
Sperling Nội tiết học Nhi khoa, Ấn bản thứ 5 – Biên dịch: Ths.Bs. Lê Đình Sáng
Sperling Pediatric Endocrinology, Fifth Edition
Tác giả: Sperling, Mark A., MD – Nhà xuất bản: Elsevier Inc.
PHẦN III. NỘI TIẾT HỌC TRẺ EM VÀ THANH THIẾU NIÊN
Chương 22. Các hội chứng Đa tuyến Tự miễn
Autoimmune Polyglandular Syndromes
Bimota Nambam; Michael J. Haller; William E. Winter; Desmond Schatz
Sperling Pediatric Endocrinology, 22, 884-903
MỞ ĐẦU
Các hội chứng đa tuyến tự miễn (APS) là những tập hợp không phổ biến của các bệnh tự miễn đặc hiệu cho từng cơ quan, được đặc trưng bởi sự xuất hiện của nhiều hơn một bệnh tự miễn ở một cá nhân bị ảnh hưởng (Bảng 22.1). Mặc dù các rối loạn nội tiết tự miễn thường ảnh hưởng đến một cơ quan duy nhất, sự liên quan tự miễn đa cơ quan của cả cơ quan nội tiết và không nội tiết, thứ phát sau sự mất dung nạp tự thân, là một đặc điểm của APS.
Dung nạp là một trạng thái sinh lý trong đó hệ thống miễn dịch của một người nhận ra các kháng nguyên tự thân và không tạo ra phản ứng miễn dịch chống lại chúng. Nếu dung nạp không được thiết lập hoặc bị mất đi, bệnh tự miễn và hậu quả là bệnh tật có thể xảy ra. Mặc dù sự phá vỡ dung nạp tự thân vẫn chưa được giải thích đầy đủ, sự hiểu biết ngày càng tăng về sự tương tác phức tạp giữa di truyền và môi trường, và các quá trình miễn dịch bất thường do đó đã xác định được một số cơ chế có thể. Để hiểu được những cơ chế này, một cái nhìn tổng quan ngắn gọn về cách dung nạp tự thân được thiết lập và duy trì là cần thiết.
CÁC CƠ CHẾ LÀM NỀN TẢNG CHO DUNG NẠP
Giới thiệu
Tuyến phòng thủ miễn dịch đầu tiên của cơ thể, sau các hàng rào sinh lý chống lại các mầm bệnh ngoại lai xâm nhập, là nhánh miễn dịch bẩm sinh. Miễn dịch bẩm sinh là một phản ứng không đặc hiệu được trung gian bởi sự biểu hiện của các gen dòng mầm, không yêu cầu phơi nhiễm trước với kháng nguyên và cung cấp sự bảo vệ tức thì. Tuy nhiên, hệ miễn dịch bẩm sinh không thể hiện trí nhớ miễn dịch và không cung cấp sự bảo vệ lâu dài. Tuyến phòng thủ tiếp theo chống lại các kháng nguyên ngoại lai là hệ miễn dịch thích ứng. Mặc dù hệ miễn dịch thích ứng giả định rằng tất cả các kháng nguyên ngoại sinh đều có khả năng gây hại, nó tạo ra các phản ứng đặc hiệu với kháng nguyên. Trong một phản ứng miễn dịch thích ứng bình thường, sinh vật chủ phải phân biệt kháng nguyên tự thân với kháng nguyên không tự thân; tạo ra một phản ứng miễn dịch; loại bỏ hoặc loại trừ kháng nguyên gây bệnh; và bảo vệ vật chủ khỏi bị tổn thương, rối loạn chức năng cơ quan, và thậm chí tử vong. Sự phân biệt tự thân/không tự thân được thực hiện bởi hệ miễn dịch thích ứng (đặc hiệu) bằng một cơ chế sử dụng các thụ thể bề mặt tế bào T và B đặc hiệu. Các thụ thể tế bào T và tế bào B nhận ra các peptide và epitope kháng nguyên riêng biệt, tương ứng, và là chìa khóa cho tính đặc hiệu của phản ứng miễn dịch thích ứng. Trong khi các tế bào B và các thụ thể của chúng nhận ra các kháng nguyên hòa tan trên bề mặt tế bào của các mầm bệnh, các tế bào T và các thụ thể của chúng (TCR) chỉ nhận biết các polypeptide ngắn khi được trình diện bởi các tế bào trình diện kháng nguyên (APC), sử dụng các phân tử bề mặt tế bào chuyên biệt được mã hóa bởi phức hợp tương hợp mô chính (MHC). MHC của người được gọi là phức hợp kháng nguyên bạch cầu người (HLA). MHC lớp I (ví dụ, các phân tử HLA-A, HLA-B, và HLA-C) được tìm thấy trên tất cả các tế bào có nhân, và trình diện các peptide nội sinh có nguồn gốc từ bào tương (chẳng hạn như từ một tế bào bị nhiễm mầm bệnh) cho các tế bào T CD8. Mặt khác, các phân tử MHC lớp II (ví dụ, HLA-DP, HLA-DQ, và HLA-DR), trình diện các kháng nguyên ngoại lai (peptide) đã được nội bào hóa và xử lý bởi các APC (chẳng hạn như tế bào B, tế bào tua, và đại thực bào) cho các tế bào T CD4.
Các tế bào T ban đầu được phân loại dựa trên các protein bề mặt cụm biệt hóa (CD) của chúng, liên kết khác nhau với các kháng nguyên được trình diện trên MHC lớp I (liên kết với CD8) và lớp II (liên kết với CD4). Ví dụ, các tế bào T CD8, còn được gọi là tế bào T gây độc tế bào khi được kích hoạt, thường nhận ra các kháng nguyên được trình diện qua MHC lớp I, và trung gian một phản ứng miễn dịch đặc hiệu với kháng nguyên đó. Ngược lại, các tế bào T CD4, đóng vai trò là tế bào T hỗ trợ hoặc điều hòa, được kích hoạt khi được trình diện với các peptide qua MHC lớp II. Sự điều hòa dung nạp tự thân của tế bào T xảy ra ở hai cấp độ riêng biệt nhưng phụ thuộc lẫn nhau: dung nạp trung ương và dung nạp ngoại vi (được mô tả sau) (Hình 22.1). Dung nạp trung ương xảy ra ở tuyến ức, trong khi dung nạp ngoại vi xảy ra ở cả các mô bạch huyết và không bạch huyết. Mặc dù nhiều cơ chế liên quan đến dung nạp tự thân vẫn chưa được hiểu rõ, gần 2 thập kỷ mô tả gen điều hòa tự miễn (AIRE) đã cải thiện sự hiểu biết của chúng ta về các con đường trong việc thiết lập và duy trì dung nạp tự thân. Gen AIRE mã hóa một yếu tố phiên mã trong tủy tuyến ức, đóng một vai trò quan trọng trong việc thiết lập dung nạp trung ương (được mô tả chi tiết ở phần sau của chương này). Sự xóa đoạn ở gen tương đồng AIRE ở chuột dẫn đến bệnh tự miễn đa cơ quan, trong khi các đột biến ở gen AIRE của người dẫn đến APS tuýp 1.
Dung nạp ban đầu được phát triển trong tử cung với các tế bào T và B đóng vai trò quan trọng. Các tế bào T và B được sản xuất liên tục trong suốt cuộc đời bởi các tế bào gốc tạo máu của tủy xương, với các tiền chất tế bào T di chuyển đến tuyến ức để trưởng thành hơn nữa. Mặc dù tuyến ức teo đi sau tuổi dậy thì, mô tuyến ức còn lại có thể cung cấp cho sự phát triển của tế bào T trong suốt cuộc đời.
Mặc dù các tế bào T yêu cầu phơi nhiễm với liều lượng nhỏ kháng nguyên để đạt được dung nạp trong quá trình phát triển ở tuyến ức, nhưng cần liều lượng kháng nguyên lớn hơn để gây ra dung nạp tế bào B, và dung nạp tế bào B thường chỉ tồn tại trong thời gian ngắn. Dung nạp là đặc hiệu về mặt miễn dịch và được gây ra ở các tế bào lympho đang phát triển sớm trong đời; tuy nhiên, nó cũng có thể được gây ra ở các tế bào lympho trưởng thành khi các tín hiệu đồng kích thích vắng mặt tại thời điểm tương tác của peptide với TCR.
Dung nạp Tế bào T Trung ương và AIRE
Các tế bào T chủ yếu được “giáo dục” để phân biệt tự thân và không tự thân khi chúng phát triển ở tuyến ức. Trong vỏ tuyến ức, các tế bào T CD4+CD8+ (dương tính kép) mang TCR α/β có khả năng liên kết với các phức hợp peptide tự thân/MHC được chọn lọc để tồn tại, trong khi các tế bào T có TCR không liên kết được sẽ trải qua quá trình apoptosis. Có tới 99% các tế bào thymo đang phát triển trải qua quá trình apoptosis và không bao giờ đến được ngoại vi (xem Hình 22.1). Điều này được gọi là chọn lọc dương tính và được thực hiện bởi các tế bào biểu mô nuôi dưỡng vỏ tuyến ức trình diện kháng nguyên (CTEC), mang MHC I và MHC II. Các tế bào T liên kết với MHC I sẽ hướng tế bào T đang phát triển theo con đường tế bào T CD8, trong khi những tế bào liên kết với MHC II sẽ phát triển thành tế bào T CD4. Các tế bào được chọn lọc dương tính di chuyển qua vùng nối vỏ-tủy vào tủy, nơi diễn ra một điểm kiểm tra thứ hai. Mặc dù các tế bào T phải có khả năng liên kết với MHC/peptide tự thân để thiết lập dung nạp, nhưng các tế bào liên kết quá chặt với các kháng nguyên tự thân có khả năng gây ra phản ứng tự miễn và do đó trải qua chọn lọc âm tính và apoptosis. Quá trình chọn lọc dương tính các tế bào T để chọn lọc các tế bào T liên kết MHC (trong vỏ tuyến ức) và chọn lọc âm tính các tế bào T liên kết chặt chẽ với các kháng nguyên tự thân (trong tủy), giải thích cho dung nạp miễn dịch trung ương (tuyến ức). Các tế bào T dương tính đơn biểu hiện CD4 hoặc CD8 sau đó di chuyển ra ngoại vi và các cơ quan bạch huyết thứ cấp.
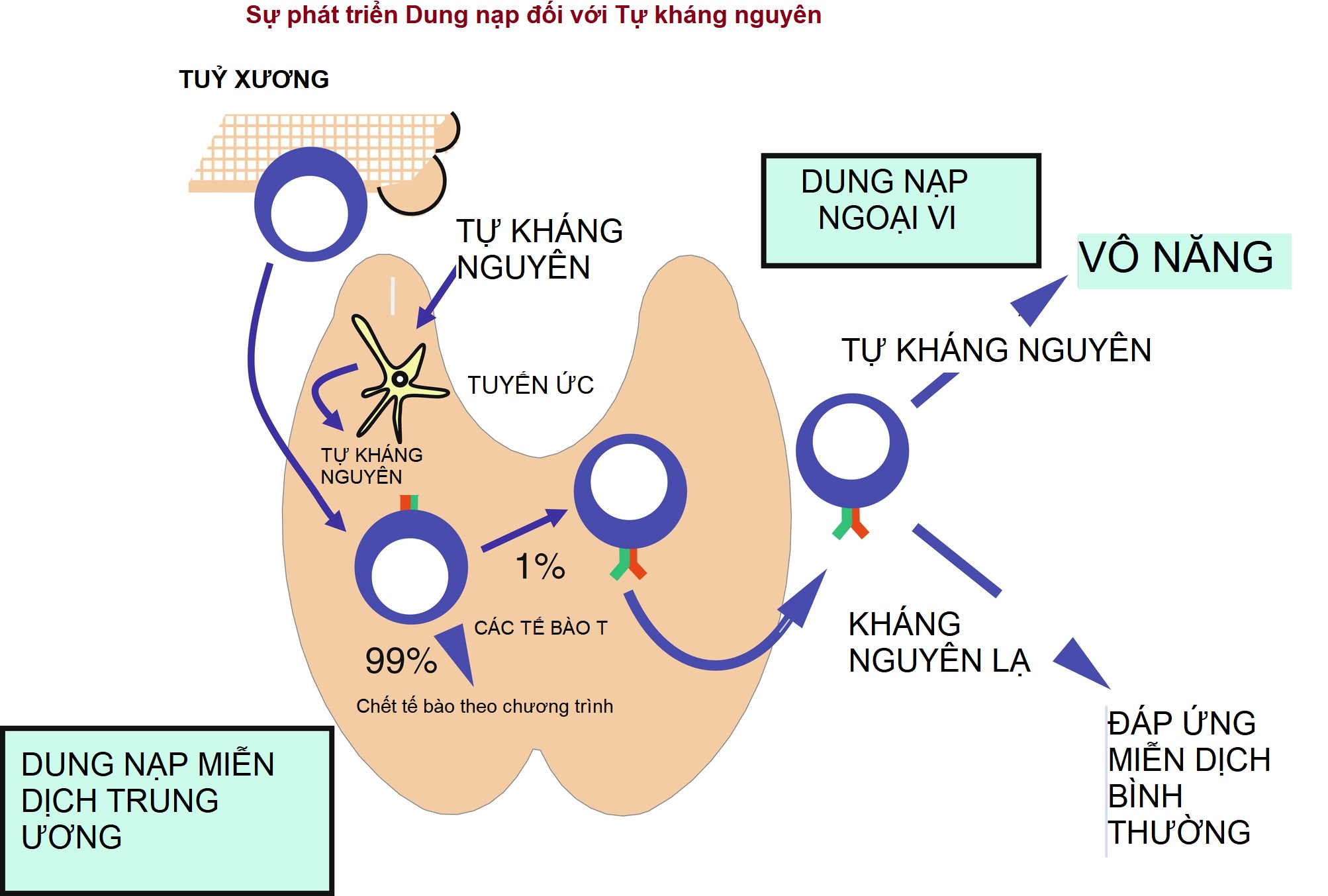
Hình 22.1: Các con đường dung nạp bình thường. Các tiền chất tế bào T ban đầu phát sinh trong tủy xương. Những tế bào tiền thân này đi vào tuyến ức, nơi các tế bào T đang phát triển gặp các kháng nguyên tự thân (SELF-AG) có nguồn gốc từ các protein lưu thông hoặc được tạo ra trong tuyến ức (ví dụ, thông qua hoạt động của các tế bào biểu mô tủy tuyến ức [mTEC]; được vẽ dưới dạng một tế bào tua màu vàng). Sự biểu hiện lạc chỗ của các protein khác nhau trong tuyến ức, bao gồm các hormone như insulin, được tạo điều kiện bởi hoạt động của gen điều hòa tự miễn (AIRE). Kích thích mạnh kháng nguyên tự thân lên các thụ thể trên các tế bào T đang phát triển gây ra apoptosis, với cuối cùng khoảng 99% tất cả các tế bào T đang phát triển chết theo cách này. Đây là “dung nạp miễn dịch trung ương” (còn được gọi là dung nạp “tuyến ức”; dưới cùng bên trái) nơi các tế bào T phản ứng mạnh chống lại tự thân bị loại bỏ. Một phần trăm tế bào T sẽ tiến triển để trở thành tế bào T CD4 hoặc tế bào T CD8. Những tế bào T nguyên bản này rời khỏi tuyến ức có thể được dung nạp với các kháng nguyên tự thân sau đó nếu chúng gặp các kháng nguyên tự thân mà không có các tín hiệu đồng kích thích bình thường (B7.1/B7.2-CD28; xem Hình 22.2). Sự gây ra dung nạp bên ngoài tuyến ức được gọi là “dung nạp ngoại vi” (trên cùng bên phải), và là một cơ chế bổ sung cho dung nạp trung ương. Dung nạp ngoại vi được biểu hiện về mặt chức năng dưới dạng vô năng: các tế bào tự phản ứng có mặt nhưng không hoạt động (trên cùng bên phải). Tuy nhiên, nếu gặp phải một kháng nguyên không tự thân, một phản ứng miễn dịch bình thường sẽ xảy ra (dưới cùng bên phải).
Các tế bào biểu mô tủy tuyến ức (mTEC) đặc biệt tham gia vào quá trình chọn lọc âm tính các tế bào T và biểu hiện một dải kháng nguyên đặc hiệu mô ngoại vi (TSA) cực kỳ đa dạng để trình diện cho các tế bào T đang phát triển. Điều này được gọi là biểu hiện gen lộn xộn (PGE) và được trung gian bởi AIRE, một yếu tố điều hòa phiên mã. Đáng chú ý, AIRE được biểu hiện trong một quần thể nhỏ các mTEC trưởng thành, với mức độ CD80 và MHC II cao. Không giống như các yếu tố điều hòa phiên mã truyền thống, nó không liên kết với các đoạn deoxyribonucleic acid (DNA) mà kích hoạt các polymerase ribonucleic acid (RNA), và kéo dài các bản phiên mã RNA của TSA. Quá trình này cũng được trung gian bởi sự tương tác của AIRE với các yếu tố điều hòa phiên mã khác. Một yếu tố điều hòa phiên mã khác có liên quan đến chọn lọc T-cell âm tính trong tuyến ức là protein ngón tay kẽm họ FEZ 2 (FEZF2), tham gia vào biểu hiện TSA độc lập với AIRE trong mTEC. Các nghiên cứu đã chỉ ra rằng AIRE và FEZF2 có chức năng bổ sung và song song trong việc thiết lập và duy trì dung nạp trung ương. Ngoài việc điều hòa biểu hiện TSA trong mTEC, AIRE còn tham gia vào việc chọn lọc và biệt hóa tế bào T CD4 tự phản ứng trong tuyến ức thành các tế bào T điều hòa (Treg), và tăng cường biểu hiện các chemokine hỗ trợ sự di chuyển của tế bào thymo. Biểu hiện ngoài tuyến ức của AIRE trong các mô, chẳng hạn như tủy xương, cũng đã được chứng minh là hỗ trợ dung nạp ngoại vi bằng cách gây ra tình trạng vô năng của các tế bào T CD4 và CD8.
Dung nạp Tế bào T Ngoại vi
Khi các tế bào T nguyên bản đi vào tuần hoàn hoặc các cơ quan bạch huyết thứ cấp (ví dụ, hạch bạch huyết và lách) và nhận ra các kháng nguyên cụ thể, chúng cần thêm các tín hiệu đồng kích thích để được kích hoạt. Tín hiệu đầu tiên liên quan đến sự tương tác của các peptide kháng nguyên liên kết với các phân tử MHC trên bề mặt APC với các TCR trên bề mặt của tế bào T CD4 và CD8. Các phân tử CD4 và CD8 trên các phân nhóm tế bào T này đóng vai trò là các đồng thụ thể không đặc hiệu kháng nguyên, liên kết với các phần không đa hình của các phân tử MHC lớp II và lớp I, tương ứng. Tín hiệu thứ hai không đặc hiệu kháng nguyên và được cung cấp bởi các phân tử B7.1 (CD80) và B7.2 (CD86) của APC tương tác với phân tử CD28 trên bề mặt tế bào T (Hình 22.2). Ngoài các tương tác CD80/CD86/CD28, còn có các tín hiệu đồng kích thích khác có vai trò quan trọng trong sự phát triển của tế bào T.
Khi các tế bào T nhận được cả hai tín hiệu (MHC-kháng nguyên với TCR và B7-CD28), một chuỗi các sự kiện truyền tín hiệu nội bào xảy ra, dẫn đến sự kích hoạt tế bào T. Các tế bào T CD8 gây độc tế bào được kích hoạt sẽ trung gian ly giải trực tiếp các tế bào đích, trong khi các tế bào T CD4 được kích hoạt dẫn đến sự biểu hiện của nhiều cytokine, thụ thể cytokine, và kháng nguyên 4 của tế bào T lympho gây độc tế bào (CTLA-4). CTLA-4 tương đồng với CD28 và cạnh tranh với CD28 để liên kết với B7.1/B7.2. Sự biểu hiện CTLA-4 bởi tế bào T được kích hoạt và sự tương tác của nó với B7.1/B7.2 cung cấp một tín hiệu ức chế miễn dịch/điều hòa miễn dịch cho tế bào T, do đó làm giảm các phản ứng của tế bào T. Do đó, CTLA-4 và CD28 mặc dù tương đồng, nhưng hoạt động đối nghịch: B7.1/B7.2-CD28 bật tế bào T, trong khi B7.1/B7.2-CTLA-4 làm giảm hoạt động của tế bào T (xem Hình 22.2).
