chính và liên quan đến các biến chứng chính của yếu cơ như suy giảm thông khí và quản lý chất tiết hoặc các biến chứng thứ phát như viêm phổi liên quan đến hít sặc.
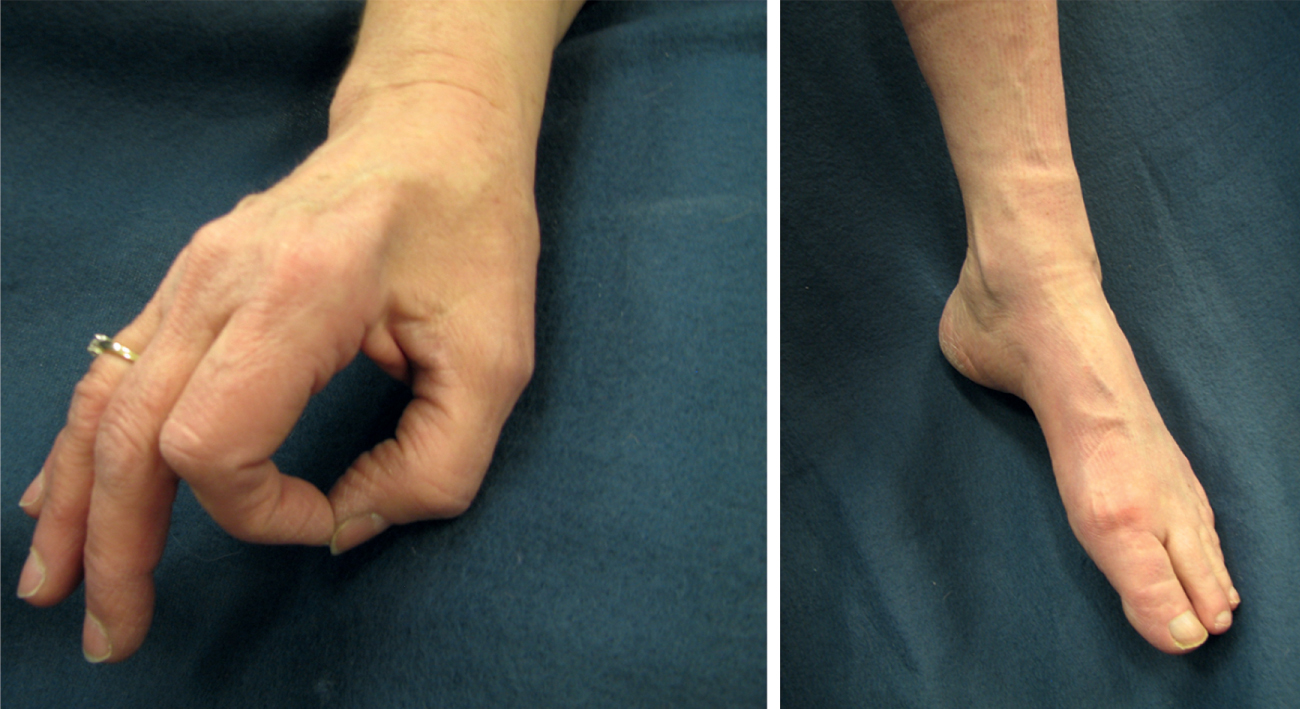
Hình 40.4. Teo cơ chi ưu thế ở ngọn chi ở một bệnh nhân bị bệnh lý thần kinh vận động di truyền ngọn chi, bắt chước bệnh Charcot-Marie-Tooth.
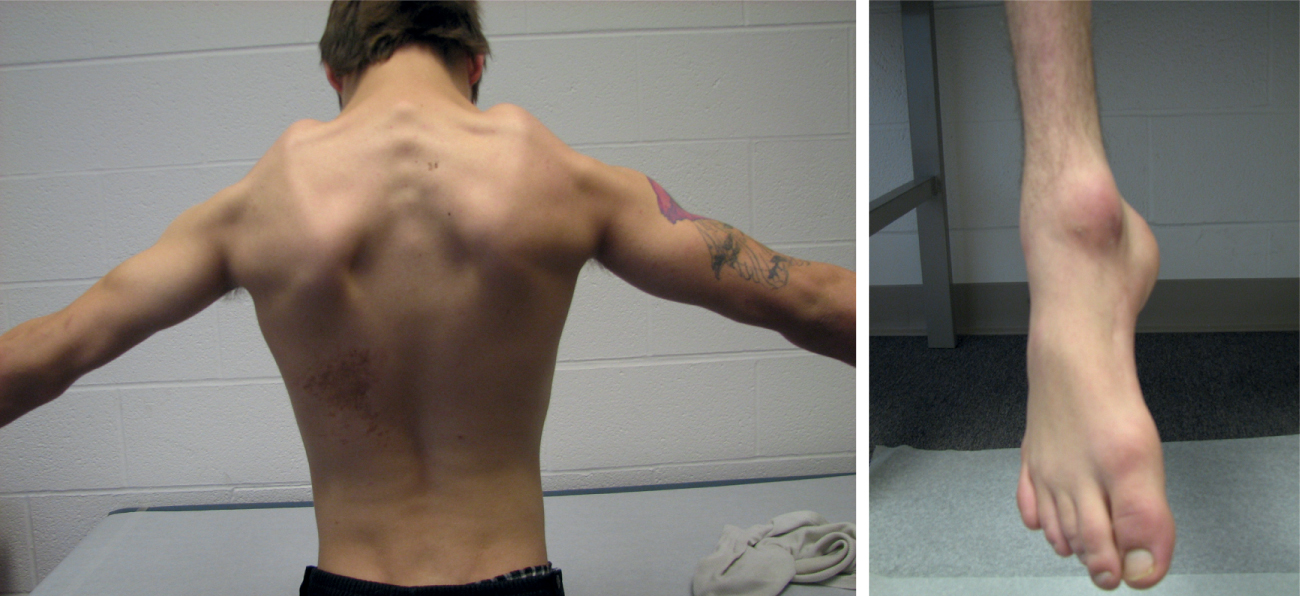
Hình 40.5. Một bệnh nhân có kiểu teo cơ quanh xương vai và cẳng chân xa bất thường liên quan đến dạng teo cơ tủy vai-mác (hội chứng Davidenkow) với sự tương đồng nổi bật với dạng thấy trong loạn dưỡng cơ mặt-vai-cánh tay.