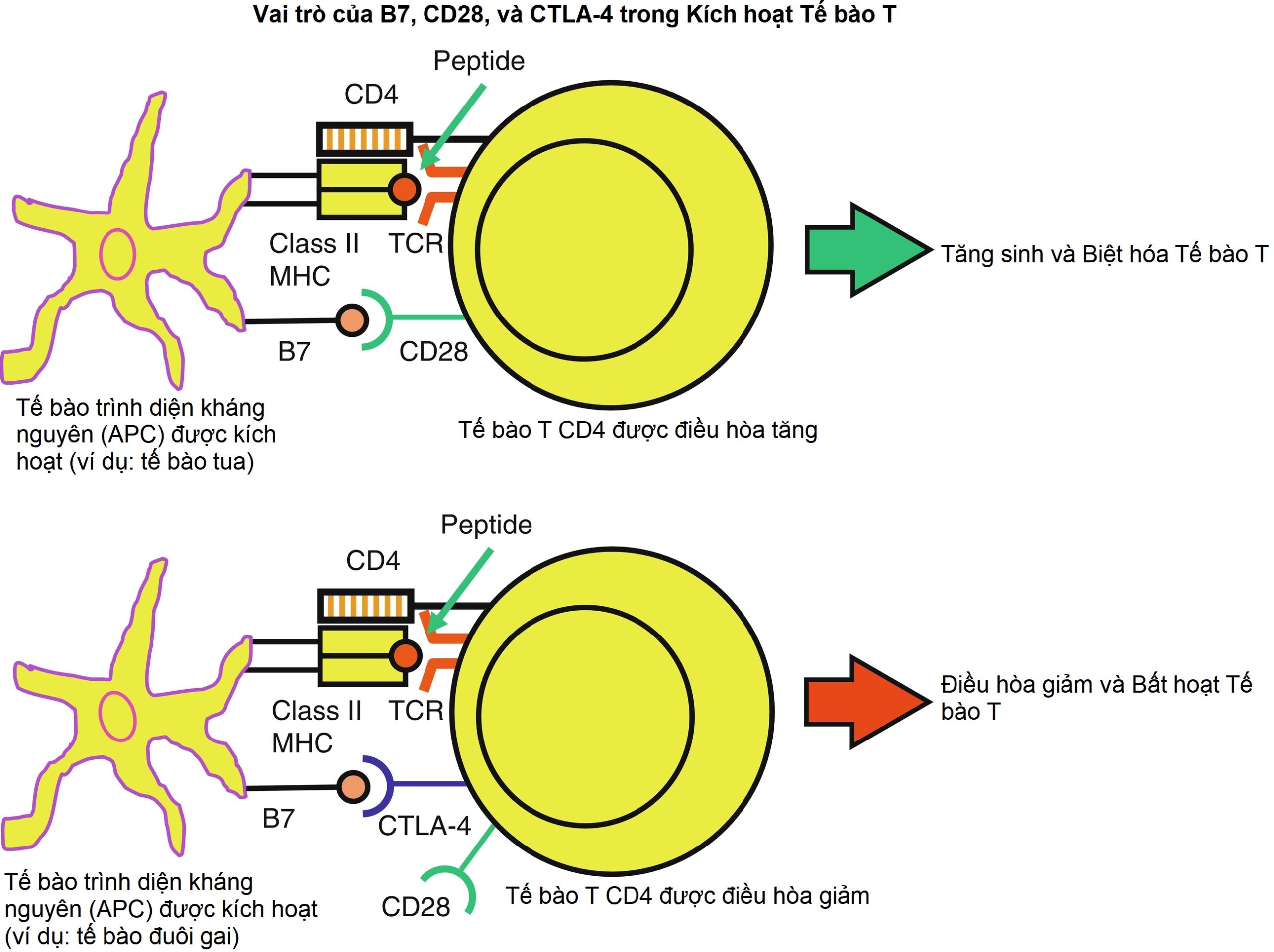
Hình 22.2: Vai trò của B7, CD28 và CTLA-4 trong Kích hoạt Tế bào T. Các tế bào trình diện kháng nguyên (APC) được kích hoạt sẽ trình diện các peptide kháng nguyên thông qua các phân tử phức hợp tương hợp mô chính (MHC) (trong hình vẽ này là MHC lớp II) và biểu hiện các đồng kích thích B7 [bao gồm B7.1 (CD80) và B7.2 (CD86)]. Khi B7.1 hoặc B7.2 được liên kết bởi CD28, và MHC cộng với peptide được liên kết bởi thụ thể tế bào T, sự tăng sinh và biệt hóa của các tế bào T nguyên bản sẽ xảy ra (hình trên). Ngược lại, các tế bào T được kích hoạt sẽ biểu hiện CTLA-4 và liên kết với B7 (hoặc B7.1 hoặc B7.2) gây ra sự điều hòa giảm và bất hoạt các tế bào T (hình dưới).
Yêu cầu về hai tín hiệu để kích hoạt các tế bào T nguyên bản chủ yếu giải thích cho dung nạp tế bào T ngoại vi. Khi tế bào T nguyên bản nhận biết peptide kháng nguyên được trình diện bởi các phân tử MHC mà không có tín hiệu đồng kích thích cần thiết (ví dụ, B7.1/B7.2-CD28), tế bào T trở nên không đáp ứng. Trạng thái không đáp ứng này được gọi là vô năng; các tế bào T vô năng thường không được tái kích thích bằng peptide kháng nguyên được hiển thị bởi các APC. Các tế bào T cũng có thể trải qua quá trình apoptosis (chết tế bào theo chương trình) để bị loại bỏ hoàn toàn khỏi vốn tế bào T. Dung nạp cũng có thể tồn tại vì TCR không gặp peptide liên quan, và điều này được gọi là sự phớt lờ của tế bào T.
Một cơ chế khác mà qua đó dung nạp tế bào T được trung gian là sự tương tác của thụ thể tử vong theo chương trình 1 (PD-1) trên các tế bào T và các phối tử của nó là PD-L1 và PD-L2. Những tương tác này gây ra sự ức chế các chức năng hiệu ứng của tế bào T một cách đặc hiệu kháng nguyên. Tín hiệu PD-1 cũng có thể trung gian sự chuyển đổi của các tế bào T nguyên bản thành các tế bào Treg.
Các tế bào T CD4 hỗ trợ được phân loại cổ điển thành hai dòng riêng biệt: (1) các tế bào Th1, kích hoạt các phản ứng qua trung gian tế bào và một số phản ứng kháng thể, và (2) các tế bào Th2, chủ yếu kích hoạt các phản ứng qua trung gian kháng thể. Tuy nhiên, các dòng tế bào T bổ sung tồn tại (tức là các tế bào Th17, các tế bào T hỗ trợ nang, và các tế bào T điều hòa) và dữ liệu gần đây đã mô tả sự linh hoạt đáng kể trong biểu hiện cytokine của chúng, cho thấy sự thay đổi chức năng của tế bào T, tùy thuộc vào các tín hiệu môi trường. Mặc dù quá đơn giản, các phân nhóm Th1 chủ yếu tiết ra các cytokine gây viêm, chẳng hạn như interleukin (IL)-2, interferon-gamma (IFN-γ), yếu tố hoại tử khối u-beta (TNF-β), gây ra sự bài tiết IL-12 từ các tế bào tua, và kích hoạt các đại thực bào và tế bào T CD8 để loại bỏ các mầm bệnh nội bào. Khi được kích hoạt, các tế bào T CD8, thường với sự giúp đỡ của các tế bào Th1 cung cấp IFN-γ để tăng cường biểu hiện B7 trên các APC, trở thành các tế bào T tiêu diệt gây độc tế bào chức năng. Ngược lại, các tế bào Th2 tạo ra IL-4, IL-5, IL-6, IL-10, và IL-13, hỗ trợ sản xuất kháng thể và bạch cầu ái toan. Cũng có sự tương tác chéo giữa các tế bào Th1 và Th2; ví dụ, IFN-γ từ các tế bào Th1 ức chế các tế bào Th2, và IL-10 từ các tế bào Th2 ức chế các tế bào Th1.
Các tế bào Treg là một phân nhóm của các tế bào T, đóng một vai trò quan trọng trong việc ức chế hoạt động của các tế bào T hiệu ứng thoát khỏi chọn lọc âm tính đối với các kháng nguyên tự thân trong tuyến ức. Các tế bào Treg chức năng có khả năng làm vô năng các tế bào T hiệu ứng tự phản ứng trước đây, dẫn đến dung nạp tự thân được cải thiện. Các tế bào Treg có nguồn gốc từ tuyến ức được gọi là các tế bào Treg trung ương hoặc tự nhiên và mang các dấu ấn bề mặt CD4+CD25+ và yếu tố phiên mã forkhead nội bào (FOXP3+), đặc hiệu cho quần thể tế bào Treg CD4+CD25+. FOXP3 là một yếu tố phiên mã cần thiết cho các tế bào T dương tính α/β TCR đang phát triển để biệt hóa thành các tế bào Treg trong tuyến ức. Lần đầu tiên được xác định ở chuột Scurfy, một mô hình chuột về rối loạn chức năng miễn dịch và bệnh đa nội tiết, sự biểu hiện FOXP3 bất thường hiện được biết là nguyên nhân gây ra sự thất bại trong dung nạp miễn dịch ở người bị ảnh hưởng bởi một bệnh đa nội tiết tương tự, như được thảo luận thêm sau đây. Sự biểu hiện FOXP3 bất thường ở người dẫn đến một bệnh tăng sinh lympho tự miễn cực kỳ hiếm gặp, di truyền lặn liên kết với nhiễm sắc thể X, và thường gây tử vong được gọi là IPEX (Rối loạn điều hòa miễn dịch, Bệnh đa nội tiết, Bệnh ruột, và Di truyền liên kết với nhiễm sắc thể X). Các khiếm khuyết trong FOXP3 gây ra IPEX được xác định trên Xp11.23-Xq13.3.
Dung nạp Tế bào B
Các tế bào B nguyên bản, trong giai đoạn phát triển sớm ở tủy xương, biểu hiện immunoglobulin (Ig)M bề mặt, đóng vai trò là các thụ thể tế bào B (BCR). Khi tương tác với các kháng nguyên tự thân, các tế bào B chưa trưởng thành nguyên bản trải qua chọn lọc âm tính, hoặc thông qua sự xóa sổ dòng hoặc tình trạng vô năng, theo đó các tế bào B đi vào trạng thái không đáp ứng và có tuổi thọ giảm. Một quá trình khác trung gian dung nạp trung ương tế bào B là chỉnh sửa thụ thể, theo đó sự tái sắp xếp di truyền của chuỗi Ig dẫn đến việc tạo ra các BCR có tính đặc hiệu kháng nguyên mới. Các tế bào B có BCR không tự phản ứng được chọn lọc dương tính và tiếp tục đi ra ngoại vi. Nếu các tế bào B tự phản ứng thoát ra ngoại vi, chúng sẽ trải qua tình trạng vô năng. Các tế bào B vô năng không chết ngay lập tức nhưng có thời gian bán thải ngắn hơn. Các tế bào B trưởng thành nguyên bản cần sự giúp đỡ của tế bào T để phát huy hết tiềm năng của chúng thông qua sự trưởng thành ái lực và chuyển lớp. Sự vắng mặt của sự giúp đỡ từ tế bào T cũng dẫn đến dung nạp tế bào B.
Các Bệnh Tự miễn
Bản chất đặc hiệu cơ quan của nhiều bệnh tự miễn là kết quả của sự nhận dạng bất thường của hệ thống miễn dịch đối với các kháng nguyên tự thân đặc hiệu mô. Trong nhiều bệnh nội tiết tự miễn, phân tử mục tiêu là một enzyme đặc hiệu mô hoặc giới hạn mô (tức là, protein không phải là duy nhất cho một mô nhưng rõ ràng bị hạn chế về sự phân bố của nó) hoặc một thụ thể bề mặt tế bào (Bảng 22.2).
Các tiêu chí để phân loại một bệnh là tự miễn không được thống nhất trên toàn cầu. Tuy nhiên, các tiêu chí chính thường được chấp nhận là bằng chứng mạnh mẽ của bệnh tự miễn bao gồm: (1) phát hiện các tự kháng thể hoặc các tế bào T tự phản ứng, bao gồm cả sự thâm nhiễm của tế bào lympho vào mô hoặc cơ quan bị nhắm mục tiêu; (2) sự truyền bệnh bằng kháng thể hoặc tế bào T; (3) sự tái phát bệnh trong mô được cấy ghép; và (4) khả năng loại bỏ quá trình bệnh bằng ức chế miễn dịch hoặc điều hòa miễn dịch. Rất ít, nếu có, các bệnh tự miễn ở người đáp ứng tất cả các tiêu chí này. Thông tin bổ sung hỗ trợ, nhưng không phải là chẩn đoán cho một bệnh tự miễn, bao gồm: (1) tần suất bệnh tăng ở phụ nữ so với nam giới, (2) sự hiện diện của các bệnh tự miễn đặc hiệu cơ quan khác ở những người bị ảnh hưởng, và (3) tần suất tăng của các alen HLA cụ thể ở những người bị ảnh hưởng.
Khiếm khuyết trong Dung nạp Gây ra Bệnh Tự miễn
Một số giả thuyết khác nhau giải thích các khiếm khuyết trong dung nạp đã được đề xuất. Bệnh tự miễn có thể phát triển vì (1) dung nạp chưa bao giờ phát triển đối với các kháng nguyên tự thân cụ thể hoặc (2) dung nạp đã được thiết lập đã bị mất (Hình 22.3). Nếu kháng nguyên tự thân không được trình diện hiệu quả trong tuyến ức, dung nạp có thể không được thiết lập trong quá trình giáo dục tế bào T trong vỏ tuyến ức. Ví dụ, các đột biến AIRE dẫn đến việc thiếu biểu hiện các kháng nguyên tự thân lạc chỗ bởi các mTEC và sự trình diện của chúng cho các tế bào T đang phát triển. Điều này dẫn đến sự thoát ra của các tế bào T tự phản ứng ra ngoại vi và cuối cùng là bệnh tự miễn đa cơ quan. Một ví dụ khác là VNTR của gen insulin (INS) (số lượng lặp lại song song thay đổi), nằm khoảng 500 cặp base ngược dòng của promoter gen INS. VNTR INS ảnh hưởng đến sự biểu hiện và giáo dục tế bào T insulin trong tuyến ức dựa trên độ dài của nó. Cụ thể, các VNTR dài hơn có liên quan đến sự biểu hiện insulin trong tuyến ức tăng lên, và do đó giảm nguy cơ phát triển đái tháo đường tuýp 1, trong khi các VNTR ngắn hơn có liên quan đến sự biểu hiện insulin trong tuyến ức giảm, không thể xóa các dòng tế bào T tự phản ứng cụ thể, và tăng nguy cơ phát triển bệnh đái tháo đường.
Nếu dung nạp chưa phát triển do sự cô lập nội bào của một kháng nguyên, và do đó không được biểu hiện trong tuyến ức trong quá trình phát sinh tế bào T, phản ứng của tế bào T ở ngoại vi sẽ không bị loại bỏ. Tuy nhiên, một số kháng nguyên ban đầu được cho là bị cô lập nội bào hiện đã được chứng minh là lưu thông ở nồng độ thấp ở những người bình thường. Thyroglobulin, một kháng nguyên tự thân trong bệnh tuyến giáp tự miễn, được biết là lưu thông với số lượng thấp nhưng đáng kể ở những người không có bằng chứng huyết thanh học về tự miễn dịch tuyến giáp. Sự phá hủy tế bào nang tuyến giáp trong viêm tuyến giáp Hashimoto chủ yếu qua trung gian tế bào và không qua trung gian các yếu tố thể dịch.
Nếu các kháng nguyên bị cô lập đóng một vai trò trong bệnh tự miễn, nhiễm virus, chấn thương, thiếu máu cục bộ, hoặc chiếu xạ đều là những cơ chế có thể làm xáo trộn tính toàn vẹn của tế bào và dẫn đến việc giải phóng các kháng nguyên nội bào. Một số kháng nguyên tự thân bị cô lập có thể không bao giờ gặp hệ thống miễn dịch, trừ khi có sự phá vỡ các hàng rào giải phẫu trong cơ thể. Một ví dụ là sự xuất hiện của tự miễn dịch đối với các protein nội nhãn, sau chấn thương ổ mắt. Mặc dù là một hậu quả hiếm gặp của tổn thương ổ mắt, việc khởi phát một phản ứng tự miễn đối với các protein nội ổ mắt bị cô lập được giải phóng trong các hạch bạch huyết lân cận có thể tạo ra các tế bào T tự phản ứng có thể xâm lấn và làm hỏng mắt đối diện (viêm mắt giao cảm). Việc loại bỏ các mô bị tổn thương gây bệnh và ức chế miễn dịch có thể được yêu cầu để duy trì thị lực ở mắt không bị tổn thương. Tương tự, phản ứng tự kháng thể thoáng qua đối với myosin tim sau nhồi máu cơ tim đã được mô tả mặc dù không có hậu quả bệnh lý nào.
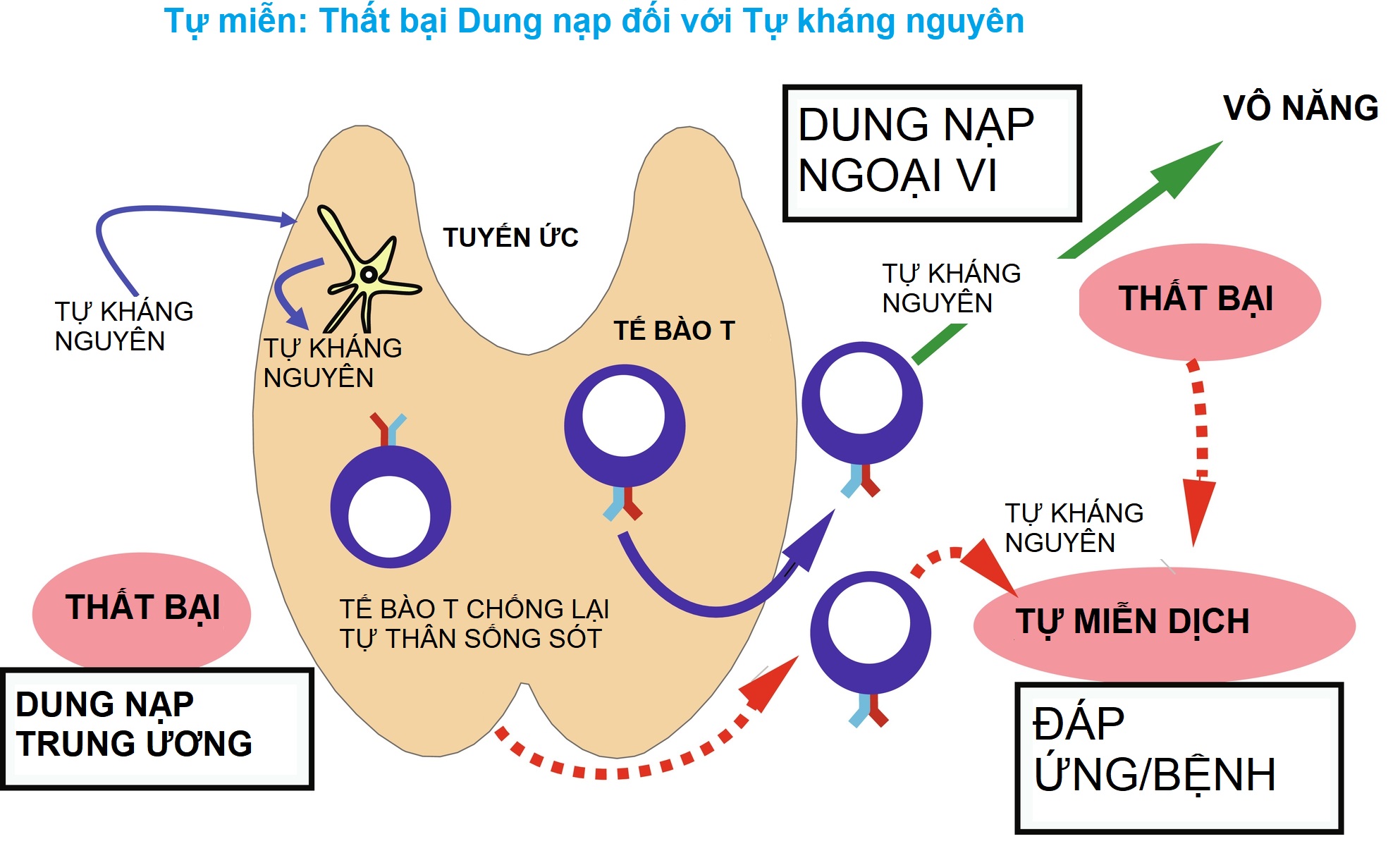
Hình 22.3: Bệnh tự miễn: Thất bại Dung nạp Kháng nguyên Tự thân. Với sự thất bại của dung nạp trung ương (dưới cùng bên trái), các tế bào T chống lại tự thân tồn tại mà bình thường không nên tồn tại. Khi các tế bào T chống lại tự thân như vậy rời khỏi tuyến ức, chúng có thể tạo ra bệnh tự miễn (mũi tên đỏ). Ngoài ra, với sự thất bại của dung nạp ngoại vi (trên cùng bên phải), nếu tình trạng vô năng (mũi tên xanh) không xảy ra sau khi tiếp xúc với kháng nguyên tự thân, một phản ứng hoặc bệnh tự miễn có thể xảy ra (mũi tên đỏ).
Sự thay đổi của các kháng nguyên tự thân do nhiễm trùng hoặc ung thư là một giả thuyết khả thi khác giải thích một số loại bệnh tự miễn. Là các tác nhân kích hoạt từ môi trường, nhiễm virus có thể dẫn đến sự biến đổi của các protein tự thân và biểu hiện tân kháng nguyên. Hoặc, một kháng nguyên tự thân có thể bị phân hủy một phần, dẫn đến một mục tiêu kháng nguyên “mới” cho hệ miễn dịch thích ứng. Kháng nguyên mới này được hệ thống miễn dịch nhận ra là ngoại lai, và phản ứng miễn dịch đối với những kháng nguyên mới này dẫn đến bệnh tự miễn. Một số tế bào/mô có thể bị tổn thương tự miễn không chủ ý khi các chất liên kết với các tế bào và gây ra một phản ứng miễn dịch ban đầu. Ví dụ, một số loại thuốc liên kết với các tế bào hồng cầu và dẫn đến thiếu máu tan máu miễn dịch. Nếu một phản ứng kháng thể đối với thuốc liên kết với hồng cầu được gây ra, phức hợp kháng nguyên-kháng thể có trên tế bào hồng cầu có thể dẫn đến sự phá hủy tế bào hồng cầu. Điều này có thể xảy ra thông qua quá trình thực bào tế bào hồng cầu bởi hệ thống đơn bào-đại thực bào hoặc thông qua sự ly giải qua trung gian bổ thể của tế bào hồng cầu. Do đó, tế bào hồng cầu trở thành một người ngoài cuộc vô tội đối với phản ứng miễn dịch thể dịch chống thuốc. Về mặt lý thuyết, điều này cũng có thể xảy ra với các virus tình cờ bám vào các mô.
Bắt chước phân tử là một cơ chế khác để giải thích sự phát triển của bệnh tự miễn. Sau khi tiếp xúc với một kháng nguyên từ chế độ ăn, virus, hoặc vi khuẩn (ví dụ, nhiễm trùng) và sự tương đồng (bắt chước phân tử) giữa kháng nguyên tự thân và kháng nguyên ngoại lai, phản ứng miễn dịch đối với kháng nguyên ngoại lai dẫn đến phản ứng chéo với kháng nguyên tự thân, tự miễn dịch, và bệnh tật. Để giả thuyết này hoạt động, dung nạp phải không tồn tại trước đó đối với kháng nguyên tự thân. Điều này có thể đúng nếu kháng nguyên tự thân thực sự bị cô lập và hệ thống miễn dịch chưa bao giờ phát triển dung nạp đối với kháng nguyên tự thân. Hoặc, các peptide kháng nguyên tự thân có thể có mặt ở nồng độ rất thấp để gây ra một phản ứng miễn dịch và sự dung nạp ban đầu chưa xảy ra. Chỉ sau khi nhiễm trùng hoặc phơi nhiễm với chế độ ăn mới sẽ có một mức độ miễn dịch hóa đủ đối với kháng nguyên ngoại sinh (tương tự như một kháng nguyên tự thân) và sau đó là phản ứng tự miễn. Nếu kháng nguyên tự thân là một kháng nguyên bề mặt tế bào, các tự kháng thể “do mầm bệnh gây ra” có thể gắn vào tự thân và gây bệnh thông qua sự cố định bổ thể, hoặc các kháng thể có thể hoạt động như các opsonin cho các tế bào thực bào cố định hoặc lưu thông (độc tế bào phụ thuộc kháng thể). Trong bệnh thấp tim, các bệnh đồng mắc như viêm tim và múa giật Sydenham được cho là các biểu hiện tự miễn thứ phát do sự tương đồng trong các thành phần cấu trúc của liên cầu khuẩn nhóm A với collagen (I và IV) và fibronectin trong các mô liên kết tim của người và tubulin trong các tế bào não người, tương ứng.
Một số trường hợp tự miễn dịch có thể là kết quả của các siêu kháng nguyên, có thể được tiết ra bởi một số vi khuẩn và virus gây bệnh. Siêu kháng nguyên là các chất kích thích tế bào T đa dòng có thể liên kết chéo các chuỗi β của TCR và các phân tử MHC, và kích hoạt tới một phần ba các tế bào T trong cơ thể. Điều này có thể khởi đầu một phản ứng miễn dịch tế bào T không đặc hiệu, bao gồm cả chống lại các kháng nguyên tự thân. Trong những trường hợp như vậy, bệnh hệ thống có thể phát triển từ việc giải phóng cytokine hàng loạt (ví dụ, hội chứng đáp ứng viêm hệ thống [SIRS]). Đây là trường hợp trong hội chứng sốc nhiễm độc, trong đó một ngoại độc tố tụ cầu hoạt động như một siêu kháng nguyên. Các kháng nguyên Mycobacterium cũng đã được đề xuất là các siêu kháng nguyên có thể có trong bệnh Crohn. Giả thuyết này yêu cầu rằng các tế bào T mang TCR chống lại tự thân chưa bị xóa sổ hoặc trở nên vô năng vĩnh viễn. Những tế bào T này có thể được kích thích và tăng sinh để phát triển một phản ứng tự miễn nếu chúng gặp phải kháng nguyên tự thân cụ thể.
Tương tự như kích hoạt tế bào T đa dòng, kích thích tế bào B đa dòng cũng đã được liên quan đến tự miễn dịch thể dịch. Các tế bào B tự phản ứng phát sinh thường xuyên như một phần của vốn tế bào B nguyên bản, và có thể được tìm thấy ở những người khỏe mạnh. Nếu một dòng tế bào B tự phản ứng gặp một kháng nguyên tự thân và một đồng kích thích (có thể không đặc hiệu, ví dụ, một virus, chẳng hạn như virus Epstein-Barr, hoặc một sản phẩm vi khuẩn, chẳng hạn như lipopolysaccharide), các tự kháng thể có thể được sản xuất, bỏ qua nhu cầu về sự giúp đỡ của tế bào T.
Bệnh tự miễn ở người có thể là kết quả của sự tương tác giữa các yếu tố môi trường và di truyền. Các yếu tố môi trường có liên quan bao gồm: ăn gliadin lúa mì và bệnh celiac, tiếp xúc với penicillamine và bệnh nhược cơ, methimazole và hạ đường huyết tự miễn do tự kháng thể insulin (được báo cáo chủ yếu ở bệnh nhân Nhật Bản), và amiodarone và viêm tuyến giáp. Ung thư cũng có liên quan đến sự phát triển của tự miễn dịch: u tuyến ức và bệnh nhược cơ, u quái buồng trứng và viêm não qua trung gian thụ thể N-methyl-D-aspartate, và ung thư vú và hội chứng người cứng. Mặc dù có những cải thiện đáng kể trong sự hiểu biết của chúng ta về miễn dịch học, các cơ chế mà qua đó sự tương tác phức tạp của gen, môi trường, và hệ thống miễn dịch dẫn đến tự miễn dịch vẫn cần được làm sáng tỏ hoàn toàn.
Các chất ức chế điểm kiểm soát và Tự miễn dịch
Các tế bào khối u có thể điều khiển cơ chế dung nạp miễn dịch vốn có để phá vỡ khả năng miễn dịch chống khối u. Một cách hiệu quả để thoát khỏi hoạt động chống khối u là bằng cách tăng các con đường điểm kiểm soát, ức chế các phản ứng của tế bào T. Một ví dụ là sự tương tác của CTLA-4 (có trên các tế bào T được kích hoạt) với B7.1/B7.2 của các APC làm giảm phản ứng của tế bào T. CILA-4 cũng có thể loại bỏ các phân tử B7 khỏi các APC, thông qua một quá trình được gọi là nội bào chuyển tiếp và ngăn chặn sự liên kết của các phân tử đồng kích thích CD28, và do đó gây ra tình trạng vô năng của tế bào T. Một ví dụ khác là sự tương tác của PD-1 trên các tế bào T với các phối tử của nó là PD-L1 và PD-L2. Các tương tác PD-1/PD-L1/PD-L2 ức chế sự tăng sinh của tế bào T và sản xuất các cytokine gây viêm (TNF-α, IFN-γ, và IL-2), cho phép các con đường điểm kiểm soát miễn dịch thúc đẩy một môi trường dung nạp.
Các chất ức chế điểm kiểm soát, hiện đang được sử dụng ngày càng nhiều trong liệu pháp chống ung thư, bao gồm các kháng thể đơn dòng nhắm vào con đường CTLA-4 và PD-1 (cả thụ thể PD-1 và phối tử PD-L1) và do đó loại bỏ sự kiềm chế đối với hoạt động chống khối u (Hình 22.4). Việc phong tỏa CTLA-4 tăng cường các tín hiệu đồng kích thích và dẫn đến việc các tế bào T nguyên bản có các phản ứng tế bào T hiệu ứng tăng lên đối với các tế bào khối u, trong khi việc phong tỏa con đường PD-1 dẫn đến sự tăng sinh của tế bào T và một môi trường gây viêm hỗ trợ hoạt động chống khối u. Ngoài ra, có các con đường miễn dịch khác bị ảnh hưởng bởi việc phong tỏa các con đường này. Việc phát hiện ra các chất ức chế điểm kiểm soát miễn dịch là một bước đột phá trong liệu pháp chống ung thư, dẫn đến giải Nobel Y học năm 2018 được trao cho Tiến sĩ James Allison và Tasuku Honjo, hai người tiên phong trong lĩnh vực này.
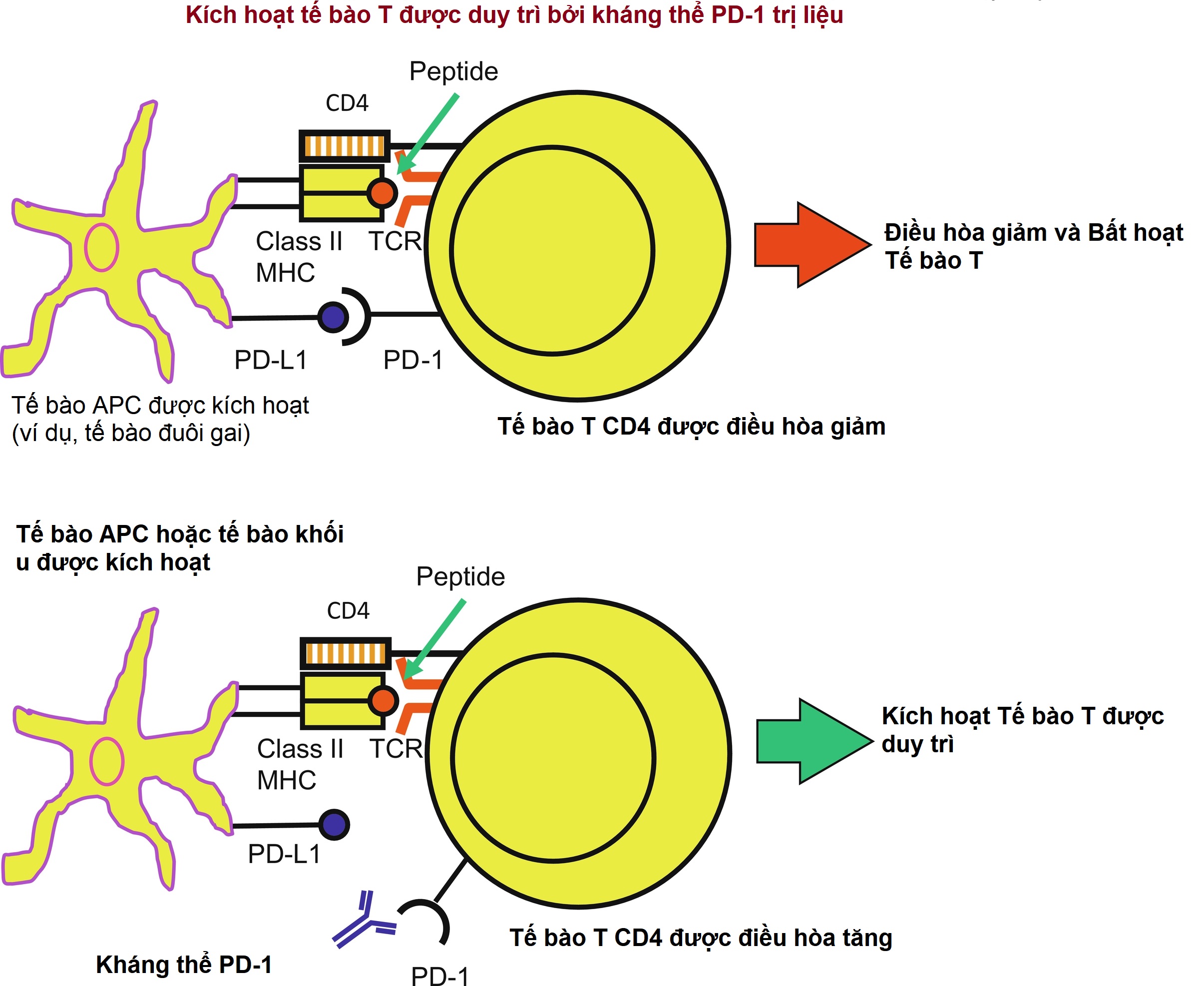
Hình 22.4: Kích hoạt tế bào T được duy trì bởi kháng thể PD-1 trị liệu. Trong khi sự liên kết của PD-L1 trên một APC với PD-1 (hình ảnh) làm giảm hoạt động của các tế bào T được kích hoạt (tế bào T CD4 trong hình vẽ này; nửa trên của hình), sự can thiệp vào tín hiệu PD-1:PD-L1 thông qua một kháng thể đơn dòng trị liệu đối với PD-1 (như hình ảnh) hoặc thông qua một kháng thể đơn dòng trị liệu đối với PD-L1 sẽ duy trì sự kích hoạt tế bào T (nửa dưới của hình). Thật vậy, các kháng thể đơn dòng trị liệu chống PD-1 và chống PD-L1 đã tạo ra những kết quả có lợi đáng kể trong điều trị một số bệnh ung thư.
Do các chất ức chế điểm kiểm soát có khả năng loại bỏ một môi trường dung nạp, không có gì ngạc nhiên khi các tác nhân điều hòa miễn dịch này có thể gây ra các tác dụng phụ liên quan đến miễn dịch (irAE), và các irAE như vậy thậm chí còn được gọi là gót chân Achilles của liệu pháp miễn dịch ung thư. Viêm tuyến yên, viêm gan, viêm da, viêm đại tràng, đái tháo đường tuýp 1, viêm tuyến giáp, viêm tuyến thượng thận, và viêm cơ tim đã được báo cáo sau khi điều trị bằng ipilimumab (kháng thể CTLA-4) và/hoặc liệu pháp kháng thể PD-1 (nivolumab). Các cơ chế tiềm năng khác dẫn đến irAES sau khi sử dụng các chất ức chế điểm kiểm soát bao gồm: (1) trình diện chéo, trong đó các kháng nguyên khối u được giải phóng sau hoạt động chống khối u được các APC tiếp nhận, khởi đầu các phản ứng miễn dịch thứ cấp, và (2) lan truyền epitope (sau khi giải phóng các kháng nguyên khối u), theo đó có sự thu nhận liên tục các tân kháng nguyên và sự huy động các tế bào T không được nhắm mục tiêu. Mặc dù sự thành công của các chất ức chế điểm kiểm soát như các loại thuốc chống ung thư, các irAE này vẫn là một mối quan tâm. Có rất ít nghiên cứu ở bệnh nhân ung thư có và không có các tình trạng tự miễn từ trước, và các tác động lâu dài của việc sử dụng chất ức chế điểm kiểm soát vẫn chưa được biết. Các phương thức mới của liệu pháp miễn dịch chống ung thư đã được nghiên cứu để giảm bớt irAE bao gồm tăng hiệu quả và sử dụng các loại vắc-xin chống lại các tân kháng nguyên khối u.
Sau khi đã xem xét các khái niệm miễn dịch học cơ bản liên quan đến dung nạp trung ương và ngoại vi, chúng ta sẽ chuyển trọng tâm của phần còn lại của chương sang các khía cạnh lâm sàng và bệnh lý của APS.
PHÂN LOẠI CÁC HỘI CHỨNG ĐA TUYẾN TỰ MIỄN
APS I, còn được gọi là bệnh đa nội tiết-nấm candida-loạn dưỡng ngoại bì tự miễn (APECED) là một rối loạn di truyền lặn trên nhiễm sắc thể thường được đặc trưng bởi một số rối loạn tự miễn với sự không đồng nhất đáng kể trong biểu hiện của nó. Hội chứng này là do một đột biến trong gen AIRE trên nhiễm sắc thể 21q22.3. Sự hiện diện của hai trong ba tình trạng sau đây là điều kiện tiên quyết để chẩn đoán APS I: (1) suy vỏ thượng thận (bệnh Addison) hoặc bằng chứng huyết thanh học về viêm tuyến thượng thận (tự kháng thể tuyến thượng thận), (2) suy tuyến cận giáp, và (3) nhiễm nấm candida niêm mạc da mạn tính. APS II được định nghĩa bởi sự cùng tồn tại của ít nhất hai trong ba bệnh nội tiết sau: (1) suy vỏ thượng thận tự miễn hoặc bằng chứng huyết thanh học về viêm tuyến thượng thận, (2) viêm tuyến giáp tự miễn, và/hoặc (3) đái tháo đường tuýp 1 hoặc bằng chứng huyết thanh học về tự miễn dịch đảo tụy. Theo truyền thống, sự xuất hiện của bệnh tuyến giáp và tuyến thượng thận tự miễn đã được gọi là hội chứng Schmidt, trong khi một bộ ba hoàn chỉnh của tự miễn dịch tuyến thượng thận, tuyến giáp, và đảo tụy (hoặc đái tháo đường tuýp 1) đã được gọi là hội chứng Carpenter (Hình 22.5). Sự hiện diện của viêm tuyến giáp, không có bệnh tuyến thượng thận, nhưng liên quan đến đái tháo đường tuýp 1, thiếu máu ác tính, bạch biến, hoặc rụng tóc đã được một số tác giả gọi là APS III, trong khi các sự kết hợp bổ sung của bệnh tự miễn đã được gọi là APS IV (tức là, bạch biến cộng với rụng tóc, đái tháo đường tuýp 1 cộng với bệnh celiac, hoặc đái tháo đường tuýp 1 và bạch biến). Do chúng có chung các gen nhạy cảm và các đặc điểm miễn dịch tương tự, các phân nhóm này thường được coi là sự mở rộng của APS II và không nhất thiết là các thực thể riêng biệt.
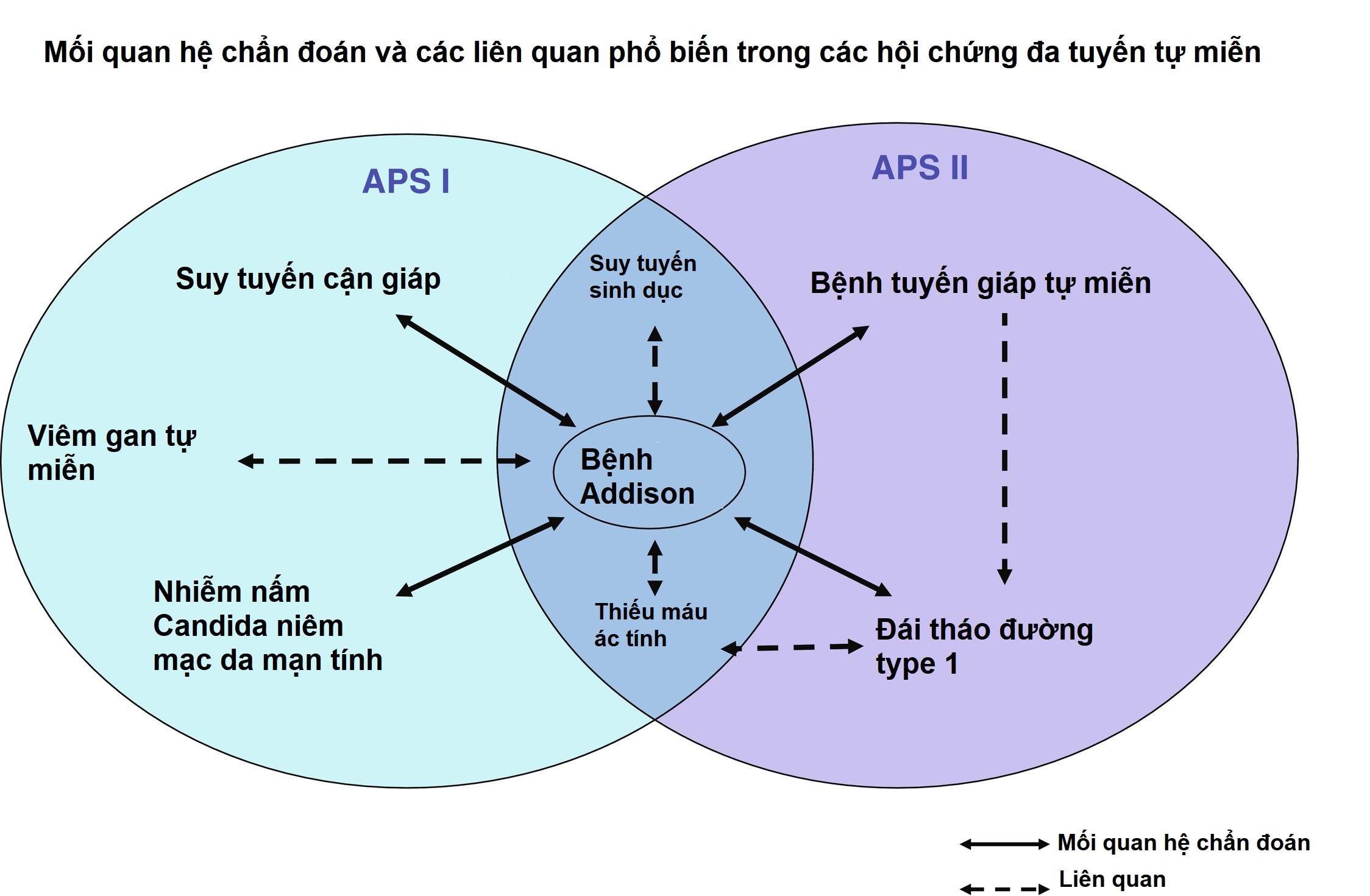
Hình 22.5: Các mối quan hệ chẩn đoán và các liên quan phổ biến trong các hội chứng đa tuyến tự miễn. Các đường liền nét chỉ ra các mối quan hệ chẩn đoán. Các đường đứt nét chỉ ra các liên quan phổ biến. Chẩn đoán APS I phụ thuộc vào sự cùng tồn tại của bệnh Addison (hoặc các tự kháng thể tuyến thượng thận) cộng với suy tuyến cận giáp hoặc nhiễm nấm candida niêm mạc da mạn tính hoặc cả hai. Chẩn đoán APS II phụ thuộc vào sự cùng tồn tại của bệnh Addison (hoặc các tự kháng thể tuyến thượng thận) cộng với bệnh tuyến giáp tự miễn hoặc đái tháo đường tuýp 1 hoặc cả hai (hoặc các tự kháng thể liên quan của chúng). Viêm gan tự miễn có thể xảy ra ở những người bị ảnh hưởng bởi APS I. Suy tuyến sinh dục hoặc thiếu máu ác tính có thể được thấy ở những người bị ảnh hưởng bởi APS I hoặc APS II. Thiếu máu ác tính cũng có liên quan mạnh mẽ với bệnh tuyến giáp tự miễn (độc lập với APS II và bệnh Addison; lưu ý: để đơn giản hóa đồ họa, không có đường nào được vẽ trong hình để chỉ ra tự miễn dịch “tuyến giáp-dạ dày” như vậy).