Bệnh teo cơ tủy-hành não liên kết X (Bệnh Kennedy)
KD là một rối loạn lặn liên kết X dẫn đến yếu chi và hành não tiến triển, teo tinh hoàn, nữ hóa tuyến vú, chuột rút cơ và rung giật bó cơ. KD thường có khởi phát các triệu chứng rõ ràng hơn vào thập kỷ thứ tư hoặc thứ năm, và nó tiến triển chậm với tuổi thọ tương đối bình thường. KD có thể có rung giật bó cơ quanh miệng (Hình 40.6).
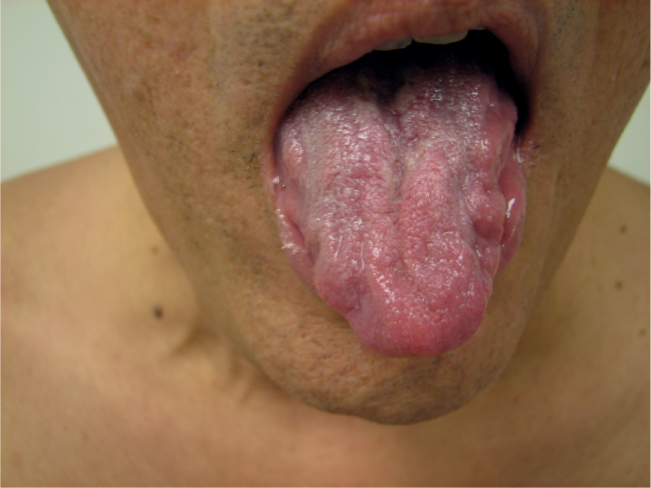
Hình 40.6. Các đặc điểm điển hình của teo thấy ở các cơ hành não và lưỡi ở một bệnh nhân mắc bệnh Kennedy.
Bệnh bại liệt và Hội chứng Sau bại liệt
Bệnh bại liệt là do poliovirus, một loại enterovirus ở người ảnh hưởng đến các tế bào sừng trước và các nhân dây thần kinh sọ. Tiêm phòng đã làm giảm đáng kể số ca mắc. Các triệu chứng là liệt mềm không đối xứng và teo sau đó ảnh hưởng đến cơ chi. Đôi khi nó có thể ảnh hưởng đến các cơ hành não. Hội chứng sau bại liệt xảy ra ở khoảng 50% bệnh nhân bại liệt, những người đã phát triển các đặc điểm muộn của sự suy giảm chức năng vận động 30 năm hoặc nhiều hơn sau lần nhiễm trùng ban đầu và có thể có khởi phát dần dần hoặc đột ngột.
Bệnh Hirayama
Bệnh Hirayama là một rối loạn tương đối lành tính liên quan đến yếu cơ và teo các cơ chi trên xa. Nó phổ biến hơn ở nam giới trong độ tuổi thiếu niên hoặc đầu những năm 20, và khởi phát yếu cơ là âm thầm và dần dần. Diễn biến tự nhiên của bệnh Hirayama có liên quan đến sự tiến triển ban đầu trong vài năm, và sự ngưng lại tự phát đã được biết là xảy ra. Sự liên quan thường nổi bật ở một chi nhưng có thể là hai bên (Hình 40.7).
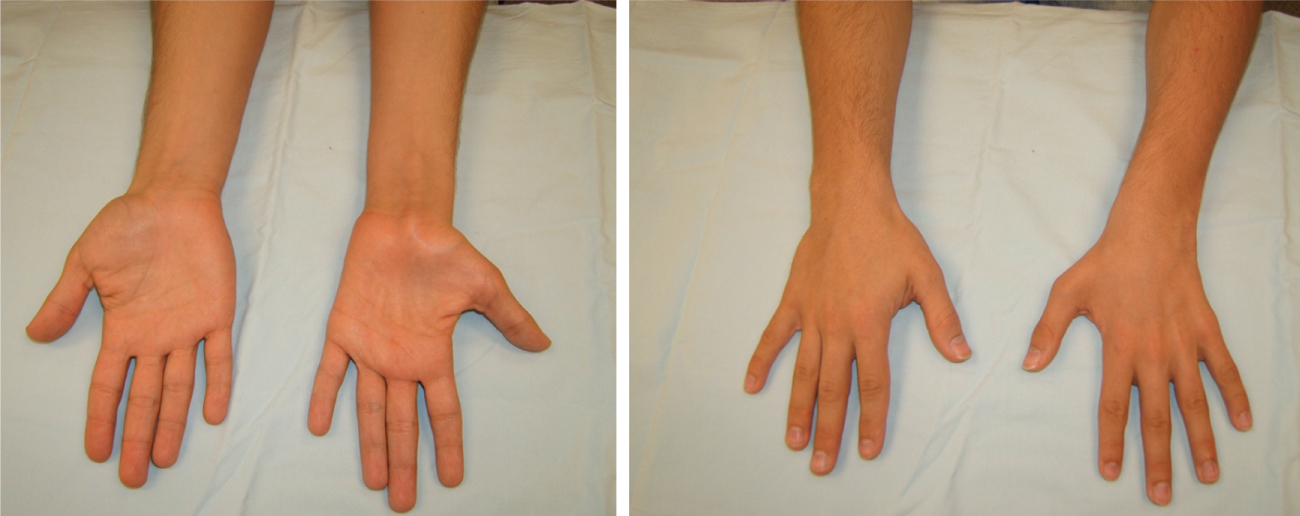
Hình 40.7. Teo cơ bàn tay và cẳng tay trái không đối xứng ở một bệnh nhân mắc bệnh Hirayama.