DI TRUYỀN CỦA APS I VÀ APS II
APS I là do các đột biến trong gen AIRE, và được di truyền theo kiểu lặn trên nhiễm sắc thể thường. Protein AIRE được biểu hiện trong tuyến ức, các hạch bạch huyết, và gan của thai nhi, cũng như trong tụy, vỏ thượng thận, và tinh hoàn. Gen này trải dài 11,9 kb, chứa 14 exon, và mã hóa một protein 545 acid amin. Protein AIRE có bốn miền chính – CARD (miền tuyển dụng caspase), SAND (SP100, AIRE1, NucP41/P75 và DEAF1), PHD1 (miền homeodomain thực vật), và PHD2. Miền CARD tham gia vào sự đa hợp của protein AIRE thành một trạng thái hoạt động, và sự gắn kết của nó với một nhiễm sắc thể đích. Miền SAND cần thiết để AIRE tương tác với một phức hợp ức chế phiên mã (ATF7ip), lần lượt là rất quan trọng cho sự biểu hiện gen phụ thuộc AIRE. Các miền PHD hoạt động như các vị trí cho nhiều sự kiện liên kết và hỗ trợ hoạt động phiên mã của AIRE.
Hơn 98% bệnh nhân APS I có các đột biến trong gen AIRE. Cho đến nay, hơn 100 đột biến trong gen AIRE đã được báo cáo, và các đột biến lặn gây bệnh được phân bố khắp gen AIRE. Mặc dù sự di truyền theo truyền thống được chấp nhận là lặn trên nhiễm sắc thể thường, có một số bệnh nhân được báo cáo có các đột biến trội dị hợp tử (trong gen mã hóa miền SAND) và các biểu hiện không điển hình hoặc không cổ điển của APS 1. Vì AIRE hoạt động ở các dạng đa hợp, những thay đổi nhỏ trong các chuỗi acid amin ở các miền quan trọng của AIRE thứ phát sau các đột biến dị hợp tử có thể dẫn đến các đa hợp bị lỗi, và do đó có một hiệu ứng trội âm, theo đó sản phẩm gen bị thay đổi ức chế hoạt động của protein kiểu dại. Một hiệu ứng như vậy cũng đã được báo cáo đối với các đột biến đơn alen trong gen AIRE mã hóa miền PHD1.
Không giống như APS I, APS II không được di truyền như một đột biến gen đơn lẻ mà thể hiện sự di truyền đa gen. APS II điển hình hơn nhiều so với các bệnh nội tiết tự miễn khác, trong đó các trường hợp có thể xảy ra một cách lẻ tẻ, hoặc trong các gia đình có APS II. Thông thường, các biểu hiện đặc hiệu cơ quan khác nhau có chung các liên kết di truyền, và hầu hết các gen này mã hóa cho các yếu tố điều hòa chính trong các con đường của hệ thống miễn dịch. Phức hợp MHC (HLA ở người) lớp II liên quan đến sự trình diện kháng nguyên là gen quan trọng nhất liên quan đến APS II.
Một nghiên cứu vào năm 1986 đã báo cáo một mối liên quan của các haplotype HLA-DR3 và/hoặc DR4 với bệnh Addison và đái tháo đường tuýp 1. Các nghiên cứu sau đó đã chỉ ra rằng các biến thể trong các haplotype DR3-DQ2 và DR4-DQ8 mang lại nguy cơ mắc bệnh đái tháo đường tuýp 1 và bệnh Addison, cũng như bệnh tuyến giáp tự miễn và bệnh celiac. Điều này có thể chỉ ra một căn nguyên sinh bệnh miễn dịch di truyền chung và làm nổi bật lý do tại sao nhiều rối loạn tự miễn có thể phát triển ở cùng một người. Các thành phần khác của APS II, chẳng hạn như bệnh Graves, đã cho thấy mối liên quan với HLA-DR3 trái ngược với viêm tuyến giáp Hashimoto, có liên quan đến HLA-DR4 hoặc DR5. Ngoài các phân tử lớp II, các phân tử lớp I (HLA-A và B) cũng đã được liên quan đến nguy cơ mắc bệnh đái tháo đường tuýp 1.
Vì mối liên quan của APS II với các alen HLA cụ thể chỉ ở mức độ khiêm tốn, vai trò của các gen nhạy cảm khác đã được nghiên cứu. Các gen không phải HLA khác mang lại nguy cơ APS II bao gồm các gen mã hóa CTLA-4, protein điều hòa phiên mã BACH2, protein tyrosine phosphatase không thụ thể loại 22 (PTPN22), và CD25 (thụ thể IL-2 ái lực cao).
CÁC KHÍA CẠNH LÂM SÀNG
Các thành phần bệnh chính, tần suất, và sự khác biệt giữa APS I và II được trình bày trong Bảng 22.1. Sự chồng chéo trong một số thành phần bệnh giữa APS I và II cũng được nêu bật trong Hình 22.5.
APS I
Mặc dù bệnh không phổ biến, với tỷ lệ hiện mắc chung dưới 1:100.000, nó phổ biến hơn ở Phần Lan (1:25.000), Sardinia (1:14.000), và trong số những người Do Thái Iran ở Israel (1:9.000). Nhóm thuần tập Phần Lan gồm 91 đối tượng là nhóm bệnh nhân APS I lớn nhất và được mô tả rõ nhất trên toàn thế giới.
Nhiễm nấm candida niêm mạc da kéo dài thường là dấu hiệu đầu tiên (60% tất cả các bệnh nhân APS I) và xuất hiện trong năm đầu hoặc hai năm đầu đời. Trong nhóm thuần tập Phần Lan, 50% phát triển bệnh nấm candida ở tuổi 5, 94% ở tuổi 20, và 100% ở tuổi 40. Hăm tã do nấm candida được quan sát thấy sớm trong đời, với viêm âm hộ-âm đạo do nấm candida thường phát triển ở tuổi dậy thì ở nữ giới. Sự xâm chiếm của nấm candida trong ruột có thể dẫn đến đau bụng và tiêu chảy từng đợt. Đau sau xương ức ở những bệnh nhân bị nấm miệng có thể gợi ý viêm thực quản do nấm candida và nên được xác nhận bằng nội soi thực quản. Nhiễm nấm móng mạn tính có thể dẫn đến sự đổi màu sẫm, dày lên, hoặc ăn mòn. Các nghiên cứu đã chỉ ra rằng các tế bào T từ những người thiếu AIRE vẫn có phản ứng tăng sinh có thẩm quyền chống lại C. albicans. Tuy nhiên, có sự suy giảm nội bào hóa qua trung gian thụ thể của các dẫn xuất thành tế bào nấm men bởi các tế bào đơn nhân trong trình diện kháng nguyên thiếu AIRE, và điều này dẫn đến việc thanh thải không hiệu quả nấm candida niêm mạc da, và do đó gây ra tình trạng mạn tính. Do đó, tất cả các bệnh nhân bị nhiễm nấm candida niêm mạc da khó chữa nên được điều tra kỹ lưỡng không chỉ về bất thường tế bào lympho T (số lượng lympho bào tuyệt đối, đếm các phân nhóm tế bào T, đánh giá chức năng tế bào T nếu có thể) mà còn về sự hiện diện của các bệnh đa nội tiết. Nấm miệng mạn tính phải được điều trị tích cực, vì có nguy cơ gia tăng ung thư biểu mô tế bào vảy (SCC) liên quan đến nấm candida của niêm mạc miệng hoặc thực quản; SCC đã được báo cáo ở bảy trong số 55 bệnh nhân APS I Phần Lan trên 25 tuổi, mặc dù năm trong số bảy người cũng là người hút thuốc.
Niêm mạc miệng phải được bảo vệ khỏi các tiếp xúc có thể làm tăng tính nhạy cảm với nhiễm nấm candida. Cụ thể, bệnh nhân nên được khuyên tránh các thực phẩm cứng, sắc, hoặc cay, cũng như các loại kem đánh răng làm trắng hoặc các chất mài mòn. Răng giả hoặc niềng răng có thể cung cấp thêm bề mặt cho nấm candida phát triển. Fluconazole có hoạt tính tốt chống lại nấm candida và là phương pháp điều trị được ưu tiên. Các azole khác đã được sử dụng bao gồm ketoconazole, và miconazole. Dữ liệu cho thấy việc sử dụng kéo dài có thể dẫn đến kháng thuốc. Với tình trạng kháng thuốc, các azole mới hơn, chẳng hạn như itraconazole, voriconazole, hoặc posaconazole có thể được sử dụng. Bệnh nhân nên theo dõi chặt chẽ với một chuyên gia về bệnh truyền nhiễm. Các bác sĩ phải nhận thức được khả năng gây ra suy thượng thận hoặc làm trầm trọng thêm tình trạng suy thượng thận đã có khi sử dụng ketoconazole; ketoconazole có thể ức chế tổng hợp cortisol. Ngoài ra, đã có các báo cáo về suy thượng thận sau khi sử dụng các azole khác (fluconazole, posaconazole) ở những bệnh nhân ốm yếu.
Suy tuyến cận giáp thường là bệnh nội tiết đầu tiên phát triển ở APS I và cuối cùng xảy ra ở hơn 85% bệnh nhân. Suy tuyến cận giáp thường xuất hiện sau khi khởi phát nhiễm nấm candida niêm mạc da nhưng trước tuổi dậy thì, với 33% bệnh nhân APS I được chẩn đoán ở tuổi 5, 66% ở tuổi 10, và gần 85% ở tuổi 30. Hạ canxi máu nặng, được biểu hiện bằng co giật, co cứng bàn tay-bàn chân, co giật cơ, và co thắt thanh quản có thể là các đặc điểm khởi phát của APS I, mặc dù những triệu chứng này có thể bị che lấp bởi tình trạng tăng canxi máu tương đối liên quan đến sự cùng tồn tại của suy thượng thận. Một số giả thuyết để giải thích sự phát triển của tăng canxi máu trong suy thượng thận bao gồm: (1) giảm lọc cầu thận do giảm thể tích, và do đó lượng canxi được lọc qua cầu thận, và (2) tăng hoạt động của 1-α hydroxylase, thường bị ức chế bởi glucocorticoid. Nồng độ hormone tuyến cận giáp (PTH) nguyên vẹn thấp hoặc bình thường không phù hợp với tình trạng hạ canxi máu và tăng phosphat máu đồng thời là chẩn đoán của suy tuyến cận giáp. Liệu pháp tiêu chuẩn bao gồm muối canxi đường uống và dùng calcitriol (xem Chương 20 để biết cách quản lý). Trong khi dữ liệu gần đây cho thấy việc tiêm hormone tuyến cận giáp tái tổ hợp dưới da một hoặc hai lần mỗi ngày (rPTH 1-34 và rPTH 1-84) có thể cung cấp liệu pháp tối ưu, đặc biệt ở những người được kiểm soát kém bằng liệu pháp tiêu chuẩn, phương pháp này chưa được Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ chấp thuận cho trẻ em ở Hoa Kỳ.
Suy vỏ thượng thận tự miễn (bệnh Addison) là thành phần chính thứ ba của APS I và thường xảy ra sau khi đã chẩn đoán nhiễm nấm candida niêm mạc da và suy tuyến cận giáp. Hơn 85% bệnh nhân APS I cuối cùng sẽ phát triển suy thượng thận. Thật không may, chẩn đoán lâm sàng về suy thượng thận thường bị bỏ sót ban đầu với chẩn đoán thường được thực hiện muộn, hoặc tại thời điểm một cơn khủng hoảng thượng thận đe dọa tính mạng. Trong nhóm thuần tập APS I của Phần Lan, 40% bị bệnh Addison ở tuổi 10 và gần 80% ở tuổi 30. Sự thiếu hụt cortisol, aldosterone, và androgen tuyến thượng thận có thể xuất hiện đồng thời hoặc có thể tiến triển trong nhiều tháng đến nhiều năm. Ngoài ra, các triệu chứng ban đầu của suy thượng thận thường không đặc hiệu, giống như bệnh tâm thần hoặc bệnh đường tiêu hóa. Chúng bao gồm mệt mỏi, sụt cân, đau cơ, đau khớp, thay đổi hành vi, buồn nôn và nôn, đau bụng, và tiêu chảy. Thiếu hụt cortisol dẫn đến tăng sản xuất proopiomelanocortin hoặc POMC ở tuyến yên, một phân tử tiền chất được cắt thành các sản phẩm bao gồm hormone vỏ thượng thận (ACTH) và hormone kích thích tế bào hắc tố (MSH). Theo thời gian, tăng sắc tố da (do MSH tăng cao) ở các vùng không tiếp xúc với ánh nắng mặt trời, cùng với hạ huyết áp tư thế, thường có thể được tìm thấy khi khám kỹ lưỡng. Mất nước nhược trương không giải thích được, với sự hiện diện đồng thời của các đặc điểm đã đề cập, nên làm dấy lên nghi ngờ về bệnh Addison. Cơn khủng hoảng thượng thận với hạ natri máu, tăng kali máu, nhiễm toan, hạ huyết áp, và hạ đường huyết có thể gây tử vong, trừ khi được nhận biết và điều trị thích hợp và kịp thời bằng glucocorticoid tiêm tĩnh mạch và dịch đẳng trương (vui lòng xem các chương cụ thể để biết thêm chi tiết). Như được thảo luận chi tiết hơn trong chương này, các tự kháng thể tuyến thượng thận được sử dụng để dự đoán suy vỏ thượng thận. Nếu xét nghiệm kháng thể dương tính, các phép đo cortisol và renin buổi sáng và/hoặc xét nghiệm kích thích ACTH có thể được sử dụng để chẩn đoán hoặc theo dõi các bệnh nhân không có triệu chứng.
Suy tuyến sinh dục tự miễn xảy ra ở hơn 50% đến 60% phụ nữ bị APS I ở tuổi 20, trong khi chưa đến 25% nam giới phát triển suy tinh hoàn. Suy tuyến sinh dục thường biểu hiện bằng vô kinh nguyên phát ở phụ nữ trẻ mặc dù kinh nguyệt không đều, buồng trứng đa nang, hoặc vô sinh có thể là các đặc điểm khởi phát. Cũng như viêm tuyến thượng thận tự miễn, suy tuyến sinh dục có thể được dự đoán bằng sự hiện diện của các tự kháng thể tế bào steroid.
Loạn dưỡng ngoại bì, không liên quan đến suy tuyến cận giáp hoặc nhiễm nấm candida niêm mạc da, đã được ghi nhận rộng rãi trong nhóm thuần tập Phần Lan. Giảm sản men răng của răng vĩnh viễn (nhưng không phải răng sữa), cũng như loạn dưỡng móng (lỗ móng 0,5-1 mm), thường được tìm thấy. Có thể có sự vắng mặt hoàn toàn của men răng hoặc các dải giảm sản ngang xen kẽ với các vùng men răng hình thành tốt. Gần một phần ba bệnh nhân Phần Lan cũng bị vôi hóa màng nhĩ và 20% đến 25% phát triển viêm giác mạc.
Như được trình bày trong Bảng 22.1, đái tháo đường tuýp 1 và tự miễn dịch tuyến giáp-dạ dày (một thuật ngữ mô tả sự kết hợp của bệnh tuyến giáp tự miễn và viêm dạ dày teo) có liên quan đến APS 1, nhưng xảy ra ít thường xuyên hơn nhiều so với ở APS II. Khi có mặt, viêm tuyến giáp thường là dạng teo hơn là dạng bướu cổ. Tự miễn dịch tế bào thành dạ dày, dẫn đến viêm dạ dày teo, với hậu quả là vô toan và thiếu yếu tố nội tại, thường biểu hiện dưới dạng thiếu máu thiếu sắt hoặc thiếu máu ác tính do thiếu vitamin B12. Viêm dạ dày teo xảy ra ở 15% đến 30% các trường hợp APS I với tuổi khởi phát trung bình là 16. Trong khi thiếu máu thiếu sắt là dạng hồng cầu nhỏ và thiếu máu do thiếu vitamin B12 là dạng hồng cầu to, sự kết hợp thiếu sắt và vitamin B12 có thể là dạng hồng cầu bình thường. Điều quan trọng cần nhận ra là các bệnh đồng mắc của tủy sống (thoái hóa kết hợp bán cấp của tủy sống) do thiếu vitamin B12 có thể xảy ra khi không có thiếu máu.
Các biểu hiện đặc hiệu cơ quan không nội tiết ít phổ biến hơn (~5%-20%) và bao gồm rụng tóc, bạch biến, viêm gan tự miễn, và kém hấp thu. Sự tiến triển thành rụng tóc toàn bộ (mất toàn bộ tóc da đầu) hoặc toàn thể (mất toàn bộ lông trên cơ thể, bao gồm lông mi, lông mày, và tóc da đầu) thường xảy ra trước tuổi dậy thì. Bạch biến ban đầu biểu hiện dưới dạng các mảng da nhỏ, nhạt màu, thiếu sắc tố. Những mảng này có thể bị bỏ sót, trừ khi được tìm kiếm đặc biệt và có thể cần khám da bằng đèn tia cực tím. Sự xuất hiện của phân màu đất sét, nước tiểu sẫm màu, và vàng da gợi ý viêm gan tự miễn hoạt động mạn tính. Viêm gan xảy ra ở 10% đến 15% bệnh nhân APS I và là nguyên nhân gây tử vong hàng đầu. Do đó, tất cả các bệnh nhân nghi ngờ mắc APS I nên được theo dõi chức năng gan thường xuyên. Viêm gan tự miễn thường được điều trị ban đầu bằng glucocorticoid, và sau đó bằng azathioprine, một khi đã kiểm soát được một phần bệnh. Kém hấp thu, có thể xảy ra từng đợt (và thường là kém hấp thu chất béo), đã được liên kết với suy tuyến cận giáp (không đủ hormone niêm mạc tá tràng và các enzyme tụy do hạ canxi máu), sự phát triển quá mức của vi khuẩn và nấm, nhạy cảm với gluten (bệnh celiac), và thiếu hụt IgA. Cũng đã có các báo cáo hiếm gặp về APS I với viêm tuyến yên (đái tháo nhạt, thiếu hụt hormone tăng trưởng, thiếu hụt ACTH), và các biểu hiện không nội tiết, chẳng hạn như viêm khớp dạng thấp, hội chứng Sjögren, viêm thận kẽ ống, viêm tiểu phế quản tự miễn, bệnh cơ, và vô lách. Các nghiên cứu gần đây trong một nhóm thuần tập châu Âu gồm 112 bệnh nhân APS 1 cũng đã phát hiện ra một loạt các biểu hiện kiểu hình và thay đổi kiểu gen trong AIRE.
Một nghiên cứu trong một nhóm thuần tập gồm 35 đối tượng Bắc và Nam Mỹ cho thấy các biểu hiện không nội tiết xảy ra sớm hơn và thường xuyên hơn nhiều so với các nhóm thuần tập châu Âu. Những biểu hiện không nội tiết này bao gồm phát ban dạng mề đay (66%), viêm gan (43%), viêm dạ dày (48%), rối loạn chức năng ruột (80%), hội chứng giống Sjögren (43%), và viêm phổi (40%), và có mặt ở 80% trước khi đáp ứng các tiêu chí chẩn đoán cặp đôi của APS. Không rõ liệu có mối tương quan giữa kiểu gen và kiểu hình có thể giải thích tần suất cao hơn của các biểu hiện không nội tiết trong nhóm thuần tập Mỹ hay không.
APS II
APS II là hội chứng phổ biến nhất trong các bệnh đa nội tiết tự miễn (không bao gồm sự trùng hợp ngẫu nhiên của đái tháo đường tuýp 1 và bệnh tuyến giáp tự miễn), với tỷ lệ hiện mắc từ 1 đến 4:100.000 và có di truyền đa gen. Không giống như APS I, APS II thường khởi phát ở tuổi trưởng thành, đặc biệt là trong thập kỷ thứ ba hoặc thứ tư và phổ biến ở nữ giới gấp ít nhất 3 lần so với nam giới, trong khi APS I phổ biến như nhau ở nam và nữ. Sự chênh lệch giới tính này ở APS II chủ yếu được giải thích bởi sự cùng tồn tại của bệnh tuyến giáp tự miễn (AITD), phổ biến hơn ở phụ nữ. Năm 1926, Schmidt lần đầu tiên mô tả mối liên quan giữa suy vỏ thượng thận và suy tuyến giáp và Carpenter đã mở rộng điều này vào năm 1964 để bao gồm cả đái tháo đường tuýp 1. Năm 1957, bản chất tự miễn của những bệnh này đã được Doniach và Roitt gợi ý khi phát hiện ra các tự kháng thể thyroglobulin ở những bệnh nhân bị viêm tuyến giáp Hashimoto.
APS II biểu hiện với suy vỏ thượng thận trong khoảng 50% các trường hợp, mặc dù nhiều thành phần bệnh có thể có mặt khi chẩn đoán. Mặc dù bệnh thường khởi phát trong độ tuổi từ 20 đến 50, nhưng không có gì lạ khi tìm thấy các trường hợp trước hoặc sau các độ tuổi này. Đái tháo đường tuýp 1 cùng tồn tại ở gần 50% bệnh nhân bị bệnh Addison, trong khi AITD cùng tồn tại ở khoảng hai phần ba bệnh nhân bị bệnh Addison. Do đó, đái tháo đường tuýp 1 và AITD phải được tìm kiếm một cách tích cực ở bất kỳ bệnh nhân nào có biểu hiện bệnh Addison.
Thành phần phổ biến nhất của APS II là AITD và được thấy ở gần 70% đến 75% bệnh nhân. AITD ảnh hưởng đến gần 4,5% dân số Hoa Kỳ, với 80% đến 90% tất cả các trường hợp xảy ra ở nữ giới. Tỷ lệ mắc AITD tăng trong những năm thiếu niên, đạt đỉnh ở thập kỷ thứ năm và thứ sáu. Viêm tuyến giáp lympho bào mạn tính (bệnh Hashimoto) cho đến nay là dạng phổ biến nhất của AITD, mặc dù bệnh Graves cũng có thể xảy ra. Viêm tuyến giáp sau sinh cũng đã được coi là một biểu hiện thoáng qua của viêm tuyến giáp tự miễn sau khi sinh và có thể biểu hiện dưới dạng suy giáp hoặc cường giáp. Một số nghiên cứu đã báo cáo về sự cùng tồn tại của tự miễn dịch đảo tụy (3%-8%) hoặc thậm chí là đái tháo đường tuýp 1 rõ rệt và AITD, trong khi chưa đến 1% bệnh nhân, với bệnh viêm tuyến giáp riêng lẻ, có bằng chứng huyết thanh học về tự miễn dịch tuyến thượng thận.
Mặc dù sự liên quan “hội chứng đa tuyến” ở những bệnh nhân bị bệnh tuyến giáp tự miễn không thường xuyên, tự miễn dịch tuyến giáp hoặc tiền sử gia đình bị viêm tuyến giáp là phổ biến ở những bệnh nhân bị thiếu máu ác tính, bạch biến, rụng tóc, bệnh nhược cơ, và hội chứng Sjögren. Gần 20% đến 40% bệnh nhân bạch biến có một thành phần khác của APS II, với tự miễn dịch tuyến giáp-dạ dày là phổ biến nhất. Hầu hết các bệnh nhân bị bạch biến đều không có triệu chứng và bằng chứng về tự miễn dịch đồng thời chỉ có thể được xác định bằng sàng lọc tự kháng thể. Bạch biến phân đoạn với sự liên quan của các vùng da không liên quan đến tự miễn dịch. Có tới 15% bệnh nhân bị rụng tóc (mảng, toàn bộ, toàn thể) và 5% người thân cấp một của họ bị bệnh tuyến giáp. Nhiều bệnh nhân APS I hơn APS II có các biểu hiện da, chẳng hạn như bạch biến hoặc rụng tóc, nhưng vì APS II phổ biến hơn nhiều, hầu hết các bệnh nhân có một trong hai biểu hiện này và một bệnh tự miễn khác được phân loại là APS II không hoàn chỉnh.
Gần 30% bệnh nhân bị bệnh nhược cơ, một bệnh tự miễn được đặc trưng bởi sự hiện diện của các tự kháng thể kháng thụ thể acetylcholine và yếu cơ trầm trọng hơn khi co cơ, có AITD. Cả viêm tuyến giáp Hashimoto và bệnh Graves đều có thể xảy ra ở những bệnh nhân bị bệnh nhược cơ. Điều thú vị là, những bệnh nhân bị bệnh nhược cơ và AITD đồng thời có xu hướng có biểu hiện nhược cơ nhẹ hơn, và tỷ lệ mắc bệnh tuyến ức và tự kháng thể chuỗi α của thụ thể acetylcholine thấp hơn. Tỷ lệ mắc bệnh nhược cơ mắt cao hơn ở những bệnh nhân bị bệnh Graves.
Đái tháo đường tuýp 1, một thành phần chẩn đoán của APS II, có tỷ lệ mắc cao nhất trong những năm thiếu niên với tỷ lệ mắc nhỏ hơn nhưng ngày càng tăng xảy ra ở những năm mẫu giáo. Tuy nhiên, bệnh có thể khởi phát ở mọi lứa tuổi. Khoảng 10% đến 15% bệnh nhân APS, bị dán nhãn sai là đái tháo đường tuýp 2 do khởi phát bệnh đái tháo đường sau 40 tuổi, thực sự mắc bệnh đái tháo đường tự miễn tiến triển chậm (còn được gọi là đái tháo đường tự miễn tiềm ẩn ở người lớn hoặc LADA). Không giống như APS II, trong đó có sự chênh lệch giới tính nữ mặc dù có sự xuất hiện của đái tháo đường tuýp 1, không có sự chênh lệch giới tính nào ở những bệnh nhân bị đái tháo đường tuýp 1 riêng lẻ. AITD (được biểu thị bằng sự hiện diện của các tự kháng thể thyroperoxidase và/hoặc thyroglobulin) xảy ra ở 20% đến 25% bệnh nhân bị đái tháo đường tuýp 1 với phụ nữ chiếm gần hai phần ba số bệnh nhân dương tính với tự kháng thể. Mặc dù tỷ lệ lưu hành cao của các tự kháng thể tuyến giáp, chưa đến 20% bệnh nhân có tự kháng thể tuyến giáp có bằng chứng về rối loạn chức năng tuyến giáp được định nghĩa là nồng độ hormone kích thích tuyến giáp (TSH) tăng cao. Tự miễn dịch vỏ thượng thận ít thường xuyên hơn nhiều ở những bệnh nhân bị đái tháo đường tuýp 1, với bằng chứng huyết thanh học được báo cáo trong 1,5% các trường hợp. Các tự kháng thể transglutaminase mô gợi ý bệnh celiac có mặt ở 3% đến 7% bệnh nhân bị đái tháo đường tuýp 1. Bệnh celiac nên được nghi ngờ ở những bệnh nhân đái tháo đường tuýp 1 bị tiêu chảy không giải thích được, sụt cân, không tăng cân, hoặc chậm phát triển, hạ đường huyết không giải thích được nên được xác nhận bằng sinh thiết ruột.
Các tự kháng thể tế bào thành dạ dày (PCA) có mặt ở khoảng 10% phụ nữ và 5% nam giới bị đái tháo đường tuýp 1. Mặc dù thiếu máu ác tính thường ảnh hưởng đến phụ nữ sau thập kỷ thứ năm, trẻ em bị PCA nên được theo dõi chặt chẽ để phát hiện sự phát triển của thiếu máu ác tính. Viêm dạ dày teo có thể dẫn đến sự phát triển của thiếu máu hồng cầu khổng lồ do thiếu yếu tố nội tại cần thiết cho sự hấp thu vitamin B12 từ ruột. Thiếu máu thiếu sắt cũng có thể xảy ra ở cả thanh thiếu niên và người lớn do suy giảm khả năng hấp thu sắt do giảm sản xuất acid (vô toan).
Khoảng 10% phụ nữ dưới 40 tuổi bị APS II phát triển suy buồng trứng. Suy buồng trứng có thể biểu hiện dưới dạng vô kinh nguyên phát hoặc thứ phát. Ở những phụ nữ bị viêm buồng trứng lympho bào đã được chứng minh bằng sinh thiết, suy vỏ thượng thận hoặc tự miễn dịch tuyến thượng thận cận lâm sàng thường có mặt. Ngược lại, sự tiến triển thành suy tuyến sinh dục rất hiếm gặp ở nam giới bị bệnh Addison.
Sự liên quan của tuyến yên đôi khi được thấy ở APS II. Viêm tuyến yên và hội chứng hố yên rỗng đã được mô tả, dẫn đến suy giảm bài tiết riêng lẻ của hormone tăng trưởng, ACTH, TSH, hormone kích thích nang trứng (FSH), hoặc hormone tạo hoàng thể (LH).
Một số tình trạng không nội tiết cũng đã được báo cáo có liên quan đến APS II. Chúng bao gồm viêm loét đại tràng, xơ gan mật nguyên phát, bệnh sarcoidosis, co thắt tâm vị, viêm cơ, và bệnh thần kinh.
Hội chứng IPEX
IPEX là một hội chứng gây ra bởi các tế bào Treg bị lỗi thứ phát sau các đột biến trong gen FOXP3. Gen FOXP3 mã hóa một yếu tố phiên mã cùng tên. Hơn 70 đột biến FOXP3 liên quan đến IPEX đã được mô tả cho đến nay. Vì FOXP3 nằm trên nhiễm sắc thể X, sự di truyền là lặn liên kết với X với nam giới bị ảnh hưởng, trong khi nữ giới là người mang mầm bệnh.
Những bệnh nhân có đột biến bất hoạt FOXP3 dẫn đến thiếu hụt FOXP3 sẽ phát triển IPEX, được đặc trưng bởi bệnh tự miễn đa cơ quan bắt đầu rất sớm trong đời và thường bao gồm bộ ba đái tháo đường tuýp 1 khởi phát ở trẻ sơ sinh, viêm da dạng chàm, và bệnh ruột (tiêu chảy nước). Các rối loạn tự miễn khác bao gồm viêm tuyến giáp, thiếu máu tan máu, giảm tiểu cầu, viêm gan, bệnh thận, viêm khớp, và bệnh phổi. Bệnh nhân mắc hội chứng IPEX thường biểu hiện các tự kháng thể sớm trong đời; những người bị đái tháo đường tuýp 1 thường có các tự kháng thể decarboxylase acid glutamic (GADA), và các tự kháng thể tế bào đảo tụy khác (ICA) trong giai đoạn sơ sinh. Các tự kháng thể khác được phát hiện sớm trong đời bao gồm các kháng thể chống lại harmonin và villin, các protein được tìm thấy trong các vi nhung mao của bờ bàn chải ruột và ống lượn gần của thận, và có thể giải thích bệnh ruột và viêm thận được tìm thấy ở những bệnh nhân này.
IPEX thường gây tử vong trong vài năm đầu đời. Cho đến nay, chỉ có ức chế miễn dịch lâu dài hoặc cấy ghép tủy xương, với mục tiêu tăng cường chức năng tế bào T điều hòa, là những liệu pháp hiệu quả cho IPEX. Sự biểu hiện bền vững của FOXP3 có thể tái lập trình các tế bào T hiệu ứng để hoạt động như các tế bào T điều hòa. Các phương pháp tiếp cận mới, bao gồm điều trị bằng các tế bào Treg được thiết kế hoặc chỉnh sửa gen nhắm mục tiêu của FOXP3, đang được nghiên cứu như các lựa chọn điều trị cho hội chứng IPEX.
PHƯƠNG PHÁP CHẨN ĐOÁN VÀ THEO DÕI
Phương pháp chẩn đoán các hội chứng đa tuyến là ba bước: (1) sàng lọc tự kháng thể để (i) xác minh bản chất tự miễn của bệnh nội tiết nghi ngờ, và (ii) kiểm tra sự liên quan của các cơ quan và mô khác; (2) đánh giá đầy đủ chức năng nội tiết ở những bệnh nhân có tự kháng thể được xác nhận và những đối tượng âm tính với tự kháng thể mà bệnh bị nghi ngờ trên lâm sàng; và (3) phân tích đột biến để xác nhận chẩn đoán, và sàng lọc anh chị em và những người thân khác về tình trạng mang mầm bệnh tiềm năng của họ.
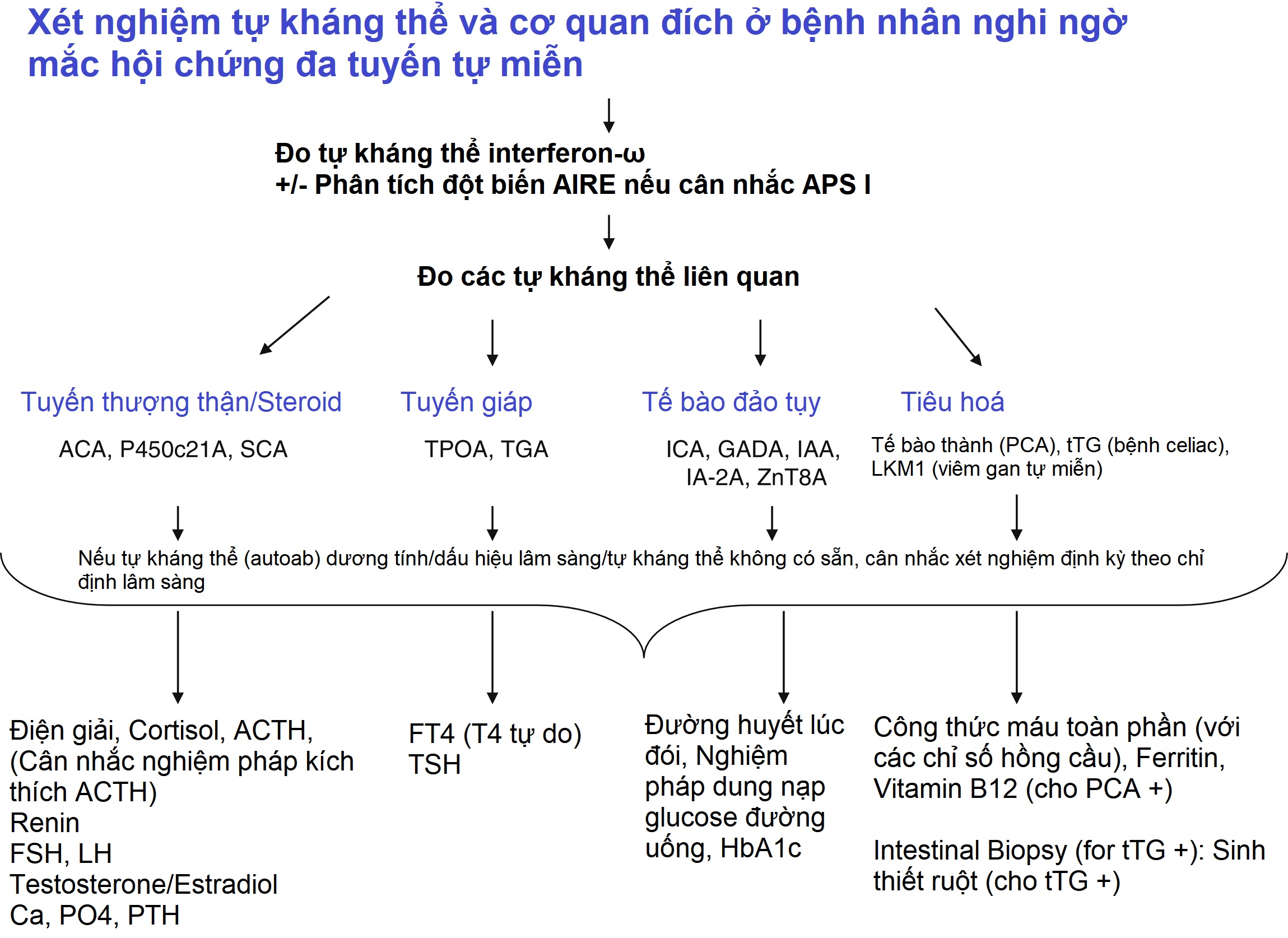
Hình 22.6: Xét nghiệm tự kháng thể và cơ quan đích ở bệnh nhân nghi ngờ mắc hội chứng đa tuyến tự miễn. Sơ đồ này chỉ ra những tự kháng thể nào nên được lấy khi nghi ngờ APS. Do độ nhạy và độ đặc hiệu của nó, sàng lọc ban đầu bằng các tự kháng thể interferon-ω (nếu có) có thể là đủ nếu nghi ngờ APS I. Tuy nhiên, chúng tôi khuyến nghị xét nghiệm tự kháng thể đặc hiệu cho cơ quan ở những đối tượng nghi ngờ mắc bệnh liên quan đến APS (viêm tuyến thượng thận, nhiễm nấm candida niêm mạc da mạn tính, hoặc suy tuyến cận giáp tự miễn) tại thời điểm chẩn đoán ban đầu. Chúng tôi cũng khuyến nghị xét nghiệm tự kháng thể ở những đối tượng có 2 hoặc nhiều bệnh tự miễn liên quan (tức là ĐTĐ tuýp 1 và viêm tuyến giáp tự miễn). Các đột biến trong gen AIRE cũng có thể được tìm kiếm. Thật không may, dữ liệu còn thiếu để đưa ra các khuyến nghị chắc chắn về sự cần thiết của việc xét nghiệm tự kháng thể lặp lại ở những bệnh nhân ban đầu âm tính với tự kháng thể. Tuy nhiên, nếu các kháng thể âm tính, chúng tôi khuyến nghị xét nghiệm tự kháng thể lặp lại ở tuổi dậy thì sớm nếu các dấu hiệu hoặc triệu chứng lâm sàng của APS vẫn tồn tại. Đối với những người dương tính với tự kháng thể, nên thực hiện xét nghiệm hàng năm về suy cơ quan đích như hình. Khi các xét nghiệm tự kháng thể không có sẵn, các bác sĩ lâm sàng phải sử dụng xét nghiệm thường xuyên các điểm cuối đặc hiệu của bệnh để xác định các bệnh đồng mắc. ACA = tự kháng thể tế bào chất tuyến thượng thận được phát hiện bằng miễn dịch huỳnh quang gián tiếp; P450c21A = Tự kháng thể chống lại enzyme 21-hydroxylase của tuyến thượng thận; SCA = tự kháng thể tế bào steroid; TPOA = tự kháng thể thyroperoxidase; TGA = tự kháng thể thyroglobulin; ICA = tự kháng thể tế bào chất đảo tụy; GADA = tự kháng thể decarboxylase acid glutamic; IAA = tự kháng thể insulin; IA-2A = tự kháng thể liên quan đến u đảo tụy-2; ZnT8A = tự kháng thể vận chuyển kẽm-8; ITG = tự kháng thể transglutaminase IgA; LKM1 = tự kháng thể microsome gan thận loại 1; ACTH = hormone vỏ thượng thận; FSH = hormone kích thích nang trứng; LH = hormone tạo hoàng thể; Ca = canxi; PO4 = phosphat; PTH = hormone tuyến cận giáp: T4 = thyroxine; TSH = hormone kích thích tuyến giáp; FPG = glucose huyết tương lúc đói, OGTT = nghiệm pháp dung nạp glucose đường uống; HbA1c = hemoglobin A1c; CBC = công thức máu toàn phần.
Việc nhận biết các bệnh tự miễn đa cơ quan trước giai đoạn có triệu chứng của chúng là rất quan trọng để giảm thiểu tỷ lệ mắc bệnh và tử vong liên quan. Cần luôn thực hiện một bệnh sử và khám thực thể kỹ lưỡng, và cần duy trì một chỉ số nghi ngờ cao. Ngoài ra, tiền sử gia đình về bệnh tự miễn đa cơ quan sẽ làm tăng nghi ngờ về APS tiềm năng.