Các nguyên nhân hiếm gặp hoặc ít được xác định rõ của Bệnh lý Nơ-ron Vận động
Các rối loạn của nơ-ron vận động là hiếm gặp. Ngoài các dạng MND phổ biến hơn là vô số các dạng bất thường và không điển hình khác ít được xác định rõ hơn và không thể mô tả chi tiết trong chương này. Một số dạng không phổ biến đã được quy cho là do các rối loạn miễn dịch cận ung thư và tự phát, các tác nhân nhiễm trùng khác, các rối loạn chuyển hóa di truyền, chấn thương điện và các quá trình tự phát.
Điểm cần lưu ý lâm sàng
|
ĐÁNH GIÁ CHẨN ĐOÁN
Bệnh sử
Một bệnh sử kỹ lưỡng là rất quan trọng khi đánh giá một bệnh nhân có khả năng bị MND. Một bệnh sử gia đình chi tiết là cần thiết để tìm kiếm bất kỳ manh mối nào về một quá trình di truyền cơ bản, và việc khám các thành viên trong gia đình đôi khi là cần thiết để xác định các đặc điểm lâm sàng tương tự nhưng trước đây không được nhận ra. Các đặc điểm liên quan, chẳng hạn như các triệu chứng cảm giác nổi bật hoặc các phàn nàn có thể quy cho rối loạn chức năng cơ thắt hoặc tự chủ, là bất thường trong các quá trình nơ-ron vận động thuần túy và nên thúc đẩy việc xem xét một chẩn đoán thay thế. Sự liên quan đến hô hấp hoặc hành não có thể tinh tế và có thể cần một số thăm dò của nhà cung cấp dịch vụ để xác định sự liên quan; các đặc điểm như khó thở khi nằm hoặc đau đầu buổi sáng có thể gợi ý liệt cơ hoành hoặc giảm thông khí về đêm, tương ứng. Tiền sử y tế và xem xét các hệ thống có thể xác định một quá trình toàn thân có thể liên quan đến rối loạn chức năng nơ-ron vận động. Bệnh sử chức năng và xã hội sẽ cung cấp thông tin quan trọng khi kê đơn các dụng cụ chỉnh hình, thiết bị hỗ trợ và trị liệu và khi đề xuất các sửa đổi cho tình trạng sống của bệnh nhân (điều này cuối cùng sẽ cần thiết khi bệnh tiến triển). Sự hiện diện của yếu và teo khu trú không đau mà không mất cảm giác nên ngay lập tức làm dấy lên một dấu hiệu cờ đỏ, và ALS là một xem xét quan trọng. Vị trí yếu ban đầu của ALS được chia gần đúng thành ba phần: một phần ba ở vùng hành não, một phần ba ở các chi trên và một phần ba ở các chi dưới. Khó nuốt là một phàn nàn biểu hiện hiếm gặp trong ALS nhưng được dự kiến trong quá trình bệnh.
Khám thực thể
Một cuộc khám thần kinh chi tiết là bắt buộc trong việc đánh giá một bệnh nhân bị nghi ngờ mắc MND. Teo khu trú, yếu và rung giật bó cơ là những đặc điểm chính của tổn thương LMN cần được tìm kiếm. Bằng chứng về sự thoái hóa UMN ở các chi trên bao gồm co cứng, vụng về, tăng phản xạ gân cơ, và các dấu hiệu UMN. Các dấu hiệu rung giật bó cơ ở một bệnh nhân bị yếu và mất phân bố thần kinh trên EMG gợi ý MND.
Các nghiên cứu Xét nghiệm
Các nghiên cứu di truyền có thể xác nhận hiệu quả chẩn đoán các MND di truyền, và các xét nghiệm khác có thể không được bảo đảm. Sinh thiết cơ thường không cần thiết ở những bệnh nhân bị nghi ngờ mắc MND. Các nghiên cứu hình ảnh có thể không được yêu cầu trong việc đánh giá bệnh nhân bị nghi ngờ mắc MND, nhưng chúng rất hữu ích và đôi khi có tầm quan trọng sống còn để giúp loại trừ các chẩn đoán bắt chước hoặc gây nhầm lẫn với sự liên quan của UMN và LMN.
Điện chẩn đoán
Xét nghiệm điện chẩn đoán là phương thức chẩn đoán chính để xác nhận sự mất mát hoặc suy giảm chức năng của hệ thần kinh ngoại biên. Các nghiên cứu ở những bệnh nhân bị nghi ngờ mắc MND được thiết kế để xác định sự liên quan của LMN và loại trừ các rối loạn bắt chước khác của hệ thần kinh ngoại biên.
Chẩn đoán và Tiêu chí cho Xơ cứng cột bên teo cơ (Bảng 40.8 và 40.9) Các tiêu chuẩn El Escorial sửa đổi với điều chỉnh Awaji tạo thành các tiêu chí chẩn đoán được sử dụng ngày nay. Ba đặc điểm lâm sàng chính được yêu cầu để chẩn đoán ALS: bằng chứng về thoái hóa LMN, bằng chứng về thoái hóa UMN, và sự liên quan của các vùng khác nhau (hành não, cổ, ngực và thắt lưng). Bằng chứng điện chẩn đoán, bệnh lý hoặc hình ảnh thần kinh về một quá trình bệnh khác loại trừ chẩn đoán ALS. Các dấu hiệu về rối loạn chức năng UMN và LMN được nhóm thành bốn vùng: hành não, cổ, ngực và thắt lưng. Chẩn đoán ALS phải được thực hiện một cách thận trọng và thường được xác nhận bởi một bác sĩ lâm sàng có chuyên môn về MND. Phương pháp tiếp cận chung đối với bệnh nhân có thể phức tạp và cần được cá nhân hóa và kịp thời. Cần phải thiết lập một mức độ chắc chắn, và một mức độ chắc chắn đặc biệt cao rằng một chẩn đoán có thể điều trị không bị bỏ sót là rất quan trọng. Tư vấn và xét nghiệm di truyền nên được xem xét cho những bệnh nhân mới được chẩn đoán ALS ngay cả ở những người không có tiền sử gia đình đã biết.
Bảng 40.8 Tiêu chuẩn El Escorial Sửa đổi với Điều chỉnh Awaji
| Chẩn đoán xơ cứng cột bên teo cơ (ALS) đòi hỏi cả hai nguyên tắc A và B.
(A) Sự hiện diện của (1) Bằng chứng về thoái hóa nơ-ron vận động dưới (LMN) bằng khám lâm sàng, điện sinh lý, hoặc bệnh lý thần kinh, (2) Bằng chứng về thoái hóa nơ-ron vận động trên (UMN) bằng khám lâm sàng, và (3) Sự lan rộng tiến triển của các triệu chứng hoặc dấu hiệu trong một vùng hoặc đến các vùng khác, được xác định bằng bệnh sử, khám thực thể, hoặc các xét nghiệm điện sinh lý (B) Sự vắng mặt của (1) Bằng chứng điện sinh lý hoặc bệnh lý về các quá trình bệnh khác có thể giải thích các dấu hiệu thoái hóa LMN và/hoặc UMN, và (2) Bằng chứng hình ảnh thần kinh về các quá trình bệnh khác có thể giải thích các dấu hiệu lâm sàng và điện sinh lý quan sát được. |
Bảng 40.9 Tiêu chuẩn El Escorial Sửa đổi với Điều chỉnh Awaji
Chẩn đoán xơ cứng cột bên teo cơ (ALS) đòi hỏi cả hai loại chẩn đoán A và B.
|
Điểm cần lưu ý lâm sàng
|
ĐIỀU TRỊ
Tổng quát
Hiện tại, không có phương pháp điều trị nào có thể chống lại đáng kể các tác động hoặc ngăn chặn sự tiến triển của hầu hết các MND. Các chiến lược điều trị được thiết kế để giảm tác động triệu chứng của MND.
Thuốc
Không có loại thuốc nào có khả năng thay đổi bệnh chính có sẵn trong lĩnh vực MND. Riluzole là loại thuốc duy nhất được chứng minh là làm chậm sự tiến triển của ALS. Riluzole làm giảm 23% tỷ lệ tử vong sau 6 tháng và 15% sau 12 tháng và dường như kéo dài thời gian sống thêm khoảng 4 tháng. Một lựa chọn điều trị mới đã là chủ đề được bệnh nhân rất phấn khích là edaravone (Radicava), một chất quét gốc tự do dành cho bệnh nhân đột quỵ. Sau đó, nó đã được chứng minh là có một số hiệu quả trong một thử nghiệm mù đôi, có đối chứng với giả dược trên 200 bệnh nhân ALS.
Phục hồi chức năng
Các chiến lược phục hồi chức năng phải được điều chỉnh cho phù hợp với nhóm bệnh nhân mục tiêu và phụ thuộc vào tác động và diễn biến tự nhiên của bệnh. Giai đoạn 1 bao gồm những bệnh nhân có thể đi lại và hoàn toàn độc lập, với yếu cơ nhẹ và vụng về. Họ có thể được hưởng lợi từ việc tăng cường sức mạnh cơ bắp không bị ảnh hưởng và duy trì tầm vận động chủ động. Giai đoạn 2 bao gồm những bệnh nhân bị yếu cơ trung bình nhưng vẫn có thể đi lại và độc lập. Suy giảm chức năng có thể nghiêm trọng ở một số khu vực mặc dù chức năng tổng thể được bảo tồn. Kê đơn dụng cụ chỉnh hình phù hợp có thể hữu ích như nẹp đối ngón (Hình 40.10 và Hình 40.11). Giai đoạn 3 bao gồm những bệnh nhân vẫn có thể đi lại nhưng bị yếu cơ nghiêm trọng ở các nhóm cơ chọn lọc. Giai đoạn 4 bao gồm những bệnh nhân không thể đi lại nhưng vẫn độc lập. Giai đoạn 5 bệnh nhân không còn độc lập. Giai đoạn 6 bệnh nhân hoàn toàn nằm liệt giường và cần sự hỗ trợ tối đa.