Như đã thảo luận sau đây, có lẽ thành tựu lớn nhất trong thập kỷ qua là việc phát hiện ra mối liên quan giữa các tự kháng thể đối với interferon loại 1 và APS I, cho phép chẩn đoán sớm bệnh nhân APS I. Mặc dù không chỉ ra sự tấn công miễn dịch đặc hiệu mô, các tự kháng thể đối với interferon (α và ω) cung cấp một xét nghiệm sàng lọc có độ nhạy và độ đặc hiệu gần 95% đến 100% cho APS 1. Sự hiện diện của các tự kháng thể đối với interferon nên được theo sau bằng xét nghiệm xác nhận các đột biến AIRE, cũng như xét nghiệm các tự kháng thể đặc hiệu mô khác (Hình 22.6). Chúng bao gồm các tự kháng thể tế bào chất của 21-hydroxylase hoặc vỏ thượng thận (đối với bệnh Addison tự miễn), GADA, các tự kháng thể liên quan đến u đảo tụy (IA-2A), các tự kháng thể insulin (IAA), các tự kháng thể vận chuyển kẽm 8 (ZnT8) (đối với đái tháo đường tuýp 1), các tự kháng thể thyroperoxidase và thyroglobulin (đối với AITD), các tự kháng thể tế bào steroid (đối với suy buồng trứng), và các tự kháng thể transglutaminase (đối với bệnh celiac). Ngoài ra, mặc dù không có sẵn, các tự kháng thể đối với 17-hydroxylase, enzyme cắt chuỗi bên, và 3-hydroxysteroid dehydrogenase có thể được sử dụng để phát hiện tự miễn dịch tuyến sinh dục và tuyến thượng thận. Các tự kháng thể đối với protein 5 lặp lại giàu leucine NACHT (NALP5) và thụ thể cảm nhận canxi cho thấy suy tuyến cận giáp tự miễn tiềm ẩn (CaSR). Tuy nhiên, độ đặc hiệu của CaSR và NALP5 để chẩn đoán suy tuyến cận giáp tự miễn lần lượt là khoảng 83% và 50%, trong khi độ nhạy đã được báo cáo lần lượt là 39% và 26%.
Các tự kháng thể khác bao gồm các kháng thể đối với enolase tuyến yên của người, và gợi ý viêm tuyến yên tự miễn.
Cần lưu ý rằng việc đo lường một số tự kháng thể này có thể không có sẵn cho tất cả các bác sĩ lâm sàng, và yêu cầu các phương pháp tiếp cận cá nhân hóa để sàng lọc và theo dõi. Ngoài ra, dữ liệu cho thấy một số tự kháng thể đối với các cytokine (IL-17A, IL-17F, và IL-22) có thể liên quan đến nhiễm nấm candida niêm mạc da. Mặc dù có một mối liên kết rõ ràng giữa sự hiện diện của các tự kháng thể đặc hiệu cơ quan và sự hiện diện của bệnh có từ trước, hoặc sự tiến triển sau đó thành bệnh, số lượng các rối loạn liên quan có thể phát triển và tuổi xuất hiện của chúng là không thể đoán trước. Do đó, cần theo dõi lâu dài ở cả những đối tượng dương tính và âm tính với tự kháng thể.
Tất cả các bệnh nhân có một bệnh tự miễn duy nhất phải được coi là có nguy cơ mắc các bệnh tự miễn khác. Việc có nên và khi nào sàng lọc các tự kháng thể khác dựa trên khả năng tìm thấy một bệnh tự miễn khác, hiệu quả chi phí, và khả năng sàng lọc sẽ ngăn ngừa tỷ lệ mắc bệnh và tử vong từ các rối loạn tiềm năng khác trong tương lai, chẳng hạn như nhiễm toan ceton do đái tháo đường, cơn khủng hoảng Addison, hoặc hạ canxi máu với co giật, v.v.
Do tỷ lệ mắc AITD cao ở bệnh nhân đái tháo đường tuýp 1, nên khuyến cáo rằng bệnh nhân đái tháo đường tuýp 1 nên được đo các tự kháng thể thyroperoxidase và thyroglobulin ngay sau khi chẩn đoán và đo thyrotropin (TSH) hai năm một lần. Việc đo cả hai tự kháng thể tuyến giáp có độ nhạy gần 90% để phát hiện AITD. Tần suất lặp lại các tự kháng thể tuyến giáp vẫn còn gây tranh cãi. Trên thực tế, một số bác sĩ lâm sàng thích theo dõi những bệnh nhân có nguy cơ chỉ bằng TSH, vì việc điều trị thường không được bắt đầu cho đến khi TSH tăng cao.
Không nên sàng lọc tất cả các thành phần của APS ở những bệnh nhân bị bệnh tuyến giáp tự miễn riêng lẻ. Tuy nhiên, một số báo cáo đã chứng minh tỷ lệ mắc PCA tăng ở những bệnh nhân trẻ tuổi bị viêm tuyến giáp tự miễn. Do đó, có thể xem xét sàng lọc PCA ở trẻ em bị AITD. Ở những bệnh nhân suy giáp có APS được xác nhận, bằng chứng về tự miễn dịch tuyến thượng thận phải được tìm kiếm trước khi bắt đầu liệu pháp thay thế hormone tuyến giáp, vì việc thay thế hormone tuyến giáp có khả năng gây ra một cơn khủng hoảng thượng thận ở những bệnh nhân có chức năng vỏ thượng thận cận biên (bằng cách tăng chuyển hóa các hormone glucocorticoid).
Chẩn đoán chậm và thậm chí tử vong có thể phòng ngừa được vẫn xảy ra ở những bệnh nhân bị suy vỏ thượng thận không được chẩn đoán. Như đã đề cập trước đây, biểu hiện thường mơ hồ và không đặc hiệu, cho đến khi một cơn khủng hoảng Addison xảy ra. Ở những bệnh nhân bị đái tháo đường tuýp 1, hạ đường huyết không giải thích được hoặc cải thiện không giải thích được trong kiểm soát đường huyết có thể là một manh mối để chẩn đoán bệnh Addison. Độ nhạy cảm được cải thiện đối với insulin được sử dụng và đường huyết được cải thiện có thể đại diện cho sự mất hoạt động đối kháng điều hòa liên quan đến thiếu hụt glucocorticoid. Tất cả các bệnh nhân bị nhiễm nấm candida niêm mạc da kéo dài hoặc không giải thích được, hoặc suy tuyến cận giáp, và phụ nữ bị suy buồng trứng sớm nên được đánh giá về APS I, đặc biệt trong bối cảnh bệnh Addison tiềm năng.
Nên đánh giá chức năng cơ quan đích ở bất kỳ bệnh nhân nào có tự kháng thể dương tính hàng năm (xem Hình 22.6). Xét nghiệm glucose máu lúc đói và/hoặc 2 giờ sau ăn, canxi, phosphat cộng với PTH, và nồng độ TSH có thể đánh giá hiệu quả chức năng của đảo tụy, tuyến cận giáp và tuyến giáp, tương ứng, ở những người không có triệu chứng. Mặc dù hemoglobin A1c có độ nhạy hạn chế như một công cụ sàng lọc cho bệnh đái tháo đường tuýp 1, các ấn phẩm trong y văn đã báo cáo rằng nó có thể dự đoán sự khởi phát lâm sàng ở những người đã có các tự kháng thể đảo tụy dương tính. Nồng độ FSH và LH tăng cao với các steroid sinh dục thấp đồng thời xác nhận suy tuyến sinh dục. Các đánh giá hàng loạt về hemoglobin, hematocrit, và các chỉ số hồng cầu có thể đánh giá sự tiến triển thành viêm dạ dày teo ở những bệnh nhân có tự miễn dịch dạ dày. Các phát hiện về thiếu máu hồng cầu khổng lồ với thể tích trung bình hồng cầu tăng cao cho thấy thiếu vitamin B12, trong khi thiếu máu nhược sắc hồng cầu nhỏ cho thấy thiếu sắt. Nồng độ vitamin B12 nên được theo dõi ở tất cả các bệnh nhân bị PCA vì bệnh thần kinh có thể phát triển mà không có thiếu máu. Trước khi bắt đầu điều trị, nên lấy cả nồng độ vitamin B12 và hồ sơ sắt. Nồng độ acid methylmalonic không thường xuyên cần thiết ở những bệnh nhân có tự miễn dịch dạ dày nhưng có thể hữu ích nếu nồng độ vitamin B12 ở mức thấp giới hạn. Các xét nghiệm chức năng gan và các tự kháng thể kháng ty thể nên được thực hiện ở những bệnh nhân bị APS I. Những bệnh nhân có các tự kháng thể transglutaminase mô hoặc kháng nội mạc cơ thường không có triệu chứng và nên được giới thiệu đến một bác sĩ chuyên khoa tiêu hóa để thảo luận về sự cần thiết của sinh thiết ruột, và xác nhận tiềm năng của bệnh celiac.
Nồng độ cortisol buổi sáng sớm thấp, các bất thường điện giải (hạ natri máu/tăng kali máu), và hạ đường huyết đại diện cho những thay đổi muộn xảy ra tại hoặc ngay trước khi khởi phát suy thượng thận. Giống như diễn biến tự nhiên của tiền đái tháo đường tuýp 1 được mô tả rõ (xem Chương 21), một mô hình tương tự được thấy trước khi phát triển suy vỏ thượng thận. Bốn giai đoạn đã được mô tả (Bảng 22.3) sau khi phát hiện các tự kháng thể tuyến thượng thận: Giai đoạn 1: tăng hoạt động renin huyết tương với aldosterone bình thường đến thấp; Giai đoạn 2: giảm đáp ứng cortisol sau khi tiêm ACTH; Giai đoạn 3: ACTH nền tăng cao; và Giai đoạn 4: cortisol nền thấp. Ở những người có tự kháng thể vỏ thượng thận, nên sàng lọc bằng cách đo nồng độ cortisol buổi sáng và ACTH. Một nồng độ cortisol buổi sáng dưới 5 μg/dL với ACTH huyết tương đồng thời cao hơn hai lần so với giới hạn tham chiếu là chẩn đoán cao của suy thượng thận nguyên phát. Nếu những giá trị này bình thường nhưng nghi ngờ lâm sàng cao, có thể thực hiện một xét nghiệm kích thích ACTH 250 μg xác nhận. Các giá trị renin nền tăng cao cũng rất gợi ý về suy vỏ thượng thận, và nên thực hiện một xét nghiệm kích thích ACTH để xác nhận.
ĐIỀU TRỊ
Các liệu pháp thay thế hormone hoặc các liệu pháp thay thế khác cho các rối loạn thành phần của APS I và APS II, chẳng hạn như suy thượng thận, suy giáp, đái tháo đường tuýp 1, thiếu máu thiếu sắt, và thiếu máu ác tính là tương tự cho dù các bệnh này xảy ra đơn lẻ hay kết hợp với các tình trạng khác. Các liệu pháp liên quan đến rối loạn nội tiết cụ thể được mô tả trong các chương riêng lẻ. Các đội ngũ đa ngành phải tham gia vào việc đánh giá và quản lý bệnh nhân APS I hoặc II, do có vô số các bệnh lý có thể biểu hiện ở cùng một cá nhân. Ngoài ra, bệnh nhân APS I đã được báo cáo là bị vô lách hoặc giảm chức năng lách, và những bệnh nhân này dễ bị nhiễm trùng bùng phát. Tiêm chủng kịp thời và, nếu cần, kháng sinh dự phòng phải là một phần của việc quản lý những bệnh nhân này.
CÁC TỰ KHÁNG THỂ TRONG CÁC HỘI CHỨNG ĐA TUYẾN TỰ MIỄN
Các tự kháng thể trong APS chủ yếu là các kháng thể IgG liên kết với các kháng nguyên tự thân. Các tự kháng thể có thể gây bệnh, như được quan sát thấy trong bệnh Graves hoặc bệnh nhược cơ. Trong bệnh Graves, các tự kháng thể chủ vận nhắm vào thụ thể TSH kích thích sản xuất quá mức hormone tuyến giáp gây ra cường giáp. Trong bệnh nhược cơ, các tự kháng thể nhắm vào thụ thể acetylcholine ở bản vận động cơ nằm trên các tế bào cơ kích thích sự nội bào hóa của thụ thể acetylcholine, gây ra yếu cơ. Tuy nhiên, các tự kháng thể thường chỉ đóng vai trò là các dấu ấn huyết thanh học của tự miễn dịch, chẳng hạn như trong đái tháo đường tuýp 1, nơi các ICA, IAA, GADA, IA-2A, và các tự kháng thể đối với ZnT8A là các chỉ số của tự miễn dịch đang diễn ra. Điều thú vị là, ở những bệnh nhân bị APS I, GADA không có khả năng dự đoán cao về sự phát triển sau đó của đái tháo đường tuýp 1.
Như đã nêu trước đây, việc phát hiện các tự kháng thể trong APS phục vụ một số chức năng quan trọng. Đầu tiên, việc phát hiện các tự kháng thể cho phép thiết lập một chẩn đoán tự miễn cụ thể. Thứ hai, việc phát hiện tự kháng thể ở những người không có triệu chứng cho thấy nguy cơ gia tăng phát triển bệnh lâm sàng sau này và bệnh nhân được theo dõi chặt chẽ hơn. Thứ ba, sự hiện diện của một tự kháng thể hoặc một bệnh tự miễn ở một cá nhân có thể gợi ý nguy cơ gia tăng đối với các bệnh tự miễn liên quan khác.
Các Tự kháng thể Không Đặc hiệu Cơ quan
1) Các Tự kháng thể Kháng Interferon
Các tự kháng thể đối với interferon (α và ω) có độ đặc hiệu và độ nhạy cao đối với APS 1, và một số tác giả đã ủng hộ việc sử dụng các kháng thể này như các xét nghiệm sàng lọc thay vì đánh giá các đột biến AIRE. Những tự kháng thể này lần đầu tiên được sử dụng trên lâm sàng như các chỉ số nguy cơ mắc bệnh nhược cơ và u tuyến ức, và các AIA có hiệu giá cao đã được tìm thấy bất ngờ ở 60/60 bệnh nhân APS I Phần Lan và 16/16 bệnh nhân APS I Na Uy. Đáng chú ý, các AIA đã được phát hiện trong các mẫu huyết thanh được lưu trữ ở những bệnh nhân APS I đã biết trước khi khởi phát các tự kháng thể đặc hiệu cơ quan. Trên thực tế, một số bệnh nhân APS I đã phát triển AIA trước khi khởi phát nhiễm nấm candida niêm mạc da hoặc các biểu hiện cơ quan khác. Để làm bằng chứng thêm về tính đặc hiệu của AIA, các đối tượng có đột biến AIRE nhưng thiếu các đặc điểm kinh điển của APSI (nhiễm nấm candida niêm mạc da, suy tuyến cận giáp, hoặc suy thượng thận) cũng đã chứng minh hiệu giá cao đối với AIA. Do đó, AIA có khả năng trở thành xét nghiệm sàng lọc chính cho APS I; AIA có thể được phát hiện bằng một xét nghiệm miễn dịch cạnh tranh. Các nghiên cứu trên động vật gần đây cũng đã chứng minh rằng các tự kháng thể IFN-ω có tác dụng trung hòa in vitro và in vivo, với khả năng cải thiện các quá trình bệnh tự miễn ở APS 1. Các tác giả của cùng một nghiên cứu cũng đặt câu hỏi liệu tỷ lệ phát triển đái tháo đường tuýp 1 thấp hơn ở bệnh nhân APS I (10%-20%), mặc dù nhiều người có các tự kháng thể GAD, có thể liên quan đến sự hiện diện của các tự kháng thể IFN-α trung hòa này hay không.
2) Các Tự kháng thể đối với IL-17A, IL-17F, và IL-22
Nhiễm nấm candida niêm mạc da thường là bất thường lâm sàng khởi phát ở APS I. Mặc dù AIA có thể dự đoán APS I, chúng không dự đoán thứ tự hoặc mức độ nghiêm trọng của các thành phần APS I đã biết. Do đó, cần có các tự kháng thể đặc hiệu bệnh hoặc cơ quan để cung cấp cho bệnh nhân và bác sĩ sự hướng dẫn dự phòng tối ưu. Một số nhóm đã báo cáo rằng các tự kháng thể đối với các cytokine, chẳng hạn như IL-17A, IL-17F, và IL-22, có thể vừa dự đoán vừa giải thích nhiễm nấm candida niêm mạc da liên quan đến APS 1. Trong điều kiện bình thường, các cytokine gây viêm này có đặc tính chống nấm và diệt khuẩn được sản xuất bởi các tế bào Th17.
Trong một nghiên cứu, 33/33 bệnh nhân bị APSI và nhiễm nấm candida niêm mạc da đã chứng minh các tự kháng thể có hiệu giá cao đối với IL-17A, IL-17F, và IL-22, trong khi 0/37 người đối chứng, và 0/103 bệnh nhân bị các bệnh tự miễn riêng lẻ dương tính với các kháng thể này. Những quan sát này đã được xác nhận trong các nghiên cứu thuần tập sau đó. Tuy nhiên, những tự kháng thể này hiện chỉ được sử dụng trong môi trường nghiên cứu.
Các Tự kháng thể Đặc hiệu Cơ quan
Các Tự kháng thể Tế bào chất Tuyến thượng thận
Các tự kháng thể tế bào chất tuyến thượng thận (ACA) lần đầu tiên được phát hiện bằng kỹ thuật cố định bổ thể với các chiết xuất nước muối của mô tuyến thượng thận và ngay sau đó bằng miễn dịch huỳnh quang gián tiếp. Thường tất cả các lớp của vỏ thượng thận (nhưng không phải tủy) đều phát huỳnh quang. Vị trí ở microsome của các tự kháng nguyên đã được xác nhận bằng cách sử dụng các thành phần tế bào được siêu ly tâm. Các xét nghiệm khác đối với các tự kháng thể tuyến thượng thận bao gồm các xét nghiệm miễn dịch phóng xạ pha rắn và các xét nghiệm miễn dịch hấp phụ liên kết với enzyme không phóng xạ.
ACA được phát hiện trong tất cả các dạng bệnh Addison tự miễn, cho dù là bệnh Addison riêng lẻ hay là một phần của APS I hoặc APS II. Khoảng 50% những người dương tính với ACA không có triệu chứng sẽ phát triển bệnh Addison trong vòng 3 năm, và có tới 75% đến 100% những người mới khởi phát hoặc gần khởi phát lâm sàng bệnh Addison có biểu hiện ACA. Trong một theo dõi 20 trẻ em dương tính với ACA trong tối đa 11 năm, nguy cơ tích lũy phát triển bệnh Addison là 100%. ACA cũng có khả năng dự đoán sự phát triển sau đó của bệnh Addison ở người lớn, mặc dù ít thường xuyên hơn so với ở trẻ em. Các hiệu giá ACA cao hơn và ACA cố định bổ thể đã được liên kết với nguy cơ gia tăng tiến triển thành bệnh lâm sàng.
Các tự kháng thể đối với bề mặt của các tế bào vỏ thượng thận đã được mô tả, và gần 90% những người bị bệnh Addison đã được báo cáo là có biểu hiện những tự kháng thể này. Tuy nhiên, những xét nghiệm này hiếm khi được thực hiện do khó khăn trong việc lấy mô tuyến thượng thận tươi của người hoặc động vật được sử dụng trong các xét nghiệm này.
Tự kháng thể kháng Enzyme tuyến thượng thận
Điển hình của nhiều bệnh tự miễn đặc hiệu cho từng cơ quan, các tự kháng nguyên chính đóng vai trò là mục tiêu của các tự kháng thể trong APS là các enzyme. Ví dụ về các bệnh tự miễn khác nhau trong đó enzyme là mục tiêu của hệ thống miễn dịch được minh họa trong Bảng 22.2. Thảo luận về tổng hợp hormone tuyến thượng thận có thể được tìm thấy trong Chương 14.
21-Hydroxylase (P450c21) là một tự kháng nguyên chính được nhận diện bởi các tự kháng thể trong huyết thanh của bệnh nhân mắc bệnh Addison. Có một mối tương quan mạnh mẽ giữa sự dương tính với ACA và tự kháng thể P450c21 (21-hydroxylase), và các tự kháng thể P450c21 dường như là một chỉ số nhạy hơn của bệnh. Gần 75% bệnh nhân APS I và APS II có tự kháng thể P450c21 dương tính. Các enzyme khác đã được xác định là tự kháng nguyên ở những bệnh nhân mắc bệnh Addison tự miễn đơn độc hoặc APS, bao gồm enzyme phân cắt chuỗi bên cholesterol P450 (P450scc), 17α-hydroxylase (P450c17), và 3ß-hydroxysteroid dehydrogenase.
Các epitope tự kháng nguyên của enzyme P450c21 nằm ở đầu C-cuối và ở vùng trung tâm của enzyme. Người ta đã báo cáo rằng hai trong bốn epitope được nhận diện bởi các tự kháng thể P450c17 có phản ứng chéo với P450c21, cho thấy rằng phản ứng với một trong những tự kháng nguyên này thực sự có thể phản ánh sự bắt chước phân tử giữa các epitope như vậy. Ngoại trừ các acid amin đầu N 1–40 và các acid amin đầu C 456–521, các epitope phản ứng miễn dịch đã được mô tả trên toàn bộ P450scc.