Hình 40.10. Yếu cơ nội tại bàn tay nghiêm trọng ở một bệnh nhân được chẩn đoán mắc bệnh xơ cứng cột bên teo cơ, người vẫn giữ được chức năng hành não và đi lại tương đối. Bệnh nhân không thể tự cầm bút để ký vào một mẫu chấp thuận.
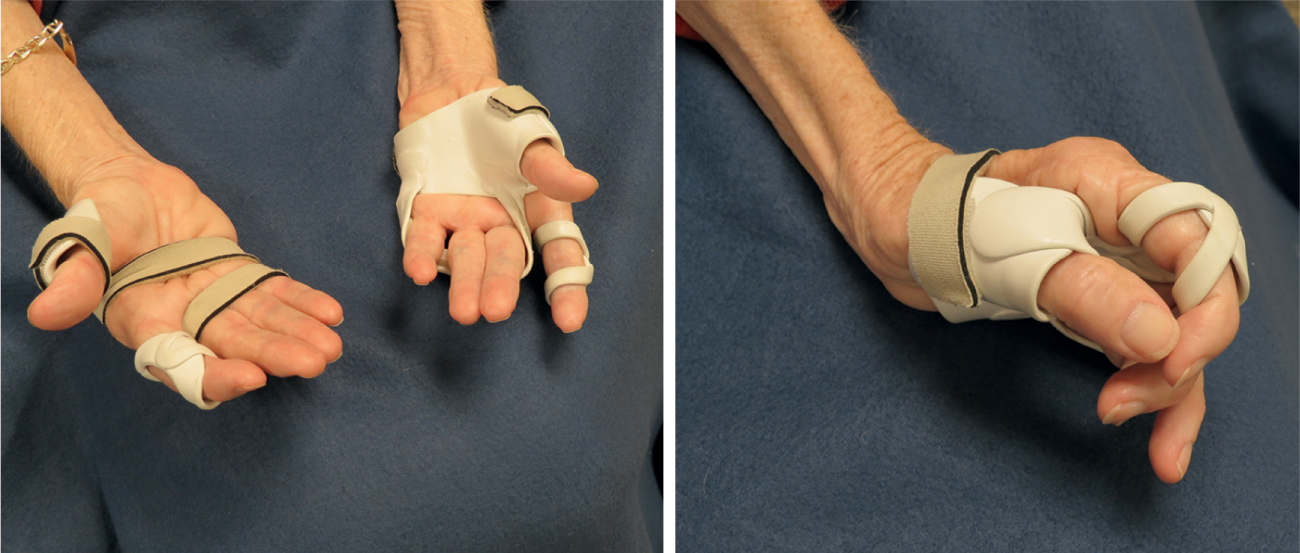
Hình 40.11. Can thiệp bằng dụng cụ chỉnh hình cho tình trạng yếu cơ nội tại bàn tay nghiêm trọng và suy giảm khả năng đối ngón cái ở một bệnh nhân mắc bệnh xơ cứng cột bên teo cơ.
Tập luyện
Tập luyện trong ALS và các bệnh thần kinh-cơ tiến triển khác là một chủ đề gây tranh cãi. Mặc dù có những lo ngại về mặt lý thuyết về việc làm nặng thêm tình trạng yếu cơ do làm việc quá sức các đơn vị vận động vốn đã đang gặp khó khăn, không có nghiên cứu kết quả có đối chứng nào cho thấy tình trạng xấu đi sau khi tập luyện ở bệnh nhân ALS. Tầm vận động và kéo giãn được coi là an toàn và nên được kê đơn cho tất cả bệnh nhân ALS. Một số hình thức tập luyện nên được thảo luận với mọi bệnh nhân ALS.
Quản lý các Tác động liên quan đến Bệnh
CO CỨNG
Điều trị co cứng trong ALS tương tự như co cứng trong các bệnh khác nhưng có một số khác biệt quan trọng. Không phải tất cả sẽ là ứng cử viên thích hợp, đặc biệt là những người có tiến triển nhanh. Độc tố botulinum có thể hữu ích nhưng có thể dẫn đến yếu cơ toàn thân.
GIAO TIẾP
Giao tiếp hiệu quả là một yếu tố chính ảnh hưởng đến chất lượng cuộc sống của bệnh nhân ALS và cần được quản lý tích cực. Các hệ thống giao tiếp tăng cường và thay thế rất hữu ích.
KHÓ NUỐT VÀ DINH DƯỠNG
Khó nuốt góp phần gây suy dinh dưỡng, có nguy cơ hít sặc, và có thể là một nguyên nhân chính gây lo âu. Dinh dưỡng được cải thiện có thể có tác động lớn đến các MND khác nhau. Các chỉ định cho ống mở dạ dày qua da bằng nội soi bao gồm sụt cân lớn hơn 10% so với ban đầu, mất nước, và các bữa ăn bị hạn chế do khó nuốt.