Nồng độ cao của cả tự kháng thể ACA và P450c21 đều liên quan đến sự suy giảm chức năng vỏ thượng thận nhiều hơn và sự phát triển sau đó của bệnh Addison. Cũng như nhiều bệnh tự miễn khác, có một mối tương quan nghịch giữa nồng độ tự kháng thể và thời gian mắc bệnh ở những bệnh nhân bị bệnh Addison. Điều này phù hợp với khái niệm rằng một khi một tự kháng nguyên bị phá hủy hoàn toàn, hệ thống miễn dịch không còn được kích thích để sản xuất tự kháng thể.
Khi có bệnh Addison lâm sàng hoặc tiền lâm sàng, không có sự kết hợp đặc trưng nào của các kháng thể tuyến thượng thận hoặc tuyến sinh dục để phân biệt APS I với APS II. Sự khác biệt giữa APS I và APS II được thực hiện trên cơ sở lâm sàng hoặc, ở những bệnh nhân không có triệu chứng, bằng cách phát hiện các tự kháng thể đồng thời liên quan đến APS II. Hơn nữa, không có epitope đặc trưng nào được nhận diện bởi các tự kháng thể P450c21 cho phép phân biệt bệnh Addison đơn độc với APS I hoặc APS II. Tuy nhiên, APS I có thể được phân biệt chắc chắn với APS II thông qua các xét nghiệm chẩn đoán phân tử để tìm đột biến trong gen AIRE và được suy ra bởi sự hiện diện (APS I) hoặc vắng mặt (APS II) của các kháng thể interferon.
Tự kháng thể Tế bào Steroid/Tuyến sinh dục
Một số cá nhân có ACA có huyết thanh phản ứng chéo với các mô sản xuất steroid sinh sản, bao gồm lớp vỏ trong của nang Graaf, tế bào Leydig của tinh hoàn, và/hoặc lớp hợp bào nuôi của nhau thai. Các huyết thanh nhận diện các kháng nguyên trong cả mô sản xuất steroid tuyến thượng thận và sinh sản mà phản ứng miễn dịch của chúng không thể bị hấp phụ bởi các chiết xuất tuyến thượng thận được gọi là kháng thể tế bào steroid/tuyến sinh dục (SCA). Sự thay đổi như vậy trong phản ứng miễn dịch có thể đại diện cho sự khác biệt về mật độ tự kháng nguyên hoặc sự khác biệt về tính khả dụng của epitope giữa các mô.
Ở những bệnh nhân không có triệu chứng, SCA có liên quan đến nguy cơ gia tăng phát triển suy tuyến sinh dục tự miễn nguyên phát. Ở phụ nữ, điều này thường biểu hiện dưới dạng vô kinh nguyên phát hoặc mãn kinh sớm; nam giới thường không có triệu chứng. SCA, tùy thuộc vào xét nghiệm được sử dụng, được tìm thấy ở 4% đến 87% phụ nữ bị suy buồng trứng sớm. SCA cũng đã được báo cáo là có khả năng dự đoán suy tuyến sinh dục ở phụ nữ mắc APS có kinh nguyệt bình thường tại thời điểm nghiên cứu ban đầu. Trong một nghiên cứu thuần tập nhỏ gồm 11 bệnh nhân nữ APS I, 100% (11/11) người dương tính với SCA đã phát triển suy buồng trứng nguyên phát trong thời gian theo dõi 12 năm. Khi có ACA mà không có SCA, suy tuyến sinh dục là hiếm gặp. Tuy nhiên, khoảng một phần ba cá nhân có ACA có SCA (sử dụng buồng trứng, tinh hoàn hoặc nhau thai làm nguồn kháng nguyên). Một nghiên cứu nhỏ trên năm phụ nữ có SCA cũng có ACA, phù hợp với APS 1, đã biểu hiện suy buồng trứng. SCA phổ biến hơn ở bệnh nhân APS I so với APS II, và ước tính rằng 60% đối tượng APS I và bệnh Addison có SCA, so với 30% bệnh nhân APS II.
Suy buồng trứng sớm độc lập với APS cũng có thể xảy ra do hậu quả của tự miễn dịch. Tuy nhiên, bệnh Addison cùng tồn tại ở khoảng 2% đến 10% phụ nữ bị suy buồng trứng sớm tự miễn, và do đó nên được tìm kiếm. Giống như các bệnh tự miễn đặc hiệu cho từng cơ quan khác, suy buồng trứng tự miễn được đặc trưng về mặt mô học bởi sự thâm nhiễm của buồng trứng bởi các tế bào viêm.
Bản chất của các tự kháng nguyên buồng trứng trong suy buồng trứng sớm còn gây tranh cãi. Các tự kháng thể phản ứng mạnh với màng trong suốt (zona pellucida) của lợn, được xác định bằng phương pháp miễn dịch huỳnh quang gián tiếp, đã được quan sát thấy ở sáu trong số 22 phụ nữ bị vô sinh. Vì vỏ thượng thận và tuyến sinh dục có chung một số con đường tổng hợp phổ biến cho các tế bào sản xuất cả steroid vỏ thượng thận và steroid sinh dục, nên có thể mong đợi sự tự miễn dịch thể dịch đối với các tự kháng nguyên chung ở những bệnh nhân bị cả bệnh Addison và suy tuyến sinh dục. Việc tổng hợp các steroid sinh dục và tuyến thượng thận đòi hỏi P450scc, 3ß-hydroxysteroid dehydrogenase, và P450c17. Tuy nhiên, dữ liệu còn mâu thuẫn. Trong một nghiên cứu, hoạt tính SCA đã được loại bỏ bằng cách tiền hấp phụ với 3ß-hydroxysteroid dehydrogenase tái tổ hợp của người, cho thấy rằng enzyme này là một tự kháng nguyên chính được phát hiện bởi các huyết thanh dương tính với SCA. Một nghiên cứu khác cho thấy SCA tương quan tốt nhất với phản ứng với P450scc và một tự kháng nguyên 51-KDa liên kết với decarboxylase acid amin L-thơm có trong các tế bào hạt, nhau thai, gan và tế bào beta của tụy.
Tự kháng thể trong Suy tuyến cận giáp
Suy tuyến cận giáp tự miễn là đặc trưng của APS I. Trong báo cáo ban đầu về các tự kháng thể tuyến cận giáp được phát hiện bằng phương pháp miễn dịch huỳnh quang gián tiếp, gần 40% bệnh nhân bị suy tuyến cận giáp tự miễn dương tính với các tự kháng thể tế bào chất tuyến cận giáp so với 6% ở nhóm chứng. Tuy nhiên, các phòng thí nghiệm khác đã không xác nhận các báo cáo ban đầu về sự tồn tại của các tự kháng thể tế bào chất tuyến cận giáp như vậy. Người ta đã chỉ ra rằng các tự kháng thể được phát hiện bằng phương pháp miễn dịch huỳnh quang gián tiếp chống lại tuyến cận giáp có thể được tiền hấp phụ bằng ty thể của người, cho thấy rằng các tự kháng thể như vậy không đặc hiệu cho mô.
Kể từ đó, một số tự kháng thể kháng tuyến cận giáp khác nhau đã được báo cáo ở những bệnh nhân bị suy tuyến cận giáp. Việc sàng lọc các thư viện DNA bổ sung của tuyến cận giáp người bằng huyết thanh từ những bệnh nhân đã biết mắc APS I và suy tuyến cận giáp đã xác định NALP5 là một tự kháng nguyên tuyến cận giáp quan trọng. Các tự kháng thể đã được phát hiện ở 49% bệnh nhân APS I và suy tuyến cận giáp, nhưng không có ở tất cả 293 người trong nhóm chứng. Mặc dù các tự kháng thể NALP5 hiếm khi (0,69%) được thấy ở những bệnh nhân bị suy tuyến cận giáp vô căn, độ đặc hiệu của chúng để chẩn đoán suy tuyến cận giáp liên quan đến APS I chỉ khoảng 50% và độ nhạy là 26%. Các tự kháng thể hoạt hóa đối với CaSR cũng được thấy ở những bệnh nhân bị suy tuyến cận giáp tự miễn. Độ đặc hiệu của các tự kháng thể CaSR để chẩn đoán suy tuyến cận giáp tự miễn là 83%, tốt hơn so với các tự kháng thể NALP. Tuy nhiên, độ nhạy của các tự kháng thể CaSR ở mức 39% vẫn còn thấp.
Các Tự kháng thể khác trong APS I và II
Ngoài các tự kháng thể đã được thảo luận trước đó, các tự kháng thể đối với tyrosine hydroxylase đã được báo cáo ở khoảng 40% bệnh nhân APS I và dường như có tương quan với rụng tóc từng mảng, trong khi các tự kháng thể đối với tryptophan hydroxylase tương quan với tình trạng kém hấp thu ở ruột. Các kháng thể đối với microsome gan/thận loại 1 (LKM1) được tìm thấy ở gần 100% bệnh nhân bị viêm gan tự miễn tuýp 2. Khoảng 40% bệnh nhân như vậy có bệnh tự miễn liên quan thường thấy trong APS I. Các tự kháng nguyên gan P450IA2 và P4502A6 dường như là mục tiêu của các kháng thể này. Các tự kháng thể đối với decarboxylase acid amin L-thơm cũng đã được nhận diện ở bệnh nhân APS I, và có liên quan đến viêm gan tự miễn và bạch biến. Các kháng thể đối với enolase tuyến yên của người có liên quan đến viêm tuyến yên tự miễn. Cuối cùng, các tự kháng thể chống lại BPIFB1 (bactericidal/permeability-increasing fold-containing B1) và chất điều hòa kênh kali KCNRG đã được liên kết với sự phát triển của viêm phổi. BPIFB1 đã được báo cáo là có độ nhạy cao, trong khi KCNRG có độ nhạy thấp, nhưng độ đặc hiệu 100% đối với viêm phổi.
Tóm tắt
APS là kết quả của sự mất dung nạp đối với các kháng nguyên tự thân. APS I, một rối loạn di truyền lặn trên nhiễm sắc thể thường được lập bản đồ trên gen AIRE, được định nghĩa bởi sự hiện diện của ít nhất hai trong ba phát hiện sau: (1) tự miễn dịch vỏ thượng thận, (2) suy tuyến cận giáp, hoặc (3) nhiễm nấm candida niêm mạc da. APS II được định nghĩa bởi sự cùng tồn tại của suy vỏ thượng thận tự miễn hoặc bằng chứng huyết thanh học về viêm tuyến thượng thận với viêm tuyến giáp tự miễn và/hoặc đái tháo đường tuýp 1. Sự xuất hiện của bất kỳ thành phần bệnh nào của một APS có thể liên quan đến sự xuất hiện của các thành phần khác thông qua các gen nền tự miễn dịch chung có liên quan đến sự mất dung nạp. Do đó, cần phải có một chỉ số nghi ngờ cao bất cứ khi nào một rối loạn tự miễn được chẩn đoán. Ví dụ, trong đái tháo đường tuýp 1, việc sàng lọc bệnh tự miễn tuyến giáp và bệnh celiac là thường quy.
Điều trị APS nên nhằm vào việc quản lý tối ưu các bệnh nền cụ thể. Nên thực hiện sàng lọc thường xuyên để phát hiện sự hiện diện của các rối loạn tự miễn liên quan. Tóm lại, sự hiểu biết được cải thiện về sự tương tác giữa các gen nhạy cảm, các tác nhân kích hoạt từ môi trường, nền tảng gen cá nhân hóa và mối quan hệ của nó với sự phát triển của suy giảm dung nạp miễn dịch, các ứng dụng miễn dịch học mới (chẳng hạn như tạo ra các tế bào biểu mô tuyến ức từ các tế bào gốc hoặc sử dụng các tế bào Treg được thiết kế) sẽ là con đường tốt nhất để cải thiện các phương thức chẩn đoán và điều trị trong việc chăm sóc bệnh nhân APS.
Đái tháo đường tuýp 1 tự miễn và các bệnh nội tiết miễn dịch khác trước 6 tháng tuổi, liên quan đến bệnh ruột ở nam giới, nên đặt ra sự cân nhắc về hội chứng IPEX và phân tích đột biến gen FOXP3; nếu không có bằng chứng về tự miễn dịch, ví dụ như kháng thể kháng đảo tụy, nên xem xét một dạng đái tháo đường sơ sinh vì các liệu pháp cụ thể cho bệnh sau bao gồm các loại thuốc uống hiệu quả và có thể không cần insulin (xem Chương 10).
Việc sử dụng ngày càng nhiều các chất ức chế điểm kiểm soát miễn dịch có liên quan đến các phản ứng có hại khác nhau, bao gồm các bệnh nội tiết, chẳng hạn như viêm tuyến yên dẫn đến suy thượng thận thứ phát, hoặc suy thượng thận nguyên phát, viêm tuyến giáp, và đôi khi là đái tháo đường tuýp 1. Những tình trạng này có thể xảy ra ở 10% đến 20% bệnh nhân người lớn từ vài tuần đến vài tháng sau khi sử dụng thuốc, mặc dù tần suất ở trẻ em và thanh thiếu niên vẫn chưa được xác định. Tuy nhiên, các bác sĩ nên nhận thức được những thực thể này và điều trị liệu pháp thay thế hormone phù hợp theo chỉ định.
BẢNG CHÚ GIẢI THUẬT NGỮ Y HỌC ANH – VIỆT. CHƯƠNG 22
| STT | Thuật ngữ tiếng Anh | Phiên âm IPA (US) | Nghĩa Tiếng Việt |
|---|---|---|---|
| 1 | Autoimmune Polyglandular Syndromes (APS) | /ˌɔːtoʊɪˈmjuːn ˌpɑːliˈɡlændʒələr ˈsɪndroʊmz/ | Hội chứng Đa tuyến Tự miễn |
| 2 | Organ-specific autoimmune diseases | /ˈɔːrɡən spəˈsɪfɪk ˌɔːtoʊɪˈmjuːn dɪˈziːzɪz/ | Bệnh tự miễn đặc hiệu cơ quan |
| 3 | Endocrine | /ˈen.də.krɪn/ | Nội tiết |
| 4 | Non-endocrine | /ˌnɑːnˈen.də.krɪn/ | Không nội tiết |
| 5 | Self-tolerance | /self ˈtɑːlərəns/ | Dung nạp tự thân |
| 6 | Tolerance | /ˈtɑːlərəns/ | Dung nạp |
| 7 | Self-antigens | /self ˈæntɪdʒənz/ | Tự kháng nguyên |
| 8 | Immune response | /ɪˈmjuːn rɪˈspɑːns/ | Đáp ứng miễn dịch |
| 9 | Autoimmunity | /ˌɔːtoʊɪˈmjuːnəti/ | Tự miễn |
| 10 | Innate immunity | /ɪˈneɪt ɪˈmjuːnəti/ | Miễn dịch bẩm sinh |
| 11 | Pathogens | /ˈpæθədʒənz/ | Mầm bệnh |
| 12 | Germline genes | /ˈdʒɜːrmlaɪn dʒiːnz/ | Gen dòng mầm |
| 13 | Immunologic memory | /ˌɪmjənəˈlɑːdʒɪk ˈmeməri/ | Trí nhớ miễn dịch |
| 14 | Adaptive immunity | /əˈdæptɪv ɪˈmjuːnəti/ | Miễn dịch thích ứng |
| 15 | T- and B-cell surface receptors | /tiː ənd biː sel ˈsɜːrfəs rɪˈseptərz/ | Thụ thể bề mặt tế bào T và B |
| 16 | Peptides | /ˈpeptaɪdz/ | Peptide |
| 17 | Epitopes | /ˈepɪtoʊps/ | Epitope (Quyết định kháng nguyên) |
| 18 | Antigen-presenting cells (APCs) | /ˈæntɪdʒən prɪˈzentɪŋ selz/ | Tế bào trình diện kháng nguyên (APC) |
| 19 | Major histocompatibility complex (MHC) | /ˈmeɪdʒər ˌhɪstoʊkəmˌpætəˈbɪləti ˈkɑːmpleks/ | Phức hợp tương hợp mô chính (MHC) |
| 20 | Human leukocyte antigen (HLA) | /ˈhjuːmən ˈluːkəsaɪt ˈæntɪdʒən/ | Kháng nguyên bạch cầu người (HLA) |
| 21 | Cytosol-derived endogenous peptides | /ˈsaɪtəˌsɔːl dɪˈraɪvd enˈdɑːdʒənəs ˈpeptaɪdz/ | Peptide nội sinh có nguồn gốc từ bào tương |
| 22 | CD8 T-cells | /siː diː eɪt tiː selz/ | Tế bào T CD8 |
| 23 | CD4 T-cells | /siː diː fɔːr tiː selz/ | Tế bào T CD4 |
| 24 | Cytotoxic T-cells | /ˌsaɪtoʊˈtɑːksɪk tiː selz/ | Tế bào T gây độc tế bào |
| 25 | Helper or regulatory T-cells | /ˈhelpər ɔːr ˈreɡjələˌtɔːri tiː selz/ | Tế bào T hỗ trợ hoặc điều hòa |
| 26 | Central tolerance | /ˈsentrəl ˈtɑːlərəns/ | Dung nạp trung ương |
| 27 | Peripheral tolerance | /pəˈrɪfərəl ˈtɑːlərəns/ | Dung nạp ngoại vi |
| 28 | Thymus | /ˈθaɪməs/ | Tuyến ức |
| 29 | Lymphoid tissues | /ˈlɪmfɔɪd ˈtɪʃuːz/ | Mô bạch huyết |
| 30 | Autoimmune regulator (AIRE) gene | /ˌɔːtoʊɪˈmjuːn ˈreɡjəleɪtər dʒiːn/ | Gen điều hòa tự miễn (AIRE) |
| 31 | Transcription factor | /trænˈskrɪpʃən ˈfæktər/ | Yếu tố phiên mã |
| 32 | Thymic medullary | /ˈθaɪmɪk ˈmedəleri/ | Thuộc tủy tuyến ức |
| 33 | Multi-organ autoimmunity | /ˈmʌlti ˈɔːrɡən ˌɔːtoʊɪˈmjuːnəti/ | Tự miễn đa cơ quan |
| 34 | Hematopoietic stem cells | /ˌhiːmətoʊpɔɪˈetɪk stem selz/ | Tế bào gốc tạo máu |
| 35 | Bone marrow | /boʊn ˈmæroʊ/ | Tủy xương |
| 36 | Thymic involution | /ˈθaɪmɪk ˌɪnvəˈluːʃən/ | Sự teo tuyến ức |