TĂNG TIẾT NƯỚC BỌT
Tăng tiết nước bọt có thể gây ảnh hưởng đến xã hội và khuyến khích nhiễm trùng miệng. Một máy hút tại nhà rất hữu ích cho hầu hết bệnh nhân. Điều trị bao gồm scopolamine đường uống hoặc qua da và benztropine. Độc tố botulinum loại A tiêm 3 đến 4 tháng một lần vào tuyến mang tai đã được sử dụng hiệu quả.
SUY HÔ HẤP
Cơ hô hấp có thể bị ảnh hưởng bởi sự mất nơ-ron vận động và là nguồn tử vong chính trong ALS và các MND khác. Bệnh nhân chọn mở khí quản và thông khí dài hạn thường sẽ sống lâu hơn nhiều. Các triệu chứng của suy hô hấp được ghi nhận khi biểu hiện ban đầu ở khoảng 25% các trường hợp ALS. Những triệu chứng này có thể bao gồm mệt mỏi, khó thở, khó thở khi nằm, và đau đầu buổi sáng. Suy hô hấp cận lâm sàng thường xuyên, và phần lớn sẽ có dung tích sống gắng sức (FVC) bất thường khi biểu hiện. Đánh giá hô hấp thường xuyên được khuyến nghị mỗi 2 đến 4 tháng. Thông khí không xâm lấn (NIV) có lợi ích cải thiện các phép đo chất lượng cuộc sống, bao gồm năng lượng, sức sống, khó thở, buồn ngủ, trầm cảm, chất lượng giấc ngủ, mệt mỏi thể chất, sự tập trung và chức năng nhận thức. Một đơn thuốc NIV ban đầu điển hình sẽ bao gồm FVC dưới 50% và áp lực hít vào tối đa dưới 60 cm H₂O. Liệu pháp oxy hầu như không bao giờ nên được sử dụng ở bệnh nhân ALS vì nó làm nặng thêm các triệu chứng hô hấp cũng như tăng CO2 máu và có thể dẫn đến hôn mê do tăng CO2 hoặc ngừng hô hấp. Quản lý chất tiết cũng rất quan trọng. Sự kết hợp giữa NIV và máy trợ ho (bơm-hút) đã được chứng minh là cải thiện kết quả.
CÁC RỐI LOẠN TÂM TRẠNG VÀ NHẬN THỨC
Các rối loạn tâm trạng phổ biến trong ALS. Khoảng 50% bệnh nhân ALS sẽ phát triển ít nhất là suy giảm nhận thức từ nhẹ đến trung bình, và khoảng 15% sẽ phát triển sa sút trí tuệ trán-thái dương.
ẢNH HƯỞNG GIẢ HÀNH NÃO
Ảnh hưởng giả hành não có thể ảnh hưởng đến 50% bệnh nhân có hoặc không có dấu hiệu hành não. Nó biểu hiện bằng việc khóc và cười và ngáp không ổn định, không phù hợp hoặc quá mức so với trạng thái cảm xúc của bệnh nhân.
ĐAU VÀ CHUỘT RÚT
Chuột rút phổ biến trong các dạng MND khác nhau. Nó là một nguồn khó chịu phổ biến ở bệnh nhân ALS và có thể xuất hiện sớm trong bệnh.
TIÊN LƯỢNG VÀ CHĂM SÓC CUỐI ĐỜI (BẢNG 40.12, 40.13 VÀ 40.14)
Các vấn đề cuối đời cần được giải quyết sớm trong việc quản lý mọi bệnh nhân ALS và các MND nghiêm trọng khác. Việc trì hoãn các cuộc trò chuyện cuối đời không thoải mái có thể hấp dẫn đối với cả nhà cung cấp dịch vụ và bệnh nhân. Sự thay thế cho một cuộc trò chuyện có khả năng không thoải mái là một quyết định quan trọng được đưa ra trong một cuộc khủng hoảng bởi các thành viên gia đình không nhận thức được mong muốn thực sự của bệnh nhân. Tiên lượng cho ALS là tàn khốc (Bảng 40.14).
Bảng 40.12 Các yếu tố ảnh hưởng tiêu cực đến Tiên lượng khi Chẩn đoán
|
Bảng 40.13 Các yếu tố Điều trị ảnh hưởng đến Tiên lượng
|
Bảng 40.14 Các số liệu Thống kê Quan trọng về Tiên lượng trong Xơ cứng cột bên teo cơ
|
Điểm cần lưu ý lâm sàng
|
TÀI LIỆU THAM KHẢO
- de Carvalho M, Dengler R, Eisen A, et al: Electrodiagnostic criteria for diagnosis of ALS, Clin Neurophysiol 119(3):497-503, 2008.