| 37 | Co-stimulatory signals | /koʊ ˈstɪmjələˌtɔːri ˈsɪɡnəlz/ | Tín hiệu đồng kích thích |
| 38 | Thymic cortex | /ˈθaɪmɪk ˈkɔːrteks/ | Vỏ tuyến ức |
| 39 | Double-positive CD4+CD8+ T-cells | /ˈdʌbəl ˈpɑːzətɪv siː diː fɔːr siː diː eɪt tiː selz/ | Tế bào T dương tính kép CD4+CD8+ |
| 40 | Apoptosis | /ˌæpəpˈtoʊsɪs/ | Chết tế bào theo chương trình |
| 41 | Positive selection | /ˈpɑːzətɪv sɪˈlekʃən/ | Chọn lọc dương tính |
| 42 | Cortical thymic epithelial cells (cTECs) | /ˈkɔːrtɪkəl ˈθaɪmɪk ˌepəˈθiːliəl selz/ | Tế bào biểu mô vỏ tuyến ức |
| 43 | Corticomedullary junction | /ˌkɔːrtɪkoʊˈmedəleri ˈdʒʌŋkʃən/ | Vùng nối vỏ-tủy |
| 44 | Medulla | /məˈdʌlə/ | Tủy |
| 45 | Negative selection | /ˈneɡətɪv sɪˈlekʃən/ | Chọn lọc âm tính |
| 46 | Medullary thymic epithelial cells (mTECs) | /ˈmedəleri ˈθaɪmɪk ˌepəˈθiːliəl selz/ | Tế bào biểu mô tủy tuyến ức |
| 47 | Peripheral tissue-specific antigens (TSAs) | /pəˈrɪfərəl ˈtɪʃuː spəˈsɪfɪk ˈæntɪdʒənz/ | Kháng nguyên đặc hiệu mô ngoại vi |
| 48 | Promiscuous gene expression (PGE) | /prəˈmɪskjuəs dʒiːn ɪkˈspreʃən/ | Biểu hiện gen lộn xộn |
| 49 | Ribonucleic acid (RNA) polymerases | /ˌraɪboʊnuːˈkliːɪk ˈæsɪd pəˈlɪməreɪsɪz/ | RNA polymerase |
| 50 | Chemokine | /ˈkiːməkaɪn/ | Chemokine |
| 51 | Anergy | /ˈænərdʒi/ | Vô năng |
| 52 | Spleen | /spliːn/ | Lách |
| 53 | Nonpolymorphic | /ˌnɑːnpɑːliˈmɔːrfɪk/ | Không đa hình |
| 54 | Cytokines | /ˈsaɪtəkaɪnz/ | Cytokine |
| 55 | Cytotoxic T-lymphocyte antigen 4 (CTLA-4) | /ˌsaɪtoʊˈtɑːksɪk tiː ˈlɪmfəsaɪt ˈæntɪdʒən fɔːr/ | Kháng nguyên 4 của tế bào lympho T gây độc tế bào (CTLA-4) |
| 56 | Immunosuppressive/immunoregulatory signal | /ˌɪmjənoʊsəˈpresɪv ˌɪmjənoʊˈreɡjələˌtɔːri ˈsɪɡnəl/ | Tín hiệu ức chế miễn dịch/điều hòa miễn dịch |
| 57 | Programmed cell death protein 1 (PD-1) | /ˈproʊɡræmd sel deθ ˈproʊtiːn wʌn/ | Protein 1 gây chết tế bào theo chương trình (PD-1) |
| 58 | Ligands | /ˈlaɪɡəndz/ | Phối tử |
| 59 | Effector functions | /ɪˈfektər ˈfʌŋkʃənz/ | Chức năng hiệu ứng |
| 60 | Th1 cells | /tiː eɪtʃ wʌn selz/ | Tế bào Th1 |
| 61 | Th2 cells | /tiː eɪtʃ tuː selz/ | Tế bào Th2 |
| 62 | Interleukin (IL) | /ˌɪntərˈluːkɪn/ | Interleukin (IL) |
| 63 | Interferon-gamma (IFN-γ) | /ˌɪntərˈfɪrɑːn ˈɡæmə/ | Interferon-gamma (IFN-γ) |
| 64 | Tumor necrosis factor-beta (TNF-β) | /ˈtuːmər nəˈkroʊsɪs ˈfæktər ˈbeɪtə/ | Yếu tố hoại tử khối u-beta (TNF-β) |
| 65 | Macrophages | /ˈmækroʊfeɪdʒɪz/ | Đại thực bào |
| 66 | Eosinophils | /ˌiːəˈsɪnəfɪlz/ | Bạch cầu ái toan |
| 67 | Regulatory T-cells (Tregs) | /ˈreɡjələˌtɔːri tiː selz/ | Tế bào T điều hòa (Treg) |
| 68 | Forkhead box transcription factor (FOXP3) | /ˈfɔːrkhed bɑːks trænˈskrɪpʃən ˈfæktər/ | Yếu tố phiên mã hộp forkhead (FOXP3) |
| 69 | Scurfy mouse | /ˈskɜːrfi maʊs/ | Chuột Scurfy |
| 70 | X-linked recessive | /eks lɪŋkt rɪˈsesɪv/ | Lặn liên kết nhiễm sắc thể X |
| 71 | IPEX syndrome | /ˈaɪpeks ˈsɪndroʊm/ | Hội chứng IPEX |
| 72 | B-cell receptors (BCRs) | /biː sel rɪˈseptərz/ | Thụ thể tế bào B (BCR) |
| 73 | Clonal deletion | /ˈkloʊnəl dɪˈliːʃən/ | Xóa sổ dòng |
| 74 | Receptor editing | /rɪˈseptər ˈedɪtɪŋ/ | Chỉnh sửa thụ thể |
| 75 | Affinity maturation | /əˈfɪnəti ˌmætʃəˈreɪʃən/ | Trưởng thành ái lực |
| 76 | Class switching | /klæs ˈswɪtʃɪŋ/ | Chuyển lớp |
| 77 | Thyroglobulin | /ˌθaɪroʊˈɡlɑːbjəlɪn/ | Thyroglobulin |
| 78 | Hashimoto’s thyroiditis | /ˌhɑːʃiˈmoʊtoʊz ˌθaɪrɔɪˈdaɪtɪs/ | Viêm tuyến giáp Hashimoto |
| 79 | Ischemia | /ɪˈskiːmiə/ | Thiếu máu cục bộ |
| 80 | Irradiation | /ɪˌreɪdiˈeɪʃən/ | Chiếu xạ |
| 81 | Sympathetic ophthalmia | /ˌsɪmpəˈθetɪk ɑːfˈθælmiə/ | Viêm mắt giao cảm |
| 82 | Myocardial infarction | /ˌmaɪəˈkɑːrdiəl ɪnˈfɑːrkʃən/ | Nhồi máu cơ tim |
| 83 | Molecular mimicry | /məˈlekjələr ˈmɪmɪkri/ | Bắt chước phân tử |
| 84 | Cross-reactivity | /krɔːs ˌriːækˈtɪvəti/ | Phản ứng chéo |
| 85 | Opsonins | /ˈɑːpsənɪnz/ | Opsonin |
| 86 | Phagocytic cells | /ˌfæɡəˈsɪtɪk selz/ | Tế bào thực bào |
| 87 | Antibody-dependent cellular cytotoxicity | /ˈæntɪbɑːdi dɪˈpendənt ˈseljələr ˌsaɪtoʊtɑːkˈsɪsəti/ | Độc tế bào phụ thuộc kháng thể |
| 88 | Rheumatic fever | /ruːˈmætɪk ˈfiːvər/ | Thấp tim |
| 89 | Chorea | /kəˈriːə/ | Múa giật |
| 90 | Superantigens | /ˌsuːpərˈæntɪdʒənz/ | Siêu kháng nguyên |
| 91 | Polyclonal | /ˌpɑːliˈkloʊnəl/ | Đa dòng |
| 92 | Systemic inflammatory response syndrome (SIRS) | /sɪˈstemɪk ɪnˈflæməˌtɔːri rɪˈspɑːns ˈsɪndroʊm/ | Hội chứng đáp ứng viêm hệ thống (SIRS) |
| 93 | Toxic shock syndrome | /ˈtɑːksɪk ʃɑːk ˈsɪndroʊm/ | Hội chứng sốc nhiễm độc |
| 94 | Crohn’s disease | /kroʊnz dɪˈziːz/ | Bệnh Crohn |
| 95 | Epstein-Barr virus | /ˈepstaɪn bɑːr ˈvaɪrəs/ | Virus Epstein-Barr |
| 96 | Lipopolysaccharide | /ˌlaɪpoʊˌpɑːliˈsækəraɪd/ | Lipopolysaccharide |
| 97 | Gliadin | /ˈɡlaɪədɪn/ | Gliadin |
| 98 | Celiac disease | /ˈsiːliæk dɪˈziːz/ | Bệnh Celiac |
| 99 | Penicillamine | /ˌpenəˈsɪləmiːn/ | Penicillamine |
| 100 | Myasthenia gravis | /ˌmaɪəsˈθiːniə ˈɡrævɪs/ | Bệnh nhược cơ |
| 101 | Methimazole | /məˈθɪməzoʊl/ | Methimazole |
| 102 | Amiodarone | /ˌæmiˈoʊdərɒn/ | Amiodarone |
| 103 | Thymoma | /θaɪˈmoʊmə/ | U tuyến ức |
| 104 | Ovarian teratoma | /oʊˈveriən ˌterəˈtoʊmə/ | U quái buồng trứng |
| 105 | Stiff-person syndrome | /stɪf ˈpɜːrsən ˈsɪndroʊm/ | Hội chứng người cứng |
| 106 | Checkpoint inhibitors | /ˈtʃekpɔɪnt ɪnˈhɪbɪtərz/ | Chất ức chế điểm kiểm soát |
| 107 | Monoclonal antibodies | /ˌmɑːnoʊˈkloʊnəl ˈæntɪbɑːdiz/ | Kháng thể đơn dòng |
| 108 | Immune-related adverse effects (irAEs) | /ɪˈmjuːn rɪˈleɪtɪd ædˈvɜːrs ɪˈfekts/ | Tác dụng phụ liên quan đến miễn dịch (irAE) |
| 109 | Hypophysitis | /ˌhaɪpoʊfɪˈsaɪtɪs/ | Viêm tuyến yên |
| 110 | Hepatitis | /ˌhepəˈtaɪtɪs/ | Viêm gan |
| 111 | Dermatitis | /ˌdɜːrməˈtaɪtɪs/ | Viêm da |
| 112 | Colitis | /kəˈlaɪtɪs/ | Viêm đại tràng |
| 113 | Thyroiditis | /ˌθaɪrɔɪˈdaɪtɪs/ | Viêm tuyến giáp |
| 114 | Adrenalitis | /əˌdriːnəˈlaɪtɪs/ | Viêm tuyến thượng thận |
| 115 | Myocarditis | /ˌmaɪəʊkɑːrˈdaɪtɪs/ | Viêm cơ tim |
| 116 | Cross-presentation | /krɔːs ˌpriːzenˈteɪʃən/ | Trình diện chéo |
| 117 | Epitope spreading | /ˈepɪtoʊp ˈspredɪŋ/ | Lan truyền epitope |
| 118 | Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) | /ˌɔːtoʊɪˈmjuːn ˌpɑːliˌendoʊkrəˈnɑːpəθi ˌkændɪˈdaɪəsɪs ˌektoʊˈdɜːrməl ˈdɪstrəfi/ | Bệnh đa nội tiết-nấm candida-loạn dưỡng ngoại bì tự miễn (APECED) |
| 119 | Adrenocortical failure (Addison disease) | /əˌdriːnoʊˈkɔːrtɪkəl ˈfeɪljər ˈædɪsənz dɪˈziːz/ | Suy vỏ thượng thận (bệnh Addison) |
| 120 | Hypoparathyroidism | /ˌhaɪpoʊˌpærəˈθaɪrɔɪdɪzəm/ | Suy tuyến cận giáp |
| 121 | Chronic mucocutaneous candidiasis | /ˈkrɑːnɪk ˌmjuːkoʊkjuːˈteɪniəs ˌkændɪˈdaɪəsɪs/ | Nhiễm nấm candida niêm mạc da mạn tính |
| 122 | Schmidt syndrome | /ʃmɪt ˈsɪndroʊm/ | Hội chứng Schmidt |
| 123 | Carpenter syndrome | /ˈkɑːrpəntər ˈsɪndroʊm/ | Hội chứng Carpenter |
| 124 | Pernicious anemia | /pərˈnɪʃəs əˈniːmiə/ | Thiếu máu ác tính |
| 125 | Vitiligo | /ˌvɪtɪˈlaɪɡoʊ/ | Bạch biến |
| 126 | Alopecia | /ˌæləˈpiːʃə/ | Rụng tóc |
| 127 | Haplotypes | /ˈhæploʊtaɪps/ | Haplotype (Kiểu đơn bội) |
| 128 | Graves’ disease | /ɡreɪvz dɪˈziːz/ | Bệnh Graves |
| 129 | Protein tyrosine phosphatase nonreceptor type 22 (PTPN22) | /ˈproʊtiːn ˈtaɪrəsiːn ˈfɑːsfəteɪs ˌnɑːnrɪˈseptər taɪp ˌtwentiˈtuː/ | Protein tyrosine phosphatase không thụ thể loại 22 (PTPN22) |
| 130 | CD25 (high-affinity IL-2 receptor) | /siː diː ˌtwentiˈfaɪv haɪ əˈfɪnəti aɪ el tuː rɪˈseptər/ | CD25 (thụ thể IL-2 ái lực cao) |
| 131 | Prevalence | /ˈprevələns/ | Tỷ lệ hiện mắc |
| 132 | Vulvovaginitis | /ˌvʌlvoʊˌvædʒəˈnaɪtɪs/ | Viêm âm hộ – âm đạo |
| 133 | Dysphagia | /dɪsˈfeɪdʒə/ | Khó nuốt |
| 134 | Esophagitis | /ɪˌsɑːfəˈdʒaɪtɪs/ | Viêm thực quản |
| 135 | Onychomycosis | /ˌɑːnɪkoʊmaɪˈkoʊsɪs/ | Nấm móng |
| 136 | Squamous cell carcinoma (SCC) | /ˈskweɪməs sel ˌkɑːrsɪˈnoʊmə/ | Ung thư biểu mô tế bào vảy (SCC) |
| 137 | Fluconazole | /fluːˈkɑːnəzoʊl/ | Fluconazole |
| 138 | Ketoconazole | /ˌkiːtoʊˈkoʊnəzoʊl/ | Ketoconazole |
| 139 | Itraconazole | /ˌɪtrəˈkoʊnəzoʊl/ | Itraconazole |
| 140 | Voriconazole | /ˌvɔːrɪˈkoʊnəzoʊl/ | Voriconazole |
| 141 | Posaconazole | /ˌpoʊsəˈkoʊnəzoʊl/ | Posaconazole |
| 142 | Tetany | /ˈtetəni/ | Co cứng (tetany) |
| 143 | Carpopedal spasm | /ˌkɑːrpoʊˈpiːdəl ˈspæzəm/ | Co cứng bàn tay-bàn chân |
| 144 | Laryngospasm | /ləˈrɪŋɡoʊspæzəm/ | Co thắt thanh quản |
| 145 | Hypercalcemia | /ˌhaɪpərkælˈsiːmiə/ | Tăng canxi máu |
| 146 | Glomerular filtration | /ɡloʊˈmerjələr fɪlˈtreɪʃən/ | Lọc cầu thận |
| 147 | Calcitriol | /ˌkælsɪˈtraɪɒl/ | Calcitriol |
| 148 | Hyperpigmentation | /ˌhaɪpərˌpɪɡmenˈteɪʃən/ | Tăng sắc tố da |
| 149 | Orthostatic hypotension | /ˌɔːrθəˈstætɪk ˌhaɪpoʊˈtenʃən/ | Hạ huyết áp tư thế |
| 150 | Hyponatremia | /ˌhaɪpoʊnəˈtriːmiə/ | Hạ natri máu |
| 151 | Hyperkalemia | /ˌhaɪpərkəˈliːmiə/ | Tăng kali máu |
| 152 | Hypoglycemia | /ˌhaɪpoʊɡlaɪˈsiːmiə/ | Hạ đường huyết |
| 153 | Gonadal failure | /ɡoʊˈnædəl ˈfeɪljər/ | Suy tuyến sinh dục |
| 154 | Primary amenorrhea | /ˈpraɪmeri əˌmenəˈriːə/ | Vô kinh nguyên phát |
| 155 | Enamel hypoplasia | /ɪˈnæməl ˌhaɪpoʊˈpleɪʒə/ | Giảm sản men răng |
| 156 | Keratitis | /ˌkerəˈtaɪtɪs/ | Viêm giác mạc |
| 157 | Atrophic gastritis | /əˈtroʊfɪk ɡæˈstraɪtɪs/ | Viêm dạ dày teo |
| 158 | Achlorhydria | /ˌeɪklɔːrˈhaɪdriə/ | Vô toan |
| 159 | Intrinsic factor | /ɪnˈtrɪnsɪk ˈfæktər/ | Yếu tố nội tại |
| 160 | Iron-deficiency anemia | /ˈaɪərn dɪˈfɪʃənsi əˈniːmiə/ | Thiếu máu thiếu sắt |
| 161 | Subacute combined degeneration of the spinal cord | /ˌsʌbəˈkjuːt kəmˈbaɪnd diːˌdʒenəˈreɪʃən əv ðə ˈspaɪnəl kɔːrd/ | Thoái hóa kết hợp bán cấp của tủy sống |
| 162 | Steatorrhea | /ˌstiːətəˈriːə/ | Tiêu phân mỡ |
| 163 | Gluten sensitivity | /ˈɡluːtən ˌsensəˈtɪvəti/ | Nhạy cảm với gluten |
| 164 | Hypophysitis | /ˌhaɪpoʊfɪˈsaɪtɪs/ | Viêm tuyến yên |
| 165 | Rheumatoid arthritis | /ˈruːmətoɪd ɑːrˈθraɪtɪs/ | Viêm khớp dạng thấp |
| 166 | Sjögren syndrome | /ˈʃoʊɡrən ˈsɪndroʊm/ | Hội chứng Sjögren |
| 167 | Tubulointerstitial nephritis | /ˌtuːbjəloʊˌɪntərˈstɪʃəl nəˈfraɪtɪs/ | Viêm thận kẽ ống |
| 168 | Autoimmune bronchiolitis | /ˌɔːtoʊɪˈmjuːn ˌbrɑːŋkiəˈlaɪtɪs/ | Viêm tiểu phế quản tự miễn |
| 169 | Myopathy | /maɪˈɑːpəθi/ | Bệnh cơ |
| 170 | Asplenia | /eɪˈspliːniə/ | Vô lách |
| 171 | Urticarial eruption | /ˌɜːrtɪˈkeriəl ɪˈrʌpʃən/ | Phát ban dạng mề đay |
| 172 | Enteropathy | /ˌentəˈrɑːpəθi/ | Bệnh ruột |
| 173 | Pneumonitis | /ˌnuːməˈnaɪtɪs/ | Viêm phổi |
| 174 | Genotype-phenotype correlations | /ˈdʒiːnoʊtaɪp ˈfiːnoʊtaɪp ˌkɔːrəˈleɪʃənz/ | Tương quan kiểu gen-kiểu hình |
| 175 | Postpartum thyroiditis | /ˌpoʊstˈpɑːrtəm ˌθaɪrɔɪˈdaɪtɪs/ | Viêm tuyến giáp sau sinh |
| 176 | Myasthenia gravis | /ˌmaɪəsˈθiːniə ˈɡrævɪs/ | Bệnh nhược cơ |
| 177 | Thymoma | /θaɪˈmoʊmə/ | U tuyến ức |
| 178 | Latent autoimmune diabetes in adults (LADA) | /ˈleɪtənt ˌɔːtoʊɪˈmjuːn ˌdaɪəˈbiːtiːz ɪn əˈdʌlts/ | Đái tháo đường tự miễn tiềm ẩn ở người lớn (LADA) |
| 179 | Gastric parietal cell autoantibodies (PCA) | /ˈɡæstrɪk pəˈraɪətəl sel ˌɔːtoʊˈæntɪbɑːdiz/ | Tự kháng thể tế bào thành dạ dày (PCA) |
| 180 | Ulcerative colitis | /ˌʌlsərətɪv kəˈlaɪtɪs/ | Viêm loét đại tràng |
| 181 | Primary biliary cirrhosis | /ˈpraɪmeri ˈbɪlieri səˈroʊsɪs/ | Xơ gan mật nguyên phát |
| 182 | Sarcoidosis | /ˌsɑːrkɔɪˈdoʊsɪs/ | Bệnh sarcoidosis |
| 183 | Achalasia | /ˌækəˈleɪʒə/ | Co thắt tâm vị |
| 184 | Myositis | /ˌmaɪəˈsaɪtɪs/ | Viêm cơ |
| 185 | Neuropathy | /nʊˈrɑːpəθi/ | Bệnh thần kinh |
| 186 | Eczematous dermatitis | /ɪɡˈzemətəs ˌdɜːrməˈtaɪtɪs/ | Viêm da dạng chàm |
| 187 | Hemolytic anemia | /ˌhiːməˈlɪtɪk əˈniːmiə/ | Thiếu máu tan máu |
| 188 | Thrombocytopenia | /ˌθrɑːmboʊˌsaɪtoʊˈpiːniə/ | Giảm tiểu cầu |
| 189 | Anti-enterocyte autoantibodies | /ˌænti ˈentəroʊsaɪt ˌɔːtoʊˈæntɪbɑːdiz/ | Tự kháng thể kháng tế bào ruột |
| 190 | Mutational analysis | /mjuːˈteɪʃənəl əˈnæləsɪs/ | Phân tích đột biến |
| 191 | Adrenal cytoplasmic autoantibodies (ACA) | /əˈdriːnəl ˌsaɪtoʊˈplæzmɪk ˌɔːtoʊˈæntɪbɑːdiz/ | Tự kháng thể tế bào chất tuyến thượng thận (ACA) |
| 192 | Interferon-ω autoantibodies | /ˌɪntərˈfɪrɑːn oʊˈmeɪɡə ˌɔːtoʊˈæntɪbɑːdiz/ | Tự kháng thể interferon-ω |
| 193 | Thyroperoxidase autoantibodies (TPOA) | /ˌθaɪroʊpərˈɒksɪdeɪs ˌɔːtoʊˈæntɪbɑːdiz/ | Tự kháng thể thyroperoxidase (TPOA) |
| 194 | Thyroglobulin autoantibodies (TGA) | /ˌθaɪroʊˈɡlɑːbjəlɪn ˌɔːtoʊˈæntɪbɑːdiz/ | Tự kháng thể thyroglobulin (TGA) |
| 195 | Tissue transglutaminase IgA autoantibodies (ITG) | /ˈtɪʃuː trænsˌɡluːˈtæməneɪs aɪ dʒiː eɪ ˌɔːtoʊˈæntɪbɑːdiz/ | Tự kháng thể transglutaminase mô IgA (ITG) |
| 196 | Liver kidney microsome type 1 (LKM1) autoantibodies | /ˈlɪvər ˈkɪdni ˈmaɪkroʊsoʊm taɪp wʌn ˌɔːtoʊˈæntɪbɑːdiz/ | Tự kháng thể microsome gan thận loại 1 (LKM1) |
| 197 | Preclinical | /ˌpriːˈklɪnɪkəl/ | Tiền lâm sàng |
| 198 | Amenorrhea | /əˌmenəˈriːə/ | Vô kinh |
| 199 | Menopause | /ˈmenoʊpɔːz/ | Mãn kinh |
| 200 | Zona pellucida | /ˈzoʊnə pəˈluːsɪdə/ | Màng trong suốt |