- Myers, Chad. (Hình 40.2)
BẢNG CHÚ GIẢI THUẬT NGỮ Y HỌC ANH – VIỆT (CHƯƠNG 40)
| STT | Thuật ngữ tiếng Anh | Phiên âm (IPA) | Nghĩa tiếng Việt |
|---|---|---|---|
| 1 | Motor Neuron Diseases (MNDs) | /ˈmoʊtər ˈnʊrɑn dɪˈzizɪz/ | Bệnh lý Nơ-ron Vận động |
| 2 | Poliomyelitis | /ˌpoʊlioʊˌmaɪəˈlaɪtɪs/ | Bệnh bại liệt |
| 3 | Amyotrophic Lateral Sclerosis (ALS) | /eɪˌmaɪəˈtroʊfɪk ˈlætərəl sklɪˈroʊsɪs/ | Xơ cứng cột bên teo cơ |
| 4 | Spinal Muscular Atrophy (SMA) | /ˈspaɪnəl ˈmʌskjələr ˈætrəfi/ | Teo cơ tủy |
| 5 | Kennedy Disease (KD) | /ˈkɛnədi dɪˈziz/ | Bệnh Kennedy |
| 6 | Lower Motor Neuron (LMN) | /ˈloʊər ˈmoʊtər ˈnʊrɑn/ | Nơ-ron vận động dưới |
| 7 | Upper Motor Neuron (UMN) | /ˈʌpər ˈmoʊtər ˈnʊrɑn/ | Nơ-ron vận động trên |
| 8 | Spasticity | /spæsˈtɪsəti/ | Co cứng |
| 9 | Dysphagia | /dɪsˈfeɪdʒə/ | Khó nuốt |
| 10 | Sialorrhea | /ˌsaɪəloʊˈriə/ | Tăng tiết nước bọt |
| 11 | Respiratory Insufficiency | /rɪˈspaɪərətɔri ˌɪnsəˈfɪʃənsi/ | Suy hô hấp |
| 12 | Pseudobulbar Affect | /ˌsudoʊˈbʌlbər əˈfɛkt/ | Ảnh hưởng giả hành não |
| 13 | Riluzole | /ˈrɪljəˌzoʊl/ | Riluzole |
| 14 | Corticobulbar Tracts | /ˌkɔrtɪkoʊˈbʌlbər trækts/ | Các bó vỏ-hành |
| 15 | Anterior Horn Cells | /ænˈtɪriər hɔrn sɛlz/ | Tế bào sừng trước tủy sống |
| 16 | Familial ALS (fALS) | /fəˈmɪliəl eɪ-ɛl-ɛs/ | ALS gia đình |
| 17 | Sporadic ALS | /spəˈrædɪk eɪ-ɛl-ɛs/ | ALS tản phát |
| 18 | Primary Lateral Sclerosis (PLS) | /ˈpraɪˌmɛri ˈlætərəl sklɪˈroʊsɪs/ | Xơ cứng cột bên nguyên phát |
| 19 | Progressive Muscular Atrophy (PMA) | /prəˈɡrɛsɪv ˈmʌskjələr ˈætrəfi/ | Teo cơ tiến triển |
| 20 | Progressive Bulbar Palsy (PBP) | /prəˈɡrɛsɪv ˈbʌlbər ˈpɔlzi/ | Liệt hành não tiến triển |
| 21 | Brachial Amyotrophic Diplegia (BAD) | /ˈbreɪkiəl ˌeɪˌmaɪəˈtroʊfɪk daɪˈpliːdʒə/ | Liệt teo cơ hai cánh tay |
| 22 | Leg Amyotrophic Diplegia (LAD) | /lɛɡ ˌeɪˌmaɪəˈtroʊfɪk daɪˈpliːdʒə/ | Liệt teo cơ hai chân |
| 23 | Transcranial Magnetic Stimulation | /trænsˈkreɪniəl mæɡˈnɛtɪk ˌstɪmjəˈleɪʃən/ | Kích thích từ xuyên sọ |
| 24 | Magnetic Resonance Spectroscopy | /mæɡˈnɛtɪk ˈrɛzənəns spɛkˈtrɑskəpi/ | Quang phổ cộng hưởng từ |
| 25 | Superoxide Dismutase 1 (SOD1) | /ˌsupərˈɑksaɪd dɪsˈmjuteɪs wʌn/ | Superoxide Dismutase 1 (SOD1) |
| 26 | C9ORF72 | /si-naɪn-oʊ-ɑr-ɛf-ˌsɛvəntiˈtu/ | C9ORF72 |
| 27 | ALS-Plus Syndrome | /eɪ-ɛl-ɛs plʌs ˈsɪnˌdroʊm/ | Hội chứng ALS-Plus |
| 28 | Parkinsonism | /ˈpɑrkɪnsəˌnɪzəm/ | Hội chứng Parkinson |
| 29 | Frontotemporal Dementia | /ˌfrʌntoʊˈtɛmpərəl dɪˈmɛnʃə/ | Sa sút trí tuệ trán-thái dương |
| 30 | Ocular Motility Abnormalities | /ˈɑkjələr moʊˈtɪləti ˌæbnɔrˈmælətiz/ | Bất thường vận động mắt |
| 31 | Autonomic Dysfunction | /ˌɔtəˈnɑmɪk dɪsˈfʌŋkʃən/ | Rối loạn chức năng tự chủ |
| 32 | Autosomal Recessive | /ˌɔtəˈsoʊməl rɪˈsɛsɪv/ | Lặn trên nhiễm sắc thể thường |
| 33 | Homozygous Deletion | /ˌhoʊmoʊˈzaɪɡəs dɪˈliʃən/ | Xóa đồng hợp tử |
| 34 | Scapuloperoneal | /ˌskæpjəloʊˌpɛrəˈniəl/ | Vai-mác |
| 35 | Davidenkow Syndrome | /dəˈvɪdənˌkɔf ˈsɪnˌdroʊm/ | Hội chứng Davidenkow |
| 36 | Fascioscapulohumeral Muscular Dystrophy | /ˌfæʃioʊˌskæpjəloʊˈhjumərəl ˈmʌskjələr ˈdɪstrəfi/ | Loạn dưỡng cơ mặt-vai-cánh tay |
| 37 | Nusinersen | /ˌn(j)uːsɪˈnɜrsɛn/ | Nusinersen |
| 38 | X-Linked Spinobulbar Muscular Atrophy | /ɛks-lɪŋkt ˌspaɪnoʊˈbʌlbər ˈmʌskjələr ˈætrəfi/ | Bệnh teo cơ tủy-hành não liên kết X |
| 39 | Testicular Atrophy | /tɛˈstɪkjələr ˈætrəfi/ | Teo tinh hoàn |
| 40 | Gynecomastia | /ˌɡaɪnɪkoʊˈmæstiə/ | Nữ hóa tuyến vú |
| 41 | Fasciculations | /fəˌsɪkjəˈleɪʃənz/ | Rung giật bó cơ |
| 42 | Perioral | /ˌpɛriˈɔrəl/ | Quanh miệng |
| 43 | Poliovirus | /ˈpoʊlioʊˌvaɪrəs/ | Poliovirus (Virus bại liệt) |
| 44 | Enterovirus | /ˌɛntəroʊˈvaɪrəs/ | Enterovirus |
| 45 | Flaccid Paralysis | /ˈflæsɪd pəˈræləsɪs/ | Liệt mềm |
| 46 | Post-Poliomyelitis Syndrome | /poʊst-ˌpoʊlioʊˌmaɪəˈlaɪtɪs ˈsɪnˌdroʊm/ | Hội chứng sau bại liệt |
| 47 | Hirayama Disease | /ˌhɪrəˈjɑmə dɪˈziz/ | Bệnh Hirayama |
| 48 | Monomelic Spinal Muscular Atrophy | /ˌmɑnoʊˈmɛlɪk ˈspaɪnəl ˈmʌskjələr ˈætrəfi/ | Teo cơ tủy một chi |
| 49 | Paraneoplastic | /ˌpærəˌnioʊˈplæstɪk/ | Cận ung thư |
| 50 | Orthopnea | /ɔrˈθɑpniə/ | Khó thở khi nằm |
| 51 | Nocturnal Hypoventilation | /nɑkˈtɜrnəl ˌhaɪpoʊˌvɛntəˈleɪʃən/ | Giảm thông khí về đêm |
| 52 | Sphincter Dysfunction | /ˈsfɪŋktər dɪsˈfʌŋkʃən/ | Rối loạn chức năng cơ thắt |
| 53 | El Escorial Criteria | /ɛl ˌɛskɔriˈæl kraɪˈtɪriə/ | Tiêu chuẩn El Escorial |
| 54 | Awaji Modification | /əˈwɑdʒi ˌmɑdɪfɪˈkeɪʃən/ | Điều chỉnh Awaji |
| 55 | Bulbar | /ˈbʌlbər/ | Hành não |
| 56 | Cervical | /ˈsɜrvɪkəl/ | Cổ |
| 57 | Thoracic | /θəˈræsɪk/ | Ngực |
| 58 | Lumbar | /ˈlʌmbɑr/ | Thắt lưng |
| 59 | Genetic Counseling | /dʒəˈnɛtɪk ˈkaʊnsəlɪŋ/ | Tư vấn di truyền |
| 60 | Edaravone (Radicava) | /ɛˈdærəˌvoʊn/ /rəˈdɪkəvə/ | Edaravone (Radicava) |
| 61 | Free Radical Scavenger | /fri ˈrædɪkəl ˈskævɪndʒər/ | Chất quét gốc tự do |
| 62 | Opponent Splint | /əˈpoʊnənt splɪnt/ | Nẹp đối ngón |
| 63 | Augmentative and Alternative Communication (AAC) | /ɔɡˈmɛntətɪv ænd ɔlˈtɜrnətɪv kəˌmjunɪˈkeɪʃən/ | Giao tiếp tăng cường và thay thế |
| 64 | Aspiration | /ˌæspəˈreɪʃən/ | Hít sặc |
| 65 | Percutaneous Endoscopic Gastrostomy (PEG) | /ˌpɜrkjuˈteɪniəs ˌɛndoʊˈskɑpɪk ɡæsˈtrɑstəmi/ | Mở dạ dày qua da bằng nội soi |
| 66 | Scopolamine | /skoʊˈpɑləˌmin/ | Scopolamine |
| 67 | Benztropine | /ˈbɛnztroʊˌpin/ | Benztropine |
| 68 | Parotid Gland | /pəˈrɑtɪd ɡlænd/ | Tuyến mang tai |
| 69 | Tracheostomy | /ˌtreɪkiˈɑstəmi/ | Mở khí quản |
| 70 | Forced Vital Capacity (FVC) | /fɔrst ˈvaɪtəl kəˈpæsəti/ | Dung tích sống gắng sức |
| 71 | Noninvasive Ventilation (NIV) | /nɑnɪnˈveɪsɪv ˌvɛntəˈleɪʃən/ | Thông khí không xâm lấn |
| 72 | Maximal Inspiratory Pressure | /ˈmæksəməl ɪnˈspaɪərətɔri ˈprɛʃər/ | Áp lực hít vào tối đa |
| 73 | Hypercapnia | /ˌhaɪpərˈkæpniə/ | Tăng CO2 máu |
| 74 | Hypercapnic Coma | /ˌhaɪpərˈkæpnɪk ˈkoʊmə/ | Hôn mê do tăng CO2 |
| 75 | Mechanical Insufflator-Exsufflator | /məˈkænɪkəl ˈɪnsəˌfleɪtər-ˈɛksəˌfleɪtər/ | Máy trợ ho (Bơm-hút) |
| 76 | End-of-life | /ɛnd-əv-laɪf/ | Cuối đời |